
Mapping and characterization of a novel powdery mildew resistance locus (PM2) in Cannabis sativa L.
Abstract
Introduction:
Breeding genetic resistance to economically important crop diseases is the most sustainable strategy for disease management and enhancing agricultural and horticultural productivity, particularly where the application of synthetic pesticides is prohibited. Powdery mildew disease, caused by the biotrophic fungal pathogen Golovinomyces ambrosiae, is one of the most prevalent threats to the cannabis and hemp industry worldwide.
Methods:
In this study, we used bulked-segregant analysis combined with high-throughput RNA sequencing (BSRSeq) to identify and map a novel single dominant resistance (R) locus (designated PM2), that strongly suppresses powdery mildew infection and sporulation in Cannabis sativa.
Results and discussion:
BSA mapped PM2 to chromosome 9. Histochemical analysis revealed that PM2-induced resistance is mediated by a highly localized hypersensitive response mainly in the epidermal cells of the host. Importantly, genetic markers capable of tracking PM2 resistance in breeding populations were developed using associated SNPs identified in this study. The ability to track PM2 will allow for successful introgression of PM resistance into elite cannabis cultivars and help move towards a more sustainable cannabis industry.
Article type: Research Article
Keywords: powdery mildew, disease resistance, genetic mapping, bulked-segregant analysis, marker assisted selection, plant breeding, sustainable agriculture
Affiliations: Breeding and Genetics Department, Aurora Cannabis, Inc., Comox, BC, Canada; Department of Botany, University of British Columbia, Vancouver, BC, Canada
License: Copyright © 2025 Seifi, Leckie, Giles, O’Brien, MacKenzie, Todesco, Rieseberg, Baute and Celedon CC BY 4.0 This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
Article links: DOI: 10.3389/fpls.2025.1543229 | PubMed: 40182551 | PMC: PMC11966446
Relevance: Relevant: mentioned in keywords or abstract
Full text: PDF (8.2 MB)
Introduction
Cannabis sativa L., commonly known as cannabis (marijuana or hemp) is a dioecious, diploid (2n=20), annual, flowering plant species belonging to the Cannabaceae family cultivated for its seed, oil, fiber, and bioactive compounds including cannabidiolic acid (CBDA) and tetrahydrocannabinolic acid (THCA) which have medicinal and psychoactive properties (ref. Pate, 1983; ref. Chandra et al., 2017; ref. Radwan et al., 2017; ref. Kumar et al., 2021). Following the legalization of medicinal and recreational uses of cannabis in many countries in the last few years, cannabis cultivation and related industries have seen a significant expansion (ref. López-Ruiz et al., 2022). Plant diseases caused by fungal, bacterial, and viral pathogens are among the most important factors that threaten cannabis production (ref. Punja, 2021). Powdery mildew (PM) is a widespread and economically damaging fungal disease affecting many indoor, greenhouse, and field grown cannabis crops around the world, including the cannabis industry in Canada and the USA (ref. Pépin et al., 2021). PM disease in cannabis is mainly caused by the obligate biotrophic fungal pathogen Golovinomyces ambrosiae, previously known as G. cichoracearum (ref. Pépin et al., 2018; ref. Scott and Punja, 2020; ref. Brochu et al., 2022). PM infection attacks the leaves, stems and flowers of cannabis, restricting photosynthesis and nutrient availability, causing premature leaf drop, poor flower quality and significant yield losses (ref. Mihalyov and Garfinkel, 2021; ref. Scott and Punja, 2020). The infection cycle in PM has three main phases: (i) conidium (spore) germination and epidermal penetration; (ii) mycelial network development on the host leaf; and (iii) conidia generation (asexual reproduction, also known as conidiation or sporulation) (ref. Hückelhoven, 2005). Under optimal conditions, i.e. high humidity and moderate temperature, and access to susceptible cannabis cultivars, PM can complete its life cycle within 1-2 weeks post inoculation (wpi).
PM control in other crops is achieved by application of chemical fungicides, biological control agents, and agricultural practices; however, use of genetically resistant cultivars has historically been the most sustainable, effective, and economical approach in important crops such as wheat, barley and tomato (ref. Agrios, 2005; ref. Jørgensen and Wolfe, 1994; ref. Seifi et al., 2014; ref. Bapela et al., 2023). In countries with legal cannabis markets, use of agrochemicals and biological products are tightly controlled by regulatory agencies, and the few available products often exhibit limited protection (ref. Mihalyov and Garfinkel, 2021; ref. Scott and Punja, 2020). Therefore, developing commercial cultivars with genetic resistance to PM remains a highly valuable and sought-after goal in the cannabis industry (ref. Sirangelo, 2023).
The plant immune system comprises several layers of constitutive and inducible defense mechanisms. Constitutive physical and biochemical barriers make up the outer layer of these defenses and can effectively suppress most pathogens. Nevertheless, those few pathogens that succeed in breaking through the preformed defenses will be dealt with by a complex defense machinery induced by the perception of the invading pathogen. Two main mechanisms are involved in the perception of an invading pathogen and the induction of an effective immune response: (i) recognition of pathogen-associated molecular patterns (PAMPs) by membrane-bound receptors leading to PAMP-triggered immunity (PTI), and (ii) detection of specific pathogen-derived effector proteins leading to effector-triggered immunity (ETI). Disease resistance (R) genes typically encode specific nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins that can interact with and detect pathogen-derived effector proteins to induce an ETI response (ref. Jones and Dangl, 2006; ref. Hammond-Kosack and Kanyuka, 2007). Triggering of ETI causes the activation of defense-associated hormonal pathways, typically salicylic acid (SA), and several downstream genes coding for defense responses, such as the hypersensitive response (HR), production of antimicrobial pathogenesis related proteins (PR proteins), and cell wall fortifications, that ultimately deprive the pathogen of the plant’s nutritional resources (ref. Hammond-Kosack and Kanyuka, 2007; ref. Shamrai, 2022). R genes are often present in tandem arrays conferring vertical or qualitative resistance in the progeny through dominant Mendelian modes of inheritance where the effect of the dominant (resistance) allele can mask the effect of the recessive (susceptible) one. Qualitative resistance to PM has been reported in hops (Humulus lupulus), which is closely related to cannabis (ref. Henning et al., 2017). The first report of a putative R gene mediated resistance against PM in cannabis (named PM1) suggested its location to be on chromosome 2 (Chr ID: NC_044375.1; GenBank acc. no. GCA_900626175.2) (ref. Mihalyov and Garfinkel, 2021). Additionally, a mutation in the susceptibility (S) gene “Mildew Locus O” (mlo-mediated loss of susceptibility) has been reported to induce strong resistance to PM in cannabis (ref. Stack et al., 2024).
Bulked-segregant analysis (BSA) coupled with high throughput sequencing has become a popular method for quantitative trait locus (QTL) mapping and is widely used for mapping disease resistance loci within economically important crops (ref. Takagi et al., 2013; ref. Win et al., 2017; ref. Imerovski et al., 2019; ref. Shen et al., 2019; ref. Liang et al., 2020), including those effected by powdery mildew (ref. Ma et al., 2021; ref. Cao et al., 2021). BSA involves the creation of a bi-parental segregating population where both parents display opposing phenotypes for a quantitative or qualitative trait of interest. Unlike traditional QTL mapping where all individuals in a segregating population are genotyped, BSA involves selecting individuals with opposing values for qualitative traits, or individuals from both tails of the distribution for quantitative traits. Selected individuals are grouped into two bulks, representing the extremes of the target trait. This makes BSA attractive for mapping disease resistance QTLs as bulks can be easily made from resistant and susceptible plants. Genotyping in BSA is limited to the two bulks of plants, drastically reducing sequencing cost. Furthermore, several methods of BSA have been developed to utilize single nucleotide polymorphisms (SNPs) called from RNA-Seq data, referred to as BSR-Seq, which provides the required read depth and gene expression data for a lower cost compared to DNA sequencing (ref. Liu et al., 2012; ref. Hill et al., 2013).
