
DNA Methylation in Autism Spectrum Disorders: Biomarker or Pharmacological Target?
Abstract
Autism spectrum disorder (ASD) is a group of heterogeneous neurodevelopmental disabilities with persistent impairments in cognition, communication, and social behavior. Although environmental factors play a role in ASD etiopathogenesis, a growing body of evidence indicates that ASD is highly inherited. In the last two decades, the dramatic rise in the prevalence of ASD has interested researchers to explore the etiologic role of epigenetic marking and incredibly abnormal DNA methylation. This review aimed to explain the current understanding of the association between changes in DNA methylation signatures and ASD in patients or animal models. We reviewed studies reporting alterations in DNA methylation at specific genes as well as epigenome-wide association studies (EWASs). Finally, we hypothesized that specific changes in DNA methylation patterns could be considered a potential biomarker for ASD diagnosis and prognosis and even a target for pharmacological intervention.
Article type: Review Article
Keywords: autism spectrum disorders, DNA methylation, epigenetics, animal model, biomarkers
Affiliations: Student Research Committee, Mashhad University of Medical Sciences, Mashhad 13131-99137, Iran; gholamah961@mums.ac.ir; Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad 91779-48564, Iran; Student Research Committee, North Khorasan University of Medical Sciences, Bojnurd 94149-75516, Iran; m.amirishahri@nkums.ac.ir (M.A.-S.); f.rasouli@nkums.ac.ir (F.R.); arinaansari80@gmail.com (A.A.); Faculty of Medicine, North Khorasan University of Medical Sciences, Bojnurd 94149-75516, Iran; Department of Cardiovascular Diseases, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad 91779-48564, Iran; baradaranrv@mums.ac.ir; Pharmacological Research Center of Medicinal Plants, Mashhad University of Medical Sciences, Mashhad 91779-48564, Iran
License: © 2024 by the authors. CC BY 4.0 Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.3390/brainsci14080737 | PubMed: 39199432 | PMC: PMC11352561
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (647 KB)
1. Introduction
Autism spectrum disorder (ASD), characterized by deficits in social communication and the presence of restricted, repetitive behaviors or interests, is a neurodevelopmental disorder affecting approximately 2.3% of 8-year-old children and 2.2% of adults in the United States [ref. 1,ref. 2]. The estimated prevalence of ASD in the United States has increased from 1.1% in 2008 to 2.3% in 2018, likely due to changes in diagnostic criteria, enhanced screening and diagnostic tools, and raised awareness among the public [ref. 1]. ASD appears to be one of the most debilitating neurodevelopmental disorders in the United States, and estimates put the annual cost at close to USD 250 billion, with the expense of caring for an individual with ASD over their lifetime falling somewhere between USD 1.5 and USD 2.5 million [ref. 3].
Noteworthy, there are currently no diagnostic biomarkers identified as specific to ASD. In the first two years of a child’s life, common early signs and symptoms of ASD include no response to the child’s name when called, no or limited use of gestures for communication, and lack of imaginative play [ref. 1]. In addition to core deficits and associated psychiatric comorbidity [ref. 4], ASD has been linked to negative non-behavioral health outcomes such as an increased risk of injury [ref. 5] and mortality [ref. 6,ref. 7].
Over time, there has been a significant shift in both the diagnostic criteria and treatment methods for ASD. The majority of current diagnostic procedures involve the use of observational screening instruments that assess a child’s cognitive and social capacities. The 5th edition of the diagnostic and statistical manual of mental disorders (DSM-5) and the modified checklist for autism in toddlers (M-CHAT) are the two most important diagnostic instruments for ASD. These instruments look for long-term problems with social interaction and communication as well as replies to “yes/no” questions that span various areas of development [ref. 8].
Multiple lines of evidence indicate that ASD is highly inherited, but environmental factors have also been associated with ASD. Monogenic traits of autism are characterized by a single DNA-level alteration or genetic modification in one gene, independently of environmental factors. These genetic abnormalities are linked to specific syndromes rather than ASD. The development of polygenic traits, conversely, involves a multitude of gene variations combined with environmental influences. As a result, they generally do not produce distinct categories but rather a wide range of phenotypic characteristics that span across a spectrum, influenced by the number and nature of the genes involved and the environmental factors [ref. 9,ref. 10].
Extensive genetic research has demonstrated a significant level of genetic variability as the underlying cause of ASD. The initial critical distinction is that ASD can manifest as either an isolated clinical phenotype or as part of a syndromic condition. In the second scenario, numerous phenotypes, including epilepsy, intellectual disability, and dysmorphic characteristics, are present in a developmental disease that manifests as ASD [ref. 9,ref. 11]. Regarding environmental factors, on the other hand, parental age, maternal medical and mental health, and perinatal environmental exposures have been suggested to play a role in the etiopathogenesis of ASD (Figure 1) [ref. 12].
In recent years, there has been a growing interest in examining epigenetic marks in ASD due to their potential mechanistic role in etiology, specifically to explain the effects of environmental exposure or gene–environment interaction associations with ASD or as a biomarker of previous exposure or disease [ref. 13]. The expression of many genes is regulated by epigenetic marks that do not change the primary DNA sequence. Both genetic mutations and environmental factors can alter epigenetic modifications, such as DNA methylation, histone methylation, and acetylation. Epigenetic dysregulation has been proven to be linked to autism based on several factors. Primarily, gene mutations involved in epigenetic regulation can cause syndromes associated with autism (Figure 1) [ref. 14].
Second, autism was linked to several areas of the chromosome that are influenced by parental imprinting. Repeated reports have shown that individuals with autism have microduplications or microdeletions of the parentally none-printable region 15qllql3 [ref. 15,ref. 16]. Several studies have indicated an association between ASD and single-nucleotide polymorphisms (SNPs) in a gene directly involved in methylation [ref. 17,ref. 18]. The expression of the retinoic acid-related orphan receptor alpha gene (RORA) and B-cell lymphoma 2 (BCL-2) was found to be reduced in the lymphoblastoid cells of autistic patients, demonstrating direct changes in the DNA methylation profile. DNA methylation regulates gene expression patterns by modifying DNA accessibility. From a molecular aspect, when DNA methyltransferases (DNMTs) add a methyl group to the fifth carbon position of cytosine at cytosine–phosphate–guanine (CpG) dinucleotides, DNA methylation takes place [ref. 19,ref. 20,ref. 21,ref. 22,ref. 23].
In the last two decades, an alarming rise in the prevalence of ASD has intensified the debate over the etiologic roles of non-genetic factors such as epigenetics (primarily abnormal DNA methylation) and environmental interaction [ref. 24,ref. 25].
This narrative review focuses on the current understanding of how DNA methylation contributes to ASD in patients or animal models. This could be of great assistance for the investigation of the molecular mechanisms underlying ASD, with the ultimate goal of identifying novel targets for the creation of innovative epigenetic drugs.
2. DNA Methylation
DNA methylation, the most studied epigenetic modification, is crucially catalyzed by DNA methyltransferases (DNMTs) that transfer a methyl group (CH3) from S-adenosyl methionine (SAM) to the fifth carbon of a cytosine residue, resulting in 5mC [ref. 26]. It is important to note that DNMT3a and DNMT3b are called de novo DNMT because they can impart a novel methylation pattern onto unmodified DNA. During DNA replication, DNMT1 transfers the DNA methylation pattern from the original DNA strand to the newly formed daughter strand [ref. 27]. Moreover, DNMT1 is capable of repairing DNA methylation [ref. 28]. DNMT3L, another member of the DNMT family, links with DNMT3a and DNMT3b and stimulates their methyltransferase activity without having the catalytic domain exist in other DNMT enzymes (Figure 1) [ref. 29,ref. 30,ref. 31].
According to the dynamics of the DNA methylation process, its reversibility can occur in two active and passive forms. The passive DNA demethylation takes place in dividing cells as the inhibition or dysfunction of DNMT1 causes newly incorporated cytosine to remain unmethylated, decreasing the overall methylation level after each cell division. Active DNA demethylation is found in both dividing and nondividing cells, but the process requires ten-eleven translocation (TeT) enzymes, which eliminate the methyl group from methylated bases via oxidation [ref. 32,ref. 33,ref. 34].
A significant amount of DNA methylation occurs at CpG sites or cytosines that precede guanine. Due to the mutagenic potential of 5mC, which can deaminate into thymine, mammalian genomes are depleted from CpG sites [ref. 35,ref. 36].
Except for CpG islands, the remaining CpG sites are widely dispersed and extensively methylated throughout the genome [ref. 36]. CpG islands are composed of a minimum of 200 base pairs, have a GC concentration exceeding 15%, and an observed-to-expected CPG ratio exceeding 60%. However, CpG islands and the nearby CpG island shores (2 kb-long regions that reside on both sides of the island) are exceptionally hypomethylated [ref. 37,ref. 38]. Many of these hypomethylated DNA regions serve as elements that regulate gene expression, including promoters and enhancers [ref. 37]. Approximately 70% of gene promoters are located within CpG islands [ref. 39]. CpG islands in the promoter regions of actively expressed genes are generally unmethylated, as DNA methylation at promoters is typically associated with gene silencing [ref. 26]. Both direct (including steric inhibition of binding with transcription factors) and indirect (using Methyl CpG binding Proteins, MeCPs, that recruit histone deacetylases) mechanisms have been involved in the repression of gene transcription due to CpG island methylation [ref. 26,ref. 40,ref. 41].
Additionally, methylation can occur at non-CpG sites, namely CpA, CpT, and CpC, with CpA being the most commonly methylated. Non-CpG methylation is significantly increased in human embryonic stem cells, neurons, and glial cells [ref. 42]. Unlike CpG methylation, which occurs early in development and fails to rise with growth, non-CpG methylation accumulates during neuron development [ref. 26,ref. 42]. Furthermore, non-CpG methylation increases concurrently with synaptic development and synaptic density. In both human and mouse brain tissue, non-CpG methylation begins during early postnatal development (in the first two years and the first 2–4 months, respectively) and decreases during adolescence. Methylation of non-CpG regions originally parallels the increase in synaptic density that occurs between birth and five years of age in humans but appears to elevate during synaptic pruning through adolescence [ref. 43].
This section mainly focused on the DNA methylation catalyzed by DNMTs as a crucial epigenetic modification. This process can be reversed through passive demethylation during cell division and active demethylation involving TeT enzymes. CpG islands, which have high CpG density, are typically hypomethylated and linked to the regulation of gene expression. Non-CpG methylation, particularly CpA, is significant in human embryonic stem cells and neurons, and it increases as synaptic development progresses, reaching its peak during early postnatal development and declining during adolescence.
3. Alterations in DNA Methylation at Specific Genes Associated with Autism Spectrum Disorders
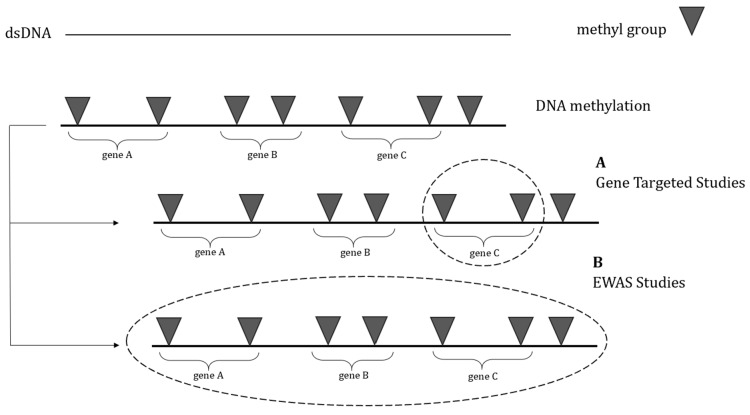
Previous researchers exploring the epigenetic alterations in ASD have mainly focused on analyzing specific genes and proteins involved in ASD. Disturbed central and peripheral hemostasis of hormones and neurotransmitters is often found in subjects with ASD [ref. 44,ref. 45,ref. 46,ref. 47,ref. 48,ref. 49,ref. 50,ref. 51,ref. 52,ref. 53]. The altered expression of several genes responsible for regulating the hemostasis was reported in ASD patients. Furthermore, DNA methylation in genes associated with the neural matrix [ref. 54,ref. 55,ref. 56,ref. 57,ref. 58], genes regulating gene expression [ref. 55,ref. 57,ref. 59,ref. 60], genes involved in syndromes with a high penetrance of ASD [ref. 61,ref. 62,ref. 63], and genes associated with environmental factors [ref. 64,ref. 65,ref. 66] has recently been recognized in ASD models. Table 1 summarizes the available studies on DNA methylation patterns of ASD patients or animal models. As shown, some paths investigated by several research groups were only preliminarily evaluated, whereas others were widely explored and revealed more consistency between different studies. Clinical data are drawn chiefly from studies in which samples were obtained from human blood cells. In fact, brain tissue analysis is based mainly on animal models. Therefore, despite the limitations of preclinical studies, they could be an essential source of evidence.
