NMR study of the interaction between MinC and FtsZ and modeling of the FtsZ:MinC complex
Abstract
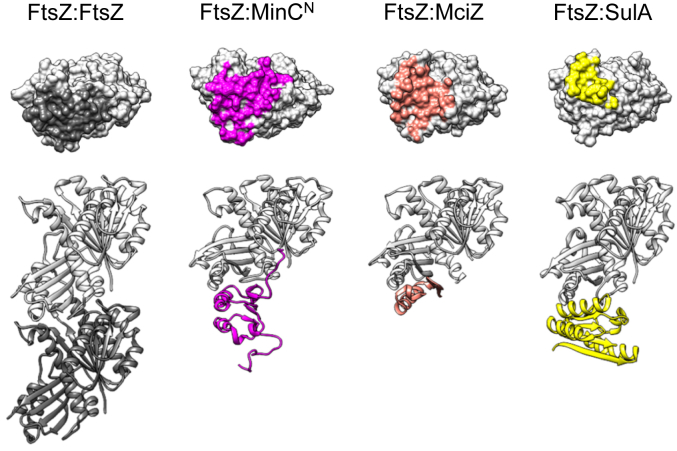
The Min system is a key spatial regulator of cell division in rod-shaped bacteria and the first FtsZ-negative modulator to be recognized. Nevertheless, despite extensive genetic and in vitro studies, the molecular mechanism used by MinC to inhibit Z-ring formation remains incompletely understood. The crystallization of FtsZ in complex with other negative regulators such as SulA and MciZ has provided important structural information to corroborate in vitro experiments and establish the mechanism of Z-ring antagonism by these modulators. However, MinC and FtsZ have so far eluded co-crystallization, probably because their complex is too unstable to be crystallized. To gain structural insight into the mechanism of action of MinC, we determined the solution structure of the N-terminal domain of Bacillus subtilis MinC, and through NMR titration experiments and mutagenesis identified the binding interfaces involved in the MinCN-FtsZ interaction. By using our experimental results as restraints in docking, we also constructed a molecular model for the FtsZ:MinCN complex and validated it by molecular dynamics. The model shows that MinCN binding overlaps with the FtsZ polymerization interface on the C-terminal globular subdomain of FtsZ and, thus, provides a structural basis for MinCN inhibition of FtsZ filament formation. Given that the C-terminal polymerization interface of FtsZ corresponds to the plus end of FtsZ filaments, we propose that capping is the main mechanism employed by MinC to antagonize FtsZ polymerization.
Article type: Research Article
Keywords: bacterial cell division, FtsZ, MinC, NMR, protein complex prediction
Affiliations: Departamento de Bioquímica, IQ, Universidade de São Paulo, São Paulo, Brazil; Brazilian Biosciences National Laboratory, LNBio, Centro Nacional de Pesquisa em Energia e Materiais (CNPEM), Campinas, Brazil; Centro de Investigaciones Biológicas Margarita Salas, Consejo Superior de Investigaciones Científicas, Madrid, Spain
License: © 2025 The Authors CC BY 4.0 This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.1016/j.jbc.2025.108169 | PubMed: 39793890 | PMC: PMC11938149
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (2.5 MB)
The Min system is a key spatial regulator of cell division in rod-shaped bacteria (ref. 1, ref. 2). It acts in concert with nucleoid occlusion proteins (SlmA in Escherichia coli and Noc in Bacillus subtilis) to restrict polymerization of the tubulin-homolog FtsZ to the proper midcell site, where a higher order assembly of FtsZ filaments known as the Z-ring will seed the formation of the bacterial divisome (ref. 3, ref. 4). Midcell division is enforced because nucleoid occlusion proteins inhibit Z-ring formation over the nucleoids, whereas the Min system prevents division from happening at the nucleoid-free regions close to cell poles (ref. 2, ref. 5, ref. 6). In the absence of Min, FtsZ rings will assemble at the cell poles and bacteria will divide asymmetrically producing anucleated minicells (ref. 7, ref. 8).
In E. coli, the Min system consists of three proteins, MinC, MinD, and MinE, which work together to generate a self-organizing oscillating protein pattern whose effect is to localize an inhibitor of Z-ring formation preferentially at the cell poles (ref. 1, ref. 2). MinC is the component of the system that interacts with FtsZ and inhibits Z ring formation and septation (ref. 9), whereas MinD and MinE are the components responsible for the oscillation (ref. 10, ref. 11). MinD is a ParA-family ATPase that oligomerizes and associates with the membrane when bound to ATP and MinE sets oscillation in motion by stimulating MinD ATP hydrolysis and membrane release (ref. 12, ref. 13). In addition to generating a time-averaged enrichment of MinC at the cell poles, the interaction of MinC with MinDE is important to concentrate MinC on the membrane, where Z-ring assembly takes place. Oscillating Min systems have also been described for other Gram-negative species (Vibrio, Xhantomonas) (ref. 14, ref. 15) and at least one cyanobacteria (ref. 16). In all cases, oscillation is associated with the existence of MinE.
An alternative paradigm to the oscillating Min system is found in B. subtilis and related rod-shaped Gram-positives. In these bacteria, the core components MinC and MinD have been conserved and execute the same functions, but they lack MinE and MinCD instead of oscillating is stably localized to the cell poles through an indirect association with DivIVA, a widely conserved pole-marking protein of Gram-positive bacteria (ref. 17, ref. 18, ref. 19). MinJ is the protein that connects MinCD to DivIVA (ref. 20, ref. 21), and the interplay between these four components generates a fixed gradient where MinCD concentrations are highest at the poles and decrease towards midcell, allowing FtsZ to polymerize there. Once a septation event occurs, DivIVA starts to accumulate at the future new pole by recognizing the negative curvature of the invaginating membrane and brings along MinCD, thus propagating the polar localization of these proteins. Because recruitment of MinCD to the new pole happens before cytokinesis is complete, the Z ring and MinCD proteins colocalize during the late part of the cell cycle in B. subtilis. Presumably, at this late stage in cytokinesis, the Z ring is no longer inhibited by MinCD. Instead, evidence suggests that MinCD is necessary at this stage for the complete disassembly of Z rings upon completion of septation (ref. 22, ref. 23, ref. 24). Z-ring disassembly has been proposed to be the main way in which MinCD prevents mini-cell formation and enforces medial division in B. subtilis (ref. 22, ref. 23, ref. 24).
Despite extensive studies, the molecular mechanism used by MinC to inhibit (or disassemble) Z-rings remains incompletely understood. MinC has two clearly independent domains connected by a flexible linker (ref. 25). The N-terminal domain (MinCN) of E. coli MinC was initially implicated as the FtsZ-inhibitory part of the protein (ref. 26, ref. 27), whereas the C-terminal domain (MinCC) mediates MinC homodimerization and interaction with MinD (ref. 27, ref. 28). Subsequently, it was recognized that MinCC is also capable of antagonizing Z-ring formation in vivo (ref. 26, ref. 29) and genetic analysis revealed that each MinC domain binds to a different portion of FtsZ. MinCN interacts with the H10 helix region at the C-terminal globular half of FtsZ, which constitutes part of FtsZ’s polymerization interface, whereas MinCC recognizes the conserved 15 residue patch in the unstructured tail—the “Conserved C-terminal peptide” or CCTP—of FtsZ (ref. 30, ref. 31). This has led to the “two-pronged model” of MinC action, in which MinC initially associates with FtsZ filaments via an interaction between MinCC and the CCTP of FtsZ, and this positions the MinCN domain to interact with its target surface (the H10 helix) on a subunit of FtsZ in the filament (ref. 30, ref. 31, ref. 32). Because the H10 helix is part of the FtsZ polymer interface, MinCN insertion between FtsZ subunits would be the ultimate cause of filament breakage. Similar, albeit less extensive, genetic data obtained with B. subtilis suggests that the two-pronged mechanism applies to this organism as well (ref. 33, ref. 34).
In vitro experiments also produced important insights into MinC’s mechanism. Initial experiments showed that MinC was capable of inhibiting the assembly of FtsZ polymers but could not determine if the effect was due to filament shortening/breakage or suppression of filament bundling (ref. 9). Importantly, MinC inhibited FtsZ assembly without affecting its GTPase activity, a property that ruled out sequestration, the mechanism employed by SulA, the SOS-induced FtsZ inhibitor of gammaproteobacteria (ref. 9, ref. 35, ref. 36). Subsequent work showed that MinC (and specifically its MinCN domain) can indeed shorten FtsZ filaments and established other general features of its inhibitory mechanism, such as the relatively weak affinity between FtsZ and MinC (Kd ∼ 1–10 μM), the inability to disassemble FtsZ filaments stabilized by non-hydrolyzable GTP analogs and its sensitivity to salt (ref. 26, ref. 30, ref. 31, ref. 33, ref. 37, ref. 38, ref. 39, ref. 40). Nevertheless, some of the results obtained by the different groups are not easily reconcilable and it is still not clear how exactly MinC shortens FtsZ filaments. Examples of contradictory observations are the works from Hernández-Rocamora and collaborators (ref. 38) and Arumugam and collaborators (ref. 37). The first described that MinC produced a homogeneous reduction of FtsZ filament size and that the protein bound preferentially to monomeric FtsZ-GDP. These observations are not compatible with a severing mechanism in which MinC would attach to filaments and break them at random positions. In contrast, Arumugam showed that MinC bound to filaments both internally and at their growing (plus) end, which corresponds to the FtsZ C-terminal polymerization face (ref. 41), and caused filament shrinking by a combination of capping and increasing the off-rate of internal subunits. Whereas some of these discrepancies may be explained by the different conditions used in the experiments (Hernández-Rocamora in solution and Arumugan in a reconstituted membrane system), it is clear that more experiments are needed before we can conclude how MinC works.
