
Interplay between endocannabinoids and dopamine in the basal ganglia: implications for pain in Parkinson’s disease
Abstract
Pain is a complex phenomenon, and basal ganglia circuitry integrates many aspects of pain including motor, emotional, autonomic, and cognitive responses. Perturbations in dopamine (DA) signaling are implicated in the pathogenesis of chronic pain due to its involvement in both pain perception and relief. Several lines of evidence support the role of endocannabinoids (eCBs) in the regulation of many electrical and chemical aspects of DAergic neuron function including excitability, synaptic transmission, integration, and plasticity. However, eCBs play an even more intricate and intimate relationship with DA, as indicated by the adaptive changes in the eCB system following DA depletion. Although the precise mechanisms underlying DA control on pain are not fully understood, given the high correlation of eCB and DAergic system, it is conceivable that eCBs may be part of these mechanisms.
In this brief survey, we describe the reciprocal regulation of eCB-DA neurotransmission with a particular emphasis on the actions of eCBs on ionic and synaptic signaling in DAergic neurons mediated by CB receptors or independent on them. Furthermore, we analyze the eCB-DA imbalance which characterizes pain condition and report the implications of reduced DA levels for pain in Parkinson’s disease. Lastly, we discuss the potential of the eCB-DA system in the development of future therapeutic strategies for the treatment of pain.
Article type: Review Article
Keywords: Dopamine, Endocannabinoids, Parkinson’s disease, Pain, Ion channels
Affiliations: https://ror.org/00s6t1f81grid.8982.b0000 0004 1762 5736Department of Brain and Behavioral Sciences, University of Pavia, c/o Mondino Foundation Via Mondino, 2, Pavia, 27100 Italy; grid.419416.f0000 0004 1760 3107IRCCS Mondino Foundation, Pavia, 27100 Italy
License: © The Author(s) 2024 CC BY 4.0 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Article links: DOI: 10.1186/s44158-024-00169-z | PubMed: 38745258 | PMC: PMC11094869
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (1.2 MB)
Introduction
The identification of the cannabinoids receptors (CB-Rs) in the early 1990s [ref. 1, ref. 2] suggested that they serve not merely as binding sites for psychotropic substances extracted from Cannabis sativa plant flowers (cannabinoids) but rather function as mediators of endogenous signaling molecules, physiologically operating as messengers in the mammalian central nervous system (CNS). This discovery prompted the search for endogenous agonists, or endocannabinoids (eCBs), which mimic the psychomotor effects of cannabinoids [ref. 3].
Since then, the eCB system has been demonstrated to play important physiological roles in the control of emotional responses, cognition, feeding, pain, motivated behaviors, and motor control. However, the precise mechanisms employed by eCBs to mediate their effects remain partially understood, due to the multiplicity and complexity of their actions. In fact, both CB1 and CB2 are G protein-coupled receptors (GPCRs), which influence the biochemical and electrical state of a cell through complex mechanisms involving kinases, phosphatases, transcription factors, and ion channels. Moreover, the interaction with other systems of neurotransmitters and the modulation of their activity can induce changes in synaptic transmission, promoting long-term changes in synapse efficacy and modifying the structural organization of neuronal circuitries.
CB-Rs are widely distributed throughout the CNS. Of note, their robust expression within the basal ganglia circuitry has stimulated investigation on the possible interplay between eCBs and dopamine (DA) system, not only in the context of movement disorders but also in pain research. Several preclinical and clinical studies have demonstrated that eCBs are crucial modulators of DAergic transmission [ref. 4], and that DA agonists may have an antinociceptive effect [ref. 5]. Conversely, DA depletion leads to enhanced nociceptive responses [ref. 6–ref. 11] and induces adaptive changes in the eCB system [ref. 12–ref. 15]. Accordingly, Parkinson’s disease (PD) patients report comorbid pain conditions and higher pain rating [ref. 16, ref. 17].
Despite such evidence, the specific mechanisms through which intracellular effectors recruited by eCBs directly or indirectly interact with DA-dependent signaling effectors to affect pain perception have been only partially identified. However, a review of all the potential cellular and molecular mechanisms is beyond the scope of this paper, and we refer the reader to a number of excellent comprehensive reviews (see [ref. 18–ref. 22]). Our brief survey will rather focus on the available evidence on the interactions between eCBs and DA. In PD, DA depletion provides an ideal clue to review and discuss the compensatory changes of the eCB system, as a potential mechanism contributing to pain in PD. Advancing our knowledge on these basic mechanisms might reveal potential molecular targets for drug development in PD-related pain.
Endocannabinoid-dopamine crosstalk
eCB and CB-Rs in the basal ganglia
The presence of eCB-Rs within the basal ganglia circuitry has been demonstrated by several neuroanatomical studies. The expression of the CB1-R has been found in the glutamatergic corticostriatal afferences in both dorsal and ventral striatum [ref. 23, ref. 24] and the projections of striatal GABAergic neurons innervating the globus pallidus (GPi and GPe) and the substantia nigra pars reticulata (SNpr) [ref. 25, ref. 26]. Some studies have also confirmed the distribution of CB1-R in the glutamatergic terminals innervating the subthalamic nucleus (STN) and the GPi/SNpr [ref. 27] (Fig. 1).
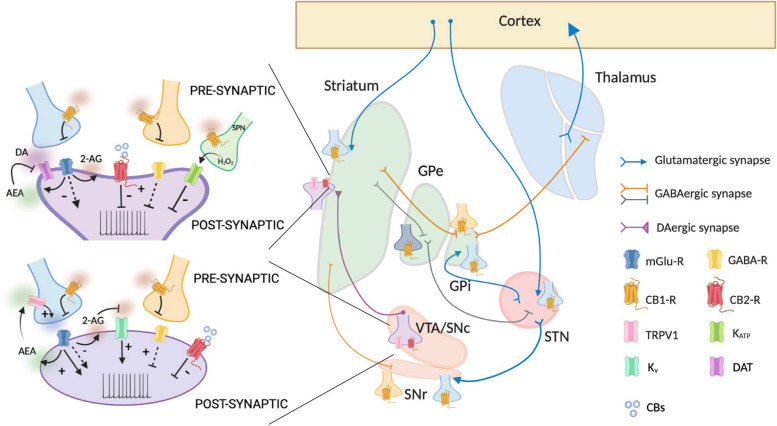
Also, CB2-R was found in the striatum, GP, DAergic neurons of the ventral tegmental area (VTA) and SN, and in the basal thalamus [ref. 28–ref. 32] (Fig. 1).
