Secreted phosphoprotein 1 as a potential prognostic and immunotherapy biomarker in multiple human cancers
Abstract
Secreted phosphoprotein 1 (SPP1) is involved in immune regulation, cell survival, and tumor progression. Studies have demonstrated that SPP1 plays an important role in certain individual tumors. However, the expression profile and oncogenic features of SPP1 in diverse cancers are remaining unknown. Therefore, we performed a comprehensive analysis using The Cancer Genome Atlas (TCGA) database. Raw data of 33 cancer types were download from the University of California Santa Cruz (UCSC) Xena website. The expression of SPP1 and its relationship with tumor prognosis, immune invasion, tumor microenvironment, and immunotherapy were analyzed using the R language. The function analysis was conducted using Gene Set Enrichment Analysis (GSEA). The oncogenic features of SPP1 was validated by wound-healing assay and EdU staining assay. SPP1 highly expressed in most cancers. The expression of SPP1 was significant related to prognosis, tumor mutation burden (TMB), microsatellite instability (MSI), and immune checkpoint genes, suggested that SPP1 plays an essential role in the tumor immune microenvironment and immune cell infiltration. The immune/stromal scores correlated positively with the SPP1 expression, and the relationship was affected by tumor heterogeneity and immunotherapy. In addition, SPP1 could predict the response of tumor immunotherapy. Functional analysis revealed the SPP1-associated terms and pathways. Finally, SPP1 significantly elevated cell proliferation and migration in A549, Huh7, HT-29, A2780 tumor cell lines. In conclusion, this study indicated that SPP1 involved in tumorigenesis, tumor progression, and regulated tumor immune microenvironment, revealing SPP1 might be a potential target for evaluating prognosis and immunotherapy in multiple cancers.
Article type: Research Article
Keywords: SPP1, prognosis, immune infiltration, tumor microenvironment
Affiliations: State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, the First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, China; Department of Hospital Infection Control, The First Affiliated Hospital of Nanchang University, Nanchang, China
License: © 2022 The Author(s). Published by Informa UK Limited, trading as Taylor & Francis Group. CC BY 4.0 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Article links: DOI: 10.1080/21655979.2021.2020391 | PubMed: 35067176 | PMC: PMC8973783
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (5.8 MB)
Introduction
Cancer is a primary cause of high morbidity and high mortality worldwide. According to the World Health Organization, More than 18 million cancer new cases and 9.6 million cancer deaths occurred in 2018 [ref. 1]. Cancer is a complex and heterogeneous disease, involving interactions between tumor and immune system, making it difficult to uncover its pathogenesis. In recent years, the pan-cancer analysis has been extensively used in the analysis and study of cancer disease [ref. 2].
The common and differential features existing in various types of cancers have been contributing to researches focusing on potential mechanisms of cancer, as well as estimating the clinical prognosis. The Cancer Genome Atlas (TCGA) is based on multiple levels of tumor data from different human cancer cell lines and tissues, focusing on molecular mutations associated with the occurrence and progression of cancer. The TCGA database contains data of 33 cancer types, which can be used for survival analysis and prognosis evaluation, such as overall survival (OS), disease-specific survival (DSS), disease-free interval (DFI), and progression-free interval (PFI). The TCGA contributed to establishing the significance of cancer genomics, helping us understanding cancer in multiple levels, and profoundly impacting the treatment concept of cancer.
As a multifunctional and secreted integrin-binding glycoprotein, secreted phosphoprotein 1 (SPP1) is found to express in different tissues and cell types. The biological functions of SPP1 are variable, specifically in some certain physiological and pathological conditions, including drug resistance, cell proliferation, invasion, survival, stem-like behavior and tumor metastasis [ref. 3]. Accumulating evidences indicated that SPP1 plays an important role in several tumor-associated processes, such as proliferation, invasion, migration, angiogenesis, and metastasis [ref. 4,ref. 5]. SPP1 promotes epithelial-mesenchymal transition during metastasis and regulates the tumor microenvironment in favor of metastasis [ref. 6,ref. 7]. In both experimental and clinical studies, SPP1 has been shown to correlated with the progression and prognosis of different tumors [ref. 8]. Recent researches demonstrated that overexpressing of SPP1 could be used as an indicator of poor prognosis in numerous human cancers, for instance, lung cancer, gastric carcinoma, colorectal cancer, and prostate cancers [ref. 8]. Studies have shown that SPP1 regulates tumorigenesis by interacting with integrins or the CD44 receptor, subsequently activating Wnt signaling [ref. 9] or focal adhesion kinase (FAK)/P-glycoprotein (P-GP) signaling transduction pathways [ref. 10].
In the present study, we aimed to explore the SPP1 expression profiles and oncogenic features among human cancers using the TCGA project for comprehensive analysis. Our findings suggested that SPP1 is a potential target for evaluating patient prognosis and immunotherapy in multiple human cancers.
Materials and Methods
TGCA data acquisition and processing
As a landmark cancer genomics program, TCGA had characterized over 20,000 primary cancer and matched normal samples from 33 cancer types. Transcriptome RNA-seq data of 33 cancers were download from the University of California Santa Cruz (UCSC) Xena website (http://xena.ucsc.edu/) [ref. 11]. 33 cancer types were included: adrenocortical carcinoma (ACC), bladder Urothelial Carcinoma (BLCA), breast invasive carcinoma (BRCA), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), cholangiocarcinoma (CHOL), colon adenocarcinoma (COAD), Lymphoid Neoplasm Diffuse Large B-cell Lymphoma (DLBC), esophageal carcinoma (ESCA), glioblastoma multiforme (GBM), head and neck squamous cell carcinoma (HNSC), kidney chromophobe (KICH), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), acute myeloid leukemia (LAML), brain lower grade glioma (LGG), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), mesothelioma (MESO), ovarian serous cystadenocarcinoma (OV), pancreatic adenocarcinoma (PAAD), pheochromocytoma and paraganglioma (PCPG), prostate adenocarcinoma (PRAD), rectum adenocarcinoma (READ), sarcoma (SARC), skin cutaneous melanoma (SKCM), stomach adenocarcinoma (STAD), testicular germ cell tumors (TGCT), thyroid carcinoma (THCA), thymoma (THYM), uterine corpus endometrial carcinoma (UCEC), uterine carcinosarcoma (UCS), and uveal melanoma (UVM).
Analysis of SPP1 expression in cancers
The Ensembl gene ID of transcriptome data was converted into Symbol ID to extract the expression of target genes in each tumor patient. Differential gene expression was analyzed based on ggpubr R package (https://www.rdocumentation.org/packages/ggpubr) in R software (version 3.6.3, 29 February 2020, R Foundation for Statistical Computing, Vienna, Austria). In addition, the gene expression was also identified via the Oncomine database (http://www.oncomine.org/resource/login.html) [ref. 12]. Setting the significant threshold: P-value of 0.001, fold change of 2, and gene ranking of 10%.
Signature score calculation
The signature score reflected the status of the studies biological process in a tumor and were calculated according to the method of Rocha et al [ref. 13]. Cutoff-high (50%) and cutoff-low (50%) values were used to split the high-expression and low-expression cohorts, patients were divided into two groups. The difference of groups was tested by Wilcoxon signed-rank, using Benjamini and Hochberg correction for multiple testing within each database.
Survival analysis and relationship with clinical stage
The survival database of 33 cancer types (survival status, time, and tumor stage) were download from UCSC Xena database. To assess the correlation between gene expression and prognosis of cancers, Kaplan-Meier (KM) curves and Cox proportional hazard regression survival analyses were performed using the survival package (https://CRAN.R-project.org/package=survival) [ref. 14]. The survival analysis included OS, DSS, DFI, PFI. Cutoff-high (50%) and cutoff-low (50%) values were used as the expression thresholds for splitting the high-expression and low-expression cohorts, patients were divided into two groups. In addition, we also analyzed the clinical correlation between gene expression and pathological stage, histological grade, age, and gender.
