
Persistent sodium currents in neurons: potential mechanisms and pharmacological blockers
Abstract
Persistent sodium current (INaP) is an important activity-dependent regulator of neuronal excitability. It is involved in a variety of physiological and pathological processes, including pacemaking, prolongation of sensory potentials, neuronal injury, chronic pain and diseases such as epilepsy and amyotrophic lateral sclerosis. Despite its importance, neither the molecular basis nor the regulation of INaP are sufficiently understood. Of particular significance is a solid knowledge and widely accepted consensus about pharmacological tools for analysing the function of INaP and for developing new therapeutic strategies. However, the literature on INaP is heterogeneous, with varying definitions and methodologies used across studies. To address these issues, we provide a systematic review of the current state of knowledge on INaP, with focus on mechanisms and effects of this current in the central nervous system. We provide an overview of the specificity and efficacy of the most widely used INaP blockers: amiodarone, cannabidiol, carbamazepine, cenobamate, eslicarbazepine, ethosuximide, gabapentin, GS967, lacosamide, lamotrigine, lidocaine, NBI-921352, oxcarbazepine, phenytoine, PRAX-562, propofol, ranolazine, riluzole, rufinamide, topiramate, valproaic acid and zonisamide. We conclude that there is strong variance in the pharmacological effects of these drugs, and in the available information. At present, GS967 and riluzole can be regarded bona fide INaP blockers, while phenytoin and lacosamide are blockers that only act on the slowly inactivating component of sodium currents.
Article type: Review Article
Keywords: Persistent sodium current, Slow inactivation, Epilepsy, Sodium channel blocker, Neuron
Affiliations: grid.428620.aDepartment Neurology and Epileptology, Hertie Institute for Clinical Brain Research, University of Tuebingen , Hoppe-Seyler-Straße 3, 72076 Tübingen, Germany; https://ror.org/038t36y30grid.7700.00000 0001 2190 4373Institute for Physiology and Pathophysiology, Medical Faculty, Heidelberg University, Im Neuenheimer Feld 326, 69120 Heidelberg, Germany
License: © The Author(s) 2024 CC BY 4.0 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Article links: DOI: 10.1007/s00424-024-02980-7 | PubMed: 38967655 | PMC: PMC11381486
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (1.8 MB)
Introduction
Voltage-gated sodium current is a fundamental component of excitable cells in all animals with active movement. It is mediated by sodium-selective cation channels, which appeared in evolution before the origin of nervous systems [ref. 137]. In mammals, the main (α) subunit of voltage-gated sodium channels (VGSC) consists of 4 identical motives, each of which containing 6 transmembrane segments, a pore-forming loop and a voltage sensor in the fourth membrane-spanning helix. A core feature of voltage-gated sodium currents is their fast inactivation following activation by membrane depolarization [ref. 91]. In most recordings, however, a small portion of the sodium current does not vanish within a few milliseconds (Fig. 1A). This persistent sodium current component (INaP) has been defined as a ‘non-inactivating or slowly inactivating sodium current’ [ref. 49, ref. 119]. Note that, although the name of the current contains the word ‘persistent’, slowly inactivating components are explicitly included. Thus, an unambiguous identification of INaP requires the demonstration of a non- or slowly inactivating component in VGSC-mediated currents.
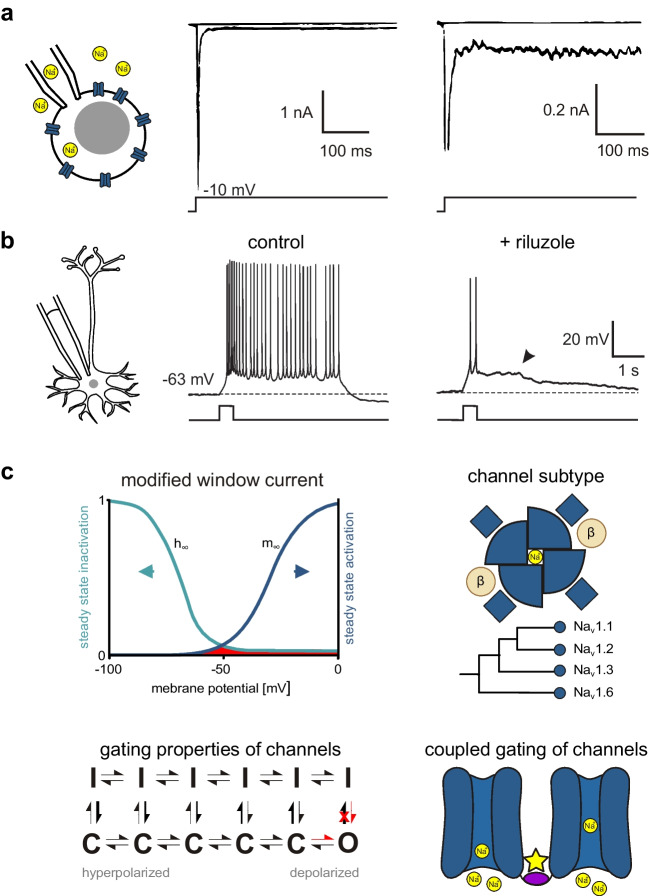
INaP is thought to contribute to multiple cellular functions. It regulates the excitability of neurons [ref. 15, ref. 69, ref. 121, ref. 130] and, specifically, of axons [ref. 160, ref. 213], it amplifies excitatory and inhibitory postsynaptic potentials (EPSP/IPSP) [ref. 38, ref. 72, ref. 174, ref. 211, ref. 212, ref. 234], and it contributes to pacemaking [ref. 27, ref. 116, ref. 120, ref. 229], resonance [ref. 99, ref. 233, ref. 249], bursting behaviour in adult [ref. 17, ref. 214] and immature neurons [ref. 196, ref. 197, ref. 201, ref. 220], place cell tuning [ref. 95] and network oscillations [ref. 114]. An example for INaP-dependent bursting behaviour in immature entorhinal cortex neuron is shown in Fig. 1B. INaP is up-regulated in several disorders, underlining its clinical importance and therapeutic potential. These pathophysiological situations include hypoxia [ref. 86, ref. 94], demyelination [ref. 85, ref. 225], neurogenic pain [ref. 126], paroxysmal extreme pain disorder [ref. 63], temporal lobe epilepsy [ref. 235], monogenetic epileptic syndromes [ref. 139, ref. 206, ref. 239], neurodegeneration [ref. 103, ref. 168, ref. 173, ref. 178, ref. 199], hemiplegic migraine [ref. 20, ref. 40], and spasticity following traumatic brain or spinal cord injury [ref. 32, ref. 136]. INaP has been described to increase with aging [ref. 140], and it is acutely modulated by protein kinase C [ref. 4, ref. 14, ref. 67], G-protein subunits [ref. 141, ref. 143], acetylcholine [ref. 156, ref. 248], dopamine [ref. 79, ref. 80, ref. 152] and endogenous polyamines [ref. 65, ref. 183]. These modulations may be of importance for the effects of drugs used in neurological or psychiatric disorders, e.g. substances affecting cholinergic or dopaminergic transmission.
Pharmacological blockers of persistent sodium current allow assessing its function in physiological experiments on living animals, brain slices, or single cells (in the latter, the current component can also be eliminated by the biophysical approach of dynamic clamp [ref. 210]). More importantly, blockers of INaP with favourable safety profile may be efficient drugs in the different clinical conditions listed above. However, there is no established standard for the use and validation of INaP blockers in different laboratory preparations, experiments on living animals or clinical treatment of humans. Previous reviews have already established that there is a large range of putative INaP blockers [ref. 206, ref. 239], in addition to the even wider range of global sodium channel blocking substances [ref. 133, ref. 200]. We will provide an overview of present knowledge on their selectivity for INaP, their potency, and their specific effects on different kinetic properties of sodium channels. The review shall provide an up-to-date basis for experimental and translational work on this important regulator of cellular excitability. It will also highlight some conceptual and semantic problems with the concept of ‘persistent sodium currents’, which are reflected in the heterogeneity of protocols used to study INaP.
INaP – Characteristics and Underlying Mechanisms
The mechanisms underlying persistent sodium currents are not completely understood and are, most likely, heterogeneous. Here, we will briefly review the dominant hypotheses about the structural or functional basis for INaP (Fig. 1C). One potential explanation for the occurrence of INaP results from the canonical kinetic model of voltage-activated sodium currents, as described in the original Hodgkin-Huxley model. The overlap of steady-state activation and inactivation curves creates a range of potentials where some Na+ channels are activated while inactivation is not complete. This forms a ‘window’ of potentials where some sodium current should be present at any time. Modulation of the voltage-dependence of activation or inactivation can alter the size of the window current and, hence, INaP (see, e.g. the discussion of carbamazepine below). However, French et al. [ref. 70] showed that the properties of INaP are not fully explained by the ‘window current’—for example, INaP conductance increases with more depolarized membrane potentials, while the window current should decrease. In addition, this ‘window current’ overestimates INaP in simulations [ref. 210]. The contradiction arises partly, because the original Hodgkin-Huxley model does consider activation and inactivation to be independent of each other and assumes complete inactivation with increasing depolarization. Taddese and Bean [ref. 216] proposed a modified version of the ‘window current’ in mammalian neurons. In this model, steady-state inactivation is dependent on steady-state activation [ref. 3, ref. 10, ref. 11, ref. 29], resulting in a small remaining current component even at highly positive voltages. This modified window current (Fig. 1C top left) can account for the presence of INaP at depolarized potentials and has been implemented in computational models [ref. 162].
A second approach derives INaP from complicated gating schemes of sodium channels using more flexible Markov models (Fig. 1C bottom left). Early models, derived from measurements in squid axons, suggested two different open states [ref. 41, ref. 46]. Later modifications of these models for mammalian cells assume only one single open state. Persistent opening does then result from one of two alternative mechanisms: i) a preference of more depolarized channels to enter inactivation even when closed [ref. 10, ref. 38, ref. 216]; ii) modal gating of sodium channels, which can enter a non-inactivating state with sustained, burst-like openings [ref. 5, ref. 171]. It has to be noted that from a kinetic standpoint the Markov model of Taddese and Bean [ref. 216] and the modified window current are identical.
The third hypothesis for the mechanism underlying INaP is the existence of a separate channel subtype with the respective kinetic properties [ref. 57]. Indeed, there are nine different known α subunits of VGSC, opening the possibility that INaP is a property of one or several specific subunits. However, evidence from the last decades supports the idea that many different α subunits can produce INaP, at least those with strong expression in the brain (for Nav1.1 see [ref. 6, ref. 112]; for Nav1.2 see [ref. 43, ref. 186]; for Nav1.3 see [ref. 60, ref. 215]; and for Nav1.6 see [ref. 186, ref. 232]); Fig. 1C top right). Amongst these subunits, Nav1.6 seems to be responsible for a major portion of INaP in the central nervous system (CNS) [ref. 186]. However, about half of the persistent sodium current remains after selective knockout of Nav1.6 in rat neocortical layer 5 pyramidal neurons, pointing towards the importance of further subunits [ref. 115]. Nevertheless, the strong contribution of Nav1.6 may be responsible for the well-known left shift of the activation curve when comparing INaP to the transient component of sodium current (INaT) [ref. 49, ref. 119]. This shift would result from the biophysical properties of Nav1.6, which activates at more hyperpolarized membrane potentials than Na+ currents mediated by the other subunits [ref. 96] (note that this left shift would increase the window current, see Fig. 1C, top left). This is supported by the right-shift of the activation curve for persistent sodium currents in hippocampal CA1 neurons of mice lacking functional Nav1.6 [ref. 182]. Conversely, though, selective knockout of Nav1.6 in cortical pyramidal neurons left the voltage-dependence of activation unchanged [ref. 115].
In dorsal root ganglion (DRG) neurons, Nav1.8 and Nav1.9 have been suggested to be responsible for INaP [ref. 119]. These subunits mediate a long-lasting, non-inactivating current component in transduction of sensory signals, which are particularly important for nociceptive stimuli [ref. 2]. A special feature of these subunits is their low sensitivity to the sodium channel blocker tetrodotoxin (TTX). Whereas Nav1.1-Nav1.4, Nav1.6 and Nav1.7 can be blocked by nanomolar concentrations of TTX, Nav1.8 and Nav1.9 require millimolar concentrations [ref. 1]. However, in measurements of INaP, TTX-resistant components are frequently regarded as leak current and, hence, subtracted before analysis. This may lead to an underestimation of the role of Nav1.8 and Nav1.9. In any case, it is unlikely that Nav1.8 and Nav1.9 are responsible for INaP in cortical neurons, as single cell transcriptomics of human and mouse cortex show no expression of both SCN10A (Nav1.8) and SCN11A (Nav1.9) [ref. 90].
The major pore forming α-subunits of sodium channels are complemented by two auxiliary β subunits. There is evidence that the presence of the β4 subunit increases persistent sodium currents, while adding the β1 subunit neutralizes this effect [ref. 6]. Knock out of β1 can lead to a paradoxical effect of sodium channel blockers, which then enhance, rather than suppress, persistent sodium current [ref. 226].
Finally, recent evidence suggests the existence of coupled gating between different individual voltage-gated sodium channels [ref. 44, ref. 101]. This supra-molecular cooperativity may also be involved in the generation of persistent sodium current (Fig. 1C bottom right) [ref. 185].
In summary, there is evidence for several different mechanisms underlying INaP, including contributions by specific molecular subtypes of α or auxiliary subunits and effects of gating kinetics. None of the explanations seems to account for all observations, and they are not mutually exclusive, suggesting convergence of several mechanisms to the generation of persistent sodium currents in many excitable cells.
Electrophysiological Isolation of INaP
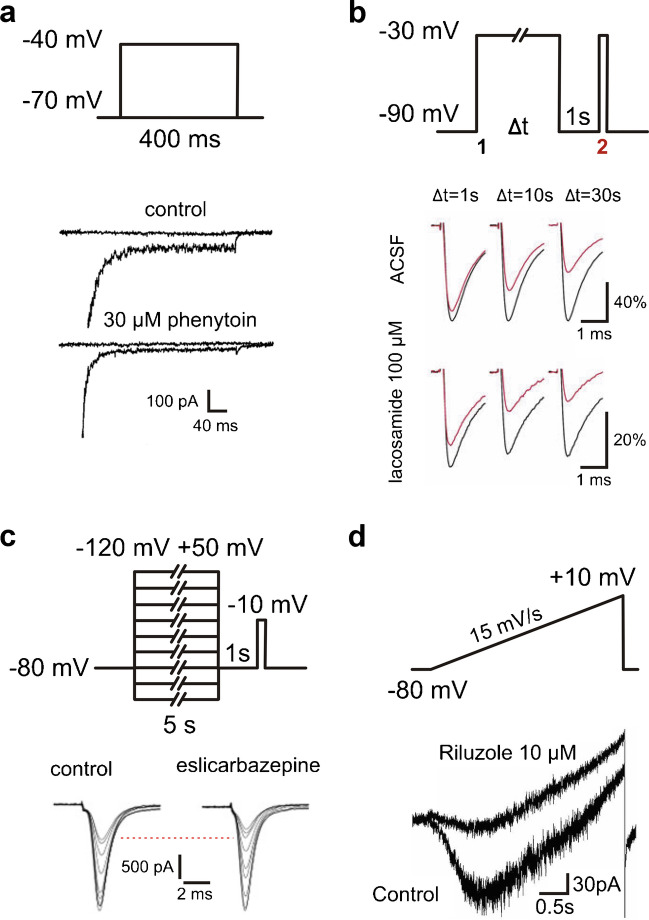
Different voltage clamp protocols are used to isolate the persistent sodium current components in cells or isolated membranes. One frequently used protocol focusses on the early sodium current component, using depolarizing voltage steps of 50—500 ms duration (Fig. 2A). The inward current that persists at the end of this step is then defined as persistent component (Fig. 1A and 2A). Historically, this protocol did underly the first description of ‘late sodium current’ in frog axons [ref. 57]. This brief ‘step pulse’ method does, however, not exclude that the apparently persistent component inactivates with a slower time course, which is not visible within the time window of the test pulse.
In addition to the well-known fast inactivation of INaT with a time constant of < 10 ms, there are intermediate inactivation with a time constant of ~ 100 ms [ref. 71] and slow inactivation with a time constant ≥ 1 s [ref. 184]. It has to be noted that intermediate inactivation is not generally embraced by the literature and many inactivation protocols with pulse lengths greater than 100 ms likely study both fast and intermediate inactivation [ref. 71]. In the following, we will use the terms fast, intermediate and slow inactivation for the three kinetic components described here. In order to test for these additional types of inactivation, especially slow inactivation, long current pulses and repetitive activation steps have been used.
