
CsMIKC1 regulates inflorescence development and grain production in Cannabis sativa plants
Abstract
Female inflorescence is the primary output of medical Cannabis. It contains hundreds of cannabinoids that accumulate in the glandular trichomes. However, little is known about the genetic mechanisms governing Cannabis inflorescence development. In this study, we reported the map-based cloning of a gene determining the number of inflorescences per branch. We named this gene CsMIKC1 since it encodes a transcription factor that belongs to the MIKC-type MADS subfamily. Constitutive overexpression of CsMIKC1 increases inflorescence number per branch, thereby promoting flower production as well as grain yield in transgenic Cannabis plants. We further identified a plant-specific transcription factor, CsBPC2, promoting the expression of CsMIKC1. CsBPC2 mutants and CsMIKC1 mutants were successfully created using the CRISPR-Cas9 system; they exhibited similar inflorescence degeneration and grain reduction. We also validated the interaction of CsMIKC1 with CsVIP3, which suppressed expression of four inflorescence development-related genes in Cannabis. Our findings establish important roles for CsMIKC1 in Cannabis, which could represent a previously unrecognized mechanism of inflorescence development regulated by ethylene.
Affiliations: Institute of Bast Fiber Crops, Chinese Academy of Agricultural Sciences, Changsa, Hunan 410205, China; State Key Laboratory of Crop Genetics & Germplasm Enhancement and Utilization, Jiangsu Engineering Research Center for Plant Genome Editing, Nanjing Agricultural University, Nanjing 210095, China; School of Pharmacy, Hunan Vocational College of Science and Technology, Changsa, Hunan 410004, China; Department of Horticulture and Landscape Architecture, Oklahoma State University, Stillwater, OK 74078, USA; College of Food Science and Biology, Hebei University of Science and Technology, Shijiazhuang, Hebei 050018, China; The College of Agriculture, Yunan University, Kunming, Yunnan 650504, China; School of Life Sciences, Nantong University, Nantong, Jiangsu 226019, China; Huazhi Biotech Co., Ltd, Changsha, Hunan 410128, China
License: © The Author(s) 2024. Published by Oxford University Press on behalf of Nanjing Agricultural University. CC BY 4.0 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited.
Article links: DOI: 10.1093/hr/uhae161 | PubMed: 39108581 | PMC: PMC11298619
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (1.7 MB)
Introduction
Cannabis sativa L. is a diploid species with a chromosome count of 2n = 20, comprising nine pairs of chromosomes and a pair of sex-determining chromosomes (XY/XX) [ref. 1]. Since early Neolithic times in East Asia, it has been utilized in diverse domains, including cosmetics, textiles, food, and medicinal purposes [ref. 2]. Cannabis female inflorescences accumulate hundreds of specialized metabolites within their glandular trichomes [ref. 3]. While Cannabis is often steeped in controversy due to the synthesis of Δ9-tetrahydrocannabinol (THC), which produces psychoactive effects in humans, it also contains phytocannabinoids with therapeutic potential. These compounds have shown promise in treating complex neurological disorders and cancer [ref. 4, ref. 5]. These phytocannabinoids include, for example, cannabidiol (CBD), cannabigerol (CBG), cannabichromene (CBC), and cannabinol (CBN) [ref. 6, ref. 7]. Cannabis seeds are recognized as a functional food due to beneficial compositions of essential fatty acids, proteins, and antioxidants [ref. 8]. The global medical Cannabis market was valued at $3.5 billion at retail prices in 2019, and significant future growth is expected, with a $20.2 billion market value forecast from 2020 to 2025 [ref. 9]. As of 2020, Cannabis-based products for medicinal use have been legalized in over 50 countries, including Australia, Canada, Germany, Israel, China (Yunnan and Heilongjiang provinces), and most US states.
During the process of domestication of wild Cannabis into cultivated Cannabis, remarkable morphological transitions have been observed, such as compact plant architecture accompanied by increased flower production resulting from increased inflorescence number per branch [ref. 10]. The inflorescence of Cannabis consists of a highly branched compound raceme, comprising several higher-order condensed branchlets. The condensed branchlet develops at the apex of the main stem, as well as on second- and third-order branches. [ref. 10]. The inflorescence number per branch identified in this article is the number of phytomers developed at second-order branches, equivalent to the number of nodes at second-order branches. Since increasing yield has been the primary goal of Cannabis improvement, understanding the genetic mechanisms underlying female flower development is crucial. However, the key genes that regulate inflorescence number had not been characterized in Cannabis, which significantly hindered progress in the genetic improvement of flower production. In this study, we mapped a quantitative trait locus (QTL) determining the number of inflorescences per branch in Cannabis and subsequently cloned the gene CsMIKC1 for the QTL. CsMIKC1 increased both inflorescence number per branch and grain production, a process regulated transcriptionally by CsBPC2. We also found that CsVIP3 interacted with CsMIKC1 and repressed expression of four genes related to flower development. Furthermore, we validate a hypothesis that ethylene functions as a key signal in Cannabis inflorescence development. Our findings not only provide insights into the molecular mechanism for increasing inflorescence number in Cannabis but also provide a basis for proposing prospective strategies for the enhancement of yield production.
Results
A major quantitative trait locus associated with inflorescence number per branch
We performed a single cross between the two Cannabis accessions, DMG12 and YMG26, with distinct inflorescence morphologies (Fig. 1a and b). Subsequently, a population of 181 F2 plants was genotyped with the genotyping-by-sequencing approach. Phenotyping was carried out in the greenhouse. A major QTL associated with inflorescence development was mapped to the long arm of chromosome 8 (hereafter referred as QId.ibfc-8 L). The LOD (log of the odds) value of this QTL was 11.8, accounting for 35.7% of the total phenotypic variation (Fig. 1c). According to the NCBI Cannabis sativa.cs10 reference genome, QId.ibfc-8 L was mapped within a 3.1-Mb genome region between markers GBS7959 and GBS3443. To fine-map QId.ibfc-8 L, we developed eight internal markers using Kompetitive Allele-Specific PCR (KASP) (Supplementary Data Table S1 and S2) to screen 431 F2:4 individuals developed from the cross DMG12 × YMG26, and identified five new crossovers between KASP1987 and KASP1993 (Fig. 1d). Compared with recombinants carrying the YMG26 allele, those recombinants carrying the DMG12 allele at QId.ibfc-8 L had a significantly increased inflorescence number per branch (Supplementary Data Fig. S1). The candidate gene was narrowed down to a 138 836-bp genomic region flanked by two markers, KASP3738 and KASP2574. Based on the NCBI Cannabis sativa.cs10 reference genome, there are two candidate genes in this genomic region: LOC115701144 and LOC115700576. To clone the gene responsible for QId.ibfc-8 L, alleles derived from DMG12 and YMG26 were sequenced for these candidate genes. The candidate gene LOC115701144 encodes a truncated transcription factor, CAULIFLOWER A, and no difference was observed in the coding region between the DMG12 and YMG26 alleles (Supplementary Data Fig. S1). The transcript levels of LOC115701144 did not show a significant difference between the DMG12 and YMG26 alleles (Supplementary Data Fig. S2). So LOC115701144 was excluded as a candidate gene for QId.ibfc-8 L, and the other one, LOC115700576, was the sole candidate gene for Qid.ibfc-8 L.
