Systematic investigation of promoter substitutions resulting from somatic intrachromosomal structural alterations in diverse human cancers
Abstract
One of the ways in which genes can become activated in tumors is by somatic structural genomic rearrangements leading to promoter swapping events, typically in the context of gene fusions that cause a weak promoter to be substituted for a strong promoter. While identifiable by whole genome sequencing, limited availability of this type of data has prohibited comprehensive study of the phenomenon. Here, we leveraged the fact that copy number alterations (CNAs) arise as a result of structural alterations in DNA, and that they may therefore be informative of gene rearrangements, to pinpoint recurrent promoter swapping at a previously intractable scale. CNA data from nearly 9500 human tumors was combined with transcriptomic sequencing data to identify several cases of recurrent activating intrachromosomal promoter substitution events, either involving proper gene fusions or juxtaposition of strong promoters to gene upstream regions. Our computational screen demonstrates that a combination of CNA and expression data can be useful for identifying novel fusion events with potential driver roles in large cancer cohorts.
Article type: Research Article
Keywords: Cancer, Computational biology and bioinformatics, Cancer genomics, Gene expression, Genomics
Affiliations: grid.8761.80000 0000 9919 9582Department Of Medical Biochemistry and Cell Biology, Institute of Biomedicine, The Sahlgrenska Academy, University of Gothenburg, 405 30 Gothenburg, Sweden
License: © The Author(s) 2020 CC BY 4.0 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Article links: DOI: 10.1038/s41598-020-74420-2 | PubMed: 33097743 | PMC: PMC7584658
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (2.9 MB)
Introduction
Copy number alterations (CNAs) significantly contribute to cancer development, usually by causing oncogene amplification or tumor suppressor deletion1–3. Well-characterized examples of cancer driver events involving CNAs are CDKN2A4 and PTEN5 deletions or MYC6, EGFR7 and ERBB22,7 amplifications. With the availability of high-resolution SNP arrays, several studies have comprehensively investigated these events in cancer, mainly focusing on gene amplitude changes8,9.
CNAs are a consequence of changes in chromosome structure10. CNAs may therefore be indicative of more complex rearrangements of genomic features such as regulatory elements that determine the transcriptional activity of genes. Recent studies have indeed uncovered that deletions and duplications may facilitate mRNA level changes by shuffling or amplifying non-coding regions in the genome including cis-regulatory elements such as enhancers11–14. Another known mechanism for a gene to be activated by genomic structural variations (SVs) is to substitute its promoter with a stronger promoter in the context of gene fusions15,16. One of the most frequent promoter substitution (PS) events in cancer involve transcriptional activation of ERG through fusion with TMPRSS2, which occurs in approximately 40% of prostate cancers as a result of a genomic deletion on chromosome 17q2217. Several other fusions involving this mechanism are known18–20. Furthermore, in a recent study based on whole genome sequencing (WGS) data from 600 tumors, we observed several non-recurrent cases of PS that arose due to intrachromosomal SVs, specifically deletions or inversions, which were associated with transcriptional activation21. Investigations based on larger cohorts could potentially give insights into whether or not such events are recurrent, suggestive of positive selection and thereby importance in cancer.
In this study, we used 9423 array-based copy number profiles made available by The Cancer Genome Atlas consortium to identify deletions and likely tandem duplications predicted to result in intrachromosomal PS events, due to either proper gene fusions or juxtaposition of strong promoters to upstream regions. We then investigated the relationship between such events and mRNA level changes. By using CNAs as a proxy of SVs, we could thus investigate this phenomenon in a cohort that is considerably larger than what is currently possible using WGS.