A simple option for testing slow inactivation employs very long stimuli to test whether activation of sodium current is impaired after them in comparison to before. A typical protocol for this purpose consists of three steps (Fig. 2B): First, a long (1 – 30 s) depolarizing pulse (e.g. -30 mV) from hyperpolarized potentials, then a brief (0.5–1 s) recovery pulse to hyperpolarized potentials and finally a short (15 ms) test pulse to a depolarized potential (e.g.—30 mV; see [ref. 92, ref. 166, ref. 195]). In theory, the recovery pulse allows fast, but not slow, inactivated channels to recover from inactivation. The test pulse then determines this fraction of channels, such that the slowly inactivated fraction can be calculated (Fig. 2B). We will call this protocol entry into slow inactivation.
Alternatively, the voltage dependence of slow inactivation was examined using a protocol in which long voltage pulses varying between -120 and + 50 mV (1–10 s duration) were followed by a recovery pulse to a hyperpolarized potential (duration 0.5–1 s) and a test pulse (10 ms duration) to depolarized potential (e.g. -10 mV). This protocol allows to analyse the voltage-dependent amount of inactivated channels and offers a fuller picture of the effects of a drug (Fig. 2C). We will call this protocol slow steady state inactivation.
A problem with both approaches lies in the theoretical assumption that there are only fast and slow inactivation: The length of the recovery pulse is inconsistent in the literature and after longer recovery pulses, channels might have already recovered from intermediate inactivation.
If sodium channels were to undergo fast, intermediate and slow inactivation, at some point there would eventually be no current and the term ‘persistent’ sodium current would obviously be misleading [ref. 45]. However, for both of the aforementioned examples, inactivation never fully completes during the depolarizing pulses. Whether or whether not there is then truly persistent, non-inactivating sodium current, remains an open question.
Another more broadly applied approach employs slow voltage ramps, typically with velocities of 10–70 mV/s ranging from -80—+ 10 mV (Fig. 2D). At these slow depolarization velocities, the transient component INaT is thought to inactivate, such that the remaining component should isolate INaP. However, depolarizing a cell at such slow speed may induce some slow inactivation, leading to a potential underestimation of the remaining INaP. This pitfall explains the hysteresis of slow current components (INaP) between ascending and descending voltage ramps, a well-known hallmark of persistent sodium current [ref. 25]. Different ramp speeds may lead to different resulting currents, including potential distortions of the ‘true’ INaP. For example, Fleidervish and Gutnick [ref. 64] found that a high depolarization speed of 233 mV/s induced action currents (a NaV-generated escape phenomenon in voltage clamp recordings) in cortical pyramidal neurons, while slower ramps of 70 mV/s did not. The velocity of voltage ramps may also affect the apparent effectiveness of INaP blockers, if these have differential effects on different inactivation components. For instance, slower ramp speeds result in larger TTX-susceptible current components and fast ramp speeds underestimate the effect of phenytoin (see Table 1, [ref. 45]). Frequently, ramps can be employed in neurons with complex morphology to reduce the space-clamp error [ref. 12, ref. 162]. In such experiments, the difference between ramp-induced currents in the absence and presence of TTX is measured and taken as a proxy for INaP, as long as TTX resistant sodium channels are absent [ref. 163]. The validation with TTX is important, as leak, potassium and calcium currents are also being evoked with this protocol. Interpretation of the resulting traces is complex, as exemplified by a study characterizing fluoxetine as an apparent INaP blocker, based on a ramp protocol that actually displays a clear potassium current block [ref. 100]. Altogether, voltage ramp measurements are less precise than step-protocols, but they are easier to implement in complex, extended cells like naturally differentiated neurons in ex vivo brain slices. Slow command voltage changes reduce the space-clamp error and help to avoid voltage-clamp escape phenomena like ‘action currents’. Arguably, they are a somewhat more physiological command than a sudden voltage step.
Table 1: List of drug effects sorted by studies. Studies included had to be performed on neuronal sodium channels or neurons. Furthermore, they needed to employ either a step protocol or ramp protocol for measuring INaP or a protocol for slow inactivation. Only voltage clamp protocols were eligible to be included in this list. Whenever possible EC50 or IC50 values are given. HP: holding potential, n.s.: not significant, V0.5:midpoint voltage, IC50: half-maximal inhibitory concentration, EC50: half-maximal effect concentration, ECmax: maximal effect size
| Drug | Preparation | Protocol for INaP | Effect on INaP | Effect on INaT | Study |
|---|---|---|---|---|---|
| Amiodarone | |||||
| 10 µM | rat neocortical neurons | 60 mV/s ramp | -77% peak amplitude | not assessed | Spadoni et al. [ref. 205] |
| Cannabidiol | |||||
| 1 µM | HEK293 cells expressing human Nav1.1 | step pulse 180 ms | n.s. effect on amplitude | n.s. shift of midpoint voltage (V0.5) of steady state inactivation 500 ms | Patel et al. [ref. 170] |
| HEK293 cells expressing human Nav1.6 | step pulse 180 ms | n.s. effect on amplitude | n.s. shift of V0.5 of steady state inactivation 500 ms | ||
| rat striatal neurons | step pulse 180 ms | -36% peak amplitude | 4 mV left shift of steady state inactivation 500 ms | ||
| 3 µM | HEK cells expressing a human Nav1.6 mutation | step pulse 100 ms | IC50 6 µM | -50% amplitude at holding potential (HP) -60 mV (IC50 3 µM) | Ghovanloo et al. [ref. 76] |
| 1 µM | HEK cells expressing human Nav1.2 | step pulse 50 ms | n.s. effect on peak amplitude | -22% average current density; n.s. effect on peak current densityn.s. shift of V0.5 of steady state inactivation 500 ms | Mason and Cummins [ref. 150] |
| Carbamazepine | |||||
| 10 µM | HEK293 cells expressing human Nav1.3 | step pulse 100 ms | -20% amplitude (EC50 16 µM Emax -46%) | 3 mV left V0.5 of steady state inactivation 500 ms (EC50 14 µM Emax 8 mV) | Sun et al. [ref. 215] |
| 100 µM | HEK293 cells expressing Nav1.3 | entry into slow inactivation 10 s | IC50 406 µM | n.s. effect on amplitude at holding potential (HP) -120 mV (IC50 2464 µM) | Sheets et al. [ref. 195] |
| 1 mM | slow steady state inactivation 10 s | n.s. effect on V0.5 | 19 mV V0.5 left shift of steady state inactivation 500 ms | ||
| 100 µM | Scn1b wildtype mouse dissociated dentate gyrus neurons | 50 mV/s ramp | -48% peak amplitude8 mV V0.5 left shift | -32% peak amplitude (HP -100)8 mV V0.5 left shift of activation | Uebachs et al. [ref. 226] |
| 100 µM | N1E-115 neuroblastoma cells | slow steady state inactivation 10 s | n.s. effect on V0.5 | not assessed | Niespodziany et al. [ref. 166] |
| 250 μM | N1E-115 neuroblastoma cells | entry into slow inactivation 30 sslow steady state inactivation 5 s | n.s. effect on amplituden.s. effect on V0.5 | n.s. V0.5 left shift of activation-24% amplitude (HP -80 mV;HP -100 mV IC50 822 µM;HP -80 mV IC50 399 µM;HP -60 mV IC50 109 µM)7 mV V0.5 left shift of steady state inactivation 500 ms | Hebeisen et al. [ref. 87] |
| 100 µM | HEK-293 cells expressing hNav1.6 perfused with ATX-II | step pulse 200 ms | -56% peak amplitude(IC50 77 µM) | -13.8% peak amplitude(IC50 2307 µM, HP -120 mV) | Kahlig et al. [ref. 111] |
| entry into slow inactivation 8 s | -63% peak amplitude(IC50 44 µM) | ||||
| 30 µM | Neuro-2a cells | 500 mV/s ramp | -22% peak amplitude | -13% peak amplitude (HP -100 mV, IC50 56 µM) | Wu et al. [ref. 245] |
| Cenobamate | |||||
| 100 µM | rat CA3 pyramidal neurons | step pulse 150 ms | -74% amplitude(IC50 53 µM) | -5% peak amplitude (HP -80 mV; IC50 > 500 µM) | Nakamura et al. [ref. 163] |
| 15 mV/s ramp | -68% peak amplitude(IC50 53 µM) | 6 mV V0.5 left shift of steady state inactivation 500 ms (EC50 48 µM ECmax 10 mV) | |||
| entry into slow inactivation 8 s | -68% peak amplitude | ||||
| 100 µM | HEK-293 cells expressing hNav1.6 perfused with ATX-II | step pulse 200 msentry into slow inactivation 8 s | -56% peak amplitude(IC50 72 µM)IC50 67 µM | amplitude: IC50 1719 µM, HP -120 mV | Kahlig et al. [ref. 111] |
| Eslicarbazepine | |||||
| 300 µM | Scn1b wildtype mouse dissociated dentate gyrus neurons | 50 mV/s ramp | -22% peak amplitude2 mV left shift of activation | not assessed | Doeser et al. [ref. 55] |
| 250 µM | N1E-115 neuroblastoma cells | entry into slow inactivation 30 sslow steady state inactivation 5 s | -41% amplitude31 mV V0.5 left shift | n.s. V0.5 shift of activation-6% amplitude (HP -80 mV;HP -100 mV IC50 15,744 µM;HP -80 mV IC50 3106 µM;HP -60 mV IC50 562 µM)n.s. V0.5 shift of steady state inactivation 500 ms | Hebeisen et al. [ref. 87] |
| 300 µM | rat dissociated dentategranule neurons | entry into slow inactivation 1 s | -4% peak amplitude | -23% peak amplitude (HP -90 mV) | Holtkamp et al. [ref. 93] |
| entry into slow inactivation 10 s | -8% peak amplitude | ||||
| entry into slow inactivation 30 s | -6% peak amplitude | ||||
| 100 µM | CHO/HEK with Nav1.1 | slow steady state inactivation 10 s | n.s. effect on V0.5 | not assessed | |
| entry into slow inactivation 10 s | n.s. effect on peak amplitude | ||||
| CHO/HEK with Nav1.2 | slow steady state inactivation 10 s | 10 mV V0.5 left shift | not assessed | ||
| entry into slow inactivation 10 s | -48% peak amplitude | ||||
| CHO/HEK with Nav1.3 | slow steady state inactivation 10 s | n.s. effect on V0.5 | not assessed | ||
| entry into slow inactivation 10 s | n.s. effect on peak amplitude | ||||
| CHO/HEK with Nav1.6 | slow steady state inactivation 10 s | 6 mV V0.5 left shift | not assessed | ||
| entry into slow inactivation 10 s | -54% peak amplitude | ||||
| 300 µM | ND7/23 cells with Nav1.6 | slow steady state inactivation 30 s | 17 mV V0.5 left shift | 3 mV V0.5 left shift of fast | Bayraktar et al. [ref. 21] |
| step pulse 100 ms | n.s. effect on amplitude | steady state inactivation 100 ms | |||
| 175 mV/S ramp | n.s. effect on amplitude | n.s. V0.5 shift of activation | |||
| Ethosuximide | |||||
| 750 µM | rat thalamocortical neurons | 200 mV/s ramp | -40% peak amplitude | n.s. effect on amplitude(HP -70 mV) | Leresche et al. [ref. 135] |
| 1 mM | rat CA1 pyramidal neurons | step pulse 250 ms | small n.s. decrease in amplitude | not assessed | Niespodziany et al. [ref. 165] |
| 10 mM | rat TC neuron | 75 mV/s ramp | -37% peak amplitude | not assessed | Broicher et al. [ref. 33] |
| Gabapentin | |||||
| 5 µM | rat DRG neurons | 20 mV/s ramp | -70% peak amplitude | n.s. effect on amplitude(HP -120 mV) | Yang et al. [ref. 250] |
| GS967 | |||||
| 1 µM | mouse hippocampal pyramidal neurons with a Nav1.2 mutation | step pulse 200 ms | -92% amplitude | not assessed | Anderson et al. [ref. 8] |
| tsA201 cells transfected with a Nav1.2 mutation | step pulse 200 ms | IC50 0.4 µM | IC50 19 µM for amplitude (HP -90 mV) | ||
| 200 nM | mouse hippocampal pyramidal neurons with a Nav1.6 mutation | step pulse 200 ms | -93% amplitude | n.s. effect on amplitude(HP -120 mV) | Baker et al. [ref. 18] |
| 5 µM | Xenopus oozytes transfected with Nav1.1 mutations | step pulse 70 ms | -70% amplitude | -10% peak amplitude(HP -90 mV) | Barbieri et al. [ref. 20] |
| 1 µM | ND7/23 cells transfected with a Nav1.6 mutation | step pulse 100 msslow steady state inactivation 1 s with 5 ms recovery pulse | -83% amplitude15 mV left shift | n.s. effect on amplitude(HP -120 mV) | Bunton-Stasyshyn et al. [ref. 35] |
| 1 µM | ND7/23 cells transfected with Nav1.6 wildtype | step pulse 80 ms | no INaP found | n.s. effect on amplitude(HP -120 mV)17 mV left shift of steady state inactivation 500 ms | Wengert et al. [ref. 240] |
| mouse subiculum pyramidal neurons with Nav1.6 wildtype | 65 mV/s ramp | -49% peak amplitude | not assessed | ||
| 1 µM | HEK cells expressing wild type human Nav1.2 channels | step pulse 50 ms | -45% current density | -35% peak current density(HP -100 mV) | Mason and Cummins [ref. 150] |
| 3 µM | mouse hippocampal fast-spiking neuron | 25 mV/s ramp | -78% area under the curve | not assessed | Auffenberg et al. [ref. 16] |
| Lacosamide | |||||
| 100 µM | mouse N1E-115 neuroblastoma cells | entry into slow inactivation 30 s | -43% peak amplitude | -28% amplitude at HP -100 mV-29% amplitude at HP -60 mVn.s. V0.5 shift of steady state inactivation 500 ms | Errington et al. [ref. 59] |
| 100 µM | HEK293 cells expressing Nav1.3 | entry into slow inactivation 10 s | IC50 415 µM | n.s. effect on amplitude at HP-120 mV (IC50 51 mM)-25% peak amplitude at HP -80 mV | Sheets et al. [ref. 195] |
| 1 mM | slow steady state inactivation 10 s | 42 mV V0.5 left shift | n.s. V0.5 shift of steady state inactivation 500 ms | ||
| 300 µM | mouse dissociated hippocampal neurons | 50 mV/s ramp | -50% peak amplitude | not assessed | Uebachs et al. [ref. 227] |
| 100 µM | N1E-115 neuroblastoma cells | slow steady state inactivation 10 s | 33 mV V0.5 left shift | not assessed | Niespodziany et al. [ref. 166] |
| 250 µM | N1E-115 neuroblastoma cells | entry into slow inactivation 10 ssteady state inactivation 5 s | -14% peak amplitude43 mV V0.5 left shift | n.s. V0.5 left shift of activation-20% peak amplitude (HP -80 mV)n.s. V0.5 shift of steady state inactivation 500 ms | Hebeisen et al. [ref. 87] |
| 100 µM | rat dissociated hippocampal granule cells | entry into slow inactivation 1 sentry into slow inactivation 10 sentry into slow inactivation 30 s | -8% peak amplitude-14% peak amplituden.s. effect on peak amplitude | 4 mV V0.5 left shift of fast steady state inactivation | Holtkamp et al. [ref. 92] |
| 100 µM | HEK-293 cells expressing hNav1.6 perfused with ATX-II | step pulse 200 msentry into slow inactivation 8 s | -9.6% amplitude(IC50 832 µM)IC50 269 µM | not assessed | Kahlig et al. [ref. 111] |
| Lamotrigene | |||||
| 100 µM | rat dissociated hippocampal neurone | slow steady state inactivation 9 sentry into slow inactivation 6 s | 11 mV V0.5 left shift-69% peak amplitude | -7% amplitude at HP -90 mV (IC50 1490 µM)-98% amplitude at HP -60 mV (IC50 7 µM) | Kuo and Lu [ref. 124] |
| 1 µM | rat neocortical pyramidal neurons | 60 mV/s ramp | n.s. effect on amplitude | n.s. effect on amplitude(HP -65 mV) | Spadoni et al. [ref. 205] |
