Multi-gradient permutation survival analysis identifies mitosis and immune signatures steadily associated with cancer patient prognosis
Abstract
The inconsistency of the association between genes and cancer prognosis is often attributed to many variables that contribute to patient survival. Whether there exist the Genes Steadily Associated with Prognosis (GEARs) and functions remains largely elusive. Here, we developed a novel method named ‘Multi-gradient Permutation Survival Analysis’ (MEMORY) to screen the GEARs by using RNA-seq data from the TCGA database. We employed a network construction approach to identify hub genes from GEARs and utilized them for cancer classification. In the case of lung adenocarcinoma (LUAD), the GEARs were found to be related to mitosis. Our analysis suggested that LUAD cell lines carrying PIK3CA mutations exhibit increased drug resistance. For breast invasive carcinoma (BRCA), the GEARs were related to immunity. Further analysis revealed that CDH1 mutation might regulate immune infiltration through the EMT process. Moreover, we explored the prognostic relevance of mitosis and immunity through their respective scores and demonstrated it as valuable biomarkers for predicting patient prognosis. In summary, our study offered significant biological insights into GEARs and highlights their potentials as robust prognostic indicators across diverse cancer types.
Article type: Research Article
Keywords: Multi-gradient Permutation Survival Analysis, MEMORY, Gene Steadily Associated with Prognosis, GEAR, hub genes, mitosis, immune, Human, Incomplete, Valuable
Affiliations: https://ror.org/05qbk4x57Key Laboratory of Systems Health Science of Zhejiang Province, School of Life Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences Hangzhou China; https://ror.org/02rrdvm96State Key Laboratory of Cell Biology, Shanghai Institute of Biochemistry and Cell Biology, Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences Shanghai China; https://ror.org/04jth1r26Zhejiang University-University of Edinburgh Institute, Zhejiang University School of Medicine Haining China; https://ror.org/00a2xv884The Second Affiliated Hospital, Zhejiang University School of Medicine Hangzhou China; https://ror.org/030bhh786School of Life Science and Technology, ShanghaiTech University Shanghai China; https://ror.org/013q1eq08Department of Pathology and Frontier Innovation Center, School of Basic Medical Sciences, Fudan University Shanghai China
License: © 2024, Cai et al CC BY 4.0 This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Article links: DOI: 10.7554/eLife.101619 | PubMed: 41404730 | PMC: PMC12711199
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (287 KB)
Introduction
Cancer is one of the leading causes of death worldwide, with over 200 types identified. Despite major advances in the field, accurately predicting cancer patient prognosis remains a significant challenge (ref. Tomczak et al., 2015; ref. Weinstein et al., 2013). Previous studies have highlighted the complexity of this task, which is influenced by various factors, including cancer types, clinical stages, therapeutic interventions, nursing care, unexpected comorbidities, and other non-cancer-related illnesses that may interplay (ref. Liu et al., 2018; ref. Smeltzer et al., 2018; ref. Miyauchi et al., 2022; ref. Teh et al., 2021). Furthermore, gene expression plays a crucial role in predicting cancer patient prognosis, such as HER2, VEGF, Ki67, etc (ref. Gimotty et al., 2005; ref. Wu et al., 2021; ref. Mouabbi et al., 2023; ref. Kang et al., 2023; ref. Cai et al., 2022; ref. Zhang et al., 2021). Nevertheless, there is still a need for further improvement in prognostic accuracy to better inform treatment decisions and patient outcomes.
Survival analysis is commonly used to assess the correlation between genes and cancer patient prognosis (ref. Lai et al., 2021). However, inconsistent findings are frequently observed, even within the same type of cancer. For instance, studies exploring the correlation between CCND1 and prognosis in non-small cell lung cancer (NSCLC) reported contrasting results, including positive correlation, inverse correlation, or negligible influence (ref. Anton et al., 2000; ref. Esposito et al., 2005; ref. Dworakowska et al., 2005). These discrepancies can be attributed to various influential factors, such as differences in sample size, cohort characteristics (including cancer subtypes and tumor staging), and variations in therapeutic approaches (ref. Freedman, 1982). Such observations raise an important question: whether there exist Genes Steadily Associated with Prognosis (hereafter referred to as GEARs) that correlate with patient outcomes across different conditions, particularly considering the varying sample sizes used in different studies? Affirmative answers to this question would have significant implications not only for the development of more accurate prognostic models, but also for enhancing our understanding of cancer biology.
The Cancer Genome Atlas (TCGA) is a comprehensive cancer genomics project initiated in the United States (https://portal.gdc.cancer.gov/). It consists of transcriptome data, genomic data, and clinical information pertaining to 33 different cancer types, making it the largest cancer clinical sample database currently available. In this study, we developed a novel method named ‘Multi-gradient Permutation Survival Analysis’ (MEMORY) with the utilization of TCGA RNA-seq database, and assessed the potential existence of GEARs across 15 cancer types, each comprising a cohort of over 200 patients. Furthermore, we evaluated the prognostic predictive power of these GEARs and explored their potential biological functions in driving cancer progression.
Results
Multi-gradient permutation survival analysis identifies GEARs associated with mitosis and immune across multiple cancer types
GEARs are a group of genes consistently and significantly correlated with cancer patient survival, independent of the sample size. To identify these GEARs, we developed a novel method called ‘Multi-gradient Permutation Survival Analysis’ (MEMORY). This method allows us to assess the correlation between a specific gene and cancer patient prognosis using available transcriptomic datasets (Figure 1A, Supplementary file 1A). Initially, we started with a sampling number at 10% of the cohort size, then gradually increased the sampling number with each 10% increment, which was analyzed with 1000 permutations. By calculating the statistical probability of each gene’s association with patient survival, we can identify a group of GEARs for further analyses.
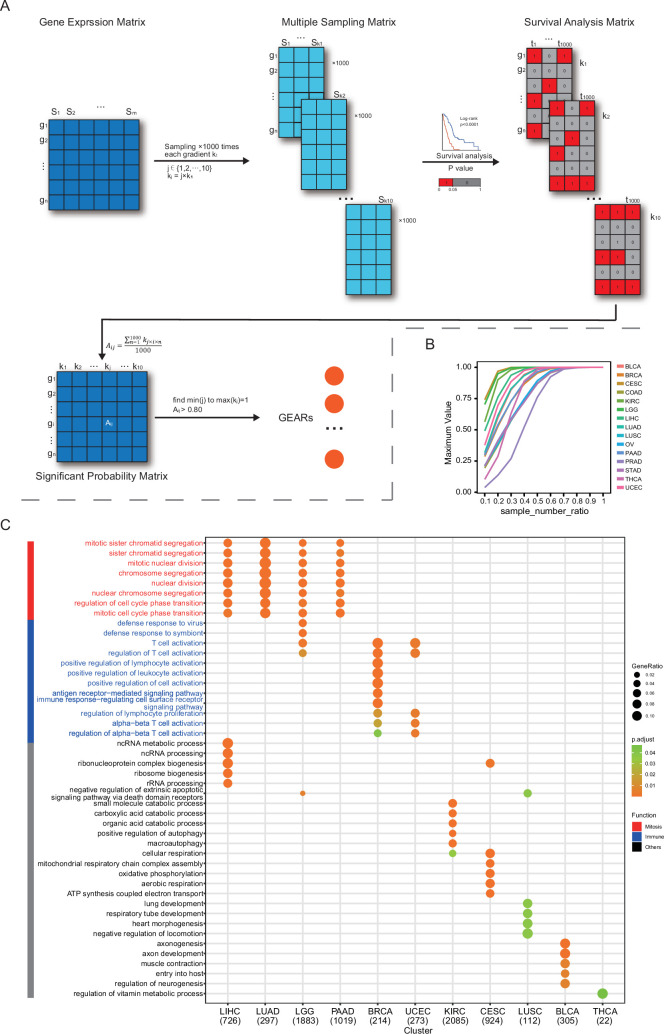
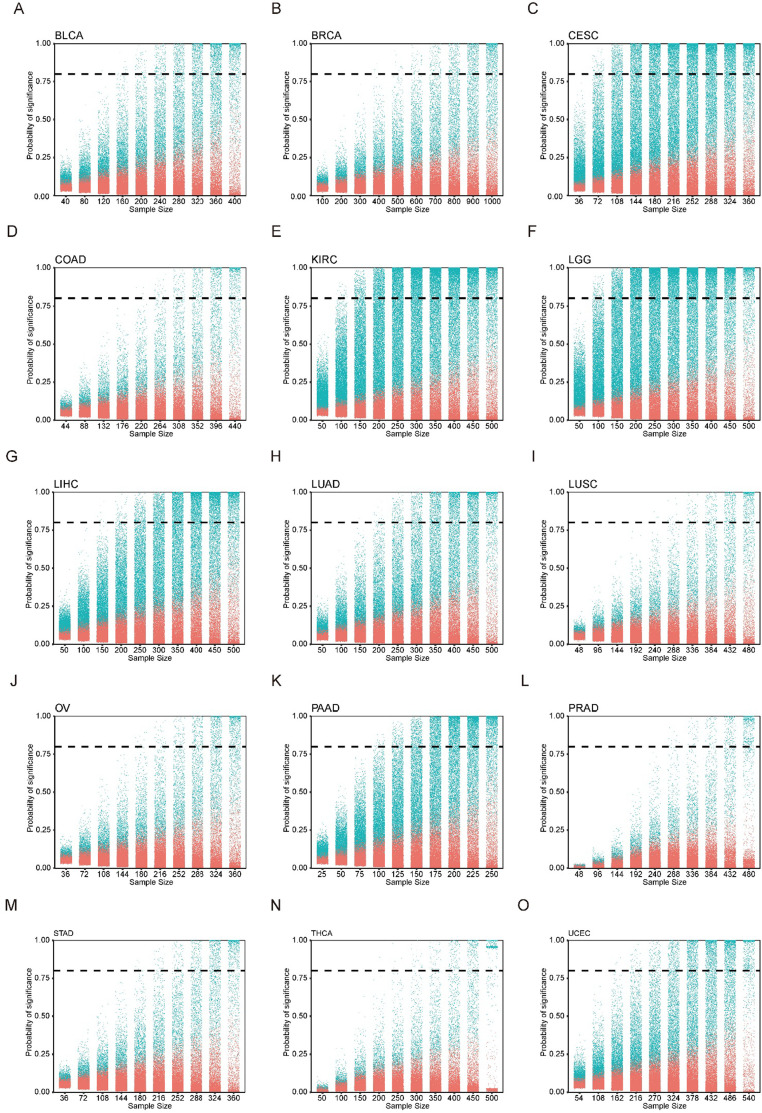
In this study, we utilized the TCGA datasets for several reasons, including the diversity of cancer types, the availability of gene expression profiles and patient prognosis information. We set a minimum cohort size of 200 and included 15 eligible cancer types for analysis. These cancer types include bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), colon adenocarcinoma (COAD), kidney renal papillary cell carcinoma (KIRC), brain lower grade glioma (LGG), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), ovarian serous cystadenocarcinoma (OV), pancreatic adenocarcinoma (PAAD), prostate adenocarcinoma (PRAD), stomach adenocarcinoma (STAD), thyroid carcinoma (THCA), and uterine corpus endometrial carcinoma (UCEC). As the number of samples increases, the significant probability of certain genes gradually approaches 1. Once this score reaches 0.8, previously employed as an empirical standard for survival analyses, and remained above this value consistent with further sample gradient increase, the gene is considered a GEAR (Figure 1B; ref. Gebski et al., 2018). Remarkably, we successfully identified a set of GEARs across all 15 cancer types (Figure 1—figure supplements 1 and 2, Supplementary file 1B). The GEAR counts in most cancer types ranged from 100 to 1000, with exceptions of CESC, KIRC, LGG, and PAAD with over 1000 GEARs, and THCA with only 22 GEARs (Supplementary file 1B). In LUAD, the top 10 GEARs with the highest significance probabilities were TLE1, GNG7, ERO1A, ANLN, DKK1, TMEM125, S100A16, KNL1, STEAP1, and BEX4. Most of these genes are known to promote LUAD malignant progression except for S100A16 (ref. Yao et al., 2014; ref. Zheng et al., 2021b; ref. Chen et al., 2022b; ref. Xu et al., 2019; ref. Zhang et al., 2018; ref. Wang et al., 2020; ref. Huo et al., 2020; ref. Zhao et al., 2018; ref. Fan et al., 2022). In other cancer types, BEX4 was identified as a common GEAR in KIRC, LGG, PAAD, and STAD (Supplementary file 1B). BEX4 is reported as a proto-oncogene promoting cancer onset and malignant progression in multiple cancers, including LUAD, glioblastoma multiforme, and oral squamous cell carcinoma (ref. Zhao et al., 2018; ref. Lee et al., 2021; ref. Gao et al., 2016). We also identified the top 1 GEAR in individual cancer types, such as TLL1 (BLCA), PGK1 (BRCA), RFXANK (CESC), DPP7 (COAD), VWA8 (KIRC), SCMH1 (LGG), HILPDA (LIHC), TLE1 (LUAD), CD151 (LUSC), ANKRD13A (OV), SOCS2 (PAAD), DRG2 (PRAD), ADAMTS6 (STAD), PSMB8 (THCA), ASS1 (UCEC) (Supplementary file 1C). Many of these genes were previously reported to be associated with tumorigenesis (ref. Yao et al., 2014; ref. He et al., 2019; ref. Matsuura et al., 2017; ref. Poplawski et al., 2023; ref. Guo et al., 2023; ref. Povero et al., 2020; ref. Erfani et al., 2021; ref. Won et al., 2022; ref. Xu et al., 2020; ref. Xu et al., 2016; ref. Mead, 2022; ref. Yang et al., 2018; ref. Keshet et al., 2020). For example, TLE1 is known as a transcriptional repressor that promotes cell proliferation, migration, and inhibits apoptosis in LUAD (ref. Palaparti et al., 1997; ref. Lin et al., 2023). Additionally, PGK1, a key enzyme in the glycolytic process, has been shown to promote cell proliferation, migration, and invasion in multiple cancers (ref. Li et al., 2016; ref. Fu and Yu, 2020). These findings demonstrate the intricate link between the functionality of GEARs and the initiation and progression of cancer.
