Progressive supranuclear palsy and corticobasal degeneration: novel clinical concepts and advances in biomarkers
Abstract
Background:
Progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) are sporadic adult-onset primary tauopathies clinically classified among the atypical parkinsonian syndromes. They are intrinsically related with regard to their clinical features, pathology, biochemistry, and genetic risk factors.
Objectives:
This review highlights the current knowledge on PSP and CBD, focusing on evolving clinical concepts, new diagnostic criteria, and advances in biomarkers.
Methods:
We performed a non-systematic literature review through the PubMed database. The search was restricted to articles written in English, published from 1964 to date.
Results:
Clinicopathologic and in vivo biomarkers studies have broadened PSP and CBD clinical phenotypes. They are now recognized as a range of motor and behavioral syndromes associated with underlying 4R-tauopathy neuropathology. The Movement Disorders Society PSP diagnostic criteria included clinical variants apart from the classical description, increasing diagnostic sensitivity. Meanwhile, imaging biomarkers have explored the complexity of symptoms and pathological processes related to corticobasal syndrome and CBD.
Conclusions:
In recent years, several prospective or clinicopathologic studies have assessed clinical, radiological, and fluid biomarkers that have helped us gain a better understanding of the complexity of the 4R-tauopathies, mainly PSP and CBD.
Article type: Review Article
Keywords: Parkinsonian Disorders, Tauopathies, Supranuclear Palsy, Progressive, Positron-Emission Tomography, Magnetic Resonance Imaging, Biomarkers, Transtornos Parkinsonianos, Tauopatias, Paralisia Supranuclear Progressiva, Tomografia por Emissão de Pósitrons, Imageamento por Ressonância Magnética, Biomarcadores
Affiliations: Universidade de São Paulo, Faculdade de Medicina, Hospital das Clínicas, Departamento de Neurologia, São Paulo, SP, Brazil.; Universidade de São Paulo, Faculdade de Medicina, Departamento de Radiologia, Laboratório de Medicina Nuclear (LIM 44), São Paulo, SP, Brazil.; Universidade de São Paulo, Faculdade de Medicina, Hospital das Clínicas, Instituto de Radiologia, Centro de Medicina Nuclear, Laboratório de Medicina Nuclear (LIM 43), São Paulo, SP, Brazil.
License: CC BY 4.0 This is an open-access article distributed under the terms of the Creative Commons Attribution License
Article links: DOI: 10.1590/0004-282X-ANP-2022-S134 | PubMed: 35976324 | PMC: PMC9491415
Relevance: Relevant: mentioned in keywords or abstract
Full text: PDF (821 KB)
INTRODUCTION
Progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) are sporadic neurodegenerative diseases clinically classified as atypical parkinsonian syndromes. After the development of levodopa to treat Parkinson’s disease (PD) in the late 1960s, diseases presenting with parkinsonism demonstrated to be wider than previously thought, and the group who had no improvement with levodopa treatment was then classified as atypical parkinsonism.
PSP is a term coined in 1964 by Steele, Richardson, and Olszewskiref. B1 to describe a progressive disease involving the basal ganglia, cerebellum, and the brainstem with neuronal loss in the substantia nigra, in which patients presented parkinsonism, supranuclear gaze palsy, axial rigidity, pseudobulbar state and dementia. Currently, the term PSP is used for a specifically defined neuropathology, whereas the clinical syndrome classically depicted is now referred to as “Richardson’s syndrome” (PSP-RS).
Corticobasal syndrome (CBS) and CBD were first described as a unique clinicopathological entity in 1967 and 1968 by Rebeiz et al.ref. B2 , ref. B3, who reported clinical and pathological findings of three patients with progressive stiffness and awkward limb movements, dystonic posturing, and gait disorder. They identified asymmetrical frontoparietal cortical atrophy, loss of neurons in the substantia nigra, and swelling of the neuronal cell bodies with achromatic cells, and called this entity “corticodentatonigral degeneration with neuronal achromasia”ref. B2 , ref. B3. Decades after the first description, the term “corticobasal degeneration” was coined by Gibb and Marsden in 1989ref. B4. Over time, it has become clear that the clinical features described by Rebeiz et al. were associated with various underlying pathologiesref. B5 – ref. B7. Thus, Boeve et al.ref. B8 introduced the term “corticobasal syndrome” to embrace the constellation of symptoms leading to the initially described clinical phenotype, while CBD currently denotes the pathological disorder.
PSP and CBD are conditions with notable overlap regarding their clinical presentation, pathological mechanisms, biochemistry, and genetic risk factorsref. B9. They are both characterized by the deposition of abnormal forms of tau protein, more specifically with the predominance of the four-repeat (4R) tau isoform and therefore classified as 4R-tauopathies.
Furthermore, PSP and CBD often pose challenges in clinical practice, primarily because patients with typical signs or symptoms might have different pathologies, and patients with the same pathology can display diverse clinical syndromes. However, although predicting underlying pathology is difficult, proper investigation of clinical features remains essential and informative to understand their progression and pathological processes. Despite being considered ‘prototype’ primary tauopathies and possibly ideal for studying neuroprotective agents, the 4R-tauopathies are still severe and untreatable diseases with no validated biomarkersref. B10.
This review aims to provide an overview of the current knowledge on PSP and CBD, mainly covering the following topics: evolving clinical concepts, new diagnostic criteria, pathology aspects, and advances in biomarkers.
SEARCH STRATEGY
In this review, we performed a non-systematic literature search restricted to articles written in English based on PubMed literature from 1964 to date, using the terms “progressive supranuclear palsy”, “corticobasal degeneration”, “corticobasal syndrome”, and “four-repeat tauopathies”. Research papers with pathologically confirmed diagnoses were preferentially sought.
NOVEL CONCEPTS: CLINICAL FEATURES AND NEW VARIANTS
PSP and CBD manifest with a broad and overlapping spectrum of clinical syndromes since the clinical presentation reflects the topographic distribution of histopathology more precisely than the specific underlying pathology. Initially considered atypical parkinsonian syndromes with mainly motor symptoms, it is recognized that they encompass a range of clinical phenotypes from movement, cognitive, and language abnormalities. PSP and CBD have a typical onset in the fifth to seventh decades and an average disease duration of 7.9 and 6.8 years, respectivellyref. B11.
