Glucocerebrosidase mutations in primary parkinsonism
Abstract
Introduction:
Mutations in the lysosomal glucocerebrosidase (GBA) gene increase the risk of Parkinson’s Disease (PD). We determined the frequency and relative risk of major GBA mutations in a large series of Italian patients with primary parkinsonism.
Methods:
We studied 2766 unrelated consecutive patients with clinical diagnosis of primary degenerative parkinsonism (including 2350 PD), and 1111 controls. The entire cohort was screened for mutations in GBA exons 9 and 10, covering approximately 70% of mutations, including the two most frequent defects, p.N370S and p.L444P.
Results:
Four known mutations were identified in heterozygous state: 3 missense mutations (p.N370S, p.L444P, and p.D443N), and the splicing mutation IVS10+1G>T, which results in the in-frame exon-10 skipping. Molecular characterization of 2 additional rare variants, potentially interfering with splicing, suggested a neutral effect. GBA mutations were more frequent in PD (4.5%, RR = 7.2, CI = 3.3–15.3) and in Dementia with Lewy Bodies (DLB) (13.8%, RR = 21.9, CI = 6.8–70.7) than in controls (0.63%). but not in the other forms of parkinsonism such as Progressive Supranuclear Palsy (PSP, 2%), and Corticobasal Degeneration (CBD, 0%). Considering only the PD group, GBA-carriers were younger at onset (52 ± 10 vs. 57 ± 10 years, P < 0.0001) and were more likely to have a positive family history of PD (34% vs. 20%, P < 0.001).
Conclusion:
GBA dysfunction is relevant for synucleinopathies, such as PD and DLB, except for MSA, in which pathology involves oligodendrocytes, and the tauopathies PSP and CBD. The risk of developing DLB is three-fold higher than PD, suggesting a more aggressive phenotype.
Article type: Research Article
Keywords: Parkinson’s disease, Parkinsonism, Association analysis, Splicing mutation, Functional characterization
Affiliations: Dipartimento di Biotecnologie Mediche e Medicina Traslazionale, Università degli Studi di Milano, Milano, Italy; Parkinson Institute, Istituti Clinici di Perfezionamento, Milan, Italy
License: © 2014 The Authors CC BY 4.0 This work is licensed under a Creative Commons Attribution 3.0 Unported License.
Article links: DOI: 10.1016/j.parkreldis.2014.09.003 | PubMed: 25249066 | PMC: PMC4228056
Relevance: Relevant: mentioned in keywords or abstract
Introduction
Mutations in the gene encoding beta-glucocerebrosidase (GBA, OMIM *606463) are an important and common risk factor for Parkinson’s disease (PD). Many studies have shown an increased frequency of GBA mutations in PD compared to controls ref. [1], ref. [2]. In particular, an odds ratio (OR) of approximately 5 has been found in a multicenter study including approximately 5000 PD patients and an equal number of controls ref. [3]. GBA mutations were repeatedly found to be increased also in Dementia with Lewy Bodies (DLB) ref. [4], ref. [5], ref. [6]. A recent multicenter study on 700 DLB patients reported a remarkable OR of 8, suggesting that GBA mutations may have an even larger role in the genetic etiology of DLB than in PD ref. [7]. Furthermore, PD carriers of the GBA mutation are more likely to progress to dementia, suggesting a significant impact on the distribution of pathology and on the resulting clinical phenotype ref. [8]. On the other hand, Multiple System Atrophy (MSA) does not appear to be associated with GBA ref. [9], ref. [10]. So far, no data are available on GBA involvement in tauopathies, such as Progressive Supranuclear Palsy (PSP) and Corticobasal Degeneration (CBD).
The frequency and distribution of GBA mutations vary among populations, hindering comparisons between different patient series. Carrier frequency is quite high among Ashkenazi Jews (about 1 person in 14), and very rare in Asia ref. [3]. In addition, studies were usually performed on series of patients with the same diagnosis often collected from many clinical centers, preventing the evaluation of the importance of GBA in the different forms of parkinsonism.
