Network medicine approaches for identification of novel prognostic systems biomarkers and drug candidates for papillary thyroid carcinoma
Abstract
Papillary thyroid carcinoma (PTC) is one of the most common endocrine carcinomas worldwide and the aetiology of this cancer is still not well understood. Therefore, it remains important to understand the disease mechanism and find prognostic biomarkers and/or drug candidates for PTC. Compared with approaches based on single‐gene assessment, network medicine analysis offers great promise to address this need. Accordingly, in the present study, we performed differential co‐expressed network analysis using five transcriptome datasets in patients with PTC and healthy controls. Following meta‐analysis of the transcriptome datasets, we uncovered common differentially expressed genes (DEGs) for PTC and, using these genes as proxies, found a highly clustered differentially expressed co‐expressed module: a ‘PTC‐module’. Using independent data, we demonstrated the high prognostic capacity of the PTC‐module and designated this module as a prognostic systems biomarker. In addition, using the nodes of the PTC‐module, we performed drug repurposing and text mining analyzes to identify novel drug candidates for the disease. We performed molecular docking simulations, and identified: 4‐demethoxydaunorubicin hydrochloride, AS605240, BRD‐A60245366, ER 27319 maleate, sinensetin, and TWS119 as novel drug candidates whose efficacy was also confirmed by in silico analyzes. Consequently, we have highlighted here the need for differential co‐expression analysis to gain a systems‐level understanding of a complex disease, and we provide candidate prognostic systems biomarker and novel drugs for PTC.
Article type: Research Article
Keywords: co‐expressed module, differential co‐expression network analysis, drug repurposing, papillary thyroid carcinoma, prognostic systems biomarkers, text mining
Affiliations: Faculty of Health Sciences Acibadem Mehmet Ali Aydinlar University İstanbul Türkiye; Department of Bioengineering Marmara University İstanbul Türkiye; Department of Bioengineering Adana Alparslan Turkes Science and Technology University Adana Türkiye
License: © 2023 The Authors. Journal of Cellular and Molecular Medicine published by Foundation for Cellular and Molecular Medicine and John Wiley & Sons Ltd. CC BY 4.0 This is an open access article under the terms of the http://creativecommons.org/licenses/by/4.0/ License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Article links: DOI: 10.1111/jcmm.18002 | PubMed: 37859510 | PMC: PMC10746936
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (682 KB)
INTRODUCTION
Recent cancer statistics show that thyroid cancer is the most common endocrine cancer encountered by mankind. In 2022, it was estimated that 43,800 patients were diagnosed with thyroid cancer, and thyroid cancer was responsible for 2230 deaths in the United States.ref. jcmm18002-bib-0001 Papillary, follicular, medullary, and undifferentiated or anaplastic thyroid carcinomas are the major histologic groups of thyroid tumours. Among the various subgroups of thyroid cancer, papillary thyroid carcinoma (PTC) accounts for the largest proportion of all thyroid carcinomas. Notably, the prevalence of PTC is approximately 85% when all thyroid carcinomas are considered.ref. jcmm18002-bib-0002 The aetiology of thyroid cancer is not well understood. Therefore, further studies are needed to uncover the underlying mechanisms of the disease, correct over‐diagnosis, and find prognostic and/or drug candidates to reduce the prevalence of PTC.
Logarithmic advances in high‐throughput sequencing and screening methods in recent decades have produced a substantial amount of X‐ome data at various molecular levels, enabling researchers to perform various bioinformatics approaches for different diseasesref. jcmm18002-bib-0003, ref. jcmm18002-bib-0004 and also for PTC.ref. jcmm18002-bib-0005 However, some of these studies take only limited account of the reality of the molecular biochemistry of the organism. To discover specific disease biomarkers or drug targets, it is essential to evaluate the entire physical and functional architecture of the organism, because the development of an abnormal phenotype (i.e. disease) is not the result of a single gene, but rather the result of complex gene interactions. One scientific field, network medicine, allows researchers to uncover these complex interactions between biomolecules of a given phenotype in a holistic view.ref. jcmm18002-bib-0006
Gene and protein networks provide valuable data for molecular interactions within the organism, and co‐expression networks represent significantly co‐regulated groups of genes (i.e. modules). Any differentiation in gene correlations between different phenotypes can provide clues to the phenotype and supports the discovery of systems biomarkers.ref. jcmm18002-bib-0007 To reveal co‐expression relationships among genes in thyroid cancer, weighted gene co‐expression network analysis (WGCNA) has been performed in a limited number of studies.ref. jcmm18002-bib-0008, ref. jcmm18002-bib-0009 WGCNA identifies gene modules by hierarchical clustering. Due to drawbacks of the algorithm, there is still no major standard for WGCNA.ref. jcmm18002-bib-0010 Therefore, in this study, instead of using the WGCNA pipeline, we used a different bioinformatics pipeline whose efficiency has also been demonstrated in various cancers, including ovarian cancer,ref. jcmm18002-bib-0011 cervical cancer,ref. jcmm18002-bib-0012 ovarian cancer stem cells,ref. jcmm18002-bib-0013 and gastric cancer.ref. jcmm18002-bib-0014 To increase the statistical power of our analysis, unlike other studies in this study, we performed a meta‐analysis based on the integration of information from multiple studies.