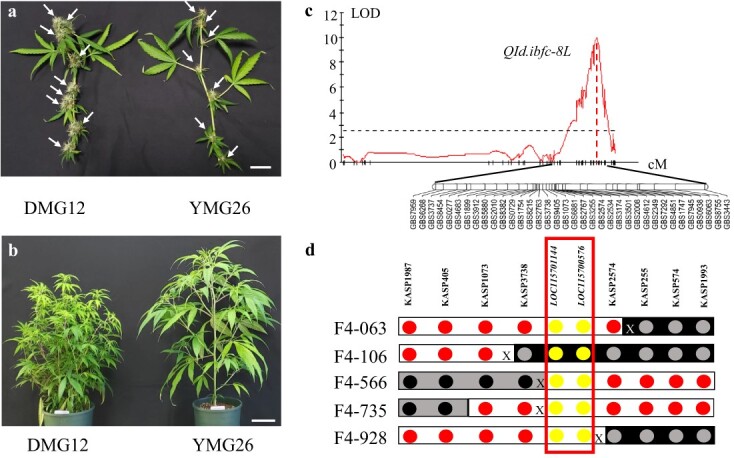
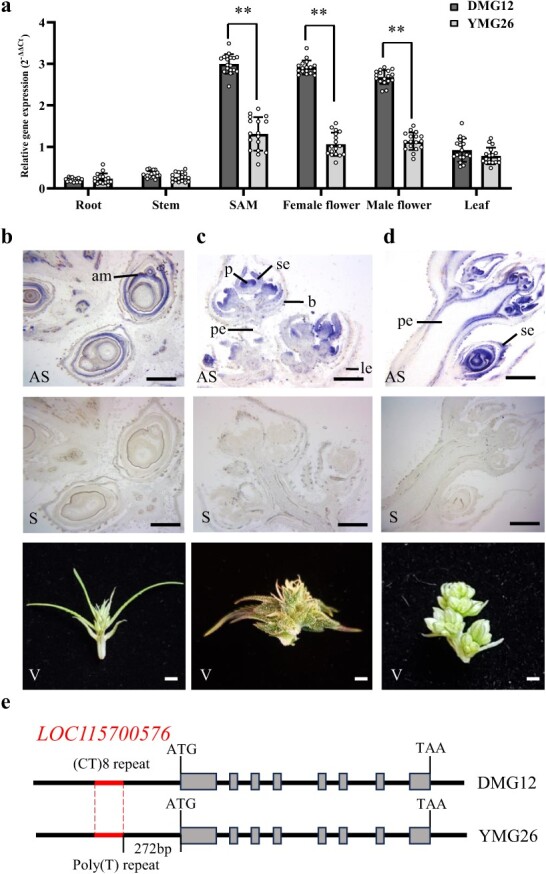
According to the NCBI Cannabis sativa.cs10 reference genome, LOC115700576 encodes a transcription factor with 269 amino acids, which belongs to the MIKC-type MADS-domain subfamily. We therefore named this gene CsMIKC1. Using the CsMIKC1 protein sequence to query the protein databases in NCBI GenBank, all the hits were hypothetical proteins (Supplementary Data Table S4). To date, CsMIKC1 or its homologous gene has not been reported to participate in the regulation of inflorescence development. The CsMIKC1 transcript levels in different tissues were evaluated using quantitative RT–PCR. In DMG12, the CsMIKC1 gene was highly expressed in shoot apical meristems (SAMs) and female and male flowers compared with the expression levels in leaf, stem, and root (Fig. 2a). Csmikc1 in YMG26 showed a similar expression pattern to CsMIKC1 in DMG12, but no significantly higher expression in the SAM or flowers compared with other organs. In situ hybridization during flower development further revealed that CsMIKC1 mRNA distribution was more abundant in floral organs and apical meristems. The signal intensity was higher on the adaxial sides of the SAM, as well as the floral meristems and young bracts, but, by contrast, the signal was weak in the peduncle, older bracts, and leaves (Fig. 2b–d). The expression patterns of CsMIKC1 suggest that this gene may have a general role in promoting cell proliferation and differentiation in floral organs and apical meristems, leading to a hypothesis that the phenotypic differences were probably determined by CsMIKC1 at the transcript level.
A key regulatory DNA element differentiated transcript levels of the two CsMIKC1 alleles
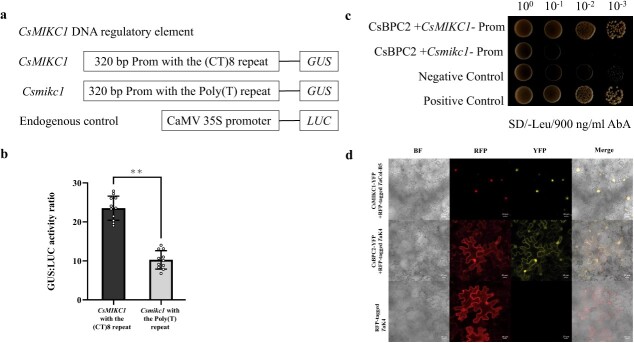
We observed that the DMG12 CsMIKC1 allele contained a (CT)8 repeat sequence in the promoter region compared with a poly(T) sequence of the YMG26 Csmikc1 allele (Fig. 2e). To test whether the (CT)8 repeat is involved in regulation of transcription, we developed two constructs to investigate the expression of the reporter gene in the GUS expression system transiently. The CsMIKC1-Prom construct includes 320 bp before the start codon from the DMG12 allele, and the Csmikc1-Prom construct includes the same region of the YMG26 allele (Fig. 3a). The two constructs had the identified promoter region (320 bp), the only difference being the (CT)8 repeat in CsMIKC1-Prom and poly(T) sequence in Csmikc1-Prom instead. As an internal control, the luciferase (LUC) gene provided an estimate of the expression efficiency. CsMIKC1-Prom and Csmikc1-Prom were co-transformed with the construct of LUC into Cannabis protoplasts. In the transformed protoplasts, CsMIKC1-Prom showed a higher expression level of GUS than Csmikc1-Prom, which lacks the (CT)8 repeat sequence (Fig. 3b). This result was the evidence that the (CT)8 repeat sequence played an essential role in regulating the transcription of CsMIKC1. To identify transcription factors that interacted with the CsMIKC1 promoter region, the promoter fragment (320 bp) was used to screen a yeast one-hybrid (Y1H) cDNA library prepared from the cultivar DMG12 (female flowers). Twenty-eight positive clones were obtained. These clones were sequenced by BLAST (Supplementary Data Table S2). A cDNA ORF encoding a putative BASIC PENTACYSTEINE2 (BPC2) protein (183 amino acids) was identified with the C-terminal conserved region of the BPC members. The interaction between CsBPC2 and the 320 bp CsMIKC1 fragment was confirmed using a full-length CsBPC2 clone (855 bp, LOC115722185) in an independent Y1H assay with the 320 bp CsMIKC1 promoter fragment (Fig. 3c). When the 320 bp Csmikc1 promoter fragment was used as a bait, CsBPC2 did not show interaction. These results indicated that CsBPC2 binds to the promoter region of CsMIKC1 but not Csmikc1 due to the presence of the (CT)8 repeat. To observe their subcellular locations, the CsBPC2 and CsMIKC1 proteins were expressed in tobacco leaf cells using pEG101-YFP (YFP, yellow fluorescent protein) vector (Fig. 3d). Significant yellow fluorescent signals of CsMIKC1 and CsBPC2 were predominantly observed on the membrane and also in the nucleus.
