
Computational drug repositioning approach to predict multi-target therapeutics for epilepsy
Abstract
Supplementary Information:
The online version contains supplementary material available at 10.1038/s41598-025-27625-2.
Article type: Research Article
Keywords: AED, Drug repurposing/Repositioning, Epilepsy, Molecular docking, Molecular dynamic simulations, Seizures, Computational biology and bioinformatics, Computational neuroscience, Virtual drug screening
Affiliations: https://ror.org/03kaab451grid.411524.70000 0004 1790 2262Toxicology and Computational Biology Group, Centre for Bioinformatics, Maharshi Dayanand University, Rohtak, Haryana 124001 India; https://ror.org/0261g6j35grid.7151.20000 0001 0170 2635Department of Bioinformatics & Computational Biology, College of Biotechnology, CCS Haryana Agricultural University, Hisar, Haryana 125004 India
License: © The Author(s) 2025 CC BY 4.0 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Article links: DOI: 10.1038/s41598-025-27625-2 | PubMed: 41402335 | PMC: PMC12708636
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (3.6 MB)
Introduction
Epilepsy is one of the most prevalent neurological disorders globally, affecting millions of individuals of all ages1,2. It encompasses a spectrum of neurological disorders characterised by abnormal electrical activity in the brain, leading to seizures. These seizures can manifest in various forms, ranging from momentary lapses of consciousness to convulsions. The underlying causes of epilepsy are diverse, including genetic predispositions, brain injuries, infections, and developmental abnormalities3,4. Epilepsy poses significant challenges to patients’ quality of life, societal integration, and healthcare systems worldwide. Numerous anti-epileptic drugs (AEDs) are available, but a substantial proportion of epilepsy patients experience inadequate seizure control or intolerable side effects with standard medications5.
Furthermore, the development of novel AEDs requires considerable time, resources, and navigates regulatory obstacles, encompassing comprehensive clinical trials, thorough safety assessments, and intricate approval procedures, which can considerably postpone their accessibility to patients, rendering it a formidable challenge. Consequently, alternative approaches, such as drug repositioning, which entails identifying new therapeutic applications for existing medications, become essential. This approach has the potential to accelerate the discovery and endorsement of efficacious treatments by circumventing numerous preliminary development phases and utilising established safety profiles.
Drug repositioning, also known as drug repurposing or reprofiling, involves identifying new therapeutic applications for existing drugs beyond their initially intended indications6. Unlike traditional drug discovery approaches, which often start from scratch, drug repositioning leverages existing pharmacological data, clinical experience, and safety profiles of approved drugs. This strategy offers several advantages, including reduced development costs, shorter timelines, and an increased likelihood of success in clinical trials7,8. Recent years have witnessed a growing interest in exploring drug repositioning strategies for epilepsy treatment, resulting in systematic screening of approved drug libraries and investigational compounds to identify candidates with anti-epileptic properties. These efforts have yielded promising findings, as the repurposed drug Lorcaserin9 demonstrates its efficacy in preclinical models and is currently under early-stage clinical trials for epilepsy.
The present study employed a virtual high-throughput screening (VHTS)-based, multi-target drug repositioning strategy to identify drug compounds with anti-epileptic activity. The DrugBank database was selected to screen approved drug compounds based on blood-brain barrier (BBB) permeability, structural similarity, molecular docking and binding site analysis. Drug(s) showing better binding affinity were evaluated for binding stability and free binding energy using molecular dynamics simulation (MDS) for 100 ns.
Materials and methodology
Retrieval of structural and physicochemical data of drug molecules
The DrugBank database is one of the most comprehensive structural databases, containing chemical, physicochemical, pharmacokinetic, pharmacodynamic, target, and metabolomic information on marketed drugs, including approved small-molecule drugs, biotech drugs, investigational drugs, and withdrawn drugs10. Due to safety concerns, experimental and withdrawn drug compounds were not considered, while approved drugs, including illicit and nutraceutical drugs, were selected for further in-silico screening. The 3D molecular files for all drug compounds were retrieved from the DrugBank database’s File Transfer Protocol (FTP) server as a single Structure Data File (SDF).
Screening of drugs for BBB permeability
The Central Nervous System (CNS) acting drug compounds must cross the blood-brain barrier (BBB) to reach the neural system for pharmacological activity. In-vivo or in-vitro BBB permeability prediction is a complex, time- and cost-intensive process. So, as an alternative, Machine Learning (ML) based in-silico BBB permeability prediction is a time-efficient and economical process with superior accuracy. Different BBB permeability prediction tools are available with great accuracy, but to gain maximum positive results, we used three different tools: (a) an in-house developed tool, BBBper (Blood Brain Barrier permeability prediction tool)11, AdmetSAR12 and LightBBB13 to screen the selected approved drug compounds for BBB permeability.
Clustering of structurally similar drugs
Most studied phenomena of drug repositioning focus on the similarity among drugs’ structural fingerprints, stating that “similar structures will have similar activity”8,14,15. So, all selected BBB-permeable drugs were further filtered for their structural similarity with available marketed AEDs (Table 1)16. The ChemMine tool17, an online molecular data analysis program supported by the R library ChemMineR18, was used to cluster selected approved and BBB-permeable drugs using binning clustering applications. The binning clustering method is used to partition a dataset into clusters by quantising the data points into bins and further assigning each bin to a cluster. Different drug clusters were generated using different similarity cutoff values ranging from 0.4 to 0.9. A lower cutoff value results in a lower similarity between compounds, leading to the clustering of less similar compounds. In contrast, a higher similarity cutoff results in the clustering of more similar compounds, leading to smaller cluster sizes. Finally, the similarity cutoff, which grouped most marketed AEDs within a single cluster, was selected for grouping drug compounds with similar structures to marketed AEDs. All compounds within the selected AEDs cluster were chosen for subsequent molecular docking analysis, based on the hypothesis that their structural conformations were analogous to those of established AEDs and, therefore, may elicit comparable therapeutic effects.
Table 1: List of marketed anti-epileptic drugs.
| Drug Name | DrugBank ID | Mechanism of action (from DrugBank) |
|---|---|---|
| Acetazolamide | DB00819 | Carbonic anhydrase inhibitor. |
| Brivaracetam | DB05541 | Synaptic vesicle glycoprotein 2 A (SV2A) agonist.VGSC alpha 1B inhibitor. |
| Cannabidiol | DB09061 | Weak partial agonist activity at Cannabinoid receptors CB1R and CB2R.Inhibits noradrenaline, dopamine, serotonin and GABA uptake19.Block T-type (low voltage-activated) Ca channel19.Antagonise mu-opioid receptor. |
| Carbamazepine | DB00564 | Inhibits VGSC alpha subuni20.Decrease dopamine turnover (dopamine antagonist) by reducing dopamine (D2) receptor density and phosphorylation.Enhance GABA synthesis.Inhibits Serotonin uptake.Decrease Norepinephrine release. |
| Clobazam | DB00349 | GABA-A receptor partial agonist (alpha and gamma 2 subunits). |
| Clonazepam | DB01068 | GABA-A receptor agonist21. |
| Diazepam | DB00829 | GABA-A receptor agonist. |
| Dronabinol | DB00470 | Cannabinoid receptors 1 and 2 agonists. |
| Eslicarbazepine acetate | DB09119 | Inhibits VGSC22.Inhibits T-type calcium channel. |
| Ethosuximide | DB00593 | Inhibits T-type VGCC alpha 1G23. |
| Ethotoin | DB00754 | Inhibits VGSC alpha 5. |
| Ezogabine | DB04953 | VGPC (Kv7.2-7.5) KCNQ2,3,4,5) agonist. |
| Felbamate | DB00949 | Antagonise Glutamate receptor (NMDA 2 A,3 A, 2B). |
| FosPhenytoin | DB01320 | Inhibits VGSC alpha 5. |
| Gabapentin | DB00996 | Structural analogue of GABA24.Inhibits VGCC subunit alpha 2, delta 1,2.Activates VGPC subfamily KQT member 3,5. |
| Lacosamide | DB06218 | Inhibits VGSC alpha 3,9,1022,25. |
| Lamotrigine (phenyl triazine) | DB00555 | Inhibits VGSC23.Inhibits adenosine A1/A2 receptors.Inhibits the K-opioid receptor. |
| Levetiracetam | DB01202 | Agonist for Synaptic vesicle glycoprotein 2 A (SV2A)26.Inhibits VGSC alpha 1B27,28. |
| Lorazepam | DB00186 | GABA-alpha receptor agonist. |
| Methsuximide | DB05246 | Inhibits VGCC T-type subunit alpha 1G. |
| Methylphenobarbital | DB00849 | GABA-alpha receptor agonist.Glutamate receptor antagonist. |
| Midazolam | DB00683 | GABA-alpha receptor agonist. |
| Nitrazepam | DB01595 | GABA R agonist.Inhibits Voltage-dependent sodium channels. |
| Oxcarbazepine | DB00776 | Inhibits VGSC20,22. |
| Perampanel | DB08883 | Glutamate receptor 1 antagonist29. |
| Phenacemide | DB01121 | Inhibits the Sodium channel protein type 1 subunit alpha |
| Phenobarbital | DB01174 | GABA R agonist.Glutamate receptor antagonist.Inhibits Calcium channels.NMDA channel antagonist. |
| Phenytoin | DB00252 | VGSC blocker alpha 1, 3 and 5 subunits. |
| Pregabalin | DB00230 | Inhibits VGCC subunit alpha 2/ delta 130,31. |
| Primidone | DB00794 | GABA alpha receptor agonist.Glutamate receptor antagonist. |
| Rufinamide | DB06201 | Inhibits VGSC32. |
| Stiripentol | DB09118 | GABA alpha receptor agonist. |
| Tiagabine | DB00906 | Inhibits GABA transferase. |
| Topiramate | DB00273 | Inhibits VGSC type 1 alpha subunit29.GABA alpha 1 receptor agonist. |
| Trimethadione | DB00347 | Inhibits Voltage-dependent T-type calcium channel subunit alpha-1G |
| Valproic acid | DB00313 | Inhibits succinic semialdehyde dehydrogenase (SSADH)33.Inhibits VGSC34,35.Inhibits GABA transferase33.Inhibits Histone deacetylase 2 and 9. |
| Vigabatrin | DB01080 | GABA analogue36.Irreversible inhibitor of 4-aminobutyrate transaminase.GABA beta receptor agonist36. |
| Zonisamide | DB00909 | Inhibits VGSC alpha 1,2,3,4,5,9,11 subunits37.Inhibits VGSC beta 1,2,3,4 subunits37.Inhibits VGCC T-type subunit alpha 1G, 1 H, 1I.Inhibits Carbonic anhydrase 1,2,3,4,5 A,5B,6,7,10,11,1213,14. |
Selection and preparation of the tertiary structure of epilepsy target receptors
Previously, we identified three first-line epilepsy therapeutic targets: the Voltage-Gated Sodium Channel (VGSC) α2 (Nav1.2), the Gamma-Aminobutyric Acid (GABA) receptor α1-β1, and the Voltage-Gated Calcium Channel (VGCC) α1G (Cav3.1)16. The tertiary protein structures of the Nav1.2 receptor, the GABA receptor α1 subunit, and the Cav3.1 channel were available in the RCSB-PDB database, with PDB IDs 6J8E38, 6HUJ39, and 6KZP40, respectively. However, the tertiary structure of the GABA receptor β1 was unavailable in the RCSB-PDB database; therefore, its 3D structure was generated by homology modelling using the SWISS-MODEL web server41. The GABA receptor β1 fasta sequence was retrieved from UniProt (UniProt ID: P18505) and subjected to homology modelling using the Swiss Model web server, with 6HUJ-B as the template. The modelled structure was further validated using the QMEAN score42, Ramachandran plot43, Verify3D44, and ProSA Z-score45. To form the functional GABA receptor α1-β1 complex, the available GABA receptor α1 chain and modelled GABA receptor β1 chain were subjected to protein-protein docking using Hex tool v8.0.046. A total of 25 searches were performed, using “shape + electro + DARS” as the correlation types and the 3D FFT mode. A side-by-side (parallel) conformation, showing head-to-head and tail-to-tail orientations for both chains, was selected, and the result was saved as a combined PDB file. The conformation of the generated GABA receptor α1-β1 file was also validated by aligning the generated structure against the GABA receptor α1-β3 (PDB ID: 6HUJ) using open-source PyMOL v2.5.047.
All the selected epilepsy target receptor proteins (Nav1.2, GABA receptor α1-β1, and Cav3.1) were subjected to energy minimisation using UCSF Chimera v1.648 for 100 steepest descent and 10 conjugate gradient steps under AMBER ff99bsc force field with a step size of 0.02Å. The energy-minimised structures were saved as PDB files for further molecular docking studies.
