
Application of Genomic Epidemiology of Pathogens to Farmed Yellowtail Fish Mycobacteriosis in Kyushu, Japan
Abstract
To investigate mycobacterial cases of farmed yellowtail fish in coastal areas of western Japan (Kagoshima, Kyushu), where aquaculture fisheries are active, Mycobacterium pseudoshottsii, the causative agent, was isolated from six neighboring fishing ports in 2012 and 2013. A phylogenetic analysis revealed that the strains isolated from one fishing port were closely related to those isolated from other regions of Japan, suggesting the nationwide spread of a single strain. However, strains from Japan were phylogenetically distinct from those from the Mediterranean and the United States; therefore, worldwide transmission was not observed based on the limited data obtained on the strains examined in this study. The present results demonstrate that a bacterial genomic analysis of infected cases, a molecular epidemiology strategy for public health, provides useful data for estimating the prevalence and transmission pathways of M. pseudoshottsii in farmed fish. A bacterial genome analysis of strains, such as that performed herein, may play an important role in monitoring the prevalence of this pathogen in fish farms and possible epidemics in the future as a result of international traffic, logistics, and trade in fisheries.
Article type: Research Article
Keywords: fish mycobacteriosis, public health, fish farm
Affiliations: Department of Microbiology, Graduate School of Human Life and Ecology, Osaka Metropolitan University, Osaka, Japan; Osaka International Research Center for Infectious Diseases, Osaka Metropolitan University, Osaka, Japan; Clinical Research Center, National Hospital Organization Kinki-chuo Chest Medical Center, Sakai, Osaka, Japan; Azuma-cho Fisheries Cooperative Association, Izumi, Kagoshima, Japan; Faculty of Human Life Sciences, Shokei University, Kumamoto, Kumamoto, Japan; Division of Bioresources, International Institute for Zoonosis Control, Hokkaido University, Sapporo, Hokkaido, Japan; Division of Research Support, Institute for Vaccine Research and Development, Hokkaido University, Sapporo, Hokkaido, Japan; Laboratory of Fish Disease, Aquaculture Course, Department of Marine Resource Science, Faculty of Agriculture and Marine Science, Kochi University, Nankoku, Kochi, Japan
License: 2024 by Japanese Society of Microbial Ecology / Japanese Society of Soil Microbiology / Taiwan Society of Microbial Ecology / Japanese Society of Plant Microbe Interactions / Japanese Society for Extremophiles. CC BY 4.0 This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Article links: DOI: 10.1264/jsme2.ME24011 | PubMed: 38897967 | PMC: PMC11220446
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (581 KB)
Mycobacterium marinum is a well-known contributor to fish mycobacteriosis (ref. Gauthier and Rhodes, 2009; ref. Aubry ; ref. Hashish ), which causes disease in both freshwater and saltwater fish, whether they are farmed or wild (ref. Jacobs ; ref. Gauthier and Rhodes, 2009). Humans may also be infected with this bacterium when they are exposed to contaminated aquatic environments or marine animals (ref. Decostere ; ref. Hashish ). Detailed genetic sequencing revealed the extensive genetic diversity of M. marinum, which has many fish disease-causing subspecies, including M. liflandii (M. ulcerans ecovar liflandii), M. shottsii, and M. pseudoshottsii (ref. Yip ; ref. Tobias ; ref. Hikima ; ref. Das ; ref. Gauthier ). M. ulcerans, which causes Buruli ulcer (ref. Guarner, 2018), a severe invasive skin disease in humans, is closely related to these species (ref. Stinear ; ref. Yip ). These species share more than 98% genome sequence identity and, thus, are collectively referred to as the M. marinum complex (ref. Stinear ; ref. Tobias ; ref. Hikima ; ref. Das ; ref. Gauthier ).