PSP is the most common form of atypical parkinsonism, yet a rare disease, with an estimated prevalence of about 5-7 cases per 100.000 peopleref. B12. However, other studies with autopsy series or different phenotypes beyond PSP-RS have found a higher prevalence of 18 cases per 100.000ref. B13 , ref. B14. Also, little is known about the epidemiology of CBD and CBS. As CBD is a rare disease with various clinical phenotypes, accurate prevalence studies are lacking. A previous study showed an estimated CBD prevalence of 4.9-7.3 cases per 100.000 individualsref. B15. Regarding CBS, a community-based Japanese study found a prevalence rate of 6 per 100.000ref. B16, while a Russian study showed an age-standardized incidence rate of 0.02 cases per 100.000 individualsref. B17.
Following hypothetical models from other neurodegenerative diseases, PSP is currently considered a clinicopathological continuum from a presymptomatic phase through a suggestive-of-PSP phase, in which individuals do not fulfill Movement Disorders Society (MDS) PSP criteria but present mild PSP symptoms, later evolving to a known clinical variantref. B18.
In the first two years of presentation, approximately two-thirds of patients with PSP pathology present different clinical variants and only years later progress to PSP-Richardson syndromeref. B19. PSP clinical variants are presently named according to their predominant clinical features and comprise seven phenotypes.
The classic PSP clinical presentation is currently referred to as Richardson syndrome (PSP-RS), presenting as a levodopa-resistant, axial-predominant, symmetrical akinetic-rigid parkinsonism with early postural instability and frequent falls and slow vertical saccades lately evolving to vertical supranuclear gaze palsy. Subtle personality changes are frequently observed, including apathy and disinhibition, cognitive executive dysfunction, pseudobulbar state, dysarthria, dysphagia, and cervical dystoniaref. B9. Notably, vertical supranuclear gaze palsy might not be present until three or four years of disease onset. In contrast, other oculomotor dysfunctions such as absent vertical optokinetic nystagmus, square wave jerks, decreased vertical saccades velocity, and apraxia of eyelid opening usually are presented earlierref. B18. PSP-RS probably accounts for 24-50% of PSP casesref. B10 , ref. B20.
PSP-parkinsonism (PSP-P) is the second most common phenotype and the most difficult to distinguish from Parkinson’s disease. Individuals with a PSP-P clinical variant often present asymmetrical or symmetrical bradykinesia, tremor, and rigidity, which initially may be responsive to levodopa and have a slower progression rate than PSP-RS patientsref. B11.
The PSP with progressive gait freezing (PSP-PGF) variant, previously named pure akinesia with gait freezing (PAGF), is a rare clinical phenotype characterized by an isolated gait disorder without other PSP classical features. There is a progressive gait disturbance with freezing of gait and starting hesitation. Worth mentioning, this clinical variant is considered highly predictive of PSP underlying pathologyref. B21.
The MDS PSP criteria also recognize clinical phenotypes that initially are mainly cognitive syndromes. One of these is the PSP-speech and language variant (PSP-SL), whose main symptoms are agrammatism and apraxia of speech, similar to the nonfluent variant primary progressive aphasia phenotype (nfvPPA). Another cortical variant is the PSP with frontal presentation (PSP-F), which presents clinical features resembling the behavioral variant of frontotemporal dementia (bvFTD), with intense personality and cognitive disturbances prior to developing motor features. Finally, another cortical variant is the corticobasal variant (PSP-CBS), a phenotype mostly linked to CBD pathology, although it might be found in cases with a PSP neuropathology. In a previous postmortem study, PSP-CBS was present in only six of 179 PSP pathological casesref. B5. Another rare and somewhat controversial phenotype not included in the MDS PSP criteria is the PSP-cerebellar ataxia (PSP-C), in which cerebellar ataxia is observed before the typical motor findings. This variant was reported in two previous autopsy studies, albeit rarely found among PSP pathological casesref. B18 , ref. B22.
Regarding CBD clinical variants, CBS (CBD-CBS) is the clinical presentation in approximately 25-50% of autopsy-confirmed casesref. B5 , ref. B23 , ref. B24. Concerning classical motor and cortical CBD features, there is notable asymmetry. It is usually characterized by akinetic-rigid parkinsonism, dystonia, and myoclonus, associated with cortical symptoms such as ideomotor apraxia, alien limb phenomena, aphasia, or cortical sensory deficits. There are many available criteria for CBS, and they differ considerablyref. B25 , ref. B26. In the current criteria, probable CBS is characterized by an asymmetric presentation with at least two extrapyramidal dysfunctions of limb rigidity/akinesia, limb dystonia, and limb myoclonus, plus two cortical dysfunctions of orobuccal or limb apraxia, cortical sensory deficits, and alien limb phenomenaref. B25. Usually, symptoms often start in one limb, which is commonly described as rigid or “clumsy”. Such rigidity is intense and of mixed nature, with aspects of rigidity, paratonia, and dystonia, associated with marked bradykinesia and commonly the most prevalent motor symptom. The typical scenario is a progressive rigidity and apraxia in one upper limb, then involvement of either the ipsilateral lower limb or the contralateral upper limb, eventually leading to severe disability several years later. Noteworthily, CBS is a clinical syndrome, not a designed pathology, and has many possible pathologies beyond CBD, including Alzheimer’s disease (AD), PSP, Pick’s disease, and othersref. B23.
Another well-described CBD phenotype is CBD-Richardson (CBD-RS) when patients develop abnormal eye movements and symmetrical parkinsonism similar to those seen in patients with PSPref. B27 – ref. B29. A previous study highlighted subtle differences in eye movement disorders in patients with CBD-RS compared to PSP-RS, such as increased saccadic latencies with preserved velocity seen in patients with CBDref. B5.
Other CBD variants are predominantly cognitive syndromes. They might show a phenotype similar to bvFTD (CBD-bvFTD) or visuospatial dysfunctions resembling the posterior cortical atrophy syndrome (CBD-PCA). The latest CBD diagnostic criteria grouped these cognitive variants into a frontal behavioral-spatial syndrome (CBD-FBS)ref. B25. CBD pathology also can manifest as nonfluent PPA and primary progressive apraxia of speech (PPAOS), named CBD-nfvPPA variant. A prior study suggested that patients with PSP pathology had more PPAOS without nfvPPA, while patients with CBD pathology showed both PPA and PPAOSref. B30. Noteworthily, although PSP and CBD usually demonstrate some preferential clinical manifestations, the width of their clinical manifestations encompasses the same heterogeneous spectrumref. B31.
