Antibacterial and Antibiofilm Activity of 8‐Hydroxyquinoline Derivatives Against Mycobacterium and Staphylococcus Species
Abstract
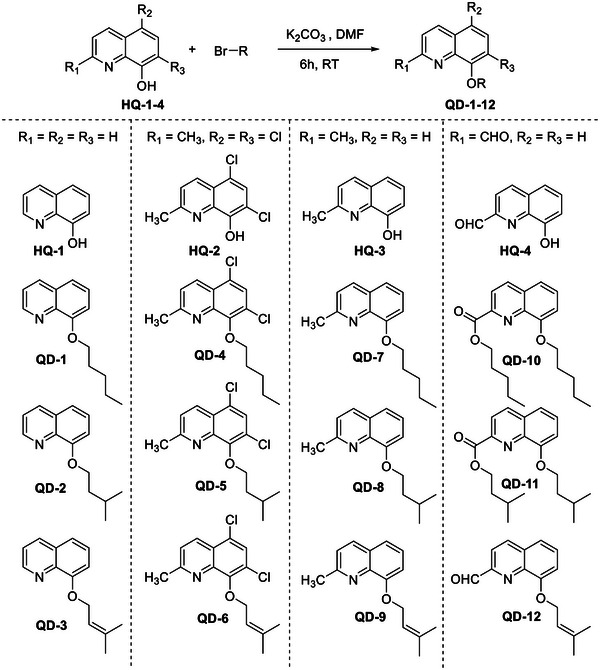
A series of 8‐alkoxyquinoline derivatives (QD‐1–12) were designed and synthesized on the basis of analogues of 8‐hydroxyquinoline (8‐HQ) (HQ 1–4). The compounds were evaluated for biofilm inhibition against Mycobacterium smegmatis and Staphylococcus aureus, including antibacterial activity against Mycobacterium tuberculosis, M. smegmatis and S. aureus. Cytotoxicity was evaluated against human monocyte (U937) and African green monkey kidney (Vero) cell lines. The 8‐O‐prenyl derivative (QD‐12) showed a minimum inhibitory concentration (MIC) of 12.5 µM, indicating an approximate 8‐fold increased selectivity for the biofilm phenotype and an increased inhibitory activity against methicillin‐resistant S. aureus (MRSA) by up to 2‐fold. 5,7‐Dichloro‐8‐hydroxy‐2‐methylquinoline (HQ‐2) showed the highest inhibitory potential with MIC values of 0.1, 1.56, 2.2 and 1.1 µM against M. tuberculosis, M. smegmatis, methicillin‐sensitive S. aureus (MSSA) and MRSA, respectively. The results indicate the importance of the 8‐OH group for antibacterial and antimycobacterial activity. Cytotoxicity revealed low‐to‐moderate toxicity of 8‐HQ (HQ‐1). All the compounds, except HQ‐1, were tested for the first time for their growth and biofilm inhibitory activity against Mycobacterium spp. and S. aureus.
Article type: Research Article
Keywords: antibiofilm, antimycobacterial, antistaphylococcal, cytotoxicity, quinolines, structure–activity relationships
Affiliations: Department of Plant and Soil Sciences, Faculty of Natural and Agricultural Sciences University of Pretoria Pretoria Gauteng South Africa; School of Natural Resources University of Missouri Columbia Missouri USA; College of Pharmacy JSS Academy of Higher Education and Research, Mysuru Bonne Terre Vacoas Mauritius; South African International Maritime Institute (SAIMI) Nelson Mandela University Gqeberha South Africa; Center for the Study of Human Health Emory University College of Arts and Sciences Atlanta Georgia USA; Department of Dermatology Emory University School of Medicine Atlanta Georgia USA
License: © 2025 The Author(s). Chemistry & Biodiversity published by Wiley‐VHCA AG. CC BY 4.0 This is an open access article under the terms of the http://creativecommons.org/licenses/by/4.0/ License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Article links: DOI: 10.1002/cbdv.202501892 | PubMed: 40768666 | PMC: PMC12716008
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (2.4 MB)
Introduction
The natural product, 8‐hydroxyquinoline (8‐HQ), is a phytotoxic component found in the roots of Centaurea diffusa Lam. [ref. 1]. It has shown potent activity against Mycobacterium tuberculosis (H37Rv) under replicating and non‐replicating conditions [ref. 2]. Its mono‐halogenated derivative, 5‐chloroquin‐8‐ol, has shown more prominent activity against standard strains and drug‐resistant clinical isolates of M. tuberculosis with a unique mode of action [ref. 3]. A dihalogenated derivative, clioquinol (5‐chloro‐7‐iodo‐quinolin‐8‐ol), was reported to have a synergistic effect, when combined with a sub‐active dose of streptomycin, on M. tuberculosis–infected guinea pigs [ref. 4]. To date, more than two hundred 8‐HQ derivatives have been evaluated for their antitubercular potential [ref. 5, ref. 6]. Furthermore, derivatization of some natural products has been shown to result in increased biological activity [ref. 7]. Recently, a quinolone‐based molecule, bedaquiline, was approved by the US Food and Drug Administration (FDA) and the European Medicine Agency as a drug for the treatment of drug‐resistant TB [ref. 8]. Bedaquiline is a novel mycobacteria‐specific adenosine triphosphate (ATP) synthase inhibitor [ref. 9]. The dihalogenated 8‐HQs (quinolin‐8‐ols), including broxyquinoline, clioquinol, chlorquinaldol and iodoquinol, were widely used to treat intestinal infection during the mid‐19th century [ref. 3]. In addition, hydroxyquinolines have been used for the treatment of viral and protozoal infections [ref. 10]. Clioquinol, an antifungal and antiprotozoal drug, has also been investigated in a Phase II clinical trial for the treatment of Alzheimer’s disease [ref. 11]. Furthermore, several 8‐HQ derivatives have shown activity against sensitive and resistant strains of bacteria and their associated persistent biofilms [ref. 12, ref. 13, ref. 14, ref. 15].