| 100 µM | rat neocortical layer 5 pyramidal neurons | 10 mV/s ramp | not quantified but similar to 100 µM phenytoin | responses ‘still inducible’ | Berger and Lüscher [ref. 26] |
| 30 µM | HEK cells expressing hNav1.2 | slow steady state inactivation 30 s | 11 mV V0.5 left shift | n.s. effect on amplitude at HP-100 mV (IC50 2.6 mM)-33% peak amplitude at HP -60 mV (IC50 172 µM)n.s. shift of activationn.s. shift of fast steady state inactivation 10 ms | Jones et al. [ref. 107] |
| 100 µM | N1E-115 neuroblastoma cells | slow steady state inactivation 10 s | 7 mV V0.5 right shift | not assessed | Niespodziany et al. [ref. 166] |
| 100 µM | HEK-293 cells expressing hNav1.6 perfused with ATX-II | step pulse 200 ms | -55.4% amplitude(IC50 78 µM) | -13.8% peak amplitude(IC50 1249 µM, HP -120 mV) | Kahlig et al. [ref. 111] |
| entry into slow inactivation 8 s | -72.8% amplitude(IC50 39 µM) | ||||
| Lidocaine | |||||
| 30 nM | rat dissociated CA1 pyramidal neurons | step pulse 500 ms | -53% amplitude | n.s. effect on amplitude(HP -100 mV) | Hammarstrom and Gage [ref. 86] |
| 10 µM | rat DRG neurons | 15 mV/s ramp | ‘obvious’ effect, not quantified | ‘little’ effect, not quantified | Dong et al. [ref. 56] |
| 100 µM | HEK293 cells expressing Nav1.3 | entry into slow inactivation 10 s | IC50 284 µM | -6% amplitude at HP -120 mV (IC50 1462 µM) | Sheets et al. [ref. 195] |
| 1 mM | slow steady state inactivation 10 s | 48 mV V0.5 left shift | 20 mV V0.5 left shift of steady state inactivation 500 ms | ||
| NBI-921352 | |||||
| 41 nM | HEK293 cells expressing a hNav1.6 mutation | step pulse 20 ms | -50% amplitude | IC50 33 µM, HP -120 mVIC50 53 nM, HP -62 mV | Johnson et al. [ref. 106] |
| Oxcarbazepine | |||||
| 10 µM | NG108-15 cells | 100 mV/s ramp | -40% peak amplitude | Huang et al. [ref. 97] | |
| 3 µM | -9 mV shift of fast steady state inactivation 30 ms-61% amplitude (HP -80 mV) | ||||
| 250 µM | N1E-115 neuroblastoma cells | entry into slow inactivation 30 sslow steady state inactivation 5 s | n.s. effect on amplitude28 mV V0.5 left shift | -24% amplitude (HP -80 mVHP -100 mV IC50 2000 µM;HP -80 mV IC50 805 µM;HP -60 mV IC50 173 µM)17 mV left V0.5 shift of steady state inactivation 500 ms | Hebeisen et al. [ref. 87] |
| 100 µM | HEK-293 cells expressing hNav1.6 perfused with ATX-II | step pulse 200 ms | -57.8% amplitude(IC50 123 µM) | IC50 1035 µM, HP -120 mV | Kahlig et al. [ref. 111] |
| entry into slow inactivation 8 s | IC50 42 µM | ||||
| Phenytoin | |||||
| 75 µM | N1E-115 | entry into slow | 20 mV V0.5 left shift | Matsuki et al. [ref. 151] | |
| 100 µM | neuroblastoma cells | inactivation 60 s | -6% amplitude at HP -100 mV-42% amplitude at HP -80 mV | ||
| 100 µM | rat CA1 neurons | slow steady state inactivation 16 s | 15 mV V0.5 left shift | -10% amplitude at HP -90 mV (IC50 600 µM)-95% amplitude at HP -50 mV (IC50 7 µM)n.s. V0.5 shift of fast steady state inactivation 100 ms | Kuo and Bean [ref. 123] |
| entry into slow inactivation 12 s | -85% peak amplitude | ||||
| 34 µM | rat neocortical and neostriatal neurons | step pulse 400 ms | -50% amplitude(EC50 34 µM) | not assessed | Chao and Alzheimer [ref. 42] |
| 50 mV/s ramp | no V0.5 shift | ||||
| 60 µM | hippocampal neurons in culture | single channel recordings in outside-out patches | -77% amplitude of late (50 – 100 ms) currents | -60% amplitude(HP -100 mV) | Segal and Douglas [ref. 192] |
| 75 µM | rat neocortical layer 5 pyramidal neurons | 7.3 mV/s ramp | -40% peak amplitude(EC50 78 µM, ECmax -90%) | not assessed | Lampl et al. [ref. 130] |
| 1 µM | rat neocortical pyramidal neurons | 60 mV/s ramp | n.s. effect on amplitude | n.s. effect on amplitude(HP -65 mV) | Spadoni et al. [ref. 205] |
| 100 µM | rat CA1 pyramidal neurons | step pulse 250 ms | -54% amplitude | not assessed | Niespodziany et al. [ref. 165] |
| 100 µM | rat CA1 pyramidal neurons | 50 mV/s ramp | -58% peak amplitude | -27% amplitude(HP -70 mV) | Yue et al. [ref. 251] |
| 100 µM | rat neocortical pyramidal neurons | 50 & 100 mV/s ramp | n.s. effect on amplitude | 9 mV V0.5 left shift of fast steady state inactivation | Colombo et al. [ref. 45] |
| 10 mV/s rampstep pulse 10 s50 mV/s ramp after 10 s slow steady state inactivation | -34% peak amplitude-26% amplitude7 mV V0.5 left shift | ||||
| 50 mV/s ramp after 200 ms depolarizing prepulse | -20% peak amplitude(EC50 28 µM) | ||||
| 50 mV/s ramp after 500 ms depolarizing prepulse | -25% peak amplitude(EC50 18 µM) | ||||
| 100 µM | N1E-115 neuroblastoma cells | slow steady state inactivation 10 s | n.s. effect on V0.5 | not assessed | Niespodziany et al. [ref. 166] |
| 100 µM | tsA201 cells transfected with a Nav1.2 mutation | step pulse 200 ms | -88% amplitude(IC50 16 µM) | -40% amplitude(IC50 143 µM, HP -120 mV) | Anderson et al. [ref. 8] |
| 50 µM | rat CA1 pyramidal neurons | step pulse 50 ms | n.s. effect on amplitude | -14% amplitude at HP -120 mV | Zeng et al. [ref. 253] |
| slow steady state inactivation 10 s | 8 mV V0.5 left shift | -24% amplitude at HP -100 mV | |||
| entry into slow inactivation 10 s | -40% amplitude | -39% amplitude at HP -80 mV (IC50 73 µM)n.s. V0.5 shift of fast steady state inactivation 50 ms7 mV V0.5 left shift of steady state inactivation 500 ms | |||
| 4 µM | mouse hippocampal pyramidal neurons with a Nav1.6 mutation | step pulse 200 ms | -45% peak amplitude | n.s. effect on amplitude(HP -120 mV) | Baker et al. [ref. 18] |
| 100 µM | mouse CA1 pyramidal cells | 50 mV/s ramp | -62.5% peak amplitude | not assessed | Kang et al. [ref. 114] |
| mouse CA1 PV + basket cells | 50 mV/s ramp | -78.9% peak amplitude | |||
| 100 µM | HEK-293 cells expressing hNav1.6 perfused with ATX-II | step pulse 200 ms | -57.8% amplitude(IC50 60 µM)IC50 48 µM | not assessed | Kahlig et al. [ref. 111] |
| entry into slow inactivation 8 s | |||||
| PRAX-562 | |||||
| 100 nM | HEK-293 cells expressing hNav1.6 perfused with ATX-II | step pulse 200 ms | -45% amplitude(IC50 141 nM) | Kahlig et al. [ref. 111] | |
| 1 µM | entry into slow inactivation 8 s | -72.3% peak amplitude(IC50 317 nM) | -13.8% peak amplitude(IC50 8.4 µM, HP -120 mV)12 mV V0.5 left shift of steady state inactivation 500 ms3 mV left shift of activation | ||
| Propofol | |||||
| 10 µM | rat neocortical pyramidal neurons | 53 mV/s ramp | -75% peak amplitude(IC50 4 µM) | n.s. effect on amplitude(HP -70 mV) | Martella et al. [ref. 149] |
| 56 µM | rat medial geniculate body neurons | step pulse 1 s | -45% amplitude | not assessed | Shi et al. [ref. 198] |
| Ranolazine | |||||
| 10 µM3 µM | NG108-15 cells | 70 mV/s ramp70 mV/s ramp | -58% peak amplitude-41% peak amplitude | 15 mV V0.5 left shift of fast steady state inactivation 100 ms | Wu et al. [ref. 244] |
| 30 µM | tsA201 cells transfected with Nav1.1 | step pulse 200 ms | -9% peak amplitude(IC50 54 µM) | n.s. effect on V0.5 of fast steady state inactivation 100 msn.s. effect on amplitude(HP -120 mV, IC50 871 µM) | Kahlig et al. [ref. 109] |
| 30 µM | nucleated somatic patches of rat CA1 pyramidal neurons | 400 mV/s ramp | n.s. effect on amplitude | n.s. effect on amplitude(HP -65 mV) | Park et al. [ref. 167] |
| 10 µM | rat cultured hippocampal neurons | slow steady state inactivation 10 sentry into slow inactivation 10 s | 10 mV V0.5 left shift-31% amplitude | 5 mV V0.5 left shift of fast steady state inactivation 100 ms | Kahlig et al. [ref. 110] |
| Riluzole | |||||
| 10 µM | rat neocortical pyramidal neurons | step pulse 250 ms | -100% amplitudeIC50 2 µM | 9 mV V0.5 left shift of fast steady state inactivation | Urbani and Belluzzi [ref. 228] |
| 14 mV/s ramp | -100% peak amplitude (identical to 0.3 µM TTX) | (EC50 50 µM; ECmax 21 mV) | |||
| 10 µM | rat preBötC respiratory pacemaker neurons | 20 mV/s ramp | -85% peak amplitude | -8% spike amplitude | Del Negro et al. [ref. 53] |
| 1 µM | rat neocortical neurons | 60 mV/s ramp | -80% peak amplitude(IC50 550 nM) | -50% amplitude(HP -65 mV) | Spadoni et al. [ref. 205] |
| 25 µM | rat isolated suprachiasmatic nucleus neurons | 100 mV/s ramp | -100% peak amplitude | not assessed | Kononenko et al. [ref. 122] |
| 10 µM | rat CA1 pyramidal neurons | step pulse 250 ms | -56% amplitude | not assessed | Niespodziany et al. [ref. 165] |
| 0.5 µM | rat G93A SOD1 mutant cultured motoneurones | 10 mV/s ramp | -56% area under the curve | not assessed | Kuo et al. [ref. 125] |
| 3 µM | rat preBötC respiratory pacemaker neurons | step pulse 50 ms | -64% amplitude at HP -100 mV-73% amplitude at HP -80 mV-80% amplitude at HP -60 mV | -23% amplitude at HP -100 mV-61% amplitude at HP -80 mV (EC50 2.4 µM)-90% amplitude at HP -60 mV10 mV V0.5 left shift of fast steady state inactivation | Ptak et al. [ref. 175] |
| 5 µM | rat mesencephalic layer V neurons | 33.3 mV/s ramp | -81% peak amplitude | -11% amplitude(HP -80 mV, EC50 51.6 µM) | Wu et al. [ref. 242] |
| 10 µM | rat CA1 pyramidal neurons | 50 mV/s ramp | -90% peak amplitude | -30% amplitude(HP -70 mV) | Yue et al. [ref. 251] |
| 5 µM10 µM | rat ventral horn neurones | 16 mV/s ramp16 mV/s ramp | -70% peak amplitude-74% peak amplitude | not assessed | Theiss et al. [ref. 221] |
| 10 µM | rat hypoglossal motor neurons | 42 mV/s ramp | not quantified in presence of Ca2+ blockers | not assessed | Lamanauskas and Nistri [ref. 128] |
| 10 µM | mouse superior cervical ganglion neurons | 10 mV/s ramp | -70% peak amplitude | -28% amplitude (HP -80 mV) | Lamas et al. [ref. 129] |
| 1 µM | cultured embryonic G93A SOD1 mouse cortical neurons | step pulse 80 ms | -48% amplitude | n.s. effect on amplitude(HP -60 mV) | Pieri et al. [ref. 173] |
| 10 µM3 µM | neuroblastoma & glioma NG108-15 cells | 70 mV/s ramp70 mV/s ramp | -55% peak amplitude-34% peak amplitude | 13 mV V0.5 left shift of fast steady state inactivation | Wu et al. [ref. 244] |
| 10 µM | compressed rat DRG neurons | 26.7 mV/s ramp | -60% peak amplitude(IC50 4 µM) | n.s. effect on amplitude(HP -80 mV) | Xie et al. [ref. 246] |
| 200 µM500 µM | -59% amplitude (HP -80 mV)-82% amplitude (HP -80 mV) | ||||
| 10 µM | nucleated somatic patches of rat CA1 pyramidal neurons | 400 mV/s ramp10 mV/s ramp | -73% peak amplitudeno INaP found | -33% amplitude13 mV V0.5 left shift of fast steady state inactivation | Park et al. [ref. 167] |
| 20 µM | rat hypoglossal motor neurons | 35 mV/s ramp | not quantified | not assessed | Bellingham [ref. 25] |
| 10 µM | rat CA3 pyramidal neurons | 15 mV/s ramp | -80% peak amplitude | n.s. effect on amplitude(HP -80 mV) | Nakamura et al. [ref. 163] |
| Rufinamide | |||||
| 100 µM | N1E-115 neuroblastoma cells | slow steady state inactivation 10 s | 9 mV V0.5 right shift | not assessed | Niespodziany et al. [ref. 166] |
| 100 µM | Xenopus oocytes with | step pulse 50 ms | n.s. effect on amplitude | n.s. V0.5 shift of fast steady state inactivation | Gilchrist et al. [ref. 78] |
| hNav1.1 | entry into slow inactivation 10 s | n.s. effect on amplitude or V0.5 | |||
| slow steady state inactivation 10 s | n.s. effect on V0.5 | 8 mV V0.5 right shift of activation | |||
| Xenopus oocytes with hNav1.2Xenopus oocytes with hNav1.3Xenopus oocytes with hNav1.6 | n.s. V0.5 shift of fast steady state inactivationn.s. V0.5 shift of fast steady state inactivation5 mV V0.5 right shift of fast steady state inactivation | ||||
| Topiramate | |||||
| 100 µM | rat neocortical layer V pyramidal neurons | 80 mV/s rampentry into slow inactivation 800 ms | -40% peak amplitude-65% peak amplitude | 7 mV V0.5 left shift of fast steady state inactivation 300 ms | Taverna et al. [ref. 218] |
| 2 µM | HEK293 cells expressing human Nav1.3 | step pulse 100 ms | -22% amplitude(EC50 61 nM Emax -30%) | 1.5 mV V0.5 left shift of steady state inactivation 500 ms(EC50 3 µM Emax 3 mV) | Sun et al. [ref. 215] |
| Valproic acid | |||||
| 200 µM | dissociated rat neocortical pyramidal neurons | 40 mV/s ramp | -80% peak amplitude(EC50 14 µM, ECmax -80%) | n.s. effect on amplitude(HP-70 mV) | Taverna et al. [ref. 217] |
| 100 µM | rat neocortical pyramidal neurons | 53 mV/s ramp | -80% peak amplitude | n.s. effect on amplitude(HP -70 mV) | Martella et al. [ref. 149] |
| 10 µM | mouse superior cervical ganglion neurons | 10 mV/s ramp | -55% peak amplitude | n.s. effect on amplitude (HP -80 mV) | Lamas et al. [ref. 129] |
| 1 mM | HEK-293 cells expressing hNav1.6 perfused with ATX-II | step pulse 200 msslow entry into slow inactivation 8 s | -2% amplitude-18% peak amplitude | -11% peak amplitude at HP-120 mV | Kahlig et al. [ref. 111] |
| Zonisamide | |||||
| 100 µM | N1E-115 neuroblastoma cells | slow steady state inactivation 10 s | n.s. effect on V0.5 | not assessed | Niespodziany et al. [ref. 166] |
Few papers have studied the effect of slow inactivation protocols on subsequent ramps. While these protocols might be one of the best ways of assessing persistent sodium current, they are very difficult to record and therefore only seldomly employed [ref. 45, ref. 123, ref. 242].
Pharmacology of INaP
Our systematic literature search identified 2586 PubMed results for persistent, slowly or late inactivating sodium currents in combination with blocking, reducing or inhibiting drug actions. Our search term was ‘(((persistent) OR (slow inactivation) OR (late)) AND (sodium current)) AND ((block) OR (inhibit) OR (reduce))’. Papers were first screened for pharmacological agents with potential clinical applications, i.e. excluding endogenous substances (e.g. acetylcholine), toxins (e.g. saxitoxin), chemicals without present or planned clinical use (e.g. insecticides or dyes) and intracellular agents (e.g. QX-314). After identification of potential blockers, a second literature search was conducted with ‘(((persistent) OR (slow inactivation) OR (late)) AND (sodium current)) AND substance name’) for each substance. Papers were considered eligible when appropriate voltage clamp protocols for either non-inactivating or slowly inactivating sodium currents were applied to CNS neurons or to cells expressing Nav1.1, Nav1.2, Nav1.3 or Nav1.6. We will now discuss the actions of the identified substances used in INaP-related research as listed in Table 1 (see also Fig. 3 for an overview). We will consider their specificity, affinity and different mechanisms of action. Where applicable, clinically relevant information such as blood concentrations and blood–brain-barrier interactions (a major problem for clinical translation) will be included. Concentrations are given as IC50 or EC50 values, respectively.