TMB and MSI analysis
TMB is the total number of mutations identified in per tumor sample [ref. 15]. We download the mutation data from UCSC Xena database, the correlation between gene expression and the TMB level is analyzed by spearman correlation. Statistical analysis and corresponding visualization were performed using the R software. MSI is due to the nucleotide insertions or deletions in tumor cells of the microsatellite loci changes in the length of repetitive microsatellite sequences [ref. 16]. The correlation between gene expression and the MSI level is also analyzed by spearman correlation.
Tumor microenvironment
We used estimate package (https://R-Forge.R-project.org/projects/estimate/) to calculate the tumor microenvironment indicators for each tumor sample and obtain the immune cell score, stromal cell content, and tumor purity [ref. 14]. The correlation between gene expression and the tumor microenvironment indicators is analyzed by spearman correlation. Statistical analysis and corresponding visualization (P < 0.001) were performed using the R software.
Relationship between SPP1 expression and immunity
To estimate relative proportion of 22 types of infiltrated immune cells in 33 cancer types, Cell-type identification by Estimating Relative Subsets of RNA Transcripts (CIBERSORT) algorithm was conducted to estimate the relationship between gene expression and 22 infiltrated immune cells based on expression file [ref. 17]. We selected the TIICs gene markers from previous research [ref. 18] and examined the association with SPP1 using R software. Spearman’s correlation coefficient and statistical significance was visualized in heatmap.
Prediction of immunotherapy response
Immunotherapy datasets were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) including GSE111636, GSE67501, GSE26383, GSE79691, and GSE100797. These cohorts were used for prediction of patient response to immunotherapy [ref. 19]. The GSE111636 cohort, a BLCA cohort receiving anti-PD-L1 antibody atezolizumab immunotherapy, was also included for prediction of immunotherapy response. The GSE67501 was an RCC cohort receiving anti-PD-L1 immunotherapy, GSE26383, GSE79691, and GSE100797 were SKCM cohorts receiving same immunotherapy. The RCC cohort was a cohort receiving immune checkpoint therapy [ref. 20]. The t-test P < 0.05 was utilized to determine the statistical significance. We calculated the correlation between two variables using the Spearman method. The threshold of P < 0.05 (Spearman’s correlation test) indicates the significance of correlation. To compare the predictive power of different genomic features for immune signatures, time-dependent receiver-operating characteristic (ROC) curves analysis were performed by using SPSS version 19.0 software package. Statistical analysis was performed by using GraphPad Prism version 7.0 or SPSS version 19.0 software package. A two-tailed P < 0.05 was considered statistically significant.
Gene set enrichment analysis
Functional analysis of gene was proceeded with GSEA [ref. 21]. We downloaded the GO (c5.all.v7.1.symboles.gml) and KEGG (c2.cp.kegg.v7.1.symboles.gml) biological process gene sets from the GSEA website (http://www.gsea-msigdb.org/gsea/index.jsp). R software was conducted to gene set enrichment analysis (P < 0.05) and exhibited the top five terms.
Analysis of SPP1 and immune-related molecules expression in liver cancers
The concentration of SPP1 in the liver cancer tissue were detected by commercial ELISA (Enzyme-linked immunosorbent assay) kits (MultiSciences) as specified by the manufacturer. The mRNA expression of SPP1 and immune-related molecules were measured by quantitative real-time PCR (qRT-PCR). Total RNA was isolated using Trizol reagent (Takara). cDNA was synthesized using a PrimeScriptTM RT Msater Mis (Takara). qRT-PCR analyses were conducted with SYBR® Premix Ex TaqTM II (Takara) with specific primers as follows:
SPP1: 5ʹ-CTCCATTGACTCGAACGACTC-3ʹ (F),
5ʹ-CAGGTCTGCGAAACTTCTTAGAT-3ʹ (R);
CD44: 5ʹ-CTGCCGCTTTGCAGGTGTA-3ʹ (F),
5ʹ-CATTGTGGGCAAGGTGCTATT-3ʹ (R);
CD80: 5ʹ-AAACTCGCATCTACTGGCAAA-3ʹ (F),
5ʹ-GGTTCTTGTACTCGGGCCATA-3ʹ (R);
LAIR1: 5ʹ-ATCGGGGTCTCAGTGGTCTTC-3ʹ (F),
5ʹ-TGCTTTATCTGATTCTGGCGATG-3ʹ (R);
NRP1: 5ʹ-GGCGCTTTTCGCAACGATAAA-3ʹ (F),
5ʹ-TCGCATTTTTCACTTGGGTGAT-3ʹ (R);
HAVCR2: 5ʹ-CTGCTGCTACTACTTACAAGGTC-3ʹ (F),
5ʹ-GCAGGGCAGATAGGCATTCT-3ʹ (R).
Immunohistochemistry
Immunohistochemistry was performed as reported elsewhere [ref. 22]. Paraffin sections of tumor and para-tumor tissues underwent dewaxing, hydration and antigen repair. Following blocking with serum, the sections were incubated with anti-SPP1 antibody (Abcam) at 4°C overnight. The slides were washed three times with PBS and incubated with a secondary antibody at 37°C for 30 min. Then, streptavidin–horseradish peroxidase was applied at 37°C for 30 min, and the slides were stained with DAB solution. Images were obtained using a direct optical microscope.
Cell migration and cell proliferation assays
The human cell lines A549 (lung carcinoma), HuH-7 (hepatoma carcinoma) and HT-29 (colorectal carcinoma) were purchased from the Cell Bank of Chinese Academy of Sciences. The human cell lines A2780 (ovarian carcinoma) was obtained from the Tumor Cell Bank of the Chinese Academy of Medical Sciences (Beijing, China). Cell migration was determined by performing a wound healing assay. Briefly, cells were inoculated into 6-well plates. Cells reached 90% confluence and were scratched using a 100 μl pipette tip to form a wound-like gap. The cells were maintained in medium, and images were captured at 0 hour and 24 hours. ImageJ software was used to quantify the area of the wound to calculate the cell migration rate. EdU staining was used for the detection of cell proliferation. Briefly, cells were seeded in 24-well plates at a density of 2 × 103 cells/well. EdU kit (Beyotime, Shanghai, China) was used for detecting cell proliferation according to the manufacturer’s instruction.
Statistical analysis
Gene differential expression from TCGA database was analyzed by Wilcoxon rank sum test. Survival data was analyzed using the Kaplan-Meier and Cox statistical methods. Spearman correlation analysis was used for the correlation analysis in this study. The difference in SPP1 expression between different tumor stages were compared suing the Kruskal-Wallis test. R software (version 3.6.3) was performed all analyses and chart visualize. Measurement data were expressed as the mean ± standard deviation (SD) from three independent experiments. The tumor and normal samples were compared using the unpaired t-tests. P-value <0.05 was considered statistically significant.
Results
SPP1, as an important gene in certain cancer types, involved in the occurrence and development of tumors. We speculated that SPP1 might be used as a potential prognostic and immune-related biomarker in human cancers. In this study, we conducted the correlation analysis between SPP1 expression and clinical characterize, survival, TMB, MSI, the tumor immune microenvironment, immunotherapy, and GSEA. Furthermore, we also performed in vivo experiments to evaluate the oncogenic effect of SPP1. These results identified that high expression of SPP1 could serve as a biomarker for prognostic and tumor immunotherapy. This study will contribute to a deeper understanding of the critical role of SPP1 in human cancers.