Table 1: Studies reported differences in DNA methylation at specific genes in ASD patients or animal models of ASD.
| Clinical Studies | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | Protein | Genomic Region | Groups of Subjects | Tissue | ASD Diagnosis | N of Subjects (in Each Group) | Age of Subjects (in Each Group) | Main DNA Methylation Finding | Ref. |
| HTR2A | Promoter | ASD, HC | Blood | DSM-4 | 90 + 101 | 5.84 ± 2.96 y | No association was proved | [ref. 44] | |
| Promoter | ASC, HC | Blood | DSM-4 | 90 + 66 | 4–45 y19–60 y | Different methylation patterns | [ref. 45] | ||
| HTR4 | Promoter | ASD, HC | Blood | DSM-5 | 61 + 66 | 4.02 ± 2.83 y5.76 ± 0.72 y | ↓ in ASD and females | [ref. 46] | |
| OXTR | Oxytocin receptor | Promoter | ASD, TD | Blood | DSM-5-TR, CARS | 27 + 39 | 39.27± 16.95 m43.69 ± 20.51 m | ↓ in ASD | [ref. 47] |
| Oxytocin receptor | Promoter | ASD, neurotypical group | Saliva | DSM-5 ASD | 35 + 64 | 27.02 ± 5.34 y28.22 ± 6.96 y | ↑ in the intron 1 area in ASD | [ref. 48] | |
| Oxytocin receptor | Promoter | HFA, HC | Blood | German guideline | 20 + 20 | 30.5 ± 7.8 y30.7 ± 7.8 y | Not reported a single significant group-dependent methylated site | [ref. 49] | |
| GAD1 | Glutamic acid decarboxylase | Regulatory | ASD, control | Brain | DSM-5 | 4 + 5 | 25.9 ± 0.8 y26.1 ± 0.8 y | More diverse in cerebral organoids from ASD subjects | [ref. 50] |
| IRS2 | Insulin receptor substrate 2 | Exon 1, Intron 1 | ASD, TD | MARBLES placenta | DSM-5 | 20 + 21 | – | ↑ in ASD | [ref. 51] |
| ESR2 | ESR2 | Proximal promoter region and an untranslated exon | ASD, HC | Blood | DSM-4 | 54 + 54 | 4.24 ± 0.98 y4.37 ± 0.80 y | Different methylation patterns | [ref. 52] |
| RELN | Reelin | Promoter | ASD, HC | Brain | – | 6 + 6 | 21.0 ± 2.9 y22.0 ± 1.8 y | Different methylation patterns | [ref. 54] |
| SHANK3 | SH3 and multiple ankyrin repeat domains 3 | Promoter | ASD, control | Brain | DSM-5 and ADI-R | 54 + 43 | – | ↑ SHANK3 CGIs | [ref. 55] |
| ST8SIA2 | ST8 alpha-N-acetyl-neuraminide | Chr. 15: 92,984,625/Chr. 15: 92998561 | ASD, TD | Blood | DSM-5, ADI-R, ADOS | 69 + 76 | 4.47 ± 1.23 y4.59 ± 1.19 y | ↑ in ASD | [ref. 56] |
| MeCP2 | Methyl CpG binding protein 2 | Promoter | ASD, control | Brain | Not mentioned | 9 + 9 | 5–56 y9–56 y | ↑ in ASD | [ref. 59] |
| NHIP | Neuronal hypoxia-inducible, placenta associated | 22q13.33 | ASD, TD | EARLI/MARBLES placenta | ADOS/ADI-R/MSEL | MARBLES:46 + 46EARLI:16 + 31 | – | ↓ in ASD | [ref. 60] |
| FMR1 | FMRP | Promoter | FXS, control | Blood | ADOS | 12 + 5 | 1–28 y1–28 y | ↑ in 4 cases (3 of which were ASD) | [ref. 61] |
| Gene | FXS + ASD, HC | Blood | DSM-5 | 18 | Male: 14.4 ±11.9 yFemale: 14.7 ± 10.9 y | FMRP levels correlate with FMR1 gene methylation | [ref. 62] | ||
| APOE | APOE | Promoter | ASD, HC | Blood | DSM-4 | 62 + 73 | – | ↑ in ASD | [ref. 63] |
| ACSF3 | Malonyl-CoA synthetase | Promoter | ASD, control | Blood | DSM-5 | 19 +18 | 6–12 y3–11 y | No DNA in the binding site of SP1 within the ACSF3 promoter | [ref. 67] |
| PPP2R2C | PP2A | Promoter | ASD, HC | Blood | DSM-5,ADOSASSQ-R | 29 + 29 | – | Hypermethylation and gene downregulation | [ref. 68] |
| CYP2E1 | Cytochrome P450 Family 2 Subfamily E Member 1 | Intron 1, Exon 2 | ASD, TD | MARBLES placenta | DSM-5 | 20 + 21 | – | ↓ in ASD | [ref. 51] |
| Preclinical Studies | |||||||||
| Gene | Protein | Genomic Region | Animal/Strain | Animal Model | Tissue | Age of subjects | Main DNA Methylation Finding | Ref. | |
| DAT | DAT | Promoter | Long-Evans rat | Midbrain and striatum | 10 w | ↑ in midbrain and striatum | [ref. 53] | ||
| RELN | Reelin | Promoter | Balb/cAnNCrlBR mice | Cerebral cortex | 4 d | Unmethylated | [ref. 57] | ||
| SHANK3 | SH3 and multiple ankyrin repeat domains 3 | Promoter | Wistar rats | PND exposure to pollution | Brain | 25 d | ↑ in exposed rats | [ref. 58] | |
| MeCP2 | MeCP2 | TSS | Adult mice | Hippocampus | 3–18 w | ↑ in hippocampus | [ref. 64] | ||
| methyl CpG binding protein 2 | Promoter | Wistar rats | PND exposure to pollution | Brain | 25 d | ↑ in exposed rats | [ref. 58] | ||
| WNT | WNT1/WNT2 | Promoter | Rat | Rat VPA ASD model | Frontal cortex and hippocampus | Offspring | Increased expression | [ref. 65] | |
| KCC2 | Potassium (K+)/chloride (Cl−) symporter | Promoter | Sprague Dawley rats | Parental exposure to sevoflurane | Hypothalamus, hippocampus | 5 d | ↑ only in male offspring | [ref. 69] | |
Abbreviations: ASD: autism spectrum disorder; TD: typical development; HC: healthy control; HFA: high-functioning autism; HTR2A: human serotonin receptor2A; DSM-4: Diagnostic and Statistical Manual of Mental Disorders, fourth edition; ASC: Autism Spectrum Condition; HTR4: human serotonin receptor 4; DSM-5-TR: Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, Text Revision; CARS: Childhood Autism Rating Scale; OXTR: oxytocin receptor; GAD1: Glutamic acid decarboxylase 1; DAT: dopamine transporter; IRS2: insulin receptor substrate 2; ESR2: estrogen receptor beta; CGIs: CpG Islands; ST8SIA2: ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 2; MeCP2: methyl-CpG binding protein 2; NHIP: neuronal hypoxia-inducible, placenta associated; APOE: apolipoprotein E; KCC2: Kþ-2ClClexporter; ADOS: Autism Diagnostic Observation Schedule; MSEL: Mullen Scales of Early Learning; ADI-R: Autism Diagnostic Interview—Revised; SP1: specificity protein 1; ACSF3: Acyl-CoA synthetase family member 3; PP2A: Protein Phosphatase type 2 A; CYP2E1: cytochrome p450 2E1; EARLI: The Early Autism Risk Longitudinal Investigation; MARBLES: Markers of Autism Risk in Babies Learning Early Signs; FMR1: fragile X mental retardation 1; FMRP: fragile X mental retardation protein; FXR: fragile X syndrome; SHANK3: SH3 and multiple ankyrin repeat domains 3; PND: Postnatal day; VPA: Valproate. y: years, m: months, w: weeks; d: days, ↑: increase, ↓: decrease.
The following section provides and compares clinical and preclinical data on DNA methylation at specific genes in ASD patients and animal models. The known clinical impact of different genes led to their classification into various groups.
3.1. Hormones and Neurotransmitter Receptor Genes
3.1.1. Serotonin
The serotonin signaling system is implicated in regulating key neurodevelopmental processes [ref. 70]. Numerous studies have reported a disruption in serotoninergic neurotransmission in ASD. Patients diagnosed with ASD showed elevated levels of serotonin in the peripheral system but had notably lower levels of serotonin in the central nervous system [ref. 71,ref. 72]. A variety of ASD symptoms, including insomnia, depression, repetitive behavior, and compulsion, were successfully treated by selective serotonin reuptake inhibitors (SSRIs) [ref. 73]. Abnormalities in genes related to serotonin signaling could explain the serotonin depletion in individuals with ASD. DNA methylation in human serotonin receptor 2A (HTR2A) and human serotonin receptor 4 (HTR4) genes were considered potential candidate genes for ASD.
HTR2A
The etiologic role of three SNPs of HTR2A (1438A/G (rs6311), 102T/C (rs6313), and 1354C/T (rs6314)) have been well documented in many neuropsychiatric disorders [ref. 74,ref. 75]. This gene’s connection to ASD has been investigated in previous studies involving Caucasian, Korean, and Indian populations. While the Korean study found a connection between the haplotype and ASD, the studies on Caucasian and Indian patients failed to identify any association [ref. 44,ref. 76,ref. 77]. However, in the Indian study, the methylation of CpG sites at the 1438A/G and 102T/C loci of the gene was detected. As reported in this study, the HTR2A gene’s promoter lacks CpG islands, which reinforces the gene’s biallelic expression pattern in peripheral blood leukocytes (PBLs). Also, in this study, G and A alleles of 1438A/G (rs6311) polymorphism did not show functional differences [ref. 44]. A more recent study by Hranilovic et al. displayed higher mean DNA methylation levels at 1438A/G (regulatory region) of the HTR2A gene in autistic subjects than in corresponding controls. However, similar to the previous study on the Indian population, no statistically significant difference was observed between AA and GG carries [ref. 45].
HTR4
As reported by recent research on ASD children, hypomethylation of the HTR4 promoter has been recognized as a possible predictive biomarker for ASD. Additionally, this study indicated the inverse association between age and DNA methylation of the HTR4 promoter in male ASD subjects [ref. 46].
3.1.2. Oxytocin
Oxytocin, a key modulator of socioemotional behaviors, acts via the oxytocin receptor (OXTR), which is enriched in subcortical emotional and reward regions [ref. 78]. In humans, it has been observed that resting state functional connectivity between frontal and ventral striatal areas was enhanced by intranasal oxytocin application [ref. 79,ref. 80]. Pivotal clinical trials demonstrated that intravenous or intranasal administration of oxytocin improves the symptoms of ASD, including repetitive behavior, speech comprehension, and emotion recognition [ref. 80,ref. 81,ref. 82]. In a recent placebo-control randomized trial, four weeks of administration of intranasal oxytocin was found to be associated with a reduction in OXTR DNA methylation and enhanced feelings of secure attachment [ref. 83]. Studies also evaluated the plasma oxytocin levels in ASD patients and their association with genetic and epigenetic signatures [ref. 84,ref. 85].