Genetic effects of CsMIKC1 on inflorescence architecture in transgenic Cannabis
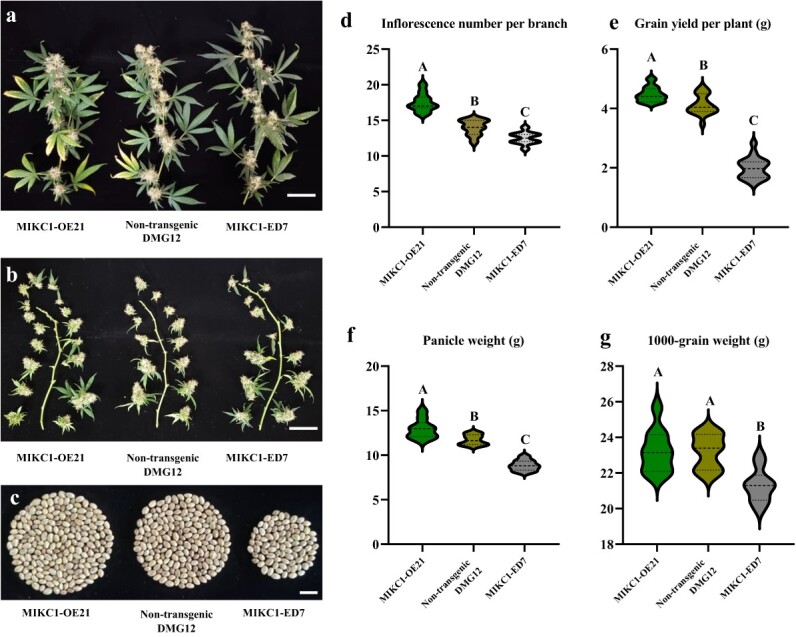
We cloned the cDNA of the CsMIKC1 allele into the pNC-Cam3304-MCS35S vector containing the constitutively expressed CaMV 35S promoter and transformed this construct into DMG12 as the host plant but not into YMG26, because it is currently not transformable. We obtained three female T0 plants, designated MIKC1-OE21, MIKC1-OE33, and MIKC1-OE56. By crossing with the male DMG12 plants, each of these T0 plants generated a T1 family. Positive T1 plants were screened with a pair of primers, OE-TEST-F1 and OE-TEST-R3. The T1 family showed a 1:1 segregation ratio between non-transgenic and transgenic plants. This result indicated the presence of CsMIKC1 expression cassette CaMV 35S-CsMIKC1-NOS integrated at a single locus. qRT–PCR was used to confirm the expression of CsMIKC1 in the transgenic Cannabis individuals (Supplementary Data Fig. S3). We also designed constructs using CRISPR-Cas9, enabling us to edit the sequence in DMG12. Two independent transgenic events (T0 plants) were obtained and designated MIKC1-ED7 and MIKC1-ED22. The MIKC1-ED7 editing event contained a 5-bp deletion. The MIKC1-ED22 editing event contained a 1-bp insertion. Each of the two editing events resulted in a frameshift (Supplementary Data Fig. S4). The MIKC1-ED7 sequence was found to have a predicted loss of 242 amino acids starting at position 27, including a loss of 51 amino acids in the MADS-MEF2-like domain, and the MIKC1-ED22 sequence had a predicted 240 amino acid loss starting at position 29, with a 49 amino acid loss in the MADS-MEF2-like domain. Transgenic plants in the greenhouse were observed with clear phenotypic segregation from the non-transgenic individuals in T1 families. Compared with the non-transgenic ones, transgenic ones overexpressing CsMIKC1 produced more inflorescences and grains, while the null CsMIKC1 mutants exhibited less compact architecture and significant reductions in inflorescence number and grain production (Fig. 4a–c). In the null CsMIKC1 mutants, the inflorescence number per branch decreased by 1.6 on average compared with the non-transgenic plants. The average weight of all the inflorescences collected from one branch (panicle weight) reduced by 24.5% in CsMIKC1 mutants (Fig. 4d–f). Plants overexpressing CsMIKC1 set 3.1 more inflorescences per branch and provided an 8.5% increase in grain yield as well as a 10.4% increase in panicle weight, compared with non-transgenic ones. The 1000-grain weight was similar between non-transgenic and transgenic plants overexpressing CsMIKC1, while the CsMIKC1 mutants exhibited a significant reduction of 8.2% compared with the non-transgenic plants (Fig. 4g).
CsBPC2 mutants exhibit inflorescence degeneration and ethylene insensitivity
To validate if CsMIKC1 expression is regulated by CsBPC2, we cloned the CsBPC2 alleles derived from DMG12 and YMG26. The sequences of the coding region and 1.5 kb before the start codon were the same in DMG12 and YMG26. Next, the transcript levels of CsBPC2 did not show significant differences in different plant tissues in DMG12 and YMG26 (Supplementary Data Fig. S5). To further examine the function of CsBPC2, we developed CsBPC2 knockout mutants in DMG12 using CRISPR-Cas9. We successfully created two CsBPC2 loss-of-function mutants (BPC2-ED2 and BPC2-ED10) using CRISPR-Cas9 genome editing. The BPC2-ED2 editing event caused a predicted loss of 163 amino acids starting at position 121, and the BPC2-ED10 editing event caused a loss of 172 amino acids starting at position 112 (Supplementary Data Fig. S6). Compared with the non-transgenic plants, the CsBPC2 mutants exhibited significant basipetal inflorescence degeneration and reduced grain production (Fig. 5a–c). Lower flower survival at the heading stage decreased the final flower and grain production in edited plants. The null CsBPC2 mutants exhibited significant reductions in inflorescence number per branch, grain yield per plant, panicle weight, and 1000-grain weight compared with the non-transgenic plants (Fig. 5d–g).