Accordingly, in the present study, five transcriptome datasets from different cohorts were statistically meta‐analysed to identify differentially expressed genes (DEGs) in PTC. By using culminated common DEGs between datasets, we identified co‐expressed gene pairs that differ between PTC and control phenotypes. After identifying the major significantly co‐expressed gene pairs, we constructed co‐expression networks and determined co‐expressed modules. We evaluated the prognostic capacity of the culminated modules, and the nodes of the modules that have high prognostic capacity were used as proxies to find drug candidates. Text mining and molecular docking analyzes were used to demonstrate the novelty and efficacy of the drug candidates.
MATERIALS AND METHODS
Selection of transcriptome datasets
Five gene expression datasets from PTC, including GSE60542,ref. jcmm18002-bib-0015 GSE3678, GSE29265, GSE35570ref. jcmm18002-bib-0016 and GSE33630,ref. jcmm18002-bib-0017 were obtained from the Gene Expression Omnibus (GEO)ref. jcmm18002-bib-0018 database. When selecting the datasets, we paid attention to the following three parameters: (i) the selected datasets were from the same platform (i.e. Affymetrix array), (ii) each phenotype contained at least three samples and (iii) the cancer samples belonged to papillary histology. The five datasets obtained included 141 PTC patients and 153 matched non‐tumour controls and were used to identify co‐expressed modules during the study. In addition, the clinical information and expression values of an independent thyroid cancer dataset (THCA) were extracted from The Cancer Genome Atlas (TGCA)ref. jcmm18002-bib-0019 to pre‐clinically validate the prognostic performance of the obtained modules. Since we focus on PTC, we extracted only papillary cancer cases from the THCA‐TCGA, representing a total number of 494 PTC samples.
Identification of DEGs and pathway over‐representation analyses
In this study, we used a well‐established statistical analysis procedureref. jcmm18002-bib-0020, ref. jcmm18002-bib-0021 to identify DEGs. Briefly, raw data from each of the five datasets were normalized by calculating the Robust Multi‐Array Average (RMA) expression measureref. jcmm18002-bib-0022 implemented in the Affy packageref. jcmm18002-bib-0023 of the R/Bioconductor platform (version 4.0.2).ref. jcmm18002-bib-0024 DEGs were identified from normalized expression values using the Linear Models for Microarray Data package (LIMMA).ref. jcmm18002-bib-0025 The Benjamini‐Hochberg method was used to control for false discovery rate (FDR). The adjusted p value <0.05 was used as a cut‐off value to determine the statistical significance of the DEGs. To determine the regulatory patterns of DEGs (i.e. up‐ and down‐regulation), the fold‐change threshold was used, with a two‐fold change accepted as statistically significant. Each data set was analysed independently, and results were comparatively analysed to identify common signatures from these independent studies.
Gene set overrepresentation analyses were performed using the functional annotation tool ConsensusPathDBref. jcmm18002-bib-0026 to reveal associated biological pathways of common DEGs. The analyses preferentially used the Kyoto Encyclopedia of Genes and Genomes (KEGG)ref. jcmm18002-bib-0027 and Reactomeref. jcmm18002-bib-0028 as data sources for the pathways. The p‐values were determined using Fisher’s Exact Test, and FDR was applied to control the p‐values. An adjusted p‐value < 0.05 was considered statistically significant in overrepresentation analyses.
Identification of co‐expressed genes in cancer and control samples
To identify co‐expressed genes between PTC and control samples, an established methodref. jcmm18002-bib-0011 was performed using culminated common DEGs. According to this method, z‐score normalization was performed to eliminate batch effects of each data set, and if the common DEG occurred more than once in the same dataset, the mean expression values were calculated. Then, the correlation patterns of each common gene pair in each condition (i.e. PTC and control) were calculated using correlation coefficients. A cut‐off value for the correlation coefficient was set, corresponding to a p value of <0.05 and was used to determine the statistical significance of the pairwise correlations.
Construction of co‐expression networks and determination of differential co‐expressed network modules
To evaluate the major significantly co‐expressed genes, p‐critical values for common DEGs were calculated by integrating the obtained pairwise gene expression correlation values with Spearman correlation coefficients (SCCs). An equation for determining the p‐critical values (Pcritic), which is the cut‐off value of the correlation coefficient, can be found in Equation 1 (Equation 1).
