
Excess cerebral TNF causing glutamate excitotoxicity rationalizes treatment of neurodegenerative diseases and neurogenic pain by anti-TNF agents
Abstract
The basic mechanism of the major neurodegenerative diseases, including neurogenic pain, needs to be agreed upon before rational treatments can be determined, but this knowledge is still in a state of flux. Most have agreed for decades that these disease states, both infectious and non-infectious, share arguments incriminating excitotoxicity induced by excessive extracellular cerebral glutamate. Excess cerebral levels of tumor necrosis factor (TNF) are also documented in the same group of disease states. However, no agreement exists on overarching mechanism for the harmful effects of excess TNF, nor, indeed how extracellular cerebral glutamate reaches toxic levels in these conditions. Here, we link the two, collecting and arguing the evidence that, across the range of neurodegenerative diseases, excessive TNF harms the central nervous system largely through causing extracellular glutamate to accumulate to levels high enough to inhibit synaptic activity or kill neurons and therefore their associated synapses as well. TNF can be predicted from the broader literature to cause this glutamate accumulation not only by increasing glutamate production by enhancing glutaminase, but in addition simultaneously reducing glutamate clearance by inhibiting re-uptake proteins. We also discuss the effects of a TNF receptor biological fusion protein (etanercept) and the indirect anti-TNF agents dithio-thalidomides, nilotinab, and cannabinoids on these neurological conditions. The therapeutic effects of 6-diazo-5-oxo-norleucine, ceptriaxone, and riluzole, agents unrelated to TNF but which either inhibit glutaminase or enhance re-uptake proteins, but do not do both, as would anti-TNF agents, are also discussed in this context. By pointing to excess extracellular glutamate as the target, these arguments greatly strengthen the case, put now for many years, to test appropriately delivered ant-TNF agents to treat neurodegenerative diseases in randomly controlled trials.
Article type: Review Article
Keywords: TNF, Glutamate, Astrocyte, Synapse, Glutaminase, Re-entry proteins, Neurodegenerative disease, Neurogenic pain
Affiliations: Biomedical Sciences and Biochemistry, Research School of Biology, Australian National University, Acton, Canberra, Australian Capital Territory 0200 Australia; Neurodegeneration Research Group, Garvan Institute, 384 Victoria Street, Sydney, New South Wales 2010 Australia
License: © The Author(s). 2016 CC BY 4.0 Open AccessThis article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Article links: DOI: 10.1186/s12974-016-0708-2 | PubMed: 27596607 | PMC: PMC5011997
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (801 KB)
Background
The amyloid theory of Alzheimer’s disease, and by extension other chronic neurodegenerative states, has dominated the field for decades. It has, however, in the face of the reality of numerous large clinical trials yielding no clinical improvement, lost momentum. A recent item on the Editor’s Blog of the webpage of the Journal of Alzheimer’s Disease entitled “Time to Dismount” (see http://www.j-alz.com/editors-blog/posts/time-dismount) eloquently again brings to the fore the long-held, widespread, and increasing unease among researchers [ref. 1–ref. 5]. Likewise, outcomes comparing in vivo human cerebral amyloid β (Aβ) deposition on Pittsburgh Compound B PET imaging have not generated optimism for the amyloid theory [ref. 6, ref. 7]. Recent key epidemiological evidence from a large population in which administering regular subcutaneous etanercept over an extended period in treatment of rheumatoid arthritis (RA) patients was reported to reduce incidence of Alzheimer’s disease (AD) [ref. 8], further reduces the likelihood of Aβ being the key to AD pathogenesis.
We have recently [ref. 9] reviewed the literature demonstrating that increased soluble Aβ does not cause direct damage but is one of the proinflammatory cytokine-induced damage-associated molecular patterns (DAMPs) recognized by toll-like receptors (TLRs). These receptors also recognize pathogen-associated molecular patterns (PAMPs) present on the surface of, for example, the microbes now widely agreed to be sometimes associated with AD [ref. 10]. Agonists of TLRs, which are on and in various types of cells, including those throughout the brain, release more of these same cytokines, including tumor necrosis factor (TNF). This is consistent with Aβ not inhibiting long-term potentiation in hippocampal slices from mice treated with anti-TNF agents, such as infliximab [ref. 11]. Clearly, from the literature we have recently quoted [ref. 9], Aβ is best regarded, along with S100 proteins and high-mobility group box 1 (HMGB1), as belonging to a class of DAMPs (secondary DAMPs) that exacerbates production of the proinflammatory cytokines responsible for their own increase, and induces them further, causing a forward feed chain reaction. Moreover, variation in levels of these other DAMPs of this same class, possessing the same TLR-mediated, TNF-generating activity in AD, may explain why normal aged patients can exhibit high Aβ plaque levels. It may also explain why removing soluble Aβ or its plaque, still the goal of the many clinical trials [ref. 12], does not retard human disease progression, since the other secondary DAMPs, S100 proteins and HMGB1, are still actively inducing TNF. In contrast, removing Aβ is successful in mouse transgenic models that have been designed to generate pathologically but artificially high Aβ [ref. 13].
Waning enthusiasm for the amyloid theory now allows many other approaches, including the last 10 years of animal studies, case reports, open trials, and off-label treatments of neurodegenerative diseases, based on neutralizing excessive levels of TNF within the brain, to receive more attention. Unaccountably, this neglected approach to neurodegenerative disease is sometimes still referred to as highly controversial [ref. 14]. This review provides the logic for increased extracellular cerebral glutamate being the central mechanism by which excessive TNF harms cerebral function and structure. TNF is the first endogenous mediator to be documented as simultaneously influencing extracellular cerebral levels of extracellular glutamate by both enhancing its release and reducing its re-uptake. Given the broad ramifications of glutamate-induced excitotoxicity in infectious and non-infectious disease, these additional layers of information about TNF provide insights with widespread therapeutic implications. In particular, it increasingly rationalizes accounts of the usefulness of neutralizing excess cerebral levels of TNF in neurodegenerative disease.
As well as providing sufficient background to enable the bigger picture of TNF in brain disease pathogenesis to be understood, we focus here on the implications of newer data, largely neglected in the world of neurodegenerative disease, on how this cytokine evidently controls levels of extracellular glutamate in the synaptic cleft. In brief, glutamate is the chief physiological excitatory neurotransmitter, essential of course in memory and learning, and indeed is functionally involved in virtually all activities of the nervous system. Glutamate’s combination of functional importance and toxicity demands tight control over its release and re-uptake. Thus, as will be discussed, control by TNF of both of these functions gives treatments based on reducing excess cerebral levels of this cytokine a solid therapeutic foundation in neurodegenerative disease, in part because of its essential effects in driving excitotoxicity. In practice, we may usefully view TNF toxicity and glutamate toxicity as two perspectives on the one pathophysiological entity.
TNF, an extremely pleiotropic cytokine
TNF was recognized, and named, as an endogenous tumor killing agent [ref. 15], and 6 years later, its wider biological importance began to be appreciated through its roles in innate immunity and the pathogenesis of infectious disease ([ref. 16], reviewed in 2004 [ref. 17]). In due course, fundamental roles for this cytokine in physiological homeostasis [ref. 18] and non-infectious disease [ref. 19] began to be explored. After being recognized as an early step in the inflammatory cytokine cascade [ref. 20], TNF began to achieve its present wide acceptance as a master cytokine in disease pathogenesis through infliximab, the first of the specific neutralizing biological anti-TNF agents, becoming a striking clinical success in treating RA [ref. 21]. Others from this research group showed that TNF is a master cytokine through observing that infliximab reduces levels of other inflammatory cytokines as well as TNF [ref. 22, ref. 23].
The extraordinarily broad relevance of TNF in biology can now be inferred by its strongly conserved state, traceable back through a remarkably ancient lineage including fish and insects, with the form generated by reef-building corals, and the TNF receptors on their cells, co-recognizing their human counterparts [ref. 24]. Unsurprisingly, therefore, every organ, including the brain, has proved to be influenced by this cytokine. By 1987, TNF had been shown to be a necessary part of the chain of events that control normal sleep [ref. 25], and a few years later, current conductance in neurons of a sea slug, Aplysia kurodai, was observed to be reduced by human TNF [ref. 26, ref. 27]. Next, physiological levels of TNF had been reasoned to be necessary for normal mammalian neuronal function, with a loss or gain of TNF beyond homeostatic limits being pathological [ref. 28]. Nevertheless, data on other proinflammatory or anti-inflammatory cytokines such as IL-1β, IL-4, IL-17, and IL-23 [ref. 29–ref. 31] continue to add to the principles behind this concept and may well generate related therapeutic avenues.
TNF excess in neurodegenerative states
Twenty years ago, the involvement of unchecked chronic TNF generation, particularly within the brain, in the pathogenesis of stroke, traumatic brain injury (TBI), and AD began to be apparent [ref. 32–ref. 34]. Refinements of these scientific arguments have accumulated to the present day [ref. 35–ref. 41]. The subtle relationship between these cytokines and the brain has been nicely put by noting that even when it appears that the nervous system is succumbing to a flared immune system, and the two systems maintain a constant dialogue in the attempt to restore homeostasis [ref. 42].
The rationale for treating chronic neurodegenerative states by reducing excess cerebral TNF extends far beyond the post-stroke syndrome, AD, and TBI noted above. Despite “belonging” to various disciplines, these cerebral states characterized by TNF excess clearly have much pathophysiology in common. They include (Table 1) Parkinson’s disease (PD) [ref. 43], neurogenic pain [ref. 44–ref. 50], Huntington’s disease [ref. 51], amyotropic lateral sclerosis [ref. 52], septic encephalopathy [ref. 53], defective post-operative cognition [ref. 54, ref. 55], defective post-irradiation [ref. 56] and post-chemotherapy [ref. 57, ref. 58] cognition, defective cognition during RA [ref. 48], epileptic seizures [ref. 59, ref. 60], viral encephalitides [ref. 61], cerebral malaria [ref. 62], and HIV dementia [ref. 63]. Moreover, recent evidence has very precisely incriminated excess brain TNF in the pathogenesis of AD [ref. 64]. The authors employed a novel multivariate regression modeling approach, termed partial least squares regression, to investigate cytokine protein concentrations in brain tissue from AD and control patients. Taking into account the order in which brain regions are known to be impacted during the development of AD, region-specific profiles were used to identify high concentrations of cytokines which, when used alone, killed neurons in vitro. Of the 48 cytokines monitored, only TNF (=TNFα in their text) met this condition. This is entirely consistent with the evidence we have previously presented [ref. 37] that increased cerebral TNF is the most logical therapeutic target for countering this disease. As we review here, the largely neglected evidence that variation in TNF, through regulating both the release and clearance of cerebral glutamate, seems destined to widen an appreciation of this cytokine within neuroscience as a mediator of plasticity and excitotoxicity.
Table 1: Association of excess TNF and glutamate in brain in neurodegenerative states. See text for references
| Disease | Excess brain TNF | Excess brain glutamate |
|---|---|---|
| Alzheimer’s disease | + | + |
| Parkinson’s disease | + | + |
| Huntington’s disease | + | + |
| Amyotropic lateral sclerosis | + | + |
| Septic encephalopathy | + | + |
| Traumatic brain injury | + | + |
| Stroke | + | + |
| Poor post-operative cognition | + | + |
| Poor post-irradiation cognition | + | + |
| Poor post-chemotherapy cognition | + | ? |
| Poor cognition in rheumatoid arthritis | + | ? |
| Epileptic seizures | + | + |
| HIV dementia | + | + |
| Cerebral malaria | + | + |
| Neurogenic pain | + | + |
| Viral encephalitides | + | + |
Glutamate in brain physiology and pathophysiology
l-glutamate, the most abundant extracellular amino acid in the brain, is, as reviewed over the decades [ref. 65–ref. 67], the chief physiological excitatory neurotransmitter, including in normal memory and learning. Cerebral glutamate is formed, in microglia and astrocytes [ref. 68], as well as neurons, by glutaminase acting on glutamine, and becomes extracellular. Homeostasis is normally maintained by a balance between this reaction and glutamate re-uptake from the synaptic cleft by a series of transport, or re-uptake, proteins that initiate its recycling back to glutamine. As discussed below, much literature associates TNF with glutamate regulation. Both too much or too little, TNF and glutamate are harmful. In brief, a plausible paired physiological role for them is low fluctuating levels of TNF determining physiological levels of glutamate in hippocampal homeostatic synaptic plasticity [ref. 69, ref. 70], as described below.
