
Mitochondrial Calcium Uniporter (MCU)-Mediated Calcium Overload in Psychoactive Drug Neurotoxicity: From Pathogenesis to Therapeutic Targets
Abstract
With rapid societal changes and increasing stress levels, the abuse of psychoactive substances has emerged as a global health crisis. Studies indicate that the mitochondrial calcium uniporter (MCU) plays a pivotal role in neurotoxic damage induced by psychoactive substances. As the primary channel for mitochondrial Ca2+ uptake, MCU dysfunction can lead to Ca2+ overload, oxidative stress, and apoptosis, representing a crucial mechanism underlying neurotoxic damage. Psychoactive substances such as 3,4-Methylenedioxymethamphetamine (MDMA), cocaine, and morphine influence MCU function through multiple pathways, resulting in excessive Ca2+ accumulation and mitochondrial dysfunction, ultimately leading to neuronal injury. Although MCU inhibitors have demonstrated potential in alleviating Ca2+ overload and improving neural function in preliminary studies, their selectivity and long-term safety require further evaluation. Future research should explore the precise regulatory mechanisms of MCU in neurotoxic damage induced by psychoactive substances and develop more effective targeted therapeutic strategies.
Article type: Review Article
Keywords: mitochondrial calcium uniporter (MCU), calcium overload, psychoactive substances, neurotoxicity, review
Affiliations: Key Laboratory of Tropical Translational Medicine of Ministry of Education, Department of Forensic Medicine, Hainan Provincial Tropical Forensic Engineering Research Center, Hainan Medical University, Haikou 571199, China; 15338917291@163.com (X.Y.); chenyinyu0728@126.com (Y.C.); 15348893351@163.com (G.Z.); Department of Pathology, School of Basic Medicine and Life Sciences, Hainan Medical University, Haikou 571199, China
License: © 2025 by the authors. CC BY 4.0 Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.3390/ijms26104732 | PubMed: 40429873 | PMC: PMC12111645
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (3.4 MB)
1. Introduction
With rapid societal changes and increasing stress levels, millions of people worldwide suffer health damage or even fatal consequences due to psychoactive substance abuse each year. These substances pose a significant public health threat due to their diverse chemical structures and regulatory lag. The abuse of psychoactive substances not only directly endangers individual health—manifesting as addiction, brain dysfunction, and psychological disorders—but also contributes to rising crime rates, family breakdowns, and increased socioeconomic burdens. Therefore, elucidating the mechanisms underlying neurotoxic damage caused by psychoactive substances and identifying effective therapeutic interventions has become an urgent research priority.
In recent years, mitochondrial calcium uniporter (MCU), an essential membrane protein, has garnered increasing research interest [ref. 1]. Emerging studies have demonstrated abnormal MCU expression in the mitochondrial inner membrane of individuals with psychoactive substance abuse, primarily involving mitochondrial Ca2+ regulation and intracellular signaling pathways. Additionally, Liu Z et al. reported that specific MCU inhibitors could mitigate neurotoxic damage by suppressing MCU activity [ref. 2]. These findings suggest that MCU holds promise as a potential therapeutic target for neurotoxic damage induced by psychoactive substances. This review systematically discusses the structure, function, regulatory mechanisms, agonists, and inhibitors of MCU, and its role in the neurotoxic damage induced by psychoactive substances. The aim is to provide insights into its mechanistic implications while offering references for developing more effective targeted therapies for individuals suffering from psychoactive substance abuse.
2. Structure and Function of MCU
2.1. Structure of MCU
MCU, also known as CCDC109A1, consists of 351 amino acids with a molecular weight of approximately 40 kDa [ref. 3]. It is present in most eukaryotes, including protozoa, plants, amoebae, fungi, and metazoans. In 2004, researchers first recorded calcium currents from the inner mitochondrial membrane (IMM) using the patch-clamp technique in isolated mitotic cells [ref. 4]. Subsequent studies confirmed that mitochondria are capable of Ca2+ uptake due to the presence of an inwardly rectifying, highly selective Ca2+ channel in the IMM, a process dependent on the MCU complex [ref. 5].
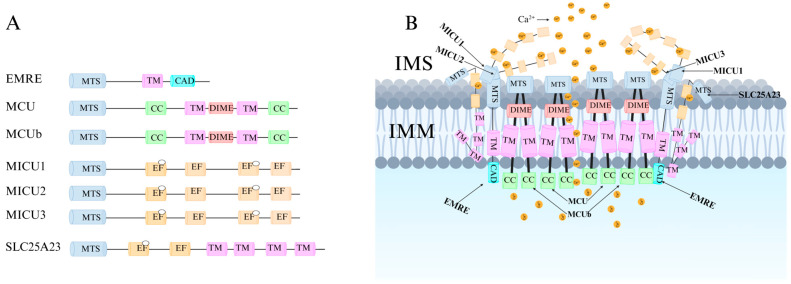
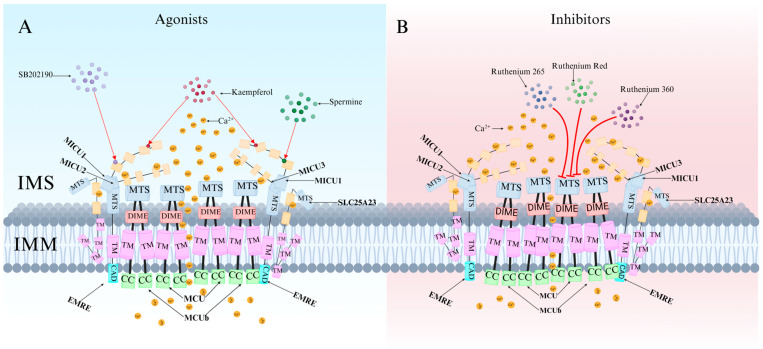
Recent research has revealed that the MCU channel exists as a heterogeneous protein complex with a molecular mass ranging between 450 and 800 kDa. This complex is structurally intricate, comprising multiple mitochondrial calcium uptake proteins (MICU1, MICU2, and MICU3), a dominant-negative subunit beta of MCU (MCUb), mitochondrial calcium uniporter regulator 1 (MCUR1), essential MCU regulator (EMRE), and solute carrier family 25 member 23 (SLC25A23) (Figure 1A) [ref. 6]. These proteins collectively regulate MCU function and control Ca2+ transport. MCU itself contains two coiled-coil domains (CC1 and CC2) and two transmembrane domains (TM1 and TM2), both embedded within the IMM. The N- and C-terminal regions of MCU are oriented toward the mitochondrial matrix, connected by a highly conserved short loop containing the DIME motif (Asp-Ile-Met-Glu). This sequence is essential for Ca2+ permeability, as mutations in its negatively charged residues significantly impact MCU function [ref. 2,ref. 6,ref. 7].
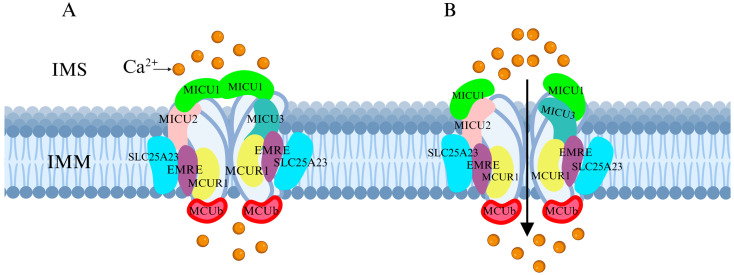
The MICU family proteins contain EF-hand domains, which function as Ca2+-binding sites, allowing them to sense Ca2+ concentration fluctuations in the cytosol or intermembrane space (IMS), thereby regulating MCU channel activity and mitochondrial Ca2+ uptake (Figure 1B) [ref. 8,ref. 9]. Additionally, other regulatory proteins modulate MCU activity through various mechanisms, influencing mitochondrial Ca2+ uptake capacity.
