
Rotenone Induces a Neuropathological Phenotype in Cholinergic-like Neurons Resembling Parkinson’s Disease Dementia (PDD)
Abstract
Supplementary Information:
The online version contains supplementary material available at 10.1007/s12640-024-00705-3.
Article type: Research Article
Keywords: Alzheimer’s disease, Alpha-synuclein, E280A, Parkinson, Presenilin, Mutation, Rotenone
Affiliations: https://ror.org/03bp5hc83grid.412881.60000 0000 8882 5269Neuroscience Research Group, Medical Research Institute, Faculty of Medicine, University of Antioquia (UdeA), Calle 70 No. 52-21, and Calle 62 # 52-59, Building 1, Room 412, Medellin, Antioquia Colombia
License: © The Author(s) 2024 CC BY 4.0 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Article links: DOI: 10.1007/s12640-024-00705-3 | PubMed: 38842585 | PMC: PMC11156752
Relevance: Relevant: mentioned in keywords or abstract
Full text: PDF (1.4 MB)
Introduction
Parkinson’s disease (PD) is a progressive chronic neurologic disorder clinically characterized by slowing of movements in the limbs, face, walking or overall body (bradykinesia), resting tremor which typically stops when the patient is active or moving, rigidity or stiffness in the arms, legs or trunk (Jankovic ref. 2008; Bloem et al. ref. 2021). Alzheimer’s disease (AD) is also a common progressive disorder beginning with mild memory loss leading to typical features of dementia such as severe impairments in thought, memory, and language (Knopman et al. ref. 2021). While PD is the consequence of dramatic loss of dopaminergic (DAergic) neurons in the ventral part of the pars compacta in the substantia nigra and deficit of dopaminergic innervation of striatum (Dickson ref. 2012), AD is caused by the losses of forebrain cholinergic neurons of the nucleus basalis of Meynert (Ch4) and cholinergic projection from the Medial septum nucleus (Ch1) to hippocampus (Liu et al. ref. 2015; Pepeu and Grazia Giovannini ref. 2017). Intriguingly, Parkinson’s disease with dementia (PDD) is a neurological disorder that overlaps with PD and AD (Irwin et al. ref. 2013). By definition, PDD is a clinical condition wherein patients with diagnosis of PD goes together with cognitive dysfunction (e.g., deficits in attention, executive functioning, visuospatial processing), neuropsychiatric symptoms (e.g., hallucinations, depression), memory loss and dementia (Goetz et al. ref. 2009; Vasconcellos and Pereira ref. 2015; Phillips et al. ref. 2023). Pathologically, PDD is characterized by severe burden of Lewy bodies, which are composed of the pathological PD-associated protein \(\mathrm{\alpha }\) -synuclein (\(\mathrm{\alpha }\)-Syn) and Lewy neurites, as well as extensive diffuse amyloid-β‑reactive plaque pathology, Tau-reactive diffuse threads and neurofibrillary tangles (NFT), which are AD-associated pathological markers (Smith et al. ref. 2019; Kouli et al. ref. 2020). Although it is supposed that \(\mathrm{\alpha }\)- Syn, A\(\upbeta\), and Tau might synergistically induce cholinergic and DAergic neuronal degeneration (Kim ref. 2023), presently the pathological mechanism of PDD remains unclear. Therefore, it is essential to delve into the cellular and molecular aspects of this neurological entity to identify potential targets for the prevention and treatment strategies (Han et al. ref. 2021).
Rotenone (ROT, PubChem CID 6758), a naturally occurring organic compound found in the roots of the Derris (Sae-Yun et al. ref. 2006; Zubairi et al. ref. 2014) and Lonchocarpus (Fang and Casida ref. 1999) plant species, is used worldwide due to its broad spectrum insecticidal (Zhang et al. ref. 2022a), acaricidal and pesticide properties (http://www.chm.bris.ac.uk/motm/rotenone/; available in November, 2023). Importantly, ROT induces specific degeneration of DAergic neurons in vitro and in vivo (Lawana and Cannon ref. 2020), which intrinsically deteriorate in PD (Giguère et al. ref. 2018). Mechanistically, ROT acts as a strong inhibitor of complex I of the mitochondrial respiratory chain (Read et al. ref. 2021) via inhibition of electron transfer from the iron-sulfur centers in complex I to ubiquinone, leading to a blockade of the IQ site (Schiller and Zickermann ref. 2022), and over-reduction of complex I causes electrons to leak and produce reactive oxygen species (ROS) such as superoxide anion radical (O2.−). This radical can dismutate into non-radical reactive hydrogen peroxide, H2O2 (Li et al. ref. 2003; Mailloux ref. 2015). In turn, this last compound, via signaling mechanisms (Marinho et al. ref. 2014; Antunes and Brito ref. 2017), induces regulated cell death apoptosis (Velez-Pardo and Jimenez-Del-Rio ref. 2020). Although, the effect of ROT appears tissue-specific, it has also been demonstrated that ROT induces cell death of non-catecholaminergic neurons such as cholinergic neurons (ChNs) in an organotypic co-culture brain slice model (Ullrich and Humpel ref. 2009). This last observation suggests that hippocampal Lewy pathology might be associated with cholinergic degeneration in PD with cognitive decline (Liu et al. ref. 2019; Aarsland et al. ref. 2021). These may explain why some brains from PDD patients present AD features such as A\(\upbeta\) plaques and tau containing NFT (Irwin et al. ref. 2013; Jellinger ref. 2023). These observations suggest that \(\mathrm{\alpha }\) -Syn, A\(\upbeta\), and Tau might together induce neuronal degeneration. In line with this view, it has been shown that ROT induced \(\mathrm{\alpha }\)-Syn and A\(\upbeta\) aggregation, as well as increased hyperphosphorylation of Tau in cultured cells from hippocampus, locus coeruleus and substantia nigra of newborn Lewis rats (Chaves et al. ref. 2010). Moreover, ROT triggered a cerebral tauopathy in rats (Höglinger et al. ref. 2005). However, the mechanism by which ROT induce co-existence of \(\mathrm{\alpha }\)-Syn, A\(\upbeta\), and Tau is unknown. Furthermore, the mechanism by which ROT produces proteinopathy in cholinergic neurons is yet unknown. Although ROT has extensively been used as model neurotoxin to disclose the molecular events of PD (Ke et al. ref. 2021), currently there are not in vitro model of PDD available.
Mesenchymal stromal cells (MSCs) derived from umbilical cord Warton’s Jelly (UC-WJ) are multipotent cells that have the potential to differentiate into neuroectodermal cell lineage (Dominici et al. ref. 2006; Viswanathan et al. ref. 2019). Our laboratory has used WJ-MSCs to recapitulate the neuropathological features of familial AD (FAD) due to a mutation in presenilin 1 (PSEN1) E280A (Soto-Mercado et al. ref. 2020). Indeed, cultured MSCs bearing the wild-type (WT) and variant PSEN1 E280A in Cholinergic-N-Run medium (Mendivil-Perez et al. ref. 2019) transdifferentiated into cholinergic-like neurons (ChLNs). Notably, PSEN1 E280A ChLNs but not WT PSEN1 ChLNs showed accumulation of intracellular amyloid precursor protein fragments (iAPP\(\upbeta\)f/ iA\(\upbeta\)), and displayed phosphorylation of protein TAU (p-TAU at residues Ser202/Thr205). Also, the variant ChLNs E280A presented oxidation of stress sensor DJ-1Cys106-SH into DJ-1Cys106-SO3, phosphorylation of c-JUN at Ser63/Ser73, and detection of dichlorofluorescein (DCF)-positive cells as evidenced of generation of ROS such as H2O2, oxidative stress (OS), concomitant loss of the \(\Delta \mathrm{\Psi m}\), DNA fragmentation, and Ca2+ flux dysregulation (Soto-Mercado et al. ref. 2020). Despite these advances, no chemical neurotoxin-induced model of FAD has yet been revealed.
Given that PDD appears neuropathologically to recapitulate AD and PD, we hypothesize that ROT can replicate both FAD and PD cellular hallmarks in ChLNs, which represent an excellent model to study PDD. We have treated ChLNs derived from WJ-MSCs with ROT (1, 5, 10 \(\upmu\)M) for 24 h. For comparative and validation purposes, we used WJ-MSC-derived PSEN1 E280A ChLNs. We found that (i) ROT induces generation of ROS and H2O2, loss of \(\Delta \mathrm{\Psi m}\), oxidized DJ-1, accumulation of iA\(\upbeta\), p-Ser202/Thr205 TAU, and cell death happens in ChLNs, as it appears in PSEN1 E280A ChLNs FAD; (ii) ROT induces concomitant p-Ser935 LRRK2 and p-Ser129-\(\mathrm{ \alpha }\)-Syn in ChLNs. Outstandingly, we report for the first time that p-LRRK2 and p-\(\mathrm{ \alpha }\)-Syn also endogenously appear in PSEN1 E280A ChLNs; (iii) ROT increases the phosphorylation of c-JUN at residues Ser63/Ser73 and the expression of TP53, PUMA and pro-apoptotic marker cleaved caspase 3 (CC3) in ChLNs. A similar profile of positive protein markers was also observed in PSEN1 E280A ChLNs; (iv) ROT impairs ACh-induced transient Ca2+ influx in ChLNs to a similar extend as observed in PSEN1 E280A ChLNs. Interestingly, ChLNs co-treated with ROT and anti-amyloidogenic and antioxidant cannabidiol, JNK inhibitor SP600125, and LRRK2 inhibitor PF-06447475 significantly blunted A\(\upbeta\), oxDJ-1, p-\(\mathrm{ \alpha }\)-Syn, p-TAU, and CC3, respectively. Given that ROT and iA\(\upbeta\) trigger mitochondrial dysfunction, and produce H2O2, they may be an important etiopathogenic factor involved not only in AD and PD (Velez-Pardo and Jimenez-Del-Rio ref. 2020) but also in PDD. Therefore, mitochondria, LRRK2, and H2O2 might be targets for therapeutic treatment for PDD (Macdonald et al. ref. 2018; Abrishamdar et al. ref. 2023).
Materials and Methods
Transdifferentiation of Mesenchymal Stromal Cell into Cholinergic-like Neurons
ChLN differentiation was performed according to (Mendivil-Perez et al. ref. 2019). Briefly, the WT (TBC# WJMSC-19) and PSEN1 E280A (TBC# WJMSC-24) were obtained from the Neuroscience Tissue Bank-UdeA. MSCs were seeded at 1–1.5 × 104 cells/cm2 in laminin-treated culture plates for 24 h in regular culture medium (RCm). The medium was removed, and cells were incubated in cholinergic differentiation medium (Cholinergic-N-Run medium, hereafter Ch–N-Rm) containing DMEM/F-12 media 1:1 Nutrient Mixture (Gibco cat# 10,565,018; 1204 N Western St, Suite C, Amarillo, TX, USA), 10 ng/mL basic fibroblast growth factor (bFGF) recombinant human protein (Gibco Cat# 13,256,029), 50 µg/mL sodium heparin (Hep, Sigma-Aldrich cat# H3393; 3050 Spruce Street, St. Louis, MO 63103, USA), 0.5 µM all-trans retinoic acid, 50 ng/mL sonic hedgehog peptide (SHH, Sigma cat# SRP3156) and 1% FBS at 37 °C for 7 days. After this process of transdifferentiation, the cells were labeled as WT PSEN1 or PSEN1 E280A ChLNs. Since Ch–N-Rm contains several factors (e.g., growth factors) that might interfere with the experiment interpretation and measurements, WT PSEN1 and PSEN1 E280A ChLNs (obtained after 7 days in Ch–N-Rm) were left in a regular culture medium (RCm) for 4 additional days of post transdifferentiation (Mendivil-Perez et al. ref. 2019).
