Fecal Metabolomics for the Diagnosis of Clostridioides difficile Infection
Abstract
Background: Clostridioides difficile infection (CDI) is the leading cause of nosocomial diarrhea. Current diagnostic tools have difficulty distinguishing between colonization and active infection. This study evaluated the utility of fecal metabolomics in diagnosing CDI in hospitalized patients with acute diarrhea. Methods: We conducted a prospective observational study involving hospitalized adults with new-onset diarrhea during admission. Participants were stratified into groups based on clinical and microbiological findings: controls, C. difficile colonized and C. difficile infected. Fecal samples were analyzed using UPLC-MS/MS and GC-MS to quantify selected short-chain fatty acids, amino acids, and bile acids. Multivariate and univariate statistical analyses included PLS-DA, sPLSDA, and tests with FDR correction. Results: Infected patients exhibited significantly higher concentrations of SCFAs and notable alterations in bile acid profiles. Key discriminative metabolites included isovalerate, propionate, isobutyrate and alpha-aminobutyric acid. ROC curve analyses showed strong diagnostic performance for these markers, with AUC values exceeding 0.85. Conclusions: Fecal metabolomic profiling could effectively differentiate between colonization and infection in CDI among hospitalized patients with diarrhea. These results highlight the potential of metabolomic signatures to enhance the diagnostic precision for CDI.
Article type: Research Article
Keywords: fecal metabolomics, diagnosis, biomarkers, bile acids, SCFAs, amino acids
Affiliations: Internal Medicine Department, Clinic University Hospital of Valencia, INCLIVA Biomedical Research Institute, 46010 València, Spain; Department of Medicine, Faculty of Medicine and Dentistry, University of València, 46010 València, Spain
License: © 2025 by the authors. CC BY 4.0 Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.3390/diagnostics15182331 | PubMed: 41008703 | PMC: PMC12468941
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (1.9 MB)
1. Introduction
Clostridioides difficile infection (CDI) is the most common cause of healthcare-associated diarrhea in high-income countries and represents a growing public health challenge due to its increasing incidence, recurrence rates, and severity [ref. 1,ref. 2]. While diagnostic techniques have evolved—from enzyme immunoassays to nucleic acid amplification tests (NAATs)—the ability to distinguish between active infection and asymptomatic colonization remains limited [ref. 3]. This distinction is clinically critical, as up to 3–21% of hospitalized patients may be colonized by toxigenic C. difficile without presenting symptoms [ref. 4,ref. 5], and overtreatment based on NAATs alone can lead to unnecessary antibiotic exposure and costs. The concept of “colonization resistance” underscores the protective role of the gut microbiota against C. difficile overgrowth [ref. 6,ref. 7]. Antibiotic-induced dysbiosis disrupts this balance, allowing spore germination, vegetative proliferation, and toxin production. However, microbial composition alone may not capture the full picture. Increasing evidence suggests that the gut metabolome—the collective metabolic output of host and microbial activity—plays a decisive role in determining susceptibility to CDI [ref. 8].
Among the most relevant microbial metabolites are bile acids and short-chain fatty acids (SCFAs). Primary bile acids, such as cholic and chenodeoxycholic acid, can promote C. difficile germination, while secondary bile acids like deoxycholic and lithocholic acid inhibit its growth and toxin production [ref. 9,ref. 10]. CDI has been consistently associated with an increased ratio of primary to secondary bile acids, reflecting impaired microbial 7α-dehydroxylation capacity [ref. 11,ref. 12]. On the other hand, SCFAs—particularly butyrate, isobutyrate, and propionate—contribute to epithelial integrity and modulate immune responses. Their depletion has been linked to gut barrier dysfunction and may facilitate disease onset and progression, although previous studies in animal and human models have shown contradictory results [ref. 13,ref. 14,ref. 15]. Despite the growing body of evidence, few studies have directly compared the fecal metabolic signatures of patients with active CDI, asymptomatic colonization, and non-colonized controls [ref. 15,ref. 16,ref. 17]. This distinction is crucial, as it mirrors the real-world diagnostic dilemma clinicians face, particularly when microbiological results and clinical presentation are discordant.
In this study, we applied targeted fecal metabolomics to characterize and compare the metabolic profiles of hospitalized patients with confirmed CDI, toxigenic C. difficile-colonized patients, and non-colonized controls with acute onset diarrhea. We aimed to identify metabolite-level alterations, especially in bile acid and SCFA pathways, that could serve as discriminatory biomarkers for CDI. By focusing on clinically relevant patterns, our goal was to explore the potential of fecal metabolomics as a complementary tool in the diagnostic work-up of CDI.
2. Materials and Methods
2.1. Study Design
This was a single-center, experimental, prospective study conducted at a tertiary hospital between January 2019 and March 2023. Fecal samples were collected from hospitalized adult patients with new-onset diarrhea for whom C. difficile diagnostic testing was requested.
2.2. Study Population and Classification
Inclusion criteria were as follows: age ≥ 18, hospitalization for more than 48 h at inclusion, acute diarrhea during admission, and availability of C. difficile diagnostic results (PCR for toxin B and toxin B antigen immunochromatographic assay). Exclusion criteria were recent CDI treatment (<14 days) or incomplete microbiological data. Patients were classified into three groups: (1) Infection: PCR-positive and antigen-positive, meeting all clinical criteria for CDI (including diarrhea with ≥3 unformed stools in 24 h, fever or abdominal pain, risk factors for CDI, and absence of alternative causes); further subdivided into first episode (I) and recurrence (R); (2) Colonized (C): PCR-positive, antigen-negative, with diarrhea but not meeting full CDI clinical criteria; (3) Control (X): Negative for both PCR and toxin, but with acute diarrhea during hospitalization.
2.3. Ethics Statement
All data were pseudonymized. Data collection followed GDPR and Spanish biomedical research regulations. The study was approved by the INCLIVA Institutional Review Board (CEIm), reference number 2020/271, on 12 November 2020, and conducted in accordance with the Declaration of Helsinki (2013).
2.4. Sample Size
This was an exploratory translational study. Based on literature and feasibility, we selected 30 CDI patients (15 first-episode infections and 15 recurrent infections), 15 colonized patients, and 15 controls.
2.5. Clinical Data Collection
Demographic, clinical, microbiological, and therapeutic variables were extracted from electronic health records. Collected variables are detailed in Supplementary Table S1.
2.6. Sample Handling and Metabolomic Processing
Fecal metabolomic profiling was conducted using a combined analytical platform incorporating both liquid chromatography–tandem mass spectrometry (LC-MS/MS) and gas chromatography–mass spectrometry (GC-MS). A total of 77 metabolites were quantified, including bile acids, amino acids, and SCFAs. Detection rates exceeded 95% for nearly all SCFAs and amino acids. Quantitative normalization followed standard internal procedures appropriate for fecal metabolomics, and concentrations were corrected for wet fecal weight.
