Identification of a JAK–STAT–miR155HG positive feedback loop in regulating natural killer (NK) cells proliferation and effector functions
Abstract
The Janus kinase/signal transducers and activators of transcription (JAK–STAT) control natural killer (NK) cells development and cytotoxic functions, however, whether long non-coding RNAs (lncRNAs) are involved in this pathway remains unknown. We found that miR155HG was elevated in activated NK cells and promoted their proliferation and effector functions in both NK92 and induced-pluripotent stem cells (iPSCs)-derived NK (iPSC-NK) cells, without reliance on its derived miR-155 and micropeptide P155. Mechanistically, miR155HG bound to miR-6756 and relieved its repression of JAK3 expression, thereby promoting the JAK–STAT pathway and enhancing NK cell proliferation and function. Further investigations disclosed that upon cytokine stimulation, STAT3 directly interacts with miR155HG promoter and induces miR155HG transcription. Collectively, we identify a miR155HG-mediated positive feedback loop of the JAK–STAT signaling. Our study will also provide a power target regarding miR155HG for improving NK cell generation and effector function in the field of NK cell adoptive transfer therapy against cancer, especially iPSC-derived NK cells.
Article type: Research Article
Keywords: NK cells, iPSC-NK cells, lncRNA, miR155HG, Competing endogenous RNA, miR-6756, JAK–STAT signaling pathway
Affiliations: Pediatrics Research Institute of Hunan Province, the Affiliated Children’s Hospital of Xiangya School of Medicine, Central South University (Hunan Children’s Hospital), Changsha 410007, China; Department of Clinical Hematology, College of Pharmacy and Laboratory Medicine Science, Army Medical University, Chongqing 400038, China; Department of Pharmacy, the General Hospital of Western Theater Command of PLA, Chengdu 610083, China; The School of Pediatrics, Hengyang Medical School, University of South China (Hunan Children’s Hospital), Changsha 410007, China; Department of Biological Sciences, Columbia University, NY 10027, USA; Department of Hepatobiliary Surgery, Hunan Provincial People’s Hospital (the First Affiliated Hospital of Hunan Normal University), Changsha 410005, China; Translational Medicine Research Center, Shanxi Medical University, Taiyuan 030001, China; Institute of Clinical Pharmacology, Anhui Medical University, Key Laboratory of Anti-inflammatory and Immune Medicine, Ministry of Education, Hefei 230032, China; State Key Laboratory of Trauma and Chemical Poisoning, Institute of Combined Injury, Chongqing Engineering Research Center for Nanomedicine, College of Preventive Medicine, Army Medical University, Chongqing 400038, China; The Fifth People’s Hospital of Chongqing, Chongqing 400062, China
License: © 2025 The Authors CC BY 4.0 This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.1016/j.apsb.2025.02.034 | PubMed: 40486832 | PMC: PMC12138116
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (1.3 MB)
Introduction
Natural killer (NK) cells are the frontline guardians in the immune response to cancer and invading pathogens, providing strong antitumor effects and exhibiting favorable safety profiles in the field of allogeneic cancer immunotherapyref. 1, ref. 2, ref. 3. Once triggered, NK cells not only unleash their cytotoxic contents, such as perforin and granzyme B, to induce direct lysis of cancer cells but also rapidly generate chemokines and cytokines like interferon-gamma (IFN-γ), thereby regulating the adaptive immune response. Additionally, enhanced NK effector activity is also triggered by CD16-mediated antibody-dependent cellular cytotoxicity signalingref. bib2,ref. bib4. However, the clinical efficacy of NK cell therapy is constrained by their shorter lifespan and diminished activity within the immunosuppressive and cytokine-deficient tumor microenvironment (TME)ref. bib1. Therefore, identifying the fundamental mechanisms that support NK cell homeostasis could deepen our comprehension of NK cell regulatory mechanisms and potentially uncover new therapeutic targets for enhancing cancer immunotherapy outcomes.
To date, the growth, longevity, and effectiveness of NK cells are thought to be significantly influenced by external cytokines and internal transcription factorsref. bib5. The synergistic effects of various cytokines, including interleukin (IL)-2, IL-12, IL-15, IL-21, and interferons, are essential for NK cell development, maturation and homeostasis. The Janus kinase/signal transducers and activators of transcription (JAK–STAT) pathway is a prototypical example that regulates distinct aspects of NK cell biology, including cell identity, survival, cytotoxicity, type I Interferon response downstream of several cytokinesref. bib6,ref. bib7. It has been shown that defective JAK–STAT signaling in NK cells contributes to NK cell dysfunction and tumorigenesis, such as loss of JAK3 and STAT5bref. bib8,ref. bib9. Although previous findings have reported that cytokine-inducible SH2-containing protein (CIS) acts in the negative feedback loops IL-15–JAK–STATs–CIS–IL-1510,11, the feedback loops of JAK–STATs are still largely unknown. Given the important role of the JAK–STAT pathway in NK cell survival and function, discovering new regulators that preserve its activity could reveal additional targets for cancer immunotherapy.
Long non-coding RNAs (lncRNAs) constitute a vast and heterogeneous group of non-coding RNAs that exceed 500 nucleotides in lengthref. bib12. LncRNAs exert important roles in various cell activities, including proliferation, metabolism and motility through RNA–RNA, RNA–DNA and RNA–protein interactionsref. 13, ref. 14, ref. 15. Some abundant lncRNAs, exemplified by the lncRNA PNUTS, bind to miRNA and act as miRNA sponges, thereby inhibiting the target-repressing function of miRNAsref. bib13. Presently, it remains to be determined whether lncRNA plays a regulatory role in the cytokine/JAK/STAT signaling pathway within NK cells.
In this study, we reveal a novel positive feedback loop in the JAK–STAT pathway, that is, cytokine/JAK/STAT signaling induces miR155HG transcription, which acts as a sponge for miR-6756, preventing the miR-6756-mediated repression of JAK3 expression and thereby enhancing the JAK–STAT signaling and thus NK cell activation and function.
Materials and methods
Reagents
The following reagents were used: IL-2 (#589106; BioLegend, San Diego, CA, USA), IL-3 (#AF-200-03-1MG; ThermoFisher Scientific, Waltham, MA, 02454, USA), IL-7 (#AF-200-07-1000UG; ThermoFisher Scientific), IL-12 (#573002; BioLegend), IL-15 (#570306; BioLegend), stem cell factor (SCF, #AF-300-07-500UG; ThermoFisher Scientific), fms-like tyrosine kinase receptor-3 ligand (FLT3L, #AF-300-19-1MG; ThermoFisher Scientific), Roswell Park Memorial Institute 1640 (RPMI 1640)/Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA), STEMdiff™ APEL™2 Medium (#05275; STEMCELL Technologies, Vancouver, Canada), TrypLE™ Select Enzyme (#12563029; STEMCELL Technologies), GlutaMAX™ DMEM (#10569010; ThermoFisher Scientific), GlutaMAX™ F12 (#31765035; ThermoFisher Scientific), fetal bovine serum (FBS; Biological Industries, Kibbutz Beit Haemek, Israel), PGM1 medium (#CA1007500; Cellapy, Beijing, China), Animal-Free Recombinant Human VEGF 165 (AF-100-20-250UG; Peprotech), Animal-Free Recombinant Human BMP4 (AF-120-05ET-250UG; Peprotech), Animal-Free Recombinant Human FGF-basic (AF-100-18B-100UG; Peprotech), Human/Murine/Rat Activin A (AF-120-14E-100UG; Peprotech), Human TPO (AF-300-18-1MG; Peprotech), Animal-Free Recombinant Human IL-6 (AF-200-06-100UG; Peprotech), Recombinant Human EPO (100-64-100UG; Peprotech), Animal-Free Recombinant Human IGF-II (AF-100-12-100UG; Peprotech), β-mercaptoethanol (#21985023; ThermoFisher Scientific), l-glutamine (#C0212; Beyotime, Shanghai, China), sodium selenite (#S5261; Sigma–Aldrich, Taufkirchen, Germany), l-ascorbic acid (#A4403; Sigma–Aldrich), cholamine (#E9508; Sigma–Aldrich), MEM Non-Essential Amino Acids Solution (#11140-050; Gibco), Ficoll (#17144003; GE Healthcare Bio-Sciences, Pittsburgh, PA, USA), Penicillin–Streptomycin (#15140122; ThermoFisher Scientific), human NK cell isolation kit (#130-098-185; Miltenyi Biotec, San Diego, CA, USA), Percoll (#17089101; GE Healthcare Bio-Sciences), Enzyme-linked immunosorbent assay (ELISA) kits (#430104; Biolegend), Protein transport inhibitor Golgi Plug (#555028; BD Bioscience, San Diego, CA, USA) and Golgi Stop (#554715; BD Bioscience), the Foxp3/transcription factor staining buffer set kit (#00-5523-00; eBioscience, San Diego, CA, USA), PMA (#00-4970-93; eBioscience), collagenase type II (#17101015; Gibco), collagenase type IV (#17104019; Gibco) and DNase I (#10104159001; Roche Diagnostics GmbH, Manheim, Germany), the apoptosis kit (#559763; BD Bioscience), CellTrace Violet Cell Proliferation Kit (#C34557; ThermoFisher Scientific), L-(+)-lactic acid (#L6402; Merck KGaA, Darmstadt, Germany), P155 (GenScript, Nanjing, China)ref. bib16, NE-PER Nuclear and Cytoplasmic Extraction Reagents (#78833; ThermoFisher Scientific), RNA antisense purification (RAP) Kit (#Bes5103; BersinBio, Guangzhou, China), Chromatin immunoprecipitation (ChIP) assay Kit (#P2078; Beyotime), Ruxolitinib (S1378; Selleckchem, Houston, TX, USA), Stattic (S7024; Selleckchem), Actinomycin D (ActD, S8964; Selleckchem).