To gain deep insights into the biological functions of these identified GEARs, we conducted Gene Ontology (GO) enrichment analysis (Figure 1C). Interestingly, we found the mitosis-related pathways were enriched in LICH, LUAD, LGG, and PAAD, and the immune-related pathways were enriched in BRCA and UCEC (ref. Waldman et al., 2020). Additionally, other cancer types exhibited enrichment in various pathways, such as organic acid metabolism pathway in KIRC, oxidative phosphorylation pathway in CESC, organ development-related pathways in LUSC, neurogenesis-related pathways in BLCA, and sustenance metabolism pathways in THCA. Given the crucial role of mitosis in cancer progression and the significance of the immune system in cancer-host interactions, we specifically focused on the mitosis and immune-related GERAs, which accounted for approximately 40% of all the analyzed cancers.
Identification of hub genes in mitosis and immune-related cancers
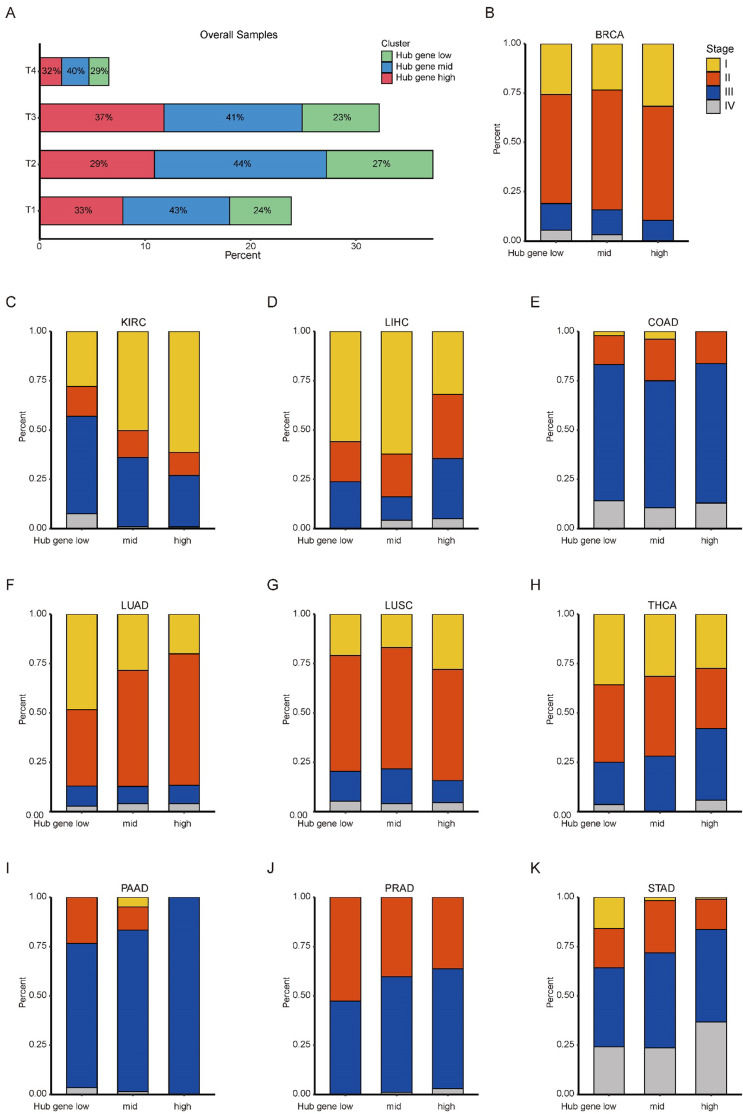
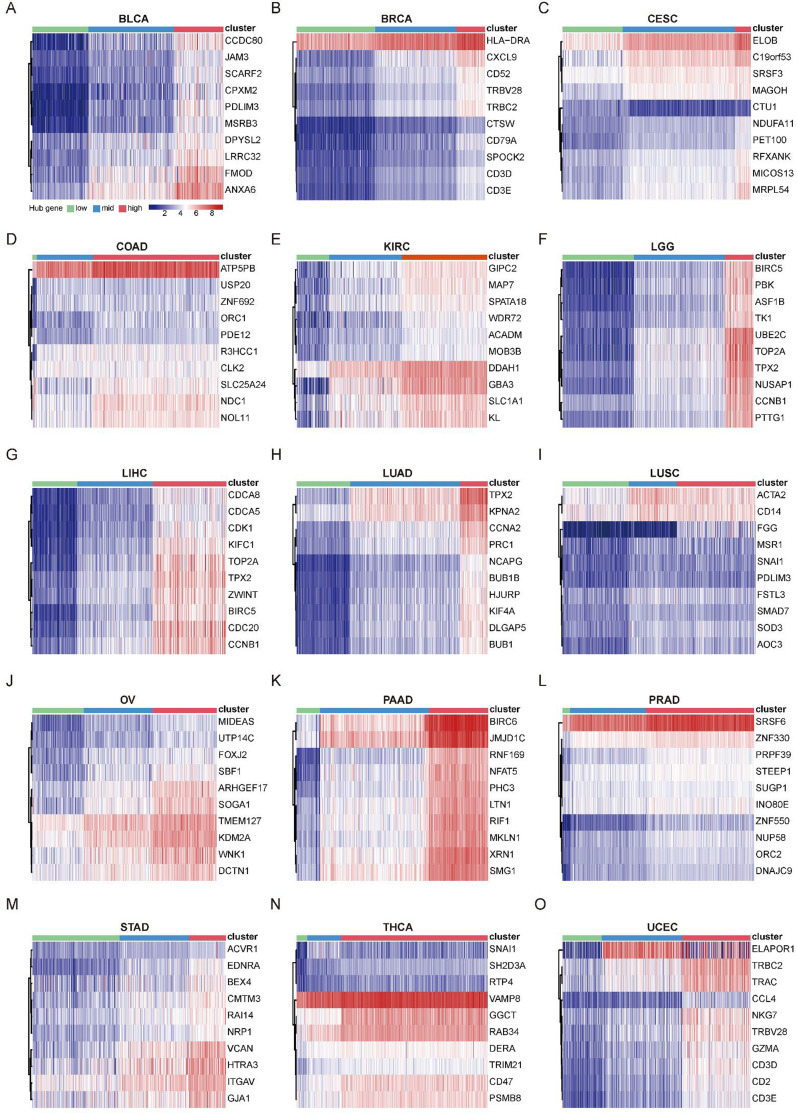
To identify crucial genes within the GEARs, we conducted and extracted the higher-ranked edges to construct the core survival network (CSN) (Figure 2A, Supplementary file 1D). The top 10 GEARs with the highest degree in these networks, selected from those with mean expression >10 TPM, were defined as hub genes (ref. Zhou et al., 2019). We then classified the samples using the hub genes derived from these networks and evaluated their clinical relevance (Figure 2—figure supplements 1–2). A significant correlation with cancer stages (TNM stages) is observed in most cancer types except for LUSC, THCA, and PAAD (Figure 2—figure supplement 3). Furthermore, we conducted GO enrichment analysis on the hub genes selected from CSNs and found that the results were consistent with the GEARs analysis (Supplementary file 1E). Specifically, the hub genes in LIHC, LGG, and LUAD were enriched in mitosis-related pathways, whereas the hub genes in BRCA and UCEC were enriched in immune-related pathways (Figure 2B). For instance, the nine hub genes in LUAD were associated with functions related to mitosis, whereas the eight hub genes of BRCA were associated with immune-related functions (Figure 2C–D).
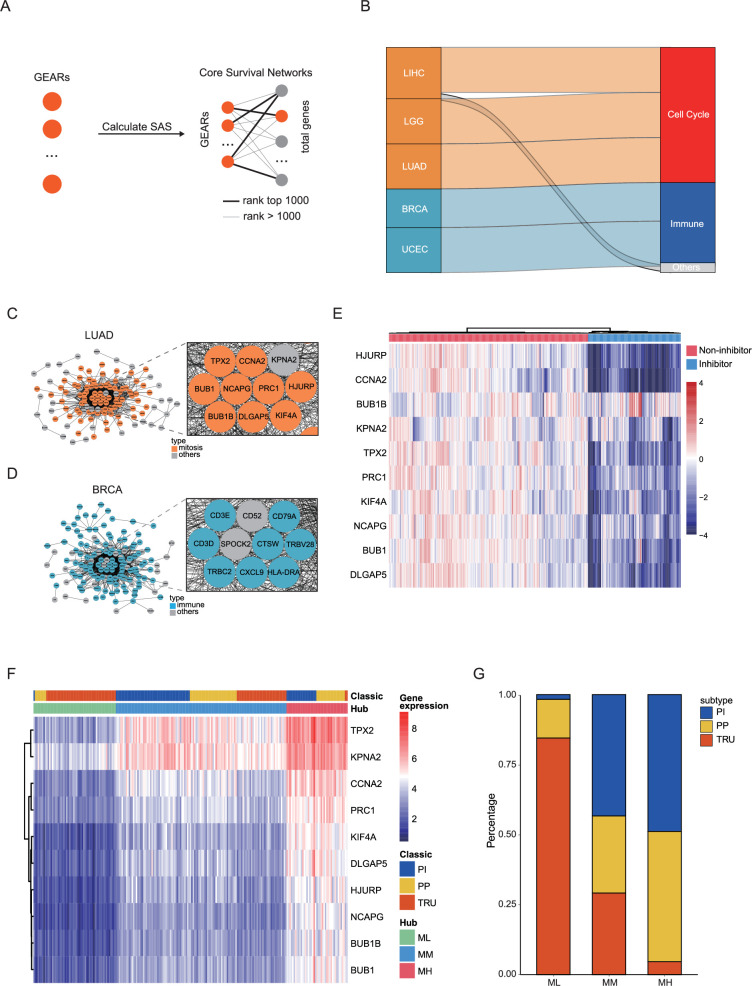
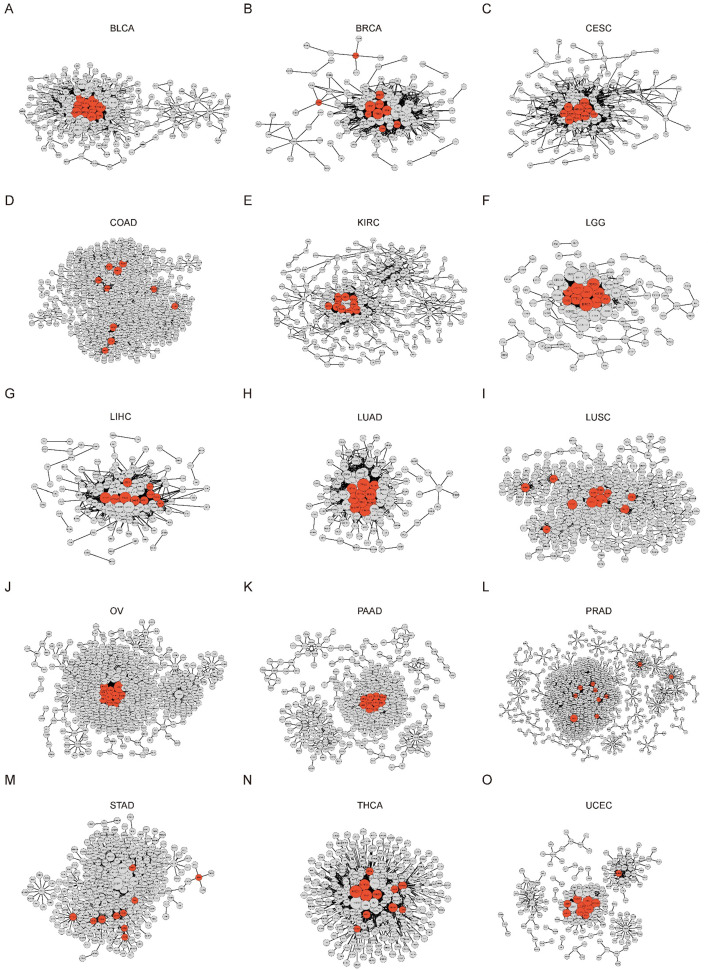
Next, we conducted survival-dependent analyses on the hub genes of LGG, LIHC, and LUAD. These analyses revealed that mitosis-related hub genes were closely associated with cancer cell viability, especially those hub genes that were correlated with multiple cancer types, such as CDC20, TOP2A, BIRC5, and TPX2 (ref. Hwang et al., 1998; ref. Uusküla-Reimand and Wilson, 2022; ref. Lamers et al., 2011; ref. Wittmann et al., 2000; Figure 2—figure supplement 4A–C). The importance of these genes is well established. For instance, TPX2, a hub gene in all three cancers, is known to play a crucial role in normal spindle assembly during mitosis and is essential for cell proliferation (ref. Kufer et al., 2002). The significance of these hub genes for the survival of cancer cells suggests that the expression levels of these hub genes could be used for inhibitors screening of tumor growth. By integrating the Genomics of Drug Sensitivity in Cancer (GDSC) and Connectivity Map (cMAP) databases, we identified a series of compounds that were able to effectively suppress the expression of the 10 hub genes in LUAD cell lines (Figure 2E, Figure 2—figure supplement 4D–E). These compounds also significantly inhibited LUAD cell growth and might serve as potential therapeutic agents for LUAD.