Results
Mapping of tandem duplications and deletions using copy number profiles in a large cancer cohort
With the ultimate aim of detecting PS events resulting from intrachromosomal SVs in a large multi-cancer cohort (Fig. 1a), we first sought to identify somatic tandem duplications and deletions using Genome-Wide Human SNP Array 6.0 (SNP6) data from The Cancer Genome Atlas (TCGA; Fig. 1b). The probe-based nature of this data limits its resolution and it is also affected by sample purity and ploidy, and we therefore applied strict filtering criteria to ensure that only events with a clear interpretation in terms of the structural basis were considered (see Methods). By comparing with WGS-based SVs from a previous study21, available for a subset of samples (600 tumors), we found that 25% of the CNA-inferred SVs had a correspondence in WGS-based SVs and of these, 97% were coherently classified as deletions or duplications in the two datasets.
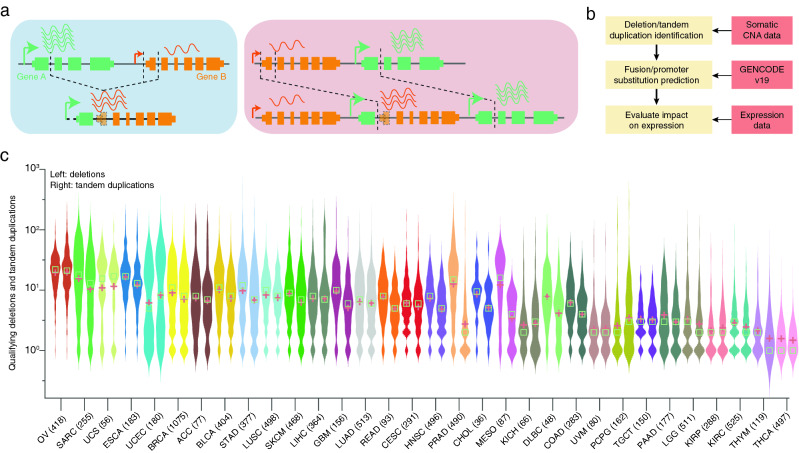
In the complete cohort, comprising of 9423 tumors from 32 different cancer types, we identified 110,463 predicted deletions and 84,052 tandem duplications that fulfilled our criteria. The number of events varied considerably between cancer types, with the highest (OV, SARC) and lowest (THYM, THCA) numbers seen in cancers previously shown to have many or few SVs based on analysis of WGS data (Fig. 1c)21. The number of deletions and tandem duplications were typically comparable in a given cancer type (both plots within twofold; Fig. 1c). However, two cancer types, prostate (PRAD) and mesothelioma (MESO), had elevated number of deletions relative to duplications (4.2 and 5.2-fold difference, respectively).
Pan-cancer analysis of SVs resulting in promoter substitutions
We next identified a subset of SVs that may result in PS (Methods), involving either gene fusions or alternatively cases where the 5′ promoter is juxtaposed to the upstream region of the 3′ partner (Fig. 1a). We found 20,715 SVs having PS potential comprising of 9754 tandem duplications and 10,961 deletions. 1925 unique gene pairs were involved in recurrent (n > 1) predicted PS events (Fig. 2a). Confirming previous reports, the most recurrent case was TMPRSS2–ERG (n = 72; Supplementary Fig. S1a), which was completely restricted to prostate cancer (n = 490 samples). The observed frequency was lower than previously reported17, and therefore we manually explored copy number profiles for the complete prostate cancer cohort, which revealed 28 additional cases with deletions potentially fusing these two genes. These were not detected by our pipeline due to presence of more complex copy number patterns in the region (Supplementary Fig. S2). A subset of 20 prostate samples had available WGS data and four of these were previously found to harbor the TMPRSS2–ERG fusion21, all of which were confirmed using the copy number pipeline. Notably, while many known functional fusions, including TMPRSS2–ERG, are restricted to specific cancer types, we observed that most recurrent cases were distributed across multiple cancers (Fig. 2a). While this does not exclude that they could be functional, further analysis was motivated.