In this frame, we decided to analyze the major mutations in the GBA gene in a large series of unrelated patients with primary parkinsonism visited in a single Italian tertiary clinic and to compare their frequency with a large control group from the same population. This design enabled the evaluation and comparison of GBA burden in various forms of parkinsonism in the Italian population.
Methods
This study was approved by the local Ethical Committee and was conducted according to the Declaration of Helsinki and to the Italian legislation on sensitive personal data recording. Written informed consent was obtained from all subjects.
Subjects
We studied 2766 unrelated consecutive patients with degenerative parkinsonism, and 1111 controls, who contributed from 2002 to 2010 to the Parkinson Institute Biobank (www.parkinsonbiobank.com), regardless of family history or age at onset. All the patients had a diagnosis of primary degenerative parkinsonism: 2350 fulfilled current criteria for probable PD, 29 for DLB, 118 for MSA, 100 for PSP, and 34 for CBD ref. [11], ref. [12]. In particular, diagnosis of DLB was made only in those fulfilling the 1-year rule between the onset of dementia and parkinsonism ref. [13]. In the remaining 135 cases, the clinical diagnosis was still uncertain and these patients are reported here as suffering from undefined primary parkinsonism (PKS). The majority of MSA cases (N = 113) had been previously reported in a multicentre collaborative study on GBA involvement in MSA ref. [10]. Patients with suspect of secondary parkinsonism were excluded. All patients were examined by neurologists expert in movement disorders. The following clinical and demographic data were collected: gender, age at onset, asymmetry of symptoms at onset, disease duration, education, cigarette smoking, and family history of PD. Among the 2350 PD patients, the mean age at onset was 56.1 years (SD ± 10.9, range 13–87), the mean disease duration was 11.6 years (SD ± 6.7, range 5–56).
Controls were recruited among spouses and caregivers and were unrelated to the patients. All subjects who reported or showed signs or symptoms of movement disorders or other neurodegenerative diseases were excluded. Among the 1111 controls, the mean age at sample collection was 62.3 years (SD ± 11; range 30–94 years). All controls denied any family history for movement disorders in first-degree relatives.
Except for 25 patients originating mainly from other European countries, all patients and controls were of Caucasian ethnicity and Italian origin.
Mutation analysis
The mutational screening of GBA exons 9 and 10 was performed by a combination of high-resolution melting (HRM) analysis (exon 9) and direct DNA sequencing (exon 10). PCR primer couples were designed on the basis of the known genomic sequence of the gene (GenBank accession number NM_000157) to amplify the two exons of interest and their exon-intron boundaries, avoiding the concomitant amplification of the highly-homologous GBA pseudogene (GBAP1) (Supplementary Table 1). A detailed description of mutation analysis methods is reported in the Supplementary material.
The 2350 PD patients were previously tested for several PD-related genes, such as LRRK2 (G2019S, R1441 C/G, I2020L), Parkin, PINK1, DJ1, and SNCA ref. [14], ref. [15], ref. [16]. The 66 patients found to be carrier of mutations in these genes were not excluded from the GBA genetic analysis.
Molecular characterization of the newly-identified splicing mutation
The effect of the IVS10+1G>T splicing mutation on GBA pre-mRNA processing was evaluated in RNA derived from whole blood of the carrier patient. Whole blood was collected in a PAXgene Blood RNA Tube (PreAnalytiX, Hombrechtikon, Switzerland) and RNA purification performed by using the PAXgene Blood miRNA kit (PreAnalytiX) following the manufacturer’s instructions. One microgram of total RNA was reverse transcribed (RT) using random nonamers and the Superscript-III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). Of a total of 20 μL, 1 μL was used as template for standard PCR reactions by means of exonic primers (Supplementary Table 1). The identity of the amplified fragments was confirmed by Sanger sequencing. To quantify the relative amount of GBA exon-10 containing vs. skipping isoforms, we performed competitive RT-PCRs by using a 6-FAM-labeled primer. Amplified fragments were separated by capillary electrophoresis on an ABI-3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) and quantitated by the GeneMapper v4.0 software.
Statistical analysis
All the following statistical procedures were performed using the R program release 2.8.0 (http://www.r-project.org/).