Assay Protocol
An initial rotenone (ROT) screening was performed at least twice in triplicate including 1, 5, and 10 μM final concentrations. Subsequently, 10 μM ROT was established as an optimal concentration for further experiments. For analyses, ChLNs were divided into two groups: (i) untreated WT PSEN1; and (ii) WT PSEN1 treated with 10 μM ROT. Chemical inhibition assay was performed by pre-incubating WT PSEN1 in absence or presence of cannabidiol (CBD; 10 μM, Mendivil-Perez et al. ref. 2023), the anthrapyrazolone JNK inhibitor SP600125 (SP; 1 μM, Soto-Mercado et al. ref. 2020), cell-permeable chemical inhibitor of p53 pifithrin-\(\mathrm{ \alpha }\)(PFT-\(\mathrm{ \alpha }\); 50 nM, Velez-Pardo et al. ref. 2002), and the potent and selective LRRK2 inhibitor PF-06447475 (PF475; 1 μM, Mendivil-Perez et al. ref. 2016) for 30 min previous to ROT exposure. The PSEN1 E280A ChLNs were used for comparative and validation purposes.
Immunofluorescence Analysis
The analysis of Alzheimer’s disease-, oxidative stress- and cell death-related markers, was exactly performed as described elsewhere (Soto-Mercado et al. ref. 2020). Briefly, the cells treated under different conditions were fixed with 4% paraformaldehyde for 20 min, followed by Triton X-100 (0.1%) permeabilization and 10% bovine serum albumin (BSA) blockage. Cells were incubated overnight with primary antibodies against the first 2 amino acids of the A\(\upbeta\) peptide amino N-terminus, namely the A\(\upbeta\)4 1E8 antibody (1:500; clone 1E8 cat# MABN639, Millipore, 3050 Spruce Street, St. Louis, MO 63304, USA), phospho-TAU (p-Tau, 1:500, Ser202/Thr205, cat# MN1020 (AT8); and primary antibodies against oxidized DJ-1 (1:500; ox(Cys106)DJ-1; spanning residue C106 of human PARK7/DJ1; oxidized to produce cysteine sulfonic (SO3) acid; cat # ab169520, Abcam). To assess cell death, we used primary antibodies against PUMA conjugated with Alexa Fluor 488 (1:500; PUMA, sc-377015 AF488, Santa Cruz Biotechnology), p53 conjugated with Alexa Fluor 594 (1:500; cat# sc-126 AF594, Santa Cruz Biotechnology), phospho-c-Jun conjugated with Alexa fluor 594 (1:500; c-Jun (S63/73) cat# sc-822 AF594, Santa Cruz Biotechnology), and caspase-3 fluorescent probe (1:500; AM- DEV-FMK- caspase 3, V35118). To evaluate α-synuclein, we used primary antibodies against total α-synuclein (1:500; clone Syn211 cat# S5566, Sigma Aldrich), and phosphorylated α-synuclein (1:500; phosphoS129; cat# ab51253, Abcam). After exhaustive rinsing, we incubated the cells with secondary fluorescent antibodies (to identify non-conjugated antibodies reactions; DyLight 488 and 594 horse anti-rabbit, -goat and -mouse, cat DI 1094, DI 3088, and DI 2488, respectively) at 1:500. The nuclei were stained with 1 μM Hoechst 33,342 (Life Technologies), and images were acquired on an Axiovert coupled to Axicam miscoscope.
Flow Cytometry Analysis
After each treatment, cells were detached using trypsin and centrifuged for 10 min at 2000 rpm. Then, cells were washed with PBS and fixed with cold ethanol (96%) overnight. Cells were washed two times with PBS and permeabilized with 0.2% Triton X-100 plus 1.5% bovine serum albumin (BSA) for 30 min. Cells were incubated overnight with primary antibodies against the first 2 amino acids of the A\(\upbeta\) peptide amino N-terminus, namely the A\(\upbeta\)4 1E8 antibody (1:500; clone 1E8 cat# MABN639, Millipore, 3050 Spruce Street, St. Louis, MO 63304, USA), phospho-TAU (AT8; 1:200), oxidized DJ-1 (1:200), PUMA (1:200), p53 (1:200), phospho-c-Jun (1:200; c-Jun (S63/73), caspase-3 (1:200), total α-synuclein (1:200)and phosphorylated α-synuclein (1:200). After exhaustive rinsing, we incubated the cells with secondary fluorescent antibodies (DyLight 488 and 594 horse anti-rabbit, -goat and -mouse, cat DI 1094, DI 3088, and DI 2488, respectively) at 1:500. Fluorescence analysis was performed on a BD LSRFortessa II flow cytometer (BD Biosciences). Cells without primary antibodies served as a negative control. For assessment, 10,000 events and quantitative data and figures were obtained using FlowJo 7.6.2 Data Analysis Software (TIBCO® Data Science). Events analysis was performed by determining the cell population (Forward Scatter analysis, Y axis) that exceeded the basal fluorescence (488 nm or 594 nm, X axis) of the negative control. Accordingly, density plots or histograms were created from event analysis.
Evaluation of Intracellular Reactive Oxygen Species (e.g., Hydrogen Peroxide, H2O2) by Fluorescence Microscopy
To assess the levels of intracellular ROS (H2O2), we used 2′,7′-dichlorofluorescein diacetate (5 μM, DCFH2-DA; Invitrogen) according to Soto-Mercado et al. ref. 2020. ChLNs were left in RCm for 4 days. Then, the cells (5 × 103) were incubated with the DCFH2-DA reagent for 30 min at 37 °C in the dark. Cells were then washed, and DCF fluorescence intensity was determined by analysis of fluorescence microscopy images (Lichtman and Conchello ref. 2005). The assessment was repeated three times in independent experiments. The nuclei were stained with 0.5 µM Hoechst 33,342 staining compound. The assessment was repeated three times in independent experiments blind to the experimenter.
Evaluation of Intracellular Hydrogen Peroxide (H2O2) by Flow Cytometry
To assess the levels of intracellular ROS (H2O2), we used 2′,7′-dichlorofluorescein diacetate (5 μM, DCFH2-DA; Invitrogen) according to Soto-Mercado et al. ref. 2020. ChLNs were left in RCm for 4 days. Then, the cells (1 × 104) were incubated with the DCFH2-DA reagent for 30 min at 37 °C in the dark. Cells were then washed, and DCF fluorescence was determined using an LSRFortessa (BD Biosciences). The assessment was repeated 3 times in independent experiments. Quantitative data and figures were obtained using FlowJo7.6.2 Data Analysis Software. The assessment was repeated three times in independent experiments blind to experimenter and flow cytometer analyst (Adan et al. ref. 2017).
Analysis of Mitochondrial Membrane Potential (ΔΨm) by Fluorescence Microscopy
ChLNs were left in a regular culture medium (RCm) for 4 days. Then, the cells (5 × 103) were incubated with the passively diffusing and active mitochondria-accumulating dye deep red MitoTracker compound (20 nM, final concentration) for 20 min at RT in the dark (cat # M22426, Invitrogen, Soto-Mercado et al. ref. 2020). Cells were then washed twice with PBS. MitoTracker fluorescence intensity was determined by analysis of fluorescence microscopy images (Lichtman and Conchello ref. 2005). The assessment was repeated three times in independent experiments. The nuclei were stained with 0.5 µM Hoechst 33,342 staining compound. The assessment was repeated three times in independent experiments blind to the experimenter.
Analysis of Mitochondrial Membrane Potential (\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\Delta \mathrm{\Psi m}$$\end{document}ΔΨm) by Flow Cytometry
ChLNs were left in a regular culture medium (RCm) for 4 days. Then, the cells (1 × 104) were incubated with the passively diffusing and active mitochondria-accumulating dye deep red MitoTracker compound (20 nM, final concentration) for 20 min at RT in the dark (cat # M22426, Invitrogen, (Soto-Mercado et al. ref. 2020). The cells were analyzed using an LSRFortessa (BD Biosciences). The experiment was performed three times in independent experiments, and 10,000 events were acquired for analysis. Quantitative data and figures were obtained using FlowJo 7.6.2 Data Analysis Software. The assessment was repeated three times in independent experiments blind to experimenter and flow cytometer analyst.
Intracellular Calcium Imaging
Intracellular calcium (Ca2+) concentration changes evoked by cholinergic stimulation were assessed according to Sekiguchi-Tonosaki et al. ref. 2009 and Pap et al. ref. 2009, with minor modifications. For the measurement, the fluorescent dye Fluo-3 (Fluo-3 AM; Thermo Fisher Scientific, cat: F1242) was employed. The dye was dissolved in DMSO (1 mM) to form a stock solution. Before the experiments, the stock solution was diluted in neuronal buffer solution (NBS buffer in mM: 137 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, pH 7.3, and 22 glucose). The working concentration of the dye was 2 μM. The WT and PSEN1 E280A ChLNs were incubated for 30 min at 37 °C with the dye containing NBS and then washed five times. Intracellular Ca2+ transients were evoked by acetylcholine (1 mM final concentration) at 4 days post differentiation. The measurements were carried out using the 20 × objective of the microscope. Several regions of interest (ROIs) were defined in the visual field of the camera. One of the ROIs was cell free, and the fluorescence intensity measured here was considered background fluorescence (Fbackgroung). The time dependence of the fluorescence emission was acquired, and the fluorescence intensities (hence the Ca2+ levels) were represented by pseudo colors. To calculate the changes of the average Ca2+-related fluorescence intensities, the Fbg value was determined from the cell-free ROI, and then the resting fluorescence intensities (Frest) of the cell-containing ROIs were obtained as the average of the points recorded during a consecutive period of 10 s before the addition of acetylcholine. The peaks of the fluorescence transients were found by calculating the average of six consecutive points and identifying those points that gave the highest average value (Fmax). The amplitudes of the Ca2+-related fluorescence transients were expressed relative to the resting fluorescence (ΔF/F) and were calculated by the following formula: ΔF/F = (Fmax − Frest)/(Frest − Fbg). For the calculation of the fluorescence intensities, ImageJ was used. The terms fluorescence intensity was used as an indirect indicator of intracellular Ca2+ concentration. The assessment was repeated three times in independent experiments blind to the experimenter.
Photomicrography and Image Analysis
Light microscopy photographs were taken using a Zeiss Axio Vert.A1 coupled to AxioCam Cm1 microscope, and fluorescence microscopy photographs were taken using a Zeiss Axio Vert.A1 Fluorescence Microscope equipped with a Zeiss AxioCam Cm1. Fluorescence images were transformed into 8-bit images and the background was subtracted (Zen 3.4 blue edition). Images were then analyzed by ImageJ software (http://imagej.nih.gov/ij/) using an in house-made macro. Briefly, the cellular measurement regions of interest (ROIs) were drawn around the nucleus (for the case of transcription factors and apoptosis effectors) or overall cells (for cytoplasmic probes), and the fluorescence intensity was subsequently determined by applying the same threshold for cells in the control and treatment conditions. Mean fluorescence intensity (MFI) was obtained by normalizing total fluorescence to the number of nuclei.
Molecular Docking
We used the x-ray diffraction crystallography protein structure of \(\upgamma\)-secretase (protein data bank, PDB, code: 5FN2, (Bai et al. ref. 2015) for molecular docking analysis. The blind molecular docking was performed with CB-Dock version 2 (Liu et al. ref. 2022), a cavity detection-guided protein–ligand blind docking web server that uses Autodock Vina (version 1.1.2, Scripps Research Institute, La Jolla, USA). The Standard Delay Format (SDF) of the chemical structure of the tested compounds (rotenone, SCH 697466, MRK560, SCH 900229, LY-374973) were downloaded from PubChem. The molecular blind docking was performed by uploading the 3D structure PDB file of listed protein into the server with the SDF file of each compound. For analysis, we selected the docking poses with the strongest Vina score in the catalytical pocket. The generated PDB files of the molecular docking of each compound were visualized with the CB-Dock2 interphase and were compared against the experimentally validated X-ray structures of the interaction with reference compounds.