Amiodarone
Amiodarone is a type III antiarrhythmic agent typically used for treatment of ventricular and supraventricular arrhythmia. It predominantly inhibits the human Ether-a-go-go-Related Gene (hERG) potassium channel that mediates a delayed rectified outward potassium current and contributes to the repolarization of cardiac myocytes. The IC50 for this effect is 9.8 µM [ref. 117]. There is only one study that assessed the effects of amiodarone on INaP in cortical neurons [ref. 205]. Here, the authors saw a > 50% inhibition of INaP with a ramp protocol at a concentration of 10 µM (Table 1). For NaV1.5, the isoform most prevalent in the heart, IC50 values were in a similar range [ref. 75, ref. 142, ref. 243]. Although amiodarone is a highly lipophilic molecule, its concentration in the brain only reaches 10% of the heart tissue concentration after intravenous administration in the rat [ref. 180]. Therefore, it is not suitable for use as an INaP blocker in neurons in in vivo approaches both in experiments or clinical settings.
Cannabidiol
Besides the well-known hallucinogenic compound tetrahydrocannabinol, cannabis sativa contains more than 100 cannabinods including cannabidiol (CBD). CBD has been used as an anti-seizure drug for patients with Dravet syndrome, a severe epileptic encephalopathy resulting from loss-of-function mutants of Nav1.1 [ref. 54]. The substance is also known for its anxiolytic and sedative effects [ref. 50]. Cannabinoids typically act via the Gi-protein-coupled cannabinoid receptors CB1 in the brain or CB2 in periphereral tissues [ref. 58]. CBD, at concentrations of 100 nM, acts as a non-competitive antagonist at CB1 receptors [ref. 222]. It also binds to the serotonin receptor 5-HT1a at the same concentration [ref. 188] and activates Kv7 channels with an EC50 of 200 nM [ref. 255]. Concerning sodium currents, CBD blocks INaP in step protocols, but seems to be even more efficient in blocking transient, rather than persistent sodium currents (Table 1). For Nav1.7 Huang et al. [ref. 98] report preferential block of fast inactivation with slow binding kinetics (see discussion of phenytoin), while for Nav1.8 CBD preferentially blocks slow inactivation [ref. 254]. Effects of CBD seem to require rather high concentrations: Ghovanloo et al. [ref. 76] report CBD to block the transient component (INaT) of all sodium channel isoforms with an IC50 of around 3 µM, while Hill et al. [ref. 89] estimate the IC50 of CBD for brain-expressed sodium channels even tenfold higher. However, this might be an artefact of using plastic instead of glass reservoirs and tubing [ref. 255]. Based in these data, CBD should not be used as an CNS INaP blocker and likely exerts its neurotherapeutic effects via non-sodium channel mediated pathways.
Carbamazepine
For a long period of time, carbamazepine has been one of the most popular anti-seizure drugs worldwide. This is due to its good efficacy in focal epilepsy and its low costs [ref. 172]. It is also a first line drug in other paroxysmal neurological diseases such as episodic ataxia type 1 [ref. 131], trigeminal neuralgia [ref. 219], paroxysmal extreme pain disorder [ref. 63] or secondary dyskinesia [ref. 68]. Carbamazepine affects voltage activated calcium channels, especially Cav2.1, at low potency with an IC50 of 452 µM [ref. 203]. In addition, at 3 µM it suppresses 70% of D-type potassium currents in NG108-15 cells [ref. 97].
The main effect of carbamazepine however, is a left shift of the fast inactivation curve of sodium currents at 10–100 µM [ref. 87, ref. 195, ref. 215, ref. 226] (Table 1). This effect leads to an earlier inactivation of sodium channels, which then already begins at more negative potentials. Therefore, a reduction of the persistent sodium current should also be expected under the window current hypothesis, which is reflected in step and ramp protocols [ref. 111, ref. 215, ref. 226]. However, multiple studies show a lack of action of carbamazepine on slowly inactivating sodium current at concentrations up to 1 mM [ref. 87, ref. 166, ref. 195] with the exception of Kahlig et al. [ref. 111]. Weighing the evidence, carbamazepine seems to preferentially target INaT, rather than INaP (Table 1).
Cenobamate
The latest anti-seizure drug being approved by the European Medicines Agency is cenobamate [ref. 61]. It is used as a third-line therapy for multi-drug-resistant focal epilepsy. Cenobamate is known for its potentiation of GABAA-mediated currents with EC50 values in the range of 42 to 194 µM [ref. 194]. At high concentrations it inhibits L-type calcium currents with an IC50 of 350 µM and has little effects on KV7.1 (IC50 1.3 mM) and KV11.1 (IC50 1.8 mM) [ref. 7].
Concerning persistent sodium current, cenobamate has been found to block both the non-inactivating and the slowly inactivating current component with an IC50 of 50–70 µM [ref. 110, ref. 163] (Table 1), which is well below the reported plasma concentration of 170 µM [ref. 34]. It also shifts the fast inactivation to the left, but this effect is much less pronounced than its suppressing effect on INaP. The substance could thus be used as a persistent sodium current blocker, but one has to keep in mind that the positive modulation of GABAA receptors occurs at similar concentrations. Thus, systemic effects may be dominated by either of these mechanisms. It is presently difficult to imagine how the substance can isolate specific effects of INaP in native brain tissue.
Eslicarbazepine
Eslicarbazepine is an anti-seizure drug that was developed in order to bypass the side effects of carbamazepine and its potentially harmful metabolite carbamazepine-10,11-epoxide [ref. 187]. Like carbamazepine, it also affects calcium channels, with a preference for Cav3.2 (a subunit mediating T-type calcium currents) at an IC50 of 62 µM [ref. 203]. The effects of eslicarbazepine on sodium currents are different from those of carbamazepine. It does not affect the amplitude or fast inactivation of transient sodium currents. Contrary to carbamazepine it shifts the slow inactivation to more hyperpolarized potentials at 300 µM [ref. 21, ref. 87, ref. 93] (Table 1). Because of its slow binding kinetics and/or its lack of action on fast inactivation, 300 µM eslicarbazepine does not affect the persistent sodium current when measured with brief voltage steps or fast ramps [ref. 21], while afflicting mild effects in slow ramps [ref. 55]. Therefore, the substance should be evaluated as a blocker of INaP, in virtue of being an enhancer of slowy inactivating sodium currents. In complex tissues or living organisms, its efficacy on Cav3.2 at similar concentrations should warrant caution.
Ethosuximide
Ethosuximide is an odd anti-seizure drug used solely to treat absence seizures, while it is not effective against other types of seizures and therefore not commonly employed. Its main mechanism of action is thought to be a partial block of T-type calcium current in thalamic neurons with an EC50 of 200 µM [ref. 48]. In addition, at 250 µM there is a partial block of the Na+/K+ ATPase [ref. 77]. At concentrations of 20–50 mM INaT is reduced by ethosuximide in the squid giant axon in a voltage-independent manner [ref. 66]. Reliable block of persistent sodium current in neurons can only be obtained at rather high concentrations of 1–10 mM [ref. 33, ref. 135] which exceed typical plasma levels of 0.3–0.7 mM [ref. 81]. Thus, while it might preferentially effect INaP, ethosuximide cannot be used as an INaP blocker, at least in complex preparations containing multiple ion channels.
Gabapentin
Gabapentin is one of the top 10 most prescribed drugs in the world, as it is commonly used for neuropathic pain in polyneuropathies and other chronic pain conditions. Its main mechanism of action is a block of N-type voltage gated calcium channels via an interaction with the α2δ-1 subunit which gabapentin binds with a Kd of 59 nM [ref. 169]. One study has shown that 5 µM gabapentin blocks INaP in dorsal root ganglion cells, using a ramp protocol [ref. 250] (Table 1). However, DRG neurons express Nav1.7, 1.8 and 1.9, i.e. isoforms with peculiar kinetics, which are not dominant in most central nervous neurons. Taken together with the well described effects on calcium channels, gabapentin is probably not a good candidate for being a general blocker of INaP.
GS967
GS967, now known as Prax330, is a novel compound initially synthesized for treating cardiac arrhythmias [ref. 23]. Until now, no clinical trials have been published for this compound, although a phase I trial has been completed (ACTRN12617001512314). Nonetheless, GS967 has been found to be effective in animal models of monogenetic epilepsy and hemiplegic migraine [ref. 9, ref. 16, ref. 18]. With regard to possible mechanisms of action, no studies have been published so far examining effects on calcium channels or other molecular targets. However, GS967 reduces INaP measured with both ramp and short step pulse protocols (Table 1) at concentrations of 0.2–1 µM [ref. 9, ref. 240]. It also shifts the fast inactivation curve to more hyperpolarized potentials (Table 1). At this point of time, there is no study evaluating the effect of the drug on slow sodium current inactivation in neurons. Evidence from cardiac slow (2 s) steady state inactivation, however, suggests that GS967 may also be effective in blocking slowly inactivating sodium current. [ref. 88]. Taken together, GS967 is a potent blocker of persistent sodium current, and might therefore be a good candidate for further translational studies.
Lacosamide
Lacosamide has been used to treat focal epilepsy and is also one of the few drugs approved for managing status epilepticus. Recently, however, there has been increased awareness concerning cardiac arrhythmia after administration of lacosamide [ref. 247]. Although lacosamide halfs calcium influx via N-type calcium channels (CaV2.2) in cortical neurons at 200 µM [ref. 157], its main mechanisms of action appears to involve slowly inactivating sodium currents and/while binding collapsin response mediator protein 2 (CRMP-2) [ref. 28], which influences trafficking of Nav1.7 [ref. 113]. Similar to eslicarbazepine, lacosamide does not affect fast and intermediate inactivation and has only very small effects on INaP voltage step protocols [ref. 111]. However, at 100–250 µM it shifts of the midpoint voltage (V0.5) of slow inactivation by 40 mV to the left [ref. 62, ref. 87]. This mechanism may underlie the 50% reduction in the ramp protocol observed at 300 µM [ref. 227]. Suggested underlying mechanisms are that lacosamide prefers fast inactivation, but has very slow binding kinetics [ref. 104] (see discussion of phenytoin), that it preferentially affects channels with slow inactivation [ref. 92] or that its interaction with CRMP-2 confers an enhancement of slow inactivation [ref. 158]. Keeping in mind the clinically used serum concentration of 20–40 µM [ref. 144] lacosamide should be considered as a potent enhancer of slow inactivation. It has, however, lacking effectiveness for INaP understood as the non-inactivating component in the brief step protocol.
Lamotrigine
Lamotrigine is currently the best drug for treating focal epilepsy [ref. 145, ref. 147], as it is well tolerated and, in some patients, even stabilizes mood. This is why lamotrigine has also entered the realm of psychiatric disease and is currently used for treating bipolar disorder or depression [ref. 47]. Another beneficial property of lamotrigine is its efficacy in generalized epilepsy, while other sodium channel blockers tend to aggravate seizures in these patients. This unique profile is linked to a broad range of mechanisms of action: Lamotrigine inhibits high-voltage-activated Ca2+ currents with an EC50 of 12 µM [ref. 209], which in turn reduces the release of glutamate [ref. 236]. It also reduces the uptake of serotonin, noradrenaline and dopamine at concentrations of 200–400 µM [ref. 204] and, at 100 µM, lamotrigine induces the expression of GABA-A β3 subunits [ref. 237]. Lamotrigine seems to bind slowly to sodium channels, which explains the modest effects on the non-inactivating current component at concentrations of 80–100 µM [ref. 111, ref. 205] (Table 1). Accordingly, lamotrigine shifts the slow inactivation to more hyperpolarized potentials and exerts inhibition on INaT amplitude at depolarized membrane potentials [ref. 107, ref. 111, ref. 124] (Table 1). These effects occur in a range of 7–40 µM which is comparable to clinical plasma concentrations of 20 µM [ref. 118]. In general, the size of these effects is comparatively low (Fig. 3 A and B) and given the effects on calcium- and GABAA-channels, it should not be regarded as a specific INaP blocker.
Lidocaine
Lidocaine is one of the most popular local anaesthetics and therefore often used to block nerve conduction during surgery. Due to its low bioavailability it is generally not administered orally [ref. 52]. In intensive care medicine, intravenous lidocaine has been used for treating ventricular arrhythmia or status epilepticus in rare cases [ref. 252]and it is recommended as an anti-arrhythmic drug during resuscitation [ref. 202].
Its mechanism of action is mainly based on the block of sodium channels, though it also blocks HCN channels at 20–50 µM [ref. 154]. Lidocaine is one of the few drugs whose binding site in VGSC has been well established: its inhibitory effect is primarily caused by disrupting the coupling between the voltage sensors of the sodium channel via long-range stabilization of the third transmembrane domain in the activated state[ref. 161].
Most of the studies concerning lidocaine and persistent sodium current have been performed in myocytes [ref. 19, ref. 22, ref. 37, ref. 102, ref. 108, ref. 238]. For neuronal channels, lidocaine at high concentration (300 µM—1 mM) does have major effects on both slow and fast sodium current inactivation [ref. 195]. However, one study in neurons shows a remarkable effect of lidocaine at 30 nM on the non-inactivating current component in a short pulse protocol [ref. 86]. Clinical use cases typically involve lidocaine levels in the range of 1–10 µM [ref. 36]. Thus, while the drug appears to be a blocker of INaP, its cardiac side effects restricts its use to preparations in the laboratory.
NBI-921352
This is a novel compound which was developed as a specific Nav1.6 inhibitor by Xenon Pharmaceuticals [ref. 106]. Potent effects on INaP appear quite possible (Table 1), but data on persistent or slowly inactivating components in neurons is yet missing.
Oxcarbazepine
Like eslicarbazepine, oxcarbazepine is a derivate of carabamazepine. It is used for focal epilepsy, neuropathic pain and sometimes also during alcohol withdrawal [ref. 191]. It is one of the few anti-seizure drugs which are not teratogenic [ref. 224]. Unlike carabamazepine and eslicarbazepine, it has been shown to also affect voltage-gated calcium channels, particularly N-type, at concentrations of 2–50 µM [ref. 208]. In addition, at 10 µM it suppresses 50% of D-type potassium currents [ref. 97]. With regard to sodium currents, oxcarbazepine seems to affect both slow and fast inactivation, with a stronger effect on slow inactivation at 10–100 µM (Table 1). It therefore bridges the gap between carbamazepine (predominantly acting on fast inactivation) and eslicarbazepine (predominantly acting on slow inactivation). The observed effects in ramp and step protocols in neuroblastoma/-glioma cells make it quite possible that the drug inhibits INaP (Table 1). However, there is no data on effects of oxcarbazepine on INaP in naturally differentiated neurons. This may be a matter of concern, as oxcarbazepine is the only antiepileptic drug which has been shown to reliably destroy glioma cells. In one study on cells derived from patients with brain tumours, the IC50 for induction of apoptosis was 45 µM [ref. 51]. Out of three studies on oxcarbazepine which are relevant for this review, two were carried out on neuroblastoma/-glioma cells [ref. 87, ref. 97], where the drug might induce apoptosis cascades [ref. 51]. Hence, more evidence is needed to classify oxcarbazepine with respect to effects on INaP.
