HEXIM1 inter-monomer autoinhibition governs 7SK RNA binding specificity and P-TEFb inactivation
Abstract
Hexim proteins are key RNA-dependent regulators of eukaryotic transcription through 7SK-dependent sequestration and inactivation of the kinase P-TEFb (Cdk9–CyclinT1/2) in the 7SK RNP. P-TEFb activity drives release of RNA polymerase II from promoter-proximal pausing for eukaryotic and HIV-1 transcription. The molecular mechanism by which 7SK binding overcomes an intrinsic Hexim autoinhibition for subsequent P-TEFb inactivation has remained unresolved. Here, using NMR and biophysical methods we demonstrate that Hexim1 homodimer engages two high-affinity sites on 7SK RNA. This dual-site binding triggers a conformational rearrangement in Hexim1’s disordered central region that unmasks the Cdk9-binding site, which is otherwise sequestered within an inter-monomer dimer interface. These findings reveal how Hexim autoinhibition dictates its specificity for 7SK RNA and prevents premature P-TEFb inhibition in the absence of 7SK, thereby providing a mechanistic understanding of Hexim/P-TEFb assembly into the 7SK RNP and further considerations for understanding Hexim–Tat competition during viral transcription.
Article type: Research Article
Keywords: Intrinsically disordered proteins, RNA, Solution-state NMR
Affiliations: https://ror.org/046rm7j60grid.19006.3e0000 0001 2167 8097Department of Chemistry and Biochemistry, University of California Los Angeles, Los Angeles, CA USA; https://ror.org/00qqv6244grid.30760.320000 0001 2111 8460Present Address: Departments of Biophysics and Obstetrics & Gynecology, Medical College of Wisconsin, Milwaukee, WI USA
License: © The Author(s) 2026 CC BY 4.0 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Article links: DOI: 10.1038/s41467-026-68285-8 | PubMed: 41540012 | PMC: PMC12901311
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (2.0 MB)
Introduction
HEXIM (HEXamethylene-bis-acetamide-Inducible protein in vascular smooth Muscle cells)1 is a unique family of RNA-dependent regulatory proteins2 whose principal target is 7SK RNA, an abundant ~332-nucleotide non-coding RNA that plays a central role in regulating RNA polymerase II (Pol II) transcription. In addition to 7SK, Hexim has been shown to interact with other RNAs, including NEAT13 and several mRNAs4, hinting at broader cellular functions. Mammals express two paralogs of Hexim (Hexim1 and Hexim2)which differ in tissue distribution and can functionally compensate for one another within the 7SK ribonucleoprotein complex (7SK RNP)5,6. Hexim suppresses the kinase activity of positive transcription elongation factor b (P-TEFb) by sequestering it within the 7SK RNP. Crucially, this sequestration depends on 7SK RNA binding to Hexim7–9. P-TEFb is a heterodimer comprising the catalytic subunit Cdk9 and a regulatory subunit, Cyclin T1 or T2 (CycT1/CycT2), which exhibits specificity for Hexim1 and Hexim2, respectively10. Treatment of live cells with a Cdk9 inhibitor mobilizes hnRNPs to bind to 7SK and consequently evict both Hexim and P-TEFb, not associated with each other, from 7SK within minutes11. Upon release from 7SK RNP, P-TEFb phosphorylates the C-terminal domain (CTD) of Pol II along with several other transcription factors (NELF, DSIF, PAF, SPT6), a process essential for Pol II to transition from promoter-proximal paused to productive elongation12,13. Beyond its host regulatory roles, 7SK RNP is also a critical target for HIV-1 replication, where Tat (Trans-activator of transcription) protein hijacks the 7SK RNP to the HIV-1 promoter and extracts P-TEFb into a viral super elongation complex via the nascent TAR (trans-activation response) RNA element14–17.
Hexim1 contains three main regions: (i) the N-terminal region (1-149), which is poorly conserved across vertebrates (Supplementary Figs. 1 and 2) and predicted to be intrinsically disordered (Supplementary Fig. 3), (ii) the central region (150-253), discussed below, and (iii) a C-terminal coiled-coil domain (CC hereafter, 254-359) that has been shown by NMR to form a stable homodimer18 and mediates Hexim1 binding to CycT119 (Fig. 1A). Within the central region only the Basic Region (BR) has been structurally characterized7,8. BR directly interacts with RNA and comprises two Arg/Lys-rich stretches separated by a conserved proline-serine and is often referred to as ARM (arginine-rich motif)7,20 due to its sequence similarity to the single ARM of HIV-1 Tat (Fig. 1B). Following the BR is a linker containing a conserved PYNT motif, known to bind Cdk97,21 and act in concert with the CC to inactivate P-TEFb9 (Fig. 1A). The linker is followed by an Acidic Region (AR). A prevailing model proposes that an internal electrostatic interaction between the BR and AR maintains Hexim in an autoinhibited state that cannot bind P-TEFb, rendering P-TEFb inhibition RNA-dependent9,22. However, the molecular mechanism by which this autoinhibition prevents PYNT–Cdk9 engagement in the absence of RNA remains unknown.
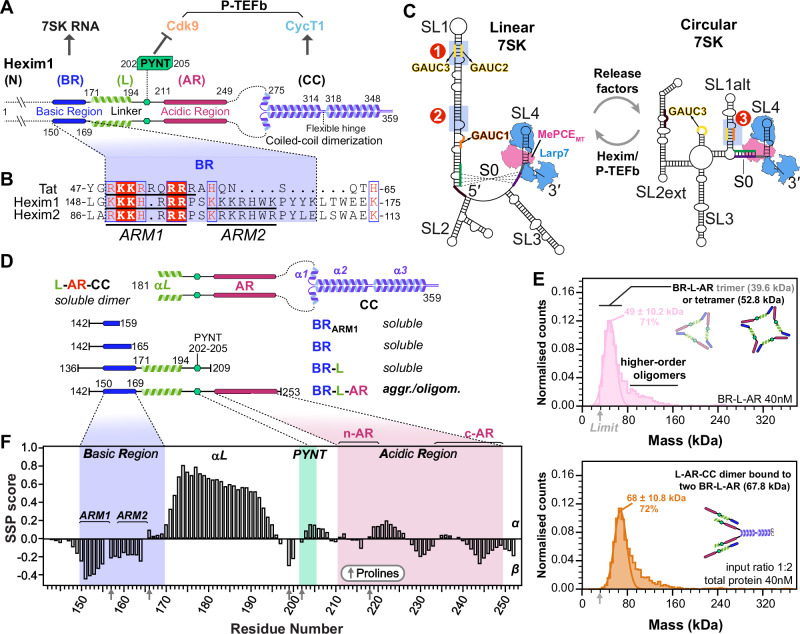
Hexim–RNA binding specificity and stoichiometry have long been subjects of debate. One study suggested Hexim1 acts as a general binder of double-stranded RNA23, whereas structural studies24–27 have focused on a specific motif—a GAUC palindrome—within stem-loop 1 (SL1) of 7SK RNA. This site (Site1 in Fig. 1C), encompassing GAUC2 and GAUC3 motifs (nts 42-45 and 64-67), is a conserved marker for 7SK genes28,29 and also serves as the RNA-binding site for HIV-1 Tat26,30. However, a biochemical study30 identified a second Hexim1 binding site in SL1 lacking a GAUC motif (Site2 in Fig. 1C). Lastly, another GAUC motif (GAUC1, nts 13-16) is present in proximal SL1 within one RNA helical turn of the binding/capping site for methylphosphate capping enzyme (MePCE)31.
The issues of Hexim binding specificity and stoichiometry are further complicated by evidence32,33, most recently from structural studies34 and RNA chemical probing by DANCE-MaP35, of at least two distinct 7SK RNA conformations in vitro and in cells: a linear form, where a long SL1 presents the Hexim binding sites discussed above, and a circular form, in which a shorter SL1alt forms a rearranged GAUC palindrome (GAUC1 and GAUC2; Site3 in Fig. 1C). Two core protein components, MePCE and Larp7, constitutively assemble with 7SK RNA36 for both linear and circular conformations, as shown by cryo-EM structures34 (Fig. 1C). Notably, switching between P-TEFb-bound and -free 7SK RNP pools, likely corresponding to the two RNA conformations, is required for stress-induced regulation of P-TEFb release34,35,37.
In this study, we used biophysical methods to characterize the central region of Hexim1, comprehensively map Hexim1 binding sites on 7SK RNA, and determine the binding stoichiometry. Paramagnetic relaxation enhancement (PRE) NMR measurements reveal that Hexim1 autoinhibition is governed by inter-monomer interaction, where the PYNT motif contacts both BR and AR regions. RNA binding to BR disrupts this interaction, unmasking the PYNT site. Complementary NMR and isothermal titration calorimetry (ITC) experiments further demonstrate that these inter-monomer interactions enhance binding specificity for linear 7SK over the circular conformation. This specificity is achieved through a cooperative recognition of dual high-affinity sites in SL1. Importantly, the sequestration of BR in the autoinhibited state weakens affinity for individual RNA sites, highlighting that both high-affinity sites are required to fully release Hexim1 autoinhibition. Together, these findings uncover an intricate network of intra-dimer interaction in Hexim1—driven by intrinsically disordered regions—that fine-tunes its RNA-binding behavior. Our results explain how RNA-mediated release of Hexim1 autoinhibition governs P-TEFb binding and inactivation, and how 7SK conformational switching regulates the partitioning of Hexim/P-TEFb in 7SK RNP.
Results
Intrinsically disordered Hexim1 BR-L-AR is prone to intermolecular interaction
To characterize the Hexim1 central region, we expressed and purified a monomeric construct (residues 142–253) encompassing BR, linker (L) containing PYNT motif, and AR (hereafter referred to as BR-L-AR). Several additionally truncated Hexim1 monomeric constructs, BRARM1, BR and BR-L (Fig. 1D), were used to compare with BR-L-AR. We observed a solubility limit of ~ 70 μM for BR-L-AR despite testing many conditions. In contrast, BRARM1, BR, and BR-L are easily soluble at ~ 0.5–0.8 mM under similar conditions. This suggests that BR-L-AR is prone to aggregation, mediated by intermolecular interactions that require both BR and AR. To detect the species formed via intermolecular interactions, we used native polyacrylamide gel electrophoresis (NativePAGE38) and mass photometry39,40 at ~ 4–25 μM and 40 nM, respectively. A clear ladder pattern of monomer and various oligomers was observed in NativePAGE for BR-L-AR, and mass photometry data captured trimer and tetramer species (Supplementary Fig. 4A–C and Fig. 1E). This indicates that the intermolecular interaction of BR-L-AR is stable enough to be observed even at 40 nM.