Herein, we report the discovery of a novel single dominant PM resistant locus (PM2) that confers strong resistance to PM disease in cannabis. We present evidence indicating that PM2 acts through the induction of a highly localized ROS accumulation in the epidermal and mesophyll layers of the host leaf tissue resulting in HR, the arrest of the pathogen growth, and suppression of its sporulation. We identified two genotypes containing PM2 within a large cannabis diversity population screened for PM resistance. Using BSR-Seq, we mapped PM2 to chromosome 9 (Chr ID: NC_083609.1, Pink Pepper reference genome) and developed genetic markers that can be used to introgress PM2 resistance into elite cannabis commercial cultivars.
Materials and methods
Plant material
A large diversity population of 510 genotypes called CanD (Cannabis Diversity), including the parental lines W03, N88, and AC, is part of Aurora’s germplasm and is maintained as mother plants to provided clones for experiments. The W03xAC and N88xAC F1 mapping populations were generated form a PM-susceptible (AC) parent and PM-resistant (W03 and N88) parents, all described as female, type-I (THC-dominant) drug-type cannabis. Selfed (S1) progeny were generated by applying a foliar spray of silver thiosulfate (Millipore Sigma) at a concentration of 0.02M to clones from W03 and N88 parental lines on days 1 and 8 of a short-day photoperiod treatment to induce male flower formation and self-pollination (ref. Mohan Ram and Sett, 1982).
PM infection assays, disease index, and plant growth conditions
All CanD genotypes were evaluated for PM susceptibility using a clone assay, following a completely randomized design (CRD) with at least 6 replicates per genotype. Plantlets were rooted for 14 days in rockwool cubes and inoculated 3 weeks after cloning. Inoculation of healthy plantlets were carried out through “dusting”, whereby fungal spores from a sporulating infected leaf are transferred by tapping the leaf and depositing fresh spores onto the surface of a non-infected leaf. PM spores were sourced from highly infected plants kept in isolation. Disease evaluation and scoring was performed at 4 wpi. Similar methods of inoculation and disease evaluation were used for seedling infection trials. To mimic a production cycle from cloning to harvest, an adult plant assay was performed over the course of a 12-week period. In all assays, plants were grown at 23°C and 80% relative humidity (RH) for the first 48 h (to ensure high levels of spore germination), and then kept at 70% RH during the rest of the infection trial. Photoperiod was 16 h of light and 8 h of dark for clonal propagation and rooting, vegetative growth during the clone assay, and the first two weeks of the adult plant assay. After 2 weeks of growth under vegetative conditions, photoperiod was changed to 12 h of light and 12 h of dark for 10 weeks to induce flowering in the adult plant assay.
Disease severity was assessed with a “disease index” following a method previously developed (ref. Seifi et al., 2013a, ref. 2021), using qualitative severity scores from zero to four according to the area covered by colonies with PM sporulation (“0”, healthy leaves with no signs of infection; “1”, less than 25% coverage; “2”, 25-50% coverage; “3”, 50-75% coverage; and “4”, 75-100% coverage). Disease index was calculated for each genotype by scoring the disease severity in the three most infected leaves from the three most infected plants per genotype using the following formula:
where DI shows the disease severity in percentage; are the number of leaves with disease severity scores of 0, 1, 2, 3 and 4 respectively; and N is the total number of leaves evaluated. Disease severity was scored at 4 wpi for clone and seedling assays, and at 12 wpi for adult plant assay.
Microscopy
The accumulation of hydrogen peroxide was visualized using 3,3-diaminobenzidine (DAB) staining following the protocol devised by ref. Thordal-Christensen et al. (1997). Briefly, samples were treated with DAB-HCl (1 mg/ml) for 3 hours before being fixed in 100% ethanol for downstream microscopy. Fungal structures were stained with 0.1% trypan blue in 10% acetic acid following the protocol described by ref. Seifi et al. (2013a). All staining protocols were followed by extensive rinsing steps in demineralized water and samples were subsequently mounted in 50% glycerol before brightfield microscopic observations using an Olympus (Tokyo, Japan) CX443 microscope.
RNA isolation
W03xAC and N88xAC F1 mapping populations were grown from seed in greenhouse conditions under 18 hours of light. At 3 weeks, plants were manually infected with PM by dusting fresh spores as described in the clone infection assay. Evaluations and tissue sampling were conducted at week 7 (4 wpi). For each F1 population, 1 g of leaf sample per plant from 25 resistant and 25 susceptible plants were collected and flash frozen in a slurry of isopropanol and dry ice. Bulks of frozen leaf samples from each population were separately ground in a mortar with liquid nitrogen and 200 mg of ground material was used for RNA isolation using the PureLink™ Plant RNA Reagent (ThermoFischer Sci.) following the manufacturer small scale protocol. RNA quality and concentration were assessed using an Agilent Technologies 2100 Bioanalyzer. Only samples with high quality RNA (RNA integrity numbers > 7) were used for RNA sequencing.
RNA-Seq
Construction of mRNA libraries and sequencing was performed at SBME-Seq center at the School of Biomedical Engineering, University of British Columbia. Each library was sequenced to a depth of ~20 million PE-reads (150 bp long), using an Illumina NextSeq2000. Library quality and presence of adaptors in raw RNA-Seq reads were analyzed using FastQC (ref. Andrews, 2010). Skewer (ref. Jiang et al., 2014) was used to trim reads to a Phred score no less than Q28. STAR (version 2.7.11a, ref. Dobin et al., 2013) was used to map processed RNA-Seq reads to the Pink Pepper (GenBank acc. no. GCA_029168945.1; ref. Ryu et al., 2024) and CBDRx (GenBank acc. no. GCA_900626175.2; ref. Grassa et al., 2021) reference genomes. Mapped RNA-Seq reads were assembled into transcripts and quantified for gene expression using Stringtie v2.1.4 (ref. Kovaka et al., 2019). To calculate the relative expression difference between susceptible and resistant bulks for PM2 gene candidates, the mean gene expression between the W03xAC and N88xAC populations was used. Gene expression was measured in Transcripts per Million (TPM), reported by Stringtie. Relative expression difference between bulks for PM2 candidate genes was calculated as:
RED is the relative expression difference, where TPMResistant Bulk and TPMSusceptible Bulk are the average gene expression between both W03xAC and N88xAC population for the resistant and susceptible bulks respectively.
Variant calling
SNP calling was performed by following The Broad Institute’s best practices for RNA-Seq short variant discovery (SNPs + Indels) using GATK (versions 4.3, ref. McKenna et al., 2010). GATK’s Picard implemented MarkDuplicates and SplitNCigarReads tools were used for marking read duplicates and splitting intron spanning reads, respectively. Read duplicates were only marked for aiding variant calling and not removed. GATK’s HaplotypeCaller was used to call variants in each bulk. Resulting variants that did not meet the following quality statistics were removed: quality (QUAL) > 30, quality by depth (QD) > 2.0, Strand Odds Ratio (SOR)< 3.0, FisherStrand (FS)< 60.0, RMS Mapping Quality (MQ) > 40.0, Mapping Quality Rank Sum Test (MQRankSum) > -12.5, and Read Position Rank Sum Test (ReadPosRankSum) > -8.0. Lastly, only bi-allelic (SNPs) variants were selected for BSR-Seq analysis.
Bulked segregant RNA-Seq
BSR-Seq was conducted using a custom in-house R program written to implement both the Euclidean distance metric developed by ref. Hill et al. (2013) and the Bayesian statistical approach developed by ref. Liu et al. (2012). Both methods rely on allele read counts from both susceptible and resistant bulks to determine genetic linkage between markers and the causal gene of a trait of interest. SNPs common to both bulks (susceptible and resistant) were used for analysis. SNPs called from read counts of less than 20 reads were filtered out as their allelic frequencies cannot be accurately measured. The empirical Bayesian approach by ref. Liu et al. (2012) estimates the conditional probability of no recombination taking place between a SNP and the causal gene in the resistance bulk, given the allelic read counts of each SNP. We incorporated the R script written by ref. Liu et al. (2012) that implements the Bayesian approach into our pipeline, adjusting two parameters: the number of plants per bulk, 25, and the total genetic length, which for Cannabis has been reported to be 818cM (ref. Grassa et al., 2021). The Euclidean distance method was implemented as described by ref. Hill et al. (2013) using the following formula:
where each letter represents the allelic frequency of its corresponding nucleotide, with subscripts Res and Sus indicating the phenotypic pool, resistant and susceptible respectively.