Phenytoin
Phenytoin is one of the oldest anti-seizure and antiarrhythmic drugs [ref. 134]. Nowadays, due to its non-linear pharmacokinetics and severe side effects its use is typically restricted to inpatients [ref. 74, ref. 179]. Serum levels of phenytoin are typically at 3–10 µM, where it has a 50% inhibitory effect on calcium channels [ref. 155]. Being one of the best characterized sodium channel blockers (Table 1), phenytoin does appear to be a prime example of a drug that affects intermediate inactivation [ref. 71, ref. 253]. This explains why the drug does not have an effect on INaP measured with short voltage steps or fast ramps. However, it strikingly inhibits later phases of currents evoked by voltage steps as well as currents evoked by slow ramps (Table 1). It has been suggested that phenytoin exhibits its effects by slowly binding sodium channels in the fast inactivated state [ref. 123]. This argument seems to be at odds, however, with the remaining sensitivity of intermediate inactivation to phenytoin after intracellular proteolysis by papain or pronase [ref. 253], which completely abolishes fast inactivation [ref. 181]. Therefore, if the effect of phenytoin would target fast inactivation after slowly binding to sodium channels, it should not be active after proteolysis. The substance remains active, however, after intracellular application of papain, suggesting that phenytoin does not inhibit sodium currents by affecting fast inactivation [ref. 253]. Thus, phenytoin does not directly interact with the fast inactivation gate. Nevertheless, the observed block can be explained by an interaction with the channel which depends on the graded movement of the activating gating charge. In this way, the drug would mimic and compete with natural fast inactivation [ref. 123]. Consequently, papain should enhance the speed of phenytoin block, as confirmed by Quandt [ref. 176] in neuroblastoma cells, but contested by Zeng et al. [ref. 253] in CA1 pyramidal cells. Is INaP then generally only a byproduct of a failure of intermediate inactivation? Probably not, as other blockers like riluzole or GS967 manage to suppress INaP measured with early steps or fast ramps. Once again, the term ‘persistent’ sodium current is misleading, because it is also applied to slow inactivation patterns at different time scales. Thus, employing phenytoin as an INaP blocker appears possible, but one has to keep these limitations in mind.
Propofol
Propofol is a sedative drug commonly used to induce loss of consciousness during narcosis, especially in patients that develop postoperative nausea from volatile anaesthetics. It is typically used at concentrations of 10–50 µM [ref. 189]. Propofol is also the last escalation step in order to suppress status epilepticus. At lower concentrations, the drug has addictive properties, like benzodiazepines. It has to be administered intravenously and can induce vasodilation and transient apnoea [ref. 189]. Thus, propofol is only used in intensive care settings. The drug predominantly acts on GABAA receptors by binding their β subunits and potentiates GABA induced currents fivefold at 2 µM or even opens the channels directly at 30 µM [ref. 84].
Regarding suppression of INaP propofol is surprisingly potent in neurons at 10–60 µM (Table 1). However, the reported effects on GABA receptors occur in a similar concentration range, again impeding causal analysis in complex preparations. The combination of all known actions of propofol may explain its efficacy in status epilepticus. In no way can it be regarded a specific INaP blocker. In addition, its low bioavailability and addictiveness limit any prolonged use in outpatients.
PRAX-562
PRAX-562 is a novel compound specifically synthesised as a persistent sodium current blocker by Praxis Precision Medicines. It was shown to be effective as an antiseizure drug in the maximal electroshock seizure model in mice [ref. 111]. Interestingly, amongst all drugs discussed in this review, it is the only substance inducing a small left shift of INaT activation. Available data from HEK-cells suggest that it acts as an INaP blocker by modulating both, fast and slow inactivation, but data from neurons are missing, as are data on potential further molecular targets.
Ranolazine
Ranolazine is a second-line drug in chronic stable angina pectoris and has shown some efficacy in microvascular coronary dysfunction as well as anti-arrhythmic activity [ref. 177]. In myocytes, it blocks persistent sodium currents (IC50 6 µM) and delayed rectifier potassium currents (IC50 12 µM) [ref. 83]. While ranolazine can cross the blood brain barrier, the CNS concentration only reaches one third of the plasma levels [ref. 109]. Ranolazine at 3–30 µM is effective in blocking INaP measured with ramps and steps [ref. 110, ref. 244], while also significantly affecting slow inactivation (Table 1). It does have a higher affinity for INaP over INaT, therefore making it an attractive persistent sodium current blocker. In systemic applications, however, the aforementioned effects on the heart and the low blood brain barrier passage must be taken into considerations.
Riluzole
For 28 years, riluzole has been the only approved drug in amyotrophic lateral sclerosis, where it prolongs life expectancy by around 2–3 months [ref. 24]. Interestingly, unlike many other drugs on this list, it is not effective in neuropathic pain [ref. 73]. This might be due to its low affinity for calcium channels, with IC50 values well above 10 µM [ref. 24]. However, at clinically used levels of 1–2 µM riluzole does enhance calcium dependent K+ currents and reduces presynaptic transmitter release [ref. 24].
Traditionally, riluzole has been the most popular blocker of INaP. The drug affects both ramp and step protocols with IC50 values well below 10 µM (Table 1). It also has effects on fast inactivation, though these are rather moderate compared to, e.g., carbamazepine (Table 1). Surprisingly, there is no data directly showing effects on slow inactivation, while indirect evidence from slow ramps and other readouts suggests an effect on intermediate inactivation [ref. 160]. Riluzole is one of the most useful INaP blockers, because it does not affect calcium currents at relevant concentrations and its effects on INaP have been shown in many different types of neurons [ref. 53, ref. 163, ref. 228].
Rufinamide
Rufinamide is a sparsely used anti-seizure drug for patients with Lennox-Gastaut syndrome. It inhibits metabotropic glutamate receptors 5 (mGluR5) at 100 µM, which can be measured by reduced quisqualate-induced phosphoinositol turnover [ref. 13]. At the same time, it inhibits slow and fast inactivation of sodium channels, especially Nav1.6, at 100 µM. However, ramp or step protocols for the assessment of effects on INaP are missing in the literature. It has been suggested that rufinamide exerts a preferential effect on intermediate inactivation, similar to phenytoin [ref. 138]. In any case, more data is required to assess its effects on INaP more completely.
Tetrodotoxin
Tetrodotoxin (TTX) is a poisonous agent found in puffer fish, where it is synthesised by bacteria [ref. 164]. It is considered the most effective sodium channel blocker, as it directly occludes sodium ion permeation through the open channel [ref. 132]. While transient sodium channels are typically blocked at concentrations of 1 µM, persistent sodium currents are efficiently blocked at lower concentrations of 20–50 nM in rodent brain slices [ref. 207, ref. 231, ref. 251]. However, Taddese and Bean [ref. 216] found the TTX at very low concentrations of 5 nM has equal effects on transient and persistent currents in tuberomammillary neurons, casting doubt on its specificity for INAP. Therefore, TTX may be a useful tool for experimental work on INaP, but careful controls for effects on INaT are required in each specific preparation. Notwithstanding, most studies use low micromolar concentrations of TTX to achieve complete absence of sodium currents [ref. 70, ref. 163, ref. 212]. Anyway, TTX has no potential for clinical use, due to its well-known capability to paralyse the diaphragm [ref. 164].
Topiramate
Topiramate is typically used in genetic generalized epilepsy, idiopathic intracranial hypertension and in the prevention of migraine episodes. However, its use is often limited by remarkable word-fluency difficulties [ref. 159]. At 10 µM, topiramate increases GABA mediated Cl− influx into neurons by 75% [ref. 241], it inhibits Cav2.3 with an IC50 of 51 µM [ref. 127] and exerts weak inhibition of carbonic anhydrases [ref. 193]. Concerning INaP, topiramate is a mildly efficient blocker in both step and ramp protocols. In one study [ref. 215] topiramate has a very potent effect on non-inactivating sodium current measured with step pulses with an EC50 of 61 nM, but the effect size is limited to a maximum of -30%. To our knowledge there is no data showing its effects on slow inactivation. The observed effects are present at clinically relevant concentrations of 2–20 µM [ref. 153], but importantly do only exert a partial block of INaP. Thus, topiramate is not a convincing candidate for use as a selective and efficient persistent sodium current blocker.
Valproic acid
Valproic acid is the first-choice drug for genetic generalized epilepsy [ref. 146, ref. 148], but is also used for stabilizing mood in bipolar disorder and preventing episodes of migraine. Due to its high teratogenicity its use in fertile women is strictly controlled in most countries [ref. 223]. This severe side-effect might be mediated by epigenomic effects through inhibition of histone deacetylases with IC50 values of 0.5–3 mM [ref. 82], well within the range of clinically used serum concentrations which range between 0.3–0.7 mM [ref. 31]. In addition, valproic acid at 0.5 mM potentiates GABAergic inhibition via a complex modulation of enzymes in GABA metabolism, and also infers with second messenger pathways [ref. 105]. Valproate is a potent blocker of persistent sodium current as assessed by ramps and step protocols in neurons (Table 1). These effects occur in the range of 10–100 µM which is well below the clinically used concentrations. Whether it does or does not affect INaT remains a controversial issue, with more evidence against (see Table 1) than for such an effect [ref. 230]. The marked effect on persistent sodium current might explain the efficacy of valproate in treating status epilepticus. However, as valproic acid has particularly many off-target effects, we do not recommend it as a pharmacological tool to isolate INaP.
Zonisamide
Zonisamide is an anti-seizure drug used in focal epilepsy, with particularly widespread application in Asia. It inhibits T-type Ca2+ currents and alters the metabolism of dopamine, 5-HT, and acetylcholine [ref. 30]. Although it has been described as a sodium channel blocker, this notion is mostly based on data from sea worm axons [ref. 190]. In the only study on mammalian channels in mouse derived neuroblastoma cells, zonisamide had no effect on slow inactivation (Table 1). Therefore, it should not be used as an INaP blocker.
Summary and recommendations
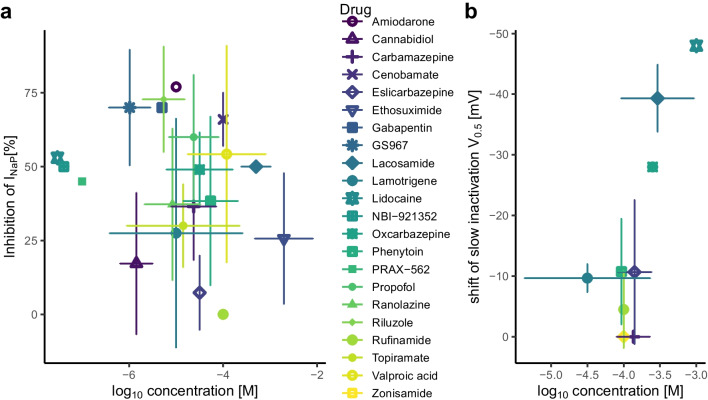
The very existence of a persistent sodium current as a separate, clearly definable entity is a controversial topic. The lack of clarity may be, at least in part, explained by the misleading word ‘persistent’, which should be understood as: ‘non-inactivating or slowly inactivating voltage-activated sodium current.’ Additional confounding issues are the poorly understood inactivation mechanisms of sodium channels, both from an electrophysiological and structural point of view. The typically employed voltage clamp protocols do not pick up all components of persistent or slowly inactivating sodium current, such that data is often incomplete. Brief voltage steps, in particular, are unable to assess slowly inactivating components. If one wants to characterise the entire effects of a drug on persistent sodium currents, one should also check for alterations of slow inactivation kinetics (notice the lack of respective of data in Fig. 3B). When using ramp protocols, only slow rates of voltage change (≤ 10 mV/s) do span over time periods of tens of seconds and are therefore able to address slow or intermediate inactivation.
Until now, the literature splits clinically employed sodium channel blockers into several types depending on the mechanism of inactivation which they enhance. Carbamazepine is considered as an archetypic fast inactivation enhancer, while lacosamide is considered to be the prototypic slow inactivation enhancer. However, most of the above-described drugs affect all three types of inactivation.
Based on our systematic literature search, we conclude that there is no pharmacological blocker of INaP that does not somehow affect transient current components. This is not surprising, since INaP is most likely a result of specific gating properties of the same sodium channels which mediate INaT, rather than a separate molecular subtype. Typically, blockers of INaP affect INaT at higher concentrations, such that they can be considered relatively specific as long as low concentrations are applied.
Another point of concern are the off-target effects of the most effective INaP blockers. Consistently, these drugs target both voltage gated sodium and calcium channels. This is not unexpected, because both channel families have a common evolutionary ancestor [ref. 137] and share a major properties of their 3D structure [ref. 39]. Taking all these caveats into account, claims that a specific physiological phenomenon is mediated by INaP should be based on similar effects of more than one drug, or on the additional use of alternative approaches, like genetic manipulation or dynamic voltage clamp [ref. 210].
In this review, we show that for CNS neurons GS967 and riluzole are the ‘best’ persistent sodium current blockers in vitro, as they significantly affect non-inactivating sodium current components measured with both ramp protocols and short voltage steps (Fig. 3A). The ‘best’ substance for enhancing intermediate inactivation is phenytoin and for slow inactivation there is lacosamide (Fig. 3B). Substances like NBI-921352 and PRAX-562 show promise for being specific blockers of INaP, but the present evidence is very limited. TTX and lidocaine are very useful tools for pharmacological isolation of INaP, but cannot be used for CNS purposes in vivo. Based on the available evidence, cannabidiol, ethosuximide, gabapentin, rufinamide and zonisamide should not be employed as bona fide persistent sodium current blockers either due to missing data or due to lacking potency (Fig. 3). Clinical translation of ranolazine and amiodarone is hindered by low penetrance of the blood–brain-barrier and simultaneous action on cardiac myocytes. High potential for confounding off target effects limits the use of cenobamate, eslicarbazepine, lamotrigine, oxcarbazepine, propofol, topiramate and valproic acid in complex preparations. And last, while still being sodium channel specific, carbamazepine exerts its main effect on fast inactivation.
Wherever possible, future studies should use a combination of brief voltage steps, voltage ramps and steady state inactivation protocols for fast, intermediate and slow inactivation. Step length for brief pulses should be around 50 ms, and they should be repeated at different voltages in order to consider voltage dependent shifts of persistent sodium currents. Ramps should be employed at around 50 mV/s (or even slower when slow inactivation component shall be directly assessed by voltage ramps; see above). TTX subtraction protocols are recommended. Slow steady state inactivation should be assessed by applying steps of 5–10 s duration, intermediate inactivation with 500 ms to 1000 ms steps and fast inactivation with 50 ms to 100 ms steps. Even if we don’t fully understand INaP, we should try to measure it with comparable parameters to facilitate comparisons and to support the further development of therapeutic strategies for conditions involving pathophysiological effects of persistent sodium currents.