2.2. Function of MCU
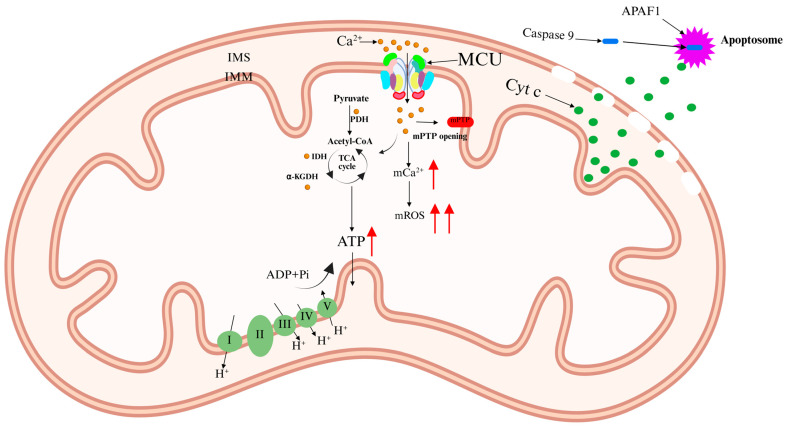
Mitochondria play essential roles in cellular physiology, including energy production, metabolic regulation, intracellular Ca2+ homeostasis, and apoptotic signaling [ref. 10]. As a crucial second messenger, Ca2+ regulates numerous cellular activities, including transmission of neuronal signal and muscle contraction. MCU-mediated Ca2+ uptake into mitochondria via the IMM is vital for these physiological processes [ref. 11].
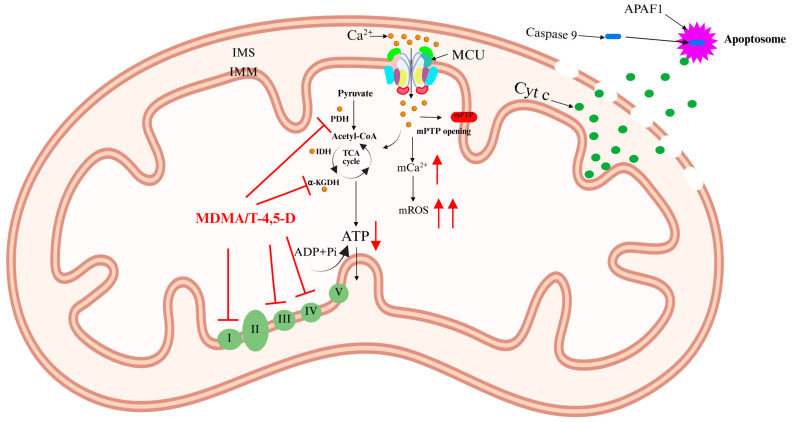
Ca2+ activates three Ca2+-sensitive dehydrogenases within the mitochondrial matrix: pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (IDH), and alpha-ketoglutarate dehydrogenase (α-KGDH) [ref. 12,ref. 13]. This activation enhances the tricarboxylic acid (TCA) cycle, increasing electron flux through the respiratory chain and boosting ATP production. However, excessive Ca2+ accumulation in the mitochondrial matrix triggers detrimental effects, including elevated mitochondrial reactive oxygen species (mROS) levels and mitochondrial membrane depolarization. This condition leads to the opening of the mitochondrial permeability transition pore (mPTP), resulting in mitochondrial swelling and the release of cytochrome c (Cyt c). Cyt c interacts with apoptotic peptidase activator 1 (APAF1) to form the apoptosome [ref. 14]. This complex recruits and activates caspase-9, initiating a proteolytic cascade that ultimately leads to apoptosis (Figure 2) [ref. 11,ref. 15]. Notably, apoptosis is a key mechanism underlying neurotoxicity induced by psychoactive substances [ref. 16].
MCU dysfunction is closely associated with various diseases. In cardiovascular disorders, reduced MCU expression contributes to Ca2+ dysregulation, whereas MCU overexpression has been found to exert protective effects by enhancing cardiovascular cell function [ref. 17,ref. 18]. In cancer, MCU is often upregulated, as observed in breast and pancreatic cancers [ref. 19,ref. 20], and suppressing MCU expression has been shown to inhibit tumor growth and metastasis. In the nervous system, neurons are highly sensitive to Ca2+ signaling perturbations, which can contribute to neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease [ref. 21]. Studies have demonstrated that excessive MCU activity leads to mitochondrial Ca2+ overload, exacerbating neurotoxicity and excitotoxicity [ref. 22,ref. 23].
3. Regulatory Mechanisms of MCU
The regulation of MCU involves multiple levels of control, including protein–protein interactions, fluctuations in Ca2+ concentration, and the influence of various regulatory factors. In particular, under the influence of psychoactive substances, MCU function may be significantly disrupted, leading to excessive mitochondrial Ca2+ accumulation and subsequent neurotoxic effects. Therefore, elucidating the precise regulatory mechanisms of MCU, especially its role in neurotoxicity, may provide new insights and therapeutic targets for neurodegenerative diseases and neurotoxicity induced by psychoactive substance abuse.
3.1. Regulation of MCU by Associated Proteins
MCU plays a crucial role in maintaining cellular homeostasis by regulating metabolism, energy production, signal transduction, and cell death. The Ca2+ uptake function of MCU is finely regulated by the MICU family, including MICU1, MICU2, and MICU3. These proteins contain EF-hand domains that allow them to sense fluctuations in cytosolic Ca2+ concentrations. When cytosolic Ca2+ levels reach a certain threshold, MICU1 and MICU2 form a heterodimer, which binds Ca2+ at the EF-hand domains, inducing a conformational change that leads to their dissociation from the MCU complex. This process regulates MCU activity and participates in cell death regulation (Figure 3A) [ref. 24,ref. 25,ref. 26,ref. 27].
MICU1 and MICU2 are widely expressed in most tissues, whereas MICU3 is particularly abundant in neurons. MICU3 enhances MCU sensitivity to Ca2+, facilitating Ca2+ uptake into mitochondria within axons, thereby accelerating ATP synthesis and maintaining the complex metabolic functions of neuronal cells [ref. 28]. Additionally, MCUb acts as a dominant-negative regulatory subunit of MCU. Structurally similar to MCU, excessive MCUb expression replaces MICU1 and MICU2 within the functional MCU complex, reducing mitochondrial Ca2+ uptake capacity [ref. 29].
The precise function of MCUR1 remains unclear. Some studies suggest that MCUR1 influences mitochondrial Ca2+ uptake by modulating interactions between MCU and EMRE [ref. 30]. As an essential subunit of MCU, MCUR1 interacts with both MCU and EMRE, and its absence results in an incomplete MCU complex, thereby inhibiting mitochondrial Ca2+ uptake. Research by Paupe V et al. indicates that MCUR1 also regulates the threshold for mPTP opening by modulating interactions between MCU and the mPTP complex [ref. 31]. However, other studies suggest that MCUR1 is not a direct regulator of MCU but instead functions as an assembly factor for Cyt c oxidase [ref. 32]. Overall, MCUR1 serves as a critical bridge connecting MCU and mPTP.
EMRE is a single-pass transmembrane protein specific to multicellular organisms and is an essential regulatory factor of the MCU complex. When MCU is expressed alone, it does not exhibit uniporter activity; only when co-expressed with EMRE does MCU become functional [ref. 33]. EMRE precisely localizes MCU to specific regions of the IMM rather than to the cristae invaginations, which may play a vital role in mitochondrial Ca2+ uptake regulation [ref. 34]. Studies indicate that the MCU-EMRE complex can dimerize [ref. 35,ref. 36], though this dimerization is not essential for channel function. Instead, it may facilitate the spatial distribution of the uniporter at contact sites between the inner and outer mitochondrial membranes, enhancing its responsiveness to intracellular Ca2+ signals and promoting efficient Ca2+ transfer from the endoplasmic reticulum to the mitochondrial matrix, leading to increased mitochondrial Ca2+ levels [ref. 33,ref. 34,ref. 37].