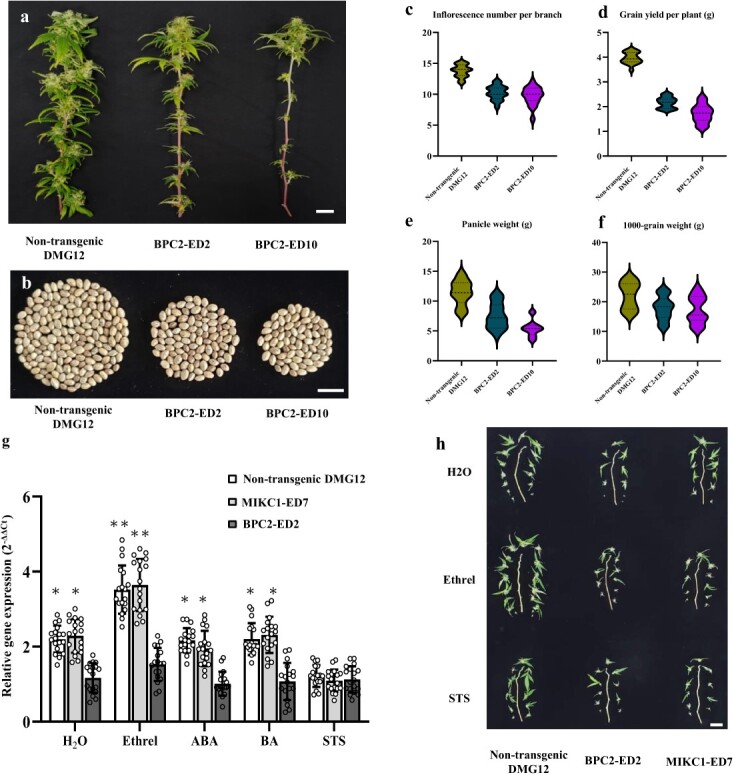
Results of previous investigations showed that BPCs are part of a complex network of transcription factors that are involved in the response to ethylene, abscisic acid (ABA), or cytokinin [ref. 11, ref. 12]. To elucidate if CsBPC2 and CsMIKC1 were linked to signaling pathways of these phytohormones, we investigated the response of CsBPC2 and CsMIKC1 to treatments with Ethrel (a kind of synthetic ethylene), silver thiosulfate (STS, an ethylene inhibitor), ABA and benzyladenine (BA, a kind of synthetic cytokinin). Solutions of these phytohormones were sprayed on the inflorescence of CsBPC2 mutants, CsMIKC1 mutants, and non-transgenic plants. Treatment with demineralized water was used as control. The qRT–PCR analysis demonstrated that CsBPC2 expression did not show significant differences after spraying these phytohormones (Supplementary Data Fig. S7). Surprisingly, after spraying Ethrel solution the transcript level of CsMIKC1 increased in the CsMIKC1 mutants and non-transgenic plants, while in the STS treatment the expression level of CsMIKC1 was significantly decreased compared with the control (Fig. 5h). The application of ABA or BA did not affect the expression level of CsMIKC1 significantly compared with the control. In the Ethrel, ABA, and BA treatments, the transcript levels of CsMIKC1 in CsBPC2 mutants were always lower than in CsMIKC1 mutants and non-transgenic plants. The application of STS mitigated the effects of ethylene on CsMIKC1 transcript in the CsMIKC1 mutants and non-transgenic plants. Compared with the water treatment, spraying Ethrel increased panicle weight by 7.96%, and the flowering dates were 4.23 days earlier, while STS greatly reduced panicle weight and caused an average delay of 3.22 days in flower initiation (Supplementary Data Table S3). In the CsBPC2 mutants and CsMIKC1 mutants, spraying phytohormones did not increase or decrease the inflorescence number compared with the water treatment. The loss of function of CsBPC2 led to reduced CsMIKC1 expression and decreased sensitivity to ethylene, and therefore supported the involvement of the CsBPC2 and CsMIKC1 genes in ethylene signaling. This study provides an example showing that spraying exogenous ethylene can be used to promote inflorescent growth and shorten the period of growing in Cannabis commercial production.
CsMIKC1 interacts with CsVIP3 in vitro and in vivo
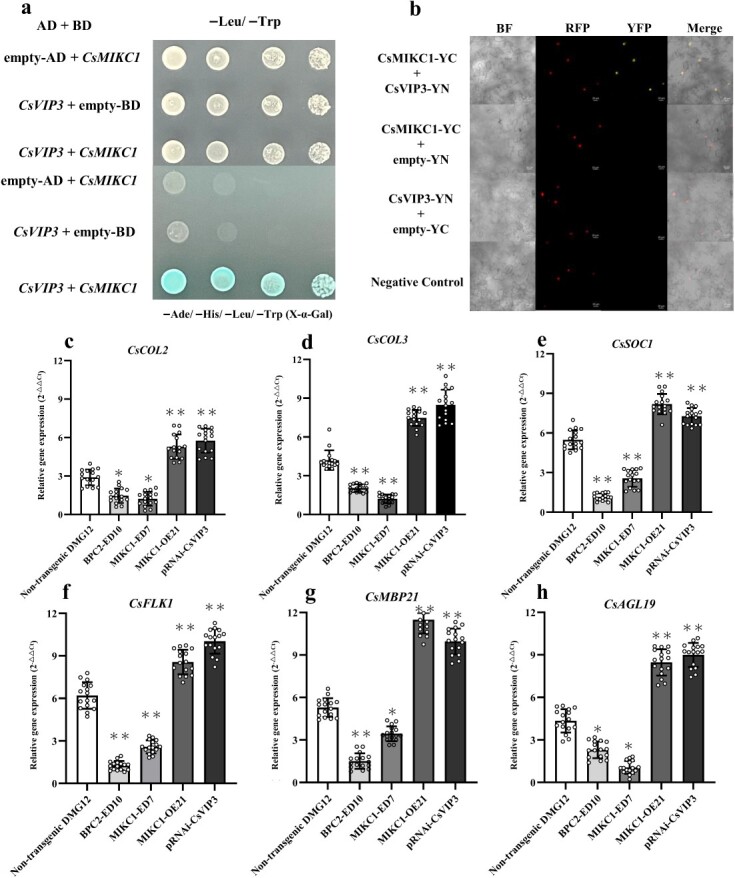
Yeast two-hybrid (Y2H) screens were performed to identify candidate genes that interact with CsMIKC1. Using a full-length CsMIKC1 cDNA as bait, 12 interacting clones were identified, rescued from yeast, and transformed into Escherichia coli. All of these clones were from the same gene, LOC115707890, located on chromosome 1, which is an ortholog of Arabidopsis VERNALIZATION INDEPENDENCE 3 (VIP3), encoding a WD40 repeat protein (GenBank accession number Q9SZQ5). The Cannabis ortholog of AtVIP3 is referred to as CsVIP3. To confirm the specificity of the observed interaction, the whole sequence of CsVIP3 cDNA was transformed back into a yeast strain containing the CsMIKC1 bait. Strains containing the CsMIKC1 bait tested positive for both X-α-Gal activity and HIS prototrophy. Strains containing the empty bait vector were negative, as they were not able to grow on plates lacking histidine and the yeast colonies were completely white in the X-α-Gal assay (Fig. 6a). We further explored the interactions between CsVIP3 and CsMIKC1 by the transient expression system in leaves of Nicotiana benthamiana (Fig. 6b). CsMIKC1 was independently fused to the C-terminal amino acid portion of YFP. CsVIP3 was fused to the N-terminal of YFP. To test their in vivo interaction, the pEarleyGate202-YC vector containing CsMIKC1 was co-transformed into the leaves of N. benthamiana with the pEarleyGate201-YN vector containing CsVIP3. Enriched yellow fluorescent signals were detected mainly in the nucleus. These results confirmed the specificity of the observed interactions between CsVIP3 and CsMIKC1.
Downstream targets regulated by CsMIKC1
Previous studies showed that MADS-box transcription factors can directly target genes by binding to the CArG box motif, and a set of genes controlling flower development were prominent members of the MADS-box network [13]. To establish a signaling network of inflorescent development, we detected which genes were affected in the MIKC1-OE21, BPC2-ED10, and MIKC1-ED7 T2 families (non-transgenic plants as control). We cloned 17 homologous genes which were commonly demonstrated to promote inflorescence development in other dicot plant species [ref. 13–15] (Supplementary Data Fig. S8), and tested their transcript levels. Compared with the expression of non-transgenic individuals, the transcript levels of CsCOL2, CsCOL3, CsSOC1, CsFLK1, CsMBP21, and CsAGL19 all increased in transgenic DMG12 plants overexpressing CsMIKC1, while the expression of these genes decreased significantly in the BPC2-ED10 and MIKC1-ED7 mutats (Fig. 6c–g). To elucidate the role of CsVIP3 in this pathway, a recombinant Agrobacterium strain carrying a RNAi construct was infiltrated in stipule segments (a component of the female flower) of DMG12 plants, which downregulated CsVIP3 expression. From the qPCR data, pRNAi-CsVIP3 treatment saw a 63% reduction in CsVIP3 transcript level compared with the plants infiltrated with disarmed Agrobacterium (Supplementary Data Fig. S9). Furthermore, qPCR results demonstrated that the expression levels of the six genes regulated by CsMIKC1 were significantly upregulated when CsVIP3 was silenced, indicating that CsVIP3 may function as a negative regulator of inflorescence development in Cannabis. This result provided experimental evidence that expressions of the six inflorescence development-related genes could be associated with the functions of CsBPC2, CsMIKC1, and CsVIP3, leading to an overall manipulation of the inflorescence architecture in Cannabis.
