The role of distinct APOBEC/ADAR mRNA levels in mutational signatures linked to aging and ultraviolet radiation
Abstract
The APOBEC/AID family is known for its mutator activity, and recent evidence also supports the potential impact of ADARs. Furthermore, the mutator impacts of APOBEC/ADAR mutations have not yet been investigated. Assessment of pancancer TCGA exomes identified enriched somatic variants among exomes with nonsynonymous APOBEC1, APOBEC3B, APOBEC3C, ADAR, and ADARB1 mutations, compared to exomes with synonymous ones. Principal component (PC) analysis reduced the number of potential players to eight in cancer exomes/genomes, and to five in cancer types. Multivariate regression analysis was used to assess the impact of the PCs on each COSMIC mutational signature among pancancer exomes/genomes and particular cancers, identifying several novel links, including SBS17b, SBS18, and ID7 mainly determined by APOBEC1 mRNA levels; SBS40, ID1, and ID2 by age; SBS3 and SBS16 by APOBEC3A/APOBEC3B mRNA levels; ID5 and DBS9 by DNA repair/replication (DRR) defects; and SBS7a-d, SBS38, ID4, ID8, ID13, and DBS1 by ultraviolet (UV) radiation/ADARB1 mRNA levels. APOBEC/ADAR mutations appeared to potentiate the impact of DRR defects on several mutational signatures, and some factors seemed to inversely affect certain signatures. These findings potentially implicate certain APOBEC/ADAR mutations/mRNA levels in distinct mutational signatures, particularly APOBEC1 mRNA levels in aging-related signatures and ADARB1 mRNA levels in UV radiation-related signatures.
Article type: Research Article
Keywords: Cancer mutation, Mutational signature, Cancer genomics, Cancer genetics, Genome, Mutation
Affiliations: https://ror.org/01c4pz451grid.411705.60000 0001 0166 0922Digestive Oncology Research Center, Digestive Disease Research Institute, Tehran University of Medical Sciences, Tehran, Iran
License: © The Author(s) 2024 CC BY 4.0 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Article links: DOI: 10.1038/s41598-024-64986-6 | PubMed: 38965255 | PMC: PMC11224270
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (6.3 MB)
Introduction
The APOBEC/AID (apolipoprotein B mRNA editing enzyme, catalytic polypeptide/activation-induced cytidine) family of cytidine deaminases, which are involved in a wide range of physiological and developmental activities, whether in DNA (reviewed in1) or RNA2,3, have also been implicated in mutagenesis across various cancers4–7. Two single base substitution mutational signatures with predominant C > T (SBS2) and C > G (SBS13) variants have been attributed to the APOBEC/AID family5,7, in addition to potentially one double base substitution signature with predominant CC > NN (DBS11) variants8. While both APOBEC3B (A3B)6,9 and APOBEC3A (A3A)9,10 are strongly linked to APOBEC mutational signatures, the implication of other family members with nuclear distribution, including APOBEC1 (A1)11, APOBEC3C (A3C)12, APOBEC3F (A3F)13, APOBEC3H (A3H)14, and AID15, cannot be excluded. AID has long been implicated in B-cell malignancies16,17. Although much attention has been given to the high mRNA levels of APOBEC/AICDA genes6,18 and their copy number9 or single nucleotide variations19, these might not fully explain all APOBEC mutagenic impacts observed in cancer genomes. For instance, Kanke et al. reported some tumors of the breast, ovary, and uterus with a predominant SBS2, which did not show high APOBEC mRNA levels20. It seems that the implication of other factors, including APOBEC somatic gene mutations, has been simply overlooked. On the other hand, there is evidence showing that known adenosine deaminases acting on RNA (ADARs) may also act on DNA. These include the DNA mutator activity of ADAR1 in MYCC and a class switch recombination region (Ig-Sμ)21; deamination of adenosine at dA-C mismatches of the DNA‒RNA hybrids by both ADAR1 and ADAR222; and some indirect evidence23,24. However, the role of APOBEC/ADAR mutations has not yet been clarified in cancer. Our pancancer analysis shows that these genes are themselves subject to somatic mutations, although at a low frequency.
A preliminary analysis showed that pancancer exomes with any of the fourteen APOBEC/ADAR genes mutated showed significantly more single nucleotide variants (SNVs) and insertions/deletions (indels). Since this could suggest either a “driver” or a “passenger” role for APOBEC/ADAR mutations in cancer hypermutation, the burden of genomic variants was assessed in exomes with nonsynonymous versus synonymous APOBEC/ADAR mutations to check their true driver impact, showing that mutations in A1, A3B, A3C, and ADAR are correlated with indel variants, while ADARB1 mutations are correlated with certain SNVs. Follow-up analyses estimated the roles of APOBEC/ADAR mutations and mRNA levels in mutational signatures among various cancer types, as adjusted for defects in DNA repair/replication (DRR) pathways.
Results
Assessment of 10,295 tumor exomes identified 3,600,963 unique somatic variants, of which 3,427,680 (95.2%) were found to be SNVs and 173,109 (4.8%) were of the indel type. Approximately 2,710,172 (75.3%) of the somatic variants were found to be nonsynonymous variants occurring within coding regions, and the remaining 24.7% (890,791) were either intronic or synonymous exonic, herein called synonymous mutations. Collectively, 874 exomes (8.5%) showed at least one nonsynonymous somatic mutation in one or more APOBEC/ADAR genes, including APOBEC1, APOBEC2, APOBEC3A, APOBEC3B, APOBEC3C, APOBEC3D, APOBEC3F, APOBEC3G, APOBEC3H, APOBEC4, AICDA, ADAR, ADARB1, and ADARB2, with 188 exomes (21.5%) showing more than one mutated gene. The mean number of total APOBEC/ADAR mutations varied between 0 per exome in PCPG, TGCT, THYM, and UVM to 0.36 and 0.62 in SKCM and UCEC, respectively (Supplementary Table S1). The most common nonsynonymous mutations were seen in ADARB2 (198; 1.92% per exome), whereas A3A (40; 0.39% per exome) was found to be the least mutated. In general, the ADAR family tended to have a higher mean number of mutations (178; 1.73% per exome) than the APOBEC family (68; 0.66% per exome). None of the nonsynonymous APOBEC/ADAR mutations reported by the TCGA-MC3 catalog were annotated as having a low Impact, 68 (5.3%) were classified as having a modifier Impact, and 1,204 (94.7%) were classified as having a moderate/high Impact. Similarly, none of the nonsynonymous DRR mutations were annotated as having a low Impact, 562 (2.8%) were classified as having a modifier Impact, and 19,292 (97.2%) were classified as having a moderate/high Impact.
Essentially, all APOBEC/ADAR-mutated exomes showed a higher number of somatic variants
The mean SNV burden was found to be enriched in those exomes with any APOBEC/ADAR mutations compared to those with wild-type ones, varying between 13.9-fold for ADARB2-mutated exomes to 25.5-fold for A2-mutated ones (Supplementary Table S2). Likewise, the mean indel burden was enriched in essentially all APOBEC/ADAR-mutated exomes compared to their wild-type counterparts, varying between 5.9-fold for AICDA-mutated exomes and 11.9-fold for A1-mutated ones.