The crystal structure of FtsZ in complex with other negative regulators (SulA, MciZ) has provided important structural information to corroborate in vitro experiments and establish their mechanism of Z-ring antagonism (ref. 42, ref. 43). However, MinC and FtsZ have so far eluded co-crystallization, probably because their complex is too unstable to be crystallized. To gain structural insight into the mechanism of MinC, we determined the solution structure of the N-terminal domain of B. subtilis MinC, and through NMR titration experiments and mutagenesis identified the binding interfaces involved in the MinCN-FtsZ interaction. Using our experimental results as restraints in docking, we also constructed a model for the MinCN-FtsZ complex and validated it by molecular dynamics. The model shows that MinCN binding overlaps with the C-terminal FtsZ polymerization interface (the plus end) and, thus, provides a structural basis for MinCN inhibition of FtsZ filament formation.
Results
NMR solution structure of MinC N-terminal domain (MinCN)
To investigate the interaction between FtsZ and MinC in atomic detail, we first solved the structure of the N-terminal domain of B. subtilis MinC (MinCN) using standard heteronuclear solution NMR techniques. We assigned 94% of MinCN (96 out of 102 residues), with the missing residues being M1, K2, K15, I91 and T92. An ensemble of the 20 lowest energy conformers was selected for analysis, and the structure calculation summary is given in Table 1. The 1H, 13C, and 15N chemical shift assignments have been deposited in the BioMagResBank database (http://www.bmrb.wisc.edu), accession number 19007, and have been published (ref. 44). The calculated structure was deposited in the Protein Data Bank with the accession code 2M4I. Note that because the protein used for solving the structure contains three extra amino acids at the N-terminus, residue numbering in the 2M4I pdb does not match the Uniprot (Q01463) numbering. To avoid confusion, we refer to MinCN residues by the UniProt numbering throughout this article.
Table 1: NMR-based restraints used for MinCN structure calculation and structure refinement statistics
| Parameters | Distance restraints |
|---|---|
| NOE | 1257 |
| Intraresidue (i, i) | 399 |
| Sequential (i, i+1) | 416 |
| Medium range (i, i+2 a i+4) | 235 |
| Long range (i, i ≥ 5) | 207 |
B. subtilis MinCN shows the same overall fold as other MinCN structures available in PDB (T. maritima, 1HF2; S. typhimurium, 3GHF; E. coli, 4L1C), being comprised of a four-stranded β-sheet packed against two α-helices in a parallel orientation (Fig. 1A). The first 11 and the last 13 residues are disordered, with the latter corresponding to part of the linker connecting MinC N-terminal and C-terminal domains (residues 1–8 and 92–102, respectively in B. subtilis MinC). This disordered region in the B. subtilis structure corresponds to a fifth strand in T. maritima’s beta-sheet, whereas the equivalent segment is a third helix in the structures of enteric bacteria. Another significant difference between B. subtilis and enteric bacteria MinCN is the fact that these proteins crystallized as swapped dimers, formed by beta-complementation using strand one of each monomer (ref. 45). The monomeric solution structure of B. subtilis MinCN is well supported by the protein molecular tumbling time (Tc) of 8.35 ns and by size exclusion chromatography, corroborating the suggestion that the swapped dimers of enteric bacteria MinCN are crystallographic artifacts (ref. 32).
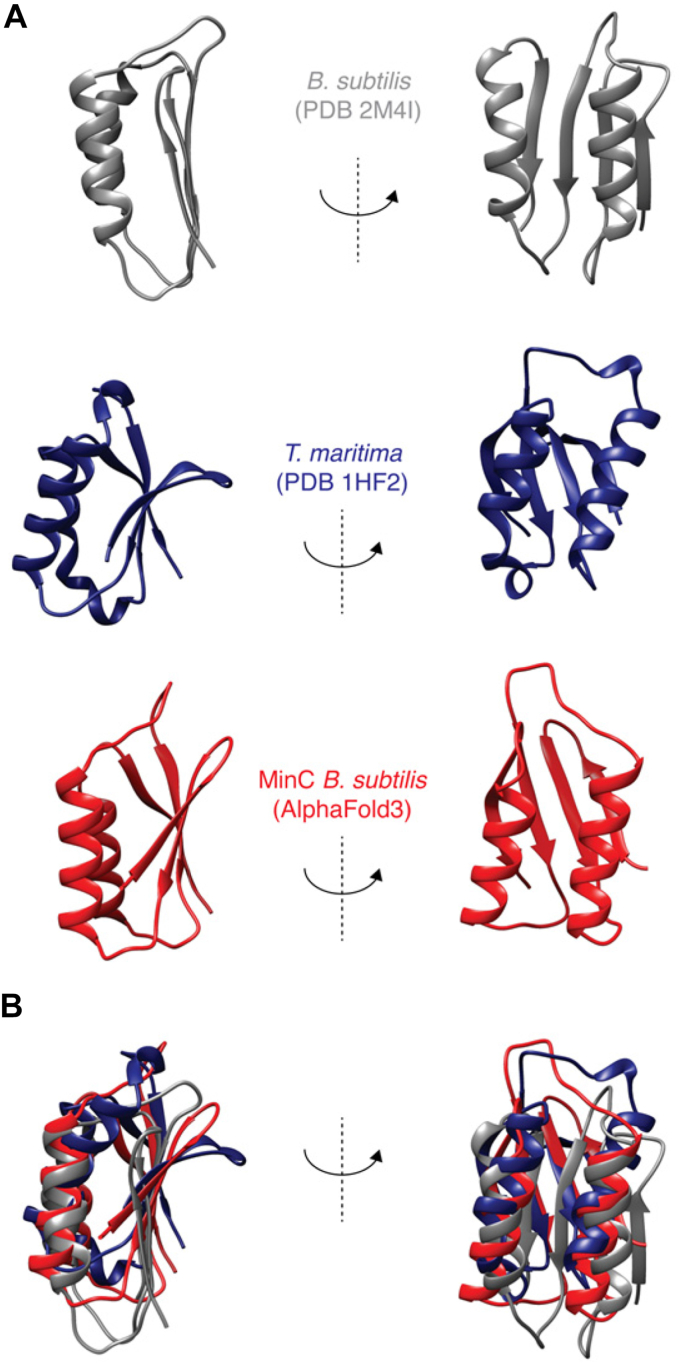
Another noteworthy difference between the B. subtilis and Gram-negative MinCN structures is the marked twist in the beta-sheet of the latter, which is absent from our structure. This twist likely affects the relative positioning of the alpha-helices H1 and H2, which are parallel to the beta strands in our structure but angled in Gram-negative MinCN. Therefore, despite their similar topology, the B. subtilis structure aligns poorly with its Gram-negative counterparts (Fig. 1B), with an RMSD of 4.9 Å when the comparison is restricted to helices H1 and H2, and 10.9 Å if the alignment includes the whole domain, with the unstructured N- and C-terminal extensions.
We also compared our structure with the Alpha Fold2 (AF2) prediction of B. subtilis MinC (https://alphafold.ebi.ac.uk/entry/Q01463) and observed that AF2′s prediction has the same beta-sheet twist and angled helices and aligns better to the Gram-negative structures than to ours (RMSD of 4.32 Å against our structure and 2.47 Å against Thermotoga) (Fig. 1B). However, the AF2 quality scores for the N-terminal domain of MinC are not very high, with the loop connecting helix H1 with strand S3, and the portion of the beta-sheet close to the loop, being regions of particularly low certainty. Reanalysis of B. subtilis MinC with Alpha Fold3 generated a similar prediction. We hypothesize reasons for the discrepancy between our experimental structure and Alpha Folds’s prediction in the discussion.
To further verify our structure, we performed auto-correlated fast timescale 15N relaxation experiments and observed that MinCN exhibits highly uniform 15N dynamics throughout its backbone with average R1 of 1.03 ± 0.01 s−1, R2 of 14.98 ± 0.7 s−1 and hetNOE of 0.7 ± 0.01 (Fig. S1). Increased dynamics are consistent with the structure, being restricted to the expected flexible regions, such as loops and in the disordered N- and C-termini. Interestingly, MinCN also showed regions—the end of helices H1 and H2 and the loop H2-S4—whose measured dynamics indicate conformational exchange. Thus, MinCN may show alternative conformations important for its interactions and function.
NMR mapping of the MinCN–FtsZ interaction
To investigate the binding site for FtsZ in MinCN, we carried out titration experiments using the ligand B. subtilis FtsZ lacking its unstructured C-terminal tail and with a mutation to keep the protein monomeric at the high concentrations necessary for NMR (A182 E, equivalent to A181 E of E. coli) (ref. 46). The monomeric state of FtsZ1-315,A182E was verified by analytical ultracentrifugation (Fig. S2). We recorded 2D [1H,15N] HSQC NMR spectra of MinCN with increasing concentrations of FtsZ1-315,A182E (1:0.12, 1:0.25, 1:0.5 M ratio) and observed two types of alterations caused by FtsZ addition: the peaks of several residues disappeared, broadened beyond detectability, whereas the peaks of another set of residues moved to new positions, i.e. showed chemical shift perturbations (CSPs), when compared to the MinCN apo spectrum (Fig. 2, A and C). Both peak shift and disappearance are indicative of an altered chemical environment but cannot distinguish between direct binding and allosteric effects (ref. 47). Peak disappearance, in particular, indicates binding in the intermediate exchange regime, which is consistent with the modest affinity between MinC and FtsZ (∼1–10 μM, depending on the pH and ionic strength) (ref. 32, ref. 38).
