
Dose-dependent effects of oral cannabidiol and delta-9-tetrahydrocannabinol on serum anandamide and related N-acylethanolamines in healthy volunteers
Abstract
Background:
The mental health benefits of cannabidiol (CBD) are promising but can be inconsistent, in part due to challenges in defining an individual’s effective dosage. In schizophrenia, alterations in anandamide (AEA) concentrations, an endocannabinoid (eCB) agonist of the eCB system, reflect positively on treatment with CBD. Here, we expanded this assessment to include eCBs alongside AEA congeners, comparing phytocannabinoids and dosage in a clinical setting.
Methods:
Liquid chromatography-tandem mass spectrometry quantified changes in serum levels of AEA, 2-arachidonoylglycerol (2-AG), alongside AEA-related compounds oleoylethanolamide (OEA) and palmitoylethanolamide (PEA), which were attained from two independent, parallel-designed, clinical trials investigating single, oral CBD (600 or 800 mg), delta-9-tetrahydrocannabinol (Δ9-THC, 10 or 20 mg) and combination administration (CBD|800 mg+Δ9-THC|20 mg) in healthy volunteers (HVs, n=75). Concentrations were measured at baseline (t=0), 65 and 160 min post administration.
Results:
CBD-led increases in AEA (1.6-fold), OEA and PEA (1.4-fold) were observed following a single 800 mg (pcorr<0.05) but not 600 mg dosage. Declining AEA was observed with Δ9-THC at 10 mg (−1.3-fold) and 20 mg (−1.4-fold) but restored to baseline levels by 160 min. CBD+Δ9-THC yielded the highest increases in AEA (2.1-fold), OEA (1.9-fold) and PEA (1.8-fold) without reaching a maximal response.
Conclusion:
CBD-administered effects towards AEA, OEA and PEA are consistent with phase II trials reporting clinical improvement for acute schizophrenia (CBD≥800 mg). Including Δ9-THC appears to enhance the CBD-induced response towards AEA and its congeners. Our results warrant further investigations into the potential of these lipid-derived mediators as metabolic measures for CBD dose prescription and co-cannabinoid administration.
Article type: Research Article
Keywords: Schizophrenia & psychotic disorders, Data Interpretation, Statistical, PSYCHIATRY
Affiliations: Brain and Mind Centre, The University of Sydney, Camperdown, New South Wales, Australia; Dept. of Psychiatry and Psychotherapy, Central Institute of Mental Health, Mannheim, Germany; Endosane Pharmaceuticals GmbH, Berlin, Germany; Dept. of Psychiatry, Psychosomatics and Psychotherapy, Goethe-Universitat Frankfurt am Main, Frankfurt am Main, Germany; Dept. of Psychiatry and Psychotherapy, University of Goettingen, Goettingen, Germany; Dept. of Cognitive and Clinical Neuroscience, Central Institute of Mental Health, Mannheim, Germany; Dept. of Psychosomatic Medicine and Psychotherapy, Central Institute of Mental Health, Mannheim, Germany
License: Copyright © Author(s) (or their employer(s)) 2024. Re-use permitted under CC BY. Published by BMJ. CC BY 4.0 This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Article links: DOI: 10.1136/bmjment-2024-301027 | PubMed: 39182921 | PMC: PMC11409355
Relevance: Relevant: mentioned in keywords or abstract
Full text: PDF (77 KB)
Introduction
Alterations to the endocannabinoid system (ECS) are implicated in the aetiology of neurological and psychiatric disorders, notably schizophrenia.ref. R1 The ECS consists of two endocannabinoids (eCBs), anandamide (AEA) and 2-arachidonoylglycerol (2-AG), which act as competing agonists for the binding of G-protein-coupled cannabinoid receptors (CB1/2-R) with delta-9-tetrahydrocannabinol (Δ9-THC), the main psychotomimetic component of Cannabis sativa (cannabis) that exacerbates schizophrenic psychoses.ref. 2ref. 4 Influences on AEA have been strongly linked to Δ9-THC and cannabis consumption. Preclinical evidence has shown that repeated treatment of Δ9-THC downregulates AEA signalling in the rat striatum.ref. R5 In humans, concentrations of AEA were notably lower in the cerebrospinal fluid (CSF) of healthy volunteers (HV)ref. R6 and antipsychotic-naïve individuals with schizophreniaref. R7 who were frequent cannabis users, compared with HVs and antipsychotic-naïve individuals with schizophrenia who were light cannabis users, respectively. Additionally, AEA exhibits protective properties early in the pathophysiology of schizophrenia.ref. R8
The principal non-psychotomimetic constituent of cannabis, cannabidiol (CBD), exerts clinical benefits either as a monotherapy or adjunctive treatment for schizophrenia,ref. R10 which are accompanied by a positive association with AEA.ref. R10 Though the mechanism is not conclusively understood, CBD’s low affinity for CB1/2-R has shifted research towards its capacity to modulate AEA degradation, alongside its congener N-acylethanolamines (NAEs), oleoylethanolamide (OEA) and palmitoylethanolamide (PEA).ref. R12 CBD’s optimal route of administration and appropriate dosage to achieve clinical efficacy remains a challenge, further complicated by the nature of the clinical syndrome(s) to be remedied.ref. R13 Hence, CBD’s clinical benefits may be underestimated due to an underappreciation of its biochemical activity. The study presented here is the first to explore effects on eCBs, alongside AEA congeners, following acute administration of CBD, Δ9-THC and combination therapy in phase I clinical trials, identifying measurable effects on these lipid mediators that appear dose dependent.
Methods
Human serum (n=75) was collected from two of our previous phase I clinical trials, exploring clinical and fMRI responses to CBD and Δ9-THC administration in HVs (LOGIN-TS4, GermanCTR: DRKS00005442; GEI-TCP-II, ClinicalTrials.gov: NCT02487381).ref. R14 Participant demographics were relatively balanced between the respective groups (table 1).