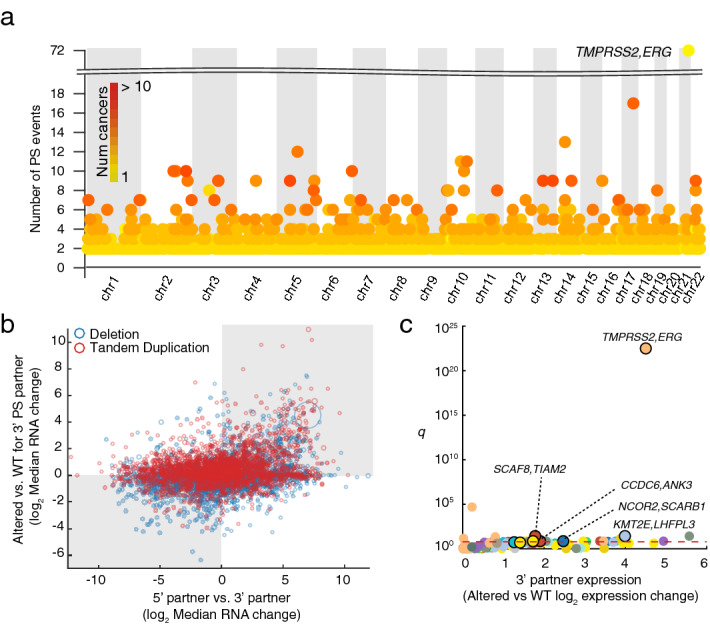
We next investigated associations between recurrently PS affected cases and gene expression changes. As expected, we found that mRNA expression of the 3′ partner increased when the 5′ partner had a stronger promoter, the latter determined by comparing the median expression levels of the two partners in a given cancer type (Fig. 2b; p = 2.51e−22, Fisher’s exact test). Additionally, we found that transcriptional induction of the 3′ partner occurred more frequently when the genes were closer together (Supplementary Fig. S3). There were 126 cases of genes being recurrently (n > = 2) affected by PS with a stronger promoter (2-fold) within an individual cancer type. In 8 of these cases, the 3′ partner gene was significantly induced in PS-affected samples (Student’s t-test at FDR 10%; Fig. 2c), although it should be noted that the statistical power was weak due to number of affected samples typically being small.
The most significant case was TMPRSS2–ERG with 22-fold increase in expression (p = 2.56e−25 uncorrected; Supplementary Fig. S1b). We also identified the previously reported ESR1–CCDC170 fusion22 in breast cancer (n = 3), associated with CCDC170 elevated expression resulting from recruiting the strong promoter of ESR1 (p = 0.02; Supplementary Fig. S4). One additional ovarian tumor harbored the same fusion, although induction of CCDC170 was not significant (p = 0.44).
Novel recurrent promoter substitution events
Notable among novel significant cases was predicted fusions between SCAF8 and TIAM2 resulting from deletions bridging these two closely positioned neighbor genes (Fig. 3a). This occurred primarily in ovarian carcinoma (n = 5), specifically in the serous histological subtype, where TIAM2 expression was increased 3.4-fold in PS-affected cases compared to remaining samples (p = 8.71e−04 uncorrected), and was also found in endometrial carcinoma (n = 1; 5.5-fold; p = 0.086 uncorrected). TIAM2 acts as an upstream regulator in the Rac pathway, and it has been shown that the overexpression of this gene promotes cell proliferation and invasion in multiple cancer types23–25. Interestingly, induction of TIAM2 in PS positive cases surpassed what was seen in cases of TIAM2 gene amplification (Fig. 3b).