The classification of genes and the significance of expression levels that differ in tumour and non‐tumour samples was determined by the defined threshold condition (parameter Ɛ). This threshold helps to identify differential co‐expression by revealing gene pairs that are significantly linked in the tumour samples but have an opposite direction of correlation in one of the cases. For an equation and consideration of the Ɛ parameter in this study, we used Equation 2 (Equation 2). SCCd represents the correlation coefficient of gene pairs in tumour tissue and SCCh represents the correlation coefficient of gene pairs in healthy tissue.
Prognostic performance analysis of differential co‐expressed modules
The prognostic abilities of significant co‐expressed modules were determined using independent RNA‐Seq data from TGCA (THCA‐TGCA).ref. jcmm18002-bib-0019 Clinical information on the samples was collected by TCGA to evaluate the prognostic performance of the modules. Because the most important prognostic factor in all cancers is the stage of the patient, we considered the stage of the patient in the analysis of prognostic performance. Of the 494 PTC samples we obtained from TCGA, 278 samples were stage I, 51 samples were stage II, 110 samples were stage III, 53 samples were stage IV (including IVA and IVC), and the stage was unknown for two samples, so these two samples were not included in the analyses of prognostic performance. The prognostic abilities of the modules were assessed using Kaplan–Meier plots and the log‐rank test. All analyses were performed using the Survival package in R/Bioconductor (version 4.0.2).ref. jcmm18002-bib-0024 Differential co‐expressed modules with a log‐rank p‐value of <0.05 were considered statistically significant and accepted as prognostic co‐expressed modules.
Drug repurposing and text mining analysis
Drug repurposing analysis was performed via L1000CDS2: the LINCS L1000 search engine for characteristic directional signaturesref. jcmm18002-bib-0029 using genes within the prognostic gene module. The analysis considered drugs that had an inverse effect on an entered gene expression signature, and the small molecules/drugs with the highest score (i.e. top 50) were selected for further investigation (i.e. text mining and molecular docking analysis).
To determine whether the identified drug candidates had been previously associated with PTC, the identified drug candidates were reviewed in the literature with Python using text mining techniques. Accordingly, the identified drug candidates were searched separately using the following four keywords ‘drug candidate’, ‘drug candidate + thyroid cancer’, ‘drug candidate + thyroid carcinoma’, and ‘drug candidate + cancer’ in the article abstracts and the results were analysed using the BioPython package.ref. jcmm18002-bib-0030 We then, calculated the term frequency (TF) and inverse document frequency (IDF). The TF‐IDF is calculated by multiplying the TF and IDF scores. Drugs with a ‘0’ TF‐IDF score were considered as novel drug candidates in the study.
Molecular docking simulations
The three‐dimensional (3D) crystal structure of the target proteins of the drug candidates was taken from the Protein Data Bank (PDB),ref. jcmm18002-bib-0031 while the structures of the drug candidates were taken from the PubChem databaseref. jcmm18002-bib-0032 when available. Molecular docking analyzes were performed using AutoDock Vina software.ref. jcmm18002-bib-0033 For docking simulations, we used previously known binding residues of the target proteins and calculated binding affinities (kcal/mol) to determine binding significance. In addition, the drugs with a higher TF‐IDF value were considered as positive controls. Molecular docking simulations were also performed for these drugs to compare the binding significance of the novel drug candidates.
RESULTS
Transcriptomic signatures of papillary thyroid cancer: DEGs and their biological insights
Five microarray gene expression datasets were analysed statistically and individually to identify DEGs. The number of DEGs in each data set showed a wide range from 955 to 1907 genes, and the highest number of DEGs was identified in GSE35570. While the number of up‐regulated genes in datasets GSE33630 and GSE35570 was lower than the number of down‐regulated genes, data sets GSE29265, GSE3678 and GSE60542 showed a profile with a higher number of down‐regulated genes (Figure 1A). Comparative analysis of the resulting DEGs showed that 534 DEGs were common in five transcriptome datasets. To ensure consistency and robustness of the analysis, further analysis was performed using these common DEGs (Figure 1B).