For each GBA mutation, standard case–control analyses on carrier frequency data were performed with the Fisher exact test; all P values are presented as non-corrected. The impact of GBA mutation burden on different clinical subtypes was evaluated by the use of the relative risk (RR) statistics.
The association of different variables with the presence of GBA mutations in PD patients was assessed by testing for differences between carriers and non-carriers by means of a chi-square test for categorical data (i.e. gender, asymmetric onset, smoking status, family history of PD), or by using the Student’s t test for continuous data (i.e. age at onset, disease duration, education; these were analyzed as a quantitative variable, after having verified that their departure from linearity was not statistically significant). The effect of each factor was expressed as the OR and 95% confidence interval (CI). Unadjusted ORs were obtained by using a logistic regression model that included only the factor of interest; adjusted ORs were obtained by using a model that included the factor of interest plus all of the factors that were significant in the first step of analysis.
Results
Screening for GBA mutations on exons 9 and 10
In the whole cohort of subjects investigated (2766 patients and 1111 controls), we identified 10 different rare genetic variants, all present in the heterozygous state (Table 1).
Table 1: GBA mutations/rare variants identified in 2766 patients with degenerative parkinsonism and 1111 healthy controls.
| Genomic positiona | Rs ID | cDNA changeb | Functionc | Alleles in cases (n) | Alleles in controls (n) |
|---|---|---|---|---|---|
| 1:155,205,659 | NA | c.1225-24T>G | IVS8-24T>G | 0 | 1 |
| 1:155,205,634 | rs76763715 | c.1226A>G | p.N370Sd | 69 | 4 |
| 1:155,205,581 | rs149171124 | c.1279G>A | p.E388K | 5 | 1 |
| 1:155,205,440 | NA | c.1388 + 32C>T | IVS9+32C>T | 1 | 0 |
| 1:155,205,138 | NA | c.1389-36C>G | IVS9–36C>G | 0 | 1 |
| 1:155,205,107 | NA | c.1389-5T>A | IVS9-5T>A | 1 | 0 |
| 1:155,205,047 | rs75671029 | c.1444G>A | p.D443Nd | 1 | 0 |
| 1:155,205,043 | rs421016 | c.1448T>C | p.L444Pd | 47 | 3 |
| 1:155,204,985 | NA | c.1505G>T | IVS10+1G>Td | 1 | 0 |
| 1:155,204,978 | rs371668537 | c.1505C>A | IVS10+8C>A | 1 | 0 |
All variants were identified in the heterozygous state. Frequent polymorphisms (minor allele frequencies >2.5%) are not reported.
Seventeen of the 47 L444P mutations found in cases and two of the 3 found in controls were associated with the p.A456P variant (indicating the presence of a complex recombinant allele).
Rs ID, refseq identification number; NA, not available [variant not reported either in the HGMD professional, or in 5400 exomes obtained from the Exome Variant Server, NHLBI GO exome Sequencing Project (ESP, v.0.0.9 data release, November 2011, http://evs.gs.washington.edu/EVS/)].
The p.N370S and p.L444P mutations were more common in patients than in controls (2.5% vs. 0.36%, P = 1.2 × 10−6 OR = 7.1, CI = 2.6–19.5; and 1.7% vs. 0.27%, P = 1.1 × 10−4 OR = 6.4, CI = 2.0–20.6, respectively) (Supplementary Table 2). In addition to these two well-known major mutations, only the p.E388K variant was found in more than one subject, with no difference between cases and controls (0.18% vs. 0.09%, P = 0.68). Therefore we considered it a probable rare polymorphism. Indeed, E388K has never been clearly associated to Gaucher Disease (GD) ref. [17], while has been found both in PD cases ref. [5], ref. [18] and controls ref. [19], ref. [20].
Other seven variants were found in individual cases (either patient or control). Among them, 2 were previously described as associated with GD: p.D443N and IVS10+1G>T [ref. [21], HGMD professional, http://www.hgmd.org/], while the other 5 rare variants were found in intronic regions (Table 1).