Differences of SPP1 gene expression in human cancers
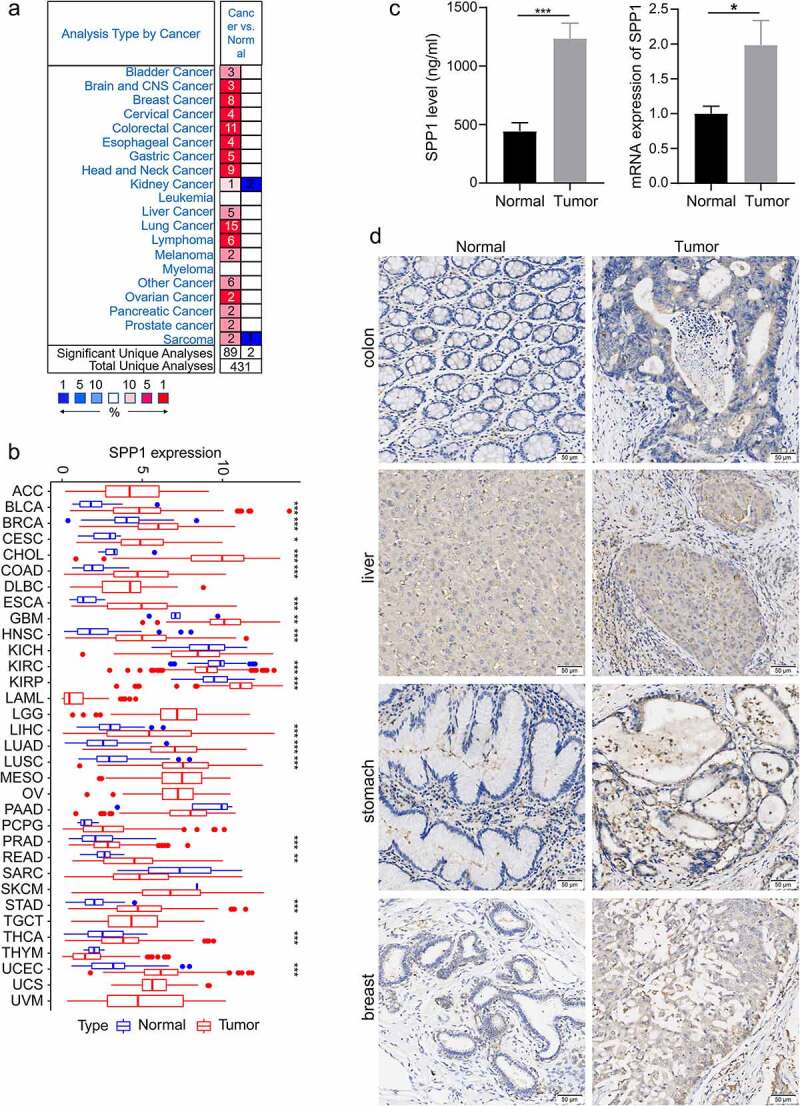
In order to investigate the SPP1 expression differences in tumor and normal samples of cancers. Initially, we used the Oncomine database to analyze the expression level of SPP1 in different tumor and normal tissues. The result showed that SPP1 was overexpressed in multiple types of cancer, including bladder, brain and central nervous system, breast, cervical, colorectal, esophageal, gastric, head and neck, kidney, liver, lung, ovarian, pancreatic, and prostate cancers, and in lymphoma, melanoma, and sarcoma in most data sets (Figure 1(a)). In some data sets, SPP1 expression was lower in kidney cancer and sarcoma compared with that in normal tissues. The details information regarding the differential expression of SPP1 in cancers versus normal tissue are summarized in Table S1. In addition, to further assess SPP1 expression levels in human cancers, we obtained the TGCA-derived transcriptome RNA-seq data of 33 cancers from the UCSC Xena database and analyzed the SPP1 expression using the R software. We found that SPP1 mRNA expression was significantly higher in BLCA, BRCA, CESC, CHOL, COAD, ESCA, GBM, HNSC, KIRP, LIHC, LUAD, LUSC, PRAD, READ, STAD, THCA, and UCEC tumor tissues compared with that in normal tissues, indicating that it might play an oncogenic role in the development of most tumors. However, SPP1 expression was lower in KIRC, KICH, PAAD, SARC, and THYM compared with that in normal tissues (Figure 1(b)). Except for that in KIRC, SPP1 expression was not statistically significant in the other tumors. To confirm our analysis results, we used liver cancer tissues and para-cancer tissues to verify the expression of SPP1 with ELISA and qRT-PCR experiment. These results supported that SPP1 expression was significantly higher in liver cancer tissues compared with para-cancer tissues (Figure 1(c)). In addition, immunohistochemical assay results showed that SPP1 highly expressed in multiple types of cancer, including breast, colon, gastric, and liver (Figure 1(d), Table S2 and S3).
The association of SPP1 expression with molecular signatures in human cancers
In order to validate the important role of SPP1 in human cancers, we studied some well-known biological processes including differentiation, proliferation, retinoblastoma (RB) pathways, TP53, and centrosome amplification. The most fundamental trait of tumor cells is differentiation and proliferation. We studied differentiation using nine genes (ZIC1, TCF7L1, KLF5, MYBL2, NFE2L3, TEAD4, ILF3, HMGA1, HMGB3), which overexpressed in poorly differentiated bladder, glioblastoma, and breast tumors [ref. 23]. 110 genes with predictive and prognostic effects were used to study proliferation [ref. 24]. Centrosome amplification were found in multiple cancer types and studied using a 20 genes signature (CA20) about poorly prognosis [ref. 25]. We explored DNA damage and apoptosis pathways using RB [ref. 26] and TP53 signature [ref. 27]. These scores were basically consistent with the expression trend of SPP1 (Figure S1). In most cancers, high expression of SPP1 means more dedifferentiation, more proliferation, more RB and TP53 mutations, and higher centrosome amplification.
Prognostic assessment and clinical correlation of SPP1 in human cancers
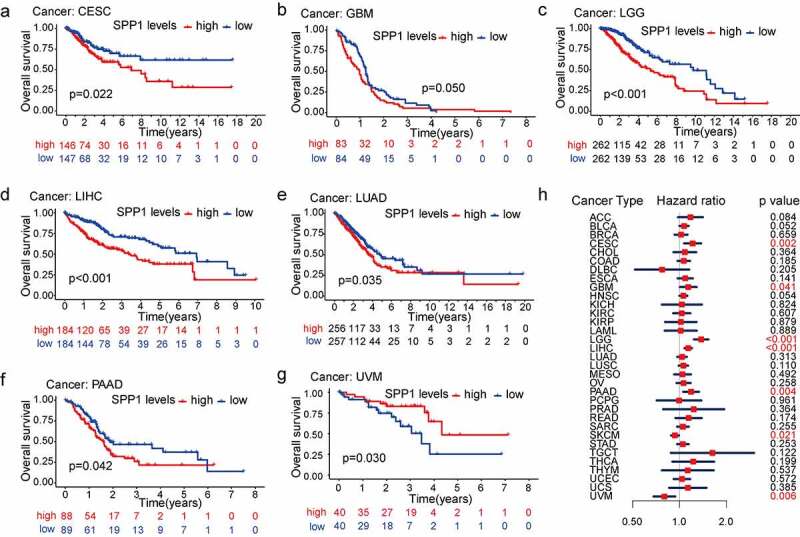
To assess the predictive value of the SPP1 expression in the prognosis of different types of cancer, we divided all patients with cancer into high-expression and low-expression groups according to the median value of their SPP1 expression and conducted survival analysis (OS, DSS, DFI, and PFI) using the Kaplan-Meier and COX methods. The Kaplan-Meier analysis indicated that the high SPP1 expression significantly decreased OS in six cancers: CESC (p = 0.022), GBM (p = 0.050), LGG (P < 0.001), LIHC (P < 0.001), LUAD (p = 0.035), and PAAD (p = 0.042) (Figure 2(a-f)). High SPP1 expression was associated with increased OS only in UVM (Figure 2(g)). In addition, we revalidated the OS analysis proposed by venet et al [ref. 28]. The results were consistent with the above analysis for four cancer types including CESC, LGG, LIHC, and UVM (Figure S2). Furthermore, we investigated the relationship between SPP1 expression, as a continuous variable, survival time and survival status using Cox analysis. In CESC, GBM, LGG, LIHC, PAAD, SKCM, and UVM (P < 0.05), SPP1 expression was associated with OS. The hazard ratio (HR) value was greater than 1, indicating that SPP1 is a high-risk gene in tumors. In other words, the higher the expression of SPP1, the worse the prognosis (Figure 2(h)). In the DSS analysis, except in UVM, patients with high SPP1 expression had a significantly worse prognosis in ESCA, LGG, LIHC, PAAD, and PRAD (Figure S3). In the DFI and PFI analysis, we observed the same phenomenon in certain types of cancer (Figure S4, S5). These results confirmed that SPP1 might act as a powerful biomarker for predicting prognosis in most cancers. Increased and decreased SPP1 expression had different prognostic values depending on the type of cancer. In addition, we investigated the relationship between SPP1 expression and clinical characteristics (pathological stage, histological grade, age, and gender) of 33 types of cancer. Increased expression of SPP1 was only positively correlated with certain tumor stages, such as COAD, ESCA, LIHC, and READ (Figure S6). SPP1 expression was correlated to pathological stages including BLCA, LGG, LGG, and LIHC. The difference was statistically significant (P < 0.05) with a higher grade being associated with a higher expression level (Figure S7). Gender difference existed in most tumors, but they were not associated with the expression level of SPP1 (Table S4). However, ESCA was more common in males, in which the expression level of SPP1 was higher, and SARC occurred more frequently in women, in which SPP1 expression was lower (Figure S8a, b). Age is associated closely with the occurrence of various tumors. Patients aged ≥ 60 years old formed a higher proportion in BLCA, COAD, LUAD, LUSC, and STAD. Patients aged < 60 years old formed a higher proportion in ACC, CESC, LGG, PCPG, TGCT, and THCA (Table S4). SPP1 expression levels were markedly higher in older (≥ 60 years old) than in younger (< 60 years old) patients in BLCA, CESC, HNSC, LIHC, PRAD, SARC, and THYM. Only in SKCM was SPP1 expression higher in younger patients (Figure S8c-j).