The largest meta-analysis investigating 16 OXTR SNPs has found that SNPs rs7632287, rs237887, rs2268491, and rs2254298 were significantly associated with ASD [ref. 86]. Also, several reports compared the methylation frequency of OXTR between autistic and non-autistic subjects. A research work on the Turkish population showed a significant reduction in the promoter methylation levels in the regions MT1 and MT3 of OXTR, but not in the MT2 and MT4 regions [ref. 47]. On the contrary, a study by Andari et al. reported higher OXTR methylation frequency in the intron 1 area of MT2 compared to neurotypical controls. Using a combined epigenetic and imaging approach, this study provided the first evidence that OXTR hypermethylation is related to the severity of clinical symptoms and brain functional connectivity between cortico-cortical areas participating in the theory of mind. In addition, methylation at a CpG site in the exon 1 area of MT2 has been shown to be positively associated with social responsiveness deficits in ASD [ref. 48]. A recent case–control study on twenty adults with high-functioning autism (HFA) revealed no apparent differences in OXTR gene sequence, except for rs918316. Also, methylation analysis did not detect a single significantly different methylated site among the 412 CpG sites of the OXTR gene in HFA patients compared to normal controls [ref. 49].
3.1.3. Dopamine
The intensity and duration of dopaminergic signaling are regulated by the dopamine transporter (DAT) [ref. 87]. In various neurodevelopmental disorders, including ASD and attention deficit hyperactivity disorder (ADHD), an altered expression of DAT has been observed [ref. 88,ref. 89]. In addition, DAT is the target of both psychotropic drugs and neurotoxicants [ref. 90,ref. 91]. In an earlier animal study, researchers investigated the epigenetic processes that control DAT expression in the midbrain of growing rats. This study showed that mRNA expression of HDAC2, DNMT1, DNMT3a, and DNMT3b decreased with age, while mRNA expression of HDAC5 and HDAC8 increased. This study confirmed that DAT protein and mRNA expression significantly increase with age. It has been mentioned that decreases in DAT promoter methylation correlate with the increased gene expression [ref. 53].
3.1.4. GABA
The processing of information by neurons and the organization of neural networks depend on maintaining the appropriate balance between excitation (E) and inhibition (I) in the brain [ref. 92,ref. 93,ref. 94]. Many neurological disorders, including ASD, take place when the modulatory mechanisms that control the E/I balance are dysfunctional [ref. 95,ref. 96]. γ-aminobutyric acid (GABA), an inhibitory neurotransmitter, plays a crucial role in maintaining the E/I balance. GAD1 is an important gene in regulating GABA levels, and it could be considered an excellent molecular biomarker in assessing E/I imbalances. DNA methylation in the regulatory region of the GAD1 gene has been compared between ASD patients and healthy controls in a previous clinical study. In the ASD group, it has been reported to exhibit a higher differential methylation pattern and also a greater CpG-wise variance [ref. 50].
3.1.5. Insulin
The effects of insulin and insulin-like growth factor 1 (IGF1) are mediated by insulin receptor substrate 2 (IRS2), which is a key downstream component of the signaling cascade [ref. 97]. Whole methylome analyses on the human placentas of high-risk pregnancies derived from the MARBLES (Markers of Autism Risk in Babies Learning Early Signs) prospective study identified 400 discriminated differentially methylated regions (DMRs) between children who were later diagnosed with ASD and typically developing controls. One of the reported DMRs is the hypermethylated DMR located at IRS2. Also, this study indicated that periconceptional maternal vitamin use modified the methylation at IRS2 [ref. 51].
3.1.6. Estrogen
It has been hypothesized that sex hormones play central roles in ASD etiopathogenesis due to the higher prevalence of ASD in the male population [ref. 98]. Increasing lines of evidence have demonstrated that estrogen receptors (ESR1 and ESR2) are involved in reproductive neuroendocrine behavior, memory, learning, and emotion regulation [ref. 99,ref. 100]. The pathogenesis of several neurologic and psychiatric disorders, including ASD, has been observed to contribute to ESR gene expression [ref. 52]. ASD patients have significantly reduced ESR2 mRNA and protein levels in their middle frontal gyrus compared with normal individuals [ref. 101]. The epigenetic regulation of the ESR2 gene in a clinical study on Chinese Han males with autism was explored. A relationship between phenotypic features of autism and DNA methylation was investigated. Indeed, in autistic individuals, a total of eight specific CpG sites showed hypermethylation, and four of these sites were found to have a positive correlation with the severity of autistic symptoms [ref. 52].
3.2. Genes Associated with Neural Matrix
3.2.1. RELN
Lintas C. and colleagues evaluated the status of RELN gene methylation in post-mortem temporocortical tissue samples from post-pubertal ASD case–control pairs and pre-pubertal ASD individuals. Although the mean percentage of methylation at each CpG position and the mean number of methylated CpGs along the whole RELN gene promoter did not differ significantly between ASD and control brains, the distribution of methylated CpG sites was noticeably different. ASD patients had a substantially greater number of methylated CpG islands and heavier methylation in the 5’ region of the RELN gene promoter. In contrast, in the 3’ region, the controls exhibited a higher number of methylated CpG positions and a more extensive level of methylation. In ASD brains, only the promoter region located furthest upstream (−458 to −364 bp) showed methylation, whereas in control brains, only the promoter region located furthest downstream (−131 to +1 bp) showed methylation. These results suggest that the pattern of methylation was distinct between ASD and control post-mortem brains. ASD-specific CpG sites in the most proximal gene promoter area might play a functional role by inhibiting RELN gene expression, potentially conferring ASD risk. It is important to mention that the findings could be affected by the limited sample size [ref. 54].
3.2.2. SHANK3
The first study analyzing altered methylation patterns in SHANK3 in ASD brain samples was published by Li Zhu et al. in 2014 This study involved DNA methylation profiling of five CpG islands (CGI-1 to CGI-5) in the SHANK3 gene using post-mortem brain tissues from both ASD patients and control subjects. In the cerebellum and cerebral cortex Brodmann region 19 (BA19), they found higher levels of DNA methylation in three intragenic CGIs (CGI-2, CGI-3, and CGI-4). In 15% of ASD brain tissues, elevated methylation was concentrated in CGI-2 and CGI-4. This indicated that SHANK3 is a useful biomarker for examining the role of gene–environment interaction in the etiology of ASD [ref. 55].
Recent investigations on mice exposed to fine particles reported alterations in SHANK3 methylation along with lower expression levels [ref. 102,ref. 103,ref. 104]. Accordingly, Kang Li and colleagues discovered that exposure to fine particles could result in an autism phenotype in early postnatal rats, which may be a result of the SHANK3 methylation level and the SHANK3 signaling pathway. The methylation level at three CpG sites in the SHANK3 gene from the brain tissues of exposed and control groups was evaluated to explore the epigenetic effects of early postnatal exposure to fine particles on the SHANK3 gene. At the epigenetic level, the reduced SHANK3 expression level in juvenile rats exposed to fine particles early in life may have occurred due to an increase in methylation [ref. 105].
3.2.3. ST8SIA2
Yang X et al. assessed the expression and methylation levels of the ST8SIA2 gene in the peripheral blood of ASD patients as compared to controls. The ASD group exhibited lower levels of ST8SIA2 gene expression compared to the control group. Additionally, these expression levels were linked to the severity of ASD in children, as well as their daily life skills, stereotype abnormalities, sensory abnormalities, and self-help ability. ASD children had elevated methylation levels at the ST8SIA2 gene’s Chr. 15: 92,984,625 and Chr. 15: 92,998,561 sites compared to the control group. The Chr. 15: 92,984,625 site was significantly associated with the stereotypical behaviors of children with ASD [ref. 56].
3.3. Genes Associated with Gene Expression
3.3.1. MeCP2
The methyl-CpG binding protein 2 (MeCP2) gene plays a key role in DNA methylation and transcriptional regulation, with the ability to activate or repress genes such as Brain-derived neurotrophic factor (BDNF) [ref. 14]. It is predominantly expressed in mature postnatal neurons, with slight variations across different brain regions, and is crucial for proper neuronal maturation [ref. 106] and function [ref. 107]. Mutations in MECP2 lead to Rett syndrome, a severe form of ASD characterized by impaired social behaviors, cognition, and coordination [ref. 108]. Despite the well-established role of MECP2 in Rett syndrome and occasional instances in autism [ref. 109], there is a lack of extensive research investigating the relationship between DNA methylation and MECP2 expression in samples of individuals with ASD.
The only clinical study examining the correlation between DNA methylation and the expression rate of the MeCP2 gene in brain samples from people with ASD was carried out by Nagarajan and colleagues in 2006. To achieve this, samples were collected from 14 frontal cortex samples from ASD patients, with six fusiform gyrus samples also being obtained. As expected, a significant reduction in MeCP2 expression was found in the experimental group. Further tests were performed to measure the level of DNA methylation across the MeCP2 gene. The promoter region, particularly at CpG site #3, showed a remarkable increase in the methylation level in cells with reduced gene expression. This site is well known as a binding target for transcription factors. However, this observation was more apparent in juvenile autism samples than in adult ones, suggesting that other indirect factors impact MeCP2 activation as the individual ages. In this study, the limited number of participants prevents making generalizations to larger populations [ref. 59].
In light of the putative role of the MeCP2 gene in ASD pathogenesis, the alteration in its expression served as a key metric for Zhou et al. [ref. 58] to evaluate the impact of air pollution (nitrogen dioxide and fine particles) on animal brains. Healthy rats were exposed to varying levels of air pollutants throughout full gestation and 12 days postnatal, after which brain samples were collected. The results revealed that rats residing in areas with higher pollution levels exhibited reduced instances of social preference and novelty (behaviors commonly associated with ASD), decreased MeCP2 content, and elevated methylation of the gene. These changes were particularly pronounced in rats exposed during postnatal days, highlighting the heightened sensitivity of DNA methylation to early-life insults. Also, in a recent study, researchers employed locus-specific epigenetic modulation to induce the methylation of the transcription start site (TTS) of MeCP2 in healthy male rats. As a result, there was a significant decrease in MeCP2 content in the hippocampus and, to a certain extent, in the parietal cortex. The alterations resulted in the development of behaviors resembling ASD in the rats, such as reduced sociability, more extended periods of self-grooming, poor memory function, and anxiety/depression. This observation places further emphasis on the causal connection between the methylation of the MeCP2 gene and disease symptoms [ref. 64].
3.3.2. NHIP
Using cutting-edge techniques to evaluate the methylome profile of genes, Zhu et al. [ref. 60] have introduced a gene formerly overlooked by prior studies on the genome of individuals with ASD. The gene in question, NHIP (neuronal hypoxia-inducible, placenta-associated), regulates other genes and optimizes neural cells’ response to environmental insults such as hypoxia and oxidative stress. NHIP expression at elevated levels acts as a protective factor, preventing neurodevelopmental defects. This explains the reduced content of NHIP in 46 placentae from ASD children in this study. However, this locus was found to be hypo-methylated instead of the usual association of higher methylation with lower gene expression. The authors interpret this as a reflection of previously or currently low levels of NHIP. Interestingly, early supplemental therapy with folic acid reversed the reduced NHIP levels and methylation, presenting NHIP as a promising therapeutic target to be considered in future studies. Given that this is the first and only ASD study involving this gene, and since it was carried out using whole-genome sequencing, further research focused on the link between NHIP expression, methylation, and ASD symptoms is warranted to draw an accurate conclusion.
3.4. Genes Involved in Disorders Associated with ASD
3.4.1. FMR1
Fragile X syndrome (FXS) is the leading monogenic cause of ASD. The most common cause of FXS is silencing of the fragile X mental retardation 1 (FMR1) gene by DNA methylation and aberrant heterochromatinization. Subsequently, the absence of the FMR1 protein (FMRP) leads to the FXS phenotype and autism [ref. 110,ref. 111]. In a clinical study on twelve males with FXS and atypical mosaicism, decreased expression levels of FMR1 mRNA and FMRP were observed in half of the study population. These reductions were correlated with the presence of ASD and lower IQ as well as the extent of DNA methylation [ref. 61]. The results align with those reported by Budimirovic et al., which confirmed the inverse association between a decrease in FMRP levels and the overall severity of the FXS phenotype. Also, two-fold lower FMRP levels were reported in FXS with ASD compared to FXS without ASD [ref. 62].
