Hybrid Molecules Containing Methotrexate, Vitamin D, and Platinum Derivatives: Synthesis, Characterization, In Vitro Cytotoxicity, In Silico ADME Docking, Molecular Docking and Dynamics
Abstract
Designing hybrid‐based drugs is one promising strategy for developing effective anticancer drugs that explore combination therapy to enhance treatment efficacy, overcome the development of drug resistance, and lower treatment duration. Bisphosphonates and Vitamin D are commonly administered drugs for the treatment of bone diseases and the prevention of bone metastases. Platinum‐based and methotrexate are widely used anticancer drugs in clinics. However, their use is hampered by adverse side effects. Hybrid‐based compounds containing either bisphosphonate, vitamin D, platinum‐based, or methotrexate were synthesized and characterized using FTIR, 1H‐,31P, 13C‐NMR, and UHPLC‐HRMS which confirmed their successful synthesis. The hydroxyapatite bone binding assay revealed a promising percentage binding affinity of the bisphosphonate hybrid compounds. In vitro cytotoxicity assays on MCF‐7 and HT‐29 cell lines revealed a promising cytotoxic effect of hybrid 19 at 50 and 100 μg/mL on HT‐29 and hybrid 15 on MCF‐7 at 100 μg/mL. Molecular docking and dynamics simulation analysis revealed a binding affinity of −9.70 kcal/mol for hybrid 15 against Human 3 alpha‐hydroxysteroid dehydrogenase type 3, showing its capability to inhibit Human 3 alpha‐hydroxysteroid dehydrogenase type 3. The Swiss ADME, ProTox‐II, GUSAR (General Unrestricted Structure‐Activity Relationships), and molecular docking and dynamics studies revealed that these compounds are promising anticancer compounds.
Article type: Research Article
Keywords: Cytotoxicity, Methotrexate, Vitamin D, Molecular docking
Affiliations: Department of Chemistry University of Fort Hare Alice Campus 5700 Alice, Eastern Cape South Africa; Department of Biochemistry University of Johannesburg Kingsway Campus, Auckland Park 2006 Johannesburg South Africa; School of Chemistry Mass Spectrometry Division University of Witwatersrand 2050 Johannesburg South Africa; Department of Chemistry University of Johannesburg Auckland Park Campus 2006 Johannesburg South Africa; Wits Advanced Drug Delivery Platform Research Unit Department of Pharmacy and Pharmacology School of Therapeutic Science Faculty of Health Sciences University of the Witwatersrand Johannesburg South Africa; Department of Chemistry and Industrial Chemistry Bowen University Iwo, Osun State Nigeria; Research Centre for Synthesis and Catalysis Department of Chemical Sciences University of Johannesburg Auckland Park 2006 Johannesburg South Africa; Laser Research Centre, Faculty of Health Sciences University of Johannesburg Doornfontein Campus 2028 Johannesburg South Africa
License: © 2024 The Author(s). Chemistry & Biodiversity published by Wiley-VHCA AG CC BY 4.0 This is an open access article under the terms of the http://creativecommons.org/licenses/by/4.0/ License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Article links: DOI: 10.1002/cbdv.202400373 | PubMed: 39278836 | PMC: PMC11741164
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (2.5 MB)
Introduction
The bone is one of the most frequent sites to which most cancers metastasize, and this is the major cause of death in cancer patients.[ref. cbdv202400373-bib-0001, ref. cbdv202400373-bib-0002] There is no cure for bone metastases, and the available treatments are administered to prevent bone metastases, improve the strength of the bones, and reduce pain. Drugs used to prevent bone metastases are bisphosphonates (BPs), vitamin D, demosumab, etc. BPs exert antitumor effects via inhibition of the cycle of increased osteolysis and tumor growth, thereby delaying bone lesion progression.ref. cbdv202400373-bib-0003 Hybrid compounds containing BPs with promising anticancer activities have been reported.[ref. cbdv202400373-bib-0004, ref. cbdv202400373-bib-0005] Vitamin D has also been reported to alter skeletal metastases.ref. cbdv202400373-bib-0006 Hybrid compounds containing vitamin D have been synthesized and are effective against cancer.[ref. cbdv202400373-bib-0007, ref. cbdv202400373-bib-0008]
Inhibiting the progression of metastases and preventing osteoclast activity improves the quality of life in cancer patients.ref. cbdv202400373-bib-0009 Thus, the development of remedies for the treatment of bone metastases remains a main clinical challenge. Platinum (Pt)‐based drugs are widely used in clinics for the treatment of cancer and are known to induce toxicity, resulting from their non‐specificity, which limits their use for long‐term treatment. Thus, targeted delivery of Pt‐based drugs has been reported to overcome the problems of their non‐specificity.ref. cbdv202400373-bib-0010 Nadar et al. prepared radioactive Pt‐bisphosphonates (195mPt‐BPs) complexes to enable targeted delivery of 195mPt to bones of high metabolic activity.ref. cbdv202400373-bib-0011 Pt‐BP demonstrated a 4.5‐fold higher affinity for bone than the Pt complexes without BPs.ref. cbdv202400373-bib-0011 On the other hand, Pt‐BP complexes formed fewer Pt‐DNA adducts than the BPs‐free platinum complexes, which indicates that the in vivo release of Pt from Pt‐BP complexes will proceed slowly, a feature useful in treating bone metastases with reduced side effects.ref. cbdv202400373-bib-0011
Methotrexate (MTX) is an anti‐metabolite derived from folic acid and is the most active remedy for t treating several types of cancer.ref. cbdv202400373-bib-0012 It has also been utilized for the management of acute leukaemia, pulmonary, osteogenic sarcoma, and epidermoid carcinoma.ref. cbdv202400373-bib-0013 The clinical application of MTX is limited due to its short half‐life, poor solubility, and rapid absorption from the gastrointestinal tract.ref. cbdv202400373-bib-0013 To prevent and reduce the abovementioned limitations, MTX is administered in combination with other drugs for the treatment of numerous neoplasms, such as acute myeloid leukaemia, bladder cancer, acute lymphoblastic leukaemia, osteosarcomas, meningeal leukaemia, lymphoma, breast cancer, etc. Hybrid compounds containing MTX have been reported to have promising anticancer activities. Kmiecik et al. synthesized MTX‐epirubicin conjugates.ref. cbdv202400373-bib-0014 However, the cytotoxic effect on FR‐positive MV‐4‐ 11, LoVo, and LoVo/Dx cell lines was promising but lower than the free drugs, MTX and epirubicin, suggesting the need for a cleavable linker between both drugs for the enhanced cytotoxic effect of the hybrid molecules.ref. cbdv202400373-bib-0014 Cai et al. synthesized MTX‐diosgenin hybrid compounds with significant cytotoxic effects on MDA‐MB‐231 cell lines than the free MTX.ref. cbdv202400373-bib-0015 The lengths of the linker influenced their cytotoxic effects and the hybrid with a disulfide linker was the most cytotoxic.ref. cbdv202400373-bib-0015 Nadhum et al. prepared an MTX‐silibinin hybrid with higher anticancer activity against the HEP‐2 cell line (Larynx carcinoma) than MTX or silibinin.ref. cbdv202400373-bib-0016 Semenenko et al. prepared MTX‐betulonic acid hybrids with different linkers. However, the hybrids’ capability to penetrate Caco‐2 cells was not significant when compared to methotrexate, resulting from the increased lipophilic nature of the hybrids.ref. cbdv202400373-bib-0017
Hybrid compounds are useful for improving therapeutic effects, decreasing unwanted side effects, reducing the risk of developing drug resistance, promoting interaction with two or more targets, lowering the occurrence of drug‐drug adverse effects, and improving patient compliance. Due to the advantages of hybrid molecules, researchers have developed hybrid compounds for the treatment and management of numerous human diseases.[ref. cbdv202400373-bib-0018, ref. cbdv202400373-bib-0019] The hybrid molecule can be transported to numerous targets involved in cancer cell proliferation and exhibit similar pharmacokinetic profiles, decreasing the occurrence of drug‐drug interactions.[ref. cbdv202400373-bib-0020, ref. cbdv202400373-bib-0021] To overcome the challenges of the current drugs and enhance their therapeutic outcomes, BPs, MTX, and Vitamin D compounds were explored as precursors for the synthesis of hybrid compounds. Hybridizing BPs with known anticancer drugs has been reported to result in synergistic anticancer effects with the potential to enhance the treatment outcomes of the BPs.ref. cbdv202400373-bib-0022 Vitamin D, which is beneficial for healthy bones, is ideal for hybridizing with other anticancer drugs for improved bone remodelling.ref. cbdv202400373-bib-0023 The hybridization of MTX with other anticancer agents was found to be effective and a promising therapeutic agent in the treatment of cancer patients.ref. cbdv202400373-bib-0024 Based on the reported therapeutic efficacy of hybrid‐based compounds, hybrid compounds containing BP, vitamin D, and methotrexate were synthesized and evaluated as potential anticancer agents against bone metastasis. The compounds were characterized using FTIR, 1H‐,31P, 13C‐NMR, UHPLC‐HRMS, and EDX, followed by in vitro hydroxyapatite bone binding assay and cytotoxicity, prediction of ADME and toxicity, molecular docking, and dynamics studies.
Experimental
Materials
All the reagents and solvents used in this work were of analytical grade and were purchased from Merck Chemicals, South Africa. They were used without further purification.
Characterization
The melting points of the synthesized compounds were determined using a Stuart melting point apparatus model SMP 11, LabFreind, South Africa. The reactions were monitored by Thin Layer Chromatography. The synthesized compounds were purified by gravity column chromatography using silica gel adsorbent with a particle size range of 40–63 μm. FTIR was performed using a Perkin Elmer spectrum FTIR spectrometer in the range of 4000–400 cm−1 to study the functional groups present in the synthesized compounds. The mass spectra of the synthesized compounds hybrid compounds were recorded on a Bruker Compact Q‐TOF mass spectrometer Bruker Daltonics. The 1H, 13C, and 31P NMR spectra of the compounds were recorded on a Bruker ultrashield 500 NMR spectrometer. The NMR analysis of the synthesized hybrid compounds was performed using either deuterium oxide (D2O), deuterated chloroform (CDCl3), or deuterated dimethyl sulfoxide (DMSO‐d6 ).
Synthesis
Synthesis of Trans‐[Pt(DACH)(Pamidronate)]
Trans‐[Pt(DACH)(H2O)2](NO3)2 (3) complex was prepared by a known protocol by first synthesizing trans‐cyclohexane‐1,2‐diamine (DACH) (1), (trans‐cyclohexane‐1,2‐diamine)platinum(II) chloride trans‐[PtCl2(DACH)] (2)ref. cbdv202400373-bib-0025 followed by reaction with silver nitrateref. cbdv202400373-bib-0026 (Scheme 1).
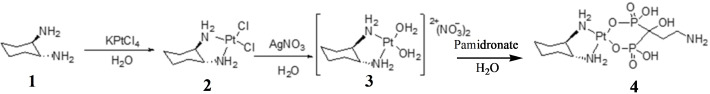
Trans‐[Pt(DACH)(Pamidronate)] (4) was prepared by a modified methodref. cbdv202400373-bib-0027 in which 3 (100 mg, 0.216 mmol, 1.00 equiv) was dissolved in 10 mL of distilled water followed by the addition of pamidronate (60.2 mg, 0.216 mmol, 1.00 equiv). The reaction mixture was stirred at room temperature for 3 days, and the flask was covered with aluminum foil to protect the reaction from light. The reaction mixture was concentrated, and acetone was added, resulting in the formation of solids which were filtered and further washed with acetone and dried under vacuum to afford yellow product (4) (amount=118 mg; % yield=73; mp=220–221 °C, HRMS: m/z calcd. for [C9H22N3O7P2Pt+H]+: 541.1, found: 541.1). FTIR (ATR, cm−1): v=2941 and 2862 sp3(C−H), 1359 (C−N), 1044–982 (P=O), 910 (P−OH), 540 (Pt−N), and 470 (Pt−O) cm−1. 1H‐NMR (500 MHz, D2O, ppm): δ=3.81–3.76 (m, 1H), 3.45 (dd, 2H), 3.35 (dd, 2H), 3.26 (s, 1H), 2.96 (t, 2H), and 1.05 (d, J=5.0 Hz, 2H). 31P‐NMR (202 MHz, D2O, ppm): δ=19.21 (Pt), 13C‐NMR (125 MHz, D2O, ppm): δ=67.93 (C5 & C4), 66.60 (C11), 39.19 (C21), 32.94 (C6 & C3), 27.23 (C2 & C1), and 17.98 (C20). The numbering of the carbons is reported in Figure S1d.