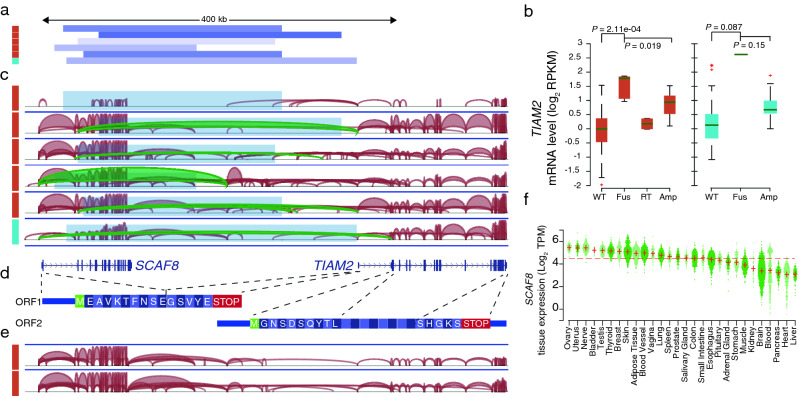
We next focused on understanding the transcript and protein structure resulting from these deletions. We found that in 5/6 cases with RNA level support, the first or second exon of SCAF8 was fused with the first noncoding exon of TIAM2 located in the 5′ UTR (Fig. 3c). This resulted in a novel transcript containing a smaller truncated open reading frame (ORF) from SCAF8 followed by the complete TIAM2 mRNA sequence including the 5′ UTR, thereby containing two possible ORFs (Fig. 3d). More work is needed to determine whether TIAM2 can be translated from this transcript.
Analysis of RNA-Seq data from all included ovarian and endometrial tumors revealed two additional samples with SCAF8–TIAM2 fusion transcripts in the absence of DNA-level support, suggesting that this could be due to read through events (Fig. 3e). Notably, TIAM2 induction was considerably lower in these cases compared to those with genomic deletions (Fig. 3b). Based on the GTEx panel of human tissues, we found that wild type SCAF8 had its highest expression in the ovary and uterus, making it an ideal fusion partner to drive high expression in ovarian and endometrial cancers (Fig. 3f). Additionally, we found that deletion breakpoints did not overlap with common fragile sites in the HumCFS database26. This, together with the tissue-restricted pattern, further supported that the reported events may be due to positive selection specifically in these cancers.
Novel recurrent cases were also found in ovarian (n = 6), endometrial (n = 2) and breast (n = 2) cancers involving CCDC6, a coiled-coil domain protein, fusing with ANK3 at the 3′ end, which encodes the ankyrin G protein that plays a key role in cell proliferation, as result of tandem duplications (Fig. 4a). While it has been shown that downregulation of ANK3 is associated with poor prognosis in multiple cancers such as prostate, ovarian, lung and breast27, a recent study also described that increased ANK3 contributes to prostate cancer progression, implying that both up and down regulation of this gene can be important at different clinical stages28. Here, we observed that fusion with CCDC6 was associated with strong overexpression of ANK3 in all three cancers (Fig. 4b). In 6/10 cases the regulatory domain of ANK3, also known as death like domain29, was retained. Furthermore, we observed that CCDC6 is normally highly expressed in ovarian, endometrial and breast compared to the other cancer types, as well as in normal ovary and uterine tissues (Supplementary Fig. S5). Further analysis of matching RNA-Seq samples showed that the fusion transcript was significantly upregulated compared to the wild type ANK3 form, consistent with ANK3 gaining the strong promoter from CCDC6 (Fig. 4c).