The following antibodies were used in Western blot: rabbit monoclonal antibodies (mAbs) against human phospho-Tyr980/981 of JAK3 (5031t), human phospho-Y705 of STAT3 (9145T), STAT3 (4904S), phospho-Y693 of STAT4 (4134S), STAT4 (2653S), phospho-Y694 of STAT5 (9359S), STAT5 (94205S), GAPDH (8884S) from Cell Signalling Technology (CST, Beverly, MA, USA); rabbit monoclonal antibodies (mAbs) against JAK3 (ab45141), phospho-Tyr1022/1023 of JAK1 (ab138005), JAK1 (ab133666) from Abcam (Cambridge, UK).
The following antibodies were used in flow cytometry analysis: CD45 (#560178; BD Biosciences; #304037; BioLegend), CD56 (#562780, BD Biosciences; #392406, BioLegend; #IM2474, Beckman Coulter, Miami, FL, USA), CD3 (#300316; BioLegend), NKp46 (#331914; BioLegend), CD16 (#302012; BioLegend), CD34 (#343516; BioLegend), CD43 (#343206; BioLegend), IFN-γ (#502530; BioLegend), Ki67 (#350530; BioLegend), granzyme B (#561142; BD Biosciences), perforin (#353314; BioLegend), CD107a (#555801; BD Biosciences), 7-AAD antibodies (#559763; BD Bioscience), NKP30 (#130-112-430; Miltenyi, Bergisch Gladbach Germany), KIR2DL5A (#566330; BD Biosciences).
Cell lines
The NK92 NK cell line was cultured in RPMI 1640 medium enriched with 10% FBS, 400 U/mL IL-2, 1% MEM Non-Essential Amino Acids Solution, along with 1% penicillin, and 1% streptomycin. HEK293T, K562 and human hepatoma cell lines HCC-LM9 were grown in DMEM supplemented with 10% FBS, penicillin and streptomycin.
Purified human primary NK (PB-NK) cells were extracted from peripheral blood following previously established methodsref. bib17. Blood samples from healthy individuals (age between 18 and 55 years) were collected from the Changsha Blood Center in China, under the approved protocol HCHLL-2023-182 by the Ethics Committee of Hunan Children’s Hospital. Peripheral blood mononuclear cells were isolated from the blood leukocytes using a standard density gradient centrifugation method with Ficoll/Isopaque. The NK cells were then purified through negative selection with a commercial human NK cell isolation kit, after which they were subjected to fluorescence-activated cell sorting using the Aria III cell sorter from BD Biosciences. The purity of the sorted CD3–CD56+ NK cells exceeded 99.0%.
iPSC cell line, hiPSC-B1, was obtained from Cellapy and iPSC-NK cells were acquired after routinely differentiated from hiPSC-B1 as described previouslyref. bib18. Briefly, when the cells reached about 70% confluence, they were detached using TrypLE™ Select Enzyme and the reaction was halted with PGM1 medium. Subsequently, the cells were enumerated and resuspended at a density of 80,000 cells per well in a round-bottom 96-well plate, with each well containing 100 μL of STEMdiff™ APEL™2 medium, which was supplemented with 40 ng/mL SCF, 50 ng/mL VEGF 165, 20 ng/mL BMP4, 20 ng/mL FGF-basic, 10 ng/mL Activin A, and 10 μmol/L Y-27632. The peripheral wells of the plate were filled with sterile water to avert evaporation of the medium. The culture medium was replenished on a thrice-weekly basis. On Day 4 of EB differentiation, the culture medium was replaced with STEMdiff™ APEL™2 medium containing 50 ng/mL SCF, 20 ng/mL BMP4, 20 ng/mL FGF-basic, 100 ng/mL VEGF 165, 100 ng/mL IGF-II, and 3 μmol/L SB 431542. On the eighth day of the spin embryoid body (EB) differentiation process, cells from 14 to 16 wells of a 96-well plate were collectively transferred into one well of a 6-well plate that had been precoated with 2% gelatin and filled with 2 mL of NK differentiation medium, which supplemented with 50 ng/mL SCF, 20 ng/mL FGF-basic, 200 ng/mL VEGF 165, 20 ng/mL IGF-II, 25 ng/mL Flt3-Ligand, 25 ng/mL TPO, 50 ng/mL IL-3, 3 U/mL EPO, and 25 ng/mL IL-6. The NK cell culture medium was then refreshed every 3–4 days. The maturation of NK cells was assessed after a 28-day culture period.
The above cells were cultivated in a humidified, 5% CO2 incubator maintained at a constant temperature of 37 °C.
Cell counting assay
For loss-of-function assays, NK92-shCtrl/shmiR155HG (5 × 104) cells were cultured in a 48-well plate for 72 h before assessment. Conversely, for gain-of-function experiments, iPSC cells were infected with lentivirus expressing the full-length miR155HG or its control sequence, then routinely differentiated into NK cells for 28 days before analysis.
Flow cytometry analysis
Cell staining was carried out as described previouslyref. 19, ref. 20, ref. 21. For surface staining of CD45, CD56, CD3, NKP46, NKP30, KIR2DL5A and CD16, NK92 or iPSC-NK cells were incubated with the specified antibodies in 1 × phosphate-buffered saline (PBS) at room temperature (RT) for 15 min in a light-protected environment. For the detection of intracellular IFN-γ, iPSC-NK cells were treated with PMA in conjunction with BD Golgi Plug™ protein transport inhibitor for 6 h. Subsequently, the cells underwent staining using the Fixation/Permeabilization Solution Kit according to the manufacturer’s protocol. For intracellular staining of Ki67, granzyme B and perforin, NK92 or iPSC-NK cells were treated with the Foxp3/Transcription Factor Staining Buffer Set to permeabilize them at 4 °C for 2 h. Subsequently, these cells were incubated with antibodies specific to Ki67, granzyme B, and perforin at RT for 30 min. For apoptosis staining of K562 cells, NK92-shCtrl/shmiR155HG cells, as well as iPSC-NK-Ctrl/miR155HG cells were harvested and stained with AnnexinV and 7-AAD antibodies suspended in 1 × binding buffer at RT for 15 min in the dark. The data were acquired on an LSRFortessa Flow Cytometer (BD Biosciences) and analyzed using FlowJo 10.5.3 software (Tree Star, Ashland, OR).
ELISA of IFN-γ secretion in cell culture medium
NK92 cell culture supernatant was harvested, and the levels of IFN-γ present in the supernatant were then quantified using a commercial ELISA kit, following the protocol provided by the manufacturer.
CD107a degranulation assay
The CD107a degranulation assay was carried out as described in previous studiesref. bib22,ref. bib23. In essence, NK92-shCtrl/shmiR155HG cells, stimulated with or without cytokines, were incubated with K562 cells at a ratio of 10:1 ratio in the presence of the anti-CD107a-PE antibodies and the protein transport inhibitor Golgi Plug and Golgi Stop. Following a 5-h co-incubation period at 37 °C, the proportion of CD107a-positive NK92 cells was examined using flow cytometry.