Data Analysis
In this experimental design, two vials of MSCs were thawed (WT PSEN1 and PSEN1 E280A), cultured and the cell suspension was pipetted at a standardized cellular density of 2.6 × 104 cells/cm2 into different wells of a 24-well plate. Cells (i.e., the biological and observational unit (Lazic et al. ref. 2018) were randomized to wells by simple randomization (sampling without replacement method), and then wells (i.e., the experimental units) were randomized to treatments by a similar method. Experiments were performed on three independent occasions (n = 3) blind to the experimenter and/or flow cytometer analyst (Lazic et al. ref. 2018). The data from the three repetitions i.e., independent experiments were averaged and a representative flow cytometry density or histogram plot from the three independent experiments was selected for illustrative purposes, whereas bar in quantification figures represent the mean ± SD and the three black dots show data point of each experimental repetition. Based on the assumption that the experimental unit (i.e., the well) data comply with the independence of observations, the dependent variable is normally distributed in each treatment group (Shapiro–Wilk test), and variances are homogeneous (Levene’s test), the statistical significance was determined by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc comparison calculated with GraphPad Prism 5.0 software. Differences between groups were only deemed significant when a p-value of < 0.05 (*), < 0.001 (**) and < 0.001 (***). All data are illustrated as the mean ± S.D.
Results
Wild Type (WT) and PSEN 1 E280 Warthon Jelly’s Mesenchymal Stromal Cells (WJ MSCs)-Derived Cholinergic-Like Cells (ChLNs) Show Typical Markers of Cholinergic Lineage
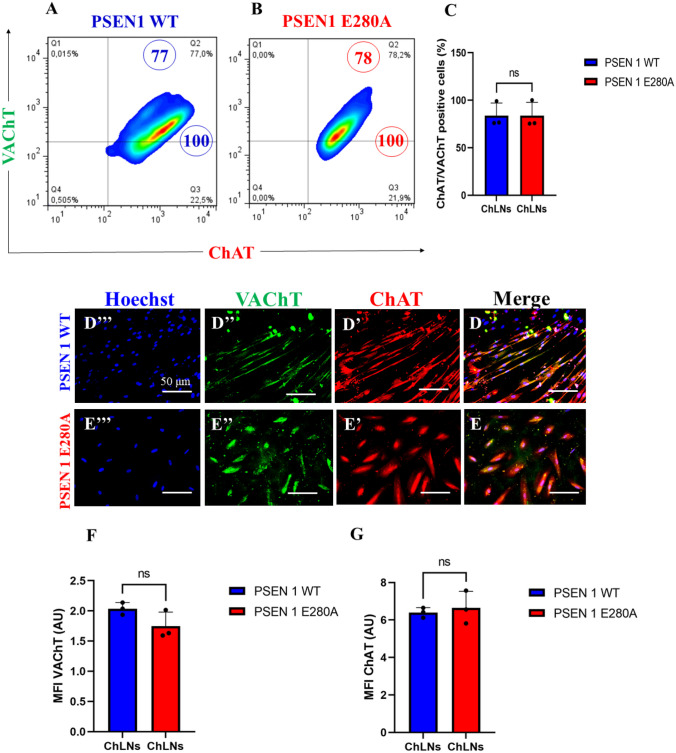
We first wanted to confirm that the mutation PSEN 1 E280A did not alter the transdifferentiation of WJ-MSCs into ChLNs cultured in Ch–N-Run medium (Soto-Mercado et al. ref. 2020; Mendivil-Perez et al. ref. 2023). Effectively, WT and mutant WJ-MSCs transdifferentiated into ChLNs in Cholinergic-N-Run yielding 75% cholinergic markers ChAT/ VAChT according to flow cytometry analysis (Fig. 1A-C). The cholinergic lineage markers were also detected by fluorescent microscopy (Fig. 1D–G). For comparative and validation purposes, we included the PSEN1 E280A ChLNs (Soto-Mercado et al. ref. 2020; Mendivil-Perez et al. ref. 2023).
Rotenone (ROT) Provokes Loss of Mitochondrial Membrane Potential (\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\Delta \mathrm{\Psi m}$$\end{document}ΔΨm) and Generation of Reactive Oxygen Species (ROS)
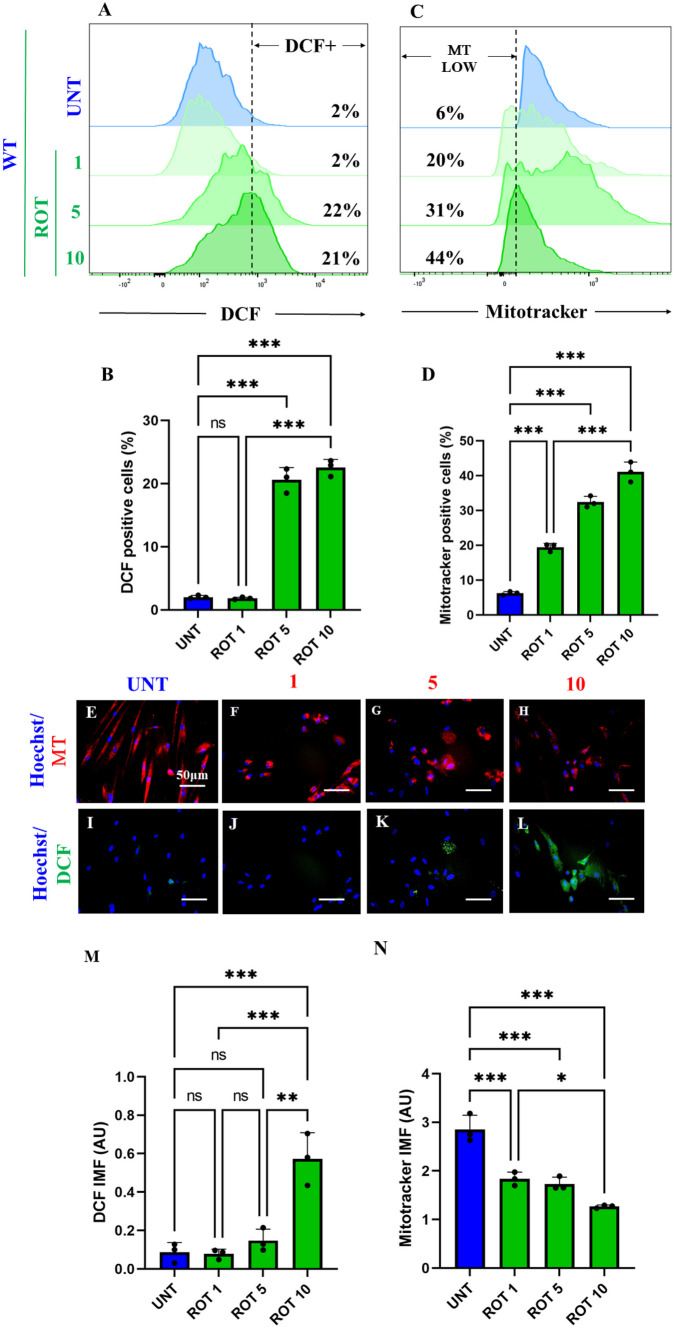
It is well-known that ROT induces ROS/H2O2 through inhibition of mitochondrial Complex I (Li et al. ref. 2003; Read et al. ref. 2021). Therefore, we determined the concentration of ROT at which generates the maximal percentage of ROS and mitochondrial damage in ChLNs. As shown in Fig. 2, ROT induces concentration-dependent loss of \(\Delta \mathrm{\Psi m}\) (Fig. 2A, B) and generation of ROS up to 5 μM (Fig. 2C, D). Similar data were observed by fluorescent microscopy (Fig. 2E–N). Therefore, we selected ROT (10 \(\upmu\)M) for further experiments.
Rotenone (ROT) Induces Phosphorylation of Leucine-Rich Repeated Kinase 2 (LRRK2) Concomitantly with Phosphorylation of \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\mathrm{\alpha }$$\end{document}α-Synuclein at Pathological Residue Ser129 in ChLNs
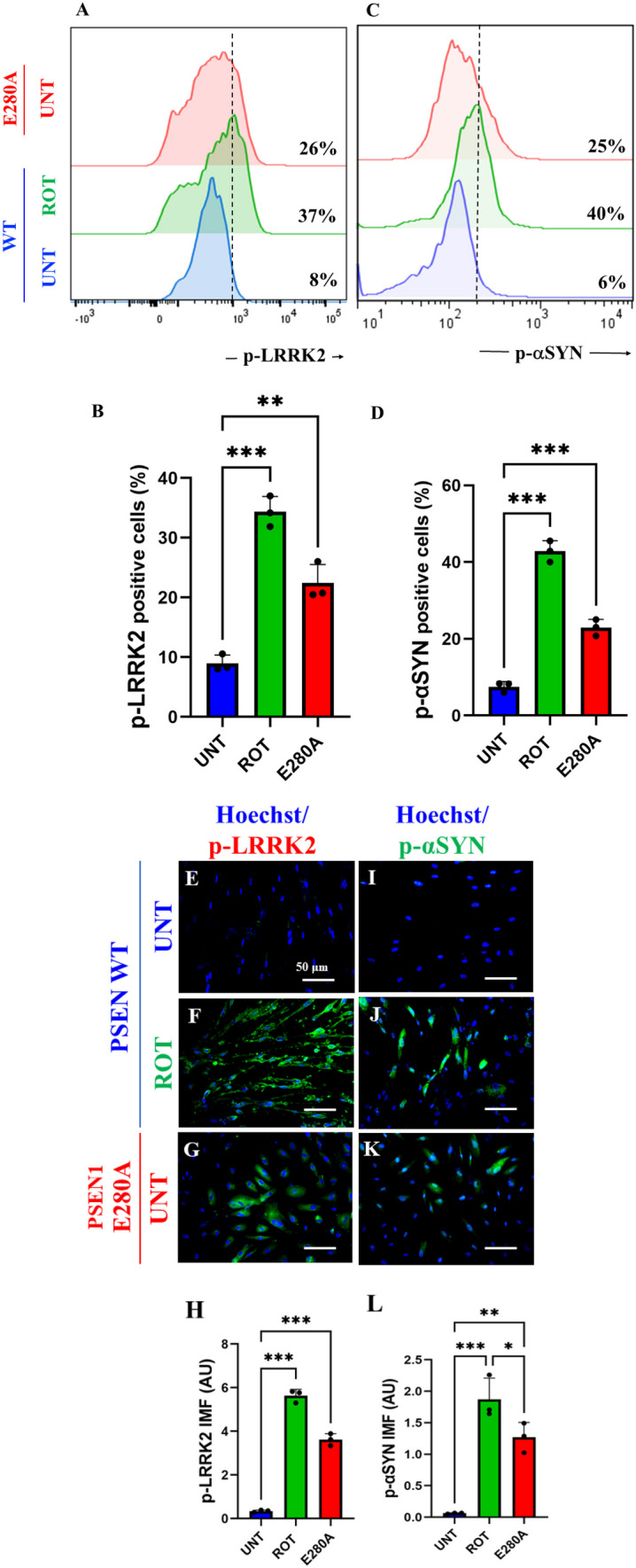
Next, we determined whether ROT induces p-LRRK2 concomitantly with p-\(\mathrm{ \alpha }\)-Syn in ChLNs. To achieve this, ChLNs were exposed to ROT (10 \(\upmu\)M) for 24 h and then p-LRRK2 at residue Ser935 and p-\(\mathrm{ \alpha }\)-Syn at residue Ser129 were evaluated. Flow cytometry analysis reveals that ROT increased the phosphorylation of both LRRK2 by + 363% (Fig. 3A, B) and \(\mathrm{\alpha }\)-Syn by + 567% (Fig. 3C, D) compared to untreated ChLNs. Surprisingly, mutant ChLNs showed an important increased in p-LRRK2 (+ 225%), and p-\(\mathrm{ \alpha }\)-Syn (+ 317%) compared to untreated ChLNs (Fig. 3A–D). Similar data were obtained by fluorescent microscopy analysis (Fig. 3E–L).