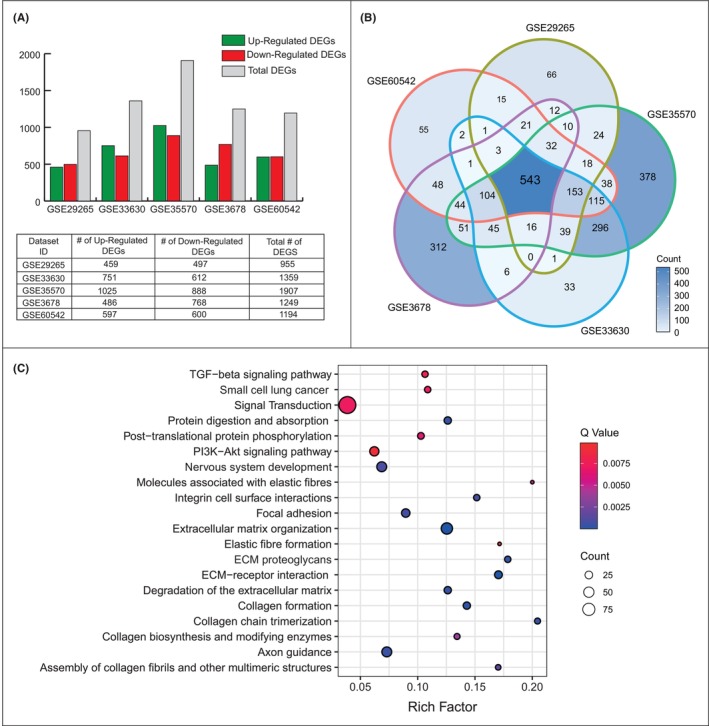
Overrepresentation analysis showed that common DEGs were associated with multiple cancer‐associated pathways (i.e. TGF‐beta pathway, PI3K‐Akt pathway and focal adhesion) and small cell lung cancer. Interestingly, we found that many collagen‐associated pathways, including collagen formation, collagen chain trimerization, collagen biosynthesis and modifying enzymes, collagen fibril assembly and other multimeric structures were significantly associated with PTC. In addition, signal transduction, extracellular matrix organization, axon guidance, and nervous system development were found to be remarkably related to the disease (Figure 1C).
Differential co‐expressed modules for papillary thyroid cancer
After identifying common DEGs in five databases, we used SCCs to identify key gene pairs that are co‐expressed and constructed networks around them. Within the constructed co‐expressed networks, a total of 12 co‐expressed modules were identified. However, considering the parameters we set for detecting highly clustered co‐expressed modules, only one module named ‘PTC‐module’ was identified within the set criteria (Figure 2A).
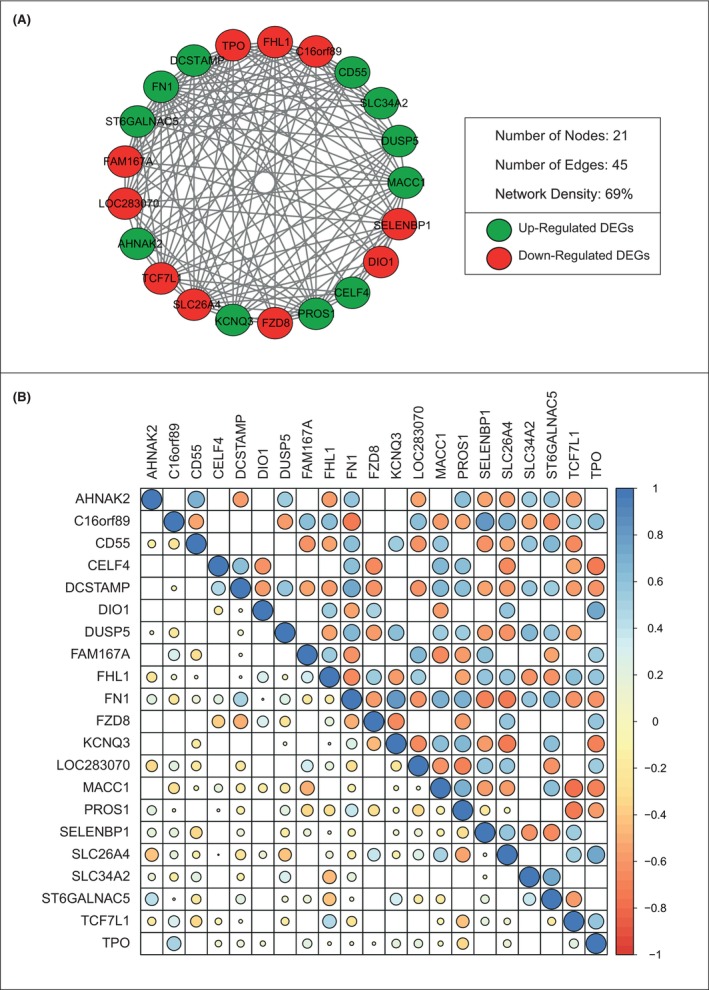
The PTC‐module consists of 21 nodes with 45 edges and has a network density of 69%. The co‐expression patterns of genes between the control and disease states showed that in the control state, 14 (9.7%) gene patterns were positively correlated and eight (5%) were negatively correlated, whereas in the disease state, 69 (48%) gene patterns were positively correlated, and 73 (51%) gene patterns were negatively correlated, indicating that disease development affects the expression of gene patterns by 83% (Figure 2B).