NK cell cytotoxicity assay
For K562 cells apoptosis assay, NK92-shCtrl/shmiR155HG cells, stimulated with or without cytokines, were incubated with CTV-labelled K562 at a ratio of 10:1 for 5 h. The cells were harvested and the frequencies of Annexin V-negative/7-AAD-negative, Annexin V-positive/7-AAD-negative, and Annexin V-positive/7-AAD-positive K562 cells were evaluated using the apoptosis kit by flow cytometry.
Model of human liver cancer and NK cell adoptive transfer
The mouse studies were approval by the Animal Ethics Committee of Hunan Children’s Hospital, with the ethical approval number being HCHDWLL-2023-07. All procedures involving animals were conducted in compliance with the Guidelines for the Care and Use of Laboratory Animals as published by the National Institutes of Health (Publication No. 80-23, revised in 1996), as well as the institutional ethical standards for animal research.
NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22Il15em1Cin(hIL15)/Gpt (NCG-IL15) mice (6 weeks of age) were utilized for the establishment of subcutaneous hepatocellular carcinoma (HCC) xenografts to evaluate the tumoricidal efficacy of NK92-shCtrl/shmiR155HG cells. Human HCC-LM9 cells (4 × 106) were suspended in 100 μL of serum-free DMEM and injected subcutaneously into the inguinal region of the mice. One week post-inoculation, mice were intravenously administered with 2 × 106 NK92-shCtrl/shmiR155HG cells via the tail vein on three separate occasions. After a total of 24 days from the initial injection, the mice were euthanized, and the resulting tumors were excised and weighed. The volume of the tumors at various time points was measured using a caliper and was determined by Eq (1):
2
RNA-sequencing and bioinformatics analysis
RNA was extracted from NK92-shCtrl/shmiR155HG cells stimulated with IL-2 (1000 U/mL) + IL-12 (10 ng/mL) for 12 h using AG RNAex Pro Reagent (AG21101; Accurate Biotechnology, Hunan, China). The quantity and purity of the RNA were assessed with BioDrop μLite equipment (Biochrom Ltd., Cambridge, UK). Poly(A) mRNA was purified using magnetic beads coated with Oligo(dT), and then the mRNA was randomly fragmented in a fragmentation buffer. Synthesis of the first strand of cDNA was carried out using the fragmented mRNA as a template and random hexamers for priming. Following end-repair and adapter ligation, RNA sequencing libraries were generated. These libraries were subsequently sequenced on an Illumina NovaSeq 6000 platform with 150 bp paired-end reads.
The raw sequencing reads were aligned to the human reference genome (hg38) using HISAT2 with default settings. StringTie (run with the “-e” parameter) and prepDE.py3 were used to calculate and merge read count information. Genes with a Counts per Million value greater than 1 in at least two samples were included in the analysis. Differential expression analysis was conducted using edgeR’s exact test, with a logarithmic fold change (LFC) threshold set at log2(1.2) and a significance cutoff at a P value of less than 0.01. Gene Ontology (GO) enrichment analysis of DEGs was performed by clusterProfiler. The data presented in the figures are accessible in the published article and the Supporting Information The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in the National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA020503) that are publicly accessible at https://ngdc.cncb.ac.cn/gsaref. bib24,ref. bib25.
Isolation of NK cells from HCC samples
Following the receipt of proper informed consent, fresh HCC tissues and their adjacent non-neoplastic liver tissues were collected from patients undergoing tumor resection at Hunan Provincial People’s Hospital. The study protocol was approved by the Institutional Review Board of the same hospital (the ethical approval number 2023-151). To isolate NK cells from these human samples, the tissues were first rinsed with PBS, minced into smaller fragments, and then treated with a mixture of collagenase type II (1 mg/mL), collagenase type IV (1 mg/mL), and DNase I (0.01 mg/mL) in DMEM with 2.5% FBS for 45 min at 37 °C, as described previouslyref. bib26. The lymphocytes were then separated using a 30%–40% Percoll gradient and rinsed twice with PBS. Subsequently, CD45+CD56+CD3−NK cells were purified using a fluorescence-activated cell sorting Aria cell sorter from BD Biosciences.
Isolation of cytoplasm and nuclear fraction
Utilizing the NE-PER Nuclear and Cytoplasmic Extraction Reagents and adhering to the manufacturer’s protocol, the cytoplasmic and nuclear RNA fractions were extracted from NK92 cells (1 × 107). The purity of non-nuclear and nuclear fractions was determined using specific RNA markers, namely GAPDH for the cytoplasmic fraction and MALAT1 for the nuclear fractionref. bib27, respectively.
Analysis of gene expression
Quantitative real-time polymerase chain reaction (RT-qPCR) and Western blot analysis were conducted to assess the expression levels of the genes of interest. The primers used for RT-qPCR are listed in Supporting Information Table S1. The clone numbers for all antibodies used in our study have been added to Supporting Information Table S2.
RAP assay
The miRNAs associated with miR155HG were identified using an RAP Kit, following the guidelines provided by the manufacturer. Biotin-labeled miR155HG probes and scramble control probes were synthesized by BersinBio, and the complete list of probe sequences utilized in the RAP assay can be found in Table S1. The process commenced with the cross-linking of NK92 cells (5 × 107) using formaldehyde, followed by lysis and sonication. Subsequently, the cell lysates were incubated with the probes for 4 h at 37 °C to facilitate hybridization. After hybridization, streptavidin beads were added to the mixture and allowed to rotate at RT for an additional h to capture the probe–miRNA complexes. The RNAs that were successfully bound to the beads were then eluted, subjected to purification, and prepared for subsequent RT-qPCR analysis to determine the miRNAs bound to miR155HG.
Transfection with miRNA mimics and inhibitors
As previously detailedref. bib28, NK92 cells were transfected with 200 nmol/L Cy3-conjugated miR-155/miR-6756 mimics or the negative control (NC), Cy3-conjugated miR-6756 inhibitor (anti-miR-6756) and a control inhibitor (anti-NC) without the use of a transfection reagent. Following a 48-h transfection period, the cells were collected by centrifugation at 200×g for 5 min, resuspended in PBS, and the Cy3-positive NK92 cells were isolated by cell sorting. The miRNA mimics and inhibitors employed in this study were procured from GenePharma (Shanghai, China), and their respective sequences are provided in Table S1.
ChIP assay
ChIP assays were conducted in accordance with the manufacturer’s protocol for a ChIP assay Kit. NK92 cells were stimulated with IL-2 (1000 U/mL) and IL-12 (10 ng/mL) for 2 h before cross-linking with a 0.5% formaldehyde solution for 15 min at RT. Subsequently, the cells were sonicated to reduce DNA to fragments ranging from 200 to 750 base pairs. The chromatin was immunoprecipitated using 2 μg of anti-STAT3 antibody or a matched isotype control IgG (2729S, CST) at 4 °C for 16 h with agitation. Protein A/G MagBeads (88802, ThermoFisher Scientific) were then used for the enrichment of DNA-protein complexes. After thorough washing, the immunocomplexes bound to the beads were extracted with 420 μL of elution buffer (0.1 mol/L NaHCO3, 1% SDS) and agitated at RT for 1 h. The supernatants were then treated with Tris–EDTA buffer to reverse the cross-links between DNA and proteins, followed by an overnight incubation at 65 °C. The resulting DNA fragments were purified and analyzed by qPCR using primers detailed in Table S1.
Luciferase reporter assay
Luciferase reporter assay was carried out as described previouslyref. bib29. Cells were cultivated in a 48-well plate, each well containing 200 μL of complete growth medium. The luciferase activity was measured 48 h after transfection using the dual-luciferase reporter assay kit from Promega. The pRL-TK vector from Promega, which encodes for Renilla luciferase, was utilized as an internal control to normalize variations in transfection efficiency and cell harvesting.
To determine the impact of STAT3 expression on the transcriptional activity of the miR155HG promoter, HEK293T cells were co-transfected with 300 ng of either pcDNA 3.1 or pcDNA 3.1-STAT3 (Public Protein/Plasmid Library, Nanjing, China), 50 ng of the p(−2.5/+0.12 k) (TsingKe Biotech, Beijing, China) promoter construct, and 25 ng of the pRL-TK plasmid.
Public sequencing database
The correlation between the expression levels of various genes was assessed using Spearman’s rank correlation coefficient, as derived from data obtained in GEPIA 2 (http://gepia2.cancer-pku.cn/#index) and the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/gds/?term=). The putative binding sites of miRNAs in miR155HG and JAK3 mRNA 3′UTR were predicted by TargetScan (https://www.targetscan.org/vert_71/). Transcription factors that may regulate miR155HG expression were predicted by ChIP-seq from the chEA3 (https://maayanlab.cloud/chea3/) website.