As reviewed [ref. 66, ref. 71], should extracellular cerebral glutamate become excessive, whether through excess release or poor clearance, or both, a harmful excitotoxicity ensues. From the 1990s, understanding the disruptions that can cause this increase has been an intense focus of interest in the pathophysiology of neurodegenerative diseases. These conditions (Table 1) came to include AD [ref. 72], PD [ref. 73], Huntington’s disease [ref. 74], amyotropic lateral sclerosis [ref. 75], stroke [ref. 76], viral encephelitides [ref. 77, ref. 78], septic encephalopathy [ref. 79], defective post-operative cognition [ref. 80], post-irradiation brain function [ref. 81], pain [ref. 82, ref. 83], bacterial meningitis [ref. 84], epileptic seizures [ref. 85], human immunodeficiency virus (HIV) dementia [ref. 86], cerebral malaria [ref. 87], and TBI [ref. 88–ref. 90]. In addition, the key studies of Jourdain and co-workers [ref. 91] convincingly combined functional and ultrastructural evidence to argue the case for glutamate from astrocytes being a key player in physiological control of synaptic strength. Increasingly, these glutamate pathways have therefore become essential background reading for those whose chief interest has been developing therapeutic drugs for treating these conditions. A recent comprehensive review [ref. 92] provides a clear account of the complexities of the control of cerebral extracellular glutamate in chronic, as distinct from acute, excitotoxicity in neurodegenerative states, and discusses amyotrophic lateral sclerosis (ALS), AD, and Huntington’s disease as examples. However, this text takes no account of the presence of excess cerebral TNF production or its influence on extracellular brain glutamate levels in these and similar diseases [ref. 34, ref. 51, ref. 93–ref. 95].
The roles of excess cerebral TNF in generating glutamate toxicity
Inhibition of re-uptake proteins
As reviewed in 2001 [ref. 96], glutamate re-uptake from the synaptic cleft noted above is controlled by fluctuations in a unique family of amino acid transport, or re-uptake, proteins that act as signal terminators. Their inhibition is intricately involved in the pathogenesis of glutamate-excess excitotoxicity diseases such as stroke, AD, epilepsy, and chronic pain syndromes. Twenty years ago, TNF was first implicated in generating excitotoxicity through its capacity to inhibit glutamate re-uptake in an HIV dementia model [ref. 97] and subsequently in cultures brain slices [ref. 98] and a Sindbis virus disease model [ref. 99]. Although outside the topic of this review, which discusses entry of glutamate into the synaptic cleft rather than its actions while there, we note that emphasis has more recently been placed on the ability of TNF to regulate the various types of glutamate receptors [ref. 100]. The details of control of these transport proteins by TNF have more recently been updated in a rat model of ALS [ref. 101]. When combined, the ideas generated in these fields of research have allowed insightful functional links of neuroinflammation and glutamate-induced excitotoxicity to be proposed [ref. 102, ref. 103].
Glutaminase upregulation
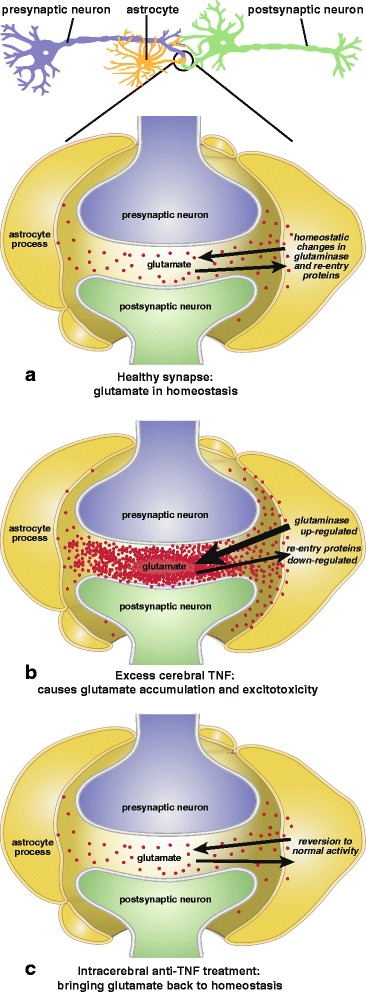
In the event, TNF became much more heavily incriminated in glutamate regulation than has been taken into consideration in the above models of excitotoxocity (Fig. 1). Ten years ago, this cytokine was reported to generate excessive glutamate levels by markedly upregulating glutaminase activity [ref. 104]. This was confirmed, as was a concomitant reduction in glutamate re-uptake, in a model of Japanese viral encephalitis [ref. 105]. The next year, with the same surprisingly little influence on mainline excitotoxicity research to date, glutaminase upregulation was reported after stimulating primary cultured human neurons with TNF or interleukin-1β [ref. 106]. Furthermore, these authors found the glutamate increase to occur in the extracellular space as well as intracellularly. The following year, this group also showed that etanercept reduces inflammation and lethality in the above model of Japanese viral encephalopathy [ref. 107]. Clearly, by increasing glutamate production while simultaneously reducing its re-uptake [ref. 97], excess TNF can be expected to readily cause glutamate to accumulate to toxic levels. This implies much more therapeutic potential for anti-TNF agents than other drugs possessing only one of these activities, such as 6-diazo-5-oxo-norleucine (DON), ceftriaxone, or riluzole, as discussed below. However, this TNF-glutaminase link, despite first being made a decade ago (above), does not yet appear to be common currency in neurodegenerative disease circles (e.g., [ref. 108]). Readers interested in the complexities of glutamate release, including its physiological control, are directed to the examples provided by references [ref. 109–ref. 111].
In passing, we note that glutamine deficiency is a long-recognized characteristic of chronic inflammatory stress and has made nutritionally motivated i.v. glutamine replacement therapy a routine, if formally untested, adjunct treatment in critical care wards [ref. 112]. However, a recent post hoc analysis of a large-scale randomized trial has shown this procedure to be of no value, perhaps even harmful [ref. 113]. This raises the possibility that chronic TNF increase present in these patients may have caused the observed glutamine depletion by the combined effects of enhancing its conversion to glutamate, plus inhibiting its reconversion from glutamate, as summarized above. Clearly, amino acids have many functions as well as providing nutrients.
Glutamate toxicity as a major manifestation of excess TNF in brain disease
The above data on TNF provide insights into the breadth of therapeutic relevance of the functional link between unbridled TNF production and glutamate neurotoxicity and how this adds immensely to the central argument of this review that TNF is a highly logical target in neurodegenerate disease. As but one example, the capacity of excess TNF to greatly increase glutamate output through activating glutaminase [ref. 104, ref. 105] casts the considerable body of work on astrocytes, glutamate, and basal ganglia excitotoxicity, in which the influence of inflammatory cytokines are not considered [ref. 114], in a new light. It also modifies the novel “glutamate grabber” approach to treating brain ischemia [ref. 115], in that etanercept is, from the above insights, likely to be much more effective than glutamate-oxaloacetate transamimase or oxaloacetate by preventing an excess of newly formed glutamate.
Importantly, intra-amygdala infusion of TNF has been reported to elevate glutamate levels in this region of the brain [ref. 116]. Likewise, etanercept, a specific anti-TNF biological in wide clinical use, lowers brain glutamate levels in experimental models (Table 2). Although etanercept is too large a molecule for all but a small amount of a subcutaneous (s.c.) dose to enter the cerebrospinal fluid (CSF), intentionally compensating for this by giving a 20-fold larger dose reduces brain glutamate in a rat model of traumatic brain injury [ref. 117]. Etanercept has also been reported, in a heart failure model in which TNF is increased [ref. 118, ref. 119], to lower rat brain glutamate dramatically when given intracerebroventricularly (i.c.v), although, again because of its high molecular weight, not when administered intraperitoneally (i.p.).
Table 2: Outcome of administering specific or non-specific anti-TNF agents in states exhibiting excess cerebral TNF and the opposing effects of TNF and anti-TNF agents of brain glutamate levels. See text for references
| Excess cerebral TNF present | Positive outcome after etanercept, etc. | Positive non-specific TNF inhibitors outcome | |||
|---|---|---|---|---|---|
| Thalid or dithio-thalid | Nilotinib | Cannabinoids | |||
| Alzheimer’s disease | + | + | + | + | + |
| Parkinson’s disease | + | ? | ? | + | + |
| Huntington’s disease | + | ? | ? | ? | ? |
| Amyotropic lateral sclerosis | + | + | ? | ? | ? |
| Septic encephalopathy | + | ? | ? | ? | ? |
| Traumatic brain injury | + | + | + | ? | + |
| Stroke | + | + | + | ? | ? |
| Poor post-operative cognition | + | + | ? | ? | ? |
| Poor post-chemother cognition | + | ? | ? | ? | ? |
| Poor post-irradiation cognition | + | ? | ? | ? | ? |
| Epileptic seizures | + | ? | ? | ? | + |
| HIV dementia | + | ? | ? | ? | + |
| Neurogenic pain | + | + | ? | ? | + |
| Viral encephalitides | + | ? | ? | ? | + |
| Elevated brain glutamate | + | ||||
| Lower brain glutamate | + | ? | ? | + |
Do these actions of TNF explain the rapid response to etanercept in neurodegenerative disease?
Control of glutamate by TNF might also explain why etanercept has often been reported to reverse a number of clinical manifestations of neurodegenerative disease surprisingly rapidly. It was shown 17 years ago [ref. 120] that turnover of cerebral extracellular glutamate is very fast, seconds to minutes in these authors’ hands. This is evolutionarily essential because of the key role of this amino acid in the synaptic cleft, where it is responsible for the fast excitatory neurotransmission necessary for the rapid brain responses demanded for survival in the real world. Thus, given the role of TNF to influence both glutaminase and re-uptake proteins described earlier, the capacity of intracerebral etanercept to lower brain glutamate, as summarized above [ref. 117–ref. 119], can be expected to act with somewhat the same degree of rapidity. It seems likely, therefore, that these data rationalize the unexpected but clearly rapid response in case reports and open trials to perispinal etanercept, initially reported in 2003 [ref. 121] and 2008 [ref. 122], and regularly confirmed since [ref. 123–ref. 127]. Awareness of this 1999 report on the rapidity of extracellular cerebral glutamate turnover [ref. 120] may now help contribute to the body of accruing evidence that should alter attitudes regarding reports of rapid responses to anti-TNF in neurodegenerative disease [ref. 126, ref. 128].
Therapeutic implications for excitotoxicity in neurodegeneration
Specific anti-TNF biologicals
Whereas infliximab and adalimumab are essentially monoclonal antibodies directed at TNF itself, etanercept, the only anti-TNF biological drug yet tested in this context, is a fusion protein consisting of the p75 TNF receptor, joined to the constant end of the IgG1 antibody [ref. 129]. An etanercept biosimilar is already in the literature [ref. 130, ref. 131], and a number of others already have approval or are being developed [ref. 132], providing the prospect of reduced treatment costs for most of the world. Recent debates on competitive pressures versus scientific rationales delaying introduction of biological biosimilars are most informative [ref. 133]. Clearly, regulating this field is a state of flux.
The use of anti-TNF agents in neurodegenerative disease has its critics who largely base their concerns on whether the functional complexities of TNF science, such as the p55 and p75 TNF receptors and membrane versus soluble location of TNF, should be more fully elucidated beforehand [ref. 134, ref. 135]. This is largely overplayed: clinical development of the specific anti-TNF biologicals in RA, psoriasis, and Crohn’s disease went ahead successfully during the past decades alongside continuing yet incomplete basic research without such reservations being aired. The case for cautious use of specific biological anti-TNF agents based on the soundness of the pathophysiological arguments in otherwise untreatable conditions, with an eye to potential concerns, has been amply made in systemic states [ref. 136]. In practice, the balance of safety versus outcomes has proved to be very much on the side of the millions of patients who have received regular treatment with these agents for many years now.