T > N SNVs were correlated with ADARB1 mutations, as were certain indels with A1, A3B, A3C, and ADAR mutations
In a more stringent assessment, the mean numbers of genomic T > N, C > N, Ind-T, and Ind-C variants were compared in exomes with nonsynonymous versus synonymous APOBEC/ADAR mutations. Assessment of the SNVs showed that the mean number of T > N variants was enriched 2.8-fold in nonsynonymous ADARB1 mutant exomes (Supplementary Table S3). On the other hand, the mean number of Ind-T variants was enriched 6.0-fold in nonsynonymous A1 mutant exomes, 5.0-fold in nonsynonymous A3B mutants, 22.2 times higher in nonsynonymous A3C mutants, and 2.3-fold in nonsynonymous ADAR mutants, and the mean number of Ind-C variants was 17.2-fold in nonsynonymous A3C mutant exomes and 2.1-fold in nonsynonymous ADAR mutant exomes (Supplementary Table S3).
Assessing the impacts of APOBEC/ADAR mutations and mRNA levels on cancer mutational signatures
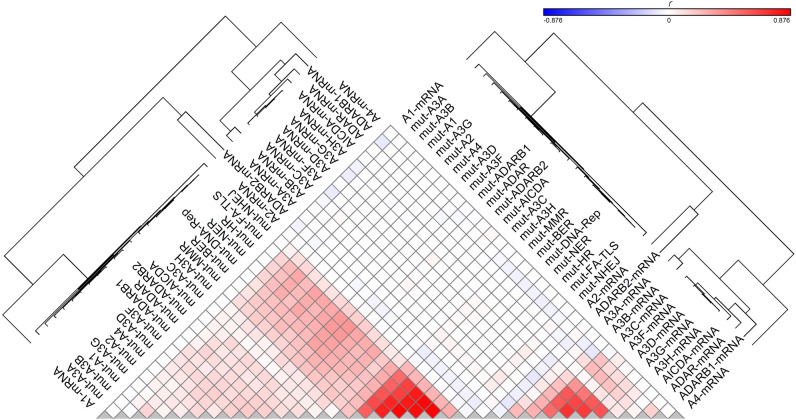
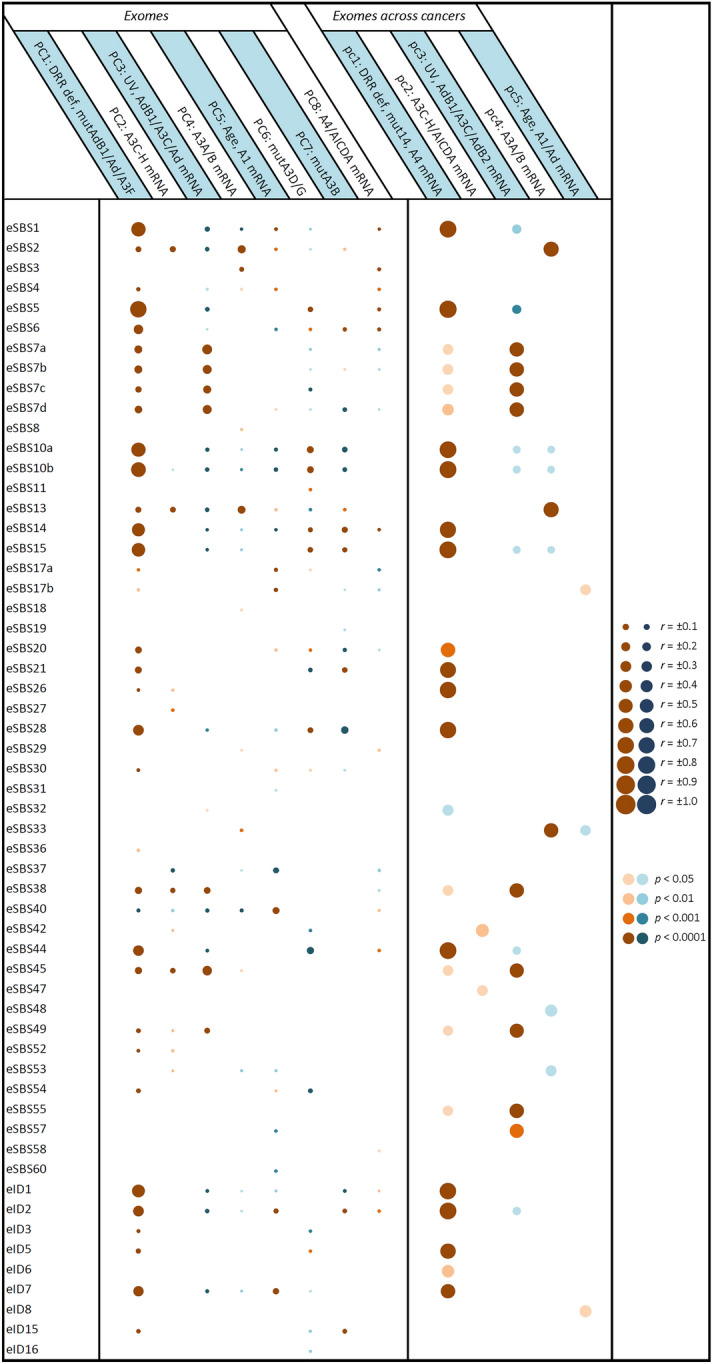
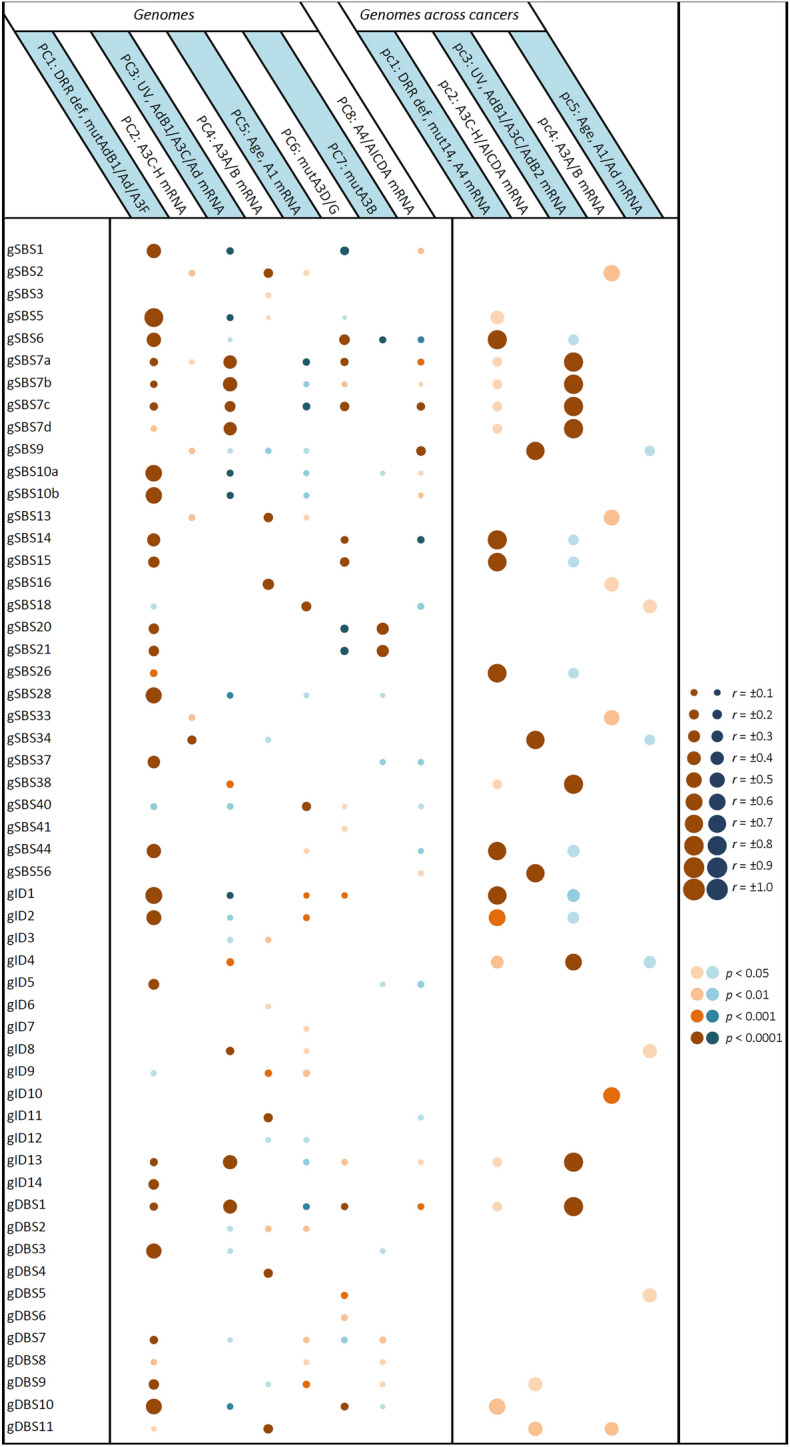
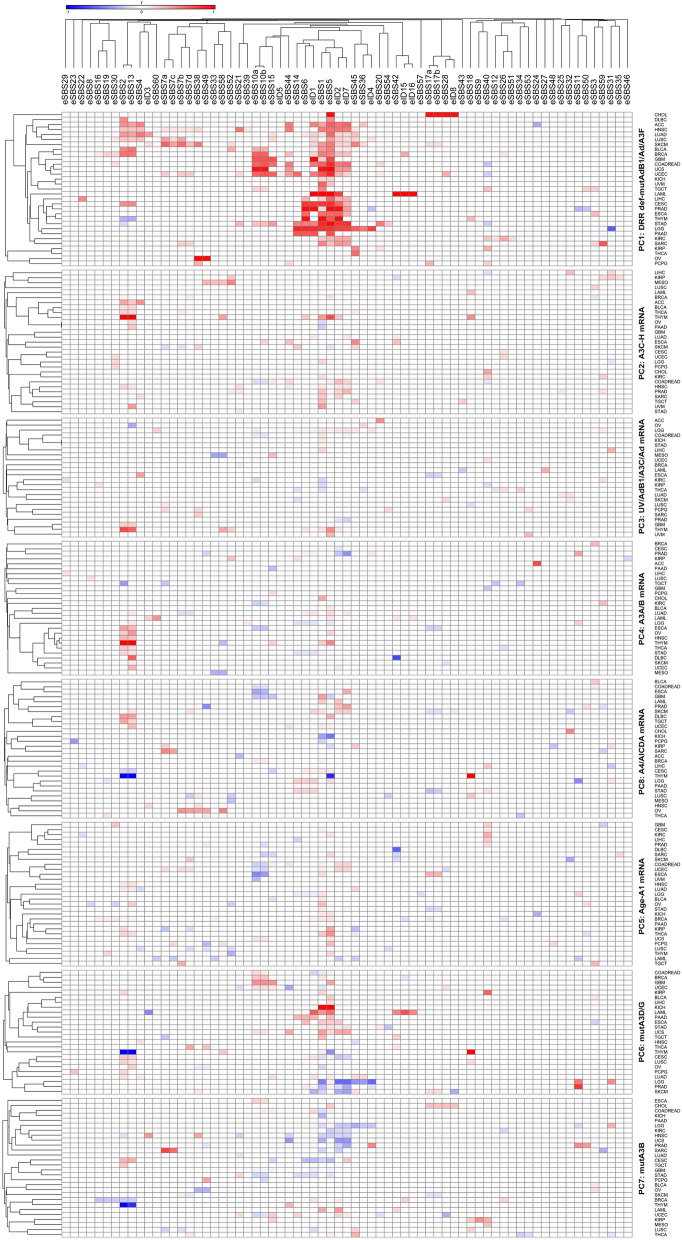
Regression followed by clustering analysis was performed using 10,126 samples with WES- and/or WGS-identified mutational signatures, identifying that many of the potential endogenous factors closely co-clustered with each other, including all DRR defects and APOBEC/ADAR mutations; A3A and A3B mRNA levels co-clustered with each other; and A3C, A3D, A3F, A3G, and A3H mRNA levels co-clustered with each other (Fig. 1). Hence, it was next attempted to reduce the number of potentially