Statistical analysis
All statistical analyses were performed utilizing GraphPad Prism version 8.0, a software package by GraphPad Software Inc., based in La Jolla, CA, USA. The results are presented as the mean ± standard error of the mean (SEM), derived from a minimum of three separate experimental trials. For comparing differences among several groups, either two-tailed unpaired or paired Student’s t-tests, or two-way ANOVA, were employed as appropriate. A statistical significance threshold was set at P < 0.05. Additionally, Spearman’s correlation coefficient was applied to evaluate the correlation between expression levels of various genes.
Results
The levels of miR155HG expression exhibit a positive correlation with the activation and effector functions of NK cells
To identify lncRNAs elevated in activated NK cells, we conducted a bioinformatic analysis utilizing the 10.13039/100000085GEO dataset (GSE110446)ref. bib30 and found that the lncRNA miR155HG fulfilled the following criteria (Fig. 1A–D; Supporting Information Fig. S1): (1) More than 2-fold upregulation in activated NK cells compared to unstimulated NK cells; (2) Found in intergenic areas of genome. (3) Coexpression with NK cells effector molecules in tumorsref. bib31. These findings were also confirmed by both human PB-NK cells and NK92 cell lines. miR155HG expression was significantly increased in human PB-NK cells treated with IL-15 alone or a combination of IL-2 and IL-12 for 12 h, compared to unstimulated NK cells (Fig. 1E, left panel). Furthermore, lactate (Fig. 1E, middle panel) and hypoxia (Fig. 1E, right panel), which are two main suppressive factors in the tumor microenvironment, suppressed the expression of miR155HG in NK92 cells. Subsequent GO enrichment analysis of genes co-expressed with miR155HG indicated that miR155HG was highly co-expressed with cytokine–cytokine receptor interaction, JAK–STAT signaling pathway, and the chemokine signaling pathway (Fig. 1F). These findings indicate that miR155HG is elevated in activated NK cells and may be positively correlated with NK cell activation and the JAK–STAT signal pathway.
miR155HG promotes NK cell proliferation and survival
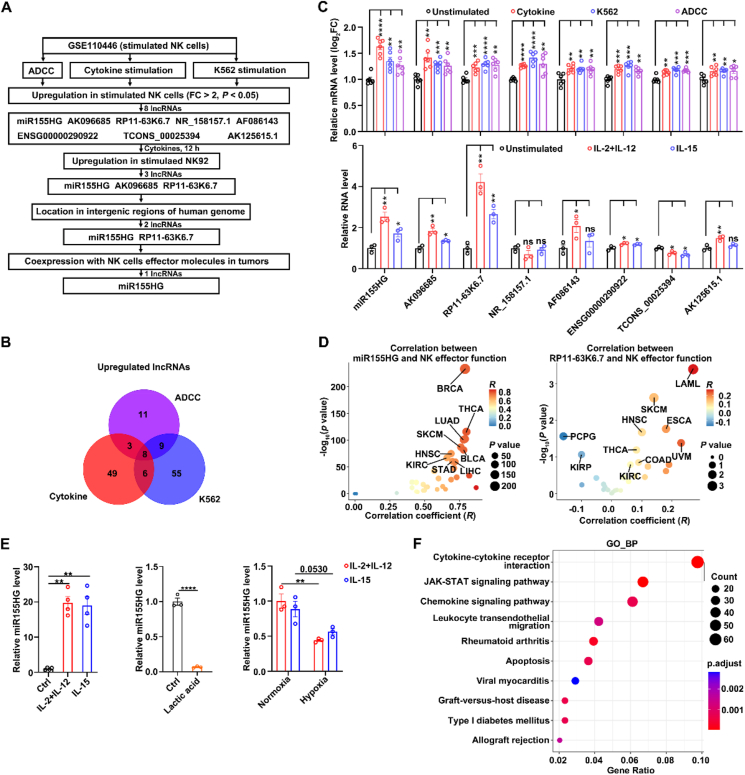
We further evaluated whether miR155HG affected NK cell proliferation, activation and effector functions, using both the NK92 cell line and iPSC-NK cells. Firstly, we analyzed the expression levels of activating and inhibitory receptors on NK cell by flow cytometry. Compared with the control (shCtrl), the stable knockdown of miR155HG (shmiR155HG) (Fig. 2A) resulted in reduced geometric mean fluorescence intensities (gMFIs) of CD16, NKP30 and NKP46, whereas the gMFIs of KIR2DL5A were increased (Fig. 2B; Supporting Information Fig. S2A). Silencing of miR155HG (shmiR155HG) led to a decrease in the cell number (Fig. 2C) and the percentages of Ki67+ cells (Fig. 2D; Fig. S2B), but an increase in the ratio of early apoptosis (Annexin V+7-AAD–) and late apoptosis (Annexin V+7-AAD+) of NK92 cells (Fig. 2E; Fig. S2C). Subsequently, we determined INF-γ secretion using an ELISA assay on the supernatants from cultured NK92 stable cell lines after stimulated with or without cytokines for 24 h. We observed that the knockdown of miR155HG reduced INF-γ secretion, regardless of cytokine stimulation (Fig. 2F). Stable knockdown of miR155HG suppressed the gMFIs of granzyme B and perforin (Fig. 2G; Fig. S2D), irrespective of cytokine treatment. Knockdown of miR155HG also decreased the expression of CD107a, a biomarker indicative of NK cell degranulation in response to stimulationref. bib32, on NK92 cells when cocultured with K562 cells, with or without cytokines stimulation for 5 h (Fig. 2H; Fig. S2E). Consistently, the overexpression of miR155HG (miR155HG) (Fig. 2I) increased the percentages of Ki67+ cells (Fig. 2J; Fig. S2F), INF-γ secretion following stimulation with IL-2 for 24 h (Fig. 2K). Overexpression of miR155HG could increase the gMFIs of granzyme B without affecting the gMFIs of perforin after stimulation with IL-2 for 24 h (Fig. 2L; Fig. S2G). Furthermore, the suppression of miR155HG reduced the cytotoxic activity of NK92 cells against K562 cells. This was evidenced by an increased proportion of viable K562 cells (Annexin V–7-AAD–) and a decreased proportion of K562 cells undergoing early apoptosis (Annexin V+7-AAD–) after a 5-h co-culture with NK92 cells. These effects were observed irrespective of whether IL-15 or a combination of IL-2 and IL-12 was present (Fig. 2M; Fig. S2H).
In vivo studies involving subcutaneous injection of human hepatoma cells HCC-LM9 into humanized IL15 transgenic NCG (NCG-hIL15) mice demonstrated that weekly adoptive transfer (intravenously) of shmiR155HG-NK92 cells, beginning on Day 7 post-injection, resulted in faster tumor growth and greater tumor weight compared to mice that received shCtrl-NK92 cell (Fig. 2N). Collectively, our data demonstrate that miR155HG enhances the antitumor capability of NK cells.
