Coexisting RET/PTC and TERT Promoter Mutation Predict Poor Prognosis but Effective RET and MEK Targeting in Thyroid Cancer
Abstract
Context:
The role of RET/PTC rearrangement in the clinical outcomes of papillary thyroid cancer (PTC) is controversial and remains to be clearly undefined.
Objective:
This work aimed to investigate the role of coexisting RET/PTC rearrangement and TERT promoter mutation in the prognosis and therapeutic targeting in PTC.
Methods:
A total of 669 PTC patients with complete clinical follow-up and genetic data were pooled from thyroid cancer data sets TCGA-THCA, MSK-MetTropism, and MSK-IMPACT, from whom 163 patients (112 women and 47 men, 4 unknown) with wild-type (WT) BRAF/RAS were identified, with a median age (interquartile range [IQR]) of 46.00 (33.00-61.00) years and a median follow-up time (IQR) of 16.13 (8.09-27.91) months for comparative genotype cohort analysis of mortality.
Results:
There was a significant concurrence index between RET/PTC and TERT promoter mutations, being 2.040 (95% CI, 1.110-3.747; P = .023). Mortality occurred in 5 of 100 (5%) patients harboring neither mutation, 2 of 18 (11.1%) patients harboring a TERT promoter mutation alone, 0 of 31 (0%) patients harboring a RET/PTC alone, and 7 of 14 (50%) patients harboring both genetic alterations, corresponding to hazard ratios (95% CI) of 1 (reference), 2.469 (0.405-14.022), 3.296e-09 (0-inf), and 9.019 (2.635-30.870), respectively, which remained essentially unchanged after adjustment for patient race, sex, and age. Similar results were observed with BRAF/RAS and TERT promoter mutations. Mechanistically, RET/PTC used the MAP kinase pathway to upregulate the mutated TERT, but not the WT TERT, and, correspondingly, targeting RET and MEK could suppress mutated TERT but not the WT TERT.
Conclusion:
Coexisting RET/PTC and TERT promoter mutation identify PTC as a unique clinical entity with high mortality, providing new implications for genetic-based prognostication and potential therapeutic targeting of RET and MEK guided by RET/PTC and TERT status.
Article type: Research Article
Keywords: thyroid cancer, mortality, prognostic biomarker
Affiliations: Thyroid Research Institute, School of Medicine, Southern University of Science and Technology, Shenzhen, Guangdong 518055, PR China
License: © The Author(s) 2024. Published by Oxford University Press on behalf of the Endocrine Society. CC BY 4.0 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited. See the journal About page for additional terms.
Article links: DOI: 10.1210/clinem/dgae327 | PubMed: 38735658 | PMC: PMC11570377
Relevance: Relevant: mentioned in keywords or abstract
Thyroid cancer is a common endocrine malignancy, with papillary thyroid cancer (PTC) being the most common type, accounting for up to more than 90% of all thyroid malignancies (ref. 1-3). The common driver genetic alterations of PTC include BRAF mutations, RAS mutations and RET/PTC rearrangements in the mitogen-activated protein kinase kinase (MAPK) pathway and telomerase reverse transcriptase (TERT) gene promoter mutations (ref. 4-6). The most common TERT promoter mutations in PTC are Chr.5:1 295 228 C > T (termed TERT C228T) and Chr.5:1 295 250 C > T (termed TERT C250T), which form the binding sites for ETS transcriptional factors, driving the expression of TERT (ref. 7). BRAF/RAS mutations and TERT promoter mutations commonly coexist to form genetic duets that drive poor clinical outcomes of PTC and hence are the main players in current genetic-based risk management of PTC (ref. 8-18) and some other cancers as well, such as melanoma (ref. 19-21). An important underlying molecular mechanism is that the mutant BRAF/RAS-activated MAPK pathway, through various downstream signaling routes/molecules, selectively activates the mutated TERT promoter, not the wild-type (WT) TERT, resulting in robust TERT expression and TERT-induced oncogenic consequences, particularly apoptosis resistance (ref. 22-27).
RET is a transmembrane glycoprotein receptor tyrosine kinase, encoded by the proto-oncogene RET and containing an intracellular catalytic tyrosine kinase domain (ref. 28-30). Activating genetic alterations of RET include mutations in familial medullary thyroid cancer and MEN2 syndrome (ref. 31-33) and, more commonly, genetic rearrangements in sporadic PTC—seen in about 25% of adult PTC and 50% of pediatric PTC (ref. 4, ref. 5, ref. 28-30).
RET rearrangement was first identified in PTC, hence designated as RET/PTC, in which the RET tyrosine kinase domain is fused to the N-terminus of a partner gene, resulting in ligand-independent constitutive kinase activation (ref. 34), which can activate both MAPK and PI3K pathways, causing oncogenesis and tumor progression (ref. 35). There are many types of RET/PTC rearrangements defined by different fusion partner genes, among which RET/PTC1 and RET/PTC3 are the most common. Unlike BRAF/RAS mutations, the clinical behaviors and oncogenic nature of RET/PTC rearrangements in PTC are controversial, although they are often shown to be nonaggressive on general analysis (ref. 4, ref. 5, ref. 28-30). This generates a dilemma in their clinical prognostic application. Inspired by the coexisting BRAF/RAS mutations and TERT promoter mutations in synergistically driving the aggressiveness of PTC, we hypothesize that the TERT promoter status may determine the nature of RET/PTC in affecting clinical outcomes. We tested this hypothesis by analyzing the effects of these genetic alterations on the mortality of PTC patients and experimentally defining the molecular mechanism.