2.6.1. Sample Preparation
Approximately 100–200 mg of each stool sample was aliquoted into 1.5 mL Eppendorf tubes and stored at −80 °C until analysis. Samples were thawed at room temperature, extracted in 600 µL CH3OH:PBS (1:1) using the PreCellys® homogenizer, and centrifuged (12,000× g, 4 °C, 12 min). Supernatants were transferred to clean vials and processed according to the analytical platform described below.
2.6.2. Bile Acids
Bile acids were quantified using a validated UHPLC-MS/MS protocol adapted from Ramos-García et al. [ref. 18]. Extracts were diluted with mobile phase (H2O:CH3CN 90:10, 0.1% HCOOH) and spiked with deuterated internal standards (2.1 µM each). Protein precipitation was achieved with cold methanol, followed by incubation (−20 °C, 20 min) and centrifugation (3500× g, 15 min, 4 °C). Methanol, propanol, acetic acid, ammonium acetate, acetonitrile, and formic acid were purchased from Merck Life Science S.L.U. (Madrid, Spain). Certified reference standards of bile acids (CA, CDCA, LCA, DCA, GCA, TCA, α-MCA, β-MCA, UDCA, HDCA, MCA, DHCA, HCA, GLCA, GUDCA, GHDCA, GCDCA, GDCA, GDHCA, GHCA, TLCA, TUDCA, THDCA, TCDCA, TDCA, TDHCA, THCA, and T-α-MCA) and deuterated internal standards (LCA-D4, CA-D4, GCDCA-D4, GCA-D4) were obtained from Steraloids (Newport, RI, USA). Sulfated bile acid standards and LCA-S-D4 were supplied by Qmx Laboratories Ltd. (Essex, UK). Analysis was performed on an ACQUITY UPLC system (Waters Ltd., Elstree, UK) coupled to a Xevo TQ-S triple quadrupole mass spectrometer (Waters, Manchester, UK) operated in negative electrospray ionization (ESI−) mode. Chromatographic separation was achieved using an ACQUITY BEH C8 column (100 × 2.1 mm, 1.7 µm; Waters) maintained at 60 °C. The mobile phases consisted of: A: Acetonitrile with 1 mM ammonium acetate (pH 4.15, adjusted with acetic acid), B: Acetonitrile:propanol (1:1, v/v). A gradient elution was applied as follows: 0–0.1 min, 10% B; 0.1–9.25 min, 10–35% B; 9.25–11.5 min, 35–85% B; 11.5–11.8 min, 85–100% B; 11.8–14.3 min, 100–10% B; and 14.3–16 min, 10% B. Injection volume: 5 µL. Detection was performed in MRM mode. Method validation followed FDA bioanalytical guidelines [ref. 19].
2.6.3. Short-Chain Fatty Acids and Branched-Chain Amino Acids
SCFAs and branched-chain amino acids (BCAAs) were analyzed using a derivatization method based on propyl chloroformate, adapted from Ramos-Garcia et al. [ref. 20]. SCFAs, branched-chain amino acids (BCAAs), propyl chloroformate, pyridine, sodium hydroxide, n-hexane, and isotopically labeled internal standard caproic acid-D3 were obtained from Sigma-Aldrich (Madrid, Spain). The AccQTag Ultra derivatization kit for amino acids was purchased from Waters (Milford, MA, USA). Ultrapure water was prepared using the Q-POD® system (Merck KGaA, Darmstadt, Germany). After derivatization, samples were analyzed using a GC 7890B system coupled to a 5977A quadrupole mass detector (Agilent Technologies, Santa Clara, CA, USA). Separation was achieved using a capillary HP-5MS column (30 m × 250 µm × 0.25 µm; Agilent J&W Scientific, Santa Clara, CA, USA).The oven program was: initial 50 °C (2 min), ramp to 70 °C at 10 °C/min, 85 °C at 3 °C/min, 110 °C at 5 °C/min, and finally 290 °C at 30 °C/min, holding for 8 min. Helium was used as the carrier gas at 1 mL/min. Data acquisition was performed in SIM mode using MassHunter B.07.01 software (Agilent, Santa Clara, CA, USA). Concentrations were normalized to wet stool weight. Analytical batches included quality control (QC) samples injected every 10 study samples, along with blanks and process controls. QC acceptance criterion: RSD < 25%. Method performance was validated for linearity, recovery, precision, and freeze–thaw stability.
2.6.4. Amino Acids
Amino acids were derivatized using the AccQTag Ultra kit (Waters, Milford, MA, USA) following the manufacturer’s protocol. 10 µL of the fecal extract was mixed with 10 µL H2O and 40 µL isopropanol (0.1% v/v formic acid). Subsequently, 10 µL of the diluted sample was combined with 70 µL borate buffer and 20 µL of the AccQTag reagent (6-aminoquinolyl-N-hydroxysuccinimidyl carbamate). After vortexing, samples were incubated at 55 °C for 10 min and transferred to a 96-well plate for analysis. Amino acids were quantified using specific MRM transitions optimized for each analyte, combined with external calibration curves prepared from pure standards. System performance and quantification accuracy were monitored through QC injections at regular intervals.
2.7. Metabolomics and Statistical Analysis
Statistical analysis of clinical variables was conducted using SPSS v29.0. Depending on the data type and distribution, group comparisons employed Student’s t-test, Mann–Whitney U test, chi-squared test, or Fisher’s exact test. Multivariate logistic regression was used to identify independent clinical predictors of CDI. Targeted metabolomics data were processed using MetaboAnalyst 6.0 [ref. 21] and MATLAB 9.14. To account for the high dimensionality and potential non-normal distribution of metabolite concentrations, non-parametric ANOVA and Mann–Whitney U tests were applied, with false discovery rate (FDR) correction to adjust for multiple comparisons. Key discriminatory metabolites were identified using Significance Analysis of Microarrays (SAM) (a feature selection method originally developed for high-dimensional data) based on delta thresholds and q-values. To evaluate the diagnostic separation between the CDI and control groups, dimensionality reduction and classification techniques were employed. Principal component analysis (PCA) was used for unsupervised pattern recognition. Partial least squares discriminant analysis (PLS-DA), a supervised multivariate technique, and its sparse variant (sPLS-DA) were applied to enhance group discrimination and pinpoint potential biomarker metabolites. Visualization tools such as clustered heatmaps and self-organizing maps (SOMs) provided intuitive representations of metabolic shifts associated with CDI. Missing values were conservatively imputed using the minimum detected positive value per metabolite, preserving biological relevance while minimizing distortion.
