CD28 hinge used in chimeric antigen receptor (CAR) T-cells exhibits local structure and conformational exchange amidst global disorder
Abstract
T-cell therapies based on chimeric antigen receptor (CAR) targeting of a tumor-specific antigen offer hope for patients with relapsed or refractory cancers. CAR hinge and transmembrane regions link antigen recognition domains to intracellular signal transduction domains. Here, we apply biophysical methods to characterize the structure and dynamic properties of the CD28 CAR hinge (CD28H) used in an FDA-approved CD19 CAR for the treatment of B-lineage leukemia/lymphoma. By using nuclear Overhauser effect spectroscopy (NOESY), which detects even transiently occupied structural motifs, we observed otherwise elusive local structural elements amidst overall disorder in CD28H, including a conformational switch from a native β-strand to a 310-helix and polyproline II helix-like structure. These local structural motifs contribute to an overall loosely formed extended geometry that could be captured by NOESY data. All FDA-approved CARs use prolines in the hinge region, which we find in CD28, and previously in CD8α, isomerize to promote structural plasticity and dynamics. These local structural elements may function in recognition and signaling events and constrain the spacing between the transmembrane and antigen recognition domains. Our study thus demonstrates a method for detecting local and transient structure within intrinsically disordered systems and moreover, our CD28H findings may inform future CAR design.
Article type: Research Article
Keywords: Intrinsically disordered proteins, Cancer immunotherapy
Affiliations: grid.94365.3d0000 0001 2297 5165Protein Processing Section, Center for Structural Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, MD USA; grid.94365.3d0000 0001 2297 5165Computational Biomolecular Magnetic Resonance Core, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD USA; grid.48336.3a0000 0004 1936 8075Macromolecular NMR Section, Center for Structural Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, MD USA; grid.94365.3d0000 0001 2297 5165Pediatric Oncology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD USA; grid.94365.3d0000 0001 2297 5165Cancer Data Science Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD USA
License: © This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply 2024 CC BY 4.0 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Article links: DOI: 10.1038/s42003-024-06770-w | PubMed: 39217198 | PMC: PMC11365992
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (3.0 MB)
Introduction
Chimeric antigen receptors (CARs) have emerged as a transformative tool in the development of novel anti-tumor T cell-based therapies. Currently, six CAR T-cell therapies have been approved by the FDA to treat cancers, including B-cell lymphomas, B-cell acute lymphoblastic leukemia, and multiple myeloma1. CAR T-cell therapies have had much success in treating hematological malignancies by targeting CD19, CD20, and CD22 molecules expressed on B-lineage leukemias and lymphomas2–4 and a CAR T-cell therapy directed against B-cell maturation antigen (BCMA) has been developed to treat multiple myeloma5,6. Despite these successes, CAR T-cell therapy treatments face ongoing challenges, including antigen loss, toxicity, and unmet needs in solid tumors7,8. Implementations of CAR therapies continue to be developed, including bi-specific9 and tri-specific CARs against CD19, CD20, and CD2210,11.
CAR constructs are engineered to target tumor-specific antigens, using single-chain variable fragments (scFv) or nanobodies. The CAR recognition domain is associated with intracellular signal transduction domains by an intervening hinge region and transmembrane domain and CARs can be monomeric or, as depicted by axicabtagene ciloleucel (Fig. 1a), dimeric12,13. Whereas structural information is available for the CAR antigen recognition, signal transduction and transmembrane domains, the hinge domain and the overall CAR structure has proven difficult to study due to their dynamic nature. Notably though, the hinge region is critical because it conditions the signaling threshold at which a CAR can respond to low antigen density tumors14,15 as well as its association with endogenous T cell receptor (TCR) components16,17.
We previously found that a CD22-targeting CAR incorporating a CD8α hinge exhibits greater cytotoxicity against CD22-low antigen density as compared to an equivalent CAR with a CD28 hinge (CD28H)18. This finding motivated us to apply biophysical methods, including nuclear magnetic resonance (NMR) spectroscopy, to study the structural and dynamic properties of the CD8α hinge region, identifying distinct conformational states undergoing dynamic exchange driven by proline isomerization18. Here, we expand these studies to CD28H and gain insights into the impact of this hinge on CAR activity. Our findings also have general implications for studies of structure within intrinsically disordered regions. In particular, we demonstrate that nuclear Overhauser effect spectroscopy (NOESY) is capable of detecting transient and otherwise elusive structural features within intrinsically disordered regions. By studying CD28H using NMR, and particularly through NOESY data, we observed local structure, including a sequence that adopts a helix in a globally disordered context despite forming a β-strand in its native context.
Results
CD28H exhibits global intrinsic disorder
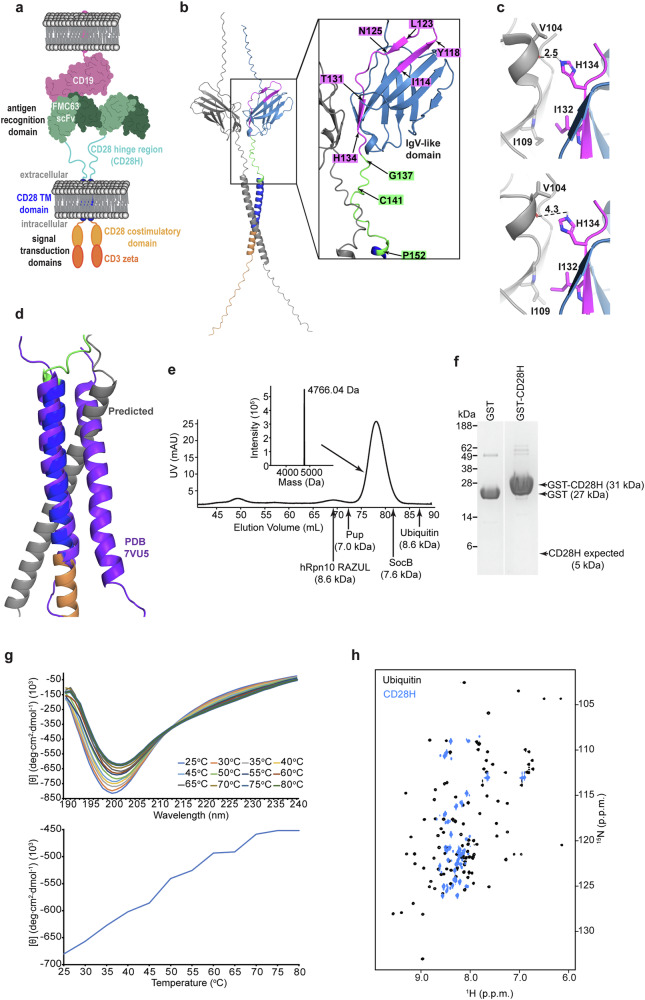
We used AlphaFold 319 to generate a model structure of the dimeric form of the CD28 protein expressed natively by human T-cells. Inputting two CD28 sequences yielded a structure with the IgV-like domain forming homodimers (Fig. 1b), similar to the experimentally determined crystal structure20. In the experimental structure, an intermolecular hydrogen bond forms between H134 and the backbone oxygen of V104 with intermolecular hydrophobic packing between I132 and I109 (Fig. 1c, upper panel). The predicted structure places these four residues at the dimeric interface, but further apart, disrupting the hydrogen bond between H134 and V104 (Fig. 1c, lower panel). The structure of the transmembrane domain was previously determined by NMR21; however, the predicted model differs from the experimental structure by an altered dimeric interface (Fig. 1d, purple versus grey)21. In the predicted structure, C141 of the hinge region is placed in proximity to form an intermolecular disulfide bond (Fig. 1b); no experimental structure exists for this region.
The CD28H in this study spans I114 – P152 (YESCARTA, Kite Pharma, Inc.), which includes residues I114 – K136 of the native IgV-like domain (Fig. 1b, magenta) and residues G137 – P152 from the native hinge region (Fig. 1b, green). Since residues I114 – Y118, L123 – D124, and T131 – H134 from the native IgV-like domain form β-strands20, we investigated how this region behaves in the CD28H context.