Molecular characterization of the identified splicing variants
Though previously described, the IVS10+1G>T splicing mutation has not yet been characterized from the molecular point of view. Hence, to evaluate the effect of IVS10+1G>T on GBA pre-mRNA processing, a cDNA region spanning exons 7–11 was amplified directly from mRNA of the carrier heterozygous patient. Results suggest that IVS10+1G>T is pathogenic through the skipping of exon 10 (Fig. 1a–b). The GBA transcript skipping exon 10 is predicted to code for a potentially non-functional protein, lacking 39 amino acids in the C-terminal portion of the enzyme.
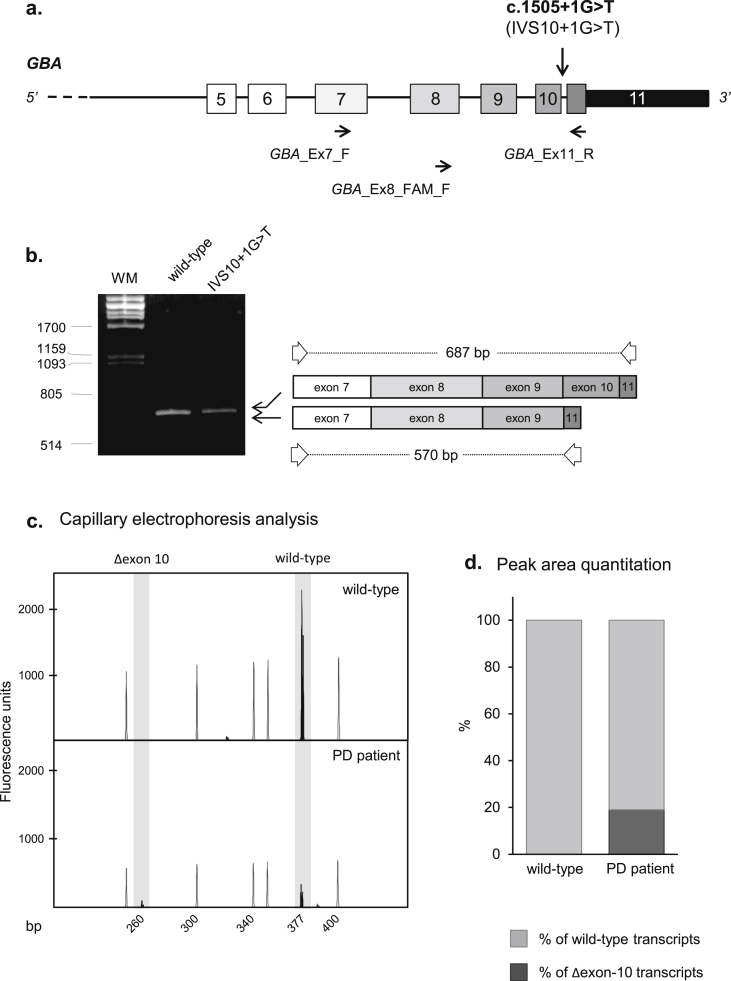
The specific quantitative measurement of the mutant transcript showed that the expression of the mutant allele amounts to only about one fifth of the wild-type allele, suggesting that the aberrantly-spliced transcript might be unstable (Fig. 1c–d).
Concerning all the remaining 5 intronic variants (IVS8-24T>G, IVS9+32C>T, IVS9–36C>G, IVS9-5T>A, IVS10+8C>A), in-silico analyses did not predict any significant alteration of the splicing process (e.g. activation of a cryptic splice site or abrogation of a physiologic site; data not shown). Nonetheless, the IVS9-5T>A and IVS10+8C>A putative splice variants, both located within 10 nucleotides from intron-exon junctions, were expressed in HeLa cells. Analysis of transcripts generated from constructs containing either IVS9-5T>A or IVS10+8C>A showed that exon 10 is correctly included into the mature mRNA, demonstrating that both variants are neutral (Supplementary Fig. 1).
Frequency of GBA mutations
The frequency of GBA mutations according to clinical diagnosis is reported in Table 2.