PSP and CBD are mainly sporadic diseases; however, rare familial forms have been described. More than 50 mutations in the microtubule-associated protein tau (MAPT) gene have been identifiedref. B32, and some of them can result in clinical phenotypes and pathological features equal to PSP and CBD. Moreover, they share similar genetic risk factors, such as the MAPT H1 haplotype, APOE e2/e2, and single nucleotide polymorphism in the MOBP generef. B33.
PSP AND CBD DIAGNOSTIC CRITERIA
Clinical diagnostic criteria were recently redefined for PSPref. B34 and CBDref. B25. However, the definitive diagnosis of PSP and CBD is still only established at postmortem examination.
The MDS-PSP criteriaref. B34 recognized suggestive forms of PSP and operationalized diagnoses of non-RS phenotypes. There were 12 core clinical features in four clinical domains: ocular motor dysfunction (O), postural instability (P), akinesia (A), and cognitive dysfunction (C). Each clinical domain includes three features graded from the more to the least specific for PSP diagnosis. The criteria also included clinical clues and imaging findings. They proposed three degrees of diagnostic certainty: probable, possible, and suggestive of PSP, and the predominant clinical variantref. B34. A diagnosis of probable PSP requires vertical gaze palsy or slow vertical saccades, with one more core clinical feature. Conversely, the last criteria from the National Institute for Neurological Disorders and Society for PSP (NINDS-SPSP)ref. B35 only considered the PSP-RS phenotype. Previous clinicopathologic studies have shown a better sensitivity for the MDS PSP criteria than for NINDS-SPSP criteriaref. B36.
Before the latest CBD diagnostic criteria, others have been proposed. However, they demonstrated low sensitivity and specificity, reflecting only the CBS phenotyperef. B37. In light of the expanding understanding of CBD clinicopathologic correlations, a specialist consensus with brain bank cases and a critical literature review developed new diagnostic criteria for CBD and CBSref. B25.
The current CBD diagnostic criteria are the first to incorporate phenotypes other than CBS into the CBD clinical continuum. They proposed two diagnostic classifications for CBD: (1) “clinical research criteria for probable sporadic CBD” (cr-CBD) and (2) “possible CBD” (p-CBD). In the most specific one, namely cr-CBD, age must be greater than or equal to 50 years, and there should not be a family history. Included phenotypes were probable CBS, nfvPPA, and frontal behavioral-spatial syndrome. Furthermore, these last two phenotypes must contain CBS clinical components. The p-CBD criteria aimed to be less restrictive with higher sensitivity. There was no minimum age, and positive family history was allowed. In addition, other phenotypes such as possible CBS and PSP phenotypes were included. In both scenarios, the progression must be gradual with insidious onset and a minimum duration of one yearref. B25.
Despite recent efforts to refine CBD clinical criteria, validation studies with clinicopathological cohorts demonstrated that there is still poor sensitivity within two years of disease onset, and patients without CBD pathology can fulfill the cr-CBD clinical criteria. Moreover, there is low specificity for distinguishing CBS due to underlying CBD pathology from others, such as Alzheimer’s diseaseref. B38 , ref. B39.
The MDS PSP criteria also proposed the novel diagnostic category “probable 4R-tauopathy” to address the phenotypic clinical overlap between PSP and CBD, thus improving their antemortem clinical recognitionref. B40. These diagnostic criteria were introduced to recognize patients with clinical syndromes predicting the underlying four-repeat tauopathy pathology with high specificity, although only moderately specific for PSP pathologyref. B40. They comprise all “probable PSP”ref. B34, possible PSP-SL, and possible PSP-CBS. These novel criteria were highly specific in a previous postmortem validation studyref. B40, and the clinical concept of 4R-tauopathies might have advantages for clinical practice and research.
PSP AND CBD PATHOLOGY ASPECTS
PSP and CBD belong to a group of diseases called tauopathies characterized by abnormal deposition of hyperphosphorylated tau protein in the brain. In contrast to Alzheimer’s disease, tau pathology is the main driver of neurodegeneration in PSP and CBD. In the brains of PSP and CBD patients, tau pathology is observed in neurons and glial cells and is predominantly comprised of four-repeat tau isoformsref. B10 , ref. B41. Tau protein is the major neuronal microtubule-associated protein (MAP) encoding by the microtubule-associated protein gene (MAPT) and promotes the assembly and stabilization of microtubules. The alternative splicing of the exons 2,3 and 10 generates six tau isoforms with four and three microtubule-binding domains (4R- and 3R-tau, respectively). An equal proportion of 3R and 4R isoforms are observed in normal human brainsref. B42.
The mechanism by which tau becomes nonfunctional is not entirely understood, but post-translational modifications may play an important role. Hyperphosphorylation is the most important and impacts microtubules’ stability and axonal transport. The decreasing tubulin-binding capacity impairs the interaction between tau and microtubules, leading to microtubule disorganization with protein self-polymerization and aggregationref. B42 , ref. B43.
According to the distribution and burden of neuronal and glial tau pathology and the involvement of different brain areas, the neuropathological phenotypes of PSP and CBD can be distinguished. Atrophy in the subthalamic nucleus, in the brainstem tegmentum, and depigmentation of substantia nigra are neuropathological features of PSP. Microscopically, a high density of globose neurofibrillary tangles, the most characteristic neuronal lesion, in basal ganglia and brainstem are found in typical PSP cases (Figure 1). However, diffuse granular cytoplasmic immunoreactivity in neurons can be present. In addition, neuropil threads are observed in cortical and subcortical areas, mainly in the striatum, globus pallidus, substantia nigra, and subthalamic nucleusref. B41. Glial inclusions are located in white and gray matter in variable amounts and distributions. Tufted-astrocytes (Figure 1) are not pathognomonic of PSP but are the most disease-specific pathological finding, mainly observed in the precentral gyrus, striatum, and superior colliculusref. B41. Oligodendroglial inclusions in the form of coiled bodies (Figure 1) are numerous in white matter tracts in the basal ganglia, thalamus, and brainstem.