Molecular Docking study
The selected epilepsy target receptor proteins (Nav1.2, GABA receptor α1-β1, and Cav3.1) were prepared for molecular docking by adding polar hydrogen and assigning Kollman and Gasteiger charges using Autodock tools. Autodock v4.2.649 was used for molecular docking of selected drug compounds against specific therapeutic target proteins. The target proteins Nav1.2 and Cav3.1 are channel proteins, and to block the channel, the Grid file parameters (GPF) were assigned around the pore region, while the grid parameters for GABA receptor α1-β1 were assigned between the α1 and β1 chains within the GABA binding area to find a GABA agonist50 (Table 2). Molecular docking was performed for 100 independent runs using the Lamarckian genetic algorithm with a population size of 150, taking a gene mutation rate of 0.02 and a crossover rate of 0.8. The dock conformation with the lowest binding energy and maximum cluster size was selected for each drug. The highest-selling AEDs, Carbamazepine, Clonazepam, and Pregabalin, were chosen as standard drugs for targeting the proteins Nav1.2, GABA receptor α1-β1, and Cav3.1, respectively16. The drugs showing better binding affinities than the corresponding standard AEDs against all three receptors were selected for further study.
Table 2: Grid box parameters for molecular Docking study of voltage-gated sodium channel 2 A (Nav1.2), GABA receptor α1-β, and voltage-gated calcium channel α1G (Cav3.1).
| Nav1.2 | GABA Receptor α1-β1 | Cav3.1 | |
|---|---|---|---|
| Size-X | 54 | 56 | 64 |
| Size-Y | 66 | 76 | 76 |
| Size-Z | 94 | 76 | 90 |
| Center-X | 129.988 | 119.412 | 176.584 |
| Center-Y | 132.695 | 134.518 | 168.642 |
| Center-Z | 135.591 | 159.123 | 192.98 |
| Grid Box |
Binding pocket analysis of selected drugs
The residues within the binding pockets are significant factors in a drug’s pharmacodynamic effect. Hence, the binding pockets of the selected drugs were compared with those of the standard drugs, hypothesising that drugs with binding pockets similar to those of the standard AEDs would produce similar therapeutic effects. Therefore, the dock complexes of standard AEDs and selected drugs against the epilepsy target proteins were generated using the Autodock tool, and the binding pockets (nearby amino acids and hydrogen bond-forming amino acids) for each dock complex were analysed using the Java-based tool LigPlot+51. Drugs showing similar binding pockets to the standards and better binding affinities than corresponding standard drugs would have similar but better therapeutic effects than the existing standard/marketed drugs, and were hence selected for further repositioning study.
Literature data mining
Selected drugs with better binding affinities and similar binding pockets to their corresponding standard drugs were analysed for previous reports regarding epilepsy/seizure or any other severe side effects related to suicidal thinking, abnormal heartbeats, etc. Drugs showing any seizure-persuading or severe side effects cannot serve as a potential repositioned drug candidate. Therefore, drugs with no report or any prior study on seizure reduction with acceptable side effects were selected for further repositioning study.
Molecular dynamics simulation study
Selected repositioned drugs with better binding energies than corresponding standard drugs against selected epilepsy target receptors were subjected to Molecular Dynamics Simulation (MDS) using GROMACS v2022.152. The charmm36m force field53 was used to generate topology files for the docked complex of the selected drugs and the epilepsy receptor(s). The entire system was solvated with TIP3P water molecules in a rectangular box, followed by the addition of sodium (Na+) and chloride (Cl-) ions to neutralise the system at a concentration of 0.15 M, to mimic physiological conditions. To relieve the system’s geometric strain, a maximum of 5,000 energy minimisation steps were performed using the steepest descent algorithm to lower the potential energy up to 1,000 KJ/mol. Then, the whole system was equilibrated under constant temperature (310 K) and pressure (1 bar) conditions for 1000 ps (1 ns) using the Nose-Hoover thermostat and Parrinello-Rahman barostat, respectively. After equilibration, MDS was performed for 100 ns (500,000,000 steps) in triplicate with a time step of 2 fs at constant temperature and pressure using Periodic Boundary Conditions (PBC). Trajectories were recorded every 100 ps. Protein-drug interactions throughout the whole simulation were monitored for stability examination.
The obtained trajectory of the simulated protein-ligand complexes was analysed based on Root Mean Square Deviation (RMSD), Root Mean Square Fluctuations (RMSF), Radius of gyration (Rg), Solvent Accessible Surface Area (SASA), Principal Component Analysis (PCA), interaction energy including Lennard-Jones potential and Coulombic interactions, and Hydrogen bond analysis. Further MMPBSA was performed using gmx_MMPBSA54 to calculate the free binding energy between the protein and drug for the entire MDS.
Results and discussion
Retrieval of approved drugs from the drugbank database
The DrugBank database v5.1.12 (accessed January 1, 2025) contained structural and physicochemical data for 12,699 drugs, including approved, illicit, nutraceuticals, investigational, experimental, veterinary, and withdrawn drugs (Supplementary File: S1). For safety purposes, only approved drugs (2,769) were considered for our drug repositioning study (Fig. 1; Supplementary File: S2; Table 3). Among these, 188 drugs were found to be withdrawn from the market after their initial approval. Hence, the remaining 2,581 approved drugs were selected for further drug repositioning screening as multitarget anti-epileptic therapeutics. The structural (SDF) and physicochemical data for the selected 2581 drugs were retrieved using the FTP service of the DrugBank database. The selected approved drugs also included 38 marketed AEDs (Table 1), which were used for structural similarity analyses.
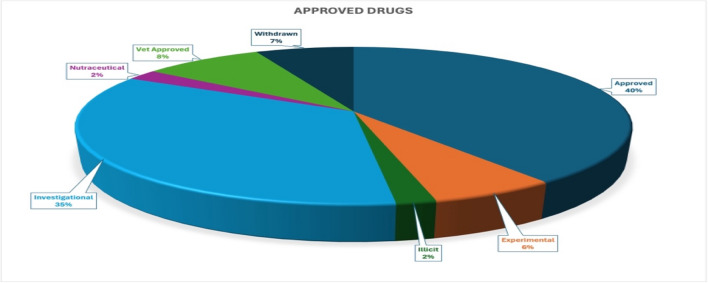
Table 3: Classification of approved drugs from the drugbank database.
| Drug groups | Number |
|---|---|
| Approved | 1099 |
| Approved; Experimental | 152 |
| Approved; Experimental; Investigational | 22 |
| Approved; Experimental; Investigational; Withdrawn | 1 |
| Approved; Experimental; Vet Approved | 2 |
| Approved; Experimental; Withdrawn | 1 |
| Approved; Illicit | 39 |
| Approved; Illicit; Investigational | 12 |
| Approved; Illicit; Investigational; Vet Approved | 4 |
| Approved; Illicit; Investigational; Withdrawn | 5 |
| Approved; Illicit; Vet Approved | 1 |
| Approved; Illicit; Withdrawn | 5 |
| Approved; Investigational | 963 |
| Approved; Investigational; Nutraceutical | 24 |
| Approved; Investigational; Nutraceutical; Vet Approved | 3 |
| Approved; Investigational; Nutraceutical; Withdrawn | 1 |
| Approved; Investigational; Vet Approved | 77 |
| Approved; Investigational; Vet Approved; Withdrawn | 8 |
| Approved; Investigational; Withdrawn | 57 |
| Approved; Nutraceutical | 36 |
| Approved; Nutraceutical; Vet Approved | 10 |
| Approved; Nutraceutical; Withdrawn | 1 |
| Approved; Vet Approved | 137 |
| Approved; Vet Approved; Withdrawn | 15 |
| Approved; Withdrawn | 94 |
| Total | 2769 |
Screening of drugs for BBB permeability
AEDs must cross the BBB to perform their action within the CNS. Three different ML algorithm-based BBB permeability prediction programs/tools were used for better accuracy. Our in-house developed tool, BBBper11, predicted 1553 BBB-permeable drugs, while admetSAR12 predicted the maximum number of 1575 drugs as BBB-permeable, and LightBBB13 predicted 1393 drugs to cross the BBB (Supplementary File: S3). All three selected programs/tools predicted 895 drugs as BBB-permeable (Fig. 2). BBBper predicted all 38 AEDs as BBB permeable, while admetSAR and LightBBB showed 37 AEDs as BBB permeable, and FosPhenytoin and Eslicarbazepine as BBB impermeable, respectively. Hence, to mitigate adverse selection, drug compounds predicted to be BBB-permeable by at least two programs/tools were selected for further study. The selection criteria resulted in 1606 drug compounds (including 38 marketed AEDs) as BBB-permeable.
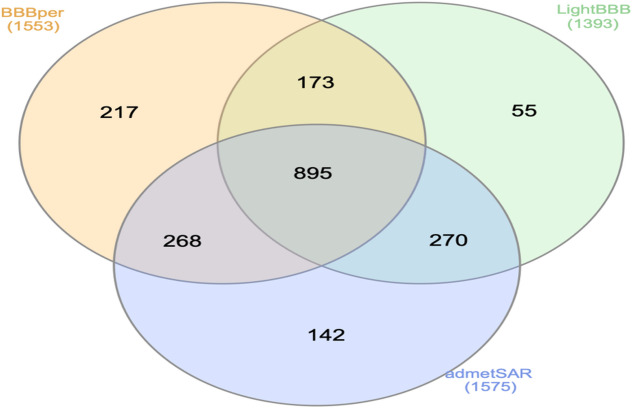
Clustering of structurally similar drugs
To identify structurally similar drugs, the selected BBB-permeable drugs were clustered based on structural similarity using the ChemMine tool17. Different similarity cutoff values, specifically 0.4, 0.5, 0.6, 0.7, 0.8, and 0.9, resulted in the formation of 645, 927, 1128, 1296, 1449, and 1492 bins, respectively (Supplementary File: S4). At a lower cutoff value, such as 0.4, the clustering produces fewer, larger bin counts, indicating broader grouping criteria; some bins include hundreds of compounds (bin-2: 360; bin-12: 185), encompassing most AEDs (bin-2: 18 AEDs). As the cutoff increases to 0.5 and 0.6, the number of bins rises, and their sizes decrease (bin-12: 133; bin-38: 92), reflecting more stringent similarity thresholds that lead to more refined subdivisions of the drug dataset. Beyond a cutoff of 0.6, minimal changes in bin sizes suggest a plateau in clustering resolution, indicating that further increasing the cutoff may not significantly improve cluster differentiation (Fig. 3).
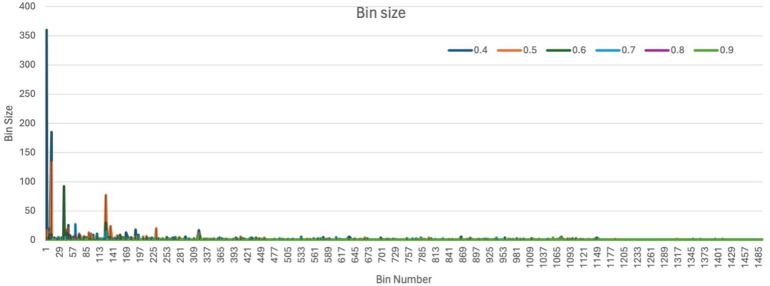
Bin-2, at the cutoff value of 0.4, was observed to have the highest number (18) of AEDs, including Carbamazepine, Clobazam, Clonazepam, Diazepam, Eslicarbazepine, Ethotoin, FosPhenytoin, Lorazepam, Methylphenobarbital, Midazolam, Methsuximide, Nitrazepam, Oxcarbazepine, Perampanel, Phenacemide, Phenobarbital, Phenytoin, and Primidone. Therefore, bin-2, with a cutoff value of 0.4, was chosen for further study because it combines a high drug count with a structural similarity of at least 40%, aligning well with marketed AED profiles. Along with bin-2, other bins at the same cutoff (0.4) contain only a single AED, highlighting a high degree of structural specificity. For example, bins like Bin-65 (Pregabalin), Bin-96 (Topiramate), Bin-120 (Valproate), Bin-286 (Ethosuximide), Bin-475 (Tiagabine), Bin-477 (Zonisamide), Bin-501 (Felbamate), Bin-531 (Gabapentin), Bin-578 (Vigabatrin), Bin-824 (Ezogabine), Bin-855 (Rufinamide), Bin-863 (Lacosamide) and Bin-1029 (Stiripentol), each contain only one drug, representing the unique structure of these 13 marketed AEDs within the dataset. Conversely, some bins, such as Bin-213 and Bin-649, contain multiple drugs, including a pair of AEDs (Bin-213: Cannabidiol and Dronabinol, and Bin-649: Brivaracetam and Levetiracetam), along with one other structurally similar drug. Additionally, Bin-140 and Bin-348 feature two drugs each, one AED (Bin-140: Trimethadione; and Bin-348: Acetazolamide) and one structurally similar drug compound, illustrating potential structural relationships. In Bin-54, Lamotrigine was observed with three other structurally similar drug compounds. Overall, 349 drug compounds with over 40% structural similarity to marketed AEDs were identified across all these bins. All these compounds were considered for further repositioning studies.