Bacterial biofilms are intrinsically resistant to conventional antibiotics and are a leading cause of microbial survival, tolerance and antibiotic resistance. Biofilms are complex structures of sessile communities of microorganisms attached to biotic or abiotic surfaces and can be free‐floating. In most biofilms, microbial cells are embedded in self‐synthesized extracellular matrices, composed of polymeric substances (EPSs), such as polysaccharides, proteins, lipids and extracellular DNA (e‐DNA), as well as molecules originating from the host, including mucus and DNA. Microbial cells in biofilms are nearly one thousand times more tolerant to antibiotic therapies than their planktonic counterparts [ref. 16, ref. 17]. Considerable research efforts have been made towards the development of new and non‐conventional antimicrobial agents suitable for the treatment of virulence factors and biofilm‐associated infections. A significant, and an often overlooked, contributor to infectious disease, including tuberculosis and Staphylococcus infections, is the ability of the bacteria to form biofilms within the host, providing a protective niche that confers tolerance to conventional antibiotic drugs and host immune responses [ref. 18, ref. 19]. The presence of these highly resilient biofilms leads to longer treatment periods, increased rates of treatment failure and relapse, and the emergence of drug‐resistance, thereby exacerbating the disease burden and necessitating the urgent discovery of novel antibiofilm agents to improve therapeutic outcomes [ref. 20, ref. 21, ref. 22].
Numerous compounds have shown interesting activities in the prophylaxis and treatment of biofilm‐related diseases [ref. 23]. Quinoline‐based compounds are one of the noteworthy classes of promising antibiofilm compounds. Recently, several halogenated hydroxyquinolines and derivatives have shown promising antibiofilm activity against several bacteria, either by inhibiting biofilm formation or by dispersing pre‐formed biofilms [ref. 13, ref. 24, ref. 25]. Although 8‐alkoxyquinolines were reported to have poor antibacterial activity against Staphylococcus aureus, several acyl derivatives of 8‐HQs have shown prominent biofilm dispersion activity [ref. 13]. No such structure–activity relationship in 8‐HQs has been reported against tuberculosis; however, a few studies have shown that increasing the lipophilicity of compounds resulted in enhanced antitubercular activity [ref. 26, ref. 27]. Therefore, the design of the 8‐HQ derivatives (QD‐1–12) was driven by the hypothesis that increasing the lipophilicity through alkylation of the C8‐hydroxyl group would enhance their antimycobacterial, antistaphylococcal and/or antibiofilm activity.
In this study, the aim was to investigate the inhibitory potential of newly designed and synthesized 8‐alkoxyquinoline derivatives for their antibiofilm and antibacterial activity against mycobacterial and staphylococcal species and to assess their structure–activity relationships. Furthermore, human monocytes (U937) and African green monkey kidney cells (Vero) were used to determine the potential cytotoxicity of the derivatives. These findings may highlight the potential for further development of this scaffold in a formal hit assessment or hit‐to‐lead and lead‐optimization phase in a drug discovery programme.
Results and Discussion
A total of 12 8‐alkoxyquinolines were synthesized on the basis of four parent 8‐HQ compounds. The synthesis was achieved by stirring quinoline with corresponding alkyl bromides in the presence of K2CO3 (prepared in dimethylformamide [DMF]) (Scheme 1). The reaction products in Entries 10 and 11 were pentyl 8‐(pentyloxy)quinoline‐2‐carboxylate (QD‐10) and isopentyl 8‐(isopentyloxy)quinoline‐2‐carboxylate (QD‐11), respectively, instead of 8‐(pentyloxy)quinoline‐2‐carbaldehyde and 8‐(isopentyloxy)quinoline‐2‐carbaldehyde, respectively (Scheme 2). The spectroscopic data are available in Figures S3–S54.
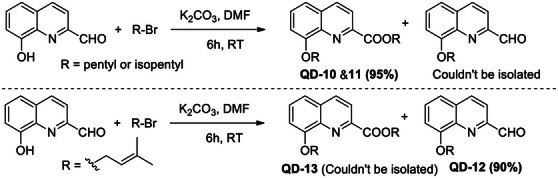
The formation of these unexpected products could be explained by the mechanism proposed in Scheme 3. The first step involved the mutual reaction of a 2‐formylquinolin‐8‐olate molecule to produce 2‐(((2‐formylquinolin‐8‐yl)oxy)oxidomethyl)quinolin‐8‐olate and concerted alkylation at oxidomethyl group to produce 2‐(alkoxy((2‐formylquinolin‐8‐yl)oxy)methyl)quinolin‐8‐olate (1). The carbonate ion then attacks the highly electrophilic intermediate product (1) to produce (8‐hydroxyquinolin‐2‐yl)(alkoxy)methyl hydrogen carbonate (2), which undergoes concerted decarboxylation and hydride transfer to release CO2 molecule to produce alkyl 8‐hydroxyquinoline‐2‐carboxylate (4) and formate ion.
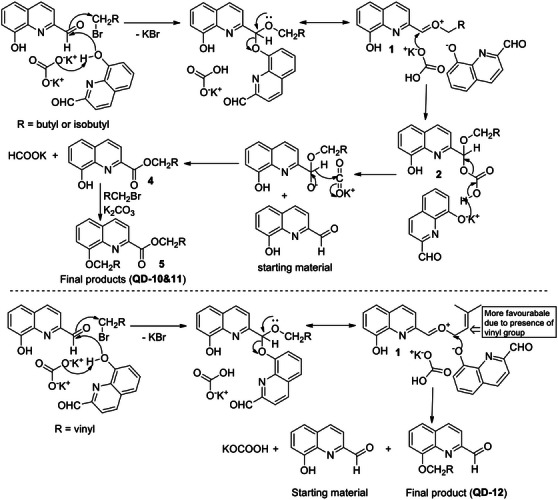
The final step involved alkylation of 8‐hydroxy group in 4 to produce alkyl 8‐(alkoxy)quinoline‐2‐carboxylate (5, QD‐10&11). The 8‐(alkoxy)quinoline‐2‐carbaldehydes could not be identified in this reaction, which indicated that the carbonate ion attack (at aldehyde group) is preferred over aryloxide ion (at alkyl group). However, in Entry 12, 8‐(alkoxy)quinoline‐2‐carbaldehyde (QD‐12) was the major product (90%) (Scheme 2). The corresponding ester (QD‐13) could not be isolated. This can be explained by the higher affinity of the prenyl group for the nucleophile than the saturated alkyl group and the preferred attack by aryloxide ion at the prenyl group over the carbonate attack at aldehyde group, which preferably gives Product 3 (Scheme 3).