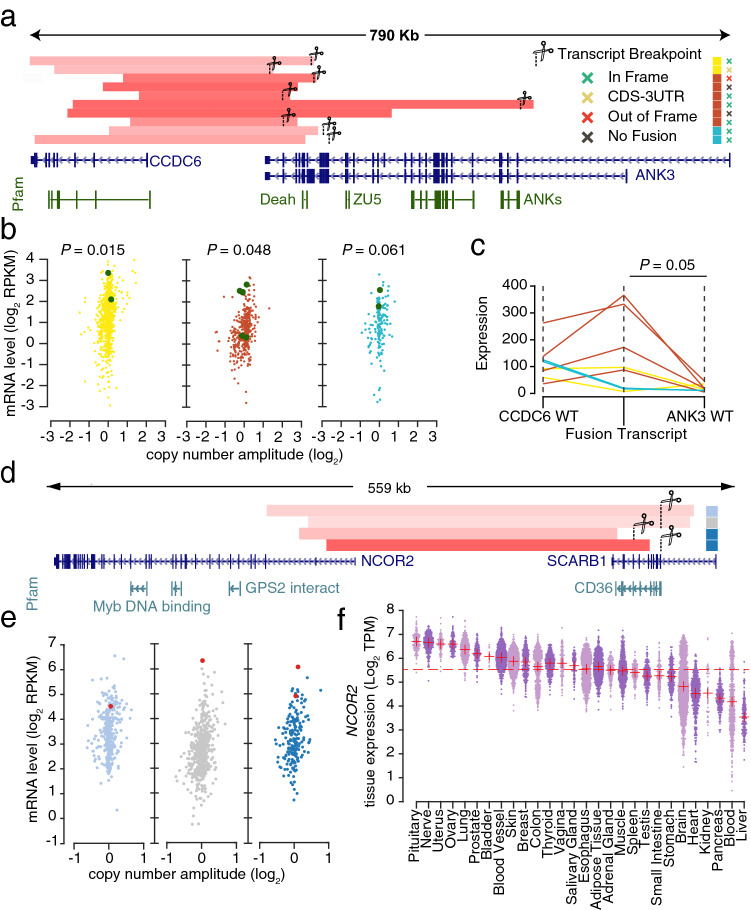
Another recurrent case (n = 4) was found in stomach, esophageal and lung adenocarcinoma, where SCARB1, a high-density lipoprotein (HDL) receptor, was overexpressed through fusion with NCOR2 due to tandem duplications on chromosome 12q24 (Fig. 4d,e). Notably, the functionally critical CD36 family domain of SCARB1, a receptor family that is crucial for cholesterol uptake, was maintained in all cases. Consistent with the elevated expression of SCARB1, we found that the NCOR2 gene is relatively highly expressed in the relevant tissue types, making it a suitable 5′ partner for activating transcription (Fig. 4f). Overexpression of SCARB1 has been associated with cancer development and shown to be inversely correlated with survival in multiple cancer types, although no molecular mechanism was proposed30,31.
Finally, we observed overexpression of LHFPL3 in four stomach tumors harboring KMT2E–LHFPL3 fusions arising due to tandem duplications on chromosome 7q22 (Supplementary Fig. S6a,b). Interestingly, in three of the four cases, a valid fusion transcript was supported by RNA-Seq, expressed at elevated levels compared to the LHFPL3 unaltered transcript (Supplementary Fig. S6c). Although more work is needed to determine the relevance of these events, it can be noted that LHFPL3 is a member of the LHPF-like gene family known to be fusion partners of HMGIC, an established tumor associated gene in lipoma32, and overexpression of this gene has been described in ovarian cancer33.
Investigation of the PS events described above in copy number profiles from the Cancer Cell Line Encyclopedia (CCLE) database confirmed all cases except TIAM2–SCAF8 (Supplementary Fig. S7a). An amplification predicted to form a NCOR2–SCARB1 fusion gene was identified in one lung cancer cell line (Supplementary Fig. S7b), CCDC6–ANK3-forming amplifications were found in two ovarian cancer cell lines (Supplementary Fig. S7c), and a LHFPL3–KMT2E-forming amplification was found in one lung cancer (Supplementary Fig. S7d). The known fusion CCDC170–ESR1 was found in three breast cancer samples (Supplementary Fig. S7e) while the well-described promoter substitution event, TMPRSS2–ERG (Supplementary Fig. S7f), was identified in one prostate cancer cell line.
Discussion
Promoter substitutions, whereby structural genomic changes lead to one gene gaining a promoter from another gene, is a known mechanism for transcriptional activation of oncogenes in cancer34,35, but the phenomenon has not previously been comprehensively investigated. Here, we took advantage of the fact the CNA profiles gives insight into structural genomic alterations, which, when combined with expression data, enabled mapping of putative PS events in a large multi-cancer cohort. CNA data have several limitations in this context, including not being informative about inversions and interchromosomal SVs. Furthermore, the array-based CNA data used in this study has limited resolution, and sensitivity may be reduced in some samples with lower sample purity. However, in return there is abundant availability of CNA profiles from human tumors, enabling detection of events that are recurrent at frequencies that are undetectable in WGS-based analysis. While only ~ 25% of CNA-based events were confirmed using WGS-based SV analysis, to a large extent this is likely to reflect of the limited the sensitivity of WGS-based SV data, and events detected using both datatypes showed a high degree of consistency (97%) in terms of deletion/duplication classification. Importantly, using our combined CNA and expression approach, we confirm several established cases and also identify new cases of recurrent PS.