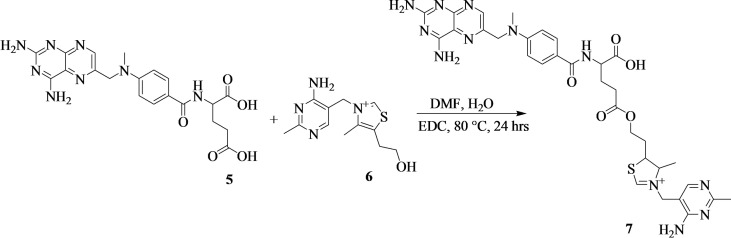
Synthesis of Methotrexate‐Vitamin B1
The synthesis of methotrexate‐vitamin B1 (7) was prepared by a modified method[ref. cbdv202400373-bib-0028, ref. cbdv202400373-bib-0029] (Scheme 2). Methotrexate (5) (500 mg, 1.10 mmol, 1.00 equiv) was dissolved in dried dimethylformamide (8 mL) and vitamin B1 (6) (292 mg, 1.10 mmol, 1.00 equiv) was dissolved in distilled water (2 mL). The mixture was heated at 80 °C followed by the addition of 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide (232 mg, 1.21 mmol, 1.10 equiv) and continuous of the reaction mixture for 24 hours. The compound was purified by column chromatography (MeOH:Hexane:H2O (3 : 2 : 1) to afford brown powder (amount=410 mg; % yield=47; mp=105–106 °C; Rf=0.29 (MeOH:Hexane:H2O (3 : 2 : 1); HRMS: m/z calcd. for [C32H39N12O5S+H]+: 703.2, found: 703.2. FTIR (ATR, cm−1): v=3346–3213 (N−H), 2830 (C−H), 1607 (C=O), 1514 (C=C/C=N), and 1210 (C−O) cm−1. 1H‐NMR (500 MHz, d6‐DMSO, ppm): δ=8.78 (s, 1H), 8.66 (s, 1H), 7.75 (d, J=5.0 Hz, 2H), 6.83 (d, J=5.0 Hz, 2H), 4.84 (s, 2H), 4.36 (s, 2H), 3.56 (t, 2H), 3.23 (s, 3H), 2.87 (t, 1H), 2.72 (s, 2H), 2.30 (s, 3H), and 0.98 (t, 3H). 13C‐NMR (125 MHz, d6‐DMSO, ppm): δ=174.36 (C27), 174.13 (C29), 166.84 (C20), 163.16 (C46), 158.88 (C44 & C1), 151.32 (C48), 150.62 (C3), 149.46 (C13), 148.97 (C5), 129.42 (C18 & C16), 128.94 (C4), 122.29 (C17), 121.75 (C19 & C15), 111.59 (C43) 61.83 (C40), 55.29 (C34), 54.97 (C23), 52.26 (C41), 42.46 (C11), 36.75 (C36), 34.54 (C14), 30.93 (C35), 29.82 (C25), 25.67 (C24), 16.03 (C49), and 15.15 (C42). The numbering of the carbons is reported in Figure S2c.
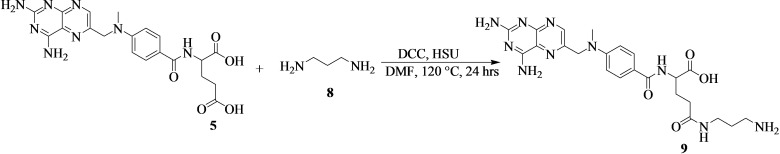
Synthesis of Methotrexate‐Propane‐1,3‐Diamine
Methotrexate‐propane‐1,3‐diamine (9) was prepared as shown in Scheme 3. A mixture of 5 (5000 mg, 11.0 mmol, 1.00 equiv) and propane‐1,3‐diamine (8) ((0.9 mL) in dried dimethylformamide (20 mL) was stirred at room temperature. N,N′‐dicyclohexylcarbodiimide (2496 mg, 12.1 mmol, 1.10 equiv), and N‐hydroxysuccinimide (1876 mg, 12.1 mmol, 1.10 equiv) were added to the mixture, and the solution was heated at 120 °C for 24 hours. The reaction mixture was monitored by thin‐layer chromatography using MeOH:Hexane (6 : 4). The reaction was cooled, followed by the purification by column chromatography (MeOH:Hexane (6 : 4) and concentration on a roti‐evaporator to afford yellow crystals (9) (4.2 g and % yield of 72 %).
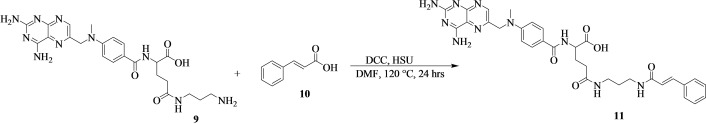
Synthesis of Methotrexate‐Propane‐1,3‐Diamine‐Cinnamic Acid
In a round bottom flask, 9 (500 mg, 0.979 mmol, 1.00 equiv) was dissolved in dried dimethylformamide (10 mL). Cinnamic acid (10) (145 mg, 0.979 mmol, 1.00 equiv) was added to the solution, followed by the addition of N,N′‐dicyclohexylcarbodiimide (222 mg, 1.08 mmol, 1.10 equiv), and N‐hydroxysuccinimide (167 mg, 1.08 mmol, 1.10 equiv). The reaction mixture was heated at 120 °C for 24 hours. The reaction was monitored by thin‐layer chromatography until the reaction was complete. The compound was purified by column chromatography (DCM:EtOAc (7 : 3) to afford a yellow powder product (11) (Scheme 4) (amount=439 mg; % yield=68; mp=100–103 °C; Rf=0.62 (DCM:EtOAc (7 : 3); HRMS: m/z calcd. for [C32H36N10O5+H]+: 641.3, found: 641.3). FTIR (ATR, cm−1): v=2933–2903 sp3(C−H), 1700 (C=O), 1623–1617 (HNC=O), 1521 cm−1 (Ar−C), 1224 (C−N), and 989 (C−H) cm−1. 1H‐NMR (500 MHz, CDCl3, ppm): δ=8.13 (s, 1H), 7.76 (d, J=15 Hz, 1H), 7.71 (s, 1H), 7.68 (s, 1H), 7.55 (d, J=10 Hz, 2H), 6.92 (d, J=10 Hz, 2H), 6.48 (d, J=15 Hz, 1H), 4.23 (s, 2H), 3.90 (s, 2H), 3.48 (t, 2H), 2.91 (s, 3H), 1.37 (q, 2H), and 1.16 (q, 2H). 13C‐NMR (125 MHz, CDCl3, ppm): δ=171.04 (C27), 165.07 (C38, C29, & C20), 146.52 (C1), 140.80 (C3), 134.98 (C13), 134.22 (C5), 130.55 (C9), 129.51 (C8), 128.91 (C42), 128.75 (C46, C44, C18 & C16), 128.27 (C45), 127.32 (C48, 43, C19 & C15), 121.12 (C17), 117.58 (C39) 48.44 (C11), 33.22 (C14), 31.02 (C36 & 34), 29.67 (C25), 25.56 (C35), and 24.84 (C24). The numbering of the carbons is reported in Figure S3c.
Synthesis of Methotrexate‐Propane‐1,3‐Diamine‐Oleanolic Acid
A mixture of 9 (500 mg, 0.979 mmol, 1.00 equiv) and oleanolic acid (12) (447 mg, 0.979 mmol, 1.00 equiv) was dissolved in 10 mL of dried dimethylformamide (Scheme 5). N,N′‐dicyclohexylcarbodiimide (222 mg, 1.08 mmol, 1.10 equiv), and N‐hydroxysuccinimide (167 mg, 1.08 mmol, 1.10 equiv) were added to the mixture. The reaction mixture was heated at 120 °C for 24 hours. The reaction mixture was purified by column chromatography (Hexane:EtOAc 7 : 3) to obtain a white powder (13) (amount=689 mg; % yield=72; mp=210–215 °C; Rf=0.63 (Hexane:EtOAc 7 : 3); HRMS: m/z calcd. for [C53H76N10O6+H]+: 948.6, found: 948.7. FTIR (ATR, cm−1) v=3586 (O−H), 3339 (RO=C−NHR), 2930 and 2899 sp3(C−H), 1466 (Ar−C/C=C), 1629–1694 (C=O), 1214 (C−N), and 1057 (C−H) cm−1. 1H‐NMR (500 MHz, CDCl3, ppm): δ=8.53 (s, 1H), 7.55 (d, J=10 Hz, 1H), 7.37 (s, 1H), 7.14 (s, J=10 Hz, 1H), 5.59 (s, 1H), 5.37 (s, 2H), 5.29 (s, 4H), 4.23 (t, 2H), and 1.62–0.78 (m, 20H). 13C‐NMR (125 MHz, CDCl3, ppm): δ=182.68 (C27), 172.74 (C38, C29, & C20), 169.31 (C1), 157.11 (C3), 143.66 (C13), 142.55 (C5), 139.24 (C9), 123.28 (C8), 122.56 (C18 & C16), 114.03 (C19 & C15), 79.02 (C64), 55.25 (C23), 47.66 (C59), 46.49 (C11), 41.64 (C43), 39.31 (C41), 38.76 (C65), 37.09 (C36 & 34), 33.76 (C56), 30.66 (C61), 29.67 (C63), 28.10 (C68), 27.20 (C42 & C40), 25.91 (C48), 25.57 (C50 & C51), 24.84 (C67), 23.48 (C58), 17.15 (C60), 15.53 (C70 & C71), and 15.30 (C69). The numbering of the carbons is reported in Figure S4c.