M. pseudoshottsii (Mps) was initially detected during a mycobacteriosis epizootic in 2007 (ref. Rhodes ). Since then, the organism’s geographical distribution range and the various fish species exposed to it have been increasingly reported. In 2009, this species was also isolated from striped bass in the New York Bight (ref. Stine ). Furthermore, cases of infection with this organism in aquaculture farms have been reported in Japan (ref. Nakanaga ), with accompanying descriptions of mass mortality. Another report was documented in 2020, describing cases of infected farmed fish in Italy (ref. Mugetti ). In 2022, the organism was detected in sardines held in aquariums in Japan (ref. Komine ). In 2020–2021, this organism caused many cases of infection in an aquaculture facility in Israel raising imported food fish from the United States (ref. Davidovich ). Consequently, there is apprehension that this pathogen may be spreading surreptitiously not only in aquaculture fisheries, but also among fish bred in different facilities worldwide, based on reports of sporadic cases of its occurrence from diverse sources.
The identification of possible sources for the transmission of Mps is critical for mitigating the damage caused by the spread of this infection. Examinations of genetic variations among strains allow for the estimation of their origins. An Italian study analyzed the partial sequences of hsp65, one of the housekeeping genes, from several strains isolated from three different aquaculture farms (ref. Mugetti ). Although these strains were classified into only three groups based on their sequences, the findings obtained indicated different sources of infection. More detailed techniques using high-throughput sequencers have recently been effectively employed to identify the source of not only M. tuberculosis cases in humans (ref. Walker , ref. 2013b) but also M. bovis in wild animals (ref. Biek ). This method is known as a molecular epidemiological analysis and has been widely used for the majority of pathogens (ref. Grad and Lipsitch, 2014; ref. Kao ), including in the COVID-19 pandemic (ref. Rockett ), as well as in mycobacteriosis.
To estimate the transmission routes of pathogens for a molecular epidemiological analysis, it is critical to sequence the full-length genomes of each strain and strictly elucidate their identity based on single-nucleotide variations (SNVs). In the case of Mps, four full-length genome sequences (the type strain L15, synonym JCM 15466T and DSM 45108, originally isolated from a striped bass in Chesapeake Bay; AR, isolated from Argyrosomus regius in Western Greece; NJB1907-Z4, isolated from an aquarium-reared Japanese sardine in Japan; YM-3, isolated from cultured yellowtail fish in Japan) are currently available in the NCBI and serve as reference sequences (ref. Imajoh ). By selecting an appropriate reference from these sequences, nucleotide variations among strains may be successfully identified, which may enable us to estimate factors of infection and/or the background of transmission.
A genome analysis of this bacterium has provided only a partial understanding of phylogenetic relationships and a detailed genome analysis has not yet been conducted. In the present study, we report the isolation of the causative organisms from an outbreak of acid-fast bacilli in cultured yellowtail fish in 2012 in Japan, demonstrating that these were cases of Mps infection, and elucidated their genome sequences. In addition, we verified genetic relatedness and geographical relationships among the strains, which serves as a molecular epidemiological approach to the setting.
Materials and Methods
Strains
The 12 strains included in the present study were isolated from fish carcasses suspected to have died due to mycobacterial infection. These carcasses were brought to Azuma-cho Fisheries Cooperative Association from six ports in the western area of Kagoshima Prefecture in 2012 and 2013. Yellow colonies of mycobacterial strains were obtained by culturing lesion samples at 25°C for 2 weeks on Ogawa’s media (Kyokuto Pharmaceutical Industrial).
To accurately identify the mycobacterial species, bacterial strains were sent to the Clinical Research Center of the National Hospital Organization Kinki-chuo Chest Medical Center. Photochromogenicity was confirmed based on Runyon’s classification. Four partial genes (16S rRNA, ITS, hsp65, and rpoB), which have been used to identify mycobacterial species (ref. Roth ; ref. Kim ), were sequenced by Eurofin Japan.
Genome sequencing
Genomic DNA samples were purified from cultured bacteria as previously described (ref. Belisle and Sonnenberg, 1998) to prepare libraries for short-read sequencing using the QIAseq FX library preparation kit (QIAGEN). MiSeq (Illumina) was used for short-read sequencing with MiSeq Sequencing Kit v3 (600-cyc) (Illumina). Raw read data were deposited in the DDBJ Sequence Read Archive (DRA) under the accession numbers DRR506146-DRR506157.