In addition to the classical CB-Rs, other putative eCB receptors exist. Among these, the transient receptor potential vanilloid type-1 (TRPV1) channel can be activated by several exogenous and endogenous stimuli. TRPV-1 has been identified in both the axonal terminals and the soma of nigrostriatal DAergic neurons [ref. 33–ref. 35] (Fig. 1).
CB-Rs and the putative receptors are activated by eicosanoids such as N-arachidonoyl-ethanolamide (AEA) or anandamide, and 2-arachidonoyl-glicerol (2-AG), and by cannabinoids and their analogues.
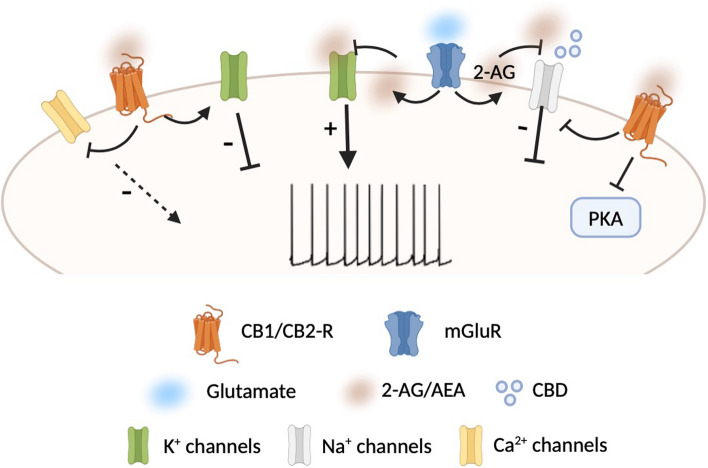
As GPCRs, both CB1-R and CB2-R activate heterotrimeric G proteins and use similar transduction systems. CB1-R is coupled to Gαi and Gαo proteins which inhibit protein kinase A (PKA). PKA mediates most of the effects of CB1-R by phosphorylating and regulating the function of a wide array of cellular substrates such as voltage-gated K+, Na+, and Ca2+ channels (Fig. 2), ionotropic glutamate, and GABA receptors and transcription factors. Noteworthy, the modulation of ion channels following CB1-R activation can either depend on its coupling with Gαolf protein or be independent of cAMP/PKA signaling. A good example is the gating of G protein-coupled inwardly rectifying K+ channels (GIRKs) at presynaptic level in the nucleus accumbens (NAc) [ref. 36]. Also, CB1-R can modulate intracellular Ca2+ levels and regulate ligand- and voltage-gated ion channels through the liberation of the Gβγ subunit upon receptor activation.
CB1-R is mainly expressed in presynaptic terminals in homo- and heterodimeric structures; however, it can also be present at postsynaptic level where it can form heterodimers with other GPCRs including DA receptors [ref. 37–ref. 39]. Depending on the DA-R bound, CB1-R can contribute in different ways to the modulation of both excitatory and inhibitory synaptic functions as well as to the decrease of neurotransmitter release in the basal ganglia circuitry (for rev., see [ref. 40]).
Similar to CB1-R, CB2-R is coupled to Gαi and Gαo proteins and its stimulation results in PKA inhibition. Although its role in the CNS is still controversial, some studies have shown its involvement in modulation of ion channels and DA levels [ref. 41–ref. 44].
CB1-Rs and CB2-Rs can also activate other intracellular signaling cascades, including the mitogen-activated protein kinase (MAPK) cascade, the phosphatidylinositol 3-kinase (PI3K) cascade, and nitric oxide production [ref. 45].
TRPV1 channels are characterized by weak voltage sensitivity and nonselective permeability to monovalent and divalent cations including Mg2+, Ca2+, and Na+. Their activation has been shown to modulate synaptic transmission and neurotransmitter release [ref. 46–ref. 48].
Dual regulation of DA/eCB neurotransmission
DA neurons are fine-tuned by eCBs, which can regulate DA neurotransmission at midbrain cell bodies, at DA axon terminal endings or by acting on DA neuron effector sites in the striatum (Fig. 1).
The modulation of DA release by eCBs in midbrain neurons occurs via different mechanisms, according to the target receptor activated.
DA neurons in midbrain do not express CB1-Rs [ref. 49]; however, many other neuronal populations connected with DAergic cells present CB1-R and are modulated by eCBs. Thus, eCBs indirectly control DA cells through modulation of local circuitry, facilitating or suppressing DAergic neuron activity [ref. 40, ref. 50, ref. 51], primarily through 2-AG (Fig. 1).
In contrast to CB1-Rs which are not expressed by midbrain DAergic neurons, CB2-Rs are present on cell bodies [ref. 29], and their activation by exogenous agonists inhibits DAergic neuronal firing [ref. 52, ref. 53] (Fig. 1); however, whether endogenous molecules driving CB2-R signaling act in a similar way to reduce DA release has not been established yet.
In the SN, an increase in the excitatory neurotransmission through a tonic modulation of the glutamatergic synaptic inputs onto DAergic neurons by the eCB AEA has been measured. Such enhancement occurs after activation of TRPV1, and the effect of AEA on DAergic transmission results in an increase in DA levels [ref. 46, ref. 47] (Fig. 1). Of note, the modulation of DA release by AEA occurs also in response to peripheral noxious stimulation in freely moving animals [ref. 47]. While TRPV1 channels have been found also on DAergic cells, it is unclear whether their activation has a direct action at postsynaptic level.
At striatal level, DA neurotransmission is modulated locally at presynaptic axonal release sites by several afferent inputs (i.e., cholinergic and glutamatergic inputs). In such context, also eCBs participate in the local modulation of axonal DA release. Specifically, it has been observed that CB1-R activation inhibits the evoked release of DA [ref. 54] (Fig. 1). The exact cellular localization of CB1-Rs responsible for such inhibition is not clear yet; however, polysynaptic and indirect mechanisms have been implicated in the decrease of DA levels.
Interestingly, also CB2-R activation on DA terminals seems responsible for causing inhibition of striatal DA release [ref. 55]. This phenomenon has been observed following exogenous activation; whether endogenous agonists do the same influencing DA tone and DA-dependent behavior is not established yet (Fig. 1).
Finally, the localization of TRPV1 channels at striatal level [ref. 34] implicates a possible involvement of these receptors in the modulation of the DA tone. These channels have already been demonstrated to be involved in the control of GABA synapses and in the regulation of excitatory transmission [ref. 48, ref. 56], thus, it is plausible that their activation indirectly may impact DAergic transmission.