Rotenone (ROT) Induces Accumulation of iA\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\upbeta$$\end{document}β, Oxidized DJ-1 (DJ-1Cys106-SO3) and Phosphorylation of TAU at Pathological Residue Ser202/Thr205 in ChLNs
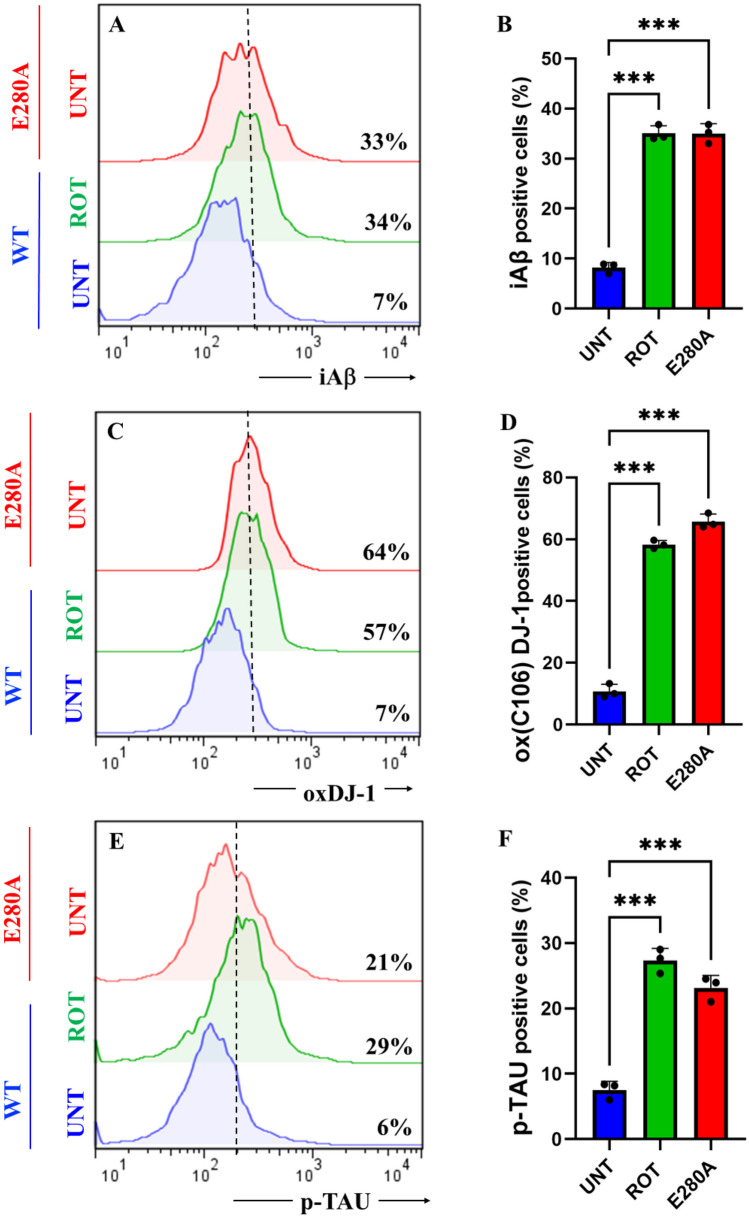
To characterize the neuronal expression of A\(\upbeta\) peptides in ChLNs under ROT exposure, we chose an antibody that has previously been shown to be specific against A\(\upbeta\)42, namely the A\(\upbeta\)4 1E8 antibody (Wiltfang et al. ref. 2001; Maler et al. ref. 2007). Thus, ChLNs were exposed to ROT (10 \(\upmu\)M) for 24 h and stained with the anti-amyloid A\(\upbeta\)4 1E8. Figure 4 shows that ROT increased the accumulation of iA\(\upbeta\) by + 386% (Fig. 4A, B), oxidized DJ-1 by + 714% (Fig. 4C, D), and p-TAU at residue Ser202/Thr205 by + 383% (Fig. 4E, F) compared to untreated ChLNs. As expected, mutant ChLNs increased accumulated iA\(\upbeta\) by + 371%, DJ-1Cys106-SO3 by + 814%, and p-TAU by + 250% (Fig. 4A–F). Similar data was obtained by fluorescent microscopy (Fig. 4G–R).
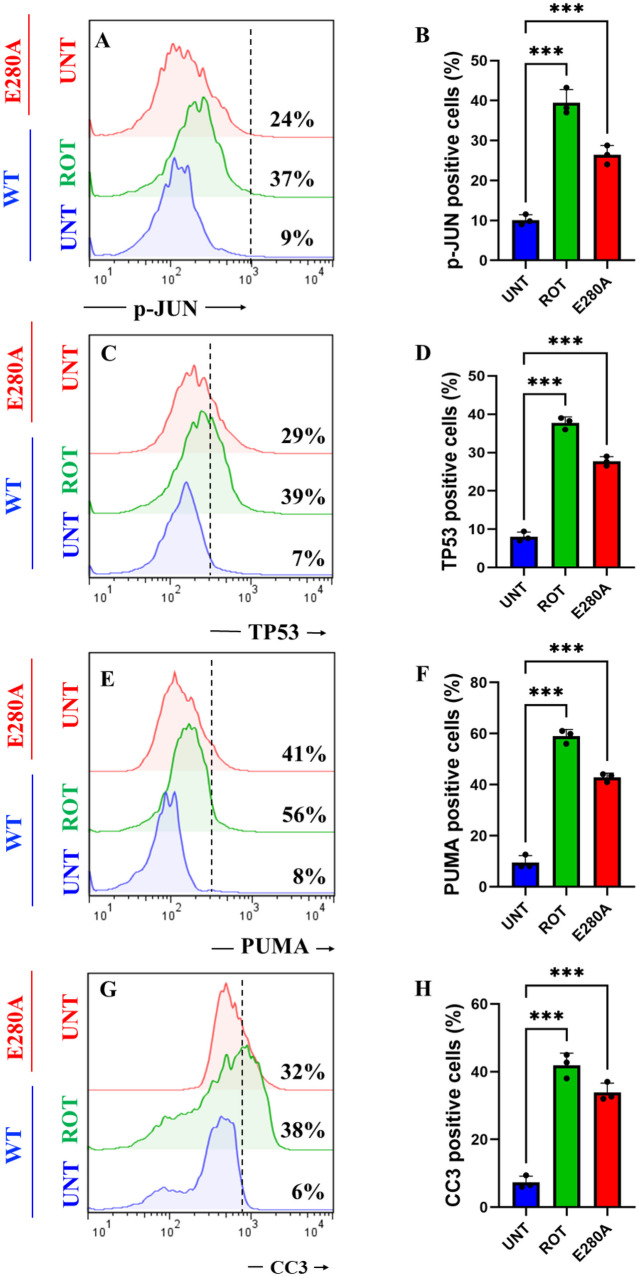
Rotenone (ROT) Increases the Phosphorylation of c-JUN at Residues Ser63/Ser73 and the Expression of TP53, PUMA and CC3 in ChLNs
We determined whether ROT induces apoptotic markers p-JUN, TP53, PUMA, and CC3 in ChLNs (Mendivil-Perez et al. ref. 2016). As shown in Fig. 6, ROT induced an important increase in the phosphorylation of p-JUN, expression TP53, PUMA, and CC3 by + 311% (Fig. 5A, B), + 457% (Fig. 5C, D), + 600% (Fig. 5E, F), and + 533% (Fig. 5G, H), respectively, in ChLNs. Compared to untreated WT, PSEN1 E280A also endogenously expressed p-JUN, TP53, PUMA, and CC3 by + 167%, + 314%, + 412%, and + 433%, respectively (Fig. 5A–H). Similar data were obtained by fluorescent microscopy (Fig. 5I–X).
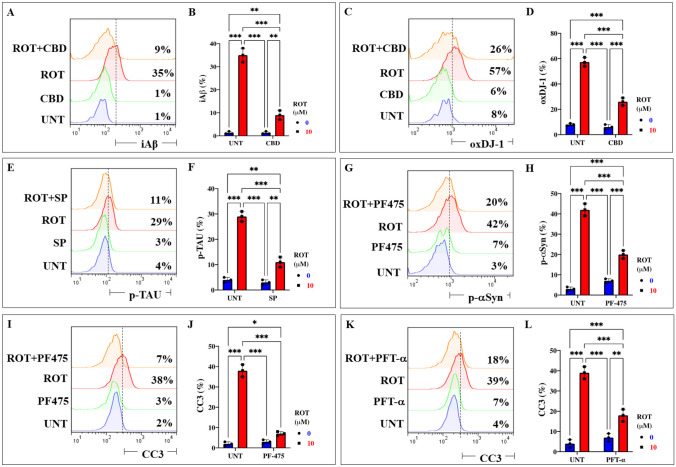
Cannabidiol (CBD), SP600125 (SP), Pifithrin-\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\mathrm{\alpha }$$\end{document}α (PFT) and PF-06447475 (PF475) Block ROT-Induced Accumulation of iA\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\upbeta$$\end{document}β, Oxidized DJ-1, p-TAU Ser202/Thr205, p-\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\mathrm{ \alpha }$$\end{document}α-Syn129, and CC3, Respectively, in ChLNs
The above observations compelled us to evaluate whether the use of antioxidant or inhibitor compounds of critical molecules (e.g., iA\(\upbeta\), DJ-1, TAU, \(\mathrm{\alpha }\)-Syn, CC3) diminish or augment ROT-induced signaling. To achieve this, ChLNs exposed to ROT alone or in combination with anti-amyloidogenic and antioxidant cannabidiol (CBD, 10 \(\upmu\)M), JNK inhibitor SP600125 (SP, 1 \(\upmu\)M), TP53 inhibitor pifithrin-\(\mathrm{ \alpha }\) (PFT-\(\mathrm{ \alpha }\), 50 nM), and LRRK2 inhibitor PF-06447475 (PF475, 1 \(\upmu\)M) were evaluated for their effect on the accumulated iA\(\upbeta\), oxDJ-1, p-\(\mathrm{ \alpha }\)-Syn, p-TAU, and CC3, respectively. Figure 6 shows that CBD reduced the accumulation of iA\(\upbeta\) by -74% (Fig. 6A, B) and DJ-1Cys106-SO3 by -54% (Fig. 6C, D); SP diminished p-TAU at Ser202/Thr205 by -62% (Fig. 6E, F), PF475 abridged p-\(\mathrm{ \alpha }\)-Syn at Ser129 by -52% (Fig. 6G, H), and blunted CC3 by -82% (Fig. 6I, J). Last, PFT-\(\mathrm{ \alpha }\) blocked CC3 by -54% (Fig. 6K, L). In addition, we assessed theoretically whether ROT binds to PSEN 1/ \(\upgamma\) -secretase. For comparative purposes, we used well-known pharmacological inhibitors of PSEN 1/ \(\upgamma\) -secretase complex e.g., SCH 697466, MRK560, SCH 900229, and GSI LY-374973 (Lee et al. ref. 2011; Wu et al. ref. 2012; Hyde et al. ref. 2013; Sogorb-Esteve et al. ref. 2018; Serneels et al. ref. 2023). Molecular in silico docking analysis reveals that ROT (PubChem CID 6758) binds to catalytic pocket of PSEN1 (RSCB protein data bank e.g., 5FN2, (Bai et al. ref. 2015)) with a binding affinity of -8.0 (kcal / mol) Vina score (Fig. 7A and Table 1) compare to the high binding affinity displayed by the \(\upgamma\) -secretase inhibitor e.g., SCH 697466 (-9.2 Vina Score, Fig. 7B and Table 1; (Hyde et al. ref. 2013). Interestingly, other binding affinities for PSEN 1 inhibitors e.g., MRK560 (Lee et al. ref. 2011), SCH 900229 (Wu et al. ref. 2012), and GSI LY-374973 (Sogorb-Esteve et al. ref. 2018) were near to the binding Vina Score of ROT (Table 1).