Materials and Methods
Clinical Cohorts and Mutational Status
This study used 3 thyroid cancer data sets—TCGA-THCA (ref. 36), MSK-MetTropism (ref. 37), and MSK-IMPACT (ref. 38). Clinical and genomic mutation data were obtained from the cBioPortal for Cancer Genomics (https://www.cbioportal.org), which are summarized in Supplementary Table S1 in the supplementary materials (ref. 39). A total of 669 PTC patients with complete clinical follow-up and genetic data were pooled from the 3 thyroid cancer data sets (Supplementary Table S2 in the Supplementary Materials (ref. 39)), from whom 163 patients (112 women and 47 men, 4 unknown sex) harboring WT BRAF/RAS were identified, with a median age (interquartile range [IQR]) of 46.00 (33.00-61.00) years and a median clinical follow-up time (IQR) of 16.13 (8.09-27.91) months. A positive mutation was designated when the PTC sample exhibited genetic changes annotated in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) or it appeared in the 3 thyroid cancer data sets (ref. 36-38). Ten RET/PTC rearrangements were identified, including RET fusions with CCDC6, NCOA4, TIMM23B, ERC1, AKAP13, BMS1, FKBP15, TRIM24, RUFY3, and SPECC1L. TERT promoter mutations included TERT C228T, TERT C250T, and TERT C242T. BRAF mutations included BRAF V600E, BRAF G469A, and BRAF fusions with AKAP9, EXOC4, and SND1. RAS mutations included G13R, Q61K, and Q61R in KRAS, HRAS, and NRAS genes.
Cohort Comparative Analysis of Patient Mortality and Survival
Given the well-established association between BRAF/RAS mutations and TERT promoter mutations as well as their synergistic oncogenic effects on the aggressiveness of PTC, our comparative analysis focused mainly on individuals with WT BRAF/RAS to avoid the influence from mutations in these genes and studied purely the genetic role of RET/PTC and TERT. For comparative cohort analyses of the effects of RET/PTC rearrangements and TERT promoter mutations on clinical outcomes, namely the overall mortality and survival of patients, this cohort with WT BRAF/RAS was divided into various genotype groups as defined in “Results.” The survival intervals were defined as the period from the time of PTC diagnosis to the patient’s death or the time of their last follow-up if alive.
In vitro Study of the Role of RET/PTC in the Regulation of TERT
TPC-1 cells harboring RET/PTC1 and TERT C228T and LC2/AD cells harboring RET/PTC1 and WT TERT were used to investigate the molecular mechanism underlying the role of RET/PTC in the regulation of TERT and hence its role in clinical outcomes of PTC. Standard cellular and molecular approaches, including cell culture, treatments with RET/PTC inhibitor LOXO292 (selpercatinib) and MEK inhibitor AZD6244 (selumetinib), RNA isolation and real-time quantitative polymerase chain reaction, Western blotting, dual-luciferase reporter assay for the hTERT promoter, and CRISPR/Cas9 editing are detailed in the supplementary materials (ref. 39).
Statistical Analysis
Categorical data were summarized as frequencies and percentages. Continuous data were summarized as medians and IQR. The χ2 test was used to analyze categorical variables and Wilcoxon-Mann-Whitney test to analyze continuous variables. The life-table method was used to determine cumulative mortality and log-rank test to construct Kaplan-Meier survival curves. Cox proportional hazard regression analysis was used to examine hazard ratios (HRs) for the effects of genetic alterations on mortality. All P values were 2-tailed and a P less than .05 was considered statistically significant. Statistical analyses were performed using R (version 4.2.3) and its appropriate packages (ref. 40). Paired t test was used to compare gene expression levels. TERT promoter activities were compared using unpaired t test.
Results
Conversion of RET/PTC by Coexisting TERT Promoter Mutation Into a Strong Driver in the Aggressiveness of Papillary Thyroid Cancer
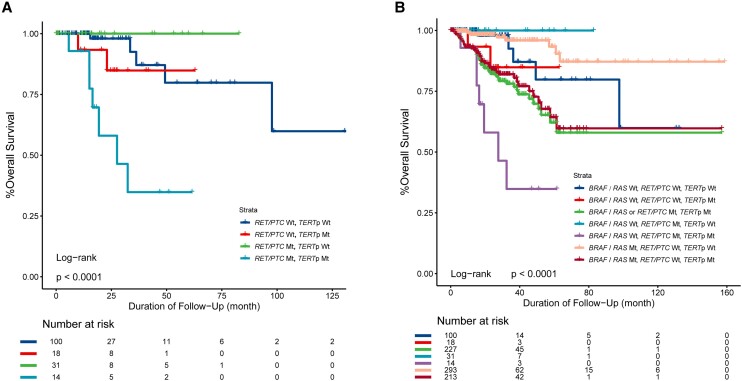
Consistent with the current knowledge, the overall analysis of the pooled data from the 3 data sets showed mutual exclusion among BRAF mutations, RAS mutations, and RET/PTC rearrangements in the MAPK pathway and coexistence of BRAF/RAS mutations with TERT promoter mutations (Supplementary Fig. S1A in the supplementary materials (ref. 39)). The most common RET/PTC rearrangements were with partner fusion genes CCDC6 and NCOA4 to form RET/PTC1 and RET/PTC3, respectively (Supplementary Fig. S1B in the supplementary materials (ref. 39)). From the 3 data sets, we identified 669 individuals with complete clinical follow-up and genetic data and their demographic characteristics are summarized in Supplementary Table S2 in the supplementary materials (ref. 39). As genetic duets of coexisting BRAF/RAS mutations and TERT promoter mutations are well known to be associated with aggressiveness of PTC, we identified a cohort of 163 patients harboring WT BRAF/RAS to study the role of RET/PTC and TERT and their demographic characteristics with respect to the RET/PTC and TERT status, which are summarized in Table 1. The genetic duet of concurrent RET/PTC and TERT promoter mutations was seen in 14 of 163 (8.59%) cases in this cohort. Concurrence test of the interactions between RET/PTC rearrangements and TERT promoter mutations revealed a concurrence index of 2.040 (95% CI, 1.110-3.747; P = .023) (Table 2), demonstrating a significant association between them.