Table 1: Demographics and eCB/NAE measurements prior to the commencement of analysis
| CBD|800 mg (n=13) | CBD|600 mg (n=10) | CBD|800 mg+Δ9-THC|20 mg (n=12) | THC|20 mg(n=12) | THC|10 mg (n=10) | Placebo (n=18) | Total (n=75) | |
|---|---|---|---|---|---|---|---|
| Age | 24.85 (1.21) | 25.50 (1.55) | 24.17 (1.00) | 23.00 (0.82) | 22.90 (1.08) | 24.11 (0.89) | 24.09 (0.44) |
| Weight (kg) | 81.80 (2.93)* | 76.50 (3.28) | 77.08 (2.11) | 76.28 (2.38) | 75.70 (2.49) | 76.09 (2.88) | 77.31 (1.14) |
| Height (cm) | 180.00 (1.56) | 180.20 (1.61) | 183.83 (1.94) | 179.83 (2.69) | 182.60 (1.98) | 178.89 (2.07) | 180.64 (0.85) |
| BMI (kg/cm2) | 25.25 (0.84) | 23.52 (0.84) | 22.80 (0.49) | 23.70 (0.94) | 22.67 (0.47) | 23.66 (0.61) | 23.68 (0.31) |
| Smoker (no/yes) | 8/5 | 9/1 | 10/2 | 10/2 | 9/1 | 12/6 | 58/17 |
| Cigarettes per day | 2.62 (1.58) | 0.10 (0.10) | 1.17 (1.00) | 1.17 (0.87) | 0.20 (0.20) | 2.06 (1.25) | 1.36 (0.46) |
| Cannabis lifetime use | 5.38 (0.73) | 0.70 (0.50) | 7.17 (0.61) | 4.67 (0.50) | 1.30 (0.50) | 3.67 (0.70) | 3.97 (0.35) |
| Prescreen eCB/NAE concentrations (pmol/mL)† | |||||||
| AEA | 1.37 (0.21) | 2.00 (0.26) | 1.03 (0.08)* | 1.24 (0.11) | 2.34 (0.25)* | 1.55 (0.10) | 1.55 (0.08) |
| 2-AG | 2.46 (0.47) | 4.49 (1.36) | 2.16 (0.23) | 3.45 (0.42) | 3.69 (0.62) | 2.86 (0.76) | 3.10 (0.30) |
| OEA | 7.18 (0.95) | 7.05 (0.58) | 6.14 (0.47) | 10.54 (1.51)* | 7.03 (0.62) | 8.07 (0.86) | 7.73 (0.40) |
| PEA | 18.22 (2.46) | 20.68 (2.84) | 16.30 (1.26) | 21.30 (1.71) | 19.78 (1.80) | 12.02 (1.25)* | 17.26 (0.84) |
* A source of variation when comparing cohorts for a particular demographic/eCB/NAE (two-way analysis of variance, p<0.05, Benjamini, Krieger and Yekutieli method) to the overall (n=75) average.
† All values (when available) are reported as the mean±SE.
AEA, anandamide; 2-AG, 2-arachidonoylglycerol; BMI, body mass index; CBD, cannabidiol; eCB, endocannabinoid; NAE, N-acylethanolamine; OEA, oleoylethanolamide; PEA, palmitoylethanolamide; Δ9-THC, delta-9-tetrahydrocannabinol.
Eligible participants included male adults aged 18–45 years with a body mass index between 18 and 30 kg/m2. Participants in their respective clinical trials received the same vegetarian meals provided by the Central Institute of Mental Health hospital kitchen. GEI-TCP-II participants (n=61) received a single dose of placebo (n=15), Δ9-THC (20 mg, n=15), CBD (800 mg, n=15) or combination (CBD|800 mg+Δ9-THC|20 mg, n=16) in a double-dummy design. A single participant dropped out during the intervention (CBD|800 mg+Δ9-THC|20 mg), and 10 participants did not complete serum collections at time points that corresponded with the LOGIN-TS4 study (figure 1), hence were omitted from our analysis.
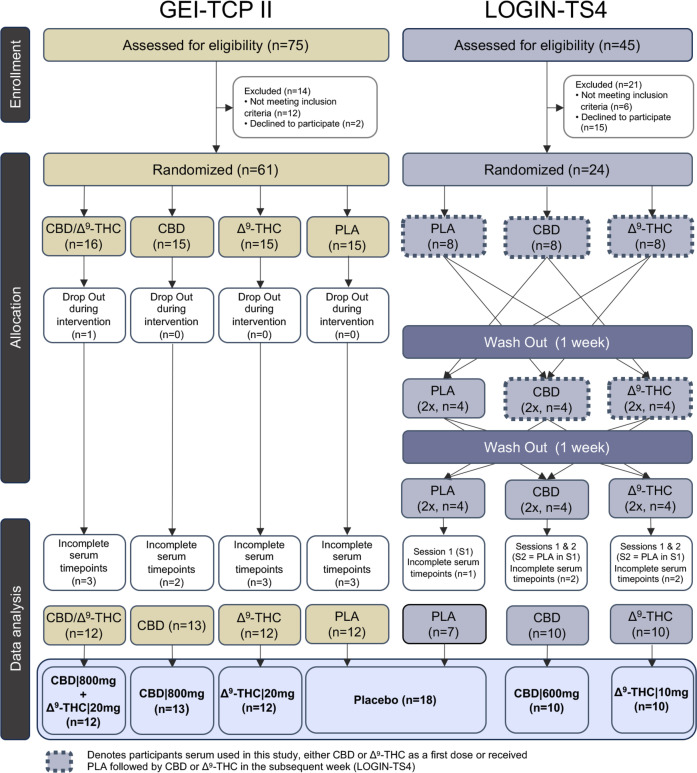
LOGIN-TS4 HVs (n=24) were part of a three-arm, cross-over, single-dose administration of placebo, Δ9-THC (10 mg) and CBD (600 mg). The order of drugs was randomised between participants, ensuring equal numbers (n=8) of drug administration before each series of measurements, with a 1-week washout period between subsequent doses (figure 1). For our study, only serum collected in weeks 1 and 2 for CBD/Δ9-THC recipients who received the placebo in week 1 was assessed. This avoided the risk of residual phytocannabinoids and potential discrepancies in eCBs/NAEs concentrations. This resulted in a total of 8 times placebo, 12 times CBD (n=4 after placebo in week 1) and 12 times Δ9-THC (n=4 after placebo in week 1) HVs for each group (figure 1). Six participants were excluded (1 time placebo, 3 times CBD, 2 times Δ9-THC) who did not complete the time collection points that mirrored GEI-TCP-II.