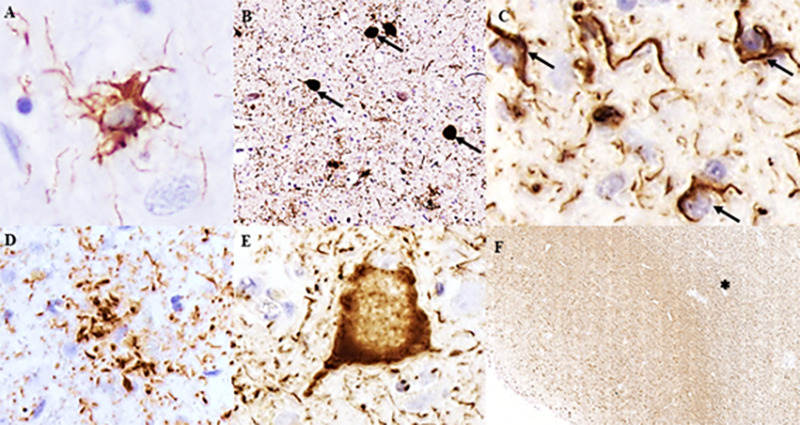
Asymmetric focal cortical atrophy in the superior frontal and parietal regions and depigmentation of the substantia nigra can be observed in CBD cases. In contrast with PSP, CBD shows more prominent neuronal tau pathology in the forebrain. Numerous neuropil threads are found in white and gray matter associated with variable amounts of coiled bodies, and astrocytic plaques, mainly in affected cortical areas and striatum, are hallmarks of CBD (Figure 1). The burden of coiled bodies is lower compared to PSP. Neuronal tau pathology differs from PSP by the presence of achromatic ballooned neurons in affected cortical areas and small globose tangles, and coiled bodies in substantia nigra and locus coeruleus (Figure 1). Besides the type of astroglial tau pathology, the severe involvement of the white matter in CBD and the presence of neurofibrillary tangles in subcortical areas in PSP are some neuropathological findings that can help distinguish PSP and CBD (Figure 1). All neuropathological lesions observed in PSP and CBD are immunoreactive for 4R-tau antibodies but negative for 3R-tauref. B41.
NEUROIMAGING AND BIOFLUIDS BIOMARKERS
There is growing interest in developing disease-specific biomarkers to aid in predicting pathology in the antemortem diagnosis of neurodegenerative diseases. Tau disease pathology-targeted therapies are currently being developed in clinical trialsref. B44. Hence, there is a need for diagnostic biomarkers to detect PSP and CBD pathology in presymptomatic individuals or early disease phases. Moreover, essential insights into the pathophysiological mechanisms and clinical symptoms have been gained by using advanced neuroimaging techniques in PSP and CBD.
The main biomarkers under study regarding PSP and CBD/CBS include structural imaging modalities such as magnetic resonance imaging (MRI), molecular imaging with functional imaging and specific ligands using positron emission tomography (PET), single-photon emission tomography (SPECT), and biofluid biomarkers such as cerebrospinal fluids (CSF) and serum components.
MRI
In PSP, the imaging hallmark is midbrain atrophy, which can be assessed through the visual identification of the “hummingbird sign”ref. B45 (specificity 99%, sensitivity 50%) on the sagittal plane, the “morning glory flower sign”ref. B46 (specificity 97%, sensitivity 37%) on the axial plane, and the superior cerebellar peduncle atrophy in the coronal plane (Figure 2)ref. B47 , ref. B48. More objective measures that can help diagnose PSP include the area, diameter, and volume of the midbrainref. B49, pons-midbrain area ratioref. B50, and the parkinsonism index MRPI (magnetic resonance parkinsonism index), calculated through the measurement of the ratios of the pons to midbrain area and middle cerebellar peduncle to superior cerebellar peduncle widthsref. B50. The latter is especially sensitive and specific for distinguishing PSP from PD, multiple system atrophy-parkinsonian type (MSA-P), and healthy controlsref. B51. Apparent diffusion coefficient (ADC) increased values in the putamen and superior cerebellar peduncle have good sensitivity and specificity in differentiating PSP-RS from PDref. B52. Also, diffusion tensor imaging (DTI) may show a degeneration pattern suggestive of PSP-RSref. B53.
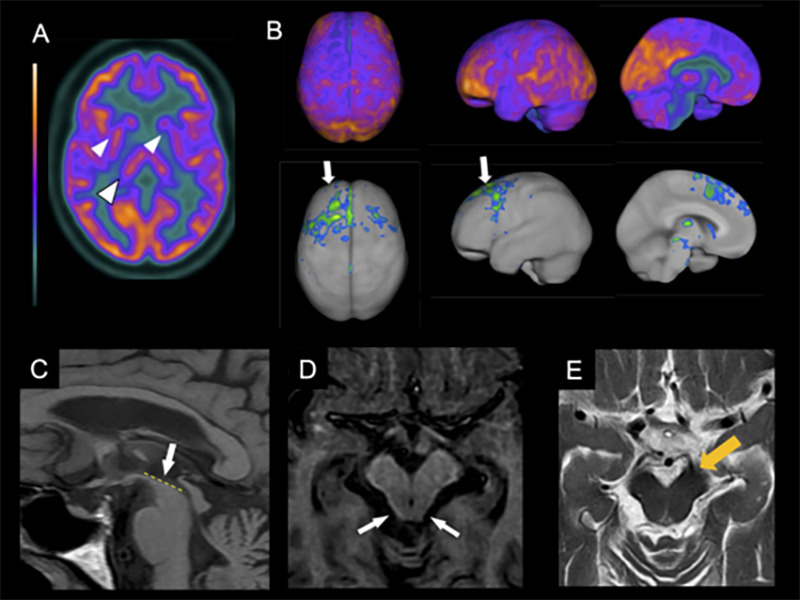
Although these MRI findings are valuable for differentiating PSP-RS from other parkinsonian syndromes with high specificity, they are less sensitive than PSP clinical diagnosis in its distinction from other degenerative parkinsonismsref. B46 , ref. B49. Another limitation is that most studies restricted their subjects to patients presenting with a PSP-RS phenotype. One of the few studies that included PSP presentations other than RS showed that cortical variants (PSP-CBS, PSP-F, PSP-SL) had more frontal atrophy than subcortical variants (PSP-RS, PSP-P, PSP-PGF)ref. B54.
Resting-state functional MRI in PSP revealed a perturbation of extensive neural networks, especially connections towards the dorsal midbrain tegmentumref. B55, as well as widespread disruption of cortical-subcortical connectivityref. B56. Frontostriatal hypoactivity can also be seen in functional MRI during vertical saccades in PSP compared with controlsref. B57. Another functional MRI study assessing speech tasks in PSP demonstrated strong activation of the lingual gyrus and reduced activation of the primary areas with the recruitment of remote areasref. B58.