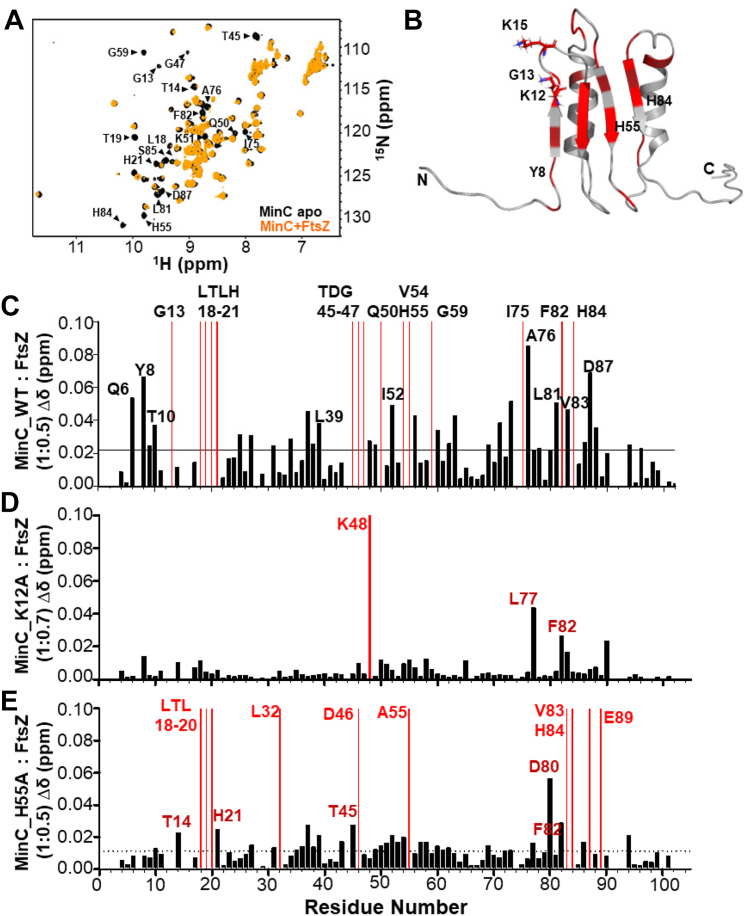
Mapping of the missing resonances and CSPs onto the MinCN structure showed that they were found predominantly on the beta-sheet and on the upper (C-terminal) part of helices H1 and H2 and the loops connecting these elements, suggesting these regions of the protein either make direct contact or are altered upon interaction with FtsZ (Fig. 2B). Interestingly, several of the residues perturbed in our titration experiments correspond to mutations identified by the Lutkenhaus and Camberg labs which disrupt MinC ability to antagonize FtsZ (Gly13, Thr19, His21, and Thr45, respectively Gly10, Ser16, Val18, and Phe42 in E. coli) (ref. 32, ref. 39). Lys12, a highly conserved lysine next to Gly13 and which is also important for MinC function in E. coli (Lys9) could not be interrogated because it was not assigned in our spectrum.
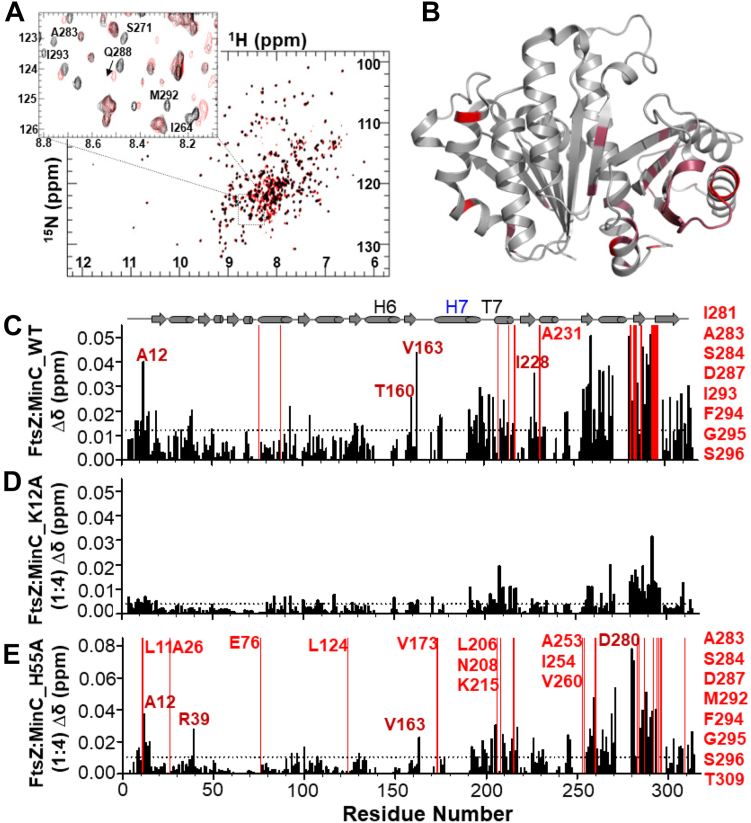
Next, to determine the binding site for MinCN in FtsZ, we carried out the reciprocal titration experiment by collecting 2D [1H,15N] TROSY NMR spectra of 2H,15N-FtsZ1-315,A182E with increasing concentrations of MinCN (1:0.25, 1:0.5, 1:1, 1:2, 1:4 M ratio). This identified CSPs and a few residues broadened beyond detectability predominantly in the C-terminal globular subdomain of FtsZ. Within this region, the majority of the altered resonances mapped to two clusters of residues between positions 250 and 300, which correspond to helix H10 and neighboring beta strands (Fig. 3, A–C). Previous genetic and biochemical experiments from the Lutkenhaus lab and our own group had already suggested that the binding site for MinCN was in the C-terminal globular half of FtsZ (ref. 30, ref. 31, ref. 33). In E. coli, mutations conferring resistance to MinC were mostly on helix H10 (L270V, R271G, E276D, N280D, I294T), whereas in B. subtilis mutations mapped to a similar but broader area, encompassing loops H9-β8 and H10-β9 in addition to helix H10 (K243, I245, D255, V260, A285, D287 and V310). Comparison of NMR and mutagenesis showed a remarkable correspondence in the affected FtsZ regions, corroborating the previous prediction that the MinC binding site in FtsZ is located in the vicinity of helix H10.
Genetic testing of the predicted MinCN interface
We tested the NMR prediction of FtsZ’s binding surface on MinCN by making mutants and assaying their function in vivo and in vitro. Because the N-terminal domain of MinC is not highly conserved between Gram+ and Gram– bacteria, instead of substituting the same residues that were mutated in E. coli MinC, we decided to choose residues to mutate based on at least two of three criteria: i) be highly conserved in Gram+ bacteria; ii) be positively charged, given that a positive charge in MinCN seems crucial for FtsZ binding (ref. 32, ref. 39), and iii) be solvent exposed and distributed over most of the interface of MinCN defined by NMR. Based on these criteria we created five substitutions (Y8A, K12A, K15A, H55A and H84A) and tested their ability to complement a minC null mutant. Mutations were constructed in the context of a GFP-MinC fusion, to allow monitoring of mutant protein stability and localization, in addition to function.
A minC null mutant exhibited frequent asymmetric division and minicell formation (16%), with consequent broadening of the cell length distribution, detectable as an increase in average cell length (from around 4.5 to almost 7 μm). Expression of the wild-type GFP-MinC fusion complemented this phenotype, as seen by the restoration of cell length and minicell formation to wild-type levels. In contrast, none of the mutant GFP-MinC fully restored proper cell division, suggesting that all the mutations perturb MinC function to some extent (Table 2). Using minicell frequency as a measure of function, the strongest mutation was K15A (7.8%), followed by Y8A and K12A (5.1 and 4.6%), and H55A and H84A (3.7 and 2.9%) being the mildest ones. Focusing on cell length, all mutations seemed similarly defective, with the exception of Y8A, whose length was closer to the wild type (Fig. S3A). All mutant proteins were expressed and accumulated to similar levels (Fig. S3C), ruling out protein instability as the explanation for their reduced function. No single mutation was capable of mimicking a null minC mutant, similar to what was observed in E. coli, where mutations that disrupt FtsZ interaction retained some function in vivo (i.e. did not have the same degree of minicelling and length distribution as the null mutant) (ref. 32, ref. 39). The fact that MinC has two binding sites on FtsZ may explain why single mutations in the N-terminal domain do not result in a null phenotype.
Table 2: Cell division phenotype of MinC WT and mutants
| Parameters | minC− | WT | Y8A | K12A | K15A | H55A | H84A |
|---|---|---|---|---|---|---|---|
| Average cell size (μm) | 6.88 | 4.75 | 4.81 | 6.02 | 5.49 | 5.88 | 5.60 |
| Minicell frequency (%) | 16.1 | 1.0 | 5.1 | 4.6 | 7.8 | 3.7 | 2.9 |
In addition to binding to FtsZ, MinC needs to interact with MinD to localize and function properly. We analyzed the subcellular localization of the mutant GFP-MinC and observed that they accumulate on cell poles and nascent septa, just like the wild-type (Fig. S3B). Given that the interaction with MinD involves MinCC and the mutations being assayed are in MinCN, this result is expected. Nevertheless, it indicates that the mutations do not destabilize the proteins or have pleiotropic effects, and is consistent with our hypothesis that the altered residues are specifically involved in the interaction between MinC and FtsZ.