3. Results
3.1. Study Population and Sample Processing
A total of 60 fecal specimens were collected using a consecutive sampling strategy stratified by each of the four clinical groups defined by microbiological and clinical criteria: 30 individuals with confirmed CDI, subdivided into 15 with a primary episode (I) and 15 with a first documented recurrence (R); 15 individuals colonized with C. difficile (C); and 15 symptomatic control patients presenting with diarrhea but testing negative for the C. difficile toxin B gene by PCR (X). Each group was defined to explore distinct pathophysiological stages along the C. difficile infection spectrum. Following sample quality control, 12 fecal samples were excluded due to excess heterogeneity, defined by visual inspection (e.g., presence of undigested food or inconsistent stool texture), which could interfere with metabolomic profiling and mass spectrometry detection. This yielded a final analytical dataset of 48 fecal specimens (Figure 1). Mean concentrations for all analyzed metabolites are provided as supplementary material in Table S2.
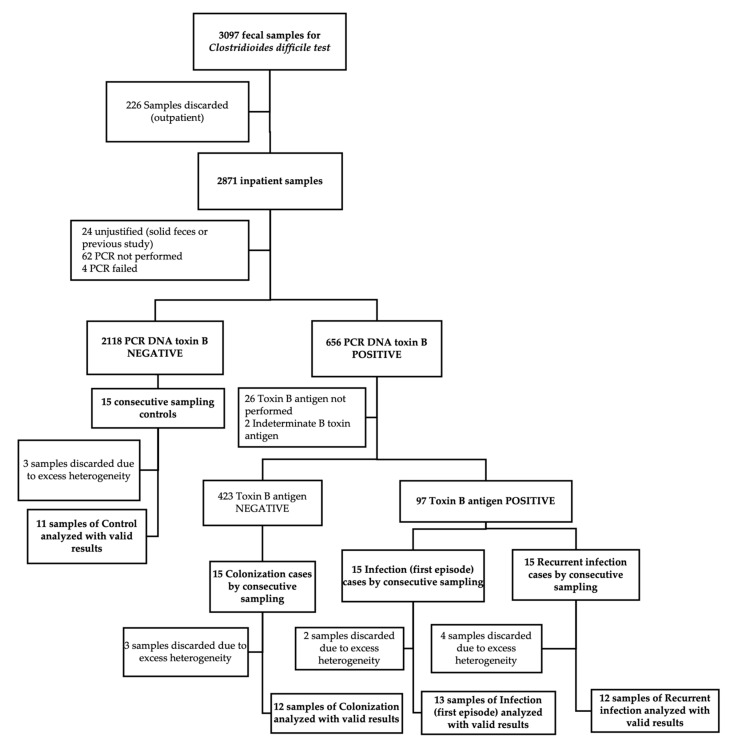
Demographic profiles, including age and sex distribution, did not differ significantly across groups (Table 1). However, patients with active CDI exhibited a slightly higher burden of chronic comorbidities, notably heart failure (40% vs. 0% in colonized and 9% in controls, p = 0.003) and advanced-stage chronic kidney disease (32% in CDI vs. 0% in colonized vs. 18% in controls, p = 0.025). Antibiotic exposure was markedly elevated in the CDI group, with 80% of patients reporting more than 10 cumulative days of antibiotic therapy in the preceding month (vs. 8.3% in colonized and 18.2% in controls, p < 0.001). While these significant clinical differences exist, subsequent analysis indicates they were not the primary drivers of the observed metabolomic signature. Other epidemiological characteristics, risk factors for CDI and clinical characteristics are summarized in Table 1 and Table 2 and Figure 2. Severity parameters and microbiological diagnostics tests results (including Ct values for PCR) among different groups at diagnosis are presented in Table A1 and Table A2, respectively.
Table 1: Epidemiological characteristics and comorbidities.
| Infection(n = 25) | Colonization(n = 12) | Control(n = 11) | p-Value | ||
|---|---|---|---|---|---|
| Female sex | 13 (52%) | 7 (58.3%) | 3 (27.3%) | 0.266 | |
| Neoplasia | Located | 3 (12.0%) | 0 (0.0%) | 1 (9.1%) | 0.560 |
| Metastatic | 6 (24.0%) | 4 (33.3%) | 2 (18.2%) | ||
| Leukemia | 1 (4.0%) | 2 (16.7%) | 1 (9.1%) | 0.444 | |
| Lymphoma | 2 (8.0%) | 2 (16.7%) | 1 (9.1%) | 0.732 | |
| Liver | Mild to moderate | 2 (8.0%) | 2 (16.7%) | 0 (0.0%) | 0.392 |
| Severe/cirrhosis | 1 (4.0%) | 0 (0.0%) | 0 (0.0%) | ||
| Moderate–severe CKD | 8 (32.0%) | 0 (0.0%) | 2 (18.2%) | 0.025 | |
| Hemodialysis | 1 (4.0%) | 0 (0.0%) | 1 (9.1%) | 0.466 | |
| IBD | 1 (4.0%) | 0 (0.0%) | 2 (18.2%) | 0.164 | |
| Diabetesmellitus | No TO damage | 1 (4.0%) | 1 (8.3%) | 4 (36.4%) | 0.024 |
| With TO damage | 4 (16.0%) | 0 (0.0%) | 0 (0.0%) | ||
| Dementia | 2 (8.0%) | 1 (8.3%) | 2 (18.2%) | 0.662 | |
| CVD | 1 (4.0%) | 1 (8.3%) | 1 (9.1%) | 0.794 | |
| Heart failure | 10 (40.0%) | 0 (0.0%) | 1 (9.1%) | 0.003 | |
| Ischemic heart disease | 3 (12.0%) | 0 (0.0%) | 1 (9.1%) | 0.288 | |
| Peripheral artery disease | 1 (4.0%) | 0 (0.0%) | 1 (9.1%) | 0.466 | |
| COPD | 1 (4.0%) | 1 (8.3%) | 0 (0.0%) | 0.510 | |
| Connective tissue disease | 1 (4.0%) | 2 (16.7%) | 0 (0.0%) | 0.199 | |
| Peptic ulcer | 2 (8.0%) | 1 (8.3%) | 1 (9.1%) | 0.994 | |
| Immunocompromised | 9 (36.0%) | 5 (41.7%) | 5 (45.5%) | 0.854 | |
| Charlson | ≤5 | 11 (44.0%) | 6 (50.0%) | 7 (63.6%) | 0.552 |
| 6 o+ | 14 (56.0%) | 6 (50.0%) | 5 (36.4%) | ||
CKD, chronic kidney disease; COPD, chronic obstructive pulmonary disease; CVD, cerebrovascular disease; IBD, inflammatory bowel disease; TO, target organ.