The eCBs, NAEs and CBD/Δ9-THC (if received) were extracted from 1 mL serum following the addition of internal standards as previously described.ref. R16 Blood draws were collected and assessed in parallel time points for each study: baseline (t=0), 65 and 160 min post administration. Bloods were also taken at a pre-screening (PS) visit the week prior. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) was performed on a TSQ Altis coupled to a Vanquish HPLC system (ThermoFisher), with analytes separated on a 4 µm Synergi Hydro-RP C18 column (150×2 mm; Phenomenex, Torrance, California, USA) over an 18 min gradient, as previous.ref. R16 Operational parameters for the specific eCBs, NAEs, CBD and Δ9-THC transitions monitored are listed in online supplemental table 1.
Peak integration and quantification were performed using Xcalibur (ThermoFisher), with peak areas normalised to their respective deuterated internal standards. All analytes quantified were expressed as pmol/mL. Endogenous effects to eCBs and NAEs following acute drug administration were analysed by two-way analysis of variance (repeated measures), corrected for multiple comparisons using the Benjamini-Krieger-Yekutieli post-test (GraphPad Prism, V.9.1.0). For all experiments, significant changes in eCBs and NAEs from baseline concentrations (t=0) were established at pcorr<0.05.
Correlations (r) on statistically significant changes to eCBs/NAEs with phytocannabinoids were performed using Spearman analyses. Phytocannabinoid concentrations were compared against serum AEA/NAEs changes, with 65 and 160 min concentrations subtracted from baseline t=0 (Δpmol/mL). To assess for concomitant changes with fatty acid amide hydrolase (FAAH)-selective analytes, associations between AEA with OEA and PEA were evaluated, both as concentrations that incorporated all time-points, including pre-screen, as well as Δpmol/mL at 65 and 160 min, with p values less than 0.05 considered significant. Outliers (ROUT method, Q=1%, GraphPad Prism, V.9.1.0) were removed prior to all correlations assessed.
Results
Baseline characteristics and CBD/Δ9-THC exposure
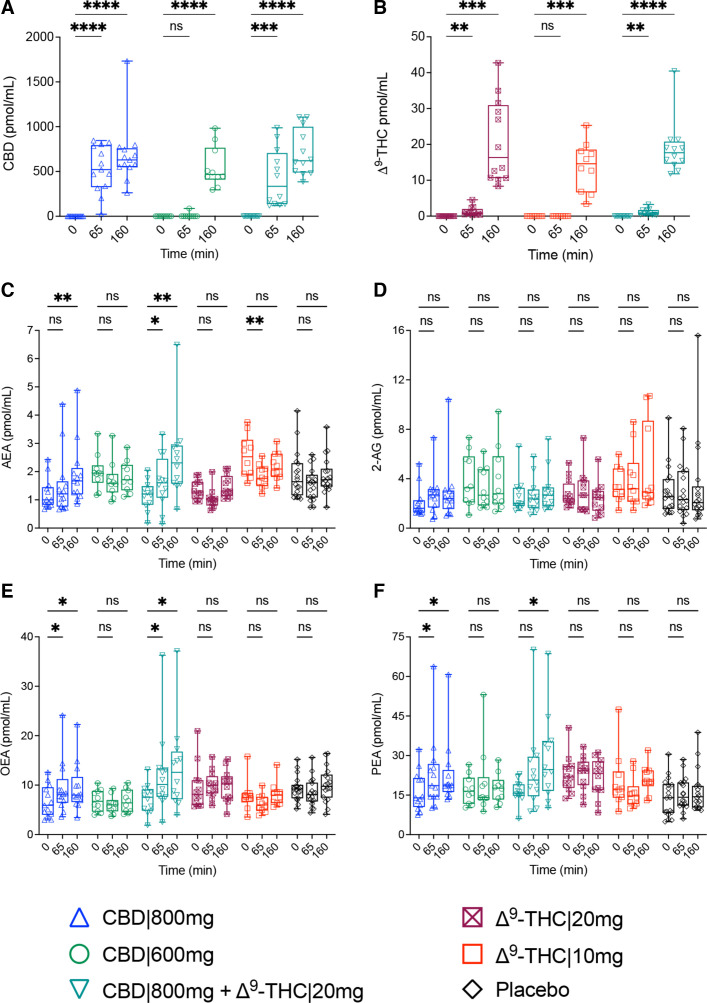
Variabilities in analyte concentrations, comparing each respective treatment group average (mean±SE, pmol/mL) to the combination of all participants, were observed for AEA (CBD|800 mg+Δ9-THC|20 mg, Δ9-THC|10 mg), OEA (Δ9-THC|20 mg) and PEA (placebo) during our PS assessments, (table 1), as well as for AEA (Δ9-THC|10 mg) at t=0 (online supplemental figure 1). However, these analytes were deemed stable at the commencement of the study as their concentrations aligned with those previously reportedref. R17 and remained analogous between PS and t=0 measurements, displaying no significant alterations between time-points prior to intervention (online supplemental figure 1). For all HVs, serum levels of exogenous CBD and Δ9-THC were sufficient by 160 min to expect physiological effects,ref. R19 with CBD and Δ9-THC administered at higher doses (800 vs 600 mg and 20 vs 10 mg, respectively) displaying a faster rate of accrual (figure 2A,B).
