
Safety of synthetic cannabidiol as a novel food pursuant to Regulation (EU) 2015/2283
Abstract
Following a request from the European Commission, the EFSA Panel on Nutrition, Novel Foods and Food Allergens (NDA) was asked to deliver an opinion on synthetic cannabidiol as a novel food (NF) pursuant to Regulation (EU) 2015/2283. The NF which is subject of the application is trans‐cannabidiol (CBD), produced by chemical synthesis and proposed to be used in food supplements at a level of 30 mg/day. The target population is the general population, excluding pregnant and lactating women. During the risk assessment, the Panel identified a number of data gaps, which needed to be addressed by the applicant. Therefore, an EFSA request for additional information was sent to the applicant. The requested data concerned the identity, the production process, the compositional data, the specifications, the genotoxicity, the reproductive and developmental toxicity and the human data of the NF. Despite being contacted several times, the applicant did not reply to EFSA’s requests for additional data. Based on the available data, the Panel concludes that the safety of the NF, i.e. synthetic cannabidiol, cannot be established.
Article type: Research Article
Keywords: CBD, food supplement, novel foods, safety, synthetic cannabidiol
License: © 2025 European Food Safety Authority. EFSA Journal published by Wiley‐VCH GmbH on behalf of European Food Safety Authority. CC BY-ND 4.0 This is an open access article under the terms of the http://creativecommons.org/licenses/by-nd/4.0/ License, which permits use and distribution in any medium, provided the original work is properly cited and no modifications or adaptations are made.
Article links: DOI: 10.2903/j.efsa.2025.9527 | PubMed: 40741368 | PMC: PMC12308208
Relevance: Relevant: mentioned in keywords or abstract
Full text: PDF (202 KB)
INTRODUCTION
Background and Terms of Reference as provided by the requestor
On 15 March 2021, the company “PureForm International Ltd./PureForm Global Inc” submitted a request to the European Commission in accordance with Article 10 of Regulation (EU) 2015/22831 to authorise placing on the market of synthetic cannabidiol as a novel food.
The application requests to authorise the use of synthetic cannabidiol in food supplements, the target population being adult population excluding pregnant and lactating women.
The applicant has requested data protection according to the provision of Article 26 of Regulation (EU) 2015/2283.
In accordance with Article 10(3) of Regulation (EU) 2015/2283, the European Commission asks the European Food Safety Authority to provide a scientific opinion on the safety of synthetic cannabidiol as a novel food.
In addition, the European Food Safety Authority is requested to include in its scientific opinion a statement as to if, and if so to what extent, the proprietary data for which the applicant is requesting data protection was used in elaborating the opinion in line with the requirements of Article 26(2)(c) of Regulation (EU) 2015/2283.
Additional information
Cannabidiol (CBD) is the active substance of Epidyolex, a medicine approved by the European Medicines Agency (EMA). Epidyolex contains nearly 100% pure CBD and it is used as adjuvant to treat patients suffering from Lennox–Gastaut syndrome, Dravet syndrome or tuberous sclerosis complex in patients from 2 years of age and above.2
On 26 April 2022, the NDA Panel adopted a Statement on the safety of CBD as a novel food which outlined data gaps and uncertainties in the risk assessment. The Panel concluded that the safety of CBD as a NF cannot be established until gaps in both the experimental animal and human data are addressed (EFSA NDA Panel, ref. 2022).
DATA AND METHODOLOGIES
Data
The safety assessment of this NF is based on data provided in the original application submitted in 2021.3 In October 2021 and June 2022, EFSA issued requests for supplementary data. However, the applicant did not reply to the additional information requested by the Panel.
Administrative and scientific requirements for NF applications referred to in Article 10 of Regulation (EU) 2015/2283 are listed in Commission Implementing Regulation (EU) 2017/2469.4
A common and structured format on the presentation of NF applications is described in the EFSA guidance on the preparation and presentation of a NF application (EFSA NDA Panel, ref. 2016). As indicated in this guidance, it is the duty of the applicant to provide all the available (proprietary, confidential and published) scientific data, (including both data in favour and not in favour) that are pertinent to the safety of the NF.
This NF application includes a request for protection of proprietary data in accordance with Article 26 of Regulation (EU) 2015/2283. The data requested by the applicant to be protected comprise stability studies, the bacterial reverse mutation test and the micronucleus test.