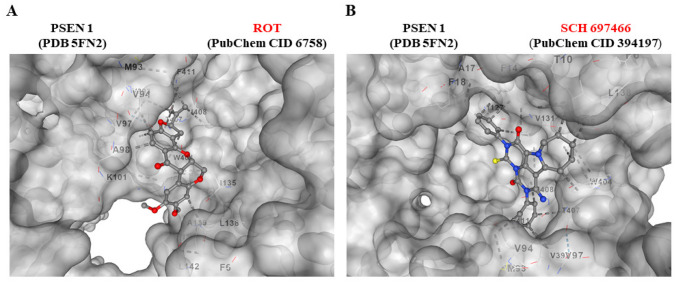
Table 1: In silico molecular docking analysis of ROT, inhibitor agents, and PSEN 1/ \(\upgamma\) secretase
| Submitted Proteina | SubmittedLigandb | Vina Scorec | Cavity Volume(A3) | Center(x, y, z) | Dockingsize(x, y, z) | Contactresidue |
|---|---|---|---|---|---|---|
| PSEN 1/\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\upgamma$$\end{document}γ secretase(5FN2) |
ROT(PubChemCID 6758) | -8.0 | 4571 | 128, 120, 115 | 35, 35, 35 | Pocket C1Chain B: MET93 VAL94 VAL97 ALA98 LYS101 VAL393 TRP404 THR407 ILE408 PHE411Chain C: PHE6 ILE135 LEU138 ALA139 LEU142 |
| PSEN 1/\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\upgamma$$\end{document}γ secretase(5FN2) |
SCH 697466(PubChemCID 394197) | -9.2 | 4571 | 128, 120, 115 | 35, 35, 35 | Pocket C1Chain B: MET93 VAL94 VAL97 VAL393 TRP404 THR407 ILE408 PHE411Chain C: PHE6 THR10 PHE14 ALA17 PHE18 ILE127 VAL131 ILE135 LEU138 |
| PSEN 1/\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\upgamma$$\end{document}γ secretase(5FN2) |
MRK560(PubChemCID 11577204) | -8.4 | 4571 | 128, 120, 115 | 35, 35, 35 | Pocket C1Chain B: THR90 MET93 VAL94 VAL97 VAL393 TRP404 THR407 ILE408 PHE411 VAL412 LEU415Chain C: PHE14 ALA17 PHE18 PHE21 LEU35 VAL36 ILE127 ILE128 VAL131 |
| PSEN 1/\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\upgamma$$\end{document}γ secretase(5FN2) |
SCH 900229(PubChemCID 25164607) | -8.3 | 4571 | 128, 120, 115 | 35, 35, 35 | Pocket C1Chain B: VAL97 VAL393 TRP404 THR407 ILE408 PHE411 VAL412 LEU415Chain C: PHE14 ALA17 PHE18 LEU35 VAL36 ALA39 ILE127 ILE128 VAL131 ILE135 |
| PSEN 1/\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\upgamma$$\end{document}γ secretase(5FN2) |
LY-374973(PubChemCID 5311272) | -8.3 | 4571 | 128, 120, 115 | 35, 35, 35 | Pocket C1Chain B: ILE143 MET146 THR147 LEU150 LEU166 SER169 SER170 LEU173 ILE229 MET233 ILE253 THR256 ASP257 ALA260 LYS265 LEU282 PHE283 LEU383 GLY384 ASP385 ILE387 PHE388 |
| \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\mathrm{\alpha }$$\end{document}α 7nAChR(7KOQ) |
ROT(PubChemCID 6758) | -7.6 | 6911 | 118, 140, 113 | 32, 34, 35 | Pocket C2Chain A: LYS45 ALA257 GLU258 MET260 ALA262Chain B: ASP41 VAL42 GLU44 GLU172 TRP173 ARG205 TYR209 TYR210 LEU214 LEU255 VAL256 GLU258 ILE259 |
| \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\mathrm{\alpha }$$\end{document}α 7nAChR(7KOQ) |
Methyllycaconitine(PubChemCID 5288811) | -7.2 | 6911 | 118, 140, 113 | 32, 34, 35 | Pocket C2Chain A: LYS45 ASN46 ALA95Chain B: MET40 ASP41 VAL42 ASP43 GLU44 LYS45 ASN46 VAL48 THR50 ILE122 LYS124 GLU258 ILE259 |
aAccording to RCSB Protein Data Base (https://www.rcsb.org/)
bAccording to PubChem database (https://pubchem.ncbi.nlm.nih.gov/)
cAccording to CB-dock2: An accurate protein-ligand bind cocking tool (https://cadd.labshare.cn/cb-dock2/php/index.php)
Rotenone (ROT) Impairs ACh-induced Transient Ca2+ Influx in ChLNs
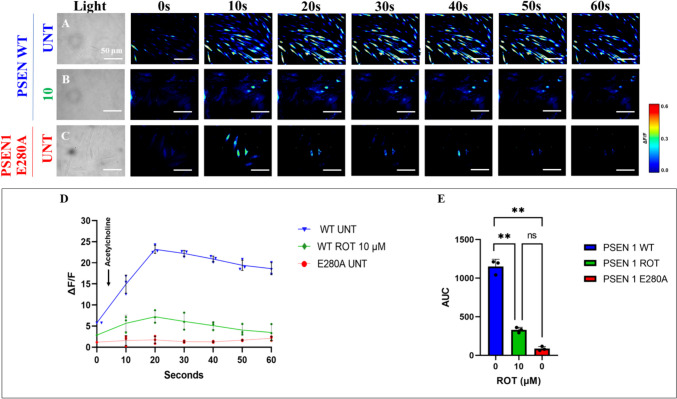
We further evaluated whether ROT alters ChLNs response to ACh stimuli as an assessment of cholinergic neuronal Ca2+ responsiveness and functionality (Deutch and Roth ref. 2014). To achieve this, ChLNs were left untreated or treated with ROT. Figure 8 shows that ACh stimulated a transient increase in intracellular Ca2+ in untreated ChLNs (Fig. 8A, ΔF/F = 23.5 ± 0.3, mean duration = 20 s; n = 20 ChLNs imaged, N = 3 dishes, Fig. 8D, E). In the presence of ROT, the Ca2+ influx was greatly reduced after ACh addition into ChLNs (Fig. 8B, D, ΔF/F = 6.00 ± 0.8, mean duration = 20 s; n = 20 ChLNs imaged, N = 3 dishes Fig. 8E). As expected, ACh did not affect intracellular dysfunctional Ca2+ influx in PSEN1 E280A ChLNs, used as control (Fig. 8C, ΔF/F = 0.50 ± 0.02, mean duration = 20 s; n = 20 ChLNs imaged, N = 3 dishes, Fig. 8D, E). We then theoretically inquired whether ROT might bind to \(\mathrm{\alpha }\) 7 nicotinic cholinergic receptors (\(\mathrm{\alpha }\)7nChR), a cation permeable ligand-gated ion channels with a high permeability to Ca2+ (Noviello et al. ref. 2021), which is implicated in AD (Singh et al. ref. 2024). Molecular in silico docking analysis reveals that ROT binds to \(\mathrm{\alpha }\) 7nChR with high affinity (-7.6 Vina score, Table 1) compared to e.g., methyllycaconitine (-7.2 Vina score, Table 1), a selective and potent antagonist of the α7nAChR at the \(\mathrm{\alpha }\)-bungarotoxin binding site (Ward et al. ref. 1990).
Discussion
In this study, we provide evidence that ROT induced the expression of the typical neuropathological hallmarks \(\mathrm{\alpha }\)-Syn, TAU, and iA\(\upbeta\) in vitro cell model ChLNs. Interestingly, those pathological markers were found in PDD humans’ brains (Smith et al. ref. 2019). Therefore, PDD is a neurological condition that recapitulates PD and AD (Goetz et al. ref. 2009; Irwin et al. ref. 2013; Jellinger ref. 2023). Although previous studies have reported that ROT induces protein aggregation containing A\(\upbeta\), p-α-Syn and hyperphosphorylated TAU in cultured cells of hippocampus, substantia nigra and locus coeruleus (Chaves et al. ref. 2010, ref. 2016), no further attempts were made to explain the mechanism by which ROT simultaneously triggers the coexistence of iA\(\upbeta\), TAU, and \(\mathrm{\alpha }\)-Syn. Here, we show for the first time that ROT-induced coexistence of iA\(\upbeta\), p-TAUSer202/Thr205, p-\(\mathrm{ \alpha }\)-SynSer129, and apoptosis signaling in ChLNs triggered by a cascade of molecular signaling, involving the oxidation of DJ-1 (DJ-Cys106-SO3), phosphorylation of LRRK2Ser935, activation of transcription factors c-JUN and TP53, expression of BH-3-only protein PUMA, loss of \(\Delta \mathrm{\Psi m}\), and activation of CASP3. Furthermore, we found that LRRK2 kinase, c-Jun N-terminal kinase (JNK) signaling, ROT itself, and TP53/c-JUN/ PUMA are implicated in the phosphorylation of \(\mathrm{\alpha }\)-Syn, p-TAU, impairment of PSEN1/ (\(\upgamma\)-secretase complex), and apoptosis, respectively, in ChLNs. These findings might be of importance for the understanding of the mechanism(s) of neuronal cell death not only in PDD, but also in PD (Dong-Chen et al. ref. 2023) and AD (Golde ref. 2009).
Mounting evidence have demonstrated that ROT generates ROS/ H2O2 (Li et al. ref. 2003) through inhibition of mitochondrial complex I (Read et al. ref. 2021). In turn, H2O2 operates via oxidation of susceptible Cys-SH residue on proteins, thereby, mediating intracellular redox-sensitive signal transduction (Marinho et al. ref. 2014; Di Marzo et al. ref. 2018). In fact, H2O2 -induced apoptotic cell death through direct or indirect activation of several pro-death signaling molecules (Velez-Pardo and Jimenez-Del-Rio ref. 2020), including kinases, transcription factors, and enzymes, among others. Here, we confirm that ROT induced concentration-dependent loss of \(\Delta \mathrm{\Psi m}\), and generation of ROS/ H2O2 according to low-MitoTracker®-positive and DFC-positive ChLNs, respectively. Indeed, H2O2 not only induces depolarization of mitochondrial membrane (Takeyama et al. ref. 2002) but also oxidizes the stress sensor protein DJ-1-Cys106-SH (sulfhydryl group) into the DJ-1-Cys106-SO3 (sulfonic acid). Actually, the Cys106-SH is the most sensitive cysteine residue in DJ-1 protein to H2O2-mediated oxidation (Kinumi et al. ref. 2004). We found that ROT significantly increase the oxidized DJ-1 in ChLNs. Given that DJ-1 plays an important role in PD (Repici and Giorgini ref. 2019), oxidized DJ-1 has been postulate as a possible biomarker of PD (Saito ref. 2017). However, whether oxidized DJ-1 might be an additional pathologic marker associated with PDD merits further investigation. Nonetheless, ROS/ H2O2 disable DJ-1 from its capacity to modulate signaling pathways related with neuroprotective actions (Neves et al. ref. 2022). Indeed, the most accepted function for DJ-1 is a neuronal protective role against OS (Biosa et al. ref. 2017). Interestingly, the antioxidant CBD (Hacke et al. ref. 2019) significantly reduced the oxDJ-1 in ChLNs exposed to ROT. Moreover, CBD has been demonstrated to inhibit the activation of CASP3 (Mendivil-Perez et al. ref. 2023). Taken together, these observations suggest that rising of ROS/ H2O2 play a critical role in the early stages of the pathophysiology of the PDD. Therefore, antioxidant therapy should not only be beneficial for PDD but also for PD (Andrade et al. ref. 2023).