Selection and Preparation of the tertiary structure of epilepsy target receptors
Three epilepsy drug targets, VGSC α2 (Nav1.2), GABA receptor α1-β1, and VGCC α1G (Cav3.1), were reportedly identified as first-line therapy targets for epilepsy16. The tertiary protein structures of the Nav1.2 receptor, the GABA receptor α1 subunit, and the Cav3.1 channel were available in the RCSB-PDB database with PDB IDs 6J8E-A, 6HUJ-A, and 6KZP, respectively. The tertiary structure of the GABA receptor β1 (UniProt ID: P18505) was not available in the RCSB-PDB database. Furthermore, a sequential similarity of 54% was observed between GABA receptor β1 and β3, with a query coverage of 78.28%, making it a suitable choice for homology modelling. Using the SwissModel web server, a homology model of the GABA receptor β1 was generated, with PDB ID 6HUJ-B of the GABA receptor β3 serving as a template. The modelled 3D structure was evaluated with a QMEAN score of -2.88, a Ramachandran Z-score of -2.818, and a pass verify-3D status. Ramachandran plot analysis revealed that 93.4% of the amino acids reside under the favoured region, and the remaining 6.6% reside in the allowed region. The overall validation criterion represented an exemplary structure of the predicted GABA receptor β1 model and was suitable for further studies.
The functional GABA receptor 3D structures are reportedly observed in parallel chains, i.e., tail-to-tail and head-to-head conformation39,55,56. Hence, to form a functional GABA receptor α1-β1 complex, the retrieved GABA receptor α1 subunit was docked against the modelled GABA receptor β1 using Hex tool 8.0.046. The functional conformation of GABA receptor α1-β1 with parallel binding (head-to-head and tail-to-tail) was selected for further study, with a binding energy of -1206.05 KJ/mol. The structural alignment of the selected GABA receptor α1-β1 docked complex against the reported tertiary structure of the GABA receptor α1-β3 (PDB ID: 6HUJ) yielded an RMSD of 0.099, indicating a high accuracy of the modelled structure. The selected structure of the GABA receptor α1-β1 was subjected to energy minimisation using UCSF Chimera for further docking studies.
Molecular docking study
The selected 349 drug molecules were energy-minimised and docked against the target proteins for epilepsy, including Nav1.2, GABA receptor α1-β1, and Cav3.1, using AutoDock v4.2.6. Top-selling AEDs, Carbamazepine, Clonazepam, and Pregabalin, were chosen as standard drugs for the studied target receptors – Nav1.2, GABA receptor α1-β1, and Cav3.1, respectively, for comparing the binding affinities of the docked drug complexes16,50. The minimum binding energies of standard medications, Carbamazepine, Clonazepam and Pregabalin, were observed to be -7.13 Kcal/mol, -6.14 Kcal/mol and − 5.76 Kcal/mol when docked against protein receptors Nav1.2, GABA receptor α1-β1, and Cav3.1, respectively.
A total of 136 drugs showed better binding affinities for the epilepsy receptor Nav1.2 compared to the standard Carbamazepine, while 72 drugs showed lower binding energies for GABA receptor α1-β1 when compared to GABA agonist Clonazepam. A total of 275 drugs, screened against Cav3.1, showed better binding affinities than the corresponding standard drug, Pregabalin (Supplementary File: S5). These 275 Cav3.1 binding drugs included all 136 Nav1.2 binding drugs and 56 out of 72 GABA receptor α1-β1 binding drugs. Overall, 46 drugs, in common, showed better binding affinities against their corresponding therapeutic target receptors (Fig. 4; Table 4). These 46 drug compounds were also predicted to be BBB permeable and showed structural similarity with already marketed AEDs; thus, they can be expected as potential candidates for AED repositioning.
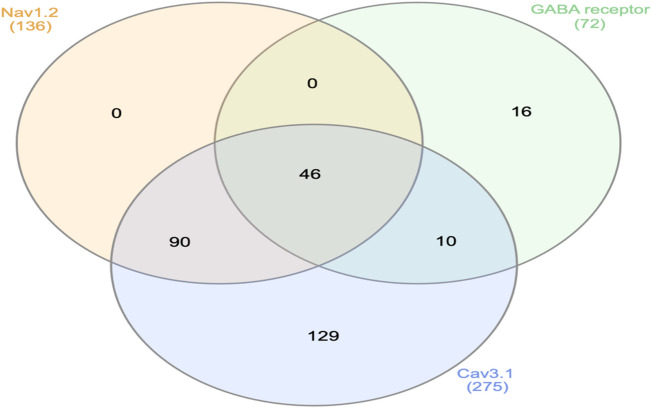
Table 4: Drugs having better binding energies (Kcal/mol) than standard drugs against selected epileptic receptors: Voltage-gated sodium channel α2 (Nav1.2), GABA receptor α1-β1, and Voltage-gated calcium channel α1G (Cav3.1).
| DrugBank ID | Name | Nav1.2 | GABA receptor α1-β1 | Cav3.1 |
|---|---|---|---|---|
| DB00564 | Carbamazepine | -7.13 | — | — |
| DB01068 | Clonazepam | — | -6.14 | — |
| DB00230 | Pregabalin | — | — | -5.76 |
| DB00192 | Indecainide | -7.84 | -6.45 | -9.73 |
| DB00321 | Amitriptyline | -7.94 | -6.53 | -9.56 |
| DB00340 | Metixene | -8.53 | -6.65 | -9.61 |
| DB00344 | Protriptyline | -7.68 | -6.38 | -8.99 |
| DB00427 | Triprolidine | -7.89 | -6.16 | -9.70 |
| DB00434 | Cyproheptadine | -8.23 | -6.63 | -8.46 |
| DB00486 | Nabilone | -8.60 | -6.50 | -8.28 |
| DB00496 | Darifenacin | -9.83 | -7.22 | -9.41 |
| DB00540 | Nortriptyline | -8.01 | -6.84 | -10.16 |
| DB00561 | Doxapram | -8.53 | -6.63 | -8.73 |
| DB00568 | Cinnarizine | -9.12 | -6.48 | -9.44 |
| DB00693 | Fluorescein | -7.70 | -6.41 | -8.01 |
| DB00719 | Azatadine | -7.81 | -6.47 | -8.58 |
| DB00850 | Perphenazine | -8.12 | -6.25 | -10.45 |
| DB00920 | Ketotifen | -7.76 | -6.67 | -8.70 |
| DB00924 | Cyclobenzaprine | -8.16 | -6.84 | -9.29 |
| DB00933 | Mesoridazine | -9.12 | -6.35 | -9.38 |
| DB00934 | Maprotiline | -8.15 | -7.06 | -9.97 |
| DB00972 | Azelastine | -9.12 | -6.62 | -10.26 |
| DB00991 | Oxaprozin | -7.61 | -6.60 | -8.11 |
| DB01009 | Ketoprofen | -7.38 | -7.26 | -7.26 |
| DB01100 | Pimozide | -9.11 | -6.80 | -10.22 |
| DB01142 | Doxepin | -7.69 | -6.37 | -9.46 |
| DB01146 | Diphenylpyraline | -8.07 | -6.32 | -8.96 |
| DB01148 | Flavoxate | -8.01 | -6.49 | -9.08 |
| DB01501 | Difenoxin | -8.70 | -7.33 | -9.46 |
| DB01544 | Flunitrazepam | -7.23 | -6.27 | -7.64 |
| DB01608 | Periciazine | -8.30 | -6.52 | -9.13 |
| DB01623 | Thiothixene | -8.83 | -6.25 | -11.47 |
| DB01624 | Zuclopenthixol | -8.17 | -6.33 | -11.30 |
| DB01628 | Etoricoxib | -7.92 | -6.63 | -7.68 |
| DB04841 | Flunarizine | -9.18 | -6.21 | -9.28 |
| DB05015 | Belinostat | -7.41 | -7.02 | -8.07 |
| DB06077 | Lumateperone | -8.84 | -7.08 | -8.73 |
| DB06153 | Pizotifen | -7.69 | -6.57 | -8.39 |
| DB06413 | Armodafinil | -7.19 | -6.23 | -6.96 |
| DB06626 | Axitinib | -8.51 | -6.40 | -9.38 |
| DB06684 | Vilazodone | -9.59 | -6.16 | -10.93 |
| DB09167 | Dosulepin | -7.96 | -6.37 | -9.42 |
| DB09488 | Acrivastine | -7.72 | -7.14 | -10.23 |
| DB11952 | Duvelisib | -9.38 | -6.34 | -8.41 |
| DB12492 | Piritramide | -9.50 | -7.08 | -11.46 |
| DB12877 | Oxatomide | -9.43 | -6.43 | -9.10 |
| DB13292 | Pimethixene | -8.36 | -6.96 | -8.24 |
| DB14033 | Acetyl sulfisoxazole | -7.58 | -6.52 | -6.50 |
| DB12792 | Boscalid | -7.37 | -6.14 | -7.72 |
Binding pocket analysis of selected drugs
The 46 drugs selected above were further evaluated for their binding interactions within the binding pockets of the target proteins. Most marketed AEDs working as VGSC inhibitors bind within the inner pore-loop (P-loop) region. The standard drug Carbamazepine forms a hydrogen bond (H-bond) with the Leu939 residue located within the P-loop region of Domain II. Amino acids Ser1461, Phe1462, and Ile1457 within the P-loop region of VGSC-Domain-III were reported to be essential for the binding of VGSC-inhibiting AEDs to generate an anti-epileptic activity57–59. 20 out of 46 selected drugs showed their binding within the binding pocket, including the P-loop region. Drugs Belinostat, Dosulepin, Flavoxate, Mesoridazine, and Oxatomide were observed to form H-bonds within the P-loop region of Domain-II. Drugs Amitriptyline, Azatadine, Azelastine, Cyproheptadine, Difenoxin, Maprotiline, Nortriptyline, Oxaprozin, and Piritramide were observed to form H-bonds within the P-loop region of Domain-III (Supplementary File: S6). Among them, Dosulepin and Flavoxate were observed to form H-bonds with Leu939, while drugs Azatadine, Azelastine, Cyproheptadine and Oxaprozin were forming H-bonds with the key amino acid Ser1461 within the P-loop region of VGSC Domain-III. Drugs Cinnarizine, Darifenacin, Flunarizine, Lumateperone, Pimozide, and Pizotifen did not show any bonded interaction but bound within the P-loop region through non-bonded interactions, while the remaining 26 drugs were not observed to bind within the binding pocket as standard drugs.
GABA and its agonist bind between the α1 and β1 chains of the GABA receptor within the aromatic box, hence creating a hydrophobic environment suitable for neurotransmitter binding. The aromatic region includes Tyrosine (β-Tyr97, β-Tyr157, β-Tyr205) and Phenylalanine (α-Phe64, α-Phe65, and β-Phe200) residues, along with Arginine (α-Arg66, α-Arg67, α-Arg131, α-Arg132, and β-Arg207), which are involved in Electrostatic interactions. Polar amino acids like Threonine (α-Thr129 and β-Thr202) and Serine (α-Ser168) form a Hydrogen bond, while Glutamic acid (β-Glu155 and β-Glu220) contributes to binding affinity by forming salt bridges60–62. The cryo-EM structures of Benzodiazepine interaction with the α1β1γ2S tri-heteromeric GABA receptor show ligand interactions with residues in the β-subunit’s Loop A/B/C (Tyr97, Glu155, Tyr157, Tyr205) and make other non-bonded interactions with nearby amino acids, such as Thr129, Thr202, and Arg6663. The selected standard drug, Clonazepam, exhibited a polar interaction with α1-Asp184, which is structurally consistent with the α-face contribution to the orthosteric pocket observed in the cryo-EM models. Along with this, several non-bonded interactions with β1-Leu124, β1-Asn125 (adjacent to β6 Thr129), β1-Met162 (neighbouring Tyr157 in Loop B), and β1-Glu178, β1-Arg187 (nearby Thr202 in Loop-C arc) were observed. These are pocket-lining positions flanking the canonical determinants and are well-placed to form auxiliary van der Waals or polar contacts. While these residues are not the classic β-face ‘signature’ triad, their proximity to Loop A–C and β2/β6 strands provides a structurally sound rationale for the observed interactions. Among selected drugs, 16 drugs (Acetyl sulfisoxazole, Acrivastine, Armodafinil, Axitinib, Azatadine, Cyclobenzaprine, Cyproheptadine, Etoricoxib, Flunitrazepam, Ketotifen, Maprotiline, Nortriptyline, Oxaprozin, Pizotifen, Protriptyline, and Vilazodone) showed their binding within the binding region of GABA receptor subunit α1 with the formation of an H-bond. Among these, Acrivastine, Doxepin, Duvelisib, Flunitrazepam, Oxaprozin, and Pizotifen showed H-bonding with Asp184 of the GABA receptor α1 subunit. Drugs Acetyl sulfisoxazole, Armodafinil, Cyclobenzaprine, Maprotiline, Nortriptyline, and Protriptyline were observed to form an H-bond with Asp63 of subunit α1. Drugs Azatadine, Belinostat, Cyproheptadine, Difenoxin, Ketotifen, Lumateperone, Nabilone, and Pizotifen were observed to form H-bonds with the amino acids Leu124, Asn125, Lys127, Met162, Asn174, and Thr176 from the GABA receptor subunit β1. In contrast, Pimethixene and Triprolidine did not show any H-bonding but were observed to bind within the binding pocket of GABA receptor α1-β1 (Supplementary File: S6).