Methodologies
The assessment follows the methodology set out in the EFSA guidance on NF applications (EFSA NDA Panel, ref. 2016) and the principles described in the relevant existing guidance documents from the EFSA Scientific Committee. The legal provisions for the assessment are laid down in Article 11 of Regulation (EU) 2015/2283 and in Article 7 of Commission Implementing Regulation (EU) 2017/2469.
This assessment concerns only the risks that might be associated with consumption of the NF under the proposed conditions of use and is not an assessment of the efficacy of the NF with regard to any claimed benefit.
ASSESSMENT
Introduction
The NF that is the subject of the application is synthetic trans‐cannabidiol (CBD).
According to Article 3 of Regulation (EU) 2015/2283, the NF falls under Article 3(2)(a)(i) ‘food with a new or intentionally modified molecular structure, where that structure was not used as, or in, a food within the Union before 15 May 1997’.
The NF is a crystalline powder produced by chemical synthesis consisting of CBD with a purity of ≥ 99.5%. It is proposed by the applicant to be used in food supplements at the use level of 30 mg/day. The proposed target population is the general adult population, excluding pregnant and lactating women.
Identity of the NF
The NF, synthetic CBD, is a highly pure chemical compound. Table 1 lists the main chemical identifiers of CBD while Figure 1 shows its chemical structure.
TABLE 1: Chemical identity of synthetic CBD (i.e. the NF) as provided by the applicant.
| Chemical substance | |
|---|---|
| Chemical (IUPAC) name | 2‐[(1R,6R)‐3‐methyl‐6‐prop‐1‐en‐2‐ylcyclohex‐2‐en‐1‐yl]‐5‐pentylbenzene‐1,3‐diol |
| Common name | (‐) trans‐cannabidiol or cannabidiol |
| Other names: Synonyms, trade names, abbreviations | CBD, Cannabidiol, (‐)‐trans‐cannabidiol, trans‐cannabidiol, (‐)‐Cannabidiol |
| CAS Number: | 13956‐29‐1 |
| Molecular formula | C21H30O2 |
| Molecular weight | 314.46 g/mol |
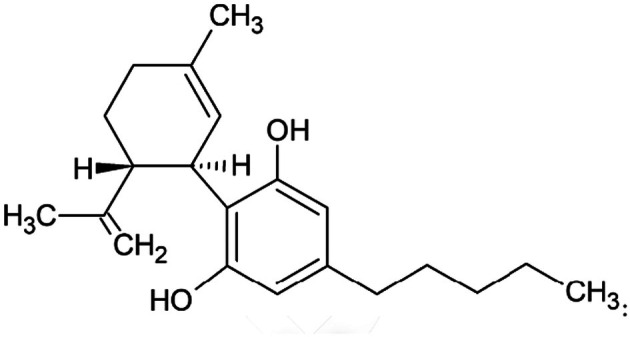
The applicant has submitted limited data regarding the chemical identity of the NF. Additional data were requested to adequately prove the chemical identity and to perform appropriate particle size analyses to detect the potential presence of nanoparticles. The applicant did not reply to these data requests.
The Panel considers that the identity of the NF has not been sufficiently demonstrated.
Production process
According to the applicant, the NF is produced in line with Good Manufacturing Practice (GMP) and International Organisation for Standardisation (ISO) certifications.
The NF is chemically synthesised, starting from olivetol and p‐menthadienol coupled with Lewis acid catalysed alkylation while heating. The final product is obtained through subsequent crystallisation, filtering, washing and drying steps.
Following EFSA’s requests for additional information to clarify and further elaborate on aspects of the production process, no response was submitted to EFSA. Therefore, the Panel considers that the described production process does not allow for a proper hazard identification.
Compositional data
The NF consists of CBD in powder form with a purity of ≥ 99.5%.
In order to confirm that the manufacturing process is reproducible and adequate for producing a product with the required characteristics on a commercial scale, the applicant provided analytical information for five independently produced batches of the NF.
According to the data submitted by the applicant, the CBD content was assessed using an in‐house method based on high‐performance liquid chromatography–ultraviolet spectroscopy (HPLC–UV) and ranged from 99.4% to 100.8% w/w. Water content was present up to 0.4% w/w while ash constituted less than 0.1% w/w of the NF.
Processing related impurities and substances (i.e. CBD regioisomers, dealkylated CBD, olivetol, THC and other impurities) were below their respective limits of detection (LODs) as analysed using an in‐house method based on HPLC–UV. Residual solvents in the NF were analysed by mean of chromatographic in‐house methods and were present at levels up to 9 ppm for petroleum ether, 17 ppm for ethanol, 1132 ppm for propylene glycol and 101 ppm for orange terpenes.