CBD, in its turn, usually presents supratentorial patterns of atrophy, mainly asymmetrical patterns in the posterior frontal, superior parietal lobe, and basal ganglia (Figure 3). Cortical thinning and subcortical volume loss prominently involve the hemisphere contralateral to the more affected limb. Also, motor severity negatively correlates with the contralateral cortical thinning in the precentral and postcentral gyri and with volumes of putamenref. B59. Moreover, multimodal MRI studies search for CBS patterns of structural lesions that may suggest underlying pathology. A clinicopathologic study suggested that patterns of gray matter loss in CBS differ according to the underlying pathologyref. B60. Individuals with CBS and a postmortem diagnosis of CBD and PSP displayed similar focal atrophy at premotor and supplementary motor areas. In contrast, patients with underlying FTD-TDP43 and AD pathology had a more widespread pattern of gray matter loss at the frontotemporal lobe and temporoparietal regions, respectivellyref. B60.
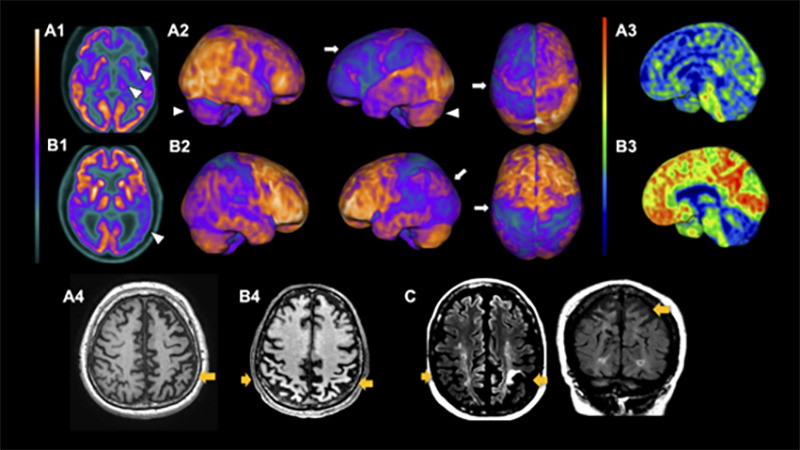
PET
Molecular neuroimaging using PET allows for quantitative visualization of functional processes in vivo. [18F]fluorodeoxyglucose (FDG) is the most commonly used radiotracer for assessing regional brain glucose metabolism as a marker of neuronal function. Additionally, specific-pathology ligands, such as the amyloid-PET and tau tracers, are leading the frontiers of neurodegenerative diseases biomarkers with their role in disclosing underlying pathology.
The brain metabolic patterns obtained from FDG-PET assist in the early diagnoses of neurodegenerative diseases and is helpful to differentiate Parkinson’s disease from atypical parkinsonismref. B61 , ref. B62. FDG-PET in PSP-RS shows a characteristic pattern of hypometabolism in the midbrain, basal ganglia, thalamus, and frontal lobes, including prefrontal, anterior cingulate, premotor, and motor regionsref. B61 , ref. B63. A typical asymmetrical hypometabolism in CBD involves the frontal and parietal lobes, basal ganglia, and thalamus (Figure 2).
Although CBD displays a characteristic frontoparietal asymmetric hypometabolismref. B64, CBS shows a more complex set of metabolic patterns due to its diverse neuropathologies. A recent study with neuropathologic examination showed that CBS underlying pathologies are associated with different metabolic degeneration patterns and described hypometabolism for CBS-CBD, CBS-AD, and CBS-PSPref. B65. Another prospective study using FDG-PET and amyloid-PET in a CBS cohort showed that individual brain metabolic patterns could distinguish with high specificity and accuracy CBS due to AD pathology from CBS non-AD pathological variantsref. B66, suggesting that it might be routinely used in the clinical workup of CBS. Accordingly, CBS-AD shows distinct metabolic signatures, such as a more posterior temporoparietal pattern and less frontal hypometabolism, compared to CBS not related to AD pathologyref. B65 , ref. B66. As neurodegeneration modulates metabolism, the metabolism also can depict its clinical features and clinical variants in CBSref. B66 , ref. B67.
Concerning PSP, however, there is still uncertainty regarding its usefulness in distinguishing PSP clinical variants. A previous study demonstrated that FDG-PET is a reproducible diagnostic tool for PSP-RS and PSP-P variantsref. B68.
Moreover, PET tracers that bind to the protein tau aggregated as neurofibrillary tangles have been developed and performed in PSP and CBD patients. Prior studies using the first generation of tau-targeting tracers, the most widely used ref. B18F-AV-1451 (also known as flortaucipir), showed good correspondence between in vivo imaging and postmortem PSP and CBD evaluationref. B54 , ref. B69 , ref. B70. Also, it was helpful to distinguish between CBS, PSP, and ADref. B69, and the uptake in globus pallidus might be a valuable measure for differentiating PSP-RS from PDref. B70. Nevertheless, as the ultrastructural characteristics of tau filaments differ across diseases, first-generation tau tracers demonstrate more affinity to paired helical filaments found in AD (tau 3R/4R) than straight filaments found in 4R-tauopathiesref. B10.
In contrast, second-generation tau PET tracers such as [18F]PI-2620 demonstrated more specificity for 4R-tauopathies. Elevated uptake has been observed in several areas in PSP-RS, such as globus pallidus, subthalamic nucleus, putamen, substantia nigra, and dentateref. B71. The same tracer has been studied in CBS and demonstrated to help diagnose underlying pathology and potentially monitor disease progressionref. B72.
The first specific tracer to amyloid-beta applied in human studies was the [ref. B11C]Pittsburgh Compound-B (PIB). Later, the second generation of amyloid tracers was developed and named florbetapir, flutemetamol, and florbetaben. Noteworthily, amyloid-PET interpretation has some limitations as the fact that it is positive in about 20-30% of cognitively normal individuals and non-AD dementias, especially when older (mostly above 70 years old)ref. B73. Currently, amyloid-PET is available for clinical use and is approved by many regulatory agencies worldwide. In CBS, it can be a valuable tool to distinguish cases related to underlying AD pathology from cases related to CBD or PSP. A prior study that used amyloid-PET in CBS patients showed that, among 14 patients, four were positive with high PIB binding (a standardized uptake ratio >1.5), indicating underlying AD pathologyref. B74. Subtle differences in the clinical presentation were noted between groups, with greater impairment of visuospatial function, more frequent deficits in sentence repetition, and more significant functional decline in PIB-positive patientsref. B74. In a more recent cohort, among 30 CBS patients, 13 had a positive PIB-PET, and they also demonstrated worse cognitive performancesref. B66 , ref. B67.