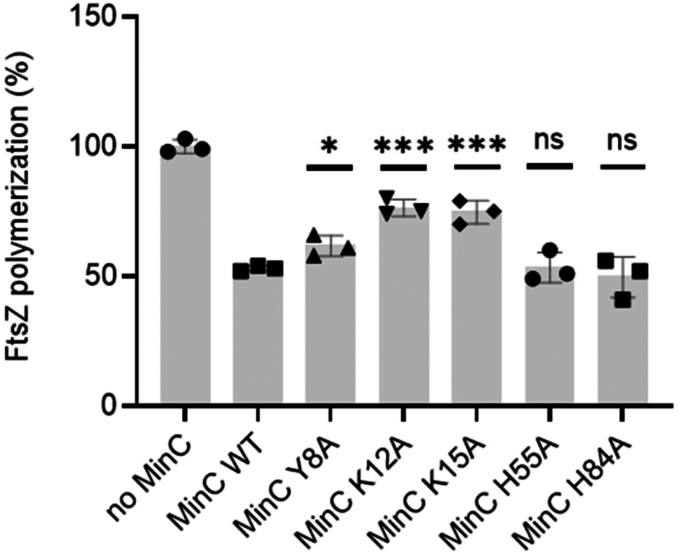
We next analyzed the ability of MinCN mutants to inhibit FtsZ polymerization in vitro, using light scattering. This time we expressed and purified wild-type and mutant MinC without a GFP fusion. In our assay condition (pH 7.7 and 100 mM potassium), wild-type MinC at a 3:1 ratio inhibited FtsZ polymerization by approximately 50%. Similar inhibition was achieved by mutants H55A and H84A, indicating that these substitutions do not significantly affect the FtsZ-MinC interaction. In contrast, MinC proteins bearing the Y8A, K12A and K15A mutations were significantly less able to inhibit FtsZ polymerization (Fig. 4). The K12A and K15A mutants were particularly impaired, inhibiting FtsZ polymerization by just 20 to 25%, thus being half as active as the wild-type protein. There was a general correlation between the mutant’s activity in vitro and its minicelling phenotype, with K12A and K15A mutations having stronger effects and H55A and H84A having the mildest effects. Y8A was harder to interpret, as it had a mild in vitro defect but minicelled as much as K12A. Nevertheless, these data suggest that not every portion of MinCN perturbed in the titration experiments is equally important for binding to FtsZ. Given that CSPs do not necessarily indicate a direct interaction of the residue perturbed, it is possible that the binding interface for FtsZ may not encompass the whole beta-sheet of MinCN, being restricted to beta strands one and two and the connecting loop, the regions where residues K12 and K15 are located. In this case, the CSPs detected for H55 and H84 could reflect a general rearrangement of MinCN‘s beta-sheet upon FtsZ interaction.
To further characterize the effects of mutations K12A and H55A, we employed NMR to directly measure the binding of these mutants to FtsZ. FtsZ titration experiments showed an almost complete loss of CSPs for MinCN K12A, even at a high MinCN-FtsZ ratio (Fig. 2D), indicating that this substitution severely impairs the MinCN-FtsZ interaction. In contrast, and in line with its mild effect in vivo and in vitro, the pattern of CSPs produced by FtsZ in MinCN H55A was similar to that observed in wild-type MinCN (compare Fig. 2, C and E), indicating that the MinCN-FtsZ interaction was generally preserved in this mutant. Reciprocal experiments titrating labeled FtsZ with unlabeled MinCN K12A and H55A produced similar conclusions: whereas K12A hardly perturbed the FtsZ spectrum, H55A induced similar CSPs as wild-type MinCN (Fig. 3, D and E). These results are consistent with the hypothesis that the binding site for FtsZ in MinCN is centered around the loop connecting strands S1 and S2, but does not seem to include contacts with the complete beta-sheet of MinCN. This conclusion is also in agreement with the mutations identified in E. coli MinCN that abolish FtsZ antagonism and which are either on the hairpin connecting strands S1 and S2 and on helix H1, but not beyond these elements (ref. 32, ref. 39).
Computational modeling of FtsZ:MinCN complex
Our NMR analysis of the MinC-FtsZ interaction, together with extensive mutagenesis work by us and others, provided an excellent set of constraints to be applied in the computational modeling of the FtsZ:MinCN complex (Fig. 5). Protein-protein docking of FtsZ monomer and MinCN was performed with the ClusPro 2.0 web server (ref. 48) and informed by NMR and mutagenesis experiments, using attraction and repulsion constraints. This generated two possible binding models of MinCN to FtsZ (Fig. S4A). Docking model A was better ranked than model B, using the cluster population criteria of ClusPro. Model A was also better when we compared the predicted interface with NMR CSP data (see “interface coverage” and “interface error” in methods). Despite the different MinCN orientation in relation to FtsZ in these models, almost in a mirrored configuration, both shared a similar interface involving the MinCN S1 and S1-S2 turn (where K12 and K15 are located) and the FtsZ final portion of H10 and S9-H10 loop (where are located residues that showed significant CSPs in NMR, as well as that conferred resistance to MinC when mutated). To investigate whether this interface was not biased by the experimental constraints applied, we also performed the same docking protocol in three other conditions: using only MinC constraints, using only FtsZ constraints, and without constraints. Importantly, all solutions produced by these alternative conditions provided an interface similar to model A (Fig. S4B).
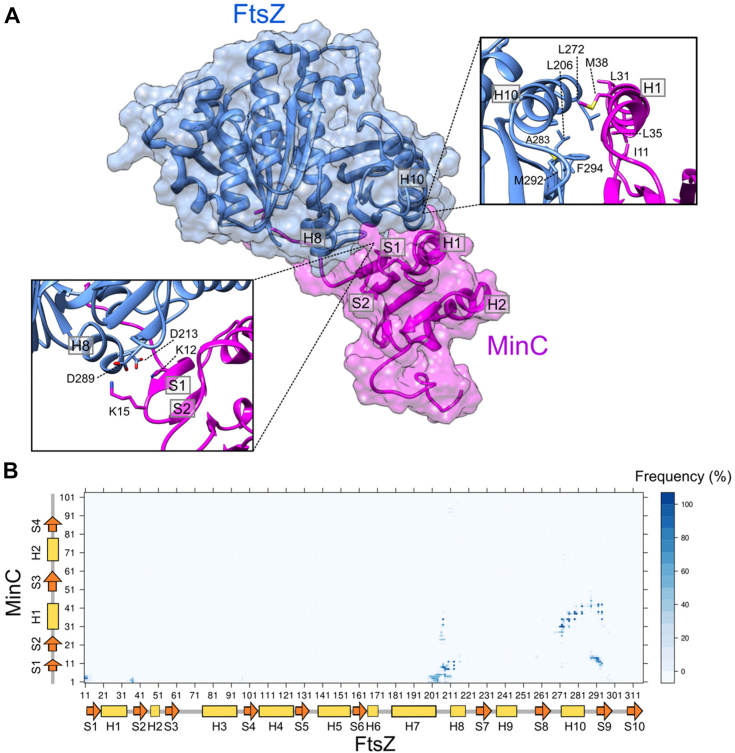
We also modeled the FtsZ:MinCN interface using AlphaFold-Multimer (ref. 49). The AlphaFold model shared a similar interface with docking model A, but with MinCN oriented perpendicularly in comparison to model A (Fig. S4C). Differently from the rigid-body docking, the AlphaFold modeling allowed the detachment of MinCN S1 from H1. This allowed MinCN S1 and S1-S2 turn in the AlphaFold model to assume a similar orientation as observed in model A, despite their orthogonal displacement (Fig. S4C, inset). In both models MinCN K12 and K15 point towards negatively charged residues in FtsZ H10-S9 loop and H8 (Fig. S4C, inset). Thus, model A and the AlphaFold model, which were obtained from different and independent computational approaches, converged to an interface region mainly involving FtsZ H10, S9, H10-S9 loop and H8.
To gain further insights on the FtsZ:MinCN interface, we submitted model A to MD simulations without constraints and evaluated the stability of the proposed FtsZ:MinCN interface by the MinCN RMSD in relation to FtsZ. MinCN remained stable during 400 ns of simulation with an average deviation of 5.7 Å from the initial configuration (Fig. S5). The contact map during the last 200 ns revealed persistent contacts of the MinCN N-terminus and S1 with the FtsZ H8 and H7-H8 loop; MinCN S1 with FtsZ S9; and MinCN H1 with FtsZ H10 and S9 (Fig. 5B). Putative hydrogen bonds (HB) were observed between side-chains of MinCN K12 or Y8 and FtsZ D213, between side-chains of MinCN K15 or K2 and FtsZ D289, and also between side-chains of MinCN E42 and FtsZ Q288 (Table S3). The refined structural model of the FtsZ:MinCN complex indicated that MinC anchors to FtsZ through two sets of contacts: electrostatic interactions between MinCN K12 and K15 with FtsZ D213 and D289, respectively (Fig. 5A, left inset), and hydrophobic helix-helix interactions between MinCN H1 and FtsZ H10 (Fig. 5A, right inset).
Discussion
We applied NMR to study the interaction between the N-terminal domain of MinC and FtsZ and used this information together with genetic data to generate a model for the MinCN-FtsZ complex. We focused on the N-terminal domain of MinC because this is the part of the protein that affects FtsZ filament assembly and length (ref. 26, ref. 27). Despite extensive work by several laboratories over many years, the mechanism of MinCN inhibition of FtsZ polymerization is still incompletely understood and controversial (ref. 26, ref. 30, ref. 32, ref. 33, ref. 37, ref. 38, ref. 39, ref. 40). One important missing link is structural information showing how MinCN and FtsZ interact, something that has eluded attack by crystallography or cryoEM because the proteins do not form a stable complex. However, NMR can still provide valuable structural insight into transient protein-protein interactions that cannot be captured by other methods (ref. 50). Our results indicate that MinCN binds to FtsZ at the bottom of its C-terminal globular domain, which corresponds to the plus end of the protein, by contacting predominantly helix H10, and strands S8 and S9 and covering a large part of the surface that FtsZ uses to interact with itself. Such binding site is in principle compatible with MinCN acting by capping or severing of FtsZ filaments, or even by sequestration of FtsZ monomers, but rules out other models in which MinCN effect would occur indirectly via inhibition of filament bundling, as previously proposed by ourselves (ref. 33). Furthermore, it provides a structural basis to inform further experimentation to fully elucidate the mechanism of this prominent yet still mysterious regulator of bacterial cell division.