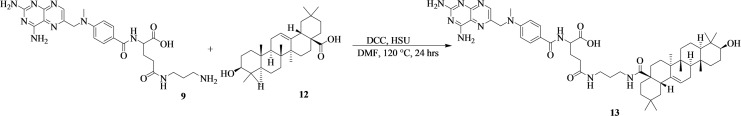
Synthesis Of Methotrexate‐Propane‐1,3‐Diamine‐Artesunate
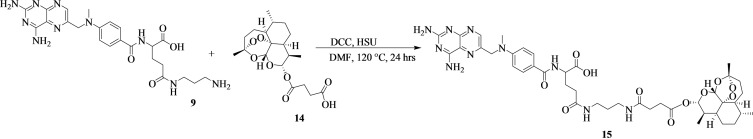
The reaction scheme showing the synthesis of methotrexate‐propane‐1,3‐diamine‐artesunate (15) is Scheme 6. 500 mg of 9 (0.979 mmol, 1.00 equiv) was dissolved in 10 mL of dried dimethylformamide. Artesunate (14) (376 mg, 0.979 mmol, 1.00 equiv) was added in a reaction followed by N,N′‐dicyclohexylcarbodiimide (222 mg, 1.08 mmol, 1.10 equiv), and N‐hydroxysuccinimide (167 mg, 1.08 mmol, 1.10 equiv). The reaction was heated at 120 °C, for 24 hours and monitored by thin‐layer chromatography. The reaction mixture was purified by column chromatography (Hexane:MeOH:EtOAc (6 : 2 : 2)) to obtain a brown viscous oil (amount=420 mg; % yield=48; Rf=0.13 (H:6; MeOH:2; EtOAc:2); HRMS: m/z calcd. for [C42H56N10O11+H]+: 876.4, found: 876.4). FTIR (ATR, cm−1) v=2904–2936 sp3(C−H), 1639 (HNC=O), 1717 (C=O), 1543 (C=C/C=N), 1218 (C−N), and 1025 (C−H) cm−1. 1H‐NMR (500 MHz, CDCl3, ppm): δ=8.11 (s, 1H), 6.49 (d, J=10 Hz, 1H), 6.01 (s, 1H), 5.71 (s, 1H), 4.25 (s, 2H), 3.41 (s, 2H), 2.71 (s, 3H), and 1.92–1.12 (m, 8H). 13C‐NMR (125 MHz, CDCl3, ppm): δ=175.06 (C27), 172.14 (C66) 171.86 (C61, C29, & C20), 164.09 (C1), 160.57 (C3), 157.96 (C13), 157.46 (C5), 109.20 (C39), 99.64 (C50), 82.41 (C41), 49.42 (C23), 47.16 (C11), 44.64 (C45), 42.42 (C14), 36.16 (C36 & 34), 34.05 (C38), 33.75 (C43), 32.97 (C47), 32.75 (C51), 25.66 (C62), 25.41 (C25), 25.00 (C48), 24.86 (C24), 24.69 (C53), 23.92 (C55), and 12.57 (C57). The numbering of the carbons is reported in Figure S5c.
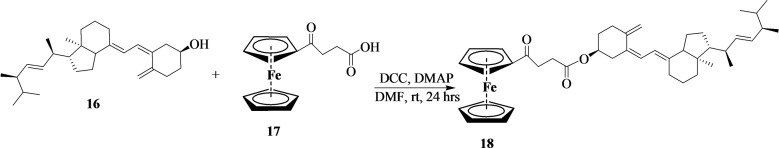
Synthesis of Vitamin D2‐4‐Ferrocene‐Ketobutanoic Acid
4‐Ferrocene ketobutanoic acid was synthesized according to our previous reports.[ref. cbdv202400373-bib-0004, ref. cbdv202400373-bib-0030, ref. cbdv202400373-bib-0031] Vitamin D2 (16) (300 mg, 0.76 mmol, 1.00 equiv) was dissolved in 10 mL of dried dimethylformamide. The solution was stirred at 0 °C for 30 minutes, and N,N′‐dicyclohexylcarbodiimide (171 mg, 0.83 mmol, 1.10 equiv) was added. 4‐Ferrocene ketobutanoic acid (17) (239 mg, 0.76 mmol, 1.00 equiv) was also added to the reaction mixture, followed by the addition of 4‐dimethylaminopyridine (92.4 mg, 0.76 mmol, 1.00 equiv). The reaction was stirred at room temperature for 24 hours and concentrated on a rotatory evaporator. The compound was then purified by column chromatography (DCM: EtOAc (7 : 3)) to obtain an orange powder (18) (Scheme 7) (amount=346 mg; % yield=64; mp=140–142 °C; Rf=0.80 (DCM:7; AtOAc:3); HRMS: m/z calcd. for [C44H62FeO3+H]+: 694.4, found: 695.5). FTIR (ATR, cm−1) v=2931 and 2910 sp3(C−H), 1625–1626 (C=O), 1577 (C=C), 1195–1244 (C−O), and 645 (C−Fe) cm−1. 1H‐NMR (500 MHz, CDCl3, ppm): δ=7.58 (s, 1H), 4.83 (s, 2H), 4.55 (s, 2H), 4.30 (s, 5H, C5H5), 4.18 (s, 2H), 3.26 (t, 2H), 2.72 (t, 2H), and 2.00–1.11 (m, 20H). 13C‐NMR (125 MHz, CDCl3, ppm): δ=204.26 (C14), 170.98 (17), 156.79 (C28 & C25), 154.43 (C35 & C34), 72.41, (C11 & C5), 70.06 (C13, C10, C9, C4, C3, C2, C1), 69.26 (C20), 54.89 (C32), 49.99 (C36), 49.13 (C42), 35,20 (C24), 33.95 (C33), 32.45 (C44), 30.69 (C38), 29.67 (C46), 28.50 (C22), 26.14 (C16), 25.63 (C30), 25.42 (C45), 24.93 (C43), 24.68 (C40 & C39), 22.66 (C41 & C37), and 14.07 (C6 & C7). The numbering of the carbons is reported in Figure S6c.
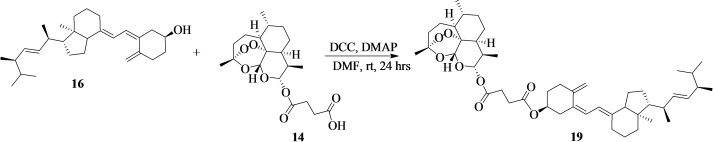
Synthesis of Vitamin D2‐Artesunate
The reaction of vitamin D2‐artesunate (19) is presented in Scheme 8. In a round bottom flask, 16 (300 mg, 0.76 mmol, 1.00 equiv) was dissolved in dried dimethylformamide (10 mL). The reaction was placed in an ice bath for 30 minutes and N,N′‐dicyclohexylcarbodiimide (171 mg, 0.83 mmol, 1.10 equiv) was added. 14 (290 mg, 0.76 mmol, 1.00 equiv) was also added, followed by the addition of 4‐dimethylaminopyridine (92.4 mg, 0.76 mmol, 1.00 equiv). The reaction mixture was stirred at room temperature overnight. The mixture was concentrated on a rotary evaporator, and the reaction mixture was purified by column chromatography (MeOH:Hexane 6 : 4) to obtain a brown viscous compound. Amount=383 mg; % yield=65; Rf=0.85 (MeOH:6; H:4); HRMS: m/z calcd. for [C47H70O8+H]+: 762.5, found: 762.5. FTIR (ATR, cm−1) v=2905–2932 sp3(C−H), 1714 (C=O), 1628–1578 (C=C), 3342 (O−H), and 1020 (C−O) cm−1. 1H‐NMR (500 MHz, CDCl3, ppm): δ=7.14 (d, J=10 Hz, 1H), 6.20 (s, 1H), 5.78 (d, J=10 Hz, 1H), 5.71 (s, 1H), 5.44 (s, 2H), 3.23–3.18 (m, 1H), 2.90 (s, 2H) 2.14 (s, 2H), and 1.92–0.86 (m, 20H). 13C‐NMR (125 MHz, CDCl3, ppm): δ=171.82 (C55 & C51), 157.08 (C28), 153.93 (C9), 147.09 (C6), 135.00 (C16), 124.45 (C15), 123.93 (C7), 119.09 (C8), 109.19 (C29), 104.47 (C31), 99.64 (C42), 91.56 (C33), 82.41 (34), 80.09 (C1), 56.52 (C10), 49.19 (C13), 45.24 (C17), 44.64 (C23), 42.44 (C5), 37.29 (C14), 35.35 (C37), 33.86 (C25), 33.47 (C30), 32.75 (C19), 31.41 (C39), 29.66 (C27), 26.19 (C4), 25.60 (C43), 24.89 (C54 & C53), 23.94 (C11), 23.52(C12), 22.02 (C26), 20.17 (C21 & C20), 18.54 (C45), 12.59 (C22), 11.98 (C18), and 11.01 (C49). The numbering of the carbons is reported in Figure S7c.
Synthesis of Vitamin D2‐Cinnamic Acid
Synthesis of vitamin D2‐cinnamic acid (20) is shown in Scheme 9. 16 (300 mg, 0.76 mmol, 1.00 equiv) was dissolved in dried dimethylformamide (10 mL). N,N′‐dicyclohexylcarbodiimide (171 mg, 0.83 mmol, 1.10 equiv) was added followed by the addition of 10 (112 mg, 0.76 mmol, 1.00 equiv), and 4‐dimethylaminopyridine (92.3 mg, 0.76 mmol, 1.00 equiv) to the reaction mixture in an ice bath for 15 minutes. The mixture was then stirred at room temperature for 48 hours. The reaction mixture was concentrated and purified by column chromatography using (DCM:EtOAc (7 : 3)) eluent to afford a cream‐white powder (amount=338 mg; % yield=82; mp=145–146 °C; Rf=0.71 (DCM:EtOAc 7 : 3); HRMS: m/z calcd. for [C37H50O2+H]+: 526.4, found: 527.4. FTIR (ATR, cm−1) v=2910–2930 sp3(C−H), 1707 (C=O), 1600–1645 (C=C), and 1233 (C−O) cm−1. 1H‐NMR (500 MHz, CDCl3, ppm): δ=7.67 (d, J=15 Hz, 2H), 7.48 (s, 2H), 6.78 (s, 1H), 6.74 (s, 1H), and 2.05–0.90 (m, 16H). 13C‐NMR (125 MHz, CDCl3, ppm): δ=166.76 (C30), 154.04 (C9), 143.38 (C28), 139,23 (C6), 134.76 (C16), 130.01 (C33), 128.88 (C37 & C35), 127.90 (C36), 119.52 (C29), 114.03 (C31), 56.23 (C10), 49.87 (C13), 32.79 (C19), 31.91 (C3), 30.97 (C27), 29.67(C11), 26.30 (C12), 25.48 (C26), 25.38 (C22), 24.67(C24), 22.66 (C21 & C20), and 14.04 (C18). The numbering of the carbons is reported in Figure S8c.
Molecular Docking Study
Eight of the synthesized compounds were selected, optimized, refined, and saved in a Molecular Operating Environment (MOE) using molecular operation environment software. The studied receptors (Human Tyrosine Phosphatase (PDB ID: 1wch)ref. cbdv202400373-bib-0032 and human 3‐alpha hydroxysteroid dehydrogenase type 3 (PDB ID: 4xo6)ref. cbdv202400373-bib-0033 were retrieved from the protein data bank. The receptors were prepared for docking and saved in MOE format. The studied compounds were optimized and saved in MOE format for further analysis using induced fit for refinement and triangle matcher for placement.
Molecular Dynamics Calculations
This study involves the use of GROMACS software for the compounds with the highest binding affinity as well as the referenced compounds. Compounds 19 and 15, with the topmost potential to inhibit Human Tyrosine Phosphatase (PDB ID: 1wch) and human 3‐alpha hydroxysteroid dehydrogenase type 3 (PDB ID: 4xo6), were subjected to a molecular dynamic simulation study. This study was accomplished using Charmm36m as a force field which was embedded in GROMACS software[34], and the simulated compounds were solvated using proper water molecules. Counter‐ions were added to the system to obtain ionic concentrations in the simulation system. The molecular dynamic simulation study was done in the NPT ensemble with constant pressure and temperature. The studied molecular dynamic simulation was carried out using 100000 ps.ref. cbdv202400373-bib-0035
Swissadme Prediction Tool
SwissADME is a reliable and open‐access online software that was used to predict some physicochemical properties of the compounds (http://www.swissadme.ch/).
In Silico Toxicity Predictions
In Silico Toxicity predictions of the compounds were studied using free online software, ProTox‐II (http://tox.charite.de/protox_II/) and GUSAR (http://www.pharmaexpert.ru/GUSAR/antitargets.html) for quantitative prediction of LD50 values, the potential routes of administration (i. e., oral, intraperitoneal, intravenous, and subcutaneous) and the environmental toxicity of the synthesized compounds. In silico cardiac toxicity of the compounds was also predicted using the free online web server, http://predherg.labmol.com.br/.