Prognostic performance of the co‐expressed module
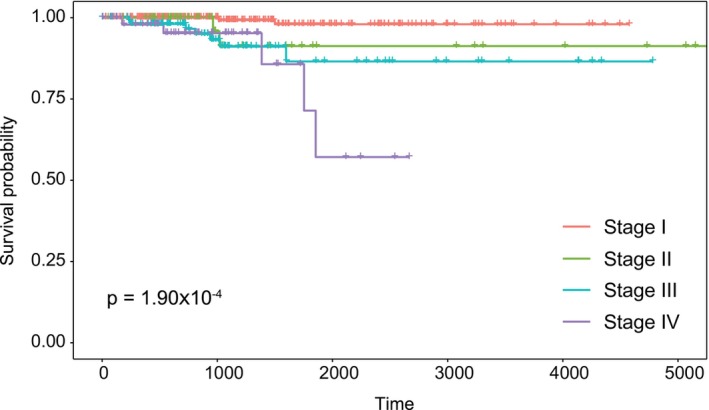
To pre‐clinically evaluate the prognostic ability of the PTC‐module, an independent RNA‐Seq dataset (THCA‐TCGA) was used, and the prognostic ability of the module was evaluated based on the disease stage of the patients using the Kaplan–Meier plot and the log‐rank test. Since we know that the most important prognostic factor in cancer patients is disease stage, we included this information in the analysis of prognostic performance. The analyzes showed that the prognostic power of the PTC‐module (log‐rank p‐value = 1.90 × 10−4) was statistically significant, so we found that this module had a high impact on the overall survival of patients according to cancer stage (Figure 3) and was considered as a prognostic systems biomarker in the study.
Identification of novel candidate repurposed therapeutic agents
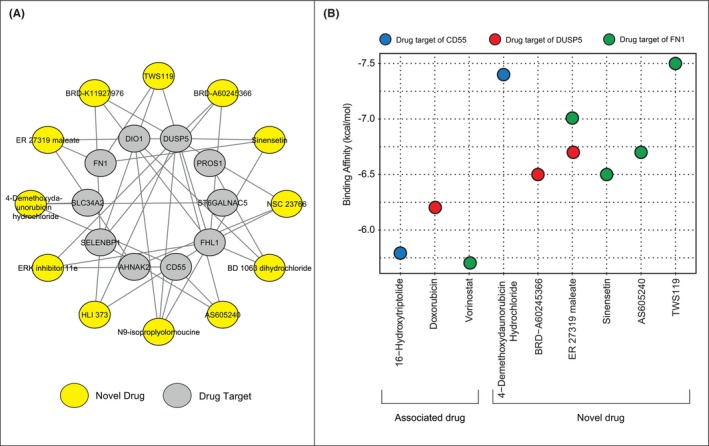
We implemented drug repurposing approach using genes in the PTC‐module using the L100CDS2 search engine. Subsequent analysis revealed that, 43 drugs had significant potential to reverse the expression profiles of differentially co‐expressed genes. To reveal the novelty of these drug candidates, the discovered drugs were examined by text‐mining analysis (Table S1). Doxorubicin hydrochloride, doxorubicin and dorsomorphin dihydrochloride yielded the higher TF‐IDF values than the other drug candidates. The drug candidates that have not yet been studied in thyroid cancer (i.e. the TF‐IDF values are ‘0’) were accepted as novel drug candidates for PTC (Figure 4A).
Molecular docking simulations
We performed molecular docking simulations to evaluate the binding affinities of the novel drugs. Of the 12 novel drugs, no 3D‐structure could be found for BD 1063 dihydrochloride, so it was not included in the docking analysis. We examined the 3D structures of PTC‐module proteins targeting 11 novel drug candidates and found suitable structures for the following proteins: (i) CD55 (PDB structure = 1H04),ref. jcmm18002-bib-0034 (ii) DUSP5 (PDB structure = 2G6Z),ref. jcmm18002-bib-0035 and (iii) FN1 (PDB structure = 6XAX).ref. jcmm18002-bib-0036 To specify the significance of affinity binding score, we used drugs with high TF‐IDF value as positive controls, namely 16‐hydroxytriptolide, doxorubicin, and vorinostat. Evaluation of molecular docking simulations revealed that the following drugs 4‐demethoxydaunorubicin hydrochloride targeting CD55 (with a binding affinity of −7.4 kcal/mol), BRD‐A60245366 targeting DUSP5 (with a binding affinity of −6.5 kcal/mol), sinensetin, AS605240, and TWS119 targeting FN1 (with a binding affinity of −6.5 kcal/mol, −6.7 kcal/mol and − 7.5 kcal/mol respectively) and ER 27319 maleate targeting DUSP5 and FN1 (with binding affinities of −6.7 kcal/mol and − 7.0 kcal/mol, respectively) have a significant docking score (kcal/mol) than the controls studied. Therefore, these six drugs represented novel drugs for the treatment of PTC in this study, and their efficiency was also demonstrated by in silico analyzes (Figure 4B).
DISCUSSION
Thyroid cancer is the most common form of endocrine cancer and PTC is the most common form of thyroid cancer. Since the true risk factors for the disease are unknown, there is still a need to identify prognostic biomarkers and improve existing treatment strategies to improve quality of life and reduce the risk of mortality. In addition, we must remember that biomolecules never function alone, but work together in a complex, interconnected network. Therefore, when discovering biomarkers or drugs, it is important not only to evaluate individual genes, but also to consider the system as a whole and to use the genes taking into account the interactions between them. Accordingly, in this study, we performed bioinformatics meta‐analyzes to identify prognostic system biomarkers and novel drug candidates for PTC.