The regulation of DA neurotransmission by eCBs at striatal sites occurs also at the level of the regulatory mechanisms that control the concentration of DA at synaptic cleft. Specifically, AEA increases extracellular DA levels [ref. 57] via inhibition of DA transporter (DAT) activity [ref. 58] (Fig. 1). The inhibition of DA uptake by eCBs and other cannabinoids has been observed in native tissue but also in heterologous expression systems [ref. 59–ref. 61]. Collectively, these data clearly show that eCBs control DA neurotransmission at multiple sites of action within the basal ganglia.
The mutual interaction between eCBs and DA is not only dependent on the ability of eCBs to modulate DA release but also vice versa. Indeed, DAergic neurons can themselves produce eCBs from their somata and dendrites [ref. 62], thereby modulating the retrograde signaling. The release of eCBs from DAergic neurons is consequent to an increase in the firing of DAergic cells which causes the mobilization of 2-AG (Fig. 1). Moreover, it has been observed that DA, following binding to D2-R, can promote the postsynaptic release of eCBs from striatal spiny projection neurons (SPNs) which, in turn, influence transmitter exocytosis through the activation of presynaptic GPCRs [ref. 63].
In general, the stimulation of D2-Rs by DA causes increases of striatal eCB levels due to an increased synthesis and an inhibited degradation [ref. 64, ref. 65]. Interestingly, D2-R and CB1-R are co-expressed, and CB1-R activation by AEA can enhance the effects of D2-R activation [ref. 66, ref. 67], thereby amplifying e-CB production.
eCB action independent of CB-Rs: focus on ion channels
Endogenous cannabinoids, as well as exogenous and synthetic congeners, have been shown to target other molecular substrates in addition to the classical CB-Rs. In particular, these molecules can modify the gating of several classes of ion channels and affect neuronal excitability and neurotransmitter release.
In fact, both eCBs and cannabidiol (CBD), a phytocannabinoid extracted from cannabis sativa, are able to interact with voltage-gated sodium channels (Nav). Specifically, 2-AG is able to induce channel block and inhibit Na+ currents or induce changes in biophysical properties of activation/inactivation of Nav channels, thereby affecting their function [ref. 68] (Fig. 2). Similarly, CBD has inhibitory effects on Na+ conductance without specificity to any of Nav subfamily [ref. 69–ref. 73] and can directly interact with Nav channels to inhibit their activity [ref. 74, ref. 75] (Fig. 2). Interestingly, CBD binds the channel in more than a single site, and the binding sites are not positioned to physically occlude the pore. Moreover, the inhibition is allosteric and determines a stabilization of the inactivated state.
In addition to Nav channels, cannabinoids have been reported to interact also with K+ channels decreasing the peak currents [ref. 76]. In this regard, it has been shown that the inhibition of native A-type K+ channels by 2-AG in DAergic neurons causes an increase in their excitability, inducing a transition from tonic firing to burst discharge and thus affecting DA release [ref. 77] (Fig. 2).
With these results in mind, and considering the close relationship between cannabinoids and DAergic system, it can be speculated that CBD and eCBs could exert their analgesic effects, at least in part, through a direct action on ion channels whose modulation causes changes in neuronal excitability and in DA release.
Endocannabinoid-DA interaction: pathological imbalance and implications for pain
eCB-DA imbalance in PD patients
Clinical studies support the evidence for a reciprocal interplay between DA and eCBs. In individuals with PD, the levels of eCBs measured in the cerebrospinal fluid (CSF) were significantly higher as compared to control subjects. This finding was observed either in drug-naive, recently diagnosed subjects, or in patients with a longer disease duration, undergoing drug washout [ref. 15]. Of interest, eCBs levels did not differ in these two subgroups of patients, indicating that elevations of CSF eCBs are independent of disease duration. However, in patients undergoing DAergic treatment, a normal eCB content was measured, not different from controls, suggesting that an adaptive, compensatory mechanism occurs during DA denervation. Evidence for a similar, adaptive change emerges from binding studies performed in post-mortem samples from PD patients. Measurement of CB1-R binding and its coupling to G proteins revealed higher stimulation by CB1 receptor agonists of [35S] GTPcS binding both in the caudate and putamen, along with an enhanced CB1 receptor binding [ref. 78]. Overall, the reported changes in eCB levels with or without DA replacement suggest that the DA/eCB interplay is highly dynamic.
The general view of this dual interaction is further confirmed by additional clinical hints and supported by preclinical findings (see below). Indeed, while DA supplementation is known to improve dramatically the motor performance observed in hypokinetic disorders, such as PD, the therapeutic use of CBs has been approved for hyperkinetic disorders, such as Tourette’s syndrome and other tic disorders [ref. 79, ref. 80], further supporting the notion of a dual interplay between DA and eCB system.
eCB-DA imbalance in models of PD
The close interaction between DA and eCBs is clearly evident not only in PD patients but also in experimental PD models. In fact, in multiple toxin-induced rodent and nonhuman primate PD models, as well as in genetic rodent PD models, eCB system is reorganized with changes measurable at level of concentration, clearance, and activity.
In particular, in 6-OHDA-denervated rats and in reserpine-treated rats, enhanced levels of eCBs have been measured in the basal ganglia circuitry [ref. 12, ref. 81]. However, the observed enhancement is a consequence of a decreased cleavage and not dependent on an increased synthesis [ref. 12]. In line with rodent data, both AEA and 2-AG levels were increased in the caudate of non-human primates treated with MPTP [ref. 82].
In contrast to toxin-induced PD models, eCB levels were unchanged in a genetic mouse model of PD carrying the recessive mutation in the PTEN-induced putative kinase 1 (PINK1) gene responsible for early-onset PD [ref. 83]. However, in spite of unaltered eCB levels, the synaptic responses to CB1-R activation were impaired, and a significant decrease of binding capacity of CB1R agonists was found in PINK1 knockout mice, again supporting the notion of a mutual interplay between DA and eCBs.
Although some differences exist among the various PD experimental models regarding the level of eCBs altered following DA depletion, the changes observed in the eCB system were in all the models reverted after DAergic therapy with levodopa or a D2-R agonist [ref. 13, ref. 15, ref. 82, ref. 83] suggesting that the upregulation of the eCB system consequent to DA depletion is an adaptive, dynamic homeostatic mechanism aimed at compensating the effects of DA loss in the basal ganglia.
Such interplay is further corroborated by a number of preclinical findings. CB-R agonists potentiated reserpine-induced hypokinesia [ref. 84] and DA receptor antagonist-induced catalepsy [ref. 85]. Conversely, they were able to reduce quinpirole-induced hyperlocomotion [ref. 86] and amphetamine-induced hyperactivity (for rev., see [ref. 87]).