To study CD28H structurally, we generated an expression plasmid in Escherichia coli in frame with an N-terminal glutathione S-transferase (GST) tag followed by a PreScission protease cleavage site. Following its expression, we purified CD28H from E. coli by affinity chromatography followed by size exclusion chromatography (SEC) on an FPLC system (Fig. 1e and Supplementary Fig. 1). The resulting CD28H sample was validated by its expected molecular weight of 4,766.04 Da by liquid chromatography-mass spectrometry (LC-MS) (Fig. 1e, top middle); however, CD28H eluted earlier than expected by SEC for its molecular weight (Fig. 1e). For example, the larger 8.6 kDa protein ubiquitin elutes later than CD28H (Fig. 1e). Early elution from SEC is characteristic of intrinsic disorder, as we previously observed for prokaryotic ubiquitin-like protein (Pup)22, hRpn10 RAZUL23 and SocB24 (Fig. 1e). By contrast, early elution could also be caused by oligomerization. Of note, this experiment was performed in a buffer that contains 2 mM dithiothreitol (DTT), causing cysteine to be reduced. To test directly whether CD28H forms an oligomer, we conducted a GST pull-down assay in the presence of dithiothreitol with GST-tagged CD28H and CD28H (Fig. 1f). GST was also incubated with CD28H as a negative control. No bands were observed at the expected molecular weight for CD28H (5 kDa) (Fig. 1f and Supplementary Fig. 1), suggesting that CD28H does not bind to GST-tagged CD28H and that CD28H does not form oligomers in these conditions.
To evaluate further whether CD28H is intrinsically disordered, we tested whether it undergoes a phase transition during thermal denaturation by measuring molar ellipticity using circular dichroism (CD) (Fig. 1g). No cooperative protein unfolding was observed, consistent with the SEC data suggesting it to be intrinsically disordered and the GST pull-down assay indicating a monomer (Fig. 1f).
The amide backbone signals of structured proteins are dispersed in NMR spectra due to the divergent chemical environments of amino acids when in different secondary and tertiary structures. We therefore recorded a 2D 1H, 15N HSQC experiment on 15N-labeled CD28H to further evaluate the structural properties of CD28H. Whereas the amide signals of ubiquitin are dispersed in the 1H dimension (Fig. 1h, black), CD28H amide 1H signals are congested (Fig. 1h, blue), further indicating that overall, CD28H is intrinsically disordered.
CD28H adopts distinct conformational states with proline cis/trans isomerization
To gain detailed structural information for each amino acid within CD28H, we assigned the signals in the 1H, 15N HSQC spectrum to the individual amide atoms of CD28H by using modern NMR methods (Fig. 2a, b), as described in Materials and Methods. This analysis revealed the presence of multiple amide signals for certain amino acids (Fig. 2b). Multiple signals occur in NMR spectra when atoms exchange slowly (>100 ms time scale) between multiple conformational states. 3D NMR datasets, which were used to correlate sequential amino acids, also indicated multiple signals for Cα and Cβ atoms, such as in 3D HNCACB (Supplementary Fig. 2a, black and green) and CBCACONH (Supplementary Fig. 2a, purple) spectra. A 3D HACAN spectrum25 was used to assign proline rich regions and also contained multiple signals for individual proline atoms (Supplementary Fig. 2b, c).
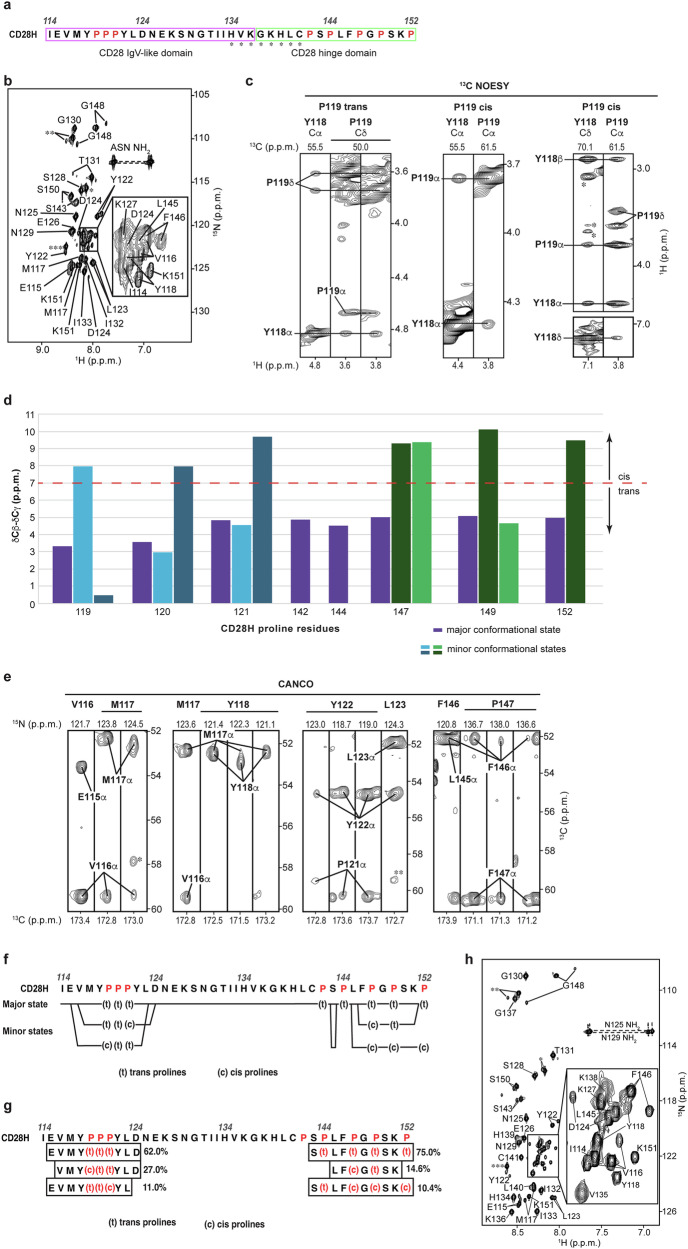
CD28H has eight prolines (Fig. 2a) and to further characterize the distinct states of CD28H, we attempted to use NOESY data to assign the isomerization state as cis or trans for each proline signal. NOESY experiments detect distance-dependent signals between hydrogens that are within 5 Å of each other. A shorter distance between Hδ and the preceding amino acid’s Hα enables detection of a corresponding inter-residue NOE interaction in trans but not cis prolines. By contrast, cis prolines are uniquely characterized by their Hα atom having an NOE interaction with the Hα atom of the preceding amino acid. A 13C-dispersed NOESY spectrum acquired on 13C-labeled CD28H indicated NOE interactions for the trans isomer of all prolines except for P120 and P121, which were precluded from this analysis by overlapping signals for Cα and Hα atoms (Fig. 2c, left, for P119). Interactions were also observed between the Hα atoms of Y118 and P119, indicating that P119 also adopts the cis isomeric state (Fig. 2c, middle). Such inter-residue Hα NOEs were not observed for other proline residues; however, signal overlap and poor signal intensity limited this analysis. The chemical shift data of proline Cβ and Cγ atoms as obtained from 3D NMR data (HNCACB, CBCACONH, 13C-dispersed NOESY, CCH-TOCSY, and HCCH-TOCSY spectra) can also be used to evaluate the proline isomeric states26,27. We found the differences observed in Cβ and Cγ chemical shifts to be consistent with the isomeric states assigned by using NOEs (Fig. 2d).