3.4.2. APOE
Abnormal methylation of the apolipoprotein E (APOE) gene has been found to be associated with Alzheimer’s disease, which might have overlapping mechanisms with ASD [ref. 112,ref. 113]. APOE hypermethylation has been observed in the peripheral blood DNA of pediatric patients with ASD. It has been documented that the percentage of methylation of a reference (PMR) of 15.4% was the most effective threshold for predicting ASD [ref. 63].
3.5. Genes Associated with Environmental Factors
3.5.1. WNT
Prenatal exposure to valproic acid, a medication used for mood stabilization and epilepsy treatment, has been shown to elevate the risk of ASD [ref. 114,ref. 115]. Preclinical studies have reported that ASD-like behaviors, including decreased socialization and activated stereotyping, were induced by the administration of VPA in pregnant rodents [ref. 116,ref. 117].
A preclinical study by Wang et al. revealed that rat exposure to VPA in early pregnancy induced demethylation in the promoter regions of WNT1 and WNT2 genes, thus increasing the mRNA and protein expression of WNT1 and WNT2 in the hippocampi and prefrontal cortexes of the offspring. This induced dysregulation in the WNT/b-catenin signaling pathway facilitates susceptibility to ASD [ref. 65].
3.5.2. KCC2
Several studies suggested that exposure to general anesthesia (GA) during cesarean section (CS) was associated with susceptibility to ASD [ref. 118]. However, a recent study did not support the role of GA in enhancing the risk of ASD [ref. 119].
The expression of the hypothalamic Kþ-2ClClexporter (KCC2) gene has been indicated to be decreased in a rat model exposed prenatally to sevoflurane, the most common anesthetic in pediatrics. This study reported increased methylation in the KCC2 promoter, concordant with the alterations in KCC2 expression. Another finding was that females were at a diminished risk compared to males of developing abnormalities in behavioral testing, memory, and the expression of KCC2 [ref. 69].
3.6. Other Genes
3.6.1. ACSF3
Recent research on Saudi autistic children found no DNA methylation in the specificity protein 1 (SP1) binding site within the Acyl-CoA synthetase family member 3 (ACSF3) promoter. However, a notable connection was reported between the gene expression of SP1 and ACSF3 in ASD patients [ref. 67].
3.6.2. PPP2R2C
Kimura et al. demonstrated that the PPP2R2C gene was downregulated and hypermethylated in ASD patients compared to the control group [ref. 68].
3.6.3. CYP2E1
An analysis of placentas from the MARBLES study showed hypomethylated DMR at the CYP2E1 gene, which is associated with ASD diagnosis (Table 2) [ref. 51].
Table 2: EWASs on ASD patients or animal models of ASD.
| Clinical Studies | |||||
|---|---|---|---|---|---|
| Subjects | ASD Diagnosis | Tissue | N of Subjects | Methods | Refs. |
| Autism cases vs. unrelated controls | ADI-R, ADOS | Post-mortem brain tissue (dorsolateral prefrontal cortex, temporal cortex, and cerebellum) | 19 autism cases, 21 unrelated controls | Infinium HumanMethylation450 BeadChip, bump hunting approach, and a permutation-based multiple testing correction method | [ref. 120] |
| autism cases vs. controls | ADI-R | Two cortical regions, prefrontal cortex (BA10), and anterior cingulate gyrus (BA24) | 13 autism cases, 12 controls | DNA was converted with sodium bisulfite and probed with the Illumina 450 K methylation array | [ref. 121] |
| ASD vs. controls | ADI-R | Anterior PFC, BA10, frontal cortex BA9 and BA8 | 16 male ASD and 15 male controls | 450 K BeadArray, targeted next-generation bisulfite sequencing | [ref. 122] |
| ASD vs. TD | ADI-R | Subventricular zone of the lateral ventricles | 17 ASD, 17 TD | ELISA, Illumina 450k Array-Based DNA Methylation Analyses | [ref. 123] |
| ASD vs. non-psychiatric control donors | Not mentioned | Post-mortem tissues samples [PFC, TC, and CBL] | 43 ASD, 38 controls | Illumina Infinium HumanMethylation450 BeadChip array of bisulfite convertedDNA | [ref. 124] |
| Case studyASD | ADI-R, ADOS | Post-mortem brain samples, SVZ, and insular cortex | 7 cases | Whole-genome Bisulfite Sequencing | [ref. 125] |
| MZ twins discordant | Not mentioned | Blood | 50 MZ twin pairs | Illumina Infinium HumanMethylation27 BeadChip array of bisulfite-converted DNA | [ref. 126] |
| ASD Monozygotic Twins | DSM-5, ADOS | Blood | 5 pairs of ASD-discordant MZ twins, 4 pairs of ASD-concordant MZ twins, and 30 pairs of sporadic patients | Illumina Infinium Human Methylation 450BeadChip array of bisulfite convertedDNA | [ref. 127] |
| MZ twins; concordant ASC vs. discordant ASC vs. control | ADI-R, ADOS | Blood | 6 concordant ASCs, 6 discordant ASCs, and 11 control pairs (total N = 46) | Illumina 27 K DNA methylation dataset;the edgeR package for differential expression analyses | [ref. 128] |
| ASD cases vs. matched controls | DPCRR, DNPR | Blood | 1316 (equal numbers of ASD cases and matched controls) | mQTL | [ref. 129] |
| MARBLESASD vs. TD | ADOS, ADI-R, MSEL, DSM-5 | Placenta | 20 ASD, 21 TD | Illumina HiSeq 2000 array of bisulfite converted DNA | [ref. 51] |
| TD vs. ASD | ADOS, MSEL | Umbilical cord blood samples | 56 TD, 50 ASD | Whole-genome bisulfite sequencing | [ref. 130] |
| Autism cases vs. controls | DSM-IV | Placental tissue | 14 cases, 10 control | Illumina HumanMethylation450 BeadChip (450K) | [ref. 131] |
| TD vs. ASD | ADOS, MSEL | Male cord blood samples | 39 TD, 35 ASD | Develop the R package Comethyl | [ref. 132] |
| ASD vs. TD (MARBLES and EARLI) | ADOS, ADI-R, MSEL, DSM-5 | Placenta | 46 ASD, 46 TD | WGBS, Illumina HiSeq X array of bisulfite converted DNA | [ref. 60] |
| ASD, FXSA, TD | ADOS, DSM-V | Peripheral blood | 23 ASD, 23 FXSA, 11 TD | Data processing and analysis were performed in R (version 4.0.2) using minfi, limma, DMRcate, ChAMP, and methylCC packages | [ref. 133] |
| Preclinical Studies | |||||
| Animal/Strain | Animal Model | Tissue | Method | Refs. | |
| Mice | autism mouse model (Cntnap2−/−) | striatum | Bioconductor package edgeR | [ref. 134] | |
| Mice | C58 mice | cerebellum, striatum, and cortex | MethylFlash Methylated DNA Quantification Kit (Epigentek) protocol | [ref. 135] | |
Abbreviations: TD: typically developing; ADOS: Autism Diagnostic Observation Schedule; MSEL: Mullen Scales of Early Learning; ADI-R: Autism Diagnostic Interview—Revised; BA10: Brodmann area 10; BA24: Brodmann area 24; PFC: prefrontal cortex; WGBS: whole-genome bisulfite sequencing; ASC: Autism Spectrum Condition; mQTL: DNA methylation quantitative trait loci; DPCRR: Danish Psychiatric Central Research Register; DNPR: Danish National Patient Register; ADOS-2: Autism Diagnostic Observation Schedule, Second Version; PFC: prefrontal cortex; TC: temporal cortex; CBL: cerebellum; FXSA: Fragile X syndrome with ASD; MZ: Monozygotic; Dup7: 7q11.23 duplication syndrome; SVZ: subventricular zone.
To conclude this section, we categorized the ASD-associated genes into various groups based on their clinical impact and discussed the current knowledge about the connection between their methylation and ASD. It is worth noting that, according to a recent meta-analysis, the phenotypical features of autism could be associated with the methylation of specific genes. For example, a connection between methylation in the ST8SIA2 gene and behavioral phenotypes of ASD has been reported. Also, severe forms of ASD showed higher methylation in the RELN gene [ref. 136].
4. Global DNA Methylation Profiling in Autism Spectrum Disorder
In recent years, due to advances in DNA methylation analysis for various applications, epigenome-wide association studies (EWASs) have become more common for assessing the relation between epigenetic alterations and disease occurrence by representing genome-wide methylation profiles.
The primary method mentioned in most epigenome-wide association studies involves assessing genome-wide DNA methylation using the Infinium HumanMethylation450K BeadChip array technology.
The fundamental benefit of global DNA methylation profiling over targeted DNA methylation studies is the ability to observe the full picture of the modifications connected to the pathology rather than just concentrating on individual genes of interest. As a result, this method enables the identification of novel potential pathways related to ASD pathophysiology mechanisms.
In this section, studies using an epigenome-wide approach method will be discussed. The studies will highlight changes in DNA methylation profiles linked to ASD or cognitive problems and repetitive behaviors in animal models. The majority of the information is derived from clinical research, which is described below. Details from the EWASs are shown in Table 2.
4.1. Clinical Evidence
This review divides clinical studies examining genome-wide DNA methylation alterations into two groups.
In the first group, the researchers assessed the DNA methylation profile in post-mortem brain tissues, while in the second group, the EWAS was conducted on peripheral tissues. In both groups, some important regions in the genome associated with ASD mechanism were found to be epigenetically altered, which suggests some new evidence for the relevance of autism and DNA methylation changes and can predict some novel insights for therapeutic approaches.
4.1.1. EWASs on Post-Mortem Brain Tissues
Ladd-Acosta C et al., in 2014, conducted a study to report the first genome-wide examination of DNA methylation outside of CpG islands and promoters in ASD among three brain regions of PFC, TC, and CBL [ref. 120]. This study compared the DNA methylation between 19 ASD cases and 21 controls of post-mortem brain samples using the Infinium Human Methylation 450 BeadChip (450K). They also adopted the “bump hunting” method to detect differentially methylated regions (DMRs) more effectively. The results showed four significant (adjusted p < 0.1) DMRs across the genome, with three DMRs found in the superior TC and one in the CBL. The DMR at the site of 3′ UTR of proline-rich transmembrane protein 1 (PRRT1), as well as DMRs within the promoter regions of tetraspanin 32 (TSPAN32) and chromosome 11 open reading frame 21 (C11orf21) were less methylated in the TC of the ASD cases in comparison to controls. Also, they identified a hypermethylated DMR in the TC of autistic cases, located in an intergenic region, with the nearest gene being ZFP57. Only one significant DMR located within SDHAP3 was identified in CBL samples, which was hypermethylated in ASD cases.
A study published in 2014 by Nardone S et al. investigated genome-wide DNA methylation patterns in 46 brain samples of 13 ASD cases and 12 controls in two brain areas, the prefrontal cortex (Brodmann Area 10; BA10) and the anterior cingulate gyrus (Brodmann Area 24; BA24) and also the significance of dysregulation in DNA methylation in the development of the disorder [ref. 121]. In this study, the Illumina 450 K methylation array identified many dysregulated CpGs in these two cortical regions of brains from ASD patients. The results revealed 5329 CpG sites that exhibited differential methylation patterns between control and autism cohorts in BA10 and 10,745 in BA24. Also, the researchers indicated that many immune response-associated genes were hypomethylated in autistic samples, and overexpression of these genes demonstrated the significant role of epigenetic alterations in the dysregulation of the immune system as an involving factor in ASD.
Nardone S and colleagues 2017 conducted another genome-wide methylation study on fluorescence-activated cell sorting-sorted neuronal nuclei from the anterior prefrontal cortex (PFC), BA10, BA9, and BA8 to assess the changes in DNA methylation patterns in cortical neurons of individuals with ASD [ref. 122]. In this study, by using next-generation bisulfite sequencing (NGBS), they validated 37 CpGs across 4 DMRs and showed a strong correlation between the methylation values detected by 450 K Bead Array and targeted NGBS. DMRs related to gamma-aminobutyric acid type B receptor subunit 1 (GABBR1) and Mir124-2 showed hypomethylation, and two other DMRs associated with family with sequence similarity 124 member B (FAM124B) and long non-coding RNA nuclear enriched abundant transcript 1 (lnNEAT1) were hypermethylated in ASD versus the control group. There is an association between Mir124 and social behavior, which has been mentioned in several studies [ref. 137,ref. 138].