Table 2: Risk factors for CDI among patients included in the study.
| Risk Factor | Infection(n = 25) | Colonization(n = 12) | Control(n = 11) | p-Value | |
|---|---|---|---|---|---|
| Previous ATB use | 25 (100%) | 9 (75.0%) | 7 (63.6%) | 0.009 | |
| Number of previous ATB | 0–2 | 14 (56.0%) | 9 (75.0%) | 6 (54.5%) | 0.435 |
| >2 | 11 (44.0%) | 3 (25.0%) | 5 (45.5%) | 0.489 | |
| Type of previous ATB | Quinolones | 12 (48.0%) | 4 (33.3%) | 4 (36.4%) | 0.641 |
| Lincosamides | 1 (4.0%) | 1 (8.3%) | 0 (0.0%) | 0.606 | |
| Cephalosporins | 20 (80.0%) | 6 (50.0%) | 5 (45.5%) | 0.065 | |
| Penicillins | 12 (48.0%) | 3 (25.0%) | 1 (9.1%) | 0.058 | |
| Carbapenems | 2 (8.0%) | 2 (16.7%) | 5 (45.5%) | 0.029 | |
| Oxazolidinones | 3 (12.0%) | 0 (0.0%) | 4 (36.4%) | 0.041 | |
| Duration of previous ATB | ≤10 days | 5 (20.0%) | 11 (91.7%) | 9 (81.8%) | <0.001 |
| >10 days | 20 (80.0%) | 1 (8.3%) | 2 (18.2%) | — | |
| Concomitant ATB use | 11 (44.0%) | 5 (41.7%) | 5 (45.5%) | 0.983 | |
| ONS | 2 (8.0%) | 1 (8.3%) | 5 (45.5%) | 0.026 | |
| Antidepressants | 10 (40.0%) | 7 (58.3%) | 1 (9.1%) | 0.032 | |
| PPI/AntiH2 | 21 (84.0%) | 8 (66.7%) | 8 (72.7%) | 0.467 |
AntiH2, H2 antihistamines; ATB, antibiotic; ONS, oral nutritional supplement; PPI, proton pump inhibitors; WBC, white blood cells.
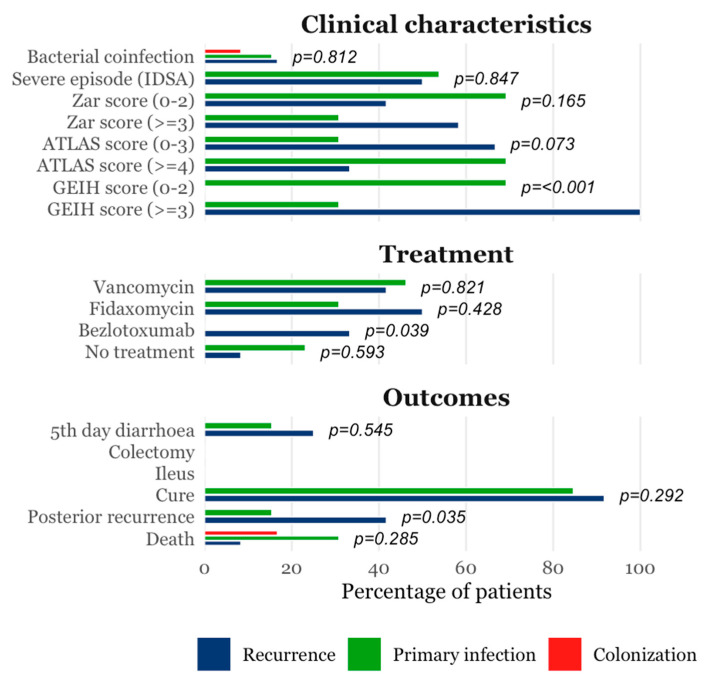
Table A1: Severity parameters among different groups at diagnosis.
| Recurrence | Infection | Colonization | Control | p-Value | |
|---|---|---|---|---|---|
| Temperature (°C) | 36.6(IQR 36.3–37.8) | 36.2(IQR 36–37.4) | 36.3(IQR 36.0–36.8) | 36.0(IQR 36.0–38.0) | 0.544 |
| WBC (cels/μL) | 14,015(IQR 7080–17,562) | 11,000(IQR 7265–24,970) | 7765(IQR 2630–9520) | 7170(IQR 5660–10,080 | 0.069 |
| Albumin (g/dL) | 3.2 (±0.6) | 3.03 (±0.7) | 3.39 (±0.7) | 2.9 (±0.7) | 0.280 |
| Creatinine (mg/dL) | 0.79(IQR 0.59–1.14) | 1.60(IQR 0.61–2.37) | 0.96(IQR 0.59–1.26) | 0.83(IQR 0.54–1.46) | 0.586 |
WBC, white blood cells.
Table A2: Microbiological diagnostic tests of CDI in the included patients.
| Test | Infection | Colonization | Control | |
|---|---|---|---|---|
| Ct PCR DNAtoxB | ≤25 | 22 (88.0%) | 2 (16.7%) | – |
| >25 | 3 (12.0%) | 10 (83.4%) | – | |
| GDH antigen | Positive | 21 (84.0%) | 5 (41.7%) | 0 (0.0%) |
| Negative | 0 (0.0%) | 5 (41.7%) | 10 (90.9%) | |
| Not performed | 4 (16.0%) | 2 (16.7%) | 1 (9.1%) | |
| Toxin B IC | Positive | 25 (100%) | 0 (0.0%) | 0 (0.0%) |
| Negative | 0 (0.0%) | 12 (100%) | 11 (100%) |
Ct, cycle threshold; GDH, glutamate dehydrogenase; IC, immunochromatographic test.