miR155HG promoted hematopoiesis and iPSC-NK cell generation
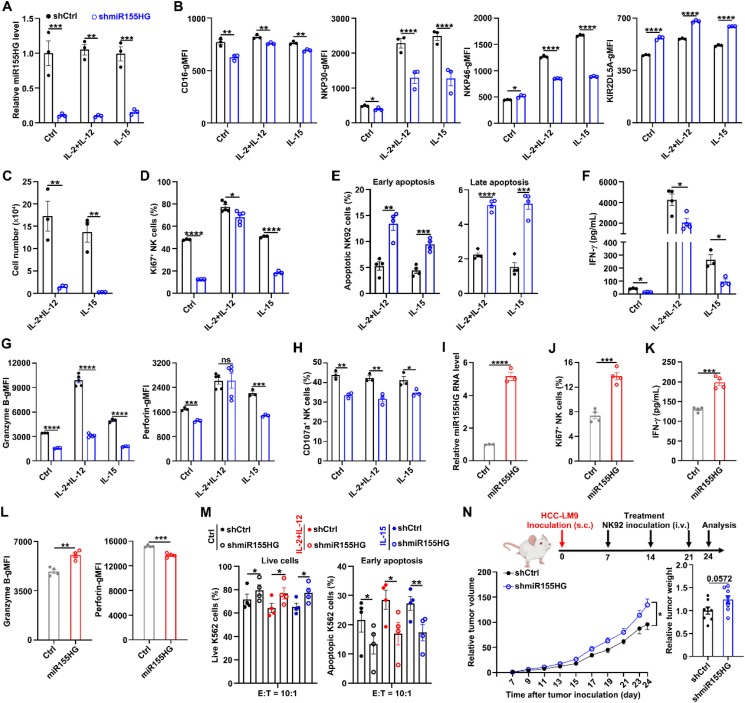
Next, we examined the effects of miR155HG on NK cell differentiation, proliferation, activation, and effector functions in iPSC-derived NK cells. Human iPSCs were infected with lentivirus expressing full-length miR155HG or shmiR155HG sequences, as well as their control sequences, and routinely differentiated into hematopoietic progenitor cells and subsequently into NK cells, as previously reportedref. bib10. To investigate the role of miR155HG during hematopoietic development, we analyzed the surface antigens of hematopoietic progenitor cells (CD34, CD43, and CD45) on Days 8 and 12 of EB formation, and on Day 7 of adherent culture differentiation of iPSCs into hematopoietic progenitor cells. Knockdown of miR155HG decreased the percentages of CD34+ cells and CD34+CD43+ HSPCs (Fig. 3A), while overexpression of miR155HG in iPSCs increased the percentages of these cells on Day 12 of EBs and this effect became more prominent on Day 7 of adherent culture (Fig. 3B). Overexpression of miR155HG in iPSCs increased the total number of iPSC-NK cells (CD45+CD56+) (Fig. 3C), as well as the percentages of CD56+NKP46+ iPSC-NK cells (Fig. 3D) and CD56+CD16+ iPSC-NK cells (Fig. 3E), and Ki67+ iPSC-NK cells (Fig. 3F), but decreased the proportions of both early and late apoptotic iPSC-NK cells (Fig. 3G) at 28 days of NK cell differentiation. These findings indicate that miR155HG can promote NK cell differentiation, proliferation, and survival.
We also examined the effect of miR155HG on the effector function of iPSC-NK cells. We found that the knockdown of miR155HG reduced and overexpression of miR155HG promoted the gMFIs of INF-γ (Fig. 3H). In the cases of miR155HG knockdown or overexpression, the gMFIs of Granzyme B remained unchanged (Fig. 3I, left panel). Knockdown of miR155HG can reduce the gMFIs of perforin, but overexpression of miR155HG does not increase the gMFIs of perforin (Fig. 3I, right panel).
miR155HG enhances JAK3 mRNA levels and the expression of downstream target genes of STAT
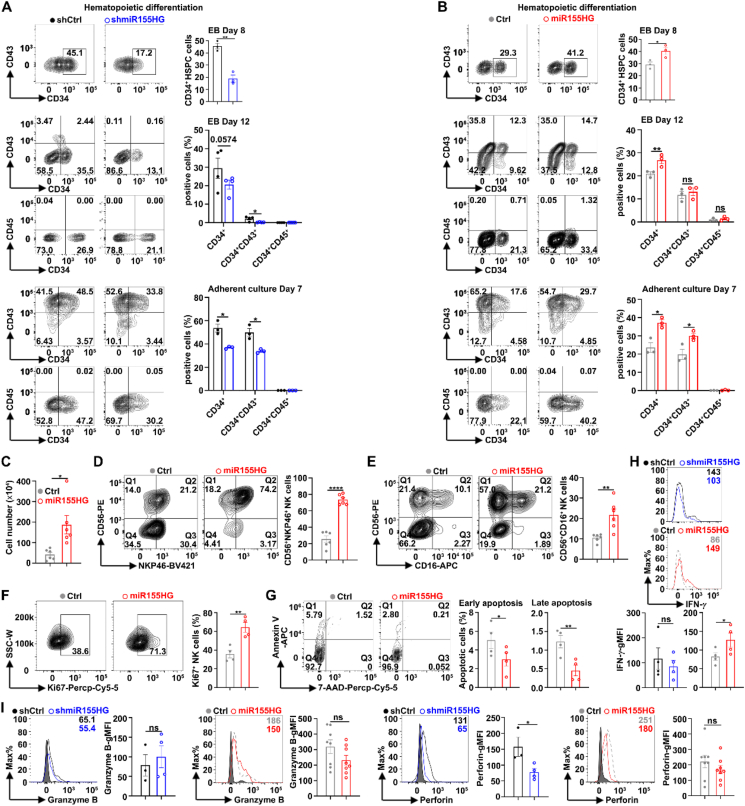
To further explore the mechanism underlying miR155HG promotes NK cell proliferation and effector function, we identified differentially expressed genes between shmiR155HG-NK92 and shCtrl-NK92 cell lines following treatment with IL-2 and IL-12 using bulk RNA-sequencing. After the suppression of miR155HG, 881 genes exhibited increased expression, while 583 genes showed decreased expression (Fig. 4A). The five most significantly downregulated KEGG pathways are cytokine–cytokine receptor interaction, the JAK–STAT signaling pathway, the p53 signaling pathway, leishmaniasis, and allograft rejection (Fig. 4B), and this finding aligns with the prior GO enrichment analysis of genes that are co-expressed with miR155HG (Fig. 1F). These results indicate that miR155HG may enhance NK cell activation and function by regulating the JAK–STAT signaling.
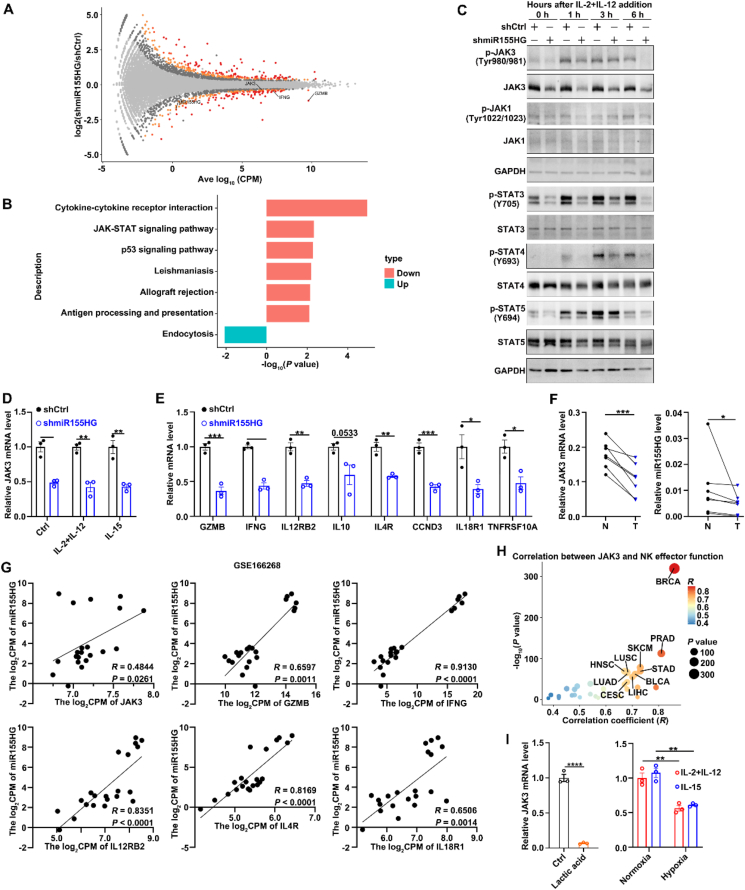
To test this hypothesis, we examined the levels of key components of the JAK–STAT pathway, including JAK1, JAK3, STAT3, STAT4, and STAT5, as well as their phosphorylation states after silencing miR155HG expression in NK92 cells. As shown, silencing miR155HG reduced the total protein level and phosphorylation level of JAK3, along with the phosphorylation levels of STAT3, STAT4, and STAT5, while the levels of JAK1 and phosphorylated JAK1 remained unchanged in NK92 cells after treatment with IL-2 plus IL-12 at different time points (Fig. 4C). The mRNA levels of JAK3 and its downstream target genes, including GZMB, IFNG, IL12RB2, CCND3, IL10, IL4R, IL18R1, and TNFRSF10A, were downregulated in shmiR155HG NK92 cells, irrespective of cytokine stimulation (Fig. 4D and E).
To find more evidence supporting the positive regulation of miR155HG on JAK3 mRNA levels, we also used several publicly available datasets as well as RT-qPCR of miR155HG and JAK3 mRNA levels in tumor tissues from HCC patients. When compared with NK cells derived from healthy liver tissues, NK cells infiltrating tumors from patients with HCC showed reduced RNA levels for both JAK3 and miR155HG (Fig. 4F). The expression of miR155HG exhibited a positive correlation with the mRNA levels of JAK3 and its downstream target genesref. bib33 (Fig. 4G). The mRNA levels of JAK3 also demonstrated a positive correlation with the functional module of NK cell effector functions across various human cancer types, as indicated by data from the TCGA and GTEx databases (Fig. 4H; Supporting Information Fig. S3). Moreover, lactate and hypoxia suppressed the mRNA expression of JAK3 in NK92 cells (Fig. 4I).