Another genome-wide methylome analysis study published in 2019 detected significant DNA methylation defects in the subventricular zone of the lateral ventricles from the post-mortem brain of 17 ASD versus TD individuals [ref. 123]. The authors discovered significant differences (p < 0.05) in DNA methylation levels in certain genes. These genes are dynamic genomic regions, presumed to be involved in transcriptional regulation due to specific histone modification states. These differences were found between ASD cases and controls in the CpG sites from the 450k array dataset. They also expressed that 5-methylcytosine (5-mC) in ASD brain was hypomethylated at transcription start sites (TSSs), gene bodies, and canonical exons in comparison to controls, which confirmed the overall hypomethylation detected in young ASD cases from the 5-mC ELISA assay.
In 2019, Wong C et al. extracted and analyzed 223 post-mortem tissue samples from 43 individuals with ASD and 38 control donors to determine genome-wide patterns of DNA methylation in three brain areas of PFC, TC, and CB by using a Illumina Infinium Human Methylation-450 BeadChip array of bisulfite converted DNA [ref. 124]. The authors expressed that many co-methylated modules strongly related to ASD were found by cortical co-methylation network analysis, and these modules were shown to be enriched for genomic regions assigned to genes involved in immune system function, synaptic signaling, and neuronal regulation.
A recent case study conducted by Takahashi E and colleagues in 2022 showed the relationship between ASD and epigenetic analyses by using MRI and diffusion tractography, whole-genome bisulfite sequencing (WGBS), flow cytometry, and RT qPCR [ref. 125]. The findings revealed that there were distinct methylation patterns in the components of three important networks associated with ASD. This was discovered through an IPA analysis of WGBS data from samples of the dorsal insula region of the brain. These networks are involved in neurological diseases, developmental disorders, as well as nervous system development and function.
4.1.2. EWASs on Peripheral Tissues
Studies on Monozygotic Twin Pairs
To the best of our knowledge, three clinical studies reported genome-wide DNA methylation alteration in monozygotic twin pairs with autism to show the relevance of ASD and epigenetic alterations. Wong C et al., in 2014, investigated the impact of environmentally driven epigenetic factors on ASD by conducting a genome-wide analysis of DNA methylation in a cohort of 50 pairs of monozygotic twins [ref. 126]. Numerous differential methylation areas linked to ASD were discovered using within-twin and between-group analysis. Their results also showed significant correlations between DNA methylation and autistic trait scores.
In another study in 2019, Liang S et al. investigated the contribution of DNA methylation to ASD etiology in discordant monozygotic twins [ref. 127]. The study involved the genome-wide analysis of DNA methylation using samples obtained from five pairs of monozygotic twins discordant for ASD. Their results showed a total of 2397 differentially methylated genes. Additionally, this gene list’s annotations in the Kyoto Encyclopedia of Genes and Genomes showed that monozygotic twins with ASD predominantly activated the neurotrophin signaling pathway. By using bisulfite-pyrosequencing, the methylation of the SH2B1 gene was further validated in monozygotic twins with ASD-discordant or ASD-concordant traits as well as a group of 30 pairs of sporadic case–control cases. The findings demonstrated that ASD-discordant monozygotic twins had a more considerable DNA methylation difference than ASD-concordant monozygotic twins. Additionally, the SH2B1 Chr.16:28856743 analysis revealed notable variations in DNA methylation between the case and control groups. These findings imply that the etiology of ASD is related to abnormal SH2B1 methylation.
Studies on ASD Patients
Hanon E et al., in 2018, was the first study to examine prenatal epigenetic variation linked to autism quantified neonatal methylomic alteration in 1263 infants with a 1:1 ratio of ASD and TD individuals [ref. 129]. Neonatal blood samples were obtained proximal to birth, and then the authors developed an enormous database of DNA methylation quantitative trait loci (mQTL), which were utilized to define the molecular implications of genetic variations linked with ASD. The results of this study demonstrated that neonatal DNA methylation did not significantly differ in ASD individuals in comparison to controls; however, methylomic variation at certain loci in blood at birth was revealed to be related to an elevated polygenic burden for autism. Another study published in 2022 assessed the global DNA methylation profiles of peripheral blood samples of 23 ASD cases, 23 cases of FXS with ASD (FXSA), and 11 TD children by using the Human Methylation EPIC Bead Chip [ref. 133]. They found out DNA methylation alteration has a key role in the pathology of these disorders by characterizing the methylome profile of each group.
Zhu Y and colleagues (2019) investigated the role of epigenetics in ASD occurrence risk by using a novel strategy to detect differentially methylated regions (DMRs) in complete methylomes from placenta samples of male children with ASD compared to the control group [ref. 51]. The researchers in this study discovered new methylation alterations at the CYP2E1 and IRS2 genes, which showed genome-wide significant differences between ASD and the control group. Both the CYP2E1 and IRS2 DMRs may be found in situations close to the transcription start site (TSS) in CpG shore intragenic regions. This finding is consistent with enriching TSS flanking regions and H3K4me3 promoter marks in the 400 ASD DMRs. This study has also shown that both of these genes are involved in protein synthesis, cell proliferation, and cell metabolism, which is the same as the results of similar studies of gene pathways in ASD showed. In 2020, Mordaunt C et al. conducted a whole-genome bisulfite sequencing of 152 umbilical cord blood samples to exhibit the association of DNA methylation and ASD at birth [ref. 130]. The autosomal ASD DMRs detected in this study were shown to be highly enriched for promoter and bivalent chromatin states in the majority of cell types. Moreover, the binding sites of methyl-sensitive transcription factors, which are important in developing the brain, were significantly found in those DMRs identified in cord blood.
Bahado-Singh R and colleagues 2021 published a study about genome-wide methylation in the placental tissue of ASD patients using the Illumina 450K array and artificial intelligence for analysis of differentially methylated loci [ref. 131]. The results showed that full-term birth ASD patients had 9655 CpGs with significantly altered methylation. Additionally, 2802 CpGs were intergenic markers, and 6853 (4129 genes) were intragenic. They found 3820 CpGs that were hypomethylated and 3033 that were hypermethylated for intragenic CpG markers. The quantity of synapse, microtubule dynamics, neuritogenesis, and abnormal morphology of neurons are four biological functions that were significantly overexpressed in ASD cases compared to controls.
Mordaunt C et al. conducted another study in 2022 about comethyl, which was a network-based methylome approach to investigate the correlation between DNA methylation and autism [ref. 132]. The researchers reported that differentially methylated genes that had important roles in developing brain and glial functions were detected in their sequencing-based analysis of cord blood samples from autism cases in comparison to controls. And at last, in 2022, Zhu Y and colleagues performed a WGBS in the placenta to investigate a previously indeterminant ASD risk gene, LOC105373085, renamed NHIP [ref. 60]. The results showed that NHIP, which had a role in cell proliferation, regulating synapses and neurogenesis directly and indirectly, was hypomethylated and had significantly increased expression in ASD vs. controls.
4.2. Preclinical Evidence
Only two preclinical studies have examined the DNA methylation changes in autism in a genome-wide aspect. In 2015, Papale L et al. assessed a genome-wide map of striatal 5-hydroxymethylcytosine (5hmC) in an autism mouse model by using chemical labeling and an affinity purification method coupled with high-throughput sequencing technology [ref. 134]. 5hmC, as an altered DNA type, is dramatically found in post-mitotic neurons and is relevant to the active transcription of neuronal genes. The results showed many differentially hydroxymethylated regions (DHMRs) in the genome profile, which were significantly located in some known ASD-associated genes. This study guided us to understand the role of 5hmC in ASD and suggest some novel ASD-associated genes. Another preclinical study published by Muehlmann A et al. in 2019 examined the hypothesis that a methyl donor-supplemented diet has a role in the occurrence of ASD in C58 mice by modifying DNA methylation [ref. 135]. The results showed that exposure to this diet affects DNA methylation increases in the brain tissues of the autistic group, which is significantly related to the frequency and patterns of repetitive behavior in the mouse model of autism.
Taken together, based on clinical studies on genome-wide DNA methylation alterations in autism, evidence of epigenetic changes was found in both post-mortem brain tissues and peripheral tissues. DMRs in genes associated with ASD were identified in various studies on post-mortem brain tissues, suggesting a potential connection between autism and DNA methylation changes [ref. 120,ref. 121,ref. 122,ref. 123,ref. 124,ref. 125]. These findings could offer new perspectives for therapeutic approaches. In other ways, conducting EWASs on peripheral tissues demonstrated that several studies have investigated DNA methylation alterations in twins with autism, showing correlations between DNA methylation and autistic traits and differential gene expression in ASD-discordant twins [ref. 126,ref. 127,ref. 128]. Additionally, studies on ASD patients revealed that neonatal DNA methylation variations are related to an elevated polygenic burden for autism, and DNA methylation alterations play a key role in the pathology of ASD and related disorders [ref. 51,ref. 60,ref. 129,ref. 130,ref. 131,ref. 132,ref. 133]. Two preclinical studies have examined DNA methylation changes in autism on a genome-wide level. One study found altered DNA type 5hmC in an autism mouse model, suggesting some novel ASD-associated genes [ref. 134]. Another study suggested that a diet supplemented with methyl donors may affect DNA methylation in the brain tissues of autistic mice, influencing repetitive behavior patterns [ref. 135]. Specific details from these studies can be found in Table 2.
5. Conclusions
This review collates the existing literature on the association between DNA methylation and ASD. Although it is generally agreed that changes in DNA methylation patterns are associated with ASD phenotype and pathology, the potential use of DNA methylation as a predictive or diagnostic biomarker for ASD is still being debated. Given the limited number of studies and the inconsistency between their findings, DNA methylation in ASD requires more thorough investigation. As represented in Table 1, DNA methylation alterations at certain genes show apparent discrepancies among studies. In other words, the available information did not concur to determine whether DNA hypomethylation or hypermethylation within each particular gene is associated with ASD. Although transcriptional expression studies currently offer valuable insights into the significance of epigenetic changes in the clinical evaluation of ASD, the available data on DNA methylation in each specific gene were limited to only one or two studies. However, in more detail, methylation at HTR2A, OXTR, and MeCP was more extensively evaluated.
Considering the growing recognition of epigenetic alterations associated with ASD, identifying the most impactful epigenetic profiles will enable researchers to concentrate their studies on the genes most likely to be relevant to disease pathogenesis. Our narrative review highlights the significance of DNA methylation among epigenetic changes. However, based on the papers reviewed here, current knowledge is insufficient to determine of which specific gene’s methylation could be used as a biomarker or drug target.
In EWAS approaches, which have the ability to identify the potential mechanisms associated with ASD pathophysiology, the full picture of modifications was manifested rather than only focusing on a single specific gene. Here, genome-wide research on post-mortem brain tissues as well as peripheral tissues suggests the significant relevance of DNA methylation in some important regions of the genome with ASD. However, further meta-analyses and more extensive controlled trials are required regarding EWAS limitations, including various techniques and statistical analysis approaches.
Several issues should be taken into consideration before accepting any final conclusion: (1) Although most of the clinical studies presented here were performed on children and the male population, significant confounding effects could be expected from differences in race, ASD diagnostic criteria, sample size, and technical perspectives. (2) Different studies concentrated on various DNA methylation sites within a single gene, which need to be highlighted. For instance, the methylation of CpG sites at 1438A/G and 102T/C loci of the HTR2A gene, MT1, and MT3 regions of the OXTR gene were proposed as candidate diagnostic biomarkers [ref. 44,ref. 47]. (3) Investigations on the post-mortem brain are the most reliable studies for comprehending the pathological processes taking place in the brains of ASD patients, which was not feasible in the majority of the clinical studies examined in this review. (4) Indeed, despite the limitations of animal studies, the mechanistic link between alternation in DNA methylation at specific genes, brain area, and behavior is taken chiefly from preclinical evidence, which is still scarce.
Eventually, in recent years, there was a rise in studies focusing on DNA methylation to understand the epigenetic mechanisms related to ASD. Clinical studies on peripheral DNA have identified specific DNA methylation markers linked to ASD. Nonetheless, further studies on animal models would aid in realizing etiopathogenetic processes involved in ASD and also in identifying putative molecular targets for innovative therapeutic approaches.