Table 1.: Demographic and genetic characteristics of the patient cohort with wild-type BRAF/RAS
| Clinicopathological characteristics | Overall (N = 163) | RET/PTC Wta | RET/PTC rearrangementb | |||
|---|---|---|---|---|---|---|
| TERTp Wtc (N = 100) | TERTp Mtd (N = 18) | TERTp Wtc (N = 31) | TERTp Mtd (14) | |||
| Age, y | Median (IQR) | 46.00 (33.00-61.00) | 46.00 (33.01-61.00) | 32.00 (27.00-46.00) | 61.53 (42.40-65.07) | 69.00 (60.62-74.59) |
| Missing Info, No. (%) | 12 (7.4) | 7 (7.0) | 0 (0) | 2 (6.5) | 3 (21.4) | |
| Sex | Female, No. (%) | 112 (68.7) | 73 (73.0) | 7 (38.9) | 22 (71.0) | 10 (71.4) |
| Male, No. (%) | 47 (28.8) | 23 (23.0) | 11 (61.1) | 9 (29.0) | 4 (28.6) | |
| Missing Info, No. (%) | 4 (2.5) | 4 (4.0) | 0 (0) | 0 (0) | 0 (0) | |
| Follow-up time, mo | Median (IQR) | 16.13 (8.09-27.91) | 14.80 (7.16-28.46) | 13.57 (3.38-24.18) | 23.84 (11.54-27.49) | 17.74 (15.42-31.19) |
| Race | White, No. (%) | 91 (55.8) | 51 (51.0) | 13 (72.2) | 19 (61.3) | 8 (57.1) |
| Asian, No. (%) | 21 (12.9) | 10 (10.0) | 3 (16.7) | 7 (22.6) | 1 (7.1) | |
| Black, No. (%) | 10 (6.1) | 8 (8.0) | 1 (5.6) | 0 (0) | 1 (7.1) | |
| Other, No. (%) | 41 (25.2) | 31 (31.0) | 1 (5.6) | 5 (16.1) | 4 (28.6) | |
| Death | Yes, No. (%) | 14 (8.6) | 5 (5.0) | 2 (11.1) | 0 (0) | 7 (50) |
| No, No. (%) | 149 (91.4) | 95 (95.0) | 16 (88.9) | 31 (100) | 7 (50) | |
Abbreviations: IQR, interquartile range; WT, wild-type.
aRET/PTC Wt, WT, that is, no RET/PTC.
bRET/PTC rearrangement: including RET fusions with CCDC6, NCOA4, TIMM23B, ERC1, AKAP13, BMS1, FKBP15, TRIM24, RUFY3, and SPECC1L.
cTERTp Wt, WT, that is, no TERT promoter mutation.
dTERT promoter mutations, including C228T, C250T, and C242T.
Table 2.: Concurrence test of interactions between RET/PTC rearrangements and TERT promoter mutations in papillary thyroid cancer
| PTC patients | Concurrence indexa | 95% CI | Pb |
|---|---|---|---|
| Overall (N = 669) | 0.840 | 0.538-1.314 | .427 |
| BRAF/RAS Wtc (N = 163) | 2.040 | 1.110-3.747 | .022 |
Abbreviations: PTC, papillary thyroid cancer; WT, wild-type.
aConcurrence index = [n(RET/PTC Mt, TERTp Mt)/n(TERTp, Mt)]/[n(RET/PTC Mt, TERTp Wt)/n(TERTp, Wt)].
bP value for chi-square test.
cBRAF/RAS Wt, WT, that is, no BRAF/RAS mutation.
When dividing the patients with WT BRAF/RAS into 4 genotype groups, mortality rates were found to be 5 of 100 (5%) in patients harboring neither mutation, 2 of 18 (11.1%) in patients harboring a TERT promoter mutation alone, 0 of 31 (0%) in patients harboring a RET/PTC alone, and 7 of 14 (50%) in patients harboring both genetic alterations (Table 3). These results demonstrated a strong association of the genetic duet of coexisting RET/PTC rearrangement and TERT promoter mutation with the highest mortality. Compared with the group with neither genetic alteration, the TERT promoter mutation alone had only a modest effect, RET/PTC alone had no effect at all, and coexisting RET/PTC and TERT promoter mutations showed a robust synergistic effect on mortality. Thus, RET/PTC alone has a limited oncogenic function, but it can be converted by the coexisting TERT promoter mutation into a strong driver for the aggressiveness of PTC.