Candidate gene selection
Candidate genes identified within the PM2 QTL region were selected based on the following criteria. Genes with annotations in the Pink Pepper reference genome known to be involved in disease response in other species were identified and selected for further analysis. To confirm the quality of gene annotations in the region of interest, we took the CDS sequence from genes having annotations related to disease response and blast them to Arabidopsis protein sequences to assess the quality of their predicted annotation. Furthermore, we searched and selected genes with conserved leucine-rich repeats (LRR) domains as probable gene candidates due to LRRs known roles in characterized R-genes. Finaly, an extensive literature review was conducted to identify homologs of candidate genes that had been shown to play a role in disease response in other plant species.
DNA isolation
Single leaflets were collected from 10-14 day old seedlings into 96-well deepwell plates containing 2mm borosilicate beads. Leaflets were lyophilized overnight, then ground into powder using a QIAGEN TissueLyser II. DNA purification was done following standard extraction protocol with a sbeadex Mini Plant DNA Purification Kit™ (Biosearch Technologies) on an Oktopure Liquid Handling system.
Genotyping
Genotyping was done using PCR Allelic Competitive Extension (PACE) 2.0 chemistry (3CR Bioscience, Essex, UK). Assays were designed by the 3CR Bioscience assay design service. All PACE reactions were performed on a QuantStudio 7 (Applied Biosystems) using the manufacturer suggested PACE reaction volumes and cycling conditions. PACE primer sequences are provided in Supplementary Table S1.
Results
Germplasm screening for PM resistance
To identify new sources of PM resistance, we screened a total of 510 genotypes in our cannabis germplasm collection, including production cultivars, landraces, and exotic lines, and called this population CanD for Cannabis Diversity. All 510 genotypes were initially evaluated for resistance/susceptibility to PM using a disease index (DI) measured using a clone infection assay (f1, see Materials and Methods). The observed distribution of DI in the CanD population was left-skewed, with more than 70% of the genotypes scoring a DI > 50 indicating a high prevalence of PM susceptibility (f1). The continuous nature of the distribution of DI in the CanD population suggests that multiple loci, most with small effects, contribute to PM resistance in cannabis.
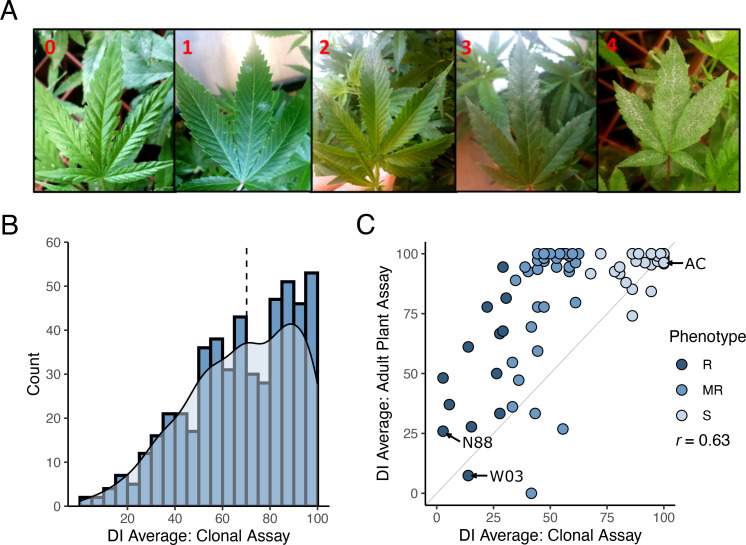
To confirm if the resistance to PM observed in clones would persist through the flowering phase of the plant’s life cycle, we performed an adult plant assay on a subset of 90 CanD genotypes. Genotypes were selected to represent the following three phenotypic groups based on their clonal infection assay score: (i) resistant (R: DI ≤ 33), (ii) moderately resistant (MR: 33< DI< 66), and (iii) susceptible (S: 66< DI ≤ 100). All resistant genotypes identified in the CanD population were included in the adult plant assay to confirm the resistance to PM observed in clones. Disease pressure in the adult-plant experiment was significantly higher than in the clone assay due to the longer duration (12 vs. 4 weeks) and repeated exposure to high inoculum levels and fresh spores. In the adult plant assay, many genotypes showed an increase in PM susceptibility compared to the clonal assay (f1, genotypes above the plot diagonal). In severe susceptibility cases, sugar leaves of the flowers, petioles, and parts of the stem tissue were also colonized by the PM pathogen. Overall, the DI in the adult plant assay positively correlated with the results of the clonal assay for the 90 genotypes tested in both experiments (r = 0.63). A final group of 12 resistant genotypes (DI< 50) were selected from the adult-plant infection trial. Within this group, 5 genotypes showed strong resistance responses to PM (DI< 33), despite the high disease pressure and length of the assay.
Mode of inheritance of the observed R locus
Twelve different F1 populations derived from highly resistant (R) and susceptible (S) genotypes were subjected to infection trials using the clone assay to determine the mode of inheritance of PM resistance (Materials and Methods). Among the populations tested, two exhibited a 1:1 ratio for R:S in their F1 progeny (f2), indicating that resistance to PM in these cases is mediated by a single dominant locus R gene, and that the resistant parents are heterozygous for that locus. The identified resistant parents, W03 and N88 were crossed with a susceptible cultivar, AC (T1). The progeny in the remaining nine F1 populations tested did not show strong PM resistance, suggesting a multigenic origin of pathogen resistance in the parents of the crosses.

Table 1: Chi-squared tests for goodness of fit for the two F1 test cross populations.
| Cross (R × S) | Resistant-progenyExp. vs. Obs. | Susceptible-progenyExp. vs. Obs. | Chi-square test | ||||
|---|---|---|---|---|---|---|---|
| χ2 | df | P-value | |||||
| N88 × AC | 33.5 | 36 | 33.5 | 31 | 0.373 | 1 | 0.5413 |
| W03 × AC | 22.5 | 24 | 22.5 | 21 | .200 | 1 | 0.6547 |
QTL mapping using bulked segregant RNA-seq analysis
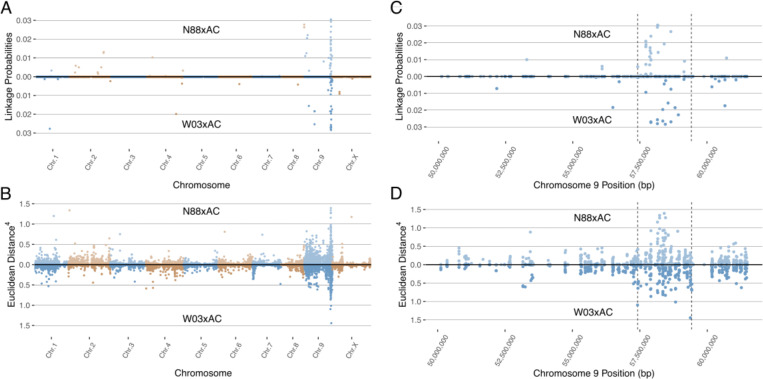
To map the observed dominant PM resistance in the cannabis genome, we performed bulked segregant analysis on the two previously described bi-parental F1 populations made from PM-resistant and PM-susceptible genotypes (N88xAC and W03xAC). For each F1 population, susceptible and resistant bulks were created consisting of 25 plants each grouped by their respective phenotype (PM susceptible and PM resistant). RNA sequencing and reference genome alignment was performed on each bulk (Materials and Methods). For the W03xAC population, bulks resulted in 25,150,091 and 17,017,509 reads uniquely aligned to the Pink Pepper Cannabis reference genome for resistant and susceptible bulks respectively and used for SNP calling. A total of 65,202 SNPs shared between both bulks were used for BSR-Seq after filtering for quality scores and read depth (see Materials and Methods). Similarly, the N88xAC population yielded 24,179,503 and 21,576,849 uniquely mapped reads in the resistant and susceptible bulks respectively, and 82,099 shared SNPs after filtering.