SLC25A23, a member of the solute carrier family, is a multi-pass transmembrane protein with three EF-hand domains [ref. 38,ref. 39]. This structure confers high Ca2+ sensitivity, and its Mg2+-ATP/Pi transporter function is similar to other Ca2+-activated channels and transporters. Although SLC25A23 can modulate mitochondrial Ca2+ influx, it does not affect the rate or total amount of mitochondrial Ca2+ efflux.
Previous studies have demonstrated that downregulation of MCU significantly reduces mitochondrial Ca2+ uptake, both in live cells treated with Ca2+-mobilizing agonists and in permeabilized cells subjected to Ca2+ buffering perfusion. However, this alteration does not affect the fundamental properties of mitochondria (Figure 3B) [ref. 6,ref. 40].
3.2. Other Regulatory Mechanisms of MCU
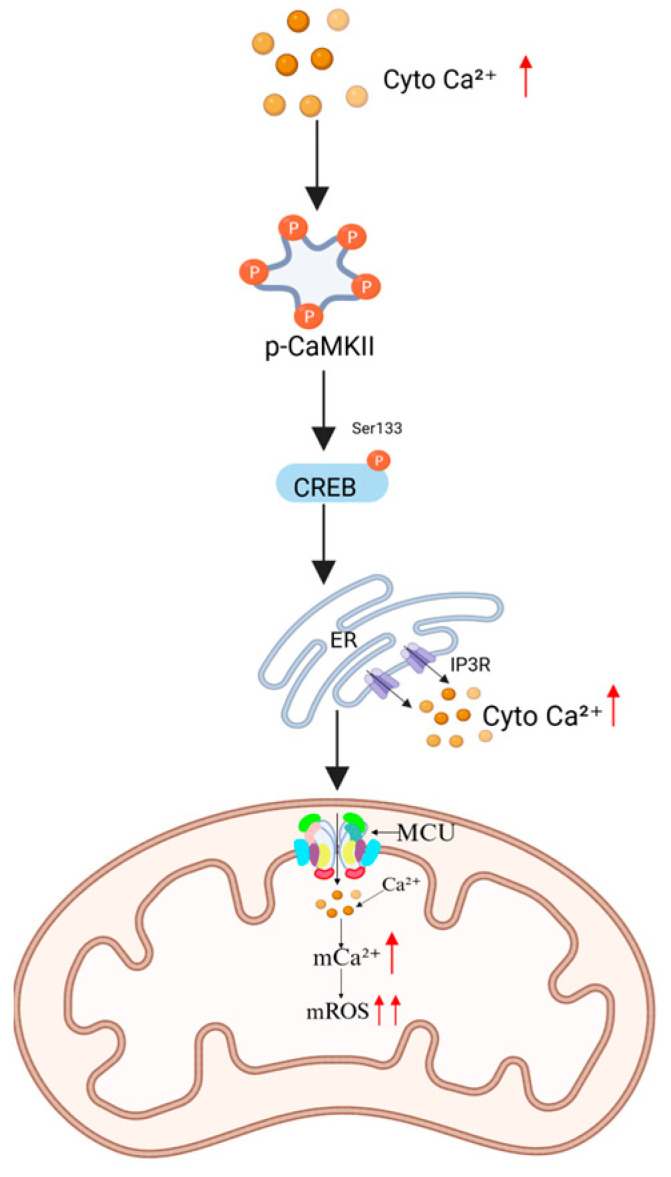
3.2.1. Regulation via the CaMKII-CREB Signaling Pathway
When intracellular Ca2+ concentrations increase, calmodulin-dependent kinase II (CaMKII) undergoes autophosphorylation and activation [ref. 41]. This activation promotes the phosphorylation of cAMP response element-binding protein (CREB) at Ser133, enhancing its DNA-binding affinity and subsequently upregulating the expression of endoplasmic reticulum (ER) Ca2+ release channels, such as inositol 1,4,5-triphosphate receptors (IP3Rs). Excessive Ca2+ release from ER stores leads to cytosolic Ca2+ overload, which in turn drives mitochondrial Ca2+ uptake Via an MCU-dependent mechanism, exacerbating mitochondrial oxidative stress (Figure 4) [ref. 42].
3.2.2. Kinase-Mediated Post-Translational Modifications of MCU
Proline-rich tyrosine kinase 2 (Pyk2) is activated under oxidative stress conditions and phosphorylates MCU at Tyr107, promoting MCU monomer oligomerization into functional calcium channels. This modification significantly enhances mitochondrial Ca2+ uptake capacity [ref. 43]. Under conditions of cellular energy deficiency, AMP-activated protein kinase (AMPK) phosphorylates MCU at Ser57, inducing a conformational change that facilitates mitochondrial Ca2+ influx. This process activates the pyruvate dehydrogenase complex, accelerating acetyl-CoA production to sustain ATP supply [ref. 44].
3.2.3. MicroRNA-Mediated Post-Transcriptional Regulatory Network
MicroRNAs (miRNAs) are critical post-transcriptional regulators involved in numerous physiological processes, including cell proliferation, differentiation, metabolism, and apoptosis. Recent studies have demonstrated that multiple miRNAs can directly target MCU mRNA, modulating its translation and thereby influencing mitochondrial Ca2+ uptake and related signaling pathways. For instance, miR-129-3p, miR-1, miR-25, miR-138-5p, and miR-340 have been shown to selectively bind to MCU mRNA, suppressing its translation and reducing mitochondrial Ca2+ uptake capacity [ref. 45,ref. 46]. This regulatory mechanism plays a crucial role under various physiological and pathological conditions, including excitotoxicity in neurons, ischemia–reperfusion injury, tumorigenesis, and drug resistance.
4. Specific Agonists and Inhibitors of MCU
4.1. MCU-Specific Agonists and Their Mechanisms of Action
Currently known MCU-specific agonists include spermine, kaempferol, and SB202190. These compounds exert their effects by modulating MICU1, potentially by binding to the EF-hand domain of MICU1 and blocking its gatekeeping activity, thereby enhancing mitochondrial Ca2+ uptake (Figure 5A) [ref. 3,ref. 30].
4.2. Classification and Characteristics of MCU-Specific Inhibitors
MCU-specific inhibitors include ruthenium red (RuRed), ruthenium 360 (Ru360), ruthenium 265 (Ru265), mitoxantrone (MTX), DS16570511, metoprolol, and KN-93. Among them, RuRed is the most commonly used MCU-specific inhibitor. It blocks MCU channels to reduce Ca2+ and Fe2+ accumulation, prevents mitochondrial depolarization, alters synaptic activity, improves mitochondrial function, and decreases DNA damage and apoptosis [ref. 47,ref. 48]. However, due to challenges in RuRed purification, commercially available RuRed formulations are not pure compounds but mixtures of several ruthenium-amine complexes [ref. 49].
Ru360 is the primary active component of RuRed mixtures. It selectively inhibits MCU without affecting Na+/Ca2+ channels or L-type Ca2+ channels and does not significantly impact the cell cycle, proliferation, or viability [ref. 50]. Studies have shown that Ru360 prevents glutamate-induced excitotoxicity in cortical neurons while mitigating oxidative stress in microglia and inhibiting amyloid-beta (Aβ)-induced apoptosis [ref. 51,ref. 52]. Compared to Ru360, Ru265 offers greater selectivity and cellular permeability, with over twice the cellular uptake efficiency of Ru360 [ref. 8]. Furthermore, Ru265 exhibits lower toxicity than many ruthenium-based compounds and provides more effective MCU inhibition than Ru360.