implicated covariates using principal component (PC) analysis. Assessment of 37 potential covariates, including age and UV factor, along with APOBEC/ADAR factors and DRR defects, reduced the number of potential covariates to eight PCs (explaining 49.5% of the variability) in pancancer exome (9,493 samples)/genome (773 samples) analyses (Supplementary Table S4). These included (1) seven DRR defects and mutations in ADARB1, ADAR, and A3F; (2) A3C, A3D, A3F, A3G, and A3H (A3C-H) mRNA levels; (3) UV factor and ADARB1/A3C/ADAR mRNA levels; (4) A3A and A3B mRNA levels; (5) age and A1 mRNA levels; (6) A3D and A3G mutations; (7) A3B mutation; and (8) A4 and AICDA mRNA levels. A similar analysis of the mean exome/genome covariates among 33 cancer types resulted in five PCs (explaining 77.6% of the variability; Supplementary Table S5), including (1) seven DRR defects, all APOBEC/ADAR mutations, and A4 mRNA levels; (2) A3C-H and AICDA mRNA levels; (3) UV factor and ADARB1/A3C/ADARB2 mRNA levels; (4) A3A and A3B mRNA levels; and (5) age and A1/ADAR mRNA levels. Multivariate correlations of the pancancer exome and genome signatures with the aforementioned PCs are shown in Figs. 2 and 3, respectively, and clustering analyses of the positive correlations are illustrated in Supplementary Figure S1. Furthermore, the correlations of the exome and genome signatures with PCs across 33 particular cancers are depicted in Figs. 4 and 5, respectively, as well as Supplementary Figures S2 and S3, respectively. Collectively, these findings can be summarized below, as illustrated in Fig. 6.