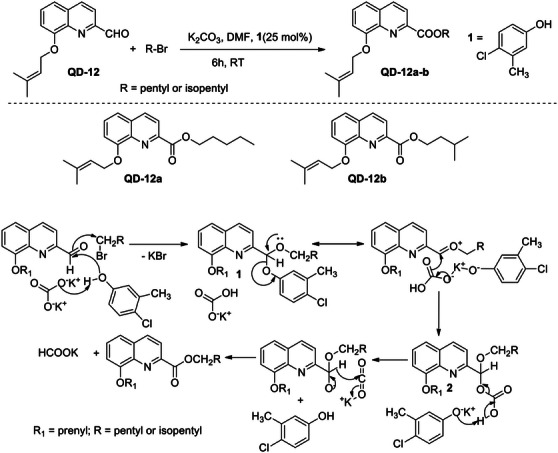
To confirm the proposed mechanism, QD‐12 was treated with pentyl and isopentyl bromide under the same reaction condition, and 25 mol% of a phenol (4‐chloro‐3‐methylphenol) was added as a catalyst, which initiated the reaction (Scheme 4). The reaction products (QD‐12a&b) were in agreement with the proposed reaction mechanism (Scheme 3), and the products formed were pentyl and isopentyl esters of QD‐12, respectively. Further standardization and validation of this synthetic method are in progress.
The quinolone‐based compounds, HQ‐1, HQ‐2, and HQ‐3, showed potent antimycobacterial activity as well as the reduction in Mycobacterium smegmatis biofilm formation (Table 1). HQ‐2 displayed the most prominent antimycobacterial activity with a minimum inhibitory concentration (MIC) of 0.1 µM and a minimum biofilm inhibitory concentration (MBIC) of 3.13 µM. However, there was no indication of a selective inhibition towards biofilm formation (biofilm selective index [BSI] ≤1), and the observed reduction in biofilm is most likely due to the bacterial growth inhibition. Although QD‐12 showed a moderate selectivity (BSI = 8) towards the inhibition of biofilm formation, this activity could be ascribed to residual inhibition of bacterial growth, and further analysis is needed to assess the specificity. The remaining derivatives showed poor activity against the mycobacterial strains.
TABLE 1: Antibacterial and antibiofilm activities and selectivity indexes (SIs) of quinolines and derivatives.
| Compounds (molecular weight in g/mol) | M. tb MIC (µM) | M. smeg MIC (µM) | M. smeg MBIC (µM) | M. smeg BSI (MIC/MBIC) | MSSA MIC (µM) | MRSA MIC (µM) | MSSA MBIC (µM) | MRSA MBIC (µM) | MRSA BSI (MIC/MBIC) |
|---|---|---|---|---|---|---|---|---|---|
| HQ‐1 (145) | 1 | 3.13 | 3.13 | 1 | 13.8 | 6.90 | 13.80 | 6.90 | 1 |
| HQ‐2 (227) | 0.1 | 1.56 | 3.13 | 0.5 | 2.2 | 1.1 | 17.6 | 35.2 | 0.03 |
| HQ‐3 (159) | 1 | 6.25 | 6.25 | 1 | 201.23 | 100.63 | 100.63 | 201.23 | 0.5 |
| HQ‐4 (173) | 1 | >100 | >100 | — | 369.94 | 369.94 | 739.88 | 739.88 | 0.5 |
| QD‐1 (215) | 100 | >100 | >100 | — | 595.35 | 595.35 | 595.35 | 595.35 | 1 |
| QD‐2 (215) | >100 | >100 | >100 | — | —cbdv70329-tbl1-note-0002 | 1190.70 | 1190.70 | 1190.70 | 1 |
| QD‐3 (213) | 100 | >100 | 100 | 1 | 300.47 | 300.47 | 600.94 | 600.94 | 0.5 |
| QD‐4 (297) | >100 | >100 | >100 | — | — | — | — | — | — |
| QD‐5 (297) | >100 | >100 | >100 | — | — | — | — | — | — |
| QD‐6 (295) | 100 | >100 | >100 | — | — | — | — | 256.0 | — |
| QD‐7 (229) | 100 | >100 | >100 | — | 1117.90 | 1117.90 | — | — | — |
| QD‐8 (229) | 100 | >100 | 100 | 1 | 558.95 | — | — | — | — |
| QD‐9 (227) | 100 | >100 | >100 | — | — | — | — | — | — |
| QD‐10 (329) | 100 | >100 | >100 | — | 389.06 | 389.06 | — | — | — |
| QD‐11 (329) | 100 | >100 | >100 | — | 389.06 | 389.06 | — | — | — |
| QD‐12 (241) | 100 | 100 | 12.5 | 8 | 265.56 | 132.78 | 531.12 | 265.56 | 0.5 |
| Isoniazidcbdv70329-tbl1-note-0003 | 0.3 | — | — | — | — | — | — | — | — |
| Ciprofloxacincbdv70329-tbl1-note-0004 | — | 0.6 | 0.325 | 2 | — | — | 2.0 | 1.0 | — |
| Oxacillincbdv70329-tbl1-note-0005 | — | — | — | — | 1.0 | 64.0 | — | — | — |
| Vancomycincbdv70329-tbl1-note-0006 | — | — | — | — | 1.0 | 1.0 | — | — | — |
Abbreviations: BSI, biofilm selective index; M. smeg, Mycobacterium smegmatis; M. tb, Mycobacterium tuberculosis; MBIC, minimum biofilm inhibitory concentration; MIC, minimum inhibitory concentration.
Not determined (due to inactivity)
Positive control against M. tuberculosis.
Positive control against Mycobacterium smegmatis.
Positive control against methicillin‐sensitive Staphylococcus aureus.
Positive control against methicillin‐resistant Staphylococcus aureus.
Two compounds (HQ‐1 and HQ‐2) exhibited noteworthy antibacterial activity against methicillin‐sensitive S. aureus (MSSA) UAMS‐1 and methicillin‐resistant S. aureus (MRSA) AH1263 or LAC. HQ‐1 showed an MIC value of 13.8 and 6.9 µM on MSSA and MRSA, respectively, whereas HQ‐2 showed an MIC value of 2.2 and 1.1 µM on MSSA and MRSA, respectively (Table 1). Both, including HQ‐4, showed a higher selectivity towards the resistant versus the susceptible strain with a 2‐fold increase in the antibacterial activity against MRSA.