Discussion
We cloned the gene CsMIKC1, and elucidated the signaling networks governing the number of inflorescences per branch in Cannabis. The dominant CsMIKC1 allele contained a (CT)8 insertion in the promoter region, which is a binding site for CsBPC2, a transcription factor involved in the ethylene signaling pathway. We successfully created null CsBPC2 mutants and null CsMIKC1 mutants, which exhibited similar inflorescence growth arrest and floral degeneration. The expression level of CsMIKC1 in CsBPC2 mutants was significantly lower compared with the non-transgenic plants, providing evidence that CsBPC2 works as an upstream regulator of CsMIKC1 expression. Furthermore, we identified a flowering repressor, CsVIP3, validated its interaction with CsMIKC1 in living cells, and identified tentative target genes whose transcription could be affected.
Numerous MADS-box transcription factors have been shown to regulate flower development and growth regulation in plant species such as in rice, maize, and Arabidopsis [ref. 16, ref. 17]. OsMADS34 mutants exhibited an altered inflorescence phenotype characterized by an increased branch number, reduced spikelet count, and changes in spikelet morphology [ref. 18]. Loss of function of maize ZAG3, a homologous gene of OsMADS6, led to spikelets producing an increased number of florets with additional sterile ovaries and lemma-like organs [ref. 19]. Overexpression of the SVP-group MADS-box genes produced floral reversion and flower deformities in Arabidopsis [ref. 20]. To date, CsMIKC1 is the first MADS-box gene that has been identified to have significant effects on Cannabis inflorescence development. The cloning of CsMIKC1 provides an alternative strategy to develop new cultivars with more inflorescences and higher grain production through transformation of CsMIKC1 as a single gene in Cannabis with various genetic backgrounds. In this study, transformation of CsMIKC1 in DMG12, which has a specific genetic background adapted to the local environments, increased the inflorescence number per branch and led to a 10.4% increase in flower production as well as an 8.5% increase in grain yield. The panicle morphology of the transgenic plants indeed showed dramatic changes, but due to the limitation of available grains the CsMIKC1 effects were characterized in a 2-year experiment under controlled environmental conditions with sufficient irrigation and fertilizers. An overall evaluation of CsMIKC1 effects on inflorescence number with various genetic backgrounds and different environments is needed in future studies.
Moreover, the results indicate that both CsBPC2 and CsMIKC1 are involved in the ethylene signaling pathway to affect inflorescence development in Cannabis. BPC genes can be directly regulated by ethylene signaling in Arabidopsis, which was observed in previous studies [ref. 12, ref. 21]. Similarly, the ethylene response is diminished in CsBPC2 frameshift mutants. Foliar spraying of exogenous ethylene promoted CsMIKC1 expression in CsMIKC1 mutants and non-transgenic plants. However, the expression level of CsMIKC1 was not significantly increased or decreased in the CsBPC2 frameshift mutants, which supported the hypothesis that loss of function of CsBPC2 led to reduced ethylene sensitivity in these mutants. The effect of applications of ethylene and its inhibitor (STS) supported the biological relevance of ethylene in Cannabis flowering and inflorescence development. Prior research has elucidated the functional role of ethylene in floral promotion in fruits, such as pineapple, mango, and lychee [ref. 22]. The data suggest that spraying exogenous ethylene promoted inflorescence growth and shortened the period of growth in Cannabis. This provides an example of how exogenous ethylene can increase flower production in commercial Cannabis cultivation.
It is worth noting that CsVIP3 may function as a negative regulator of inflorescence development in Cannabis. VIP genes define a mechanism involved in multiple developmental processes, including flowering and floral development [ref. 23]. In Arabidopsis, the VIP loci encoded a group of flowering repressors previously unreported [ref. 24]. While the relationships among these genes remain largely unclear, the evidence suggested that these genes encode defined components of a protein complex. It has been demonstrated that the VIP3 gene encodes an SKI (cytoplasmic Superkiller) complex component that affects the stability of mRNA and leads to late flowering and aberrant flower development in Arabidopsis [ref. 25, ref. 26]. Our results also confirmed the specificity of the observed interactions between CsVIP3 and CsMIKC1 in vivo and in vitro, and hence CsVIP3 and CsMIKC1 may function as components of a protein complex to regulate inflorescence development in Cannabis. If this were the case, then loss of CsVIP3 function would not be expected to suppress the inflorescence development-related genes. To test this, we evaluated the expression of 17 homologous genes related to floral development in other plant species. Meanwhile, we investigated their expression in CsVIP3-silenced plants, CsMIKC1-overexpressing transgenic plants, CsMIKC1 mutants, and CsBPC2 mutants.
Notably, the expression of six genes (CsCOL2, CsCOL3, CsSOC1, CsFLK1, CsMBP21, and CsAGL19) was promoted in the lines overexpressing CsMIKC1 or silencing CsVIP3, while their expression was suppressed significantly in the CsMIKC1 mutants and CsBPC2 mutants. These genes may function in the same network to prevent or induce inflorescence development, regulated by CsBPC2, CsMIKC1, and CsVIP3 (Fig. 7). The CONSTANS-like (COL) gene family was predicted to play a core role in regulating flowering time in Cannabis [ref. 27]. As a member of the MADS-box gene family, SUPPRESSOR OF OVEREXPRESSION OF CO 1 (SOC1) homologs in Arabidopsis are known to play significant roles in regulating floral development and controlling flowering time [ref. 28]. In Rosa odorata, SOC1 represses the expression of GID1B, a gibberellin (GA) receptor involved in regulating flower development, while activating expression of FRUITFULL (FUL) and poly(A) binding (PAB), and enhancing flower initiation and seed production [ref. 29]. It is worth mentioning that FLOWERING LOCUS C (FLC), a suppressor of flower initiation, can directly repress SOC1 in the inflorescence meristem, while FLOWERING LOCUS K (FLK) primarily acts as an inhibitor of FLC expression, and hence promotes SOC1 expression [ref. 30], which explain the significant upregulation of both CsSOC1 and CsFLK1 in plants overexpressing CsMIKC1 or with silenced CsVIP3. In Arabidopsis, the genes exhibiting reduced expression in the flk mutant, such as FLOWERING LOCUS T (FT), PSEUDO-RESPONSE REGULATOR5 (PRR5), and PRR7, are recognized as positive regulators of floral initiation [ref. 31]. Silencing of MADS-box protein 21 (MBP21) can activate the expression of ten 1-aminocyclopropane-1-carboxylate synthase (ACS) genes and eight 1-aminocyclopropane-1-carboxylate oxidase (ACO) genes, and affect the ethylene and auxin levels in tomato sepals [ref. 32]. Moreover, the chloroplast content and Rubisco activity in SlMBP21 mutants are detected dramatically higher than in the wild type the Chl contents, which can improve photosynthetic efficiency in sepals [ref. 33]. AGAMOUS-LIKE 19 (AGL19), together with AGL24 and SOC1, coordinately activates the expression of floral meristem identity genes, including LEAFY (LFY), APETALA1 (AP1), and FT, to promote flowering [ref. 34]. AGL19 participates in the regulation of the flowering process through the HISTONE DEACETYLASE 9 (HDA9)-AGL19-FT model [ref. 35]. We hypothesize that the inflorescence development-related genes mentioned above could be potential components in the ethylene signaling pathway to regulate inflorescence development in Cannabis and finally contribute to diversity in yield production. Verification of their functions will be done to further understand the molecular mechanisms and regulatory network of inflorescence development in Cannabis.