The TIAM gene family is part of the Rac signaling pathway, and has been shown to contribute to tumor development in multiple cancer types36–38. Genomic alterations involving the TIAM1 gene have been previously described39,40, and TIAM2 has been shown to be upregulated in lung and liver tumors23,25, but little is known about the underlying mechanism for this activation. Here, we describe a novel mechanism leading to TIAM2 overexpression in ovarian and endometrial carcinoma, that involves formation of a new fusion transcript transcribed from a nearby promoter that is highly active in these tissue types. More work is needed to determine if the resulting mRNA, which has an unusual structure, can serve as a template for TIAM2 translation, but the fact that the transcript is abundant suggests avoidance of nonsense-mediated decay and hence proper translation. The functional consequences of increased TIAM2 protein levels in these tumor types will need to be determined in future experimental studies.
Several studies have shown that the cholesterol plays a role in development of cancer41,42. SCARB1 is a protein that is involved in transporting HDL cholesterol in the body, and overexpression of this gene is known to facilitate this mechanism. Although the activation of SCARB1 has been shown to be associated with tumor size and worse overall survival in cancer43, the underlying mechanism by which this gene becomes active is poorly understood. Here, we show that PS can activate SCARB1 in three cancer types, although more work is needed to determine whether these are driving events.
In summary, we leveraged CNA and expression profiles available for nearly 10,000 tumors to screen for cases where genes were transcriptionally activated due to fusion with nearby genes having strong promoters, pinpointing several events with potential importance for cancer development. While the extent to which these events are due to positive selection remains an open question, it should be noted that they occur recurrently, sometimes in a tissue-restricted manner, and affect genes previously implicated in cancer. Future experimental studies should aim to investigate the functional consequences of these events in cancer.
Methods
Copy number and gene expression data processing
SNP6 segmented copy number profiles from 9423 tumors in 32 cancer types were obtained from the TCGA data portal. We classified segments into 5 copy number state categories in regards to their log2 amplitude provided in the raw seg files. Segments with seg_mean < − 1 were classified in homozygous deletions, − 1 < = seg_mean < − 0.2 hemizygous deletions, − 0.2 < = seg_mean < − 0.3 neutral, 0.3 < = seg_mean < 0.7 gain and, seg_mean = > 0.7 amplifications. Nearby segments with the same copy number state were merged to a bigger segment. Segments adjacent to a no-data regions bigger that 100 kb were removed for further analysis.
SV deletions were defined as (1) hemizygous deleted region where neither of adjacent segments were homozygous deletions and (2) homozygous deleted segments where both adjacent segments were hemizygous deletions. Gained segments with no adjacent “amplified segments” were considered as SV tandem duplications. SVs with breakpoints within 2 Mb range of telomeres and centromeres, or smaller than 15 Kb were removed for further analysis. The breakpoints were annotated against GENCODE v19 gene annotation with the following priority: overlapping coding gene, overlapping lincRNA, and closest upstream gene.
Matching RNA-Seq data were downloaded from the TCGA portal and used to quantify gene expression as described previously44. Normal tissue expression was obtained from the GTEx portal. Fusion transcripts were detected using ericScript45. WGS-based SV data for a subset of samples (600) was obtained from Alaei-Mahabadi, et al.21.