By using a 3D CANCO experiment28 in combination with a 15N-dispersed NOESY experiment, we were able to assign NMR chemical shift values to the various conformational states of the residues in CD28H. Residues I114, N125 – P142, and P144 yielded a single peak each, indicating a single conformational state, while the spectrum indicated that other amino acids occupy up to three distinct conformational states (Fig. 2e, f, and Supplementary Table 1). Proline cis-trans isomerization can occur on a 400 ms time scale, which allows distinct NMR signals to be recorded for each state29. Careful evaluation of the 3D datasets indicated that except for P142 and P144, all prolines showed multiple sets of signals (Fig. 2f and Supplementary Fig. 3). For each proline, a set of signals associated with a trans isomeric state was observed, which was consistent with the NOESY analyses. Prolines 119, 121, and 149 exhibited an additional cis and trans isomeric state, while P152 exhibited only one additional set of cis isomer signals (Fig. 2f). Two additional sets of signals were observed for trans and cis isomers respectively for P120 and P147 (Fig. 2f).
To define the relative population of each conformational state, we integrated the signals of the 3D NMR spectra (including HACAN, HNCACB, and CANCO experiments) to obtain an averaged value (Fig. 2g). The major conformational state contains only trans proline isomers and represents 62% and 75% of the population respectively for the N-terminal stretch that includes 119PPP121 and the extreme C-terminal stretch. The lesser populated states contain different mixtures of cis and trans prolines. Only one conformational state is observable at I114 and for the region spanning N125 – P142 (Fig. 2f); however, this latter region includes H134 – C141, for which the amide signals are not observable at 25 °C due to conformational exchange at the so-called intermediate exchange time scale (Fig. 2a, b)30. We note that aliphatic signals were observed in this region at 25 °C and because of the inclusion of C141, we tested whether omission of DTT or inclusion of 20 μM ZnSO4 at induction causes the appearance of the amide signals. No changes were observed however with either of these alterations (Supplementary Fig. 4a, b). We tested and found however that amide signals for H134 – C141 do appear in spectra recorded at 11 °C (Fig. 2h). We therefore also collected 15N-dispersed and 13C-dispersed NOESY experiments at 11 °C and found greater signal-to-noise and additional NOE interactions throughout the sequence. In addition, the 13C-dispersed NOESY at 11 °C was optimized to record pyrrolidine-aromatic CH-π interactions and detected an NOE between Y118 Hδ and P119 Hα of the P119 cis isomer (Fig. 2c, right). Altogether, our data indicate that CD28H undergoes temperature-dependent conformational exchange that includes, but is not limited to, proline cis-trans isomerization.
Local structural elements are formed in CD28H that diverge from native CD28
To further investigate the structural properties of CD28H, we evaluated a CD spectrum recorded at 25 °C as in Fig. 1g for secondary structure by DichroWeb31. This analysis predicted 25% β-strand and 15% helicity, with 61% of the sequence predicted to be disordered (Fig. 3a). Although no long-range NOE interactions were observed in NOESY spectra recorded on CD28H, several medium-range inter-residue NOE correlations were observed at 11 °C (Fig. 3b). For example, within the region spanning L123 – N129, NOEs were detected from the Hβ and methyl groups of L123 to E126 HN, from D124 Hα and Hβ to K127 HN, and from N125 Hα to S128 HN (Fig. 3c). These NOEs suggest helicity for the region spanning L123 – N129, consistent with the CD prediction of 15% helicity (Fig. 3a).
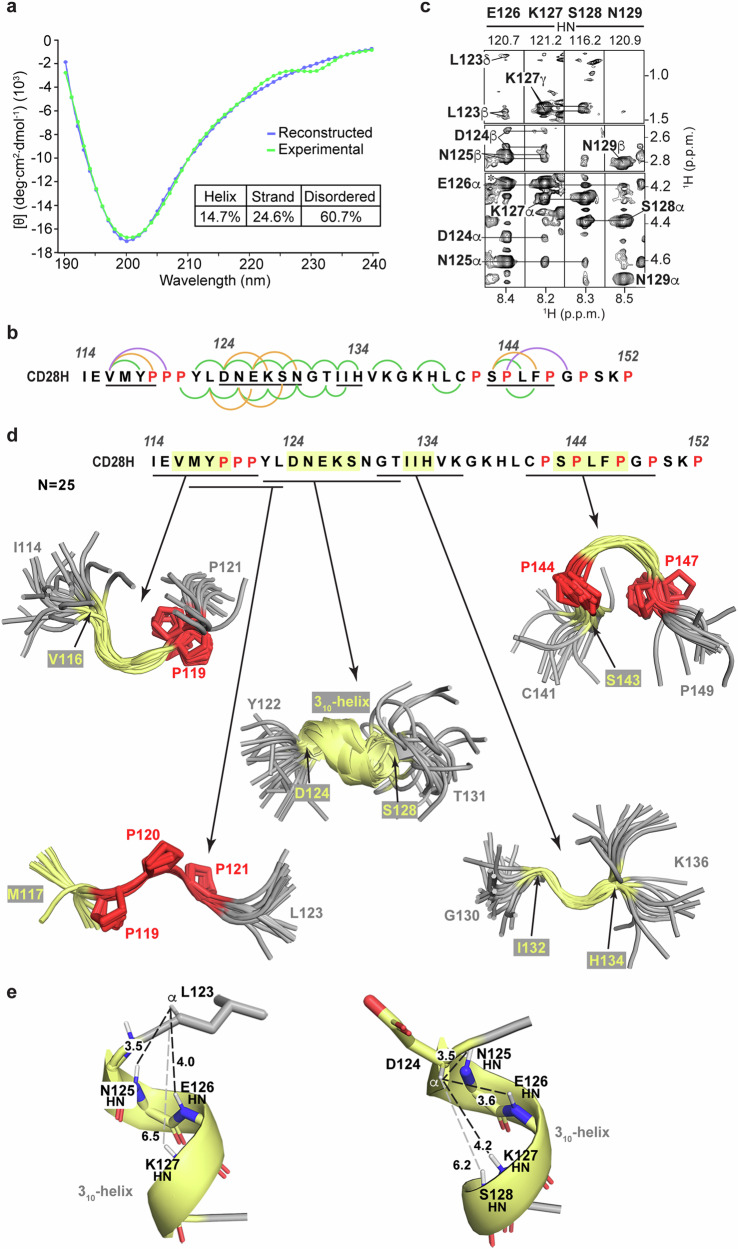
We used our sparse NOESY data at 11 °C (Fig. 3b) to calculate structures for the major conformational state of CD28H by using Xplor-NIH 3.732, with the structural statistics summarized in Table 1. The signals detected for the minor conformations were too weak for a comparable analysis or structure calculations. Backbone ϕ and ψ torsion angle restraints were also incorporated into the structure calculations – these restraints were defined by TALOS+ based on NMR chemical shift information at 11 °C. Although the calculated structures did not converge to a single solution overall, several short stretches formed loosely converged backbone structures (Fig. 3d). In the initial rounds of structure calculations, a 310-helix was formed spanning D124 – S128, consistent with the NOE (Fig. 3b and c) and CD (Fig. 3a) data. Rather than observing an α-helical hydrogen bonding pattern between the carbonyl group of a residue i and the amide group of residue i + 4, CD28H exhibited a 310-helix hydrogen bonding pattern between i and i + 3 residues (Fig. 3e). Based on these observations, long-range HNCO-COSY experiments33,34 were collected on CD28H to determine experimentally whether hydrogen bonds can be detected within D124 – S128. Only intra-residue hydrogen bonds involving sidechain atoms were observed, however. The detected signals were between the backbone amide atoms of D124 and N125 and their respective sidechain oxygen atoms (Supplementary Fig. 5). These two hydrogen bonds were added to the final structure calculations, however no other hydrogen bonds were included in the structure calculations.