HQ‐1 and HQ‐2 also showed the highest inhibitory activity against biofilm formation of susceptible and resistant S. aureus with MBIC values of 6.90 and 35.2 µM, respectively. However, this observed activity is likely due to the potent growth inhibitory effects, rather than true antibiofilm activity. This is evident in the calculated BSI against MRSA, with no biofilm selectivity observed. The alkyl derivatives showed poor activity against bacterial strains and biofilm formation. However, similar to the antimycobacterial activity, the prenyl derivative (QD‐12) exhibited increased antibacterial activity compared to the parent compound HQ‐4. The staphylococcal growth and biofilm inhibitory curves are available in Figures S1 and S2.
A similar trend to the antibacterial activity was observed when the cytotoxicity of the quinoline compounds was investigated, where the parent compounds (HQ‐1, HQ‐2 and HQ‐4) showed significant toxicity when compared to the derivatives, with HQ‐1, HQ‐2 and HQ‐4 displaying 50% inhibitory concentrating (IC50) values <100 µM against the U937 cells and HQ‐1 and HQ‐2 showing IC50 values <100 µM against the Vero cells (Table 2). Most of the derivatives had IC50 values above 100 μ M, indicative of moderate‐to‐low toxicity [ref. 28].
TABLE 2: Cytotoxicity and selective indexes (SIs) of quinoline derivatives against human monocytes (U937) and monkey kidney (Vero) cells.
| Compounds | U937 | Vero | ||
|---|---|---|---|---|
| IC50 cbdv70329-tbl2-note-0002 ± SD (µM) | SI | IC50 ± SD (µM) | SI | |
| HQ‐1 | 32.82 ± 11.43 | 10.5 | 47.81 ± 6.59 | 15.3 |
| HQ‐2 | 2.70 ± 0.31 | 1.7 | 82.77 ± 7.09 | 53.1 |
| HQ‐3 | >100 | — | >100 | — |
| HQ‐4 | 4.97 ± 1.93 | 0.05 | >100 | — |
| QD‐1 | >100 | — | >100 | — |
| QD‐2 | >100 | — | >100 | — |
| QD‐3 | 83.83 ± 0.80 | 0.8 | >100 | — |
| QD‐4 | >100 | — | >100 | — |
| QD‐5 | >100 | — | >100 | — |
| QD‐6 | >100 | — | >100 | — |
| QD‐7 | >100 | — | >100 | — |
| QD‐8 | >100 | — | >100 | — |
| QD‐9 | >100 | — | >100 | — |
| QD‐10 | 69.48 ± 0.16 | 0.7 | 84.77 ± 3.42 | 0.8 |
| QD‐11 | >100 | — | 70.44 ± 3.99 | 0.7 |
| QD‐12 | >100 | 1 | >100 | 1 |
| Actinomycin D | 2.0 × 10−3 ± 6.1 × 10−4 | — | 2.0 × 10−2 ± 8.0 × 10−3 | — |
Abbreviations: SD, standard deviation; SI, selective index (IC50/MIC against Mycobacterium smegmatis).
aFifty per cent inhibitory concentration.
From Table 1, it is evident that the 8‐HQs showed an overall increased inhibitory activity against mycobacterial species compared to the staphylococcal bacterial strains. The substitution at the 2‐position did not affect the activity of 8‐HQs against M. tuberculosis; however, in the case of QD‐12, it reduced the growth and biofilm inhibitory potential of both M. smegmatis and S. aureus. On the contrary, substitution with halides in ring‐A, significantly improved the antibacterial activity. This is also evident from a previous study conducted against S. aureus [ref. 12]. Unfortunately, the alkylation of 8‐hydroxy group resulted in a significant reduction in the activity of almost all the 8‐HQs against each of the tested mycobacterial and staphylococcal strains. Alkylation in 8‐hydroxy‐2‐quinoline carboxaldehyde (HQ‐4/QD‐12) improved the growth inhibitory activity against M. smegmatis, MSSA and MRSA by 2‐ to 4‐fold. Enhanced inhibitory activity was observed in M. smegmatis biofilm growth with a reduction from >100 to 12.5 µM. Although a promising finding, the observed increased biofilm selectivity could also be explained due to residual bacterial growth inhibition, and further optimization and analysis are required. The structure of the prenyl group at the C8 position of QD‐12 appears to be important for this biofilm selectivity. Unlike the saturated pentyl or isopentyl groups, the prenyl moiety may have an optimal balance of lipophilicity and molecular shape that enhances specific interactions with components of the biofilm matrix or modulates bacterial quorum‐sensing pathways, rather than directly targeting essential metabolic processes. This suggests a distinct SAR for antibiofilm activity compared to direct antibacterial effects. On the basis of the observations, it may be concluded that the 8‐hydroxy is crucial to increase activity, whereas alkylation of the group may not always produce results that show increased antimycobacterial, antistaphylococcal or antibiofilm activity.
Acylhydroxyquinolines have shown equivalent or better antibacterial activity compared to the corresponding hydroxyquinolines in several previously reported studies [ref. 13]. It has been observed, from literature, that increasing the lipophilicity of the molecules generally resulted in increased antimycobacterial activity [ref. 26, ref. 27]. Several alkyl hydroxyquinoline/phenazine molecules have also been reported to possess significant antibacterial/antibiofilm activity [ref. 24, ref. 25, ref. 29, ref. 30]. Although a reduction in antibacterial activity against S. aureus after alkylation of 8‐OH group of a few 8‐HQs has also been reported by Lam et al., there were only a few examples, and therefore, it was difficult to conclude that alkylation at 8‐OH position results in reduced antibacterial activity of diversely substituted quinolines [ref. 12]. This study was designed to assess the effect of alkylation of 8‐OH groups in diversely substituted 8‐HQs, resulting in increased lipophilicity of the molecules and may result in increased antistaphylococcal and antimycobacterial activity. However, this hypothesis should be rejected, as most of the synthesized derivatives resulted in the reduction of antimycobacterial and antibacterial activity, whereas only QD‐12 showed enhanced antibiofilm activity against M. smegmatis. It can be concluded that the 8‐OH group is crucial for antibacterial and antimycobacterial activity. This indicates that simple C8‐alkylation, despite increasing lipophilicity, does not typically lead to improved direct antibacterial activity for these quinoline scaffolds; instead, it suggests that the free 8‐OH group is critical for the antibacterial mechanism of action.