To identify the region associated with PM resistance in these populations, we used two separate methods developed for BSR-Seq: the Bayesian implementation by ref. Liu et al. (2012) to estimate the probability of SNPs in linkage disequilibrium with the casual gene of a trait of interest, and the Euclidean distance metric developed by ref. Hill et al. (2013), a robust measure of allele frequency differences between bulks. The Bayesian approach identified a cluster of SNPs with high probability of being linked to PM resistance on chromosomes 9 in both the W03xAC and N88xAC populations (f3). Using the Euclidean distance metric, a strong signal was observed in the same region of chromosome 9 in both populations (f3). Both the ED and Bayesian probability metrics identify an overlapping region of approximately 2.0Mbp. The boundaries of this region were defined between positions 57,417,178 and 59,418,457 based on SNPs with elevated ED scores and increased posterior probabilities to being linked with PM resistance (f3). To further confirm that the identified region is associated with PM resistance, we repeated the BSR-Seq analysis on a second publicly available reference genome, CBDRx. Using both the Bayesian probability and ED metrics, a cluster of SNPs highly associated with PM resistance was observed on chromosome 9 (NC_044376.1) corresponding to the same region observed in the Pink Pepper reference genome (Supplementary Figure S1). These results strongly suggest that both F1 populations are segregating for the same single dominant PM resistance locus, hereafter named PM2.
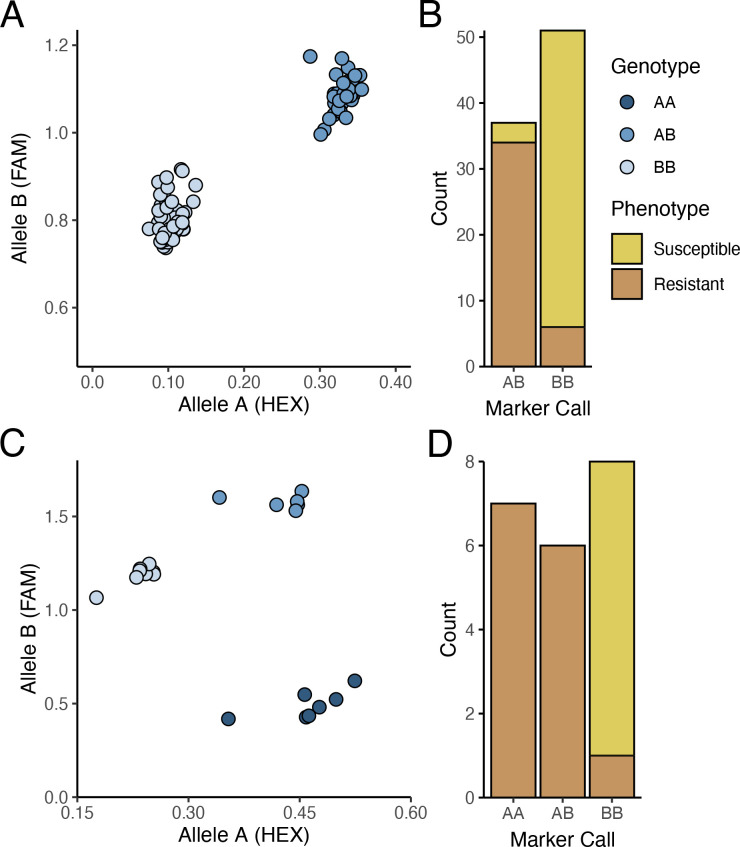
Development and validation of genetic markers for breeding PM2 resistance
The PM2 QTL defined region contains 2,342 SNPs for the W03xAC population and 2,208 SNPs for the N88xAC population. We developed PACE genotyping assays to validate five SNPs as genetic markers for PM2 resistance in W03xAC and N88xAC F1, and W03 and N88 selfed (S1) populations (Supplementary Table S2). The accuracy of the markers was tested by comparing their genotype calls to the populations phenotypes as determined by clonal-infection assay. For the W03xAC and N88xAC F1 populations, markers had a prediction accuracy between 92 and 99% (T2). Marker accuracies were slightly lower when tested on W03 and N88 selfed populations, ranging between 86-93%. The number of genotyped plants in the S1 populations were however smaller compared to the number tested in the F1 populations (T2). To evaluate allelic segregation in the S1 populations, a chi-squared test for deviation from Mendelian ratios of inheritance was used and showed that all populations fell within accepted ranges (T3). Marker 110 was extensively tested in both F1 and S1 populations, with its genotype calls and marker accuracy depicted in f4.
Table 2: Marker accuracy results and flanking sequence.
| Marker Assay | Flanking Sequence | Validated population | Marker accuracy |
|---|---|---|---|
| MR110 | ATCACCAGCCAAGAATCCCACCACWGGAACCACATCCTTAACCTCTCCAAGCCTCCCCATGGGGCACTCATCCACCACTTTCTTCAAGTCATCCTCACT[C/T]TTCCCCGAAAAGAACATATCSGTCGCGATTGGTCCCGGKGCCAMGCAGTTGGCCGTGATCCCCRTCCCCTTYAGCTCCTTAGCAAGTATCTTGATCATCG | W03xAC | 95% (n=216) |
| N88xAC | 98% (n=187) | ||
| W03 S1 | 90% (n=21) | ||
| N88 S1 | 86% (n=28) | ||
| MR121 | ATACTATGATGGCCGTGAAAATGTCCACACAGATTATCCTGTCGCGGATCTGTTGCAGATGATGGGTCATGCTAGTCGGCCATTGCTAGATAATTCCGG[T/G]AAATGTGTGATCCTCTGCCATGCACCTCGTAAAGAATATTACAAGGAGTTCTTATATGAATCATTCCCAGTTGAAAGCCATTTACACCACTACTTGCATG | W03xAC | 92% (n=176) |
| W03 S1 | 90% (n=21) | ||
| MR131 | GAGAAAGCACAGTGAGACTGTGGTGGTGGCACCAGTGAAGAAGGATGATACAGCTGCAGAGAGGCCTMAAAGGACWCTTTTGKGTTGGAAAGAGAAGAA[G/A]AATGAAGCAGAMSCAGAAACTGAATCAACTCCGTTTTTCAGGAACAAAGAGAAGRTTTTRGTTACTTGTTCTCGAAGAATTAATTACAGGTTTGWAAAAA | N88xAC | 99% (n=171) |
| N88 S1 | 93% (n=28) | ||
| MR124 | TGAGACTGTGGTGGTGGCACCAGTGAAGAAGGATGATACAGCTGCAGAGAGGCCTMAAAGGACWCTTTTGKGTTGGAAAGAGAAGAARAATGAAGCAGA[A/C]SCAGAAACTGAATCAACTCCGTTTTTCAGGAACAAAGAGAAGRTTTTRGTTACTTGTTCTCGAAGAATTAATTACAGGTTTGWAAAAAKAATCRATCAAA | N88xAC | 97% (n=184) |
| N88 S1 | 93% (n=28) | ||
| MR125 | TGTGAGAAAATTCARAAAAATGACACGGTTRAAAGGATAAGAASTCGGAAAAGAAACAATACCCARTCCAAGATCAACAAGATATAGTTTCTTCTCATC[G/A]ACTGTTCCAGGTTGACCAAGTAAAAAATTCTCGGGCTTTACATCTCCATGCACAAACCTATTTAGAATTAGAAAATTTGAAAATAATCAACAATGCAWTT | N88xAC | 99% (n=180) |
| N88 S1 | 93% (n=28) |
Marker accuracy for F1 populations are the combined results from two independent qPCR runs for each population, resulting in the higher total N value compared to S1 populations, which were single runs per populations. Bold letters within brackets indicate the polymorphic site of the KASP markers with allelic variants.
Table 3: Chi-squared test for deviation from mendelian patterns of inheritance (df=2).
| Marker | Population | AA | AB | BB | χ2 | p-value | |||
|---|---|---|---|---|---|---|---|---|---|
| O | E | O | E | O | E | ||||
| MR 110 | W03 S1 | 7 | 5.25 | 6 | 10.5 | 8 | 5.25 | 3.9524 | 0.1386 |
| MR 110 | N88 S1 | 8 | 7 | 12 | 14 | 8 | 7 | 0.5714 | 0.7515 |
| MR 121 | W03 S1 | 8 | 5.25 | 6 | 10.5 | 7 | 5.25 | 3.9524 | 0.1386 |
| MR 124 | N88 S1 | 8 | 7 | 11 | 14 | 9 | 7 | 1.3571 | 0.5074 |
| MR 125 | N88 S1 | 8 | 7 | 12 | 14 | 8 | 7 | 0.5714 | 0.7515 |
| MR 131 | N88 S1 | 8 | 7 | 11 | 14 | 9 | 7 | 1.3571 | 0.5074 |
O, observed, E, expected.