3.2. Global Metabolomic Trends
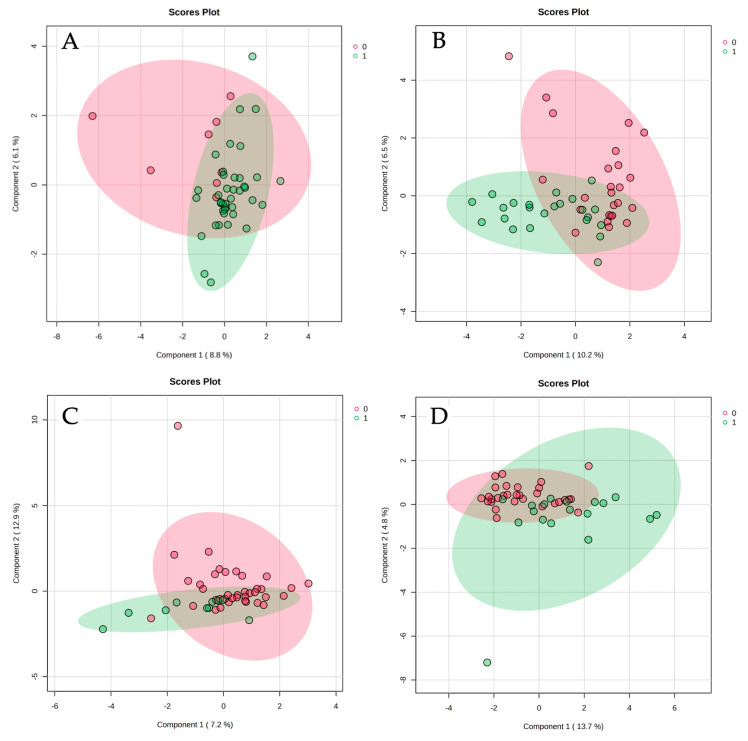
PCA revealed partial segregation between groups, with controls showing the most distinct separation from CDI patients. The first two principal components captured approximately 31% of the total variance. SCFAs contributed prominently to PC1, though their levels were notably elevated in C. difficile-positive samples. PLS-DA supported this trend, showing clear separation between controls and C. difficile-positive patients, with substantial overlap between colonized and infected groups. These data suggest that C. difficile colonization—irrespective of symptomatology—is also associated with a metabolically distinct fecal profile (Figure 3).
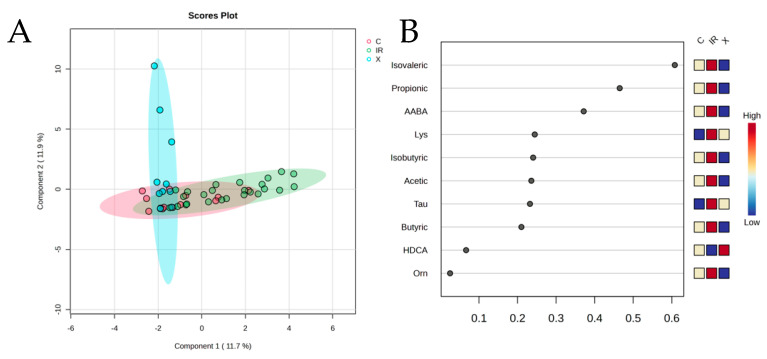
3.3. Differential Metabolites and Diagnostic Stratification
Several metabolites, particularly SCFAs and select amino acids, were significantly elevated in C. difficile-positive patients compared to controls. Propionate, isovalerate, and alpha-aminobutyric acid (AABA) showed the most consistent increases in infected patients. Boxplots and heatmaps revealed that these metabolites were markedly enriched in infection states, particularly in cases with recurrence (Figure 4).
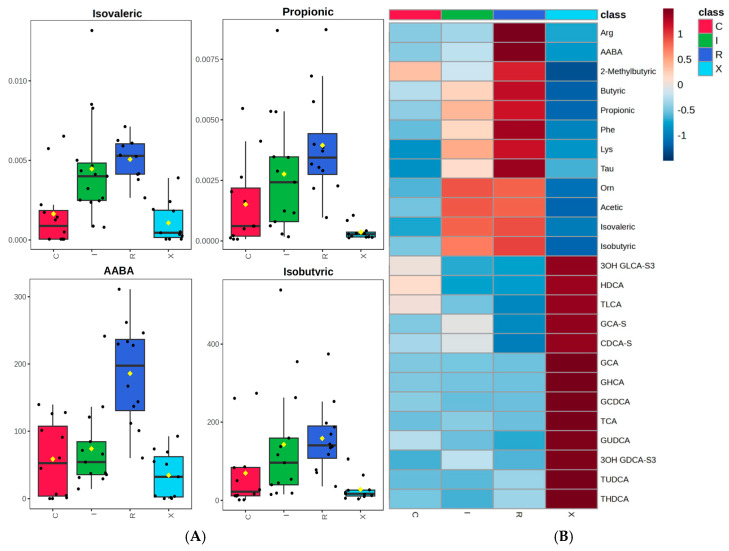
When comparing active CDI (first episode + recurrent infection) to non-infected patients (colonized + controls), isovalerate, propionate, and AABA were also significantly higher in the CDI group (p < 0.001; FDR < 0.01). Hierarchical clustering using these top metabolites provided good separation of infected from non-infected samples. Notably, SCFA elevation in CDI was observed in a much smaller proportion of colonized individuals, reinforcing its link with active infection.
Receiver operating characteristic (ROC) analysis confirmed the diagnostic potential of these analytes. Isovalerate and propionate achieved AUCs of 0.892 (0.786–0.979) and 0.849 (0.731–0.947) for identifying CDI, respectively. Metabolite ratios improved discriminative capacity further: aminoadipic acid (AAA)/propionate yielded an AUC of 0.929 (0.818–1.000) in 100-fold MCCV (Figure 5). Restricting analysis to CDI versus strictly non-colonized controls resulted in even higher diagnostic precision. Isovalerate alone produced an AUC of 0.941 (0.875–1.000). The AUC, sensitivity, and specificity of the main metabolites and ratios with biomarker potential for identifying CDI were determined through logistic regression with 10-fold cross-validation (Table 3).
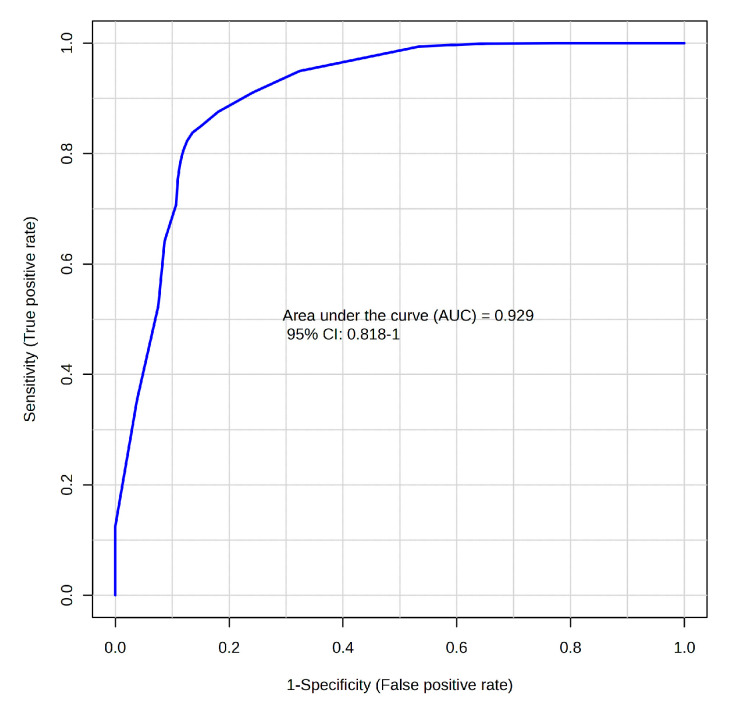
Table 3: Main metabolites and ratios with the highest potential as biomarkers for CDI and for recurrent infection, identified through logistic regression with 10-fold cross-validation.