AEA and congener changes in response to phytocannabinoid dosage and combinations
An initial effect towards AEA was observed on administration of Δ9-THC, leading to a decrease in concentration at 65 min (Δ9-THC|10 mg, −1.4-fold, pcorr=0.0014; THC|20 mg, −1.3-fold, pcorr=0.1160). However, by 165 min, levels had returned to t=0 levels (figure 2C). In contrast, CBD administered at 800 mg demonstrated a continued increase in AEA concentration (65 min, 1.3-fold, pcorr=0.0514; 160 min, 1.6-fold pcorr=0.0030), with the combination treatment (CBD|800mg+Δ9-THC|20 mg) inducing an even greater response (65 min, 1.4-fold, pcorr=0.0328; 160 min, 2.1-fold, pcorr=0.0080) (figure 2C). No reported differences in AEA concentrations were observed with CBD|600 mg (figure 2C). Neither CBD nor Δ9-THC significantly influenced 2-AG at any time or dosage (figure 2D).
OEA and PEA concentrations increased, in concert with their structural analogue AEA, following the intake of CBD|800 mg (65 min: OEA, 1.4-fold, pcorr=0.0132; PEA, 1.4-fold, pcorr=0.0478) and CBD|800mg+Δ9-THC|20 mg (65 min: OEA, 1.7-fold, pcorr=0.0303; PEA, 1.5-fold pcorr=0.0520). CBD|800 mg mediated changes appeared to have reached their maximal response (165 min: OEA, 1.4-fold pcorr=0.0132; PEA, 1.4-fold pcorr=0.0405) while effects following CBD|800mg+Δ9-THC|20 mg (OEA: 1.9-fold, pcorr=0.0234; PEA, 1.8-fold pcorr=0.0190) continued over the course of the analysis (figure 2E,F). The mean concentrations (pmol/mL) for eCBs and NAEs at each time-point and treatment group are provided alongside the individual and adjusted p values (pcorr) in online supplemental table 2.
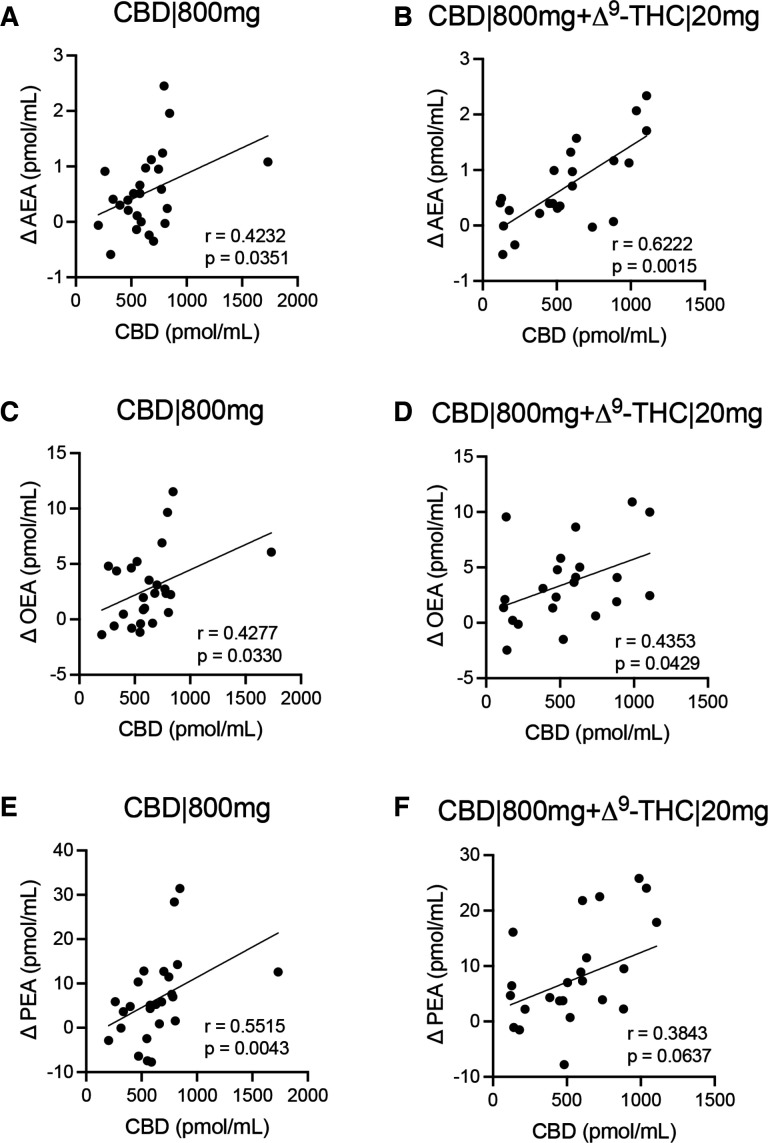
Spearman analyses confirmed that increasing concentrations of CBD at 65 and 160 min positively associated with changes (Δpmol/mL) in AEA (CBD|800 mg, r=0.4232, p=0.0351; CBD|800mg+Δ9-THC|20 mg, r=0.6222, p=0.0015), OEA (CBD|800 mg, r=0.4277, p=0.0330; CBD|800mg+Δ9-THC|20 mg, r=0.4353, p=0.0429) and PEA (CBD|800 mg, r=0.5515, p=0.0043; CBD|800mg+Δ9-THC|20 mg, r=0.3843, p=0.0637), when administered at 800 mg CBD, with or without the addition of Δ9-THC (figure 3). Positive associations between AEA and NAEs were also consistent across participants in both CBD|800 mg and CBD|800mg+THC|20 mg treatment arms, both as change from t=0 (Δpmol/mL) and when comparing concentrations across all time-points measured, including PS values (pmol/mL) for all participants (online supplemental figure 2). Concentrations of Δ9-THC|10 mg were below the limit of detection to yield values for association with AEA at 65 min (data not shown). However, we did demonstrate a negative association for Δ9-THC|20 mg with AEA (r=−0.4098, p=0.1859), with OEA and PEA not displaying any directed association towards Δ9-THC|20 mg (r<0.1, online supplemental figure 3A,C,E). In contrast, Δ9-THC levels were positively associated with AEA, OEA and PEA when coadministered with CBD|800 mg (online supplemental figure 3D,E,F).