In our analyzes based on five independent transcriptome datasets, we first attempted to detect differences between the PTC and control phenotypes (i.e. DEG analysis). To increase the reliability of the study and the robustness of the DEG results, we integrated the resulting DEGs in each dataset and focused only on the common 543 DEGs. Analysis of the overrepresentation of common DEGs revealed that several signalling pathways are already associated with thyroid cancer. Indeed, overexpression of TGF‐β1 has been reported to promote PTC cell invasion and migration.ref. jcmm18002-bib-0037 In thyroid carcinogenesis, it became clear that the PI3K/Akt pathway plays an essential role.ref. jcmm18002-bib-0038 In the progression of PTC, deregulation of focal adhesion kinase (FAK) is observed at both post‐transcriptional and translational levels.ref. jcmm18002-bib-0039 Collagen is one of the most abundantly expressed proteins in humans, and collagens are involved at every level of cancer development and establish interactions with cancer cells.ref. jcmm18002-bib-0040 One of the recent studies has shown that collagen family genes (especially COL18A1 overexpression) are associated with poor prognosis and can be used as prognostic biomarkers for PTC.ref. jcmm18002-bib-0041
After finding co‐expressed gene pairs, we created a differential co‐expression network and identified differential co‐expressed gene modules. According to certain criteria, one module, called the ‘PTC‐module,’ was found to be highly clustered and also to have high prognostic significance. When we evaluate the compromising 21 nodes of this PTC‐module, we find that several of these genes have already been individually associated with thyroid cancer, which further strengthens our confidence in our observations when we consider them as systems biomarkers. The following genes: PROS1,ref. jcmm18002-bib-0042 DCSTAMP,ref. jcmm18002-bib-0043 DUSP5,ref. jcmm18002-bib-0044 AHNAK2,ref. jcmm18002-bib-0045 SLC34A2,ref. jcmm18002-bib-0046 FN1,ref. jcmm18002-bib-0047 C16orf89,ref. jcmm18002-bib-0048 FHL1,ref. jcmm18002-bib-0049 TPO,ref. jcmm18002-bib-0050 DIO1ref. jcmm18002-bib-0051 has been associated with thyroid cancer and/or PTC.
In this study, to find novel treatment strategies for PTC, we used PTC‐module nodes as proxies and performed drug repurposing analysis, revealed the novelty of drugs by text mining and demonstrated their efficiency by molecular docking simulations. We identified a total of six novel drug candidates (4‐demethoxydaunorubicin hydrochloride, AS605240, BRD‐A60245366, ER 27319 maleate, sinensetin and TWS119) for the treatment of PTC, whose potential efficacy was also demonstrated by molecular docking simulations. 4‐Demethoxydaunorubicin hydrochloride, also known as idarubicin hydrochloride, is a type of anthracycline antibiotic used to treat acute myeloid leukaemia. To execute its anticancer effects, the drug inhibits topoisomerase II, which this resulting in impaired DNA replication.ref. jcmm18002-bib-0052 A PI3Kγ‐selective inhibitor, AS605240, has shown associations with glomerulonephritis, rheumatoid arthritis,ref. jcmm18002-bib-0053 and Alzheimer’s diseaseref. jcmm18002-bib-0054 in mouse models. BRD‐A60245366, also known as AS601245, is a JNK inhibitor and has anti‐inflammatory properties. It has been associated with colon cancer. It has been reported that the combined treatment of BRD‐A60245366 and clofibrate has an effect on colon cancer cell lines (i.e. induces cell responses).ref. jcmm18002-bib-0055 ER 27319 maleate is a selective inhibitor of Syk kinase and has been associated with head and neck cancer.ref. jcmm18002-bib-0056 Sinensetin has multiple pharmacological activities, including antineoplastic, anti‐inflammatory and antimicrobial activities. It has been associated with gastric, colon, and breast cancer.ref. jcmm18002-bib-0057 TWS119 is a potent inhibitor of GSK‐3β and it has been reported that TWS119 increases the replication and cytolytic activity of human γδT cells against human colon cancer cells. These γδT cells represent unique T cell subpopulations and can be used in cancer immunotherapy. Therefore, TWS119 has been used as a complementary agent to enhance γδT‐cell‐based immunotherapy for colorectal cancer.ref. jcmm18002-bib-0058
In conclusion, the distribution of possible significant changes in gene pairs during the transition from a normal to a tumour phenotype is of great importance. Moreover, the evaluation of the modular structures around these co‐expressed gene pairs will provide us with much more effective results in the development of diagnostic, prognostic biomarkers, and/or treatment strategies. Therefore, in this study, we investigated the different co‐expression profiles between PTC and control phenotypes. Based on this information, we proposed a module (i.e. the PTC‐module) with a high prognostic capacity and suggested it as a system biomarker. In addition, six novel drug candidates for PTC were presented in this study, which deserve to be investigated with experimental studies. The benefits of integrative bioinformatics approaches are also highlighted to develop a network medicine approach for thyroid cancer prognosis and treatment in the future.