Heavy metals such as cadmium, lead, arsenic and mercury were analysed via spectrometry by an external laboratory. However, the validation of the method was not provided. All these parameters met the specifications set by the applicant.
Limited microbiological analyses were also conducted following USP <61> and USP <62>.5 The results showed that total aerobes counts and yeasts and moulds counts were below 500 CFU/g and 10 CFU/g, respectively. Escherichia coli was not detected in 1 g of the NF.
Information on the accreditation was provided for the laboratories that performed the analyses. While all the in‐house analytical methods were described, validation data was only provided for the HPLC–UV method and particle size distribution.
In the course of the safety assessment, the Panel requested the applicant to provide further information on methods of analysis and perform additional analyses for specific parameters (e.g. the individual quantification of Δ8‐THC and Δ9‐THC, as well as the residual starting materials and reagents that could potentially remain in the final NF).
The requested information and data were not submitted to EFSA. The Panel considers that the information provided on composition is not sufficient to adequately characterise the NF.
Stability
The applicant performed the following stability tests with the NF.
- Two batches were tested at 25 ± 2°C and at 60 ± 5% relative humidity (RH) for a period of 12 months. The batches were analysed for chemical stability, total impurities and water content. In one batch, impurities were detected after 1, 3, 6 and 12 months of storage.
- Three batches were tested for 18 months to assess the chemical and microbiological stability of the NF, as well as the particle size distribution. The Panel noted that the original report did not include the certificate of analysis for the initial time point nor information on the temperature and RH at which the samples were stored.
- Two batches were kept at 5 ± 3°C for 12 months and at 40 ± 2°C and at 75 ± 5% RH for 6 months and checked for chemical stability, total impurities and water content. All the parameters assessed remained within the specification limits.
- Five batches of the NF diluted in medium‐chain triglyceride (MCT) oil (in a formulation that the applicant claims to be the ‘typical’ formulation) were stored at 20 ± 3°C for 3 months. Stability was assessed by evaluating a broad spectrum of cannabinoids, peroxide value, water activity and the presence of microorganisms. The Panel noted that the CBD content decreased in three samples by 7% to 11% compared to the initial content, while the percentage of free fatty acids and peroxide values increased in all samples throughout the 3‐month storage period. The content of other cannabinoids remained unchanged throughout the storage period, and no microbial growth was observed.
Additional data were requested from the applicant (e.g. the shelf‐life of the NF). However, the applicant did not reply to these data requests. The Panel considers that the data provided are not sufficient to assess the stability of the NF.
Specifications
The applicant proposed specifications for the NF. During the risk assessment, the Panel requested the applicant to revise the proposed limits and to provide specifications for additional parameters.
The applicant did not reply to these data requests.
Owing to the conclusions made about the identity and the composition of the NF, the Panel considers that specifications cannot be established.
History of use of the NF and/or of its source
CBD (synthetically produced or plant‐derived) has no history of consumption as food in the EU.
CBD is a naturally occurring compound produced in the trichomes, which are primarily located on the flowering tops and leaves of Cannabis sativa L. plant. Traces of CBD can also be found on the surface of the seeds. In hemp used for fibre production, the concentration of CBD typically ranges between 0.5% and 2.0%.
Traces of CBD in hempseed oil can be detected at levels of 10–200 mg/kg. Cleaning of hempseed is not sufficient to eliminate CBD from hempseed oil.
History of use of the NF
As reported above (see Section 1.2) Epidyolex is the only CBD containing product authorised for oral consumption in EU.
Proposed uses and use levels and anticipated intake
The NF is intended to be used as a food supplement.
Target population
The target population proposed by the applicant is the general adult population, excluding pregnant and lactating women.
Proposed uses and use levels
The applicant intends to market the NF for use in food supplements, at a maximum use level of 30 mg per day (i.e. 0.43 mg/kg body weight per day for a 70‐kg adult).
Absorption, distribution, metabolism and excretion (ADME)
The applicant presented information from a literature search for the assessment of ADME.
Nutritional information
The Panel considers that, taking into account the limited data on the composition of the NF, it cannot be established whether or not the consumption of the NF is nutritionally disadvantageous.
Toxicological information
The applicant provided the studies listed in Table 2.