Discussion
This study sought to observe relative changes to eCBs and NAEs in the serum of HVs following oral phytocannabinoid administration in a retrospective investigation that measured matched time-points from two completed clinical trials (LOGIN-TS4, GEI-TCP-II). We postulate that the observed changes for CBD|800 mg (AEA, OEA and PEA), Δ9-THC (AEA) and in-combination (CBD|800mg+Δ9-THC|20 mg; AEA, OEA and PEA) represent mechanistically relevant responses, given no significant influences in their endogenous levels were detected prior to phytocannabinoid administration.
Increased levels of AEA from CBD|800 mg were consistent with our prior findings for CBD treatment of schizophrenia, leading to increased sera AEA associated with improved clinical symptoms.ref. R10 Reports suggest CBD-induced increase is caused by inhibiting FAAH, AEA’s principal metabolising enzyme, verified in rat brain membranes,ref. R10 mouse brain microsomesref. R12 and COS-7 cells expressing rodent FAAH.ref. R12 However, CBD has not been reported to inhibit FAAH in humans, rather blocking fatty acid binding proteins (FABPs) intracellular transport of AEA to FAAH at the endoplasmic reticulum.ref. R12 Although the relevance of this mechanism remains conjectural, CBD’s capacity to block FABPs-mediated transport to FAAH would also lead to increases in OEA and PEA, given these endogenous lipid mediators share this conserved hydrolysis pathway with AEA.ref. R12 Significant increases in OEA and PEA were observed following CBD administration, aligning with the proposed FABPs–FAAH mechanism of inhibition (figure 2E,F).
Though evidently more complex, it is widely acknowledged that NAEs follow a canonical biosynthesis pathway initiated by the N-acylation of membrane glycerophospholipids.ref. 20ref. 22 This process results in the formation of N-acyl-phosphatidylethanolamine (NAPE) precursors, which undergoes specific phospholipase D type (NAPE-PLD) hydrolysis, with N-arachidonoyl-phosphatidylethanolamine, N-oleoyl-phosphatidylethanolamine and N-palmitoyl-phosphatidylethanolamine each being converted into AEA, OEA and PEA, respectively.ref. R23 Given that NAPE-PLD is integral to the NAE metabolic process, an alternative, although speculative, viewpoint is that CBD induces concomitant increases in AEA and its congeners through increased biosynthesis. Indeed, CBD has been shown to promote the upregulation of NAEs in multiple brain regions of rats via an NAPE-PLD-dependent mechanism,ref. R25 as well as enhance mRNA expression of NAPE-PLD in syncytiotrophoblasts.ref. R26
Increases in AEA, OEA and PEA occurred only from the administration of CBD at 800 mg, alone or in combination with Δ9-THC. This metabolic response aligns with the dose-dependent clinical efficacy of CBD. In our previous report, 800 mg of CBD significantly improved schizophrenia symptoms,ref. R10 while clinical trials reported CBD at 600 mg, as an add-on therapy, was insufficient to improve cognitive function or attenuate psychotic symptoms,ref. R27 though the latter study did observe increased connectivity during resting state.ref. R28 In support of a ≥800 mg threshold, CBD as an add-on to conventional antipsychotics does exhibit clinical benefit at 1000 mg,ref. R11 while displaying no positive effects when given as a CBD/Δ9-THC mix (Bedrolite, with CBD ca. 300 mg/day) to patients with a psychotic disorder and comorbid cannabis use.ref. R29
CBD’s poor oral bioavailability (estimated at 4%–6%ref. R30) could explain why benefits and induced effects on AEA/NAEs require the high dosage, as 800 mg CBD exhibits a faster accrual, over 600 mg (figure 2A). This same rationale may also account for comparable effects towards AEA following Δ9-THC at different doses, where their accumulation rates and subsequent impact on AEA were similar (figure 2B). Analogous declines in AEA concentrations of the CSF have been reported in studies comparing heavy (>10 times/month) to light (<10 times/month) cannabis consumers without any diagnosis of psychopathology according to Diagnostic and Statistical Manual of Mental Disorders, version IV (DSM-IV) criteria,ref. R6 as well as in individuals diagnosed with schizophrenia and higher consumption of cannabis in their lifetime (>20 times/life) compared with those with no or sporadic use (<5 occasions in their lifetime).ref. R7 However, it should be noted that in the latter study, a subgroup of high-frequency cannabis users with schizophrenia tested positive for urinary Δ9-THC still displayed elevated AEA in the CSF, though statistical significance was not reached. Therefore, the overall lower AEA levels in high-frequency cannabis users diagnosed with schizophrenia may not be attributed to recent cannabis use.
Previous reports on alterations in circulating AEA due to Δ9-THC and cannabis consumption have shown considerable variability. The aforementioned studies found no effects on human sera,ref. R6 while others reported increases in AEA alongside mixed effects on OEA and PEA levels in participants undertakingref. R31 or undergoing cessationref. R32 from high-cannabis use. Lower AEA concentrations have been observed in the plasma of individuals with psychosis who self-reported high-cannabis use,ref. R33 whereas higher AEA levels were found in individuals with cannabis use disorder, accompanied by concomitant increases in OEA.ref. R34 Clinical evidence supports elevated eCB/NAE response following acute, oral administration of Δ9-THC at a dosage identical (20 mg) to a treatment arm used in this study, with results displaying higher levels of AEA alongside 2-AG and OEA in plasma 2–3 hours post in-take.ref. R35 Conversely, a longer clinical trial reported a decline in plasma AEA over 28 days in participants undergoing cessation for their cannabis use disorder, with stable AEA levels in participants that received 800 mg of CBD, while this effect was absent in those receiving 400 mg of CBD.ref. R36 The contradictory reports of AEA/NAE levels may be a consequence of their biphasic response towards Δ9-THC, which is postulated to be mediated by synthesis and/or degradation processesref. R37 or through catecholaminergic and glucocorticoid signalling that, in turn, promote eCB/NAE concentrations.ref. 37ref. 41 Thieme et al reported that plasma concentrations of AEA and 2-AG undergo repeated changes (both upregulation and downregulation) at multiple time points up to 48 hours post intravenous injections of Δ9-THC.ref. R38 This infers that those acute investigations, including this study, may have limitations in interpreting the effects of Δ9-THC on AEA and its congeners. Additionally, formulation and mode of administration could also impact the response to Δ9-THCref. R16 and its combination with CBD,ref. R37 as discussed below.