Blood-brain barrier (BBB) passage by specific anti-TNF biologicals
As other therapeutic molecules, the biological anti-TNF agent etanercept, though employed widely with great success systemically, is, as often noted, too large to cross the BBB in significant amounts unaided. Two BBB-crossing techniques now exist side by side in the literature, the earliest and simplest from a small group, the later technically complex, and not yet tried in patients. Here, we summarize their origins and rationale.
Perispinal delivery of etanercept
Understanding the perispinal delivery of large molecular weight drugs into the central nervous system requires an appreciation of the cerebrospinal venous system. As recently reviewed in detail [ref. 137], this route of cerebral venous drainage has had, since its discovery well over a century and a half ago, a most complex and interesting history, and more recently application, in medical advances. Contemporary awareness of the potential of this route began when researchers in aviation medicine were exploring an animal model of the effects of gravity and body position on pilots of high-performance aircraft [ref. 138]. They noted that restraining anesthetized rabbits on a tilt board and rotating them to a head-down position considerably increased CSF levels of the plasma protein albumin within 5 min. The authors noted, in passing, that as well as aiding their branch of science, their data had implications for getting large molecular weight therapeutics into the brain. As discussed [ref. 139, ref. 140], the principle behind this approach—drug delivery to the brain by retrograde venous flow—began to be used off-label the early 2000s to get etanercept into the brain in patients with neurogenic pain [ref. 121] and AD [ref. 141]. Although these open trial observations (Table 2) continue to be reported post-stroke [ref. 125] and TBI [ref. 125], and the principles they embrace now have a solid foundation in animal models [ref. 50, ref. 55, ref. 142–ref. 144], remarkably they have not, as did parallels in inflammatory states in other organs earlier, attracted funding for randomized human trials. As noted above, a wider awareness of the rapid rate of glutamine to glutamate kinetics [ref. 120] may well, through rationalizing the reported rapid response [ref. 121, ref. 122], reduce skepticism.
Trojan Horse delivery of etanercept
An alternative method of delivery of large molecules into the brain exists [ref. 145–ref. 147], but it ignores the above input from aviation medicine and, as has been discussed [ref. 148–ref. 150], remains fraught with technical difficulties. In 2011, the UCLA/Armagen group reported that a re-engineered version of etanercept, in which the IgG part of the fusion protein is a chimeric monoclonal antibody against the mouse transferrin receptor, could be delivered into the mouse brain in this way [ref. 151]. The following year, they reported that this re-engineered etanercept reduced the harmful effects of experimental stroke in a mouse model [ref. 152]. It cannot, however, be tested in humans until etanercept is re-engineered so that it recognized human, as distinct from mouse, transferrin. Moreover, re-engineering is essential for each large molecule under consideration, whereas they can be expected to function in their original form when introduced perispinally. A recent review [ref. 153] discusses a number of further complexities that need addressing before Trojan Horse delivery could become routinely used.
Non-specific inhibitors of TNF
3,6 Dithio-thalidomides
Thirty years after being removed from the market in 1961 because of its disastrous effects on fetal development, thalidomide had begun to be explored to treat a number of intractable conditions in patients other than child-bearing age women. It was shown to selectively inhibit TNF production by stimulated human monocytes [ref. 154], and to do so by enhancing degradation of the mRNA for this cytokine [ref. 155]. A decade later, a series of thio-thalidomides with higher anti-TNF effects than the parent compound were synthesized [ref. 156], and the outcomes of their use on TNF mRNA generation closely compared [ref. 157]. A considerable literature now exists on some compounds of this class, which are orally active, pass the blood-brain barrier (BBB) and improve outcome in neurodegenerative disease models by inhibiting TNF. They are widely efficacious, by various behavioral and cognitive criteria, in models of lipopolysaccharide-induced neuroinflammation [ref. 158], AD [ref. 159–ref. 161], TBI [ref. 55], and stroke [ref. 142]. Recently, the parent compound has been reported to reduce a form of neurogenic pain by repressing the inflammatory response [ref. 162]. We are unaware of any literature on dithio-thalidomides influencing glutaminase, but the parent compound also has been reported to prevent hypoxia-induced TNF from inhibiting one of the glutamate re-uptake proteins [ref. 163].
Nilotinib
Nilotinib, a tyrosine kinase inhibitor, is 30 times more potent than imatinib, which it is replacing for treating certain leukemias [ref. 164, ref. 165]. A small open trial of 6-month duration performed at Georgetown University Hospital was reported at the most recent Annual Meeting of the American Society for Neuroscience. Daily oral nilotinib showed promise, in an initial uncontrolled trial, of reversing clinical aspects of PD with or without dementia, as well Lewy Body dementia [ref. 166]. Phosphorylated tau (P-tau), α-synuclein, and Aβ were noted to have been significantly reduced [ref. 166] in nilotinib-treated patients. Previously, nilotinib had been reported, by this group and others, to be successful in controlled studies in mouse PD models [ref. 167–ref. 169]. One of these groups [ref. 169] demonstrated that a clinically useful proportion of orally administered nilotinib, as used in this new open trial [ref. 166], passes through the BBB. Prior animal studies also show that using nilotinib and the closely related dasatinib was useful in models of AD [ref. 170–ref. 172].
The capacity of nilotinib (and indeed dasatinib [ref. 170]) to inhibit TNF generation in vivo [ref. 173, ref. 174], and the observations that blocking TNF duplicates this effect of nilotinib in PD models [ref. 43, ref. 175], appear to have not yet been considered as a plausible mechanism of these new clinical observations with this agent [ref. 166]. Nevertheless, as an anti-TNF agent, nilotinab can be expected, from the activity of TNF in these contexts [ref. 97, ref. 104], to inhibit glutaminase activity as well as enhance glutaminate re-uptake proteins. In addition to nilotinib [ref. 176] another selective Src tyrosine kinase, pyrazolopyrimidine-2 (PP-2), inhibits production of TNF [ref. 177]. Also, given that insulin resistance is a common direct consequence of chronically increased TNF [ref. 178], further evidence for anti-TNF effects being central to these observations with nilotinib comes from the ability of these tyrosine kinase inhibitors to treat type 2 diabetes mellitus (T2DM) by decreasing insulin resistance [ref. 179].
How might nilotinib reduce TNF levels? Tyrosine phosphorylation is central to TLR stimulation and subsequent activation of NF-kappaB [ref. 180] that generates cytokines such as TNF. Endotoxin tolerance is associated with inhibited phosphorylation Src, a non-receptor tyrosine kinase protein [ref. 181]. It is therefore plausible that agents such as nilotinib, which inhibit Srcs, reduce TNF production [ref. 173, ref. 174] by mimicking tolerance to TLR agonists such as endotoxin.
Cannabinoids
As reviewed [ref. 182], the therapeutic and pharmacological secrets of Cannabis sativa have fascinated researchers for about two centuries. About 90 phytocannabinoids (i.e., compounds present in the plant) have been identified, the two with the largest literatures being tetrahydrocannabinol (THC) and cannabidiol (CBD). The former is psychotropic and thus under a legal cloud, although a synthetic trans-9-delta isomer, termed dronabinol, is an example of forms of THC nowadays undergoing limited investigation [ref. 183]. CBD, in contrast, does not cause significant behavioral change and is researched much more widely. These phytocannabinoids, self-evidently BBB permeable, proved to be ligands for two previously unsuspected receptors, mainly found on cells of the immune system, and whose presence led to the prediction and discovery of endogenous cannabinoids, or endocannabinoids. In physiological terms, these may be considered as part neurotransmitter, part cytokine, and part hormone and have been identified and studied at length (see [ref. 184, ref. 185] for reviews.)
Both endocannabinoids and CBD have been shown to be active in models for pain [ref. 186–ref. 190], AD [ref. 191–ref. 195], epileptic seizures [ref. 196–ref. 199], PD [ref. 200–ref. 202], HIV dementia [ref. 203–ref. 205], viral encephalitis [ref. 206], and TBI [ref. 207]. Clearly, this list parallels the conditions, discussed earlier, with which excessive cerebral levels of TNF are associated. These agents are also active in hypoxic encephalopathy, a stroke parallel in newborns [ref. 208, ref. 209], a condition associated with raised inflammatory cytokines and glutamate [ref. 210]. Not surprisingly, therefore, cannabinoids, whether synthetic, endogenous, or of plant origin, have proven to be established anti-TNF agents in vitro and in vivo, in the sense that they reduce its production by the usual recognized stimuli [ref. 211–ref. 214]. This list includes treating the murine malarial encephalopathy (cerebral malaria) [ref. 214], a condition in which, as discussed below, 6-diazo-5-oxo-norleucine is also efficacious for a related and predictable reason concerned with lowering extracellular cerebral glutamate [ref. 215].
Again, the list of model conditions investigated for therapeutic use of cannabinoids in the previous paragraph remarkably mirrors the list of previously discussed conditions associated with excessive cerebral extracellular levels of glutamate. Moreover, treatment with cannabinoids or altering the function of their cellular receptors [ref. 185, ref. 191, ref. 216–ref. 221] has been reported to lower the levels, or function, of brain glutamate (Table 2). This is entirely consistent with their activity as anti-TNF agents [ref. 211–ref. 214].
Agents that do not influence TNF but still reduce extracellular brain glutamate
6-Diazo-5-oxo-norleucine (DON), a glutaminase inhibitor
DON, a glutamine analogue, is studied largely with a view to reduce extracellular glutamate, and thus treat glutamate toxicity, through inhibiting glutaminase. Having been earlier shown [ref. 222] to possess anti-tumor properties, nearly 40 years later, DON was reported to inhibit glutaminase and thus reduce the release of glutamate in the rat cerebral cortex [ref. 223]. Through the last decade, DON has been a useful, albeit often toxic [ref. 224], experimental tool to demonstrate that glutamate-mediated excitotoxicity is a significant component of the pathogenesis of various neurodegenerative states, including brain ischemia [ref. 225]. Cerebral glutamate homeostasis is disrupted in mouse models of both the neurological sequelae of Sindbis virus infection [ref. 226] and malarial encephalopathy caused by Plasmodium berghei ANKA [ref. 87], and DON has been successfully used therapeutically in experimental versions of both conditions [ref. 215, ref. 227]. It has also been useful in an in vitro HIV dementia model [ref. 228] and in both in vitro and ex vivo experimental autoimmune encephalitis, a mouse model of multiple sclerosis [ref. 229].
These new data on DON and the relationship between TNF and glutamate excess through glutaminase enhancement put historic observations on malarial encephalopathy into clearer focus. As has been reviewed [ref. 17], malaria was the first disease, infectious or otherwise, for which TNF was argued to be central to its pathogenesis, and it set the pattern for the rest. Yet a large trial of a specific anti-TNF antibody, injected intravenously, failed to show evidence, in a large trial in West Africa, of any protective effect in children with cerebral malaria [ref. 230]. At that time, however, ideas on malarial disease were predicated on harmful levels of TNF being produced intravascularly, where the parasites that stimulate its production reside, so it was considered logical to administer the antibody into this compartment. Not until 8 years ago was the excess TNF in cerebral malaria shown to originate in the brain [ref. 62]. These authors predicted that interventions to decrease TNF production in the brain might be required in order to improve outcomes. Thus, treating human cerebral malaria with perispinal etanercept, evidently an equivalent to administering it i.c.v. [ref. 125], will have at least as good and theoretically better ability—since it would also enhance re-uptake proteins—as DON to reduce excessive levels of glutamate, and therefore improving clinical outcome. DON, a molecule not known to affect re-uptake proteins, but which inhibits glutaminase [ref. 223], as well as passes the BBB when given i.p. [ref. 231], has recently also been effective in treating mice infected with Sindbis virus [ref. 227] as well as the cerebral malaria model discussed above [ref. 215].