In Vitro Cytotoxicity Evaluation on MCF‐7and HT‐29 Cell Lines
Cell viability was determined using tetrazolium salt (3‐(4, 5‐dimethylthiazol‐2‐yl)‐2,5‐ diphenyltetrazolium bromide) (MTT) assay. Briefly, MCF‐7 cells were seeded in 35 mm cell culture plates at a density of 3×105 cells and incubated for 4 h prior to experimentation. Incubation was performed for 20 h after treatment, in which the medium was replaced with a fresh medium containing tetrazolium salt. The presence of the mitochondrial enzyme succinate dehydrogenase cleaved the tetrazolium salt to give a blue product (formazan). The optical density was read at 590 nm wavelength using Perkin‐Elmer, VICTOR3TM plate reader.
The colon cell line was grown in an incubator until 80 % of cell confluence was reached. Trypan blue was used to check the viability of the cells. The desired concentration of cells used was 1.0×105 for the cell lines, and a total of 100 μL of cells/mL was seeded in the 96 well plates for 24 h. After 24 h of incubation, the cell line was treated with different concentrations of the compound, 100 μg/mL, 50 μg/mL, 25 μg/mL, 12.5 μg/mL, and 6.25 μg/mL. The positive control used was cisplatin at the concentration of 200 μg/mL. The negative control was DMSO with cells (0.1 %) and cells with media only. After 24 h of treatment, 10 μL of AlamarBlue reagent was added in each well in the dark, and the plates were wrapped with aluminium foil as the dye is sensitive to light. It was incubated for 2 h, after which the plates were read by a fluorescence microplate reader (Synergy HT).
Results
FTIR, 1H‐, 31P‐ and 13C‐NMR, Spectrum OF Trans‐[Pt(DACH)(Pamidronate)] (4)
The complex 4 is synthesized for the first time in this study. Table 1 depicts the physical properties of the complex.
Table 1: Physical properties and structure of the complexes.
| Compound | Mass(g) | %Yield | Solubility | Colour | Melting Point (°C) | Structure |
|---|---|---|---|---|---|---|
| [Pt(DACH)(Pamidronate)] | 0.118 | 73 | H2O | yellow | 220–221 |
The FTIR spectrum of 4 (Figure S1a) exhibited a characteristic band pattern, confirming the appearance of the corresponding bisphosphonate ligands in the synthesized platinum complex. The important peaks observed at 2941 and 2862 cm−1 are attributed to C−H stretching. The prominent peak at 1359 cm−1 region is attributed to the bending modes of C−N of the amine. The peaks in the range of 1044–982 cm−1 result from the P=O stretch and the peak at 910 cm−1 is due to the P−OH stretch. Similar findings for bisphosphonates were observed by Alvarez‐Valdes et al., Dlamini et al., and Qiu et al.[ref. cbdv202400373-bib-0027, ref. cbdv202400373-bib-0036, ref. cbdv202400373-bib-0037] The C−O stretch was visible at 1153 cm−1. The appearance of the bands at 540 and 470 cm−1 region is ascribed to Pt−N and Pt−O stretching, confirming the bisphosphonate ligand is coordinated to the platinum complex. Similar findings were reported by Ray et al. for platinum‐based complexes.ref. cbdv202400373-bib-0038 In the 1H‐NMR spectrum of 4, the signals of amino groups were visible at 3.80–3.77 ppm, the multiplet signals due to the aliphatic protons of the phosphonate ligand range between 3.46–3.44 ppm, the signals corresponding to amine and hydroxyl group were at 3.36–3.26 ppm, the protons of diaminocyclohexane ring were visible between 2.96–1.04 ppm (Figure S1b). The coordination of bisphosphonate ligand to platinum via the oxygen atoms of amino bisphosphonate was further confirmed by 31PNMR spectroscopy. The 31PNMR spectrum displayed a characteristic peak at 19.21 ppm (Figure S1c). These chemical shifts confirm the formation of the complexes. 13C‐NMR spectra confirmed the number of carbons in the complex. Carbon signals due to the phosphonates were displayed in a range of 67.93–66.60 ppm, and the carbons due to diaminocyclohexane were visible at 39.19–17.98 ppm (Figure S1d). The UHPLC‐HRMS spectrum of complex 4 exhibited a peak at 541.1 (Figure S1e), which further confirmed the successful synthesis of the compound.
Spectroscopic Analysis of the Hybrid Compounds Containing Methotrexate
The FTIR spectrum of 7 revealed significant peaks for C−H stretch at 2981 cm−1, H−C= stretch at 3191 cm−1, C=C stretch at 1551–1400 cm−1, N−H stretch at 3314 cm−1, C=O stretch at 1611 cm−1, O−H stretch at 3500–2500 cm−1, C−O stretch at 1304–1044 cm−1, and C−S stretch of the thiazole group at 774 cm−1, confirming the successful synthesis of the compound (Figure S2a). El‐Megharbel reported a similar finding for C−S stretching at 773 cm−1 for the thiazole group in a vitamin‐B1 complex.ref. cbdv202400373-bib-0039 The 1H‐NMR spectrum showed a signal at 4.84 ppm ascribed to NH2, the signals at the range of 3.23–0.96 ppm are due to three CH3 (one from MTX and two from Vitamin B1), the signal at 8.66 ppm is attributed to amide N−H and the signals at 7.75–6.82 ppm are ascribed to aromatic protons (Figure S2b). The 13CNMR spectrum revealed the ester O−C=O peak at 174.13 ppm, a crucial functional group on the linker of the hybrid molecule (Figure S2c).
The FTIR spectrum of 11 displayed the following absorption peaks: C−H stretch at 2924 and 2853 cm−1, H−C= stretch at 3068 cm−1, N−H stretch at 3274 cm−1, C=C stretch at 1600–1400 cm−1, C=N stretch at 1329 cm−1, C=O stretch at 1618 cm−1, and C−O stretch at 1218–1082 cm−1 (Figure S3a). Similar bands were visible on MTX derivatives reported by Nemat et al.ref. cbdv202400373-bib-0040
The 1H‐NMR spectra of 11 showed the appearance of the singlet signal at 8.13 ppm (s, 1H) which is ascribed to the C9 pyrazine proton. The presence of the doublet signals at 7.77–7.74 ppm is attributed to the aromatic protons (Figure S3b). The 13CNMR spectrum displayed signals of the carboxylic and amide carbons at 171.04 ppm and 165.07 ppm, respectively (Figure S3c).
The FTIR spectrum of 13 showed characteristic bands for C−H stretch at 2931 and 2832 cm−1, N−H stretch at 3597 and 3288 cm−1, C−O stretch 1205–1056 cm−1, C=N stretch at 1377 cm−1, C=C stretch at 1611–1427 cm−1, and C=O stretch at 1760 and 1674 cm−1 (Figure S4a). 1H‐NMR spectrum of 13 showed signals between 0.79–1.27 ppm which are attributed to the CH3 groups. The presence of a singlet signal at 8.53 ppm (s, 1H) is ascribed to the pyrazine proton, and the doublet signals at 7.56–7.54 ppm and 7.15–7.13 ppm are attributed to the aromatic protons (Figure S4b). The 13C‐NMR spectrum of 13 showed a carboxylic signal at 182.68 ppm and the amide carbon signals at 172.74 ppm, 169.31 ppm, and 157.11 ppm. The signals assigned to the (C=N and C=C) were found at 143.66–139.24 ppm, and the signals of the aromatic carbons were visible between 123.28 and 114.03 ppm (Figure S4c). Similar signals were reported by Tang et al. for oleanolic acid‐based hybrid compounds.ref. cbdv202400373-bib-0041
On the FTIR spectrum of 15, important peaks confirming the successful synthesis of the hybrid molecules were significant for C−H stretch at 2931 and 2858 cm−1, N−H stretch, C=C stretch at 1637–1452 cm−1, C=N stretch at 1377 cm−1, C=O stretch at 1698 cm−1, C−O stretch at 1230–1008 cm−1, O−H stretch at 3314 cm−1, and −COO stretch at 872 cm−1 (Figure S5a). Lawal et al reported COO stretching for the endoperoxide bridge of artesunate at 890–820 cm−1.ref. cbdv202400373-bib-0042 The 1H‐NMR spectrum of 15 showed the presence of a singlet signal at 8.11 ppm ascribed to the pyrazine proton (Figure S5b). The 13C‐NMR spectrum of 15 exhibited a carboxylic signal at 175.06 ppm, a ketone carbon signal at 172.14 ppm, and amide carbon at 171.86, 164.09, and 160.57 ppm. The CH3 and CH2 signals were significant at 1.12–1.17 ppm (Figure S5c). Similar findings were reported by Munnik et al. for ferrocenyl‐artesunate hybrid compounds.ref. cbdv202400373-bib-0043 The UHPLC‐HRMS spectra of the hybrid compounds showed peaks at 703.2, 641.3, 948.7, 876.4, respectively, for 7, 11, 13, and 15, which further confirmed the successful synthesis of the compounds (Figures S2d, S3d, S4d, and S5d).
Spectroscopic Analysis of the Hybrid Compounds Containing Vitamin D2
The FTIR spectra of hybrid compounds containing Vitamin D2 are reported in the Supplementary Figures. The FTIR spectrum of 18 displayed peaks for C=O stretch at 1748 and 1674 cm−1, C−O ester stretch 1266–1056 cm−1, C−H stretch at 2931 and 2844 cm−1, C=C stretch at 1644–1440 cm−1, and C−Fe stretch at 650–527 cm−1 (Figure S6a). On the FTIR spectrum of 19, the absorption peaks of O−H, −C−H, C=C, C=O, C−O ester, and −COO stretching were visible at 3327, 2931–2832, 1597–1476, 1710–1649, 1230–1020, and 872 cm−1, respectively (Figure S7a). The FTIR spectrum of 20 displayed characteristic peaks of O−H, −C−H, =C−H, C=C, C=O, and C−O ester stretching at 3264, 2931–2844, 3042, 1599–1464, 1698, 1266–1155 cm−1, respectively (Figure S8a).
In the 1H‐NMR spectrum of 18, the singlet signals at 4.30 ppm and 4.18 ppm are attributed to the cyclopentadiene ring (Figure S6b). The 1H‐NMR spectrum of 19 showed the signals of CH3 at 0.86–1.54 ppm (Figure S7b). The 1H‐NMR spectrum of 20 revealed characteristic signals at 7.69–7.66 ppm for aromatic protons, multiplets between 2.05 and 1.62 ppm are attributed to the aliphatic protons and the signals at 1.28 ppm, and 0.90 ppm is assigned to the methyl protons of the compound (Figure S8b). Similar findings were reported by Pietraszek et al. for 1,25‐dihydroxyergocalciferol analogues.ref. cbdv202400373-bib-0044 The 13C NMR spectrum of 18 showed signals for ketone carbon at 204.26 ppm, C=O ester carbon was found at 170.98 ppm, and the carbons due to the (C=C) were found at 156.79–154.43 ppm. The signal between 72.41 and 69.26 ppm was assigned to the cyclopentadiene carbon (Figure S6c). The 13C NMR spectrum of 19 displayed ester carbon, C=O at 171.82 ppm, and C=C at 157.08–109.19 ppm (Figure S7c). The 13C‐NMR spectrum of 20 showed a carboxylic C=O peak at 174.24 ppm, a signal of an ester carbon C=O at 170.38 ppm and an amide carbon at 162.86 ppm (Figure S8c). The 13C NMR spectra of the hybrid compounds containing vitamin D2 revealed characteristic signals of C=O stretch, resulting from the ester linker at 170.98, 171.82, and 166.76 ppm for 18, 19, and 20, respectively. The UHPLC‐HRMS spectra of the hybrid compounds showed peaks at 695.5, 762.5, and 527.4, which also confirmed the successful synthesis of the hybrid compounds, 18, 19, and 20, respectively (Figures S6d, S7d, and S8d).