OEA and PEA concentrations appeared to plateau with CBD-alone treatment, suggesting that the maximal affinity for inhibiting their degradation may have been reached. This is postulated to be due to either a higher affinity of FAAH for AEAref. R43 or the metabolism of OEA and PEA via alternative pathways, such as N-acylethanolamine acid amidase (NAAA),ref. R44 which is not primarily responsible for the hydrolytic deactivation of AEA,ref. R45 nor inhibited by CBD.ref. R46 Additionally, NAPE-PLD is not exclusively responsible for the formation of NAEs.ref. R47 Alternative pathways involve phospholipase C, phospholipase A or α/β Hydrolase Domain-Containing Protein 4 catalytic hydrolysis, as reviewed by Simard et al.ref. R48 While the importance of these multiple pathways remains unknown, they may facilitate the differential synthesis of AEA, OEA and PEA, potentially varying based on the specific tissue or physiological process involved.ref. R49 Differential effects have also been noted across the overall pathway, with preclinical models recognising FAAH as a critical step for regulating NAEs in the brain but not heart tissue.ref. R50 Hence, identifying the source(s) of our circulatory AEA/NAE alterations could elucidate relevant aspects or overlapping mechanisms of the metabolic pathway contributing to the dose-dependent changes observed with CBD and Δ9-THC. However, resolving the origin of circulating eCBs/NAEs presents a challenge, owing to their lipophilic nature, which means they are identifiable in almost every tissue and produced by nearly all cell types.ref. R51
In this study, NAEs continued to increase, alongside a greater upturn of AEA, from the combination of CBD+Δ9-THC, leading us to speculate a positive contribution towards CBD-derived mechanisms occurs with the inclusion of Δ9-THC (‘entourage effect’).ref. R52 Though beyond this project’s scope, prior evidence of CBD+Δ9-THC therapy supports analgesic efficacyref. R53; however, safety and tolerance of Δ9-THC remain debated.ref. R54 Furthermore, single-dose administration of CBD+Δ9-THC through inhalation yielded no effect on eCB/NAE levels.ref. R37 It is important to note that doses administered by Chester et al were designed to reflect recreational cannabis use, with CBD doses of only up to 30 mg. Additionally, comparisons between formulations are challenging due to the delayed absorption and metabolism from the first-pass effect of oral phytocannabinoid administration.ref. R55 Refinement of Δ9-THC dosage and pharmacokinetics to support CBD efficacy for certain indications while minimising adverse effects warrants future investigation.
Our study has several limitations. Though endogenous effects were observed, our sample size remains relatively small. This may account for eCB/NAE changes higher in dosage (e.g., Δ9-THC 20|mg effects on AEA) or magnitude (CBD|800 mg+Δ9-THC|20 mg response on PEA at 65 min) did not reach statistical significance. The exploratory nature of combining randomised clinical trials, along with the increased likelihood of variation in results when working with small sample sizes, constrains our comparative measures to within-treatment arms. This underscores the necessity of addressing these limitations in expanded studies to authenticate our observations. In particular, clinical trials that allow for more controllable comparisons between CBD and Δ9-THC (including combined effects), increased sampling times that better encapsulate endogenous alterations or validate non-apparent responses observed herein (eg, CBD|600 mg) and, if permissible, higher concentrations or longer administrations to clarify whether endogenous effects are dose-dependent or reflect a pre-existing state. Influences on endogenous eCBs/NAEs, and the required dosage to achieve them, may also have been affected by food intake. Phytocannabinoid bioavailability increases with high-fat meals, likely due to their lipophilicity requiring intestinal lumen solubilisation before absorption.ref. R56 Although participants in both studies received standardised meals, they were not identical. This may have slightly belated the absorption of CBD and Δ9-THC between the studies (GEI-TCP-II and LOGIN-TS4), delaying any subsequent effects on AEA and NAEs. However, this would not account for differences between CBD-alone versus combination (CBD+Δ9-THC), which were controlled in the same study and administered with an identical dosage of CBD. Finally, we also acknowledge that this study does not address the question of whether the observed peripheral changes are indicative of a cerebral response. Although our observations are consistent with the reported clinical benefits of CBDref. R10 and Δ9-THC effects on the CSF,ref. R6 response in peripheral systems may differ from cerebral effects, as there is evidence of a lack of association between these two biofluids.ref. R57 To the best of our knowledge, no comparative investigations have yet to assess CBD’s effects on the eCBs and NAEs in the CSF alongside paired peripheral fluids.
In summary, our data supports both previous clinical and mechanistic evidence for AEA’s and NAEs’ response to phytocannabinoids. Our results could reflect a dose-dependent metabolic signature for CBD, with potential enhancement from Δ9-THC, encouraging future investigations into these endogenous lipid mediators as indicative markers of CBD efficacy, both as a stand-alone or co-administered cannabinoid therapy.