Effects of soluble products of inflammation on eCB-DA signaling in PD
Neuroinflammation is a well-established feature of PD, and a growing body of evidence suggests that both peripheral and central immune dysregulation can upregulate cytokines such as IL-1β, IL-6, and TNF-α [ref. 88], which participate in the development and persistence of pain states during PD. The increased levels of these cytokines in PD patients’ serum and CSF can affect different neural networks and influence synaptic transmission with significant changes in eCB-DA signaling as well as behavioral implications. Intriguingly, a modulatory effect on both excitatory and inhibitory neurotransmissions at striatal level has been reported for IL-1β with involvement of eCB and DAergic systems. As a matter of fact, the exposure to this cytokine causes enhancement of spontaneous excitatory currents via the activation of presynaptic TRPV1 [ref. 89] and a reduced sensitivity of CB1-R [ref. 90]. In addition, IL-1β reduces GABAergic inhibition inducing SPN hyperactivation [ref. 91]. Analogously, TRPV1 and CB1-R seem to be implicated in such inhibitory modulation [ref. 92, ref. 93]. The striatal excitatory/inhibitory imbalance induced by IL-1β is coupled to an alteration of DAergic transmission as suggested by the finding that evoked axonal DA release is reduced when IL-1β is upregulated and restored following administration of IL-1 receptor antagonist (IL-1ra) [ref. 94]. Moreover, IL-1ra application also rescues striatal CB1-R sensitivity and DA-dependent behavior [ref. 95]. Although not exhaustive, these observations clearly confirm the strong connection between eCB and DAergic system and their sensitivity to inflammatory mediators suggesting a contribution for these molecules in causing the eCB-DA imbalance. In addition to IL-1β, other pro-inflammatory cytokines have been proven to affect DA neurotransmission [ref. 96]; however, whether this impairment also exerts influence on eCB system and has implications for pain process is still unknown and deserves to be investigated.
eCB-DA imbalance during pain
The role of DA in pain can be better appreciated in conditions of DA loss such as in PD. Indeed, compelling evidence indicates that up to 80% of individuals with PD manifest pain during the disease course [ref. 17]. The degeneration of nigrostriatal DA pathway in PD patients causes hyperalgesia [ref. 97], and the administration of levodopa induces an increase in the pain threshold [ref. 98, ref. 99]. Intriguingly, toxin-induced rodent models of PD (i.e., MPTP and 6-OHDA) have provided a valuable contribution in understanding the role of DA in pain process. Similar to humans, DA-depleted animals show an increased nociception and decreased nociceptive threshold [ref. 10, ref. 100–ref. 104] suggesting that the loss of DA in the basal ganglia is involved in the reduction of pain threshold [ref. 105]. Of note, the elevation of striatal DA following electrical stimulation of the SN or the direct striatal administration of apomorphine, a D1/D2-R agonist, induces pain inhibition [ref. 106]. Similarly, systemic administration of DA agonists determines hypoalgesia by modulating D2 receptors in dorsolateral striatum [ref. 107]. In spite of such evidence, both in humans and in models of PD, replacement with DA therapy does not promote recovery from pain, thereby suggesting that other neurotransmitters may play a role in the pain signaling process.
Although the great therapeutic potential of eCB system in the modulation of pain has been widely recognized [ref. 108, ref. 109], the involvement of DA/eCB system within basal ganglia circuitry in chronic pain has been less extensively explored [ref. 110]. eCBs are elevated at various sites in nociceptive pathways in chronic pain [ref. 19] highlighting their role as endogenous analgesics. Based on the existence of a concurrent decrease in DA concentrations observed during pain, the increase in eCB levels might compensate DA decrease. The imbalance in eCB-DA system during pain condition is somehow reminiscent of the alteration observed during PD. However, eCB effects on nociception and pain following DA depletion have been scarcely investigated. Also, whether DAergic drugs are able to restore resting levels of eCBs as in animal models of PD [ref. 13] needs to be clarified.
Recently, Nascimento and colleagues [ref. 111] analyzed the effect of CBD on nociception in experimental PD, highlighting the ability of this drug to exert antinociception via activation of CB1-R. Moreover, the authors suggest that the analgesic effect induced by CBD is also dependent on an increase in AEA which likely potentiates the activation of CB1. An elevation in AEA levels has been found in 6-OHDA-denervated animals and is dependent on a decreased cleavage [ref. 12]; thus, it is plausible that this drug may cause an increase in DA levels acting at the same sites used by eCBs to directly modulate somatodendritic or axonal DA release in the basal ganglia.
Conclusions
Although the role of DA in pain perception in PD has not been fully elucidated, it is now evident that an altered DA neurotransmission correlates with impaired eCB levels. Hence, the eCB system would represent, in principle, a promising target for the treatment of pain in PD. Additional work is needed to clarify the functional meaning of these interactions, in order to gain a better understanding of the mechanisms through which eCBs affect DA neurotransmission, which may translate into novel opportunities for improving pain treatment.