Organic molecules such as MTX and DS16570511 lack MCU-specific selectivity, often producing off-target biological effects [ref. 53,ref. 54]. DS16570511 inhibits both MCU and MICU1, alleviating mitochondrial Ca2+ overload while increasing cytosolic Ca2+ concentration and muscle contraction frequency.
KN-93 effectively attenuates bupivacaine-induced neurotoxicity by inhibiting the CaMKIIα-MCU signaling pathway [ref. 2]. Additionally, two small-molecule compounds, MCU-i4 and MCU-i11, have been identified through high-throughput screening. These compounds reduce mitochondrial Ca2+ influx by directly binding to MICU1-specific sites (Figure 5B). Notably, their inhibitory effects are observed only in cellular contexts [ref. 55].
5. Research on MCU in Neurotoxic Damage Induced by Psychoactive Substances
Psychoactive substances refer to compounds that alter brain function, influencing cognition, emotions, consciousness, behavior, or perception. Recent studies have revealed that psychoactive substances can disrupt MCU function, leading to neuronal Ca2+ overload and mitochondrial dysfunction, thereby inducing oxidative stress, apoptosis, and neurodegeneration. Understanding the role of MCU in these processes not only provides insights into the mechanisms of neurotoxic damage caused by psychoactive substances but also offers potential directions for drug interventions and neuroprotection. This section systematically examines the regulatory effects of different psychoactive substances on MCU and their associated neurotoxic damage, providing a theoretical foundation for targeted therapeutic strategies (Table 1).
Table 1: Drugs–MCU interaction pathways leading to mitochondrial neurotoxicity.
| Drugs | MCU-Related Mechanisms | Final Effects |
|---|---|---|
| MDMA | Inhibits mitochondrial complexes I/III → ROS↑, Ca2+ overload → ATP depletion → apoptosis. Metabolite T-4,5-D inhibits PDH/α-KGDH and inactivates complexes I/IV. | Mitochondrial Ca2+ overload → apoptosis |
| Cocaine | Astrocyte acetyl-CoA↑ → GPCR activation → ↑MCU-mediated mitochondrial Ca2+ uptake → mROS↑→ neurodegeneration. MCU knockdown activates AMPK-dependent glycolysis. | Metabolic imbalance → neurodegeneration |
| Morphine | Epigenetic MCU upregulation Via pCREB/CPEB1 → mitochondrial Ca2+ overload → tolerance-related neurotoxicity. CPEB1 stabilizes polyadenylation complexes. | Morphine tolerance → neuronal damage |
| Cannabis | CB1 activation → AKT-mediated MICU1 phosphorylation → ↑MCU-driven Ca2+ influx → mitochondrial depolarization → mPTP opening → apoptosis. | Mitochondrial Ca2+ overload → apoptosis |
| METH | MCU overactivation → mitochondrial Ca2+ overload → Bax activation → mPTP opening → Cyt c/AIF release → caspase-dependent/independent apoptosis. | Mitochondrial stress → caspase-dependent/independent apoptosis |
| Ketamine | ROS↑ Via CaMKII/CaM/ERK → MCU activation → mitochondrial Ca2+ overload → Cyt c release → apoptosis. NMDAR NR1 subunit upregulation → glutamate excitotoxicity → Tau hyperphosphorylation. | Oxidative phosphorylation uncoupling → apoptosis |
“↑” Indicates increase/upregulation.
5.1. 3,4-Methylenedioxymethamphetamine (MDMA)
MDMA, commonly known as “ecstasy”, is a widely abused novel psychoactive substance with both stimulant and hallucinogenic properties. MDMA is frequently misused in nightlife and party settings. A single low-dose administration of MDMA produces effects such as euphoria, wakefulness, relaxation, enhanced sociability, and increased emotional connection with others [ref. 56]. However, animal studies have shown that chronic MDMA abuse leads to central nervous system (CNS) damage, particularly neurotoxicity, which may progressively manifest over time [ref. 57]. Mitochondrial dysfunction is one of the key mechanisms underlying MDMA-induced neurotoxic damage. Disrupted Ca2+ homeostasis and ATP depletion play crucial roles in MDMA-induced neurotoxicity. Previous studies have reported that MDMA inhibits mitochondrial complexes I and III, significantly increasing ROS levels and inducing oxidative stress. This oxidative damage impairs cellular components such as Ca2+ pumps, ATPase, and Na+-K+-ATPase, leading to abnormal intracellular Ca2+ accumulation, MCU dysfunction, and ultimately cell death [ref. 58,ref. 59]. Additionally, MDMA’s metabolite, tryptamine-4,5-dione (T-4,5-D), inhibits PDH and α-KGDH and inactivates mitochondrial complexes I and IV, exacerbating mitochondrial membrane depolarization and ATP depletion [ref. 60]. This process indirectly activates MCU, resulting in mitochondrial Ca2+ overload and ultimately neuronal apoptosis (Figure 6). However, direct evidence demonstrating the ability of MCU inhibitors to reverse MDMA-induced neurotoxic damage is still lacking. Future studies should explore this hypothesis through gene knockout models or specific inhibitor intervention experiments.
5.2. Cocaine
Cocaine, an alkaloid extracted from coca leaves, has potent anesthetic properties, high permeability, and strong CNS stimulant effects, making it one of the most widely abused psychoactive substances. Cocaine acts on the brain’s reward pathways, leading to addiction, and chronic use results in severe CNS dysfunction. Moreover, genetic alterations in cocaine abusers may affect subsequent generations. Astrocytes, a subtype of glial cells in the CNS, regulate synaptic transmission and plasticity through intracellular Ca2+ signaling. Recent studies indicate that cocaine induces neurotoxicity by modulating MCU function in astrocytes. In the brain tissue of cocaine abusers, acetyl-CoA expression is elevated in astrocytes [ref. 61]. Increased acetyl-CoA activates G protein-coupled receptors (GPCRs), leading to elevated mitochondrial Ca2+ levels [ref. 62], which enhance MCU-mediated Ca2+ uptake, subsequently increasing mROS production. Excessive mROS impairs neuronal health and exacerbates neurotoxic damage. Since mitochondria serve as the primary site for fatty acid oxidation (FAO) and mitochondrial Ca2+ is a key regulator of mitochondrial bioenergetics, MCU knockdown has been shown to activate AMPK-dependent lipid metabolic reprogramming, inhibiting FAO while shifting energy metabolism toward anaerobic glycolysis. This metabolic shift maintains neuronal energy supply through lactate production [ref. 61]. These findings suggest that cocaine-induced neurotoxicity is closely related to metabolic imbalance driven by excessive MCU activation. Thus, targeting MCU inhibition or AMPK activation may serve as a potential therapeutic strategy to alleviate cocaine addiction and mitigate neurodegeneration.