Supplementary Materials
References
- FM Leweke, JK Mueller, B Lange. Role of the Endocannabinoid System in the Pathophysiology of Schizophrenia: Implications for Pharmacological Intervention. CNS Drugs, 2018. [DOI | PubMed]
- HM Emrich, FM Leweke, U Schneider. Towards a cannabinoid hypothesis of schizophrenia: cognitive impairments due to dysregulation of the endogenous cannabinoid system. Pharmacol Biochem Behav, 1997. [DOI | PubMed]
- L Cristino, T Bisogno, V Di Marzo. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat Rev Neurol, 2020. [DOI | PubMed]
- DC D’Souza, WM Abi-Saab, S Madonick. Delta-9-tetrahydrocannabinol effects in schizophrenia: implications for cognition, psychosis, and addiction. Biol Psychiatry, 2005. [DOI | PubMed]
- V Di Marzo, F Berrendero, T Bisogno. Enhancement of anandamide formation in the limbic forebrain and reduction of endocannabinoid contents in the striatum of delta9-tetrahydrocannabinol-tolerant rats. J Neurochem, 2000. [DOI | PubMed]
- CJA Morgan, E Page, C Schaefer. Cerebrospinal fluid anandamide levels, cannabis use and psychotic-like symptoms. Br J Psychiatry, 2013. [DOI | PubMed]
- FM Leweke, A Giuffrida, D Koethe. Anandamide levels in cerebrospinal fluid of first-episode schizophrenic patients: impact of cannabis use. Schizophr Res, 2007. [DOI | PubMed]
- FM Leweke. Anandamide dysfunction in prodromal and established psychosis. Curr Pharm Des, 2012. [DOI | PubMed]
- D Koethe, A Giuffrida, D Schreiber. Anandamide elevation in cerebrospinal fluid in initial prodromal states of psychosis. Br J Psychiatry, 2009. [DOI | PubMed]
- FM Leweke, D Piomelli, F Pahlisch. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry, 2012. [DOI | PubMed]
- P McGuire, P Robson, WJ Cubala. Cannabidiol (CBD) as an Adjunctive Therapy in Schizophrenia: A Multicenter Randomized Controlled Trial. Am J Psychiatry, 2018. [DOI | PubMed]
- MW Elmes, M Kaczocha, WT Berger. Fatty acid-binding proteins (FABPs) are intracellular carriers for Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD). J Biol Chem, 2015. [DOI | PubMed]
- C Larsen, J Shahinas. Dosage, Efficacy and Safety of Cannabidiol Administration in Adults: A Systematic Review of Human Trials. J Clin Med Res, 2020. [DOI | PubMed]
- O Grimm, M Löffler, S Kamping. Probing the endocannabinoid system in healthy volunteers: Cannabidiol alters fronto-striatal resting-state connectivity. Eur Neuropsychopharmacol, 2018. [DOI | PubMed]
- T Woelfl, C Rohleder, JK Mueller. Effects of Cannabidiol and Delta-9-Tetrahydrocannabinol on Emotion, Cognition, and Attention: A Double-Blind, Placebo-Controlled, Randomized Experimental Trial in Healthy Volunteers. Front Psychiatry, 2020. [DOI | PubMed]
- TA Couttas, C Boost, F Pahlisch. Simultaneous Assessment of Serum Levels and Pharmacologic Effects of Cannabinoids on Endocannabinoids and N-Acylethanolamines by Liquid Chromatography-Tandem Mass Spectrometry. Cannabis Cannabinoid Res, 2023. [DOI | PubMed]
- MGJ Balvers, KCM Verhoeckx, RF Witkamp. Development and validation of a quantitative method for the determination of 12 endocannabinoids and related compounds in human plasma using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 2009. [DOI]
- D Schreiber, S Harlfinger, BM Nolden. Determination of anandamide and other fatty acyl ethanolamides in human serum by electrospray tandem mass spectrometry. Anal Biochem, 2007. [DOI | PubMed]
- P Fusar-Poli, JA Crippa, S Bhattacharyya. Distinct effects of {delta}9-tetrahydrocannabinol and cannabidiol on neural activation during emotional processing. Arch Gen Psychiatry, 2009. [DOI | PubMed]
- V Di Marzo, A Fontana, H Cadas. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature New Biol, 1994. [DOI]
- N Wellner, TA Diep, C Janfelt. N-acylation of phosphatidylethanolamine and its biological functions in mammals. Biochim Biophys Acta, 2013. [DOI | PubMed]
- HS Hansen, B Moesgaard, HH Hansen. N-Acylethanolamines and precursor phospholipids – relation to cell injury. Chem Phys Lipids, 2000. [DOI | PubMed]
- Y Okamoto, J Morishita, K Tsuboi. Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem, 2004. [DOI | PubMed]
- Z Hussain, T Uyama, K Tsuboi. Mammalian enzymes responsible for the biosynthesis of N-acylethanolamines. Biochim Biophys Acta Mol Cell Biol Lipids, 2017. [DOI | PubMed]
- E Leishman, M Manchanda, R Thelen. Cannabidiol’s Upregulation of N-acyl Ethanolamines in the Central Nervous System Requires N-acyl Phosphatidyl Ethanolamine-Specific Phospholipase D. Cannabis Cannabinoid Res, 2018. [DOI | PubMed]
- T Podinic, L Limoges, C Monaco. Cannabidiol Disrupts Mitochondrial Respiration and Metabolism and Dysregulates Trophoblast Cell Differentiation. Cells, 2024. [DOI | PubMed]
- DL Boggs, T Surti, A Gupta. The effects of cannabidiol (CBD) on cognition and symptoms in outpatients with chronic schizophrenia a randomized placebo controlled trial. Psychopharmacology (Berl), 2018. [DOI | PubMed]
- R van Boxel, SS Gangadin, H Janssen. The impact of cannabidiol treatment on resting state functional connectivity, prefrontal metabolite levels and reward processing in recent-onset patients with a psychotic disorder. J Psychiatr Res, 2023. [DOI | PubMed]