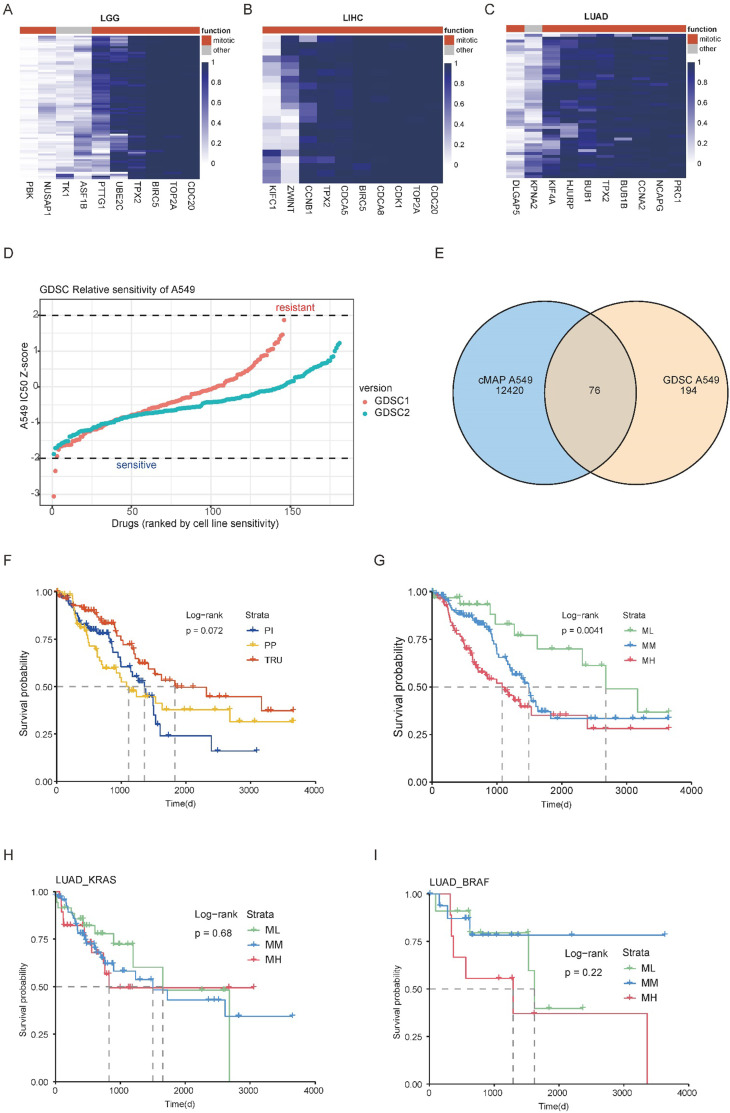
We then focused our analysis on the LUAD dataset, which had a larger sample size compared to LGG and LIHC. According to the expression of the hub genes, we classified the LUAD samples into three subgroups: mitosis low (ML), mitosis medium (MM), and mitosis high (MH; Figure 2F, Supplementary file 1F). A previous study has reported three classic subgroups of LUAD known as the terminal respiratory unit (TRU), the proximal-proliferative (PP), and the proximal-inflammatory (PI) subgroups (ref. Cancer Genome Atlas Research, 2014). Our analysis revealed that the ML subgroup was primarily enriched with the TRU subgroup and a small number of samples from the PP subgroup, but not the PI subgroup. The MH subgroup showed high expression of mitosis-related genes and predominantly encompassed PI and PP subgroups, whereas the MM group exhibited intermediate levels of mitosis-related gene expression (Figure 2F–G). This suggests that the new categorization method may help identify new factors that influence patient prognosis (Figure 2—figure supplement 2F-G).
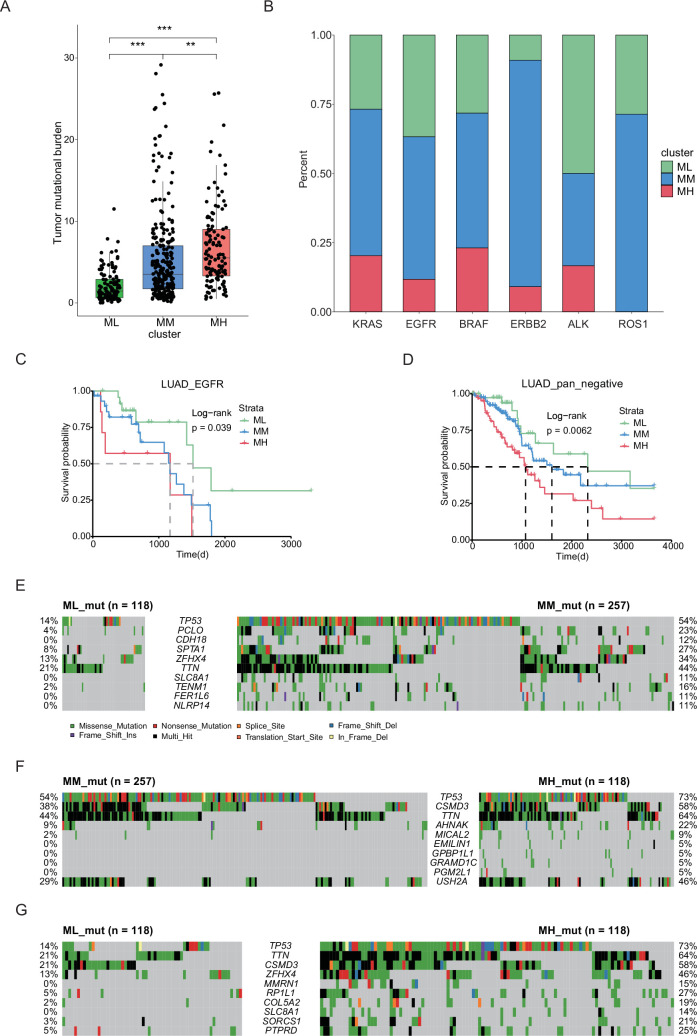
Distinct genetic mutation landscapes characterize different clusters of LUAD
The expression of hub genes provided crucial indicators for distinguishing various subgroups of LUAD. However, the underlying mechanisms driving these variations in expression patterns remain unknown. In our subsequent research, we aimed to explore the genomic differences, particularly in terms of gene mutations, among different LUAD subgroups. Initially, we analyzed the variations of genetic mutation landscapes of different LUAD subgroups by assessing the tumor mutation burden (TMB). Interestingly, we found significant differences in TMB among the various subgroups of LUAD (Figure 3A). Common driver genes were heterogeneously distributed across the three molecular subgroups. ALK and ROS1 fusions were most enriched in the prognosis-favorable ML and MM clusters, while KRAS, EGFR, BRAF, and ERBB2 mutations were more evenly distributed or enriched in the less prognosis-favorable MH group (Figure 3B). Due to limited counts for some fusion events, formal statistical comparison was not performed, but the distributional patterns suggested distinct subtype preferences across different driver events. Moreover, low tumor mitotic activity was associated with better prognosis in LUAD subgroups with EGFR mutations or pan-negative (no oncogenic alteration in genes including KRAS, EGFR, BRAF, ERBB2, PIK3CA, ALK, and ROS1) but not in those with KRAS or BRAF mutations (Figure 3C–D, Figure 2—figure supplement 4H–I). Besides, we observed unique mutation characteristics in each of these three subgroups (Figure 3E–G). For example, TP53 mutation, a prevalent tumor suppressor gene mutation, was frequently observed in the MH subgroup with a mutation rate of 73%, compared to mutation rates of 14% in the ML subgroup and 39% in the MM subgroup. While genes, such as CSMD3, RP1L1, and ZFHX4, exhibited similar trends in mutation frequency across the three subgroups. These findings indicate that substantial genomic differences among these three LUAD subgroups are based on the hub genes.
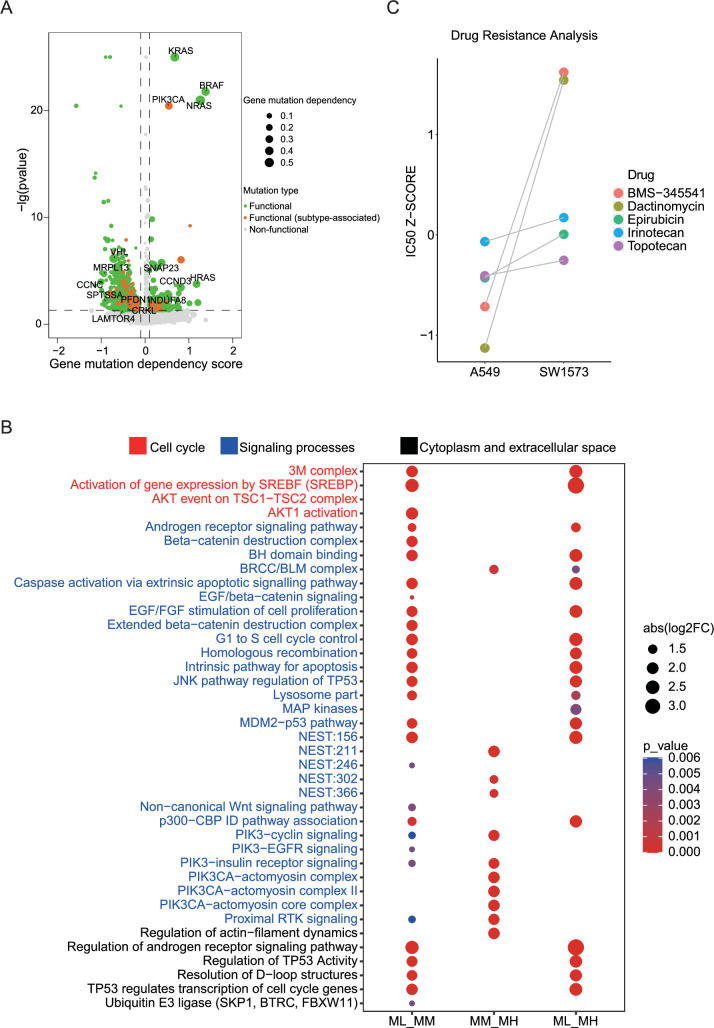
We identified gene mutations that showed significant changes through gene dependence analysis (Figure 4A). To further explore the functional implications of these mutations, we enriched them using a pathway system called Nested Systems in Tumors (NeST; https://idekerlab.ucsd.edu/nest/) (ref. Zheng et al., 2021a). The results revealed notable differences in multiple functional pathways, including androgen receptor, cell cycle, TP53, EGFR, IL-6, and PIK3CA-related pathways (Figure 4B). To gain further insights into the functional implications of these different mutations, we assessed the gene dependency of cells with these mutations by using the DepMap database. Importantly, we observed that the pathway differences between the MM and MH clusters were primarily associated with PIK3CA-related pathway. Previous studies have reported that PIK3CA mutation confers resistance in colorectal cancer, lung cancer, and breast cancer (ref. Wang et al., 2018; ref. Shibata et al., 2009; ref. Hanker et al., 2013). Therefore, we aimed to validate the role of PIK3CA mutations in drug resistance by using public databases. We analyzed two lung adenocarcinoma cell lines, A549 cells and SW1573, harboring KRAS mutation and both KRAS and PIK3CA mutations, respectively, and identified five potentially effective inhibitors (BMS-345541, Dactinomycin, Epirubicin, Irinotecan, and Topotecan) from the previously screened set of 76 LUAD cell growth inhibitors by using GDSC data. Strikingly, SW1573 cells exhibited increased resistance to all these five inhibitors when compared to A549 cells (Figure 4C). In line with this, clinical data supported the notion that concurrent PIK3CA mutation is a poor prognostic factor for LUAD patients (ref. Eng et al., 2015). These findings suggest that PIK3CA mutation may contribute to drug resistance in LUAD.