Correlation between SPP1 expression and gene alterations
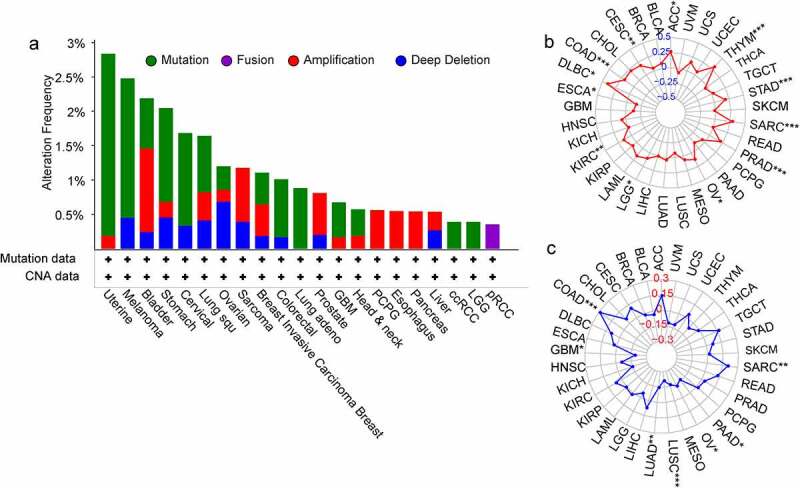
It is widely acknowledged that tumorigenesis is mainly caused by mutations in certain genes. We assessed the genetic alterations of the SPP1 genes in patients with cancer using the cBioPortal database. The SPP1 mutation frequency was the primary alteration in uterine tumors, melanoma, stomach tumors, cervical tumors, lung squamous cell carcinoma, colorectal tumors, lung adenocarcinoma, GBM, HNSC, ccRCC, and LGG. In bladder tumors, sarcoma, breast tumors, prostate tumors, PCPG, esophagus tumors, and pancreas tumors, gene amplification was the most important genetic alteration (Figure 3(a)). In addition, we analyzed the correlation between SPP1 expression and the TMB and MSI (Table S5). SPP1 expression correlated positively with the TMB in ACC, CESC, COAD, KIRC, LGG, OV, PRAD, SARC, STAD, and THYM, and correlated negatively in ESCA (Figure 3(b)). SPP1 expression correlated positively with MSI in COAD, SARC, and correlated negatively in GBM, LUAD, LUSC, MESO, OV, and PAAD (Figure 3(c)).
Correlation between SPP1 expression and the tumor immune microenvironment
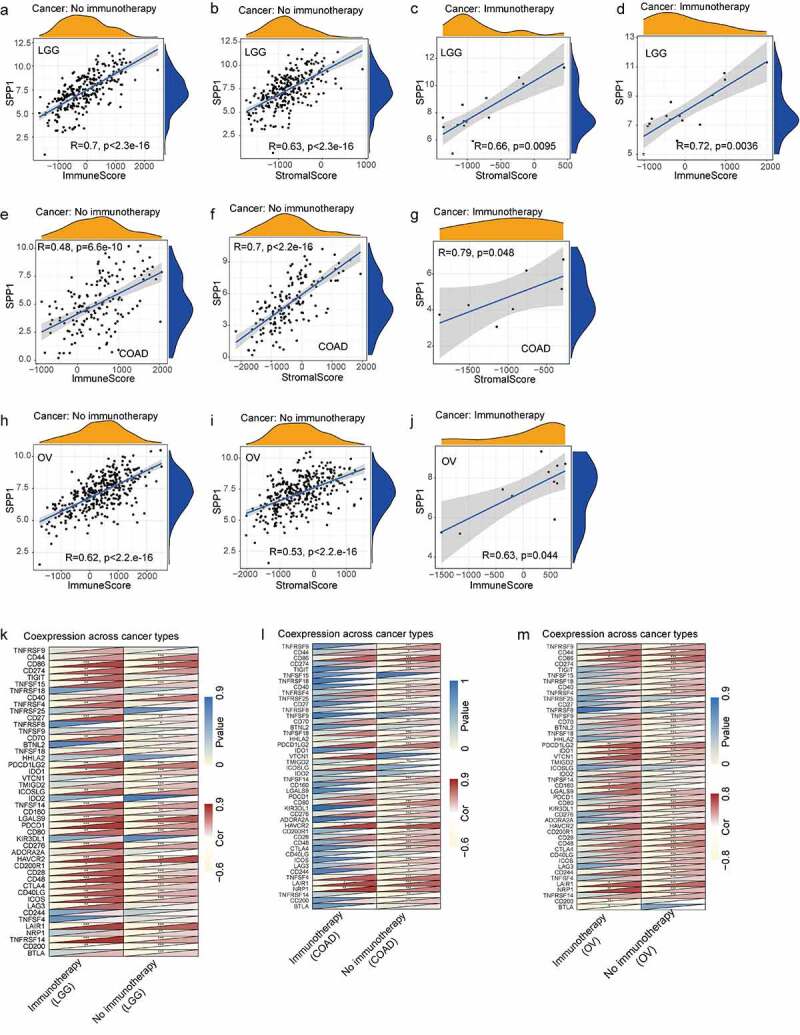
The tumor microenvironment is a complex milieu of stromal cells, tumor cells, and immune cells. Stromal cells support tumor growth [ref. 29], and immune cells promote or inhibit tumor growth [ref. 30]. To better understand the relevance and underlying mechanism of SPP1 expression in cancer, we evaluated the relationship between immune and stromal scores and SPP1 expression using the CIBERSORT analysis. SPP1 expression correlated positively with the tumor immune and stromal scores at P < 0.001 (Table S6). The top three tumors that correlated most significantly with SPP1 were LGG, OV, and THCA (immune score) (Figure 4(a-c)), and COAD, LGG, and READ (stromal score) (Figure 4(d-f)). We further performed a subgroup analysis of SPP1 expression with immune and stromal scores based on different stages and grades of the tumors, including COAD, LGG, OV, READ, and THCA. SPP1 was significantly positive correlated with immune and stromal scores in different stages of COAD and OV. In the histological grade of LGG and OV (G2 and G3), SPP1 also showed the same trend. This result indicated that tumor grades and stages did not affect the relationship between SPP1 and immune/stromal score. However, in READ and THCA, SPP1 was not correlated with stage І of READ and stage Ⅳ of THCA, only correlated with other stages (Table S7). This result indicated that tumor grades and stages affected the relationship between SPP1 and immune/stromal score.