The experiments and model described here employed B. subtilis proteins, but we expect that their conclusions should be generally applicable, including to E. coli where most work with MinC has been carried out. Despite the low conservation of the amino acid sequence, the structure of B. subtilis MinCN has the same topology and is quite similar to MinCN of other bacteria whose experimental structures are available (T. maritima – 1HF2; E. coli – 4L1C; S. typhymurium – 3GHF). Moreover, mutations that perturb the MinC-FtsZ interaction identified in E. coli and B. subtilis map to similar regions and, in some cases, identical or equivalent residues in both species. Examples are the mutations in the H10 helix of FtsZ that make it resistant to MinC (ref. 30, ref. 33, ref. 34), and the conserved lysine in MinC’s first beta-strand/loop (K9 in E. coli and K12 in B. subtilis), whose substitution strongly diminishes MinCN inhibitory activity (ref. 32, ref. 39) and this work). Lastly, the biochemical properties of B subtilis MinC, despite being less thoroughly explored, are in general agreement with E. coli‘s, including the shortening of filaments without inhibition of FtsZ GTPase, the salt sensitivity of the interaction with FtsZ and the rescuing effect of ZapA (ref. 33, ref. 40).
Evaluation of the modeled FtsZ:MinCN complex
The docking model of the FtsZ:MinCN complex was constructed using information from CSPs of MinC and FtsZ, as well as mutations in each protein that disrupt their interaction. Neither CSPs or mutant data are free of ambiguity by themselves, but the combination of both should increase the chance that our model is of high quality and closely reflects the real structure of the complex. Independent evidence of the quality of the model can be obtained by checking how its predicted interface matches with mutations that disrupt the MinCN-FtsZ interaction in E. coli and which were not used to inform the construction of our model. As seen in Fig. S6 and Table S5, there is an excellent correlation between the phenotype of a mutation and its presence (or not) at the interface of the complex. Among the cases that disagree, there are two residues on strand S2 (T19 and H21) that are too distant to make direct contact but are close and likely influence the residues on strand S1 that contact FtsZ.
Another evidence that our model is correct is the similarity to the one predicted by AlphaFold Multimer, with the same surface of FtsZ being used in both cases, strengthening the conclusion that the S8, S9 and H10 elements in the plus end of FtsZ are the binding site for MinCN. Interestingly, however, there was a difference in the orientation of MinCN in the two models, with MinCN in Alpha Fold’s rotated around 90 degrees compared to ours. The altered orientation also translated into different contacts made by MinCN, which included helix H1 in our model but not in AlphaFold’s. The reason for this disagreement between the models seems to be a difference in the structure of MinCN itself, which has alternative orientations of the helices H1 and H2 relative to the beta-sheet formed by S1-4, when we compare our experimental structure with AlphaFold’s (Fig. 1, A and B). A recent survey of the accuracy of AlphaFold models showed that even very high-confidence predictions can differ from experimental maps on a global scale through distortion and domain orientation, as observed here (ref. 51). The N-terminal domain of MinC may pose a special challenge to Alpha Fold, as it is a particularly poorly conserved protein. This is evident from the existence of two different PFAM entries for this domain, one that recognizes the N-terminal domain of Gram-negative bacteria (PF05209, MinC_N) and an alternative one that recognizes Gram-positive proteins and which was named “MinC, N-terminal domain-like” (PF22642, MinC_N_1) (https://www.ebi.ac.uk/interpro/set/pfam/CL0802/). The poor conservation of MinCN may be the cause of the low confidence of the Alpha Fold model of this domain. We also wonder whether the similarity of the Alpha Fold prediction to Gram-negative MinC (Fig. 1B) could be the result of the predominance of these structures in its training set.
To seek further evidence of which model better reflects reality, we also evaluated their general interface features (buried area, polar versus nonpolar contacts, etc.) and how the Alpha Fold interface matched with the E. coli mutations that disrupt the MinCN-FtsZ interaction. The interfaces in both complexes have similar overall features, with a slightly larger buried area and frequency of nonpolar contacts in the case of our complex (Table S4). However, there was a better correlation between E. coli mutations and the predicted interface in our model than in Alpha Fold’s, with five out of eight mutations corresponding to predicted contacts in our model versus three out of eight for Alpha Fold’s (Table S5 and Fig. S6). Importantly, there were two mutations on helix H1 (E42, T45 in B. subtilis), which is part of the MinCN-FtsZ interface only in our model. A third mutation on the same helix which did not affect MinC binding (A41) faced away from the interface, consistent with the contacts predicted for H1 in our model. The discrepancy between the AlphaFold model and E. coli mutations reinforces that the AF2 prediction of MinC structure is problematic, and our proposed MinCN orientation in the complex is likely correct. However, regardless of which model is more accurate, they are equivalent in the way FtsZ is affected and hence the mechanistic implications will be the same, as described below.
FtsZ:MinCN structure is compatible with MinCN acting as a capper
How does the proposed structure of the FtsZ:MinCN complex advances our understanding of MinC’s mechanism? A widely accepted view of how MinC shortens FtsZ filaments is the “two-pronged model”, put forth by Lutkenhaus and collaborators (ref. 30, ref. 31). This model proposes that MinC gets recruited to FtsZ filaments via an interaction between FtsZ’s CTP and MinCC and this, in turn, positions MinCN so that it can break the nearest Z-Z bond. Such breaking of filaments at internal bonds is known as severing and has been shown to occur for actin and microtubules and require some form of active splitting of subunits, often by imposing mechanical stress on the filament (ref. 52, ref. 53). Because MinC only affects FtsZ filaments that hydrolyze GTP, one assumption is that MinCN can only break bonds that are occupied by GDP and which are flexible enough to allow MinCN to wedge itself in between FtsZ subunits to find its binding surface. Even though this model is consistent with genetic and some biochemical data, our prediction that MinCN and FtsZ share a large interaction surface (2353 Å2 buried area) and its location far inside the FtsZ polymerization interface suggests that it is unlikely that MinCN would be able to reach its binding site by “sneaking in” between FtsZ subunits of a filament, even if these are connected via a GDP bond. Both molecular dynamics as well as structures of filaments in GDP-bound state have shown that the elements recognized by MinCN in FtsZ (H10, S9, S8) remain in intimate contact with the neighboring FtsZ subunit when GDP is at the interface (ref. 54, ref. 55, ref. 56, ref. 57, ref. 58). In fact, the opening of the interface upon GTP hydrolysis has been proposed to take place at the opposite side recognized by MinCN (see (ref. 56) and MovieS2 in (ref. 55). Thus, given that MinCN can only access its binding surface on FtsZ after the filament is broken, we conclude that for severing to occur another part of MinC (MinCC?) must be in charge of breaking the filament to expose MinCN‘s binding site. However, data from several laboratories showed that MinCC or full length MinC with mutations in MinCN are not capable of shortening or breaking FtsZ filaments (ref. 26, ref. 32, ref. 39). Another argument against severing is the observation that MinC-treated FtsZ filaments maintain a narrow size distribution, instead of the broader one expected if filament breakage occurred randomly along their lengths (ref. 38). Thus, our structural predictions, together with previously published biochemical data, are inconsistent with MinC acting as a severing protein. Instead, the binding site for MinCN seems more consistent with MinC acting by capping or sequestration. Capping the plus end of FtsZ filaments is a highly effective way to promote filament shortening by blocking the addition of subunits to its growing end, as shown for MciZ, and more recently for MipZ (ref. 42, ref. 59, ref. 60). Sequestration is also an effective way to shorten filaments and the mechanism employed by SulA (ref. 35, ref. 36, ref. 59), and probably also by the less well-characterized modulators OpgH (ref. 61) and Kil (ref. 62), but usually requires high affinity for FtsZ, much higher than the affinity displayed by MinC. Capping and sequestration can have similar effects on filament length distribution and it is not uncommon that cappers will also display sequestration activity depending on the experimental regimen (ref. 42). However, cappers and sequesterers differ in one critical way: whereas cappers are capable of increasing the GTPase activity of FtsZ (via increased filament ends/turnover), sequesterers always reduce FtsZ’s GTPase (ref. 59). Although no systematic studies have been carried out to investigate MinC effect on FtsZ GTPase under different stoichiometries (something necessary to reveal increases in GTPase activity), the fact that MinC shortens filaments without reducing FtsZ’s GTPase strongly suggests that this negative modulator functions primarily as a capper.
FtsZ’s plus end is the main target of negative modulators
Another noteworthy conclusion from this work is that the binding site recognized by MinCN in FtsZ overlaps substantially with the ones employed by SulA and MciZ, other negative modulators that inhibit filament formation by sequestration and capping respectively, and the only ones with a crystal structure in complex with FtsZ (ref. 42, ref. 43) (Fig. 6). In addition, recent data indicates that MipZ, a C. crescentus negative modulator responsible for Z ring positioning also interacts with the H10 region of FtsZ (ref. 60). Thus, the polymerization surface on the C-terminal globular domain of FtsZ – the plus end – seems to be a hotspot for inhibitors of filament formation and we can speculate why this is so. One possibility is that the plus end surface of FtsZ has structural features that make it easier to accommodate new protein-protein interactions. More likely, however, inhibitors evolved to hit FtsZ on the plus end, as this is the most effective way of promoting the disassembly of filaments that have kinetic polarity and undergo treadmilling like actin and FtsZ (ref. 42, ref. 59, ref. 63). Binding to the N-terminal polymerization face – the minus end – would instead promote filament nucleation and stabilization (ref. 64). Targeting the N-terminal face would only inhibit filament formation if it caused a concurrent conformational change which prevented the subunit from associating with a filament, i.e., a sequestration mechanism.
In conclusion, we have applied a combination of NMR and modeling to produce the first atomic description of the FtsZ-MinC interaction, and the information obtained strongly suggests that MinC inhibits FtsZ polymerization by a capping mechanism. A similar strategy could be employed to advance our knowledge of other FtsZ negative regulators that are unsuitable for crystallography and whose mechanisms remain poorly understood.