Distinct clusters of BRCA exhibit different immune infiltration landscapes
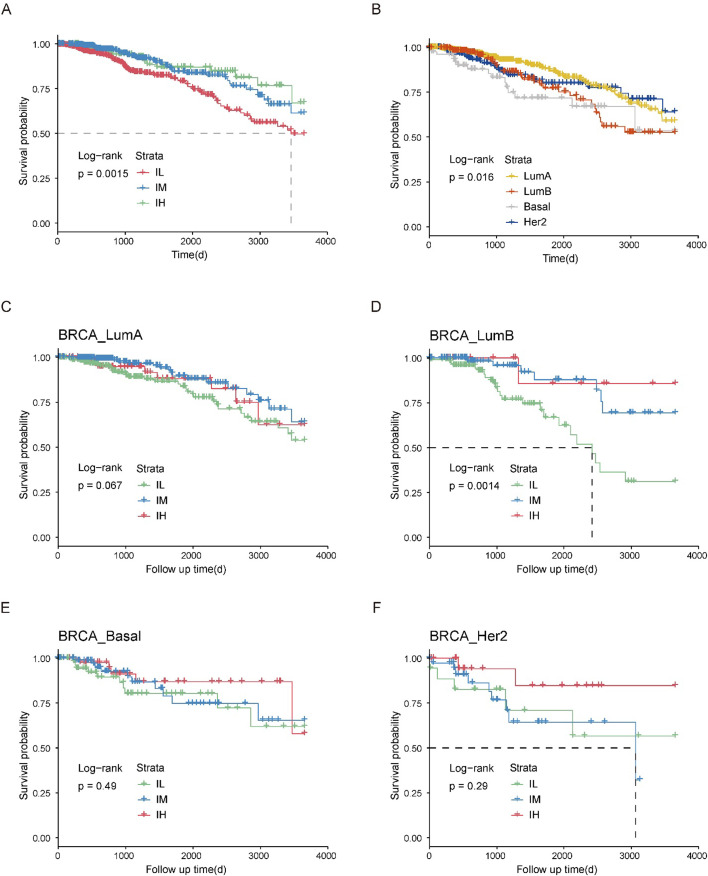
In the meantime, we conducted a comprehensive analysis on BRCA, which has the largest sample size among immune-related cancers (Figure 1C). We hierarchically grouped the BRCA into three distinct immune subgroups based on the expression of hub genes: immune low (IL), immune medium (IM), and immune high (IH; Figure 5A, Supplementary file 1G). Interestingly, each PAM50 subtype included tumors from the IL, IM, and IH immune subgroups, with only minor differences in their relative frequencies (ref. Thorsson et al., 2018; Figure 5B, Figure 5—figure supplement 1A-B). Next, we investigated the relationship among PAM50 classification, immune subtypes, and patient prognosis. Our data revealed significant prognostic differences among immune subgroups of the luminal B subtypes but not the luminal A, basal, or HER2 subtypes (Figure 5—figure supplement 1C–F), highlighting that integrating our method with traditional classification may enable a more detailed stratification of breast cancer samples. Mutation analysis showed that distinct patterns of gene mutations among the three subgroups had significant differences in mutation frequencies of genes such as TP53, CDH1, and LRP2 (Figure 5C–E). We conducted a comparison of the proportions of immune cells across these three subgroups and observed a strong association between the overall immune cell proportion and the clustering results based on hub genes (Figure 5F, Figure 5—figure supplement 2A–J). Specifically, the IH subgroup exhibited significantly higher proportions of CD8+ T cells and Treg cells, with median percentages at 3.9% and 5.5%, respectively, compared to 1.2% and 2.8% in the IM subgroup, and 0.2% and 1.6% in the IL subgroup. Together, these findings suggest that distinct immune responses across the three subgroups potentially link genetic mutation patterns to differences in the immune microenvironment.
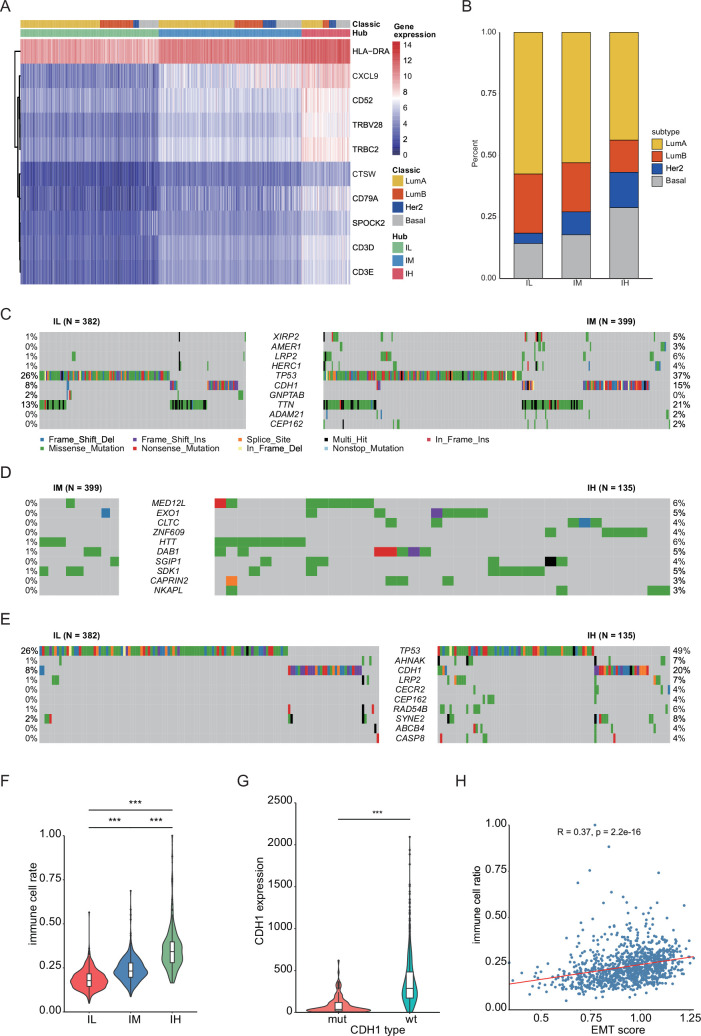
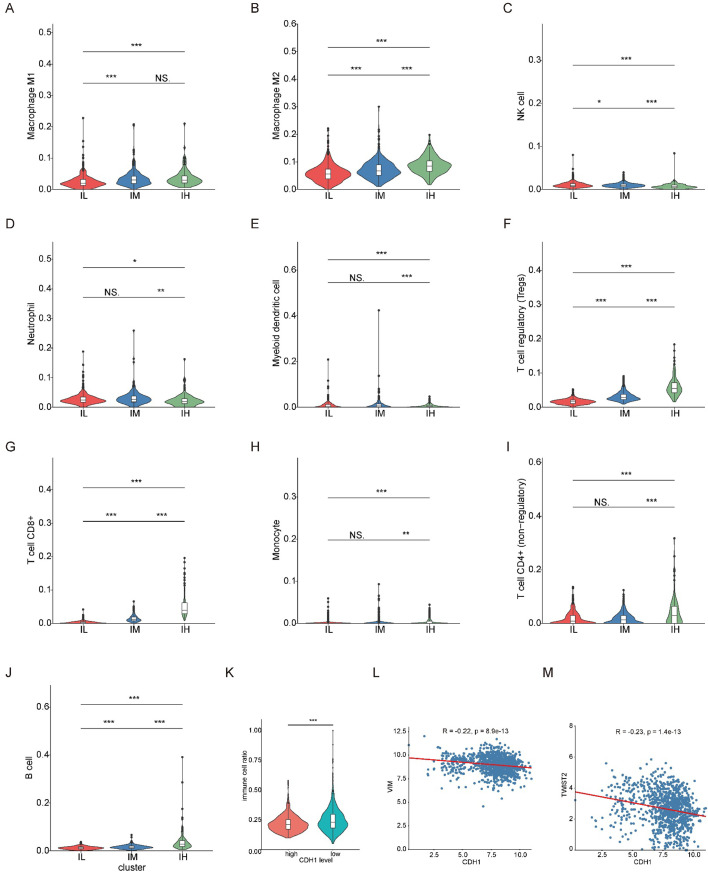
Next, we explored the specific genomic factors influencing immune infiltration in BRCA. Mutation analysis revealed that CDH1 gene mutation ranked as the top two genetic alteration in BRCA samples, following TP53 mutation (Figure 5C–E). A previous study has reported that CDH1 is involved in the mechanisms regulating cell-cell adhesions, mobility, and proliferation of epithelial cells (ref. Meigs et al., 2002). We found that CDH1-mutant samples exhibited significantly lower CDH1 expression compared to CDH1-wildtype samples, and CDH1 expression showed a close correlation with immune cell infiltration (Figure 5G, Figure 5—figure supplement 2K). These results were consistent with clinical observations of CDH1 mutation and high immune infiltration in invasive lobular carcinoma of the breast (ref. An et al., 2018).
The above result encouraged us to investigate how CDH1 mutation influenced immune infiltration. In BRCA, we observed a significant correlation between the expression of CDH1 and EMT marker genes VIM and TWIST2 (Figure 5—figure supplement 2L–M; ref. Zeisberg and Neilson, 2009; ref. Yang et al., 2004). This suggests that the regulation of CDH1 is intricately linked with the EMT process. EMT score analysis showed that there’s a positive correlation between the EMT score and the proportion of immune infiltration (Figure 5H), which suggests that CDH1 might influence immune infiltration through the EMT process.
Mitotic and immune signatures predict patient prognosis at pan-cancer level
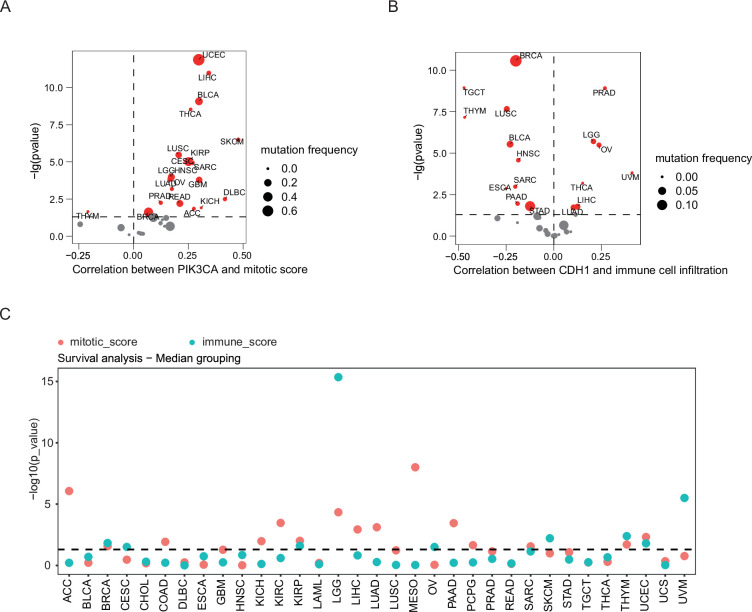
Finally, we analyzed the association of CDH1 and PIK3CA with specific biological processes at pan-cancer level. Our data showed that PIK3CA level was positively correlated with mitotic scores in most of the cancer types, whereas the correlation between CDH1 expression and the proportion of immune cell infiltration was positive in PRAD, LGG, OV, Uveal Melanoma (UVM), THCA, LIHC, and LUAD but negative in BRCA, Testicular Germ Cell Tumors (TGCT), Thymoma (THYM), LUSC, BLCA, Head and Neck squamous cell carcinoma (HNSC), Sarcoma (SARC), Esophageal carcinoma (ESCA), PAAD, and STAD (Figure 6A–B). Eventually, we sought to explore the prognostic relevance of mitosis and immune to patient outcomes at the pan-cancer level. The scores for these biological processes were computed using RNA-seq data from the TCGA database, and the median score was used as a cut-off to categorize patients into different subgroups. Out of 33 cancer types, 19 showed a statistically significant correlation (p<0.05) with at least one pathway score (Figure 6C). Specifically, 10 cancer types were exclusively associated with the mitosis score, 4 cancer types were exclusively associated with the immune score, and 5 cancer types exhibited a correlation with both mitosis and immune scores simultaneously. Overall, the identification of mitosis and immune-related biological processes as significant prognostic factors at the pan-cancer level suggests their potential utility as valuable biomarkers for predicting patient prognosis.