Table 2: GBA mutation carriers according to clinical diagnosis.
| Diagnosis | Subjects (n) | Carriers (n) | % Of subjects | RR (95% CI) | P valuea |
|---|---|---|---|---|---|
| PD | 2350 | 106 | 4.5% | 7.2 (3.3–15.3) | 2.2 × 10−11 |
| DLB | 29 | 4 | 13.8% | 21.9 (6.8–70.7) | 9.9 × 10−6 |
| MSA | 118 | 1 | 0.85% | 1.3 (0.17–10.8) | 0.56 |
| PSP | 100 | 2 | 2.0% | 3.2 (0.67–15.1) | 0.17 |
| CBD | 34 | 0 | 0% | 2.1 (0.12–36.4) | 0.99 |
| Undefined PKS | 135 | 5 | 3.7% | 5.9 (1.9–18.3) | 5.9 × 10−3 |
| Healthy controls | 1111 | 7 | 0.63% | / | / |
All the pathogenic mutations (p.N370S, p.D443N, p.L444P, IVS10+1G>T) considered together. RR was calculated considering healthy controls as reference group.
Significant results are in bold.
Parkinson’s disease (PD); Dementia with Lewy Bodies (DLB); Multiple System Atrophy (MSA); Progressive Supranuclear Palsy (PSP); Corticobasal Degeneration (CBD); Parkinsonisms (PKS).
Pathogenic GBA mutations (p.N370S, p.D443N, p.L444P, and IVS10+1G>T) were significantly increased in patients with PD, DLB, and undefined PKS, but not in those with MSA, PSP or CBD. The highest RR was in DLB (RR = 21.9, CI = 6.8–70.7, P = 9.9 × 10−6).
Five carriers (3.7%) occurred in the heterogeneous group of patients with an undefined PKS, containing all cases with a primary parkinsonism whose clinical features did not fulfill currently established consensus criteria ref. [11], ref. [12], ref. [13]. All of them had parkinsonism (bradykinesia and rigidity) and co-occurring dementia, while three out of 5 had poor response to levodopa.
Genotype-phenotype correlations
We compared clinical phenotype between GBA-carriers and non-carriers in the PD subgroup only, given the relatively low number of carriers with other diagnoses (Table 3). We excluded from this analysis those patients who were carriers of mutations in other PD-related genes: 62 in the non-carrier group, and 4 in the GBA-carrier group (LRRK2-p.G2019S, n = 2; LRRK2-p.R1441C, n = 1; and homozygous deletion of exon 3 in parkin gene, n = 1). An extensive clinical description of these 4 PD patients is reported in the online Supplementary material.
Table 3: Cross-sectional analysis of demographic and general clinical features of 2284 consecutive patients with PD according to GBA mutation status.
| Feature | GBA carriers (n = 102) | Non-carriers (n = 2182) | Unadjusted analysis | Adjusted analysisa | ||
|---|---|---|---|---|---|---|
| P value | Or (95% CI) | P value | Or (95% CI) | |||
| Male, n (%) | 58 (56.9%) | 1322 (60.6%) | 0.45 | 1.17 (0.78–1.74) | – | – |
| Age at onset (y, mean ± SD) | 51.54 ± 10.63 | 56.69 ± 10.51 | 1.97 × 10−6 | 1.04 (1.03–1.06) | 5.29 × 10−6 | 1.04 (1.03–1.07) |
| Asymmetric onset, n (%) | 78 (85.7%)b | 1675 (85.1%)b | 0.87 | 1.05 (0.58–1.91) | – | – |
| Disease duration (y, mean ± SD) | 11.35 ± 7.315 | 11.87 ± 6.48 | 0.44 | 0.99 (0.96–1.02) | – | – |
| Education (y, mean ± SD) | 10.70 ± 4.72c | 9.80 ± 4.39c | 0.049 | 1.05 (1.00–1.09) | 0.31 | 1.02 (0.98–1.07) |
| Cigarette smoking, n (%) | 39 (41.1%)d | 797 (38.4%)d | 0.60 | 1.12 (0.74–1.70) | – | – |
| Positive family history for PD, n (%) | 21 (20.6%) | 250 (11.6%)e | 7.4 × 10−3 | 1.97 (1.20–3.25) | 0.019 | 1.87 (1.11–3.15) |
Values are expressed as counts (and %) or as mean ± standard deviation (SD). Significant values are in bold.