How does ROT link \(\mathrm{\alpha }\)-Syn, iA\(\upbeta\), and Tau in ChLNs as model of PDD? Our findings suggest that ROT triggers three alternative and complementary mechanisms, involving the putative interaction between ROT and PSEN1/ \(\upgamma\) -secretase, ROT-induced activation of JNK, and phosphorylation of LRRK2 kinase, which eventually converge on p-\(\mathrm{ \alpha }\)-Syn, iA\(\upbeta\), p-TAU, and apoptosis. Several observations support this. First, H2O2 might activate LRRK2 kinase activity by directly enhancing its autophosphorylation, e.g., at Tyr1967 (Kamikawaji et al. ref. 2009), Ser2032, and Tyr2035 (West et al. ref. 2007; Li et al. ref. 2010), or indirectly, via phosphorylation of Ser910 and Ser935 via the inhibitor of nuclear factor-κB (IκB) kinase (IKK) complex (Dzamko et al. ref. 2012). In line with this, we found a significant increase p-Ser935 LRRK2 concomitant with an important increase of p-Ser129 α-Syn in ChLNs exposed to ROT. These results suggest that, once active, the LRRK2 kinase phosphorylates α-Syn at Ser129 (Qing et al. ref. 2009), which is the major component of pathological deposits in PD (Fujiwara et al. ref. 2002; Du et al. ref. 2021). Of note, the inhibitor LRRK2 kinase PF-06447475 almost completely abolished the p-Ser129-\(\mathrm{ \alpha }\)-Syn. Taken together, these results suggest that p-Ser935 LRRK2 is implicated in the phosphorylation of \(\mathrm{\alpha }\)-Syn at residue Ser129 in ChLNs treated with ROT (Qing et al. ref. 2009). Second, H2O2 indirectly activates JNK kinase through activation of ASK-1 (Nadeau et al. ref. 2009). In turn, JNK kinase phosphorylates TAU protein at Ser202/Thr205 (Reynolds et al. ref. 2000), two amino acid residues implicated in TAU protein aggregation (Neddens et al. ref. 2018). Interestingly, phosphorylation of TAU at residue Ser208, identified with antibody AT8 used in this work, also promotes aggregation and reveals neuropathologic diversity in Alzheimer’s disease and other tauopathies (Xia et al. ref. 2020). Indeed, the combined phosphorylation at the Ser202/Thr205/Ser208 sites produces a Tau sample that readily forms fibers (Despres et al. ref. 2017). Inhibition of the JNK signaling pathway might avoid TAU hyperphosphorylation and aggregation. Accordingly, we found that JNK inhibitor SP600125 significantly reduced p-TAU in ChLNs treated with ROT. Therefore, JNK is a potential therapeutic target for PDD, PD and AD (Hepp Rehfeldt et al. ref. 2020; Usmani et al. ref. 2021; Zhu et al. ref. 2022). Nonetheless, LRRK2 may also contribute to TAU hyperphosphorylation and aggregation (Shanley et al. ref. 2015; Hamm et al. ref. 2015). Third, ROT induces high levels of accumulated iA\(\upbeta\) in ChLNs. However, the molecular mechanism by which ROT induces iA\(\upbeta\) is not yet fully established. A possible explanation is that ROT binds to PSEN 1/ \(\upgamma\) -complex. Indeed, a molecular in silico docking analysis shows that ROT binds to a putative binding pocket in the PSEN1/ \(\upgamma\) secretase with nearly similar binding Vina scores as those found with typical reference synthetic inhibitor PSEN1/ \(\upgamma\) secretase MRK560, SCH 900229, and LY-374973 (Table 1). Although not yet experimentally confirmed, it is predicted that ROT might be able to affect dynamic conformational changes that control progressive catalysis by PSEN 1/ γ-secretase towards intracellular overproduction and accumulation of A\(\upbeta\) (Svedružić et al. ref. 2023). Indeed, the enzymatic activity of PSEN1/ \(\upgamma\) -secretase is highly sensitive to structural changes induced by either PSEN1 mutations (Do et al. ref. 2023) or \(\upgamma\) secretase modulators GSM (Xia ref. 2019). Interestingly, ROT displays mitochondrial off-target (e.g., binds to tubulin, Srivastava and Panda ref. 2007) contributing to its toxic effects (Ren et al. ref. 2005). Additionally, we found that CBD diminished accumulation of iA\(\upbeta\) in ChLNs exposed to ROT. These observations suggest that CBD, in addition to operate as antioxidant, it also works as anti-amyloidogenic agent. Given that CBD possesses several pharmacological effects (Castillo-Arellano et al. ref. 2023), this cannabinoid might be a potential therapeutic agent for the treatment of PDD (Ferreira-Junior et al. ref. 2020; Zhang et al. ref. 2022b; Mendivil-Perez et al. ref. 2023). Finally, we confirm that ROT-induced apoptosis involving mitochondria depolarization and activation of CASP3 (Li et al. ref. 2003) reflected as CC3 in ChLNs. Indeed, ROT increased the expression of TP53 and PUMA. Interestingly, both transcription factors TP53 and p–c-JUN transcribed PUMA (Nakano and Vousden ref. 2001; Yu et al. ref. 2001; Lu et al. ref. 2014), a BH-3-only proapoptotic protein directly involved in mitochondrial depolarization and apoptosis (Roufayel et al. ref. 2022; Czabotar and Garcia-Saez ref. 2023), thereby contributing to the release of apoptogenic cytochrome C and activation of CASP3 (Dorstyn et al. ref. 2018). Taken together, these observations imply that mitochondrial up-and downstream signaling are critical to trigger apoptosis in ChLNs. This last assumption is further supported by significant reduction of apoptosis in ChLNs when co-treated with inhibitor TP53 pifithrin-\(\alpha\) or LRRK2 inhibitor PF-06447475 and ROT.
Additionally, ROT almost abolishes the ACh-induced transient intracellular Ca2+ flux in ChLNs. This suggests that ROT, similar to extracellular (e) A\(\upbeta\) (Wang et al. ref. 2000), somehow blocks the binding of ACh to ligand-gated Ca2+ ion channels (Uteshev ref. 2012; Brown ref. 2019). Moreover, docking analysis support the view that ROT can bind to nAChRs, e.g., \(\mathrm{\alpha }\)7nAChR with high affinity (Table 1). These observations might explain why ROT impairs learning and memory in mice (Guo et al. ref. 2022). However, further investigation is needed to clarify these issues. We conclude that ROT-induced apoptosis, co-existence of p-\(\mathrm{ \alpha }\)-Syn, iA\(\upbeta\), and p-TAU and neuronal Ca2+ influx dysfunction in ChLNs through OS-dependent and -independent mechanisms (Fig. 9).
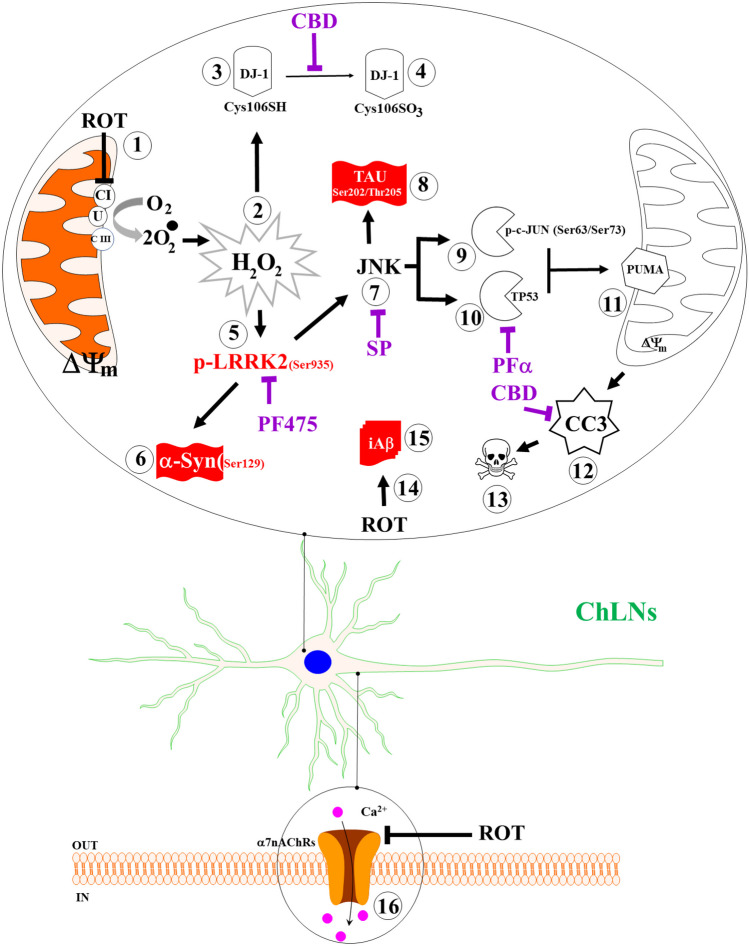
For comparative purposes, we used PSEN 1 E280A (Soto-Mercado et al. ref. 2020). Effectively, mutant ChLNs reproduced the pathological markers of Alzheimer’s disease, involving iA\(\upbeta\), p-TAU, and oxDJ-1. Additionally, mutant ChLNs showed loss of \(\Delta \mathrm{\Psi m}\), up regulation of p–c-JUN, TP53, PUMA, and CC3, leading to apoptosis and eA\(\upbeta\)-induced Ca2+ influx impairments (Soto-Mercado et al. ref. 2020). Therefore, ROT and iA\(\upbeta\) seem to be mechanistically homologous. Similar to ROT (this work), iA\(\upbeta\) disrupts mitochondria bioenergetics (Sinclair et al. ref. 2021), thereby generating ROS/H2O2, activates ASK-1 (Kadowaki et al. ref. 2005), JNK/c-JUN axis, and p-TAU (Solas et al. ref. 2023), and induces apoptosis dependent on CASP3 (Eimer and Vassar ref. 2013). However, in contrast to ROT, A\(\upbeta\) selectively inhibit Complex IV (Canevari et al. ref. 1999). Surprisingly, we found for the first time an important endogenously increase in p-Ser935 LRRK 2 and p-Ser129 \(\mathrm{\alpha }\)-Syn in PSEN 1 E280A ChLNs compared to wild type ChLNs. Given that LRRK2 is an indispensable pro-apoptotic kinase (Quintero-Espinosa et al. ref. 2017, ref. 2023; Perez-Abshana et al. ref. 2023), these results indicate that LRRK2 may play a major role in both DAergic and ChLNs demise. Furthermore, from a neuropathologic perspective, our findings might explain why some brains from demented patients show a mixed pathology of dementia with Lewy bodies (DLB) and AD (Kantarci et al. ref. 2020). We report for the first time that PSEN 1 E280A ChLNs display the typical p-Ser129 \(\mathrm{\alpha }\)-Syn, a pathological feature found in PD, DLB, and PDD. However, whether p-Ser129 \(\mathrm{\alpha }\)-Syn and p-Ser935 LRRK2 are positive markers in human brains from FAD PSEN 1 E280A patients need further investigation.
Here, we show that ChLNs derived from WJ-MSCs treated with ROT might be an excellent system to model PDD. Our findings have one major implication for PD and PDD. It is known that the most significant nonmotor symptom in PD is progressive cognitive impairment (Aarsland et al. ref. 2021), and PDD may affect 80% of PD patients long-term (Russell et al. ref. 2014). Unfortunately, this recognition has not translated into significant treatment advances. One possible explanation for this drawback is the lack of proper in vitro or in vivo models. Here, we provide an in vitro model that accounts for the molecular mechanism by which ROT-induced cell death occurs in ChLNs, the major neuronal group involved in early AD and late PD. Furthermore, we found that LRRK2 is a key master kinase that links both PD through p-Ser129 \(\mathrm{\alpha }\)-Syn and FAD through apoptotic cell death signaling. This adds another layer of molecular complexity to the understanding of cell demise in both PD (Michel et al. ref. 2016; Quintero-Espinosa et al. ref. 2017, ref. 2023; Velez-Pardo and Jimenez-Del-Rio ref. 2020; Perez-Abshana et al. ref. 2023) and FAD (Soto-Mercado et al. ref. 2020; Brokaw et al. ref. 2020). However, provided that LRRK2 is a druggable target (Thakur et al. ref. 2022), it should be implemented in clinical trials including not only PDD but also PD as well as FAD patients. Our model system offers an understanding of the mechanisms of neurodegeneration in PD and AD. Nonetheless, our model has limitations. Although the data suggested that ROT impairs PSEN1 /\(\upgamma\) -secretase towards increased accumulation of A\(\upbeta\), there is no reasonable explanation for the endogenous production of iA\(\upbeta\) in PDD brains. It is worth mentioning that a reformulation of the amyloid cascade hypothesis (ACH), now known as ACH 2.0, provides enlightenment on this issue (Volloch and Rits-Volloch ref. 2023). Accordingly, it is proposed that AD is triggered by a first benign stage, wherein an APP-derived iA\(\upbeta\) accumulated to sufficient levels in both sporadic (SAD) and FAD, and is driven by a second deleterious stage, wherein iA\(\upbeta\) generated dependently of APP in FAD and independently of APP in SAD induced mitochondrial dysfunction and apoptosis (Volloch and Rits-Volloch ref. 2023). Therefore, the role of iA\(\upbeta\) and \(\mathrm{\alpha }\)-Syn pathology in the development of cognitive deficits in PD suggests that emerging disease-modifying therapies for AD may be beneficial for PDD patients.