In another study, 8‐hydroxy‐2‐quinolinecarbaldehyde has been reported to possess in vitro cytotoxicity against several human cancer cell lines, including breast cancer (MDA‐MB‐231, T‐47D, Hs578t), osteosarcoma (SaoS2), leukaemia (K562), hepatocellular carcinoma (SKHep1) (with a 50% reduction of MTS assay signal compared to the control (MTS50) range of 12.5−25 µg/mL) and hepatoma (Hep3B) (with an MTS50 of 6.25 ± 0.034 µg/mL). It was observed that the cytotoxicity increased with the alkylation of the 8‐OH group in other quinoline molecules. An increase in the alkyl chain length also resulted in increased cytotoxicity [ref. 31]. Thus, on the basis of this observation, it was hypothesized that 8‐alkoxy‐2‐quinolinecarbaldehyde might have more cytotoxic potential than 8‐hydroxy‐2‐quinolinecarbaldehyde. The compounds were evaluated for their cytotoxicity against U937 and Vero cells. Almost all the derivatives were found to have IC50 values >100 µM; therefore, they could potentially be considered moderately toxic (if the IC50 value was <300 µM); however, they could also be considered to have low toxicity if the IC50 values were >300 µM against the tested non‐tumorigenic cell lines. Compounds with IC50 <100 µM are considered significantly toxic, as shown with HQ‐1, HQ‐2 and HQ‐4. This result confirms that QD‐12 could potentially be used as an antibiofilm agent against mycobacterial biofilms. However, further analyses through a formal hit assessment and additional toxicity studies are required.
Furthermore, the 8‐alkoxyquinoline‐2‐carboxylates, similar in structure to QD‐10, QD‐11 and QD‐12, have been previously reported to have anti‐obesity and mitochondrial uncoupling activities [ref. 32]. Thus, the compounds in this study, along with similar compounds, should be considered for evaluation for their anti‐obesity and mitochondrial uncoupling potential. Synthesis of the other quinoline‐2‐carboxylate compounds for the validation of our synthetic method as well as for the evaluation of their anti‐obesity potential is in progress. The synthetic method developed for the synthesis of symmetrical and unsymmetrical 8‐alkoxyquinoline‐2‐carboxylates (QD‐10, QD‐11 and QD‐12) is a simple and cost‐effective technique, which produces high yields and involves fewer reaction steps compared to the method followed by Kikuchi et al. [ref. 32]. Furthermore, this is the first reported method for the direct conversion of quinolinecarbaldehyde into quinolinecarboxylates using alkyl halide and K2CO3. Conversion of heterocyclic aldehydes to esters using alcohols has been reported by Goswami et al. [ref. 33]. However, this method is limited to the conversion of aldehydes to esters only and cannot be applied for the direct synthesis of alkoxy quinoline carboxylates. Furthermore, this method utilizes hazardous sodium cyanoborohydride (Na(CN)BH3) as a catalyst, and the yield is low in comparison to the method described in the current study. Hence, the method described in this study is advantageous over previously reported methods, as it can be applied for the conversion of quinolinecarbaldehydes to corresponding carboxylates and hydroxyquinoline carbaldehydes to alkoxy quinoline carboxylates using mild reaction conditions.
Conclusions
From Table 1, it is evident that the 8‐HQs showed an overall increased inhibitory activity against mycobacterial species compared to the staphylococcal bacterial strains. The substitution at the 2‐position did not affect the activity of 8‐HQs against M. tuberculosis; however, in the case of QD‐12, it reduced the growth and biofilm inhibitory potential of both M. smegmatis and S. aureus. On the contrary, substitution with halides in ring‐A significantly improved the antibacterial activity. This is also evident from a previous study conducted against S. aureus [ref. 12]. Unfortunately, the alkylation of 8‐hydroxy group resulted in a significant reduction in the activity of almost all the 8‐HQs against each of the tested mycobacterial and staphylococcal strains. Alkylation in 8‐hydroxy‐2‐quinoline carboxaldehyde (HQ‐4/QD‐12) improved the growth inhibitory activity against M. smegmatis, MSSA and MRSA by 2‐ to 4‐fold. Enhanced inhibitory activity was observed in M. smegmatis biofilm growth with a reduction from >100 to 12.5 µM. Although a promising finding, the observed increased biofilm selectivity could also be explained due to residual bacterial growth inhibition, and further optimization and analysis are needed. On the basis of the observations, it may be concluded that the 8‐hydroxy is crucial to increase activity, whereas alkylation of the group may not always produce results that show increased antimycobacterial, antistaphylococcal or antibiofilm activity.
Experimental Section
General
All reagents used in this study were purchased from Sigma‐Aldrich (Johannesburg, South Africa) unless otherwise stated. The NMR spectra were recorded on the Bruker Avance 400 MHz NMR spectrometer using tetramethylsilane (TMS) as an internal standard and CDCl3 as a solvent. The HR‐ESIMS data were obtained using a Waters UPLC‐MS system with PDA detector and Waters Synapt G2 QTOF mass spectrometer. The 8‐HQ compounds (HQ1–4) were purchased from Sigma Aldrich.
Synthesis
A total of 12 derivatives were synthesized on the basis of the four parent quinoline compounds, namely, 8‐HQ (HQ‐1), 5,7‐dichloro‐8‐hydroxy‐2‐methylquinoline (HQ‐2), 2‐methyl‐8‐hydroxyquinoline (HQ‐3) and 8‐hydroxyquinoline‐2‐carbaldehyde (HQ‐4) using three different alkyl bromides (pentyl bromide, isopentyl bromide and prenyl bromide). The quinolines (1 mM) were prepared in 10 mL of dried DMF in a round‐bottom flask. The corresponding alkyl bromide and potassium carbonate (3 mM each) were added to the quinoline solution and stirred for 6 h at room temperature.
The progress of the reaction was monitored by thin layer chromatography (TLC). After the disappearance of the quinoline spot, the reaction was quenched by the addition of distilled water. The reaction mixture was neutralized with 4% HCl solution, and the product was extracted with ethyl acetate. The ethyl acetate layer was washed four times with water to remove residual DMF. Finally, it was washed with a concentrated salt solution, dried over anhydrous sodium sulphate (Na2SO4) and concentrated under vacuum to yield the crude product. The desired product was purified using column chromatography. The yield of the reactions was between 60% and 95%. Compound structures were confirmed on the basis of NMR and mass spectroscopic data. The synthetic scheme and final products are presented in Scheme 1.