TABLE 2: List of toxicological studies performed with the NF.
| Reference | Type of study | Test system | Dose |
|---|---|---|---|
| Study No. 873‐0003‐GT (Unpublished, ref. 2018a) | Bacterial reverse mutation test (GLP, OECD TG 471) | S. Typhimurium TA98, TA100, TA1535 and TA1537 and Escherichia coli WP2 uvrA | Up to 5000 μg/plate (absence and presence of S9 mix) |
| Study No. 873‐0004‐GT (Unpublished, ref. 2018b) | In vitro mammalian cells micronucleus test (GLP, OECD TG 487) | Chinese hamster ovary (CHO) cells | 1–22 μg/mL (absence and presence of S9 mix) |
Owing to the lack of a correct characterisation of the fraction of small particles, including nanoparticles, of the NF (EFSA Scientific Committee, ref. 2021), the Panel cannot confirm whether the toxicological testing strategy proposed by the applicant is appropriate to assess the safety of the NF.
Genotoxicity
A bacterial reverse mutation test was performed with the NF in S. Typhimurium strains TA1535, TA1537, TA98, TA100 and Escherichia coli WP2 uvrA (Unpublished, ref. 2018a). According to the applicant, this assay was performed in compliance with Principles of GLP of 1997 and OECD TG 471 (OECD, ref. 1997).
Based on the findings of a dose range‐finding assay, the main experiment was performed with the plate incorporation method. Four Salmonella strains were tested without S9 at concentrations from 2.5 to 5000 μg/plate, and with S9 at concentrations from 5 to 5000 μg/plate. Escherichia coli WP2 uvrA was tested without S9 at levels from 40 to 5000 μg/plate, and with S9 at levels from 15 to 5000 μg/plate. Precipitation was observed at concentrations ≥ 2500 μg/plate in all tested strains with and without S9. In the absence of S9, toxicity was detected at concentrations ≥ 320 μg per plate in TA1537; in the presence of S9, at ≥ 2500 μg per plate in TA100 and TA1537.
The NF did not cause an increase in the mean number of revertant colonies compared to the negative control (DMSO) in any of the strains tested, both in the absence and in the presence of S9.
Neither confirmation of the negative results nor a justification for not conducting the confirmatory test was provided. This is not in accordance with OECD Guideline 471.
The NF was also evaluated for its potential to induce micronuclei (MN) in Chinese Hamster Ovary (CHO) cells in a study in compliance with the Principles of GLP of 1998 and with OECD TG 487 (OECD, ref. 2016) (Unpublished, ref. 2018b). A main experiment was performed in duplicate with concentrations selected based on a concentration range‐finding assay. The cells were incubated with the NF at concentrations from 2 to 28 μg/mL for 3 h treatment in the presence of S9 and from 2 to 15 μg/mL in the absence of S9, further incubated for 21 h (short‐term treatment) and from 1 to 12 μg/mL for 24 h in the absence of S9 (long‐term treatment).
Precipitation was not observed at any concentration tested. Short‐term treatment with 22 μg/mL of the NF in the presence of S9 induced 55% cytotoxicity, and the MN were scored for the samples at concentrations of 6, 18, 20 and 22 μg/mL. Short‐term treatment with 13 μg/mL of the NF in the absence of S9 induced 54% cytotoxicity and the MN were scored for the samples at concentrations of 6, 11, 12, 13 μg/mL. Long‐term treatment with 6 μg/mL of the NF induced 50% cytotoxicity and the MN were scored for the samples at concentrations of 1, 4, 5 and 6 μg/mL.
A concentration‐related and statistically significant increase in micronucleated cells was observed at 13 μg/mL after treatment for 3 h in the absence of S9 when compared to the negative control (DMSO). However, the number of MN detected was within the historical control range.
In consideration of these equivocal results and taking into account the trend of increased micronucleated cells observed in the 3 h treatment in the presence of S9 and 24 h treatment in the absence of S9 at the highest concentration, the applicant was requested to repeat the in vitro micronucleus study, using cytochalasin B.
The applicant did not reply to this data request.
Subchronic toxicity
No studies on the NF were provided. The applicant presented information from a literature search for toxicological studies.
Considering a risk assessment conducted by the NDA Panel based on the body of evidence provided in other CBD dossiers and the data gaps identified in the Statement on safety of cannabidiol as a novel food (EFSA NDA Panel, ref. 2022), the applicant was requested to provide reproductive and developmental toxicity studies and human data on consumption of CBD (see Sections 3.10.3 and 3.10.4).