Genome analysis with M. marinum complex strains
After filtering low-quality or contamination reads using the ‘quality trimming’ and ‘removal of mapped reads’ functions in the CLC Genomics Workbench v20.0.4 (QIAGEN), the sequence reads of each strain were assembled using SPAdes genome assembler v3.15.4 (ref. Prjibelski ). The contigs obtained were used for a core-genome analysis in combination with 19 complete genome sequences of the M. marinum complex registered in the NCBI database (Table S1). Short-read sequences obtained from three Mps strains reported from Israel (ref. Davidovich ) were also downloaded from the database, assembled in the same manner as described above, and integrated into this analysis. The core SNVs were retrieved using kSNP4.0, an upgraded version of kSNP3 (ref. Gardner ). The k-mer was 19 and covered >99% of the genome sequences using Kchooser4, a program bundled with kSNP4.0. The fraction of core k-mers (FCK) was also checked using this program. The matrix of SNV data was used as an alignment to construct maximum likelihood trees using IQ-TREE ver. 1.6.12 (ref. Nguyen ). The substitution model was selected using the option “-m MFP+ASC” in the command line, which allowed for model selection under the Bayesian information criterion with ascertainment bias correction. The tree files constructed using this tool were visualized using iTOL ver. 6.7.6 (ref. Letunic and Bork, 2021).
Detection of SNVs with a close reference
Based on the core genome phylogenetic tree, YM-3 was selected as the reference strain for further analyses. The short reads of the 12 Mps strains and NJB1907-Z4 (accession no. DRR337915) were mapped to the complete genome sequence of YM-3 (ref. Imajoh ) using the CLC Genomics Workbench v20.0.4 (QIAGEN) to detect SNVs. The SNVs were called only from reference regions with a mapping coverage >10 and not containing non-specific mapping reads.
After the filtration of heterogeneous SNVs (<80% variant calls), the alleles of the SNVs were compared between the 12 strains and the reference YM-3. All nucleotides corresponding to the SNVs were concatenated to each nucleotide sequence for each strain and used to construct a median-joining tree (ref. Bandelt ) with POPART ver. 1.7 (ref. Leigh and Bryant, 2015). All SNV alleles of JCM 15466 were also confirmed by comparisons with the YM-3 genome sequence using the Nucmer program in Mummer3 (ref. Kurtz ).
Results
Occurrence and symptoms of Mps infection
A part of the Amakusa archipelago in the western area of Kagoshima Prefecture, located in southern Kyushu, Japan (Fig. 1), is known for its active food fish aquaculture, particularly for yellowtail and red snapper. During yellowtail farming in these areas, primarily during summer (July to September), fatalities and a high number of sick individuals among cultured fish have been observed (up to ~5% of the total number of fish in a fish tank), particularly in zero-year-old fish, due to infection with acid-fast bacilli. White granulomas are commonly found on the gills and internal organs, primarily the liver and kidneys. Ulcerative lesions are occasionally observed on the skin surface, which are diagnosed as mycobacterial infections through the acid-fast staining of tissue sections.