5.3. Morphine
Morphine is a potent opioid widely used for pain management, particularly in postoperative pain and cancer-related pain treatment. However, long-term morphine use leads to adaptive changes in the nervous system, resulting in tolerance and drug dependence. Morphine tolerance necessitates increasing doses to achieve the same analgesic effect, which may exacerbate neuronal damage and neurotoxicity. Studies have shown that morphine tolerance induces spinal neuronal neurotoxicity, a process involving MCU [ref. 63]. Recent research has demonstrated that CREB regulates neuroplasticity, contributing to hyperalgesia and chronic pain, and plays a crucial role in synaptic plasticity, learning, and memory [ref. 64]. Both short-term and long-term morphine exposure modulates phosphorylated CREB (pCREB) expression across different brain regions, mediating epigenetic regulation of MCU transcription. In vertebrates, the cytoplasmic polyadenylation element binding (CPEB) mRNA protein family consists of four members (CPEB1-4). Among them, CPEB1 is essential for mitochondrial energy production in neurons and stabilizes the activated cytoplasmic polyadenylation-specific factor complex [ref. 65]. CPEB1 recruits poly (A) polymerase, catalyzing poly (A) tail elongation, which participates in translational activation. As a result, MCU overexpression enhances mitochondrial Ca2+ uptake, leading to mitochondrial Ca2+ overload and neurotoxicity. Animal studies have shown that MCU inhibitors such as Ru360, or pharmacological blockers targeting pCREB or CPEB1 upregulation, reduce morphine tolerance-related behavioral responses. These findings suggest that MCU inhibition may help prevent opioid tolerance [ref. 66]. Future research should explore epigenetic regulatory mechanisms, such as histone modifications and non-coding RNA involvement in MCU expression, to develop multi-target intervention strategies.
5.4. Cannabis
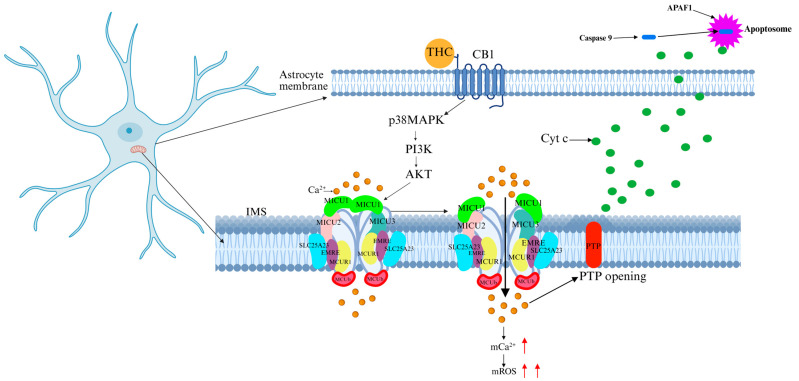
Cannabis is the most widely abused psychoactive substance globally, encompassing the largest variety of compounds and accounting for the highest sales volume. Due to its hallucinogenic and addictive properties, cannabis is classified as a strictly controlled substance in most countries. The primary psychoactive component of cannabis, Δ9-tetrahydrocannabinol (Δ9-THC), exerts potent effects on the CNS. Studies have revealed that THC alters mitochondrial function in brain and muscle tissues [ref. 67,ref. 68]. By binding to cannabinoid receptors (CB1 and CB2, both G-protein-coupled receptors), THC disrupts neurotransmitter systems in the brain. In particular, CB1 receptor activation induces neurotransmitter imbalances, increases oxidative stress and ROS production, and impairs mitochondrial function [ref. 69]. CB1 receptor activation has been shown to promote mitochondrial Ca2+ accumulation in both cultured astrocytes and in vivo animal models [ref. 70]. CB1 modulates soluble adenylyl cyclase (sAC) activity, which regulates mitochondrial oxygen consumption in neurons and astrocytes [ref. 69,ref. 71]. Additionally, under chronic or excessive cannabis use, CB1 activation triggers the protein kinase B/AKT pathway, leading to AKT-mediated phosphorylation of MICU1, which increases Ca2+ influx into the mitochondrial matrix. This process results in mitochondrial Ca2+ overload, mitochondrial membrane potential disruption, and mPTP opening, ultimately causing neuronal apoptosis (Figure 7) [ref. 72]. Cannabis abuse is associated with cognitive impairment, mood disturbances, and neurodegeneration. Previous clinical studies have shown that selective CB1 antagonists partially reverse THC-induced neurotoxicity; however, these agents may interfere with the physiological functions of the endocannabinoid system [ref. 73]. Therefore, targeting MCU-specific modulators rather than cannabinoid receptor antagonists may provide a safer therapeutic approach.
5.5. Methamphetamine (METH)
METH, commonly known as “crystal meth”, is a highly addictive CNS stimulant. While short-term use induces euphoria, heightened alertness, and increased energy, it also leads to aggressiveness, anxiety, and paranoia. Chronic METH use, however, causes severe neurotoxicity, manifesting as brain damage, cognitive and memory impairments, cardiovascular complications, and immune suppression. Due to its high addiction potential and profound effects on CNS function, METH is classified as an illicit substance in most countries. The neurotoxic effects of METH are primarily linked to oxidative stress and mitochondrial dysfunction. Studies have demonstrated that prolonged METH exposure leads to MCU overactivation, resulting in excessive mitochondrial Ca2+ accumulation, triggering mitochondrial stress and ROS production [ref. 74]. These changes destabilize the mitochondrial membrane, causing alterations in membrane permeability and loss of membrane potential. Ca2+ overload, in conjunction with pro-apoptotic protein Bax, induces mPTP opening, facilitating the release of Cyt c and apoptosis-inducing factor (AIF). This process activates both caspase-3-dependent and caspase-3-independent apoptotic pathways, promoting cell death and neurotoxicity [ref. 75]. Given that MCU serves as the central regulatory hub for mitochondrial Ca2+ influx, its dysfunction plays a critical role in METH-induced mitochondrial-dependent cell death. Thus, targeting MCU may represent a novel therapeutic strategy for mitigating METH-induced neurotoxicity.
5.6. Ketamine
Ketamine, commonly known as ‘K’, is a derivative of phencyclidine and acts as a non-competitive N-methyl-D-aspartate receptor (NMDAR) antagonist. It was first synthesized by Calvin Stevens in 1962. Due to its rapid absorption, fast onset, mild respiratory suppression, and limited cardiovascular effects, ketamine was introduced as a human anesthetic in 1970. However, in recent years, ketamine has been misused as a hallucinogen due to its dissociative and euphoric effects, particularly in club and party settings. Several recent studies have demonstrated that ketamine disrupts Ca2+ signaling, induces DNA fragmentation, increases ROS production, impairs mitochondrial function, and triggers apoptosis [ref. 76,ref. 77]. Excessive glutamate release is a major mechanism underlying excitotoxic cell damage. Chronic ketamine abuse leads to compensatory upregulation of the NMDAR NR1 subunit, a process in which persistent or prolonged NMDAR blockade triggers adaptive regulation, making neurons overly responsive to normal extracellular glutamate levels. This phenomenon drives excessive Ca2+ influx, elevating intracellular Ca2+ concentrations and inducing ROS production Via the CaMKII/CaM/ERK signaling pathway. Subsequently, ROS activates MCU, leading to mitochondrial Ca2+ overload, respiratory suppression, and uncoupling of oxidative phosphorylation, ultimately promoting Cyt c release from the mitochondrial membrane into the cytoplasm, triggering apoptosis [ref. 78]. Additionally, cytosolic Ca2+ stimulates nitric oxide synthase (NOS) to produce NO, which inhibits complex IV, further increasing ROS generation and exacerbating oxidative stress [ref. 79]. Animal studies have shown that ketamine abuse induces hyperphosphorylation of Tau protein at Ser202, Ser396, and Trp205, impairing synaptic function in hippocampal neurons and increasing neurotoxicity [ref. 80]. However, the precise mechanisms underlying ketamine-induced neurotoxicity and neuronal apoptosis remain unclear. Whether MCU inhibitors can mitigate ketamine-induced neurotoxicity requires further investigation.