Table 1: NMR and refinement statistics for CD28H
| Protein | |
|---|---|
| NMR distance and dihedral restraints | |
| Distance restraints | |
| Total NOE | 479 |
| Intra-residue | 266 |
| Inter-residue | 213 |
| Sequential (|i – j| = 1) | 159 |
| Medium-range (|i – j| < 4) | 54 |
| Long-range (|i – j| > 5) | 0 |
| Hydrogen bonds | 2 |
| Total dihedral angle restraints | |
| ϕ | 13 |
| ψ | 13 |
| Structure statistics* | |
| Violations (mean and s.d.) | |
| Distance restraints (Å) | 0.18 ± 0.06 |
| Dihedral angle restraints (°) | 1.11 ± 0.10 |
| Max. dihedral angle violation (°) | <5 |
| Max. distance restraint violation (Å) | <0.5 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.003 ± 0.000 |
| Bond angles (°) | 0.390 ± 0.028 |
| Impropers (°) | 0.212 ± 0.024 |
| Average pairwise r.m.s. deviation** (Å) | |
| Heavy | 1.10 ± 0.63 |
| Backbone | 2.35 ± 0.87 |
*Statistics for the 25 lowest energy structures without NOE, dihedral or torsion angle violations for CD28H (I114-P152).
**Pairwise r.m.s.d. for the 25 lowest energy structures for D124-S128.
Among the 25 calculated lowest energy structures, 23 form a 310-helix that spans D124 – S128, and the ϕ and ψ torsion angles of all 25 structures are more consistent with a 310-helix than an α-helix (Fig. 4a, top panel). The divergence from helicity for two of the structures may reflect transient exchange between a helical and disordered state, such that NOEs consistent with helicity are recorded when the helix is formed. As mentioned above, in the context of the intact IgV-like domain, L123 – N125 forms a β-strand20 stabilized by hydrogen bonds from L123 to V116, D124 to I21, and N125 to I114 (Fig. 4b, top panel). L123 is also incorporated into a hydrophobic patch that includes I21, I114, and V116, which cluster on one side of the β-sheet (Fig. 4b, bottom panel). Loss of the surrounding amino acids in CD28H causes a reconfiguration of the secondary structure to allow formation of the helix (Fig. 3e).
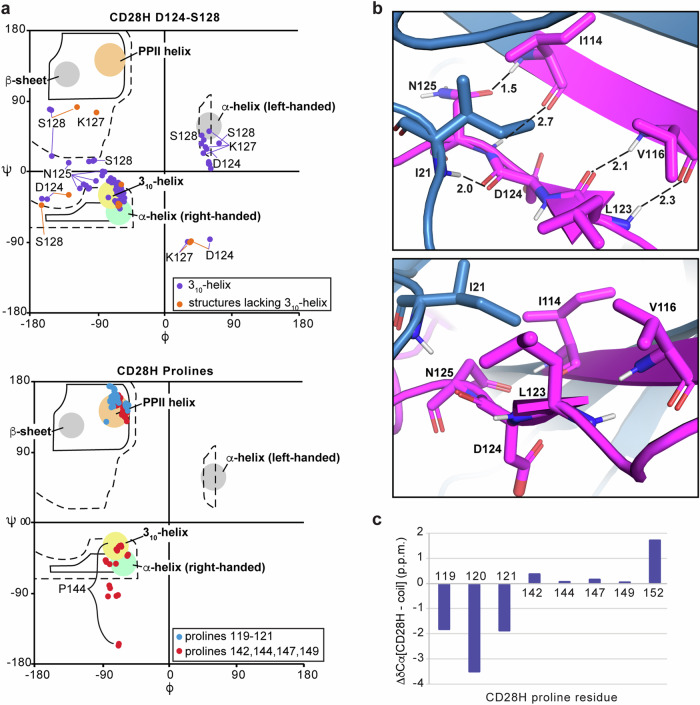
The structure calculations yielded a polyproline II (PPII) helix-like structure for the region spanning 119PPP121 (Fig. 3d), with characteristic torsion angles (Fig. 4a, bottom panel indicated in blue). By contrast, the ϕ and ψ angles of prolines 142, 144, 147, and 149 distribute to the multiple regions allowed for proline residues (Fig. 4a, bottom panel in red). This structure is supported by the Cα chemical shift values of 119PPP121, which are shifted downfield compared to the other prolines present in CD28H (Fig. 4c).
NMR reveals local structure driven by interactions involving CD28H methyl groups
Three additional regions in CD28H show converged backbone structures spanning V116 – P119, I132 – H134, and S143 – P147 (Fig. 3d). Whereas I114 – Y118 forms a β-strand in the intact IgV domain as part of a β-sheet (Fig. 1b), V116 – P119 in CD28H is structurally defined by NOE interactions between the V116 methyl groups and P119 Hα and Hδ atoms (Fig. 5a). These interactions are weak but sufficient to restrict the overall backbone geometry of this local region and to induce a slight bend (Fig. 3d), providing van der Waals contacts for the methyl group (Fig. 5b). This bending is distinct from the native β-strand (Fig. 5c).
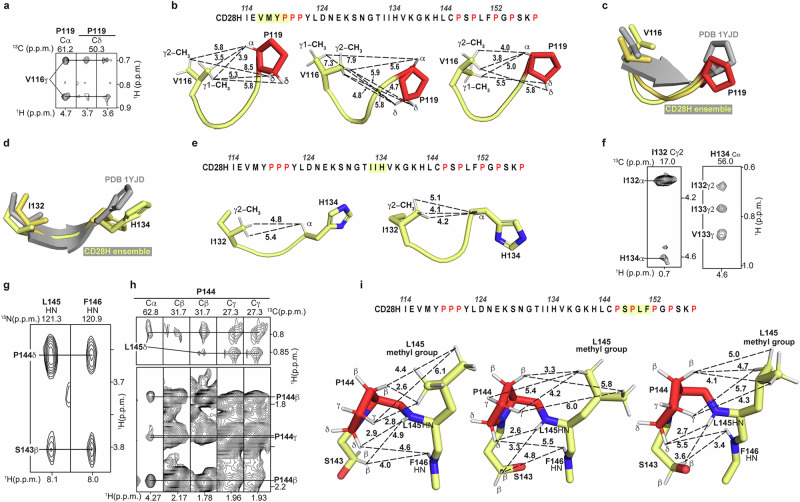
T131 – H134 forms a β-strand at the dimeric interface of the IgV-like domain in the intact CD28 dimeric protein (Fig. 1b). This region in CD28H mimics the native CD28 dimeric protein (Figs. 3d and 5d), with a bend that promotes van der Waals contacts to the I132 methyl group (Fig. 5e). This structure in CD28H is supported by weak NOE correlations between I132 and H134 (Fig. 5f). Variances in the sidechain conformations fit the sparse data, causing the distances between the I132 methyl groups and H134 Hα atom to differ (Fig. 5e) and for slight dispersion of the backbone geometry (Fig. 3d).
In the region spanning S143 – F146, NOE interactions were recorded from S143 Hβ to L145 and F146 HN atoms (Fig. 5g), and from the L145 methyl groups to P144 atoms (Fig. 5h). These data allow for some variance in sidechain conformations (Fig. 5i) but restrict the backbone geometry of S143 – F146 to an arched structure (Fig. 3d) with hydrophobic interactions between P144 and L145 (Fig. 5i). Altogether, these data indicate that CD28H has stretches of local structure, some of which diverge from the native CD28 dimeric protein, that is driven by van der Waals interactions, primarily involving methyl groups.
Axicabtagene ciloleucel CARs place the transmembrane domain of CD28 C-terminally adjacent to CD28H (Fig. 1a). To gain insight into the consistency of our structural findings in the context of the full CAR construct, we tested whether CD28H has affinity for a phospholipid membrane. We added 35 μM MSPΔH5 nanodiscs to 100 μM 15N-labeled CD28H and acquired 1H, 15N HSQC experiments to test for interaction. No spectral changes were induced for CD28H however by MSPΔH5 nanodiscs (Supplementary Fig. 6). This result suggests that CD28H is not likely to interact with the C-terminal transmembrane region of the intact CAR. Moreover, AlphaFold 3 did not predict an interaction to occur between CD28H and the N-terminally adjacent FMC63 antigen recognition domain. Altogether these data suggest that CD28H may act largely as an independent structural entity within the intact CAR.