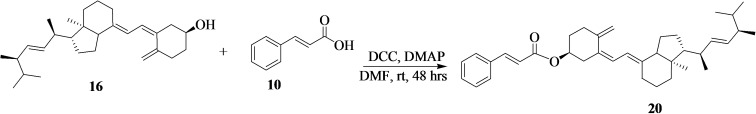
In Vitro Hydroxyapatite Binding Assay of Trans‐[ Pt(DACH)(Pamidronate)] (4)
Hydroxyapatite (HA) is the main component of bone mineral. The HA binding assay was conducted to evaluate the affinity of the complex for bone (Figure 1). The HA binding assay was conducted at 20 min, 40 min, and 60 min time intervals by known procedure.[ref. cbdv202400373-bib-0045, ref. cbdv202400373-bib-0046] Within 20 min of incubation, 41 % of 4 was bound to HA, and as the time increased to 40 min, the percentage increased to 45 %. After 60 min, the amount bound to HA increased to 55 %. The results indicate that the complex has a moderate affinity for HA which increases with an increase in the reaction time.[ref. cbdv202400373-bib-0045, ref. cbdv202400373-bib-0046] In an earlier study, the binding affinity of pamidronate was reported to be 60 %, which was the highest when compared to risedronate.ref. cbdv202400373-bib-0047 Bisphosphonates with P−C‐P backbone structure have a high affinity for bone. In this study, the modification of the hydroxyl groups on bisphosphonates did not significantly reduce the binding affinity of the compound. The compound displayed promising bone binding affinity, revealing that preparing bisphosphonate/platinum‐based complexes is a promising approach to developing therapeutics that target the bones.
Swissadme Prediction of the Complex and Hybrid Compounds
The complexes were analyzed for their Absorption, distribution, metabolism, and excretion (ADME) properties using SwissADME software. The physicochemical properties, lipophilicity, water solubility, pharmacokinetics, and drug‐likeness of the complexes were also predicted using SwissADME, as shown in (Tables S1 and S2). The molar refractivity of 4, 7, 11, 13, 15, 18, 19, and 20, was 82.24, 194.55, 176.28, 269.82, 225.63, 190.87, 218.08, 168.72, respectively, and showed topological polar surface area (TPSA) of 210.97, 279.65, 231.44, 251.67, 294.66, 43.37, 89.52, and 23.60 Å2, respectively (Table 3 and Supplementary Table). The hybrid compounds containing Vitamin D2 pharmacophore displayed TPSA values of less than 140 Å2, revealing good absorption potential in the intestines. TPSA values are used to predict drug absorption, such as absorption in the intestines, drug bioavailability, and blood‐brain barrier penetration.ref. cbdv202400373-bib-0048
Table 3: Anticancer activity of the selected compound (11, 13, 15, 18, 20) against MCF‐7 cell line.
| Compounds | % Viability | ||
|---|---|---|---|
| 10 μg/mL | 50 μg/mL | 100 μg/mL | |
| 11 | 87.98 | 102.5 | 78.74 |
| 13 | 82.40 | 89.44 | 70.53 |
| 15 | 90.76 | 77.86 | 59.53 |
| 18 | 81.82 | 99.71 | 78.23 |
| 20 | 84.89 | 66.13 | 68.91 |
| MCF‐7 ctrl cells | 100 | 100 | 100 |
The TPSA values of most of the hybrid compounds were in the range of 210.97–294.66 Å2, which is attributed to their high molecular weight (Tables S1 and S2). The lipophilicity of the compounds was also predicted using SwissADME, a vital parameter for drug development to predict their solubility, capability to penetrate via cell barriers, and uptake to the molecular target.ref. cbdv202400373-bib-0049 The Log P o/w (XLOGP3), (WLOGP), (MLOGP), (SILICOS‐IT), and Consensus Log P o/w values of the complex and hybrid compounds were evaluated. The compounds’ Log P lipophilicity values were less than three except in the case of 13 and the hybrid compounds containing Vitamin D2 pharmacophore. The software was unable to predict the ADME properties of Vitamin D2 hybrid compounds. A lipophilicity value of greater than 5 contributes to features such as strong drug plasma protein binding, drug accumulation in the tissues, rapid drug metabolic turnover, and poor water solubility.ref. cbdv202400373-bib-0050 The hybrid compounds were mostly water soluble except 13. Good water solubility of drugs is important in drug pharmacokinetics and pharmacodynamics.ref. cbdv202400373-bib-0051 Furthermore, the pharmacokinetic properties of the platinum complex and the hybrid compounds containing methotrexate were studied, and the BOILED‐Egg models are shown in Figures S9–S16. Cytochrome P450 isoforms, CYP1 A2, CYP2 C19, CYP2 C9, CYP2D6, and CYP3 A4 play a crucial role in drug bioactivation and safety. CYP3 A4 is one major important drug‐metabolizing enzyme found in the gastrointestinal tract and liver of the human body. It metabolizes most clinically approved drugs. The inhibition of CYP3 A4 can result in drug‐drug interactions, drug toxicity, and side effects. However, in some cases, its inactivation can enhance the drug therapeutic effectiveness of rapidly metabolized drugs by increasing their plasma levels.ref. cbdv202400373-bib-0052 The drugs, 7, 11, and 13 were predicted to be inhibitors of CYP3 A4. The compounds displayed a low degree of GI absorption and were not permeable via the BBB. The compounds 4, 7, 11, 13, and 15 were predicted to be P‐gp substrates. P‐glycoprotein (P‐gp) influences the uptake and efflux of drugs. It is expressed in tissues, such as the liver, small intestine, kidney, and the blood‐tissue barriers.ref. cbdv202400373-bib-0053 Substrates of P‐glycoprotein can act as inhibitors or inducers of its function. The inhibition of P‐gp results in increased drug bioavailability.ref. cbdv202400373-bib-0053 P‐glycoprotein is believed to be important against potentially toxic substances. It promotes drug elimination into the bile and urine and protects tissues such as the brain, placenta, testis, etc.[ref. cbdv202400373-bib-0053, ref. cbdv202400373-bib-0054] The bioavailability score of the compound was either 0.11 or 0.17. A bioavailability score of 0.17 suggests a 17 % chance of having greater than 10 % bioavailability in rats.[ref. cbdv202400373-bib-0055, ref. cbdv202400373-bib-0056] The drug‐likeness was studied using Lipinski, Ghose, Veber, Egan, and Muegge rules. However, the high molecular weight and TPSA values influenced their violation of drug‐likeness prediction. Based on Lipinski’s assumptions, drug molecules that violate these rules are likely not to possess desirable features. It is important to indicate that many approved and existing drugs do not follow the drug‐likeness rules but are orally bioavailable.ref. cbdv202400373-bib-0057
The predicted targets of compounds 4, 7, 11, 13, 15, 18, 19, and C20 are cytochrome P450, lyase, oxidoreductase, protease, kinase, transferase, family AG protein‐coupled receptor, and phosphatase (Figures S17–S24). These are promising drug targets explored in cancer therapies. Serine‐threonine protein phosphatase 2 A regulates some signaling pathways and its modulation has been reported to result in potent antitumor activity, indicating that it is a therapeutic target for treating cancer.ref. cbdv202400373-bib-0058 Cytochrome enzymes are involved in multi‐organ metastasis and cancer progression. They are overexpressed in the tumour microenvironment. Targeting Cytochrome enzymes is beneficial in cancer therapy.ref. cbdv202400373-bib-0059 Transferase enzymes, such as Carnitine palmitoyl transferase I, are involved in breast cancer survival and invasion. Carnitine palmitoyl transferase I have been identified as a tumour target for anticancer therapeutics.ref. cbdv202400373-bib-0060 G protein‐coupled receptors are potential targets for the treatment of colorectal cancer, and they control different cellular functions, such as tumorigenesis, etc.ref. cbdv202400373-bib-0061 Oxidoreductase enzymes such as NAD(P)H: quinone oxidoreductase 1 are overexpressed in different types of tumours and are a target for cancer therapy.ref. cbdv202400373-bib-0062 Proteases promote tumour growth and progression, which is dependent on the supply of nutrients and oxygen. High levels of proteases in tumours at an early stage have been reported. They promote proliferation, tumour invasion, inflammatory cell recruitment, angiogenesis, metastasis, etc.ref. cbdv202400373-bib-0063
Toxicity Prediction of the Complex and Hybrid Compounds
The ProTox‐II web server was used in the study for the prediction of oral toxicity, organ toxicity (hepatotoxicity), and toxicological endpoints (carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity). The organ toxicity (hepatotoxicity, mutagenicity, and cytotoxicity) of the compounds were all predicted as inactive, revealing that they are not capable of causing damage to the liver, genetic mutation, or cell death, respectively. However, 15, 18, 19, and 20 were predicted to be immunotoxic. The predicted toxicity of the compounds is shown in Table S3. GUSAR‐free online software was used for quantitative in silico toxicity prediction of LD50 values, the four possible routes of administration (i. e., oral, intraperitoneal, intravenous, and subcutaneous), and environmental toxicity of the synthesized compounds (Table S4). The hybrid compounds were mostly predicted to belong to classes 4 and 5 and are suitable for intraperitoneal administration, suggesting the non‐toxic nature of the drugs. In most cases, the compounds were within the applicability domain of models.
The bioaccumulation factor (BCF) is the ratio of the chemical concentration in an aquatic organism to that in water at a condition of a steady state.ref. cbdv202400373-bib-0064 The BCF values of the hybrid compounds ranged from −0.042–0.945 when compared to the precursors in the range of −0.620–0.684 (Table S5). The Daphnia magna LC50 represents the concentration in water that can kill half of a population of Daphnia magna (a water flea). The range LC50 values of the hybrid compounds were in the range of 5.239–7.472, and the precursors were in the range of 3.222–6.279. The fathead minnow LC50 is the concentration in water that can kill half of a population of fathead minnows (Pimephales promelas). The fathead minnow values of all compounds varied from −1.776–−8.891. Tetrahymena pyriformis IGC50 is a 50 % growth inhibitory concentration of T. pyriformis organism. The compounds’ IGC50 values ranged from 0.449–3.142. Most of the compound falls in the applicability domain of models.
The antitarget, hERG K+ channels are important and should be considered in drug development. The blockage of the hERG K+ channels results in cardiotoxicity, such as lethal cardiac arrhythmia.ref. cbdv202400373-bib-0065 Some drugs have been withdrawn from clinical use due to severe side effects.ref. cbdv202400373-bib-0066 The human ether‐a‐go‐go related gene (hERG) channel is expressed in the nervous system and the heart. It is crucial to predict a drug candidate that can block the hERG potassium ion channel at the early stage of drug discovery.ref. cbdv202400373-bib-0066 The hybrid compounds were evaluated to predict their capability to block hERG K+ channels (Table S6). Four outcomes were obtained namely: binary prediction Consensus applicability domain model), multiclass prediction (Consensus applicability domain model), probability maps extracted from the binary models, and fragment Contribution Maps for the Regression Model together with predicted IC50 values.ref. cbdv202400373-bib-0067 The fragment Contribution Maps for the Regression Model and the probability maps extracted from the binary models are shown in Table S7. In the predicted probability maps, the green atoms/fragments indicate a contribution towards the blockage of hERG, the pink atoms/fragments contribute to the decrease of hERG blockage, and the grey means no contribution.ref. cbdv202400373-bib-0067 The hybrid compounds containing vitamin D2, such as 18, 19, and 20, were predicted to block hERG K+ channels. However, the compounds, 4, 7, 11, 13, and 15 were predicted to be non‐blockers of hERG K+ channels.