References
- LA Matsuda, SJ Lolait, MJ Brownstein, AC Young, TI Bonner. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature, 1990. [DOI | PubMed]
- S Munro, KL Thomas, M Abu-Shaar. Molecular characterization of a peripheral receptor for cannabinoids. Nature, 1993. [DOI | PubMed]
- V Di Marzo, A Fontana, H Cadas, S Schinelli, G Cimino, JC Schwartz, D Piomelli. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature, 1994. [DOI | PubMed]
- M Melis, M Pistis. Endocannabinoid signaling in midbrain dopamine neurons: more than physiology?. Curr Neuropharmacol, 2007. [DOI | PubMed]
- SG Dennis, R Melzack. Effects of cholinergic and dopaminergic agents on pain and morphine analgesia measured by three pain tests. Exp Neurol, 1983. [DOI | PubMed]
- NE Saadé, SF Atweh, NB Bahuth, SJ Jabbur. Augmentation of nociceptive reflexes and chronic deafferentation pain by chemical lesions of either dopaminergic terminals or midbrain dopaminergic neurons. Brain Res, 1997. [DOI | PubMed]
- W Dieb, O Ouachikh, F Durif, A Hafidi. Lesion of the dopaminergic nigrostriatal pathway induces trigeminal dynamic mechanical allodynia. Brain Behav, 2014. [DOI | PubMed]
- M Ogata, K Noda, H Akita, H Ishibashi. Characterization of nociceptive response to chemical, mechanical, and thermal stimuli in adolescent rats with neonatal dopamine depletion. Neuroscience, 2015. [DOI | PubMed]
- AMW Taylor, S Beccker, P Schweinhardt, C Cahill. Mesolimbic dopamine signaling in acute and chronic pain: implications for motivation, analgesia, and addiction. Pain, 2016. [DOI | PubMed]
- KA Charles, F Naudet, R Bouali-Banazzouz, M Landry, P De Deurwaerdère, P Fossat, A Banazzouz. Alteration of nociceptive integration in the spinal cord of a rat model of Parkinson’s disease. Mov Disord, 2018. [DOI | PubMed]
- B Ziółkowska. Corticostriatal glutamatergic transmission in chronic pain. Brain Sci, 2021. [DOI | PubMed]
- P Gubellini, B Picconi, M Bari, N Battista, P Calabresi, D Centonze, G Bernardi, A Finazzi-Agrò, M Maccarone. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci, 2002. [DOI | PubMed]
- M Maccarrone, P Gubellini, M Bari, B Picconi, N Battista, D Centonze, G Bernardi, A Finazzi-Agrò, P Calabresi. Levodopa treatment reverses endocannabinod system abnormalities in experimental parkinsonism. J Neurochem, 2003. [DOI | PubMed]
- A Pisani, F Fezza, S Galati, N Battista, S Napolitano, A Finazzi-Agrò, G Bernardi, L Brusa, M Pirantozzi, P Stanzione, M Maccarrone. High endogenous cannabinoid levels in the cerebrospinal fluid of untreated Parkinson’s disease patients. Ann Neurol, 2005. [DOI | PubMed]
- V Pisani, G Madeo, A Tassone, G Sciamanna, M Maccarrone, P Stanzione, A Pisani. Homeostatic changes of the endocannabinoid system in Parkinson’s disease. Mov Disord, 2011. [DOI | PubMed]
- A Nebe, G Ebersbach. Pain intensity on and off levodopa in patients with Parkinson’s disease. Mov Disord, 2009. [DOI | PubMed]
- P Ghosh, P Imbriani, N Caputi, S Natoli, T Schirinzi, G Di Lazzaro, L Covington, AD Sparks, Y Salnikova, K Rukaniva, KR Chaudhuri, A Pisani. A dual centre study of pain in Parkinson’s disease and its relationship with other non-motor symptoms. J Parkinsons Dis, 2020. [DOI | PubMed]
- R Maldonado, JE Baños, D Cabañero. The endocannabinoid system and neuropathic pain. Pain, 2016. [DOI | PubMed]
- SG Woodhams, V Chapman, DP Finn, AG Hohmann, V Neugebauer. The cannabinoid system and pain. Neuropharmacology, 2017. [DOI | PubMed]
- G Donvito, SR Nass, JL Wilkerson, ZA Curry, LD Schurman, SG Kinsey, AH Lichtman. The endogenous cannabinoid system: a budding source of targets for treating inflammatory and neuropathic pain. Neuropsychopharmacology, 2018. [DOI | PubMed]
- L Cristino, T Bisogno, V Di Marzo. Cannabinoids and the expanded endocannabinoids system in neurological disorders. Nat Rev Neurol, 2020. [DOI | PubMed]
- DP Finn, S Haroutounian, AG Hohmann, E Krane, N Soliman, ASC Rice. Cannabinoids, the endocannabinoid system, and pain: a review of preclinical studies. Pain, 2021. [DOI | PubMed]
- G Gerdeman, DM Lovinger. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol, 2001. [DOI | PubMed]
- A Köfalvi, RJ Rodrigues, C Ledent, K Mackie, E Sylvester Vizi, RA Cunha, B Sperlágh. Involvement of cannabinoid receptors in the regulation of neurotransmitter release in the rodent striatum: a combined immunochemical and pharmacological analysis. J Neurosci, 2005. [DOI | PubMed]
- M Herkenham, AB Lynn, BR de Costa, EK Richfield. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res, 1991. [DOI | PubMed]
- K Tsou, S Brown, MC Sañudo-Peña, K Mackie, JM Walker. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience, 1998. [DOI | PubMed]
- MC Sañudo-Peña, JM Walker. Role of the subthalamic nucleus in cannabinoid actions in the substantia nigra of the rat. J Neurophysiol, 1997. [DOI | PubMed]
- S Sierra, N Luquin, AJ Rico, V Gómez-Bautista, E Roda, IG Dopeso-Reyes, A Vázquez, E Martínez-Pinilla, JL Labandeira-García, R Franco, JL Lanciego. Detection of cannabinoid receptors CB1 and CB2 within basal ganglia output neurons in macaques: changes following experimental parkinsonism. Brain Struct Funct, 2015. [DOI | PubMed]
- HY Zhang, GH Bi, X Li, J Li, H Qu, SJ Zhang, CY Li, ES Onaivi, EL Gardner, ZX Xi, QR Liu. Species differences in cannabinoid receptor 2 and receptor responses to cocaine self-administration in mice and rats. Neuropsychopharmacology, 2015. [DOI | PubMed]
- R Sánchez-Zavaleta, H Cortés, JA Avalos-Fuentes, U García, J Segovia Vila, D Erlij, B Florán. Presynaptic cannabinoid CB2 receptors modulate [3 H]-glutamate release at subthalamo-nigral terminals of the rat. Synapse, 2018. [DOI | PubMed]