Conclusion
We provide an in vitro model recreating the neuropathology of PDD induced by ROT in ChLNs. Indeed, ROT induces p-\(\mathrm{ \alpha }\)-Syn, A\(\upbeta\), p-Tau, and cell death in ChLNs. Furthermore, we identify the LRRK2 as master kinase that link both PD and AD via apoptotic cell death signaling. The “ChLNs plus ROT” approach provides an excellent platform to test for potential therapeutic strategies against PDD. Our data suggest that ROT induces a neuropathologic phenotype in ChLNs similar to that caused by the mutation PSEN1 E280A.
Supplementary Materials
References
- D Aarsland, L Batzu, GM Halliday. Parkinson disease-associated cognitive impairment. Nat Rev Dis Primers, 2021. [DOI | PubMed]
- M Abrishamdar, MS Jalali, Y Farbood. Targeting Mitochondria as a Therapeutic Approach for Parkinson’s Disease. Cell Mol Neurobiol, 2023. [DOI | PubMed]
- A Adan, G Alizada, Y Kiraz. Flow cytometry: basic principles and applications. Crit Rev Biotechnol, 2017. [DOI | PubMed]
- S Andrade, D Nunes, M Dabur. Therapeutic Potential of Natural Compounds in Neurodegenerative Diseases: Insights from Clinical Trials. Pharmaceutics, 2023. [DOI | PubMed]
- F Antunes, PM Brito. Quantitative biology of hydrogen peroxide signaling. Redox Biol, 2017. [DOI | PubMed]
- XC Bai, E Rajendra, G Yang, Y Shi, SH Scheres. Sampling the conformational space of the catalytic subunit of human γ-secretase. Elife, 2015. [DOI | PubMed]
- A Biosa, F Sandrelli, M Beltramini. Recent findings on the physiological function of DJ-1: Beyond Parkinson’s disease. Neurobiol Dis, 2017. [DOI | PubMed]
- BR Bloem, MS Okun, C Klein. Parkinson’s disease. The Lancet, 2021. [DOI]
- DL Brokaw, IS Piras, D Mastroeni. Cell death and survival pathways in Alzheimer’s disease: an integrative hypothesis testing approach utilizing -omic data sets. Neurobiol Aging, 2020. [DOI | PubMed]
- DA Brown. Acetylcholine and cholinergic receptors. Brain Neurosci Adv, 2019. [DOI]
- L Canevari, JB Clark, TE Bates. β-Amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett, 1999. [DOI | PubMed]
- J Castillo-Arellano, A Canseco-Alba, SJ Cutler, F León. The Polypharmacological Effects of Cannabidiol. Molecules, 2023. [DOI | PubMed]
- RS Chaves, TQ Melo, SA Martins, MF Ferrari. Protein aggregation containing beta-amyloid, alpha-synuclein and hyperphosphorylated tau in cultured cells of hippocampus, substantia nigra and locus coeruleus after rotenone exposure. BMC Neurosci, 2010. [DOI | PubMed]
- RS Chaves, AI Kazi, CM Silva. Presence of insoluble Tau following rotenone exposure ameliorates basic pathways associated with neurodegeneration. IBRO Rep, 2016. [DOI | PubMed]
- PE Czabotar, AJ Garcia-Saez. Mechanisms of BCL-2 family proteins in mitochondrial apoptosis. Nat Rev Mol Cell Biol, 2023. [DOI | PubMed]
- C Despres, C Byrne, H Qi. Identification of the Tau phosphorylation pattern that drives its aggregation. Proc Natl Acad Sci, 2017. [DOI | PubMed]
- AY Deutch, RH Roth. Pharmacology and Biochemistry of Synaptic Transmission. Introduction to Cellular and Molecular Neuroscience, 2014. [DOI]
- N Di Marzo, E Chisci, R Giovannoni. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells, 2018. [DOI | PubMed]
- DW Dickson. Parkinson’s Disease and Parkinsonism: Neuropathology. Cold Spring Harb Perspect Med, 2012. [DOI | PubMed]
- HN Do, S Devkota, A Bhattarai. Effects of presenilin-1 familial Alzheimer’s disease mutations on γ-secretase activation for cleavage of amyloid precursor protein. Commun Biol, 2023. [DOI | PubMed]
- M Dominici, K Le Blanc, I Mueller. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy, 2006. [DOI | PubMed]
- X Dong-Chen, C Yong, X Yang. Signaling pathways in Parkinson’s disease: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther, 2023. [DOI | PubMed]
- L Dorstyn, CW Akey, S Kumar. New insights into apoptosome structure and function. Cell Death Differ, 2018. [DOI | PubMed]
- T Du, L Wang, W Liu. Biomarkers and the Role of α-Synuclein in Parkinson’s Disease. Front Aging Neurosci, 2021. [DOI | PubMed]
- N Dzamko, F Inesta-Vaquera, J Zhang. The IkappaB Kinase Family Phosphorylates the Parkinson’s Disease Kinase LRRK2 at Ser935 and Ser910 during Toll-Like Receptor Signaling. PLoS ONE, 2012. [DOI | PubMed]
- WA Eimer, R Vassar. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ42 accumulation and Caspase-3 activation. Mol Neurodegener, 2013. [DOI | PubMed]
- N Fang, JE Casida. Cubé Resin Insecticide: Identification and Biological Activity of 29 Rotenoid Constituents. J Agric Food Chem, 1999. [DOI | PubMed]
- NC Ferreira-Junior, AC Campos, FS Guimarães. Biological bases for a possible effect of cannabidiol in Parkinson’s disease. Braz J Psychiatry, 2020. [DOI | PubMed]
- H Fujiwara, M Hasegawa, N Dohmae. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol, 2002. [DOI | PubMed]
- N Giguère, S Burke Nanni, L-E Trudeau. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front Neurol, 2018. [DOI | PubMed]
- CG Goetz, M Emre, B Dubois. Parkinson’s disease dementia: Definitions, guidelines, and research perspectives in diagnosis. Ann Neurol, 2009. [DOI]
- TE Golde. The therapeutic importance of understanding mechanisms of neuronal cell death in neurodegenerative disease. Mol Neurodegener, 2009. [DOI | PubMed]
- Z Guo, Z Ruan, D Zhang. Rotenone impairs learning and memory in mice through microglia-mediated blood brain barrier disruption and neuronal apoptosis. Chemosphere, 2022. [DOI | PubMed]
- ACM Hacke, D Lima, F De Costa. Probing the antioxidant activity of Δ9-tetrahydrocannabinol and cannabidiol in Cannabis sativa extracts. Analyst, 2019. [DOI | PubMed]
- M Hamm, R Bailey, G Shaw. Physiologically relevant factors influence tau phosphorylation by leucine-rich repeat kinase 2. J Neurosci Res, 2015. [DOI | PubMed]
- J Han, Y Fan, P Wu. Parkinson’s Disease Dementia: Synergistic Effects of Alpha-Synuclein, Tau, Beta-Amyloid, and Iron. Front Aging Neurosci, 2021. [DOI | PubMed]
- SC Hepp Rehfeldt, F Majolo, MI Goettert, S Laufer. c-Jun N-Terminal Kinase Inhibitors as Potential Leads for New Therapeutics for Alzheimer’s Diseases. Int J Mol Sci, 2020. [DOI | PubMed]
- GU Höglinger, A Lannuzel, ME Khondiker. The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J Neurochem, 2005. [DOI | PubMed]
- LA Hyde, Q Zhang, RA Del Vecchio. In Vivo Characterization of a Novel -Secretase Inhibitor SCH 697466 in Rodents and Investigation of Strategies for Managing Notch-Related Side Effects. Int J Alzheimers Dis, 2013. [DOI]
- DJ Irwin, VM-Y Lee, JQ Trojanowski. Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci, 2013. [DOI | PubMed]
- J Jankovic. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry, 2008. [DOI | PubMed]
- KA Jellinger. Morphological characteristics differentiate dementia with Lewy bodies from Parkinson disease with and without dementia. J Neural Transm, 2023. [DOI | PubMed]
- H Kadowaki, H Nishitoh, F Urano. Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ, 2005. [DOI | PubMed]
- S Kamikawaji, G Ito, T Iwatsubo. Identification of the Autophosphorylation Sites of LRRK2. Biochemistry, 2009. [DOI | PubMed]
- K Kantarci, VJ Lowe, Q Chen. β-Amyloid PET and neuropathology in dementia with Lewy bodies. Neurology, 2020. [DOI | PubMed]
- Ke M, Chong C-M, Zhu Q et al (2021) Comprehensive Perspectives on Experimental Models for Parkinson’s Disease. Aging Dis 12:223. 10.14336/AD.2020.0331
- JR Kim. Oligomerization by co-assembly of β-amyloid and α-synuclein. Front Mol Biosci, 2023. [DOI | PubMed]
- T Kinumi, J Kimata, T Taira. Cysteine-106 of DJ-1 is the most sensitive cysteine residue to hydrogen peroxide-mediated oxidation in vivo in human umbilical vein endothelial cells. Biochem Biophys Res Commun, 2004. [DOI | PubMed]
- DS Knopman, H Amieva, RC Petersen. Alzheimer Disease Nat Rev Dis Primers, 2021. [DOI | PubMed]
- A Kouli, M Camacho, K Allinson, CH Williams-Gray. Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol Commun, 2020. [DOI | PubMed]
- V Lawana, JR Cannon. Rotenone neurotoxicity: Relevance to Parkinson’s disease. Adv Neurotoxicol, 2020. [DOI]
- SE Lazic, CJ Clarke-Williams, MR Munafò. What exactly is ‘N’ in cell culture and animal experiments?. PLoS Biol, 2018. [DOI | PubMed]
- J Lee, L Song, G Terracina. Identification of Presenilin 1-Selective γ-Secretase Inhibitors with Reconstituted γ-Secretase Complexes. Biochemistry, 2011. [DOI | PubMed]
- N Li, K Ragheb, G Lawler. Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis through Enhancing Mitochondrial Reactive Oxygen Species Production. J Biol Chem, 2003. [DOI | PubMed]
- X Li, DJ Moore, Y Xiong. Reevaluation of Phosphorylation Sites in the Parkinson Disease-associated Leucine-rich Repeat Kinase 2. J Biol Chem, 2010. [DOI | PubMed]
- JW Lichtman, J-A Conchello. Fluorescence microscopy. Nat Methods, 2005. [DOI | PubMed]
- AKL Liu, RC-C Chang, RKB Pearce, SM Gentleman. Nucleus basalis of Meynert revisited: anatomy, history and differential involvement in Alzheimer’s and Parkinson’s disease. Acta Neuropathol, 2015. [DOI | PubMed]
- AKL Liu, TW Chau, EJ Lim. Hippocampal CA2 Lewy pathology is associated with cholinergic degeneration in Parkinson’s disease with cognitive decline. Acta Neuropathol Commun, 2019. [DOI | PubMed]
- Y Liu, X Yang, J Gan. CB-Dock2: improved protein–ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res, 2022. [DOI | PubMed]
- H Lu, G Hou, Y Zhang. c-Jun transactivates Puma gene expression to promote osteoarthritis. Mol Med Rep, 2014. [DOI | PubMed]
- R Macdonald, K Barnes, C Hastings, H Mortiboys. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: can mitochondria be targeted therapeutically?. Biochem Soc Trans, 2018. [DOI | PubMed]
- RJ Mailloux. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol, 2015. [DOI | PubMed]
- JM Maler, H-W Klafki, S Paul. Urea-based two-dimensional electrophoresis of beta-amyloid peptides in human plasma: Evidence for novel Aβ species. Proteomics, 2007. [DOI | PubMed]
- HS Marinho, C Real, L Cyrne. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol, 2014. [DOI | PubMed]