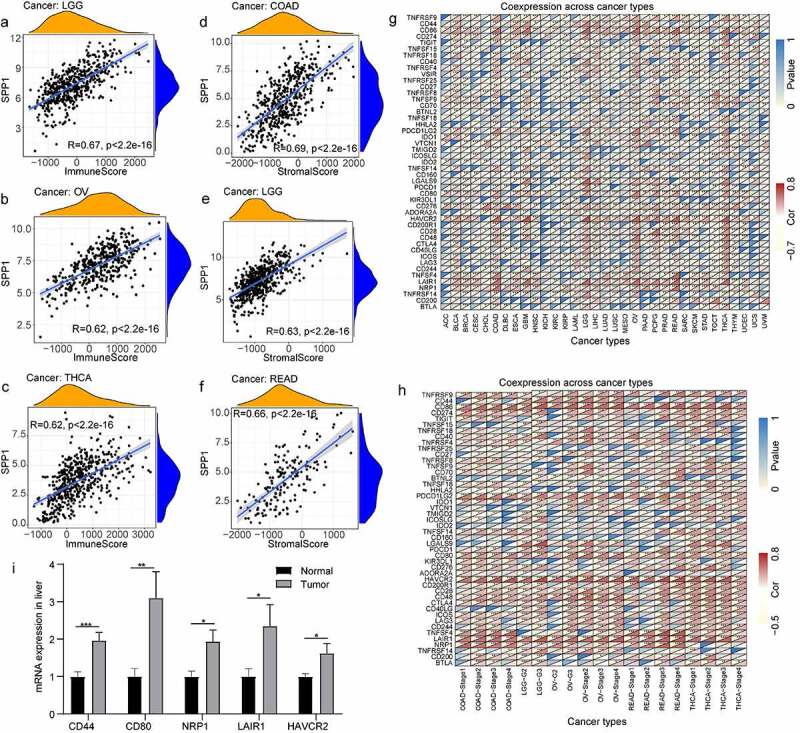
In addition, we analyzed the co-expression between SPP1 and 47 genes associated with immune infiltrating cell types in these tumors, including those encoding the tumor necrosis factor (TNF) superfamily (TNFSF), the TNF receptor superfamily (TNFRSF), multiple CD molecules (CD40, CD44, CD86, CD27, CD70, CD160, CD80, CD28, CD48, CD244, CD274, and CD200), and markers of exhausted T cells, such as PDCD1 (programmed cell death 1) and PDCD1LG2 (programmed cell death 1 ligand 2), CTLA4 (cytotoxic T-lymphocyte associated protein 4), LAG3 (lymphocyte activating 3), and HAVCR2 (Hepatitis A virus cellular receptor 2). In most tumors, SPP1 was co-expressed with markers of immune cells and showed significant positive correlations with them. For example, except for KIR3DL1 (encoding killer cell immunoglobulin like receptor, three Ig domains and long cytoplasmic tail 1), HHLA2 (encoding HERV-HLTR-associating 2), TNFSF9, TNFRSF25, and TNFRSF18, SPP1 expression correlated significantly and positively with other immune cells in LGG (Figure 4(g)). In LIHC, SPP1 was correlated with more than half of these genes including NRP1 (neuropilin 1), LAIR1 (leukocyte associated immunoglobulin like receptor 1), CTLA4, HAVCR2, and CD80. Similarly, we further performed correlation analysis of SPP1 expression with these immune checkpoint genes based on different stages and grades of the tumor. Heatmap showing most genes positively correlated with SPP1, especially HAVCR2, NRP1 (neuropilin 1), LAIR1 (leukocyte associated immunoglobulin like receptor 1), CD80, PDLD1LG2, and CD86 (Figure 4(h)). SPP1 expression was observed to correlated positively with immune checkpoint genes in multiple types of cancer. Therefore, we hypothesized that SPP1 might associate with immunotherapy. To further validate the analysis results, we examined some immune checkpoint genes expression in liver cancer. The results showed that these genes were highly expressed in liver cancer including CD80, CD44, HAVCR2, NRP1, and LAIR1 (Figure 4(i)). Together, these data indicated that high SPP1 expression was widely associated with immunity in cancers.
We performed a correlation analysis of SPP1 expression with immune and stromal scores in COAD, LGG, and OV patients treated with or without immunotherapy. In LGG, SPP1 was significantly positive correlated with immune and stromal scores regardless of whether patients received immunotherapy or not (Figure 5(a-d)). The heatmap also showed that SPP1 and immune checkpoint genes were co-expression in immunotherapy and no immunotherapy group (Figure 5(k)). The results indicated that immunotherapy did not affect the correlation between SPP1 and immune/stromal score in LGG. In COAD, SPP1 was correlated with stromal score in immunotherapy and no immunotherapy groups, but only correlated with immune score in no immunotherapy group (Figure 5(e-g)). In OV, SPP1 was correlated with immune score in immunotherapy and no immunotherapy groups, but only correlated with stromal score in no immunotherapy group (Figure 5(h-j)). The heatmap displayed that the expression of SPP1 in COAD and OV was only related to immune checkpoint genes in no immunotherapy group (Figure 5(l,m)). These results indicated that immunotherapy affected immune/stromal score in COAD and OV.
Tumor-infiltrating immune cells (TIICs) are a part of the complex microenvironment that regulates tumor development and progression [ref. 31]. Therefore, we calculated the relative abundance of 22 types of immune cells in each tumor, and analyzed their correlation with SPP1 expression. The results indicated that SPP1 expression had significant correlations with resting mast cells in 7 types of cancer, with macrophages in 21 types of cancer, with neutrophils in 10 types of cancer, with B cell in 6 types of cancer, with DC in 5 types of cancer, with CD8 in 5 types of cancer, and with monocytes in 5 types of cancer (Table S8). The results also revealed that at the level of immune cell infiltration, BRCA, COAD, GBM, LUAD, SARC and STAD correlated most strongly with SPP1 expression.