Ceftriaxone and riluzole, glutamate re-uptake transporter enhancers
Ceftriaxone is a broad-spectrum beta-lactam antibiotic, largely reserved, in this context, for use against otherwise resistant bacteria. In contrast to cannabinoids and nilotinib, it has been shown not to reduce TNF release from LPS-treated human monocytes [ref. 232], implying it does not act against excitotoxicity by inhibiting TNF-mediated glutaminase enhancement [ref. 97] or enhancing TNF-mediated reduced glutamate re-uptake [ref. 104]. In 2007, ceftriaxone was shown to independently enhance glutamate re-uptake and thus reduce the glutamate-dependent portion of morphine-dependent hyperthermia [ref. 233]. This activity of ceftriaxone was soon shown, in primary fetal human astrocytes, to operate through increased expression of excitatory amino acid transporter 2 (EAAT2) promoter activity, allowing it to inhibit glutamate-induced excitotoxicity of its own accord [ref. 234]. As these authors noted, this implies that ceftriaxone could have therapeutic activity in a range of neurodegenerative conditions, essentially the examples we discussed earlier as exhibiting excitotoxicity. With this mechanism in mind, ceftriaxone is nowadays under active consideration as a therapy in models of AD [ref. 235], stroke [ref. 236], TBI [ref. 237–ref. 239], and PD [ref. 240–ref. 243]. The most complete evidence consistent with this approach to date is a very recent extensive report on ceftriaxone rescuing brain function in Toxoplasma gondii-infected mice [ref. 244]. The authors documented high brain glutamate, although how this arose remains uncertain. T. gondii is a well-known TNF inducer. Being a pathogen, it possesses the PAMP activity discussed earlier.
Riluzole (6-(trifluoromethoxy)benzothiazol-2-amine), a relatively toxic material nevertheless approved for treatment of ALS, has for some time been known to be a glutamate release inhibitor and thus affecting the glutamate functions discussed above. This has been reported to include enhancing levels of glutamate re-uptake transporters [ref. 245], including in astrocytes [ref. 246, ref. 247]. This principle is entirely consistent with findings in a mouse AD model [ref. 248] and has been extended in a recent study in which riluzole proved to reverse the same array of human gene changes in AD and aging [ref. 249]. Both research groups suggest the effects of riluzole as a possible mechanism underlying its improvement in cognitive function in their studies.
Relative effectiveness of these treatments
As discussed earlier, neurodegenerative diseases are characterized by excessive levels of extracellular cerebral glutamate that can be expected to have accumulated through its too rapid formation as well as its slowed re-uptake and conversion back to glutamine. The ideal therapy would be able to reverse both of these changes. So far as we are aware, TNF is the only endogenous mediator that, when in excess, as in the brain in these diseases, enhances cerebral glutaminase [ref. 104] and also inhibits glutamate re-uptake proteins [ref. 97]. Since anti-TNF agents, both specific and non-specific, can be predicted to simultaneously reverse both of these TNF-induced changes, their efficacy in reversing this excitotoxicity can be expected, from first principles, to be higher than agent such as DON, ceftriaxone, or riluzole, which correct only one of these two pathways.
Specific anti-TNF biologicals are expensive, but can be very effective through neutralizing a precise target, in this case excessive cerebral TNF, known to be central to the disease in question. As discussed earlier, their large size need not be a problem. In contrast, pharmaceuticals such as dithio-thalidomides, nilotinib, cannabinoids, DON, ceftriaxone, and riluzole, although less expensive, may prove to be burdened with unknown targets that generate greater side effects than anti-TNF biologicals can. They have, however, a considerable advantage in neurodegenerative disease in that when administered orally or systemically, they can traverse the BBB and get to where they are needed [ref. 143, ref. 157, ref. 169, ref. 231].
Conclusions
We propose that the excess levels of TNF, and glutamate in the brain across a range of neurodegenerative diseases are crucially linked, high TNF causing extracellular glutamate to accumulate to levels high enough to inhibit synaptic activity and kill neurons by two synergistic mechanisms. As described, these are increasing glutamate production by enhancing glutaminase and simultaneously reducing glutamate clearance by inhibiting re-uptake proteins, thus causing it to accumulate in the synaptic cleft. The shared efficacy of specific anti-TNF biologicals and non-specific anti-TNF agents (thio-thalidomides, nilotinib and cannabinoids) on this superficially diverse range of conditions can thus be understood. The usefulness of DON, ceftriaxone, and riluzole, agents without apparent anti-TNF activity, but each possessing separate activities that counter one of these two influences of high TNF on glutamate accumulation, are similarly rationalized.
References
- HG Lee, X Zhu, RJ Castellani, A Nunomura, G Perry, MA Smith. Amyloid-beta in Alzheimer disease: the null versus the alternate hypotheses. J Pharmacol Exp Ther, 2007. [DOI | PubMed]
- RJ Castellani, X Zhu, HG Lee, MA Smith, G Perry. Molecular pathogenesis of Alzheimer’s disease: reductionist versus expansionist approaches. Int J Mol Sci, 2009. [DOI | PubMed]
- K Mullane, M Williams. Alzheimer’s therapeutics: continued clinical failures question the validity of the amyloid hypothesis-but what lies beyond?. Biochem Pharmacol, 2013. [DOI | PubMed]
- MA Castello, JD Jeppson, S Soriano. Moving beyond anti-amyloid therapy for the prevention and treatment of Alzheimer’s disease. BMC Neurol, 2014. [DOI | PubMed]
- GP Morris, IA Clark, B Vissel. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun, 2014. [PubMed]
- HJ Aizenstein, RD Nebes, JA Saxton, JC Price, CA Mathis, ND Tsopelas, SK Ziolko, JA James, BE Snitz, PR Houck. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol, 2008. [DOI | PubMed]
- CC Rowe, KA Ellis, M Rimajova, P Bourgeat, KE Pike, G Jones, J Fripp, H Tochon Danguy, L Morandeau, G O’Keefe. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging, 2010. [DOI | PubMed]
- 8.Chou RC, Kane M, Ghimire S, Gautam S, Gui J. Treatment for rheumatoid arthritis and risk of Alzheimer’s disease: a nested case-control analysis. CNS Drugs. 2016
- IA Clark, B Vissel. Amyloid beta: one of three danger-associated molecules that are secondary inducers of the proinflammatory cytokines that mediate Alzheimer’s disease. Br J Pharmacol, 2015. [DOI | PubMed]
- 10.Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, Bullido MJ, Carter C, Clerici M, Cosby SL, et al. Microbes and Alzheimer’s disease. J Alzheimers Dis. 2016;51:979-84.
- MJ Rowan, I Klyubin, Q Wang, NW Hu, R Anwyl. Synaptic memory mechanisms: Alzheimer’s disease amyloid beta-peptide-induced dysfunction. Biochem Soc Trans, 2007. [DOI | PubMed]
- S Salloway, R Sperling, NC Fox, K Blennow, W Klunk, M Raskind, M Sabbagh, LS Honig, AP Porsteinsson, S Ferris. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med, 2014. [DOI | PubMed]
- C Janus, J Pearson, J McLaurin, PM Mathews, Y Jiang, SD Schmidt, MA Chishti, P Horne, D Heslin, J French. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature, 2000. [DOI | PubMed]
- J Butchart, L Brook, V Hopkins, J Teeling, U Puntener, D Culliford, R Sharples, S Sharif, B McFarlane, R Raybould. Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology, 2015. [DOI | PubMed]
- EA Carswell, LJ Old, RL Kassel, S Green, N Fiore, B Williamson. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A, 1975. [DOI | PubMed]
- IA Clark, J-L Virelizier, EA Carswell, PR Wood. Possible importance of macrophage-derived mediators in acute malaria. Infect Immun, 1981. [PubMed]
- IA Clark, LE Alleva, AC Mills, WB Cowden. Pathogenesis of malaria and clinically similar conditions. Clin Microbiol Rev, 2004. [DOI | PubMed]
- MG Tovey. The expression of cytokines in the organs of normal individuals: role in homeostasis. A review. J Biol Regul Homeost Agents, 1988. [PubMed]
- MA Marano, LL Moldawer, Y Fong, H Wei, J Minei, R Yurt, A Cerami, SF Lowry. Cachectin/TNF production in experimental burns and Pseudomonas infection. Arch Surg, 1988. [DOI | PubMed]
- PP Nawroth, I Bank, D Handley, J Cassimeris, L Chess, D Stern. Tumor necrosis factor/cachectin interacts with endothelial cell receptors to induce release of interleukin 1. J Exp Med, 1986. [DOI | PubMed]
- MJ Elliott, RN Maini, M Feldmann, JR Kalden, C Antoni, JS Smolen, B Leeb, FC Breedveld, JD Macfarlane, H Bijl. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet, 1994. [DOI | PubMed]
- FM Brennan, D Chantry, A Jackson, R Maini, M Feldmann. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet, 1989. [DOI | PubMed]
- P Charles, MJ Elliott, D Davis, A Potter, JR Kalden, C Antoni, FC Breedveld, JS Smolen, G Eberl, K deWoody. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J Immunol, 1999. [PubMed]
- SD Quistad, A Stotland, KL Barott, CA Smurthwaite, BJ Hilton, JA Grasis, R Wolkowicz, FL Rohwer. Evolution of TNF-induced apoptosis reveals 550 My of functional conservation. Proc Natl Acad Sci U S A, 2014. [DOI | PubMed]
- S Shoham, D Davenne, AB Cady, CA Dinarello, JM Krueger. Recombinant tumor necrosis factor and interleukin-1 enhance slow-wave sleep. Am J Physiol, 1987. [PubMed]
- M Sawada, N Hara, T Maeno. Extracellular tumor necrosis factor induces a decreased K+ conductance in an identified neuron of Aplysia kurodai. Neurosci Lett, 1990. [DOI | PubMed]
- M Sawada, N Hara, T Maeno. Analysis of a decreased Na + conductance by tumor necrosis factor in identified neurons of Aplysia kurodai. J Neurosci Res, 1991. [DOI | PubMed]
- TA Ignatowski, RN Spengler. Tumor necrosis factor-alpha: presynaptic sensitivity is modified after antidepressant drug administration. Brain Res, 1994. [DOI | PubMed]
- SP Gadani, JC Cronk, GT Norris, J Kipnis. IL-4 in the brain: a cytokine to remember. J Immunol, 2012. [DOI | PubMed]
- JM Benson, CW Sachs, G Treacy, H Zhou, CE Pendley, CM Brodmerkel, G Shankar, MA Mascelli. Therapeutic targeting of the IL-12/23 pathways: generation and characterization of ustekinumab. Nat Biotechnol, 2011. [DOI | PubMed]
- CL Langrish, Y Chen, WM Blumenschein, J Mattson, B Basham, JD Sedgwick, T McClanahan, RA Kastelein, DJ Cua. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med, 2005. [DOI | PubMed]
- B Arvin, LF Neville, FC Barone, GZ Feuerstein. The role of inflammation and cytokines in brain injury. Neurosci Biobehav Rev, 1996. [DOI | PubMed]
- FC Barone, B Arvin, RF White, A Miller, CL Webb, RN Willette, PG Lysko, GZ Feuerstein. Tumor necrosis factor-alpha—a mediator of focal ischemic brain injury. Stroke, 1997. [DOI | PubMed]
- E Tarkowski, N Andreasen, A Tarkowski, K Blennow. Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Psychiatry, 2003. [DOI | PubMed]
- MM Esiri. The interplay between inflammation and neurodegeneration in CNS disease. J Neuroimmunol, 2007. [DOI | PubMed]
- FE McAlpine, JK Lee, AS Harms, KA Ruhn, M Blurton Jones, J Hong, P Das, TE Golde, FM LaFerla, S Oddo. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis, 2009. [DOI | PubMed]
- IA Clark, LM Alleva, B Vissel. The roles of TNF in brain dysfunction and disease. Pharmacol Ther, 2010. [DOI | PubMed]
- P Eikelenboom, R Veerhuis, E van Exel, JJ Hoozemans, AJ Rozemuller, WA van Gool. The early involvement of the innate immunity in the pathogenesis of late-onset Alzheimer’s disease: neuropathological, epidemiological and genetic evidence. Curr Alzheimer Res, 2011. [DOI | PubMed]
- K Bhaskar, N Maphis, G Xu, NH Varvel, ON Kokiko-Cochran, JP Weick, SM Staugaitis, A Cardona, RM Ransohoff, K Herrup, BT Lamb. Microglial derived tumor necrosis factor-alpha drives Alzheimer’s disease-related neuronal cell cycle events. Neurobiol Dis, 2014. [DOI | PubMed]
- MT Heneka, MP Kummer, E Latz. Innate immune activation in neurodegenerative disease. Nat Rev Immunol, 2014. [DOI | PubMed]
- M Lyman, DG Lloyd, X Ji, MP Vizcaychipi, D Ma. Neuroinflammation: the role and consequences. Neurosci Res, 2014. [DOI | PubMed]
- I Marin, J Kipnis. Learning and memory…the immune system. Learn Mem, 2013. [DOI | PubMed]
- AS Harms, CJ Barnum, KA Ruhn, S Varghese, I Trevino, A Blesch, MG Tansey. Delayed dominant-negative TNF gene therapy halts progressive loss of nigral dopaminergic neurons in a rat model of Parkinson’s disease. Mol Ther, 2011. [DOI | PubMed]
- TA Ignatowski, WC Covey, PR Knight, CM Severin, TJ Nickola, RN Spengler. Brain-derived TNFalpha mediates neuropathic pain. Brain Res, 1999. [DOI | PubMed]
- WC Covey, TA Ignatowski, PR Knight, RN Spengler. Brain-derived TNFalpha: involvement in neuroplastic changes implicated in the conscious perception of persistent pain. Brain Res, 2000. [DOI | PubMed]
- E Tobinick, S Davoodifar. Efficacy of etanercept delivered by perispinal administration for chronic back and/or neck disc-related pain: a study of clinical observations in 143 patients. Curr Med Res Opin, 2004. [DOI | PubMed]
- SP Cohen, N Bogduk, A Dragovich, CC Buckenmaier, S Griffith, C Kurihara, J Raymond, PJ Richter, N Williams, TL Yaksh. Randomized, double-blind, placebo-controlled, dose-response, and preclinical safety study of transformational epidural etanercept for the treatment of sciatica. Anesthesiology, 2009. [DOI | PubMed]
- YM Chen, HH Chen, JL Lan, DY Chen. Improvement of cognition, a potential benefit of anti-TNF therapy in elderly patients with rheumatoid arthritis. Joint Bone Spine, 2010. [DOI | PubMed]
- A Hess, R Axmann, J Rech, S Finzel, C Heindl, S Kreitz, M Sergeeva, M Saake, M Garcia, G Kollias. Blockade of TNF-alpha rapidly inhibits pain responses in the central nervous system. Proc Natl Acad Sci U S A, 2011. [DOI | PubMed]
- 50.Gerard E, Spengler RN, Bonoiu AC, Mahajan SD, Davidson BA, Ding H, Kumar R, Prasad PN, Knight PR, Ignatowski TA. Chronic constriction injury-induced nociception is relieved by nanomedicine-mediated decrease of rat hippocampal tumor necrosis factor. Pain. 2015;156:1320-33.