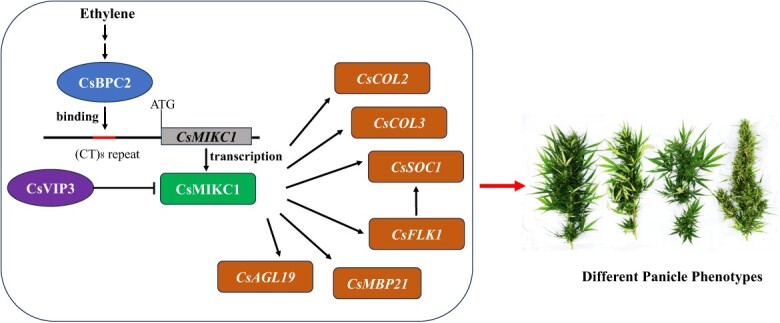
In conclusion, our findings reveal the molecular mechanisms driving the development of female flowers in Cannabis. The cloning of CsMIKC1 serves as a starting point for elucidating the functions of numerous orthologous genes involved in inflorescence development. This understanding can facilitate the modification of inflorescence architecture and maximize plant productivity in Cannabis. In addition, our findings suggest that ethylene plays a role in positively regulating Cannabis inflorescence production, and could be widely used in its commercial cultivation.
Materials and methods
Mapping and cloning of QId.Ibfc-8 L
DMG12 and YMG26 grains were provided by the national germplasm bank, Chinese Academy of Agriculture Sciences (CAAS). By inducing sex change and self-pollination, we have purified the two cultivars in eight generations separately. In addition, we have randomly genotyped 1000 individuals of each cultivar using a 20 K SNP chip that includes 20 626 SNPs (Huazhi Biotech, Changsha, China). Data showed that, in each cultivar, the heterozygosity ratio of 20 626 SNPs was <0.3%, which provided strong evidence that DMG12 and YMG26 are not highly heterozygous plants. The F1 hybrid was created by crossing DMG12 as the male parent with YMG26 as the female parent. DMG12 parental line hermaphrodites were generated by GA treatment. The concentration of GA solution was 50 μg/l. Female plants were sprayed three times before flowering and the whole treatment lasted 10 days [ref. 36] . GA solutions were applied to the F1 female plants on the first, fifth, and tenth days, after which the plants began producing male flowers at the newly formed nodes. Self-pollination of the resulting F1 plants generated 181 F2 plants, which were used for phenotyping and genotyping. The phenotypic traits of the plants were correlated with genotyping-by-sequencing (GBS) markers, leading to the mapping of QId.ibfc-8 L to the long arm of chromosome 8. This region had not been previously associated with inflorescence development. Based on the primers for KASP markers (Supplementary Data Table S1), the positional cloning approach was used to clone QId.ibfc-8 L. DMG12 and YMG26 were auto-flowering Cannabis varieties. Under long-day photoperiod conditions, DMG12 and YMG26 automatically switched from vegetative growth to the flowering stage ~70 days after planting. To benefit vegetative growth, the plants were cultivated under long-day conditions with day/night temperatures of 26/23°C and a photoperiod of 16 h light followed by 8 h of darkness prior to anthesis. Since Cannabis inflorescence can develop under short days after anthesis and cultivation under long days can get quite expensive, it was changed to short-day conditions – a photoperiod of 8 h light followed by 16 h of darkness after flowering – which was also the preferable choice in Cannabis commercial production. Plants were individually potted in a greenhouse, with each pot measuring 10 cm in diameter and 12 cm in height. We characterized the productivity and traits associated with yield components of these individual plants, as well as the number of flower nodes on each second-order branch. Since producing seeds can decrease cannabinoid contents, plants for panicle weight measurement were grown in another greenhouse to prevent cross-pollination, and the flowers at each branch were collected and weighed. Panicle weight was represented by the mean of the weight of the dried flowers in each branch plus 15% standard moisture. We calculated grain weight per plant as the average weight of dried grains adjusted to 15% standard moisture content. The 1000-grain weight was determined three times for each treatment. Morphological traits were assessed manually.
Generation of transgenic plants
The cDNA of CsMIKC1 was cloned using the primers CsMIKC1-CDNAF1 and CsMIKC1-CDNAR1 (Supplementary Data Table S1), and then transferred into the pNC-Cam3304-MCS35S vector from the laboratory of Dr Yan (ITBB, CATAS) [ref. 37]. The construct was transformed into DMG12 as the host plant but not into YMG26, because it has a low regeneration rate and is currently not transformable [ref. 9]. The expression level of CsMIKC1 in the positive transgenic plants was measured using qRT–PCR with primers named MIKC1-rt-F1 and MIKC1-rt-R1 in Supplementary Data Table S1. The pG41sg (harboring the developmental regulator genes CsGRF3 and CsGIF1) constructs were used for genome editing [ref. 9]. The gRNAs targeting CsMIKC1 and CsBPC2 were designed with CHOPCHOP (online software, https://chopchop.cbu.uib.no/). The gRNA primers were designed based on Cannabis genomic characters as well as the scores of potential off-target sites (Supplementary Data Table S1). Two pairs of primers (CsCAS9F2/R2 and CAS-TEST-F6/R6; Supplementary Data Table S1) were employed to detect positive plants harboring the genome-editing construct integrated into the Cannabis genome. Subsequently, positive plants underwent sequencing to identify deletions or insertions in the targeted region. Considering that developmental regulators may have some effect on inflorescence morphology, we modified the methodology and selected successful mutants without developmental regulators in the progeny. In the generation of transgenic plants overexpressing CsMIKC1, developmental regulators were not combined into the vector, and hence the positive individuals did not contain these developmental regulators. T2 transgenic plants of Cas9-mutants were stabilized as homozygotes by inducing sex change and self-pollination, and these homozygotes were used in relevant analyses. The genomic sequences of CsMIKC1 and CsBPC2 were amplified using PCR. Mutations were detected in the two alleles by deep sequencing. In addition, T2 transgenic plants overexpressing CsMIKC1 were confirmed by qRT–PCR and evaluated in the analysis.