References
- CA Ahern, J Payandeh, F Bosmans, B Chanda. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J Gen Physiol, 2016. [DOI | PubMed]
- AN Akopian, V Souslova, S England, K Okuse, N Ogata, J Ure. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci, 1999. [DOI | PubMed]
- RW Aldrich, DP Corey, CF Stevens. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature, 1983. [DOI | PubMed]
- G Alroy, H Su, Y Yaari. Protein kinase C mediates muscarinic block of intrinsic bursting in rat hippocampal neurons. J Physiology, 1999. [DOI]
- C Alzheimer, P Schwindt, W Crill. Modal gating of Na+ channels as a mechanism of persistent Na+ current in pyramidal neurons from rat and cat sensorimotor cortex. J Neurosci, 1993. [DOI | PubMed]
- TK Aman, TM Grieco-Calub, C Chen, R Rusconi, EA Slat, LL Isom. Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J Neurosci, 2009. [DOI | PubMed]
- 7.Amuzescu BC, Dan Corlan A, Radu BM (2023) Inhibitory effects of cenobamate on multiple human cardiac ion channels and possible arrhythmogenic consequences. PREPRINT (Version 1) available at Research Square. 10.21203/rs.3.rs-3735338/v1
- LL Anderson, CH Thompson, NA Hawkins, RD Nath, AA Petersohn, S Rajamani. Antiepileptic activity of preferential inhibitors of persistent sodium current. Epilepsia, 2014. [DOI | PubMed]
- LL Anderson, NA Hawkins, CH Thompson, JA Kearney, AL George. Unexpected Efficacy of a Novel Sodium Channel Modulator in Dravet Syndrome. Sci Rep, 2017. [DOI | PubMed]
- CM Armstrong. Na channel inactivation from open and closed states. Proc Natl Acad Sci, 2006. [DOI | PubMed]
- CM Armstrong, F Bezanilla. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol, 1977. [DOI | PubMed]
- 12.Armstrong CM, Gilly WF. [5] Access resistance and space clamp problems associated with whole-cell patch clamping. Methods Enzymol. 207: Academic Press; 1992. p. 100–122 10.1016/0076-6879(92)07007-B
- S Arroyo. Rufinamide. Neurotherapeutics, 2007. [DOI | PubMed]
- N Astman, MJ Gutnick, IA Fleidervish. Activation of protein kinase C increases neuronal excitability by regulating persistent Na+ current in mouse neocortical slices. J Neurophysiol, 1998. [DOI | PubMed]
- N Astman, MJ Gutnick, IA Fleidervish. Persistent sodium current in layer 5 neocortical neurons is primarily generated in the proximal axon. J Neurosci, 2006. [DOI | PubMed]
- E Auffenberg, UB Hedrich, R Barbieri, D Miely, B Groschup, TV Wuttke. Hyperexcitable interneurons trigger cortical spreading depression in an Scn1a migraine model. J Clin Invest, 2021. [DOI | PubMed]
- R Azouz, MS Jensen, Y Yaari. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol, 1996. [DOI | PubMed]
- EM Baker, CH Thompson, NA Hawkins, JL Wagnon, ER Wengert, MK Patel. The novel sodium channel modulator GS-458967 (GS967) is an effective treatment in a mouse model of SCN8A encephalopathy. Epilepsia, 2018. [DOI | PubMed]
- JR Balser, HB Nuss, DN Romashko, E Marban, GF Tomaselli. Functional consequences of lidocaine binding to slow-inactivated sodium channels. J Gen Physiol, 1996. [DOI | PubMed]
- R Barbieri, S Bertelli, M Pusch, P Gavazzo. Late sodium current blocker GS967 inhibits persistent currents induced by familial hemiplegic migraine type 3 mutations of the SCN1A gene. J Headache Pain, 2019. [DOI | PubMed]
- E Bayraktar, Y Liu, L Sonnenberg, UBS Hedrich, Y Sara, A Eltokhi. In vitro effects of eslicarbazepine (S-licarbazepine) as a potential precision therapy on SCN8A variants causing neuropsychiatric disorders. Br J Pharmacol, 2022. [DOI | PubMed]
- BP Bean, CJ Cohen, RW Tsien. Lidocaine block of cardiac sodium channels. J Gen Physiol, 1983. [DOI | PubMed]
- L Belardinelli, G Liu, C Smith-Maxwell, W-Q Wang, N El-Bizri, R Hirakawa. A Novel, Potent, and Selective Inhibitor of Cardiac Late Sodium Current Suppresses Experimental Arrhythmias. J Pharmacol Exp Ther, 2013. [DOI | PubMed]
- MC Bellingham. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade?. CNS Neurosci Ther, 2011. [DOI | PubMed]
- MC Bellingham. Pre- and postsynaptic mechanisms underlying inhibition of hypoglossal motor neuron excitability by riluzole. J Neurophysiol, 2013. [DOI | PubMed]
- T Berger, HR Lüscher. Associative somatodendritic interaction in layer V pyramidal neurons is not affected by the antiepileptic drug lamotrigine. Eur J Neurosci, 2004. [DOI | PubMed]
- MD Bevan, CJ Wilson. Mechanisms underlying spontaneous oscillation and rhythmic firing in rat subthalamic neurons. J Neurosci, 1999. [DOI | PubMed]
- BK Beyreuther, J Freitag, C Heers, N Krebsfänger, U Scharfenecker, T Stöhr. Lacosamide: a review of preclinical properties. CNS Drug Rev, 2007. [DOI | PubMed]
- F Bezanilla, CM Armstrong. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol, 1977. [DOI | PubMed]
- V Biton. Clinical Pharmacology and Mechanism of Action of Zonisamide. Clin Neuropharmacol, 2007. [DOI]
- CL Bowden, PG Janicak, P Orsulak, AC Swann, JM Davis, JR Calabrese. Relation of serum valproate concentration to response in mania. Am J Psychiatry, 1996. [DOI | PubMed]
- C Brocard, V Plantier, P Boulenguez, S Liabeuf, M Bouhadfane, A Viallat-Lieutaud. Cleavage of Na(+) channels by calpain increases persistent Na(+) current and promotes spasticity after spinal cord injury. Nat Med, 2016. [DOI | PubMed]
- T Broicher, T Seidenbecher, P Meuth, T Munsch, SG Meuth, T Kanyshkova. T-current related effects of antiepileptic drugs and a Ca2+ channel antagonist on thalamic relay and local circuit interneurons in a rat model of absence epilepsy. Neuropharmacology, 2007. [DOI | PubMed]
- 34.Brown PC. 212839Orig1s000. NON-CLINICAL REVIEW(S), U.S. Food and Drug Administration
- RKA Bunton-Stasyshyn, JL Wagnon, ER Wengert, BS Barker, A Faulkner, PK Wagley. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain, 2019. [DOI | PubMed]
- KJ Butterwick, MP Goldman, S Sriprachya-Anunt. Lidocaine levels during the first two hours of infiltration of dilute anesthetic solution for tumescent liposuction: rapid versus slow delivery. Dermatol Surg, 1999. [DOI | PubMed]
- E Carmeliet, T Saikawa. Shortening of the action potential and reduction of pacemaker activity by lidocaine, quinidine, and procainamide in sheep cardiac purkinje fibers. An effect on Na or K currents?. Circ Res, 1982. [DOI | PubMed]
- BC Carter, AJ Giessel, BL Sabatini, BP Bean. Transient sodium current at subthreshold voltages: activation by EPSP waveforms. Neuron, 2012. [DOI | PubMed]
- 39.Catterall WA, TM, Swanson (2015) Structural Basis for Pharmacology of Voltage-Gated Sodium and Calcium Channels. Mol Pharmacol 88(1):141. 10.1124/mol.114.097659
- 40.Cestèle S, Scalmani P, Rusconi R, Terragni B, Franceschetti S, Mantegazza M (2008) Self-Limited Hyperexcitability: Functional Effect of a Familial Hemiplegic Migraine Mutation of the Nav1.1 (SCN1A) Na+ Channel. J Neurosci 28(29):7273–7283. 10.1523/jneurosci.4453-07.2008
- WK Chandler, H Meves. Evidence for two types of sodium conductance in axons perfused with sodium fluoride solution. J Physiol, 1970. [DOI | PubMed]
- TI Chao, C Alzheimer. Effects of phenytoin on the persistent Na+ current of mammalian CNS neurones. NeuroReport, 1995. [DOI | PubMed]
- Y Chen, FH Yu, EM Sharp, D Beacham, T Scheuer, WA Catterall. Functional properties and differential neuromodulation of Na(v)1.6 channels. Mol Cell Neurosci, 2008. [DOI | PubMed]
- J Clatot, M Hoshi, X Wan, H Liu, A Jain, K Shinlapawittayatorn. Voltage-gated sodium channels assemble and gate as dimers. Nat Commun, 2017. [DOI | PubMed]
- E Colombo, S Franceschetti, G Avanzini, M Mantegazza. Phenytoin inhibits the persistent sodium current in neocortical neurons by modifying its inactivation properties. PLoS One, 2013. [DOI | PubMed]
- AM Correa, F Bezanilla. Gating of the squid sodium channel at positive potentials: II. Single channels reveal two open states. Biophys J, 1994. [DOI | PubMed]
- B Costa, N Vale. Understanding Lamotrigine’s Role in the CNS and Possible Future Evolution. Int J Mol Sci, 2023. [DOI | PubMed]
- DA Coulter, JR Huguenard, DA Prince. Characterization of ethosuximide reduction of low-threshold calcium current in thalamic neurons. Ann Neurol, 1989. [DOI | PubMed]
- WE Crill. Persistent Sodium Current in Mammalian Central Neurons. Annu Rev Physiol, 1996. [DOI | PubMed]
- JA Crippa, FS Guimarães, AC Campos, AW Zuardi. Translational Investigation of the Therapeutic Potential of Cannabidiol (CBD): Toward a New Age. Front Immunol, 2018. [DOI | PubMed]
- P Dao Trong, G Jungwirth, A Unterberg, C Herold-Mende, R Warta. The Antiepileptic Drug Oxcarbazepine Inhibits the Growth of Patient-Derived Isocitrate Dehydrogenase Mutant Glioma Stem-like Cells. Cells, 2023. [DOI | PubMed]
- AG de Boer, DD Breimer, H Mattie, J Pronk, JM Gubbens-Stibbe. Rectal bioavailability of lidocaine in man: Partial avoidance of “first-pass” metabolism. Clin Pharmacol Ther, 1979. [DOI | PubMed]
- CA Del Negro, C Morgado-Valle, JL Feldman. Respiratory Rhythm: An Emergent Network Property?. Neuron, 2002. [DOI | PubMed]
- O Devinsky, JH Cross, L Laux, E Marsh, I Miller, R Nabbout. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. N Engl J Med, 2017. [DOI | PubMed]
- 55.Doeser A, Soares-da-Silva P, Beck H, Uebachs M (2014) The effects of eslicarbazepine on persistent Na+ current and the role of the Na+ channel β subunits. Epilepsy Res 108(2):202–211. 10.1016/j.eplepsyres.2013.11.022
- H Dong, YH Fan, YY Wang, WT Wang, SJ Hu. Lidocaine suppresses subthreshold oscillations by inhibiting persistent Na(+) current in injured dorsal root ganglion neurons. Physiol Res, 2008. [DOI | PubMed]
- JM Dubois, C Bergman. Late sodium current in the node of Ranvier. Pflugers Arch, 1975. [DOI | PubMed]
- MR Elphick, M Egertova. The neurobiology and evolution of cannabinoid signalling. Philos Trans R Soc Lond B Biol Sci, 2001. [DOI | PubMed]
- AC Errington, T Stöhr, C Heers, G Lees. The Investigational Anticonvulsant Lacosamide Selectively Enhances Slow Inactivation of Voltage-Gated Sodium Channels. Mol Pharmacol, 2008. [DOI | PubMed]
- M Estacion, A Gasser, SD Dib-Hajj, SG Waxman. A sodium channel mutation linked to epilepsy increases ramp and persistent current of Nav1.3 and induces hyperexcitability in hippocampal neurons. Exp Neurol, 2010. [DOI | PubMed]
- 61.European-Medicines-Agency. Ontozry 2021. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/ontozry
- YC Feng, DP Howrigan, LE Abbott, K Tashman, F Cerrato, T Singh, H Heyne, A Byrnes, C Churchhouse, N Watts, M Solomonson. Ultra-Rare Genetic Variation in the Epilepsies: A Whole-Exome Sequencing Study of 17, 606 Individuals. Am J Hum Genet, 2019. [DOI | PubMed]
- CR Fertleman, MD Baker, KA Parker, S Moffatt, FV Elmslie, B Abrahamsen. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron, 2006. [DOI | PubMed]
- IA Fleidervish, MJ Gutnick. Kinetics of slow inactivation of persistent sodium current in layer V neurons of mouse neocortical slices. J Neurophysiol, 1996. [DOI | PubMed]
- 65.Fleidervish IA, Libman L, Katz E, Gutnick MJ (2008) Endogenous polyamines regulate cortical neuronal excitability by blocking voltage-gated Na+ channels. Proc Natl Acad Sci 105(48):18994–18999. 10.1073/pnas.0803464105
- JF Fohlmeister, WJ Adelman, JJ Brennan. Excitable channel currents and gating times in the presence of anticonvulsants ethosuximide and valproate. J Pharmacol Exp Ther, 1984. [PubMed]
- S Franceschetti, S Taverna, G Sancini, F Panzica, R Lombardi, G Avanzini. Protein kinase C-dependent modulation of Na+ currents increases the excitability of rat neocortical pyramidal neurones. J Physiology, 2000. [DOI]
- J Freiha, N Riachi, MA Chalah, R Zoghaib, SS Ayache, R Ahdab. Paroxysmal Symptoms in Multiple Sclerosis-A Review of the Literature. J Clin Med, 2020. [DOI | PubMed]
- CR French, PW Gage. A threshold sodium current in pyramidal cells in rat hippocampus. Neurosci Lett, 1985. [DOI | PubMed]
- CR French, P Sah, KJ Buckett, PW Gage. A voltage-dependent persistent sodium current in mammalian hippocampal neurons. J Gen Physiol, 1990. [DOI]
- CR French, Z Zeng, DA Williams, EL Hill-Yardin, TJ O’Brien. Properties of an intermediate-duration inactivation process of the voltage-gated sodium conductance in rat hippocampal CA1 neurons. J Neurophysiol, 2015. [DOI | PubMed]
- D Fricker, R Miles. EPSP amplification and the precision of spike timing in hippocampal neurons. Neuron, 2000. [DOI | PubMed]
- BS Galer, LL Twilling, J Harle, RS Cluff, E Friedman, MC Rowbotham. Lack of efficacy of riluzole in the treatment of peripheral neuropathic pain conditions. Neurology, 2000. [DOI | PubMed]
- K Gallop. Review article: phenytoin use and efficacy in the ED. Emerg Med Australas, 2010. [DOI | PubMed]
- 75.Ghovanloo MR, Abdelsayed M, Ruben PC (2016) Effects of amiodarone and n-desethylamiodarone on cardiac voltage-gated sodium channels. Front Pharmacol 7. 10.3389/fphar.2016.00039
- M-R Ghovanloo, NG Shuart, J Mezeyova, RA Dean, PC Ruben, SJ Goodchild. Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J Biol Chem, 2018. [DOI | PubMed]
- JC Gilbert, AK Scott, MG Wyllie. Proceedings: Effects of ethosuximide on adenosine triphosphatase activities of some subcellular fractions prepared from rat cerebral cortex. Br J Pharmacol, 1974
- J Gilchrist, S Dutton, M Diaz-Bustamante, A McPherson, N Olivares, J Kalia. Nav1.1 modulation by a novel triazole compound attenuates epileptic seizures in rodents. ACS Chem Biol, 2014. [DOI | PubMed]
- 79.Gorelova N, Seamans JK (2015) Cell-attached single-channel recordings in intact prefrontal cortex pyramidal neurons reveal compartmentalized D1/D5 receptor modulation of the persistent sodium current. Front Neural Circuits 9. 10.3389/fncir.2015.00004
- NA Gorelova, CR Yang. Dopamine D1/D5 receptor activation modulates a persistent sodium current in rat prefrontal cortical neurons in vitro. J Neurophysiol, 2000. [DOI | PubMed]
- MZ Gören, F Onat. Ethosuximide: From Bench to Bedside. CNS Drug Rev, 2007. [DOI | PubMed]
- M Göttlicher, S Minucci, P Zhu, OH Krämer, A Schimpf, S Giavara. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J, 2001. [DOI | PubMed]
- T Gupta, S Khera, D Kolte, WS Aronow, S Iwai. Antiarrhythmic properties of ranolazine: A review of the current evidence. Int J Cardiol, 2015. [DOI | PubMed]
- TG Hales, JJ Lambert. The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. Br J Pharmacol, 1991. [DOI | PubMed]
- MS Hamada, MH Kole. Myelin loss and axonal ion channel adaptations associated with gray matter neuronal hyperexcitability. J Neurosci, 2015. [DOI | PubMed]
- AK Hammarstrom, PW Gage. Inhibition of oxidative metabolism increases persistent sodium current in rat CA1 hippocampal neurons. J Physiol, 1998. [DOI | PubMed]
- S Hebeisen, N Pires, AI Loureiro, MJ Bonifácio, N Palma, A Whyment. Eslicarbazepine and the enhancement of slow inactivation of voltage-gated sodium channels: a comparison with carbamazepine, oxcarbazepine and lacosamide. Neuropharmacology, 2015. [DOI | PubMed]