6. Conclusions and Future Perspectives
This review systematically highlights the crucial role of MCU in neurotoxic damage induced by psychoactive substances. With the worsening global crisis of psychoactive substance abuse, it is imperative to clarify the underlying neurotoxic mechanisms. As the core channel regulating mitochondrial Ca2+ uptake in the inner mitochondrial membrane, MCU dysfunction is closely linked to Ca2+ homeostasis imbalance, mitochondrial dysfunction, oxidative stress, and apoptosis. Studies have demonstrated that various psychoactive substances—including MDMA, cocaine, morphine, cannabis, METH, and ketamine—disrupt MCU function via different mechanisms, leading to mitochondrial Ca2+ overload and neurodegeneration. For instance, MDMA inhibits mitochondrial complex activity, increasing ROS levels, while morphine upregulates MCU expression via the CREB signaling pathway, exacerbating Ca2+-dependent neurotoxicity. Additionally, MCU-specific inhibitors (e.g., Ru360, Ru265) have demonstrated potential in alleviating Ca2+ overload, restoring mitochondrial function, and exerting neuroprotective effects in in vitro and animal models, providing new directions for drug intervention.
Despite significant progress, further research is required in several key areas. The precise mechanisms by which MCU mediates psychoactive substance-induced neurotoxicity remain unclear, necessitating single-cell sequencing and gene-editing approaches to elucidate these pathways. Given that current MCU inhibitors suffer from low purity and off-target effects, future research should focus on developing highly selective, low-toxicity small molecules or gene therapies for precise MCU regulation and neuroprotection. Furthermore, the functional heterogeneity of MCU across different tissues and diseases, particularly in comorbid models of neurodegenerative diseases and drug addiction, remains to be fully elucidated. Additionally, the long-term inhibition of MCU must be evaluated for its potential impact on physiological Ca2+ signaling. Advanced models, such as organoids and non-human primates, should be utilized to assess safety and efficacy before clinical translation. In conclusion, MCU serves as a critical node in psychoactive substance-induced neurotoxicity, making it a promising target for both mechanistic research and therapeutic development. Future studies should integrate multi-omics and interdisciplinary approaches to translate fundamental discoveries into clinical applications.
References
- D. De Stefani, A. Raffaello, E. Teardo, I. Szabò, R. Rizzuto. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature, 2011. [DOI | PubMed]
- Z. Liu, W. Zhao, P. Yuan, P. Zhu, K. Fan, Z. Xia, S. Xu. The mechanism of CaMK2α-MCU-mitochondrial oxidative stress in bupivacaine-induced neurotoxicity. Free Radic. Biol. Med., 2020. [DOI | PubMed]
- J. Wang, J. Jiang, H. Hu, L. Chen. MCU complex: Exploring emerging targets and mechanisms of mitochondrial physiology and pathology. J. Adv. Res., 2025. [DOI | PubMed]
- Y. Kirichok, G. Krapivinsky, D.E. Clapham. The mitochondrial calcium uniporter is a highly selective ion channel. Nature, 2004. [DOI | PubMed]
- T. Pathak, M. Trebak. Mitochondrial Ca2+ signaling. Pharmacol. Ther., 2018. [DOI | PubMed]
- J.M. Baughman, F. Perocchi, H.S. Girgis, M. Plovanich, C.A. Belcher-Timme, Y. Sancak, X.R. Bao, L. Strittmatter, O. Goldberger, R.L. Bogorad. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature, 2011. [DOI | PubMed]
- R. Baradaran, C. Wang, A.F. Siliciano, S.B. Long. Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nature, 2018. [DOI | PubMed]
- J.J. Woods, J.J. Wilson. Inhibitors of the mitochondrial calcium uniporter for the treatment of disease. Curr. Opin. Chem. Biol., 2020. [DOI | PubMed]
- Y. Xing, M. Wang, J. Wang, Z. Nie, G. Wu, X. Yang, Y. Shen. Dimerization of MICU Proteins Controls Ca2+ Influx through the Mitochondrial Ca2+ Uniporter. Cell Rep., 2019. [DOI | PubMed]
- M.J. Twyning, R. Tufi, T.P. Gleeson, K.M. Kolodziej, S. Campesan, A. Terriente-Felix, L. Collins, F. De Lazzari, F. Giorgini, A.J. Whitworth. Partial loss of MCU mitigates pathology in vivo across a diverse range of neurodegenerative disease models. Cell Rep., 2024. [DOI | PubMed]
- D. D’Angelo, R. Rizzuto. The Mitochondrial Calcium Uniporter (MCU): Molecular Identity and Role in Human Diseases. Biomolecules, 2023. [DOI | PubMed]
- J.G. McCormack, R.M. Denton. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J., 1979. [DOI | PubMed]
- G.E. Hansen, G.E. Gibson. The α-Ketoglutarate Dehydrogenase Complex as a Hub of Plasticity in Neurodegeneration and Regeneration. Int. J. Mol. Sci., 2022. [DOI | PubMed]
- Z. Zhou, T. Arroum, X. Luo, R. Kang, Y.J. Lee, D. Tang, M. Hüttemann, X. Song. Diverse functions of cytochrome c in cell death and disease. Cell Death Differ., 2024. [DOI | PubMed]
- R. Rizzuto, D. De Stefani, A. Raffaello, C. Mammucari. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol., 2012. [DOI | PubMed]
- Y. Qin, Y. Huang, W. Lin, R. Huang, K. Li, X. Han, Y. Ren. Neurotoxic effects induced by flunitrazepam and its metabolites in zebrafish: Oxidative stress, apoptosis, and histone hypoacetylation. Sci. Total Environ., 2024. [DOI | PubMed]
- Z. Hong, K.H. Chen, A. DasGupta, F. Potus, K. Dunham-Snary, S. Bonnet, L. Tian, J. Fu, S. Breuils-Bonnet, S. Provencher. MicroRNA-138 and MicroRNA-25 Down-regulate Mitochondrial Calcium Uniporter.; Causing the Pulmonary Arterial Hypertension Cancer Phenotype. Am. J. Respir. Crit. Care Med., 2017. [DOI | PubMed]
- T. Liu, N. Yang, A. Sidor, B. O’Rourke. MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca2+ Leak. Circ. Res., 2021. [DOI | PubMed]
- A. Tosatto, R. Sommaggio, C. Kummerow, R.B. Bentham, T.S. Blacker, T. Berecz, M.R. Duchen, A. Rosato, I. Bogeski, G. Szabadkai. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol. Med., 2016. [DOI | PubMed]
- X. Wang, Y. Li, Z. Li, S. Lin, H. Wang, J. Sun, C. Lan, L. Wu, D. Sun, C. Huang. Mitochondrial Calcium Uniporter Drives Metastasis and Confers a Targetable Cystine Dependency in Pancreatic Cancer. Cancer Res., 2022. [DOI | PubMed]
- P. Sukumaran, V. Nascimento Da Conceicao, Y. Sun, N. Ahamad, L.R. Saraiva, S. Selvaraj, B.B. Singh. Calcium Signaling Regulates Autophagy and Apoptosis. Cells, 2021. [DOI | PubMed]