Experimental procedures
MinCN NMR assignment and structure determination
15N-13C-labeled MinCN was expressed from plasmid pAT6 as a 6-His-tagged protein. Cells were grown in M9 minimal medium at 37°C with 1 g/L of 15NH4Cl and 2 g/L of 13C glucose. The MinCN protein expression was induced at OD600 = 0.8 using 0.5 mM IPTG, after induction cells were incubated at 25 °C for 8h. Following collection by centrifugation, cells were resuspended in lysis buffer (50 mM sodium phosphate buffer pH 7.4 and 50 mM KCl) and lysed by sonication. The supernatant of the cell lysate was loaded onto a HisTrap column (5 ml-GE Healthcare) and the bound protein was eluted with an imidazole gradient (0–1 M). The fractions containing MinCN were collected, concentrated and cleaved with thrombin (1 U/ml) for 16h at 4 °C. After cleavage, the protein was purified by size exclusion chromatography using a Sephadex 200 16/60 (GE Healthcare) column with the lysis buffer.
NMR spectra were collected from 1H,15N,13C-labeled MinCN at 450 μM in 50 mM KCl and 50 mM Na2HPO4, pH 7.4 (10% D2O/90% H2O). All data were acquired at 293 K on an Agilent 600 MHz spectrometer, equipped with an inverse detection triple resonance cryoprobe, processed with NMRPipe (ref. 65) and visualized with NMRView (ref. 66).
To solve the solution structure of MinCN the sequential backbone resonance assignments were obtained using 2D 15N-HSQC, 3D CBCA(CO)NH/HNCACB and 3D HNCO/HN(CA)CO experiments. Side chains for both aliphatic and/or aromatics were assigned using 3D HCCH- and CCH-TOCSY, 13C NOESY-HSQC and 13C HSQC experiments. Distance restraints for structure calculations were derived from cross-peaks in 15N-edited NOESY-HSQC (τm = 80 ms), 13C-edited aliphatic and aromatic NOESY-HSQC in H2O (τm = 80 ms) respectively. Peak picking was performed manually using NMRView (ref. 66).
NOE assignment and structure calculations were performed using CYANA (version 2.1) (ref. 67) in a semi-automated interactive manner, using 100 starting conformers. CYANA protocol was applied to calibrate and assign NOE cross peaks. After the first rounds of automatic calculations, the NOESY spectra was analyzed again to identify additional cross peaks consistent with the structural model and to correct misidentified NOEs. Slowly exchanging amides were identified by lyophilizing the protein from water and then dissolving it in 100% D2O. The final 20 lowest-energy structures were refined with the CNS package. The resulting structures were analyzed using PROCHEK validation software (ref. 68). The final refined ensemble of 20 structures and resonance assignments for MinCN domain were deposited into the Protein Data Bank (PDB ID 2M4I) and BioMagRes DB (BMRB accession number 19007), respectively.
Expression and purification of FtsZ and MinC for NMR titration
FtsZ1-315,A182E and MinC1-102 (hereafter referred to as MinCN) were subcloned into RP1B vector to generate N-terminal His-tagged proteins (plasmids and strains used in this work are listed in Tables S1 and S2). Site-directed mutagenesis was used to create variants of MinC. For protein expression, plasmid DNAs were transformed into E. coli BL21(DE3) cells. Cells were grown in Luria broth in the presence of selective antibiotics at 37 °C to an O.D.600nm of ∼ 0.8, and expression was induced by the addition of 0.5 to 1 mM isopropyl thio-β-D-galactoside. Induction proceeded overnight at 18 °C prior to harvesting by centrifugation at 7647 × g (15 min, 4 °C). Cell pellets were stored at −80 °C until purification. For NMR measurements, expression of uniformly 2H,15N or 2H,15N,13C-labeled FtsZ was accomplished by growing cells in D2O-based M9 minimal medium. 1H,15N or 1H,15N- labeled MinC was obtained by growing cells in M9 minimal medium. Both media contained 1 g/L of 15NH4Cl and/or 3 g/L of D-[2H,13C]-glucose (CIL) as the sole nitrogen and carbon sources, respectively. Multiple rounds (25, 50, 70, and 100% of D2O) of adaptation were necessary for the high yield expression of FtsZ.
Cells pellets were resuspended in Buffer A (50 mM Tris-HCl, pH 8.0, 500 mM KCl, 5 mM imidazole, 5% glycerol) with 0.1% Triton X-100, 1 mM PMSF and 100 μg/ml lysozyme for both proteins, and 20 μM GDP for FtsZ. After resuspension, cells were lysed using high-pressure homogenization (Avestin C3 EmulsiFlex). The lysate was cleared by centrifugation (40,905 × g, 45 min, 4 °C). The supernatant was filtered and loaded onto a His-Trap 5 ml column equilibrated in Buffer A. Protein was eluted in a step gradient of Buffer B (50 mM Tris-HCl, pH 8.0, 500 mM KCl, 500 mM imidazole, 5% glycerol). Protein came out in the fraction of 30% of Buffer B. The pooled eluted protein was incubated with tobacco etch virus (TEV) protease overnight at 4 °C in dialysis buffer C (50 mM Tris-HCl, pH 8.0, 500 mM KCl, 5% glycerol). The next day, a subtraction His-Trap purification was performed to remove TEV and the cleaved His tag. Final purification was achieved using SEC (Superdex 75 16/60, GE Healthcare) equilibrated in Buffer D (20 mM Hepes, pH 7.4, 500 mM KCl, 1 mM EDTA). The eluted protein was dialyzed against the Analysis Buffer (20 mM Hepes, pH 7.4, 150 mM KCl, 1 mM EDTA).
NMR analysis of FtsZ-MinC interaction and MinCN dynamics
NMR data were collected using Bruker Avance III 800 MHz spectrometer equipped with a TCI Z-gradient cryoprobe at 298 K. NMR measurements of FtsZ or MinC were recorded using 2H,15N-labeled FtsZ or 1H,15N-labeled MinC at a final concentration of 0.1 mM in presence of ligands, in Analysis Buffer and 90% H2O, 10% D2O. Chemical shift perturbation (Δδ) of 2H,15N-FtsZ in the presence of unlabeled MinC or 1H,15N-MinC in the presence of unlabeled FtsZ, were calculated using the following equation (Equation 1):
The backbone assignment of FtsZ was obtained using 2H,15N,13C-labeled FtsZ (0.2 mM) and through the analysis of 3D TROSY-HNCA, 3D TROSY-HN(CO)CA and 3D TROSY-HNCACB experiments. All NMR data were processed using NMRpipe (ref. 65) and analyzed using CcpNMR version 2.4 (ref. 69) or CARA (http://www.nmr.ch).
MinCN backbone 15N longitudinal (R1) and transverse (R2) relaxation rates were obtained by using inversion recovery (R1) and CPMG (R2) HSQC based Bruker pulse sequences. T1 and T2 were acquired with a recycle delay of 3 s between experiments and the following relaxation delays for T1: 150, 300, 450, 600, 900, 1250, 1500, 1800, 2000 and 2225 ms; and T2: 16.32, 32.64, 48.96, 65.28, 81.60, 97.92, 114.24, 130.56 and 163.20 ms. Systematic error in both T1 and T2 experiments were estimated from the variance of repetition experiments, which were acquired with delays of 600 ms for T1 and 81.60 ms for T2. 1H,15N-NOE (hetNOE) values were determined from a pair of interleaved spectra acquired with or without pre-saturation and a recycle delay of 5 s. Fittings of the intensity decay curves to calculate R1 and R2 relaxation rates and the hetNOE were carried out using CCPN software (ref. 69).
Analytical ultracentrifugation
Sedimentation velocity experiments were carried out in a Beckman Optima XL-I analytical ultracentrifuge equipped with interference optics, as described (ref. 70), in Analysis Buffer with 1.5 mM GDP or 1.5 mM GTP at 25 C.
Light scattering
For light scattering experiments, FtsZ1-382 and MinC1-224 WT and mutants were used. pET28a-FtsZ1-382 (pAB20) and pET24b-MinC1-224 (pAT30) WT and mutants were expressed in E. coli Bl21(DE3)-RIL (Stratagene) upon induction by 0.1 mM IPTG. FtsZ1-382 was purified by two-step precipitation: 30% ammonium sulfate, followed by the addition of 1 mM GTP and 20 mM Ca2+. MinC1-224 WT and mutants were purified by nickel affinity chromatography. His-MinC were eluted from the column applying an imidazole gradient (100 mM to 1 M) in HMK buffer (50 mM Hepes, pH 7.7, 5 mM Mg(CH3COO)2, 100 mM KCH3COO).
Light scattering by FtsZ polymers was measured using a Hitachi F-4500 fluorimeter. Excitation and emission wavelengths were set to 350 nm, with slit widths of 3.5 nm, and the photomultiplier tube at 950 V. Protein was incubated in 150 μl of HMK buffer at 25 °C until baseline stabilization. GTP was added to 2 mM to start polymerization and the change in scattering was followed for the next 30 min. MinC was added to reactions either before or after the addition of GTP with similar results. Buffer effects were excluded by the inclusion of MinC storage buffer to a similar volume as the maximum amount of MinC added.
Fluorescence microscopy
Microscopy was performed on a Nikon Eclipse Ti microscope, equipped with GFP BrightLine and mCherry BrightLineFilter Sets (Semrock), a Plan APO VC Nikon 100X objective (NA = 1.4), a 25 mm SmartShutter and Andor EMCCD i-Xon camera. Exposure times varied from 0.3 to 1 s. Cells were grown to the exponential phase and incubated in chambers with LB plus 1% agarose. Membrane stain FM5-95 (final concentration of 5 μg/ml; Molecular Probes) and 0.5 mM IPTG were added to the solidified LB, where indicated. All images were captured using NIS software version 3.07 (Nikon) and processed with ImageJ software (http://rsb.info.nih.gov/ij/).