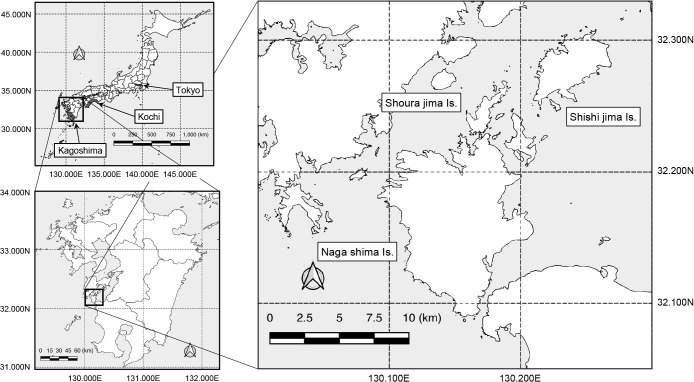
All yellowtail fish carcasses collected for examination (377 cases from 2012 and 337 from 2013) from six ports were found to be anally open with no skin lesions. Autopsy revealed the presence of jaundice and kidney nodules in 26 cases from 2012 and 53 from 2013, which strongly indicated mycobacterial infection. Twelve slow-growing acid-fast bacilli were isolated from these carcasses (Table 1), all of which were classified as Runyon I, including M. marinum, based on their photochromogenicity (data not shown). These strains were culturable at room temperature (approximately 25°C), but did not grow at 37°C. Of the 12 strains, seven were isolated from samples provided by Port A on Shishijima Island, where mycobacteriosis cases were monitored for two years. The remaining five strains were selected from different fishing ports (Ports B to F) on Moroshojima Island and Nagashima Island, where cases were monitored in 2012 only. To anonymize each port, only the approximate distances between ports are shown (Table 2).
Table 1.: Clinical strains of Mycobacterium pseudoshottsii isolated from yellowtail fish in aquaculture facilities of Japan
| Strain | Date of Isolation | Isolation port | Age |
|---|---|---|---|
| MPSJQ12-A1 | Sep. 5, 2012 | A | zero-years-old |
| MPSJQ12-A2 | Sep. 6, 2012 | A | zero-years-old |
| MPSJQ12-A3 | Oct. 15, 2012 | A | one-year-old |
| MPSJQ13-A4 | Aug. 28, 2013 | A | zero-years-old |
| MPSJQ13-A5 | Sep. 13, 2013 | A | one-year-old |
| MPSJQ13-A6 | Sep. 30, 2013 | A | one-year-old |
| MPSJQ13-A7 | Nov. 8, 2013 | A | zero-years-old |
| MPSJQ12-B1 | Aug. 21, 2012 | B | zero-years-old |
| MPSJQ12-C1 | Oct. 15, 2012 | C | zero-years-old |
| MPSJQ12-D1 | Sep. 13, 2012 | D | zero-years-old |
| MPSJQ12-E1 | Aug. 29, 2012 | E | zero-years-old |
| MPSJQ12-F1 | Sep. 14, 2012 | F | one-year-old |
Table 2.: Geographic relationship of fishing ports from which mycobacteriosis cases were analyzed in the present study
| Port | Island | Approx. distance to another port (km) | |||||
|---|---|---|---|---|---|---|---|
| Port A | Port B | Port C | Port D | Port E | Port F | ||
| A | Shishijima Is. | 0.0 | 3.8 | 5.2 | 6.2 | 6.2 | 8.0 |
| B | Shourajima Is. | 3.8 | 0.0 | 2.0 | 2.7 | 3.7 | 5.2 |
| C | Shourajima Is. | 5.2 | 2.0 | 0.0 | 1.1 | 1.8 | 3.2 |
| D | Shourajima Is. | 6.2 | 2.7 | 1.1 | 0.0 | 2.2 | 2.8 |
| E | Nagashima Is. | 6.2 | 3.7 | 1.8 | 2.0 | 0.0 | 1.7 |
| F | Nagashima Is. | 8.0 | 5.2 | 3.2 | 2.8 | 1.7 | 0.0 |
The partial nucleotide sequences of four genes (16S rRNA, ITS, hsp65, and rpoB), which are essential housekeeping genes, were also identified in these strains. The results obtained showed that all these strains belonged to the M. marinum complex (data not shown). Notably, all 12 strains showed c.637C>T in hsp65, which is a characteristic feature of Mps (ref. Nakanaga ).