| Metabolite/Ratio | Condition | AUC | Sensitivity | Specificity |
|---|---|---|---|---|
| AAA/propionate | CDI (I+R vs. X+C) | 0.883(0.785–0.982) | 0.783(0.783–0.951) | 0.880(0.753–1.000) |
| AAA/isovalerate | CDI (I+R vs. X+C) | 0.893(0.776–1.000) | 0.880(0.880–1.000) | 0.833(0.622–1.000) |
| GCDCA/isobutyrate | CDI (I+R vs. X) | 0.942(0.868–1.000) | 0.995(0.950–1.000) | 0.880(0.753–1.000) |
| AABA | Recurrent infection (R vs. I) | 0.888(0.700–1.000) | 0.917(0.917–1.000) | 0.769(0.540–0.998) |
AAA, aminoadipic acid; AABA, alpha-aminobutyric acid; GCDCA, glycochenodeoxycholic acid.
3.4. Recurrent Infection and Group-Specific Trends
The overall recurrence rate in our cohort was 28%, comparable to rates reported in previous studies (Seekatz et al., 34% [ref. 22]; Dawkins et al., 29% [ref. 23]; Khanna et al., 28.5% [ref. 24]) and higher than the 15% reported by Guh et al. [ref. 25]. Notably, none of our patients received metronidazole, which has been associated with increased recurrence risk; all were treated with vancomycin or fidaxomicin. When comparing recurrent CDI cases (R) to primary infections (I), modest differences in fecal metabolite concentrations were observed. Recurrent cases tended to exhibit higher levels of AABA and select SCFAs such as isovalerate and propionate. However, only AABA consistently reached statistical significance in the Mann–Whitney U test (FDR 0.045). To further explore this observation, we examined the metabolomic profiles of individuals who experienced a recurrence after their sample had been collected during the primary episode. Individuals in their first infection episode clustered more closely with the recurrent infection group in both unsupervised and supervised analyses, reinforcing the potential of fecal metabolomics to capture features associated with recurrence risk even prior to clinical relapse.
3.5. Influence of Comorbidities and Risk Factors
To assess potential confounding effects, we analyzed the fecal metabolomic profiles stratified by the presence or absence of major comorbidities and known risk factors for CDI, including heart failure, advanced chronic kidney disease, and recent antibiotic use. No significant differences in metabolite abundance or composition were observed between patients with and without these risk factors (Figure A1). This suggests that the metabolic alterations identified are primarily associated with infection status rather than underlying host factors, strengthening the specificity of the metabolomic signature for CDI.
4. Discussion
This study demonstrates that targeted fecal metabolomics can effectively differentiate between CDI, colonization, and non-infectious diarrhea in hospitalized patients. Elevated levels of specific SCFAs—notably isovalerate, propionate, and isobutyrate—along with AABA, emerged as robust metabolic signatures of active CDI. Diagnostic accuracy improved further through the construction of metabolite ratios, such as AAA/SCFAs and GCDCA/SCFAs, achieving AUC values exceeding 0.85. AAA was included in several metabolite ratios that achieved high AUC values, given its relatively stable concentrations across all study groups, providing a consistent reference point for comparative analysis. These results highlight the potential of metabolomic signatures to differentiate not only between infected and control individuals but also to distinguish infection from colonization—an enduring diagnostic challenge.
These findings are consistent with and extend previous research, particularly the landmark study by Robinson et al. [ref. 15], which also identified 4-methylpentanoic acid (4-MPA), an SCFA product of Stickland fermentation, an anaerobic amino acid fermentation pathway used by certain gut bacteria, as a potential CDI biomarker. More recently, a large multi-omics study of hospital-acquired diarrhea by Bosnjak et al. [ref. 26] likewise reported elevated levels of several Stickland by-products in CDI, including 4-MPA, isovalerate, and isobutyrate, further corroborating our observations. Unlike these cohorts, our study incorporated a more granular clinical characterization and stratification into first infection, recurrence, colonization, and control groups. The observed enrichment of SCFAs in infected patients aligns with the anaerobic fermentative metabolism of C. difficile, particularly via the Stickland reaction. This contrasts with prior assumptions that SCFA depletion is a hallmark of dysbiosis and, therefore, was associated with CDI [ref. 13,ref. 27]. Instead, our data suggest that certain SCFAs may accumulate specifically in the setting of C. difficile overgrowth, likely due to bacterial metabolic activity rather than host or commensal dysfunction. Notably, in our study, isovalerate exhibited excellent discriminatory capacity, reinforcing the role of leucine fermentation pathways in CDI pathogenesis. In addition to SCFAs, AABA showed strong discriminatory performance, especially in recurrent infections. While previously underappreciated, AABA is a non-proteinogenic amino acid linked to nitrogen export during bacterial amino acid metabolism [ref. 28] and may reflect intensified metabolic flux in recurrent disease. This supports its potential role as a recurrence-specific biomarker.
The profile of bile acids also revealed group-specific patterns. As expected from prior studies, infected patients showed relatively lower levels of secondary bile acids, consistent with a loss of microbial dehydroxylation activity [ref. 12,ref. 29]. Though these differences were less pronounced than those observed for SCFAs, ratios involving GCDCA and secondary bile acids added diagnostic value, particularly when distinguishing between infection and colonization. This is in line with experimental data showing that primary bile acids promote C. difficile germination, while secondary bile acids inhibit its vegetative growth and toxin activity [ref. 10,ref. 30].
It is worth noting that in our exploratory PCA, the first two components explained approximately 31% of the total variance. While this percentage may seem modest, it is consistent with the inherent complexity of fecal metabolomic datasets, which capture highly multidimensional information influenced by numerous host and microbial factors. Despite this, both PCA and PLS-DA revealed clear separation between controls and C. difficile-positive patients, supporting the biological relevance of the metabolomic differences observed.
Importantly, we found no significant influence of major comorbidities (e.g., DM, heart failure), antibiotic duration, or common medications (e.g., PPIs, antidepressants) on metabolomic patterns within clinical groups. Although the use of oral nutritional supplements (ONS) was more frequent in controls—possibly contributing to diarrhea in some of these patients—it did not significantly affect fecal metabolomic profiles. This supports the hypothesis that the metabolic shifts observed are primarily attributable to infection status, enhancing their potential utility as specific diagnostic tools.
While not designed for prediction, exploratory analyses suggested that metabolomic fingerprints may offer early indications of recurrence risk. In particular, samples from patients who later relapsed showed a profile more similar to recurrent cases at baseline. This echoes observations from Dawkins et al. [ref. 23], who highlighted metabolomic delays in post-treatment microbiome recovery as potential predictors of recurrence.