In contrast, freshly purified Hexim1 and L-AR-CC dimeric proteins both migrated as a single major band in NativePAGE, and mass photometry showed a homogeneous particle distribution corresponding to dimers (Fig. 1A, D and Supplementary Fig. 4A, D, E). However, when we combined BR-L-AR with either dimeric protein, we observed a stable intermolecular complex by mass photometry (Fig. 1E and Supplementary Fig. 4D, E). Together, these data suggest that in full-length Hexim1, the BR-L-AR from each monomer interacts with each other, consistent with the previous model that interaction between BR and AR maintains Hexim in an autoinhibited state9,22. These interactions are robust enough to prevent oligomerization of the dimeric Hexim1, but are readily competed by adding BR-L-AR monomeric protein in trans, suggesting relatively fast exchange dynamics, consistent with the nature of interactions formed by charged disordered protein regions (IDR)41.
To determine the extent of disorder and secondary structure in BR-L-AR, we used multi-dimensional heteronuclear NMR experiments on uniformly 15N,13C-labeled protein (see “Methods”). Despite the low BR-L-AR concentration (< 70 μM) we could achieve due to intermolecular interactions, we were able to assign 1HN, 15N, 1Hα, 13Cα, and 13Cβ chemical shifts and calculate secondary structure propensity (SSP) scores42 using Cα and Cβ chemical shifts (Fig. 1F and Supplementary Fig. 5A). The SSP scores, ranging from + 1 to − 1, reflect the fraction of α- or β- structure formed by each residue. From the SSP scores, we identified an α-helix (aa 171–194) in the linker region, between BR and PYNT motif (Fig. 1F). We also defined the two Arg/Lys-rich regions in BR as ARM1 and ARM2 (Fig. 1C): of these, ARM1 (residues 151–154) shows a moderate propensity to form β-structure, with a minimum SSP reaching − 0.45. The rest of BR-L-AR is largely disordered, with SSP scores within + 0.3 to − 0.3 (Fig. 1F).
In summary, BR-L-AR is largely intrinsically disordered, contains a short α-helix in the linker following the BR, and has a strong tendency to form intermolecular interactions with other monomers and with Hexim1 dimer.
Characterization of Hexim monomer–7SK RNA Site1 interaction
To look at the interaction of BR-L-AR with the previously characterized single 7SK RNA binding site (SL1 Site1 in Fig. 1C)24–27,43,44, we first investigated the structure of two designed RNA constructs from the distal end of linear 7SK SL1 (SL1-dI and SL1-dIΔU) using NMR (Fig. 2A, C). We assigned N-H…N imino (base-paired U/G) and non-exchangeable base and H1′ proton (all nucleotides) resonances for SL1-dI (Supplementary Figs. 6–8). Our analysis confirmed its complex architecture, which consists of a base-paired stem flanked by two dynamic U-rich bulges, consistent with previous structural studies24–27,43,44 (Fig. 2A). Those studies had variously reported that the two flanking U-rich bulges (U40U41 and U63) can form base triples with the A43-U66 and U44-A65 or G42-C67 base pairs, respectively; however, which of these base triples were observed varied with sample conditions including pH25,26,43,44. For SL1-dI, at 150 mM KCl and pH 6.2, we observed a stable U40…A43-U66 base-triple and a dynamic U63…A65-U44, where the U63 bulge samples at least two states, likely between stacking on G64 and Hoogsteen paring with A65 (Fig. 2A and Supplementary Fig. 6A, C). To investigate the relative dynamics of these nucleotides, the non-exchangeable resonance intensities from non-constant time 13C-1H HQSC spectra were acquired, and normalized 13C intensities were plotted, where higher normalized intensity generally reflects faster dynamics on the pico-to-nanosecond timescale45 (Fig. 2A). These measurements confirmed that U40 is stable (low intensities of 0.1 as helical stem) while U41, U63, and A65 are much more flexible (elevated intensities) consistent with our observation of one stable and one dynamic base triple (Fig. 2A). At pH 5.2, both these base triples were stably observed26.
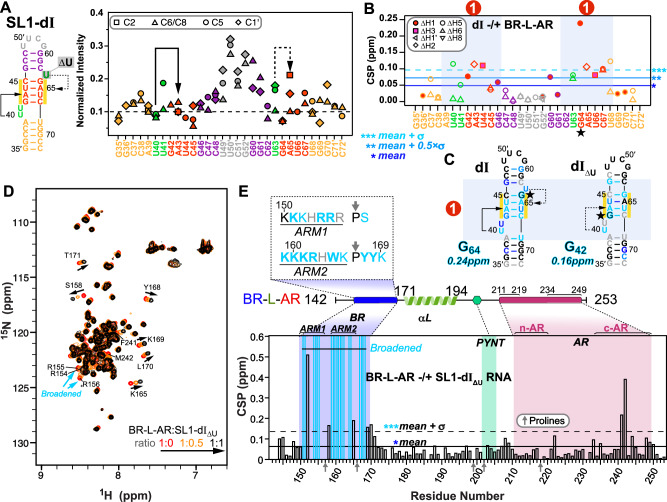
Next, we titrated BR-L-AR into the RNAs and monitored chemical shift perturbations (CSP). On the RNA side, for SL1-dI, the entire GAUC2-GAUC3 stem, U40U41 and U63 bulges are significantly shifted, with G64 affected the most (Fig. 2B, C and Supplementary Fig. 6). For SL1-dIΔU, although the same overall Site1 was used for binding, the distal half of GAUC2-GAUC3 stem exhibits smaller CSP (Fig. 2C and Supplementary Fig. 8), indicating a moderate contribution of U63 bulge (deleted in SL1-dIΔU) to Hexim1 binding, consistent with an EMSA binding assay44.
On the protein side, 15N-1H HSQC spectra of BR-L-AR titrated with either SL1-dIΔU or SL1-dI showed significant chemical shift changes and severe peak broadening for protein residues 151-170 spanning the entire BR (including ARM1, ARM2, and surrounding sequence) (Fig. 2D, E and Supplementary Fig. 9). Less line broadening is observed for BR-L-AR when SL1-dIΔU is added than for SL1-dI, indicative of a shift towards fast exchange on the NMR timescale likely due to a faster off-rate46. This allowed better tracing of the peak migration trajectories and observation of the BR residue S158 in the RNA-bound spectrum to confirm the binding site (Fig. 2D, E). Interestingly, in both cases, the C-terminal region of AR (c-AR) exhibited chemical shift changes at residues Phe241-Met242. To verify that the c-AR shift is indirect, we monitored NMR titration of SL1-dIΔU into the shorter BR-L or BR and found that it bound the same way as BR-L-AR (Supplementary Fig. 10A–E). These results indicate 7SK RNA binding to BR induces an allosteric change in c-AR.
In summary, 7SK SL1 Site1 features a stem with two flanking bulges that can form one stable and one dynamic U…A-U base-triple, and both base-triples are required for specific BR binding that engages the entire GAUC-palindrome. Hexim1 uses both ARM1 and ARM2 of BR for RNA binding, and this binding induces a conformational change in BR-L-AR. In contrast to Hexim, HIV-1 Tat protein has only ARM1 that is necessary and sufficient for binding Site1 (Fig. 1C)25,26,30, indicating a significant difference in Hexim and Tat recognition of 7SK RNA.
Characterization of Hexim1 autoinhibited conformation by PRE
To characterize the autoinhibitory interactions within the BR-L-AR region, we used Paramagnetic Relaxation Enhancement (PRE)47 NMR. This method can map weak and transient interactions48,49 common in IDRs50 by measuring distance-dependent line broadening from a covalently attached MTSL nitroxide spin label up to 25 Å away. A key challenge is distinguishing intra-monomer interactions from inter-monomer interactions. To dissect these, we established two distinct experimental setups using three individual cysteine substitution sites (S226C, S233C, S237C): (i) “total interaction” experiment: 15N-labeled and MTSL-tagged BR-L-AR, capturing both intra- and inter-monomer contacts (Fig. 3A, B and Supplementary Fig. 11); and (ii) “inter-only” experiment: 1:1 mixture of the reporter 15N-labeled, untagged BR-L-AR with the broadener 14N-labeled and MTSL-tagged BR-L-AR (Fig. 3C–E). In the latter, only inter-monomer interactions will result in line-broadening, however to a lesser extent than the “total” experiments, since only 50% of the “dimer” population is the combination of 15N and 14N-MTSL labeled proteins. To visualize and identify interactions between structural segments, we defined regions of primary broadening (red) and secondary broadening (orange) as stretches harboring 3 or more residues (either contiguous or non-contiguous) with intensity ratios lower than [mean – σ] and lower than [mean – 0.5xσ], respectively (Fig. 3B–E).