For the full synthetic and characterization details, please review the Supporting Information section.
Antimycobacterial and Biofilm Inhibitory Activity
Mycobacterium Species
The microorganisms, M. smegmatis (MC2 155) and a drug‐susceptible strain of M. tuberculosis, H37Rv (ATCC 27264), were kindly donated by the South African Medical Research Council, Pretoria, South Africa. Cultures of M. smegmatis were maintained on Middlebrook 7H11 agar base plates and allowed to grow for 24 h at 37°C, followed by sub‐culturing for an additional 24 h. Mycobacterium tuberculosis was plated onto Löwenstein–Jensen medium and allowed to grow for 3–4 weeks at 37°C.
Antimycobacterial Activity
The antimycobacterial activity of the quinoline compounds and derivatives was tested using the microtitre Alamar Blue assay method, as described by Franzblau et al., with minor modifications with the use of the viability reagent, Presto Blue [ref. 34, ref. 35, ref. 36]. Briefly, 100 µL of 7H9 broth (supplemented with 0.4% glycerol, 0.5% Tween 80 and OADC) was dispensed in each well of a sterile flat‐bottom 96‐well plate. The compounds were dissolved to a stock concentration of 20 mM (100% DMSO) and further diluted in media, whereafter 100 µL of the prepared compounds and the positive controls, ciprofloxacin (M. smegmatis) and isoniazid (M. tuberculosis), were added to the first row of wells, followed by a 2‐fold serial dilution yielding a test concentration range of 0.8–100 µM for the compounds (10‐fold serial dilution for M. tuberculosis [0.0001–100 µM] and 0.04–5 µg/mL for ciprofloxacin [0.2–15 µM] and isoniazid [0.3–36.5 µM]). Negative untreated bacteria, sterility and vehicle (1% DMSO) controls were included in the assay. The bacterial inoculum was prepared to a 0.5 McFarland and diluted to yield a test concentration of 1.5 × 106 CFU/mL. The inoculum of 100 mL was added to all the wells, excluding the sterility controls, to yield a final assay volume of 200 µL. After a 24‐h (5 days for M. tuberculosis) incubation period at 37°C, 20 µL of Presto Blue (Thermo Fisher Scientific, Massachusetts, USA) was added to all the wells and incubated for an additional 3–4 h. The samples were tested in triplicate in two or more independent experiments. The MIC values were defined as the lowest concentration where no colour change from blue to pink could be observed.
Mycobacterial Biofilm Inhibition
A microtitre antibiofilm assay was conducted in a sterile flat‐bottomed 96‐well plate for the determination of the biofilm inhibitory activity. Briefly, 100 µL of basic 7H9 media (without Tween 80) was added to the plates. The subsequent addition of 100 µL of the samples was added to the first row of the wells, followed by a 2‐fold serial dilution yielding the same concentration range as mentioned above. The M. smegmatis inoculum was prepared to a 0.5 McFarland and diluted 100‐fold to yield a test concentration of 1.5 × 106 CFU/mL. Negative untreated bacteria, sterility and vehicle (1% DMSO) controls were included in the assay [ref. 37, ref. 38]. The plates were incubated for 2–4 days at 37°C. The MBIC was observed and determined as the lowest concentration at which no visible biofilm formation could be observed. The experiment was repeated in three independent experiments and tested in triplicate.
Antistaphylococcal Biofilm Inhibitory Activity
Staphylococcus aureus Strains
To determine the antibacterial activity of the compounds, two S. aureus strains were used. An MSSA identified as S. aureus UAMS‐1 was provided by Mark Smeltzer, and an MRSA identified as AH1263 or LAC was provided by Alexander Horswill. The same strains used for MIC determination were employed for the MBIC assays. Staphyllococcus aureus UAMS‐929, the isogenic sarA mutant of UAMS‐1 (biofilm‐deficient phenotype), was used as a positive control (Table 3).
TABLE 3: Description of the Staphylococcus aureus bacterial strains used in this study.
| Strain ID | Characteristics | Resistance profile | Source | Ref. |
|---|---|---|---|---|
| UAMS‐1, ATCC 49230 | Osteomyelitis clinical isolate; prototype biofilm isolate. Methicillin‐sensitive | BEN | MS | [ref. 36] |
| LAC, AH1263 | USA300. Methicillin‐resistant | BEN, CIP, LEV, MET, MOX, OXA, PIP | AH | [ref. 37] |
Note: “MS” denotes M. Smeltzer, UAMS.3. “AH” denotes A. Horswill.
Abbreviations: BEN, benzylpenicillin; CIP, ciprofloxacin; LEV, levofloxacin; MET, methicillin; MOX, moxifloxacin; OXA, oxacillin; PIP, piperacillin.
Antistaphylococcal Activity
A broth microdilution assay in a 96‐well plate was used to determine the MIC as described by the CLSI guidelines [ref. 39]. The overnight culture was standardized to 5 × 105 CFU/mL in CAMHB media with a Cytation 3 multimode plate reader (BioTek, Winooski, VT, USA) with optical density OD600 nm, as previously described. Vehicle (100% DMSO) and positive controls (antibiotics: oxacillin and vancomycin) were included in each experiment. The final total per cent DMSO in the well volume was <5% for all assays. Compounds were dissolved in DMSO at a stock concentration of 20 mg/mL prior to dispensing in 96‐well plates to reach final test concentrations ranging from 0.125 to 256 µg/mL (approximately 0.5–1000 µM) by serial dilution. Plates were read at 0 h and after 18 h of static incubation with humidity at 37°C. Per cent inhibition of bacterial growth was calculated using the formula: (1 − (ΔODsample/ΔODvehicle)) × 100 [ref. 40]. All the compounds were tested in triplicate in two independent experiments.