The pore-forming intracellular region of S5 and S6 in VGCC has been reportedly to be necessary for the trafficking of Ca+ 2 ions64; hence, drug binding within this region would generate a therapeutic response for epileptic seizures. The standard drug, Pregabalin, showed three H-bonded interactions with amino acids Glu354 (2.72Å), Gln922 (2.77Å), and Glu923 (2.7Å) within the S6 helical region of repeats I and II. All the selected drugs were observed to bind within the binding pocket of Cav3.1, similar to the standard drug. However, the H-bond analysis, in comparison to the standard drug Pregabalin, revealed that 25 drugs share common amino acids. Among those, Amitriptyline, Dosulepin, Doxepin, Flavoxate, Maprotiline, Metixene, Nortriptyline, Pizotifen and Protriptyline showed H-bond with Glu354; Boscalid, Doxapram, Etoricoxib, Fluorescein, Ketotifen, Nabilone, Oxaprozin, Piritramide and Thiothixene showed H-bond with Gln922, while drugs Indecainide and Pimozide were observed to form H-bond with Glu923. Drugs Acrivastine, Cyproheptadine, and Ketotifen formed hydrogen bonds with Glu354 and Gln922. In comparison, Cyclobenzaprine and Triprolidine were observed to form H-bonds with Glu354 and Glu923 (Supplementary File: S6).
The binding pocket comparison of selected drugs revealed six drugs (Azatadine, Cyproheptadine, Maprotiline, Nortriptyline, Oxaprozin, and Pizotifen) with binding pockets similar to those of standard drugs for their respective target receptors. The binding pocket analysis of these drugs confirms their ability to mimic the pharmacodynamics of the standard drugs. These six drug hits were further evaluated for their role in seizure management, using exhaustive text mining.
Literature data mining
The selected marketed drugs (Azatadine, Cyproheptadine, Maprotiline, Nortriptyline, Oxaprozin, and Pizotifen) belong to different classes of drugs and were analysed for their previous reports in the context of epileptic seizures and other toxicity assays.
Azatadine
Azatadine is a first-generation antihistamine that primarily functions as an H1 receptor antagonist, demonstrating efficacy in treating allergic reactions such as rhinitis, conjunctivitis, and urticaria65. Its chemical structure is closely related to tricyclic compounds, contributing to its ability to cross the blood-brain barrier. As a result, Azatadine can cause significant CNS effects, such as drowsiness and sedation66. The sedative effects of Azatadine may lower the seizure threshold, potentially increasing the risk of seizures, especially in individuals with a history of epilepsy or other neurological conditions. Additionally, its anticholinergic effects, such as dry mouth, blurred vision, and urinary retention, may interfere with the normal neurotransmitter balance in the brain, thereby complicating seizure management and other neurological functions67. Azatadine is usually taken orally and may interact with other CNS depressants like alcohol, sedatives, or tranquillisers, enhancing its sedative and central effects. Considering these factors, particularly in patients with seizure disorders, Azatadine is generally contraindicated and has been excluded from our study outcomes to avoid confounding effects related to neurological safety.
Cyproheptadine
Cyproheptadine is also classified as a first-generation antihistamine with anticholinergic, antiserotonergic, and sedative properties. It was initially developed to treat allergies by blocking the H1 histamine receptors; furthermore, it is also widely used to stimulate appetite, especially in patients experiencing cachexia or malnutrition68,69. It is also prescribed for the treatment of serotonin syndrome, a potentially life-threatening condition caused by excessive serotonergic activity in the CNS. The serotonin receptors, particularly the 5-HT2A receptor, are implicated in seizure activity. In some cases of intractable epilepsy, where patients do not respond well to conventional anticonvulsants, Cyproheptadine has been used off-label to help control seizures by modulation of serotonergic pathways, which can influence neuronal excitability and seizure thresholds. Animal studies have also shown a reduction in seizure frequency and neural cell death following Cyproheptadine treatment70. Additionally, the drug’s antiserotonergic effects may influence other serotonergic-related conditions, including migraine prevention and specific psychiatric disorders. Its sedative properties are also utilised in managing symptoms of anxiety and sleep disturbances.
Maprotiline
Maprotiline is a tetracyclic antidepressant (TeCA) primarily used to treat depression, particularly major depressive disorder and dysthymia. It functions by inhibiting the reuptake of norepinephrine, a neurotransmitter, thereby increasing its levels in the brain and alleviating depressive symptoms71,72. Unlike tricyclic antidepressants (TCAs), which affect multiple neurotransmitters, Maprotiline is more selective for norepinephrine, which reduces some of the side effects typically associated with TCAs. Additionally, it has strong sedative properties, which makes it worthwhile for patients with anxiety or insomnia, but potentially problematic for those who are sensitive to sedatives71. Maprotiline has also been explored for its potential effects on epilepsy, but it is not a first-line treatment. The clinical studies suggest that Maprotiline can lower the seizure threshold; however, at higher doses or in patients with a predisposition to seizures, it behaves as a proconvulsant73,74. Therefore, Maprotiline use in epileptic patients is generally approached with caution, often requiring close monitoring and consideration of alternative therapies.
Nortriptyline
Nortriptyline is another TCA, primarily used to treat major depressive disorder; however, it is also prescribed for conditions such as chronic pain, neuropathic pain, and particular anxiety disorders75–77. It works by inhibiting the reuptake of neurotransmitters, particularly norepinephrine and, to a lesser extent, serotonin, in the brain, which enhances the mood-stabilising effects of these chemicals. This enhancement may serve to alleviate depressive symptoms and improve pain management. Furthermore, it demonstrates sedative properties, rendering it advantageous in cases involving anxiety and insomnia. The medication is available in a generic formulation and typically represents a cost-effective alternative to newer antidepressants, although it may be associated with a higher incidence of adverse effects. These adverse effects include dry mouth, blurred vision, dizziness, urinary retention, and constipation. Due to its anticholinergic properties, caution is recommended when prescribing to elderly patients to prevent adverse events such as confusion, falls, and cognitive decline. Nortriptyline is reported to lower the seizure threshold, which can increase the risk of seizures in susceptible individuals78,79. Consequently, its use should be carefully considered in patients with a history of seizures or predisposition thereto. Nonetheless, in certain circumstances, it may be appropriate where an epileptic patient suffers from comorbid conditions like depression or chronic pain, where the benefits of its mood-stabilising effects might outweigh the seizure risks. Hence, the use of nortriptyline in patients with epilepsy involves a careful risk-benefit analysis, considering the individual’s seizure history, the severity of comorbid conditions, and the potential for drug interactions.
Oxaprozin
Oxaprozin is a non-steroidal anti-inflammatory drug (NSAID) commonly used to treat inflammation or pain caused by osteoarthritis or rheumatoid arthritis. It inhibits the cyclooxygenase (COX) enzyme non-selectively, thereby reducing the synthesis of prostaglandins and lipid compounds; consequently, inflammation, pain and fever are reduced80. Oxaprozin is distinguished by its prolonged half-life, which allows for once-daily administration, thereby enhancing patient compliance and adherence. It is generally well-tolerated; however, potential adverse effects include gastrointestinal discomfort, cardiovascular complications, and alterations in renal function, particularly with prolonged usage. Although it is widely utilised for inflammatory conditions. However, Oxaprozin has not been widely studied or recommended for use in individuals with epilepsy. Public databases and clinical trials have not documented seizure-related side effects associated with its administration. Notably, recent research indicates a potential new therapeutic application for Oxaprozin. Specifically, an in vivo study involving seizure models in rats demonstrated that Oxaprozin may possess anticonvulsive properties during the behavioural assessment81. This finding paves the way for further investigation into its potential use as an adjunctive treatment for epilepsy or seizure disorders, representing a promising frontier for future pharmacological innovation.
Pizotifen
Pizotifen is a tricyclic drug primarily used as a preventive treatment for migraine headaches and cluster headaches. It functions as a serotonin receptor antagonist, explicitly targeting the 5-HT2A and 5-HT2C receptors, and exhibits antihistamine and anticholinergic properties82,83. Inhibiting serotonin, a neurotransmitter believed to play a role in the dilation of blood vessels in the brain, results in reduced frequency and severity of migraines. Pizotifen is often prescribed when other treatments, such as beta-blockers or anti-epileptic drugs, are either ineffective or unsuitable for the patient. Its role in epilepsy is less prominent and more complex, as it has been observed to exhibit some anticonvulsant properties in adult zebrafish by modulating serotonin levels within the brain84. However, its efficacy in epilepsy is not well-established, and it is generally not considered a standard anti-epileptic drug. The use of Pizotifen in epilepsy might be explored in patients with comorbid conditions, such as migraines, where the dual action of preventing headaches and potentially reducing seizures could be beneficial. Nonetheless, more research is required to better understand its exact mechanism and effectiveness in managing epilepsy.
Based on their pharmacological profiles and existing evidence, Cyproheptadine, Oxaprozin, and Pizotifen are promising candidates for epilepsy repurposing, with Cyproheptadine’s antiserotonergic effects and prior off-label success in seizure management emphasising its potential. Oxaprozin has demonstrated anticonvulsant activity in animal models, warranting further investigation, while Pizotifen’s antagonism of serotonin receptors suggests therapeutic benefits, particularly for patients experiencing migraines and seizures. Conversely, Azatadine, Maprotiline, and Nortriptyline pose safety concerns, primarily due to their propensity to lower seizure thresholds and induce CNS complications, which leads to their exclusion from future studies. Therefore, we propose Oxaprozin, Pizotifen, and the antihistamine Cyproheptadine as potential multitarget AEDs for various types of epileptic seizures, regardless of their origin—whether genetic, congenital, due to physical injury, or environmental.
Molecular dynamics simulation study
The docked complex of selected potential multi-targeted AED candidates—Cyproheptadine, Oxaprozin, and pizotifen—with the epilepsy target receptors Nav1.2, GABA receptor α1-β1, and Cav3.1—was subjected to molecular dynamics studies to evaluate their stability in physiological conditions. All the receptor-drug complexes showed a minimised energy configuration within a 3000 ps run, followed by equilibration at an average temperature and pressure of 310 K and one psi, respectively. The docked complexes of the selected drugs were finally simulated for a 100-ns runtime in triplicate.
Root mean square deviation (RMSD)
The RMSD analysis measures the structural stability (average distance between the atoms of superimposed molecules) and conformational shifts of protein-ligand complexes throughout MDS. For all simulated complexes, the RMSD graph appeared to stabilise within the initial 10-ns run and remained stable until the end of the simulation (Fig. 5).
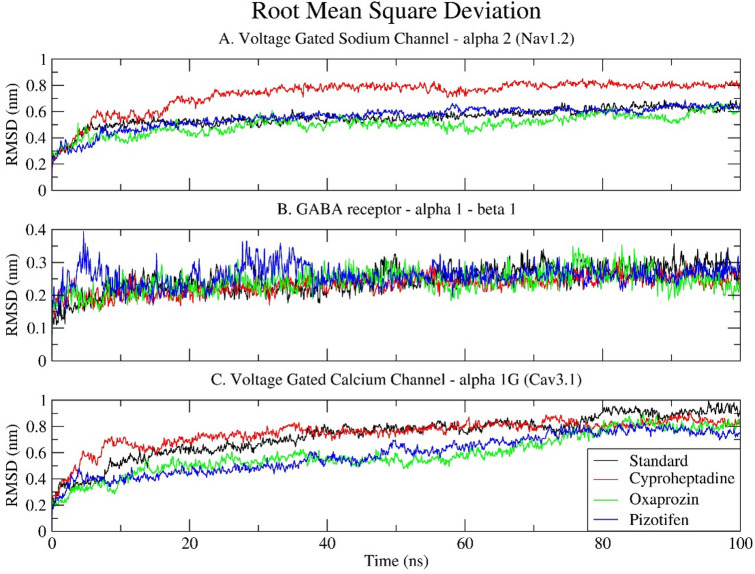
Regarding the Nav1.2 sodium channel, all ligand-bound complexes demonstrated an upward trend in RMSD during the initial 20–30 ns, indicative of structural adjustments and system equilibration. The standard drug, Carbamazepine, forms a comparatively stable complex, with RMSD values stabilising around 0.5 to 0.6 nm following an initial equilibration phase. Pizotifen and Oxaprozin exhibit marginally lower deviations, stabilising within the range of 0.45 to 0.55 nm, thereby suggesting an enhancement in structural stability relative to the reference. Conversely, Cyproheptadine results in significantly higher deviations, with RMSD values gradually rising to approximately 0.7 to 0.8 nm. This implies that Cyproheptadine may induce destabilisation of Nav1.2 or facilitate increased conformational flexibility, potentially attributable to suboptimal binding interactions or alterations in key structural regions. Despite the RMSD reaching a plateau, the consistently elevated values associated with Cyproheptadine suggest a less stable complex formation compared to the other ligands (Fig. 5A).