The applicant did not reply to this data request.
Reproductive and developmental toxicity
No studies on the NF were provided. The applicant presented information from a literature search for reproductive and developmental toxicity studies.
As reported above, to fill the data gaps identified in the Statement on safety of cannabidiol as a novel food (EFSA NDA Panel, ref. 2022), the applicant was requested to provide reproductive and developmental toxicity studies.
The applicant did not reply to this data request.
Human data
No studies on the NF were provided. The applicant presented information from a literature search for human data.
As reported above, to fill the data gaps identified in the Statement on safety of cannabidiol as a novel food (EFSA NDA Panel, ref. 2022), the applicant was requested to provide a human study on consumption of CBD.
The applicant did not reply to this data request.
Allergenicity
The applicant presented information from a literature search on preclinical information on the allergenic potential of CBD. The Panel considers that, owing the synthetic nature of the NF, it is unlikely that the NF will trigger allergic reactions in the target population under the proposed conditions of use.
DISCUSSION
The NF which is the subject of the application is synthetic CBD.
The NF is intended to be used as food supplement in the general adult population, excluding pregnant and lactating women.
During the risk assessment, the Panel identified several data gaps needed to be addressed by the applicant.
Therefore, EFSA requests for additional information/data were sent to the applicant on 15/10/2021 and on 7/6/2022. The requested data concerned the identity, the production process, compositional data, specifications, genotoxicity, reproductive and developmental toxicity, human data regarding the NF. However, the applicant did not reply to any of these requests.
CONCLUSIONS
Based on the available data, the Panel concludes that the safety of the NF synthetic CBD cannot be established.
ABBREVIATIONS
ADME
absorption, distribution, metabolism and excretion
bw
body weight
CAS
Chemical Abstracts Service
CBD
trans‐cannabidiol
CFU
colony forming units
CHO
Chinese hamster ovary
DMSO
dimethyl sulfoxide
EMA
European Medicines Agency
GLP
Good Laboratory Practice
GMP
Good Manufacturing Practice
HPLC–UV
High‐Performance Liquid Chromatography–ultraviolet spectroscopy
ISO
International Organisation for Standardisation
IUPAC
International Union of Pure and Applied Chemistry
LOD
limit of detection
MCT
medium‐chain triglyceride
MN
micronuclei
NDA
Panel on Nutrition, Novel Foods and Food Allergens
NF
Novel Food
OECD
Organisation for Economic Co‐Operation and Development
RH
relative humidity
TG
Test Guideline
THC
tetrahydrocannabinol
USA
United States of America
USP
United States Pharmacopeia
w/w
weight for weight
REQUESTOR
European Commission
QUESTION NUMBER
EFSA‐Q‐2021‐00243
COPYRIGHT FOR NON‐EFSA CONTENT
EFSA may include images or other content for which it does not hold copyright. In such cases, EFSA indicates the copyright holder and users should seek permission to reproduce the content from the original source.
PANEL MEMBERS
Dominique Turck, Torsten Bohn, Montaña Cámara, Jacqueline Castenmiller, Stefaan de Henauw, Karen‐Ildico Hirsch‐Ernst, Ángeles Jos, Alexandre Maciuk, Inge Mangelsdorf, Breige McNulty, Androniki Naska, Kristina Pentieva, Alfonso Siani and Frank Thies.
References
- Guidance on the preparation and presentation of an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283.. EFSA Journal,, 2016. [DOI]
- Statement on safety of cannabidiol as a novel food: Data gaps and uncertainties.. EFSA Journal,, 2022. [DOI]
- Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles.. EFSA Journal,, 2021. [DOI]
- OECD . (1997). Test no. 471: Bacterial reverse mutation test. OECD guidelines for the testing of chemicals, section 4. OECD Publishing. 10.1787/9789264071247-en
- OECD . (2016). Test no. 487: In vitro mammalian cell micronucleus test. OECD guidelines for the testing of chemicals, section 4. OECD Publishing (Corrected 2023). 10.1787/9789264264861-en
- Bacterial reverse mutation test (GLP, OECD TG 471). Study no. 873‐0003‐GT (claimed as proprietary by the applicant).. 2018a
- In vitro mammalian cells micronucleus test (GLP, OECD TG 487). Study no. 873‐0004‐GT (claimed as proprietary by the applicant).. 2018b