AUTHOR CONTRIBUTIONS
Medi Kori: Data curation (equal); formal analysis (equal); methodology (equal); writing – original draft (equal); writing – review and editing (equal). Kubra Temiz: Data curation (equal); formal analysis (equal); writing – original draft (equal). Esra Gov: Conceptualization (equal); methodology (equal); supervision (equal); writing – review and editing (equal).
CONFLICT OF INTEREST STATEMENT
The authors confirm that there are no conflicts of interest.
Supplementary Materials
References
- Cancer statistics, 2022.. CA Cancer J Clin., 2022. [DOI | PubMed]
- Papillary thyroid cancer‐aggressive variants and impact on management: a narrative review.. Adv Ther., 2020. [DOI | PubMed]
- Bioinformatics prediction and machine learning on gene expression data identifies novel gene candidates in gastric cancer.. Genes., 2022. [PubMed]
- HPV16 status predicts potential protein biomarkers and therapeutics in head and neck squamous cell carcinoma.. Virology., 2023. [DOI | PubMed]
- Systems biomarkers for papillary thyroid cancer prognosis and treatment through multi‐Omics networks.. Arch Biochem Biophys., 2022. [DOI | PubMed]
- Network medicine: a network‐based approach to human disease.. Nat Rev Genet., 2011. [DOI | PubMed]
- Differential Co‐expression analyses allow the identification of critical Signalling pathways altered during tumour transformation and progression.. Int J Mol Sci., 2020. [DOI | PubMed]
- Identifying gene modules of thyroid cancer associated with pathological stage by weighted gene Co‐expression network analysis.. Gene., 2019. [DOI | PubMed]
- Research on a weighted gene Co‐expression network analysis method for mining pathogenic genes in thyroid cancer.. PloS One., 2022. [DOI | PubMed]
- K‐module algorithm: an additional step to improve the clustering results of WGCNA Co‐expression networks.. Genes., 2021. [DOI | PubMed]
- Differential Co‐expression analysis reveals a novel prognostic gene module in ovarian cancer.. Sci Rep., 2017. [PubMed]
- novel genomic biomarker candidates for cervical cancer As identified by differential Co‐expression network analysis.. Integr Biol., 2019. [DOI]
- Co‐expressed functional module‐related genes in ovarian cancer stem cells represent novel prognostic biomarkers in ovarian cancer.. Syst Biol Reprod Med., 2020. [DOI | PubMed]
- forecasting gastric cancer diagnosis, prognosis, and drug repurposing with novel gene expression signatures.. Integr Biol., 2022
- Revisiting the transcriptional analysis of primary tumours and associated nodal metastases with enhanced biological and statistical controls: application to thyroid cancer.. Br J Cancer., 2015. [DOI | PubMed]
- Gene signature of the post‐Chernobyl papillary thyroid cancer.. Eur J Nucl Med Mol Imaging., 2016. [DOI | PubMed]
- A general method to derive robust organ‐specific gene expression‐based differentiation indices: application to thyroid cancer diagnostic.. Oncogene., 2012. [DOI | PubMed]
- NCBI GEO: Archive for Functional Genomics Data Sets—Update.. Nucleic Acids Res., 2013. [DOI | PubMed]
- The cancer genome atlas (TCGA): an immeasurable source of knowledge.. Contemp Oncol (Pozn)., 2015. [DOI | PubMed]
- Multiomics data integration identifies new molecular signatures for abdominal aortic aneurysm and aortic occlusive disease: implications for early diagnosis, prognosis, and therapeutic targets.. Omics., 2022. [DOI | PubMed]
- Drug repurposing for rheumatoid arthritis: identification of new drug candidates via bioinformatics and text mining analysis.. Autoimmunity., 2022. [PubMed]
- A comparison of normalization methods for high density oligonucleotide Array data based on variance and bias.. Bioinformatics (Oxford, England)., 2003. [DOI | PubMed]
- Affy—Analysis of Affymetrix GeneChip Data at the Probe Level.. Bioinformatics (Oxford, England)., 2004. [DOI | PubMed]
- Orchestrating high‐throughput genomic analysis with Bioconductor.. Nat Methods., 2015. [DOI | PubMed]
- Limma powers differential expression analyses for RNA‐sequencing and microarray studies.. Nucleic Acids Res., 2015. [DOI | PubMed]
- ConsensusPathDB—a Database for Integrating Human Functional Interaction Networks.. Nucleic Acids Res., 2009. [DOI | PubMed]
- KEGG: Kyoto encyclopedia of genes and genomes.. Nucleic Acids Res., 2000. [DOI | PubMed]
- The Reactome pathway knowledgebase.. Nucleic Acids Res., 2016. [DOI | PubMed]