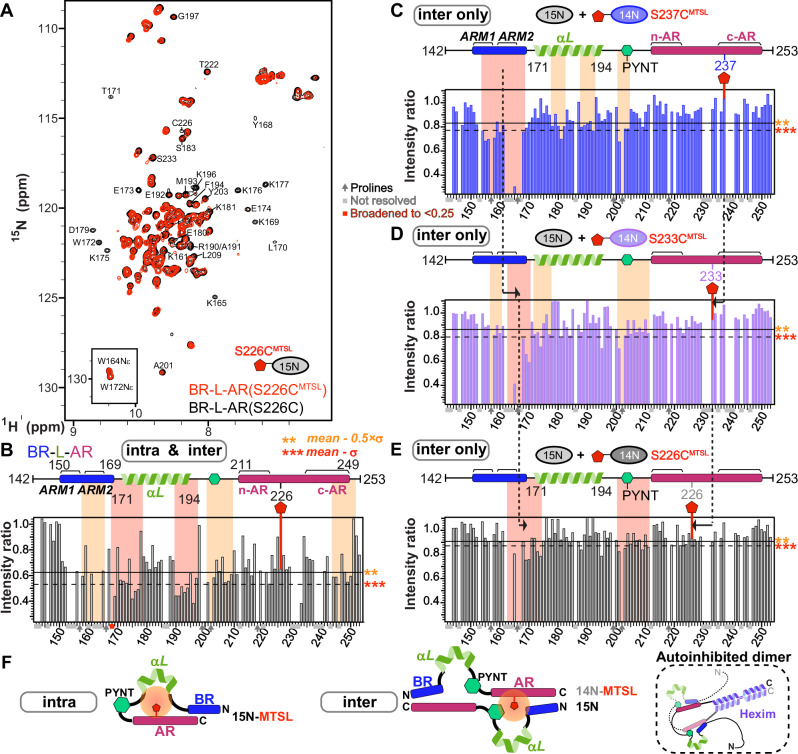
In the inter-only experiments, we observed a distinct pattern: S237CMTSL primarily broadens the entire BR (154-168), with ARM2 broadened more severely than ARM1 (Fig. 3C); S233CMTSL broadens ARM2-PYYK (Fig. 3D); and S226CMTSL broadens ARM2-PYYK, beginning of αL, and PYNT motif (Fig. 3E). This pattern indicates an overall head-to-tail orientation for BR-L-AR inter-monomer interaction (Fig. 3F), where the primary broadened region (red) shifts in the N- to C-terminal direction when we move the MTSL-tag from the C- to N-terminal direction (dashed lines in Fig. 3C–E).
We then analyzed the “total interaction” experiment by comparing this “total” profile to the “inter-only” S226C profile (compare Figs. 3B–E) to deconvolve the two interaction types. The secondary broadening (orange) observed in the “total” experiment at ARM2-PYYK and PYNT motif matched the “inter-only” experiment, confirming this as the inter-monomer component. Therefore, the remaining primary broadening, which was observed at both the beginning and the end of the αL, represents the intra-monomer component (Fig. 3B, F). This indicates that helix αL is bent or kinked, allowing its two ends to approach S226C. In the S237CMTSL “total” experiment, almost all the resonances are broadened more severely than S226CMTSL (Supplementary Fig. 11), indicating a significant role of c-AR in the autoinhibition, consistent with Phe241-Met242 exhibiting allosteric CSP upon RNA binding (Fig. 2E).
In summary, PRE NMR experiments distinguished and mapped intra- and inter-monomer interactions in Hexim1 BR-L-AR. Based on these results, the inter-monomer interaction adopts a head-to-tail orientation (N→C to C→N), which in full-length Hexim1 would correspond to a self-hugging topology between the two monomers (Fig. 3F).
PYNT motif is sequestered by autoinhibition and is unmasked upon RNA binding
The PYNT motif (conserved residues 202PYNTTQFLM210) plays a critical role in directly binding and inactivating the Cdk9 kinase subunit of P-TEFb7,21. We further investigated the PYNT interactions in BR-L-AR using an A201CMTSL substitution which positions the MTSL-tag immediately N-terminal to the P202YNT motif (Fig. 4A). The PRE profile of 15N BR-L-AR(A201CMTSL) showed significant broadening of the PYNT motif itself, BR and c-AR (Fig. 4A and Supplementary Fig. 12). This result recapitulated the findings from BR-L-AR(S226CMTSL) (Fig. 3B orange) and confirms that the PYNT motif is a central node in the BR–PYNT–AR inter-monomer network. Based on data suggesting that αL is involved (Fig. 3C), we next probed from its central region using a S183CMTSL tag (Supplementary Fig. 13). The PRE profile of 15N BR-L-AR(S183CMTSL) showed significant broadening extending from the αL through the PYNT motif excluding the C-terminal end of αL, which along with BR and the two ends of AR shows secondary broadening (Fig. 4B). This indicates that the inter-monomer BR–PYNT–AR interactions partially involve the central region of αL.
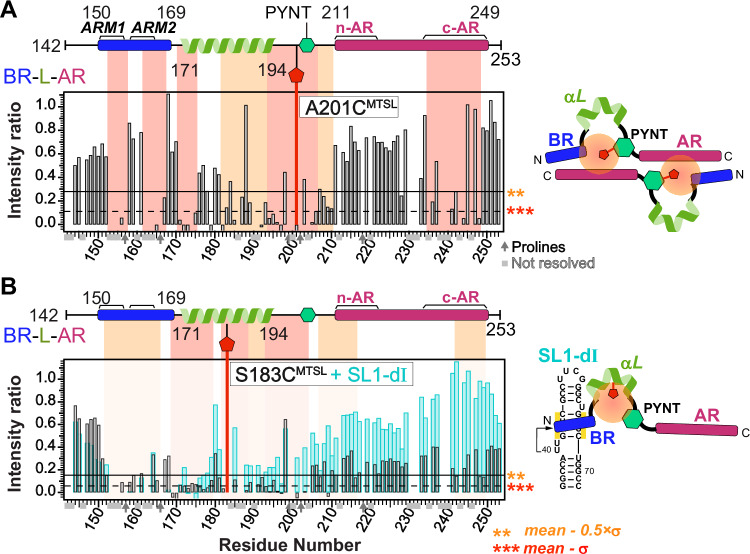
Lastly, we investigated the effect of 7SK RNA binding on the inter-monomer interactions. Addition of SL1-dI to 15N BR-L-AR(S183CMTSL) resulted in significant peak intensity recovery across the entire protein, except for BR (Fig. 4B, cyan bars). This recovery signifies that the PRE-induced broadening has disappeared, meaning the inter-monomer network has been disrupted and the S183CMTSL tag is no longer in proximity to the rest of the protein. Note that the lack of intensity recovery for the BR is expected. As shown previously (Supplementary Fig. 9), BR residues are independently and severely broadened by the direct binding of SL1-dI RNA, masking any PRE-related effects. Taken together, the PRE data show that the PYNT motif directly participates in inter-monomer interactions, where it simultaneously contacts both BR and AR. This autoinhibitory sequestration is released upon BR binding to RNA, resulting in the unmasking of PYNT, making it available to bind and inactivate Cdk9 in the 7SK RNP.
Linear 7SK SL1 contains two high-affinity BR binding sites
While binding of Hexim1 BR to SL1 Site1 has been well documented, other potential binding sites have been proposed but not characterized24–26,30,43,44. To resolve the outstanding questions of Hexim1–7SK RNA binding site location(s), stoichiometry, and specificity, we systematically investigated the full 7SK SL1 domain in linear versus circular conformations. Linear 7SK SL1 folds into a minimum 36-bp long stem containing eight internal bulges or loops (Fig. 5A). We employed a “divide-and-conquer” strategy, designing a series of RNA stem-loop segments from the distal (d), middle (m) and proximal (p) regions of linear SL1, as well as circular SL1alt. All constructs include a minimum 4-bp stem with one flanking bulge, based on the Hexim1 binding requirements for Site1 described above. We then used NMR chemical shift perturbations (CSPs) to map binding sites (Fig. 5) and EMSA to determine stoichiometry (Supplementary Figs. 14, 15). For NMR mapping, BRARM1 was used in order to reduce the line broadening in the longer RNA constructs (SL1-d, dIIm, p and mp) when bound to protein. We verified that the same nucleotides in Site1 were perturbed for BRARM1 as for BR-L-AR (Fig. 5 and Supplementary Fig. 17D). NMR assignments, CSP plots and 1D spectra of titration series for the additional RNA constructs studied (Supplementary Figs. 16–22) were acquired as for Site1. Using the RNA CSPs (Fig. 5A–D), we mapped binding Sites 1, 2, 4 and 5 in linear SL1 and Site3 in circular SL1alt. The non-specific site at SL1-pII exhibits minimum CSP upon binding BRARM1, providing a negative control. The binding sites are numbered based on the relative order of binding affinities observed by NMR, where Site1 (not Site4) shifted by adding 1:1 BRARM1 to SL1-d, Site2 (not Site4) shifted in SL1-dIIm, and Site2 (not Site5) shifted in SL1-mp (Fig. 5A); and by EMSA experiments, where two sequential shifts were observed for SL1-dΔU and SL1-d, corresponding to the high-affinity Site1 followed by low-affinity Site4 (Supplementary Fig. 14D, E and Notes). The largest CSP values and the exchange behaviors on the NMR timescale also agreed well with the order of binding affinities determined from combined NMR and EMSA analysis (Fig. 5A stars and Supplementary Fig. 17C).
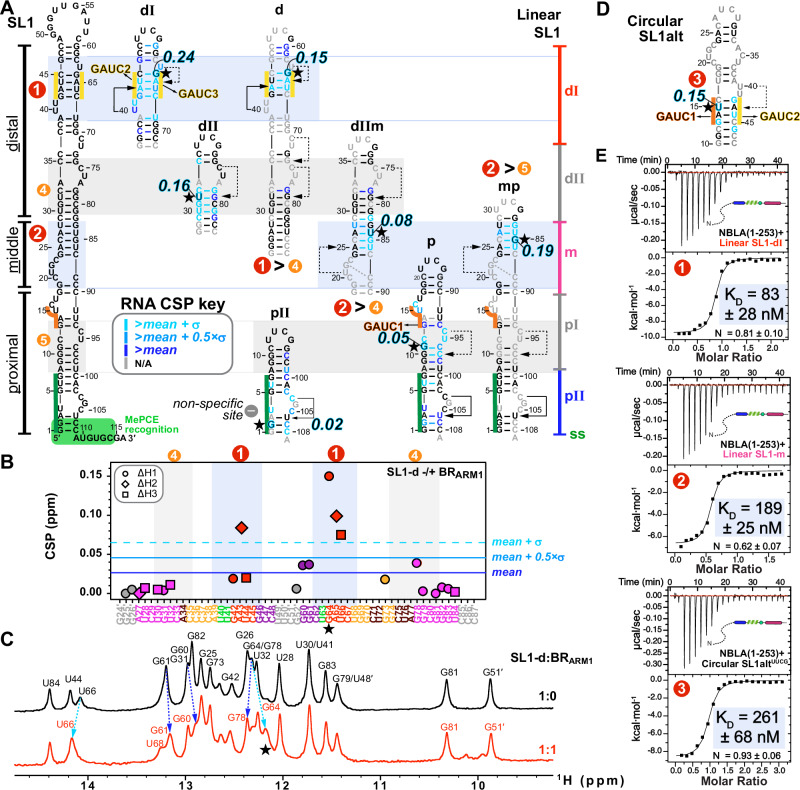
To quantitatively compare the binding affinities of these RNAs, we acquired thermodynamic binding parameters by ITC (Figs. 5E, 6). Due to sample precipitation that occurs during ITC titration of RNA into BR-L-AR, potentially due to stirring, we instead used N-BR-L-AR (NBLA hereafter) that includes the N-terminal region. ITC measurements yielded KD values of 83, 189, and 261 nM for Sites 1, 2 and 3, respectively. Together, these findings indicate that linear 7SK SL1 contains two high-affinity sites and two low-affinity sites for BR. In contrast, circular 7SK SL1alt contains only one binding site for one BR, at the alternative GAUC1-GAUC2 palindrome (Fig. 5D). Comparison of these 5 binding sites indicates that the determining factor for high-affinity Hexim1 binding appears to be an A-U rich stem with at least one flanking 5′ U-containing bulge that participates in a U…A-U base triple, a common feature for Sites 1 and 2, but not for Sites 4 and 5, or the non-specific site SL1-pII. Site3 can form a U40…A43-U15 base triple, but the U40U41 bulge is now on the 3′ side of the GAUC1-GAUC2 stem instead of on the 5′ side in Site1, geometrically analogous to the U63 bulge of Site1, consistent with a lower affinity than Sites 1 and 2.