The GABA receptor α1-β1 exhibits notably stable behaviour across all ligand-bound forms. Throughout the 100 ns simulation, the RMSD values for the four ligand systems remain consistently low, typically ranging from 0.15 to 0.3 nm. This minimal variation indicates a high degree of structural rigidity, suggesting that the GABA receptor maintains its native conformation irrespective of the ligand bound. The RMSD trajectories of the standard drugs, Oxaprozin, Cyproheptadine, and Pizotifen, nearly coincide, with only minor and transient deviations, particularly with Pizotifen. This observation implies that the binding of any of these compounds does not substantially modify the receptor’s backbone architecture, thereby underscoring the receptor’s intrinsic stability and robustness (Fig. 5B). The lower RMSD in this case may also be attributed to the smaller size of the receptor complex and the limited flexibility of its core structural regions.
Regarding Cav3.1, the RMSD patterns bear a close resemblance to those observed in Nav1.2. The standard drug, Pregabalin, maintains the receptor stability within a range of 0.65 to 0.75 nm. Pizotifen and Oxaprozin demonstrate marginally lower deviations, sustaining RMSD values of approximately 0.55–0.65 nm, thereby implying that they facilitate stable receptor-ligand interactions. Cyproheptadine exhibits the most considerable deviation, approaching 0.85 nm, which suggests it may induce greater conformational alterations or flexibility within the calcium channel. Although all examined systems appear to attain equilibrium after roughly 40 nanoseconds, the elevated RMSD associated with Cyproheptadine indicates a comparatively less stable interaction, potentially affecting the channel’s structural integrity or functionality (Fig. 5C).
The relatively high RMSD values observed in the Nav1.2 and Cav3.1 complexes, particularly with Cyproheptadine, do not necessarily indicate instability. Instead, they are likely attributable to the substantial size and intricate structure of these transmembrane ion channels, which encompass multiple domains and flexible loops. These proteins inherently exhibit greater conformational flexibility, especially in extracellular and intracellular loops or unresolved terminal regions that may have been modelled during structure preparation. RMSD values ranging from 0.6 to 0.8 nm are typical in such large membrane proteins and are deemed acceptable, particularly when the RMSD plateaus, indicating that the system has achieved equilibrium without substantial structural compromise.
In summary, all three receptor–ligand systems reach equilibrium during the simulation. Oxaprozin and Pizotifen generally maintain or enhance structural stability compared to standard drugs. Although Cyproheptadine causes higher RMSD in both voltage-gated ion channels, these values remain within acceptable limits, indicating dynamic rearrangements rather than destabilisation. The GABA receptor complexes, being smaller and more rigid, show minimal deviation across all ligands, demonstrating strong stability. Overall, these findings suggest that Oxaprozin and Pizotifen are more structurally advantageous binders and could offer improved therapeutic stability.
Root mean square fluctuation (RMSF)
The RMSF measures the average deviation, flexibility, and dynamic behaviour of a particle (e.g., protein residue) over time from a reference position (typically the time-averaged position of the particle) during molecular dynamics (MD) simulations. It analyses the portions of the structure that fluctuate from their mean structure the most or least. The Cyproheptadine drug showed the lowest RMSF, followed by Pizotifen and the standard drugs, while Oxaprozin exhibited the maximum RMS fluctuation upon binding to all three selected epilepsy target receptors (Fig. 6).
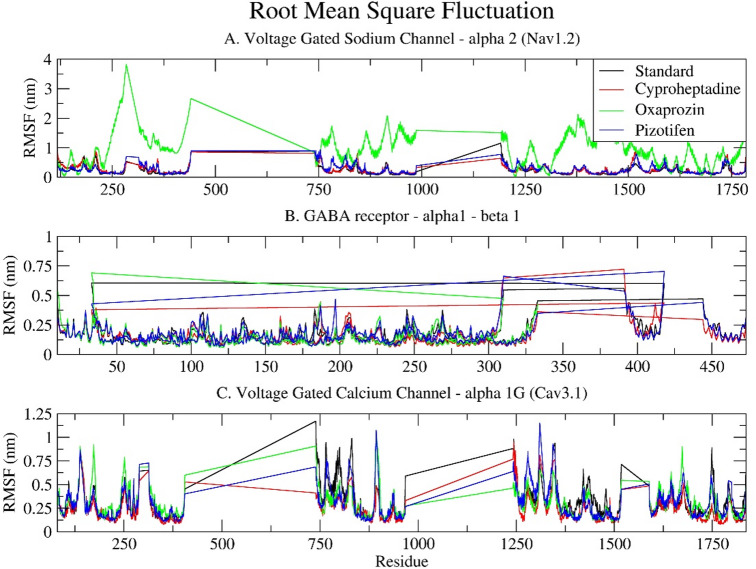
Regarding Nav1.2, the standard drug Carbamazepine exhibits low to moderate fluctuations along the polypeptide chain, indicative of a stable receptor-ligand interaction. Cyproheptadine and Pizotifen demonstrate RMSF profiles analogous to the standard, suggesting their binding does not significantly alter the conformational stability of Nav1.2. Conversely, Oxaprozin induces notable fluctuations in specific regions, particularly residues 150–300, 700–900, and 1000–1300, with RMSF values reaching up to 3.16 nm, which might be due to bonding and non-bonding interactions. These fluctuations suggest increased local flexibility or destabilisation, which could potentially affect the gating function of the sodium channel. Furthermore, the RMSF plot displays flat, horizontal lines between the fluctuating regions, representing unresolved residues in the experimental model and are thus excluded from the molecular dynamics simulations. These lines are visual artefacts indicating missing segments, rather than actual data (Fig. 6A).
The standard drug Clonazepam exhibits relatively low RMSF values for the GABA receptor α1-β1, with most values being below 0.5 nm, indicating a stable complex. Cyproheptadine and Pizotifen display slightly higher flexibility in certain regions, particularly around residues 280–320 and 380–420, likely corresponding to extracellular or cytoplasmic loops. Oxaprozin exhibits a slight fluctuation near residue 50 but generally resembles the standard closely. This suggests all three drugs maintain the structural integrity of the GABA receptor, with only localised, non-disruptive flexibility variations. The RMSF plot distinctly shows the α1 and β1 regions, with separate plots and no connecting lines, highlighting their physical separation. (Fig. 6B).
In the Voltage Gated Calcium Channel alpha 1G (Cav3.1), all four ligands, including the Standard, exhibit highly similar fluctuation patterns, with minor peaks near residues 800 and 1400. Cyproheptadine and Pizotifen closely follow the Standard, suggesting they have little effect on the receptor’s overall flexibility. Conversely, Oxaprozin shows slightly increased fluctuations across several regions, especially between residues 200–400, 750–900, and 1200–1500, corresponding to Domains I and IV. Though less severe than in Nav1.2, these fluctuations suggest moderate destabilisation. As with the sodium channel, the RMSF plot for Cav3.1 displays straight lines, indicating gaps in the resolved structural data (see Fig. 6C).
Compared to the standard drug, Cyproheptadine and Pizotifen exhibit similar structural behaviours across all three receptors, as evidenced by their comparable RMSF profiles, which denote the preservation of native conformational dynamics. This correlates with their moderate RMSD values, indicating stable overall structures with only localised fluctuations. Conversely, Oxaprozin displays a distinct pattern characterised by increased residue-level flexibility, particularly in the loop and inter-domain regions of Nav1.2 and Cav3.1 channels. This heightened flexibility is demonstrated by its higher RMSD values, suggesting potential ligand-induced conformational changes or allosteric effects. Such deviations may indicate a different binding mechanism, which could subsequently influence receptor modulation.
Radius of gyration (Rg)
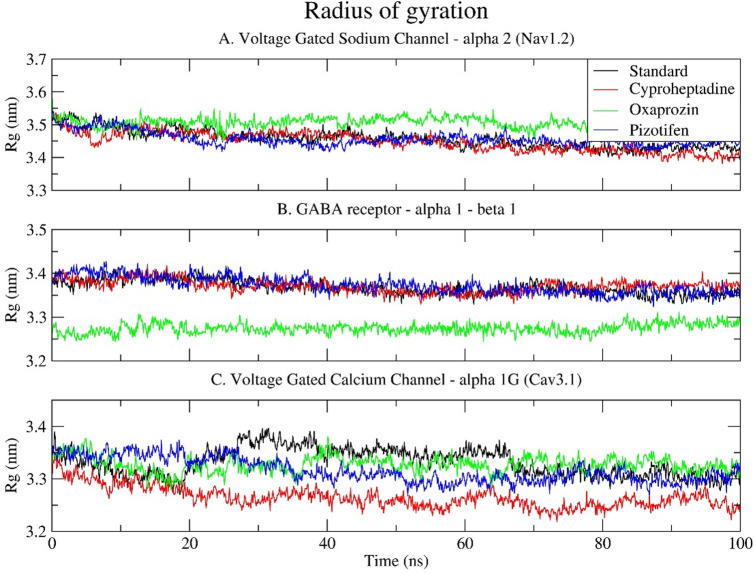
Rg measures the distribution of protein atoms around its centre of mass, giving key insights into the overall compactness and stability of receptor-ligand complexes during simulation. All three target receptors exhibited stable atomic distributions, ranging from 3.2 to 3.6 nm (Fig. 7). For Nav1.2, complexes maintained consistent Rg values over 100 ns, indicating no significant unfolding (Fig. 7A). The Oxaprozin-bound complex had slightly higher Rg values than the standard and other ligands, hinting at a mild expansion of the protein structure, which aligns with its higher RMSD and RMSF. In the GABA receptor α1–β1 complex, the standard, Cyproheptadine, and Pizotifen complexes exhibited similar Rg profiles, ranging from 3.35 to 3.4 nm, indicating consistent compactness. Oxaprozin showed a lower Rg (~ 3.25–3.3 nm), possibly reflecting tighter packing or structural constriction from its binding mode (Fig. 7B). In the calcium channel (Cav3.1), standard and Pizotifen complexes maintained stable, compact Rg trajectories. Cyproheptadine’s Rg slightly decreased over time, indicating increased compactness, while Oxaprozin’s Rg stayed intermediate but stable. These results align with earlier RMSD and RMSF data, where Cyproheptadine and Pizotifen had moderate dynamics, and Oxaprozin showed more variation depending on the receptor (Fig. 7C). Overall, Rg analysis supports the RMSD and RMSF findings, suggesting that Oxaprozin may influence receptor conformations by causing slight expansion (Nav1.2) or compaction (GABA receptor), while Cyproheptadine and Pizotifen tend to maintain structures similar to those of the standard ligand.
Solvent accessible surface area (SASA)
SASA is a critical parameter that describes the area surrounding a macromolecule accessible to solvent molecules. It aids in understanding protein folding, ligand binding, and the conformational flexibility and solvent exposure of protein-ligand complexes, which are fundamental for examining molecular interactions at the atomic level. For all drug-receptor complexes, the SASA graphs remained stable, indicating the overall size of the protein (see Fig. 8). The higher SASA values for Nav1.2 (580 nm² – 620 nm²) and Cav3.1 (510 nm² – 550 nm²) in comparison to the GABA receptor α1-β1 (290 nm² – 340 nm²) reflect their larger structures, with Nav1.2 and Cav3.1 containing more than 1000 amino acids, whereas the GABA receptor α1-β1 comprises 602 amino acids.