The age at which the patient noticed the first PD symptom was considered to be the age at onset of disease. Disease duration was calculated on the basis of the last examination. Education and smoking data are based on patient self-reporting. Current and former smokers were aggregated into the single category of smokers. The definition of positive family history was restricted to patients having at least one 1st degree relative with a formal diagnosis of PD.
GBA-carriers had an earlier onset than non-carriers, with a mean age of 52 and 57 years, respectively. In particular, a total of 14/152 (9%) carriers had early onset (<40 years) compared to 88/2132 non-carriers (4%). Moreover, carriers were more likely to have a positive family history of PD (20.6% vs 11.6%).
Discussion
In this study we analyzed a large case series of patients consecutively collected at a single site, together with a large control group, to evaluate the impact of the most frequent GBA mutations in various forms of primary parkinsonism. GBA mutations contributed to PD and even more so to DLB, whereas they did not increase the risk for developing tauopathies (PSP and CBD).
Consistently with other European studies ref. [3], ref. [19], ref. [20], the frequency of GBA mutations in PD among our Italian patients is 4.5%, confirming that GBA mutations are the most common genetic determinant of both familial and sporadic PD. However, in a previous study in the South of Italy, a lower frequency of mutations was found in PD patients (11/395, 2.8%) and in controls (1/483, 0.2%), and the most common mutation was p.L444P ref. [22]. Conversely, we found that the most frequent genetic defect is p.N370S, as previously reported for Italian GD patients ref. [23]. Our Institute is located in the North of Italy; however, it is a tertiary referral center and attending patients come from all parts of the country. Therefore, the difference may be due to a particular frequency of GBA mutations in Southern Italy or to the relatively small size of the case series analyzed by DeMarco and colleagues ref. [22].
A limitation of our study is that we screened only exons 9 and 10, and therefore we lost the carriers of rare mutations in other regions of GBA. However, this screening strategy covers the vast majority of GBA mutations in our population ref. [20], ref. [23], and was chosen to be able to analyze a large number of individuals.
The p.L444P mutation, causing a more severe GD phenotype ref. [23], does not appear to be associated with a greater risk compared to the less severe p.N370S (Supplementary Table 2), in contrast to what previously reported ref. [24], ref. [25]. This is relevant for genetic counseling. Considering the p.L444P and the recombinant p.L444P + p.A456P alleles separately, only the non-recombinant allele is significantly associated with a higher PD risk. However, this is probably due to the lower frequency of the p.L444P + p.A456P variant.
Moreover, our study provides a precise assessment of GBA mutation “carrier frequency” in the Italian population. Indeed, considering that the 2 major mutations account for around 70% of disease alleles, GBA mutation carriers including rare mutations should be 0.9% of the general population, consistently with a prevalence of GD in Italy of 1/40,000 ref. [26].
Finally, we confirm that in GBA-carriers the disease onset occurs 5 years earlier than in non-carriers ref. [3], at difference with what observed in our case series of LRRK2-mutated patients where age at onset was similar between carriers and non-carriers ref. [16]. It remains to investigate the reason that why, although both genetic factors operate in a multifactorial context, their effect on the phenotype is expressed in a different way.
Considering other forms of parkinsonism, GBA mutations were most common in DLB, where the highest RR was found. Although our DLB cohort is too small to allow generalizations, this observation seems to confirm the trend towards more widespread pathological damage, even including the cortex and, consequently, the increased risk for dementia in GBA mutation carriers ref. [1]. Consistently, the GBA-carriers amongst the patients categorized as undefined PKS had not only parkinsonism, but also dementia. This suggests that these patients are likely part of the Diseases with Lewy Bodies spectrum, a continuum of clinic-pathologic entities spanning from PD, PD and Dementia (PDD), to DLB ref. [13]. In the future, it will be interesting to verify whether our GBA mutation carriers develop dementia more often than non-carriers, as suggested by other studies ref. [3], ref. [8].