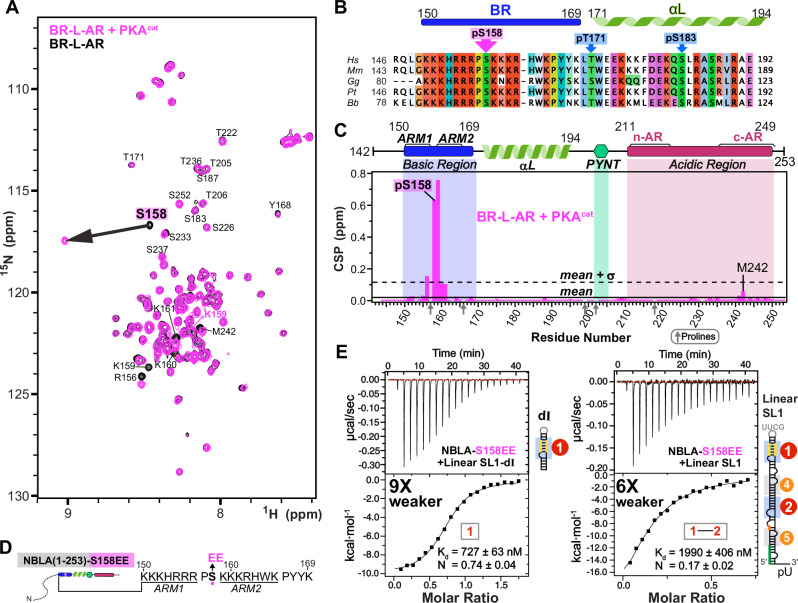
S158 phosphorylation minimally affects autoinhibition and S158EE impairs RNA binding
Multiple phosphorylation sites have been identified in Hexim1 unstructured regions and suggested to play roles in releasing P-TEFb from the 7SK RNP complex51–54, including phosphorylation of BR residue Ser158 (pSer158) by protein kinase C (PKC)55 or protein kinase A (PKA)56. To investigate the effect of pSer158 on Hexim1 conformation, we phosphorylated BR-L-AR in vitro using PKA catalytic subunit (PKAcat) and monitored phosphorylation by NMR 1H-15N HSQC (Fig. 6A, B). A large shift of S158 amide resonance was observed due to pS158, with induced shifts at adjacent lysines 159–161 and R156 (P157 lacks an amide) (Fig. 6A, C). Of note, c-AR residues F241 and M242 also show small chemical shift changes, although to a much smaller extent (CSP ~ 10-fold smaller than in Fig. 2C). This suggests that pS158 only minimally induces the same allosteric change in c-AR as RNA binding does, largely maintaining autoinhibition.
Longer incubation with PKA resulted in phosphorylation of additional sites (T171 and S183) and destabilization of the entire αL (Supplementary Fig. 23A). We note that αL does contribute to RNA binding affinity, as shown by our EMSA (compare Supplementary Fig. 14C to 14A). To avoid potential destabilization of αL and assure a homogenous sample, as well as to avoid a predicted pS49 by PKA (Supplementary Fig. 23B), we made a phosphomimetic variant NBLA-S158EE to look at the effect of two additional negative charges in BR on RNA binding by ITC, in the context of the longer soluble monomeric construct. An increase in KD of 9- and 6-fold was observed for NBLA-S158EE with SL1-dI and full-length linear SL1, respectively, compared to wild-type NBLA (Fig. 6D, E), indicating that S158EE significantly reduces RNA binding. Together, these observations suggest that in vivo S158 phosphorylation would reduce RNA binding without significantly disrupting Hexim autoinhibition, thereby triggering release of P-TEFb as a net result.
Both monomers in Hexim1 dimer engage in high-affinity RNA binding simultaneously
Since full-length Hexim1 is a stable homodimer, the above results with Hexim1 monomeric constructs suggest that a dimer could simultaneously bind two sites on linear SL1, one for each monomer. Indeed, EMSAs using full-length SL1 or the minimum functional 7SK (SL1-SL4, that has been shown to assemble in cells with MePCE–Larp7–Hexim–P-TEFb57) show that full-length Hexim1 dimer binds first to Site1 and Site2. At higher Hexim1:RNA ratios, another dimer subsequently binds to Sites4 and Site5 (Supplementary Fig. 14F,G). These Site4 and Site5 are likely not primary binding sites for Hexim1 in cells, rather they may serve as docking sites during the Hexim1 searching process for Sites 1 and 2.
We next used the same short SL1 constructs described above versus full-length SL1, titrated into Hexim1 NBLA monomer versus full-length Hexim1 dimer, to determine binding stoichiometry by ITC (Fig. 7 and Supplementary Fig. 24). Systematic comparison of the N values shows that the monomeric NBLA binds as one molecule per RNA to individual Sites 1, 2, and 3 (N = 0.62–0.93) and binds as two molecules per full-length linear SL1 (N = 0.36) (Fig. 7A, B). Full-length Hexim1 (dimer) also binds as one monomer per individual Sites 1, 2, and 3 (N = 0.68–0.79, using monomer concentrations when fitting titration curves), and binds as two monomers, i.e., one dimer, per linear SL1 (N = 0.39) (Fig. 7A, C). We further verified the stoichiometry by reversing the titration order (i.e., titrated Hexim1 from the syringe to RNA in the cell), and again observed one monomer per single-site RNAs (N = 0.87–1.12) and two monomers (one dimer) for linear SL1 (N = 1.75). Together with the NMR and EMSA results, we conclude that both monomers within Hexim1 dimer are engaged in SL1 RNA binding, to Sites 1 and 2, simultaneously (Fig. 7C).
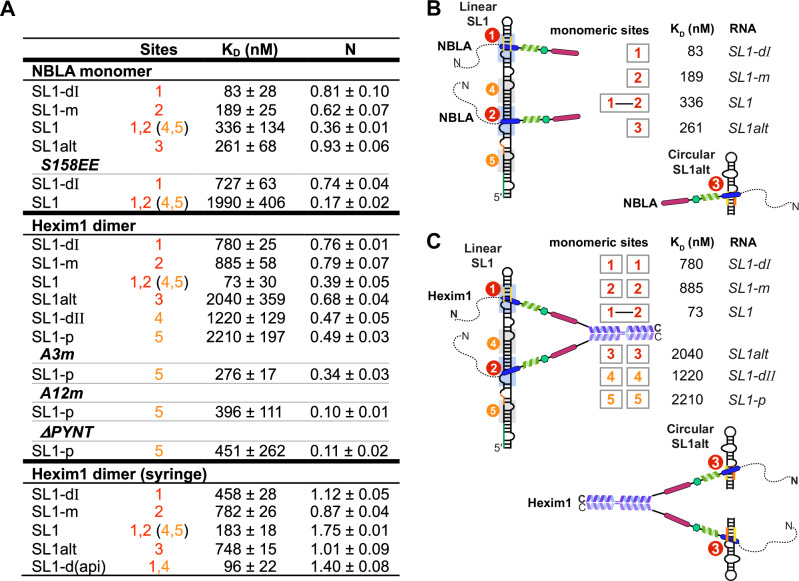
Hexim1 autoinhibition renders specificity for linear 7SK SL1
After mapping the individual RNA binding sites with their relative order of affinities and determining the stoichiometry of Hexim1 dimer binding with SL1, we next compared the ITC-determined binding affinities between NBLA and Hexim1 dimer, where inter-monomer autoinhibition is enforced by dimerization in the full-length Hexim1 but not in the monomer, to quantitatively determine the effect of Hexim1 autoinhibition on RNA binding. Comparison of the dissociation constants KD shows that the Hexim1 dimer binds 10-, 5- and 8-fold weaker than the monomer (NBLA) to the individual Site1, Site2, and Site3, respectively, suggesting that autoinhibition of Hexim1 dimer weakens binding to single sites (Fig. 7). However, Hexim1 dimer binding to linear SL1 shows cooperative binding with a > 10-fold lower KD (73 nM) than the single Site1 and Site2 (780 and 885 nM). In contrast, no cooperativity was observed for NBLA monomer, with a KD value for SL1 (336 nM) slightly larger than those of Site1 and Site2 (83 and 189 nM). This cooperative effect that is exclusively observed for the dimer is recapitulated in experiments where the titration order is reversed, albeit with a smaller effect likely due to increased aggregation when Hexim1 is concentrated in the syringe (Fig. 7A bottom). These results indicate that the autoinhibition of Hexim1 dimer is required for the specificity towards full-length linear SL1 over single RNA sites, whereas NBLA monomer lacks this specificity.