In Vitro Cytotoxicity Assay of the Compounds
The cell viability for 4, 7, and 19 revealed toxic effects against the colorectal cancer cells. They were conducted in varying concentrations of 100 μg/mL, 50 μg/mL, 25 μg/mL, 12.5 μg/mL, and 6.25 μg/mL as shown in Table 2. The growth inhibitory effects at 100 μg/mL displayed good concentrations for all selected compounds. However, 19 displayed a significant inhibition effect than compounds 4 and 7 at 50 μg/mL and 100 μg/mL when compared to the parent drug, vitamin D. The IC50 value of 19 was 62.3 μg/mL, which was significant when compared to 4 and 7 (Table 2). Colorectal cancer metastasis to the bone is not common and has been reported in only 6–10 % of cases. However, recently, there has been a reported increase in cases of bone metastasis from colorectal cancer.[ref. cbdv202400373-bib-0068, ref. cbdv202400373-bib-0069] Bone metastasis resulting from colorectal cancer affects the survival rate of the patients, with a median survival time of five to thirteen months.ref. cbdv202400373-bib-0069 Bone metastasis due to colorectal cancer occurs mostly in the spine and the pelvis or hips.ref. cbdv202400373-bib-0068 Bisphosphonate has been reported to be effective when administered to cancer patients with bone metastases, resulting in delayed skeletal‐related events.ref. cbdv202400373-bib-0070 The anti‐metastatic effects of oxythiamine, an analogue of thymine (Vitamin B1) have been reported.ref. cbdv202400373-bib-0071 Vitamin D has an antiproliferative effect and suppresses tumour metastasis and angiogenesis.[ref. cbdv202400373-bib-0072, ref. cbdv202400373-bib-0073] The design of hybrid compounds containing a combination of anticancer drugs and pamidronate, vitamin D2, or Vitamin B1 is a promising approach to developing effective anticancer drugs to treat cancer that can metastasize to the bone.
Table 2: Anticancer activity of the selected compound (4, 7, 19) against HT‐29 cell lines.
| Compounds | % Viability | 1 C50 (μg/mL) | ||||
|---|---|---|---|---|---|---|
| 6.25 μg/mL | 12.5 μg/mL | 25 μg/mL | 50 μg/mL | 100 μg/mL | – | |
| 4 | 75.45838 | 66.89329 | 138.6372 | 65.36956 | 58.2559 | 118 |
| 7 | 111.4532 | 87.97642 | 112.3503 | 74.63331 | 59.89005 | 125.02 |
| 19 | 77.19797 | 75.49189 | 112.8483 | 51.26488 | 43.89068 | 62.3 |
| Alendronate | 145.1377 | 111.1793 | 231.5063 | 90.39642 | 88.98565 | – |
| Vitamin D | 74.78048 | 72.75136 | 84.40247 | 73.54453 | 56.96499 | – |
| Cisplatin (Positive control) | 44.97 | |||||
The cell viability studies of selected hybrid using MCF‐7 cell lines showed that 15 was cytotoxic at a concentration of 100 μg/mL, revealing a promising anticancer activity. However, more studies are needed to fully understand the mode of action of the hybrid compounds (Table 3). The morphology of the cell lines after treatment with the hybrid 20 at the concentration of 100 μg/mL revealed apoptotic cell features, confirming the cytotoxic effect (Figure 2). Similar findings were reported by Majidzadeh et al. for cancer cell lines treated with a combination of two anticancer agents.ref. cbdv202400373-bib-0074

Molecular Docking Studies
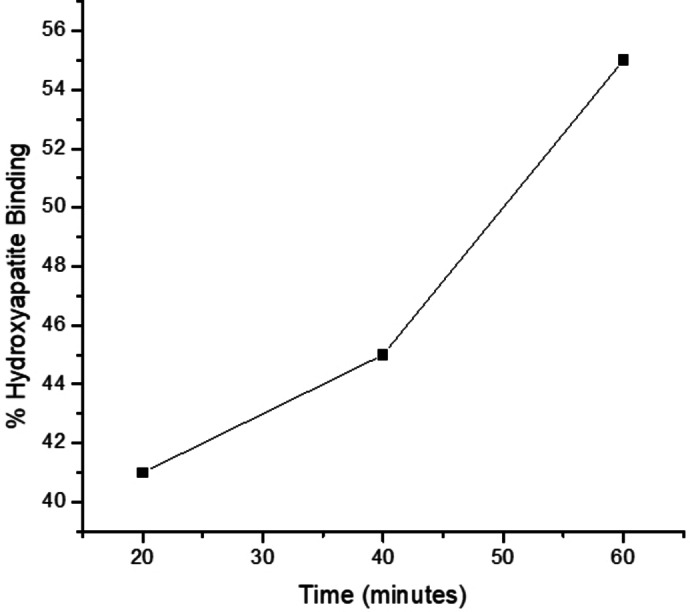
In this study, selected molecular compounds that were experimentally viable were docked against human Tyrosine Phosphatase and Human 3 alpha‐hydroxysteroid dehydrogenase type 3 to investigate the amino acid residues and the type of non‐bonding interactions that occurred between the studied complexes. Representative 3D structure of the receptor: Human 3 alpha‐hydroxysteroid dehydrogenase type 3 and the selected molecular compound are shown in Figure S25. Compounds 4, 7, and 19 were docked against human Tyrosine Phosphatase, and the calculated scoring were −3.35 kcal/mol, −8.42 kcal/mol and −8.64 kcal/mol, respectively (Table 4). The calculated binding affinity for the docked 11, 13, 15, 18, and 20 against Human 3 alpha‐hydroxysteroid dehydrogenase type 3 was −8.36 kcal/mol, −9.28 kcal/mol, −9.70 kcal/mol, −7.02 kcal/mol and −7.13 kcal/mol, respectively (Table 4). The reference compounds proved to possess a higher ability to inhibit human Tyrosine Phosphatase than compound 4. Oyebamiji et al., 2023 reported that a compound’s capability to inhibit receptors is attributed to a low binding affinity value.ref. cbdv202400373-bib-0075 Compounds 7 and 19 showed a greater tendency to inhibit the target than compound 4 and the reference compounds. Compound 19 exhibited the highest tendency to inhibit human Tyrosine Phosphatase than other studied compounds (Figure 3) and (Table 4).
Table 4: Calculated binding affinity for the studied compounds against human Tyrosine Phosphatase.
| 4 | ALA 2354 (A) H‐donorALA 2354 (A)SER 2171 (A)ALA 2354 (A) H‐Acceptor | −3.35 |
| 7 | Ligand Receptor Interaction Distance E (kcal/mol)N47 49 NZ LYS 2301 (A) H‐acceptor 3.01 −11.8O28 30 NH2 ARG 2400 (A) Ionic 3.07 −4.0 | −8.42 |
| 19 | – | −8.64 |
| Ref1 | Ligand Receptor Interaction Distance E (kcal/mol)O7 7 NZ LYS 2185 (A) H‐acceptor 3.40 −0.5 | −6.18 |
| Cisplatin | −3.86 |
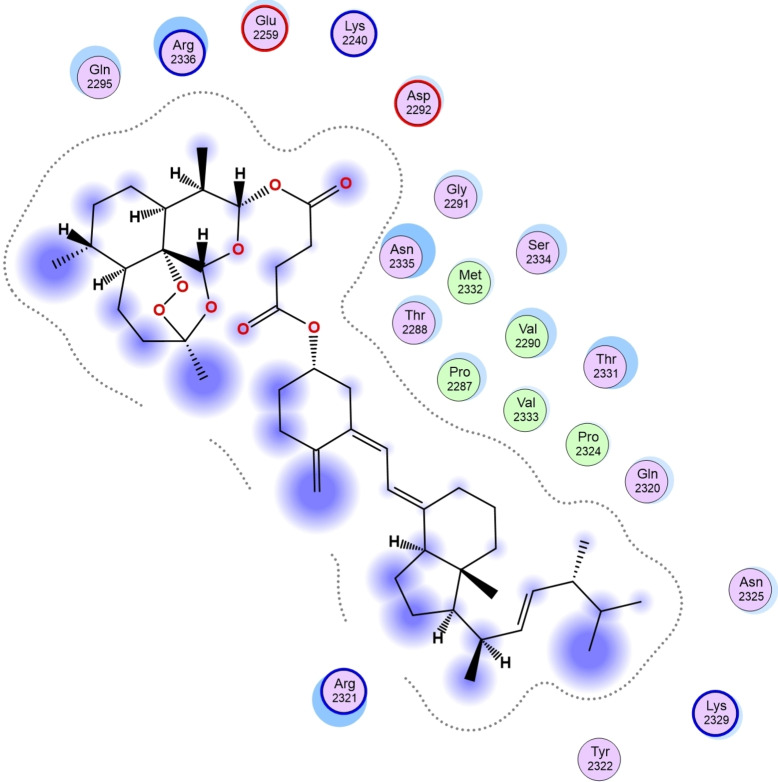
Furthermore, the calculated scoring from the docking of compounds 11, 13, 15, 18 and 20 against Human 3 alpha‐hydroxysteroid dehydrogenase type 3 revealed the compound with the highest tendency to inhibit the studied receptor. As shown in Table 5, compound 15 with a calculated binding affinity of −9.70 kcal/mol proved to have the greatest capability to inhibit Human 3 alpha‐hydroxysteroid dehydrogenase type 3 than other studied compounds as well as the referenced compounds used in this research (Figure 4).
Table 5: Calculated binding affinity for the studied compounds against Human 3 alpha‐hydroxysteroid dehydrogenase type 3.
| 11 | Ligand Receptor Interaction Distance E (kcal/mol)O27 29 NZ LYS 270 (A) Ionic 3.56 −1.7O28 30 NZ LYS 270 (A) Ionic 3.00 −4.56‐ring CB ALA 27 (A) pi‐H 3.68 −1.06‐ring CA HIS 222 (A) pi‐H 4.46 −0.76‐ring N LYS 270 (A) pi‐H 4.53 −2.76‐ring CB LYS 270 (A) pi‐H 4.05 −0.5 | −8.36 |
| 13 | Ligand Receptor Interaction Distance E (kcal/mol)N40 40 OG SER 217 (A) H‐donor 2.78 −1.4O23 23 NE ARG 276 (A) H‐acceptor 3.14 −3.0O23 23 NH2 ARG 276 (A) H‐acceptor 3.37 −1.0O27 27 NZ LYS 270 (A) H‐acceptor 3.02 −0.9O27 27 NZ LYS 270 (A) Ionic 3.02 −4.46‐ring CB HIS 222 (A) pi‐H 4.22 −0.6 | −9.28 |
| 15 | Ligand Receptor Interaction Distance E (kcal/mol)N11 13 O GLN 275 (A) H‐donor 2.95 −2.1N31 33 OE2 GLU 285 (A) H‐donor 2.95 −1.5O27 29 NH2 ARG 250 (A) H‐acceptor 3.11 −6.3O28 30 NH1 ARG 250 (A) H‐acceptor 2.79 −8.2O43 45 N GLN 6 (A) H‐acceptor 3.00 −4.2O27 29 NH1 ARG 250 (A) Ionic 3.41 −2.2O27 29 NH2 ARG 250 (A) Ionic 3.11 −3.8O28 30 NH1 ARG 250 (A) Ionic 2.79 −6.0O28 30 NH2 ARG 250 (A) Ionic 3.97 −0.6 | −9.70 |
| 18 | −7.02 | |
| 20 | Ligand Receptor Interaction Distance E (kcal/mol)6‐ring N LYS 270 (A) pi‐H 4.00 −0.7 | −7.13 |
| [Ref1] | −7.21 | |
| Cisplatin | Ligand Receptor Interaction Distance E (kcal/mol)N 1 O GLN 275 (A) H‐donor 2.86 −3.7N 18 O ARG 276 (A) H‐donor 2.91 −2.9O 6 N GLN 282 (A) H‐acceptor 2.86 −1.7OE1 17 OG1 THR 251 (A) H‐acceptor 2.78 −1.5O 23 N VAL 283 (A) H‐acceptor 3.20 −1.5 | −4.384 |
Molecular Dynamics Simulation Analysis
The compounds with the highest binding affinity were subjected to a molecular dynamics simulation study to obtain the actual binding energy between the studied complexes. Compounds 19 and 15 proved potent in inhibiting human Tyrosine Phosphatase and Human 3 alpha‐hydroxysteroid dehydrogenase type 3, respectively. As shown in Figures 5 and 6, the predicted Root Mean Square Deviation (RMSD) using 100 ns simulation time revealed that compounds 19 and 15 were steady compared to the studied reference compounds. This is an indication that 19 and 15 interacted well with the receptor, thereby inhibiting the target than the referenced molecules.