Contrasting data on the frequency of GBA mutations in DLB compared to PD have been reported: lower for some authors ref. [6], ref. [18] and higher for others ref. [4], ref. [5]. In our analysis the frequency of GBA mutations was clearly higher in DLB than in PD, with an RR (21.9), which was considerably greater than in most studies. This may be due to the low number of cases with diagnosis of DLB in our study (note that the CI of the RR was 6.8–70.7), as well as to a selection bias at our Institute, which focuses on PD. Indeed, the DLB patients who have prominent dementia or dementia almost free from parkinsonism probably do not come to our attention. GBA may be more involved in forms with dementia with more important signs of parkinsonism. Future research to explore this hypothesis may focus on differences in cognitive and motor symptoms between PDD and LBD carriers. Furthermore it may be interesting to evaluate GBA mutation frequency within the LBD spectrum.
GBA mutations do not seem to play a role in the predisposition both to MSA, as previously evidenced by us ref. [10] and others ref. [9], and to tauopathies. This may underlie a different involvement of the GBA-mediated lysosomal impairment in different forms of parkinsonism. Indeed, in neurons, the connection between beta-glucocerebrosidase (GCase) impairment and Lewy bodies formation has been proposed to rely on GCase dysfunction leading to α-synuclein accumulation in the lysosome, which in turns exacerbates loss of GCase activity ref. [27]. Unlike other synucleinopathies, the histopathologic hallmark of MSA is accumulation of α-synuclein within glial cytoplasmic inclusions (GCI) instead of within neurons. At the beginning of the pathogenic process, α-synuclein accumulates in the GCIs, located mainly within the oligodendroglial cells, leading to neurodegeneration and, ultimately, neuronal death ref. [28]. The evidence that GBA mutations are not associated with the risk of developing MSA is in line with the hypothesis that the GCase impairment does not play an important role in oligodendrocytes.
Similar conclusions can be drawn for PSP and CBD tauopathies, in which to our knowledge GBA mutations have been sought for the first time. Indeed, although the mechanisms that lead to deposit of tau protein are unclear ref. [29], the GBA-mediated lysosomal impairment does not seem to have a major role. This is of considerable interest, in view of the close correlation among α-synuclein, tau and amyloid β pathologies ref. [30].
References
- M. Swan, R. Saunders-Pullman. The association between ß-glucocerebrosidase mutations and parkinsonism. Curr Neurol Neurosci Rep, 2013. [DOI | PubMed]
- J. Liu, H.X. Zhang. Significant study of population stratification, sensitivity analysis and trim and fill analyses on GBA mutation and Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet, 2014
- E. Sidransky, M.A. Nalls, J.O. Aasly, J. Aharon-Peretz, G. Annesi, E.R. Barbosa. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med, 2009. [PubMed]
- O. Goker-Alpan, B.I. Giasson, M.J. Eblan, J. Nguyen, H.I. Hurtig, V.M. Lee. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology, 2006. [PubMed]
- L.N. Clark, L.A. Kartsaklis, R. Wolf Gilbert, B. Dorado, B.M. Ross, S. Kisselev. Association of glucocerebrosidase mutations with dementia with lewy bodies. Arch Neurol, 2009. [PubMed]
- D. Tsuang, J.B. Leverenz, O.L. Lopez, R.L. Hamilton, D.A. Bennett, J.A. Schneider. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology, 2012. [PubMed]
- M.A. Nalls, R. Duran, G. Lopez, M. Kurzawa-Akanbi, I.G. McKeith, P.F. Chinnery. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol, 2013. [PubMed]
- S.E. Winder-Rhodes, J.R. Evans, M. Ban, S.L. Mason, C.H. Williams-Gray, T. Foltynie. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain, 2013. [PubMed]