References
- T. Hirota, B.H. King. Autism Spectrum Disorder: A Review. JAMA, 2023. [DOI | PubMed]
- T. Kodak, S. Bergmann. Autism Spectrum Disorder: Characteristics, Associated Behaviors, and Early Intervention. Pediatr. Clin. N. Am., 2020. [DOI | PubMed]
- A.V. Buescher, Z. Cidav, M. Knapp, D.S. Mandell. Costs of autism spectrum disorders in the United Kingdom and the United States. JAMA Pediatr., 2014. [DOI | PubMed]
- B.H. King. Psychiatric comorbidities in neurodevelopmental disorders. Curr. Opin. Neurol., 2016. [DOI | PubMed]
- A. Jain, D. Spencer, W. Yang, J.P. Kelly, C.J. Newschaffer, J. Johnson, J. Marshall, F. Azocar, L.P. Tabb, T. Dennen. Injuries among children with autism spectrum disorder. Acad. Pediatr., 2014. [DOI | PubMed]
- T. Hirvikoski, E. Mittendorfer-Rutz, M. Boman, H. Larsson, P. Lichtenstein, S. Bölte. Premature mortality in autism spectrum disorder. Br. J. Psychiatry, 2016. [DOI | PubMed]
- D.E. Schendel, M. Overgaard, J. Christensen, L. Hjort, M. Jørgensen, M. Vestergaard, E.T. Parner. Association of Psychiatric and Neurologic Comorbidity with Mortality Among Persons with Autism Spectrum Disorder in a Danish Population. JAMA Pediatr., 2016. [DOI | PubMed]
- R. Lordan, C. Storni, C.A. De Benedictis. Autism Spectrum Disorders: Diagnosis and Treatment. Autism Spectrum Disorders, 2021
- W. Weuring, J. Geerligs, B.P. Koeleman. Gene therapies for monogenic autism spectrum disorders. Genes, 2021. [DOI | PubMed]
- D. Antaki, J. Guevara, A.X. Maihofer, M. Klein, M. Gujral, J. Grove, C.E. Carey, O. Hong, M.J. Arranz, A. Hervas. A phenotypic spectrum of autism is attributable to the combined effects of rare variants, polygenic risk and sex. Nat. Genet., 2022. [DOI | PubMed]
- Y. Yasuda, J. Matsumoto, K. Miura, N. Hasegawa, R. Hashimoto. Genetics of autism spectrum disorders and future direction. J. Hum. Genet., 2023. [DOI | PubMed]
- P. Karimi, E. Kamali, S.M. Mousavi, M. Karahmadi. Environmental factors influencing the risk of autism. J. Res. Med. Sci., 2017. [PubMed]
- C. Ladd-Acosta, M.D. Fallin. The role of epigenetics in genetic and environmental epidemiology. Epigenomics, 2016. [DOI | PubMed]
- M. Chahrour, S.Y. Jung, C. Shaw, X. Zhou, S.T.C. Wong, J. Qin, H.Y. Zoghbi. MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science, 2008. [DOI | PubMed]
- J.A.S. Vorstman, W.G. Staal, E. van Daalen, H. van Engeland, P.F.R. Hochstenbach, L. Franke. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol. Psychiatry, 2006. [DOI]
- A. Bremer, M. Giacobini, M. Nordenskjöld, K. Brøndum-Nielsen, M. Mansouri, N. Dahl, B. Anderlid, J. Schoumans. Screening for copy number alterations in loci associated with autism spectrum disorders by two-color multiplex ligation-dependent probe amplification. Am. J. Med. Genet. Part B Neuropsychiatr. Genet., 2010. [DOI | PubMed]
- N.S. Mohammad, J.M.N. Jain, K.P. Chintakindi, R.P. Singh, U. Naik, R.R.D. Akella. Aberrations in folate metabolic pathway and altered susceptibility to autism. Psychiatr. Genet., 2009. [DOI | PubMed]
- X. Liu, F. Solehdin, I.L. Cohen, M.G. Gonzalez, E.C. Jenkins, M.E.S. Lewis, J.J.A. Holden. Population- and Family-Based Studies Associate the MTHFR Gene with Idiopathic Autism in Simplex Families. J. Autism Dev. Disord., 2011. [DOI | PubMed]
- A. Smith, F. Kaufman, M.S. Sandy, A. Cardenas. Cannabis Exposure During Critical Windows of Development: Epigenetic and Molecular Pathways Implicated in Neuropsychiatric Disease. Curr. Environ. Health Rep., 2020. [DOI | PubMed]
- B. Abd-Nikfarjam, A. Dolati-Somarin, V. Baradaran Rahimi, V.R. Askari. Cannabinoids in neuroinflammatory disorders: Focusing on multiple sclerosis, Parkinsons, and Alzheimers diseases. Biofactors, 2023. [DOI | PubMed]
- V.R. Askari, V. Baradaran Rahimi, R. Shafiee-Nick. Low Doses of β-Caryophyllene Reduced Clinical and Paraclinical Parameters of an Autoimmune Animal Model of Multiple Sclerosis: Investigating the Role of CB(2) Receptors in Inflammation by Lymphocytes and Microglial. Brain Sci., 2023. [DOI | PubMed]
- V.R. Askari, R. Shafiee-Nick. Promising neuroprotective effects of β-caryophyllene against LPS-induced oligodendrocyte toxicity: A mechanistic study. Biochem. Pharmacol., 2019. [DOI | PubMed]
- V.R. Askari, R. Shafiee-Nick. The protective effects of β-caryophyllene on LPS-induced primary microglia M(1)/M(2) imbalance: A mechanistic evaluation. Life Sci., 2019. [DOI | PubMed]
- N.Q.V. Tran, K. Miyake. Neurodevelopmental Disorders and Environmental Toxicants: Epigenetics as an Underlying Mechanism. Int. J. Genom., 2017. [DOI]
- K.P. Keil, P.J. Lein. DNA methylation: A mechanism linking environmental chemical exposures to risk of autism spectrum disorders?. Environ. Epigenetics, 2016. [DOI | PubMed]
- L.D. Moore, T. Le, G. Fan. DNA Methylation and Its Basic Function. Neuropsychopharmacology, 2013. [DOI | PubMed]
- J. Feng, Y. Zhou, S.L. Campbell, T. Le, E. Li, J.D. Sweatt, A.J. Silva, G. Fan. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci., 2010. [DOI | PubMed]
- O. Mortusewicz, L. Schermelleh, J. Walter, M.C. Cardoso, H. Leonhardt. Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. USA, 2005. [DOI | PubMed]
- U. Aapola, R. Lyle, K. Krohn, S.E. Antonarakis, P. Peterson. Isolation and initial characterization of the mouse Dnmt3l gene. Cytogenet. Cell Genet., 2001. [DOI | PubMed]
- K. Hata, M. Okano, H. Lei, E. Li. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development, 2002. [DOI | PubMed]
- D. Jia, R.Z. Jurkowska, X. Zhang, A. Jeltsch, X. Cheng. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature, 2007. [DOI | PubMed]
- W. Mayer, A. Niveleau, J. Walter, R. Fundele, T. Haaf. Demethylation of the zygotic paternal genome. Nature, 2000. [DOI | PubMed]
- Z. Paroush, I. Keshet, J. Yisraeli, H. Cedar. Dynamics of demethylation and activation of the alpha-actin gene in myoblasts. Cell, 1990. [DOI | PubMed]
- H. Gujar, D.J. Weisenberger, G. Liang. The Roles of Human DNA Methyltransferases and Their Isoforms in Shaping the Epigenome. Genes, 2019. [DOI | PubMed]
- C. Coulondre, J.H. Miller, P.J. Farabaugh, W. Gilbert. Molecular basis of base substitution hotspots in Escherichia coli. Nature, 1978. [DOI | PubMed]
- A.P. Bird. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res., 1980. [DOI | PubMed]
- A. Bird, M. Taggart, M. Frommer, O.J. Miller, D. Macleod. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell, 1985. [DOI | PubMed]
- R.A. Irizarry, C. Ladd-Acosta, B. Wen, Z. Wu, C. Montano, P. Onyango, H. Cui, K. Gabo, M. Rongione, M. Webster. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet., 2009. [DOI | PubMed]
- S. Saxonov, P. Berg, D.L. Brutlag. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA, 2006. [DOI | PubMed]
- A. Menke, E.B. Binder. Epigenetic alterations in depression and antidepressant treatment. Dialogues Clin. Neurosci., 2014. [DOI | PubMed]
- K. Skvortsova, N. Iovino, O. Bogdanović. Functions and mechanisms of epigenetic inheritance in animals. Nat. Rev. Mol. Cell Biol., 2018. [DOI | PubMed]
- H.S. Jang, W.J. Shin, J.E. Lee, J.T. Do. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes, 2017. [DOI | PubMed]
- R. Lister, E.A. Mukamel, J.R. Nery, M. Urich, C.A. Puddifoot, N.D. Johnson, J. Lucero, Y. Huang, A.J. Dwork, M.D. Schultz. Global epigenomic reconfiguration during mammalian brain development. Science, 2013. [DOI | PubMed]
- S. Guhathakurta, A.S. Singh, S. Sinha, A. Chatterjee, S. Ahmed, S. Ghosh, R. Usha. Analysis of serotonin receptor 2A gene (HTR2A): Association study with autism spectrum disorder in the Indian population and investigation of the gene expression in peripheral blood leukocytes. Neurochem. Int., 2009. [DOI | PubMed]
- D. Hranilovic, S. Blazevic, J. Stefulj, P. Zill. DNA methylation analysis of HTR2A regulatory region in leukocytes of autistic subjects. Autism Res., 2016. [DOI | PubMed]
- Z. Hu, X. Ying, L. Huang, Y. Zhao, D. Zhou, J. Liu, J. Zhong, T. Huang, W. Zhang, F. Cheng. Association of human serotonin receptor 4 promoter methylation with autism spectrum disorder. Medicine, 2020. [DOI | PubMed]
- M. Elagoz Yuksel, B. Yuceturk, O.F. Karatas, M. Ozen, B. Dogangun. The altered promoter methylation of oxytocin receptor gene in autism. J. Neurogenet., 2016. [DOI | PubMed]
- E. Andari, S. Nishitani, G. Kaundinya, G.A. Caceres, M.J. Morrier, O. Ousley, A.K. Smith, J.F. Cubells, L.J. Young. Epigenetic modification of the oxytocin receptor gene: Implications for autism symptom severity and brain functional connectivity. Neuropsychopharmacology, 2020. [DOI | PubMed]
- J. Wieting, K. Jahn, S. Bleich, H. Frieling, M. Deest. A targeted long-read sequencing approach questions the association of OXTR methylation with high-functioning autism. Clin. Epigenetics, 2023. [DOI | PubMed]
- G. Pearson, C. Song, S. Hohmann, T. Prokhorova, T.M. Sheldrick-Michel, T. Knöpfel. DNA Methylation Profiles of GAD1 in Human Cerebral Organoids of Autism Indicate Disrupted Epigenetic Regulation during Early Development. Int. J. Mol. Sci., 2022. [DOI | PubMed]
- Y. Zhu, C.E. Mordaunt, D.H. Yasui, R. Marathe, R.L. Coulson, K.W. Dunaway, J.M. Jianu, C.K. Walker, S. Ozonoff, I. Hertz-Picciotto. Placental DNA methylation levels at CYP2E1 and IRS2 are associated with child outcome in a prospective autism study. Hum. Mol. Genet., 2019. [DOI | PubMed]
- X. Wang, S. Liang, Y. Sun, H. Li, F. Endo, M. Nakao, N. Saitoh, L. Wu. Analysis of estrogen receptor β gene methylation in autistic males in a Chinese Han population. Metab. Brain Dis., 2017. [DOI | PubMed]