Discussion
Due to the multifaceted nature of variables affecting cancer patient prognosis, it remains uncertain whether there exists a set of genes steadily associated with cancer prognosis, regardless of sample size and other factors. Here, we utilized the MEMORY method to address this question and discovered that all the cancer types have GEARs. We observed significant variation in the number of GEARs among different cancer types, indicative of cancer type-specific patterns. The substantial heterogeneity in driver genes and mortality rates among various cancer types could potentially explain this phenomenon. For example, THCA, known for its low malignancy, displays the fewest genetic expression alterations among all the studied cancer types. This observation could be correlated with the relatively high five-year survival rate in THCA, which exceeds 50% even in advanced stage (ref. Egner, 2010). In contrast, PRAD, despite a favorable prognosis similar to THCA, exhibits a significantly higher number of genetic expression alterations. We hypothesize that the positive prognosis in PRAD cases might be largely attributed to early diagnosis, which enables timely treatment for the majority of PRAD patients (ref. Rebello et al., 2021). This discrepancy between the number of genetic alterations and prognosis in THCA and PRAD highlights the intricate nature of cancer genetics and emphasizes the importance of personalized considerations in cancer treatment and prognosis evaluation. Furthermore, certain genes are commonly found in the GEARs of various cancer types, indicating their potential significance in cancer development. For instance, BEX4 is present in the GEARs of LUAD, KIRC, LGG, PAAD, and STAD and is known to play an oncogenic role in inducing carcinogenic aneuploid transformation via modulating the acetylation of α-tubulin (ref. Lee et al., 2016; ref. Lee et al., 2018). Aneuploidy is a hallmark characteristic of cancer and can lead to alterations in the dosage of oncogenes or tumor suppressor genes, thereby influencing tumor initiation and progression (ref. Beroukhim et al., 2010; ref. Davoli et al., 2013; ref. Girish et al., 2023; ref. Bosco et al., 2023). The regulation of aneuploid transformation by BEX4 may represent a common mechanism through which this gene impacts prognosis across different cancer types. Subsequently, we discovered that GEARs across different cancers displayed distinct functional characteristics. Notably, a recurrent theme was the prominence of mitosis-related and immune-related features. Specifically, the GEARs of LGG, LIHC, and LUAD were enriched in mitosis-related pathways, whereas BRCA and UCEC showed the enrichment of immune-related pathways. Mitosis and immune processes have been widely recognized as having a significant impact on patient prognosis (ref. Ha et al., 2016; ref. Zhou et al., 2018; ref. Mäkinen et al., 2017; ref. Byrne et al., 2020; ref. Liu et al., 2021). However, current clinical guidelines do not yet recommend the use of transcriptomic data to assess scores related to mitotic and immune pathways for predicting patient outcomes, despite the well-established association between these pathways and prognosis in cancer (ref. Ducreux et al., 2015; ref. Cardoso et al., 2019; ref. Oaknin et al., 2022; ref. Vogel et al., 2018; ref. Stupp et al., 2014; ref. Postmus et al., 2017). Cancers enriched with mitosis pathways often exhibit heterogeneous tumor growth kinetics across individuals, with tumor size being one of the crucial factors influencing patient survival (ref. Gui et al., 2018; ref. Alvarez et al., 2022; ref. Infante et al., 2013). For instance, in the case of LGG, the growth rate of tumors directly impacts the patient’s prognosis due to the primary location in the brain. Consequently, alterations in the expression levels of genes related to mitosis are often indicative of the prognosis of LGG (ref. Hoshino, 1984). Cancer types that are enriched in immune-related pathways, such as BRCA and UCEC, are closely associated with deficiencies in DNA mismatch repair (MMR; ref. Sajjadi et al., 2021; ref. Doghri et al., 2019). Cancers enriched in immune-related pathways often exhibit heterogeneous tumor growth kinetics across individuals, where tumor size is closely correlated with patient survival.
To explore the underlying differences between samples associated with distinct mitotic and immune-related pathways, we employed GEAR analysis to identify hub genes for further molecular classification and mutation analysis. Specifically, we focused on LUAD and BRCA, two representative cancer types exhibiting enriched mitosis and immune signatures in GEARs. By classifying the hub gene, we divided LUAD into three subgroups: ML, MM, and MH, which displayed significant differences in survival outcomes. Subsequently, we utilized NeST to analyze the mutations within these subgroups. Notably, there were significant differences observed in cell cycle and signal transduction-related pathways among ML, MM, and MH subgroups. Of particular importance, the PIK3CA-related pathway emerged as a key differentiating pathway between the MH and MM subgroups. Our analyses suggest that LUAD cell lines carrying PIK3CA mutations may exhibit increased drug resistance. This aligns with previous studies demonstrating that targeting the PI3K pathway can overcome drug resistance (ref. Zhang et al., 2016; ref. Donev et al., 2011). Of course, future research is warranted to elucidate the exact functional role of PIK3CA mutations in LUAD.
To investigate the factors influencing immune infiltration in BRCA, we classified the samples based on hub genes and analyzed the differentially mutated genes. Interestingly, we found that CDH1 mutation occurred at a high frequency in the IH subgroups. This led us to speculate that CDH1 plays a crucial role in the regulation of immune infiltration in BRCA. Furthermore, previous studies have reported that CDH1 inactivation promotes immune infiltration in breast cancer (ref. An et al., 2018). CDH1 is a vital gene associated with EMT, and various studies have demonstrated the close relationship between EMT and the tumor immune microenvironment in different cancers (ref. Wang et al., 2021). Consistently, our results also supported the correlation between EMT score and immune infiltration in BRCA. These findings suggest that the mutations in PIK3CA and CDH1, identified through GEAR analysis, have significant impacts on cancer development and hold potential value in improving clinical therapies. This further emphasizes the importance of GEARs in understanding cancer biology and guiding treatment strategies.
Lastly, we investigated the prognostic predictive capabilities of the mitosis score and immune score at the pan-cancer level. Surprisingly, we found that approximately half of the cancer types exhibited significant correlations between these two scores and patient prognosis. Interestingly, even for cancers originating from the same primary location, their correlations with these scores could differ, indicating the potential diversity of mechanisms underlying cancer-related mortality. For instance, both LGG and GBM are brain tumors, but the primary risk factor for patient prognosis in LGG is closely linked to tumor diameter, whereas the main risk factor for GBM patients lies in its high invasiveness and challenge of surgical resection (ref. Brown et al., 2019; ref. Chen et al., 2022a).
Although the present study defined a preliminary catalog of GEARs and supported their relevance with extensive biological data, we recognize several outstanding methodological limitations at both the algorithmic and analytical levels. Chief among them is the need for rigorous, large-scale multiple-testing adjustment before any GEAR list can be considered clinically actionable. Because this work was conceived as an exploratory screen, such correction was intentionally deferred; nonetheless, forthcoming versions of MEMORY will incorporate a dedicated false-positive–control module that will be applied to the consolidated GEAR catalogue prior to any translational use. Although GEARs show robust prognostic associations, the CSN edges are undirected, and hub genes serve primarily as stable biomarkers. Future work will explore causality and therapeutic implications. As a result, the hub genes identified from GEAR in the CSN may primarily serve as stable and effective biological markers. Additionally, through multi-omics analysis, we obtained some functional mutations, but the therapeutic significance of these mutations remains to be elucidated. For example, further study is needed to understand the exact role of PIK3CA mutations in promoting tumor cell proliferation and drug resistance. Similarly, the association of CDH1 mutations with the infiltration of multiple immune cell types also warrants additional experimental investigation. In our future studies, we plan to utilize protein-protein interaction networks and pathway databases to construct a novel network based on the CSN. This approach will allow us to directly screen genes from GEARs that could potentially serve as therapeutic targets. Undoubtedly, future efforts are still required to utilize protein-protein interaction networks and pathway databases to construct a new network based on CSN.
In conclusion, our study utilized the MEMORY algorithm to identify GEARs in 15 cancer types, highlighting the significance of mitosis and immunity in cancer prognosis. Our findings demonstrate that GEARs possess substantial biological significance beyond their role as prognostic biomarkers. This study provides valuable guidance for establishing standards for survival analysis evaluation and holds potential for the development of novel therapeutic strategies.
Methods
Datasets
The gene expression profiles and clinical information of 33 cancers were obtained from the TCGA database and downloaded by the GDC data website (https://portal.gdc.cancer.gov/). All gene expression data and survival data were integrated and normalized.
Multi-gradient permutation survival analysis
RNA-seq count data were normalized by TPM from the gene expression matrix (genes × samples) for each cancer type. We randomly sampled the gene expression data according to the gradient. The sampling strategy was as follows: Ten gradient increases in sample size were pre-set, ranging from approximately 10% to about 100%, with intervals of 10%. Random samples were taken from total samples of each cancer 1000 times, based on each pre-set sample size. A multiple sampling matrix was obtained after the sampling strategy was performed in each gradient. Then survival analysis was performed using the R package ‘survival’. Gene expression values were dichotomized based on the median expression level of each gene in the sampled matrix. The survival analysis method was as follows according to the median expression value of every gene, every sampling matrix was divided into a high and low expression group. This approach divides the samples into high and low expression groups with equal size, providing a standardized grouping strategy for survival analysis. We used 1 for significant survival analysis results (p<0.05) and 0 for non-significant results (p>0.05). The survival analysis matrices were obtained after these processes. This generated a binary matrix of shape (genes ×1000) per gradient, representing the significance profile across permutations. The binary matrix was integrated to a significant-probability matrix by a formula:
\[
\displaystyle A_{ij}=\frac{\sum \limits_{n=1}^{1000}k_{j\times i\times n}}{1000}
\]
where the Aij is the value from row i and column j in the significant-probability matrix. We defined the sampling size kj reached saturation when the max value of column j was equal to 1 in a significant-probability matrix. The least value of kj was selected, and the genes with their corresponding Aij greater than 0.8 were extracted as GEAR.
Construction of core survival network
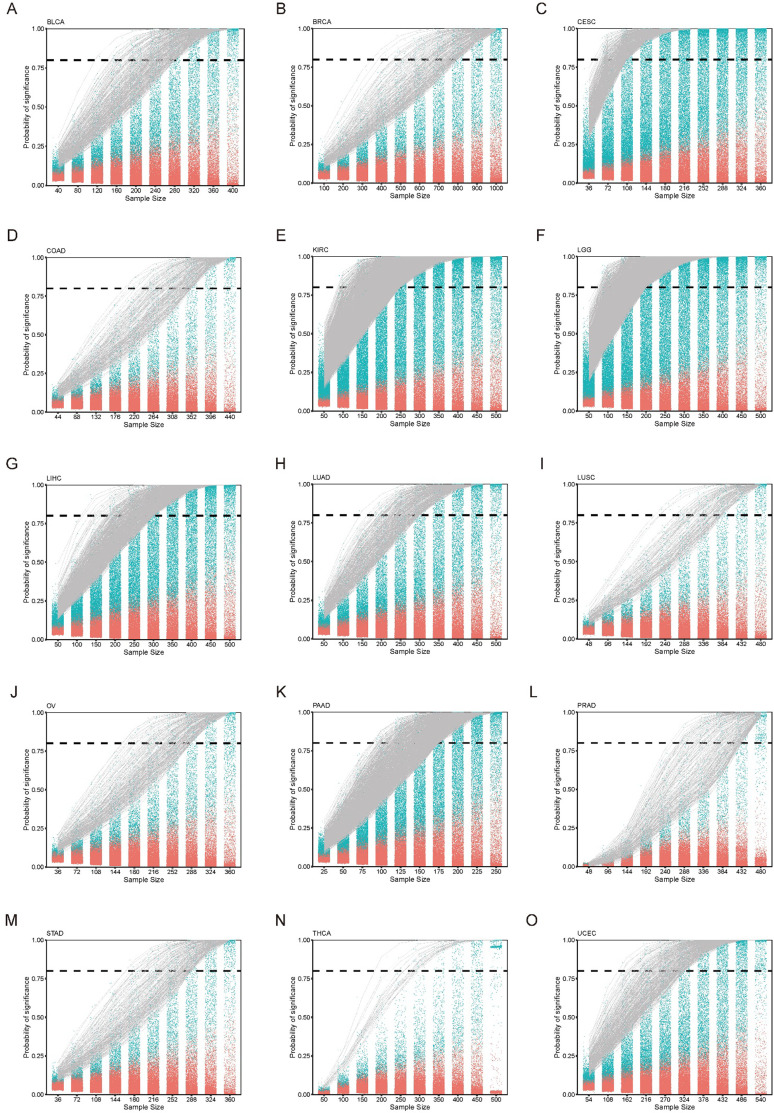
We also defined survival analysis similarity (SAS) as the similarity of the effect on patient prognosis in two genes. For each gene, the 1 000 permutation tests performed at the first sampling gradient yield a binary vector \(A=\left (A_{1},\, \cdots ,A_{1000}\right)\),\(B=\left (B_{1},\, \cdots ,B_{1000}\right)\) (1=significant, 0=non-significant). Vectors from the first sampling gradient (\(k_1\), approximately 10% of the cohort) were used as input. At this sample size, significance patterns exhibited high variability, with many genes not yet consistently significant. This variability was utilized to enable SAS to differentiate gene pairs with concordant versus discordant behavior.