Potential of SPP1 as an indicator of response to immunotherapy
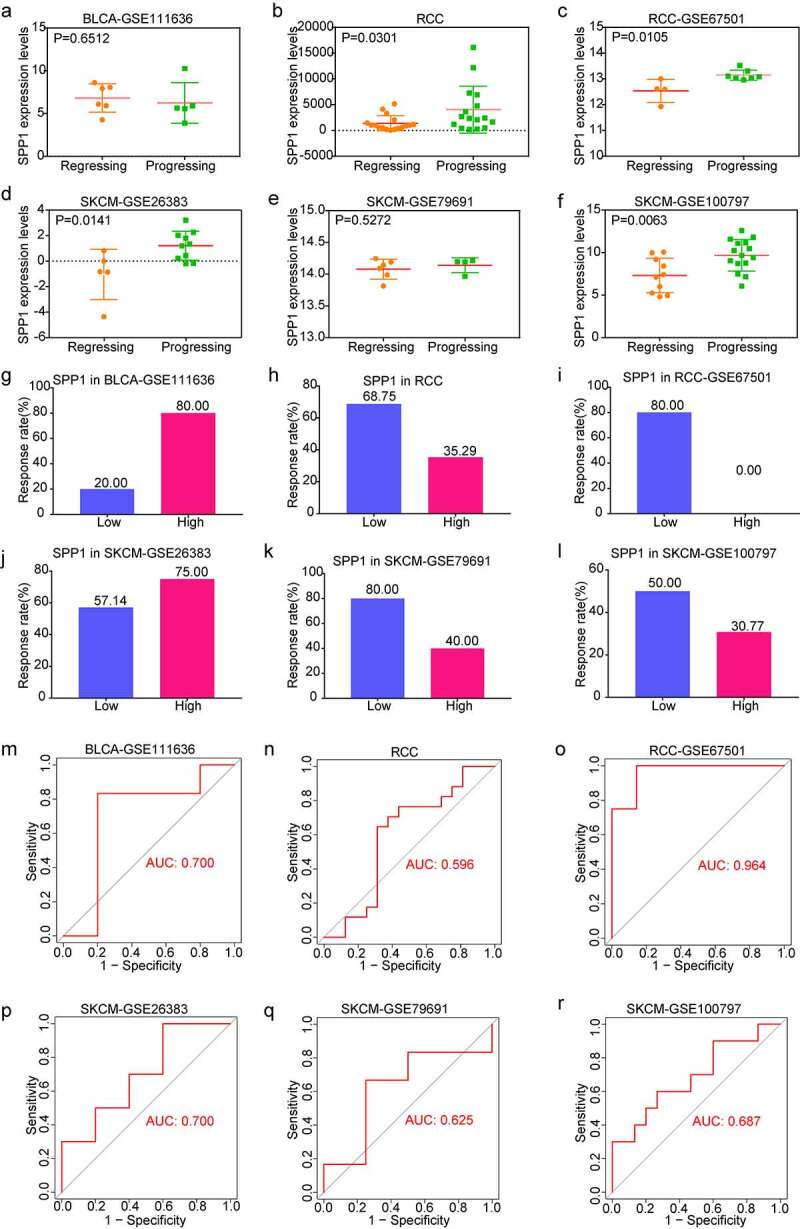
To investigate the role of SPP1 in tumor immunotherapy, we downloaded and analyzed immunotherapy datasets from the GEO database including GSE111636, GSE67501, GSE26383, GSE79691, and GSE100797. The RCC cohort was derived from data which was given in the literature with receiving immune checkpoint therapy [ref. 20]. In order to analyze the relationship between SPP1 expression and the effect of immunotherapy, we divided patients into two groups according immunotherapeutic effect. With the except of GSE111363 and GSE79691 cohorts, SPP1 expression showed a high level in progressing group and a low level in regressing group including RCC, GSE67501, GSE26383, and GSE100797 cohorts (Figure 6(a-f)). Furthermore, on the basis of SPP1 expression, we divided patients into two groups (low-expression and high-expression) compared the immunotherapy response rate. We found that the lower SPP1 was related to a better response rate to immunotherapy than the high-expression group (68.75% versus 35.29% in RCC; 80% versus 0% in GSE6701; 80% versus 20% in GSE79691; 50% versus 30.77% in GSE100797) (Figure 6(h, i, k, l)). In another two cohorts, the high-expression group had a higher response rate than the low-expression group in the GSE111636 and GSE26383 cohorts (80% versus 20% in the GSE111636 cohort; 75% versus 57.14% in the GSE26383 cohort) (Figure 6(g, j)). In addition, we analyzed the predictive value of SPP1 for immunotherapy using the ROC curve. From the value of the area under curve (AUC), SPP1 has significant prediction power for tumor immunotherapy (AUC: 0.700 in GSE111636; AUC: 0.596 in RCC; AUC: 0.964 in GSE67501; AUC: 0.700 in GSE26383; AUC: 0.625 in GSE79691; AUC: 0.687 in GSE100797) (Figure 6(m-r)). Collectively, these results revealed that SPP1 has the potential as a predictive indicator for the effectiveness of immunotherapy.
Functional analysis
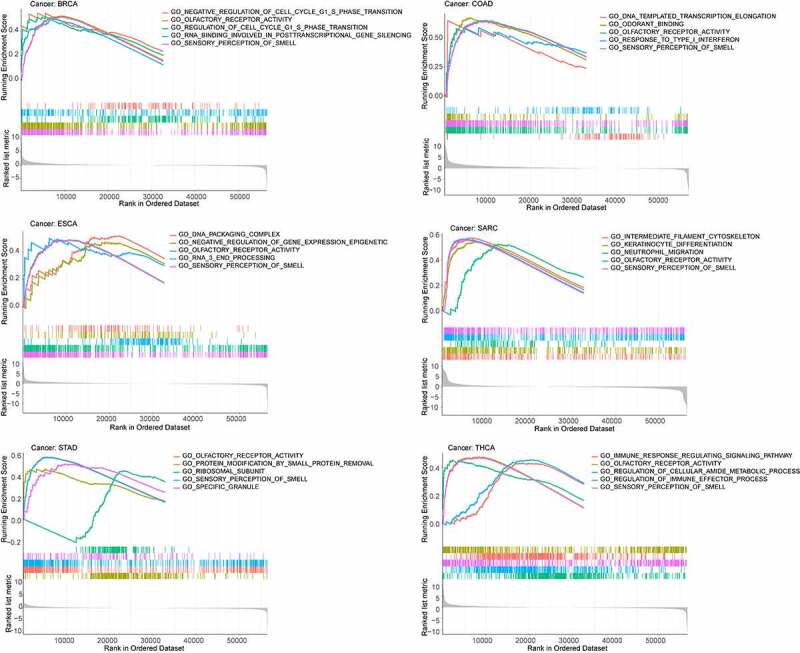
To determine which signaling pathways associated with SPP1 contribute to tumorigenesis, GSEA was performed, including functional analysis using gene ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Table S9 and Table S10). We identified the top five signaling pathways in GO analysis and found that SPP1 had a major impact on sensory perception of smell and olfactory receptor activity in most tumors. The higher the expression of SPP1, the more active these two pathways were in BRCA, COAD, ESCA, SRAC, STAD, and UCEC (Figure 7). The lower the expression of SPP1, the more active these two pathways were in BLCA, DLBC, OV, PRAD, READ, and THCA (Figure S9). The KEGG analysis showed that SPP1 primary affects the olfactory transduction pathway in most tumors.
Knockdown SPP1 inhibit the proliferation and migration of tumor cells
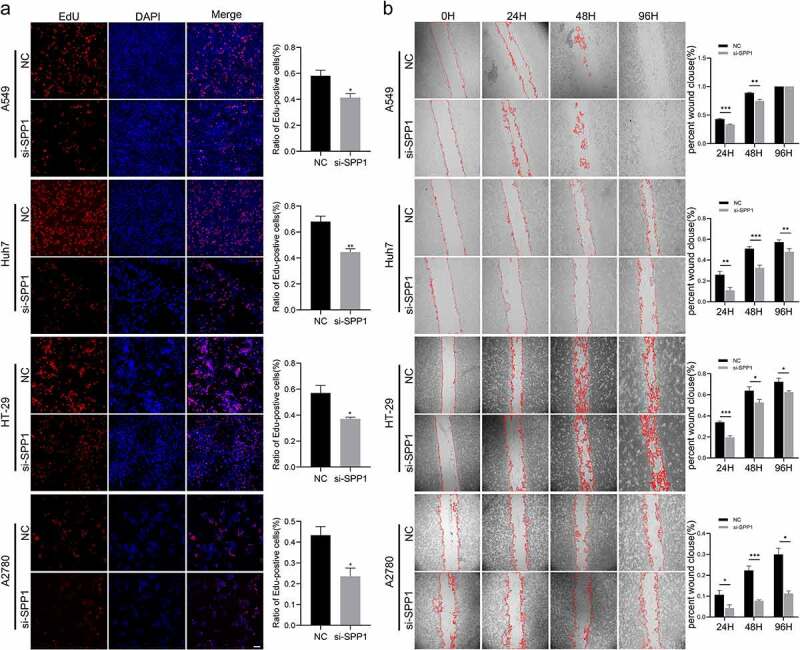
To further verify the oncogene features of SPP1, we evaluate the ability of SPP1 in cell proliferation and migration. We used siRNA to knockdown SPP1 expression in tumor cell lines. Following transfection, cell migration and proliferation were evaluated by EdU staining assay and wounding healing assay. The results showed that si-SPP1 also decreased cell proliferation of these tumor cell lines (Figure 8(a)). We further investigated the effect of SPP1 silencing on the cell migration. The results indicated that si-SPP1 significantly decreased cell migration of A549, Huh-7, HT-29, and A2780 cell lines (Figure 8(b)).