These data indicate that miR155HG may promote NK cell activation and function by increasing JAK3 mRNA levels and its downstream STAT signals.
miR155HG functions as a miR-6756 sponge to upregulate JAK3 mRNA levels
We next explored how miR155HG enhances JAK3 mRNA and protein levels. miR155HG is situated on chromosome 21q21 and comprises three exons encompassing a region of approximately 1.5 kilobasesref. bib16. It is reported that miR155HG encodes a 17-amino acid micropeptide (P155)ref. bib16 as well as a precursor RNA of miRNA-155ref. bib34. Our data show that P155 neither abrogated the inhibitory effect of miR155HG silencing on JAK3 expression nor affected the expression of molecules related to NK cell proliferation and effector function (Fig. 5A–G). miR-155, a derivative of miR155HG, is reported to be upregulated during NK cell activation and induced IFN-γ production by inhibiting the expression of the inositol phosphatase SHIP1ref. bib35. However, our data show that miRNA-155 overexpression did not rescue JAK3 protein levels in miR155HG knockdown NK92 cells (Fig. 5H).
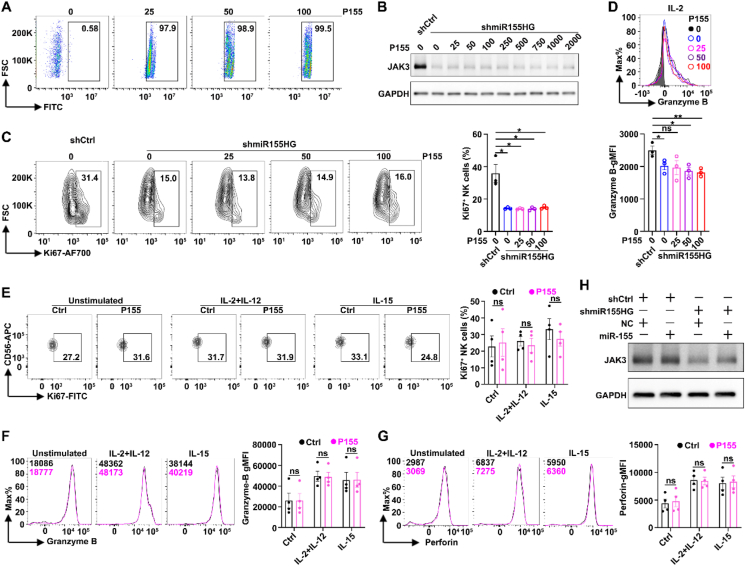
We next determined the cellular location of miR155HG and found it predominantly localized in the cytoplasm (Fig. 6A), suggesting that miR155HG may act as a miRNA sponge. Subsequent analysis with the TargetScan prediction algorithm identified 10 miRNAs with potential binding sites on both miR155HG and JAK3 mRNA 3′UTR (Fig. 6B). To verify the direct interaction between miR155HG and miRNAs, biotinylated miR155HG and control probes were used to perform RAP assay with streptavidin beads. The data showed that, compared to the control probes, miR-6756, but not the other nine miRNAs, was significantly enriched in the miR155HG probes-precipitates (Fig. 6C). Analysis utilizing the TargetScan prediction algorithm revealed that miR-6756 potentially targets a single binding site within both the miR155HG and JAK3 mRNA 3′UTR (1811–1823-nt) (Fig. 6D), and we then examined whether miR155HG upregulates JAK3 expression by working as a miR-6756 sponge. Overexpression of miR-6756 significantly suppressed both the mRNA and protein levels of JAK3 in NK92 cells (Fig. 6E), whereas miR-6756 inhibitors rescued the reduced JAK3 protein levels in miR155HG-silenced NK92 cells (Fig. 6F). Moreover, we found that overexpression of miR-6756 could decrease the INF-γ secretion (Fig. 6G), the percentage of Ki67+ NK cells (Fig. 6H), and the gMFIs of granzyme B and perforin (Fig. 6I).
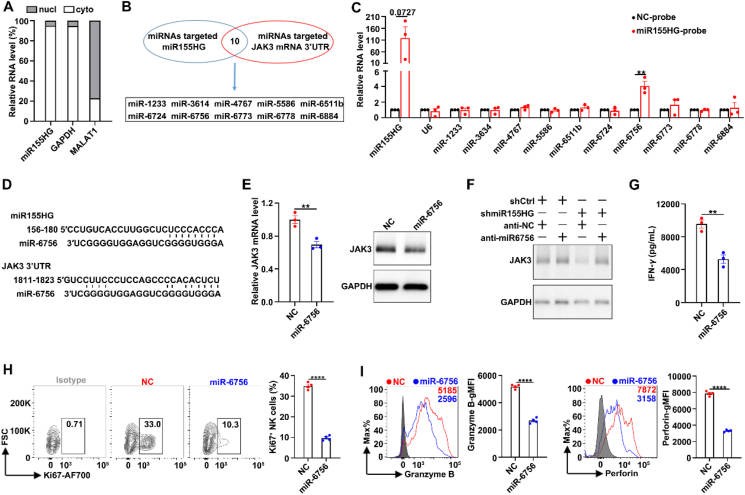
These results suggest that miR155HG enhances JAK3 protein expression by sequestering miR-6756, thereby reducing miR-6756’s inhibitory effect on JAK3 expression and effector function of NK cells.
The transcription of miR155HG is induced by the cytokines–JAK–STAT3 axis
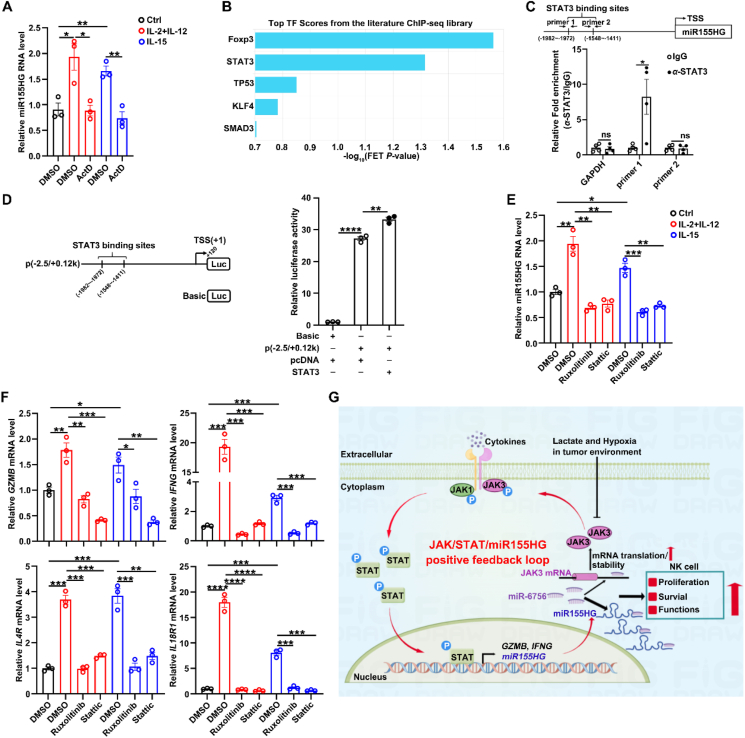
We then explored the regulatory mechanism responsible for the upregulation of miR155HG in activated NK cells. It is widely recognized that cytokines, including IL-2, IL-12, IL-15, and IL-21, trigger NK cell activation through the JAK–STAT signaling pathwayref. bib6. To examine whether cytokines promoted the expression of miR155HG at the transcriptional level, the expression of miR155HG after treatment of transcription inhibitor ActD with cytokines stimulated NK cells. We found that the role of cytokines in increasing miR155HG level was abrogated by blocking gene transcription with ActD (Fig. 7A), implying that cytokines may enhance miR155HG transcription. By using bioinformatics analysis based on ChIP-sequencing data, we identified potential binding sites for STAT3 within the core promoter region of miR155HG (Fig. 7B). Our ChIP assays revealed that the segment from −1981 to −1785 base pairs of the miR155HG promoter, but not the segment from −1735 to −1598 base pairs, was significantly enriched in the DNA precipitated by anti-STAT3 antibodies (Fig. 7C). To further validate the transcription factor STAT3 promoted miR155HG transcription, we constructed the promoter reporter p(−2.5/+0.12 k) of miR155HG (Fig. 7D, left panel) that exhibited much higher activity than the control plasmid pGL3-basic, suggesting that this segment contains the miR155HG promoter. Moreover, the promoter activity of p(−2.5/+0.12 k) was increased by overexpressing STAT3 (Fig. 7D, right panel). Furthermore, both the inhibitors of JAK and STAT3 significantly attenuated the upregulation of miR155HG (Fig. 7E) and its downstream target genes (Fig. 7F) induced by cytokines.