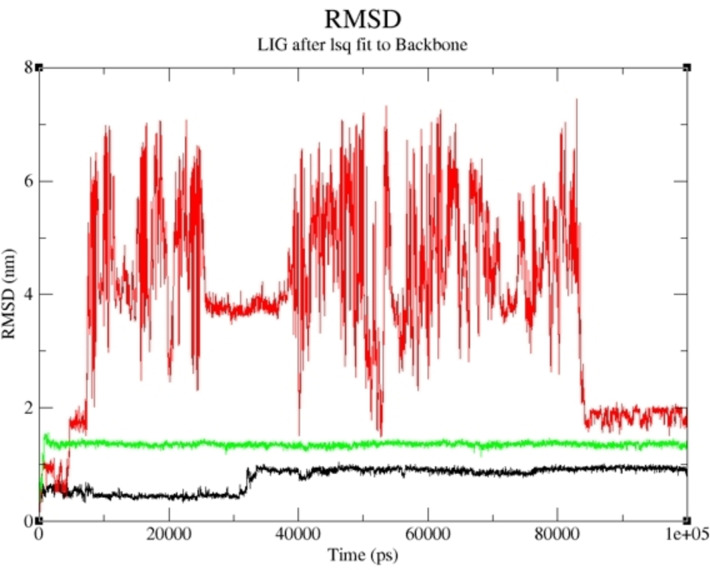
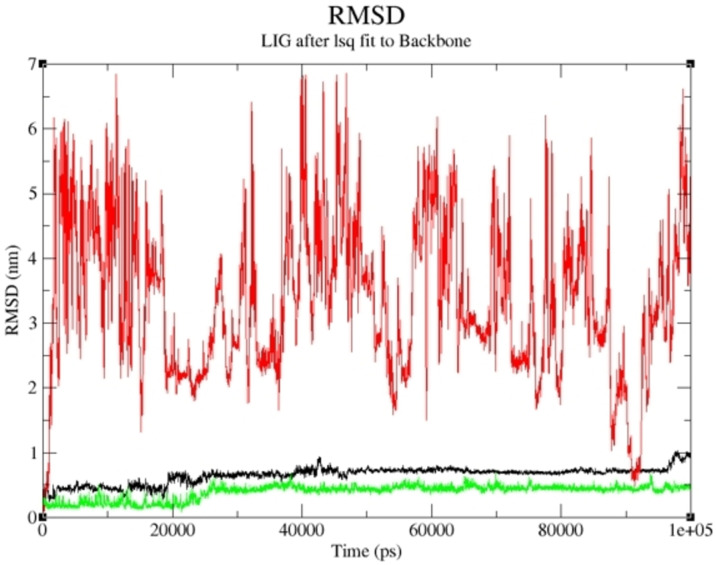
More so, the suppleness of the residues involved in the linking of the selected complexes was investigated via Root Mean Square Fluctuation (RMSF). As shown in Figures 7 and 8, a similar configuration was observed for the pattern formed by the selected compounds and the studied receptor except for little deviation; thus, a greater degree of affinity of 19 and 15 toward the studied targets supports their potential greater capacity as anti‐cancer agents. The lower the binding free energy of a compound, the better the capacity of such compound to interact and inhibit the targetref. cbdv202400373-bib-0075; thus, 19 and 15 proved to be better inhibitors of human Tyrosine Phosphatase and Human 3 alpha‐hydroxysteroid dehydrogenase type 3, respectively. As reported in Tables 6 and 7, the calculated value for van der Waal energy, electrostatic energy, and gas‐phase components were more favourable towards the inhibiting ability compounds 19 and 15 than the reference molecules used in this work.
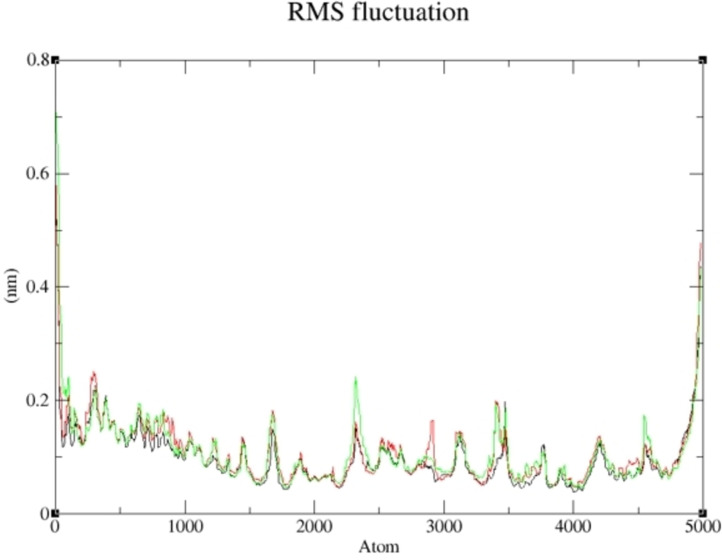
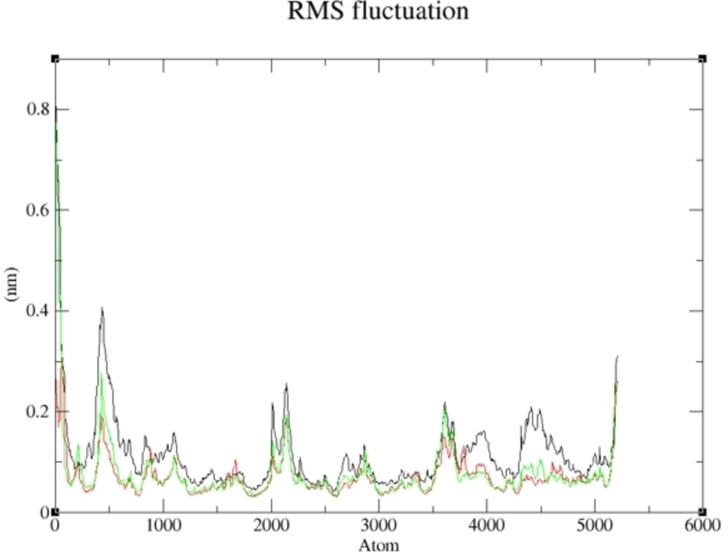
Table 6: Calculated binding Energy Components for 19 and the reference compounds against human Tyrosine Phosphatase.
| Complexes | Binding Energy Components (kcal/mol) | ||||
|---|---|---|---|---|---|
| ΔEvdw | ΔEele | ΔGgas | ΔGsol | ΔGbind | |
| Comp19‐1wch | −37.69± 0.69 | −11.8 1.02 | −49.49± 1.22 | 60.21± 1.45 | −10.71± 1.09 |
| Cisplatin‐1wch | −5.06±0.55 | −6.06± 2.3 | −11.12±1.98 | 13.29± 1.62 | −2.17± 0.86 |
| REF‐1wch | −9.0±0.43 | −3.14±0.86 | −12.15±1.22 | 15.99±1.3 | 3.85±0.54 |
Table 7: Calculated binding Energy Components for 15 and the reference compounds against Human 3 alpha‐hydroxysteroid dehydrogenase type 3.
| Complexes | Binding Energy Components (kcal/mol) | ||||
|---|---|---|---|---|---|
| ΔEvdw | ΔEele | ΔGgas | ΔGsol | ΔGbind | |
| Comp15‐4xo6 | −85.47± 2.98 | −39.02± 2.14 | −124.49± 1.51 | 140.75± 1.82 | −16.26± 3.07 |
| Cisplatin‐4xo6 | −10.53±0.85 | −18.86± 3.3 | −29.39±2.95 | 29.68± 1.72 | 0.29± 1.75 |
| REF‐4xo6 | −36.52±1.15 | −1.74±0.94 | −38.27±0.82 | 39.26± 1.03 | 1.0± 1.63 |
Conclusions
Hybrid Molecules containing Methotrexate, Vitamin D, and bisphosphonates Derivatives are promising compounds for the treatment of cancer that metastasize to the bone. The compounds were synthesized with good yield. They were predicted to be non‐toxic and non‐blockers of hERG K+ channels. Their capability to cause environmental toxicity was not significant. Docking studies revealed compounds 19 and 15 have potent capability to inhibit human Tyrosine Phosphatase and Human 3 alpha‐hydroxysteroid dehydrogenase type 3, respectively. Molecular dynamics studies further showed that compounds 19 and 15 interact well with the receptor, thereby inhibiting the target than the referenced molecules. The Root Mean Square Fluctuation showed a greater degree of affinity of 19 and 15 toward the studied targets, indicating their greater capacity as anti‐cancer agents. The van der Waal energy, electrostatic energy, and gas‐phase components were more favourable towards the inhibiting capability of 19 and 15 than the reference molecules used in this work. Compounds 19 and 15 are promising compounds that require further studies.
Conflict of Interests
The authors declare no conflict of interest, financial or otherwise.
Supplementary Materials
References
- 1 G. Ren , M. Esposito , Y. Kang , J. Mol. Med. 2015, 93, 1203–1212.26275789
- 2 R. Cathomas , Z. Bajory , M. Bouzid , A. El Ghoneimy , S. Gillessen , F. Goncalves , G. Kacso , G. Kramer , P. Milecki , D. Pacik , W. Tantawy , Urol. Int. 2014, 92, 377–386.24802278
- 3 R. Aft , J. R. Perez , N. Raje , V. Hirsh , F. Saad , Crit. Rev. Oncol. Hematol. 2012, 82, 233–248.21683613
- 4 C. G. Anusionwu , B. A. Aderibigbe , S. A. Adeyemi , P. Ubanako , S. O. Oselusi , Y. E. Choonara , X. Y. Mbianda , Bioorg. Med. Chem. 2022, 58, 116652.35180594
- 5 C. Chen , Y. Li , X. Yu , Q. Jiang , X. Xu , Q. Yang , Z. Qian , Chin. Chem. Lett. 2018, 29, 1609–1612.
- 6 J. Welsh , Arch. Biochem. Biophys. 2012, 523, 107–114.22085499
- 7 A. Pietraszek , M. Malińska , M. Chodyński , M. Krupa , K. Krajewski , P. Cmoch , K. Woźniak , A. Kutner , Steroids. 2013, 78, 1003–1014.23811018
- 8 M. A. Maestro , F. Molnar , C. Carlberg , J. Med. Chem. 2019, 62, 6854–6875.30916559
- 9 R. Fernandes , P. Siegel , S. Komarova , J. Hilton , C. Addison , M. F. Ibrahim , J. Werier , K. Dennis , G. Singh , E. Amir , V. Jarvis , J. Bone. Oncol. 2016, 5, 57–62.27335772
- 10 K. Krüger , J. Thomale , N. Stojanović , M. Osmak , C. Henninger , S. Bormann , G. Fritz , Biochim. Biophys. Acta (BBA)-Mol. Cell. Res. 2015, 1853, 685–698.
- 11 R. A. Nadar , K. Farbod , K. C. der Schilden , L. Schlatt , B. Crone , N. Asokan , A. Curci , M. Brand , M. Bornhaeuser , M. Iafisco , N. Margiotta , Sci. Rep. 2020, 10, 1–2.31913322
- 12 P. Koźmiński , P. K. Halik , R. Chesori , E. Gniazdowska , Int. J. Mol. Sci. 2020, 21, 3483.32423175
- 13 B. Álvarez-González , M. Rozalen , M. Fernández-Perales , M. A. Álvarez , M. Sánchez-Polo , Molecules 2020, 25, 6049.33371436
- 14 S. W. Kmiecik , M. A. Krzyścik , B. Filip-Psurska , J. Wietrzyk , J. Boratyński , T. M. Goszczyński , Adv. Hyg. Exp. Med. 2017, 71, 618–623.