Some experts have proposed that C. difficile colonization and infection should be understood as a pathophysiological continuum rather than as two distinct clinical entities [ref. 31]. This spectrum-based model considers the interplay of multiple host and microbial factors that modulate disease progression. In our study, the proximity of metabolomic profiles observed between some colonized individuals and patients with active infection supports this concept and suggests that a subset of colonized patients may represent a preclinical stage of CDI. Identifying such high-risk individuals early could have important preventive implications, although the central clinical challenge remains determining when targeted treatment is truly warranted.
Although the comprehensive targeted fecal metabolomics approach used in this study relies on LC-MS/MS and GC-MS platforms that are currently confined to research settings, our findings highlight the translational potential of specific metabolites as diagnostic biomarkers. By improving the ability to distinguish between C. difficile colonization and true infection, these biomarkers could complement NAATs, which have high sensitivity but limited specificity and may lead to overtreatment. In the future, once validated in larger multicenter cohorts, simplified assays quantifying a small set of key metabolites directly from stool samples could enable practical implementation in clinical laboratories without requiring complex mass spectrometry workflows.
A distinguishing feature of our work is the comprehensive and systematic assessment of clinical characteristics and potential risk factors, which was conducted in far greater detail than in any previous metabolomic study of C. difficile infection, thereby allowing a more robust adjustment for possible confounders. Another strength of this study is the rigorous methodology applied in fecal sampling, storage, and analysis. Sample handling was tightly standardized to minimize variability related to collection, homogenization, and freeze–thaw cycles—critical issues in fecal metabolomics. Dual-platform analysis using GC-MS and UPLC-MS/MS enabled broad coverage of both volatile and non-volatile compounds, ensuring reliable quantification of key metabolite classes.
Despite these promising findings, several limitations should be noted. First, the study was exploratory, with a relatively small sample size and a cross-sectional design, which limit the statistical power, subgroup analyses, and the generalizability of our results. Consequently, the associations observed between specific metabolites and infection status should be interpreted with caution and considered hypothesis-generating. Although formal power calculations were not feasible due to the lack of prior effect size estimates for most metabolites, we acknowledge the potential for type I and type II errors as well as model overfitting. To mitigate these risks, we employed repeated cross-validation and multiple testing correction strategies; nevertheless, independent validation in larger, multicenter prospective studies is required to confirm the diagnostic potential of the identified biomarkers. Second, although all patients received standardized hospital diets and detailed information on antibiotic and other relevant drug use was collected, residual confounding from unmeasured dietary variability and concomitant medications cannot be ruled out. Given the well-known impact of both factors on the fecal metabolome, the lack of full control over these variables represents a significant limitation of our study and should be considered when interpreting our findings. Third, the lack of concurrent microbiome sequencing limits our ability to link metabolic shifts with specific microbial taxa. Future work integrating metagenomics with metabolomics will be critical to clarify host–microbiota–metabolite interactions.
5. Conclusions
This study adds to the growing evidence that fecal metabolomics may help refine the diagnostic approach to CDI. Our findings suggest that SCFAs, AABA, and bile acid ratios could complement existing microbiological testing by improving specificity—particularly in distinguishing colonization from infection, a current clinical challenge. These metabolites may therefore represent the basis for the development of adjunct diagnostic tools, although this hypothesis requires rigorous validation. Prospective studies in larger, multicenter cohorts with longitudinal follow-up and integration of microbiome sequencing will be essential to confirm these associations and assess their clinical utility. Ultimately, if validated, the incorporation of metabolomic biomarkers into clinical workflows has the potential to enable earlier and more accurate diagnoses, optimize treatment decisions, and reduce unnecessary antibiotic use.
References
- D.N. Gerding, S. Johnson, L.R. Peterson, M.E. Mulligan, J. Silva. Clostridium Difficile-Associated Diarrhea and Colitis. Infect. Control. Hosp. Epidemiol., 1995. [DOI | PubMed]
- F.C. Lessa, Y. Mu, W.M. Bamberg, Z.G. Beldavs, G.K. Dumyati, J.R. Dunn, M.M. Farley, S.M. Holzbauer, J.I. Meek, E.C. Phipps. Burden of Clostridium Difficile Infection in the United States. N. Engl. J. Med., 2015. [DOI | PubMed]
- L.C. McDonald, D.N. Gerding, S. Johnson, J.S. Bakken, K.C. Carroll, S.E. Coffin, E.R. Dubberke, K.W. Garey, C.V. Gould, C. Kelly. Clinical Practice Guidelines for Clostridium Difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis., 2018. [DOI | PubMed]
- C.R. Clabots, S. Johnson, M.M. Olson, L.R. Peterson, D.N. Gerding. Acquisition of Clostridium Difficile by Hospitalized Patients: Evidence for Colonized New Admissions as a Source of Infection. J. Infect. Dis., 1992. [DOI | PubMed]
- F. Alasmari, S.M. Seiler, T. Hink, C.-A.D. Burnham, E.R. Dubberke. Prevalence and Risk Factors for Asymptomatic Clostridium Difficile Carriage. Clin. Infect. Dis., 2014. [DOI | PubMed]
- M.C. Abt, P.T. McKenney, E.G. Pamer. Clostridium Difficile Colitis: Pathogenesis and Host Defence. Nat. Rev. Microbiol., 2016. [DOI | PubMed]