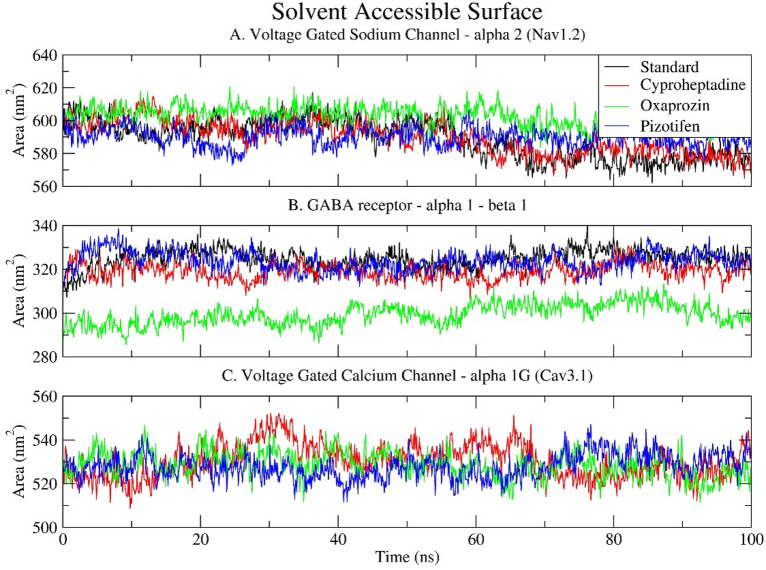
In Nav1.2, all ligand-bound complexes exhibit relatively stable SASA values, ranging from approximately 590 to 610 nm². Oxaprozin, however, demonstrates a marginally higher SASA (608 nm²) in comparison to the typical 590 nm² observed for Cyproheptadine and Pizotifen. This indicates a more expanded or loosely packed structure, consistent with its elevated Rg and RMSF values, which signify increased flexibility and conformational diversity (Fig. 8A).
In the GABA receptor α1–β1, SASA trends are more noticeable. Oxaprozin exhibits a notable decrease in solvent accessibility, averaging about 302 nm², in contrast to the standard and other drugs, which range from 320 to 330 nm². This suggests a more compact protein conformation, consistent with the lower Rg observed in this complex, indicating possible structural tightening or different folding when Oxaprozin binds. This also highlights Oxaprozin’s unique interaction pattern with GABA receptor residues (see Fig. 8B).
All Cav3.1 complexes show moderate SASA fluctuations between 520 and 540 nm², with no significant changes. Cyproheptadine and Pizotifen have slightly higher and more stable SASA values compared to Oxaprozin, indicating better solvent exposure while maintaining stability, which is also reflected in their consistent Rg and RMSD measures. Conversely, Oxaprozin displays marginally lower SASA, suggesting more internalised or compact areas, possibly due to changes in binding-induced topology (Fig. 8C).
Along with the Rg and RMSD data, the SASA analysis confirms that Cyproheptadine and Pizotifen preserve solvent-exposed, compact, and stable receptor conformations. Conversely, Oxaprozin induces receptor-specific conformational modifications, either expansion in Nav1.2 or contraction in the GABA receptor, that may influence ligand binding and receptor functionality.
Principal Component Analysis (PCA).
PCA was employed to analyse the overall motions and essential dynamics of the protein-ligand complexes throughout the simulation. This technique reduces complex atomic movements into principal components (PCs), with the initial few PCs accounting for the majority of conformational variability. In the case of Nav1.2, the standard drug Carbamazepine, alongside Cyproheptadine and Pizotifen, cluster closely within the PC1 versus PC2 space, indicating a limited conformational range and suggesting stabilised dynamics with minimal large-scale structural alterations. Conversely, Oxaprozin occupies a broader, more dispersed region across the PCs, signifying greater structural flexibility and dynamic variability (Fig. 9A). This observation aligns with elevated RMSD, Rg, and SASA values for the same receptor, thereby reinforcing the conclusion that Oxaprozin binding results in a more flexible or destabilised conformation.
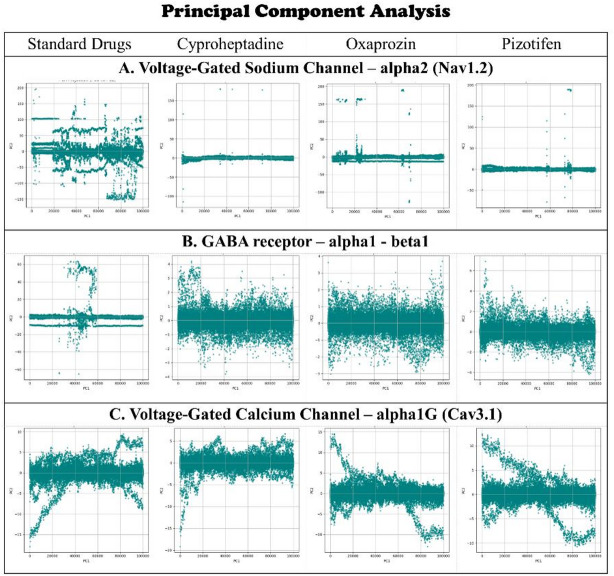
The PCA plot for the GABA receptor α1-β1 shows tighter clustering for Cyproheptadine and Pizotifen, suggesting limited movement and stable complex formation. Oxaprozin also exhibits restricted mobility in PCA space, aligned with its lower SASA and Rg values, which indicate a more compact or rigid structure. This suggests that Oxaprozin binds in a manner that restricts the motion of key domains, potentially impacting its functional dynamics (Fig. 9B). In Cav3.1, PCA patterns display moderate clustering across all ligands, with Cyproheptadine and Pizotifen forming slightly more cohesive groups than Oxaprozin, hinting at more uniform dynamics. However, these differences are less marked than in Nav1.2 or the GABA receptor, suggesting Oxaprozin does not significantly destabilise Cav3.1 but does influence its motion more than the standard drug and other compounds (Fig. 9C). Overall, PCA provides a clear view of the complexes’ dynamical behaviour, showing Cyproheptadine and Pizotifen promote stable, functionally consistent dynamics across all three receptors. In contrast, Oxaprozin causes receptor-specific changes in key motions by either increasing flexibility or rigidity, thereby setting its interaction profile apart from that of the standard drug.
Interaction energy
The interaction energy analysis provides valuable insights into the non-bonding forces that influence the stability of the ligand–receptor complex during Molecular Dynamics Simulation (MDS). This analysis evaluates Lennard-Jones (van der Waals) and Coulombic (electrostatic) interactions, enabling an assessment of the ligand’s binding affinity and potency within the active site. In all three receptor complexes (Nav1.2, GABA receptor α1–β1, and Cav3.1), selected drugs demonstrated stronger interaction energies than their respective standard drugs, indicating potentially more stable and favourable binding (Fig. 10; Table 5).
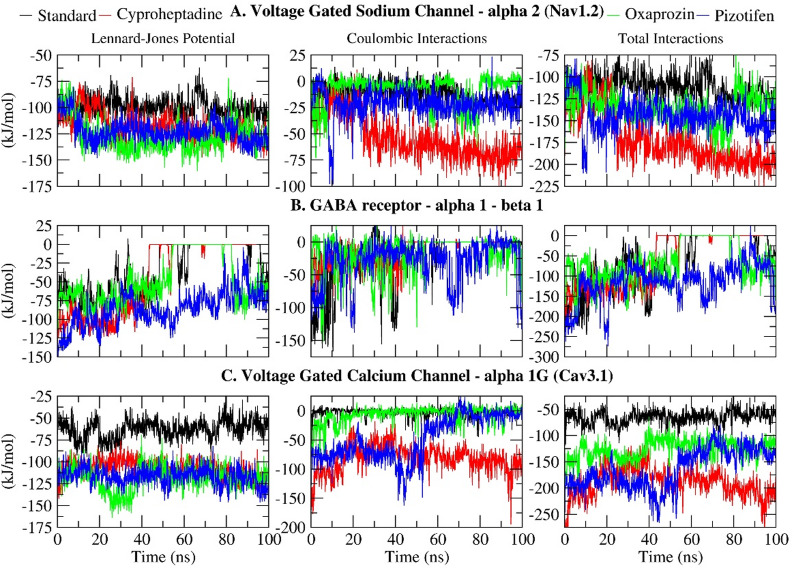
Table 5: Interaction energy (KJ/mol) between selected drugs and epilepsy receptor Nav1.2, GABA receptor alpha1-beta1 and Cav3.1.
| Lennard-Jones Potential (KJ/mol) | Coulombic Interaction (KJ/mol) | Total interactions (KJ/mol) | |
|---|---|---|---|
| Voltage-Gated Sodium Channel – α2 (Nav1.2) | |||
| Standard (Carbamazepine) | -99.8047 | -11.793 | -111.5977 |
| Cyproheptadine | -117.832 | -53.279 | -171.111 |
| Oxaprozin | -127.217 | -8.5828 | -135.7998 |
| Pizotifen | -122.406 | -21.6646 | -144.0706 |
| GABA receptor – α1 β1 | |||
| Standard (Clonazepam) | -35.405 | -21.1496 | -56.5546 |
| Cyproheptadine | -41.7388 | -14.1175 | -55.8563 |
| Oxaprozin | -47.8904 | -16.6913 | -64.5817 |
| Pizotifen | -58.7435 | -29.3292 | -115.0727 |
| Voltage-Gated Calcium Channel – α1G (Cav3.1) | |||
| Standard (Pregabalin) | -62.8288 | -2.68518 | -65.51398 |
| Cyproheptadine | -107.54 | -81.9922 | -189.5322 |
| Oxaprozin | -118.768 | -7.07558 | -125.84350 |
| Pizotifen | -117.041 | -47.3968 | -158.4378 |
For Nav1.2, the standard drug Carbamazepine had a total interaction energy of -111.5977 kJ/mol, while Cyproheptadine showed the most favourable energy at -171.111 kJ/mol, mainly due to stronger van der Waals (-117.832 kJ/mol) and Coulombic (-53.279 kJ/mol) interactions. Both Pizotifen (-144.0706 kJ/mol) and Oxaprozin (-135.7998 kJ/mol) also demonstrated better binding than the standard. In the GABA α1–β1 receptor system, Pizotifen again was the most stable with an interaction energy of -115.0727 kJ/mol, followed by Oxaprozin (-64.5817 kJ/mol). Cyproheptadine showed a slightly weaker interaction (-55.8563 kJ/mol) compared to the standard drug Clonazepam (-56.5546 kJ/mol), indicating receptor-specific affinity differences. Regarding Cav3.1, Pregabalin, the standard drug, had the weakest total interaction energy (-65.51398 kJ/mol), whereas Cyproheptadine had the most favourable at -189.5322 kJ/mol, mainly due to a strong Coulombic contribution (-81.9922 kJ/mol). Pizotifen (-158.4378 kJ/mol) and Oxaprozin (-125.8435 kJ/mol) also bound more strongly than Pregabalin. These results align with previous analyses showing Pizotifen and Oxaprozin exhibited more stable RMSD and radius of gyration values, suggesting tight, persistent binding during the simulation.
The comprehensive interaction energy profile designates Pizotifen as the most promising ligand across all receptors, owing to its sustained, high-affinity interactions, with Oxaprozin ranking subsequently. Although Cyproheptadine demonstrated lower affinity within the GABA complex, its elevated interaction energies with Nav1.2 and Cav3.1 render it a compelling candidate for drug repurposing. In conclusion, these interaction energies underscore the potential of all three pharmaceuticals to serve as efficacious modulators of vital neurological targets implicated in epilepsy.
Hydrogen bond (H-bond)
The analysis of hydrogen bonding provided critical insights into the stability and specificity of ligand–receptor interactions throughout the 100 ns molecular dynamics simulation. Among the compounds examined, Oxaprozin consistently demonstrated the highest number of hydrogen bonds across all three target receptors, indicating robust and persistent polar interactions within the binding sites. Specifically, with Nav1.2, Oxaprozin averaged 1.342 hydrogen bonds, reaching a peak of three bonds around 22 ns, although it predominantly maintained a single hydrogen bond during the simulation. Conversely, the standard drug Carbamazepine exhibited a significantly lower average of 0.199 hydrogen bonds, with sporadic formation of up to two hydrogen bonds during the mid-phase of the simulation. Both Cyproheptadine and Pizotifen formed negligible hydrogen bonds with Nav1.2 (0.007 and 0.027, respectively), which correlates with their lower interaction energies and suggests a primarily hydrophobic or non-polar binding mode (Fig. 11A).