Let \(a=\sum \limits_{i=1}^{1000}A_{i}\), \(b=\sum \limits_{i=1}^{1000}B_{i}\), \(c=\sum \limits_{i=1}^{1000}A_{i}B_{i}\) where c is the number of permutations in which both genes are significant and a, b are the total significant counts for each gene.
SAS is calculated as
\[
\displaystyle SAS\left (A,B\right)=\frac{c}{a+b-2c+1}
\]
a Jaccard-like metric bounded between 0 (no overlap) and 1 (complete overlap); the “+1” term prevents division by zero.
Pair-wise SAS values were computed between every GEAR and all other genes. The 1 000 SAS were selected to construct a core survival network (CSN) in Cytoscape (ref. Shannon et al., 2003). Node degrees were calculated, and the top 10 GEARs with the highest degree in these networks, selected from those with mean expression >10 TPM, were defined as hub genes. We constructed the Core Survival Network (CSN) using the top 1000 gene pairs ranked by SAS values. As the CSN was designed as a heuristic similarity network to prioritize genes for exploration, edge selection was based on empirical ranking rather than formal statistical thresholds.
Gene ontology enrichment analysis
Gene ontology has been used to classify genes based on functions. The gene functions were divided into three types, including molecular function (MF), biological process (BP), and cellular component (CC). ClusterProfiler is an R package for gene set enrichment analysis (ref. Yu et al., 2012). The gene ontology (GO) enrichment analysis was processed in GEARs and hub genes by ClusterProfiler.
Hub gene classification
Tumor samples were genotyped using RNA-seq data. The following steps outline the genotyping and clustering methods used in this study. The expression matrix of hub genes corresponding to various cancer types was extracted from the RNA-seq data. This involved filtering the RNA-seq data to retain only the expression levels of the identified hub genes. To normalize the expression data and reduce the impact of extreme values, a pseudo count of 1 was added to each expression value, and the resulting matrix \(M\) was log-transformed using the formula: \(M^{‘}=log_{2}\left (M+1\right)\). The log-transformed matrix \(M^{‘}\) was then subjected to hierarchical clustering. This clustering was performed in R (v4.3) using the Ward’s method (hclust, method = "ward.D2") to minimize the variance within clusters. The distance metric used was the Euclidean distance (dist, method = "euclidean"). The number of clusters was set to 3 using the cutree function (cutree(model, k=3)), based on visual inspection of the dendrogram structure.
Calculation of tumor mutation burden (TMB) and differential mutation
The TMB and differential mutation gene analysis were carried out to explore the differences between different genotyped samples at the genomic level. The data are from the gene mutation data of TCGA tumor samples, and the grouping information is from the hub gene hierarchical clustering results. Maftools is an R package for the analysis of somatic variant data, which can export results in the form of charts and graphs (ref. Mayakonda et al., 2018). The calculation of TMB and difference mutation analysis was used by the maftools.
Quantifying the effect of gene mutations for tumor cell viability
Gene dependence refers to the extent to which genes are essential for cell proliferation and survival. The Cancer Dependency Map (Depmap, https://depmap.org/portal/) database provides genome-wide gene dependence data for a large number of tumor cell lines (ref. Tsherniak et al., 2017). Gene mutation data of cell lines was obtained from the CCLE database (https://sites.broadinstitute.org/ccle). The genes appearing in the gene mutation data of cell lines were extracted and sorted into mutation lists. Each gene in the list was then analyzed for survival-dependent differences. For each gene in the mutation list, we compared the gene dependence score (S) between cell lines with and without the gene mutation. Specifically, cell lines from the CCLE mutation dataset harboring a mutation in a given gene were classified into one group, while cell lines without the mutation constituted the control group. S for the mutation (Sm) and wild-type (Swt) cell lines were then extracted from the DepMap database.
A two-sample t-test was used to assess the statistical significance of differences between Sm and Swt. The test yielded a p-value, with an alpha level set at 0.05, to determine the significance of the difference in means. Dm was defined as gene mutation dependence, and the computational formula was as follows:
\[
\displaystyle D_{m}=|\bar{s}_{m}-\bar{s}_{wt}|
\]
Subsequently, we conducted a standardization assessment of Dm, employing the following formula:
\[
\displaystyle S_{dm}=\frac{2\left (\bar{s}_{m}-\bar{s}_{wt}\right)}{\bar{s}_{m}+\bar{s}_{wt}}
\]
In this study, we categorized mutations identified in cell lines into three groups based on their impact on gene dependency. Functional mutations refer to the mutations that exhibit significant changes in gene dependency scores, with a p-value <0.05 and Sdm >0.1. Non-functional mutations were those without significant changes in gene dependency scores compared to wild type. Subtype-associated functional mutations were a subset of functional mutations that show statistically significant differences in occurrence frequency across different LUAD subtypes.
Identification of drugs downregulating hub gene expression
The IC50 data of 76 compounds of LUAD cell lines were obtained from Genomics of Drug Sensitivity in Cancer (GDSC) database (ref. Yang et al., 2013). The hub gene expression data of A549 cell lines after treatment with 76 compounds was obtained from Connectivity MAP (cMAP, https://clue.io/) database, and the data were grouped by hierarchical clustering (ref. Subramanian et al., 2017). The drugs that were presented only in the hub gene expression suppression group were considered to be effective against the LUAD cell.
Immune infiltration analysis
Immune infiltration data was calculated by quantiseq method (ref. Finotello et al., 2019). Immunedeconv was an R package for unified access to computational methods for estimating immune cell fractions from bulk RNA-seq data (ref. Sturm et al., 2020). The data were from the gene expression data of TCGA tumor samples after TPM was standardized. Then the immune cell fractions of tumor samples were calculated by the quantiseq method invoking immunedeconv.
Biological function score calculation
GSVA is an R package for calculating biological function score based on a single sample (ref. Hänzelmann et al., 2013). The data was from TCGA in the calculation process, and the software package was GSVA. The immune score was T-cell infiltration rate of that calculated by quantiseq (ref. Finotello et al., 2019). The gene sets that calculated the mitosis score were obtained from gene ontology (GO:0140014).
References
- M Alvarez, JN Benhammou, N Darci-Maher, SW French, SB Han, JS Sinsheimer, VG Agopian, JR Pisegna, P Pajukanta. Human liver single nucleus and single cell RNA sequencing identify a hepatocellular carcinoma-associated cell-type affecting survival. Genome Medicine, 2022. [DOI | PubMed]
- Y An, JR Adams, DP Hollern, A Zhao, SG Chang, MS Gams, PED Chung, X He, R Jangra, JS Shah, J Yang, LA Beck, N Raghuram, KJ Kozma, AJ Loch, W Wang, C Fan, SJ Done, E Zacksenhaus, CJ Guidos, CM Perou, SE Egan. Cdh1 and Pik3ca mutations cooperate to induce immune-related invasive lobular carcinoma of the breast. Cell Reports, 2018. [DOI | PubMed]
- RC Anton, DM Coffey, MM Gondo, MA Stephenson, RW Brown, PT Cagle. The expression of cyclins D1 and E in predicting short-term survival in squamous cell carcinoma of the lung. Modern Pathology, 2000. [DOI | PubMed]
- R Beroukhim, CH Mermel, D Porter, G Wei, S Raychaudhuri, J Donovan, J Barretina, JS Boehm, J Dobson, M Urashima, KT Mc Henry, RM Pinchback, AH Ligon, Y-J Cho, L Haery, H Greulich, M Reich, W Winckler, MS Lawrence, BA Weir, KE Tanaka, DY Chiang, AJ Bass, A Loo, C Hoffman, J Prensner, T Liefeld, Q Gao, D Yecies, S Signoretti, E Maher, FJ Kaye, H Sasaki, JE Tepper, JA Fletcher, J Tabernero, J Baselga, M-S Tsao, F Demichelis, MA Rubin, PA Janne, MJ Daly, C Nucera, RL Levine, BL Ebert, S Gabriel, AK Rustgi, CR Antonescu, M Ladanyi, A Letai, LA Garraway, M Loda, DG Beer, LD True, A Okamoto, SL Pomeroy, S Singer, TR Golub, ES Lander, G Getz, WR Sellers, M Meyerson. The landscape of somatic copy-number alteration across human cancers. Nature, 2010. [DOI | PubMed]
- N Bosco, A Goldberg, X Zhao, JC Mays, P Cheng, AF Johnson, JJ Bianchi, C Toscani, E Di Tommaso, L Katsnelson, D Annuar, S Mei, RE Faitelson, IY Pesselev, KS Mohamed, A Mermerian, EM Camacho-Hernandez, CA Gionco, J Manikas, YS Tseng, Z Sun, S Fani, S Keegan, SM Lippman, D Fenyö, S Giunta, S Santaguida, T Davoli. KaryoCreate: a CRISPR-based technology to study chromosome-specific aneuploidy by targeting human centromeres. Cell, 2023. [DOI | PubMed]
- TJ Brown, DA Bota, MJ van Den Bent, PD Brown, E Maher, D Aregawi, LM Liau, JC Buckner, M Weller, MS Berger, M Glantz. Management of low-grade glioma: a systematic review and meta-analysis. Neuro-Oncology Practice, 2019. [DOI | PubMed]
- A Byrne, P Savas, S Sant, R Li, B Virassamy, SJ Luen, PA Beavis, LK Mackay, PJ Neeson, S Loi. Tissue-resident memory T cells in breast cancer control and immunotherapy responses. Nature Reviews Clinical Oncology, 2020. [DOI | PubMed]
- C Cai, X Wang, Q Fu, A Chen. The VEGF expression associated with prognosis in patients with intrahepatic cholangiocarcinoma: a systematic review and meta-analysis. World Journal of Surgical Oncology, 2022. [DOI | PubMed]
- X Cai. Software Heritage, 2024
- N Cancer Genome Atlas Research. Comprehensive molecular profiling of lung adenocarcinoma. Nature, 2014. [DOI | PubMed]
- F Cardoso, S Kyriakides, S Ohno, F Penault-Llorca, P Poortmans, IT Rubio, S Zackrisson, E Senkus. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology, 2019. [DOI | PubMed]
- L Chen, J Ma, Z Zou, H Liu, C Liu, S Gong, X Gao, G Liang. Clinical characteristics and prognosis of patients with glioblastoma: a review of survival analysis of 1674 patients based on SEER database. Medicine, 2022a. [DOI | PubMed]
- G Chen, Q Wang, K Wang. MicroRNA-218-5p affects lung adenocarcinoma progression through targeting endoplasmic reticulum oxidoreductase 1 alpha. Bioengineered, 2022b. [DOI | PubMed]
- T Davoli, AW Xu, KE Mengwasser, LM Sack, JC Yoon, PJ Park, SJ Elledge. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell, 2013. [DOI | PubMed]