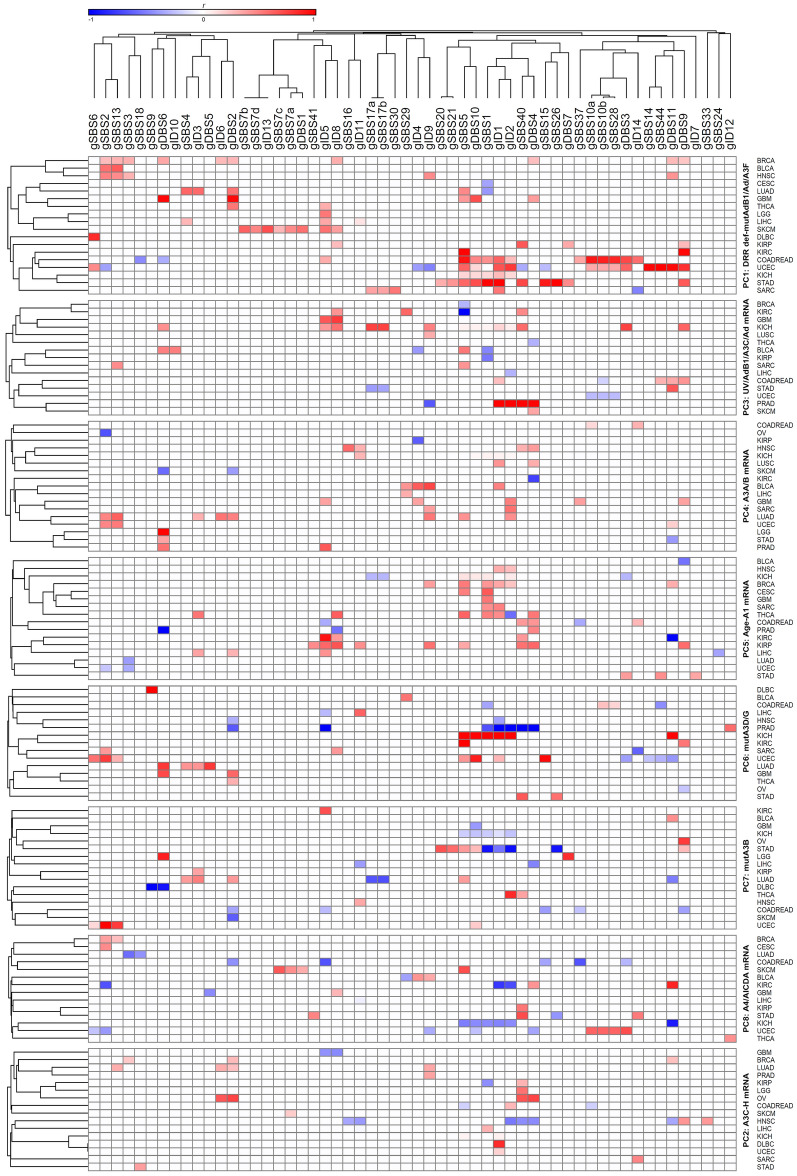
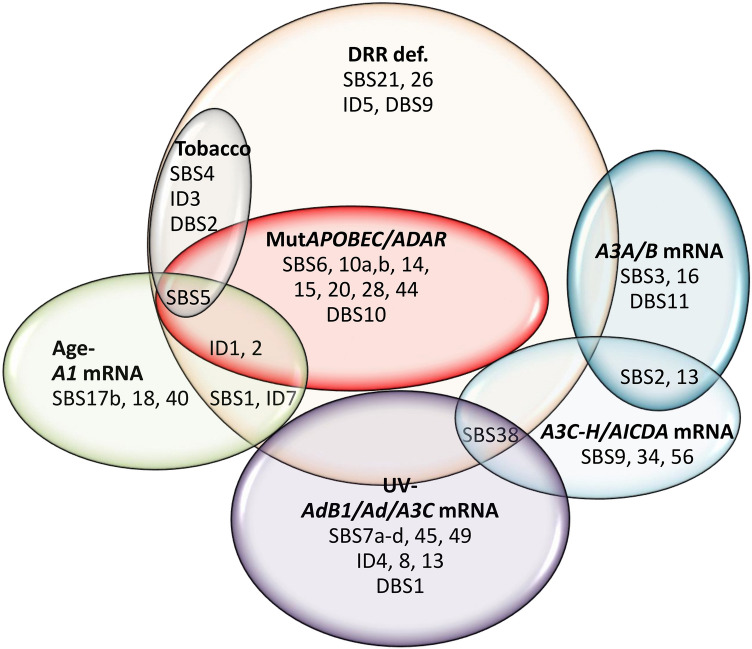
UV-ADARB1 mRNA cluster
Seven signatures are known to be UV radiation-related, which were also found to be correlated with PC3 representing UV factor and ADARB1/A3C/ADAR mRNA levels (Fig. 6; Supplementary Figure S4). These include SBS7a,b,c,d, SBS38, ID13, and DBS1. Furthermore, the unknown genome ID4 (gID4) and the so-called artifact exome signatures SBS45 (eSBS45) and SBS49 (eSBS49) were mainly determined by this component. Not surprisingly, these signatures were primarily associated with significant DRR defects as well. More detailed analysis showed that SBS7a-d, SBS38, ID13, and DBS1 were merely or mainly determined by UV factor, while ID4 was determined by only the ADARB1 mRNA level, and ID8 was determined by both ADARB1 and ADAR mRNA.
A3A/B mRNA cluster
Pancancer analysis across both samples and cancer types identified both exome and genome signatures SBS2 and SBS13 highly determined by A3A/A3B (A3A/B) mRNA levels (Fig. 6; Supplementary Figure S5). These findings were consistent with analyses of the exome mutational signatures across particular cancers, showing that eSBS2 was determined by A3A/B mRNA (THYM, OV, ESCA, THCA, STAD, HNSC) as well as A3C-H mRNA (THYM, ACC, HNSC, THCA, BLCA, BRCA), age-A1 mRNA (KIRP, THCA, HNSC, BRCA, LUAD), and DRR defects in various cancers (Fig. 4). eSBS13 was also determined by A3A/B mRNA (THYM, OV, DLBC, ESCA, THCA, STAD, HNSC, UCEC, SKCM, LGG, LUAD), A3C-H mRNA (THYM, HNSC, THCA, BLCA, PAAD, ACC, OV), age-A1 mRNA (OV, THCA, HNSC, BRCA, LUAD), and DRR defects in various cancers. However, the genome signatures SBS2 (gSBS2) and SBS13 (gSBS13) were found to be determined solely by DRR defects in particular cancers (Fig. 5). As expected, gDBS11 was also determined by the A3A/B mRNA level. Furthermore, HR-related eSBS3 and alcohol-related gSBS16 were found to be highly determined by A3A/B mRNA levels.
Age-APOBEC1 (A1) mRNA cluster
The so-called 5FU-related eSBS17b and the ROS-related gSBS18 were found to be determined by age-A1 mRNA level, as did the unknown SBS40 (Fig. 6; Supplementary Figure S6). Assessment of individual cancers (CESC, GBM, KIRC, LIHC, PRAD) across exomes; KIRP, KIRC, COADREAD across genomes) corroborated the correlation of SBS40 with age-A1 mRNA level. A combined impact of the DRR-ADARB1/ADAR/A3F mutations and age-A1 mRNA levels (BLCA, BRCA, CESC, COADREAD, ESCA, KICH, LGG, OV, STAD, THCA, UCEC across exomes; BRCA, CESC, GBM, KICH, SARC, THCA across genomes) was seen in aging-related SBS1. Likewise, the combined impact of the DRR-ADARB1/ADAR/A3F mutations and age-A1 mRNA levels was also seen for indel signatures ID1, ID2, and ID7 in pancancer analyses as well as in certain cancers. A more detailed analysis of PC1 covariates identified SBS40, ID1, and ID2 to be determined mainly by age, while SBS17b, SBS18, and ID7 were determined mainly by A1 mRNA level.