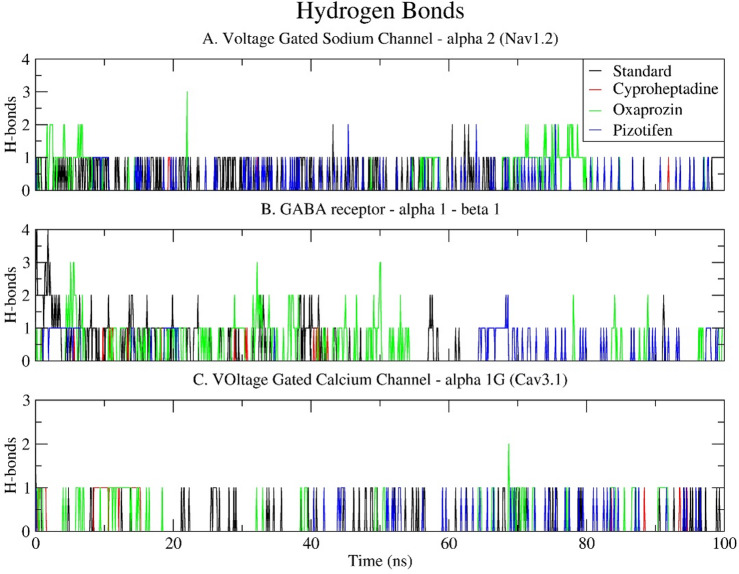
The GABA receptor α1–β1 complex further corroborated the superior hydrogen bonding capacity of Oxaprozin, which averaged 1.532 hydrogen bonds, reaching a peak of three hydrogen bonds during the initial and middle stages of the simulation. The standard drug Clonazepam demonstrated a moderate average of 0.296 hydrogen bonds, initially forming up to four bonds that gradually diminished to one or two bonds towards the end. Pizotifen established a single hydrogen bond only after 60 nanoseconds, with an overall average of 0.155, while Cyproheptadine was observed to form sporadic and infrequent hydrogen bonds (0.024 on average), thereby indicating its lower polar affinity and reduced binding stability (Fig. 11B). Regarding Cav3.1, hydrogen bond formation was generally less frequent across all ligands. Nonetheless, Oxaprozin exhibited better performance, with an average of 0.821 hydrogen bonds, typically maintaining one bond throughout the simulation. The standard drug Pregabalin, in conjunction with Cyproheptadine, formed an average of 0.086 hydrogen bonds, with these interactions being transient. Pizotifen demonstrated the least hydrogen bond activity, averaging 0.021, consistent with its fewer polar contacts despite possessing a favourable interaction energy profile (Fig. 11C).
Overall, the hydrogen bond analysis highlights Oxaprozin’s robust polar interactions with all target proteins, corroborating the findings from interaction energy and RMSF analyses. This consistent hydrogen-bond engagement may contribute to maintaining stable binding and potentially modulate receptor conformation allosterically, thereby enhancing its potential as a compelling drug repurposing candidate for neurological targets.
Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA)
The MMPBSA method was employed to estimate the binding free energies of the selected drug–receptor complexes, offering insights into the thermodynamic stability and affinity of each ligand. The binding free energy components: van der Waals energy (ΔVDW), electrostatic energy (ΔEEL), gas-phase energy (ΔG_GAS = ΔBonds + ΔAngle + ΔDihedral + ΔVDW + ΔEEL), and solvation energy (ΔG_SOLV = ΔEGB + ΔESURF) were carefully evaluated. For the Nav1.2 receptor, Oxaprozin achieved the lowest ΔG_Total of − 23.98 kcal/mol, outperforming the standard drug Carbamazepine (-18.10 kcal/mol) and the investigational drugs Pizotifen (-17.78 kcal/mol) and Cyproheptadine (-16.35 kcal/mol). Notably, Oxaprozin showed the most favourable ΔVDW and ΔG_SOLV values, reflecting strong hydrophobic and solvent-based interactions (Fig. 12A; Table 6). In the GABA receptor α1–β1, Oxaprozin again demonstrated the most favourable ΔG_Total (-13.40 kcal/mol), followed by Clonazepam (-12.65 kcal/mol), Pizotifen (-9.32 kcal/mol), and Cyproheptadine (-3.41 kcal/mol). While Clonazepam exhibited the lowest ΔEEL and ΔG_GAS, Oxaprozin maintained the lowest ΔVDW and ΔG_SOLV, indicating its balanced contribution from both gas-phase and solvation components (Fig. 12B). For the Cav3.1, Oxaprozin recorded the lowest ΔG_Total of -22.94 kcal/mol, significantly outperforming Pizotifen (-17.93 kcal/mol) and Cyproheptadine (-13.13 kcal/mol). The standard drug Pregabalin showed the weakest interaction, with a ΔG_Total of only − 9.64 kcal/mol, highlighting Oxaprozin’s superior affinity for this calcium channel (Fig. 12C). Taken together, these findings strongly indicate that Oxaprozin demonstrates the most stable and energetically favourable binding profile across all three neuroreceptors, primarily driven by its robust van der Waals and solvation contributions. This reinforces its potential as a promising repurposed candidate for neurological targeting compared to the standard and other test drugs.
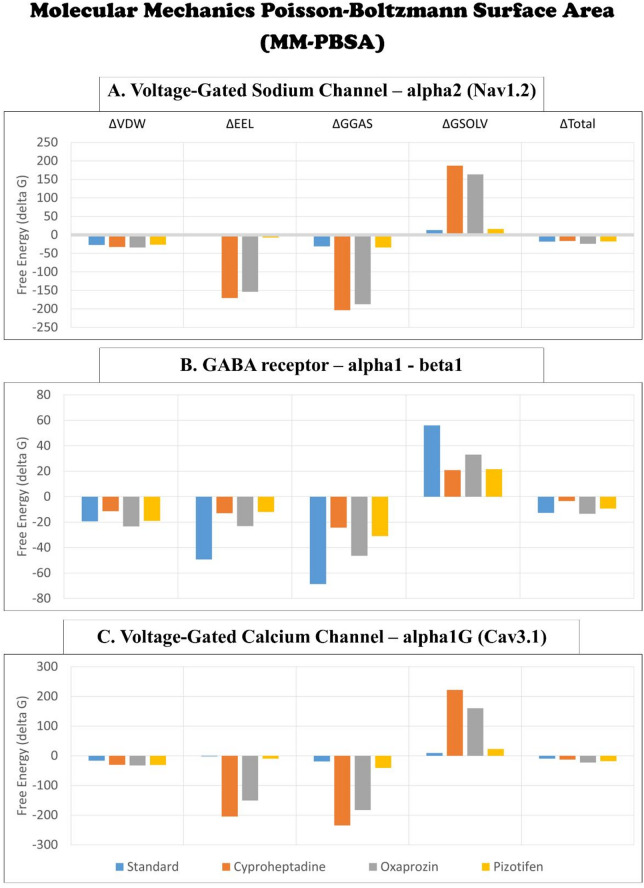
Table 6: The free binding energy (kcal/mol) between selected epilepsy receptor proteins and drugs using MMPBSA analysis.
| Poisson Boltzmann | ΔVDW | ΔEEL | ΔG GAS | ΔG SOLV | ΔG Total |
|---|---|---|---|---|---|
| Voltage-Gated Sodium Channel – α2 (Nav1.2) | |||||
| Standard (Carbamazepine) | -27.38 | -3.59 | -30.97 | 12.87 | -18.10 |
| Cyproheptadine | -32.62 | -170.84 | -203.46 | 187.11 | -16.35 |
| Oxaprozin | -33.79 | -153.70 | -187.50 | 163.52 | -23.98 |
| Pizotifen | -26.53 | -7.40 | -33.93 | 16.15 | -17.78 |
| GABA receptor – α1 β1 | |||||
| Standard (Clonazepam) | -19.40 | -49.29 | -68.68 | 56.04 | -12.65 |
| Cyproheptadine | -11.37 | -12.95 | -24.32 | 20.92 | -3.41 |
| Oxaprozin | -23.39 | -23.09 | -46.47 | 33.07 | -13.40 |
| Pizotifen | -18.98 | -11.98 | -30.97 | 21.65 | -9.32 |
| Voltage-Gated Calcium Channel – α1G (Cav3.1) | |||||
| Standard (Pregabalin) | -16.79 | -2.15 | -18.94 | 9.30 | -9.64 |
| Cyproheptadine | -30.40 | -204.51 | -234.91 | 221.78 | -13.13 |
| Oxaprozin | -32.44 | -150.56 | -183.01 | 160.07 | -22.94 |
| Pizotifen | -30.95 | -9.99 | -40.94 | 23.00 | -17.93 |
Molecular dynamics simulations of the three target receptors —VGSC–α2 (Nav1.2), GABA receptor α1β1, and VGCC–α1G (Cav3.1) —indicated distinct dynamic behaviours and binding efficacies of the repurposed drugs Oxaprozin, Cyproheptadine, and Pizotifen. Initial structural assessments, such as RMSD, RMSF, Rg, and SASA, demonstrated that Oxaprozin induced greater fluctuations and flexibility, particularly within Nav1.2 and Cav3.1, signifying moderate destabilisation, in contrast to the more stable conformations associated with Cyproheptadine and Pizotifen. Subsequent analyses involving hydrogen bonding, interaction energy profiles, and MMPBSA calculations revealed that Oxaprozin established more stable and energetically favourable interactions with all three receptors, exhibiting the strongest non-bonded interaction energies and the lowest binding free energies. These findings suggest that, although Oxaprozin may provoke local conformational changes or increased flexibility, such effects enhance its overall binding affinity, potentially via deeper pocket penetration or allosteric engagement. Pizotifen, characterised by stable dynamics and favourable energetic profiles, ranked second overall, effectively balancing structural stability with consistent receptor binding. Cyproheptadine, while displaying dynamic stability, exhibited weaker hydrogen bonding and less favourable binding energies, particularly within the GABA complex, thereby ranking third. In conclusion, despite moderate fluctuations, Oxaprozin emerged as the most effective ligand, followed by Pizotifen and Cyproheptadine, underscoring their varied potentials as repurposed agents for modulating neuronal ion channels and receptors.
Conclusion
A comprehensive in-silico drug repositioning approach was applied to screen the entire DrugBank database of molecules for identifying a new multi-targeted therapeutic option for epilepsy. A total of 2769 FDA-approved drugs were primarily screened for blood-brain permeability and structural similarity with currently marketed AEDs. The Voltage-Gated Sodium channel – α2, GABA receptor α1 β1, and Voltage-Gated Calcium channel – α1G, were selected as therapeutic target proteins for molecular docking studies using Carbamazepine, Clonazepam, and Pregabalin, as standard reference drugs, respectively. Only 46 marketed drugs, in common, showed higher binding affinities than the selected standard drugs. The binding pocket analyses and text mining studies narrowed our findings to three drug compounds, namely, Oxaprozin, Pizotifen, and Cyproheptadine, as potential candidates for drug repurposing as a multitarget treatment for epilepsy. The molecular dynamics simulation study revealed that all three selected drug compounds exhibit stable and strong binding to all three studied therapeutic target proteins. Oxaprozin was observed to show the strongest binding affinity and stability, followed by Pizotifen and Cyproheptadine. We believe that the in-silico simulation studies in our study provide robust hypothesis-generating mechanistic evidence for ligand–pocket engagement. Furthermore, structure refinement—encompassing pharmacophore modelling, scaffold modification, and fragment-based design—along with experimental validation, will be crucial in improving efficacy and reducing adverse effects, thereby supporting the clinical translation of multitarget therapies for epilepsy.
Supplementary Materials
References
- P Klein. Repurposed molecules for antiepileptogenesis: missing an opportunity to prevent epilepsy?. Epilepsia, 2020. [DOI | PubMed]
- RD Thijs, R Surges, TJ O’Brien, JW Sander. Epilepsy in adults. Lancet, 2019. [DOI | PubMed]
- C Lossin, DW Wang, TH Rhodes, CG Vanoye, AL George. Molecular basis of an inherited epilepsy. Neuron, 2002. [DOI | PubMed]
- 4.Annegers, J. F. & Rocca, W. A. Causes of epilepsy: Contributions of the rochester epidemiology project. in Mayo Clin Proc 71, 570–575 (Elsevier, 1996).