- R Doghri, Y Houcine, N Boujelbène, M Driss, L Charfi, I Abbes, K Mrad, R Sellami. Mismatch repair deficiency in endometrial cancer: immunohistochemistry staining and clinical implications. Applied Immunohistochemistry & Molecular Morphology, 2019. [DOI | PubMed]
- IS Donev, W Wang, T Yamada, Q Li, S Takeuchi, K Matsumoto, T Yamori, Y Nishioka, S Sone, S Yano. Transient PI3K inhibition induces apoptosis and overcomes HGF-mediated resistance to EGFR-TKIs in EGFR mutant lung cancer. Clinical Cancer Research, 2011. [DOI | PubMed]
- M Ducreux, AS Cuhna, C Caramella, A Hollebecque, P Burtin, D Goéré, T Seufferlein, K Haustermans, JL Van Laethem, T Conroy, D Arnold. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology, 2015. [DOI | PubMed]
- D Dworakowska, E Jassem, J Jassem, C Boltze, KH Wiedorn, R Dworakowski, J Skokowski, K Jaśkiewicz, E Czestochowska. Prognostic value of cyclin D1 overexpression in correlation with pRb and p53 status in non-small cell lung cancer (NSCLC). Journal of Cancer Research and Clinical Oncology, 2005. [DOI | PubMed]
- JR Egner. AJCC cancer staging manual. JAMA, 2010. [DOI]
- J Eng, KM Woo, CS Sima, A Plodkowski, MD Hellmann, JE Chaft, MG Kris, ME Arcila, M Ladanyi, A Drilon. Impact of concurrent PIK3CA mutations on response to EGFR tyrosine kinase inhibition in EGFR-Mutant lung cancers and on prognosis in oncogene-driven lung adenocarcinomas. Journal of Thoracic Oncology, 2015. [DOI | PubMed]
- S Erfani, H Hua, Y Pan, BP Zhou, XH Yang. The context-dependent impact of integrin-associated CD151 and other tetraspanins on cancer development and progression: a class of versatile mediators of cellular function and signaling, tumorigenesis and metastasis. Cancers, 2021. [DOI | PubMed]
- V Esposito, A Baldi, A De Luca, G Tonini, B Vincenzi, D Santini, P Persichetti, A Mancini, G Citro, F Baldi, AM Groeger, M Caputi. Cell cycle related proteins as prognostic parameters in radically resected non-small cell lung cancer. Journal of Clinical Pathology, 2005. [DOI | PubMed]
- T Fan, Y Liu, H Liu, L Wang, H Tian, Y Zheng, B Zheng, L Xue, C Li, J He. Transmembrane protein-based risk model and H3K4me3 modification characteristics in lung adenocarcinoma. Frontiers in Oncology, 2022. [DOI | PubMed]
- F Finotello, C Mayer, C Plattner, G Laschober, D Rieder, H Hackl, A Krogsdam, Z Loncova, W Posch, D Wilflingseder, S Sopper, M Ijsselsteijn, TP Brouwer, D Johnson, Y Xu, Y Wang, ME Sanders, MV Estrada, P Ericsson-Gonzalez, P Charoentong, J Balko, NF da Cunha Carvalho de Miranda, Z Trajanoski. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Medicine, 2019. [DOI | PubMed]
- LS Freedman. Tables of the number of patients required in clinical trials using the logrank test. Statistics in Medicine, 1982. [DOI | PubMed]
- Q Fu, Z Yu. Phosphoglycerate kinase 1 (PGK1) in cancer: a promising target for diagnosis and therapy. Life Sciences, 2020. [DOI | PubMed]
- W Gao, JZ-H Li, S-Q Chen, C-Y Chu, JY-W Chan, T-S Wong. Decreased brain-expressed X-linked 4 (BEX4) expression promotes growth of oral squamous cell carcinoma. Journal of Experimental & Clinical Cancer Research, 2016. [DOI | PubMed]
- V Gebski, V Garès, E Gibbs, K Byth. Data maturity and follow-up in time-to-event analyses. International Journal of Epidemiology, 2018. [DOI | PubMed]
- PA Gimotty, P Van Belle, DE Elder, T Murry, KT Montone, X Xu, S Hotz, S Raines, ME Ming, P Wahl, D Guerry. Biologic and prognostic significance of dermal Ki67 expression, mitoses, and tumorigenicity in thin invasive cutaneous melanoma. Journal of Clinical Oncology, 2005. [DOI | PubMed]
- V Girish, AA Lakhani, SL Thompson, CM Scaduto, LM Brown, RA Hagenson, EL Sausville, BE Mendelson, PK Kandikuppa, DA Lukow, ML Yuan, EC Stevens, SN Lee, KM Schukken, SM Akalu, A Vasudevan, C Zou, B Salovska, W Li, JC Smith, AM Taylor, RA Martienssen, Y Liu, R Sun, JM Sheltzer. Oncogene-like addiction to aneuploidy in human cancers. Science, 2023. [DOI | PubMed]
- C Gui, SE Kosteniuk, JC Lau, JF Megyesi. Tumor growth dynamics in serially-imaged low-grade glioma patients. Journal of Neuro-Oncology, 2018. [DOI | PubMed]
- M Guo, F Li, L Zhao, Z Fang, H Yu, Z Songyang, Y Xiong. Pan-cancer investigation of C-to-U editing reveals its important role in cancer development and new targets for cancer treatment. Frontiers in Oncology, 2023. [DOI | PubMed]
- SY Ha, M Choi, T Lee, CK Park. The prognostic role of mitotic index in hepatocellular carcinoma patients after curative hepatectomy. Cancer Research and Treatment, 2016. [DOI | PubMed]
- AB Hanker, AD Pfefferle, JM Balko, MG Kuba, CD Young, V Sánchez, CR Sutton, H Cheng, CM Perou, JJ Zhao, RS Cook, CL Arteaga. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. PNAS, 2013. [DOI | PubMed]
- S Hänzelmann, R Castelo, J Guinney. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics, 2013. [DOI | PubMed]
- Y He, Y Luo, D Zhang, X Wang, P Zhang, H Li, S Ejaz, S Liang. PGK1-mediated cancer progression and drug resistance. American Journal of Cancer Research, 2019. [PubMed]
- T Hoshino. A commentary on the biology and growth kinetics of low-grade and high-grade gliomas. Journal of Neurosurgery, 1984. [DOI | PubMed]
- S-F Huo, W-L Shang, M Yu, X-P Ren, H-X Wen, C-Y Chai, L Sun, K Hui, L-H Liu, S-H Wei, X-X Wang, Y Wang, Y-X Tian. STEAP1 facilitates metastasis and epithelial-mesenchymal transition of lung adenocarcinoma via the JAK2/STAT3 signaling pathway. Bioscience Reports, 2020. [DOI | PubMed]
- LH Hwang, LF Lau, DL Smith, CA Mistrot, KG Hardwick, ES Hwang, A Amon, AW Murray. Budding yeast Cdc20: a target of the spindle checkpoint. Science, 1998. [DOI | PubMed]
- M Infante, T Berghmans, MA Heuvelmans, G Hillerdal, M Oudkerk. Slow-growing lung cancer as an emerging entity: from screening to clinical management. The European Respiratory Journal, 2013. [DOI | PubMed]
- YJ Kang, SJ Oh, SY Bae, EK Kim, YJ Lee, EH Park, J Jeong, HK Park, YJ Suh, YS Kim. Predictive biological factors for late survival in patients with HER2-positive breast cancer. Scientific Reports, 2023. [DOI | PubMed]
- R Keshet, JS Lee, L Adler, M Iraqi, Y Ariav, LQJ Lim, S Lerner, S Rabinovich, R Oren, R Katzir, H Weiss Tishler, N Stettner, O Goldman, H Landesman, S Galai, Y Kuperman, Y Kuznetsov, A Brandis, T Mehlman, S Malitsky, M Itkin, SE Koehler, Y Zhao, K Talsania, T-W Shen, N Peled, I Ulitsky, A Porgador, E Ruppin, A Erez. Targeting purine synthesis in ASS1-expressing tumors enhances the response to immune checkpoint inhibitors. Nature Cancer, 2020. [DOI | PubMed]
- TA Kufer, HHW Silljé, R Körner, OJ Gruss, P Meraldi, EA Nigg. Human TPX2 is required for targeting Aurora-A kinase to the spindle. The Journal of Cell Biology, 2002. [DOI | PubMed]
- Y Lai, G OuYang, L Sheng, Y Zhang, B Lai, M Zhou. Novel prognostic genes and subclasses of acute myeloid leukemia revealed by survival analysis of gene expression data. BMC Medical Genomics, 2021. [DOI | PubMed]
- F Lamers, I van der Ploeg, L Schild, ME Ebus, J Koster, BR Hansen, T Koch, R Versteeg, HN Caron, JJ Molenaar. Knockdown of survivin (BIRC5) causes apoptosis in neuroblastoma via mitotic catastrophe. Endocrine-Related Cancer, 2011. [DOI | PubMed]
- JK Lee, J Lee, H Go, CG Lee, S Kim, HS Kim, H Cho, KS Choi, GH Ha, CW Lee. Oncogenic microtubule hyperacetylation through BEX4-mediated sirtuin 2 inhibition. Cell Death & Disease, 2016. [DOI | PubMed]
- JK Lee, GH Ha, HS Kim, CW Lee. Oncogenic potential of BEX4 is conferred by Polo-like kinase 1-mediated phosphorylation. Experimental & Molecular Medicine, 2018. [DOI | PubMed]
- S Lee, H Kang, E Shin, J Jeon, H Youn, B Youn. BEX1 and BEX4 induce GBM progression through regulation of actin polymerization and activation of YAP/TAZ signaling. International Journal of Molecular Sciences, 2021. [DOI | PubMed]
- X Li, Y Jiang, J Meisenhelder, W Yang, DH Hawke, Y Zheng, Y Xia, K Aldape, J He, T Hunter, L Wang, Z Lu. Mitochondria-Translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA Cycle in tumorigenesis. Molecular Cell, 2016. [DOI | PubMed]
- W Lin, J Wang, J Ge, R Zhou, Y Hu, L Xiao, Q Peng, Z Zheng. The activity of cuproptosis pathway calculated by AUCell algorithm was employed to construct cuproptosis landscape in lung adenocarcinoma. Discover Oncology, 2023. [DOI | PubMed]
- J Liu, T Lichtenberg, KA Hoadley, LM Poisson, AJ Lazar, AD Cherniack, AJ Kovatich, CC Benz, DA Levine, AV Lee, L Omberg, DM Wolf, CD Shriver, V Thorsson, H Hu. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell, 2018. [DOI | PubMed]
- W Liu, L Sun, J Zhang, W Song, M Li, H Wang. The landscape and prognostic value of immune characteristics in uterine corpus endometrial cancer. Bioscience Reports, 2021. [DOI | PubMed]
- JM Mäkinen, K Laitakari, S Johnson, R Mäkitaro, R Bloigu, P Pääkkö, E Lappi-Blanco, R Kaarteenaho. Histological features of malignancy correlate with growth patterns and patient outcome in lung adenocarcinoma. Histopathology, 2017. [DOI | PubMed]
- K Matsuura, H Sawai, K Ikeo, S Ogawa, E Iio, M Isogawa, N Shimada, A Komori, H Toyoda, T Kumada, T Namisaki, H Yoshiji, N Sakamoto, M Nakagawa, Y Asahina, M Kurosaki, N Izumi, N Enomoto, A Kusakabe, E Kajiwara, Y Itoh, T Ide, A Tamori, M Matsubara, N Kawada, K Shirabe, E Tomita, M Honda, S Kaneko, S Nishina, A Suetsugu, Y Hiasa, H Watanabe, T Genda, I Sakaida, S Nishiguchi, K Takaguchi, E Tanaka, J Sugihara, M Shimada, Y Kondo, Y Kawai, K Kojima, M Nagasaki, K Tokunaga, Y Tanaka. Genome-Wide association study identifies TLL1 variant associated with development of hepatocellular carcinoma after eradication of hepatitis C virus infection. Gastroenterology, 2017. [DOI | PubMed]
- A Mayakonda, DC Lin, Y Assenov, C Plass, HP Koeffler. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Research, 2018. [DOI | PubMed]
- TJ Mead. ADAMTS6: emerging roles in cardiovascular, musculoskeletal and cancer biology. Frontiers in Molecular Biosciences, 2022. [DOI | PubMed]
- TE Meigs, M Fedor-Chaiken, DD Kaplan, R Brackenbury, PJ Casey. Galpha12 and Galpha13 negatively regulate the adhesive functions of cadherin. The Journal of Biological Chemistry, 2002. [DOI | PubMed]
- E Miyauchi, S Morita, A Nakamura, Y Hosomi, K Watanabe, S Ikeda, M Seike, Y Fujita, K Minato, R Ko, T Harada, K Hagiwara, K Kobayashi, T Nukiwa, A Inoue. Updated analysis of NEJ009: gefitinib-alone versus gefitinib plus chemotherapy for non-small-cell lung cancer with mutated EGFR. Journal of Clinical Oncology, 2022. [DOI | PubMed]
- JA Mouabbi, A Singareeka Raghavendra, RL Bassett, A Hassan, D Tripathy, RM Layman. Survival outcomes in patients with hormone receptor-positive metastatic breast cancer with low or no ERBB2 expression treated with targeted therapies plus endocrine therapy. JAMA Network Open, 2023. [DOI | PubMed]