- HY Hsiao, FL Chiu, CM Chen, YR Wu, HM Chen, YC Chen, HC Kuo, Y Chern. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum Mol Genet, 2014. [DOI | PubMed]
- MK McCoy, MG Tansey. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation, 2008. [DOI | PubMed]
- K Belarbi, C Arellano, R Ferguson, T Jopson, S Rosi. Chronic neuroinflammation impacts the recruitment of adult-born neurons into behaviorally relevant hippocampal networks. Brain Behav Immun, 2012. [DOI | PubMed]
- N Terrando, C Monaco, D Ma, BM Foxwell, M Feldmann, M Maze. Tumor necrosis factor-alpha triggers a cytokine cascade yielding postoperative cognitive decline. Proc Natl Acad Sci U S A, 2010. [DOI | PubMed]
- R Baratz, D Tweedie, V Rubovitch, W Luo, JS Yoon, BJ Hoffer, NH Greig, CG Pick. Tumor necrosis factor-alpha synthesis inhibitor, 3,6’-dithiothalidomide, reverses behavioral impairments induced by minimal traumatic brain injury in mice. J Neurochem, 2011. [DOI | PubMed]
- CM Wilson, MW Gaber, OM Sabek, JA Zawaski, TE Merchant. Radiation-induced astrogliosis and blood-brain barrier damage can be abrogated using anti-TNF treatment. Int J Radiat Oncol Biol Phys, 2009. [DOI | PubMed]
- S Kesler, M Janelsins, D Koovakkattu, O Palesh, K Mustian, G Morrow, FS Dhabhar. Reduced hippocampal volume and verbal memory performance associated with interleukin-6 and tumor necrosis factor-alpha levels in chemotherapy-treated breast cancer survivors. Brain Behav Immun, 2013. [DOI | PubMed]
- PA Ganz, JE Bower, L Kwan, SA Castellon, DH Silverman, C Geist, EC Breen, MR Irwin, SW Cole. Does tumor necrosis factor-alpha (TNF-alpha) play a role in post-chemotherapy cerebral dysfunction?. Brain Behav Immun, 2013. [DOI | PubMed]
- C Savin, J Triesch, M Meyer-Hermann. Epileptogenesis due to glia-mediated synaptic scaling. J R Soc Interface, 2008
- J Yang, F He, Q Meng, Y Sun, W Wang, C Wang. Inhibiting HIF-1alpha decreases expression of TNF-alpha and caspase-3 in specific brain regions exposed kainic acid-induced status epilepticus. Cell Physiol Biochem, 2016. [DOI | PubMed]
- KA Kulcsar, VK Baxter, R Abraham, A Nelson, DE Griffin. Distinct immune responses in resistant and susceptible strains of mice during neurovirulent alphavirus encephalomyelitis. J Virol, 2015. [DOI | PubMed]
- CC John, A Panoskaltsis Mortari, RO Opoka, GS Park, PJ Orchard, AM Jurek, R Idro, J Byarugaba, MJ Boivin. Cerebrospinal fluid cytokine levels and cognitive impairment in cerebral malaria. Am J Trop Med Hyg, 2008. [PubMed]
- NA Brabers, HS Nottet. Role of the pro-inflammatory cytokines TNF-alpha and IL-1beta in HIV-associated dementia. Eur J Clin Invest, 2006. [DOI | PubMed]
- LB Wood, AR Winslow, EA Proctor, D McGuone, DA Mordes, MP Frosch, BT Hyman, DA Lauffenburger, KM Haigis. Identification of neurotoxic cytokines by profiling Alzheimer’s disease tissues and neuron culture viability screening. Sci Rep, 2015. [DOI | PubMed]
- WJ McEntee, TH Crook. Glutamate: its role in learning, memory, and the aging brain. Psychopharmacology (Berl), 1993. [DOI | PubMed]
- RJ Thomas. Excitatory amino acids in health and disease. J Am Geriatr Soc, 1995. [DOI | PubMed]
- Y Zhou, NC Danbolt. Glutamate as a neurotransmitter in the healthy brain. J Neural Transm (Vienna), 2014. [DOI | PubMed]
- P Bezzi, M Domercq, L Brambilla, R Galli, D Schols, E De Clercq, A Vescovi, G Bagetta, G Kollias, J Meldolesi, A Volterra. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci, 2001. [DOI | PubMed]
- BC Albensi, MP Mattson. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse, 2000. [DOI | PubMed]
- C Bonansco, A Couve, G Perea, CA Ferradas, M Roncagliolo, M Fuenzalida. Glutamate released spontaneously from astrocytes sets the threshold for synaptic plasticity. Eur J Neurosci, 2011. [DOI | PubMed]
- DW Choi. Glutamate neurotoxicity and diseases of the nervous system. Neuron, 1988. [DOI | PubMed]
- DE Miulli, DY Norwell, FN Schwartz. Plasma concentrations of glutamate and its metabolites in patients with Alzheimer’s disease. J Am Osteopath Assoc, 1993. [PubMed]
- PA Loschmann, KW Lange, H Wachtel, L Turski. MPTP-induced degeneration: interference with glutamatergic toxicity. J Neural Transm Suppl, 1994. [PubMed]
- PF Behrens, P Franz, B Woodman, KS Lindenberg, GB Landwehrmeyer. Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain, 2002. [DOI | PubMed]
- O Spreux-Varoquaux, G Bensimon, L Lacomblez, F Salachas, PF Pradat, N Le Forestier, A Marouan, M Dib, V Meininger. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: a reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J Neurol Sci, 2002. [DOI | PubMed]
- H Benveniste, J Drejer, A Schousboe, NH Diemer. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem, 1984. [DOI | PubMed]
- JL Nargi-Aizenman, DE Griffin. Sindbis virus-induced neuronal death is both necrotic and apoptotic and is ameliorated by N-methyl-D-aspartate receptor antagonists. J Virol, 2001. [DOI | PubMed]
- J Darman, S Backovic, S Dike, NJ Maragakis, C Krishnan, JD Rothstein, DN Irani, DA Kerr. Viral-induced spinal motor neuron death is non-cell-autonomous and involves glutamate excitotoxicity. J Neurosci, 2004. [DOI | PubMed]
- HZ Toklu, MK Uysal, L Kabasakal, S Sirvanci, F Ercan, M Kaya. The effects of riluzole on neurological, brain biochemical, and histological changes in early and late term of sepsis in rats. J Surg Res, 2009. [DOI | PubMed]
- Y Li, S Wang, K Ran, Z Hu, Z Liu, K Duan. Differential hippocampal protein expression between normal aged rats and aged rats with postoperative cognitive dysfunction: a proteomic analysis. Mol Med Rep, 2015. [PubMed]
- F Alaoui, J Pratt, S Trocherie, L Court, JM Stutzmann. Acute effects of irradiation on the rat brain: protection by glutamate blockade. Eur J Pharmacol, 1995. [DOI | PubMed]
- ME Fundytus. Glutamate receptors and nociception: implications for the drug treatment of pain. CNS Drugs, 2001. [DOI | PubMed]
- M Valdes, A Collado, N Bargallo, M Vazquez, L Rami, E Gomez, M Salamero. Increased glutamate/glutamine compounds in the brains of patients with fibromyalgia: a magnetic resonance spectroscopy study. Arthritis Rheum, 2010. [DOI | PubMed]
- M Spranger, S Schwab, S Krempien, M Winterholler, T Steiner, W Hacke. Excess glutamate levels in the cerebrospinal fluid predict clinical outcome of bacterial meningitis. Arch Neurol, 1996. [DOI | PubMed]
- BS Meldrum. The role of glutamate in epilepsy and other CNS disorders. Neurology, 1994. [PubMed]
- C Ferrarese, A Aliprandi, L Tremolizzo, L Stanzani, A De Micheli, A Dolara, L Frattola. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology, 2001. [DOI | PubMed]
- AS Miranda, LB Vieira, N Lacerda-Queiroz, AH Souza, DH Rodrigues, MC Vilela, MV Gomez, FS Machado, MA Rachid, AL Teixeira. Increased levels of glutamate in the central nervous system are associated with behavioral symptoms in experimental malaria. Braz J Med Biol Res, 2010. [DOI | PubMed]
- JF Stover, B Schoning, TF Beyer, C Woiciechowsky, AW Unterberg. Temporal profile of cerebrospinal fluid glutamate, interleukin-6, and tumor necrosis factor-alpha in relation to brain edema and contusion following controlled cortical impact injury in rats. Neurosci Lett, 2000. [DOI | PubMed]
- RA Ruppel, PM Kochanek, PD Adelson, ME Rose, SR Wisniewski, MJ Bell, RS Clark, DW Marion, SH Graham. Excitatory amino acid concentrations in ventricular cerebrospinal fluid after severe traumatic brain injury in infants and children: the role of child abuse. J Pediatr, 2001. [DOI | PubMed]
- 90.Fontana AC, Fox DP, Zoubroulis A, Mortensen OV, Raghupathi R. Neuroprotective effects of the glutamate transporter activator (R)-(-)-5-methyl-1-nicotinoyl-2-pyrazoline (MS-153) following traumatic brain injury in the adult rat. J Neurotrauma. 2015, in press.