- A Canseco-Alba, N Schanz, B Sanabria, J Zhao, Z Lin, QR Liu, ES Onaivi. Behavioral effects of psychostimulants in mutant mice with cell-type specific deletion of CB2 cannabinoid receptors in dopamine neurons. Behav Brain Res, 2019. [DOI | PubMed]
- CJ Jordan, ZX Xi. Progress in brain cannabinoid CB2 receptor research: from genes to behavior. Neurosci Biobehav Rev, 2019. [DOI | PubMed]
- E Mezey, ZE Tóth, DN Cortright, MK Arzubi, JE Krause, R Elde, A Guo, PM Blumberg, A Szallasi. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci U S A, 2000. [DOI | PubMed]
- L Cristino, L de Petrocellis, G Pryce, D Baker, V Guglielmotti, V Di Marzo. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience, 2006. [DOI | PubMed]
- S Marinelli, V Di Marzo, F Florenzano, F Fezza, MT Viscomi, M van der Stelt, G Bernardi, M Molinari, M Maccarrone, NB Mercuri. N-arachidonoyl-dopamine tunes synaptic transmission onto dopaminergic neurons by activating both cannabinoid and vanilloid receptors. Neuropsychopharmacology, 2007. [DOI | PubMed]
- D Robbe, G Alonso, F Duchamp, J Bockaert, OJ Manzoni. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci, 2001. [DOI | PubMed]
- JA Przybyla, VJ Watts. Ligand-induced regulation and localization of cannabinoid CB1 and dopamine D2L receptor heterodimers. J Pharmacol Exp Ther, 2010. [DOI | PubMed]
- SS Khan, FJS Lee. Delineation of domains within the cannabinoid CB1 and dopamine D2 receptors that mediate the formation of the heterodimer complex. J Mol Neurosci, 2014. [DOI | PubMed]
- AM Bagher, RB Laprairie, ME Kelly, EM Denovan-Wright. Antagonism of dopamine receptor 2 long affects cannabinoid receptor 1 signaling in a cell culture model of striatal medium spiny projection neuros. Mol Pharmacol, 2016. [DOI | PubMed]
- C Garcia, C Palomo-Garo, Y Gómez-Gálvez, J Fernández-Ruiz. Cannabinoid-dopamine interactions in physiology and physiopathology of the basal ganglia. Br J Pharmacol, 2016. [DOI | PubMed]
- DG Demuth, A Molleman. Cannabinoid signaling. Life Sci, 2006. [DOI | PubMed]
- ZX Xi, XQ Peng, X Li, R Song, HY Zhang, QR Liu, HJ Yang, GH Bi, J Li, EL Gardner. Brain cannabinoid CB2 receptors modulate cocaine’s actions in mice. Nat Neurosci, 2011. [DOI | PubMed]
- Z Ma, F Gao, B Larsen, M Gao, Z Luo, D Chen, X Ma, S Qiu, Y Zhou, J Xie, ZX Xi, J Wu. Mechanisms of cannabinoid CB2 receptor-mediated reduction of dopamine neuronal excitability in mouse ventral tegmental area. EBioMedicine, 2019. [DOI | PubMed]
- E Galaj, GH Bi, HJ Yang, ZX Xi. Cannabidiol attenuates the rewarding effects of cocaine in rats by CB2, 5-HT1A and TRPV1 receptor mechanisms. Neuropharmacology, 2020. [DOI | PubMed]
- A Tadijan, I Vlašić, J Vlainić, D Dikić, N Oršlić, M Jazvinšćak Jembrek. Intracellular molecular targets and signaling pathways involved in antioxidative and neuroprotective effects of cannabinoids in neurodegenerative conditions. Antioxidant (Basel), 2022. [DOI]
- S Marinelli, V Di Marzo, N Berretta, I Matias, M Maccarrone, G Bernardi, NB Mercuri. Presynaptic facilitation of glutamatergic synapses to dopaminergic neurons of the rat substantia nigra by endogenous stimulation of vanilloid receptors. J Neurosci, 2003. [DOI | PubMed]
- S Marinelli, T Pascucci, G Bernardi, S Puglisi-Allegra, NB Mercuri. Activation of TRPV1 in the VTA excites dopaminergic neurons and increases chemical- and noxious-induced dopamine release in the nucleus accumbens. Neuropsychopharmacology, 2005. [DOI | PubMed]
- M Maccarrone, S Rossi, M Bari, V De Chiara, F Fezza, A Musella, V Gasperi, C Prosperetti, G Bernardi, A Finazzi-Agrò, BF Cravatt, D Centonze. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat Neurosci, 2008. [DOI | PubMed]
- MD Julian, AB Martin, B Cuellar, F Rodriguez De Fonseca, M Navarro, R Moratalla, LM Garcia-Segura. Neuroanatomical relationship between type 1 cannabinoid receptors and dopaminergic systems in the rat basal ganglia. Neuroscience, 2003. [DOI | PubMed]
- M Melis, S Perra, AL Muntoni, G Pillolla, B Lutz, G Marsicano, V Di Marzo, GL Gessa, M Pistis. Prefrontal cortex stimulation induces 2-arachidonoyl-glycerol-mediated suppression of excitation in dopamine neurons. J Neurosci, 2004. [DOI | PubMed]
- B Szabo, S Siemes, I Wallmichrath. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci, 2002. [DOI | PubMed]
- HY Zhang, M Gao, QR Liu, GH Bi, X Li, HJ Yang, EL Gardner, J Wu, ZX Xi. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc Natl Acad Sci U S A, 2014. [DOI | PubMed]
- HY Zhang, M Gao, H Shen, GH Bi, HJ Yang, QR Liu, J Wu, EL Gardner, A Bonci, ZX Xi. Expression of functional cannabinoid CB2 receptor in VTA dopamine neurons in rats. Addict Biol, 2017. [DOI | PubMed]
- Z Sidló, PH Reggio, ME Rice. Inhibition of striatal dopamine release by CB1 receptor activation requires nonsynaptic communication involving GABA, H2O2, and KATP channels. Neurochem Int, 2008. [DOI | PubMed]
- DJ Foster, JM Wilson, DH Remke, MS Mahmood, MJ Uddin, J Wess, S Patel, LJ Marnett, CM Niswender, CK Jones, Z Xiang, CW Lindsley, JM Rook, PJ Conn. Antipsychotic-like effects of M4 positive allosteric modulators are mediated by CB2 receptor-dependent inhibition of dopamine release. Neuron, 2016. [DOI | PubMed]
- A Musella, V De Chiara, S Rossi, C Prosperetti, G Bernardi, M Maccarrone, D Centonze. TRPV1 channels facilitate glutamate transmission in the striatum. Mol Cell Neurosci, 2009. [DOI | PubMed]
- M Solinas, Z Justinova, SR Goldberg, G Tanda. Anandamide administration alone and after inhibition of fatty acid amide hydrolase (FAAH) increases dopamine levels in the nucleus accumbens shell in rats. J Neurochem, 2006. [DOI | PubMed]