Screening for association between SVs and RNA levels resulting from promoter switching
SVs resulting in a valid PS cases were identified using the following logic: We considered two different cases: (1) SVs predicted to produce a viable fusion between two genes, i.e. where both breakpoints fell within annotated genes and where both genes were transcribed in the same direction. In this case, the gene on the 3′ side will have gained the promoter from the 5′ partner gene. (2) SVs predicted to fuse the 5′ part of a gene (including the promoter) with a position somewhat upstream of another gene transcribed in the same direction. This may lead to the promoter of the 5′ partner gene driving expression of the 3′ partner due to transcriptional readthrough. In this case, the 3′ partner gene was required to be located no further than 200 kb downstream of the breakpoint. Only coding genes were considered in the analyses. Read count based estimation of the expression levels of the WT 5′ gene, the WT 3′ gene and the predicted chimeric gene was based on ericScript45. These values were used to visualize the transcriptional consequences of the predicted fusion events.
Confirming fusions in cancer cell line encylopedia CNA data
In order to confirm the presence of fusions we obtained copy number profiles from the CCLE https://portals.broadinstitute.org/ccle. Fusion genes were identified and samples sorted by breakpoint frequency to identify samples with CNVs at the known fusion sites in Integrative Genomics Viewer (IGV).
Supplementary Materials
References
- PB Illei, VW Rusch, MF Zakowski, M Ladanyi. Homozygous deletion of CDKN2A and codeletion of the methylthioadenosine phosphorylase gene in the majority of pleural mesotheliomas. Clin. Cancer Res., 2003. [PubMed]
- OP Kallioniemi. ERBB2 amplification in breast cancer analyzed by fluorescence in situ hybridization. Proc. Natl. Acad. Sci. USA, 1992. [DOI | PubMed]
- TI Zack. Pan-cancer patterns of somatic copy number alteration. Nat. Genet., 2013. [DOI | PubMed]
- A Kamb. A cell cycle regulator potentially involved in genesis of many tumor types. Science, 1994. [DOI | PubMed]
- J Li. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science, 1997. [DOI | PubMed]
- CE Nesbit, JM Tersak, EV Prochownik. MYC oncogenes and human neoplastic disease. Oncogene, 1999. [DOI | PubMed]
- N Vogt. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc. Natl. Acad. Sci. USA, 2004. [DOI | PubMed]
- R Beroukhim. The landscape of somatic copy-number alteration across human cancers. Nature, 2010. [DOI | PubMed]
- TM Kim. Functional genomic analysis of chromosomal aberrations in a compendium of 8000 cancer genomes. Genome Res., 2013. [DOI | PubMed]
- L Feuk, AR Carson, SW Scherer. Structural variation in the human genome. Nat. Rev. Genet., 2006. [DOI | PubMed]
- F Haller. Enhancer hijacking activates oncogenic transcription factor NR4A3 in acinic cell carcinomas of the salivary glands. Nat. Commun., 2019. [DOI | PubMed]
- D Hnisz. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science, 2016. [DOI | PubMed]
- PA Northcott. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature, 2014. [DOI | PubMed]
- J Weischenfeldt. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet., 2017. [DOI | PubMed]
- KH Nord. GRM1 is upregulated through gene fusion and promoter swapping in chondromyxoid fibroma. Nat. Genet., 2014. [DOI | PubMed]
- AM Oliveira. Aneurysmal bone cyst variant translocations upregulate USP6 transcription by promoter swapping with the ZNF9, COL1A1, TRAP150, and OMD genes. Oncogene, 2005. [DOI | PubMed]
- The molecular taxonomy of primary prostate cancer. Cell, 2015. [DOI | PubMed]
- FP Duhoux. PRDM16 (1p36) translocations define a distinct entity of myeloid malignancies with poor prognosis but may also occur in lymphoid malignancies. Br. J. Haematol., 2012. [DOI | PubMed]