Defective DRR cluster
This constituted the largest cluster by far, contributing to many mutational signatures. In addition to SBS21 and SBS26, which are currently attributed to some MMR defects, both unknown ID5 and DBS9 were also determined by DRR defects across exomes and/or genomes (Fig. 6). One subcluster consisted of signatures that were also determined by tobacco exposure, including the known signatures SBS4, ID3, and DBS2, as well as the so-called aging-related SBS5. Another subcluster consisted of SBS6, SBS10a,b, SBS14, SBS15, SBS20, SBS28, SBS44, ID1, ID2, and DBS10, which are currently known or predicted to be related to DRR defects, but they were also found to be determined by APOBEC/ADAR mutations (Fig. 6; Supplementary Figure S7). As discussed earlier, the mutational signatures SBS1, ID1, ID2, and ID7 were also determined by age-A1 mRNA levels. Intriguingly, mutA3B (PC7) was also seen to inversely impact several mutational signatures across various cancers, particularly SBS1, SBS5, ID1, ID2, and ID7 (Fig. 4). Likewise, an apparently inverse impact was seen for other covariates in certain cancers, particularly those of mutA3D/G (most commonly affecting SBS1, SBS44, and ID2) and UV-ADARB1/A3C/ADAR mRNA levels (most commonly affecting SBS10a,b and SBS28).
A3C-A3H/AICDA mRNA cluster
Although the variations in A3C, A3D, A3F, A3G, and A3H (A3C-H) and AICDA mRNA levels tended to be separate across particular exomes/genomes, they were very close across different cancer types. In light of this, three mutational signatures, including PolH-related SBS9, unknown SBS34, and the so-called artifact SBS56, were found to be determined by A3C-H/AICDA mRNA levels across both genomes and cancer types (Fig. 6). As mentioned earlier, both SBS2 and SBS13 were also determined by A3C-H mRNA levels in certain cancers.
Discussion
In this study, DRR defects were found to be a major determinant of various mutational signatures, sometimes even for those primarily known to be related to other factors such as aging, A3A/B mRNA levels, or UV exposure. Since the high mRNA levels of A1, A3A/B, and ADARB1 were all found to determine distinct mutational signatures in both wild-type and mutated DRR subgroups, it is reasonable to believe that these factors can act independently of the DRR defects as well. This is not surprising, considering the independent regulation of the expression levels of the aforementioned genes. However, this was not the case for the APOBEC/ADAR mutations, which almost always occurred in the presence of a DRR defect. Therefore, it can be concluded that while the APOBEC/ADAR mutations have arisen from a DRR defect, they potentiate the impact of the latter on specific mutational signatures. Several mutational signatures were found to be solely determined by DRR defects, including SBS21, SBS26, ID5, and DBS9. Boichard et al. showed that mutations in the A1, A4, AICDA, and A3 subfamily predicted the mutational burden regardless of MMR or POLD1/POLE defects, with no assessment of individual APOBEC mutations or other DRR defects25. Our preliminary analysis showed that rather infrequent APOBEC/ADAR mutations occur more commonly in hypermutated exomes. Since synonymous mutations are supposed to be of no functional impact and occur rather constantly over time26, in this study the burden of genomic variants was assessed among those exomes with nonsynonymous APOBEC/ADAR mutations compared to those with synonymous ones, showing that T > N SNVs were enriched in ADARB1-mutant exomes, while all indels were found to be enriched in A3C and ADAR-mutant exomes and Ind-T variants were enriched in A1 and A3B-mutant exomes.
The high number of potentially implicated covariates made a PC analysis inevitable, although complicating the attribution of distinct mutational signatures to individual APOBEC/ADAR aberrations, particularly infrequent APOBEC/ADAR mutations. PC analysis reduced the number of potentially implicated factors, and multivariate models using the measured principal components proposed novel correlations for the so-called artifact/unknown mutational signatures, including SBS34 and SBS56 (determined by A3C-H/AICDA mRNA level), SBS40 (determined by age), ID5 and DBS9 (determined by DRR defects), and SBS45, SBS49, and ID4 (determined by UV-ADARB1/A3C/ADAR mRNA level). SBS34 is commonly seen among DLBC, STAD, and PAAD and shows asymmetry toward the intergenic regions, as does SBS927. SBS40 has been proposed to be related to environmental factors because of its accumulation with age in some cancer types28. SBS45 and SBS49 are claimed to be possible sequencing artifacts, the former due to 8-oxo-guanines introduced during sequencing27. SBS56 has been reported to be a marker of AKT inhibitor sensitivity in some cancer cell lines29, further undermining an artifact nature.
Moreover, some findings were apparently discrepant from the current literature, including SBS3, SBS16, SBS17b, SBS18, ID4, and ID8. SBS16 has been suggested to be alcohol-related in ESCC30, while it was found here to be determined by A3A/A3B expression levels. Whether this discrepancy is due to the genomic hypomethylating impact of ethanol, which increases A3C-H mRNA levels31, or because of a shift in A3A function from so-called physiological 5hmC demethylation to potentially oncogenic C demethylation32 needs to be investigated in more detail. The so-called 5FU-related SBS17b33 and ROS-related SBS1827 were also found to be determined by A1 mRNA levels. In addition, the clock-like signature SBS134 was found to be determined by DRR defects and A1 mRNA levels, while the other clock-like signature SBS534, which has also been reported to be smoking-related35, was determined by DRR defects, tobacco use, APOBEC/ADAR mutations, and age. Intriguingly, at least three AID/APOBEC proteins, including AID, A3A, and A3B, have been reported to efficiently deaminate dC neighboring DNA damage induced by oxidation or alkylation36, a function that might be implicated in this case as well. A1 mRNA levels have already been linked to cancer indels, in particular Ind-T ones18, and it was shown here that at least ID2 (Del-T) and ID7 (Del-C/T) signatures were determined by A1 mRNA levels in addition to the known DRR defect impact37,38, further characterizing the A1 mRNA impact on cancer genomes. The impacts of age on ID1 (Ins-T) and ID8 (long Del) also seem to be mediated through A1 mRNA. Unknown ID4, which has been suggested to be TOP1-related39, was determined by ADARB1 mRNA level, and the so-called radiation-related ID8 was determined by ADARB1 and ADAR mRNA levels across genomes. Recent studies have shown that ADAR1 (encoded by ADAR) can edit DNA:RNA hybrids that form during transcription and DNA replication40,41. Specifically, ADAR2 (encoded by ADARB1) has been reported to play a role in editing such hybrids needed for dsDNA break repair and genomic stability40. This is the function which might be implicated in indel mutational signatures that are supposed to be UV-related, including ID4, ID8, and ID13. Since both AID and A3A have also been proposed to mediate skin cancer through chronic inflammation and mutational events, respectively42,43, more detailed studies might be warranted to explore the true endogenous UV mediator. The supposedly HR-related SBS3 was determined by A3A/B mRNA levels. Whether this is a real link or A3A/B mRNA is a proxy for other factors (Fig. 1) needs to be determined by further investigation.