- A Oaknin, TJ Bosse, CL Creutzberg, G Giornelli, P Harter, F Joly, D Lorusso, C Marth, V Makker, MR Mirza, JA Ledermann, N Colombo. Endometrial cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Annals of Oncology, 2022. [DOI | PubMed]
- A Palaparti, A Baratz, S Stifani. The Groucho/transducin-like enhancer of split transcriptional repressors interact with the genetically defined amino-terminal silencing domain of histone H3. The Journal of Biological Chemistry, 1997. [DOI | PubMed]
- P Poplawski, S Alseekh, U Jankowska, B Skupien-Rabian, R Iwanicka-Nowicka, H Kossowska, A Fogtman, B Rybicka, J Bogusławska, A Adamiok-Ostrowska, K Hanusek, J Hanusek, M Koblowska, AR Fernie, A Piekiełko-Witkowska. Coordinated reprogramming of renal cancer transcriptome, metabolome and secretome associates with immune tumor infiltration. Cancer Cell International, 2023. [DOI | PubMed]
- PE Postmus, KM Kerr, M Oudkerk, S Senan, DA Waller, J Vansteenkiste, C Escriu, S Peters, EG Committee. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology, 2017. [DOI | PubMed]
- D Povero, SM Johnson, J Liu. Hypoxia, hypoxia-inducible gene 2 (HIG2)/HILPDA, and intracellular lipolysis in cancer. Cancer Letters, 2020. [DOI | PubMed]
- RJ Rebello, C Oing, KE Knudsen, S Loeb, DC Johnson, RE Reiter, S Gillessen, T Van der Kwast, RG Bristow. Prostate cancer. Nature Reviews Disease Primers, 2021. [DOI | PubMed]
- E Sajjadi, K Venetis, R Piciotti, M Invernizzi, E Guerini-Rocco, S Haricharan, N Fusco. Mismatch repair-deficient hormone receptor-positive breast cancers: Biology and pathological characterization. Cancer Cell International, 2021. [DOI | PubMed]
- P Shannon, A Markiel, O Ozier, NS Baliga, JT Wang, D Ramage, N Amin, B Schwikowski, T Ideker. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research, 2003. [DOI | PubMed]
- T Shibata, A Kokubu, K Tsuta, S Hirohashi. Oncogenic mutation of PIK3CA in small cell lung carcinoma: a potential therapeutic target pathway for chemotherapy-resistant lung cancer. Cancer Letters, 2009. [DOI | PubMed]
- MP Smeltzer, NR Faris, MA Ray, RU Osarogiagbon. Association of pathologic nodal staging quality with survival among patients with non-small cell lung cancer after resection with curative intent. JAMA Oncology, 2018. [DOI | PubMed]
- R Stupp, M Brada, MJ van den Bent, JC Tonn, G Pentheroudakis. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology, 2014. [DOI | PubMed]
- G Sturm, F Finotello, M List. Immunedeconv: an R package for unified access to computational methods for estimating immune cell fractions from bulk RNA-Sequencing data. Methods in Molecular Biology, 2020. [DOI | PubMed]
- A Subramanian, R Narayan, SM Corsello, DD Peck, TE Natoli, X Lu, J Gould, JF Davis, AA Tubelli, JK Asiedu, DL Lahr, JE Hirschman, Z Liu, M Donahue, B Julian, M Khan, D Wadden, IC Smith, D Lam, A Liberzon, C Toder, M Bagul, M Orzechowski, OM Enache, F Piccioni, SA Johnson, NJ Lyons, AH Berger, AF Shamji, AN Brooks, A Vrcic, C Flynn, J Rosains, DY Takeda, R Hu, D Davison, J Lamb, K Ardlie, L Hogstrom, P Greenside, NS Gray, PA Clemons, S Silver, X Wu, WN Zhao, W Read-Button, X Wu, SJ Haggarty, LV Ronco, JS Boehm, SL Schreiber, JG Doench, JA Bittker, DE Root, B Wong, TR Golub. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell, 2017. [DOI | PubMed]
- SH Teh, S Uong, TY Lin, S Shiraga, Y Li, IY Gong, LJ Herrinton, RA Li. Clinical outcomes following regionalization of gastric cancer care in a US integrated health care system. Journal of Clinical Oncology, 2021. [DOI | PubMed]
- V Thorsson, DL Gibbs, SD Brown, D Wolf, DS Bortone, T-H Ou Yang, E Porta-Pardo, GF Gao, CL Plaisier, JA Eddy, E Ziv, AC Culhane, EO Paull, IKA Sivakumar, AJ Gentles, R Malhotra, F Farshidfar, A Colaprico, JS Parker, LE Mose, NS Vo, J Liu, Y Liu, J Rader, V Dhankani, SM Reynolds, R Bowlby, A Califano, AD Cherniack, D Anastassiou, D Bedognetti, Y Mokrab, AM Newman, A Rao, K Chen, A Krasnitz, H Hu, TM Malta, H Noushmehr, CS Pedamallu, S Bullman, AI Ojesina, A Lamb, W Zhou, H Shen, TK Choueiri, JN Weinstein, J Guinney, J Saltz, RA Holt, CS Rabkin, AJ Lazar, JS Serody, EG Demicco, ML Disis, BG Vincent, I Shmulevich. The immune landscape of cancer. Immunity, 2018. [DOI | PubMed]
- K Tomczak, P Czerwińska, M Wiznerowicz. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemporary Oncology, 2015. [DOI | PubMed]
- A Tsherniak, F Vazquez, PG Montgomery, BA Weir, G Kryukov, GS Cowley, S Gill, WF Harrington, S Pantel, JM Krill-Burger, RM Meyers, L Ali, A Goodale, Y Lee, G Jiang, J Hsiao, WFJ Gerath, S Howell, E Merkel, M Ghandi, LA Garraway, DE Root, TR Golub, JS Boehm, WC Hahn. Defining a cancer dependency map. Cell, 2017. [DOI | PubMed]
- L Uusküla-Reimand, MD Wilson. Untangling the roles of TOP2A and TOP2B in transcription and cancer. Science Advances, 2022. [DOI | PubMed]
- A Vogel, A Cervantes, I Chau, B Daniele, JM Llovet, T Meyer, J-C Nault, U Neumann, J Ricke, B Sangro, P Schirmacher, C Verslype, CJ Zech, D Arnold, E Martinelli. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology, 2018. [DOI | PubMed]
- AD Waldman, JM Fritz, MJ Lenardo. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nature Reviews. Immunology, 2020. [DOI | PubMed]
- Q Wang, Y Shi, K Zhou, L Wang, Z Yan, Y Liu, L Xu, S Zhao, H Chu, T Shi, Q Ma, J Bi. PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death & Disease, 2018. [DOI | PubMed]
- T Wang, R Zhai, X Lv, K Wang, J Xu. LINC02418 promotes malignant behaviors in lung adenocarcinoma cells by sponging miR-4677-3p to upregulate KNL1 expression. BMC Pulmonary Medicine, 2020. [DOI | PubMed]
- G Wang, D Xu, Z Zhang, X Li, J Shi, J Sun, HZ Liu, X Li, M Zhou, T Zheng. The pan-cancer landscape of crosstalk between epithelial-mesenchymal transition and immune evasion relevant to prognosis and immunotherapy response. NPJ Precision Oncology, 2021. [DOI | PubMed]
- JN Weinstein, EA Collisson, GB Mills, KRM Shaw, BA Ozenberger, K Ellrott, I Shmulevich, C Sander, JM Stuart. The cancer genome atlas pan-cancer analysis project. Nature Genetics, 2013. [DOI | PubMed]
- T Wittmann, M Wilm, E Karsenti, I Vernos. TPX2, A novel xenopus MAP involved in spindle pole organization. The Journal of Cell Biology, 2000. [DOI | PubMed]
- M Won, KA Park, S Kim, E Ju, Y Ko, H Yoo, H Ro, J Lee, J Oh, EG Lee, SY Kim, SW Nam, H-M Shen, M-K Yeo, JM Kim, GM Hur. ANKRD13a controls early cell-death checkpoint by interacting with RIP1 independent of NF-κB. Cell Death and Differentiation, 2022. [DOI | PubMed]
- Y Wu, M Lv, T Qian, Y Shen. Correlation analysis of Ki67 and CK7 expression with clinical characteristics and prognosis of postoperative cervical adenocarcinoma patients. Annals of Palliative Medicine, 2021. [DOI | PubMed]
- C Xu, H Li, L Zhang, T Jia, L Duan, C Lu. MicroRNA‑1915‑3p prevents the apoptosis of lung cancer cells by downregulating DRG2 and PBX2. Molecular Medicine Reports, 2016. [DOI | PubMed]
- J Xu, H Zheng, S Yuan, B Zhou, W Zhao, Y Pan, D Qi. Overexpression of ANLN in lung adenocarcinoma is associated with metastasis. Thoracic Cancer, 2019. [DOI | PubMed]
- J Xu, Q Chen, K Tian, R Liang, T Chen, A Gong, NW Mathy, T Yu, X Chen. m6A methyltransferase METTL3 maintains colon cancer tumorigenicity by suppressing SOCS2 to promote cell proliferation. Oncology Reports, 2020. [DOI | PubMed]
- J Yang, SA Mani, JL Donaher, S Ramaswamy, RA Itzykson, C Come, P Savagner, I Gitelman, A Richardson, RA Weinberg. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell, 2004. [DOI | PubMed]
- W Yang, J Soares, P Greninger, EJ Edelman, H Lightfoot, S Forbes, N Bindal, D Beare, JA Smith, IR Thompson, S Ramaswamy, PA Futreal, DA Haber, MR Stratton, C Benes, U McDermott, MJ Garnett. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Research, 2013. [DOI | PubMed]
- B-Y Yang, J-W Song, H-Z Sun, J-C Xing, Z-H Yang, C-Y Wei, T-Y Xu, Z-N Yu, Y-N Zhang, Y-F Wang, H Chang, Z-P Xu, M Hou, M-J Ji, Y-S Zhang. PSMB8 regulates glioma cell migration, proliferation, and apoptosis through modulating ERK1/2 and PI3K/AKT signaling pathways. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie, 2018. [DOI | PubMed]
- X Yao, SK Ireland, T Pham, B Temple, R Chen, MHG Raj, H Biliran. TLE1 promotes EMT in A549 lung cancer cells through suppression of E-cadherin. Biochemical and Biophysical Research Communications, 2014. [DOI | PubMed]
- G Yu, LG Wang, Y Han, QY He. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS, 2012. [DOI | PubMed]
- M Zeisberg, EG Neilson. Biomarkers for epithelial-mesenchymal transitions. The Journal of Clinical Investigation, 2009. [DOI | PubMed]
- Q Zhang, T Sun, P Kang, K Qian, B Deng, J Zhou, R Wang, B Jiang, K Li, F Liu, S Wu, Q Tan. Combined analysis of rearrangement of ALK, ROS1, somatic mutation of EGFR, KRAS, BRAF, PIK3CA, and mRNA expression of ERCC1, TYMS, RRM1, TUBB3, EGFR in patients with non-small cell lung cancer and their clinical significance. Cancer Chemotherapy and Pharmacology, 2016. [DOI | PubMed]
- P Zhang, S Li, C Lv, J Si, Y Xiong, L Ding, Y Ma, Y Yang. BPI-9016M, a c-Met inhibitor, suppresses tumor cell growth, migration and invasion of lung adenocarcinoma via miR203-DKK1. Theranostics, 2018. [DOI | PubMed]
- C Zhang, L Wang, C Xiong, R Zhao, H Liang, X Luo. The role of vascular endothelial growth factor as a prognostic and clinicopathological marker in osteosarcoma: a systematic review and meta-analysis. Journal of Orthopaedic Surgery and Research, 2021. [DOI | PubMed]
- Z Zhao, J Li, F Tan, S Gao, J He. mTOR up-regulation of BEX4 promotes lung adenocarcinoma cell proliferation by potentiating OCT4. Biochemical and Biophysical Research Communications, 2018. [DOI | PubMed]
- F Zheng, MR Kelly, DJ Ramms, ML Heintschel, K Tao, B Tutuncuoglu, JJ Lee, K Ono, H Foussard, M Chen, KA Herrington, E Silva, SN Liu, J Chen, C Churas, N Wilson, A Kratz, RT Pillich, DN Patel, J Park, B Kuenzi, MK Yu, K Licon, D Pratt, JF Kreisberg, M Kim, DL Swaney, X Nan, SI Fraley, JS Gutkind, NJ Krogan, T Ideker. Interpretation of cancer mutations using a multiscale map of protein systems. Science, 2021a. [DOI | PubMed]
- H Zheng, H Tian, X Yu, P Ren, Q Yang. G protein gamma 7 suppresses progression of lung adenocarcinoma by inhibiting E2F transcription factor 1. International Journal of Biological Macromolecules, 2021b. [DOI | PubMed]
- C Zhou, Y Wang, X Liu, Y Liang, Z Fan, T Jiang, Y Wang, L Wang. Molecular profiles for insular low-grade gliomas with putamen involvement. Journal of Neuro-Oncology, 2018. [DOI | PubMed]
- Z Zhou, Y Li, H Hao, Y Wang, Z Zhou, Z Wang, X Chu. Screening hub genes as prognostic biomarkers of hepatocellular carcinoma by bioinformatics analysis. Cell Transplantation, 2019. [DOI | PubMed]