- T Hézső, M Naveed, C Dienes, D Kiss, J Prorok, T Árpádffy-Lovas. Mexiletine-like cellular electrophysiological effects of GS967 in canine ventricular myocardium. Sci Rep, 2021. [DOI | PubMed]
- AJ Hill, NA Jones, I Smith, CL Hill, CM Williams, GJ Stephens. Voltage-gated sodium (NaV) channel blockade by plant cannabinoids does not confer anticonvulsant effects per se. Neurosci Lett, 2014. [DOI | PubMed]
- RD Hodge, TE Bakken, JA Miller, KA Smith, ER Barkan, LT Graybuck. Conserved cell types with divergent features in human versus mouse cortex. Nature, 2019. [DOI | PubMed]
- AL Hodgkin, AF Huxley. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol, 1952. [DOI | PubMed]
- D Holtkamp, T Opitz, I Niespodziany, C Wolff, H Beck. Activity of the anticonvulsant lacosamide in experimental and human epilepsy via selective effects on slow Na(+) channel inactivation. Epilepsia, 2017. [DOI | PubMed]
- D Holtkamp, T Opitz, S Hebeisen, P Soares-da-Silva, H Beck. Effects of eslicarbazepine on slow inactivation processes of sodium channels in dentate gyrus granule cells. Epilepsia, 2018. [DOI | PubMed]
- EM Horn, TG Waldrop. Hypoxic augmentation of fast-inactivating and persistent sodium currents in rat caudal hypothalamic neurons. J Neurophysiol, 2000. [DOI | PubMed]
- CL Hsu, X Zhao, AD Milstein, N Spruston. Persistent Sodium Current Mediates the Steep Voltage Dependence of Spatial Coding in Hippocampal Pyramidal Neurons. Neuron, 2018. [DOI | PubMed]
- W Hu, C Tian, T Li, M Yang, H Hou, Y Shu. Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nat Neurosci, 2009. [DOI | PubMed]
- C-W Huang, C-C Huang, M-W Lin, J-J Tsai, S-N Wu. The synergistic inhibitory actions of oxcarbazepine on voltage-gated sodium and potassium currents in differentiated NG108–15 neuronal cells and model neurons. Int J Neuropsychopharmacol, 2008. [DOI | PubMed]
- J Huang, X Fan, X Jin, S Jo, HB Zhang, A Fujita. Cannabidiol inhibits Nav channels through two distinct binding sites. Nat Commun, 2023. [DOI | PubMed]
- B Hutcheon, Y Yarom. Resonance, oscillation and the intrinsic frequency preferences of neurons. Trends Neurosci, 2000. [DOI | PubMed]
- KM Igelström, PM Heyward. The antidepressant drug fluoxetine inhibits persistent sodium currents and seizure-like events. Epilepsy Res, 2012. [DOI | PubMed]
- V Ilin, A Malyshev, F Wolf, M Volgushev. Fast Computations in Cortical Ensembles Require Rapid Initiation of Action Potentials. J Neurosci, 2013. [DOI | PubMed]
- N Inomata, T Ishihara, N Akaike. Different time courses of the blockade of sodium current by lignocaine and SUN 1165 in single myocytes isolated from guinea-pig atrium. Br J Pharmacol, 1989. [DOI | PubMed]
- Y Iwai, K Shibuya, S Misawa, Y Sekiguchi, K Watanabe, H Amino. Axonal Dysfunction Precedes Motor Neuronal Death in Amyotrophic Lateral Sclerosis. PLoS One, 2016. [DOI | PubMed]
- S Jo, BP Bean. Lacosamide Inhibition of Nav1.7 Voltage-Gated Sodium Channels: Slow Binding to Fast-Inactivated States. Mol Pharmacol, 2017. [DOI | PubMed]
- CU Johannessen. Mechanisms of action of valproate: a commentatory. Neurochem Int, 2000. [DOI | PubMed]
- 106.Johnson JP, Focken T, Khakh K, Tari PK, Dube C, Goodchild SJ, et al (2022) NBI-921352, a first-in-class, Na(V)1.6 selective, sodium channel inhibitor that prevents seizures in Scn8a gain-of-function mice, and wild-type mice and rats. Elife 11 10.7554/eLife.72468
- PJ Jones, EC Merrick, TW Batts, NJ Hargus, Y Wang, JP Stables. Modulation of sodium channel inactivation gating by a novel lactam: implications for seizure suppression in chronic limbic epilepsy. J Pharmacol Exp Ther, 2009. [DOI | PubMed]
- YK Ju, DA Saint, PW Gage. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol, 1996. [DOI | PubMed]
- KM Kahlig, I Lepist, K Leung, S Rajamani, AL George. Ranolazine selectively blocks persistent current evoked by epilepsy-associated Naν1.1 mutations. Br J Pharmacol, 2010. [DOI | PubMed]
- KM Kahlig, R Hirakawa, L Liu, AL George, L Belardinelli, S Rajamani. Ranolazine Reduces Neuronal Excitability by Interacting with Inactivated States of Brain Sodium Channels. Mol Pharmacol, 2014. [DOI | PubMed]
- KM Kahlig, L Scott, RJ Hatch, A Griffin, G Martinez Botella, ZA Hughes. The novel persistent sodium current inhibitor PRAX-562 has potent anticonvulsant activity with improved protective index relative to standard of care sodium channel blockers. Epilepsia, 2022. [DOI | PubMed]
- F Kalume, FH Yu, RE Westenbroek, T Scheuer, WA Catterall. Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci, 2007. [DOI | PubMed]
- AH Kanellopoulos, J Koenig, H Huang, M Pyrski, Q Millet, S Lolignier, T Morohashi, SJ Gossage, M Jay, JE Linley, G Baskozos. Mapping protein interactions of sodium channel Na V 17 using epitope-tagged gene-targeted mice. EMBO J, 2018. [DOI | PubMed]
- YJ Kang, EM Clement, SL Sumsky, Y Xiang, IH Park, S Santaniello. The critical role of persistent sodium current in hippocampal gamma oscillations. Neuropharmacology, 2020. [DOI | PubMed]
- E Katz, O Stoler, A Scheller, Y Khrapunsky, S Goebbels, F Kirchhoff. Role of sodium channel subtype in action potential generation by neocortical pyramidal neurons. Proc Natl Acad Sci USA, 2018. [DOI | PubMed]
- ZM Khaliq, BP Bean. Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J Neurosci, 2010. [DOI | PubMed]
- J Kiehn, D Thomas, CA Karle, W Schöls, W Kübler. Inhibitory effects of the class III antiarrhythmic drug amiodarone on cloned HERG potassium channels. Arch Pharmacol, 1999. [DOI]
- ES Kilpatrick, G Forrest, MJ Brodie. Concentration–effect and concentration–toxicity relations with lamotrigine: a prospective study. Epilepsia, 1996. [DOI | PubMed]
- T Kiss. Persistent Na-channels: origin and function. A review. Acta Biol Hung, 2008. [DOI | PubMed]
- H Koizumi, JC Smith. Persistent Na+ and K+-dominated leak currents contribute to respiratory rhythm generation in the pre-Bötzinger complex in vitro. J Neurosci, 2008. [DOI | PubMed]
- MH Kole. First node of Ranvier facilitates high-frequency burst encoding. Neuron, 2011. [DOI | PubMed]
- NI Kononenko, LR Shao, FE Dudek. Riluzole-sensitive slowly inactivating sodium current in rat suprachiasmatic nucleus neurons. J Neurophysiol, 2004. [DOI | PubMed]
- CC Kuo, BP Bean. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol Pharmacol, 1994. [PubMed]
- CC Kuo, L Lu. Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurones. Br J Pharmacol, 1997. [DOI | PubMed]
- JJ Kuo, T Siddique, R Fu, CJ Heckman. Increased persistent Na(+) current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol, 2005. [DOI | PubMed]
- S Kuwabara, S Misawa. Pharmacologic intervention in axonal excitability: in vivo assessment of nodal persistent sodium currents in human neuropathies. Curr Mol Pharmacol, 2008. [DOI | PubMed]
- JB Kuzmiski, W Barr, GW Zamponi, BA MacVicar. Topiramate Inhibits the Initiation of Plateau Potentials in CA1 Neurons by Depressing R-type Calcium Channels. Epilepsia, 2005. [DOI | PubMed]
- N Lamanauskas, A Nistri. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur J Neurosci, 2008. [DOI | PubMed]
- JA Lamas, M Romero, A Reboreda, E Sánchez, SJ Ribeiro. A riluzole- and valproate-sensitive persistent sodium current contributes to the resting membrane potential and increases the excitability of sympathetic neurones. Pflugers Arch, 2009. [DOI | PubMed]
- I Lampl, P Schwindt, W Crill. Reduction of cortical pyramidal neuron excitability by the action of phenytoin on persistent Na+ current. J Pharmacol Exp Ther, 1998. [PubMed]
- 131.Lauxmann S, Sonnenberg L, Koch NA, Bosselmann C, Winter N, Schwarz N et al (2021) Therapeutic Potential of Sodium Channel Blockers as a Targeted Therapy Approach in KCNA1-Associated Episodic Ataxia and a Comprehensive Review of the Literature. Front Neurol 12. 10.3389/fneur.2021.703970
- CH Lee, PC Ruben. Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels, 2008. [DOI | PubMed]
- N Lenkey, R Karoly, P Lukacs, ES Vizi, M Sunesen, L Fodor. Classification of Drugs Based on Properties of Sodium Channel Inhibition: A Comparative Automated Patch-Clamp Study. PLoS One, 2010. [DOI | PubMed]
- WA Leonard. The use of diphenylhydantoin (dilantin) sodium in the treatment of ventricular tachycardia. AMA Arch Intern Med, 1958. [DOI | PubMed]
- N Leresche, HR Parri, G Erdemli, A Guyon, JP Turner, SR Williams. On the Action of the Anti-Absence Drug Ethosuximide in the Rat and Cat Thalamus. J Neurosci, 1998. [DOI | PubMed]
- Y Li, MA Gorassini, DJ Bennett. Role of persistent sodium and calcium currents in motoneuron firing and spasticity in chronic spinal rats. J Neurophysiol, 2004. [DOI | PubMed]
- BJ Liebeskind, DM Hillis, HH Zakon. Evolution of sodium channels predates the origin of nervous systems in animals. Proc Natl Acad Sci USA, 2011. [DOI | PubMed]
- Y-C Lin, Y-C Lai, T-H Lin, Y-C Yang, C-C Kuo. Selective stabilization of the intermediate inactivated Na+ channel by the new-generation anticonvulsant rufinamide. Biochem Pharmacol, 2022. [DOI | PubMed]
- LF Lopez-Santiago, Y Yuan, JL Wagnon, JM Hull, CR Frasier, HA O’Malley. Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc Natl Acad Sci U S A, 2017. [DOI | PubMed]
- O Lunko, D Isaev, O Maximyuk, G Ivanchick, V Sydorenko, O Krishtal. Persistent sodium current properties in hippocampal CA1 pyramidal neurons of young and adult rats. Neurosci Lett, 2014. [DOI | PubMed]
- JY Ma, WA Catterall, T Scheuer. Persistent sodium currents through brain sodium channels induced by G protein betagamma subunits. Neuron, 1997. [DOI | PubMed]
- VA Maltsev, HN Sabbah, AI Undrovinas. Late Sodium Current is a Novel Target for Amiodarone: Studies in Failing Human Myocardium. J Mol Cell Cardiol, 2001. [DOI | PubMed]
- M Mantegazza, FH Yu, AJ Powell, JJ Clare, WA Catterall, T Scheuer. Molecular determinants for modulation of persistent sodium current by G-protein betagamma subunits. J Neurosci, 2005. [DOI | PubMed]
- S Markoula, R Teotonio, N Ratnaraj, JS Duncan, JW Sander, PN Patsalos. Lacosamide serum concentrations in adult patients with epilepsy: the influence of gender, age, dose, and concomitant antiepileptic drugs. Ther Drug Monit, 2014. [DOI | PubMed]
- AG Marson, AM Al-Kharusi, M Alwaidh, R Appleton, GA Baker, DW Chadwick. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet, 2007. [DOI | PubMed]
- AG Marson, AM Al-Kharusi, M Alwaidh, R Appleton, GA Baker, DW Chadwick. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet, 2007. [DOI | PubMed]
- A Marson, G Burnside, R Appleton, D Smith, JP Leach, G Sills. The SANAD II study of the effectiveness and cost-effectiveness of levetiracetam, zonisamide, or lamotrigine for newly diagnosed focal epilepsy: an open-label, non-inferiority, multicentre, phase 4, randomised controlled trial. Lancet, 2021. [DOI | PubMed]
- A Marson, G Burnside, R Appleton, D Smith, JP Leach, G Sills. The SANAD II study of the effectiveness and cost-effectiveness of valproate versus levetiracetam for newly diagnosed generalised and unclassifiable epilepsy: an open-label, non-inferiority, multicentre, phase 4, randomised controlled trial. Lancet, 2021. [DOI | PubMed]
- G Martella, C De Persis, P Bonsi, S Natoli, D Cuomo, G Bernardi. Inhibition of Persistent Sodium Current Fraction and Voltage-gated L-type Calcium Current by Propofol in Cortical Neurons: Implications for Its Antiepileptic Activity. Epilepsia, 2005. [DOI | PubMed]
- ER Mason, TR Cummins. Differential Inhibition of Human Nav1.2 Resurgent and Persistent Sodium Currents by Cannabidiol and GS967. Int J Mol Sci, 2020. [DOI | PubMed]
- N Matsuki, FN Quandt, RE Ten Eick, JZ Yeh. Characterization of the block of sodium channels by phenytoin in mouse neuroblastoma cells. J Pharmacol Exp Ther, 1984. [PubMed]
- N Maurice, T Tkatch, M Meisler, LK Sprunger, DJ Surmeier. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci, 2001. [DOI | PubMed]
- TW May, B Rambeck, U Jürgens. Serum Concentrations of Topiramate in Patients With Epilepsy: Influence of Dose, Age, and Comedication. Ther Drug Monit, 2002. [DOI | PubMed]
- QT Meng, ZY Xia, J Liu, DA Bayliss, X Chen. Local anesthetic inhibits hyperpolarization-activated cationic currents. Mol Pharmacol, 2011. [DOI | PubMed]
- RO Messing, CL Carpenter, DA Greenberg. Mechanism of calcium channel inhibition by phenytoin: comparison with classical calcium channel antagonists. J Pharmacol Exp Ther, 1985. [PubMed]
- T Mittmann, C Alzheimer. Muscarinic inhibition of persistent Na+ current in rat neocortical pyramidal neurons. J Neurophysiol, 1998. [DOI | PubMed]
- A Moutal, L François-Moutal, S Perez-Miller, K Cottier, LA Chew, SK Yeon. (S)-Lacosamide Binding to Collapsin Response Mediator Protein 2 (CRMP2) Regulates CaV2.2 Activity by Subverting Its Phosphorylation by Cdk5. Mol Neurobiol, 2016. [DOI | PubMed]
- A Moutal, LA Chew, X Yang, Y Wang, SK Yeon, E Telemi, S Meroueh, KD Park, R Shrinivasan, KB Gilbraith, C Qu, JY Xie, A Patwardhan, TW Vanderah, M Khanna, F Porreca, R Khanna. (S)-lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. Pain, 2016. [DOI | PubMed]
- M Mula. Topiramate and cognitive impairment: evidence and clinical implications. Ther Adv Drug Saf, 2012. [DOI | PubMed]
- P Müller, A Draguhn, AV Egorov. Persistent sodium current modulates axonal excitability in CA1 pyramidal neurons. J Neurochem, 2018. [DOI | PubMed]
- Y Muroi, B Chanda. Local anesthetics disrupt energetic coupling between the voltage-sensing segments of a sodium channel. J Gen Physiol, 2009. [DOI | PubMed]
- R Murphy, H Alle, JRP Geiger, JF Storm. Estimation of persistent sodium-current density in rat hippocampal mossy fibre boutons: Correction of space-clamp errors. J Physiol, 2024. [DOI | PubMed]
- M Nakamura, JH Cho, H Shin, IS Jang. Effects of cenobamate (YKP3089), a newly developed anti-epileptic drug, on voltage-gated sodium channels in rat hippocampal CA3 neurons. Eur J Pharmacol, 2019. [DOI | PubMed]
- T Narahashi. Pharmacology of tetrodotoxin. J Toxicol Toxin Rev, 2001. [DOI]
- I Niespodziany, H Klitgaard, DG Margineanu. Is the persistent sodium current a specific target of anti-absence drugs?. NeuroReport, 2004. [DOI | PubMed]
- I Niespodziany, N Leclère, C Vandenplas, P Foerch, C Wolff. Comparative study of lacosamide and classical sodium channel blocking antiepileptic drugs on sodium channel slow inactivation. J Neurosci Res, 2013. [DOI | PubMed]
- YY Park, D Johnston, R Gray. Slowly inactivating component of Na+ current in peri-somatic region of hippocampal CA1 pyramidal neurons. J Neurophysiol, 2013. [DOI | PubMed]
- SB Park, MC Kiernan, S Vucic. Axonal Excitability in Amyotrophic Lateral Sclerosis : Axonal Excitability in ALS. Neurotherapeutics, 2017. [DOI | PubMed]
- R Patel, AH Dickenson. Mechanisms of the gabapentinoids and α 2 δ-1 calcium channel subunit in neuropathic pain. Pharmacol Res Perspect, 2016. [DOI | PubMed]