- B Galindo-Mendez, LC Mayor, F Velandia-Hurtado, C Calderon-Ospina. Failure of antiepileptic drugs in controlling seizures in epilepsy: what do we do next?. Epilepsy Behav. Case Rep., 2015. [DOI | PubMed]
- Z Liu. In Silico drug repositioning-what we need to know. Drug Discov Today, 2013. [DOI | PubMed]
- TT Ashburn, KB Thor. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov, 2004. [DOI | PubMed]
- JP Jourdan, R Bureau, C Rochais, P Dallemagne. Drug repositioning: a brief overview. J. Pharm. Pharmacol., 2020. [DOI | PubMed]
- P Tolete. Lorcaserin therapy for severe epilepsy of childhood onset. Neurology, 2018. [DOI | PubMed]
- DS Wishart. DrugBank 5.0: A major update to the drugbank database for 2018. Nucleic Acids Res., 2018. [DOI | PubMed]
- P Kumar. A machine Learning-based online tool for Blood-Brain barrier (BBB) permeability prediction. CNS Neurol. Disord Drug Targets, 2024. [DOI]
- F Cheng. AdmetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model., 2012. [DOI | PubMed]
- B Shaker. LightBBB: computational prediction model of blood-brain-barrier penetration based on LightGBM. Bioinformatics, 2021. [DOI | PubMed]
- MR Hurle. Computational drug repositioning: from data to therapeutics. Clin. Pharmacol. Ther., 2013. [DOI | PubMed]
- S Ekins, AJ Williams, MD Krasowski, JS Freundlich. In Silico repositioning of approved drugs for rare and neglected diseases. Drug Discov Today, 2011. [DOI | PubMed]
- P Kumar, D Sheokand, V Saini, A Kumar. In-silico identification and analysis of hub proteins for designing novel First-line Anti-seizure medications. Lett. Drug Des. Discov, 2022
- TWH Backman, Y Cao, T Girke. ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic Acids Res., 2011. [DOI | PubMed]
- Y Cao, A Charisi, LC Cheng, T Jiang, T Girke. ChemmineR: A compound mining framework for R. Bioinformatics, 2008. [DOI | PubMed]
- S Silvestro, S Mammana, E Cavalli, P Bramanti, E Mazzon. Use of Cannabidiol in the treatment of epilepsy: efficacy and security in clinical trials. Molecules, 2019. [DOI | PubMed]
- A Beydoun. Current role of carbamazepine and oxcarbazepine in the management of epilepsy. Seizure, 2020. [DOI | PubMed]
- V Dokkedal-Silva. Indications, side Effects, and potential for nonmedical use. Harv. Rev. Psychiatry, 2019. [DOI | PubMed]
- S Hebeisen. Eslicarbazepine and the enhancement of slow inactivation of voltage-gated sodium channels: A comparison with carbamazepine, oxcarbazepine and lacosamide. Neuropharmacology, 2015. [DOI | PubMed]
- 23.Brigo, F. & Igwe, S. C. Ethosuximide, sodium valproate or lamotrigine for absence seizures in children and adolescents. Cochrane Database of Systematic Reviews (2017).
- M Markota, RJ Morgan. Treatment of generalized anxiety disorder with Gabapentin. Case Rep. Psychiatry, 2017
- MA Rogawski, A Tofighy, HS White, A Matagne, C Wolff. Current Understanding of the mechanism of action of the antiepileptic drug lacosamide. Epilepsy Res., 2015. [DOI | PubMed]
- C Stockburger. A mitochondrial role of SV2a protein in aging and alzheimer’s disease: studies with Levetiracetam. J. Alzheimer’s Disease, 2016. [DOI | PubMed]
- GL Krauss. Levetiracetam treatment of idiopathic generalised epilepsy. Seizure, 2003. [DOI | PubMed]
- AL Mruk, KL Garlitz, NR Leung. Levetiracetam in neonatal seizures: A review. J. Pediatr. Pharmacol. Ther., 2015. [PubMed]
- 29.Hansen, C. C., Ljung, H., Brodtkorb, E. & Reimers, A. Mechanisms underlying aggressive behavior induced by antiepileptic drugs: Focus on topiramate, levetiracetam, and perampanel. Behav. Neurol. (2018).
- SRA Alles, SM Cain, TP Snutch. Pregabalin as a pain therapeutic: beyond calcium channels. Front. Cell. Neurosci., 2020. [DOI | PubMed]
- 31.Manjushree, N., Chakraborty, A., Shashidhar, K. & Narayanaswamy, S. A review of the drug Pregabalin. Int. J. Basic. Clin. Pharmacol. 601–605. 10.18203/2319-2003.ijbcp20150359 (2015).
- E Perucca, J Cloyd, D Critchley, E Fuseau. Rufinamide: Clinical pharmacokinetics and concentration-response relationships in patients with epilepsy. Epilepsia, 2008. [DOI | PubMed]
- S Piplani, PK Verma, A Kumar. Neuroinformatics analyses reveal GABAt and SSADH as major proteins involved in anticonvulsant activity of valproic acid. Biomed. Pharmacotherapy, 2016. [DOI]
- M Romoli. Valproic acid and epilepsy: from molecular mechanisms to clinical evidences. Curr. Neuropharmacol., 2018. [DOI]
- G Zanatta. Valproic acid interactions with the NavMs voltage-gated sodium channel. Proc. Natl. Acad. Sci. U S A, 2019. [DOI | PubMed]
- JJ Luszczki, N Ratnaraj, PN Patsalos, SJ Czuczwar. Isobolographic and behavioral characterizations of interactions between Vigabatrin and Gabapentin in two experimental models of epilepsy. Eur. J. Pharmacol., 2008. [DOI | PubMed]
- V Biton. Clinical Pharmacology and mechanism of action of Zonisamide. Clin. Neuropharmacol., 2007. [DOI | PubMed]
- X Pan. Molecular basis for pore Blockade of human Na + channel Na v 1.2 by the m-conotoxin KIIIA. Sci. (1979), 2019
- S Masiulis. GABAA receptor signalling mechanisms revealed by structural Pharmacology. Nature, 2019. [DOI | PubMed]
- Y Zhao. Cryo-EM structures of Apo and antagonist-bound human Cav3.1. Nature, 2019. [DOI | PubMed]
- K Arnold, L Bordoli, J Kopp, T Schwede. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics, 2006. [DOI | PubMed]
- P Benkert, SCE Tosatto, D Schomburg. QMEAN: A comprehensive scoring function for model quality assessment. Proteins: Struct. Function Genet., 2008. [DOI]
- 43.Agnihotry, S., Pathak, R. K., Singh, D. B., Tiwari, A. & Hussain, I. Protein structure prediction. Bioinformatics: Methods Appl. 177–188. 10.1016/B978-0-323-89775-4.00023-7 (2021).
- D Eisenberg, R Lüthy, JU Bowie. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol., 1997. [DOI | PubMed]
- M Wiederstein, MJ Sippl. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res., 2007. [DOI | PubMed]
- 46.Macindoe, G., Mavridis, L., Venkatraman, V., Devignes, M. D. & Ritchie, D. W. HexServer: an FFT-based protein Docking server powered by graphics processors. Nucleic Acids Res38 (2010).
- LW Delano. Pymol: an open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr., 2002
- EF Pettersen. UCSF Chimera – A visualization system for exploratory research and analysis. J. Comput. Chem., 2004. [DOI | PubMed]
- GM Morris. Software news and updates AutoDock4 and AutoDockTools4: automated Docking with selective receptor flexibility. J. Comput. Chem., 2009. [DOI | PubMed]
- P Kumar, D Sheokand, A Grewal, V Saini, A Kumar. Clinical side-effects based drug repositioning for anti-epileptic activity. J. Biomol. Struct. Dyn., 2024. [DOI | PubMed]
- RA Laskowski, MB Swindells, LigPlot+. Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model., 2011. [DOI | PubMed]
- MJ Abraham, Gromacs. High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 2015. [DOI]
- J Huang. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods, 2016. [DOI | PubMed]
- MS Valdés-Tresanco, ME Valdés-Tresanco, PA Valiente, E Moreno. Gmx_MMPBSA: A new tool to perform End-State free energy calculations with GROMACS. J. Chem. Theory Comput., 2021. [DOI | PubMed]
- 55.Ghit, A., Assal, D., Al-Shami, A. S. & Hussein, D. E. E. GABAA receptors: structure, function, pharmacology, and related disorders. J. Genet. Eng. Biotechnol.19, 1–15 (2021).
- E Sigel, ME Steinmann. Structure, function, and modulation of GABAA receptors. J. Biol. Chem., 2012. [DOI | PubMed]
- M Abdelsayed, S Sokolov. Voltage-gated sodium channels: pharmaceutical targets via anticonvulsants to treat epileptic syndromes. Channels, 2013. [DOI | PubMed]
- RJ Docherty, CE Farmer. The Pharmacology of voltage-gated sodium channels in sensory neurones. Handb. Exp. Pharmacol., 2009. [DOI]
- GM Lipkind, HA Fozzard. Molecular model of anticonvulsant drug binding to the voltage-gated sodium channel inner pore. Mol. Pharmacol., 2010. [DOI | PubMed]
- 60.Ghit, A., Assal, D., Al-Shami, A. S. & Hussein, D. E. E. GABAA receptors: structure, function, pharmacology, and related disorders. J. Genet. Eng. Biotechnol.19, 123 (2021).
- MP Goldschen-Ohm, DA Wagner, MV Jones. Three arginines in the GABAA receptor binding pocket have distinct roles in the formation and stability of Agonist- versus Antagonist-Bound complexes. Mol. Pharmacol., 2011. [DOI | PubMed]
- CL Padgett, AP Hanek, HA Lester, DA Dougherty, SCR Lummis. Unnatural amino acid mutagenesis of the GABAA receptor binding site residues reveals a novel Cation–π interaction between GABA and β2Tyr97. J. Neurosci., 2007. [DOI | PubMed]
- 63.Phulera, S. et al. Cryo-EM structure of the benzodiazepine-sensitive α1β1γ2S tri-heteromeric GABAA receptor in complex with GABA. Elife 7 (2018).
- KL Cunningham, JT Littleton. Mechanisms controlling the trafficking, localization, and abundance of presynaptic Ca2 + channels. Front. Mol. Neurosci., 2023. [DOI]
- C Katelaris. Comparative effects of Loratadine and Azatadine in the treatment of seasonal allergic rhinitis. Asian Pac. J. Allergy Immunol., 1990. [PubMed]
- 66.Welch, M. J., Meltzer, E. O., Estelle, F. & Simons, R. H1-antihistamines and the central nervous system. Histamine and H1-Antihistamines in allergic disease. Second Ed. Revis. Expanded 337–388. 10.3109/9780203910375-16 (2002).
- 67.Hill, T. et al. Antidepressant use and risk of epilepsy and seizures in people aged 20 to 64 years: cohort study using a primary care database. BMC Psychiatry15 (2015).
- V Bertrand. Safety of Cyproheptadine, an orexigenic Drug. Analysis of the French National pharmacovigilance Data-Base and systematic review. Front. Pediatr., 2021. [DOI | PubMed]
- F Kalpaklioglu, A Baccioglu. Efficacy and safety of H1-Antihistamines: an update. Antiinflamm Antiallergy Agents Med. Chem., 2013. [DOI]
- JL Winkler, JW Skovira, RK Kan. Anticonvulsant efficacy of antihistamine Cyproheptadine in rats exposed to the chemical warfare nerve agent Soman. Neurotoxicology, 2017. [DOI | PubMed]
- PA Baumann, L Maitre. Neurobiochemical aspects of maprotiline (Ludiomil ®) action. J. Int. Med. Res., 1979. [DOI | PubMed]
- ML Barbaccia, L Ravizza, E Costa, Maprotiline. An antidepressant with an unusual Pharmacological profile. J. Pharmacol. Exp. Ther., 1986. [DOI | PubMed]
- C Johannessen Landmark, O Henning, SI Johannessen. Proconvulsant effects of antidepressants — What is the current evidence?. Epilepsy Behav., 2016. [DOI | PubMed]
- BF Hoffman, R Wachsmuth. Maprotiline and seizures. J. Clin. Psychiatry, 1982. [PubMed]
- 75.Derry, S., Wiffen, P. J., Aldington, D. & Moore, R. A. Nortriptyline for neuropathic pain in adults. Cochrane Database of Systematic Reviews (2017).
- JP Reinert, MA Veronin, C Medina. Tricyclic antidepressants in nociceptive and neuropathic pain: A review of their analgesic properties in combination with opioids. J. Pharm. Technol., 2023. [DOI | PubMed]
- 77.Wiffen, P. J., Moore, R. A., Aldington, D. & Derry, S. Nortriptyline for neuropathic pain in adults. Cochrane Database of Systematic Reviews (2014).
- DJ Luchins, AP Oliver, RJ Wyatt. Seizures with antidepressants: an in vitro technique to assess relative risk. Epilepsia, 1984. [DOI | PubMed]
- B Lipper, A Bell, B Gaynor. Recurrent hypotension immediately after seizures in Nortriptyline overdose. Am. J. Emerg. Med., 1994. [DOI | PubMed]
- JP Peesa. Oxaprozin prodrug as safer nonsteroidal anti-inflammatory drug: synthesis and Pharmacological evaluation. Arch. Pharm. (Weinheim), 2018. [DOI]
- P Khatami, N Mirazi, M Khosravi, M Bananej. Nonsteroidal Anti-inflammatory drug Oxaprozin is beneficial against Seizure-induced memory impairment in an experimental model of seizures in rats: impact on oxidative stress and Nrf2/HO-1 signaling pathway. J. Mol. Neurosci., 2022. [DOI | PubMed]
- MS Robbins, R Burch. Preventive migraine treatment. CONTINUUM Lifelong Learn. Neurol., 2021. [DOI]
- E Tronvik, S Giri, W Young. Preventive treatment of migraine: Non-specific oral agents. Handb. Clin. Neurol., 2024. [DOI | PubMed]
- FLA Batista. Anticonvulsant and anxiolytic-like potential of the essential oil from the ocimum Basilicum Linn leaves and its major constituent Estragole on adult zebrafish (Danio rerio). Neurochem Int., 2024. [DOI | PubMed]