- K Kas. Promoter swapping between the genes for a novel zinc finger protein and beta-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nat. Genet., 1997. [DOI | PubMed]
- MP Simon. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat. Genet., 1997. [DOI | PubMed]
- B Alaei-Mahabadi, J Bhadury, JW Karlsson, JA Nilsson, E Larsson. Global analysis of somatic structural genomic alterations and their impact on gene expression in diverse human cancers. Proc. Natl. Acad. Sci. USA, 2016. [DOI | PubMed]
- J Veeraraghavan. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat. Commun., 2014. [DOI | PubMed]
- JS Chen, IJ Su, YW Leu, KC Young, HS Sun. Expression of T-cell lymphoma invasion and metastasis 2 (TIAM2) promotes proliferation and invasion of liver cancer. Int. J. Cancer, 2012. [DOI | PubMed]
- RWJ Wong. Enhancer profiling identifies critical cancer genes and characterizes cell identity in adult T-cell leukemia. Blood, 2017. [DOI | PubMed]
- ZY Zhao. TIAM2 enhances non-small cell lung cancer cell invasion and motility. Asian Pac. J. Cancer Prev., 2013. [DOI | PubMed]
- R Kumar. HumCFS: A database of fragile sites in human chromosomes. BMC Genomics, 2019. [DOI | PubMed]
- GV Glinsky, O Berezovska, AB Glinskii. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Invest., 2005. [DOI | PubMed]
- T Wang. Ankyrin G expression is associated with androgen receptor stability, invasiveness, and lethal outcome in prostate cancer patients. J. Mol. Med. (Berl.), 2016. [DOI | PubMed]
- JJ Ipsaro, L Huang, A Mondragon. Structures of the spectrin–ankyrin interaction binding domains. Blood, 2009. [DOI | PubMed]
- JL Gutierrez-Pajares, C Ben Hassen, S Chevalier, PG Frank. SR-BI: Linking cholesterol and lipoprotein metabolism with breast and prostate cancer. Front. Pharmacol., 2016. [DOI | PubMed]
- LK Mooberry, NA Sabnis, M Panchoo, B Nagarajan, AG Lacko. Targeting the SR-B1 receptor as a gateway for cancer therapy and imaging. Front. Pharmacol., 2016. [DOI | PubMed]
- MM Petit. LHFP, a novel translocation partner gene of HMGIC in a lipoma, is a member of a new family of LHFP-like genes. Genomics, 1999. [DOI | PubMed]
- Y Collins. Identification of differentially expressed genes in clinically distinct groups of serous ovarian carcinomas using cDNA microarray. Int. J. Mol. Med., 2004. [PubMed]
- G Klein, E Klein. Conditioned tumorigenicity of activated oncogenes. Cancer Res., 1986. [PubMed]
- P Leder. Translocations among antibody genes in human cancer. Science, 1983. [DOI | PubMed]
- GA Cardama, N Gonzalez, J Maggio, PL Menna, DE Gomez. Rho GTPases as therapeutic targets in cancer (review). Int. J. Oncol., 2017. [DOI | PubMed]
- B Chen. Tiam1, overexpressed in most malignancies, is a novel tumor biomarker. Mol. Med. Rep., 2012. [DOI | PubMed]
- TN Gaitanos, J Koerner, R Klein. Tiam-Rac signaling mediates trans-endocytosis of ephrin receptor EphB2 and is important for cell repulsion. J. Cell Biol., 2016. [DOI | PubMed]
- P De. RAC1 GTP-ase signals Wnt-beta-catenin pathway mediated integrin-directed metastasis-associated tumor cell phenotypes in triple negative breast cancers. Oncotarget, 2017. [DOI | PubMed]
- JJ Molenaar. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature, 2012. [DOI | PubMed]
- C Danilo, PG Frank. Cholesterol and breast cancer development. Curr. Opin. Pharmacol., 2012. [DOI | PubMed]
- G Llaverias. Role of cholesterol in the development and progression of breast cancer. Am. J. Pathol., 2011. [DOI | PubMed]
- J Li. Up-regulated expression of scavenger receptor class B type 1 (SR-B1) is associated with malignant behaviors and poor prognosis of breast cancer. Pathol. Res. Pract., 2016. [DOI | PubMed]
- NJ Fredriksson. Recurrent promoter mutations in melanoma are defined by an extended context-specific mutational signature. PLoS Genet., 2017. [DOI | PubMed]
- M Benelli. Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics, 2012. [DOI | PubMed]