Staphylococcal Biofilm Inhibition
A microtitre test in a 96‐well plate was performed to determine the antibiofilm activity of the quinolone derivatives. Overnight culture was standardized to 5 × 105 CFU/mL in biofilm media (TSB + 3% NaCl + 0.5% dextrose) by using a Cytation 3 multimode plate reader (BioTek, Winooski, VT, USA) with optical density OD600 nm, as previously described [ref. 41, ref. 42]. Human plasma was added to the media at 2% of the total volume of biofilm media at the time of the experimental setup. Samples were dissolved in DMSO at a stock concentration of 20 mg/mL. The test concentration range was from 0.125 to 256 µg/mL (approximately 0.5–1000 µM) by serial dilution, and a vehicle control was included. Plates were incubated for 22 h of static incubation with humidity at 37°C. Following incubation, the wells were gently washed with phosphate‐buffered saline (PBS), fixed with ethanol, stained with 0.2% crystal violet, rinsed in tap water and the stain eluted into 33% acetic acid and transferred to a new plate prior to quantification of the eluent at OD595 nm. All the compounds were tested in triplicate in two independent experiments.
Cytotoxicity
The human monocyte (U937) and African green monkey kidney (Vero) cell lines were used in this study to determine the cytotoxic potential of the quinoline derivatives. The cell lines were purchased from Separation Scientific SA (Pty) Ltd. (Johannesburg, South Africa) and maintained in Roswell Park Memorial Institute (RPMI) 1640 and Eagle’s Minimal Essential Medium (EMEM), supplemented with 10% heat‐inactivated foetal bovine serum (FBS), 1% antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin) and 1% antifungal agent (250 µg/mL amphotericin B) as previously described [ref. 28]. The cells were cultured at 37°C, 5% CO2 in a humidified incubator to 80% confluency. Detachment was achieved by rinsing the cells with phosphate buffer saline followed by the addition of trypsin–EDTA (0.25% trypsin containing 0.01% EDTA). After 5 min, the reaction was inhibited through the addition of supplemented media.
Cell viability was assessed through the method described by Lall et al., using Presto Blue as the viability reagent [ref. 35]. The cells were seeded in a flat‐bottom 96‐well plate (100 µL) at a concentration of 1.0 × 106 cells/mL and allowed to adhere for 24 h. The samples were prepared in DMSO at a stock concentration of 20 mM, serially diluted in complete media and added to the plate at a final concentration range of 0.78–100 µM. Controls included a vehicle control (2% DMSO), an untreated cell control, a 0% control (no cells) and actinomycin D, as the positive control, at a final test concentration range of 3.9 × 10−4 to 0.05 µg/mL. Cells were incubated for 72 h, followed by the addition of 20 µL of Presto Blue reagent to all the wells and an additional 2 h incubation. Cell viability was measured using a VICTOR Nivo Multimode Microplate Reader (PerkinElmer) at an excitation/emission wavelength of 540/590 nm. The 50% inhibitory concentration (IC50) was calculated using GraphPad Prism 4.
Statistical Analysis
All results are presented as the mean ± standard deviation (where applicable) of experiments done in triplicate and in two or three independent experiments (as stated in each methods section). Descriptive analysis was used to directly compare and rank the compounds and derivatives based on their activity in the biological assays. Additionally, by comparing the antibiofilm activity to the antibacterial activity and the antibacterial activity to the cytotoxicity, selectivity indices were used to assess the preferential activity of the compounds. Overall, 50% inhibitory concentrations (absolute IC50) were calculated using a four‐parameter logistic equation with constraints on the top (100) and bottom (0) parameters.
Author Contributions
Namrita Lall: conceptualization, funding acquisition, project administration, resources, validation, writing – review and editing, supervision. Anna‐Mari Kok: writing – original draft preparation, writing – review and editing. Carel B. Oosthuizen: conceptualization, data curation, investigation, methodology, visualization, formal analysis, writing – original draft preparation, writing – review and editing. Surjeet Verma: conceptualization, data curation, investigation, methodology, visualization, formal analysis, writing – original draft preparation, writing – review and editing. François Chassagne: data curation, investigation, methodology, visualization, formal analysis, writing – review and editing. Phuc H. Vo: data curation, investigation, methodology, visualization, formal analysis. Khanh‐Van Ho: data curation, investigation, methodology, visualization, formal analysis. Chung‐Ho Lin: funding acquisition, project administration, resources, validation, writing – review and editing, supervision. Cassandra L. Quave: funding acquisition, project administration, resources, validation, writing – review and editing, supervision. Danielle Twilley: data curation, investigation, methodology, visualization, formal analysis, writing – original draft preparation, writing – review and editing. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Supplementary Materials
References
- Biogeographical Variation in Community Response to Root Allelochemistry: Novel Weapons and Exotic Invasion,”. Ecology Letters, 2004. [DOI]
- Killing of Non‐Replicating Mycobacterium tuberculosis by 8‐Hydroxyquinoline,”. Journal of Antimicrobial Chemotherapy, 2010. [DOI | PubMed]
- In Vitro Activities of Cloxyquin (5‐Chloroquinolin‐8‐ol) Against Mycobacterium tuberculosis ,”. Antimicrobial Agents and Chemotherapy, 2007. [DOI | PubMed]
- The Remarkable Effect of a Combination of Iodochloroxyquinoline With a Subactive Dose of Streptomycin on Experimental Tuberculosis in guinea Pigs,”. Annales De L Institut Pasteur, 1952. [PubMed]
- High‐Throughput Screening for Inhibitors of Mycobacterium tuberculosis H37Rv,”. Tuberculosis, 2009. [DOI | PubMed]
- Quinoline: A Promising Antitubercular Target,”. Biomedicine & Pharmacotherapy, 2014. [DOI | PubMed]