The N-terminal half of CD28H has a loosely converged extended global shape
Intrinsically disordered proteins are commonly described in terms of their radius of gyration, which has previously been found to increase linearly with sequence length in randomly coiled proteins35. The average radius of gyration of the 39-residue CD28H calculated ensemble representing the major state was computed to be 16.03 ± 2.87 Å, which falls within the range expected for an intrinsically disordered protein35. While CD28H has regions containing local structure, this protein sequence is overall globally disordered.
Alignment of the whole CD28H sequence indicated overall disorder; however, two overlapping loosely ordered regions were apparent in the CD28H structural ensemble that were somewhat spatially restricted (Fig. 6a). The region spanning V116 – S128 contains small stretches of high convergence that includes the PPII helix-like structure and 310-helix (Fig. 3d). When the broader region is aligned, the ensemble appears to have a slightly expanded global shape, driven by the 119PPP121 stretch (Fig. 6a). This extended shape is indicated by the comparison of Cα chemical shifts of CD28H to random coil values36,37, an analysis that predicts CD28H to have strand-like structure in the N-terminal portion spanning residues I114 – L123 (Fig. 6b), similar to the extended global shape observed in structure calculations (Fig. 6a). Downfield shifts support the helical structure of D124 – G130 (Fig. 6b), consistent with the calculated 310-helical structure (Fig. 3d). Disorder was suggested by this analysis in the C-terminal portion of CD28H spanning residues T131 – P152 (Fig. 6b).
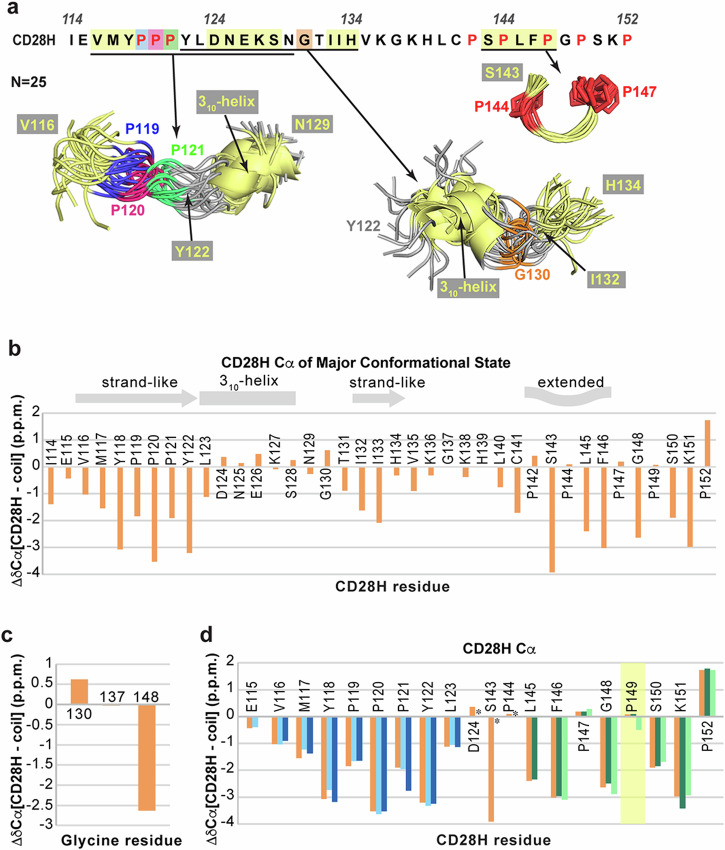
The region containing Y122 – H134 also exhibits a loose global shape, driven by the local structure of D124 – S128 and I132 – H134 (Fig. 6a). This larger portion features a bend at residue G130, where no medium-range inter-residue NOE interactions were observed (Fig. 6a). The bend observed in the region spanning Y122 – H134 forms due to G130 backbone ϕ (92.4o ± 15.5o) and ψ (−0.3o ± 12.7o) torsion angle restraints, which were derived by TALOS+ based on chemical shift information, including a downfield-shifted Cα signal (Fig. 6c). Altogether, our data indicate that although CD28H is disordered with only short regions of convergence, a loosely defined global shape exists in the N-terminal half, which in the native CD28 protein, is part of an IgV-like domain.
We further evaluated the minor conformational states of CD28H by similarly examining the chemical shift information. Overall, the values were similar to the dominant state (Fig. 6d, orange) except at residue P149 (Fig. 6d, highlighted in yellow), for which the Cα chemical shift value is shifted more upfield in one of the minor conformers (Fig. 6d, light green). This divergence suggests a different (trans) configuration for the backbone of P149 in a less populated state, however overall, the trends in Cα chemical shift values resemble the dominant state.
Discussion
Our previous study, which found that the hinge region in the CD22 CAR conditions its cytotoxicity against CD22low leukemia18 combined with findings that hinges influence signaling thresholds14,15 and association with endogenous cellular machinery16,17, motivated our structural characterization of the CD28H. The N-terminal half of CD28H was taken from the well-structured IgV-like domain with regions that form β-strands. We observed local structural features throughout the CD28H sequence; however, we find that when out of the full protein context, the β-strand region spanning D124 to E126 switches to a 310-helix. This conversion can be rationalized by the loss of inter-strand interactions, such as with I21, which is missing from CD28H. Rather than such long-range interactions that can drive β-sheet formation, we detect shorter hydrophobic interactions involving methyl groups. Our study of CD28H indicates methyl groups to be a key driving force for local structure within CD28H. This finding is likely generally true for intrinsically disordered proteins. We note that NOE interactions are recorded only when atoms are within 6 Å of each other and signals are detected even for transiently populated structures. Therefore, it is plausible that the local structures we observe in CD28H are only transiently populated.
All hinges (CD28, CD8α, and IgG4) incorporated into the six FDA-approved CARs contain proline residues. Hinges from CD28 and CD8α are highly dynamic and form multiple conformational states in part due to proline isomerization18. The effect of prolines on CAR hinge plasticity and dynamics may be generalized to other hinge domains; however, future studies are needed to discern whether these effects can be promoted by other amino acids and/or combinations such as glycine, which also has the capacity to adopt a large range of backbone dihedral angles. For example, exchange broadening is observed for CD28H H134 – C141 at 25°C, a region that lacks proline but includes glycine.
We find considerable heterogeneity within CD28H, including proline isomerization, and such dynamics may contribute to CAR T-cell signaling. A limitation of our study is that we were unable to record NOE interactions for the lesser populated states; however, Cα chemical shift values were found to be similar in these states compared to the dominant population, suggesting similar structural features.
Intrinsically disordered proteins (IDPs) are very difficult to study by methods involving crystallography and microscopy, which are unable to capture dynamic structural features. Even NMR studies are stymied by the lack of chemical shift dispersion exhibited by IDPs, leading to potential signal overlap. In this study, chemical shift indices similarly struggled to capture the dynamic local structures present in CD28H (Fig. 6b). Nevertheless, NMR data is useful for studying the structure of IDPs, as demonstrated by a study of the structure of intrinsically disordered Tau in the context of varying phosphorylation states, leading to the discovery of helicity rather than a previously suggested β-turn structure38,39.
In our study, we used NMR data in Xplor-NIH 3.7 to calculate a structural ensemble for CD28H. Collecting NOESY data was key to studying the local structures present in CD28H, which otherwise we would not have been detected. The software platform IDPConformerGenerator was developed to predict structures of intrinsically disordered proteins based on protein structures in the Protein Data Bank40. We inputted the CD28H sequence into IDPConformerGenerator to find similarities in the predicted ensemble compared to the Xplor-NIH structures. In particular, the predicted ensemble showed convergence for the regions spanning V116 – P119, P119 – P121, D124 – S128, I132 – H134, and P144 – P147 (Fig. 7a), with similar structural features to those observed experimentally (Fig. 3d). Some notable differences are observed, however. The NMR data support a 310-helix for D124-S128 (Fig. 3e), as no NOE interactions are observed from Hα atoms to amide groups of residues C-terminal by four amino acids (Fig. 3b). IDPConformerGenerator predicts helicity in this region (Fig. 7a), but with 310 (Supplementary Fig. 7a) and α helical (Supplementary Fig. 7b) configurations (Supplementary Fig. 7a, b). Moreover, in the region spanning P119 – P121, IDPConformerGenerator predicted 36% of conformers to form long-range contacts between M117/Y118 and Y122/L123 (Fig. 7b). Such contacts are not supported by NOE data (Fig. 3b). We also evaluated predictions from AlphaFold 3 for CD28H to find 60% of the outputted structures were predicted to be helical in the region spanning L123 – K138 (Fig. 7c). Thus, the predictions from IDPConformerGenerator more closely matched our experimental data. Our findings imply that IDPConformerGenerator can offer reasonable predictions for intrinsically disordered regions and moreover, that local structural features can be experimentally detected by using NOESY data.