Fluid biomarkers
Several fluid biomarkers have been assessed in neurodegenerative diseases. It is now established that the increase of total tau (T-tau) and phosphorylated tau (p-tau) with low levels of the 42 aminoacids beta-amyloid isoform (Aß42) are highly sensitive and specific for predicting AD pathologyref. B75. In contrast, there is not yet an accurate set of fluid biomarkers for differentiating underlying pathologies for parkinsonian disorders. Some studies demonstrated that the neurofilament light chain protein (NfL) is increased in patients with atypical parkinsonian syndromes and may help differentiate them from Parkinson’s diseaseref. B76.
A previous study assessing 160 AD, PD, CBS, PSP, FTD, and MSA patients and 30 controls tested nine potential CSF biomarkers: T-tau, p-tau, Aß42, NfL, ?-synuclein, YKL-40, MCP-1, and soluble amyloid precursor protein α and β (sAPPα and sAPPβ)ref. B77. In this study, NfL, ?-synuclein, and sAPP? were the ones that better discriminated PD from atypical parkinsonism, but no biomarkers combination could discriminate between atypical parkinsonism subtypes (PSP, CBD, MSA).
NfL, despite its limited role in differential diagnosis, appears to be helpful as a neuronal lesion marker and to predict disease progressionref. B78. Several longitudinal studies assessing biomarkers and clinical progression in PSP showed that NfL increases with time and correlates with disease progressionref. B78.
CSF NfL may increase up to 30% in a one-year interval in PSP patientsref. B79, and NfL increase accompanies disease severity (as measured by Hoen and Yahr and PSP rating scale) and the decrease of superior cerebellar peduncles’ volumeref. B77. Serum NfL correlates with CSF NfL levels and may predict worse outcomes at higher levelsref. B79. NfL has been included in recent clinical trials as part of exploratory endpoints and is promising as a surrogate outcome for future trialsref. B80.
In conclusion, in recent years, several prospective or clinicopathologic studies have assessed clinical, radiological, and fluid biomarkers that have helped better understand the heterogeneity and complexity of 4R-tauopathies. Currently, it is acknowledged that their heterogeneous clinical phenotypes are led by pathology distribution and affected neural networks that comprise a primary motor, extrapyramidal, cognitive, and behavioral pathways. Although several diagnosing methods are useful in clinical practice, more studies are needed so the clinician may depend less on pathological findings to make a definitive diagnosis of these disorders.
References
- JC Steele, JC Richardson, J Olszewski. Progressive supranuclear palsy: a heterogeneous degeneration involving the brain stem, Basal Ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol, 1964. [DOI | PubMed]
- JJ Rebeiz, EH Kolodny, EP Richardson. Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc, 1967. [PubMed]
- JJ Rebeiz, EH Kolodny, EP Richardson. Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol, 1968. [DOI | PubMed]
- WR Gibb, PJ Luthert, CD Marsden. Corticobasal degeneration. Brain, 1989. [DOI | PubMed]
- H Ling, SS O’Sullivan, JL Holton, T Revesz, LA Massey, DR Williams. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain, 2010. [DOI | PubMed]
- DS Horoupian, PH Wasserstein. Alzheimer’s disease pathology in motor cortex in dementia with Lewy bodies clinically mimicking corticobasal degeneration. Acta Neuropathol, 1999. [DOI | PubMed]
- BF Boeve, DM Maraganore, JE Parisi, JE Ahlskog, N Graff-Radford, RJ Caselli. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology, 1999. [DOI | PubMed]
- BF Boeve, AE Lang, I Litvan. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol, 2003. [DOI | PubMed]
- H Ling, A Macerollo. Is it useful to classify PSP and CBD as different disorders? Yes. Mov Disord Clin Pract, 2018. [DOI | PubMed]
- M Stamelou, G Respondek, N Giagkou, JL Whitwell, GG Kovacs, GU Höglinger. Evolving concepts in progressive supranuclear palsy and other 4-repeat tauopathies. Nat Rev Neurol, 2021. [DOI | PubMed]
- G Respondek, C Kurz, T Arzberger, Y Compta, E Englund, LW Ferguson. Which ante mortem clinical features predict progressive supranuclear palsy pathology?. Mov Disord, 2017. [DOI | PubMed]
- IT Coyle-Gilchrist, KM Dick, K Patterson, P Vázquez Rodríquez, E Wehmann, A Wilcox. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology, 2016. [DOI | PubMed]
- K Yoshida, Y Hata, K Kinoshita, S Takashima, K Tanaka, N Nishida. Incipient progressive supranuclear palsy is more common than expected and may comprise clinicopathological subtypes: a forensic autopsy series. Acta Neuropathol, 2017. [DOI | PubMed]
- H Takigawa, M Kitayama, K Wada-Isoe, H Kowa, K Nakashima. Prevalence of progressive supranuclear palsy in Yonago: change throughout a decade. Brain Behav, 2016. [DOI | PubMed]
- DM Togasaki, CM Tanner. Epidemiologic aspects. Adv Neurol, 2000. [PubMed]
- Y Osaki, Y Morita, T Kuwahara, I Miyano, Y Doi. Prevalence of Parkinson’s disease and atypical parkinsonian syndromes in a rural Japanese district. Acta Neurol Scand, 2011. [DOI | PubMed]
- Y Winter, Y Bezdolnyy, E Katunina, G Avakjan, JP Reese, J Klotsche. Incidence of Parkinson’s disease and atypical parkinsonism: Russian population-based study. Mov Disord, 2010. [DOI | PubMed]
- AL Boxer, JT Yu, LI Golbe, I Litvan, AE Lang, GU Höglinger. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol, 2017. [DOI]