- B. Segarane, A. Li, R. Paudel, S. Scholz, J. Neumann, A. Lees. Glucocerebrosidase mutations in 108 neuropathologically confirmed cases of multiple system atrophy. Neurology, 2009. [PubMed]
- K. Srulijes, A.K. Hauser, I. Guella, R. Asselta, K. Brockmann, C. Schulte. No association of GBA mutations and multiple system atrophy. Eur J Neurol, 2013. [DOI | PubMed]
- A.J. Hughes, S.E. Daniel, L. Kilford, A.J. Lees. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry, 1992. [PubMed]
- I. Litvan, K.P. Bhatia, D.J. Burn, C.G. Goetz, A.E. Lang, I. McKeith. Movement disorders society scientific issues committee report: SIC task force appraisal of clinical diagnostic criteria for parkinsonian disorders. Mov Disord, 2003. [PubMed]
- I.G. McKeith, D.W. Dickson, J. Lowe, M. Emre, J.T. O’Brien, H. Feldman. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology, 2005. [PubMed]
- F. Sironi, P. Primignani, M. Zini, S. Tunesi, C. Ruffmann, S. Ricca. Parkin analysis in early onset Parkinson’s disease. Park Relat Disord, 2008
- F. Sironi, P. Primignani, S. Ricca, S. Tunesi, M. Zini, S. Tesei. DJ.1 analysis in a large cohort of Italian early onset Parkinson disease. Neurosci Lett, 2013. [DOI | PubMed]
- R. Cilia, C. Siri, D. Rusconi, R. Allegra, A. Ghiglietti, G. Sacilotto. LRRK2 mutations in Parkinson’s disease: confirmation of a gender effect in the Italian population. Park Relat Disord, 2014
- S. Paciotti, E. Persichetti, S. Pagliardini, M. Deganuto, C. Rosano, C. Balducci. First pilot newborn screening for four lysosomal storage diseases in an Italian region: identification and analysis of a putative causative mutation in the GBA gene. Clin Chim Acta, 2012. [PubMed]
- K. Nishioka, O.A. Ross, C. Vilariño-Güell, S.A. Cobb, J.M. Kachergus, D.M. Mann. Glucocerebrosidase mutations in diffuse Lewy body disease. Park Relat Disord, 2011
- J. Bras, C. Paisan-Ruiz, R. Guerreiro, M.H. Ribeiro, A. Morgadinho, C. Januario. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Portugal. Neurobiol Aging, 2009. [PubMed]
- S. Lesage, M. Anheim, C. Condroyer, P. Pollak, F. Durif, C. Dupuits. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum Mol Genet, 2011. [PubMed]
- M. Siebert, H. Bock, K. Michelin-Tirelli, J.C. Coelho, R. Giugliani, M.L. Saraiva-Pereira. Novel mutations in the glucocerebrosidase gene of brazilian patients with Gaucher disease. JIMD Rep, 2013. [PubMed]
- E.V. De Marco, G. Annesi, P. Tarantino, F.E. Rocca, G. Provenzano, D. Civitelli. Glucocerebrosidase gene mutations are associated with Parkinson’s disease in southern Italy. Mov Disord, 2008. [PubMed]
- M. Filocamo, R. Mazzotti, M. Stroppiano, M. Seri, F. Giona, G. Parenti. Analysis of the glucocerebrosidase gene and mutation profile in 144 Italian gaucher patients. Hum Mutat, 2002
- Z. Gan-Or, N. Giladi, U. Rozovski, C. Shifrin, S. Rosner, T. Gurevich. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology, 2008. [PubMed]
- M.J. Barrett, P. Giraldo, J.L. Capablo, P. Alfonso, P. Irun, B. Garcia-Rodriguez. Greater risk of parkinsonism associated with non-N370S GBA1 mutations. J Inherit Metab Dis, 2013. [PubMed]
- C. Dionisi-Vici, C. Rizzo, A.B. Burlina, U. Caruso, G. Sabetta, G. Uziel. Inborn errors of metabolism in the Italian pediatric population: a national retrospective survey. J Pediatr, 2002 Mar. [PubMed]
- J.R. Mazzulli, Y.H. Xu, Y. Sun, A.L. Knight, P.J. McLean, G.A. Caldwell. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell, 2011. [PubMed]
- L. Fellner, N. Stefanova. The role of glia in α-synucleinopathies. Mol Neurobiol, 2013. [PubMed]
- M.G. Spillantini, M. Goedert. Tau pathology and neurodegeneration. Lancet Neurol, 2013. [PubMed]
- D.J. Irwin, V.M. Lee, J.Q. Trojanowski. Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci, 2013. [PubMed]