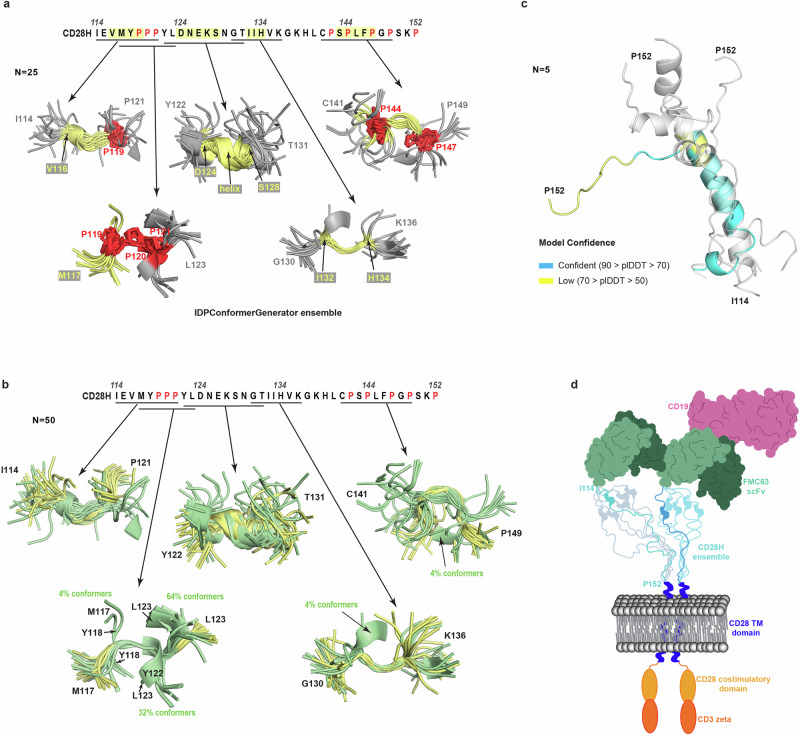
In summary, we find CD28H to adopt local structural elements and undergo dynamic exchange between different configurations that include cis-trans proline isomerization. These properties resemble those of the CD8α hinge18, suggesting that dynamics and local structure may play a role in CAR T-cell signaling. In the context of CD28H, we also find a reconfiguration of a β-strand into a 310-helix based on the omission of amino acids that form inter-strand interactions. Formation of the local structures observed in CD28H may be associated with recognition events, as intrinsically disordered proteins have previously been reported to adopt local structural elements in complexes with binding partners23,41,42. It is possible that these structures in the intrinsically disordered proteins are selected from transiently populated structures present in the free protein. Such flipping of secondary structure based on context has important implications for structural prediction algorithms, and even single amino acid substitutions can stymie structural prediction by AlphaFold243. The intrinsic dynamics and resulting heterogeneity of CARs make detailed structural study of the intact CAR T-cell/tumor cell interaction challenging. A common feature of this intercellular interaction is induced dimerization at each end by the transmembrane domain and antigen recognition domain, adding a key structural restraint on the hinge region21,44 (Fig. 7d). Modulation of the intermembrane distance between CAR T-cell and tumor cells has been linked to CAR T-cell killing efficacy13,45. We find the structure of CD28H to greatly differ from the hinge region in the context of native CD28. This difference is important to the CAR T-cell because CD28H loses the highly structured β-strand observed in CD28 and instead, forms a 310-helix within its N-terminal portion. This structural difference may affect the spacing imposed by the hinge on the transmembrane domain relative to the antigen recognition domain. Future studies taking advantage of cryo-electron tomography, aimed at dissecting the importance of global structural features for CAR T-cell signaling in combination with local structural data, such as that which we have obtained for CD28H, is likely to inform the development of new CAR-based immunotherapies.
Methods
Protein sample preparation
CD28H (I114 – P152) was subcloned into the pGEX-6P-1 vector between BamHI and XhoI restriction sites in a frame with an N-terminal glutathione S-transferase (GST) tag and a PreScission protease cleavage site. The construct was purchased through GenScript and contains codons optimized for expression in Escherichia coli. The plasmid was transformed into Escherichia coli strain BL21 (DE3) (Thermo Fisher Scientific C600003) with selection by ampicillin. The transformed colonies were grown in 10 mL of Luria-Bertani Broth (LB) medium (ampicillin 100 μg/ml) overnight at 37 °C with shaking and centrifuged for 10 minutes at 2000 g. Bacterial pellets containing CD28H construct were gently resuspended and diluted at 1:100 ratio into 1 L of M9 minimal media supplemented with 1 g/L 15N ammonium chloride (Sigma-Aldrich) and 3 g/L 13C glucose (Sigma-Aldrich) as the sole nitrogen or carbon source respectively. Cells were grown at 37 °C with shaking until they reached an OD600 of 0.5-0.6 at which point isopropyl β-D-1-thiogalactopyranoside (UBPBio) was added to a final concentration of 0.4 mM to induce protein expression at 17 °C overnight. The cells were pelleted by centrifugation at 4000 rpm and 4 °C for 45 minutes by using a Beckman Coulter J6-M1 centrifuge with a JS-4.2 rotor and then stored at −80 °C until purification.
Following resuspension in buffer 1 (20 mM HEPES at pH 7.5, 300 mM NaCl, 2 mM dithiothreitol, and an EDTA-free protease inhibitor cocktail tablet (Roche Diagnostics 11836170001)), cells were lysed via sonication and centrifuged at 27,000 g and 4 °C for 30 minutes. The supernatant was incubated with pre-washed glutathione sepharose beads (Cytiva 17-0756-05) for three hours at 4 °C. The beads were then washed extensively with buffer 1. Additional washes were conducted using buffer 2 (20 mM HEPES at pH 7.0, 50 mM NaCl, and 2 mM DTT). CD28H was cleaved from the GST tag by incubation overnight with PreScission protease. GST-tagged CD28H was eluted in buffer 2 with the addition of 20 mM reduced L-glutathione. CD28H was further purified by size exclusion chromatography (SEC) on an ÄKTA pure FPLC system (Cytiva) using a HiLoad 16/600 Superdex 75 prep grade column in buffer 3 (20 mM NaPO4 at pH 6.5, 50 mM NaCl, 2 mM DTT, and 20 μM ZnSO4) supplemented with an EDTA-free protease inhibitor cocktail tablet. GST-tagged CD28H was similarly purified by SEC in buffer 4 (20 mM HEPES pH 7.5, 50 mM NaCl, 2 mM DTT, 20 μM ZnSO4, EDTA-free protease inhibitor).
SDS-PAGE
Protein lysates were subjected to SDS-PAGE on 12% NuPAGE Bis-Tris gels (Thermo Fisher Scientific NP0342) with MES SDS running buffer (Thermo Fisher Scientific NP0002) and visualized by Coomassie staining.
Electrospray ionization mass spectrometry
Mass spectrometry was performed with CD28H (10 μM) in buffer 3 and GST-tagged CD28H (2 μM) in buffer 4 with 10% acetonitrile on a 6100 Series Quadrupole LC mass spectrometer (Agilent Technologies, Inc.), equipped with an electrospray source and operated in the positive ion mode. Data acquisition and analyses were performed using OpenLAB CDS ChemStation Edition software (version C.01.05, Agilent Technologies, Inc.).