Table 3.: Hazard ratios of effects of various genotypes in papillary thyroid cancer on patient mortality in comparison with the genotype of wild-type BRAF and RAS and no RET/PTC rearrangement in Cox regression model
| Genetic alterations | Mortality rate n/N (%) | HRs | |||||
|---|---|---|---|---|---|---|---|
| BRAF or RAS mutationa | RET/PTC rearrangementb | TERTp mutationc | Univariable model | Multivariable modeld | |||
| HR (95% CI) | P | HR (95% CI) | P | ||||
| − | − | − | 5/100 (5.0) | 1 (reference) | N/A | 1 (reference) | N/A |
| − | − | + | 2/18 (11.1) | 2.469 (0.405-14.022) | .308 | 3.110 (0.332-29.113) | .320 |
| − | + | − | 0/31 (0) | 3.296e-09 (0-Inf) | .999 | 3.206e-11 (0-Inf) | .999 |
| − | + | + | 7/14 (50) | 9.019 (2.635-30.870) | <.001 | 13.061 (2.338-72.953) | .003 |
| + | − | − | 9/293 (3.1) | 0.532 (0.179-1.578) | .255 | 0.465 (0.317-8.641) | .550 |
| + | − | + | 37/213 (17.4) | 3.073 (1.239-7.621) | .015 | 2.605 (1.073-6.323) | .034 |
| +e | +e | + | 44/227 (19.4) | 3.375 (1.386-8.220) | .007 | 2.817 (1.189-6.673) | .019 |
Abbreviations: HR, hazard ratio; N/A, not available; PTC, papillary thyroid cancer.
aBRAF or RAS alternation, including BRAF mutations of BRAF V600E and BRAF G469A, and BRAF fusions with AKAP9, EXOC4, and SND1; and RAS mutations of G13R, Q61K, and Q61R in KRAS, HRAS, and NRAS genes.
bRET/PTC rearrangements, including RET fusions with CCDC6, NCOA4, TIMM23B, ERC1, AKAP13, BMS1, FKBP15, TRIM24, RUFY3, and SPECC1L.
cTERT promoter mutations, including C228T, C250T, and C242T.
dAdjusted for patient race, sex, and age at the diagnosis of PTC.
ePatients with PTC harboring BRAF mutation or RAS mutation or RET/PTC rearrangement.
Synergized Effects of RET/PTC and TERT Promoter Mutations on Poor Patient Survival in Papillary Thyroid Cancer on Kaplan-Meier Analyses
On the Kaplan-Meier survival analysis of patients with WT BRAF/RAS, the survival curve was flat in patients harboring RET/PTC alone, slightly declined in patients harboring the TERT promoter mutation alone, and sharply declined in patients harboring both genetic alterations (Fig. 1A). Cox regression analysis comparing with patients harboring no genetic alterations (no BRAF, RAS, RET/PTC, or TERT promoter mutations) showed survival HRs (95% CI) of 2.469 (0.405-14.022) for TERT promoter mutation alone, 3.296e-09 (0-inf) for RET/PTC alone, and 9.019 (2.635-30.870) for their coexistence (see Table 3); the former 2 were both insignificant, the latter was robustly significant, and these HRs remained essentially unchanged after adjustment for patient race, sex, and age, demonstrating a strong synergistic effect of the 2 genetic alterations on poor patient survival (see Table 3). To compare the effects of coexisting RET/PTC and TERT promoter mutations with those of coexisting BRAF/RAS mutation and TERT promoter mutation on clinical outcomes of PTC, we divided the 669 patients with complete clinical follow-up and genetic data into 7 groups according to the genetic status of BRAF, RAS, RET/PTC, and TERT (Fig. 1B). Consistent with previous reports (ref. 9, ref. 12), the survival curve of patients with a coexisting BRAF/RAS mutation and TERT promoter mutation declined sharply. The survival curve decline was also sharp when patients with a coexisting BRAF/RAS mutation or RET/PTC rearrangement and TERT promoter mutation were grouped into one entity (Fig. 1B). Cox regression analysis comparing with patients harboring no genetic alterations (no BRAF, RAS, RET/PTC, or TERT promoter mutations) showed mortality HRs (95% CI) of 3.073 (1.239-7.621) for a coexisting BRAF/RAS mutation and TERT promoter mutation; and 3.375 (1.386-8.220) for a coexisting BRAF/RAS mutation or RET/PTC rearrangement and TERT promoter mutation (see Table 3).
Selective Regulation of the Mutated TERT but not the Wild-Type TERT by RET/PTC
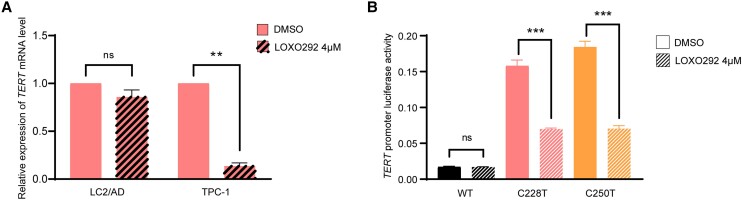
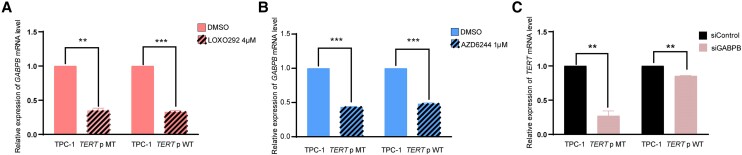
Treatment with the RET/PTC inhibitor LOXO292 dramatically suppressed TERT gene expression in TPC-1 cells harboring RET/PTC and TERT C228T, but not in LC2/AD cells harboring RET/PTC and WT TERT (Fig. 2A). Luciferase report assay using TPC-1 cells demonstrated that mutated TERT promoter C228T and C250T activities were significantly suppressed by treatment with the RET/PTC inhibitor LOXO292, while the WT TERT promoter activity was not affected (Fig. 2B). Thus, RET/PTC regulated the TERT gene in a TERT promoter mutation-dependent manner; it robustly upregulated the mutated TERT.