The expression of APOBEC3 family members is regulated by different factors, including B-cell receptor signaling, noncanonical NF-κB signaling, and SF3B1 inhibition44,45. Additionally, the expression of APOBEC3 genes can be regulated by epigenetic modifications, such as DNA methylation and histone acetylation46. However, there is much evidence supporting some links between APOBEC/ADAR activation and environmental mutagens, including viruses, tobacco, and UV radiation. Not unexpectedly, HPV-positive HNSCs show higher mRNA levels of A3A, A3B, and A3H than HPV-negative HNSCs47. It has been reported that increased expression of A3B in response to ionizing radiation could contribute to the acquisition of radiation resistance in cancer cells48, and radiotherapy is followed by APOBEC mutagenesis49. A3G has also been shown to be activated by UV radiation and rescue cells from its detrimental DNA effects50,51, as well as enhancing double-strand break (DSB) repair in leukemia and lymphoma cells16, making them radioresistant. Likewise, assessment of tobacco-associated cancers suggested that the cellular machinery underlying APOBEC signatures was activated by tobacco smoke35, and APOBEC rather than tobacco-associated mutagenesis predominated in two series of bladder cancers52. On the other hand, smoking has been shown to repress ADAR expression, enhancing intracellular oxidative stress53,54.
One biologically plausible explanation for the inverse impacts of certain factors might be the competitive actions of homologous proteins, including those of mutA3B on A1 mRNA-determined signatures SBS1, SBS5, ID1, ID2, and ID7. Certain APOBEC/ADAR proteins have been known to modulate each other through heterodimerization or coexpression. These include A2 dimerizing with and inhibiting A155, ADAR1 (encoded by ADAR) dimerizing with and sequestering ADAR2 (encoded by ADARB1)56, and ADAR3 (encoded by ADARB2) downregulating ADAR257. Likewise, gain- or loss-of-function variants of APOBEC/ADAR proteins have been reported, including an A3C variant (S188I) with increased dimerization of the protein and hypermutation of target sequences58 and an ADAR variant (P193A) destabilizing the protein-Z-DNA complex and affecting tumor cell proliferation59. Although ADAR3 has not been shown to have any catalytic activity thus far60, some SNVs in ADARB2 show a consistent link to longevity across populations61, suggesting a functional role.
One limitation of the present study is that PC2 does not differentiate between the distinct roles of the co-regulated A3C-H and AICDA genes. Given the cellular location of the gene products, however, it is reasonable to believe that A3H and AID are the potential players among A3C-H/AID proteins. I also acknowledge that some proposed novel links might seem unexpected by currently known mechanisms. Whether or not the reciprocal nucleic acid changes described elsewhere3 are implicated in these novel links would be an intriguing field of future studies, including the unexpected T > C changes in SBS16 found to be linked to A3A/B mRNA levels. The fact that several highly significant associations were observed only in the much smaller genome rather than the exome subgroup (i.e., gSBS16, gSBS18, gSBS34, gSBS56, gID4, gID8, gID13, gDBS1, gSBS9, gDBS10, and gDBS11) might implicate some truly differential mutagenic mechanisms acting among intergenic versus genic regions.
In conclusion, this pancancer approach links several exome/genome mutational signatures with novel factors among APOBEC and ADAR families, including SBS17b, SBS18, and ID7 (A1 mRNA level); SBS3 and SBS16 (A3A/B mRNA level); ID4 and ID8 (ADARB1 mRNA level); SBS40, ID1, and ID2 (age); and ID5 and DBS9 (DRR defects). While some APOBEC/ADAR mutations potentiate the impact of DRR defects on certain mutational signatures, the impact of high expression levels of A1, A3A/B, A3C-H/AICDA, and ADARB1 on distinct signatures can be independent of other mutagenic factors, while still modulating them. It is proposed that the mutagenic impacts of aging are at least partly mediated through the A1 mRNA level, while the UV impacts are mainly mediated through ADARB1 mRNA levels. The precise mechanisms that are involved need to be investigated in detail.
Methods
Patients and samples
The findings of the current study are based on data generated by the TCGA Research Network. A catalog of all somatic variants identified by whole-exome sequencing of 10,295 paired tumor-normal samples was obtained from sftp://tcgaftps.nci.nih.gov/tcgajamboree/mc3/pancan.merged.v0.2.7.PUBLIC.maf.gz, comprising a total of 3,600,963 variants from 33 tumor types. In brief, the MC3 version of the TCGA variants had been consistently called using seven methods with proven performance including MuTect (https://github.com/broadinstitute/mutect), Pindel (https://github.com/genome/pindel), Radia (https://github.com/aradenbaugh/radia), VarScan2 (http://dkoboldt.github.io/varscan/), SomaticSniper (https://github.com/genome/somatic-sniper), MuSE (https://github.com/danielfan/MuSE), and Indelocator (http://archive.broadinstitute.org/cancer/cga/indelocator), as described before62. Non-synonymous APOBEC/ADAR variants used for analysis included all variants but the silent, intronic, and flanking ones. The same rule was applied for the mutations in DRR pathway genes. The list of key DRR pathway genes was obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG)63, as follows: base excision repair (BER; hsa03410; 33 genes), nucleotide excision repair (NER) and transcription-coupled NER (hsa03420; 45 genes), mismatch repair (MMR; hsa03430; 22 genes), homologous recombination (HR; hsa03440; 41 genes), nonhomologous end-joining (NHEJ; hsa03450; 10 genes), and Fanconi anemia-translesion synthesis (FATLS; hsa03460; 54 genes), as well as DNA replication pathway (DNA-Rep; hsa03030; 35 genes), collectively 168 unique genes. All nonsynonymous exome variants were included for analysis of the potentially implicated pathways. A list of APOBEC/AICDA/ADAR gene mutations was extracted, in which mutations were classified into synonymous or nonsynonymous mutations, with the latter including missense, nonsense, splice site, and indel mutations.
The following open access (Level 3) databases were obtained from the Broad Institute GDAC Firehose (http://gdac.broadinstitute.org/): normalized RNA-Seq by Expectation–Maximization (RSEM) for genes (RSEM_genes_normalized_data, v.20160128) and clinical data, including vital status and days to death/last follow-up (Clinical_Pick_Tier1, v.20171224). The normalized RSEM data resulting from RNA sequencing were used as an estimate of TCGA gene expression profiles, and they were further normalized to TATA-binding protein (TBP) mRNA levels and then scaled to 1. Indels longer than one nucleotide were classified based on their 5’-end nucleotide. The absolute number of variants attributed to each mutational signature as detected by whole-exome or whole-genome sequencing (COSMIC release V89, May 2019) was obtained from Rosenthal’s study62 and used for assessment of the correlation with APOBEC/ADAR aberrations as well as DRR defects. This version includes 93 mutational signatures: SBS1-SBS60, ID1-ID17, and DBS1-DBS11. Three signatures including SBS7, SBS10, and SBS17 were represented by four, two, and two sub-signatures, respectively.