- M Oz, V Jaligam, S Galadari, G Petroianu, YM Shuba, TS Shippenberg. The endogenous cannabinoid, anandamide, inhibits dopamine transporter function by a receptor-independent mechanism. J Neurochem, 2010. [DOI | PubMed]
- N Chen, M Appell, JL Berfield, ME Reith. Inhibition by arachidonic acid and other fatty acids of dopamine uptake at the human dopamine transporter. Eur J Pharmacol, 2003. [DOI | PubMed]
- M Steffens, TJ Feuerstein. Receptor-independent depression of DA and 5-HT uptake by cannabinoids in rat neocortex–involvement of Na(+)/K(+)-ATPase. Neurochem Int, 2004. [DOI | PubMed]
- DA Price, WA Owens, GG Gould, A Frazer, JL Roberts, LC Daws, A Giuffrida. CB1-independent inhibition of dopamine transporter activity by cannabinoids in mouse dorsal striatum. J Neurochem, 2007. [DOI | PubMed]
- V Seutin. Dopaminergic neurones: much more than dopamine?. Br J Pharmacol, 2005. [DOI | PubMed]
- W Wang, D Dever, J Lowe, GP Storey, A Bhansali, EK Eck, I Nitulescu, J Weimer, NS Bamford. Regulation of prefrontal excitatory neurotransmission by dopamine in the nucleus accumbens core. J Physiol, 2012. [DOI | PubMed]
- A Giuffrida, LH Parsons, TM Kerr, F Rodríguez de Fonseca, M Navarro, D Piomelli. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci, 1999. [DOI | PubMed]
- M Beltramo, FR de Fonseca, M Navarro, A Calignano, MA Gorriti, G Grammatikopoulos, AG Sadile, A Giuffrida, D Piomelli. Reversal of dopamine D(2) receptor responses by an anandamide transport inhibitor. J Neurosci, 2000. [DOI | PubMed]
- R Alonso, B Voutsinos, M Fournier, C Labie, R Steinberg, J Souilhac, G Le Fur, P Soubrié. Blockade of cannabinoid receptors by SR141716 selectively increases Fos expression in rat mesocorticolimbic areas via reduced dopamine D2 function. Neuroscience, 1999. [DOI | PubMed]
- JP Meschler, TJ Conley, AC Howlett. Cannabinoid and dopamine interaction in rodent brain: effects on locomotor activity. Pharmacol Biochem Behav, 2000. [DOI | PubMed]
- Y Okada, KG Imendra, T Miyazaki, H Hotokezaka, R Fujiyama, JL Zeredo, T Miyamoto, K Toda. Biophysical properties of voltage-gated Na+ channels in frog parathyroid cells and their modulation by cannabinoids. J Exp Biol, 2005. [DOI | PubMed]
- AJ Hill, NA Jones, I Smith, CL Hill, CM Williams, GJ Stephens, BJ Whalley. Voltage-gated sodium (NaV) channel blockade by plant cannabinoids does not confer anticonvulsant effects per se. Neurosci Lett, 2014. [DOI | PubMed]
- RR Patel, C Barbosa, T Brustovetsky, N Brustovetsky, T Cummins. Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain, 2016. [DOI | PubMed]
- MR Ghovanloo, NG Shuart, J Mezeyova, RA Dean, PC Ruben, SJ Goodchild. Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J Biol Chem, 2018. [DOI | PubMed]
- ER Mason, TR Cummins. Differential inhibition of human Nav1.2 resurgent and persistent sodium currents by cannabidiol and GS967. Int J Mol Sci, 2020. [DOI | PubMed]
- CJ Milligan, LL Anderson, MT Bowen, SD Banister, IS McGregor, JC Arnold, S Petrou. A nutraceutical product, extracted from Cannabis sativa, modulates voltage-gated sodium channel function. J Cannabis Res, 2022. [DOI | PubMed]
- LG Sait, A Sula, MR Ghovanloo, D Hollingworth, PC Ruben, BA Wallace. Cannabidiol interactions with voltage-gated sodium channels. Elife, 2020. [DOI | PubMed]
- J Huang, X Fan, X Jin, S Jo, HB Zhang, A Fujita, BP Bean, N Yan. Cannabidiol inhibits Nav channels through two distinct binding sites. Nat Commun, 2023. [DOI | PubMed]
- AR Watkins. Cannabinoid interactions with ion channels and receptors. Channels (Austin), 2019. [DOI | PubMed]
- SC Gantz, BP Bean. Cell-autonomous excitation of midbrain dopamine neurons by endocannabinoid-dependent lipid signaling. Neuron, 2017. [DOI | PubMed]
- I Lastres-Becker, M Cebeira, ML de Ceballos, BY Zeng, P Jenner, JA Ramos, JJ Fernández-Ruiz. Increased cannabinoid CB1 receptor binding and activation of GTP-binding proteins in the basal ganglia of patients with Parkinson’s syndrome and of MPTP-treated marmosets. Eur J Neurosci, 2001. [DOI | PubMed]
- BS Koppel. Cannabis in the treatment of dystonia, dyskinesias, and tics. Neurotherapeutics, 2015. [DOI | PubMed]
- V Roessner, H Eichele, JS Stern, L Skov, R Rizzo, NM Debes, P Nagy, AE Cavanna, C Termine, C Ganos, A Münchau, N Szejko, D Cath, KR Müller-Vahl, C Verdellen, A Hartmann, A Rothenberger, PJ Hoekstra, KJ Plessen. European clinical guidelines for Tourette syndrome and other tic disorders-version 2.0. Part III: pharmacological treatment. Eur Child Adolesc Psychiatry, 2022. [DOI | PubMed]
- V Di Marzo, MP Hill, T Bisogno, AR Crossman, JM Brotchie. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson’s disease. FASEB J, 2000. [DOI | PubMed]
- M van der Stelt, SH Fox, M Hill, AR Crossman, S Petrosino, V Di Marzo, JM Brotchie. A role for endocannabinoids in the generation of parkinsonism and levodopa-induced dyskinesia in MPTP-lesioned non-human primate models of Parkinson’s disease. FASEB J, 2005. [DOI | PubMed]
- G Madeo, T Schirinzi, M Maltese, G Martella, C Rapino, F Fezza, N Mastrangelo, P Bonsi, M Maccarrone, A Pisani. Dopamine-dependent CB1 receptor dysfunction at corticostriatal synapses in homozygous PINK1 knockout mice. Neuropharmacology, 2016. [DOI | PubMed]
- DE Moss, SB McMaster, J Rogers. Tetrahydrocannabinol potentiates reserpine-inuced hypokinesia. Pharmacol Biochem Behav, 1981. [DOI | PubMed]
- JJ Anderson, AM Kask, TN Chase. Effects of cannabinoi receptor stimulation and blockade on catalepsy produced by dopamine receptor antagonists. Eur J Pharmacol, 1996. [DOI | PubMed]