- P Jourdain, LH Bergersen, K Bhaukaurally, P Bezzi, M Santello, M Domercq, C Matute, F Tonello, V Gundersen, A Volterra. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci, 2007. [DOI | PubMed]
- J Lewerenz, P Maher. Chronic glutamate toxicity in neurodegenerative diseases—what is the evidence?. Front Neurosci, 2015. [DOI | PubMed]
- M Kiaei, S Petri, K Kipiani, G Gardian, DK Choi, J Chen, NY Calingasan, P Schafer, GW Muller, C Stewart. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci, 2006. [DOI | PubMed]
- HY Hsiao, YC Chen, HM Chen, PH Tu, Y Chern. A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington’s disease. Hum Mol Genet, 2013. [DOI | PubMed]
- 95.Haroon E, Fleischer CC, Felger JC, Chen X, Woolwine BJ, Patel T, Hu XP, Miller AH. Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol Psychiatry. 2016, in press.
- NC Danbolt. Glutamate uptake. Prog Neurobiol, 2001. [DOI | PubMed]
- SM Fine, RA Angel, SW Perry, LG Epstein, JD Rothstein, S Dewhurst, HA Gelbard. Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes—implications for pathogenesis of HIV-1 dementia. J Biol Chem, 1996. [DOI | PubMed]
- JY Zou, FT Crews. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res, 2005. [DOI | PubMed]
- J Carmen, JD Rothstein, DA Kerr. Tumor necrosis factor-alpha modulates glutamate transport in the CNS and is a critical determinant of outcome from viral encephalomyelitis. Brain Res, 2009. [DOI | PubMed]
- D Stellwagen, EC Beattie, JY Seo, RC Malenka. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci, 2005. [DOI | PubMed]
- AO Dumont, S Goursaud, N Desmet, E Hermans. Differential regulation of glutamate transporter subtypes by pro-inflammatory cytokine TNF-alpha in cortical astrocytes from a rat model of amyotrophic lateral sclerosis. PLoS One, 2014. [DOI | PubMed]
- G Olmos, J Llado. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators Inflamm, 2014. [DOI | PubMed]
- B Viviani, M Boraso, N Marchetti, M Marinovich. Perspectives on neuroinflammation and excitotoxicity: a neurotoxic conspiracy?. Neurotoxicology, 2014. [DOI | PubMed]
- H Takeuchi, S Jin, J Wang, G Zhang, J Kawanokuchi, R Kuno, Y Sonobe, T Mizuno, A Suzumura. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem, 2006. [DOI | PubMed]
- CJ Chen, YC Ou, CY Chang, HC Pan, SL Liao, SY Chen, SL Raung, CY Lai. Glutamate released by Japanese encephalitis virus-infected microglia involves TNF-alpha signaling and contributes to neuronal death. Glia, 2012. [DOI | PubMed]
- L Ye, Y Huang, L Zhao, Y Li, L Sun, Y Zhou, G Qian, JC Zheng. IL-1beta and TNF-alpha induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem, 2013. [DOI | PubMed]
- J Ye, R Jiang, M Cui, B Zhu, L Sun, Y Wang, A Zohaib, Q Dong, X Ruan, Y Song. Etanercept reduces neuroinflammation and lethality in mouse model of Japanese encephalitis. J Infect Dis, 2014. [DOI | PubMed]
- 108.Osborn LM, Kamphuis W, Wadman WJ, Hol EM. Astrogliosis: an integral player in the pathogenesis of Alzheimer’s disease. Prog Neurobiol. 2016
- KL Moulder, JP Meeks, S Mennerick. Homeostatic regulation of glutamate release in response to depolarization. Mol Neurobiol, 2006. [DOI | PubMed]
- EB Malarkey, Y Ni, V Parpura. Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia, 2008. [DOI | PubMed]
- MS Santos, H Li, SM Voglmaier. Synaptic vesicle protein trafficking at the glutamate synapse. Neuroscience, 2009. [DOI | PubMed]
- LR Weitzel, PE Wischmeyer. Glutamine in critical illness: the time has come, the time is now. Crit Care Clin, 2010. [DOI | PubMed]
- DK Heyland, G Elke, D Cook, MM Berger, PE Wischmeyer, M Albert, J Muscedere, G Jones, AG Day. Glutamine and antioxidants in the critically ill patient: a post hoc analysis of a large-scale randomized trial. JPEN J Parenter Enteral Nutr, 2015. [DOI | PubMed]
- I Morales, M Rodriguez. Self-induced accumulation of glutamate in striatal astrocytes and basal ganglia excitotoxicity. Glia, 2012. [DOI | PubMed]
- J Castillo, MI Loza, D Mirelman, J Brea, M Blanco, T Sobrino, F Campos. A novel mechanism of neuroprotection: blood glutamate grabber. J Cereb Blood Flow Metab, 2015. [DOI | PubMed]
- H Jing, Y Hao, Q Bi, J Zhang, P Yang. Intra-amygdala microinjection of TNF-alpha impairs the auditory fear conditioning of rats via glutamate toxicity. Neurosci Res, 2015. [DOI | PubMed]
- CC Chio, JW Lin, MW Chang, CC Wang, CZ Yang, CP Chang. Therapeutic evaluation of etanercept in a model of traumatic brain injury. J Neurochem, 2010. [DOI | PubMed]
- YM Kang, RL He, LM Yang, DN Qin, A Guggilam, C Elks, N Yan, Z Guo, J Francis. Brain tumour necrosis factor-alpha modulates neurotransmitters in hypothalamic paraventricular nucleus in heart failure. Cardiovasc Res, 2009. [DOI | PubMed]
- YM Kang, Y Wang, LM Yang, C Elks, J Cardinale, XJ Yu, XF Zhao, J Zhang, LH Zhang, ZM Yang, J Francis. TNF-alpha in hypothalamic paraventricular nucleus contributes to sympathoexcitation in heart failure by modulating AT1 receptor and neurotransmitters. Tohoku J Exp Med, 2010. [DOI | PubMed]
- D Jabaudon, K Shimamoto, Y Yasuda-Kamatani, M Scanziani, BH Gahwiler, U Gerber. Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci U S A, 1999. [DOI | PubMed]
- EL Tobinick, S Britschgi-Davoodifar. Perispinal TNF-alpha inhibition for discogenic pain. Swiss Med Wkly, 2003. [PubMed]
- EL Tobinick, H Gross. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. J Neuroinflammation, 2008. [DOI | PubMed]
- WS Griffin. Perispinal etanercept: potential as an Alzheimer therapeutic. J Neuroinflammation, 2008. [DOI | PubMed]
- E Tobinick. Rapid improvement of chronic stroke deficits after perispinal etanercept: three consecutive cases. CNS Drugs, 2011. [DOI | PubMed]
- E Tobinick, NM Kim, G Reyzin, H Rodriguez-Romanacce, V DePuy. Selective TNF inhibition for chronic stroke and traumatic brain injury: an observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs, 2012. [DOI | PubMed]
- TA Ignatowski, RN Spengler, KM Dhandapani, H Folkersma, RF Butterworth, E Tobinick. Perispinal etanercept for post-stroke neurological and cognitive dysfunction: Scientific rationale and current evidence. CNS Drugs, 2014. [DOI | PubMed]
- E Tobinick, H Rodriguez-Romanacce, A Levine, TA Ignatowski, RN Spengler. Immediate neurological recovery following perispinal etanercept years after brain injury. Clin Drug Investig, 2014. [DOI | PubMed]
- E Tobinick. Deciphering the physiology underlying the rapid clinical effects of perispinal etanercept in Alzheimer’s disease. Current Alzheimers Dis, 2012. [DOI]
- K Peppel, D Crawford, B Beutler. A tumor necrosis factor (TNF) receptor-IgG heavy chain chimeric protein as a bivalent antagonist of TNF activity. J Exp Med, 1991. [DOI | PubMed]
- 130.Emery P, Vencovsky J, Sylwestrzak A, Leszczynski P, Porawska W, Baranauskaite A, Tseluyko V, Zhdan VM, Stasiuk B, Milasiene R, et al. A phase III randomised, double-blind, parallel-group study comparing SB4 with etanercept reference product in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2015, in press.
- 131.Lee YJ, Shin D, Kim Y, Kang JW, Gauliard A, Fuhr R. A randomised phase l pharmacokinetic study comparing SB4 and etanercept reference product (Enbrel(R)) in healthy subjects. Br J Clin Pharmacol. 2016;82: 64-73.
- VF Azevedo, N Galli, A Kleinfelder, J D’Ippolito, PC Urbano. Etanercept biosimilars. Rheumatol Int, 2015. [DOI | PubMed]
- K Chapman, A Adjei, P Baldrick, A da Silva, K De Smet, R DiCicco, SS Hong, D Jones, MW Leach, J McBlane. Waiving in vivo studies for monoclonal antibody biosimilar development: national and global challenges. MAbs, 2016. [DOI | PubMed]
- L Probert. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience, 2015. [DOI | PubMed]
- BT Baune. Inflammation and neurodegenerative disorders: is there still hope for therapeutic intervention?. Curr Opin Psychiatry, 2015. [PubMed]
- D Baeten, PM van Hagen. Use of TNF blockers and other targeted therapies in rare refractory immune-mediated inflammatory diseases: evidence-based or rational?. Ann Rheum Dis, 2010. [DOI | PubMed]
- EL Tobinick. Perispinal delivery of CNS drugs. CNS Drugs, 2016. [DOI | PubMed]
- TS Wen, DC Randall, JF Zolman. Protein accumulation in cerebrospinal fluid during −90 degrees head-down tilt in rabbit. J Appl Physiol, 1994. [PubMed]
- E Tobinick, CP Vega. The cerebrospinal venous system: anatomy, physiology, and clinical implications. Med Gen Med, 2006. [DOI]
- EL Tobinick. Perispinal etanercept for neuroinflammatory disorders. Drug Discov Today, 2009. [DOI | PubMed]
- EL Tobinick, H Gross, A Weinberger, H Cohen. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. Medscape Gen Med Neurol Neurosurg, 2006
- JS Yoon, JH Lee, D Tweedie, MR Mughal, S Chigurupati, NH Greig, MP Mattson. 3,6’-dithiothalidomide improves experimental stroke outcome by suppressing neuroinflammation. J Neurosci Res, 2013. [DOI | PubMed]
- GA Cabral, M Jamerson. Marijuana use and brain immune mechanisms. Int Rev Neurobiol, 2014. [DOI | PubMed]
- CC Chio, CH Chang, CC Wang, CU Cheong, CM Chao, BC Cheng, CZ Yang, CP Chang. Etanercept attenuates traumatic brain injury in rats by reducing early microglial expression of tumor necrosis factor-alpha. BMC Neurosci, 2013. [DOI | PubMed]
- WM Pardridge. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab, 2012. [DOI | PubMed]
- 146.Armagen: http://www.prnewswire.com/news-releases/armagen-technologies-inc-announces-17-million-series-a-financing-181460481.html. 2012.