- A.L. Green, A. Eid, L. Zhan, H. Zarbl, G.L. Guo, J.R. Richardson. Epigenetic regulation of the ontogenic expression of the dopamine transporter. Front. Genet., 2019. [DOI | PubMed]
- C. Lintas, R. Sacco, A.M. Persico. Differential methylation at the RELN gene promoter in temporal cortex from autistic and typically developing post-puberal subjects. J. Neurodev. Disord., 2016. [DOI | PubMed]
- L. Zhu, X. Wang, X.-L. Li, A. Towers, X. Cao, P. Wang, R. Bowman, H. Yang, J. Goldstein, Y.-J. Li. Epigenetic dysregulation of SHANK3 in brain tissues from individuals with autism spectrum disorders. Hum. Mol. Genet., 2014. [DOI | PubMed]
- X. Yang, L. Li, X. Chai, J. Liu. The association between ST8SIA2 gene and behavioral phenotypes in children with autism spectrum disorder. Front. Behav. Neurosci., 2022. [DOI | PubMed]
- E. Blumkin, T. Levav-Rabkin, O. Melamed, D. Galron, H.M. Golan. Gender-specific effect of Mthfr genotype and neonatal vigabatrin interaction on synaptic proteins in mouse cortex. Neuropsychopharmacology, 2011. [DOI | PubMed]
- Q. Zhou, Y. Tian, C. Xu, J. Wang, Y. Jin. Prenatal and postnatal traffic pollution exposure, DNA methylation in Shank3 and MeCP2 promoter regions, H3K4me3 and H3K27me3 and sociability in rats’ offspring. Clin. Epigenetics, 2021. [DOI | PubMed]
- R. Nagarajan, A. Hogart, Y. Gwye, M.R. Martin, J.M. LaSalle. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics, 2006. [DOI]
- Y. Zhu, J.A. Gomez, B.I. Laufer, C.E. Mordaunt, J.S. Mouat, D.C. Soto, M.Y. Dennis, K.S. Benke, K.M. Bakulski, J. Dou. Placental methylome reveals a 22q13. 33 brain regulatory gene locus associated with autism. Genome Biol., 2022. [DOI | PubMed]
- P. Jiraanont, M. Kumar, H.-T. Tang, G. Espinal, P.J. Hagerman, R.J. Hagerman, N. Chutabhakdikul, F. Tassone. Size and methylation mosaicism in males with Fragile X syndrome. Expert. Rev. Mol. Diagn., 2017. [DOI | PubMed]
- D.B. Budimirovic, A. Schlageter, S. Filipovic-Sadic, D.D. Protic, E. Bram, E.M. Mahone, K. Nicholson, K. Culp, K. Javanmardi, J. Kemppainen. A genotype-phenotype study of high-resolution FMR1 nucleic acid and protein analyses in fragile X patients with neurobehavioral assessments. Brain Sci., 2020. [DOI | PubMed]
- Z. Hu, Y. Yang, Y. Zhao, H. Yu, X. Ying, D. Zhou, J. Zhong, Z. Zheng, J. Liu, R. Pan. APOE hypermethylation is associated with autism spectrum disorder in a Chinese population. Exp. Ther. Med., 2018. [DOI | PubMed]
- Z. Lu, Z. Liu, W. Mao, X. Wang, X. Zheng, S. Chen, B. Cao, S. Huang, X. Zhang, T. Zhou. Locus-specific DNA methylation of Mecp2 promoter leads to autism-like phenotypes in mice. Cell Death Dis., 2020. [DOI | PubMed]
- Z. Wang, L. Xu, X. Zhu, W. Cui, Y. Sun, H. Nishijo, Y. Peng, R. Li. Demethylation of Specific Wnt/β-Catenin Pathway Genes and its Upregulation in Rat Brain Induced by Prenatal Valproate Exposure. Anat. Rec. Adv. Integr. Anat. Evol. Biol., 2010. [DOI]
- M.A. Konopko, A.L. Densmore, B.K. Krueger. Sexually dimorphic epigenetic regulation of brain-derived neurotrophic factor in fetal brain in the valproic acid model of autism spectrum disorder. Dev. Neurosci., 2017. [DOI | PubMed]
- K. Algothmi, A. Alqurashi, A. Alrofaidi, M. Alharbi, R. Farsi, N. Alburae, M. Ganash, S. Azhari, F. Basingab, A. Almuhammadi. DNA Methylation Level of Transcription Factor Binding Site in the Promoter Region of Acyl-CoA Synthetase Family Member 3 (ACSF3) in Saudi Autistic Children. Pharmgenomics Pers. Med., 2022. [DOI | PubMed]
- R. Kimura, M. Nakata, Y. Funabiki, S. Suzuki, T. Awaya, T. Murai, M. Hagiwara. An epigenetic biomarker for adult high-functioning autism spectrum disorder. Sci. Rep., 2019. [DOI | PubMed]
- L.S. Ju, J.J. Yang, T.E. Morey, N. Gravenstein, C.N. Seubert, J.L. Resnick, J.Q. Zhang, A.E. Martynyuk. Role of epigenetic mechanisms in transmitting the effects of neonatal sevoflurane exposure to the next generation of male, but not female, rats. Br. J. Anaesth., 2018. [DOI | PubMed]
- A. Bonnin, P. Levitt. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience, 2011. [DOI | PubMed]
- E.H. Cook, B.L. Leventhal. The serotonin system in autism. Curr. Opin. Pediatr., 1996. [DOI | PubMed]
- J.B. Vincent, A. Noor, C. Windpassinger, P.J. Gianakopoulos, T. Schwarzbraun, S.E. Alfred, B. Stachowiak, S.W. Scherer, W. Roberts, K. Wagner. Characterization of a de novo translocation t (5; 18)(q33. 1; q12. 1) in an autistic boy identifies a breakpoint close to SH3TC2, ADRB2, and HTR4 on 5q, and within the desmocollin gene cluster on 18q. Am. J. Med. Genet. Part B Neuropsychiatr. Genet., 2009. [DOI]
- K. Williams, A. Brignell, M. Randall, N. Silove, P. Hazell. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database Syst. Rev., 2013. [DOI]
- G. Spurlock, A. Heils, P. Holmans, J. Williams, U. D’Souza, A. Cardno, K. Murphy, L. Jones, P.R. Buckland, P. McGuffin. A family based association study of T102C polymorphism in 5HT2A and schizophrenia plus identification of new polymorphisms in the promoter. Mol. Psychiatry, 1998. [DOI | PubMed]
- J.T. Warren, M.L. Peacock, L.C. Rodriguez, J.K. Fink. An MspI polymorphism in the hyman serotonin receptor gene (HTR2): Detection by DGGE and RFLP analysis. Hum. Mol. Genet., 1993. [DOI]
- I.H. Cho, H.J. Yoo, M. Park, Y.S. Lee, S.A. Kim. Family-based association study of 5-HTTLPR and the 5-HT2A receptor gene polymorphisms with autism spectrum disorder in Korean trios. Brain Res., 2007. [DOI | PubMed]
- J. Veenstra-VanderWeele, S.J. Kim, C. Lord, R. Courchesne, N. Akshoomoff, B.L. Leventhal, E. Courchesne, E.H. Cook. Transmission disequilibrium studies of the serotonin 5-HT2A receptor gene (HTR2A) in autism. Am. J. Med. Genet., 2002. [DOI | PubMed]
- D.S. Quintana, J. Rokicki, D. van der Meer, D. Alnæs, T. Kaufmann, A. Córdova-Palomera, I. Dieset, O.A. Andreassen, L.T. Westlye. Oxytocin pathway gene networks in the human brain. Nat. Commun., 2019. [DOI | PubMed]
- R.A. Bethlehem, M.V. Lombardo, M.-C. Lai, B. Auyeung, S.K. Crockford, J. Deakin, S. Soubramanian, A. Sule, P. Kundu, V. Voon. Intranasal oxytocin enhances intrinsic corticostriatal functional connectivity in women. Transl. Psychiatry, 2017. [DOI | PubMed]
- A.J. Guastella, S.L. Einfeld, K.M. Gray, N.J. Rinehart, B.J. Tonge, T.J. Lambert, I.B. Hickie. Intranasal oxytocin improves emotion recognition for youth with autism spectrum disorders. Biol. Psychiatry, 2010. [DOI | PubMed]
- E. Hollander, S. Novotny, M. Hanratty, R. Yaffe, C.M. DeCaria, B.R. Aronowitz, S. Mosovich. Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger’s disorders. Neuropsychopharmacology, 2003. [DOI | PubMed]
- H. Kosaka, T. Munesue, M. Ishitobi, M. Asano, M. Omori, M. Sato, A. Tomoda, Y. Wada. Long-term oxytocin administration improves social behaviors in a girl with autistic disorder. BMC Psychiatry, 2012. [DOI | PubMed]
- M. Moerkerke, N. Daniels, L. Tibermont, T. Tang, M. Evenepoel, S. Van der Donck, E. Debbaut, J. Prinsen, V. Chubar, S. Claes. Chronic oxytocin administration stimulates the oxytocinergic system in children with autism. Nat. Commun., 2024. [DOI | PubMed]
- S.K. Siecinski, S.N. Giamberardino, M. Spanos, A.C. Hauser, J.R. Gibson, T. Chandrasekhar, M.D.P. Trelles, C.M. Rockhill, M.L. Palumbo, A.W. Cundiff. Genetic and epigenetic signatures associated with plasma oxytocin levels in children and adolescents with autism spectrum disorder. Autism Res., 2023. [DOI | PubMed]
- M. Evenepoel, M. Moerkerke, N. Daniels, V. Chubar, S. Claes, J. Turner, B. Vanaudenaerde, L. Willems, J. Verhaeghe, J. Prinsen. Endogenous oxytocin levels in children with autism: Associations with cortisol levels and oxytocin receptor gene methylation. Transl. Psychiatry, 2023. [DOI | PubMed]
- D. LoParo, I. Waldman. The oxytocin receptor gene (OXTR) is associated with autism spectrum disorder: A meta-analysis. Mol. Psychiatry, 2015. [DOI | PubMed]
- B.K. Gorentla, R.A. Vaughan. Differential effects of dopamine and psychoactive drugs on dopamine transporter phosphorylation and regulation. Neuropharmacology, 2005. [DOI | PubMed]
- T.J. Spencer, J. Biederman, S.V. Faraone, B.K. Madras, A.A. Bonab, D.D. Dougherty, H. Batchelder, A. Clarke, A.J. Fischman. Functional genomics of attention-deficit/hyperactivity disorder (ADHD) risk alleles on dopamine transporter binding in ADHD and healthy control subjects. Biol. Psychiatry, 2013. [DOI | PubMed]
- K. Nakamura, Y. Sekine, Y. Ouchi, M. Tsujii, E. Yoshikawa, M. Futatsubashi, K.J. Tsuchiya, G. Sugihara, Y. Iwata, K. Suzuki. Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch. Gen. Psychiatry, 2010. [DOI | PubMed]
- E. Rodríguez-Traver, O. Solís, E. Díaz-Guerra, Ó. Ortiz, E. Vergaño-Vera, H.R. Méndez-Gómez, P. García-Sanz, R. Moratalla, C. Vicario-Abejón. Role of Nurr1 in the generation and differentiation of dopaminergic neurons from stem cells. Neurotox. Res., 2016. [DOI | PubMed]
- K.C. Schmitt, M.E. Reith. Regulation of the dopamine transporter: Aspects relevant to psychostimulant drugs of abuse. Ann. N. Y. Acad. Sci., 2010. [DOI | PubMed]
- S. Zhou, Y. Yu. Synaptic EI balance underlies efficient neural coding. Front. Neurosci., 2018. [DOI | PubMed]
- V.R. Askari, V. Baradaran Rahimi, A. Ghorbani, H. Rakhshandeh. Hypnotic Effect of Ocimum basilicum on Pentobarbital-Induced Sleep in Mice. Iran. Red. Crescent Med. J., 2016. [DOI | PubMed]
- V. Baradaran Rahimi, V.R. Askari, A.S. Tajani, A. Hosseini, H. Rakhshandeh. Evaluation of the Sleep-Prolonging Effect of Lagenaria vulgaris and Cucurbita pepo Extracts on Pentobarbital-Induced Sleep and Possible Mechanisms of Action. Medicina, 2018. [DOI | PubMed]