- Activity of 7‐Methyljuglone Derivatives Against Mycobacterium tuberculosis and as Subversive Substrates for Mycothiol Disulfide Reductase,”. Bioorganic & Medicinal Chemistry, 2007. [DOI | PubMed]
- Global Introduction of New Multidrug‐Resistant Tuberculosis Drugs—Balancing Regulation With Urgent Patient Needs,”. Emerging Infectious Diseases, 2016. [DOI | PubMed]
- Structure of the Mycobacterial ATP Synthase Fo Rotor Ring in Complex With the Anti‐TB Drug Bedaquiline,”. Science Advances, 2015. [DOI | PubMed]
- Hydroxyquinolines Inhibit Ribonucleic Acid‐Dependent Deoxyribonucleic Acid Polymerase and Inactivate Rous Sarcoma Virus and Herpes Simplex Virus,”. Antimicrobial Agents and Chemotherapy, 1976. [DOI | PubMed]
- Metal‐Protein Attenuation with Iodochlorhydroxyquin (Clioquinol) Targeting Aβ Amyloid Deposition and Toxicity in Alzheimer Disease,”. Archives of Neurology, 2003. [DOI | PubMed]
- Preparation of 8‐Hydroxyquinoline Derivatives as Potential Antibiotics Against Staphylococcus aureus ,”. Bioorganic & Medicinal Chemistry Letters, 2014. [DOI | PubMed]
- Discovery of Quinoline Small Molecules With Potent Dispersal Activity Against Methicillin‐Resistant Staphylococcus aureus and Staphylococcus epidermidis Biofilms Using a Scaffold Hopping Strategy,”. Bioorganic & Medicinal Chemistry Letters, 2014. [DOI | PubMed]
- Derivatives of 8‐Hydroxyquinoline—Antibacterial Agents That Target Intra‐ and Extracellular Gram‐Negative Pathogens,”. Bioorganic & Medicinal Chemistry Letters, 2012. [DOI | PubMed]
- Ethnobotany and the Role of Plant Natural Products in Antibiotic Drug Discovery,”. Chemical Reviews, 2020. [DOI | PubMed]
- Biofilm Dispersion in Pseudomonas aeruginosa ,”. Journal of Microbiology (Seoul, Korea), 2016. [DOI | PubMed]
- Antibiofilm Agents: A New Perspective for Antimicrobial Strategy,”. Journal of Microbiology 2017 5510, 2017. [DOI]
- Growth of Mycobacterium tuberculosis Biofilms Containing Free Mycolic Acids and Harbouring Drug‐Tolerant Bacteria,”. Molecular Microbiology, July 2008. [DOI | PubMed]
- Autopsy Identification of Viable Mycobacterium tuberculosis in the Lungs of a Markedly Decomposed Body,”. Journal of Forensic Science, November 2020. [DOI]
- Piperine‐Coated Zinc Oxide Nanoparticles Target Biofilms and Induce Oral Cancer Apoptosis via BCl‐2/BAX/P53 Pathway,”. BMC Oral Health, June 2024. [DOI | PubMed]
- Adaptation of Mycobacterium tuberculosis to Biofilm Growth Is Genetically Linked to Drug Tolerance,”. Antimicrobial Agents and Chemotherapy, October 2019. [DOI | PubMed]
- Mycobacterial Biofilm: Mechanisms, Clinical Problems, and Treatments,”. International Journal of Molecular Sciences, July 2024. [DOI | PubMed]
- Synthetic Small Molecules as Anti‐Biofilm Agents in the Struggle Against Antibiotic Resistance,”. European Journal of Medicinal Chemistry, 2019. [DOI | PubMed]
- Halogenated Quinolines Discovered Through Reductive Amination With Potent Eradication Activities Against MRSA, MRSE and VRE Biofilms,”. Organic & Biomolecular Chemistry, 2015. [DOI | PubMed]
- Synthetically Tuning the 2‐Position of Halogenated Quinolines: Optimizing Antibacterial and Biofilm Eradication Activities via Alkylation and Reductive Amination Pathways,”. Chemistry: A European Journal, 2016. [DOI | PubMed]
- Synthesis and Antitubercular Activity of Lipophilic Moxifloxacin and Gatifloxacin Derivatives,”. Bioorganic & Medicinal Chemistry Letters, 2007. [DOI | PubMed]
- Antibacterial and Antitubercular Activity of Fosmidomycin, FR900098, and Their Lipophilic Analogs,”. Bioorganic & Medicinal Chemistry Letters, 2011. [DOI | PubMed]
- African Flora Has the Potential to Fight Multidrug Resistance of Cancer,”. BioMed Research International, 2015
- Regioselective Synthesis, Antimicrobial Evaluation and Theoretical Studies of 2‐Styryl Quinolines,”. Organic & Biomolecular Chemistry, 2015. [DOI | PubMed]
- Halogenated Phenazines That Potently Eradicate Biofilms, MRSA Persister Cells in Non‐Biofilm Cultures, and Mycobacterium tuberculosis ,”. Angewandte Chemie (International ed in English), 2015. [DOI | PubMed]
- Synthesis of 8‐Hydroxyquinoline Derivatives as Novel Antitumor Agents,”. ACS Medicinal Chemistry Letters, 2013. [DOI | PubMed]
- Synthesis of Prenylated Quinolinecarboxylic Acid Derivatives and Their Anti‐Obesity Activities,”. Bioorganic & Medicinal Chemistry, 2015. [DOI | PubMed]
- Sodium Cyanoborohydride–Mediated Direct Conversion of Some Heterocyclic Aldehydes to Esters,”. 2011. [DOI]
- Rapid, Low‐Technology MIC Determination With Clinical Mycobacterium tuberculosis Isolates by Using the Microplate Alamar Blue Assay,”. Journal of Clinical Microbiology, 1998. [DOI | PubMed]
- Viability Reagent, Prestoblue, in Comparison With Other Available Reagents, Utilized in Cytotoxicity and Antimicrobial Assays,”. International Journal of Microbiology, 2013. [DOI]
- Activity of 7‐Methyljuglone in Combination With Antituberculous Drugs Against Mycobacterium tuberculosis ,”. Phytomedicine, 2006. [DOI | PubMed]
- Inhibitory Effect of Cyclic Trihydroxamate Siderophore, Desferrioxamine E, on the Biofilm Formation of Mycobacterium Species,”. Biological & Pharmaceutical Bulletin, 2011. [DOI | PubMed]
- Inhibition of Mycothione Disulphide Reductase and Mycobacterial Biofilm by Selected South African Plants,”. South African Journal of Botany, 2019. [DOI]
- Performance Standards for Antimicrobial Susceptibility Testing,”. 35th ed. CLSI supplement M100. Clinical and Laboratory Standards Institute, Wayne, Pennsylvania, USA (. 2017
- Effects of Extracts From Italian Medicinal Plants on Planktonic Growth, Biofilm Formation and Adherence of Methicillin‐Resistant Staphylococcus aureus ,”. Journal of Ethnopharmacology, 2008. [DOI | PubMed]
- American Civil War Plant Medicines Inhibit Growth, Biofilm Formation, and Quorum Sensing by Multidrug‐Resistant Bacteria,”. Scientific Reports 2019 91, 2019. [DOI]
- Ellagic Acid Derivatives From Rubus ulmifolius Inhibit Staphylococcus aureus Biofilm Formation and Improve Response to Antibiotics,”. PLoS ONE, 2012. [DOI | PubMed]