- A.O. Egunlusi, S.F. Malan, V.A. Palchykov, J. Joubert. Calcium Modulating Effect of Polycyclic Cages: A Suitable Therapeutic Approach Against Excitotoxic-induced Neurodegeneration. Mini Rev. Med. Chem., 2024. [DOI | PubMed]
- M. Maiolino, N. O’Neill, V. Lariccia, S. Amoroso, S. Sylantyev, P.R. Angelova, A.Y. Abramov. Inorganic Polyphosphate Regulates AMPA and NMDA Receptors and Protects Against Glutamate Excitotoxicity via Activation of P2Y Receptors. J. Neurosci., 2019. [DOI | PubMed]
- T.L. Stevens, H.M. Cohen, J.F. Garbincius, J.W. Elrod. Mitochondrial calcium uniporter channel gatekeeping in cardiovascular disease. Nat. Cardiovasc. Res., 2024. [DOI | PubMed]
- D. Tomar, M. Thomas, J.F. Garbincius, D.W. Kolmetzky, O. Salik, P. Jadiya, S.K. Joseph, A.C. Carpenter, G. Hajnóczky, J.W. Elrod. MICU1 regulates mitochondrial cristae structure and function independently of the mitochondrial Ca2+ uniporter channel. Sci. Signal., 2023. [DOI | PubMed]
- C. Wang, A. Jacewicz, B.D. Delgado, R. Baradaran, S.B. Long. Structures reveal gatekeeping of the mitochondrial Ca2+ uniporter by MICU1-MICU2. eLife, 2020. [DOI | PubMed]
- W. Wu, J. Zheng, Z. Jia. Structural characterization of the mitochondrial Ca2+ uniporter provides insights into Ca2+ uptake and regulation. iScience, 2021. [DOI | PubMed]
- G. Ashrafi, J. de Juan-Sanz, R.J. Farrell, T.A. Ryan. Molecular Tuning of the Axonal Mitochondrial Ca2+ Uniporter Ensures Metabolic Flexibility of Neurotransmission. Neuron, 2020. [DOI | PubMed]
- J.P. Lambert, T.S. Luongo, D. Tomar, P. Jadiya, E. Gao, X. Zhang, A.M. Lucchese, D.W. Kolmetzky, N.S. Shah, J.W. Elrod. MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation, 2019. [DOI | PubMed]
- M. Rodríguez-Prados, K.T. Huang, K. Márta, M. Paillard, G. Csordás, S.K. Joseph, G. Hajnóczky. MICU1 controls the sensitivity of the mitochondrial Ca2+ uniporter to activators and inhibitors. Cell Chem. Biol., 2023. [DOI | PubMed]
- V. Paupe, J. Prudent, E.P. Dassa, O.Z. Rendon, E.A. Shoubridge. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab., 2015. [DOI | PubMed]
- D. Chaudhuri, D.J. Artiga, S.A. Abiria, D.E. Clapham. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl. Acad. Sci. USA, 2016. [DOI | PubMed]
- T. Yamamoto, R. Yamagoshi, K. Harada, M. Kawano, N. Minami, Y. Ido, K. Kuwahara, A. Fujita, M. Ozono, A. Watanabe. Analysis of the structure and function of EMRE in a yeast expression system. Biochim. Biophys. Acta, 2016. [DOI | PubMed]
- Y. Wang, N.X. Nguyen, J. She, W. Zeng, Y. Yang, X.C. Bai, Y. Jiang. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell, 2019. [DOI | PubMed]
- Y. Wang, Y. Han, J. She, N.X. Nguyen, V.K. Mootha, X.C. Bai, Y. Jiang. Structural insights into the Ca2+-dependent gating of the human mitochondrial calcium uniporter. eLife, 2020. [DOI | PubMed]
- A. Watanabe, K. Maeda, A. Nara, M. Hashida, M. Ozono, A. Nakao, A. Yamada, Y. Shinohara, T. Yamamoto. Quantitative analysis of mitochondrial calcium uniporter (MCU) and essential MCU regulator (EMRE) in mitochondria from mouse tissues and HeLa cells. FEBS Open Bio, 2022. [DOI]
- B.R. Alevriadou, A. Patel, M. Noble, S. Ghosh, V.M. Gohil, P.B. Stathopulos, M. Madesh. Molecular nature and physiological role of the mitochondrial calcium uniporter channel. Am. J. Physiol. Cell Physiol., 2021. [DOI | PubMed]
- N.E. Hoffman, H.C. Chandramoorthy, S. Shanmughapriya, X.Q. Zhang, S. Vallem, P.J. Doonan, K. Malliankaraman, S. Guo, S. Rajan, J.W. Elrod. SLC25A23 augments mitochondrial Ca2+ uptake.; interacts with MCU.; and induces oxidative stress-mediated cell death. Mol. Biol. Cell., 2014. [DOI | PubMed]
- M.T. Bassi, M. Manzoni, R. Bresciani, M.T. Pizzo, M.A. Della, S. Barlati, E. Monti, G. Borsani. Cellular expression and alternative splicing of SLC25A23, a member of the mitochondrial Ca2+-dependent solute carrier gene family. Gene, 2005. [DOI | PubMed]
- S. Feno, R. Rizzuto, A. Raffaello, R.D. Vecellio. The molecular complexity of the Mitochondrial Calcium Uniporter. Cell Calcium, 2021. [DOI | PubMed]
- M.M. Kreusser, J. Backs. Integrated mechanisms of CaMKII-dependent ventricular remodeling. Front. Pharmacol., 2014. [DOI | PubMed]
- D. Faccenda, G. Gorini, A. Jones, C. Thornton, A. Baracca, G. Solaini, M. Campanella. The ATPase Inhibitory Factor 1 (IF1) regulates the expression of the mitochondrial Ca2+ uniporter (MCU) via the AMPK/CREB pathway. Biochim. Biophys. Acta Mol. Cell Res., 2021. [DOI | PubMed]
- J. O-Uchi, B.S. Jhun, S. Xu, S. Hurst, A. Raffaello, X. Liu, B. Yi, H. Zhang, P. Gross, J. Mishra. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid. Redox Signal., 2014. [DOI | PubMed]
- H. Zhao, T. Li, K. Wang, F. Zhao, J. Chen, G. Xu, J. Zhao, T. Li, L. Chen, L. Li. AMPK-mediated activation of MCU stimulates mitochondrial Ca2+ entry to promote mitotic progression. Nat. Cell Biol., 2019. [DOI | PubMed]
- B. Wang, Y. Li, C. You. miR-129-3p Targeting of MCU Protects Against Glucose Fluctuation-Mediated Neuronal Damage via a Mitochondrial-Dependent Intrinsic Apoptotic Pathway. Diabetes Metab. Syndr. Obes., 2021. [DOI | PubMed]
- T. Zaglia, P. Ceriotti, A. Campo, G. Borile, A. Armani, P. Carullo, V. Prando, R. Coppini, V. Vida, T.O. Stølen. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. USA, 2017. [DOI | PubMed]
- Y. Sharma, D. Garabadu. Ruthenium red, mitochondrial calcium uniporter inhibitor, attenuates cognitive deficits in STZ-ICV challenged experimental animals. Brain Res. Bull., 2020. [DOI | PubMed]
- Y. Zhao, P. Wang, T. Liu, Y. Yang, J. Guo, Y. He, J. Xi. Zn2+ protect cardiac H9c2 cells from endoplasmic reticulum stress by preventing mPTP opening through MCU. Cell. Signal., 2022. [DOI | PubMed]
- K.M. Broekemeier, R.J. Krebsbach, D.R. Pfeiffer. Inhibition of the mitochondrial Ca2+ uniporter by pure and impure ruthenium red. Mol. Cell Biochem., 1994. [DOI | PubMed]
- C. Sun, L. Zhu, H. Qin, H. Su, J. Zhang, S. Wang, X. Xu, Z. Zhao, G. Mao, J. Chen. Inhibition of mitochondrial calcium uptake by Ru360 enhances the effect of 1800 MHz radio-frequency electromagnetic fields on DNA damage. Ecotoxicol. Environ. Saf., 2023. [DOI | PubMed]
- A.Y. Abramov, M.R. Duchen. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim. Biophys. Acta., 2008. [DOI | PubMed]