The weaker single sites SL1-dII (Site4) and SL1-p (Site5) exhibited larger KD values (1221 and 2208 nM, respectively) than Site1 or Site2 for Hexim1 dimer. Interestingly, the N values for these weaker binding sites are slightly smaller (0.47 and 0.49), indicating a partial release of autoinhibition such that only one monomer is available for binding within a subpopulation of the dimer. In contrast, SL1alt (Site3) in circular 7SK conformation has weak binding affinity to Hexim1 dimer (2035 nM), albeit with a similar N value (0.68) to Sites 1 and 2 (0.76 and 0.79). This Hexim1 dimer–SL1alt affinity is unexpectedly much lower than the NBLA monomer–SL1alt, suggesting significantly different binding kinetics for dimer–Site3 from monomer–Site3. Consistent with these results, a SL1-d(api)27 (identical to SL1-d except for the apical loop sequence) has an enhanced binding affinity (96 nM) but a smaller N value (1.40) than SL1, indicating two binding events overall (Sites 1,4 vs 1,2) but with a moderately reduced release of Hexim1 autoinhibition.
Finally, to further test the role of Hexim1 autoinhibition in RNA-binding specificity, we disrupted the autoinhibition with three Hexim1 dimer variants, A3m and A12m, that neutralized negatively charged residues in c-AR and n-AR with alanines7, and ΔPYNT, and titrated them with the low-affinity binder SL1-p (Site5) monitored by ITC (Fig. 7 and Supplementary Fig. 25). These three variants showed 8-, 6- and 5-fold tighter binding affinities than wild-type Hexim1, respectively, suggesting that disruption of autoinhibition turned Hexim1 into a non-specific RNA binder.
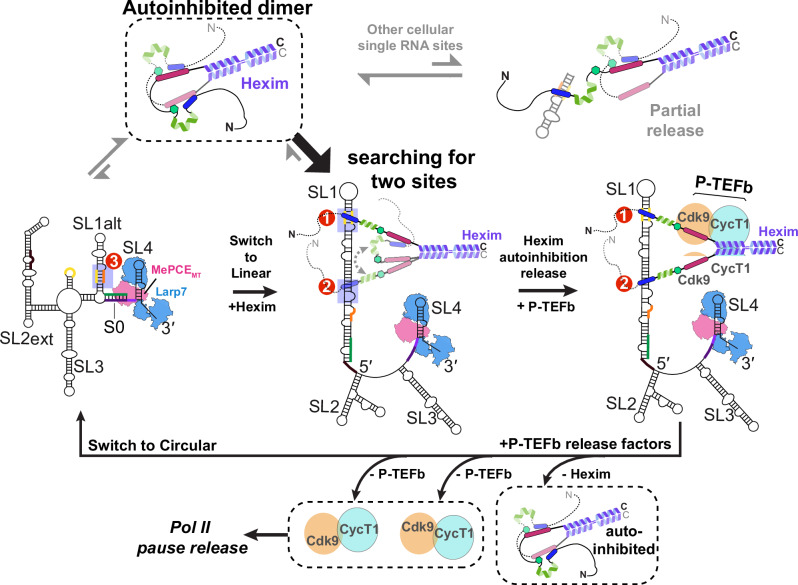
Together, these results show that Hexim1 dimer preferentially and cooperatively binds Sites 1 and 2 in linear SL1 with each monomer, respectively, and the inter-monomer autoinhibition within the dimer renders the specificity for linear SL1 over isolated RNA binding sites, including the circular SL1alt. This is achieved through two sides of the same coin: cooperative binding to the dual high-affinity sites in SL1 and decreased affinity for RNAs with only a single binding site. Together with the PRE results, we conclude that Hexim1 binding to two sites in linear SL1 provides an effective allosteric switch to fully release Hexim1 autoinhibition in the dimer, which subsequently exposes two PYNT motifs for Cdk9 binding and inactivation from two P-TEFb heterodimers (Fig. 8).
Discussion
RNA-binding intrinsically disordered protein regions (IDRs) are increasingly recognized as important and versatile regulators of RNA metabolism, RNP phase separation and cellular signaling, yet their mechanistic roles remain elusive compared to their well-structured counterparts58,59. Recent advances have also uncovered that such IDRs can modulate the RNA- and DNA-binding of an adjacent folded domain60,61. Our studies shed light on this growing paradigm by dissecting how the Hexim1 disordered BR-L-AR enables finely tuned interaction with the conformationally dynamic non-coding RNA, 7SK.
We show that Hexim1 employs a stand-alone disordered basic region (BR) to selectively recognize A-U-rich RNA stems with flanking U-rich bulge(s), whereas the disordered acidic region (AR) serves as a specificity checkpoint. In the absence of RNA, the Hexim1 central region adopts a conformational ensemble wherein the key regulatory PYNT motif, required for Cdk9 binding and inactivation, becomes masked through a BR–PYNT–AR interaction network—specifically in a head-to-tail inter-monomer configuration (Fig. 8). Strikingly, 7SK RNA binding to the BR disrupts this autoinhibition, effectively unmasking PYNT and enabling subsequent interaction with Cdk9. Using a monomeric BR-L-AR construct, we demonstrate that linear 7SK SL1 harbors two high-affinity and two low-affinity binding sites (Sites 1,2 versus 4,5). Notably, Hexim1 dimer binds linear SL1 with a 1-to-1 stoichiometry, with each monomer BR engaged with a high-affinity site. These findings, together with previous studies9,22, support a model in which Hexim1 CC dimerization enables BR-L-AR mediated inter-monomer dual autoinhibition (Fig. 8): BR sequestration restricts RNA binding, while PYNT masking prevents premature Cdk9 interaction, and both are reversed upon specific RNA recognition of two high-affinity sites.
The regulatory role of AR is thus two-fold towards RNA binding: (1) autoinhibition enhances specificity towards linear 7SK SL1 over its circular SL1alt; and (2) autoinhibition prevents Hexim1 from being trapped by non-specific binding to RNAs, accelerating the search for functional 7SK targets—an RNA-analog to the “facilitated search” role recently described in disordered D/E patches for DNA-binding protein62. In light of this particular feature of Hexim1, our study illustrates the importance of considering Hexim1 dimer interaction with full-length linear SL1 and circular SL1alt, in contrast to previous foundational studies that primarily focused on a single site (Site1)25–27,44. Our results provide a more complete picture for 7SK RNA engagement, highlighting how 7SK conformational dynamics influence the regulatory architecture of 7SK RNP. We show that Hexim1 dimer binds circular 7SK SL1alt, which has a single binding site, with micromolar affinity, but binds linear 7SK SL1 with nanomolar affinity (73 nM; Fig. 7C), reinforcing RNA conformational switching as a central P-TEFb release mechanism35.
Building on our comprehensive binding studies and prior structural studies of MePCE–7SK–Larp7 core RNP31,34,63, we propose a refined stoichiometry model for the P-TEFb-inhibitory 7SK RNP: one linear 7SK RNA, one MePCE, one Larp7, one Hexim dimer bound to two RNA sites, and two P-TEFb heterodimers64 (Fig. 8). The ability of Hexim1 to simultaneously inactivate two P-TEFb complexes suggests a signal-amplifying module, where two P-TEFb units can be activated per unit cellular signaling event. This model also implies a cooperative assembly mechanism, in which 7SK RNA, Hexim and P-TEFb mutually stabilize each other in 7SK RNP, contributing to a fast and efficient on- and off-switch during the Pol II pause release and the proposed transcription termination via 7SK65, respectively.
For protein-centric P-TEFb release pathways, we show here that S158 phosphorylation acts as an RNA-binding off-switch, while maintaining Hexim1 autoinhibition, thereby releasing P-TEFb. These results highlight the need for further investigations of Hexim posttranslational modifications in both RNA binding and autoinhibition. Comparison of Hexim1 with its paralog Hexim2 shows that Hexim2 has a much shorter AR, which lacks the c-AR found in Hexim1 (Supplementary Fig. 2). Our data suggest that c-AR plays a critical role in the BR–PYNT–AR interaction network. Thus, Hexim2 may exhibit reduced autoinhibition and altered P-TEFb regulatory dynamics, particularly in tissues where Hexim2 is preferentially expressed.
In addition to linear-to-circular 7SK conformational switching, another RNA-centric pathway for regulated P-TEFb release was recently shown, i.e., N6-methylationadenosine (m6A) modifications of 7SK RNA by METTL3-METTL14 alters Hexim binding. Specifically, m6A43 and m6A65 in GAUC2 and GAUC3 motifs, which are more prominent in cancer cells66,67, likely disrupt Site1 base-triples essential for Hexim binding44. This would diminish Site1 affinity and effectively convert linear SL1 into a one-site binder (Site2), reducing Hexim1 dimer binding and promoting P-TEFb release. A similar strategy is adopted by HIV-116,68, where Tat protein only binds Site1 but not Site230, and lacks an analogous acidic patch that could inhibit its single ARM (Supplementary Fig. 26). However, details of the molecular mechanism of Hexim–Tat competition to form the intermediate Tat–7SK RNP at HIV-1 promoter remain elusive14,69. Notably, past studies of Tat–Hexim competition focused only on Site126,27,43; our results suggest that inclusion of Site2, along with consideration of the autoinhibitory state of Hexim dimer, is crucial for understanding the molecular basis of this competition.
In summary, this work resolves three major longstanding questions about Hexim function in 7SK RNP: how autoinhibition enforces Hexim1 specificity towards linear 7SK RNA; the precise stoichiometry and architecture of Hexim–7SK binding and the minimal components of the P-TEFb-inhibitory 7SK RNP; and the mechanism by which Hexim1–7SK binding unmasks the PYNT motif, enabling Cdk9 sequestration and inactivation in the 7SK RNP. These insights provide a mechanistic foundation for understanding the regulation of P-TEFb binding and release by 7SK RNP, which is fundamental to its role in eukaryotic transcription. They also open previously unexplored avenues for exploring how various P-TEFb release factors and HIV-1 Tat could interact with and/or remodel 7SK RNA conformational dynamics.