- L1000CDS2: LINCS L1000 characteristic direction signatures search engine.. Npj Systems Biology and Applications., 2016. [DOI]
- Biopython: freely available python tools for computational molecular biology and bioinformatics.. Bioinformatics., 2009. [DOI | PubMed]
- Trendspotting in the protein data Bank.. FEBS Lett., 2013. [DOI | PubMed]
- PubChem 2019 update: improved access to chemical data.. Nucleic Acids Res., 2019. [DOI | PubMed]
- AutoDock Vina: improving the Speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading.. J Comput Chem., 2010. [DOI | PubMed]
- Mapping CD55 function.. J Biol Chem., 2003. [DOI | PubMed]
- Crystal structure of the catalytic domain of human DUSP5, a dual specificity MAP kinase protein phosphatase.. Proteins: Structure, Function, and Bioinformatics., 2006. [DOI]
- Structural insights into fibronectin type III domain‐mediated signaling.. J Mol Biol., 2007. [DOI | PubMed]
- TGF‐Β1 promotes the invasion and migration of papillary thyroid carcinoma cells by inhibiting the expression of LncRNA‐NEF.. Oncol Lett., 2019. [DOI | PubMed]
- The PI3K/Akt pathway in tumors of endocrine tissues.. Front Endocrinol., 2015. [DOI]
- Focal adhesion kinase splicing and protein activation in papillary thyroid carcinoma progression.. Histochem Cell Biol., 2022. [DOI | PubMed]
- Targeting type I collagen for cancer treatment.. Int J Cancer., 2022. [DOI | PubMed]
- Identification of the collagen family as prognostic biomarkers in papillary thyroid carcinoma.. Endocrine., 2022. [DOI | PubMed]
- Aberrant expression of PROS1 correlates with human papillary thyroid cancer progression.. PeerJ., 2021. [DOI | PubMed]
- Upregulation of Dendrocyte‐expressed seven transmembrane protein is associated with unfavorable outcomes in differentiated thyroid cancer.. Endocrine., 2023. [DOI | PubMed]
- DUSP5 and DUSP6, two ERK specific phosphatases, are markers of a higher MAPK signaling activation in BRAF mutated thyroid cancers.. PloS One., 2017. [DOI | PubMed]
- AHNAK2 promotes thyroid carcinoma progression by activating the NF‐ΚB pathway.. Life Sci., 2021. [PubMed]
- SLC34A2 simultaneously promotes papillary thyroid carcinoma growth and invasion through distinct mechanisms.. Oncogene., 2020. [DOI | PubMed]
- FN1 promotes thyroid carcinoma cell proliferation and metastasis by activating the NF‐Κb pathway.. Protein Pept Lett., 2023. [DOI | PubMed]
- Initial characterization of C16orf89, a novel thyroid‐specific gene.. Thyroid: Official Journal of the American Thyroid Association., 2010. [DOI | PubMed]
- Four and a half LIM domains protein 1 can Be as a double‐edged sword in cancer progression.. Cancer Biol Med., 2020. [DOI | PubMed]
- The Value of Thyroperoxidase as a Prognostic Factor for Differentiated Thyroid Cancer—a Long‐Term Follow‐up Study.. Thyroid Res., 2015. [DOI | PubMed]
- A study of the role of DIO1 and DIO2 polymorphism in thyroid cancer and drug response to therapy in the Saudi population.. Saudi Pharmaceutical Journal: SPJ: The Official Publication of the Saudi Pharmaceutical Society., 2019. [DOI | PubMed]
- Conventional and high‐dose daunorubicin and idarubicin in acute myeloid leukaemia remission induction treatment: a mixed treatment comparison meta‐analysis of 7258 patients.. Hematol Oncol., 2015. [DOI | PubMed]
- Phosphatidylinositol 3‐kinase inhibitors: promising drug candidates for cancer therapy.. Cancer Sci., 2008. [DOI | PubMed]
- Phosphoinositide 3‐kinase inhibitor AS605240 ameliorates Streptozotocin‐induced Alzheimer’s disease like sporadic dementia in experimental rats.. EXCLI J., 2020. [DOI | PubMed]
- AS601245, an anti‐inflammatory JNK inhibitor, and Clofibrate have a synergistic effect in inducing cell responses and in affecting the gene expression profile in CaCo‐2 colon cancer cells.. PPAR Research., 2012. [DOI | PubMed]
- Spleen tyrosine kinase expression is correlated with human papillomavirus in head and neck cancer.. Oral Oncol., 2020. [DOI | PubMed]
- Therapeutic effectiveness of Sinensetin against cancer and other human complications: a review of biological potential and pharmacological activities.. Cardiovasc Hematol Disord Drug Targets., 2022. [DOI]
- Wnt pathway activator TWS119 enhances the proliferation and Cytolytic activity of human ΓδT cells against colon cancer.. Exp Cell Res., 2018. [DOI | PubMed]