Regulation of the Mutated TERT by RET/PTC Through the Mitogen-Activated Protein Kinase Pathway
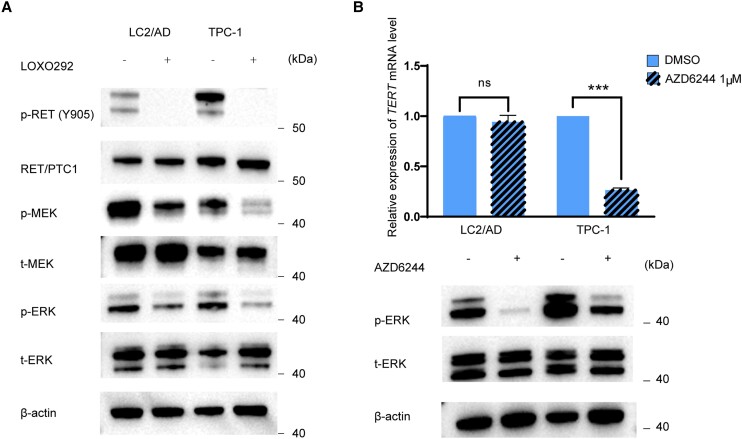
As shown in Fig. 3A, inhibition of RET/PTC using LOXO292 completely suppressed RET phosphorylation, accompanied by the dramatic reduction of the phosphorylation of MEK and ERK both in TPC-1 and LC2/AD cells, consistent with the known role of RET/PTC in the activation of the MAPK pathway. Similar to the treatment with RET/PTC inhibitor LOXO292 (see Fig. 2A), treatment with the MEK inhibitor AZD6244 also dramatically suppressed TERT gene expression in TPC-1 cells harboring TERT C228T, but not in LC2/AD cells harboring WT TERT (Fig. 3B). Thus, RET/PTC selectively regulated the mutated TERT gene through activating the MAPK pathway.
Conversion of Mutant TERT to Wild Type by CRISPR/Cas9 Editing Results in Loss of Effect of RET/PTC
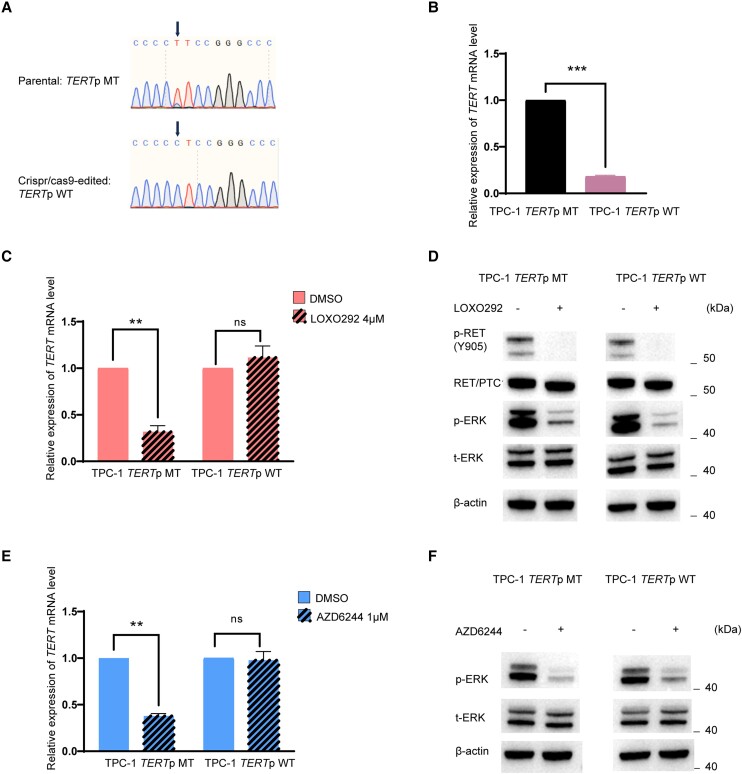
Conversion of TERT C228T to WT by CRISPR/Cas9 editing in TPC-1 cells (Fig. 4A) induced a dramatic downregulation of TERT gene expression (Fig. 4B). The RET/PTC inhibitor LOXO292 dramatically suppressed TERT expression in parental TPC-1 cells harboring RET/PTC and TERT C228T but had no effect on CRISPR/Cas9-edited TPC-1 cells harboring RET/PTC and WT TERT (Fig. 4C), while RET/PTC and MAPK pathway were inhibited in both cells (Fig. 4D). Similarly, the MEK inhibitor AZD6244 dramatically suppressed TERT expression in parental TPC-1 cells but not in CRISPR/Cas9-edited TPC-1 cells (Fig. 4E), although it inhibited the MAP kinase pathway in both cells (Fig. 4F). These results demonstrated again the TERT promoter mutation-dependence of the action of RET/PTC.