Putative candidate genes at the PM2 locus
Analysis of gene annotations in the PM2 QTL region revealed 13 coding sequences with known functions in disease resistance in other plant species which represent putative candidate genes that could explain PM2 resistance (f5; T4). The ratio of expression between resistant and susceptible bulks in some of the selected genes indicated differences that provide additional evidence supporting a putative role in PM2 resistance (Supplementary Table S3).
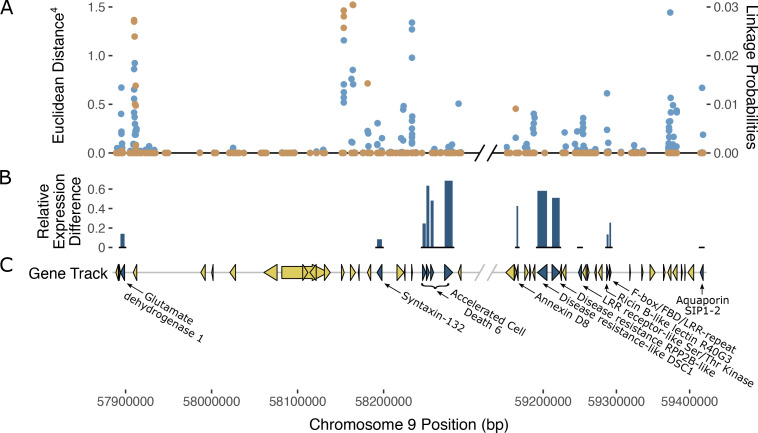
Table 4: Putative candidate genes within the PM2 region associated with defense signaling and pathogen response.
| Gene name | Start (bp) | End (bp) | Annotation | Mechanism | Pathosystem | Reference |
|---|---|---|---|---|---|---|
| LOC115722251 | 57894037 | 57898806 | glutamate dehydrogenase 1 | TCA cycle exhaustion – cell death | Tobacco – response to biotrophic pathogens | ref. Seifi et al., 2013b |
| LOC115722780 | 58192366 | 58198073 | syntaxin-132 | SA defense pathway – PR1 expression | Tomato – Oidium lycopersicum (powdery mildew) | ref. Bracuto et al., 2017 |
| LOC115723470 LOC133031067 LOC133031066 LOC133031533 | 58249744 58270938 58245244 58254683 | 58253111 58280233 58248979 58258108 | ACCELERATED CELL DEATH 6ACCELERATED CELL DEATH 6ACCELERATED CELL DEATH 6ACCELERATED CELL DEATH 6 | SA defense signaling, cell deathSA defense signaling, cell deathSA defense signaling, cell deathSA defense signaling, cell death | Arabidopsis – response to PM (G. cichoracearum) and Pseudomonas syringae | ref. Dong, 2004; ref. Todesco et al., 2010 |
| LOC115723381 | 59163552 | 59165815 | annexin D8 | ROS accumulation, callose formation | Wheat – puccinia striiformis (yellow rust) | ref. Shi et al., 2023 |
| LOC133030743 | 59192095 | 59205193 | disease resistance-like protein DSC1 | Potential R gene | Arabidopsis – root-knot nematode | ref. Warmerdam et al., 2020 |
| LOC133030744 | 59212063 | 59222527 | disease resistance protein RPP2B-like | Potential R gene | Arabidopsis – Hyaloperonospora arabidopsidis (downy mildew) | ref. Sinapidou et al., 2004 |
| LOC115722098 | 59248198 | 59252239 | putative LRR receptor-like protein kinase At2g24130 | Potential R gene | ||
| LOC115722788 | 59286614 | 59289221 | ricin B-like lectin R40G3 | Defense signaling | Wheat – Fusarium graminearum (head blight) | ref. Song et al., 2021 |
| LOC115722789 | 59290403 | 59292733 | F-box/FBD/LRR-repeat protein At3g26920-like | Potential R gene | ||
| LOC115722965 | 59414491 | 59418457 | aquaporin SIP1-2 | ROS accumulation, defense signaling | Arabidopsis – Pseudomonas syringae (bacterial blight) | ref. Tian et al., 2016 |
ROS, reactive oxygen species; SA, salicylic acid; PR1, pathogenesis related protein 1.
Microscopy analysis of PM2-mediated resistance response
To investigate the defense mechanisms underlying PM2 resistance, DAB staining was employed to detect hydrogen peroxide (H2O2), a key indicator of the hypersensitive response in plants (ref. Asselbergh et al., 2007; ref. Seifi et al., 2013a). After infecting clones with PM pathogen, PM2-resistant genotypes showed strong and localized DAB staining in the epidermis and mesophyll cells of infected leaves (f6; Supplementary Figure S2) while no staining could be observed in the leaves of the susceptible genotype (f6).
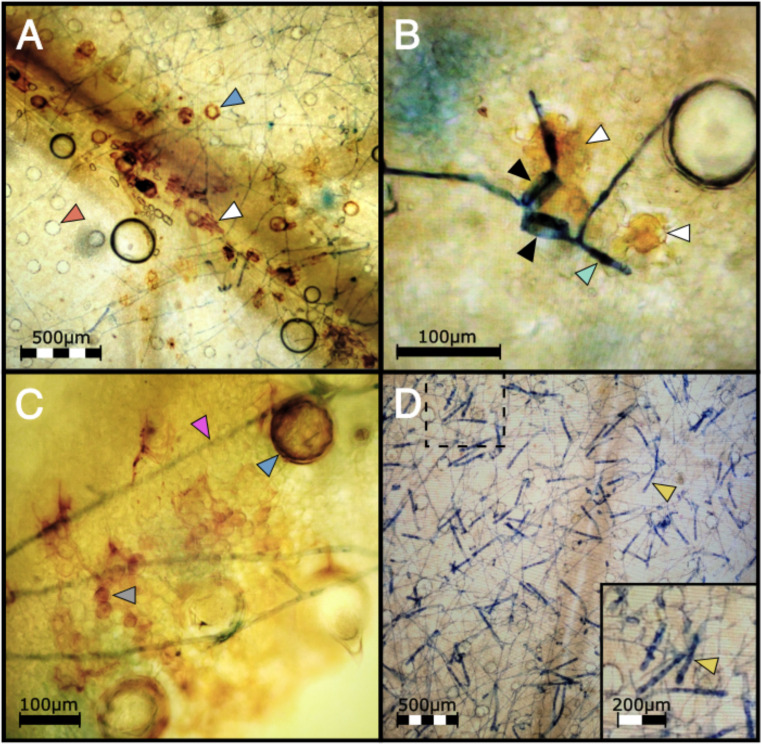
In susceptible cannabis genotypes, the PM fungal pathogen G. ambrosiae fully develops its mycelial network and conidiophores within 2 wpi, completing its infectious life cycle. Disease development was compared between W03, AC, and a third genotype, P04, having an unknown form of PM resistance (PMU) identified in our clonal infection assay (f7). Clear differences in infection symptoms, mycelia growth, and pathogen reproduction were observed between PM2-mediated resistance and PMU. PM2-mediated resistance in W03 and N88 showed restricted mycelial growth and strong suppression of the conidiophore formation, which is a key developmental stage for the sporulation phase of the pathogen (f7, and Supplementary Figure S2). In contrast, susceptible genotype AC under identical conditions showed dense mycelial growth with high amounts of conidiophore formation (f7). While conidiophore formation and sporulation were largely suppressed in the PM2-mediated response, normal conidiophore formation and sporulation occurred in sporadic patches of restricted PM colonies in the PMU-induced response (f7). It should be noted that P04 scored a disease index value of 16.77 in our clonal infection assay and was therefore deemed resistant by our criteria (DI< 33). The reduced level of infection observed in W03 and N88 compared to P04 highlights the efficacy of PM2-mediated resistance.