Discussion
Increasing evidence indicates that SPP1 is an oncogene in many cancers [ref. 32–34]. Therefore, it is necessary to pay more attention to the role of SPP1 in different aspects of tumor biology. In the present study, we analyzed the expression of SPP1 in multiple tumors using the Oncomine and TCGA databases, revealing significantly differences of SPP1 expression in various types of cancer. Compared with that in normal tissues, the SPP1 mRNA level was significantly higher in most tumor tissues, including bladder, brain and central nervous system, cervical, colorectal, esophageal, gastric, head and neck, liver, lung, ovarian, pancreatic, and prostate cancer, and melanoma, while the expression trend of SPP1 was inconsistent kidney cancer and sarcoma. In the TCGA database, SPP1 expression was significant higher in most tumor tissues, but showed lower expression in KICH, PAAD, SKCM, and THYM, however, without statistical significance. The analysis results in the two databases were broadly similar. However, there were also some differences that might have been caused by the data collection and analysis methods. In addition, according to the Kaplan-Meier and COX survival analyses, we found that high expression of SPP1 in most cancers is indicative of a poor prognosis, such as in CESC, ESCA, GBM, LGG, LIHC, LUAD, MESO, PAAD, PRAD, and STAD. Studies have also found that SPP1 could promote the proliferation, migration, and invasion of malignant tumor cells and inhibit cell apoptosis, leading to a poor prognosis in certain tumors [ref. 35]. Increasing evidences indicate that SPP1 is an important prognostic biomarker for many cancers [ref. 36,ref. 37].
The immune system normally recognizes and clears tumor cells in the tumor microenvironment. However, the growth of tumor cells also inhibits the immune system. When tumor cells are killed, the immune system is also suppressed. The biological function of SPP1 has been observed in various immune cells and is involved in initiating immune responses [ref. 38]. Notably, in this study, we demonstrated that the SPP1 expression was related to cancer immunity. SPP1 expression was significantly associated with the degree of infiltration in macrophages (M0, M1, and M2), resting mast cells, B cells, and neutrophils in at least six types of cancer according to CIBERSORT analysis.
Previous studies suggested that SPP1 promoted glioma progression by upregulating GBM-infiltrating neutrophils and macrophages, and was associated with infiltration of these cells within tumor specimens [ref. 39,ref. 40]. SPP1 was also involved in neutrophil activation [ref. 41], and neutralization of SPP1 attenuated neutrophil migration [ref. 42]. Currently, pan-cancer analysis employs the widely used ESTIMATE method to obtain immune and stromal scores in the tumor microenvironment, and to understand tumor prognosis [ref. 43,ref. 44]. In the present study, SPP1 expression was observed to correlated positively with immune and stromal scores. Immune checkpoint inhibitors have revolutionized cancer treatment [ref. 45,ref. 46]. Therefore, we analyzed the expression relationship between SPP1 and 47 common immune checkpoint genes. SPP1 expression was observed to correlated positively with immune checkpoint gene expression in multiple types of cancer. Studies have demonstrated that SPP1 could interact with CD44 in prostate cancer, GBM, and breast cancer [ref. 47,ref. 48]. Moreover, SPP1 might trigger pro-inflammatory stimuli, such as that mediated by TNF [ref. 49]. Almost all members of the TNFSF have proinflammatory activity, which is partially activated by the nuclear factor kappa B (NF-κB). TNFRSFs are primarily transmembrane proteins involved in physiological processes, such as host defense, inflammation, apoptosis, and immune process [ref. 50]. As a checkpoint immunotherapies targets, the CTLA4 has shown remarkable success in the treatment of certain cancer types [ref. 51]. This study provided the basis for a deeper understanding of the potential mechanism of SPP1 tumor immunity and its related cancer biomarkers. In recent years, immunotherapies have shown gradually increasing efficacy in treating tumors. Studies have demonstrated that immune checkpoint genes have a critical influence on immune cell infiltration and immunotherapy [ref. 52]. SPP1 was likely correlated with immune cell infiltration and might contribute to the immunotherapy in hepatocellular carcinoma [ref. 53].This work found a strong relationship between SPP1 and immune checkpoint genes, which provided a theoretical basis for combined molecular targeting immunotherapy in the future. We also found that SPP1 was associated with immunotherapy response in BLCA, RCC, and SKCM. Therefore, SPP1 might be a potential indicator in immunotherapy.
Genetic mutation is the major cause of tumorigenesis. Mutation and amplification were observed to be the main alteration frequency with mutation types of SPP1, especially in uterine cancer and melanoma. The TMB and MSI are emerging biomarkers that have predictive value in cancers [ref. 2,ref. 54,ref. 55]. The TMB has been widely studied as a predictive biomarker of the response to immune checkpoint inhibitors. To some extent, the TMB reflected the immunogenicity of the tumor, thus affecting the patient’s response to immune checkpoint inhibitors [ref. 56]. For example, the TMB determined the immune-related survival results of patients with breast cancer [ref. 57]. The high-quality and matched data from the TCGA contributed to a thorough investigation of the general prognostic impact of the TMB in patients newly diagnosed with cancer [ref. 56]. Previous studies showed that a high TMB was associated with better prognosis in STAD [ref. 58]; however a high TMB was associated with poor prognosis in non-small cell lung cancer [ref. 59]. Thus, the relationship between a high TMB and tumor prognosis might depend on the interaction between the tumor and the microenvironment. Recent studies showed that MSI was closely related to the occurrence and progression of many tumors. Microsatellite mutation can cause normal cells to transform into malignant cells. MSI was reported to be increased significantly in a variety of cancer tissues [ref. 60,ref. 61]. Therefore, MSI has become an important diagnostic index to screen malignant tumors. Lu et al have already found that olfactory transduction pathway could affect apoptosis of lung cancer cells and might be new hallmark of lung cancer [ref. 62]. The olfactory transduction is the main signaling pathway enriched from the unique subset of genes identified in esophageal squamous cell carcinoma [ref. 63]. Our study also found that SPP1 had a major impact on olfactory transduction. However, the underlying mechanisms requires further investigation.
Conclusions
In summary, our findings suggested that SPP1 could regulate tumorigenesis, tumor progression, and prognosis. The expression of SPP1 was significantly different in diverse human cancers. Therefore, SPP1 expression could be a valuable prognostic biomarker, because SPP1 upregulation resulted in a significant decrease in patient survival in certain cancer types. In addition, we revealed that the SPP1 expression was related to cancer immunity. Immunotherapy did not affect the immune/stromal score in LGG, and
affected immune/stromal score in COAD and OV. Therefore, SPP1 may serve as an attractive target in cancer mechanistic research and in treatment.