Methods
Plasmids and mutagenesis
Full-length human Hexim1 protein (aa 1–359) and monomeric Hexim1 BR-L-AR (aa 142–253) were cloned into pETDuet-1 vector with an N-terminal His6-tagged maltose binding protein (MBP) fusion, followed by Tobacco Etch Virus (TEV) protease cleavage site between MBP and Hexim1. Further truncated monomeric Hexim1 constructs, BR-L (aa 136–209) and BRext (aa 149–179) were cloned into pETDuet-1 vector with N-terminal His6-tagged 56-residue B1 domain of Streptococcal protein G (GB1) fusion70 followed by Tobacco Etch Virus (TEV) protease cleavage site between GB1 and Hexim1 monomers. BR (aa 141–165) and BRARM1 (aa 141–159) constructs were cloned by site-directed mutagenesis using the pETDuet-1 GB1-ts-BR-L plasmid as template. A non-native glycine remains at the N-terminus of the final full-length Hexim1 protein, BR-L-AR, BR-L, BR and BRARM1 after TEV protease cleavage, whereas a native Glycine residue from BRext (aa 149) was a part of the TEV cleavage site (ts). Cysteine substitutions of BR-L-AR variants for PRE (S183C, A201C, S226C, S233C and S237C) were cloned by site-directed mutagenesis using the pETDuet1 MBP-ts-BR-L-AR as template.
Minimum linear 7SK (SL1-SL4ms2) and linear SL1 RNA genes were cloned into the pUC19 vector with a Hammerhead ribozyme gene at the 3′ end followed by a BamHI restriction enzyme cleavage sequence. All site-directed mutagenesis was performed using the Q5 kit (New England Biolabs).
Protein expression and purification
Hexim1 protein genes in either pETDuet-MBP or pETDuet-GB1 vectors were transformed into BL21-GOLD(DE3) competent cells for protein expression. Bacterial culture was grown at 37 °C in minimum media to O.D.600 of 0.6, then transferred to 18 °C for 1 hr prior to induction by 0.5 mM IPTG for 18–20 hrs. For uniformly [15N, 13C] (or [15N]) enriched Hexim1 proteins, M9 minimal media containing 1 g/L of 15NH4Cl and 3 g/L of [13C-6]-D-glucose (or natural abundance glucose) (Cambridge Isotope Laboratories) were used. Cells were pelleted by centrifugation, resuspended in lysis buffer (20 mM Tris, pH 7.5, 1 M NaCl, 5 mM imidazole and 1 mM PMSF) supplemented with protease inhibitor cocktail tablet (ThermoScientific Pierce A32965) and lysozyme. Cells were lysed by sonication on ice and clarified by centrifugation and filtration with 0.45 μm syringe filter. Lysates were loaded onto a 5-mL Ni Sepharose affinity column (HisTrap-HP; GE Healthcare), and a linear gradient of 5–500 mM imidazole was used to elute the His6-tagged proteins. The eluted protein was added 1 mg of TEV protease, dialyzed (20 mM Tris, pH 7.5, 300 mM NaCl, 5 mM βME) and further purified with a second Ni affinity column to removed His6-tagged fusion proteins (MBP or GB1) and TEV protease. For the full-length Hexim1 protein, an additional purification step was carried out with size-exclusion chromatography (SEC; HiLoad 26/600 Superdex 200 or HiLoad 16/600 Superdex 75; GE Healthcare) in binding buffer (20 mM HEPES, pH 7.5, 150 mM KCl, 1 mM TCEP). Proteins were concentrated using Amicon devices (Millipore), and concentration was measured by absorbance at 280 nm. For monomeric Hexim1 BR-L-AR, concentrating and buffer exchanging using Amicon devices into a final buffer (50 mM sodium phosphate, pH 6.2, 0/150/300 mM KCl, 1 mM TCEP, 0.02% NaN3, 5% D2O added post buffer exchange for NMR) was performed with caution to prevent precipitation at higher local protein concentration. For even shorter monomeric proteins, 1 kDa MWCO dialysis tubing (for BRext) or desalting column (HiTrap Desalting 5 mL, GE Healthcare; for BR and BRARM1) were used for buffer exchange into 5 mM NH4HCO3 buffer, freeze dried with a lyophilizer and dissolved directly in NMR buffer (50 mM sodium phosphate, pH 6.2, 150 mM KCl, 0.02% NaN3, 5% D2O).
In vitro phosphorylation by PKA catalytic subunit
Phosphorylation of Hexim1 BR-L-AR was performed utilizing cAMP-dependent protein kinase A catalytic subunit (PKAcat; NEB P6000S). The protein was first buffer-exchanged into NMR buffer (50 mM sodium phosphate, pH 6.2, 150 mM KCl, 0.02% NaN3, 5% D2O), then concentrated with constant mixing using Amicon devices (Millipore). The final phosphorylation reaction (500 µL total volume) contained 35 µM BR-L-AR, 2 mM ATP, 10 mM MgCl2, I mM EDTA, 2 mM DTT, and 4 µL PKA (10,000 units). Two separate reactions were incubated in a water bath at 37 °C for 3 and 24 h, respectively, before being analyzed using NMR.
NativePAGE and mass photometry characterization of Hexim1 constructs
Prior to loading, freshly purified protein samples of full-length Hexim1, L-AR-CC, and BR-L-AR were centrifuged at 14,000 × g for 10 minutes at 4 °C to remove potential contaminants and aggregates. Samples were prepared with NativePAGETM Sample Buffer (4X) (Invitrogen BN2003). For full-length Hexim1 and L-AR-CC, 5 µg was loaded per lane. For BR-L-AR, 2.5 µg and 5 µg were loaded on separate lanes. Samples were run on NativePAGETM 3–12% Bis-Tris Mini Gels (InvitrogenTM BN1003BOX) at room temperature at 150 V using a cathode buffer supplemented with Coomassie Blue G-250 at a final concentration of 0.02% (InvitrogenTM BN2007).
Mass photometry measurements of full-length Hexim1, L-AR-CC, and BR-L-AR were performed on a Refeyn TwoMP mass photometer. Freshly purified protein samples were prepared as 4 µM for monomers and 2 µM for dimers, diluted (or mixed as complexes and diluted) with SEC buffer to 400 nM stocks, and then added by the drop-dilution method into phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) to achieve final total protein concentrations of 40 nM. Interferograms were recorded for 60 seconds with Refeyn AcquireMP, and data were analyzed with Refeyn DiscoverMP using three molecular weight standards for calibration: Bovine Serum Albumin (BSA, ThermoFisher 23209) 66 kDa/132 kDa, beta-amylase (BAM, Sigma A8781) 112 kDa/224 kDa and Bovine Thyroglobulin (TG, Sigma 609310) 670 kDa (R2 = 1.000, Max. mass error 3.0%). The percentages associated with each peak fitting are derived from the DiscoverMP analysis, which calculates the proportion of total particle counts represented by the Gaussian-fitted peaks.
Nitroxide spin labeling of proteins
Protein modification with nitroxide spin label was carried out using the established protocol71. Briefly, each purified protein solution, containing about 50 nmole of Hexim1 BR-L-AR cysteine variants, was buffer exchanged using a PD-10 desalting column with the gravity protocol to remove the reducing agent into PRE buffer (50 mM sodium phosphate, pH 6.2, 150 mM KCl, 0.02% NaN3). The PD-10 elution was collected into a falcon tube, containing 10-fold molar excess of S-(1-oxyl-2,2,5,5,-tetramethyl-2,5,-dihydro-1H-pyrrol-3-yl) methylmethane-sulfonothiolate (MTSL; Toronto Research Chemicals TRC-O875000) dissolved in 0.5 mL of PRE-buffer, covered with aluminum foil to avoid light. The reaction was kept at room temperature under gentle stirring in the dark for > 15 min. Subsequently, an additional aliquot of MTSL was added to the reaction mixture to reach a final ratio of 1:20 protein:MTSL. The reaction was further incubated at room temperature under gentle stirring overnight in the dark.
The unreacted MTSL molecule was removed by running through a PD-10 desalting column equilibrated in PRE buffer twice using the gravity protocol. The MTSL-tagged protein was concentrated with caution using Amicon devices, and 5% v/v D2O was added to prepare the final NMR sample.
In vitro transcription of RNA
RNA samples were prepared by in vitro transcription with T7 RNA polymerase P266L variant34,72 and are described briefly below. Short RNA constructs, including linear 7SK SL1 dissected constructs SL1-dI, SL1-dIΔU, SL1-dII, SL1-d, SL1-dΔU SL1-dIIm, SL1-m, SL1-mp, SL1-pII, SL1-pIIo, SL1-p, and circular 7SK SL1alt (wt or UUCG apical loop), were transcribed from chemically synthesized DNA oligonucleotides (Integrated DNA Technologies). Transcription mixtures (40 mM Tris, pH 8.0, 1 mM spermidine, 0.01% Triton-X100, 2.5 mM DTT, 25 mM MgCl2, 0.5 μM oligonucleotide DNA template, T7 RNAP P266L, 4 mM each rATP, rUTP, rGTP and rCTP) were incubated at 37 °C for 4–6 hrs. Longer RNA constructs, minimum linear 7SK (SL1-SL4ms2) and linear 7SK SL1, were transcribed from plasmid DNA templates prepared with QIAGEN Plasmid Maxi Kit and linearized with BamHI-HF restriction enzyme (New England Biolabs). Transcription mixtures are the same as the short RNA constructs above, except from lower MgCl2 concentration (15-20 mM) and overnight incubation at 37 °C.
Transcription mixtures were loaded onto 10–20% denaturing polyacrylamide gel electrophoresis (Urea-PAGE; 19:1 crosslinking ratio) for RNA purification. Purified RNA molecules were eluted out of gel pieces either with an Elutrap device (GE Whatman) or by crush-and-soak in 1X TBE buffer (90 mM Tris-borate, 2 mM EDTA, pH 8.3). Collected RNA eluents were concentrated and buffer exchanged into sterilized nanopure water using 3 kDa or 10 kDa MWCO Amicon devices (Millipore), supplied with counterion in high salt buffer (10 mM sodium phosphate, pH 7.6, 1 mM EDTA, 1.5 M KCl) and buffer exchanged back into sterilized nanopure water. Dilute RNA samples < 100 μM were heated at 95 °C for 5 min and snap cooled on ice for 1 hr prior to final buffer exchanging and concentrating with Amicon devices (Millipore). Stock RNA solutions were exchanged into sterilized nanopure water and stored in − 20 °C freezer.