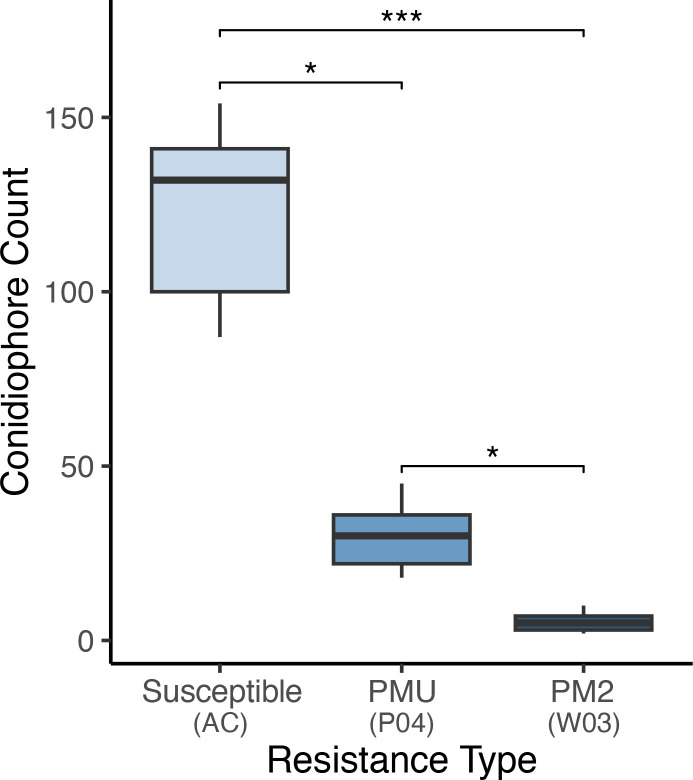
To validate the repression of conidiophore formation observed in PM2-mediated resistance, number of conidiophores per 3.8 mm2 of leaf surface were counted at 9 randomly selected positions on leaves of W03, P04, and susceptible genotype AC (f8). Significant differences in mean conidiophore count were observed in all three genotypes [Kruskal-Wallis test, X 2 (2, 27) = 23.15, p< 0.001]. Mean conidiophore counts from W03 and P04 were significantly lower compared to susceptible AC, p< 0.001 and p< 0.05 respectively (Dunn’s post-hoc test with Bonferroni correction). When comparing PM2-mediated resistance to PMU, W03 had significantly lower counts of conidiophores compared to P04 (p< 0.05), with mean number of observed conidiophores at 5.33 compared to 35.44 for P04, and 118.33 for AC. These results further demonstrate the robust level of PM resistance conferred by PM2.
Discussion
Powdery mildew (PM) is the most prevalent fungal disease in indoor cannabis growing operations, causing significant losses to the industry (ref. Pépin et al., 2021). Qualitative resistance to PM has been reported in hops (Humulus lupulus), a species closely related to cannabis (ref. Henning et al., 2017). Recently, the first report of a single dominant resistance locus against PM was reported in cannabis, named PM1, with a suggested location of the causal R gene on chromosome 2 (CS10 Chr ID: NC_044375.1; GenBank acc. no. GCA_900626175.2) (ref. Mihalyov and Garfinkel, 2021). In this study, using BSR-seq, we identified a new single dominant PM resistance locus, named PM2, located on chromosome 9 (CS10cChr ID: NC_044376.1). No macroscopic chlorotic spots were detected on PM2 leaves. However, DAB staining revealed H2O2 accumulation in epidermal and mesophyll cells beneath the pathogen’s mycelial growth, indicating a highly localized HR reaction in PM2-mediated resistance. Furthermore, the reproduction phase of the fungal pathogen was strongly suppressed, resulting in a significant reduction of conidia generation (>90%) in PM2 genotypes. This could be explained by the inhibition of penetration through HR by the resistant genotype, leading to the observed long ‘wandering’ mycelial growth with delayed and minimal reproduction. Such a phenomenon has been previously reported in an interaction between the biotrophic rust pathogen Puccinia striiformis and a resistant cultivar of wheat harboring the YR15 R gene (ref. Seifi et al., 2021).
Further exploration focused on defense-associated genes flanking PM2 revealed several candidate genes (T4). Notably, these genes fall into three main functional categories: 1) hormonal regulation, particularly SA signaling pathway; 2) genes involved in ROS accumulation and cell death induction; and 3) genes predicted to encode potential R proteins, including one (LOC115722098) exhibiting LLR and Ser/Thr-kinase domains. Studies in other plant species have highlighted the role of annexin and aquaporin proteins in defense responses against biotrophic pathogens. For example, annexin proteins regulate SA-dependent defense responses and influence ROS generation and callose deposition (ref. Shi et al., 2023), while aquaporin proteins link extracellular ROS accumulation to intracellular defense signaling in Arabidopsis challenged by pathogens (ref. Tian et al., 2016). RPP2B, an R gene with a TIR-NB-LRR domain, plays a crucial role in race-specific recognition and defense signaling against downy mildew (ref. Sinapidou et al., 2004). Similarly, the TIR-NB-LRR DSC1 protein is implicated in basal immunity against root-knot nematodes in Arabidopsis (ref. Warmerdam et al., 2020). Research on the TCA cycle through glutamate dehydrogenase suggests its role in defense responses against biotrophic pathogens, including the induction of programmed cell death (ref. Seifi et al., 2013b). Accelerated Cell Death 6 (ACD6), involved in SA-mediated defense responses in Arabidopsis, triggers programmed cell death and expression of PR genes against bacterial pathogens (ref. Dong, 2004). Notably, ACD6 belongs to the transmembrane-ankyrin-repeat protein family with a key regulating role in tradeoffs between vegetative growth and general pathogen defense, conferring broad-spectrum disease resistance to pathogens including PM in Arabidopsis (ref. Todesco et al., 2010). Syntaxin proteins have been identified as crucial players in plant defense responses to PM. Studies on barley and Arabidopsis suggest that syntaxin genes regulate fungal penetration and enhance resistance against PM pathogens (ref. Bracuto et al., 2017). Notably, silencing of the syntaxin gene SlPEN compromised resistance in a mlo-resistant tomato line against Oidium neolycopersici, highlighting their importance in PM resistance (ref. Bracuto et al., 2017).
Originally developed using restriction fragment length polymorphisms (RFLPs) and random amplified polymorphic DNAs (RAPDs) markers (ref. Michelmore et al., 1991), BSA now employs the use of next-generation sequencing (NGS) technologies which have greatly improved its power of detection in mapping phenotypic traits. Algorithms designed for detecting marker associations to target traits have been developed for NGS-based BSA (also referred to as BSA-Seq). These algorithms are primarily based on detecting differences in allele frequencies between bulks, paired with smoothing techniques that help identify positive signals from noise. Popular BSA-Seq statistics include the G’ value (ref. Magwene et al., 2011), SNP-index/InDel-index (ref. Takagi et al., 2013; ref. Singh et al., 2017), Euclidean distance (ED, ref. Hill et al., 2013), and smoothLOD (ref. Zhang et al., 2019), among others (see ref. Li and Xu, 2022). In addition to whole genome resequencing, bulked segregant analysis using SNP markers called from transcriptome data (BSR-Seq) has been successfully used to map QTLs in maize (ref. Liu et al., 2012), zebrafish (ref. Hill et al., 2013), pacific white shrimp (ref. Dai et al., 2018), pea (ref. Wu et al., 2022) and wheat (ref. Trick et al., 2012; ref. Li et al., 2018; ref. Hao et al., 2019). Several BSA statistics have been developed specifically to handle the challenges inherent to RNA-Seq data, such as allele-specific expression and variable coverage. These include a Bayesian approach to estimate marker association (ref. Liu et al., 2012) and the ED metric (ref. Hill et al., 2013), both of which were successfully used in the present study to map PM2 mediated resistance in cannabis. Both the Bayesian and Euclidean distance methods identified the same 2Mb region within chromosome 9, containing the PM2 QTL. While the Bayesian method required considerably longer computational times, the signal to noise ratio was superior to ED, leaving an easily identified signal peak associated with PM resistance. The comparison of two mapping populations segregating for PM2 provided further strength to our analysis. Both populations showed clear signals for PM2 mediated resistance in the same location on chromosome 9. A minor secondary peak was observed on chromosome 7, however this was not investigated in this study as it was observed in the W03xAC F1 population only.