Core genome comparison with the M. marinum complex
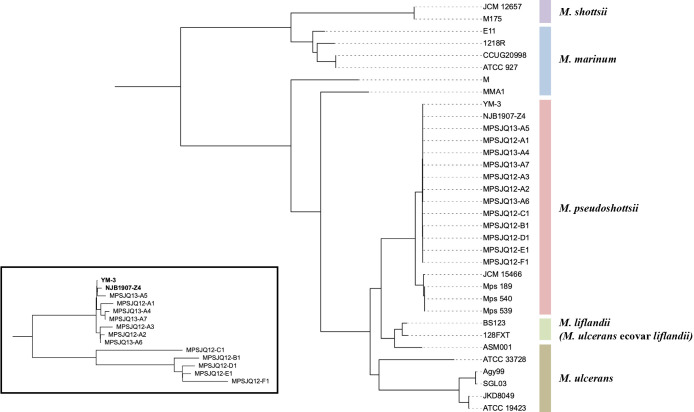
All bacterial strains were genomically sequenced, and contigs of approximately 6.0 Mb were assembled. These contig sequences were incorporated into the genome sequences of M. marinum complex strains available from the nucleotide database for a core genome analysis, along with the contig sequences of three Mps strains (named as isolates 189, 539, and 540) isolated from the East Mediterranean Sea of Israel (ref. Davidovich ). The core genome phylogenetic tree showed that the sequences of the two registered Mps strains (YM-3 and NJB1907-Z4) and those of the 12 strains in the present study were almost identical, whereas those of three Mediterranean strains were similar to that of the reference strain JCM 15466 (Fig. 2). An enlargement of the tree of these 14 strains showed that strains isolated from Port A were very closely related to the two well-known strains, while strains isolated from other ports belonged to another branch (Fig. 2, inset).
Comparison based on single-locus variants (SLVs)
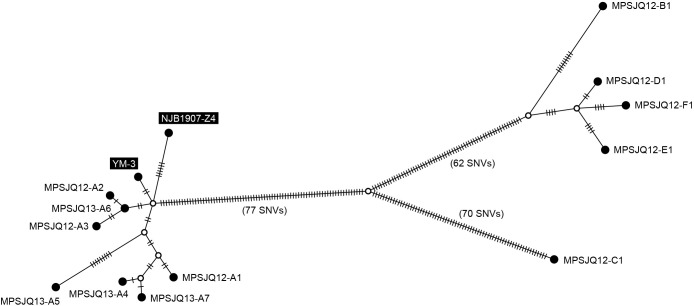
The 12 Mps strains obtained in the present study were very closely related to the strains isolated in Japan (YM-3 and NJB1907-Z4, isolated in 1986 and 2019, respectively), which prompted us to integrate them for a detailed genetic linkage analysis. A median-joining tree was constructed based on all SNVs (264 positions, listed in Table S2) detected through a mapping analysis of the 13 strains, using the YM-3 complete genome sequence as a reference (Fig. 3). These strains were divided into two major subgroups and a separated unique strain (MPSJQ12-C1; isolated from Port C). All strains from Port A were highly similar to the two strains (YM-3 and NJB1907-Z4) isolated from a separate area of Japan, implying the recent transmission of these strains across a wide area of the country. In contrast, the four isolates from the respective ports (B, D, E, and F) were also closely related to each other, indicating local transmission, in contrast to Port A. Although these four ports are located on two geographically close islands, MPSJQ12-C1 was unique from the other strains because it had a higher number of SNVs even though it originated from Port C, which is located on the same island as Ports B and D.
Discussion
This is the first study to introduce a molecular epidemiological approach to investigate the source of fish disease caused by Mps, a closely related species of M. marinum, which is etiological in aquatic animals. Phylogenetically, this species is distinct from M. ulcerans and its ecovar liflandii, which are also derived subspecies (Fig. 2). In contrast to these two subspecies, the number of cases of infection owing to Mps has been increasing in industrial fisheries, and there are concerns that its economic impact may be severe (ref. Stine ; ref. Mugetti ; ref. Davidovich ). This situation led us to anticipate that controlling infection by this species in fisheries will be required in the future. In the present study, strains from only 12 cases were analyzed, out of the many cases that would have occurred within the nearshore area of Kagoshima Prefecture, Japan over a 2-year period, of which one port was involved in the collection of strains over a 2-year period. ref. Mugetti also collected strains in a similar setting, and a more detailed analysis with additional strains will provide a more accurate estimation of transmission pathways. The introduction of a genomic epidemiological analysis, which is used in human public health, to these cases may lead to the more effective management of farmed fish and the possibility of mitigating or preventing economic damage caused by infectious disease.