Supplementary Materials
References
- Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries.. CA Cancer J Clin., 2018. [PubMed]
- An integrative pan-cancer analysis revealing LCN2 as an oncogenic immune protein in tumor microenvironment.. Front Oncol., 2020. [PubMed]
- Role of osteopontin in the pathophysiology of cancer.. Matrix Biol., 2014. [PubMed]
- SPP1 promotes ovarian cancer progression via Integrin beta1/FAK/AKT signaling pathway.. Onco Targets Ther., 2018. [PubMed]
- Follistatin-like Protein 1 inhibits lung cancer metastasis by preventing proteolytic activation of osteopontin.. Cancer Res., 2019. [PubMed]
- Osteopontin-A master regulator of epithelial-mesenchymal transition.. J Clin Med., 2016. [DOI]
- Evaluation of SPP1/osteopontin expression as predictor of recurrence in tamoxifen treated breast cancer.. Sci Rep., 2020. [PubMed]
- Human osteopontin: potential clinical applications in cancer (Review).. Int J Mol Med., 2017. [PubMed]
- OPN is a promising serological biomarker for hepatocellular carcinoma diagnosis.. J Med Virol., 2020. [DOI]
- Abnormally activated OPN/integrin alphaVbeta3/FAK signalling is responsible for EGFR-TKI resistance in EGFR mutant non-small-cell lung cancer.. J Hematol Oncol., 2020. [PubMed]
- The UCSC Xena platform for public and private cancer genomics data visualization and interpretation.. bioRxiv., 2019. [DOI]
- Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles.. Neoplasia., 2007. [PubMed]
- Pan-cancer molecular patterns and biological implications associated with a tumor-specific molecular signature.. Cells., 2020. [DOI]
- High Level of METTL7B indicates poor prognosis of patients and is related to immunity in glioma.. Front Oncol., 2021. [PubMed]
- Comprehensive analysis of hypermutation in human cancer.. Cell., 2017. [PubMed]
- A molecular portrait of microsatellite instability across multiple cancers.. Nat Commun., 2017. [PubMed]
- Robust enumeration of cell subsets from tissue expression profiles.. Nat Methods., 2015. [PubMed]
- Gene expression markers of tumor infiltrating leukocytes.. J Immunother Cancer., 2017. [PubMed]
- Immune cell infiltration-based signature for prognosis and immunogenomic analysis in breast cancer.. Brief Bioinform., 2021. [PubMed]
- Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma.. Science., 2018. [PubMed]
- Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles.. Proc Natl Acad Sci U S A., 2005. [PubMed]
- PADI6 regulates trophoblast cell migration-invasion through the Hippo/YAP1 pathway in hydatidiform moles.. J Inflamm Res., 2021. [PubMed]
- An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors.. Nat Genet., 2008. [PubMed]
- Robust prognostic value of a knowledge-based proliferation signature across large patient microarray studies spanning different cancer types.. Br J Cancer., 2008. [PubMed]
- Prognostic value of CA20, a score based on centrosome amplification-associated genes, in breast tumors.. Sci Rep., 2017. [PubMed]
- Pan-cancer molecular analysis of the RB tumor suppressor pathway.. Commun Biol., 2020. [PubMed]
- Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas.. Cell Rep., 2019. [PubMed]
- Most random gene expression signatures are significantly associated with breast cancer outcome.. PLoS Comput Biol., 2011. [PubMed]
- Extracellular vesicles shedding promotes melanoma growth in response to chemotherapy.. Sci Rep., 2019. [PubMed]
- Major histocompatibility complex genes as therapeutic opportunity for immune cold molecular cancer subtypes.. J Immunol Res., 2020. [PubMed]
- Downexpression of HSD17B6 correlates with clinical prognosis and tumor immune infiltrates in hepatocellular carcinoma.. Cancer Cell Int., 2020. [PubMed]
- Analysis of expression and its clinical significance of the secreted phosphoprotein 1 in lung adenocarcinoma.. Front Genet., 2020. [PubMed]
- Identification of SPP1 as a promising biomarker to predict clinical outcome of lung adenocarcinoma individuals.. Gene., 2018. [PubMed]
- Squalene synthase promotes the invasion of lung cancer cells via the osteopontin/ERK pathway.. Oncogenesis., 2020. [PubMed]
- Upregulation of secreted phosphoprotein 1 affects malignant progression, prognosis, and resistance to cetuximab via the KRAS/MEK pathway in head and neck cancer.. Mol Carcinog., 2020. [PubMed]
- A novel prognostic models for identifying the risk of hepatocellular carcinoma based on epithelial-mesenchymal transition-associated genes.. Bioengineered., 2020. [PubMed]
- Association between the expression of secreted phosphoprotein – related genes and prognosis of human cancer.. BMC Cancer., 2019. [PubMed]
- The role of osteopontin in the progression of solid organ tumour.. Cell Death Dis., 2018. [PubMed]
- Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target.. J Clin Invest., 2019. [PubMed]
- ADAM8 affects glioblastoma progression by regulating osteopontin-mediated angiogenesis.. Biol Chem., 2020. [DOI]
- Biomarkers of macrophage activation and immune danger signals predict clinical outcomes in alcoholic hepatitis.. Hepatology., 2019. [PubMed]
- Neutralization of osteopontin attenuates neutrophil migration in sepsis-induced acute lung injury.. Crit Care., 2015. [PubMed]
- Tumor microenvironment differences between primary tumor and brain metastases.. J Transl Med., 2020. [PubMed]
- Immune cell and stromal signature associated with progression-free survival of patients with resected pancreatic ductal adenocarcinoma.. Gastroenterology., 2018. [PubMed]
- AGI-134: a fully synthetic alpha-Gal glycolipid that converts tumors into in situ autologous vaccines, induces anti-tumor immunity and is synergistic with an anti-PD-1 antibody in mouse melanoma models.. Cancer Cell Int., 2019. [PubMed]
- Intratumoral expression levels of PD-L1, GZMA, and HLA-A along with oligoclonal T cell expansion associate with response to nivolumab in metastatic melanoma.. Oncoimmunology., 2016. [PubMed]
- Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth.. Cell Stem Cell., 2014. [PubMed]
- Osteopontin as a multifaceted driver of bone metastasis and drug resistance.. Pharmacol Res., 2019. [PubMed]
- Persistently elevated osteopontin serum levels predict mortality in critically ill patients.. Crit Care., 2015. [PubMed]
- Differential immune transcriptome and modulated signalling pathways in rainbow trout infected with Viral Haemorrhagic Septicaemia Virus (VHSV) and Its Derivative Non-Virion (NV) gene deleted.. Vaccines (Basel)., 2020. [DOI | PubMed]
- The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy.. Nat Rev Cancer., 2019. [PubMed]
- Immune checkpoint blockade: a common denominator approach to cancer therapy.. Cancer Cell., 2015. [PubMed]
- Seven immune-related genes prognostic power and correlation with tumor-infiltrating immune cells in hepatocellular carcinoma.. Cancer Med., 2020. [PubMed]
- The Role of Molecular Profiling to Predict the Response to Immune Checkpoint Inhibitors in Lung Cancer.. Cancers (Basel)., 2019. [DOI]
- Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the Phase II KEYNOTE-158 study.. J Clin Oncol., 2020. [PubMed]
- Tumor mutational and indel burden: a systematic pan-cancer evaluation as prognostic biomarkers.. Ann Transl Med., 2019. [PubMed]
- Tumor mutational burden is a determinant of immune-mediated survival in breast cancer.. Oncoimmunology., 2018. [PubMed]
- Association of MUC16 mutation with tumor mutation load and outcomes in patients with gastric cancer.. JAMA Oncol., 2018. [PubMed]
- Prognostic impact of tumor mutation burden in patients with completely resected non-small cell lung cancer: brief report.. J Thorac Oncol., 2018. [PubMed]
- Burden of unique and low prevalence somatic mutations correlates with cancer survival.. Sci Rep., 2019. [PubMed]
- Rational application of the first-line chemotherapy and immune checkpoint inhibitors in advanced nonsmall cell lung cancer: a meta-analysis.. Cancer Med., 2019. [PubMed]
- Genome-wide transcriptional analysis of apoptosis-related genes and pathways regulated by H2AX in lung cancer A549 cells.. Apoptosis., 2013. [PubMed]
- Heterogeneity analysis of esophageal squamous cell carcinoma in cell lines, tumor tissues and patient-derived xenografts.. J Cancer., 2021. [PubMed]