Electrophoretic mobility shift assay
1 μL of 50 μM RNA and stock solutions of 25–50 μM Hexim1 proteins were used to achieve varying protein-to-RNA molar ratios in a total volume of 5.5 μL in binding buffer, and added 1 μL loading dye (30% glycerol and bromophenol blue). The binding mixtures were incubated at room temperature for 15 min prior to gel electrophoresis on 5.7% non-denaturing polyacrylamide gel (PAGE; 37.5:1 crosslinking ratio) or 0.8% Agarose gel (for minimum linear 7SK SL1-SL4ms2) in 0.5X TBE buffer at room temperature. Polyacrylamide gels (BioRad Mini-PROTEAN handcast) and agarose gel (BioRad Mini-Sub 7 × 7 cm) were run at 70 V. Polyacrylamide gels were subsequently stained with Toluidine blue solution and destained in water, whereas agarose gels were made with SYBR™ Safe stain (Invitrogen) added prior to solidifying. Gels were imaged with a Pharos FX Plus scanner (Bio-Rad), and band intensities were extracted using ImageJ and fit to binding models (see Supplementary Fig. 14 notes) with Igor-Pro 9 software.
NMR spectroscopy
For NMR assignments of SL1-dIΔU RNA bound BR-L-AR, dilute BR-L-AR uniformly enriched with 15N and 13C were combined with dIΔU in a 1:1.2 molar ratio, and the dilute complex was buffer exchanged in NMR buffer (50 mM sodium phosphate, pH 6.2, 150 mM KCl, 0.02% NaN3, 5% D2O) and concentrated down to a final concentration of 0.22 mM BR-L-AR and 0.25 mM SL1-dIΔU. Backbone assignments were carried out using the following NMR experiments: HNCACB, CBCA(CO)NH, HNCA and C(CO)NH. For NMR assignments of Hexim1 BR-L-AR, 70 μM sample uniformly enriched with 15N and 13C in the NMR buffer was used. Backbone assignments were carried out using the CBCA(CO)NH and HNCA NMR experiments, which were collected with non-uniform sampling and processed with iterative soft thresholding reconstruction73, and comparison to the RNA-bound BR-L-AR assignments.
For NMR assignments of further truncated Hexim1 constructs, BR-L, BR, BRext and BRARM1, samples uniformly enriched with 15N and 13C in the range of 0.5-0.8 mM concentration in NMR buffer were used. Backbone assignments were carried out using the following NMR experiments: HNCACB, CBCA(CO)NH and C(CO)NH. Additional side-chain assignments were acquired for BR-L and BR using HCCH-TOCSY. BRARM1 15N-1H HSQC resonances were assigned based on almost complete overlapping with the BR 15N-1H HSQC peaks. For 1H imino sequential assignments of RNA samples in the absence and presence of monomeric Hexim1 proteins, the two-dimensional NOESY spectra were recorded using 5% D2O samples at 283.15 K with NOE mixing time of 150 ms. For PRE NMR experiments, 15N-1H HSQC spectra were recorded at 290.15 K with Avance III HD 600 MHz.
For NMR assignments of 7SK RNA constructs, natural abundance samples in the range of 0.15–1.24 mM concentration in NMR buffer (50 mM sodium phosphate, pH 6.2, 150 mM KCl, 0.02% NaN3, 5% D2O, with the exception of 0 mM KCl NMR buffer for SL1-dIΔU, SL1-dII and SL1-d, and 10 mM sodium phosphate, 50 mM KCl, pH6.3, 5% D2O for SL1altUUCG) were used. For secondary structure determination and N-H…N imino proton assignments (exchangeable-proton, here H1 for Gua and H3 for Ura), NOESY and TOCSY spectra were recorded using 95% H2O/5% D2O samples at both 283 K and 298 K. For nonexchangeable proton assignments (here H2/H8 for Ade, H8 for Gua, H5/H6 for Cyt/Ura, and H1′ for all nucleotides), NOESY and TOCSY spectra were recorded using 100% D2O samples at both 283 K and 298 K. The 13C chemical shifts from the nonexchangeable C-H vector were assigned by mapping proton assignments on 13C-1H HSQC spectra individually recorded for the aromatic region (C8-H8, C6-H6 and C2-H2) and for the remaining vectors (C1′-H1′ and C5-H5). Protein-RNA binding experiments were all performed in NMR buffer of 50 mM sodium phosphate, pH 6.2, 150 mM KCl, 0.02% NaN3, 5% D2O.
For the chemical shift perturbation (CSP) analysis of RNA constructs, observed imino chemical shift values (H3 from Us and H1 from Gs) together with H2 of Ade from A-U base pairs were used to calculate the change in chemical shift Δ between the free and protein-bound states. For Site1, Site4 and Site5 constructs, additionally assigned nonexchangeable protons were included in the CSP analyses. Protein-bound RNA spectra of NOESY and TOCSY were assigned by tracing peak trajectories in the corresponding titration series, and only assignments of high confidence were included in the final analyses. Severely broadened and highly overlapping residues were excluded from the CSP calculations.
For the CSP analysis for Hexim1 proteins, the overall change in chemical shift Δ were calculated for protein 15N-1H HSQC resonances between the free and RNA-bound states using the following equation:
\Delta=\sqrt{{{\Delta \delta }_{H}}^{2}+{{0.152\times \Delta \delta }_{N}}^{2}}
\]
where ΔδH and ΔδN are the differences between the 1HN and 15N chemical shifts of the two states being compared.
All the NMR spectra were collected at 298 K with an Avance Neo 800 MHz spectrometer equipped with TCI H&F cryoprobe, an Avance III HD 600 MHz spectrometer equipped with QCI HCNP cryoprobe or a Bruker DRX 500 MHz with cryoprobe. Data were collected with TopSpin (Bruker), processed with NMRPipe74, and analyzed in NMRFAM-Sparky75. Hexim1 protein CSP values and PRE intensity ratios were plotted and visualized with Igor-Pro 9 software.
Isothermal titration calorimetry
The dissociation constants (KD) for binding between Hexim1 proteins and 7SK RNA constructs were determined using a MicroCal 200 ITC instrument (GE). RNA and protein were buffer exchanged using separate 3 kDa MWCO dialysis tubings into to the same bucket of ITC buffer (20 mM HEPES, pH 7.5, 150 mM KCl, 1 mM TCEP). Dilute dialyzed RNA solutions < 20 μM were heated at 95 °C for 5 min and snap cooled on ice for 1 hr right before the first set of titration experiments. RNA final concentrations were determined by UV absorbance at 90 °C at 260 nm with the Lambert-Beer law, using a Hewlett-Packard HP8453 diode-array UV/Visible spectrophotometer with Peltier temperature control. Extinction coefficients of melt RNA were calculated by the OligoAnalyzer Tool from Integrated DNA Technologies. RNA stock solutions were kept at room temperature to avoid cold-induced oligomerization for the 2-3 days duration of one complete titration set. RNA at concentration 63-195 μM in the syringe was titrated into 7–40 μM protein in the sample cell at 295 K, or for reversed titration order protein at concentrations of 102–120 μM in the syringe was titrated into 6–12 μM RNA in the sample cell at 295 K. Calorimetric data was fit using ORIGIN 7 (Micro-Cal). The binding parameters stoichiometry (N), entropy(ΔS), enthalpy (ΔH) and association constant (KA) were kept as floating variables during each fit. ITC experiments were performed in biological triplicate or quadruplicate (n = 3 or 4) to verify reproducibility and estimate experimental error. No statistical methods were used to predetermine sample sizes, as n = 3 is standard for quantitative biophysical measurements of purified systems. Full thermodynamics parameters and individual replicate numbers are listed in Supplementary Table 1.