The comparative genomic analysis of the strains in this study allowed us to arrive at the following three inferences. The strains isolated from cases that occurred in Port A were highly similar to strains previously isolated from other distant regions of Japan (Fig. 1: YM-3 from Kochi and NJB1907-Z4 from Tokyo), suggesting that nationwide transmission routes exist. Furthermore, local transmission in several ports (B, D, E, and F) may be inferred from the similarities of strains from the four ports, which were distinct from those isolated from Port A. Moreover, the strain isolated from Port C was unique, suggesting that the source of infection differed from that of the other ports or that it was port-specific. Genomic data on strains from cases within Port A analyzed in this study indicate nationwide spread, as mentioned earlier; however, no correlation was found between reports from other regions, indicating uncertainty regarding the source of the infection. Yellowtails are sometimes subject to “intermediate rearing”, in which young fish are reared for a year and then transferred to other ports, which may be a possible route of transmission owing to traffic among ports. On the other hand, the group of strains was isolated from four different ports (B, D, E, and F), suggested that a genome-based study will provide a regional basis for the spread of this pathogen.
It is important to note that transmission routes among these clonal strains may be overestimated because the mutation rate of this species may be slow, similar to that in other mycobacteria (e.g., in M. tuberculosis, <0.5 SNP/year) (ref. Walker ; ref. Guerra-Assunção ). Therefore, phylogenetic clusters comprising strains from Port A and from the references may be interpreted as originating from unrelated subgroups that had branched independently from a common ancestor. However, even under such an assumption, it may be argued that the evidence obtained also supports nationwide spread. However, more strains need to be collected from each port for multiple cases for a comparative genomic analysis.
The worldwide Mps epidemic, which may occur because of trade and logistics, may be monitored through genome comparisons of strains accumulated internationally from cases of Mps infection. Mps strains from Mediterranean farmed fish in Italy and Israel were genome-phylogenetically distinct from Japanese strains (Fig. 2). The strains from Israel were phylogenetically close to the type strain JCM 15466T isolated in the United States; therefore, the possibility of international fish trade being responsible for the spread of infection has been noted (ref. Mugetti ). The strains from Japan analyzed in the present study were distinct from these strains, indicating that the dissemination of these strains may be limited to Japan, whereas cross-transmission among Western countries has not yet been examined. The future distribution of these distinct strains may lead to a more global mixture and genome-based monitoring may be able to accurately characterize the outbreak. Since the cases we analyzed occurred in 2012, constant monitoring may be required for prompt implementation.
In summary, the present study demonstrated that strain comparisons based on a genomic analysis of Mps outbreaks in breeding fish may provide valuable insights for epidemic monitoring. The results obtained also indicate that the accumulation of detailed genomic data on a pathogen may generate a public health contribution to the search for the origin of mycobacterial diseases in aquaculture fisheries.
Citation
Wada, T., Yoshida, S., Yamamoto, T., Nonaka, L., Fukushima, Y., Nakajima, C., et al. (2024) Application of Genomic Epidemiology of Pathogens to Farmed Yellowtail Fish Mycobacteriosis in Kyushu, Japan. Microbes Environ 39: ME24011.