Statistical analysis
Unpaired two-tailed Student’s t test (IBM SPSS, v.22) was used to compare the mean number of indel variants and SNVs among wild-type versus mutant APOBEC/ADAR exomes, as well as nonsynonymous versus synonymous APOBEC/ADAR mutant exomes, and the Benjamini–Hochberg false discovery rate (FDR) was used to adjust for multiple testing, with an acceptable FDR of up to 0.05. Principal component (PC) analysis was performed using the Varimax method with Kaiser Normalization (IBM SPSS, v.22) across 10,126 samples with WES (9,493)/WGS (773)-identified mutational signatures (140 having both) to reduce the number of 37 covariates, including 14 APOBEC/ADAR mutations, 14 APOBEC/ADAR mRNA levels, seven DRR pathway defects, age, and UV factor (positive in SKCM and UVM). An acceptable eigenvalue cutoff of 1.0 was used to select the PCs and a cutoff of 0.4 for significant rotated components. A similar PC analysis was performed using the mean exome/genome values of the aforementioned 37 covariates across 33 cancer types, with an eigenvalue cutoff of 1.5 used to select the PCs and a cutoff of 0.4 for significant rotated components. Next, forward linear regression analysis (IBM SPSS, v.22) was used to estimate the correlation of the mutational signatures with PCs (eight PCs for pancancer exome/genome analysis, and five for mean exome/genome values across 33 cancers), with an acceptable P value of up to 0.05. For mutational signatures that were found to be correlated with PC3 (UV-ADARB1/A3C/ADAR mRNA level) and PC5 (age-A1 mRNA level), repeat detailed analysis was performed to identify the exact factor correlated with each signature. For those mutational signatures whose univariate Pearson regression analysis (IBM SPSS, v.22) suggested a potential link to tobacco use, this factor was also included in multivariate regression analysis. The third kind of multivariate regression analysis involving mutational signatures was performed using varying PCs (aforementioned eight) in particular cancers, including adrenocortical carcinoma (ACC; 91 exomes/0 genomes), bladder carcinoma (BLCA; 389/22), breast carcinoma (BRCA; 930/86), cervical/endocervical cancer (CESC; 271/20), cholangiocarcinoma (CHOL; 36/0), colon and rectal adenocarcinoma (COADREAD; 496/52), diffuse large B-cell lymphoma (DLBC; 31/6), esophageal carcinoma (ESCA; 182/0), glioblastoma multiforme (GBM; 362/35), head and neck squamous cell carcinoma (HNSC; 464/43), kidney chromophobe (KICH; 50/45), kidney clear cell carcinoma (KIRC; 334/27), kidney papillary cell carcinoma (KIRP; 251/33), acute myeloid leukemia (LAML; 139/0), low-grade glioma (LGG; 512/18), hepatocellular carcinoma (LIHC; 311/52), lung adenocarcinoma (LUAD; 528/37), lung squamous cell carcinoma (LUSC; 427/48), mesothelioma (MESO; 82/0), ovarian serous cystadenocarcinoma (OV; 385/27), pancreatic adenocarcinoma (PAAD; 174/0), pheochromocytoma/paraganglioma (PCPG; 182/0), prostate adenocarcinoma (PRAD; 484/19), sarcoma (SARC; 208/30), skin melanoma (SKCM; 412/37), stomach adenocarcinoma (STAD; 401/38), testicular germ cell tumor (TGCT; 149/0), thyroid carcinoma (THCA; 478/48), thymoma (THYM; 123/0), uterine corpus endometrial carcinoma (UCEC; 474/50), uterine carcinosarcoma (UCS; 57/0), and uveal melanoma (UVM; 80/0). Those correlations that were found in at least 2 out of 6 analyses (pancancer exomes/genomes, pancancer mean exomes/genome values across 33 cancers, and cancer-specific exomes//genomes) were considered to be consensus, and the data were used to cluster mutational signatures based on potential underlying factors. The median of the mean normalized A3A and A3B mRNA levels was used to classify A3A/B mRNA into low and high levels, and APOBEC/ADAR mutational status was considered to be positive when at least one of the corresponding genes was mutated. An unpaired two-tailed t test with Welch’s correction (IBM SPSS, v.22) was used to compare the mean number of mutational signatures among paired subgroups, including wild type versus mutated DRR, wild type versus mutated A3D/G, wild type versus mutated ADARB1/ADAR/A3F, wild type versus mutated APOBEC/ADAR, low- versus high-A1 mRNA level, low- versus high-A3A/B mRNA level, and low- versus high-ADARB1 mRNA level. Gene-E (https://software.broadinstitute.org/GENE-E/) was used to cluster r heat maps.
Supplementary Materials
References
- SG Conticello. The AID/APOBEC family of nucleic acid mutators. Genome Biol., 2008. [PubMed]
- LM Powell. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell, 1987. [DOI | PubMed]
- A Niavarani. APOBEC3A is implicated in a novel class of G-to-A mRNA editing in WT1 transcripts. PloS ONE, 2015. [DOI | PubMed]
- RS Harris, SK Petersen-Mahrt, MS Neuberger. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. cell, 2002. [DOI | PubMed]
- LB Alexandrov. Signatures of mutational processes in human cancer. Nature, 2013. [DOI | PubMed]
- MB Burns, NA Temiz, RS Harris. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat.re Gene., 2013. [DOI]
- SA Roberts. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Gene., 2013. [DOI]
- 8.Alexandrov, L. B. et al. The Repertoire of Mutational Signatures in Human Cancer. bioRxiv, 322859, 10.1101/322859 (2018).
- S Nik-Zainal. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat. Gene., 2014. [DOI]
- K Chan. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Gene., 2015. [DOI]
- PP Lau, WJ Xiong, HJ Zhu, SH Chen, L Chan. Apolipoprotein B mRNA editing is an intranuclear event that occurs posttranscriptionally coincident with splicing and polyadenylation. J. Biol. Chem., 1991. [DOI | PubMed]
- HP Bogerd. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Nat. Acad. Sci. United States Am., 2006. [DOI]
- RC Burdick, WS Hu, VK Pathak. Nuclear import of APOBEC3F-labeled HIV-1 preintegration complexes. Proc. Nat. Acad. Sci. United States Am., 2013. [DOI]
- M OhAinle, JA Kerns, MM Li, HS Malik, M Emerman. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe, 2008. [DOI | PubMed]
- S Ito. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc. Nat. Acad. Sci. United States of Am., 2004. [DOI]
- L Pasqualucci. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature, 2001. [DOI | PubMed]
- F Forconi. Hairy cell leukemia: at the crossroad of somatic mutation and isotype switch. Blood, 2004. [DOI | PubMed]
- 18.Niavarani, A., Shahrabi Farahani, A., Sharafkhah, M. & Rassoulzadegan, M. Pancancer Analysis Identifies Prognostic High-APOBEC1 Expression Level Implicated in Cancer In-Frame Insertions and Deletions. Carcinogenesis (2018).