Transient promoter activity assays
The CsMIKC1-Prom construct includes 320 bp before the start codon of the DMG12 allele. The Csmikc1-Prom sequence included 320 bp of promoter sequence with a poly(T) sequence instead of the (CT)8 repeat from the YMG26 allele (Fig. 3a). The CsMIKC1-Prom and Csmikc1-Prom constructs contained the 320 bp sequence, differing only in a 16-bp sequence. The CaMV 35S promoter was used to drive the LUC gene, serving as a control to estimate transient expression efficiency. The 320-bp fragments from DMG12 and YMG26 promoters were fused separately to the uidA gene, which encodes GUS as a reporter. These constructs were then cloned into the pNC-Cam3304-MCS35S vector. Primers used for cloning are listed in Supplementary Data Table S1. The CsMIKC1 promoter includes the (CT)8 repeat from the DMG12 allele and the Csmikc1 promoter includes a poly(T) sequence from the YMG26 allele, which was the only difference between the two fragments. Internal control for the CsMIKC1::GUS assay comprised the pNC-Cam3304-MCS35S construct. This construct contained the 35S promoter fused to the LUC gene. The LUC gene was cloned using primers LUC-F1/LUC-R1. We transformed the 35S::LUC construct together with the CsMIKC1-GUS or Csmikc1-GUS construct into Cannabis protoplasts. These protoplasts were isolated from SAMs using an enzyme solution that consisted of 30 mM 2-(N-morpholino) ethanesulfonic acid (Sigma), 3% cellulose (R-10, Yakult), 20 mM CaCl2 (Sigma), 15 mM KCl (Sigma), and 0.6 M d-mannitol (Sigma). The meristems were incubated with shaking at 160 rpm for 10 h at 26°C. PEG solutions were prepared for transfection, and consisted of 0.3 M mannitol, 35% PEG, and 200 mM CaCl2 [ref. 38]. The ratios of GUS to LUC were utilized to determine relative promoter activities. After transformation, the protoplasts were incubated at 26°C for 36 h in a 2-ml centrifuge tube containing lysis buffer with 0.8 mM 4-methylumbelliferyl-β-d-glucuronide (Thermo Fisher Scientific Inc.). To terminate the reaction, 0.15 M Na2CO3 was added to the reaction after 40 min. LUC activity was assessed using the Luciferase Assay System E4550 (Promega), while GUS activity was measured with a Synergy H1 reader (Agilent Technologies, CA, USA). The GUS/LUC ratio was calculated as (GUS40 min − GUS0 min) × 10/LUC.
Quantification of gene transcript levels
We extracted RNA samples from transgenic and non-transgenic plants cultivated in the greenhouse, including root, stem, SAM, female flower, male flower, and leaf tissue from adult plants. Samples were processed to extract total RNA using the EASYspin Plus RNA kit (Aidlab Biotech, Beijing, China). The extracted RNA was utilized to synthesize cDNA employing the SuperScript II Reverse kit (Thermo Fisher Scientific, CA, USA). The gene transcript levels were determined by qRT–PCR with specific primers (Supplementary Data Table S1) in a CFX Opus 384 Real-Time PCR System (Bio-Rad, CA, USA). The level of TUB expression was measured as an endogenous control for normalization of qRT–PCR data. The primers to detect the expression of the Cannabis TUB gene can be found in Supplementary Data Table S1 [ref. 1]. Gene transcript levels were quantified utilizing the 2−ΔΔCT method, with CT representing the threshold cycle [ref. 39]. The cycle difference between the target gene and TUB gene was first calculated and then the second difference of the first calculated ΔCT value between a sample and the selected control sample was calculated, which was the ΔΔCT value.
In situ RNA hybridization
The cDNA encoding the complete CsMIKC1 protein was amplified with primers CsMIKC1-CDNAF1 and CsMIKC1-CDNAR1 and cloned into pGEM-T Easy vector. This plasmid was sequenced to verify identity. For in situ hybridization, digoxigenin-labeled RNA was produced based on the instructions (Roche). The plant tissues were fixed in 0.15 M sodium phosphate buffer containing 0.15% Tween-20, 0.15% Triton X-100, 4% paraformaldehyde and 0.25% glutaraldehyde. SAM, female flower, and male flower tissues were fixed and embedded in Paraplast Plus (Sigma), sectioned at a thickness of 15 μm, and mounted on poly-l-lysine-treated slides (Thermo Fisher Scientific Inc.). Hybridization together with immunological detection were conducted following previously described methods [ref. 40].
Yeast one-hybrid screen
The tests were conducted following the method described in reference [ref. 41]. Promoter fragments of CsMIKC1 and Csmikc1 were amplified from the genomic DNA samples prepared from DMG12 and YMG26 (Supplementary Data Table S1). We fused these fragments into the pAbAi vector to generate the bait constructs. The plasmids were then digested with the restriction enzyme BstBI and integrated into the yeast strain Y1H Gold using Yeast One-Hybrid kits (Clontech). Bait constructs were used to screen the Y1H prey library constructed from the Chinese commercial cultivar YUNMA 8, which has been widely cultivated in the Yunnan Province in China. The transformants were cultured on selective medium (SD/−Lue + 1100 ng/ml AbA). The growth ability on medium can be judged by detecting the positive binding between prey and bait colonies. The positive colonies were confirmed and sequenced following previously described procedures [ref. 41]. Positive control colonies were generated by transforming yeast cells with both pAbAi-p53 and pGADT7-p53 vectors (Clontech). Negative control colonies were established by transforming the yeast cells with the pAbAi-CsMIKC1-Prom vector together with the empty pGADT7 vector.
Yeast two-hybrid screen
The full-length cDNA which encoded the complete CsMIKC1 protein was amplified by primers CsMIKC1-CDNAF1 and CsMIKC1-CDNAR1 (Supplementary Data Table S1), then cloned into the pGBKT7 vector. The vector was utilized in screening the Y2H library. The cDNA sequence was used to test autoactivation. We transformed the MIKC1-Y2H construct into the yeast strain Y187, which was used to screen a Y2H prey library constructed from YUNMA 8 as a bait, then incubated for 4 days at 32°C. Positive colonies on the culture medium were screened, confirmed, and sequenced. Fragments of the positive cDNA clones were queried against the NCBI database using BLAST to identify the proteins interacting with CsMIKC1. The interaction between CsMIKC1 and CsVIP3 was confirmed through co-transformation three times in the Y2H system [ref. 42].
In vivo protein interaction between CsMIKC1 and CsVIP3
The subcellular locations of CsMIKC1 and CsVIP3 proteins were observed in tobacco leaf cells by expressing them using a pEG101-YFP vector. To analyze subcellular localization, cDNAs were cloned into the pDONR207 vector and then transferred into pEarleygate101 vector (pEG101) using the BP and LR cloning kits (Thermo Fisher Scientific). CsMIKC1 was cloned from the pDONR207 vector and fused into the pEG202-YC vector. pEG202-YC encoded the C-terminal region of YFP. CsVIP3 was fused to the sequence in the pEG201-YN vector encoding the N-terminal portion of YFP. We co-transformed the CsMIKC1-fused pEG202-YC vector with CsVIP3-fused pEG201-YN vector into tobacco leaves to test the in vivo interaction between the two proteins. Primers for amplifying cDNAs are listed in Supplementary Data Table S1. Tobacco leaf disks were prepared and imaged following a reported protocol [ref. 42].
Silencing of CsVIP3 via transient RNAi expression
The pNC-Cam1304-RNAi vector from the laboratory of Dr Yan (ITBB, CATAS) was used within this study. The cDNA fragment of CsVIP3 was amplified with CsVIP3-RNAi-F1/CsVIP3-RNAi-R1 and cloned into the pNC-Cam1304-RNAi vector following the protocols reported previously [37]. We transformed these vectors into Agrobacterium tumefaciens strain AGL1 (Biomed®, Beijing, China) using a previously reported protocol [9]. Stipule segments (a component of the female flower) were taken from fully expanded flowers and immersed in an AGL1 suspension for 4 min at 500 mbar under vacuum pressure. The stipule material was washed with sterile water and placed on moist filter paper in a Petri dish. The Petri dish was then placed in a controlled environment room at 26°C with a 16-h photoperiod for 2 days. We extracted total RNA from the stipules and synthesized cDNA following established protocols [ref. 1].