https://doi.org/10.1264/jsme2.ME24011
Supplementary Materials
References
- Mycobacterium marinum.. Microbiol Spectrum, 2017
- Median-joining networks for inferring intraspecific phylogenies.. Mol Biol Evol, 1999. [PubMed]
- Isolation of genomic DNA from mycobacteria.. Methods Mol Biol, 1998. [PubMed]
- Whole genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations.. PLoS Pathog, 2012. [PubMed]
- Extensive genomic diversity among Mycobacterium marinum strains revealed by whole genome sequencing.. Sci Rep, 2018. [PubMed]
- Identification of Mycobacterium pseudoshottsii in the Eastern Mediterranean.. Microbiol Spectrum, 2023
- Piscine mycobacteriosis: a literature review covering the agent and the disease it causes in fish and humans.. Vet Microbiol, 2004. [PubMed]
- kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome.. Bioinformatics, 2015. [PubMed]
- Mycobacteriosis in fishes: a review.. Vet J, 2009. [PubMed]
- Genomic degeneration and reduction in the fish pathogen Mycobacterium shottsii.. Microbiol Spectrum, 2022
- Epidemiologic data and pathogen genome sequences: a powerful synergy for public health.. Genome Biol, 2014. [PubMed]
- Buruli ulcer: review of a neglected skin mycobacterial disease.. J Clin Microbiol, 2018. [PubMed]
- Large-scale whole genome sequencing of M. tuberculosis provides insights into transmission in a high prevalence area.. eLife, 2015. [PubMed]
- Mycobacterium marinum infection in fish and man: epidemiology, pathophysiology and management; a review.. Vet Q, 2018. [PubMed]
- Draft genome sequence of the fish pathogen Mycobacterium pseudoshottsii strain JCM15466, a species closely related to M. marinum.. Genome Announc, 2016. [PubMed]
- Retrospective identification of pathogenic mycobacterial species in fish: Mycobacterium pseudoshottsii YM-3, isolated from a yellowtail fish in 1986 in Kochi, Japan.. Microbiol Resour Announc, 2023. [PubMed]
- A review of mycobacteriosis in marine fish.. J Fish Dis, 2009. [PubMed]
- Supersize me: how whole-genome sequencing and big data are transforming epidemiology.. Trends Microbiol, 2014. [PubMed]
- Differentiation of mycobacterial species by hsp65 duplex PCR followed by duplex-PCR-based restriction analysis and direct sequencing.. J Clin Microbiol, 2006. [PubMed]
- A case of mycobacteriosis associated with Mycobacterium pseudoshottsii in aquarium-reared fish in Japan.. J Vet Med Sci, 2022. [PubMed]
- Versatile and open software for comparing large genomes.. Genome Biol, 2004. [PubMed]
- POPART: full-feature software for haplotype network construction.. Methods Ecol Evol, 2015
- Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation.. Nucleic Acids Res, 2021. [PubMed]
- Mycobacterium pseudoshottsii in Mediterranean fish farms: new trouble for European aquaculture?. Pathogens, 2020. [PubMed]
- Mycobacterium pseudoshottsii isolated from 24 farmed fishes in western Japan.. J Vet Med Sci, 2012. [PubMed]
- IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies.. Mol Biol Evol, 2015. [PubMed]
- Using SPAdes de novo assembler.. Curr Protoc Bioinf, 2020
- Mycobacterium pseudoshottsii sp. nov., a slowly growing chromogenic species isolated from Chesapeake Bay striped bass (Morone saxatilis).. Int J Syst Evol Microbiol, 2005. [PubMed]
- Revealing COVID-19 transmission in Australia by SARS-CoV-2 genome sequencing and agent-based modeling.. Nat Med, 2020. [PubMed]
- Differentiation of phylogenetically related slowly growing mycobacteria based on 16S-23S rRNA gene internal transcribed spacer sequences.. J Clin Microbiol, 1998. [PubMed]
- Expanded range and new host species of Mycobacterium shottsii and M. pseudoshottsii.. J Aquat Anim Health, 2009. [PubMed]
- Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer.. Genome Res, 2007. [PubMed]
- Complete genome sequence of the frog pathogen Mycobacterium ulcerans ecovar liflandii.. J Bacteriol, 2013. [PubMed]
- Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study.. Lancet Infect Dis, 2013a. [PubMed]
- Contact investigations for outbreaks of Mycobacterium tuberculosis: advances through whole genome sequencing.. Clin Microbiol Infect, 2013b. [PubMed]
- Evolution of Mycobacterium ulcerans and other mycolactone-producing mycobacteria from a common Mycobacterium marinum progenitor.. J Bacteriol, 2007. [PubMed]