RET/PTC Regulates Mutant TERT Through Mitogen-Activated Protein Kinase Pathway Involving GABPB
Since the BRAF V600E/MAP kinase pathway regulated mutant TERT through regulating GABPB (ref. 22-27), we investigated whether this was also the case with RET/PTC. Treatment with the RET/PTC inhibitor LOXO292 dramatically suppressed the GABPB gene expression both in parental TPC-1 cells and CRISPR/Cas9-edited cells (Fig. 5A). Similarly, treatment with the MEK inhibitor AZD6244 also dramatically suppressed the GABPB gene expression both in parental TPC-1 cells and CRISPR/Cas9-edited cells (Fig. 5B). In contrast, knockdown of GABPB resulted in suppression of TERT gene expression dramatically in parental TPC-1 cells but only minimally in CRISPR/Cas9-edited TPC-1 cells (Fig. 5C). Thus, RET/PTC selectively regulated the mutant TERT through the MAP kinase pathway involving GABPB as BRAF V600E did (ref. 22-27).
PI3K/AKT Pathway Is not Involved in the Regulation of Mutant TERT by RET/PTC
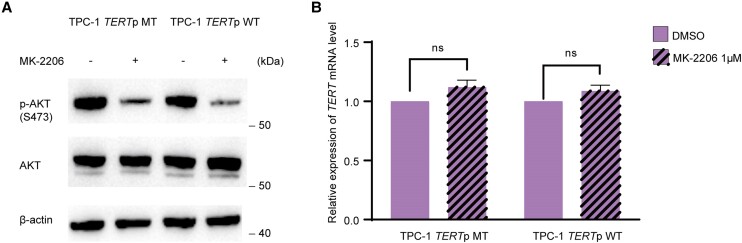
Considering that RET/PTC can activate both MAPK and PI3K/AKT pathways (ref. 35), we investigated whether the PI3K/AKT pathway was also involved in the regulation of mutant TERT expression by RET/PTC. Treatment with the protein kinase B (AKT) inhibitor MK-2206 inhibited the PI3K/AKT pathway both in parental TPC-1 cells (harboring RET/PTC and TERT C228T) and CRISPR/Cas9-edited TPC-1 cells (harboring RET/PTC and WT TERT) (Fig. 6A) but did not affect TERT expression in either cell (Fig. 6B). These results suggest that the regulation of mutant TERT by RET/PTC does not involve the PI3K/AKT pathway.
Discussion
RET/PTC rearrangements are among the most common genetic alterations in the MAPK pathway in PTC (ref. 4, ref. 5, ref. 28-30), but, unlike BRAF/RAS mutations, their role in the clinical outcomes of PTC is controversial and remains to be clearly undefined. Numerous studies have demonstrated a robust oncogenic function of the genetic duets of BRAF/RAS mutations and TERT promoter mutations in driving tumor aggressiveness and poor clinical outcomes of PTC (ref. 8-18). The underlying molecular mechanism is that the BRAF/RAS mutation-activated MAPK pathway robustly and selectively upregulates the mutated TERT gene, but not the WT TERT (ref. 22-27). TERT, at aberrantly elevated levels, is a strong oncoprotein that promotes tumorigenesis and cancer aggressiveness involving its noncanonical telomerase-independent functions, including, prominently, the suppression of apoptosis (ref. 41). In these studies on the role of BRAF/RAS mutations and TERT promoter mutations, RET/PTC rearrangements have not been considered. Given that BRAF/RAS mutations and RET/PTC rearrangements are mutually exclusive among themselves in the MAPK pathway and they each can independently activate the pathway, we speculated that RET/PTC, when coexisting with TERT promoter mutations, could, like BRAF/RAS mutations, also selectively upregulate the mutant TERT through activating the MAPK pathway, thus promoting tumor aggressiveness and poor clinical outcomes of PTC.
Indeed, the present study proved this hypothesis to be true. Specifically, we demonstrated that, like the BRAF mutation, RAS mutation, and TERT prompter mutation, each alone had little effect in previous studies, the RET/PTC and TERT promoter mutation each alone also had little effect in the present study; in fact, RET/PTC alone had no effect at all on patient survival. In striking contrast, a robust synergistic effect between RET/PTC and TERT promoter mutation on the mortality of PTC patients was observed. A previous study demonstrated that GABP in the form of complex GABPA and GABPB could directly bind the mutant TERT promoter to activate and upregulate the TERT gene (ref. 7). Our group demonstrated that the BRAF V600E-activated MAPK pathway upregulated the mutant TERT through upregulating the GABPB gene and promoting the formation of the GABP complex (ref. 23). In the present study, we demonstrated that RET/PTC used a similar mechanism to regulate mutant TERT. Specifically, our in vitro studies showed that the action of RET/PTC in the genetic duet with TERT promoter mutations, like that of BRAF/RAS mutations in their genetic duet with TERT promoter mutations, was mediated through activating the MAPK pathway and GABPB, leading to the selective activation of the mutated TERT, but not the WT TERT. Thus, the normally ineffective RET/PTC can be functionally converted by TERT promoter mutations into a strong oncogenic genetic driver promoting the aggressiveness of PTC. Based on these results, it is reasonable to propose that PTC harboring only RET/PTC can be treated in a relatively conservative manner while PTC harboring coexisting RET/PTC and TERT promoter mutations should be treated in a relatively aggressive manner. The present study may explain the previous findings of mild oncogenic behaviors of RET/PTC in general but aggressive behaviors in a portion of cases, as RET/PTC alone is much more common than its coexistence with TERT promoter mutations as shown in the present study (Supplementary Fig. S1 in the supplementary materials (ref. 39)). The present study is also consistent with the long-time knowledge that pediatric PTC has a far better prognosis than adult PTC even though RET/PTC is far more common in the former than the latter (ref. 4, ref. 5, ref. 42), probably because TERT promoter mutations are very uncommon, hence with very rare coexistence of RET/PTC and TERT promoter mutations, in the former (ref. 42). The present study also demonstrated that the genetic duet of RET/PTC and TERT promoter mutations had similar effects to those of the genetic duet of BRAF/RAS and TERT promoter mutations on clinical outcomes of PTC. Therefore, PTCs with a coexisting BRAF/RAS mutation or RET/PTC rearrangement and TERT promoter mutation could be clinically considered as one disease entity with common poor clinical outcomes.