- N. Xie, C. Wu, C. Wang, X. Cheng, L. Zhang, H. Zhang, Y. Lian. Inhibition of the mitochondrial calcium uniporter inhibits Aβ-induced apoptosis by reducing reactive oxygen species-mediated endoplasmic reticulum stress in cultured microglia. Brain Res., 2017. [DOI | PubMed]
- D. Shehwar, S. Barki, A. Aliotta, L. Veuthey, C.D. Bertaggia, L. Alberio, M.R. Alam. Inhibition of mitochondrial calcium transporters alters adp-induced platelet responses. Mol. Biol. Rep., 2024. [DOI | PubMed]
- N. Kon, M. Murakoshi, A. Isobe, K. Kagechika, N. Miyoshi, T. Nagayama. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov., 2017. [DOI | PubMed]
- M.G. Di, F. Vallese, B. Jourde, C. Bergsdorf, M. Sturlese, M.A. De, V. Techer-Etienne, D. Haasen, B. Oberhauser, S. Schleeger. A High-Throughput Screening Identifies MICU1 Targeting Compounds. Cell Rep., 2020. [PubMed]
- F. Holze, P. Vizeli, F. Müller, L. Ley, R. Duerig, N. Varghese, A. Eckert, S. Borgwardt, M.E. Liechti. Distinct acute effects of LSD.; MDMA.; and D-amphetamine in healthy subjects. Neuropsychopharmacology, 2020. [DOI | PubMed]
- G. Costa, K. Gołembiowska. Neurotoxicity of MDMA: Main effects and mechanisms. Exp. Neurol., 2022. [DOI | PubMed]
- J.P. Capela, F.D. Carvalho. A review on the mitochondrial toxicity of “ecstasy” (3, 4-methylenedioxymethamphetamine, MDMA). Curr. Res. Toxicol., 2022. [DOI | PubMed]
- D.J. Barbosa, J.P. Capela, R. Feio-Azevedo, A. Teixeira-Gomes, M. Bastos, F. Carvalho. Mitochondria: Key players in the neurotoxic effects of amphetamines. Arch. Toxicol., 2015. [DOI | PubMed]
- X.R. Jiang, G. Dryhurst. Inhibition of the alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase complexes by a putative aberrant metabolite of serotonin, tryptamine-4,5-dione. Chem. Res. Toxicol., 2002. [DOI | PubMed]
- K. Natarajaseenivasan, B. Cotto, S. Shanmughapriya, A.A. Lombardi, P.K. Datta, M. Madesh, J.W. Elrod, K. Khalili, D. Langford. Astrocytic metabolic switch is a novel etiology for Cocaine and HIV-1 Tat-mediated neurotoxicity. Cell Death Dis., 2018. [DOI | PubMed]
- D. Balu, J. Ouyang, R.A. Parakhia, S. Pitake, R.S. Ochs. Ca2+ effects on glucose transport and fatty acid oxidation in L6 skeletal muscle cell cultures. Biochem. Biophys. Rep., 2016. [PubMed]
- J. Mao, B. Sung, R.R. Ji, G. Lim. Neuronal apoptosis associated with morphine tolerance: Evidence for an opioid-induced neurotoxic mechanism. J. Neurosci., 2002. [DOI | PubMed]
- S.W. Ko, Y. Jia, H. Xu, S.J. Yim, D.H. Jang, Y.S. Lee, M.G. Zhao, H. Toyoda, L.J. Wu, T. Chatila. Evidence for a role of CaMKIV in the development of opioid analgesic tolerance. Eur. J. Neurosci., 2006. [DOI | PubMed]
- M. Ivshina, P. Lasko, J.D. Richter. Cytoplasmic polyadenylation element binding proteins in development.; health.; and disease. Annu. Rev. Cell Dev. Biol., 2014. [DOI | PubMed]
- K. Takahashi, H. Yi, J. Gu, D. Ikegami, S. Liu, T. Iida, Y. Kashiwagi, C. Dong, T. Kunisawa, S. Hao. The mitochondrial calcium uniporter contributes to morphine tolerance through pCREB and CPEB1 in rat spinal cord dorsal horn. Br. J. Anaesth., 2019. [DOI | PubMed]
- P. Chiu, R. Karler, C. Craven, D.M. Olsen, S.A. Turkanis. The influence of delta9-tetrahydrocannabinol.; cannabinol and cannabidiol on tissue oxygen consumption. Res. Commun. Chem. Pathol. Pharmacol., 1975. [PubMed]
- B. Hempel, Z.X. Xi. Receptor mechanisms underlying the CNS effects of cannabinoids: CB1 receptor and beyond. Adv. Pharmacol., 2022. [PubMed]
- D. Jimenez-Blasco, A. Busquets-Garcia, E. Hebert-Chatelain, R. Serrat, C. Vicente-Gutierrez, C. Ioannidou, P. Gómez-Sotres, I. Lopez-Fabuel, M. Resch-Beusher, E. Resel. Glucose metabolism links astroglial mitochondria to cannabinoid effects. Nature, 2020. [DOI | PubMed]
- R. Serrat, A. Covelo, V. Kouskoff, S. Delcasso, A. Ruiz-Calvo, N. Chenouard, C. Stella, C. Blancard, B. Salin, F. Julio-Kalajzić. Astroglial ER-mitochondria calcium transfer mediates endocannabinoid-dependent synaptic integration. Cell Rep., 2021. [DOI | PubMed]
- E. Soria-Gomez, A.C.P. Zottola, Y. Mariani, T. Desprez, M. Barresi, I. Bonilla-Del Río, C. Muguruza, M. Le Bon-Jego, F. Julio-Kalajzić, R. Flynn. Subcellular specificity of cannabinoid effects in striatonigral circuits. Neuron, 2021. [DOI | PubMed]
- S. Marchi, M. Corricelli, A. Branchini, S. Missiroli, G. Morciano, M. Perrone, M. Ferrarese, C. Giorgi, M. Pinotti, L. Galluzzi. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca2+ levels and tumor growth. EMBO J., 2019. [DOI | PubMed]
- A. Flavin, P. Azizi, N. Murataeva, K. Yust, W. Du, R. Ross, I. Greig, T. Nguyen, Y. Zhang, K. Mackie. CB1 Receptor Negative Allosteric Modulators as a Potential Tool to Reverse Cannabinoid Toxicity. Molecules, 2024. [DOI | PubMed]
- I.N. Krasnova, J.L. Cadet. Methamphetamine toxicity and messengers of death. Brain Res. Rev., 2009. [DOI | PubMed]
- B. Shen, R. Zhang, G. Yang, Y. Peng, Q. Nie, H. Yu, W. Dong, B. Chen, C. Song, Y. Tian. Cannabidiol prevents methamphetamine-induced neurotoxicity by modulating dopamine receptor D1-mediated calcium-dependent phosphorylation of methyl-CpG-binding protein 2. Front. Pharmacol., 2022. [DOI | PubMed]
- J. Bustamante, L. Acosta, A.G. Karadayian, S. Lores-Arnaiz. Ketamine induced cell death can be mediated by voltage dependent calcium channels in PC12 cells. Exp. Mol. Pathol., 2019. [DOI | PubMed]
- A. Czerniczyniec, A.G. Karadayian, J. Bustamante, S. Lores-Arnaiz. Ketamine treatment affects hippocampal but not cortical mitochondrial function in prepubertal rats. Int. J. Dev. Neurosci., 2020. [DOI | PubMed]
- W.M. Han, X.B. Hao, Y.X. Hong, S.S. Zhao, X.C. Chen, R. Wang, Y. Wang, G. Li. NMDARs antagonist MK801 suppresses LPS-induced apoptosis and mitochondrial dysfunction by regulating subunits of NMDARs via the CaM/CaMKII/ERK pathway. Cell Death Discov., 2023. [DOI | PubMed]
- V. Haynes, C. Giulivi. Calcium-Dependent Interaction of Nitric Oxide Synthase with Cytochrome c Oxidase: Implications for Brain Bioenergetics. Brain Sci., 2023. [DOI | PubMed]
- Y. Li, R. Ding, X. Ren, G. Wen, Z. Dong, H. Yao, Y. Tan, H. Yu, X. Wang, X. Zhan. Long-term ketamine administration causes Tau protein phosphorylation and Tau protein-dependent AMPA receptor reduction in the hippocampus of mice. Toxicol. Lett., 2019. [DOI | PubMed]