The use of RNA-Seq data for SNP calling provides the added benefit of gene expression data from both resistant and susceptible bulks. Several gene candidates for PM2 mediated resistance identified by BSR-Seq showed increased gene expression in resistant bulks compared to susceptible bulks, including ACD6, Annexin D8, RPP2B, and DSC1. This gene expression data, however, comes from pooled samples with no biological replicates, and therefore should not be used to draw conclusions, but to guide follow-up studies.
Populations frequently used in BSA include bi-parental populations segregating for a trait of interest. Common types include F2, F2:3, BC1, and recombinant inbred lines (ref. Zou et al., 2016). However, any population containing individuals with contrasting traits can in theory be used in a bulked sample analysis (ref. Li and Xu, 2022). For species that have long life cycles, or are not amenable to selfing, F1 populations have been used (ref. Shen et al., 2019; ref. Dougherty et al., 2018). These populations require parents to be highly heterozygous and have observable phenotypic variation in the F1 generation, as is the case with dominant traits (ref. Li and Xu, 2022). Both our N88xAC and W03xAC F1 populations exhibited clear segregation in PM resistance; employing these populations saved a considerable amount of time, by removing the need to proceed to the F2 generation.
The development of resistant cultivars is an effective practice for the management of PM disease control. Successful introgression of PM2 mediated resistance into elite cannabis cultivars will reduce the reliance of chemical pesticides, which are heavily regulated in cannabis as in most other crops. As the cannabis industry expands by developing new markets and applications, there will be an increased need for resistant cannabis cultivars, highlighting the relevance of studies, such as the one presented here, identifying the genetic basis of resistance and susceptibility to pathogens in this species.
References
- G. N. Agrios. Plant diseases caused by fungi,” in. Plant Pathology (, 2005. [DOI]
- S. Andrews. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at:, 2010
- B. Asselbergh, K. Curvers, S. C. França, K. Audenaert, M. Vuylsteke, F. van Breusegem. Resistance to Botrytis cinerea in sitiens, an Abscisic Acid-Deficient Tomato Mutant, Involves Timely Production of Hydrogen Peroxide and Cell Wall Modifications in the Epidermis.. Plant Physiol., 2007. [DOI | PubMed]
- T. Bapela, H. Shimelis, T. Terefe, S. Bourras, J. Sánchez-Martín, D. Douchkov. Breeding wheat for powdery mildew resistance: Genetic resources and methodologies—A review.. Agronomy, 2023. [DOI]
- V. Bracuto, M. Appiano, Z. Zheng, A.-M. A. Wolters, Z. Yan, L. Ricciardi. Functional characterization of a syntaxin involved in tomato (Solanum lycopersicum) resistance against powdery mildew.. Front. Plant Sci., 2017. [DOI]
- A. S. Brochu, C. Labbe, R. Belanger, E. Perez-Lopez. First report of powdery mildew caused by Golovinomyces ambrosiae on Cannabis sativa (Marijuana) in Quebec, Canada.. Plant Dis., 2022. [DOI]
- Y. Cao, Q. Diao, Y. Chen, H. Jin, Y. Zhang, H. Zhang. Development of KASP markers and identification of a QTL underlying powdery mildew resistance in melon (Cucumis melo L.) by bulked segregant analysis and RNA-Seq.. Front. Plant Sci., 2021. [DOI]
- S. Chandra, H. Lata, I. A. Khan, M. A. El Sohly. Cannabis sativa L.: Botany and horticulture,” In:. Cannabis sativa L. – Botany and Biotechnology (, 2017. [DOI]
- P. Dai, J. Kong, S. Wang, X. Lu, K. Luo, B. Cao. Identification of SNPs associated with residual feed intake from the muscle of Litopenaeus vannamei using bulk segregant RNA-seq.. Aquaculture, 2018. [DOI]
- A. Dobin, C. A. Davis, F. Schlesinger, J. Drenkow, C. Zaleski, S. Jha. STAR: ultrafast universal RNA-seq aligner.. Bioinformatics, 2013. [DOI | PubMed]
- X. Dong. The role of membrane-bound ankyrin-repeat protein ACD6 in programmed cell death and plant defense.. Sci. STKE, 2004. [DOI | PubMed]
- L. Dougherty, R. Singh, S. Brown, C. Dardick, K. Xu. Exploring DNA variant segregation types in pooled genome sequencing enables effective mapping of weeping trait in Malus.. J. Exp. Bot., 2018. [DOI | PubMed]
- C. J. Grassa, G. D. Weiblen, J. P. Wenger, C. Dabney, S. G. Poplawski, S. Timothy Motley. A new Cannabis genome assembly associates elevated cannabidiol (CBD) with hemp introgressed into marijuana.. New Phytol., 2021. [DOI | PubMed]
- K. E. Hammond-Kosack, K. Kanyuka. Resistance genes (R genes) in plants,” in. Encyclopedia of Life Sciences (, 2007. [DOI]
- Z. Hao, M. Geng, Y. Hao, Y. Zhang, L. Zhang, S. Wen. Screening for differential expression of genes for resistance to sitodiplosis mosellana in bread wheat via BSR-seq analysis.. Theor. Appl. Genet., 2019. [DOI | PubMed]
- J. A. Henning, D. H. Gent, M. S. Townsend, J. L. Woods, S. T. Hill, D. Hendrix. QTL analysis of resistance to powdery mildew in hop (Humulus lupulus L.).. Euphytica, 2017. [DOI]
- J. T. Hill, B. L. Demarest, B. W. Bisgrove, B. Gorsi, Y. C. Su, H. J. Yost. MMAPPR: Mutation mapping analysis pipeline for pooled RNA-seq.. Genome Res., 2013. [DOI | PubMed]
- R. Hückelhoven. Powdery mildew susceptibility and biotrophic infection strategies.. FEMS Microbiol. Lett., 2005. [DOI | PubMed]
- I. Imerovski, B. Dedić, S. Cvejić, D. Miladinović, S. Jocić, G. L. Owens. BSA-seq mapping reveals major QTL for broomrape resistance in four sunflower lines.. Mol. Breed., 2019. [DOI]
- H. Jiang, R. Lei, S.-W. Ding, S. Zhu. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads.. BMC Bioinf., 2014. [DOI]
- J. D. G. Jones, J. L. Dangl. The plant immune system.. Nature, 2006. [DOI | PubMed]
- J. H. Jørgensen, M. P. Wolfe. Genetics of powdery mildew resistance in barley.. Crit. Rev. Plant Sci., 1994. [DOI]
- S. Kovaka, A. V. Zimin, G. M. Pertea, R. Razaghi, S. L. Salzberg, M. Pertea. Transcriptome assembly from long-read RNA-seq alignments with StringTie2.. Genome Biol., 2019. [DOI | PubMed]
- P. Kumar, D. K. Mahato, M. Kamle, R. Borah, B. Sharma, S. Pandhi. Pharmacological properties, therapeutic potential, and legal status of Cannabis sativa L.: An overview.. Phytother. Res., 2021. [DOI | PubMed]
- M. Li, B. Li, G. Guo, Y. Chen, J. Xie, P. Lu. Mapping a leaf senescence gene els1 by BSR-seq in common wheat.. Crop J., 2018. [DOI]
- Z. Li, Y. Xu. Bulk segregation analysis in the NGS era: a review of its teenage years.. Plant J., 2022. [DOI | PubMed]
- T. Liang, W. Chi, L. Huang, M. Qu, S. Zhang, Z. Q. Chen. Bulked segregant analysis coupled with whole-genome sequencing (BSA-seq) mapping identifies a novel pi21 haplotype conferring basal resistance to rice blast disease.. Int. J. Mol. Sci., 2020. [DOI | PubMed]
- S. Liu, C. T. Yeh, H. M. Tang, D. Nettleton, P. S. Schnable. Gene mapping via bulked segregant RNA-seq (BSR-Seq).. PloS One, 2012. [DOI | PubMed]