The RET/PTC inhibitor LOXO292 (selpercatinib) has been approved by the US Food and Drug Administration to treat RET-altered cancers, including advanced thyroid cancer harboring RET/PTC, with good therapeutic responses in some patients but not in others (ref. 43). The strong inhibitory effects of LOXO292 on RET, the MAPK pathway, and the mutated TERT gene, but not the WT TERT gene in cells harboring RET/PTC observed in the present study, suggest that it is possible that the patients that responded well to LOXO292 harbored the genetic duet of RET/PTC and TERT promoter mutations while those that did not respond well harbored only RET/PTC. In a recent in vitro and animal study we demonstrated that BRAF/MEK inhibitors could induce strong therapeutic responses in cancer cells harboring both BRAF V600E and mutant TERT promoter through causing robust apoptosis but not cells harboring WT TERT (ref. 24). A subsequent German clinical trial on patients with melanoma demonstrated that BRAF/MEK inhibitors could induce much superior therapeutic responses in patients harboring a coexisting BRAF mutation and TERT promoter mutation compared with patients harboring only a BRAF mutation (ref. 25). We proposed that BRAF mutation-induced activation of the MAPK pathway and hence elevated TERT in mutated TERT cancers, which is a strong suppressor of apoptosis, made such cancer cells evolutionarily become dependent on this molecular signaling system for powerful survival (through apoptosis resistance); once this system was disrupted, as achieved by the inhibition of the MAPK pathway, expression of the mutated TERT gene suddenly shut off, removing the suppression of apoptosis and resulting in robust cancer cell apoptosis (ref. 24). We speculate that this is the same mechanism that the genetic duet of RET/PTC and mutated TERT may confer on PTC’s particularly good responsiveness to the treatment with RET/PTC inhibitors. In this context and given that a MEK inhibitor could strongly suppress TERT in RET/PTC-harboring cancer cells in the present study, it is possible that MEK inhibitors, like RET/PTC inhibitors, may also be therapeutically effective in PTC harboring the genetic duet of RET/PTC and TERT promoter mutations.
A limitation of the present study was the relatively small number of cases with the genetic duet of RET/PTC and TERT promoter mutations, being 8.59% in patients with PTC harboring the WT BRAF/RAS. However, the robust role of this genetic duet demonstrated in the present study suggests that PTC with it is a distinct clinical entity, making its identification important for genetic-guided precision management. Given the large patient population with PTC globally, a considerable number of patients are expected to harbor the genetic duet of RET/PTC and TERT promoter mutations and thus potentially benefit from treatment with MEK inhibitors. RET fusion genes are also seen in other human cancers, such as non–small cell lung cancer (ref. 44). The findings in PTC in the present study may likely be applicable also to the management of such cancers. Another limitation of this study is the limited availability of RET/PTC-positive cell lines, which are rare in the field. Nevertheless, the 2 precious cell lines of this type used in the present study showed robust results, strongly supporting our conclusions. Moreover, this limitation was mitigated by CRISPR/Cas9 editing to generate a new genotype of the TPC-1 cell that was equivalent to a new RET/PTC cell line.
In summary, we for the first time report a robust, synergistic oncogenic role of the genetic duet of coexisting RET/PTC and TERT promoter mutations with no role of the RET/PTC alone in poor clinical outcomes of PTC. Like the genetic duet of BRAF/RAS mutations and TERT promoter mutations, the genetic duet of RET/PTC and TERT promoter mutation also identifies PTC with high mortality, making this PTC clinically unique. As such, PTCs harboring coexisting a BRAF/RAS mutation or RET/PTC rearrangement and TERT promoter mutation can be clinically considered as one disease entity sharing a common poor prognosis. This study provides new implications for genetic-based prognostication as well as the potential of effective therapeutic targeting of RET and MEK guided by RET/PTC and TERT status in PTC patients. It is therefore advisable that genetic patterns of RET/PTC and TERT promoter be added to the current genetic-based risk stratification and precision management of PTC.