NMR samples and experiments
Five NMR samples were prepared including 15N, 13C-labeled CD28H at 500 μM (sample 1), 600 μM (sample 2), 560 μM (sample 3), 230 μM (sample 4), or 450 μM (sample 5) and 15N-labeled CD28H at 400 μM (sample 6), 280 μM (sample 7), 100 μM (sample 8), or 100 μM with MSPΔH5 nanodiscs (sample 9). 2D 1H-15N HSQC, 2D CON, 3D HNCACB/CBCA(CO)NH, 3D HNCO/HN(CA)CO, and 3D HACAN spectra were collected on sample 1. 2D 1H-13C HSQC, 3D 13C-dispersed NOESY (120 ms mixing time), 3D CCH-TOCSY, and 3D HCCH-TOCSY spectra were collected on sample 2. 3D 13C-dispersed NOESY (250 ms mixing time) spectra were collected on sample 3. 3D long-range HNCO-COSY spectra were collected on sample 4. 2D CACO and 3D CANCO spectra were collected on sample 5. 3D 15N-dispersed NOESY (120 ms mixing time) spectrum was collected on sample 6. 2D 1H-15N HSQC and 3D HNHB spectra were collected on sample 7. 2D 1H-15N HSQC spectra were collected on samples 8 and 9. The mixing time of the 3D 13C-dispersed NOESY spectrum was optimized for sample 2 using 2D 1H, 1H planes with mixing times varying from 80 to 300 ms (Supplementary Fig. 8).
All experiments were collected in buffer 3 (20 mM NaPO4 at pH 6.5, 50 mM NaCl, 2 mM DTT, 20 μM ZnSO4) with 1 mM pefabloc, 0.1% NaN3, and 5% 2H2O / 95% 1H2O, except for the 2D 1H-13C HSQC, 3D 13C-dispersed NOESY, 3D CCH-TOCSY, and 3D HCCH-TOCSY experiments, which were collected on samples that were lyophilized in buffer 3 containing 1 mM pefabloc and 0.1% NaN3, and resuspended in 100% 2H2O. All experiments were collected at 25°C except for a 3D 13C-dispersed NOESY (250 ms mixing time), 3D 15N-dispersed NOESY (120 ms mixing time), 3D HNCO/HN(CA)CO, 3D HNCACB/CBCA(CO)NH, 2D and 3D long-range HNCO-COSY spectra, which were acquired at 11°C. The spectra were recorded on Bruker AvanceIII 700, 800, 850, or 900 MHz spectrometers equipped with cryogenically cooled probes and operating with TopSpin 3.6.
IPAP processing was applied to CON, CANCO, and CACO experiments in TopSpin prior to further processing. All NMR data processing was performed using NMRpipe46 and spectra were visualized and analyzed with XEASY47. Backbone ϕ and ψ torsion angle restraints were assessed by the TALOS+ program using HN, Hα, Cα, Hβ, C’, and N chemical shifts48. The structure of CD28H was calculated with Xplor-NIH 3.732 using NOE and hydrogen bond distance restraints along with backbone ϕ and ψ torsion angle restraints determined using TALOS+. The structural refinement used an extended initial structure generated by Xplor-NIH32 and included TorsionDB and HBPot energy terms. Hydrogen bond restraints were included only in the final rounds of structure calculations. The major conformational state consisting of all trans prolines was used for the structure calculations. Distance restraints were obtained by integrating the intensities of the NOE signals by using the peakint script in XEASY47. All detected NOE signals were able to be assigned and were used in the structure calculations. Center averaging was used to define distances involving non-stereospecifically assigned methyl, methylene, and aromatic atoms, with a pseudo-atom correction applied32,49. Calibration of the 15N-dispersed NOESY experiment was done by assigning the average intra-residue HN to Hα NOE for residues E115 – Y118, Y122 – E126, and N129 to a distance of 3 Å. The 13C-edited NOESY experiment was similarly calibrated by assigning a 3 Å distance to the average of the intra-residue Cβ to Cδ NOE of residues P119 – P121, P142, and P144 and Cβ to Cγ NOEs of residues P119 – P121, P142, P144, P147, P149, and the proline residue from the non-native amino acid sequence GPLGS that remains following cleavage of the GST tag. The distance restraints (in Å) for the other NOE signals were determined relative to the 3 Å-associated NOEs by using the ratio 1/r6 with minimum van der Waals distances and maximum distance of 3.0, 3.5, 4.5, 5.5, or 6.5 Å. Due to steric clashes or violations of the distance restraints determined from NOE data, 12% (6 of 50) of the structures calculated by Xplor-NIH were excluded from the study, and the 25 lowest energy structures were chosen for visualization and statistical analyses. PyMOL (PyMOL Molecular Graphics System, https://www.pymol.org) was used to visualize structures, generate figures, and calculate their radii of gyration50 and torsion angles. Chemical shift index (CSI) values for CD28H Cα atoms were calculated by comparison to random coil values of the same amino acid type with sequence-dependent correction factors36,37. A CD28H structural ensemble was generated by inputting the CD28H sequence into the software IDPConformerGenerator40 in NMRbox (https://nmrbox.nmrhub.org)51.
GST pull-down analysis
10 nmol of purified GST-tagged CD28H or GST (Fisher Scientific LLC, PI20237), as a negative control, were bound to 40 μL of prewashed glutathione sepharose resin and incubated with 20 nmol CD28H. Unbound protein was removed by extensive washing in buffer 4 (20 mM HEPES pH 7.5, 50 mM NaCl, 2 mM DTT, 20 μM ZnSO4, EDTA-free protease inhibitor). Proteins that were retained on the resin were resolved by SDS-PAGE.
CD experiments
Far-UV range CD spectra of CD28H were recorded in buffer 5 (20 mM NaPO4 at pH 6.5, 10 mM NaCl, and 1 mM 2‑mercaptoethanol) on a Jasco J-1500CD spectrometer using a quartz cuvette with 1.0 mm path length. Thermostability measurements were collected for wavelengths 190 – 240 nm on CD28H (30 μM) following one minute of incubation at temperatures ranging from 25 °C to 80 °C. To collect secondary structure information, additional CD spectra were recorded in the far-UV range (190 – 250 nm) on CD28H (10 μM) with temperature controlled at 25 ± 0.1 °C. Buffer 5 was used as a control. All spectra were collected continuously at a scan speed of 20 nm/min and averaged over accumulation of three spectra. The buffer spectrum was subtracted from the protein spectra during data analyses. Molar ellipticity θ (in deg cm2 dmol−1) was calculated from the measured machine units m° in millidegrees at wavelength λ using Eq. (1).
\[
\theta =\frac{{{\rm{m}}}^{\circ }}{(10\cdot C\cdot L)}\,
\]
C is the concentration of the sample in mol\(\cdot\)L-1 and L is the path length of the cell (cm). Secondary structure analysis was conducted with the program CONTIN52 by the DichroWeb server31 using reference dataset SP175t (190–240 nm)52.
AlphaFold 3 prediction
AlphaFold 3 prediction was utilized through the AlphaFold Server (https://alphafoldserver.com)19. Structures were analyzed and figures generated by using PyMOL (PyMOL Molecular Graphics System, http://www.pymol.org).