Discussion
The control of JAK–STAT signaling homeostasis is critical for maintaining both immune cells resting and immune responseref. bib36,ref. bib37. A previous study reported a negative feedback loop of JAK–STAT signaling in NK cells, where the IL-15–JAK–STAT signal axis induces upregulation of CIS, which interacts with JAK1, resulting in its enzymatic activity inhibition and proteasomal degradationref. bib11. In our research, we identified miR155HG as an activator of the JAK–STAT pathway in NK cells. JAK–STAT signaling triggered by cytokine stimulation led to the induction of miR155HG expression in NK cells. Mechanistically, upregulated miR155HG acts as a competing endogenous RNA partner of JAK3 mRNA to maintain JAK3 levels in NK cells. Suppression of miR155HG led to decreased proliferation and survival of NK cells, as well as reduced IFN-γ secretion and cytotoxic capacity both in vitro and in vivo. Notably, overexpression of miR155HG promoted iPSC-NK cell expansion more than threefold, along with improved survival. These results strongly suggest that enhancing miR155HG expression provides a new target for NK cell adoptive transfer therapy, including improving the in vitro expansion efficiency and anti-tumor effect of NK cells.
Unlike other members of the JAK family, JAK3 is predominantly found in cells of hematopoietic origin and is recognized as the sole signaling component that directly engages with the γc receptorref. bib38. Individuals with mutations in JAK3, whether human or murine, exhibit severe combined immunodeficiency, a condition defined by the absence of NK cellsref. bib7,ref. bib39,ref. bib40. Prior research on mice carrying a spontaneous JAK3 mutation has unveiled a link between compromised JAK3 signaling and an arrest in the differentiation of NK cells and innate lymphoid cells 1 at the precursor NK and innate lymphoid cell precursor stagesref. bib41. Downstream of the signal transduction of γc cytokines, JAK3 phosphorylates STAT1/3/4/5/6, thereby regulating the downstream target genes. We found that miR155HG promotes the crucial target genes of JAK–STAT, including GZMB, IFNG, IL12RB2, CCND3, IL10, IL4R, IL18R1 and TNFRSF10A, which contributed to NK cell expansion and effector function. Thus, our study identified miR155HG as a gatekeeper for the homeostatic expression of JAK3 in NK cells based on the following evidence: (1) Through bioinformatics analysis, RAP assay, and biochemical assays, it was discovered that miR155HG functions as an absorbent for miR-6756, thereby inhibiting the suppressive effects of miR-6756 on JAK3 expression. (2) In NK cells, the expression levels of miR155HG were found to be positively associated with the mRNA levels of JAK3 and its target genes. In tumor-infiltrating NK cells derived from HCC patients, the RNA levels of both JAK3 and miR155HG were observed to be downregulated. (3) According to data from the TCGA and GTEx project, the RNA levels of JAK3 and miR155HG exhibited a positive correlation with the effector function module of NK cells across various human cancer types.
miR155HG exerts its biological function in a highly cell- and disease-dependent manner. In glioblastoma multiforme, miR155HG is overexpressed and works as a competing endogenous RNA for the tumor suppressor miR-185 to promote ANXA2 expression and tumor growthref. bib42. miR155HG exhibits elevated expression in dendritic cells during inflammation and encodes a 17-amino acid micropeptide, P155, that was previously unknown. This micropeptide P155 interacts directly with HSC70, thereby significantly enhancing autoinflammatory responses driven by dendritic cells in miceref. bib16. In this study, we found that the regulatory mechanism of miR155HG on JAK3 expression is independent of its derivative miR-155 and P155, as both failed to abrogate the inhibitory effect of silencing miR155HG on JAK3 expression. Furthermore, P155 did not influence the proliferation or the levels of effector molecules in NK cells. In this context, we discovered that miR155HG potentially functions as a sponge for miR-6756, inhibiting miR-6756’s suppressive effect on JAK3 transcription, which in turn promotes the activation and effector functions of NK cells.
The blockade of SOCS proteins exerts a significant effect on strengthening NK cell-mediated tumor immunity, highlighting the potential of SOCS proteins as favorable targets for immunotherapeutic interventions. Lactate, a main suppressive factor in the TME, could suppress the expression of miR155HG and JAK3, attenuate the miR155HG/JAK/STAT positive feedback circuitry, and thereby reduce NK cell expansion and effector functions in the TME. Besides being a metabolite, lactate can induce gene expression through its receptor, which may be exploited for the induction of miR155HG expression in the TME by knocking in lactate response element in miR155HG promoter using gene editing. Lactate-induced-miR155HG-expressing NK cells may display more resistance to TME and enhanced tumor-killing ability.
Conclusions
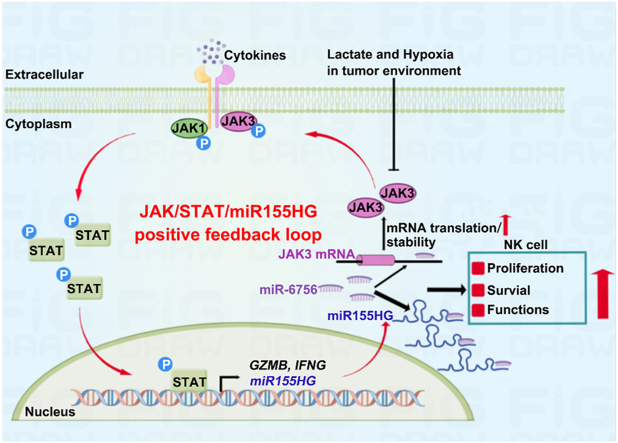
In conclusion, we disclose that upon cytokine stimulation, STAT3 directly interacts with the miR155HG promoter and induces miR155HG transcription, and in turn, miR155HG functions by sequestering miR-6756, thereby preventing miR-6756 from repressing JAK3 expression. This sequence of events bolsters the JAK–STAT pathway, which is essential for NK cell development, activation, and function (Fig. 7G). Our findings underscore the crucial role of lncRNAs in modulating the cytokine/JAK/STAT signaling pathways and in driving the initiation and execution of NK cell responses. Furthermore, they indicate that these lncRNAs could serve as potential therapeutic targets for optimizing the outcomes of NK cell adoptive transfer treatments.
Author contributions
Songyang Li: conceived and designed the experiments, curated and analyzed the data, acquired the funding, wrote original draft and revised the manuscript. Yongjie Liu, Peiwen Xiong: Data analysis. Xiaofeng Yin, Yao Yang, Jiaxing Qiu, Qinglan Yang, Xinjia Liu, Yana Li, Zhiguo Tan, Hongyan Peng, Shuting Wu, Xiangyu Wang and Lanlan Huang performed the experiments and analyzed the data. Sulai Liu, Yuxing Gong, Yuan Gao, Lingling Zhang, Junping Wang: analyzed the data, review and revised the manuscript. Yafei Deng and Zhaoyang Zhong: designed the research, supervised the study, and revised the manuscript. Youcai Deng devised the concept, designed the research, acquired the funding, supervised the study, wrote and revised the manuscript.
Conflicts of interest
Songyang Li, Xiaofeng Yin, and Youcai Deng are beneficiaries of a pending patent for the recombinant plasmids and cells for iPSC-NK cell generation described in this manuscript.