Western blotting
Cell lysates were resolved on SDS PAGE gels, transferred to PVDF membrane and probed with anti-GFP rabbit polyclonal serum (kind gift of D. Rudner) diluted to 1:3000. After washing, secondary anti-IgG conjugated with HRP (Pierce) was added at 1:10,000. Immunoblots were revealed with ECL Prime Western Blotting Detection Reagent (GE Healthcare) in High Performance Chemiluminescence Film (GE Healthcare).
Molecular docking and modeling
Protein-protein docking employed the ClusPro 2.0 web server (ref. 48). B. subtilis FtsZ and MinCN structures were obtained from PDB with IDs 2VAM and 2M4I, respectively. To avoid bias in the rigid-body docking approach, we removed the flexible unstructured N and C-terminal tails of MinCN, (residues 1–10 and 94–105, respectively) from the structure prior to docking. Protein-protein contact information obtained from NMR and mutagenesis experiments were used to optimize docking solutions. Attraction constraints were applied to FtsZ residues 255, 256, 257, 258, 284, 285, 286, 287, 288, 289, 290, and 291 and MinCN residues 12 and 15. The top 10 ranked structures provided by ClusPro were further analyzed and the solutions that caused obstruction of the FtsZ GTP binding site or the FtsZ unstructured C-terminal tail, which is not present in crystallographic structure, were discarded. Two FtsZ:MinCN models that successfully met the structural criteria were further evaluated. Three other conditions were alternatively applied in the docking approach: only MinCN attraction, only FtsZ attraction or no constraints for both proteins.
To assess which model better described the FtsZ:MinCN complex interface, we compared the predicted interface with the interface identified by NMR. We used a distance cut-off of 5 Å, including hydrogen atoms, to define interface residues and considered only the residues that were common to the ClusPro and Alphafold-Multimer models. We then calculated an interface coverage as the ratio of MinCN or FtsZ residues from the predicted interface that presented chemical shift perturbation in NMR, as follows (Equation 2):
Where Ipred is the set of FtsZ or MinC residues from the predicted interface of the model and INMR is the set of FtsZ or MinC residues that presented chemical shift perturbation in NMR experiments. The number of residues in each set is defined by the cardinality of the set.
We also determined an interface error as the ratio of the number of MinCN or FtsZ residues from the predicted interface that did not show chemical shift perturbation by the total of residues from the predicted interface, as follows (Equation 3):
Where Ipred is the set of FtsZ or MinC residues from the predicted interface of the model and INMR is the set of FtsZ or MinC residues that presented chemical shift perturbation in NMR experiments. The number of residues in each set is defined by the cardinality of the set. The best model was the one with the highest ratio between coverage and error percentage, according to Equations 2 and 3.
In addition to an algorithm based on physical energy functions, such as ClusPro, we also modeled the FtsZ:MinCN complex by Alphafold-Multimer (ref. 49). AlphaFold-Multimer (v2.1.1) was performed with a local installation from (https://github.com/deepmind/alphafold). The FtsZ and MinC FASTA sequences were used as input and obtained from PDB (PDB ID: 2VAM and 2M4I, respectively). AlphaFold was executed in multimer mode with a prokaryote flag. The maximum template data was set to 2021-01 to 27. The analysis and comparisons with docking models were performed using the best ranked relaxed predicted structure generated by the algorithm.
Molecular dynamics simulations
Molecular dynamics (MD) simulations of the selected FtsZ:MinCN model were performed to evaluate the stability of the complex and the interface contacts. The simulations used the YASARA software (ref. 71) with AMBER14 force field. Prior to simulations, we restored the MinCN N- and C-terminal tails that were previously removed from the NMR structure and added ACE and NME caps to FtsZ N- and C-terminus. The FtsZ:MinCN complex was solvated with water in a 15 Å cubic cell with periodic boundaries, neutralized and submitted to energy minimization. Hydrogen-containing bonds were constrained using the LINCS algorithm. Long-range electrostatic interactions were calculated using the Particle Mesh Ewald algorithm and short-range non-bonded interactions were calculated with a 8 Å cutoff. The production run was performed at 298 K and pH 7.4 with a multiple 2 × 2.5 fs time step. Simulation snapshots were saved each 250 ps.
The stability of MinCN in relation to FtsZ was assessed by first aligning the simulation trajectory by FtsZ and then calculating the root-mean-square deviation (RMSD) of the MinCN backbone, not considering the N- and C-terminal tails, using VMD software. The last 200 ns of the simulation, in which the system achieved RMSD convergence, was used for further analysis. Putative hydrogen bonds between FtsZ and MinCN during the simulation were computed in VMD, using default criteria of donor-acceptor distance of 3 Å and angle cut-off of 20°. The trajectory was submitted to a hierarchical cluster analysis and the structure closest (minimum RMSD) to the average structure of the most populated cluster was selected as a representative model of the simulation. This structure was further refined by energy minimization.
Data availability
MinCN structure is available as PDB accession 2M4I and assignments under BMRB 19007. FtsZ backbone assignments have been deposited in BMRB under accession number 52358. The 3D structural models presented in this study are available in https://github.com/LBC-LNBio/MinC-FtsZ_models.
Supplementary Materials
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- J. Lutkenhaus. Assembly dynamics of the bacterial MinCDE system and spatial regulation of the Z ring. Annu. Rev. Biochem., 2007. [PubMed]
- V.W. Rowlett, W. Margolin. The Min system and other nucleoid-independent regulators of Z ring positioning. Front. Microbiol., 2015. [DOI]
- S. Du, J. Lutkenhaus. At the heart of bacterial cytokinesis: the Z ring. Trends Microbiol., 2019. [PubMed]
- R. McQuillen, J. Xiao. Insights into the structure, function, and dynamics of the bacterial cytokinetic FtsZ-ring. Annu. Rev. Biophys., 2020. [PubMed]
- D.W. Adams, L.J. Wu, J. Errington. Cell cycle regulation by the bacterial nucleoid. Curr. Opin. Microbiol., 2014. [PubMed]
- M. Bramkamp, S. Van Baarle. Division site selection in rod-shaped bacteria. Curr. Opin. Microbiol., 2009. [PubMed]
- P.A. de Boer, R.E. Crossley, L.I. Rothfield. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell, 1989. [PubMed]
- P.A. Levin, J.J. Shim, A.D. Grossman. Effect of minCD on FtsZ ring position and polar septation in Bacillus subtilis. J. Bacteriol., 1998. [PubMed]
- Z. Hu, A. Mukherjee, S. Pichoff, J. Lutkenhaus. The MinC component of the division site selection system in Escherichia coli interacts with FtsZ to prevent polymerization. Proc. Natl. Acad. Sci. U A., 1999
- D.M. Raskin, P.A. de Boer. Rapid pole-to-pole oscillation of a protein required for directing division to the middle of Escherichia coli. Proc. Natl. Acad. Sci. U A., 1999
- D.M. Raskin, P.A. de Boer. MinDE-dependent pole-to-pole oscillation of division inhibitor MinC in Escherichia coli. J. Bacteriol., 1999. [PubMed]
- B. Ramm, T. Heermann, P. Schwille. The E. coli MinCDE system in the regulation of protein patterns and gradients. Cell. Mol. Life Sci., 2019. [PubMed]
- L. Wettmann, K. Kruse. The Min-protein oscillations in Escherichia coli: an example of self-organized cellular protein waves. Philos. Trans. R. Soc. B Biol. Sci., 2018
- E. Galli, M. Poidevin, R. Le Bars, J.-M. Desfontaines, L. Muresan, E. Paly. Cell division licensing in the multi-chromosomal Vibrio cholerae bacterium. Nat. Microbiol., 2016
- A.S.G. Lorenzoni, G.C. Dantas, T. Bergsma, H. Ferreira, D.-J. Scheffers. Xanthomonas citri MinC oscillates from Pole to Pole to ensure proper cell division and shape. Front. Microbiol., 2017. [DOI | PubMed]
- J.S. MacCready, J. Schossau, K.W. Osteryoung, D.C. Ducat. Robust M in-system oscillation in the presence of internal photosynthetic membranes in cyanobacteria. Mol. Microbiol., 2017. [PubMed]
- L.R. Hammond, M.L. White, P.J. Eswara. ¡vIVA la DivIVA!. J. Bacteriol., 2019. [DOI]
- A.L. Marston, H.B. Thomaides, D.H. Edwards, M.E. Sharpe, J. Errington. Polar localization of the MinD protein of Bacillus subtilis and its role in selection of the mid-cell division site. Genes Dev., 1998. [PubMed]
- H. Strahl, L.W. Hamoen. Finding the corners in a cell. Curr. Opin. Microbiol., 2012. [PubMed]
- M. Bramkamp, R. Emmins, L. Weston, C. Donovan, R.A. Daniel, J. Errington. A novel component of the division site selection system of Bacillus subtilis and a new mode of action for the division inhibitor MinCD. Mol. Microbiol., 2008. [PubMed]