References
- Recent incidences and differential trends of thyroid cancer in the USA.. Endocr Relat Cancer., 2016. [PubMed]
- Trends in thyroid cancer incidence and mortality in the United States, 1974-2013.. JAMA., 2017. [PubMed]
- Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries.. CA Cancer J Clin., 2021. [PubMed]
- BRAF mutation in thyroid cancer.. Endocr Relat Cancer., 2005. [PubMed]
- Molecular pathogenesis and mechanisms of thyroid cancer.. Nat Rev Cancer., 2013. [PubMed]
- TERT promoter mutations in thyroid cancer.. Endocr Relat Cancer., 2016. [PubMed]
- Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer.. Science., 2015. [PubMed]
- Highly prevalent TERT promoter mutations in aggressive thyroid cancers.. Endocr Relat Cancer., 2013. [PubMed]
- BRAF v600e and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence.. J Clin Oncol., 2014. [PubMed]
- Prognostic effects of TERT promoter mutations are enhanced by coexistence with BRAF or RAS mutations and strengthen the risk prediction by the ATA or TNM staging system in differentiated thyroid cancer patients.. Cancer., 2016. [PubMed]
- Mortality risk stratification by combining BRAF V600E and TERT promoter mutations in papillary thyroid cancer: genetic duet of BRAF and TERT promoter mutations in thyroid cancer mortality.. JAMA Oncol., 2017. [PubMed]
- A six-genotype genetic prognostic model for papillary thyroid cancer.. Endocr Relat Cancer., 2017. [PubMed]
- Effects of coexistent BRAF(V600E) and TERT promoter mutations on poor clinical outcomes in papillary thyroid cancer: a meta-analysis.. Thyroid., 2017. [PubMed]
- Genomic characterization of high-recurrence risk papillary thyroid carcinoma in a southern Chinese population.. Diagn Pathol., 2020. [PubMed]
- Co-occurrence of TERT promotor mutations with BRAF or NRAS alterations correlates with worse prognosis in melanoma.. Br J Dermatol., 2021. [PubMed]
- The predictive value of coexisting BRAFV600E and TERT promoter mutations on poor outcomes and high tumour aggressiveness in papillary thyroid carcinoma: a systematic review and meta-analysis.. Clin Endocrinol (Oxf)., 2021. [PubMed]
- The genetic duet of BRAF V600E and TERT promoter mutations predicts the poor curative effect of radioiodine therapy in papillary thyroid cancer.. Eur J Nucl Med Mol Imaging., 2022. [PubMed]
- Is molecular testing in patients with low risk papillary thyroid carcinoma justified? A Markovian model.. Am J Otolaryngol., 2022. [PubMed]
- Coexistence of TERT promoter and BRAF mutations in cutaneous melanoma is associated with more clinicopathological features of aggressiveness.. Virchows Arch., 2015. [PubMed]
- TERT promoter mutations in melanoma survival.. Int J Cancer., 2016. [PubMed]
- Clinical, environmental and histological distribution of BRAF, NRAS and TERT promoter mutations among patients with cutaneous melanoma: a retrospective study of 563 patients.. Br J Dermatol., 2021. [PubMed]
- TERT promoter mutations in melanoma render TERT expression dependent on MAPK pathway activation.. Oncotarget., 2016. [PubMed]
- Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer.. Nat Commun., 2018. [PubMed]
- TERT promoter mutation determines apoptotic and therapeutic responses of BRAF-mutant cancers to BRAF and MEK inhibitors: Achilles Heel.. Proc Natl Acad Sci USA., 2020. [PubMed]
- TERT promoter mutations are associated with longer progression-free and overall survival in patients with BRAF-mutant melanoma receiving BRAF and MEK inhibitor therapy.. Eur J Cancer., 2022. [PubMed]
- Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer.. Endocr Relat Cancer., 2019. [PubMed]
- TERT expression induces resistance to BRAF and MEK inhibitors in BRAF-mutated melanoma in vitro.. Cancers (Basel)., 2023. [PubMed]
- The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults.. J Clin Endocrinol Metab., 2000. [PubMed]
- Rearrangement of the RET gene in papillary thyroid carcinoma.. Wiad Lek., 2001
- RET/PTC rearrangement in thyroid tumors.. Endocr Pathol., 2002. [PubMed]
- Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis.. J Med Genet., 2000. [PubMed]
- Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A.. Nature., 1993. [PubMed]
- Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours.. Hum Mol Genet., 1994. [PubMed]
- PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas.. Cell., 1990. [PubMed]
- Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer.. Thyroid., 2010. [PubMed]
- Integrated genomic characterization of papillary thyroid carcinoma.. Cell., 2014. [PubMed]
- Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients.. Cell., 2022. [PubMed]
- Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients.. Nat Med., 2017. [PubMed]
- 39 Zhang W , LinS, WangZ, ZhangW, XingM. Supplementary materials for “Coexisting RET/PTC and TERT promoter mutation predict poor prognosis but effective RET and MEK targets in thyroid cancer”. figshare. doi:10.6084/m9.figshare.25567905.v1. Date of deposit 2024.
- 40 Therneau T . A Package for Survival Analysis in R. 2023; R package version 3.5-7. https://CRAN.R-project.org/package=survival
- Multiple actions of telomerase reverse transcriptase in cell death regulation.. Biomedicines., 2023. [PubMed]
- Genetic alterations landscape in paediatric thyroid tumours and/or differentiated thyroid cancer: systematic review.. Rev Endocr Metab Disord., 2023. [PubMed]
- Progress and challenges in RET-targeted cancer therapy.. Front Med., 2023. [PubMed]
- RET fusions in solid tumors.. Cancer Treat Rev., 2019. [PubMed]