- WM Pardridge. Blood-brain barrier drug delivery of IgG fusion proteins with a transferrin receptor monoclonal antibody. Expert Opin Drug Deliv, 2015. [DOI | PubMed]
- JA Couch, YJ Yu, Y Zhang, JM Tarrant, RN Fuji, WJ Meilandt, H Solanoy, RK Tong, K Hoyte, W Luk. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med, 2013. [DOI]
- N Bien-Ly, YJ Yu, D Bumbaca, J Elstrott, CA Boswell, Y Zhang, W Luk, Y Lu, MS Dennis, RM Weimer. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J Exp Med, 2014. [DOI | PubMed]
- YJ Yu, JK Atwal, Y Zhang, RK Tong, KR Wildsmith, C Tan, N Bien-Ly, M Hersom, JA Maloney, WJ Meilandt. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med, 2014. [DOI | PubMed]
- QH Zhou, RJ Boado, EK Hui, JZ Lu, WM Pardridge. Brain-penetrating tumor necrosis factor decoy receptor in the mouse. Drug Metab Dispos, 2011. [DOI | PubMed]
- RK Sumbria, RJ Boado, WM Pardridge. Brain protection from stroke with intravenous TNFalpha decoy receptor-Trojan horse fusion protein. J Cereb Blood Flow Metab, 2012. [DOI | PubMed]
- NQ Shi, XR Qi, B Xiang, Y Zhang. A survey on “Trojan Horse” peptides: opportunities, issues and controlled entry to “Troy”. J Control Release, 2014. [DOI | PubMed]
- EP Sampaio, EN Sarno, R Galilly, ZA Cohn, G Kaplan. Thalidomide selectively inhibits tumor necrosis factor-alpha production by stimulated human monocytes. J Exp Med, 1991. [DOI | PubMed]
- AL Moreira, EP Sampaio, A Zmuidzinas, P Frindt, KA Smith, G Kaplan. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J Exp Med, 1993. [DOI | PubMed]
- X Zhu, T Giordano, QS Yu, HW Holloway, TA Perry, DK Lahiri, A Brossi, NH Greig. Thiothalidomides: novel isosteric analogues of thalidomide with enhanced TNF-alpha inhibitory activity. J Med Chem, 2003. [DOI | PubMed]
- NH Greig, T Giordano, X Zhu, QS Yu, TA Perry, HW Holloway, A Brossi, JT Rogers, K Sambamurti, DK Lahiri. Thalidomide-based TNF-alpha inhibitors for neurodegenerative diseases. Acta Neurobiol Exp, 2004
- K Belarbi, T Jopson, D Tweedie, C Arellano, W Luo, NH Greig, S Rosi. TNF-alpha protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J Neuroinflamm, 2012. [DOI]
- D Tweedie, RA Ferguson, K Fishman, KA Frankola, H Van Praag, HW Holloway, W Luo, Y Li, L Caracciolo, I Russo. Tumor necrosis factor-alpha synthesis inhibitor 3,6’-dithiothalidomide attenuates markers of inflammation. Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J Neuroinflammation, 2012. [DOI | PubMed]
- SP Gabbita, MK Srivastava, P Eslami, MF Johnson, NK Kobritz, D Tweedie, NH Greig, FP Zemlan, SP Sharma, ME Harris-White. Early intervention with a small molecule inhibitor for tumor necrosis factor-alpha prevents cognitive deficits in a triple transgenic mouse model of Alzheimer’s disease. J Neuroinflammation, 2012. [DOI | PubMed]
- I Russo, L Caracciolo, D Tweedie, SH Choi, NH Greig, S Barlati, F Bosetti. 3,6’-Dithiothalidomide, a new TNF-alpha synthesis inhibitor, attenuates the effect of Abeta1-42 intracerebroventricular injection on hippocampal neurogenesis and memory deficit. J Neurochem, 2012. [DOI | PubMed]
- T Song, X Ma, K Gu, Y Yang, L Yang, P Ma, W Wang, J Zhao, R Yan, J Guan. Thalidomide represses inflammatory response and reduces radiculopathic pain by inhibiting IRAK-1 and NF-kappaB/p38/JNK signaling. J Neuroimmunol, 2016. [DOI | PubMed]
- HE Boycott, JA Wilkinson, JP Boyle, HA Pearson, C Peers. Differential involvement of TNF alpha in hypoxic suppression of astrocyte glutamate transporters. Glia, 2008. [DOI | PubMed]
- T Maekawa, E Ashihara, S Kimura. The Bcr-Abl tyrosine kinase inhibitor imatinib and promising new agents against Philadelphia chromosome-positive leukemias. Int J Clin Oncol, 2007. [DOI | PubMed]
- M Breccia, G Alimena. Nilotinib: a second-generation tyrosine kinase inhibitor for chronic myeloid leukemia. Leuk Res, 2010. [DOI | PubMed]
- F Pagan. Society for Neuroscience (SfN) 2015 Annual Meeting. Abstract 12.01. Presented October 18, 2015, 2015
- ML Hebron, I Lonskaya, CE Moussa. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson’s disease models. Hum Mol Genet, 2013. [DOI | PubMed]
- A Tanabe, Y Yamamura, J Kasahara, R Morigaki, R Kaji, S Goto. A novel tyrosine kinase inhibitor AMN107 (nilotinib) normalizes striatal motor behaviors in a mouse model of Parkinson’s disease. Front Cell Neurosci, 2014. [DOI | PubMed]
- SS Karuppagounder, S Brahmachari, Y Lee, VL Dawson, TM Dawson, HS Ko. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci Rep, 2014. [DOI | PubMed]
- G Dhawan, CK Combs. Inhibition of Src kinase activity attenuates amyloid associated microgliosis in a murine model of Alzheimer’s disease. J Neuroinflammation, 2012. [DOI | PubMed]
- G Dhawan, AM Floden, CK Combs. Amyloid-beta oligomers stimulate microglia through a tyrosine kinase dependent mechanism. Neurobiol Aging, 2012. [DOI | PubMed]
- I Lonskaya, ML Hebron, NM Desforges, JB Schachter, CE Moussa. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J Mol Med (Berl), 2014. [DOI | PubMed]
- LM Ocuin, S Zeng, MJ Cavnar, EC Sorenson, ZM Bamboat, JB Greer, TS Kim, R Popow, RP DeMatteo. Nilotinib protects the murine liver from ischemia/reperfusion injury. J Hepatol, 2012. [DOI | PubMed]
- DS El-Agamy. Nilotinib ameliorates lipopolysaccharide-induced acute lung injury in rats. Toxicol Appl Pharmacol, 2011. [DOI | PubMed]
- MK McCoy, TN Martinez, KA Ruhn, DE Szymkowski, CG Smith, BR Botterman, KE Tansey, MG Tansey. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J Neurosci, 2006. [DOI | PubMed]
- SJ Blake, AB Lyons, TP Hughes. Nilotinib inhibits the Src-family kinase LCK and T-cell function in vitro. J Cell Mol Med, 2009. [DOI | PubMed]
- M Ferlito, OG Romanenko, K Guyton, S Ashton, F Squadrito, PV Halushka, JA Cook. Implication of Galpha i proteins and Src tyrosine kinases in endotoxin-induced signal transduction events and mediator production. J Endotoxin Res, 2002. [PubMed]
- A Chawla, KD Nguyen, YP Goh. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol, 2011. [DOI | PubMed]
- D Mokhtari, N Welsh. Potential utility of small tyrosine kinase inhibitors in the treatment of diabetes. Clin Sci (Lond), 2010. [DOI | PubMed]
- S Chattopadhyay, GC Sen. Tyrosine phosphorylation in Toll-like receptor signaling. Cytokine Growth Factor Rev, 2014. [DOI | PubMed]
- 181.Xiong Y, Murphy M, Manavalan TT, Pattabiraman G, Qiu F, Chang HH, Ho IC, Medvedev AE. Endotoxin tolerance inhibits lyn and c-Src phosphorylation and association with toll-like receptor 4 but increases expression and activity of protein phosphatases. J Innate Immun. 2015
- R Mechoulam, L Hanus. A historical overview of chemical research on cannabinoids. Chem Phys Lipids, 2000. [DOI | PubMed]
- M de Vries, DC van Rijckevorsel, OH Wilder-Smith, H van Goor. Dronabinol and chronic pain: importance of mechanistic considerations. Expert Opin Pharmacother, 2014. [DOI | PubMed]
- D Piomelli. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci, 2003. [DOI | PubMed]
- JY Xu, C Chen. Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist, 2015. [DOI | PubMed]
- SG Kinsey, JZ Long, ST O’Neal, RA Abdullah, JL Poklis, DL Boger, BF Cravatt, AH Lichtman. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther, 2009. [DOI | PubMed]
- JR Johnson, M Burnell-Nugent, D Lossignol, ED Ganae-Motan, R Potts, MT Fallon. Multicenter, double-blind, randomized, placebo-controlled, parallel-group study of the efficacy, safety, and tolerability of THC:CBD extract and THC extract in patients with intractable cancer-related pain. J Pain Symptom Manage, 2010. [DOI | PubMed]
- J Guindon, A Guijarro, D Piomelli, AG Hohmann. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br J Pharmacol, 2011. [DOI | PubMed]
- BK Lau, CW Vaughan. Targeting the endogenous cannabinoid system to treat neuropathic pain (some toxicity, anti-TNF at least specific). Front Pharmacol, 2014. [DOI | PubMed]
- M Serpell, S Ratcliffe, J Hovorka, M Schofield, L Taylor, H Lauder, E Ehler. A double-blind, randomized, placebo-controlled, parallel group study of THC/CBD spray in peripheral neuropathic pain treatment. Eur J Pain, 2014. [DOI | PubMed]
- X Chen, J Zhang, C Chen. Endocannabinoid 2-arachidonoylglycerol protects neurons against beta-amyloid insults. Neuroscience, 2011. [DOI | PubMed]
- D Cheng, JK Low, W Logge, B Garner, T Karl. Chronic cannabidiol treatment improves social and object recognition in double transgenic APPswe/PS1E9 mice. Psychopharmacology (Berl), 2014. [DOI | PubMed]
- D Cheng, AS Spiro, AM Jenner, B Garner, T Karl. Long-term cannabidiol treatment prevents the development of social recognition memory deficits in Alzheimer’s disease transgenic mice. J Alzheimers Dis, 2014. [PubMed]
- T Karl, D Cheng, B Garner, JC Arnold. The therapeutic potential of the endocannabinoid system for Alzheimer’s disease. Expert Opin Ther Targets, 2012. [DOI | PubMed]
- BG Ramirez, C Blazquez, T Gomez del Pulgar, M Guzman, ML de Ceballos. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci, 2005. [DOI | PubMed]
- MJ Wallace, RE Blair, KW Falenski, BR Martin, RJ DeLorenzo. The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. J Pharmacol Exp Ther, 2003. [DOI | PubMed]
- RE Blair, LS Deshpande, S Sombati, KW Falenski, BR Martin, RJ DeLorenzo. Activation of the cannabinoid type-1 receptor mediates the anticonvulsant properties of cannabinoids in the hippocampal neuronal culture models of acquired epilepsy and status epilepticus. J Pharmacol Exp Ther, 2006. [DOI | PubMed]
- MD Bhaskaran, BN Smith. Cannabinoid-mediated inhibition of recurrent excitatory circuitry in the dentate gyrus in a mouse model of temporal lobe epilepsy. PLoS One, 2010. [DOI | PubMed]
- I Soltesz, BE Alger, M Kano, SH Lee, DM Lovinger, T Ohno-Shosaku, M Watanabe. Weeding out bad waves: towards selective cannabinoid circuit control in epilepsy. Nat Rev Neurosci, 2015. [DOI | PubMed]
- DA Price, AA Martinez, A Seillier, W Koek, Y Acosta, E Fernandez, R Strong, B Lutz, G Marsicano, JL Roberts, A Giuffrida. WIN55,212-2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Eur J Neurosci, 2009. [DOI | PubMed]