- R Schipper, M Dekker, L de Haan. Medicinal cannabis (Bedrolite) substitution therapy in inpatients with a psychotic disorder and a comorbid cannabis use disorder: A case series. J Psychopharmacol, 2018. [DOI | PubMed]
- A Ohlsson, JE Lindgren, S Andersson. Single-dose kinetics of deuterium-labelled cannabidiol in man after smoking and intravenous administration. Biomed Environ Mass Spectrom, 1986. [DOI | PubMed]
- F Zufferey, E Buitrago, R Rahban. Gonadotropin axis and semen quality in young Swiss men after cannabis consumption: Effect of chronicity and modulation by cannabidiol. Andrology (Los Angel), 2024. [DOI]
- D Muhl, M Kathmann, C Hoyer. Increased CB2 mRNA and anandamide in human blood after cessation of cannabis abuse. Naunyn Schmiedebergs Arch Pharmacol, 2014. [DOI | PubMed]
- A Bassir Nia, CL Gibson, SA Spriggs. Cannabis use is associated with low plasma endocannabinoid Anandamide in individuals with psychosis. J Psychopharmacol, 2023. [DOI | PubMed]
- N Boachie, E Gaudette, RP Bazinet. Circulating Endocannabinoids and N-Acylethanolamines in Individuals with Cannabis Use Disorder-Preliminary Findings. Brain Sci, 2023. [DOI]
- C Walter, N Ferreirós, P Bishay. Exogenous delta⁹-tetrahydrocannabinol influences circulating endogenous cannabinoids in humans. J Clin Psychopharmacol, 2013. [DOI | PubMed]
- DY-H Hua, C Hindocha, G Baio. Effects of cannabidiol on anandamide levels in individuals with cannabis use disorder: findings from a randomised clinical trial for the treatment of cannabis use disorder. Transl Psychiatry, 2023. [DOI | PubMed]
- LA Chester, A Englund, E Chesney. Effects of Cannabidiol and Delta-9-Tetrahydrocannabinol on Plasma Endocannabinoid Levels in Healthy Volunteers: A Randomized Double-Blind Four-Arm Crossover Study. Cannabis Cannabinoid Res, 2024. [DOI | PubMed]
- U Thieme, G Schelling, D Hauer. Quantification of anandamide and 2-arachidonoylglycerol plasma levels to examine potential influences of tetrahydrocannabinol application on the endocannabinoid system in humans. Drug Test Anal, 2014. [DOI]
- A Dlugos, E Childs, KL Stuhr. Acute stress increases circulating anandamide and other N-acylethanolamines in healthy humans. Neuropsychopharmacology, 2012. [DOI | PubMed]
- M Ranganathan, G Braley, B Pittman. The effects of cannabinoids on serum cortisol and prolactin in humans. Psychopharmacology (Berl), 2009. [DOI | PubMed]
- MAP Bloomfield, AH Ashok, ND Volkow. The effects of Δ9-tetrahydrocannabinol on the dopamine system. Nature New Biol, 2016. [DOI]
- T Kearney-Ramos, ES Herrmann, I Belluomo. The Relationship Between Circulating Endogenous Cannabinoids and the Effects of Smoked Cannabis. Cannabis Cannabinoid Res, 2023. [DOI | PubMed]
- BQ Wei, TS Mikkelsen, MK McKinney. A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem, 2006. [DOI | PubMed]
- N Ueda, K Yamanaka, S Yamamoto. Purification and characterization of an acid amidase selective for N-palmitoylethanolamine, a putative endogenous anti-inflammatory substance. J Biol Chem, 2001. [DOI | PubMed]
- M Migliore, S Pontis, AL Fuentes de Arriba. Second‐Generation Non‐Covalent NAAA Inhibitors are Protective in a Model of Multiple Sclerosis. Angew Chem Int Ed, 2016. [DOI]
- L De Petrocellis, A Ligresti, AS Moriello. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol, 2011. [DOI | PubMed]
- D Leung, A Saghatelian, GM Simon. Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry, 2006. [DOI | PubMed]
- M Simard, A-S Archambault, J-PC Lavoie. Biosynthesis and metabolism of endocannabinoids and their congeners from the monoacylglycerol and N-acyl-ethanolamine families. Biochem Pharmacol, 2022. [DOI | PubMed]
- DR Sagar, AG Gaw, BN Okine. Dynamic regulation of the endocannabinoid system: implications for analgesia. Mol Pain, 2009. [DOI | PubMed]
- A Kilaru, G Isaac, P Tamura. Lipid profiling reveals tissue-specific differences for ethanolamide lipids in mice lacking fatty acid amide hydrolase. Lipids, 2010. [DOI | PubMed]
- CJ Hillard. Circulating Endocannabinoids: From Whence Do They Come and Where are They Going?. Neuropsychopharmacology, 2018. [DOI | PubMed]
- E Russo, GW Guy. A tale of two cannabinoids: the therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med Hypotheses, 2006. [DOI | PubMed]
- JR Johnson, M Burnell-Nugent, D Lossignol. Multicenter, double-blind, randomized, placebo-controlled, parallel-group study of the efficacy, safety, and tolerability of THC:CBD extract and THC extract in patients with intractable cancer-related pain. J Pain Symptom Manage, 2010. [DOI | PubMed]
- KP Hill, MS Gold, CB Nemeroff. Risks and Benefits of Cannabis and Cannabinoids in Psychiatry. Am J Psychiatry, 2022. [DOI | PubMed]
- MA Huestis. Human cannabinoid pharmacokinetics. Chem Biodivers, 2007. [DOI | PubMed]
- L Taylor, B Gidal, G Blakey. A Phase I, Randomized, Double-Blind, Placebo-Controlled, Single Ascending Dose, Multiple Dose, and Food Effect Trial of the Safety, Tolerability and Pharmacokinetics of Highly Purified Cannabidiol in Healthy Subjects. CNS Drugs, 2018. [DOI | PubMed]
- A Minichino, M Senior, N Brondino. Measuring Disturbance of the Endocannabinoid System in Psychosis: A Systematic Review and Meta-analysis. JAMA Psychiatry, 2019. [DOI | PubMed]
- A Giuffrida, FM Leweke, CW Gerth. Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. Neuropsychopharmacology, 2004. [DOI | PubMed]