- 15 B. Cai , A. Liao , K. K. Lee , J. S. Ban , H. S. Yang , Y. J. Im , C. Chun , Steroids. 2016, 116, 45–51.27770617
- 16 S. A. Nadhum , M. H. Mohammed , Iraqi. J. Pharm. Sci. 2015, 24, 74–84.
- 17 O. M. Semenenko , V. V. Lipson , A. O. Sadchenko , O. V. Vashchenko , N. A. Kasian , L. V. Sviechnikova , L. M. Lisetski , M. L. Babak , V. M. Vakula , O. V. Borysov , Y. V. Holota , Steroids. 2024, 201, 109332.37939980
- 18 Y. Li , C. Zhao , J. Zhang , S. Zhai , B. Wei , L. Wang , J. Chem. Inf. Model. 2019, 59, 4063–4069.31524396
- 19 J. M. Delou , A. S. Souza , L. C. Souza , H. L. Borges , Cells 2019, 8, 1013.31480389
- 20 S. A. Desai , A. Manjappa , P. Khulbe , J. Egypt. Natl. Canc. Inst. 2021, 33, 1–4.33415591
- 21 P. M. Njogu , J. Gut , P. J. Rosenthal , K. Chibale , ACS Med. Chem. Lett. 2013, 4, 637–641.24900723
- 22 L. Pasqua , I. E. De Napoli , M. De Santo , M. Greco , E. Catizzone , D. Lombardo , Nanoscale. Adv. 2019, 1, 3269–3278.36133588
- 23 H. Nakatake , H. Ekimoto , M. Aso , A. Ogawa , A. Yamaguchi , H. Suemune , Chem. Pharm. Bull. 2011, 59, 710–713.
- 24 C. W. Wu , H. C. Liu , Y. L. Yu , Y. T. Hung , C. W. Wei , G. T. Yiang , Oncol. Rep. 2017, 37, 2177–2184.28259996
- 25 B. A. Aderibigbe , A. Mugogodi , M. Nwamadi , S. S. Ray , V. Steenkamp , M. O. Balogun , W. M. Matshe , J. Inorg. Organomet. Polym. Mater. 2020, 30, 1503–1518.
- 26 H. E. Mukaya , R. L. Van Zyl , N. J. Van Vuuren , X. Y. Mbianda , Polym. Bull. 2017, 74, 3161–3178.
- 27 A. Alvarez-Valdes , A. I. Matesanz , J. Perles , C. Fernandes , J. D. Correia , F. Mendes , A. G. Quiroga , J. Inorg. Biochem. 2019, 191, 112–118.30496946
- 28 D. Gupta , S. V. Gupta , K. D. Lee , G. L. Amidon , Mol. Pharm. 2009, 6, 1604–1011.19566080
- 29 J. K. Laha , K. V. Patel , K. S. Tummalapalli , M. K. Hunjan , ACS Omega 2018, 3, 8787–8793.31459011
- 30 V. Khwaza , O. O. Oyedeji , S. O. Oselusi , E. Morifi , M. Nwamadi , D. T. Ndinteh , P. Ramushu , T. Matsebatlela , B. A. Aderibigbe , Chem. Biodivers. 2023, 20, e202300034.36920086
- 31 Z. Mbese , M. Nell , Y. T. Fonkui , D. T. Ndinteh , V. Steenkamp , B. A. Aderibigbe , Recent. Adv. Anti-Infect. Drug. Discov. 2022, 17, 54–68.
- 32 F. Villa , M. Deak , G. B. Bloomberg , D. R. Alessi , D. M. F. Van Aalten , J. Biol. Chem. 2005, 280, 8180.15611135
- 33 B. Zhang , X. J. Hu , X. Q. Wang , J. F. Theriault , D. W. Zhu , P. Shang , F. Labrie , S. X. Lin , Biochem. J. 2016, 473, 1037–1046.26929402
- 34 U. A. Cevik , I. Celik , A. Isik , R. R. Pillai , T. E. Tallei , R. Yadav , Y. Ozkay , Z. A. Kaplancikli , J. Mol. Struct. 2022, 15, 1252.
- 35 D. R. Roe , T. E. Cheatham III , J. Chem. Theory. Comput. 2013, 9, 3084–3095.26583988
- 36 N. L. Dlamini , H. E. Mukaya , R. L. Van Zyl , N. C. Jansen van Vuuren , X. Y. Mbianda , Artif. Cells. Nanomed. Biotechnol. 2018, 46, 287–296.
- 37 L. Qiu , G. Lv , Y. Cao , L. Chen , H. Yang , S. Luo , M. Zou , J. Lin , J. Biol. Inorg. Chem. 2015, 20, 1263–1275.26531104
- 38 S. Ray , R. Sarkar , A. Chattopadhyay , A. K. Ghosh , Prog. React. Kinet. Mech 2014, 39, 122–136.
- 39 S. M. El-Megharbel , R. Z. Hamza , A. A. Gobouri , M. S. Refat , Appl. Organomet. Chem. 2019, 33, e4892.
- 40 A. Nemat , I. N. Khan , S. Kalsoom , S. A. Malik , S. Ayub , F. Adnan , M. A. Kamal , M. Iqbal , J. Biomol. Struct. Dyn. 2022, 40, 2865–2877.33183168
- 41 L. Tang , Y. Zhang , J. Xu , Q. Yang , F. Du , X. Wu , M. Li , J. Shen , S. Deng , Y. Zhao , Z. Xiao , Molecules 2023, 28, 1414.36771080
- 42 A. Lawal , R. A. Umar , M. G. Abubakar , U. Z. Faruk , U. Wali , J. Pharm. Biomed. Sci. 2012, 24, 6–14.
- 43 B. L. Munnik , C. H. Kaschula , C. R. Harding , P. Chellan , Dalton. Trans. 2023, 52, 15786–15797.37681434
- 44 A. Pietraszek , M. Malińska , M. Chodyński , M. Krupa , K. Krajewski , P. Cmoch , K. Woźniak , A. Kutner , Steroids. 2013, 78, 1003–1014.23811018
- 45 G. Bonzi , S. Salmaso , A. Scomparin , A. Eldar-Boock , R. Satchi-Fainaro , P. Caliceti , Bioconjug. Chem. 2015, 26, 489–501.25613006
- 46 M. Karacivi , B. Sumer Bolu , R. Sanyal , Mol. Pharm. 2017, 14, 1373–1383.28358515
- 47 W. Jahnke , C. Henry , ChemMedChem. 2010, 5, 770–776.20209564
- 48 M. Janicka , A. Śliwińska , M. Sztanke , K. Sztanke , Int. J. Mol. Sci. 2022, 23, 15887.36555527
- 49 B. Morak-Młodawska , M. Jeleń , E. Martula , R. Korlacki , Int. J. Mol. Sci. 2023, 24, 6970.37108135
- 50 K. Tsaioun , B. J. Blaauboer , T. Hartung , ALTEX-Altern. Anim. Exp 2016, 33, 343–358.
- 51 M. Rodriguez-Aller , D. Guillarme , J. L. Veuthey , R. Gurny , J. Drug. Deliv. Sci. Technol. 2015, 30, 342–351.
- 52 E. R. Samuels , I. Sevrioukova , Mol. Pharm. 2018, 15, 279–288.29232137
- 53A. Finch, P. Pillans, ‘Aust. Prescr. 2014, 37, 137–139.
- 54 S. K. Dewanjee , T. Dua , N. Bhattacharjee , A. Das , M. Gangopadhyay , R. Khanra , S. Joardar , M. Riaz , V. De Feo , M. Zia-Ul-Haq , Molecules 2017, 22, 871.28587082
- 55 P. A. Akinnusi , S. O. Olubode , A. A. Alade , A. A. Ashimi , O. L. Onawola , A. O. Agbolade , A. P. Emeka , S. A. Shodehinde , O. Y. Adeniran , Bioinform. Biol. Insights. 2023, 17, 11779322231167970.37124131
- 56 P. J. Ameji , A. Uzairu , G. A. Shallangwa , S. Uba , J. Taibah. Univ. Med. Sci. 2023, 18, 1417–1437.38162870
- 57 M. D. Shultz , J. Med. Chem. 2018, 62, 1701–1714.30212196
- 58 P. Kalev , A. Sablina , Anti-Cancer. Agents. Med. Chem. 2011, 11, 38–46.
- 59 Z. Guo , V. Johnson , J. Barrera , M. Porras , D. Hinojosa , I. Hernández , P. McGarrah , D. A. Potter , Cancer. Metastasis Rev. 2018, 37, 409–423.30066055
- 60 S. Pucci , M. J. Zonetti , T. Fisco , C. Polidoro , G. Bocchinfuso , A. Palleschi , G. Novelli , L. G. Spagnoli , P. Mazzarelli , Oncotarget 2016, 7, 19982.26799588
- 61 A. Hermawan , H. Putri , Egypt. J. Med. Hum. Genet. 2022, 23, 1–7.37521842
- 62 K. Zhang , D. Chen , K. Ma , X. Wu , H. Hao , S. Jiang , J. Med. Chem. 2018, 61, 6983–7003.29712428
- 63 S. D. Mason , J. A. Joyce , Trends. Cell. Biol. 2011, 21, 228–237.21232958
- 64 S. S. Hassan , W. D. Zhang , H. Z. Jin , S. H. Basha , S. S. Priya , J. Biomol. Struct. Dyn. 2022, 40, 484–498.32876526
- 65 A. Garrido , A. Lepailleur , S. M. Mignani , P. Dallemagne , C. Rochais , Eur. J. Med. Chem. 2020, 195, 112290.32283295
- 66 S. Kalyaanamoorthy , K. H. Barakat , Med. Res. Rev. 2018, 38, 525–555.28467598
- 67 R. C. Braga , V. M. Alves , M. F. Silva , E. Muratov , D. Fourches , L. M. Lião , A. Tropsha , C. H. Andrade , Mol. Inform. 2015, 34, 698–701.27490970
- 68 L. Holladay , J. Luu , V. Balendra , K. Kmetz , Cancer. Treat. Res. Commun. 2023, 37, 100763.37839182
- 69 F. Liu , J. Zhao , J. Xie , L. Xie , J. Zhu , S. Cai , H. Zheng , Y. Xu , Tumor. Biol. 2016, 37, 16127–16134.
- 70 T. L. Ng , M. M. Tu , M. F. Ibrahim , B. Basulaiman , S. F. McGee , A. Srikanthan , R. Fernandes , L. Vandermeer , C. Stober , M. Sienkiewicz , A. Jeong , Support. Care. Cancer. 2021, 29, 925–943.32535678
- 71 C. M. Yang , Y. Z. Liu , J. W. Liao , M. L. Hu , Clin. Exp. Metastasis. 2010, 27, 341–349.20449639
- 72 J. Xu , W. Li , J. Ma , J. Liu , H. Sha , S. Zhou , F. Wang , Q. Ma , Curr. Med. Chem. 2013, 20, 4109–4120.23895682
- 73 S. Y. Na , K. B. Kim , Y. J. Lim , H. J. Song , J. Cancer. Prev. 2022, 27, 147.36258716
- 74 H. Majidzadeh , M. Araj-Khodaei , M. Ghaffari , A. Jafari , D. Shanehbandi , M. Torbati , J. E. Dolatabadi , Pharm. Sci. 2021, 27, 536–542.
- 75A. K. Oyebamiji, G. F. Tolufashe, O. M. Oyawoye, T. A. Oyedepo, B. Semire, J. Chem. 2020, 1–10