References
- N.K. Wolf, D.U. Kissiov, D.H. Raulet. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat Rev Immunol, 2023. [PubMed]
- N.A. Maskalenko, D. Zhigarev, K.S. Campbell. Harnessing natural killer cells for cancer immunotherapy: dispatching the first responders. Nat Rev Drug Discov, 2022. [PubMed]
- X. Sun, X. Xu, J. Wang, X. Zhang, Z. Zhao, X. Liu. Acid-switchable nanoparticles induce self-adaptive aggregation for enhancing antitumor immunity of natural killer cells. Acta Pharm Sin B, 2023. [PubMed]
- N.K. Björkström, B. Strunz, H.G. Ljunggren. Natural killer cells in antiviral immunity. Nat Rev Immunol, 2022. [PubMed]
- S.Y. Wu, T. Fu, Y.Z. Jiang, Z.M. Shao. Natural killer cells in cancer biology and therapy. Mol Cancer, 2020. [PubMed]
- G. Scarno, G. Pietropaolo, C. Di Censo, M. Gadina, A. Santoni, G. Sciumè. Transcriptional, epigenetic and pharmacological control of JAK/STAT pathway in NK Cells. Front Immunol, 2019. [PubMed]
- D. Gotthardt, J. Trifinopoulos, V. Sexl, E.M. Putz. JAK/STAT cytokine signaling at the crossroad of NK cell development and maturation. Front Immunol, 2019. [PubMed]
- A. Bottos, D. Gotthardt, J.W. Gill, A. Gattelli, A. Frei, A. Tzankov. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat Commun, 2016
- A. Vargas-Hernández, L.R. Forbes. JAK/STAT proteins and their biological impact on NK cell development and function. Mol Immunol, 2019. [PubMed]
- H. Zhu, R.H. Blum, D. Bernareggi, E.H. Ask, Z. Wu, H.J. Hoel. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell, 2020
- R.B. Delconte, T.B. Kolesnik, L.F. Dagley, J. Rautela, W. Shi, E.M. Putz. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol, 2016. [PubMed]
- J. Ferrer, N. Dimitrova. Transcription regulation by long non-coding RNAs: mechanisms and disease relevance. Nat Rev Mol Cell Biol, 2024. [PubMed]
- K. Nemeth, R. Bayraktar. Non-coding RNAs in disease: from mechanisms to therapeutics. Nat Rev Genet, 2024. [PubMed]
- S. He, G. Huang, R. Lei, R. Jia, Z. He, J. Chen. LncRNA ZFPM2-AS1 promotes phyllodes tumor progression by binding to CDC42 and inhibiting STAT1 activation. Acta Pharm Sin B, 2024. [PubMed]
- J. Sun, T. Jin, Z. Niu, J. Guo, Y. Guo, R. Yang. LncRNA DACH1 protects against pulmonary fibrosis by binding to SRSF1 to suppress CTNNB1 accumulation. Acta Pharm Sin B, 2022. [PubMed]
- L. Niu, F. Lou, Y. Sun, L. Sun, X. Cai, Z. Liu. A micropeptide encoded by lncRNA MIR155HG suppresses autoimmune inflammation via modulating antigen presentation. Sci Adv, 2020
- B. Yao, Q. Yang, Y. Yang, Y. Li, H. Peng, S. Wu. Screening for active compounds targeting human natural killer cell activation identifying daphnetin as an enhancer for IFN-γ production and direct cytotoxicity. Front Immunol, 2021
- D.A. Knorr, Z. Ni, D. Hermanson, M.K. Hexum, L. Bendzick, L.J. Cooper. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cell Transl Med, 2013
- S. Wu, H. Peng, S. Li, L. Huang, X. Wang, Y. Li. The ω-3 Polyunsaturated fatty acid docosahexaenoic acid enhances NK-cell antitumor effector functions. Cancer Immunol Res, 2024. [PubMed]
- Y.F. Deng, S.T. Wu, H.Y. Peng, L. Tian, Y.N. Li, Y. Yang. mTORC2 acts as a gatekeeper for mTORC1 deficiency-mediated impairments in ILC3 development. Acta Pharmacol Sin, 2023. [PubMed]
- J.Z. Jiao, Y. Zhang, W.J. Zhang, M.D. He, M. Meng, T. Liu. Radiofrequency radiation reshapes tumor immune microenvironment into antitumor phenotype in pulmonary metastatic melanoma by inducing active transformation of tumor-infiltrating CD8+ T and NK cells. Acta Pharmacol Sin, 2024. [PubMed]
- M. Meng, Z. Zhong, L. Song, Z. Zhang, X. Yin, X. Xie. mTOR signaling promotes rapid m6A mRNA methylation to regulate NK-cell activation and effector functions. Cancer Immunol Res, 2024. [PubMed]
- Y. Deng, Y. Kerdiles, J. Chu, S. Yuan, Y. Wang, X. Chen. Transcription factor Foxo1 is a negative regulator of natural killer cell maturation and function. Immunity, 2015. [PubMed]
- Database resources of the national Genomics data center, China national center for bioinformation in 2022. Nucleic Acids Res, 2022. [PubMed]
- T. Chen, X. Chen, S. Zhang, J. Zhu, B. Tang, A. Wang. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics Proteomics Bioinformatics, 2021. [PubMed]
- X. Zheng, Y. Qian, B. Fu, D. Jiao, Y. Jiang, P. Chen. Mitochondrial fragmentation limits NK cell-based tumor immunosurveillance. Nat Immunol, 2019. [PubMed]
- D. Kumar, S. Gurrapu, Y. Wang, S.Y. Bae, P.R. Pandey, H. Chen. LncRNA Malat1 suppresses pyroptosis and T cell-mediated killing of incipient metastatic cells. Nat Cancer, 2024. [PubMed]
- M. Lenart, E. Działo, A. Kluczewska, K. Węglarczyk, A. Szaflarska, M. Rutkowska-Zapała. miRNA regulation of nk cells antiviral response in children with severe and/or recurrent herpes simplex virus infections. Front Immunol, 2020
- S.Y. Li, Y. Zhu, R.N. Li, J.H. Huang, K. You, Y.F. Yuan. LncRNA Lnc-APUE is repressed by HNF4α and promotes G1/S phase transition and tumor growth by regulating miR-20b/E2F1 axis. Adv Sci (Weinh), 2021
- M.C. Costanzo, D. Kim, M. Creegan, K.G. Lal, J.A. Ake, J.R. Currier. Transcriptomic signatures of NK cells suggest impaired responsiveness in HIV-1 infection and increased activity post-vaccination. Nat Commun, 2018. [PubMed]
- H. Song, J. Song, M. Cheng, M. Zheng, T. Wang, S. Tian. METTL3-mediated m6A RNA methylation promotes the anti-tumour immunity of natural killer cells. Nat Commun, 2021. [PubMed]
- G. Alter, J.M. Malenfant, M. Altfeld. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods, 2004. [PubMed]
- C.C. Cubitt, E. McClain, M. Becker-Hapak, J.A. Foltz, P. Wong, J.A. Wagner. A novel fusion protein scaffold 18/12/TxM activates the IL-12, IL-15, and IL-18 receptors to induce human memory-like natural killer cells. Mol Ther Oncolytics, 2022. [PubMed]
- B. Cui, L. Chen, S. Zhang, M. Mraz, J.F. Fecteau, J. Yu. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood, 2014. [PubMed]
- R. Trotta, L. Chen, D. Ciarlariello, S. Josyula, C. Mao, S. Costinean. miR-155 regulates IFN-γ production in natural killer cells. Blood, 2012. [PubMed]
- N. Fortelny, M. Farlik, V. Fife, A.D. Gorki, C. Lassnig, B. Maurer. JAK–STAT signaling maintains homeostasis in T cells and macrophages. Nat Immunol, 2024. [PubMed]
- J. Tan, J. Zhang, C. Hu, G. Wang, Q. Ren, C. Wang. Pharmacokinetic enhancement of oncolytic virus M1 by inhibiting JAK‒STAT pathway. Acta Pharm Sin B, 2024. [PubMed]
- W.J. Leonard, J.X. Lin, J.J. O’Shea. The γc family of cytokines: basic biology to therapeutic ramifications. Immunity, 2019. [PubMed]
- T. Nosaka, J.M. van Deursen, R.A. Tripp, W.E. Thierfelder, B.A. Witthuhn, A.P. McMickle. Defective lymphoid development in mice lacking JAK3. Science, 1995. [PubMed]
- J.L. Roberts, A. Lengi, S.M. Brown, M. Chen, Y.J. Zhou, J.J. O’Shea. Janus kinase 3 (JAK3) deficiency: clinical, immunologic, and molecular analyses of 10 patients and outcomes of stem cell transplantation. Blood, 2004. [PubMed]
- M.L. Robinette, M. Cella, J.B. Telliez, T.K. Ulland, A.D. Barrow, K. Capuder. Jak3 deficiency blocks innate lymphoid cell development. Mucosal Immunol, 2018. [PubMed]
- W. Wu, T. Yu, Y. Wu, W. Tian, J. Zhang, Y. Wang. The miR155HG/miR-185/ANXA2 loop contributes to glioblastoma growth and progression. J Exp Clin Cancer Res, 2019. [PubMed]