- R. López-Ruiz, J. Marín-Sáez, A. Garrido Frenich, R. Romero-González. Recent applications of chromatography for analysis of contaminants in cannabis products: A review.. Pest Manage. Sci., 2022. [DOI]
- P. Ma, L. Wu, Y. Xu, H. Xu, X. Zhang, W. Wang. Bulked segregant RNA-Seq provides distinctive expression profile against powdery mildew in the wheat genotype YD588.. Front. Plant Sci., 2021. [DOI]
- P. M. Magwene, J. H. Willis, J. K. Kelly. The statistics of bulk segregant analysis using next-generation sequencing.. PloS Comput. Biol., 2011. [DOI | PubMed]
- A. McKenna, M. Hanna, E. Banks, A. Sivachenko, K. Cibulskis, A. Kernytsky. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data.. Genome Res., 2010. [DOI | PubMed]
- R. W. Michelmore, I. Paran, R. V. Kesseli. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations.. Proc. Natl. Acad. Sci. U.S.A., 1991. [DOI | PubMed]
- P. D. Mihalyov, A. R. Garfinkel. Discovery and genetic mapping of PM1, a powdery mildew resistance gene in Cannabis sativa L.. Front. Agron., 2021. [DOI]
- H. Y. Mohan Ram, R. Sett. Induction of fertile male flowers in genetically female Cannabis sativa plants by silver nitrate and silver thiosulphate anionic complex.. Theor. Appl. Genet., 1982. [DOI | PubMed]
- D. W. Pate. Possible role of ultraviolet radiation in evolution of Cannabis chemotypes.. Economic Bot., 1983. [DOI]
- N. Pépin, F. O. Hebert, D. L. Joly. Genome-wide characterization of the MLO gene family in Cannabis sativa reveals two genes as strong candidates for powdery mildew susceptibility.. Front. Plant Sci., 2021. [DOI]
- N. Pépin, Z. K. Punja, D. L. Joly. Occurrence of powdery mildew caused by Golovinomyces cichoracearum sensu lato on Cannabis sativa in Canada.. Plant Dis., 2018. [DOI]
- Z. K. Punja. Emerging diseases of Cannabis sativa and sustainable management.. Pest Manage. Sci., 2021. [DOI]
- M. M. Radwan, A. S. Wanas, S. Chandra, M. A. ElSohly. Natural cannabinoids of Cannabis and methods of analysis. In:. Cannabis sativa L. – Botany and Biotechnology (, 2017. [DOI]
- B. R. Ryu, G. J. Gim, Y. R. Shin, M. J. Kang, M. J. Kim, T. H. Kwon. Chromosome-level haploid assembly of Cannabis sativa L. cv. Pink Pepper.. Sci. Data, 2024. [DOI | PubMed]
- C. Scott, Z. K. Punja. Evaluation of disease management approaches for powdery mildew on Cannabis sativa L. (marijuana) plants.. Canadian Journal of Plant Pathology, 2020. [DOI]
- H. S. Seifi, K. Curvers, D. de Vleesschauwer, I. Delaere, A. Aziz, M. Höfte. Concurrent overactivation of the cytosolic glutamine synthetase and the GABA shunt in the ABA-deficient sitiens mutant of tomato leads to resistance against Botrytis cinerea .. New Phytol., 2013. [DOI | PubMed]
- A. Seifi, D. Gao, Z. Zheng, S. Pavan, L. Faino, R. G. F. Visser. Genetics and molecular mechanisms of resistance to powdery mildews in tomato (Solanum lycopersicum) and its wild relatives.. Eur. J. Plant Pathol., 2014. [DOI]
- H. S. Seifi, M. Serajazari, M. Kaviani, P. Pauls, H. Booker, A. Navabi. Immunity to stripe rust in wheat: A case study of a hypersensitive-response (HR)-independent resistance to Puccinia striiformis f. sp. tritici in Avocet-Yr15.. Can. J. Plant Pathol., 2021. [DOI]
- H. S. Seifi, J. van Bockhaven, G. Angenon, M. Höfte. Glutamate metabolism in plant disease and defense: Friend or foe?. Mol. Plant-Microbe Interact. (MPMI), 2013. [DOI | PubMed]
- S. M. Shamrai. Recognition of pathogen attacks by plant immune sensors and induction of plant immune response.. Cytology Genet., 2022. [DOI]
- F. Shen, Z. Huang, B. Zhang, Y. Wang, X. Zhang, T. Wu. Mapping gene markers for apple fruit ring rot disease resistance using a multi-omics approach.. G3 Genes|Genomes|Genetics, 2019. [DOI | PubMed]
- B. Shi, W. Liu, Q. Ma. The wheat annexin TaAnn12 plays positive roles in plant disease resistance by regulating the accumulation of reactive oxygen species and callose.. Int. J. Mol. Sci., 2023. [DOI | PubMed]
- E. Sinapidou, K. Williams, L. Nott, S. Bahkt, M. Tör, I. Crute. Two TIR: NB : LRR genes are required to specify resistance to Peronospora parasitica isolate Cala2 in Arabidopsis .. Plant J., 2004. [DOI | PubMed]
- V. K. Singh, A. W. Khan, R. K. Saxena, P. Sinha, S. M. Kale, S. Parupalli. Indel-seq: A fast-forward genetics approach for identification of trait-associated putative candidate genomic regions and its application in pigeonpea (Cajanus cajan).. Plant Biotechnol. J., 2017. [DOI | PubMed]
- T. M. Sirangelo. NLRs and mlo-based resistance mechanisms against Powdery mildew.. Plants, 2023. [DOI | PubMed]
- P. Song, L. Zhang, L. Wu, H. Hu, Q. Liu, D. Li. A ricin b-like lectin protein physically interacts with TaPFT and is involved in resistance to fusarium head blight in wheat.. Phytopathology®, 2021. [DOI | PubMed]
- G. M. Stack, A. R. Cala, M. A. Quade, J. A. Toth, L. A. Monserrate, D. G. Wilkerson. Genetic mapping, identification, and characterization of a candidate susceptibility gene for powdery mildew in Cannabis sativa .. Mol. Plant-Microbe Interact., 2024. [DOI | PubMed]
- H. Takagi, A. Abe, K. Yoshida, S. Kosugi, S. Natsume, C. Mitsuoka. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations.. Plant J., 2013. [DOI | PubMed]
- H. Thordal-Christensen, Z. Zhang, Y. Wei, D. B. Collinge. Subcellular localization of H2O2 in plants: H2O2 accumulation in papillae and hypersensitive response during the barley-powdery mildew interaction.. Plant J., 1997
- S. Tian, X. Wang, P. Li, H. Wang, H. Ji, J. Xie. Plant aquaporin AtPIP1;4 links apoplastic H2O2 induction to disease immunity pathways.. Plant Physiol., 2016. [DOI | PubMed]
- M. Todesco, S. Balasubramanian, T. T. Hu, M. B. Traw, M. Horton, P. Epple. Natural allelic variation underlying a major fitness trade-off in Arabidopsis thaliana.. Nature, 2010. [DOI]
- M. Trick, N. M. Adamski, S. G. Mugford, C. C. Jiang, M. Febrer, C. Uauy. Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat.. BMC Plant Biol., 2012. [DOI | PubMed]
- S. Warmerdam, M. G. Sterken, O. C. A. Sukarta, C. C. van Schaik, M. E. P. Oortwijn, J. L. Lozano-Torres. The TIR-NB-LRR pair DSC1 and WRKY19 contributes to basal immunity of Arabidopsis to the root-knot nematode Meloidogyne incognita .. BMC Plant Biol., 2020. [DOI | PubMed]
- K. T. Win, J. Vegas, C. Zhang, K. Song, S. Lee. QTL mapping for downy mildew resistance in cucumber via bulked segregant analysis using next-generation sequencing and conventional methods.. Theor. Appl. Genet., 2017. [DOI | PubMed]
- L. Wu, R. Fredua-Agyeman, S. E. Strelkov, K. F. Chang, S. F. Hwang. Identification of novel genes associated with partial resistance to Aphanomyces root rot in field pea by BSR-Seq analysis.. Int. J. Mol. Sci., 2022. [DOI | PubMed]
- H. Zhang, X. Wang, Q. Pan, P. Li, Y. Liu, X. Lu. QTG-Seq accelerates QTL fine mapping through QTL partitioning and whole-genome sequencing of bulked segregant samples.. Mol. Plant, 2019. [DOI | PubMed]
- C. Zou, P. Wang, Y. Xu. Bulked sample analysis in genetics, genomics, and crop improvement.. Plant Biotechnol. J., 2016. [DOI | PubMed]