- M Mendivil-Perez, C Velez-Pardo, M Jimenez-Del-Rio. Neuroprotective Effect of the LRRK2 Kinase Inhibitor PF-06447475 in Human Nerve-Like Differentiated Cells Exposed to Oxidative Stress Stimuli: Implications for Parkinson’s Disease. Neurochem Res, 2016. [DOI | PubMed]
- M Mendivil-Perez, C Velez-Pardo, M Jimenez-Del-Rio. Direct transdifferentiation of human Wharton’s jelly mesenchymal stromal cells into cholinergic-like neurons. J Neurosci Methods, 2019. [DOI | PubMed]
- M Mendivil-Perez, AA Felizardo-Otalvaro, M Jimenez-Del-Rio, C Velez-Pardo. Cannabidiol Protects Dopaminergic-like Neurons against Paraquat- and Maneb-Induced Cell Death through Safeguarding DJ-1CYS 106 and Caspase 3 Independently of Cannabinoid Receptors: Relevance in Parkinson’s Disease. ACS Chem Neurosci, 2023. [DOI | PubMed]
- PP Michel, EC Hirsch, S Hunot. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron, 2016. [DOI | PubMed]
- PJ Nadeau, SJ Charette, J Landry. REDOX Reaction at ASK1-Cys250 Is Essential for Activation of JNK and Induction of Apoptosis. Mol Biol Cell, 2009. [DOI | PubMed]
- K Nakano, KH Vousden. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol Cell, 2001. [DOI | PubMed]
- J Neddens, M Temmel, S Flunkert. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol Commun, 2018. [DOI | PubMed]
- M Neves, M Grãos, SI Anjo, B Manadas. Modulation of signaling pathways by DJ-1: An updated overview. Redox Biol, 2022. [DOI | PubMed]
- CM Noviello, A Gharpure, N Mukhtasimova. Structure and gating mechanism of the α7 nicotinic acetylcholine receptor. Cell, 2021. [DOI | PubMed]
- P Pap, Á Kőszeghy, G Szűcs, Z Rusznák. Cytoplasmic Ca2+ concentration changes evoked by cholinergic stimulation in primary astrocyte cultures prepared from the rat cochlear nucleus. Hear Res, 2009. [DOI | PubMed]
- G Pepeu, M Grazia Giovannini. The fate of the brain cholinergic neurons in neurodegenerative diseases. Brain Res, 2017. [DOI | PubMed]
- LP Perez-Abshana, M Mendivil-Perez, C Velez-Pardo, M Jimenez-Del-Rio. Rotenone Blocks the Glucocerebrosidase Enzyme and Induces the Accumulation of Lysosomes and Autophagolysosomes Independently of LRRK2 Kinase in HEK-293 Cells. Int J Mol Sci, 2023. [DOI | PubMed]
- O Phillips, D Ghosh, HH Fernandez. Parkinson Disease Dementia Management: an Update of Current Evidence and Future Directions. Curr Treat Options Neurol, 2023. [DOI]
- H Qing, W Wong, EG McGeer, PL McGeer. Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem Biophys Res Commun, 2009. [DOI | PubMed]
- D Quintero-Espinosa, M Jimenez-Del-Rio, C Velez-Pardo. Knockdown transgenic Lrrk Drosophila resists paraquat-induced locomotor impairment and neurodegeneration: A therapeutic strategy for Parkinson’s disease. Brain Res, 2017. [DOI | PubMed]
- DA Quintero-Espinosa, S Sanchez-Hernandez, C Velez-Pardo. LRRK2 Knockout Confers Resistance in HEK-293 Cells to Rotenone-Induced Oxidative Stress, Mitochondrial Damage, and Apoptosis. Int J Mol Sci, 2023. [DOI | PubMed]
- AD Read, RET Bentley, SL Archer, KJ Dunham-Snary. Mitochondrial iron–sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox Biol, 2021. [DOI | PubMed]
- Y Ren, W Liu, H Jiang. Selective Vulnerability of Dopaminergic Neurons to Microtubule Depolymerization. J Biol Chem, 2005. [DOI | PubMed]
- M Repici, F Giorgini. DJ-1 in Parkinson’s Disease: Clinical Insights and Therapeutic Perspectives. J Clin Med, 2019. [DOI | PubMed]
- CH Reynolds, JC Betts, WP Blackstock. Phosphorylation Sites on Tau Identified by Nanoelectrospray Mass Spectrometry. J Neurochem, 2000. [DOI | PubMed]
- R Roufayel, K Younes, A Al-Sabi, N Murshid. BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis. Life, 2022. [DOI | PubMed]
- A Russell, A Drozdova, W Wang, M Thomas. The Impact of Dementia Development Concurrent with Parkinson’s Disease: A New Perspective. CNS Neurol Disord Drug Targets, 2014. [DOI | PubMed]
- A Sae-Yun, C Ovatlarnporn, A Itharat, R Wiwattanapatapee. Extraction of rotenone from Derris elliptica and Derris malaccensis by pressurized liquid extraction compared with maceration. J Chromatogr A, 2006. [DOI | PubMed]
- Y Saito. DJ-1 as a Biomarker of Parkinson’s Disease. Adv Exp Med Biol, 2017. [DOI | PubMed]
- J Schiller, V Zickermann. Binding of Natural Inhibitors to Respiratory Complex I. Pharmaceuticals, 2022. [DOI | PubMed]
- M Sekiguchi-Tonosaki, M Obata, A Haruki. Acetylcholine induces Ca 2+ signaling in chicken retinal pigmented epithelial cells during dedifferentiation. Am J Physiol-Cell Phys, 2009. [DOI]
- L Serneels, R Narlawar, L Perez-Benito. Selective inhibitors of the PSEN1-gamma-secretase complex. J Biol Chem, 2023. [DOI | PubMed]
- MR Shanley, D Hawley, S Leung. LRRK2 Facilitates tau Phosphorylation through Strong Interaction with tau and cdk5. Biochemistry, 2015. [DOI | PubMed]
- P Sinclair, A Baranova, N Kabbani. Mitochondrial Disruption by Amyloid Beta 42 Identified by Proteomics and Pathway Mapping. Cells, 2021. [DOI | PubMed]
- S Singh, N Agrawal, A Goyal. Role of Alpha-7-Nicotinic Acetylcholine Receptor in Alzheimer’s Disease. CNS Neurol Disord Drug Targets, 2024. [DOI | PubMed]
- C Smith, N Malek, K Grosset. Neuropathology of dementia in patients with Parkinson’s disease: a systematic review of autopsy studies. J Neurol Neurosurg Psychiatry, 2019. [DOI | PubMed]
- A Sogorb-Esteve, M-S García-Ayllón, M Llansola. Inhibition of γ-Secretase Leads to an Increase in Presenilin-1. Mol Neurobiol, 2018. [DOI | PubMed]
- M Solas, S Vela, C Smerdou. JNK Activation in Alzheimer’s Disease Is Driven by Amyloid β and Is Associated with Tau Pathology. ACS Chem Neurosci, 2023. [DOI | PubMed]
- V Soto-Mercado, M Mendivil-Perez, C Velez-Pardo. Cholinergic-like neurons carrying PSEN1 E280A mutation from familial Alzheimer’s disease reveal intraneuronal sAPPβ fragments accumulation, hyperphosphorylation of TAU, oxidative stress, apoptosis and Ca2+ dysregulation: Therapeutic implications. PLoS ONE, 2020. [DOI | PubMed]
- P Srivastava, D Panda. Rotenone inhibits mammalian cell proliferation by inhibiting microtubule assembly through tubulin binding. FEBS J, 2007. [DOI | PubMed]
- ŽM Svedružić, V Šendula Jengić, L Ostojić. The Binding of Different Substrate Molecules at the Docking Site and the Active Site of γ-Secretase Can Trigger Toxic Events in Sporadic and Familial Alzheimer’s Disease. Int J Mol Sci, 2023. [DOI | PubMed]
- N Takeyama, S Miki, A Hirakawa, T Tanaka. Role of the Mitochondrial Permeability Transition and Cytochrome c Release in Hydrogen Peroxide-Induced Apoptosis. Exp Cell Res, 2002. [DOI | PubMed]
- G Thakur, V Kumar, KW Lee, C Won. Structural Insights and Development of LRRK2 Inhibitors for Parkinson’s Disease in the Last Decade. Genes (basel), 2022. [DOI | PubMed]
- C Ullrich, C Humpel. Rotenone Induces Cell Death of Cholinergic Neurons in an Organotypic Co-Culture Brain Slice Model. Neurochem Res, 2009. [DOI | PubMed]
- A Usmani, F Shavarebi, A Hiniker. The Cell Biology of LRRK2 in Parkinson’s Disease. Mol Cell Biol, 2021. [DOI | PubMed]
- VV Uteshev. α7 Nicotinic ACh Receptors as a Ligand-Gated Source of Ca2+ Ions: The Search for a Ca2+ Optimum. Adv Exp Med Biol, 2012. [DOI | PubMed]
- LFR Vasconcellos, JS Pereira. Parkinson’s disease dementia: Diagnostic criteria and risk factor review. J Clin Exp Neuropsychol, 2015. [DOI | PubMed]
- C Velez-Pardo, G Garcia Ospina, M Jimenez del Rio. Aβ[25–35] Peptide and Iron Promote Apoptosis in Lymphocytes by an Oxidative Stress Mechanism: Involvement of H2O2, Caspase-3, NF-κB, p53 and c-Jun. Neurotoxicology, 2002. [DOI | PubMed]
- C Velez-Pardo, M Jimenez-Del-Rio. Oxidative stress signaling and regulated cell death in Parkinson’s disease. In Genetics, Neurology, Behavior, and Diet in Parkinson’s Disease. Neurosci Parkinson, 2020. [DOI]
- S Viswanathan, Y Shi, J Galipeau. Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT®) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy, 2019. [DOI | PubMed]
- V Volloch, S Rits-Volloch. The Amyloid Cascade Hypothesis 2.0: Generalization of the Concept. J Alzheimers Dis Rep, 2023. [DOI | PubMed]
- H-Y Wang, DHS Lee, MR D’Andrea. β-Amyloid1–42 Binds to α7 Nicotinic Acetylcholine Receptor with High Affinity. J Biol Chem, 2000. [DOI | PubMed]
- JM Ward, VB Cockcroft, GG Lunt. Methyllycaconitine: a selective probe for neuronal α-bungarotoxin binding sites. FEBS Lett, 1990. [DOI | PubMed]
- AB West, DJ Moore, C Choi. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet, 2007. [DOI | PubMed]
- J Wiltfang, H Esselmann, P Cupers. Elevation of β-Amyloid Peptide 2–42 in Sporadic and Familial Alzheimer’s Disease and Its Generation in PS1 Knockout Cells. J Biol Chem, 2001. [DOI | PubMed]
- W-L Wu, M Domalski, DA Burnett. Discovery of SCH 900229, a Potent Presenilin 1 Selective γ-Secretase Inhibitor for the Treatment of Alzheimer’s Disease. ACS Med Chem Lett, 2012. [DOI | PubMed]
- W Xia. γ-Secretase and its modulators: Twenty years and beyond. Neurosci Lett, 2019. [DOI | PubMed]
- Y Xia, S Prokop, K-MM Gorion. Tau Ser208 phosphorylation promotes aggregation and reveals neuropathologic diversity in Alzheimer’s disease and other tauopathies. Acta Neuropathol Commun, 2020. [DOI | PubMed]
- J Yu, L Zhang, PM Hwang. PUMA Induces the Rapid Apoptosis of Colorectal Cancer Cells. Mol Cell, 2001. [DOI | PubMed]
- P Zhang, M Zhang, TA Mellich. Variation in Rotenone and Deguelin Contents among Strains across Four Tephrosia Species and Their Activities against Aphids and Whiteflies. Toxins (basel), 2022. [DOI | PubMed]
- X-B Zhang, J Li, J Gu, Y-Q Zeng. Roles of Cannabidiol in the Treatment and Prevention of Alzheimer’s Disease by Multi-target Actions. Mini-Reviews in Med Chem, 2022. [DOI]
- Y Zhu, W Shuai, M Zhao. Unraveling the Design and Discovery of c-Jun N-Terminal Kinase Inhibitors and Their Therapeutic Potential in Human Diseases. J Med Chem, 2022. [DOI | PubMed]
- SI Zubairi, MR Sarmidi, RA Aziz. A Study of Rotenone from Derris Roots of Varies Location, Plant Parts and Types of Solvent Used. Adv Environ Biol, 2014