- J.E. Patrick, D.B. Kearns. MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol. Microbiol., 2008. [PubMed]
- J.A. Gregory, E.C. Becker, K. Pogliano. Bacillus subtilis MinC destabilizes FtsZ-rings at new cell poles and contributes to the timing of cell division. Genes Dev., 2008. [PubMed]
- S. Van Baarle, M. Bramkamp. The MinCDJ system in Bacillus subtilis prevents Minicell Formation by promoting divisome disassembly. PLoS One, 2010
- Y. Yu, J. Zhou, F. Dempwolff, J.D. Baker, D.B. Kearns, S.C. Jacobson. The min system disassembles FtsZ foci and inhibits polar peptidoglycan remodeling in Bacillus subtilis. mBio, 2020
- S.C. Cordell, R.E. Anderson, J. Lowe. Crystal structure of the bacterial cell division inhibitor MinC. EMBO J., 2001. [PubMed]
- A. Dajkovic, G. Lan, S.X. Sun, D. Wirtz, J. Lutkenhaus. MinC spatially controls bacterial cytokinesis by antagonizing the scaffolding function of FtsZ. Curr. Biol., 2008. [PubMed]
- Z. Hu, J. Lutkenhaus. Analysis of MinC reveals two independent domains involved in interaction with MinD and FtsZ. J. Bacteriol., 2000. [PubMed]
- T.H. Szeto, S.L. Rowland, G.F. King. The dimerization function of MinC resides in a structurally autonomous C-terminal domain. J. Bacteriol., 2001. [PubMed]
- D. Shiomi, W. Margolin. The C-terminal domain of MinC inhibits assembly of the Z ring in Escherichia coli. J. Bacteriol., 2007. [PubMed]
- B. Shen, J. Lutkenhaus. Examination of the interaction between FtsZ and MinCN in E. coli suggests how MinC disrupts Z rings. Mol. Microbiol., 2010. [PubMed]
- B. Shen, J. Lutkenhaus. The conserved C-terminal tail of FtsZ is required for the septal localization and division inhibitory activity of MinC(C)/MinD. Mol. Microbiol., 2009. [PubMed]
- K.-T. Park, A. Dajkovic, M. Wissel, S. Du, J. Lutkenhaus. MinC and FtsZ mutant analysis provides insight into MinC/MinD-mediated Z ring disassembly. J. Biol. Chem., 2018. [PubMed]
- V. Blasios, A.W. Bisson-Filho, P. Castellen, M.L. Nogueira, J. Bettini, R.V. Portugal. Genetic and biochemical characterization of the MinC-FtsZ interaction in Bacillus subtilis. PLoS One, 2013
- I.F. de Oliveira, A. de Sousa Borges, V. Kooij, J. Bartosiak-Jentys, J. Luirink, D.J. Scheffers. Characterization of ftsZ mutations that render Bacillus subtilis resistant to MinC. PLoS One, 2010
- Y. Chen, S.L. Milam, H.P. Erickson. SulA inhibits assembly of FtsZ by a simple sequestration mechanism. Biochemistry, 2012. [PubMed]
- A. Dajkovic, A. Mukherjee, J. Lutkenhaus. Investigation of regulation of FtsZ assembly by SulA and development of a model for FtsZ polymerization. J. Bacteriol., 2008. [PubMed]
- S. Arumugam, Z. Petrášek, P. Schwille. MinCDE exploits the dynamic nature of FtsZ filaments for its spatial regulation. Proc. Natl. Acad. Sci., 2014. [DOI]
- V.M. Hernández-Rocamora, C. García-Montañés, B. Reija, B. Monterroso, W. Margolin, C. Alfonso. MinC protein shortens FtsZ protofilaments by preferentially interacting with GDP-bound subunits. J. Biol. Chem., 2013. [PubMed]
- C.J. LaBreck, J. Conti, M.G. Viola, J.L. Camberg. MinC N- and C-domain interactions modulate FtsZ assembly, division site selection, and MinD-dependent oscillation in Escherichia coli. J. Bacteriol., 2019. [DOI]
- D.J. Scheffers. The effect of MinC on FtsZ polymerization is pH dependent and can be counteracted by ZapA. FEBS Lett., 2008. [PubMed]
- S. Du, S. Pichoff, K. Kruse, J. Lutkenhaus. FtsZ filaments have the opposite kinetic polarity of microtubules. Proc. Natl. Acad. Sci., 2018. [PubMed]
- A.W. Bisson-Filho, K.F. Discola, P. Castellen, V. Blasios, A. Martins, M.L. Sforça. FtsZ filament capping by MciZ, a developmental regulator of bacterial division. Proc. Natl. Acad. Sci. U. S. A., 2015. [PubMed]
- S.C. Cordell, E.J. Robinson, J. Lowe. Crystal structure of the SOS cell division inhibitor SulA and in complex with FtsZ. Proc. Natl. Acad. Sci. U A., 2003
- P. Castellen, M.L. Sforça, F.J. Gueiros-Filho, A.C. De Mattos Zeri. Backbone and side chain NMR assignments for the N-terminal domain of the cell division regulator MinC from Bacillus subtilis. Biomol. NMR Assign., 2015. [PubMed]
- J.Y. An, T.G. Kim, K.R. Park, J.-G. Lee, H.-S. Youn, Y. Lee. Crystal structure of the N-terminal domain of MinC dimerized via domain swapping. J. Synchrotron Radiat., 2013. [PubMed]
- Y. Li, J. Hsin, L. Zhao, Y. Cheng, W. Shang, K.C. Huang. FtsZ protofilaments use a hinge-opening mechanism for constrictive force generation. Science, 2013. [PubMed]
- M.P. Williamson. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc., 2013. [PubMed]
- D. Kozakov, D.R. Hall, B. Xia, K.A. Porter, D. Padhorny, C. Yueh. The ClusPro web server for protein–protein docking. Nat. Protoc., 2017. [PubMed]
- R. Evans, M. O’Neill, A. Pritzel, N. Antropova, A. Senior, T. Green. Protein Complex Prediction with AlphaFold-Multimer (Preprint). bioRxiv, 2021. [DOI]
- G.M. Clore, V. Venditti. Structure, dynamics and biophysics of the cytoplasmic protein–protein complexes of the bacterial phosphoenolpyruvate: sugar phosphotransferase system. Trends Biochem. Sci., 2013. [PubMed]
- T.C. Terwilliger, D. Liebschner, T.I. Croll, C.J. Williams, A.J. McCoy, B.K. Poon. AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination. Nat. Methods, 2024. [PubMed]
- F.J. McNally, A. Roll-Mecak. Microtubule-severing enzymes: from cellular functions to molecular mechanism. J. Cell Biol., 2018. [PubMed]
- S. Ono. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int. Rev. Cytol., 2007. [PubMed]
- J. Fujita, R. Harada, Y. Maeda, Y. Saito, E. Mizohata, T. Inoue. Identification of the key interactions in structural transition pathway of FtsZ from Staphylococcus aureus. J. Struct. Biol., 2017. [PubMed]
- E. Ramírez-Aportela, J.R. López-Blanco, J.M. Andreu, P. Chacón. Understanding nucleotide-regulated FtsZ filament dynamics and the monomer assembly switch with large-scale atomistic simulations. Biophys. J., 2014. [PubMed]
- F.M. Ruiz, S. Huecas, A. Santos-Aledo, E.A. Prim, J.M. Andreu, C. Fernández-Tornero. FtsZ filament structures in different nucleotide states reveal the mechanism of assembly dynamics. PLoS Biol., 2022
- M.A. Schumacher, T. Ohashi, L. Corbin, H.P. Erickson. High-resolution crystal structures of Escherichia coli FtsZ bound to GDP and GTP. Acta Crystallogr. Sect. F Struct. Biol. Commun., 2020. [PubMed]
- T. Yoshizawa, J. Fujita, H. Terakado, M. Ozawa, N. Kuroda, S. Tanaka. Crystal structures of the cell-division protein FtsZ from Klebsiella pneumoniae and Escherichia coli. Acta Crystallogr. Sect. F Struct. Biol. Commun., 2020. [PubMed]
- L.C. Corbin, H.P. Erickson. A unified model for treadmilling and nucleation of single-stranded FtsZ protofilaments. Biophys. J., 2020. [PubMed]
- L. Corrales-Guerrero, W. Steinchen, B. Ramm, J. Mücksch, J. Rosum, Y. Refes. MipZ caps the plus-end of FtsZ polymers to promote their rapid disassembly. Proc. Natl. Acad. Sci., 2022
- N.S. Hill, P.J. Buske, Y. Shi, P.A. Levin. A moonlighting enzyme links Escherichia coli cell size with central metabolism. PLoS Genet., 2013
- V.M. Hernández-Rocamora, C. Alfonso, W. Margolin, S. Zorrilla, G. Rivas. Evidence that bacteriophage λ Kil peptide inhibits bacterial cell division by disrupting FtsZ protofilaments and sequestering protein subunits. J. Biol. Chem., 2015. [PubMed]
- J.M. Wagstaff, M. Tsim, M.A. Oliva, A. García-Sanchez, D. Kureisaite-Ciziene, J.M. Andreu. A polymerization-associated structural switch in FtsZ that Enables treadmilling of model filaments. mBio, 2017. [DOI]
- R.D. Mullins, J.A. Heuser, T.D. Pollard. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci., 1998. [PubMed]
- F. Delaglio, S. Grzesiek, Geerten W. Vuister, G. Zhu, J. Pfeifer, A. Bax. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR, 1995. [DOI]
- B.A. Johnson, R.A. Blevins. NMR View: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR, 1994. [PubMed]
- P. Güntert. Protein NMR Techniques, 2004
- R.A. Laskowski, J.A.C. Rullmann, M.W. MacArthur, R. Kaptein, J.M. Thornton. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR, 1996. [DOI]
- W.F. Vranken, W. Boucher, T.J. Stevens, R.H. Fogh, A. Pajon, M. Llinas. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins Struct. Funct. Bioinforma., 2005
- S. Huecas, E. Ramírez-Aportela, A. Vergoñós, R. Núñez-Ramírez, O. Llorca, J.F. Díaz. Self-organization of FtsZ polymers in solution reveals spacer role of the disordered C-terminal tail. Biophys. J., 2017. [PubMed]
- E. Krieger, G. Koraimann, G. Vriend. Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins Struct. Funct. Bioinforma., 2002
- B.J. Grant, A.P.C. Rodrigues, K.M. ElSawy, J.A. McCammon, L.S.D. Caves. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics, 2006. [PubMed]