- M.B. Francis, C.A. Allen, R. Shrestha, J.A. Sorg. Bile Acid Recognition by the Clostridium Difficile Germinant Receptor, CspC, Is Important for Establishing Infection. PLoS Pathog., 2013. [DOI | PubMed]
- C.L. Ross, J.K. Spinler, T.C. Savidge. Structural and Functional Changes within the Gut Microbiota and Susceptibility to Clostridium Difficile Infection. Anaerobe, 2016. [DOI | PubMed]
- R. Thanissery, J.A. Winston, C.M. Theriot. Inhibition of Spore Germination, Growth, and Toxin Activity of Clinically Relevant C. Difficile Strains by Gut Microbiota Derived Secondary Bile Acids. Anaerobe, 2017. [DOI | PubMed]
- C.M. Theriot, V.B. Young. Microbial and Metabolic Interactions between the Gastrointestinal Tract and Clostridium Difficile Infection. Gut Microbes, 2013. [DOI | PubMed]
- L.-J. Palmieri, D. Rainteau, H. Sokol, L. Beaugerie, M. Dior, B. Coffin, L. Humbert, T. Eguether, A. Bado, S. Hoys. Inhibitory Effect of Ursodeoxycholic Acid on Clostridium Difficilegermination Is Insufficient to Prevent Colitis: A Study in Hamsters and Humans. Front. Microbiol., 2018. [DOI | PubMed]
- J.R.-M. Brown, B. Flemer, S.A. Joyce, A. Zulquernain, D. Sheehan, F. Shanahan, P.W. O’Toole. Changes in Microbiota Composition, Bile and Fatty Acid Metabolism, in Successful Faecal Microbiota Transplantation for Clostridioides Difficile Infection. BMC Gastroenterol., 2018. [DOI | PubMed]
- R.D. Rolfe. Role of Volatile Fatty Acids in Colonization Resistance to Clostridium Difficile. Infect. Immun., 1984. [DOI | PubMed]
- J.A. Ferreyra, K.J. Wu, A.J. Hryckowian, D.M. Bouley, B.C. Weimer, J.L. Sonnenburg. Gut Microbiota-Produced Succinate Promotes C. Difficile Infection after Antibiotic Treatment or Motility Disturbance. Cell Host Microbe, 2014. [DOI | PubMed]
- J.I. Robinson, W.H. Weir, J.R. Crowley, T. Hink, K.A. Reske, J.H. Kwon, C.-A.D. Burnham, E.R. Dubberke, P.J. Mucha, J.P. Henderson. Metabolomic Networks Connect Host-Microbiome Processes to Human Clostridioides Difficile Infections. J. Clin. Investig., 2019. [DOI | PubMed]
- P. Zhou, N. Zhou, L. Shao, J. Li, S. Liu, X. Meng, J. Duan, X. Xiong, X. Huang, Y. Chen. Diagnosis of Clostridium Difficile Infection Using an UPLC-MS Based Metabolomics Method. Metabolomics, 2018. [DOI | PubMed]
- M. Patel, D. Fowler, J. Sizer, C. Walton. Faecal Volatile Biomarkers of Clostridium Difficile Infection. PLoS ONE, 2019. [DOI | PubMed]
- V. Ramos-Garcia, I. Ten-Doménech, M. Vento, C. Bullich-Vilarrubias, M. Romaní-Pérez, Y. Sanz, A. Nobili, M. Falcone, M. Di Stefano, G. Quintás. Fast Profiling of Primary, Secondary, Conjugated, and Sulfated Bile Acids in Human Urine and Murine Feces Samples. Anal. Bioanal. Chem., 2023. [DOI | PubMed]
- Office of Clinical Pharmacology in the Center for Drug Evaluation and Research and the Center for Veterinary Medicine at the Food and Drug Administration Bioanalytical Method Validation Guidance for Industry Biopharmaceutics Bioanalytical Method Validation Guidance for Industry Biopharmaceutics Contains Nonbinding Recommendations. 2018
- V. Ramos-Garcia, I. Ten-Doménech, A. Moreno-Giménez, L. Campos-Berga, A. Parra-Llorca, Á. Solaz-García, I. Lara-Cantón, A. Pinilla-Gonzalez, M. Gormaz, M. Vento. GC-MS Analysis of Short Chain Fatty Acids and Branched Chain Amino Acids in Urine and Faeces Samples from Newborns and Lactating Mothers. Clin. Chim. Acta, 2022. [DOI | PubMed]
- Z. Pang, Y. Lu, G. Zhou, F. Hui, L. Xu, C. Viau, A.F. Spigelman, P.E. Macdonald, D.S. Wishart, S. Li. MetaboAnalyst 6.0: Towards a Unified Platform for Metabolomics Data Processing, Analysis and Inter-pretation. Nucleic Acids Res., 2024. [DOI | PubMed]
- A.M. Seekatz, K. Rao, K. Santhosh, V.B. Young. Dynamics of the Fecal Microbiome in Patients with Recurrent and Nonrecurrent Clostridium Difficile Infection. Genome Med., 2016. [DOI | PubMed]
- J.J. Dawkins, J.R. Allegretti, T.E. Gibson, E. McClure, M. Delaney, L. Bry, G.K. Gerber. Gut Metabolites Predict Clostridioides Difficile Recurrence. Microbiome, 2022. [DOI | PubMed]
- S. Khanna, E. Montassier, B. Schmidt, R. Patel, D. Knights, D.S. Pardi, P.C. Kashyap. Gut Microbiome Predictors of Treatment Response and Recurrence in Primary Clostridium Difficile Infection. Aliment. Pharmacol. Ther., 2016. [DOI | PubMed]
- A.Y. Guh, Y. Mu, L.G. Winston, H. Johnston, D. Olson, M.M. Farley, L.E. Wilson, S.M. Holzbauer, E.C. Phipps, G.K. Dumyati. Trends in U.S. Burden of Clostridioides Difficile Infection and Outcomes. N. Engl. J. Med., 2020. [DOI | PubMed]
- M. Bosnjak, A.V. Karpe, T.T.H. Van, D. Kotsanas, G.A. Jenkin, S.P. Costello, P. Johanesen, R.J. Moore, D.J. Beale, Y.N. Srikhanta. Multi-Omics Analysis of Hospital-Acquired Diarrhoeal Patients Reveals Biomarkers of Enterococcal Proliferation and Clostridioides Difficile Infection. Nat. Commun., 2023. [DOI | PubMed]
- C.M. Theriot, A.A. Bowman, V.B. Young. Antibiotic-Induced Alterations of the Gut Microbiota Alter Secondary Bile Acid Production and Allow for Clostridium Difficile Spore Germination and Outgrowth in the Large Intestine. mSphere, 2016. [DOI]
- M. Neumann-Schaal, J.D. Hofmann, S.E. Will, D. Schomburg. Time-Resolved Amino Acid Uptake of Clostridium Difficile 630Δerm and Concomitant Fermentation Product and Toxin Formation. BMC Microbiol., 2015. [DOI | PubMed]
- A.M. Seekatz, C.M. Theriot, K. Rao, Y.-M. Chang, A.E. Freeman, J.Y. Kao, V.B. Young. Restoration of Short Chain Fatty Acid and Bile Acid Metabolism Following Fecal Microbiota Transplantation in Patients with Recurrent Clostridium Difficile Infection. Anaerobe, 2018. [DOI | PubMed]
- A.R. Weingarden, C. Chen, A. Bobr, D. Yao, Y. Lu, V.M. Nelson, M.J. Sadowsky, A. Khoruts. Microbiota Transplantation Restores Normal Fecal Bile Acid Composition in Recurrent Clostridium Difficile Infection. Am. J. Physiol. Gastrointest Liver Physiol., 2014. [DOI | PubMed]
- B.W. Hurley, C.C. Nguyen. The Spectrum of Pseudomembranous Enterocolitis and Antibiotic-Associated Diarrhea. Arch. Intern. Med., 2002. [DOI | PubMed]