- G Respondek, M Stamelou, C Kurz, LW Ferguson, A Rajput, WZ Chiu. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord, 2014. [DOI | PubMed]
- DR Williams, JL Holton, C Strand, A Pittman, R Silva, AJ Lees. Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain, 2007. [DOI | PubMed]
- DR Williams, AJ Lees. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol, 2009. [DOI | PubMed]
- S Koga, KA Josephs, K Ogaki, C Labbé, RJ Uitti, N Graff-Radford. Cerebellar ataxia in progressive supranuclear palsy: an autopsy study of PSP-C. Mov Disord, 2016. [DOI | PubMed]
- JB Parmera, RD Rodriguez, A Studart, R Nitrini, SMD Brucki. Corticobasal syndrome: a diagnostic conundrum. Dement Neuropsychol, 2016. [DOI | PubMed]
- SE Lee, GD Rabinovici, MC Mayo, SM Wilson, WW Seeley, SJ DeArmond. Clinicopathological correlations in corticobasal degeneration. Ann Neurol, 2011. [DOI | PubMed]
- MJ Armstrong, I Litvan, AE Lang, TH Bak, KP Bhatia, B Borroni. Criteria for the diagnosis of corticobasal degeneration. Neurology, 2013. [DOI | PubMed]
- R Mathew, TH Bak, JR Hodges. Diagnostic criteria for corticobasal syndrome: a comparative study. J Neurol Neurosurg Psychiatry, 2012. [DOI | PubMed]
- N Kouri, JL Whitwell, KA Josephs, R Rademakers, DW Dickson. Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat Rev Neurol, 2011. [DOI | PubMed]
- N Kouri, ME Murray, A Hassan, R Rademakers, RJ Uitti, BF Boeve. Neuropathological features of corticobasal degeneration presenting as corticobasal syndrome or Richardson syndrome. Brain, 2011. [DOI | PubMed]
- E Bayram, DW Dickson, SG Reich, I Litvan. Pathology-proven corticobasal degeneration presenting as Richardson’s syndrome. Mov Disord Clin Pract, 2020. [DOI | PubMed]
- KA Josephs, JR Duffy, EA Strand, JL Whitwell, KF Layton, JE Parisi. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain, 2006. [DOI | PubMed]
- GU Höglinger. Is it useful to classify progressive supranuclear palsy and corticobasal degeneration as different disorders?. Mov Disord Clin Pract, 2018. [DOI | PubMed]
- B Ghetti, AL Oblak, BF Boeve, KA Johnson, BC Dickerson, M Goedert. Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol, 2015. [DOI | PubMed]
- N Kouri, OA Ross, B Dombroski, CS Younkin, DJ Serie, A Soto-Ortolaza. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun, 2015. [DOI | PubMed]
- GU Höglinger, G Respondek, M Stamelou, C Kurz, KA Josephs, AE Lang. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord, 2017. [DOI | PubMed]
- I Litvan, Y Agid, D Calne, G Campbell, B Dubois, RC Duvoisin. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology, 1996. [DOI | PubMed]
- F Ali, PR Martin, H Botha, JE Ahlskog, JH Bower, JY Masumoto. Sensitivity and specificity of diagnostic criteria for progressive supranuclear palsy. Mov Disord, 2019. [DOI | PubMed]
- GM Saranza, JL Whitwell, GG Kovacs, AE Lang. International review of neurobiology, 2019
- H Ouchi, Y Toyoshima, M Tada, M Oyake, I Aida, I Tomita. Pathology and sensitivity of current clinical criteria in corticobasal syndrome. Mov Disord, 2014. [DOI | PubMed]
- SK Alexander, T Rittman, JH Xuereb, TH Bak, JR Hodges, JB Rowe. Validation of the new consensus criteria for the diagnosis of corticobasal degeneration. J Neurol Neurosurg Psychiatry, 2014. [DOI | PubMed]
- G Respondek, MJ Grimm, I Piot, T Arzberger, Y Compta, E Englund. Validation of the movement disorder society criteria for the diagnosis of 4-repeat tauopathies. Mov Disord, 2020. [DOI | PubMed]
- GG Kovacs. Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol, 2015. [DOI | PubMed]
- MG Spillantini, M Goedert. Tau pathology and neurodegeneration. Lancet Neurol, 2013. [DOI]
- F Clavaguera, F Grueninger, M Tolnay. Intercellular transfer of tau aggregates and spreading of tau pathology: implications for therapeutic strategies. Neuropharmacology, 2014. [DOI | PubMed]
- GU Höglinger, I Litvan, N Mendonca, D Wang, H Zheng, B Rendenbach-Mueller. Safety and efficacy of tilavonemab in progressive supranuclear palsy: a phase 2, randomised, placebo-controlled trial. Lancet Neurol, 2021. [DOI]
- N Kato, K Arai, T Hattori. Study of the rostral midbrain atrophy in progressive supranuclear palsy. J Neurol Sci, 2003. [DOI | PubMed]
- LA Massey, C Micallef, DC Paviour, SS O’Sullivan, H Ling, DR Williams. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord, 2012. [DOI | PubMed]
- C Mueller, A Hussl, F Krismer, B Heim, P Mahlknecht, M Nocker. The diagnostic accuracy of the hummingbird and morning glory sign in patients with neurodegenerative parkinsonism. Park Relat Disord, 2018. [DOI]
- DC Paviour, SL Price, JM Stevens, AJ Lees, NC Fox. Quantitative MRI measurement of superior cerebellar peduncle in progressive supranuclear palsy. Neurology, 2005. [DOI | PubMed]
- JL Whitwell, GU Höglinger, A Antonini, Y Bordelon, AL Boxer, C Colosimo. Radiological biomarkers for diagnosis in PSP: where are we and where do we need to be?. Mov Disord, 2017. [DOI | PubMed]
- M Cosottini, R Ceravolo, L Faggioni, G Lazzarotti, MC Michelassi, U Bonuccelli. Assessment of midbrain atrophy in patients with progressive supranuclear palsy with routine magnetic resonance imaging. Acta Neurol Scand, 2007. [DOI | PubMed]
- A Quattrone, G Nicoletti, D Messina, F Fera, F Condino, P Pugliese. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology, 2008. [DOI | PubMed]