Statistics and reproducibility
To calculate the protein structure, 50 random linear structures were used as starting structures. After simulated annealing and energy minimization steps, the 25 lowest energy structures were chosen for visualization and statistical analyses. The violations and deviations from idealized geometry in Table 1 were obtained by XPLOR-NIH and average pairwise root-mean-square deviation was calculated by MOLMOL. Mean values, standard deviation, and standard error were calculated by using Microsoft Excel. OpenLAB CDS ChemStation Edition software (version C.01.05, Agilent Technologies, Inc.) was used to deconvolute the mass spectrum in Fig. 1e. Biochemical experiments, including protein purification and SDS-PAGE were done in biological replicates of greater than 10 times. CD experiments were done in triplicate. 2D NMR, LC-MS, and AlphaFold 3 prediction were repeated at least once. All replications were consistent.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary Materials
References
- KM Cappell, JN Kochenderfer. Long-term outcomes following CAR T cell therapy: what we know so far. Nat. Rev. Clin. Oncol., 2023. [DOI | PubMed]
- TJ Fry. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med., 2018. [DOI | PubMed]
- NN Shah. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat. Med., 2020. [DOI | PubMed]
- BG Till. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood, J. Am. Soc. Hematol., 2012
- JN Brudno. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J. Clin. Oncol., 2018. [DOI | PubMed]
- L Mikkilineni, JN Kochenderfer. CAR T cell therapies for patients with multiple myeloma. Nat. Rev. Clin. Oncol., 2021. [DOI | PubMed]
- K Kheyrolahzadeh. Theranostic chimeric antigen receptor (CAR)-T cells: Insight into recent trends and challenges in solid tumors. Life Sci., 2023. [DOI | PubMed]
- X Zhang, L Zhu, H Zhang, S Chen, Y Xiao. CAR-T cell therapy in hematological malignancies: current opportunities and challenges. Front. Immunol., 2022. [DOI | PubMed]
- JY Spiegel. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat. Med., 2021. [DOI | PubMed]
- D Schneider. Trispecific CD19-CD20-CD22-targeting duoCAR-T cells eliminate antigen-heterogeneous B cell tumors in preclinical models. Sci. Transl. Med., 2021. [DOI | PubMed]
- H Shalabi. CD19/22 CAR T cells in children and young adults with B-ALL: phase 1 results and development of a novel bicistronic CAR. Blood J. Am. Soc. Hematol., 2022
- AH Long. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med., 2015. [DOI | PubMed]
- N Singh. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat. Med., 2021. [DOI | PubMed]
- K Fujiwara. Hinge and transmembrane domains of chimeric antigen receptor regulate receptor expression and signaling threshold. Cells, 2020. [DOI | PubMed]
- RG Majzner. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov., 2020. [DOI | PubMed]
- YD Muller. The CD28-Transmembrane domain mediates chimeric antigen receptor heterodimerization with CD28. Front Immunol., 2021. [DOI | PubMed]
- MC Ramello. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal, 2019. [DOI | PubMed]
- X Chen. The CD8α hinge is intrinsically disordered with a dynamic exchange that includes proline cis-trans isomerization. J. Magn. Reson, 2022. [DOI | PubMed]
- J Abramson. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 2024. [DOI | PubMed]
- EJ Evans. Crystal structure of a soluble CD28-Fab complex. Nat. Immunol., 2005. [DOI | PubMed]
- H Wu, R Cao, M Wen, H Xue, B OuYang. Structural characterization of a dimerization interface in the CD28 transmembrane domain. Structure, 2022. [DOI | PubMed]
- X Chen. Prokaryotic ubiquitin-like protein pup is intrinsically disordered. J. Mol. Biol., 2009. [DOI | PubMed]
- GR Buel. Structure of E3 ligase E6AP with a proteasome-binding site provided by substrate receptor hRpn10. Nat. Commun., 2020. [DOI | PubMed]
- U Nowicka. Mycobacterium tuberculosis copper-regulated protein SocB is an intrinsically disordered protein that folds upon interaction with a synthetic phospholipid bilayer. Proteins, 2016. [DOI | PubMed]
- V Kanelis. Sequential assignment of proline-rich regions in proteins: application to modular binding domain complexes. J. Biomol. NMR, 2000. [DOI | PubMed]
- W Haar, S Fermandjian, J Vicar, K Blaha, P Fromageot. 13C-nuclear magnetic resonance study of [85% 13C-enriched proline]thyrotropin releasing factor: 13C-13C vicinal coupling constants and conformation of the proline residue. Proc. Natl Acad. Sci. USA, 1975. [DOI | PubMed]
- M Schubert, D Labudde, H Oschkinat, P Schmieder. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on 13C chemical shift statistics. J. Biomol. NMR, 2002. [DOI | PubMed]
- W Bermel. Complete assignment of heteronuclear protein resonances by protonless NMR spectroscopy. Angew. Chem. Int. Ed. Engl., 2005. [DOI | PubMed]
- C Grathwohl. & Wüthrich, K. NMR studies of the rates of proline cis–trans isomerization in oligopeptides. Biopolym. Orig. Res. Biomol., 1981
- KJ Walters, H Matsuo, G Wagner. A simple method to distinguish intermonomer nuclear Overhauser effects in homodimeric proteins with C 2 symmetry. J. Am. Chem. Soc., 1997. [DOI]
- AJ Miles, SG Ramalli, BA Wallace. DichroWeb, a website for calculating protein secondary structure from circular dichroism spectroscopic data. Protein Sci., 2022. [DOI | PubMed]
- CD Schwieters, JJ Kuszewski, N Tjandra, GM Clore. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson, 2003. [DOI | PubMed]
- F Cordier, L Nisius, AJ Dingley, S Grzesiek. Direct detection of N-H[…]O=C hydrogen bonds in biomolecules by NMR spectroscopy. Nat. Protoc., 2008. [DOI | PubMed]
- F Cordier, S Grzesiek. Direct observation of hydrogen bonds in proteins by interresidue 3hJNC‘ scalar couplings. J. Am. Chem. Soc., 1999. [DOI]
- JE Kohn. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl Acad. Sci. USA, 2004. [DOI | PubMed]
- DS Wishart, BD Sykes. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR, 1994. [DOI | PubMed]
- S Schwarzinger. Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc., 2001. [DOI | PubMed]
- M Schwalbe. Structural impact of tau phosphorylation at threonine 231. Structure, 2015. [DOI | PubMed]
- NL Daly, R Hoffmann, L Otvos, DJ Craik. Role of phosphorylation in the conformation of T peptides implicated in Alzheimer’s disease. Biochemistry, 2000. [DOI | PubMed]
- JM Teixeira. IDPConformerGenerator: a flexible software suite for sampling the conformational space of disordered protein states. J. Phys. Chem. A, 2022. [DOI | PubMed]
- MV Staller. Directed mutational scanning reveals a balance between acidic and hydrophobic residues in strong human activation domains. Cell Syst., 2022. [DOI | PubMed]
- JM Rogers. Interplay between partner and ligand facilitates the folding and binding of an intrinsically disordered protein. Proc. Natl Acad. Sci. USA, 2014. [DOI | PubMed]
- GR Buel, KJ Walters. Can AlphaFold2 predict the impact of missense mutations on structure?. Nat. Struct. Mol. Biol., 2022. [DOI | PubMed]
- J Seigner. Solving the mystery of the FMC63-CD19 affinity. Sci. Rep., 2023. [DOI | PubMed]
- J Mirazee. 401 | Hinge length: A novel method of predicting cytotoxicity of CAR constructs against antigen-low leukemia. J. Immunother. Cancer, 2022
- F Delaglio. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR, 1995. [DOI | PubMed]
- C Bartels, TH Xia, M Billeter, P Güntert. & Wüthrich, K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR, 1995. [DOI | PubMed]
- Y Shen, F Delaglio, G Cornilescu, A Bax. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR, 2009. [DOI | PubMed]
- K Wuthrich, M Billeter, W Braun. Pseudo-structures for the 20 common amino acids for use in studies of protein conformations by measurements of intramolecular proton-proton distance constraints with nuclear magnetic resonance. J. Mol. Biol., 1983. [DOI | PubMed]
- 50.Holder, T. Radius of gyration, https://pymolwiki.org/index.php/Radius_of_gyration (2011).
- MW Maciejewski. NMRbox: A resource for biomolecular NMR computation. Biophys. J., 2017. [DOI | PubMed]
- N Sreerama, RW Woody. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem, 2000. [DOI | PubMed]