- YC Chung, E Bok, SH Huh, JY Park, SH Yoon, SR Kim, YS Kim, S Maeng, SH Park, BK Jin. Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J Immunol, 2011. [DOI | PubMed]
- D Fernandez-Suarez, M Celorrio, JI Riezu-Boj, A Ugarte, R Pacheco, H Gonzalez, J Oyarzabal, CJ Hillard, R Franco, MS Aymerich. Monoacylglycerol lipase inhibitor JZL184 is neuroprotective and alters glial cell phenotype in the chronic MPTP mouse model. Neurobiol Aging, 2014. [DOI | PubMed]
- TS Lu, HK Avraham, S Seng, SD Tachado, H Koziel, A Makriyannis, S Avraham. Cannabinoids inhibit HIV-1 Gp120-mediated insults in brain microvascular endothelial cells. J Immunol, 2008. [DOI | PubMed]
- M Bari, C Rapino, P Mozetic, M Maccarrone. The endocannabinoid system in gp120-mediated insults and HIV-associated dementia. Exp Neurol, 2010. [DOI | PubMed]
- HJ Kim, AH Shin, SA Thayer. Activation of cannabinoid type 2 receptors inhibits HIV-1 envelope glycoprotein gp120-induced synapse loss. Mol Pharmacol, 2011. [DOI | PubMed]
- MV Solbrig, Y Fan, P Hazelton. Prospects for cannabinoid therapies in viral encephalitis. Brain Res, 2013. [DOI | PubMed]
- D Panikashvili, C Simeonidou, S Ben-Shabat, L Hanus, A Breuer, R Mechoulam, E Shohami. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature, 2001. [DOI | PubMed]
- D Fernandez-Lopez, MR Pazos, RM Tolon, MA Moro, J Romero, I Lizasoain, J Martinez-Orgado. The cannabinoid agonist WIN55212 reduces brain damage in an in vivo model of hypoxic-ischemic encephalopathy in newborn rats. Pediatr Res, 2007. [DOI | PubMed]
- S Carloni, D Alonso-Alconada, S Girelli, A Duranti, A Tontini, D Piomelli, E Hilario, A Alvarez, W Balduini. Pretreatment with the monoacylglycerol lipase inhibitor URB602 protects from the long-term consequences of neonatal hypoxic-ischemic brain injury in rats. Pediatr Res, 2012. [DOI | PubMed]
- MR Pazos, N Mohammed, H Lafuente, M Santos, E Martinez-Pinilla, E Moreno, E Valdizan, J Romero, A Pazos, R Franco. Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: role of 5HT(1A) and CB2 receptors. Neuropharmacology, 2013. [DOI | PubMed]
- R Gallily, A Breuer, R Mechoulam. 2-Arachidonylglycerol, an endogenous cannabinoid, inhibits tumor necrosis factor-alpha production in murine macrophages, and in mice (endogenous). Eur J Pharmacol, 2000. [DOI | PubMed]
- F Facchinetti, E Del Giudice, S Furegato, M Passarotto, A Leon. Cannabinoids ablate release of TNFalpha in rat microglial cells stimulated with lypopolysaccharide (both synthetic and endogenous). Glia, 2003. [DOI | PubMed]
- CG Haj, PF Sumariwalla, L Hanus, NM Kogan, Z Yektin, R Mechoulam, M Feldmann, R Gallily. HU-444, a novel, potent anti-inflammatory, nonpsychotropic cannabinoid (puts TNF down). J Pharmacol Exp Ther, 2015. [DOI | PubMed]
- AC Campos, F Brant, AS Miranda, FS Machado, AL Teixeira. Cannabidiol increases survival and promotes rescue of cognitive function in a murine model of cerebral malaria. Neuroscience, 2015. [DOI | PubMed]
- EB Gordon, GT Hart, TM Tran, M Waisberg, M Akkaya, AS Kim, SE Hamilton, M Pena, T Yazew, CF Qi. Targeting glutamine metabolism rescues mice from late-stage cerebral malaria. Proc Natl Acad Sci U S A, 2015. [DOI | PubMed]
- M Shen, TM Piser, VS Seybold, SA Thayer. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci, 1996. [PubMed]
- N Hajos, C Ledent, TF Freund. Novel cannabinoid-sensitive receptor mediates inhibition of glutamatergic synaptic transmission in the hippocampus. Neuroscience, 2001. [DOI | PubMed]
- G Gerdeman, DM Lovinger. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol, 2001. [PubMed]
- MR Domenici, SC Azad, G Marsicano, A Schierloh, CT Wotjak, HU Dodt, W Zieglgansberger, B Lutz, G Rammes. Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci, 2006. [DOI | PubMed]
- KA Takahashi, PE Castillo. The CB1 cannabinoid receptor mediates glutamatergic synaptic suppression in the hippocampus. Neuroscience, 2006. [DOI | PubMed]
- S Barzegar, A Komaki, S Shahidi, A Sarihi, N Mirazi, I Salehi. Effects of cannabinoid and glutamate receptor antagonists and their interactions on learning and memory in male rats. Pharmacol Biochem Behav, 2015. [DOI | PubMed]
- GL Coffey, J Ehrlich, MW Fisher, AB Hillegas, DL Kohberger, HE Machamer, WA Rightsel, FR Roegner. 6-Diazo-5-oxo-L-norleucine, a new tumor-inhibitory substance. I. Biologic studies. Antibiot Chemother (Northfield), 1956. [PubMed]
- F Conti, A Minelli. Glutamate immunoreactivity in rat cerebral cortex is reversibly abolished by 6-diazo-5-oxo-L-norleucine (DON), an inhibitor of phosphate-activated glutaminase. J Histochem Cytochem, 1994. [DOI | PubMed]
- RH Earhart, JM Koeller, HL Davis. Phase I trial of 6-diazo-5-oxo-L-norleucine (DON) administered by 5-day courses. Cancer Treat Rep, 1982. [PubMed]
- H Takeuchi, S Jin, H Suzuki, Y Doi, J Liang, J Kawanokuchi, T Mizuno, M Sawada, A Suzumura. Blockade of microglial glutamate release protects against ischemic brain injury. Exp Neurol, 2008. [DOI | PubMed]
- NA Prow, DN Irani. The inflammatory cytokine, interleukin-1 beta, mediates loss of astroglial glutamate transport and drives excitotoxic motor neuron injury in the spinal cord during acute viral encephalomyelitis. J Neurochem, 2008. [DOI | PubMed]
- MC Potter, VK Baxter, RW Mathey, J Alt, C Rojas, DE Griffin, BS Slusher. Neurological sequelae induced by alphavirus infection of the CNS are attenuated by treatment with the glutamine antagonist 6-diazo-5-oxo-l-norleucine. J Neurovirol, 2015. [DOI | PubMed]
- J Zhao, AL Lopez, D Erichsen, S Herek, RL Cotter, NP Curthoys, J Zheng. Mitochondrial glutaminase enhances extracellular glutamate production in HIV-1-infected macrophages: linkage to HIV-1 associated dementia. J Neurochem, 2004. [DOI | PubMed]
- J Shijie, H Takeuchi, I Yawata, Y Harada, Y Sonobe, Y Doi, J Liang, L Hua, S Yasuoka, Y Zhou. Blockade of glutamate release from microglia attenuates experimental autoimmune encephalomyelitis in mice. Tohoku J Exp Med, 2009. [DOI | PubMed]
- MB Van Hensbroek, A Palmer, E Onyiorah, G Schneider, S Jaffar, G Dolan, H Memming, J Frenkel, G Enwere, S Bennett. The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J Infect Dis, 1996. [DOI | PubMed]
- KR Vogel, E Arning, BL Wasek, T Bottiglieri, KM Gibson. Non-physiological amino acid (NPAA) therapy targeting brain phenylalanine reduction: pilot studies in PAHENU2 mice. J Inherit Metab Dis, 2013. [DOI | PubMed]
- F Meloni, P Ballabio, L Bianchi, FA Grassi, GG Gialdroni Grassi. Cefodizime modulates in vitro tumor necrosis factor-alpha, interleukin-6 and interleukin-8 release from human peripheral monocytes. Chemotherapy, 1995. [DOI | PubMed]
- SM Rawls, R Tallarida, W Robinson, M Amin. The beta-lactam antibiotic, ceftriaxone, attenuates morphine-evoked hyperthermia in rats. Br J Pharmacol, 2007. [DOI | PubMed]
- SG Lee, ZZ Su, L Emdad, P Gupta, D Sarkar, A Borjabad, DJ Volsky, PB Fisher. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem, 2008. [DOI | PubMed]
- J Zumkehr, CJ Rodriguez-Ortiz, D Cheng, Z Kieu, T Wai, C Hawkins, J Kilian, SL Lim, R Medeiros, M Kitazawa. Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer’s disease. Neurobiol Aging, 2015. [DOI | PubMed]
- T Inui, B Alessandri, A Heimann, F Nishimura, K Frauenknecht, C Sommer, O Kempski. Neuroprotective effect of ceftriaxone on the penumbra in a rat venous ischemia model. Neuroscience, 2013. [DOI | PubMed]
- J Wei, X Pan, Z Pei, W Wang, W Qiu, Z Shi, G Xiao. The beta-lactam antibiotic, ceftriaxone, provides neuroprotective potential via anti-excitotoxicity and anti-inflammation response in a rat model of traumatic brain injury. J Trauma Acute Care Surg, 2012. [DOI | PubMed]
- GS Goodrich, AY Kabakov, MQ Hameed, SC Dhamne, PA Rosenberg, A Rotenberg. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J Neurotrauma, 2013. [DOI | PubMed]
- C Cui, Y Cui, J Gao, L Sun, Y Wang, K Wang, R Li, Y Tian, S Song, J Cui. Neuroprotective effect of ceftriaxone in a rat model of traumatic brain injury. Neurol Sci, 2014. [DOI | PubMed]
- TC Leung, CN Lui, LW Chen, WH Yung, YS Chan, KK Yung. Ceftriaxone ameliorates motor deficits and protects dopaminergic neurons in 6-hydroxydopamine-lesioned rats. ACS Chem Neurosci, 2012. [DOI | PubMed]
- T Chotibut, RW Davis, JC Arnold, Z Frenchek, S Gurwara, V Bondada, JW Geddes, MF Salvatore. Ceftriaxone increases glutamate uptake and reduces striatal tyrosine hydroxylase loss in 6-OHDA Parkinson’s model. Mol Neurobiol, 2014. [DOI | PubMed]
- CK Huang, YT Chang, TG Amstislavskaya, MA Tikhonova, CL Lin, CS Hung, TJ Lai, YJ Ho. Synergistic effects of ceftriaxone and erythropoietin on neuronal and behavioral deficits in an MPTP-induced animal model of Parkinson’s disease dementia. Behav Brain Res, 2015. [DOI | PubMed]
- JN Weng, MA Tikhonova, JH Chen, MS Shen, WY Meng, YT Chang, KH Chen, KC Liang, CS Hung, TG Amstislavskaya, YJ Ho. Ceftriaxone prevents the neurodegeneration and decreased neurogenesis seen in a Parkinson’s disease rat model: an immunohistochemical and MRI study. Behav Brain Res, 2016. [DOI | PubMed]
- CN David, ES Frias, JI Szu, PA Vieira, JA Hubbard, J Lovelace, M Michael, D Worth, KE McGovern, IM Ethell. GLT-1-dependent disruption of CNS glutamate homeostasis and neuronal function by the protozoan parasite Toxoplasma gondii. PLoS Pathog, 2016. [DOI | PubMed]
- E Fumagalli, M Funicello, T Rauen, M Gobbi, T Mennini. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol, 2008. [DOI | PubMed]
- M Carbone, S Duty, M Rattray. Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochem Int, 2012. [DOI | PubMed]
- OP Dall’Igna, LD Bobermin, DO Souza, A Quincozes-Santos. Riluzole increases glutamate uptake by cultured C6 astroglial cells. Int J Dev Neurosci, 2013. [DOI | PubMed]
- HC Hunsberger, DS Weitzner, CC Rudy, JE Hickman, EM Libell, RR Speer, GA Gerhardt, MN Reed. Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. J Neurochem, 2015. [DOI | PubMed]
- 249.Pereira AC, Gray JD, Kogan JF, Davidson RL, Rubin TG, Okamoto M, Morrison JH, McEwen BS. Age and Alzheimer’s disease gene expression profiles reversed by the glutamate modulator riluzole. Mol Psychiatry. 2016