- G Nicoletti, C Tonon, R Lodi, F Condino, D Manners, E Malucelli. Apparent diffusion coefficient of the superior cerebellar peduncle differentiates progressive supranuclear palsy from Parkinson’s disease. Mov Disord, 2008. [DOI | PubMed]
- JL Whitwell, CG Schwarz, RI Reid, K Kantarci, CR Jack, KA Josephs. Diffusion tensor imaging comparison of progressive supranuclear palsy and corticobasal syndromes. Parkinsonism Relat Disord, 2014. [DOI]
- JL Whitwell, N Tosakulwong, H Botha, F Ali, HM Clark, JR Duffy. Brain volume and flortaucipir analysis of progressive supranuclear palsy clinical variants. Neuroimage Clin, 2020. [DOI | PubMed]
- RC Gardner, AL Boxer, A Trujillo, JB Mirsky, CC Guo, ED Gennatas. Intrinsic connectivity network disruption in progressive supranuclear palsy. Ann Neurol, 2013. [DOI | PubMed]
- J Rosskopf, M Gorges, HP Müller, D Lulé, I Uttner, AC Ludolph. Intrinsic functional connectivity alterations in progressive supranuclear palsy: Differential effects in frontal cortex, motor, and midbrain networks. Mov Disord, 2017. [DOI | PubMed]
- J Lemos, D Pereira, L Almendra, D Rebelo, M Patrício, J Castelhano. Cortical control of vertical and horizontal saccades in progressive supranuclear palsy: an exploratory fMRI study. J Neurol Sci, 2017. [DOI | PubMed]
- S Sachin, SS Kumaran, S Singh, V Goyal, G Shukla, H Mahajan. Functional mapping in PD and PSP for sustained phonation and phoneme tasks. J Neurol Sci, 2008. [DOI | PubMed]
- N Upadhyay, A Suppa, MC Piattella, F Di Stasio, N Petsas, C Colonnese. Gray and white matter structural changes in corticobasal syndrome. Neurobiol Aging, 2016. [DOI | PubMed]
- JL Whitwell, CR Jack, BF Boeve, JE Parisi, JE Ahlskog, DA Drubach. Imaging correlates of pathology in corticobasal syndrome. Neurology, 2010. [DOI | PubMed]
- LK Teune, AL Bartels, BM de Jong, AT Willemsen, SA Eshuis, JJ de Vries. Typical cerebral metabolic patterns in neurodegenerative brain diseases. Mov Disord, 2010. [DOI | PubMed]
- S Hellwig, F Amtage, A Kreft, R Buchert, OH Winz, W Vach. IBZM-SPECT for the differential diagnosis of parkinsonism. Neurology, 2012. [DOI | PubMed]
- T Eckert, A Barnes, V Dhawan, S Frucht, MF Gordon, AS Feigin. FDG PET in the differential diagnosis of parkinsonian disorders. Neuroimage, 2005. [DOI | PubMed]
- M Niethammer, CC Tang, A Feigin, PJ Allen, L Heinen, S Hellwig. A disease-specific metabolic brain network associated with corticobasal degeneration. Brain, 2014. [DOI | PubMed]
- M Pardini, ED Huey, S Spina, WC Kreisl, S Morbelli, EM Wassermann. FDG-PET patterns associated with underlying pathology in corticobasal syndrome. Neurology, 2019. [DOI | PubMed]
- JB Parmera, AM Coutinho, MR Aranha, A Studart-Neto, C de Godoi Carneiro, IJ de Almeida. FDG-PET Patterns Predict Amyloid Deposition and Clinical Profile in Corticobasal Syndrome. Mov Disord, 2021. [DOI | PubMed]
- JB Parmera, IJ de Almeida, MCB de Oliveira, ML Silagi, C de Godoi Carneiro, A Studart-Neto. Metabolic and structural signatures of speech and language impairment in corticobasal syndrome: a multimodal PET/MRI study. Front Neurol, 2021. [DOI | PubMed]
- G Martí-Andrés, L van Bommel, SK Meles, M Riverol, R Valentí, RV Kogan. Multicenter Validation of Metabolic Abnormalities Related to PSP According to the MDS-PSP Criteria. Mov Disord, 2020. [DOI | PubMed]
- KA Josephs, JL Whitwell, P Tacik, JR Duffy, ML Senjem, N Tosakulwong. AV-1451 tau-PET uptake does correlate with quantitatively measured 4R-tau burden in autopsy-confirmed corticobasal degeneration. Acta Neuropathol, 2016. [DOI | PubMed]
- H Cho, JY Choi, MS Hwang, SH Lee, YH Ryu, MS Lee. Subcortical 18F-AV-1451 binding patterns in progressive supranuclear palsy. Mov Disord, 2017. [DOI | PubMed]
- M Brendel, H Barthel, T van Eimeren, K Marek, L Beyer, M Song. Assessment of 18F-PI-2620 as a Biomarker in Progressive Supranuclear Palsy. JAMA Neurol, 2020. [DOI | PubMed]
- C Palleis, M Brendel, A Finze, E Weidinger, K Bötzel, A Danek. PI-2620 binding differentiates corticobasal syndrome subtypes. Mov Disord, 2021. [DOI | PubMed]
- R Laforce, JP Soucy, L Sellami, C Dallaire-Théroux, F Brunet, D Bergeron. Molecular imaging in dementia: past, present, and future. Alzheimers Dement, 2018. [DOI | PubMed]
- JR Burrell, M Hornberger, VL Villemagne, CC Rowe, JR Hodges. Clinical profile of PiB-positive corticobasal syndrome. PLoS One, 2013. [DOI | PubMed]
- K Blennow, H Zetterberg. The past and the future of Alzheimer’s disease CSF biomarkers-a journey toward validated biochemical tests covering the whole spectrum of molecular events. Front Neurosci, 2015. [DOI | PubMed]
- N Magdalinou, AJ Lees, H Zetterberg. Cerebrospinal fluid biomarkers in parkinsonian conditions: an update and future directions. J Neurol Neurosurg Psychiatry, 2014. [DOI | PubMed]
- NK Magdalinou, RW Paterson, JM Schott, NC Fox, C Mummery, K Blennow. A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry, 2015. [DOI | PubMed]
- E Jabbari, H Zetterberg, HR Morris. Tracking and predicting disease progression in progressive supranuclear palsy: CSF and blood biomarkers. J Neurol Neurosurg Psychiatry, 2017. [DOI | PubMed]
- JC Rojas, A Karydas, J Bang, RM Tsai, K Blennow, V Liman. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol, 2016. [DOI | PubMed]
- AL Boxer, I Qureshi, M Ahlijanian, M Grundman, LI Golbe, I Litvan. Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: a randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet Neurol, 2019. [DOI]