- CD Middlebrooks. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat. Gene., 2016. [DOI]
- Y Kanke. Gene aberration profile of tumors of adolescent and young adult females. Oncotarget, 2018. [DOI | PubMed]
- N Tsuruoka. ADAR1 protein induces adenosine-targeted DNA mutations in senescent Bcl6 gene-deficient cells. J. Biol. Chem., 2013. [DOI | PubMed]
- Y Zheng, C Lorenzo, PA Beal. DNA editing in DNA/RNA hybrids by adenosine deaminases that act on RNA. Nucl. Acids Res., 2017. [DOI | PubMed]
- RA Lindley, P Humbert, C Larner, EH Akmeemana, CR Pendlebury. Association between targeted somatic mutation (TSM) signatures and HGS-OvCa progression. Cancer Med., 2016. [DOI | PubMed]
- RA Lindley, NE Hall. APOBEC and ADAR deaminases may cause many single nucleotide polymorphisms curated in the OMIM database. Mutation Res., 2018. [DOI | PubMed]
- A Boichard, IF Tsigelny, R Kurzrock. High expression of PD-1 ligands is associated with kataegis mutational signature and APOBEC3 alterations. Oncoimmunology, 2017. [DOI | PubMed]
- M Kimura. Evolutionary rate at the molecular level. Nature, 1968. [DOI | PubMed]
- SG Jin, Y Meng, J Johnson, PE Szabo, GP Pfeifer. Concordance of hydrogen peroxide-induced 8-oxo-guanine patterns with two cancer mutation signatures of upper GI tract tumors. Sci. Adv., 2022. [DOI]
- P Karihtala, K Porvari, O Kilpivaara. Mutational signatures associate with survival in gastrointestinal carcinomas. Cancer Genom. Proteom., 2022. [DOI]
- J Levatic, M Salvadores, F Fuster-Tormo, F Supek. Mutational signatures are markers of drug sensitivity of cancer cells. Nat. Commun., 2022. [DOI | PubMed]
- J Chang. Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nat. Commun., 2017. [DOI | PubMed]
- C Liu. A DNA methylation biomarker of alcohol consumption. Mol. Psych., 2018. [DOI]
- EK Schutsky, CS Nabel, AKF Davis, JE DeNizio, RM Kohli. APOBEC3A efficiently deaminates methylated, but not TET-oxidized, cytosine bases in DNA. Nucl. Acids Res., 2017. [DOI | PubMed]
- S Christensen. 5-Fluorouracil treatment induces characteristic T>G mutations in human cancer. Nat. Commun., 2019. [DOI | PubMed]
- LB Alexandrov. Clock-like mutational processes in human somatic cells. Nat. Gene., 2015. [DOI]
- LB Alexandrov. Mutational signatures associated with tobacco smoking in human cancer. Science, 2016. [DOI | PubMed]
- CP Diamond. AID, APOBEC3A and APOBEC3B efficiently deaminate deoxycytidines neighboring DNA damage induced by oxidation or alkylation. Biochim. et Biophys. Acta. General Subj., 2019. [DOI]
- EA Sia, RJ Kokoska, M Dominska, P Greenwell, TD Petes. Microsatellite instability in yeast: dependence on repeat unit size and DNA mismatch repair genes. Mol. Cell. Biol., 1997. [DOI | PubMed]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature, 2012. [DOI | PubMed]
- MAM Reijns. Signatures of TOP1 transcription-associated mutagenesis in cancer and germline. Nature, 2022. [DOI | PubMed]
- S Jimeno. ADAR-mediated RNA editing of DNA:RNA hybrids is required for DNA double strand break repair. Nat. Commun., 2021. [DOI | PubMed]
- EJ Steele, RA Lindley. ADAR deaminase A-to-I editing of DNA and RNA moieties of RNA:DNA hybrids has implications for the mechanism of Ig somatic hypermutation. DNA Repair, 2017. [DOI | PubMed]
- T Nonaka. Involvement of activation-induced cytidine deaminase in skin cancer development. J. Clin. Invest., 2016. [DOI | PubMed]
- P Pham, A Landolph, C Mendez, N Li, MF Goodman. A biochemical analysis linking APOBEC3A to disparate HIV-1 restriction and skin cancer. J. Biol. Chem., 2013. [DOI | PubMed]
- K Butler, AR Banday. APOBEC3-mediated mutagenesis in cancer: causes, clinical significance and therapeutic potential. J. Hematol. Oncol., 2023. [DOI | PubMed]
- Z Wang. B cell receptor signaling drives APOBEC3 expression via direct enhancer regulation in chronic lymphocytic leukemia B cells. Blood Cancer J., 2022. [DOI | PubMed]
- D Menendez, TA Nguyen, J Snipe, MA Resnick. The cytidine deaminase APOBEC3 family is subject to transcriptional regulation by p53. Mol. Cancer Res. MCR, 2017. [DOI | PubMed]
- S Kondo. APOBEC3A associates with human papillomavirus genome integration in oropharyngeal cancers. Oncogene, 2017. [DOI | PubMed]
- Y Saito. Involvement of APOBEC3B in mutation induction by irradiation. J. Radiat. Res., 2020. [DOI | PubMed]
- E Kocakavuk. Radiotherapy is associated with a deletion signature that contributes to poor outcomes in patients with cancer. Nat. Gene., 2021. [DOI]
- A Botvinnik. APOBEC3G rescues cells from the deleterious effects of DNA damage. FEBS J., 2021. [DOI | PubMed]
- Y Tong, S Kikuhara, T Onodera, L Chen, AB Myat, S Imamichi, Y Sasaki, Y Murakami, T Nozaki, H Fujimori, M Masutani. Radiosensitization to γ-ray by functional inhibition of APOBEC3G. Int. J. Mol. Sci., 2022. [DOI | PubMed]
- MT Chang. Small-cell carcinomas of the bladder and lung are characterized by a convergent but distinct pathogenesis. Clin. Cancer Res. Offic. J. Am. Assoc. Cancer Res., 2018. [DOI]
- M Takizawa, M Nakano, T Fukami, M Nakajima. Decrease in ADAR1 expression by exposure to cigarette smoke enhances susceptibility to oxidative stress. Toxicol. Lett., 2020. [DOI | PubMed]
- S Koutros. Targeted deep sequencing of bladder tumors reveals novel associations between cancer gene mutations and mutational signatures with major risk factors. Clin. Cancer Res. Offic. J. Am. Assoc. Cancer Res., 2021. [DOI]
- S Anant. ARCD-1, an apobec-1-related cytidine deaminase, exerts a dominant negative effect on C to U RNA editing. Am. J. Physiol. Cell Physiol., 2001. [DOI | PubMed]
- C Cenci. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J. Biol. Chem., 2008. [DOI | PubMed]
- CX Chen. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA, 2000. [DOI | PubMed]
- CJ Wittkopp, MB Adolph, LI Wu, L Chelico, M Emerman. A single nucleotide polymorphism in human APOBEC3C enhances restriction of lentiviruses. PLoS Pathog., 2016. [DOI | PubMed]
- U Beyer. Rare ADAR and RNASEH2B variants and a type I interferon signature in glioma and prostate carcinoma risk and tumorigenesis. Acta Neuropathol., 2017. [DOI | PubMed]
- E Eisenberg, EY Levanon. A-to-I RNA editing – immune protector and transcriptome diversifier. Nat. Rev. Gene., 2018. [DOI]
- P Sebastiani. RNA editing genes associated with extreme old age in humans and with lifespan in C. elegans. PloS ONE, 2009. [DOI | PubMed]
- R Rosenthal, N McGranahan, J Herrero, BS Taylor, C Swanton. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol., 2016. [DOI | PubMed]
- M Kanehisa, S Goto. KEGG: kyoto encyclopedia of genes and genomes. Nucl. Acids Res., 2000. [DOI | PubMed]