- V.S. Sohal, J.L. Rubenstein. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry, 2019. [DOI | PubMed]
- J.P. Hegarty, D.J. Weber, C.M. Cirstea, D.Q. Beversdorf. Cerebro-cerebellar functional connectivity is associated with cerebellar excitation–inhibition balance in autism spectrum disorder. J. Autism Dev. Disord., 2018. [DOI | PubMed]
- H.J. Park, S.K. Kim, W.S. Kang, J.K. Park, Y.J. Kim, M. Nam, J.W. Kim, J.-H. Chung. Association between IRS 1 Gene Polymorphism and Autism Spectrum Disorder: A Pilot Case-Control Study in Korean Males. Int. J. Mol. Sci., 2016. [DOI | PubMed]
- S. Baron-Cohen, M.V. Lombardo, B. Auyeung, E. Ashwin, B. Chakrabarti, R. Knickmeyer. Why are autism spectrum conditions more prevalent in males?. PLoS Biol., 2011. [DOI | PubMed]
- H. Östlund, E. Keller, Y.L. Hurd. Estrogen receptor gene expression in relation to neuropsychiatric disorders. Ann. N. Y. Acad. Sci., 2003. [DOI | PubMed]
- G.J. ter Horst. Estrogen in the limbic system. Vitam. Horm., 2010. [PubMed]
- A. Crider, R. Thakkar, A.O. Ahmed, A. Pillai. Dysregulation of estrogen receptor beta (ERβ), aromatase (CYP19A1), and ER co-activators in the middle frontal gyrus of autism spectrum disorder subjects. Mol. Autism, 2014. [DOI | PubMed]
- H. Wei, F. Liang, G. Meng, Z. Nie, R. Zhou, W. Cheng, X. Wu, Y. Feng, Y. Wang. Redox/methylation mediated abnormal DNA methylation as regulators of ambient fine particulate matter-induced neurodevelopment related impairment in human neuronal cells. Sci. Rep., 2016. [DOI | PubMed]
- P.-Y. Shih, Y.-L. Fang, S. Shankar, S.-P. Lee, H.-T. Hu, H. Chen, T.-F. Wang, K.-C. Hsia, Y.-P. Hsueh. Phase separation and zinc-induced transition modulate synaptic distribution and association of autism-linked CTTNBP2 and SHANK3. Nat. Commun., 2022. [DOI | PubMed]
- Y. Wang, S. Chiola, G. Yang, C. Russell, C.J. Armstrong, Y. Wu, J. Spampanato, P. Tarboton, H.A. Ullah, N.U. Edgar. Modeling human telencephalic development and autism-associated SHANK3 deficiency using organoids generated from single neural rosettes. Nat. Commun., 2022. [DOI | PubMed]
- K. Li, X. Liang, X. Xie, L. Tian, J. Yan, B. Lin, H. Liu, W. Lai, X. Liu, Z. Xi. Role of SHANK3 in concentrated ambient PM2. 5 exposure induced autism-like phenotype. Heliyon, 2023. [DOI | PubMed]
- M.K. Singleton, M.L. Gonzales, K.N. Leung, D.H. Yasui, D.I. Schroeder, K. Dunaway, J.M. LaSalle. MeCP2 is required for global heterochromatic and nucleolar changes during activity-dependent neuronal maturation. Neurobiol. Dis., 2011. [DOI | PubMed]
- M.V. Nguyen, F. Du, C.A. Felice, X. Shan, A. Nigam, G. Mandel, J.K. Robinson, N. Ballas. MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci., 2012. [DOI | PubMed]
- P. Moretti, H.Y. Zoghbi. MeCP2 dysfunction in Rett syndrome and related disorders. Curr. Opin. Genet. Dev., 2006. [DOI | PubMed]
- Z. Wen, T.L. Cheng, G.Z. Li, S.B. Sun, S.Y. Yu, Y. Zhang, Y.S. Du, Z. Qiu. Identification of autism-related MECP2 mutations by whole-exome sequencing and functional validation. Mol. Autism, 2017. [DOI | PubMed]
- R.J. Hagerman, E. Berry-Kravis, H.C. Hazlett, D.B. Bailey, H. Moine, R.F. Kooy, F. Tassone, I. Gantois, N. Sonenberg, J.L. Mandel. Fragile X syndrome. Nat. Rev. Dis. Primers, 2017. [DOI | PubMed]
- M. Pieretti, F. Zhang, Y.-H. Fu, S.T. Warren, B.A. Oostra, C.T. Caskey, D.L. Nelson. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell, 1991. [DOI | PubMed]
- J. Foraker, S.P. Millard, L. Leong, Z. Thomson, S. Chen, C.D. Keene, L.M. Bekris, C.-E. Yu. The APOE gene is differentially methylated in Alzheimer’s disease. J. Alzheimer’s Dis., 2015. [DOI | PubMed]
- E. Napoli, C. Ross-Inta, S. Wong, C. Hung, Y. Fujisawa, D. Sakaguchi, J. Angelastro, A. Omanska-Klusek, R. Schoenfeld, C. Giulivi. Mitochondrial dysfunction in Pten haplo-insufficient mice with social deficits and repetitive behavior: Interplay between Pten and p53. PLoS ONE, 2012. [DOI | PubMed]
- K.L. Wisner, E. Leckman-Westin, M. Finnerty, S.M. Essock. Valproate prescription prevalence among women of childbearing age. Psychiatr. Serv., 2011. [DOI | PubMed]
- E.E. Gerard, K.J. Meador. An update on maternal use of antiepileptic medications in pregnancy and neurodevelopment outcomes. J. Pediatr. Genet., 2015. [PubMed]
- R. Mychasiuk, S. Richards, A. Nakahashi, B. Kolb, R. Gibb. Effects of rat prenatal exposure to valproic acid on behaviour and neuro-anatomy. Dev. Neurosci., 2012. [DOI | PubMed]
- T. Schneider, R. Przewłocki. Behavioral alterations in rats prenatally exposed to valproic acid: Animal model of autism. Neuropsychopharmacology, 2005. [DOI | PubMed]
- M. Huberman Samuel, G. Meiri, I. Dinstein, H. Flusser, A. Michaelovski, A. Bashiri, I. Menashe. Exposure to general anesthesia may contribute to the association between cesarean delivery and autism spectrum disorder. J. Autism Dev. Disord., 2019. [DOI | PubMed]
- M.L. Laporta, J. Sprung, C.A. Fejedelem, D.T. Henning, A.L. Weaver, A.C. Hanson, D.R. Schroeder, S.M. Myers, R.G. Voigt, T.N. Weingarten. Association between exposure of children to general anesthesia and autism spectrum disorder. J. Autism Dev. Disord., 2021. [DOI | PubMed]
- C. Ladd-Acosta, K.D. Hansen, E. Briem, M.D. Fallin, W.E. Kaufmann, A.P. Feinberg. Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry, 2014. [DOI | PubMed]
- S. Nardone, D.S. Sams, E. Reuveni, D. Getselter, O. Oron, M. Karpuj, E. Elliott. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry, 2014. [DOI | PubMed]
- S. Nardone, D.S. Sams, A. Zito, E. Reuveni, E. Elliott. Dysregulation of Cortical Neuron DNA Methylation Profile in Autism Spectrum Disorder. Cereb. Cortex, 2017. [DOI | PubMed]
- M.J. Corley, N. Vargas-Maya, A.P.S. Pang, A. Lum-Jones, D. Li, V. Khadka, R. Sultana, D.C. Blanchard, A.K. Maunakea. Epigenetic Delay in the Neurodevelopmental Trajectory of DNA Methylation States in Autism Spectrum Disorders. Front. Genet., 2019. [DOI | PubMed]
- C.C.Y. Wong, R.G. Smith, E. Hannon, G. Ramaswami, N.N. Parikshak, E. Assary, C. Troakes, J. Poschmann, L.C. Schalkwyk, W. Sun. Genome-wide DNA methylation profiling identifies convergent molecular signatures associated with idiopathic and syndromic autism in post-mortem human brain tissue. Hum. Mol. Genet., 2019. [DOI | PubMed]
- E. Takahashi, N. Allan, R. Peres, A. Ortug, A.J.W. van der Kouwe, B. Valli, E. Ethier, J. Levman, N. Baumer, K. Tsujimura. Integration of structural MRI and epigenetic analyses hint at linked cellular defects of the subventricular zone and insular cortex in autism: Findings from a case study. Front. Neurosci., 2022. [DOI | PubMed]
- C.C. Wong, E.L. Meaburn, A. Ronald, T.S. Price, A.R. Jeffries, L.C. Schalkwyk, R. Plomin, J. Mill. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol. Psychiatry, 2014. [DOI | PubMed]
- S. Liang, Z. Li, Y. Wang, X. Li, X. Yang, X. Zhan, Y. Huang, Z. Gao, M. Zhang, C. Sun. Genome-Wide DNA Methylation Analysis Reveals Epigenetic Pattern of SH2B1 in Chinese Monozygotic Twins Discordant for Autism Spectrum Disorder. Front. Neurosci., 2019. [DOI | PubMed]
- A. Saffari, M. Arno, E. Nasser, A. Ronald, C.C.Y. Wong, L.C. Schalkwyk, J. Mill, F. Dudbridge, E.L. Meaburn. RNA sequencing of identical twins discordant for autism reveals blood-based signatures implicating immune and transcriptional dysregulation. Mol. Autism, 2019. [DOI | PubMed]
- E. Hannon, D. Schendel, C. Ladd-Acosta, J. Grove, C.S. Hansen, S.V. Andrews, D.M. Hougaard, M. Bresnahan, O. Mors, M.V. Hollegaard. Elevated polygenic burden for autism is associated with differential DNA methylation at birth. Genome Med., 2018. [DOI | PubMed]
- C.E. Mordaunt, J.M. Jianu, B.I. Laufer, Y. Zhu, H. Hwang, K.W. Dunaway, K.M. Bakulski, J.I. Feinberg, H.E. Volk, K. Lyall. Cord blood DNA methylome in newborns later diagnosed with autism spectrum disorder reflects early dysregulation of neurodevelopmental and X-linked genes. Genome Med., 2020. [DOI | PubMed]
- R.O. Bahado-Singh, S. Vishweswaraiah, B. Aydas, U. Radhakrishna. Placental DNA methylation changes and the early prediction of autism in full-term newborns. PLoS ONE, 2021. [DOI | PubMed]
- C.E. Mordaunt, J.S. Mouat, R.J. Schmidt, J.M. LaSalle. Comethyl: A network-based methylome approach to investigate the multivariate nature of health and disease. Brief. Bioinform., 2022. [DOI | PubMed]
- M. Jasoliya, J. Gu, R.R. AlOlaby, B. Durbin-Johnson, F. Chedin, F. Tassone. Profiling Genome-Wide DNA Methylation in Children with Autism Spectrum Disorder and in Children with Fragile X Syndrome. Genes, 2022. [DOI | PubMed]
- L.A. Papale, Q. Zhang, S. Li, K. Chen, S. Keleş, R.S. Alisch. Genome-wide disruption of 5-hydroxymethylcytosine in a mouse model of autism. Hum. Mol. Genet., 2015. [DOI | PubMed]
- A.M. Muehlmann, N. Bliznyuk, I. Duerr, T.P. Yang, M.H. Lewis. Early exposure to a methyl donor supplemented diet and the development of repetitive motor behavior in a mouse model. Dev. Psychobiol., 2020. [DOI | PubMed]
- A. Stoccoro, E. Conti, E. Scaffei, S. Calderoni, F. Coppedè, L. Migliore, R. Battini. DNA Methylation Biomarkers for Young Children with Idiopathic Autism Spectrum Disorder: A Systematic Review. Int. J. Mol. Sci., 2023. [DOI | PubMed]
- E. Gascon, K. Lynch, H. Ruan, S. Almeida, J.M. Verheyden, W.W. Seeley, D.W. Dickson, L. Petrucelli, D. Sun, J. Jiao. Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med., 2014. [DOI | PubMed]
- Y. Yang, X. Shu, D. Liu, Y. Shang, Y. Wu, L. Pei, X. Xu, Q. Tian, J. Zhang, K. Qian. EPAC null mutation impairs learning and social interactions via aberrant regulation of miR-124 and Zif268 translation. Neuron, 2012. [DOI | PubMed]