- D Marcellino, P Carriba, M Filip, A Borgkvist, M Frankowska, I Bellido, S Tanganelli, CE Müller, G Fisone, C Lluis, LF Agnati, R Franco, K Fuxe. Antagonistic cannabinoid CB1/dopamine D2 receptor interactions in striatal CB1/D2 heteromers. A combined neurochemical and behavioral analysis. Neuropharmacology, 2008. [DOI | PubMed]
- J Fernández-Ruiz. The endocannabinoid system as a target for the treatment of motor dysfunction. Br J Pharmacol, 2009. [DOI | PubMed]
- MN Karpenko, AA Vasilishina, EA Gromova, ZM Muruzheva, A Bernadotte. Interleukin-1beta, interleukin-1 receptor antagonist, interleukin-6, interleukin-10, and tumor necrosis factor-alpha levels in CSF and serum in relation to the clinical variety of Parkinson’s disease. Cell Immunol, 2018. [DOI | PubMed]
- S Rossi, R Furlan, V De Chiara, C Motta, V Studer, F Mori, A Musella, A Bergami, L Muzio, G Bernardi, L Battistini, G Martino, D Centonze. Interleukin-1β causes synaptic hyperexcitability in multiple sclerosis. Ann Neurol, 2012. [DOI | PubMed]
- V De Chiara, C Motta, S Rossi, V Studer, F Barbieri, D Lauro, G Bernardi, D Centonze. Interleukin-1β alters the sensitivity of cannabinoid CB1 receptors controlling glutamate transmission in the striatum. Neuroscience, 2013. [DOI | PubMed]
- S Rossi, V Studer, C Motta, V De Chiara, F Barbieri, G Bernardi, D Centonze. Inflammation inhibits GABA transmission in multiple sclerosis. Mult Scler, 2012. [DOI | PubMed]
- G Musumeci, G Grasselli, S Rossi, V De Chiara, A Musella, C Motta, V Studer, G Bernardi, N Haji, H Sepman, D Fresegna, M Maccarrone, G Mandolesi, D Centonze. Transient receptor potential vanilloid 1 channels modulate the synaptic effects of TNF-α and of IL-1β in experimental autoimmune encephalomyelitis. Neurobiol Dis, 2011. [DOI | PubMed]
- S Rossi, L Sacchetti, F Napolitano, V De Chiara, C Motta, V Studer, A Musella, F Barbieri, M Bari, G Bernardi, M Maccarrone, A Usiello, D Centonze. Interleukin-1b causes anxiety by interacting with the endocannabinoid system. J Neurosci, 2012. [DOI | PubMed]
- A Gentile, D Fresegna, M Federici, A Musella, FR Rizzo, H Sepman, S Bullitta, F De Vito, N Haji, S Rossi, NB Mercuri, A Usiello, G Mandolesi, D Centonze. Dopaminergic dysfunction is associated with IL-1β-dependent mood alterations in experimental autoimmune encephalomyelitis. Neurobiol Dis, 2015. [DOI | PubMed]
- A Gentile, D Fresegna, A Musella, H Sepman, S Bullitta, F De Vito, R Fantozzi, A Usiello, M Maccarrone, NB Mercuri, B Lutz, G Mandolesi, D Centonze. Interaction between interleukin1b and type-1 cannabinoid receptor is involved in anxiety-like behavior in experimental autoimmune encephalomyelitis. J Neuroinflammation, 2016. [DOI | PubMed]
- M Mancini, S Natoli, F Gardoni, M Di Luca, A Pisani. Dopamine transmission imbalance in neuroinflammation: perspectives on long-term COVID-19. Int J Mol Sci, 2023. [DOI | PubMed]
- A Fil, R Cano-de-la-Cuerda, E Muñoz-Hellín, L Vela, M Ramiro-González, C Fernández-de-Las-Peñas. Pain in Parkinson disease: a review of the literature. Parkinsonism Relat Disord, 2013. [DOI | PubMed]
- C Brefel-Courbon, P Payoux, C Thalamas, F Ory, I Quelven, F Chollet, JL Montastruc, O Rascol. Effect of levodopa on pain threshold in Parkinson’s disease: a clinical and positron emission tomography study. Mov Disord, 2005. [DOI | PubMed]
- A Gerdelat-Mas, M Simonetta-Moreau, C Thalamas, F Ory-Magne, T Slaoui, O Rascol, C Brefel-Courbon. Levodopa raises objective pain threshold in Parkinson’s disease: a RIII reflex study. J Neurol Neurosurg Psychiatry, 2007. [DOI | PubMed]
- Y Zengin-Toktas, J Ferrier, F Durif, PM Llorca, N Authier. Bilateral lesions of the nigrostriatal pathways are associated with chronic mechanical pain hypersensitivity in rats. Neurosci Res, 2013. [DOI | PubMed]
- LE Gee, N Chen, A Ramirez-Zamora, DS Shin, JG Pilitsis. The effects of subthalamic deep brain stimulation on mechanical and thermal thresholds in 6OHDA-lesioned rats. Eur J Neurosci, 2015. [DOI | PubMed]
- BC Kaszuba, I Walling, LE Gee, DS Shin, JG Pilitsis. Effects of subthalamic deep brain stimulation with duloxetine on mechanical and thermal thresholds in 6OHDA lesioned rats. Brain Res, 2017. [DOI | PubMed]
- A Gómez-Paz, R Drucker-Colín, D Milán-Aldaco, M Palomero-Rivero, M Ambriz-Tututi. Intrastriatal chromospheres’ transplant reduces nociception in hemiparkinsonian rats. Neuroscience, 2018. [DOI | PubMed]
- GC Nascimento, K Bariotto-Dos-Santos, CRA Leite-Panissi, EA Del-Bel, M Bortolanza. Nociceptive response to L-DOPA-induced dyskinesia in hemiparkinsonian rats. Neurotox Res, 2018. [DOI | PubMed]
- G Wasner, G Deuschl. Pains in Parkinson disease–many syndromes under one umbrella. Nat Rev Neurol, 2012. [DOI | PubMed]
- MT Lin, JJ Wu, A Chandra, BL Tsay. Activation of striatal dopamine receptors induces pain inhibition in rats. J Neural Transm, 1981. [DOI | PubMed]
- JE Magnusson, K Fisher. The involvement of dopamine in nociception: the role of D(1) and D(2) receptors in the dorsolateral striatum. Brain Res, 2000. [DOI | PubMed]
- J Guindon, A Hohmann. The endocannabinoid system and pain. CNS Neurol Disord Drug Targets, 2009. [DOI | PubMed]
- MS McDonagh, BJ Morasco, J Wagner, AY Ahmed, R Fu, D Kansagara, R Chou. Cannabis-based products for chronic pain: a systematic review. Ann Intern Med, 2022. [DOI | PubMed]
- J Mlost, A Wąsik, K Starowicz. Role of endocannabinoid system in dopamine signalling within the reward circuits affected by chronic pain. Pharmacol Res, 2019. [DOI | PubMed]
- G Crivelaro do Nascimento, DP Ferrari, FS Guimaraes, EA Del Bel, M Bortolanza, NC Ferreira-Junior. Cannabidiol increases the nociceptive threshold in a preclinical model of Parkinson’s disease. Neuropharmacology, 2020. [DOI | PubMed]