Design of the phytohormone experiment
One week after anthesis, the BPC2-ED2 mutation line, the MIKC1-ED7 mutation line, and non-transgenic DMG12 plants were selected for the phytohormone experiment. Eighteen plants were selected for each phytohormone treatment (six plants from each line). Solutions of Ethrel (synthetic ethylene, 100 ppm), ABA (100 ppm), and BA (100 ppm) were prepared with demineralized water. Plants treated with demineralized water served as the control group. Spraying was performed in the early morning, with each plant receiving three sprays of equal solution volume at 8-day intervals. Three flowers from each plant were sampled randomly, and the relative expression level of CsMIKC1 in each data set contained a total of 18 biological samples. The flowers of each DMG12 plant were harvested 4 weeks after anthesis and panicle weight was represented by the mean of the weight of the dried flowers in each branch plus 13% standard moisture. Flowering time was defined as the time the first solitary flower developed in the axils of leaf petioles. Phytohormones were applied when the first foliar bud emerged, which was 8–10 days earlier than flowering for DMG12 and YMG26.
Supplementary Materials
References
- Selection and validation of reference genes for normalization of qRT-PCR data to study the cannabinoid pathway genes in industrial hemp.. PLoS One., 2021
- Large-scale whole-genome resequencing unravels the domestication history of Cannabis sativa.. Sci Adv., 2021
- A cell-free platform for the prenylation of natural products and application to cannabinoid production.. Nat Commun., 2009
- Screening microbially produced Δ9-tetrahydrocannabinol using a yeast biosensor workflow.. Nat Commun., 2022. [PubMed]
- A GPCR-based yeast biosensor for biomedical, biotechnological, and point-of-use cannabinoid determination.. Nat Commun., 2022. [PubMed]
- The effect of transplant date and plant spacing on biomass production for floral hemp (Cannabis sativa L.).. Agronomy., 2022
- Cannabis: chemistry, extraction and therapeutic applications.. Chemosphere., 2022
- Fiber hemp (Cannabis sativa L.) yield and its response to fertilization and planting density in China.. Ind Crop Prod., 2022
- Establishment of an Agrobacterium-mediated genetic transformation and CRISPR/Cas9-mediated targeted mutagenesis in hemp (Cannabis sativa L.).. Plant Biotechnol J., 2021. [PubMed]
- Architecture and florogenesis in female Cannabis sativa plants.. Front Plant Sci., 2019. [PubMed]
- Comparative RNA-Seq analysis reveals genes associated with masculinization in female Cannabis sativa.. Planta., 2021
- BASIC PENTACYSTEINE proteins repress ABSCISIC ACID INSENSITIVE4 expression via direct recruitment of the polycomb-repressive complex 2 in Arabidopsis root development.. Plant Cell Physiol., 2017. [PubMed]
- Molecular and genetic pathways for optimizing spikelet development and grain yield.. aBIOTECH., 2020. [PubMed]
- Dissecting the role of MADS-box genes in monocot floral development and diversity.. J Exp Bot., 2018. [PubMed]
- Beyond the genetic pathways, flowering regulation complexity in Arabidopsis thaliana.. Int J Mol Sci., 2021. [PubMed]
- Comparative genome-wide analysis of WRKY, MADS-box and MYB transcription factor families in Arabidopsis and rice.. Sci Rep., 2021. [PubMed]
- Evolutionary variation in MADS box dimerization affects floral development and protein abundance in maize.. Plant Cell., 2020. [PubMed]
- Rice SEPALLATA genes OsMADS5 and OsMADS34 cooperate to limit inflorescence branching by repressing the TERMINAL FLOWER1-like gene RCN4.. New Phytol., 2022. [PubMed]
- The wheat AGL6-like MADS-box gene is a master regulator for floral organ identity and a target for spikelet meristem development manipulation.. Plant Biotechnol J., 2022. [PubMed]
- The roles of MADS-box genes from root growth to maturity in Arabidopsis and rice.. Agronomy., 2022
- BASIC PENTACYSTEINE2 negatively regulates osmotic stress tolerance by modulating LEA4-5 expression in Arabidopsis thaliana.. Plant Physiol Biochem., 2022
- Foliar application of ethephon induces bud dormancy and affects gene expression of dormancy-and flowering-related genes in ‘Mauritius’ litchi (Litchi chinensis Sonn.).. J Plant Physiol., 2022
- A mechanism related to the yeast transcriptional regulator Paf1c is required for expression of the Arabidopsis FLC/MAF MADS box gene family.. Plant Cell., 2004. [PubMed]
- Genetic analysis of early flowering mutants in Arabidopsis defines a class of pleiotropic developmental regulator required for expression of the flowering-time switch Flowering Locus C.. Genetics., 2003. [PubMed]
- Context-dependent dual role of SKI8 homologs in mRNA synthesis and turnover.. PLoS Genet., 2012
- Transcriptional and post-transcriptional regulation of heading date in rice.. New Phytol., 2021. [PubMed]
- Genome-wide identification, expression, and sequence analysis of CONSTANS-like gene family in Cannabis reveals a potential role in plant flowering time regulation.. BMC Plant Biol., 2021. [PubMed]
- Dormancy-associated MADS-box (DAM) genes influence chilling requirement of sweet cherries and co-regulate flower development with SOC1 gene.. Int J Mol Sci., 2020. [PubMed]
- Developmental transcriptome analysis of floral transition in Rosa odorata var. gigantea.. Plant Mol Biol., 2018. [PubMed]
- Gene regulatory networks controlled by FLOWERING LOCUS C that confer variation in seasonal flowering and life history.. J Exp Bot., 2021. [PubMed]
- Photoperiodic flowering in Arabidopsis: multilayered regulatory mechanisms of CONSTANS and the florigen FLOWERING LOCUS T.. Plant Commun., 2023
- Genome editing for tomato improvement. In: Zhao. K, Mishra R, Joshi RK (eds). Genome Editing Technologies for Crop Improvement., 2022
- The MADS-box gene SlMBP21 regulates sepal size mediated by ethylene and auxin in tomato.. Plant Cell Physiol., 2017. [PubMed]
- The MADS-domain factors AGAMOUS-LIKE15 and AGAMOUS-LIKE18, along with SHORT VEGETATIVE PHASE and AGAMOUS-LIKE24, are necessary to block floral gene expression during the vegetative phase.. Plant Physiol., 2014. [PubMed]
- HISTONE DEACETYLASE 9 promotes hypocotyl-specific auxin response under shade.. Plant J., 2023. [PubMed]
- Induction of male flowers on female plants of Cannabis sativa by gibberellins and its inhibition by abscisic acid.. Planta., 1972. [PubMed]
- Nimble cloning: a simple, versatile, and efficient system for standardized molecular cloning.. Front Bioeng Biotechnol., 2020
- A versatile protoplast system and its application in Cannabis sativa L.. Botany., 2022
- Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method.. Methods., 2001. [PubMed]
- The expression of cell proliferation-related genes in early developing flowers is affected by a fruit load reduction in tomato plants.. J Exp Bot., 2006. [PubMed]
- Spatial auxin signaling controls leaf flattening in Arabidopsis.. Curr Biol., 2017. [PubMed]
- Vernalization requirement duration in winter wheat is controlled by TaVRN-A1 at the protein level.. Plant J., 2013. [PubMed]
- TaCol-B5 modifies spike architecture and enhances grain yield in wheat.. Science., 2022. [PubMed]