Multiple sequence alignment
Initial list of orthologs of Hexim1 and Hexim2 genes were extracted from NCBI gene cards (Gene ID: 10614 and 124790). Truncated and low-confidence genes were removed by manual inspection, resulting in 294 genes for Hexim1 and 210 genes for Hexim2. Sequence alignments were performed by COBALT (NCBI) and visualized using Jalview76. Five sequences for each Hexim1 and Hexim2 were selected for the final display, while the sequence placements were kept from the full alignments. A list of HIV-1 Tat protein sequences and alignment were acquired from UniProt, visualized using Jalview and one long isoform from each group and subtype of virus were selected for the final display.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary Materials
Supplementary Materials
References
- M Kusuhara. Cloning of hexamethylene-bis-acetamide-inducible transcript, HEXIM1, in human vascular smooth muscle cells. Biomed. Res., 1999. [DOI]
- AA Michels, O Bensaude. Hexim1, an RNA-controlled protein hub. Transcription, 2018. [DOI | PubMed]
- M Morchikh. HEXIM1 and NEAT1 Long non-coding RNA form a multi-subunit complex that regulates DNA-mediated innate immuneresponse. Mol. Cell, 2017. [DOI | PubMed]
- Y Fujimoto, Y Nakamura, S Ohuchi. HEXIM1-binding elements on mRNAs identified through transcriptomic SELEX and computational screening. Biochimie, 2012. [DOI | PubMed]
- JH Yik, R Chen, AC Pezda, Q Zhou. Compensatory contributions of HEXIM1 and HEXIM2 in maintaining the balance of active and inactive positive transcription elongation factor b complexes for control of transcription. J. Biol. Chem., 2005. [DOI | PubMed]
- SA Byers, JP Price, JJ Cooper, Q Li, DH Price. HEXIM2, a HEXIM1-related protein, regulates positive transcription elongation factor b through association with 7SK. J. Biol. Chem., 2005. [DOI | PubMed]
- AA Michels. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J., 2004. [DOI | PubMed]
- JH Yik, R Chen, AC Pezda, CS Samford, Q Zhou. A human immunodeficiency virus type 1 Tat-like arginine-rich RNA-binding domain is essential for HEXIM1 to inhibit RNA polymerase II transcription through 7SK snRNA-mediated inactivation of P-TEFb. Mol. Cell. Biol., 2004. [DOI | PubMed]
- N Czudnochowski, CA Bösken, M Geyer. Serine-7 but not serine-5 phosphorylation primes RNA polymerase II CTD for P-TEFb recognition. Nat. Commun., 2012. [DOI | PubMed]
- N Czudnochowski, F Vollmuth, S Baumann, K Vogel-Bachmayr, M Geyer. Specificity of Hexim1 and Hexim2 complex formation with cyclin T1/T2, importin alpha and 7SK snRNA. J. Mol. Biol., 2010. [DOI | PubMed]
- TGW Graham. Single-molecule live imaging of subunit interactions and exchange within cellular regulatory complexes. Mol. Cell, 2025. [DOI | PubMed]
- NF Marshall, DH Price. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem., 1995. [DOI | PubMed]
- SM Vos. Structure of activated transcription complex Pol II-DSIF-PAF-SPT6. Nature, 2018. [DOI | PubMed]
- I D’Orso, AD Frankel. RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat. Struct. Mol. Biol., 2010. [DOI | PubMed]
- H Lu. AFF1 is a ubiquitous P-TEFb partner to enable Tat extraction of P-TEFb from 7SK snRNP and formation of SECs for HIV transactivation. Proc. Natl. Acad. Sci. USA, 2014. [DOI | PubMed]
- I D’Orso. Transition step during assembly of HIV Tat:P-TEFb transcription complexes and transfer to TAR RNA. Mol. Cell. Biol., 2012. [DOI | PubMed]
- M Barboric. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res., 2007. [DOI | PubMed]
- SA Dames. Structure of the Cyclin T binding domain of Hexim1 and molecular basis for its recognition of P-TEFb. Proc. Natl. Acad. Sci. USA, 2007. [DOI | PubMed]
- A Schulte. Identification of a cyclin T-binding domain in Hexim1 and biochemical analysis of its binding competition with HIV-1 Tat. J. Biol. Chem., 2005. [DOI | PubMed]
- TS Bayer, LN Booth, SM Knudsen, AD Ellington. Arginine-rich motifs present multiple interfaces for specific binding by RNA. RNA, 2005. [DOI | PubMed]
- L Kobbi. An evolutionary conserved Hexim1 peptide binds to the Cdk9 catalytic site to inhibit P-TEFb. Proc. Natl. Acad. Sci. USA, 2016. [DOI | PubMed]
- M Barboric. Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. EMBO J., 2005. [DOI | PubMed]
- Q Li. HEXIM1 is a promiscuous double-stranded RNA-binding protein and interacts with RNAs in addition to 7SK in cultured cells. Nucleic Acids Res., 2007. [DOI | PubMed]
- I Lebars. HEXIM1 targets a repeated GAUC motif in the riboregulator of transcription 7SK and promotes base pair rearrangements. Nucleic Acids Res., 2010. [DOI | PubMed]
- S Bourbigot. Solution structure of the 5’-terminal hairpin of the 7SK small nuclear RNA. RNA, 2016. [DOI | PubMed]
- VV Pham. HIV-1 Tat interactions with cellular 7SK and viral TAR RNAs identifies dual structural mimicry. Nat. Commun., 2018. [DOI | PubMed]
- VV Pham, M Gao, JL Meagher, JL Smith, VM D’Souza. A structure-based mechanism for displacement of the HEXIM adapter from 7SK small nuclear RNA. Commun. Biol., 2022. [DOI | PubMed]
- AR Gruber. Arthropod 7SK RNA. Mol. Biol. Evol., 2008. [DOI | PubMed]
- AR Gruber. Invertebrate 7SK snRNAs. J. Mol. Evol., 2008. [DOI | PubMed]
- L Muniz, S Egloff, B Ughy, BE Jády, T Kiss. Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog., 2010. [DOI | PubMed]
- Y Yang, CD Eichhorn, Y Wang, D Cascio, J Feigon. Structural basis of 7SK RNA 5’-γ-phosphate methylation and retention by MePCE. Nat. Chem. Biol., 2019. [DOI | PubMed]
- M Marz. Evolution of 7SK RNA and its protein partners in metazoa. Mol. Biol. Evol., 2009. [DOI | PubMed]
- JE Brogie, DH Price. Reconstitution of a functional 7SK snRNP. Nucleic Acids Res., 2017. [DOI | PubMed]
- Y Yang. Structural basis of RNA conformational switching in the transcriptional regulator 7SK RNP. Mol. Cell, 2022. [DOI | PubMed]
- SW Olson. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Mol. Cell, 2022. [DOI | PubMed]
- Y Xue, Z Yang, R Chen, Q Zhou. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res., 2010. [DOI | PubMed]
- E Van Herreweghe. Dynamic remodelling of human 7SK snRNP controls the nuclear level of active P-TEFb. EMBO J., 2007. [DOI | PubMed]
- H Schägger, G von Jagow. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem., 1991. [DOI | PubMed]
- A Sonn-Segev. Quantifying the heterogeneity of macromolecular machines by mass photometry. Nat. Commun., 2020. [DOI | PubMed]
- G Young. Quantitative mass imaging of single biological macromolecules. Science, 2018. [DOI | PubMed]
- K Bugge. Role of charges in a dynamic disordered complex between an IDP and a folded domain. Nat. Commun., 2025. [DOI | PubMed]
- JA Marsh, VK Singh, Z Jia, JD Forman-Kay. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci., 2006. [DOI | PubMed]
- K Brillet. Different views of the dynamic landscape covered by the 5’-hairpin of the 7SK small nuclear RNA. RNA, 2020. [DOI | PubMed]
- D Martinez-Zapien. The crystal structure of the 5′ functional domain of the transcription riboregulator 7SK. Nucleic Acids Res., 2017. [PubMed]
- Q Zhang, X Sun, ED Watt, HM Al-Hashimi. Resolving the motional modes that code for RNA adaptation. Science, 2006. [DOI | PubMed]
- B Meyer, T Peters. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem. Int. Ed. Engl., 2003. [DOI | PubMed]
- JL Battiste, G Wagner. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry, 2000. [DOI | PubMed]
- GM Clore, J Iwahara. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev., 2009. [DOI | PubMed]
- C Tang, R Ghirlando, GM Clore. Visualization of transient ultra-weak protein self-association in solution using paramagnetic relaxation enhancement. J. Am. Chem. Soc., 2008. [DOI | PubMed]
- OM Morris, JH Torpey, RL Isaacson. Intrinsically disordered proteins: modes of binding with emphasis on disordered domains. Open Biol., 2021. [DOI | PubMed]
- CQ AJ, A Bugai, M Barboric. Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res., 2016. [DOI | PubMed]
- X Contreras, M Barboric, T Lenasi, BM Peterlin. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog., 2007. [DOI | PubMed]
- YK Kim, U Mbonye, J Hokello, J Karn. T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J. Mol. Biol., 2011. [DOI | PubMed]
- UR Mbonye, B Wang, G Gokulrangan, MR Chance, J Karn. Phosphorylation of HEXIM1 at Tyr271 and Tyr274 promotes release of P-TEFb from the 7SK snRNP complex and enhances proviral HIV gene expression. Proteomics, 2015. [DOI | PubMed]
- K Fujinaga. PKC phosphorylates HEXIM1 and regulates P-TEFb activity. Nucleic Acids Res., 2012. [DOI | PubMed]
- Y Sun. Activation of P-TEFb by cAMP-PKA signaling in autosomal dominant polycystic kidney disease. Sci. Adv., 2019. [DOI | PubMed]
- S Egloff, E Van Herreweghe, T Kiss. Regulation of polymerase II transcription by 7SK snRNA: two distinct RNA elements direct P-TEFb and HEXIM1 binding. Mol. Cell Biol, 2006. [DOI | PubMed]
- A Zeke. Deep structural insights into RNA-binding disordered protein regions. Wiley Interdiscip. Rev. RNA., 2022. [DOI | PubMed]
- MW Hentze, A Castello, T Schwarzl, T Preiss. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol., 2018. [DOI | PubMed]
- C Qiu. Intra- and inter-molecular regulation by intrinsically-disordered regions governs PUF protein RNA binding. Nat. Commun., 2023. [DOI | PubMed]
- PA Chong, RM Vernon, JD Forman-Kay. RGG/RG Motif Regions in RNA Binding and Phase Separation. J. Mol. Biol., 2018. [DOI | PubMed]
- X Wang. Negatively charged, intrinsically disordered regions can accelerate target search by DNA-binding proteins. Nucleic Acids Res., 2023. [DOI | PubMed]
- CD Eichhorn, Y Yang, L Repeta, J Feigon. Structural basis for recognition of human 7SK long noncoding RNA by the La-related protein Larp7. Proc. Natl. Acad. Sci. USA, 2018. [DOI | PubMed]
- Q Li. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J. Biol. Chem., 2005. [DOI | PubMed]
- KV Prasanth. Nuclear organization and dynamics of 7SK RNA in regulating gene expression. Mol. Biol. Cell, 2010. [DOI | PubMed]
- Y Wang. N(6)-methyladenosine in 7SK small nuclear RNA underlies RNA polymerase II transcription regulation. Mol. Cell, 2023. [DOI | PubMed]
- M Perez-Pepe. 7SK methylation by METTL3 promotes transcriptional activity. Sci. Adv., 2023. [DOI | PubMed]
- U Mbonye, F Kizito, J Karn. New insights into transcription elongation control of HIV-1 latency and rebound. Trends Immunol., 2023. [DOI | PubMed]
- B Sobhian. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell, 2010. [DOI | PubMed]
- Y Cheng, DJ Patel. An efficient system for small protein expression and refolding. Biochem. Biophys. Res. Commun., 2004. [DOI | PubMed]
- 71.Sjodt, M. & Clubb, R. T. Nitroxide labeling of proteins and the determination of paramagnetic relaxation derived distance restraints for NMR studies. Bio Protoc. 7, 10.21769/BioProtoc.2207 (2017).
- J Guillerez, PJ Lopez, F Proux, H Launay, M Dreyfus. A mutation in T7 RNA polymerase that facilitates promoter clearance. Proc. Natl. Acad. Sci. USA, 2005. [DOI | PubMed]
- SG Hyberts, AG Milbradt, AB Wagner, H Arthanari, G Wagner. Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J. Biomol. NMR, 2012. [DOI | PubMed]
- F Delaglio. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR, 1995. [DOI | PubMed]
- W Lee, M Tonelli, JL Markley. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics, 2015. [DOI | PubMed]
- M Clamp, J Cuff, SM Searle, GJ Barton. The Jalview Java alignment editor. Bioinformatics, 2004. [DOI | PubMed]