- RR Patel, C Barbosa, T Brustovetsky, N Brustovetsky, TR Cummins. Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain, 2016. [DOI | PubMed]
- JB Patlak, M Ortiz. Two modes of gating during late Na+ channel currents in frog sartorius muscle. J Gen Physiol, 1986. [DOI | PubMed]
- E Perucca. Treatment of epilepsy in developing countries. BMJ, 2007. [DOI | PubMed]
- M Pieri, I Carunchio, L Curcio, NB Mercuri, C Zona. Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol, 2009. [DOI | PubMed]
- M Prakriya, S Mennerick. Selective Depression of Low-Release Probability Excitatory Synapses by Sodium Channel Blockers. Neuron, 2000. [DOI | PubMed]
- K Ptak, GG Zummo, GF Alheid, T Tkatch, DJ Surmeier, DR McCrimmon. Sodium Currents in Medullary Neurons Isolated from the Pre-Bötzinger Complex Region. J Neurosci, 2005. [DOI | PubMed]
- FN Quandt. Modification of slow inactivation of single sodium channels by phenytoin in neuroblastoma cells. Mol Pharmacol, 1988. [PubMed]
- 177.Rayner‐Hartley E, Sedlak T (2016) Ranolazine: A Contemporary Review. J Am Heart Assoc 5(3):e003196. 10.1161/JAHA.116.003196
- S-c Ren, P-z Chen, H-h Jiang, Z Mi, F Xu, B Hu. Persistent sodium currents contribute to Aβ1-42-induced hyperexcitation of hippocampal CA1 pyramidal neurons. Neurosci Lett, 2014. [DOI | PubMed]
- A Richens. Clinical pharmacokinetics of phenytoin. Clin Pharmacokinet, 1979. [DOI | PubMed]
- E Riva, M Gerna, P Neyroz, R Urso, I Bartosek, A Guaitani. Pharmacokinetics of Amiodarone in Rats. J Cardiovasc Pharmacol, 1982. [DOI | PubMed]
- E Rojas, B Rudy. Destruction of the sodium conductance inactivation by a specific protease in perfused nerve fibres from Loligo. J Physiol, 1976. [DOI | PubMed]
- M Royeck, M-T Horstmann, S Remy, M Reitze, Y Yaari, H Beck. Role of Axonal NaV1.6 Sodium Channels in Action Potential Initiation of CA1 Pyramidal Neurons. J Neurophysiol, 2008. [DOI | PubMed]
- M Royeck, T Kelly, T Opitz, DM Otte, A Rennhack, A Woitecki. Downregulation of Spermine Augments Dendritic Persistent Sodium Currents and Synaptic Integration after Status Epilepticus. J Neurosci, 2015. [DOI | PubMed]
- B Rudy. Slow inactivation of the sodium conductance in squid giant axons. Pronase resistance. J Physiol, 1978. [DOI | PubMed]
- AH Rühlmann, J Körner, R Hausmann, N Bebrivenski, C Neuhof, S Detro-Dassen. Uncoupling sodium channel dimers restores the phenotype of a pain-linked Nav1.7 channel mutation. Br J Pharmacol, 2020. [DOI | PubMed]
- AM Rush, SD Dib-Hajj, SG Waxman. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol, 2005. [DOI | PubMed]
- JL Russell, HA Spiller, DD Baker. Markedly Elevated Carbamazepine-10,11-epoxide/Carbamazepine Ratio in a Fatal Carbamazepine Ingestion. Case Report Med, 2015. [DOI]
- EB Russo, A Burnett, B Hall, KK Parker. Agonistic Properties of Cannabidiol at 5-HT1a Receptors. Neurochem Res, 2005. [DOI | PubMed]
- MM Sahinovic, MMRF Struys, AR Absalom. Clinical Pharmacokinetics and Pharmacodynamics of Propofol. Clin Pharmacokinet, 2018. [DOI | PubMed]
- CL Schauf. Zonisamide enhances slow sodium inactivation in Myxicola. Brain Res, 1987. [DOI | PubMed]
- G Schik, FR Wedegaertner, J Liersch, L Hoy, HM Emrich, U Schneider. Oxcarbazepine versus carbamazepine in the treatment of alcohol withdrawal. Addict Biol, 2005. [DOI | PubMed]
- MM Segal, AF Douglas. Late sodium channel openings underlying epileptiform activity are preferentially diminished by the anticonvulsant phenytoin. J Neurophysiol, 1997. [DOI | PubMed]
- RP Shank, JF Gardocki, JL Vaught, CB Davis, JJ Schupsky, RB Raffa. Topiramate: preclinical evaluation of structurally novel anticonvulsant. Epilepsia, 1994. [DOI | PubMed]
- R Sharma, M Nakamura, C Neupane, BH Jeon, H Shin, SM Melnick. Positive allosteric modulation of GABAA receptors by a novel antiepileptic drug cenobamate. Eur J Pharmacol, 2020. [DOI | PubMed]
- PL Sheets, C Heers, T Stoehr, TR Cummins. Differential block of sensory neuronal voltage-gated sodium channels by lacosamide [(2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide], lidocaine, and carbamazepine. J Pharmacol Exp Ther, 2008. [DOI | PubMed]
- MG Sheroziya, AV Egorov. Effects of extracellular calcium on the volley activity of entorhinal cortex neurons in neonatal rats: computer simulation. Neurosci Behav Physiol, 2010. [DOI | PubMed]
- MG Sheroziya, OV Halbach, K Unsicker, AV Egorov. Spontaneous bursting activity in the developing entorhinal cortex. J Neurosci, 2009. [DOI | PubMed]
- QQ Shi, X Sun, H Fang. A mechanism study on propofol’s action on middle latency auditory evoked potential by neurons in ventral partition of medial geniculate body in rats. Eur Rev Med Pharmacol Sci, 2014. [PubMed]
- K Shibuya, S Misawa, H Kimura, Y-i Noto, Y Sekiguchi, Y Iwai. Increased motor axonal persistent sodium currents predict rapid functional declines in amyotrophic lateral sclerosis. Neurol Clin Neurosci, 2016. [DOI]
- KS Silver, Y Du, Y Nomura, EE Oliveira, VL Salgado, BS Zhorov. Voltage-Gated Sodium Channels as Insecticide Targets. Adv In Insect Phys, 2014. [DOI | PubMed]
- ST Sipilä, K Huttu, J Voipio, K Kaila. Intrinsic bursting of immature CA3 pyramidal neurons and consequent giant depolarizing potentials are driven by a persistent Na+ current and terminated by a slow Ca2+ -activated K+ current. Eur J Neurosci, 2006. [DOI | PubMed]
- J Soar, BW Böttiger, P Carli, K Couper, CD Deakin, T Djärv. European Resuscitation Council Guidelines 2021: Adult advanced life support. Resuscitation, 2021. [DOI | PubMed]
- P Soares-da-Silva, N Pires, MJ Bonifácio, AI Loureiro, N Palma, LC Wright. Eslicarbazepine acetate for the treatment of focal epilepsy: an update on its proposed mechanisms of action. Pharmacol Res Perspect, 2015. [DOI | PubMed]
- E Southam, D Kirkby, GA Higgins, RM Hagan. Lamotrigine inhibits monoamine uptake in vitro and modulates 5-hydroxytryptamine uptake in rats. Eur J Pharmacol, 1998. [DOI | PubMed]
- F Spadoni, AH Hainsworth, NB Mercuri, L Caputi, G Martella, F Lavaroni. Lamotrigine derivatives and riluzole inhibit INa, P in cortical neurons. NeuroReport, 2002. [DOI | PubMed]
- CE Stafstrom. Persistent sodium current and its role in epilepsy. Epilepsy Curr, 2007. [DOI | PubMed]
- CE Stafstrom, PC Schwindt, MC Chubb, WE Crill. Properties of persistent sodium conductance and calcium conductance of layer V neurons from cat sensorimotor cortex in vitro. J Neurophysiol, 1985. [DOI | PubMed]
- A Stefani, A Pisani, M De Murtas, NB Mercuri, MG Marciani, P Calabresi. Action of GP 47779, the Active Metabolite of Oxcarbazepine, on the Corticostriatal System. II. Modulation of High-Voltage-Activated Calcium Currents. Epilepsia, 1995. [DOI | PubMed]
- A Stefani, F Spadoni, A Siniscalchi, G Bernardi. Lamotrigine inhibits Ca2+ currents in cortical neurons: functional implications. Eur J Pharmacol, 1996. [DOI | PubMed]
- 210.Storm J, Vervaeke K, Hu H, Graham L. Functions of the Persistent Na+ Current in Cortical Neurons Revealed by Dynamic Clamp. Dynamic-Clamp. New York, NY: Springer; 2009. p. 165–197 10.1007/978-0-387-89279-5_8
- G Stuart. Voltage–activated sodium channels amplify inhibition in neocortical pyramidal neurons. Nat Neurosci, 1999. [DOI | PubMed]
- G Stuart, B Sakmann. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron, 1995. [DOI | PubMed]
- PK Stys, H Sontheimer, BR Ransom, SG Waxman. Noninactivating, tetrodotoxin-sensitive Na+ conductance in rat optic nerve axons. Proc Natl Acad Sci U S A, 1993. [DOI | PubMed]
- H Su, G Alroy, ED Kirson, Y Yaari. Extracellular calcium modulates persistent sodium current-dependent burst-firing in hippocampal pyramidal neurons. J Neurosci, 2001. [DOI | PubMed]
- G-c Sun, TR Werkman, A Battefeld, JJ Clare, WJ Wadman. Carbamazepine and Topiramate Modulation of Transient and Persistent Sodium Currents Studied in HEK293 Cells Expressing the Nav1.3 α–Subunit. Epilepsia, 2007. [DOI | PubMed]
- A Taddese, BP Bean. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron, 2002. [DOI | PubMed]
- S Taverna, M Mantegazza, S Franceschetti, G Avanzini. Valproate selectively reduces the persistent fraction of Na+ current in neocortical neurons. Epilepsy Res, 1998. [DOI | PubMed]
- S Taverna, G Sancini, M Mantegazza, S Franceschetti, G Avanzini. Inhibition of transient and persistent Na+ current fractions by the new anticonvulsant topiramate. J Pharmacol Exp Ther, 1999. [PubMed]
- JC Taylor, S Brauer, ML Espir. Long-term treatment of trigeminal neuralgia with carbamazepine. Postgrad Med J, 1981. [DOI | PubMed]
- S Tazerart, L Vinay, F Brocard. The persistent sodium current generates pacemaker activities in the central pattern generator for locomotion and regulates the locomotor rhythm. J Neurosci, 2008. [DOI | PubMed]
- RD Theiss, JJ Kuo, CJ Heckman. Persistent inward currents in rat ventral horn neurones. J Physiol, 2007. [DOI | PubMed]
- A Thomas, GL Baillie, AM Phillips, RK Razdan, RA Ross, RG Pertwee. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol, 2007. [DOI | PubMed]
- T Tomson, D Battino, E Bonizzoni, J Craig, D Lindhout, E Perucca. Dose-dependent teratogenicity of valproate in mono- and polytherapy: an observational study. Neurology, 2015. [DOI | PubMed]
- T Tomson, D Battino, E Bonizzoni, J Craig, D Lindhout, E Perucca. Comparative risk of major congenital malformations with eight different antiepileptic drugs: a prospective cohort study of the EURAP registry. Lancet Neurol, 2018. [DOI | PubMed]
- BD Trapp, PK Stys. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol, 2009. [DOI | PubMed]
- M Uebachs, T Opitz, M Royeck, G Dickhof, M-T Horstmann, LL Isom. Efficacy Loss of the Anticonvulsant Carbamazepine in Mice Lacking Sodium Channel β Subunits via Paradoxical Effects on Persistent Sodium Currents. J Neurosci, 2010. [DOI | PubMed]
- M Uebachs, C Albus, T Opitz, L Isom, I Niespodziany, C Wolff. Loss of β1 accessory Na+ channel subunits causes failure of carbamazepine, but not of lacosamide, in blocking high-frequency firing via differential effects on persistent Na+ currents. Epilepsia, 2012. [DOI | PubMed]
- A Urbani, O Belluzzi. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci, 2000. [DOI | PubMed]
- V Uteshev, DR Stevens, HL Haas. A persistent sodium current in acutely isolated histaminergic neurons from rat hypothalamus. Neuroscience, 1995. [DOI | PubMed]
- RJ Van den Berg, P Kok, RA Voskuyl. Valproate and sodium currents in cultured hippocampal neurons. Exp Brain Res, 1993. [DOI | PubMed]
- W van Drongelen, H Koch, FP Elsen, HC Lee, A Mrejeru, E Doren. Role of Persistent Sodium Current in Bursting Activity of Mouse Neocortical Networks In Vitro. J Neurophysiol, 2006. [DOI | PubMed]
- KR Veeramah, JE O’Brien, MH Meisler, X Cheng, SD Dib-Hajj, SG Waxman. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet, 2012. [DOI | PubMed]
- J Vera, J Alcayaga, M Sanhueza. Competition between Persistent Na(+) and Muscarine-Sensitive K(+) Currents Shapes Perithreshold Resonance and Spike Tuning in CA1 Pyramidal Neurons. Front Cell Neurosci, 2017. [DOI | PubMed]
- K Vervaeke, H Hu, LJ Graham, JF Storm. Contrasting effects of the persistent Na+ current on neuronal excitability and spike timing. Neuron, 2006. [DOI | PubMed]
- M Vreugdenhil, G Hoogland, CW van Veelen, WJ Wadman. Persistent sodium current in subicular neurons isolated from patients with temporal lobe epilepsy. Eur J Neurosci, 2004. [DOI | PubMed]
- SJ Wang, TS Sihra, PW Gean. Lamotrigine inhibition of glutamate release from isolated cerebrocortical nerve terminals (synaptosomes) by suppression of voltage-activated calcium channel activity. NeuroReport, 2001. [DOI | PubMed]
- J-F Wang, X Sun, B Chen, LT Young. Lamotrigine Increases Gene Expression of GABA-A Receptor β3 Subunit in Primary Cultured Rat Hippocampus Cells. Neuropsychopharmacology, 2002. [DOI | PubMed]
- JA Wasserstrom, JJ Salata. Basis for tetrodotoxin and lidocaine effects on action potentials in dog ventricular myocytes. Am J Physiol Heart Circ Physiol, 1988. [DOI]
- ER Wengert, MK Patel. The Role of the Persistent Sodium Current in Epilepsy. Epilepsy Curr, 2020. [DOI | PubMed]
- ER Wengert, AU Saga, PS Panchal, BS Barker, MK Patel. Prax330 reduces persistent and resurgent sodium channel currents and neuronal hyperexcitability of subiculum neurons in a mouse model of SCN8A epileptic encephalopathy. Neuropharmacology, 2019. [DOI | PubMed]
- HS White, SD Brown, JH Woodhead, GA Skeen, HH Wolf. Topiramate enhances GABA-mediated chloride flux and GABA-evoked chloride currents in murine brain neurons and increases seizure threshold. Epilepsy Res, 1997. [DOI | PubMed]
- N Wu, A Enomoto, S Tanaka, CF Hsiao, DQ Nykamp, E Izhikevich. Persistent sodium currents in mesencephalic v neurons participate in burst generation and control of membrane excitability. J Neurophysiol, 2005. [DOI | PubMed]
- L Wu, S Rajamani, JC Shryock, H Li, J Ruskin, C Antzelevitch. Augmentation of late sodium current unmasks the proarrhythmic effects of amiodarone. Cardiovasc Res, 2007. [DOI | PubMed]
- S-N Wu, B-S Chen, T-I Hsu, H Peng, Y-H Wu, Y-C Lo. Analytical studies of rapidly inactivating and noninactivating sodium currents in differentiated NG108–15 neuronal cells. J Theor Biol, 2009. [DOI | PubMed]
- P-M Wu, H-Y Cho, C-W Chiang, T-H Chuang, S-N Wu, Y-F Tu. Characterization in Inhibitory Effectiveness of Carbamazepine in Voltage-Gated Na+ and Erg-Mediated K+ Currents in a Mouse Neural Crest-Derived (Neuro-2a) Cell Line. Int J Mol Sci, 2022. [DOI | PubMed]
- R-G Xie, D-W Zheng, J Xing, X-J Zhang, Y Song, Y-B Xie. Blockade of Persistent Sodium Currents Contributes to the Riluzole-Induced Inhibition of Spontaneous Activity and Oscillations in Injured DRG Neurons. PLoS ONE, 2011. [DOI | PubMed]
- R Yadav, E Schrem, V Yadav, A Jayarangaiah, S Das, P Theetha Kariyanna. Lacosamide-Related Arrhythmias: A Systematic Analysis and Review of the Literature. Cureus, 2021. [DOI | PubMed]
- J Yamada-Hanff, BP Bean. Persistent sodium current drives conditional pacemaking in CA1 pyramidal neurons under muscarinic stimulation. J Neurosci, 2013. [DOI | PubMed]
- J Yamada-Hanff, BP Bean. Activation of Ih and TTX-sensitive sodium current at subthreshold voltages during CA1 pyramidal neuron firing. J Neurophysiol, 2015. [DOI | PubMed]
- R-H Yang, W-T Wang, J-Y Chen, R-G Xie, S-J Hu. Gabapentin selectively reduces persistent sodium current in injured type-A dorsal root ganglion neurons. Pain, 2009. [DOI | PubMed]
- C Yue, S Remy, H Su, H Beck, Y Yaari. Proximal persistent Na+ channels drive spike afterdepolarizations and associated bursting in adult CA1 pyramidal cells. J Neurosci, 2005. [DOI | PubMed]
- FA Zeiler, KJ Zeiler, CJ Kazina, J Teitelbaum, LM Gillman, M West. Lidocaine for status epilepticus in adults. Seizure, 2015. [DOI | PubMed]
- Z Zeng, EL Hill-Yardin, D Williams, T O’Brien, A Serelis, CR French. Effect of phenytoin on sodium conductances in rat hippocampal CA1 pyramidal neurons. J Neurophysiol, 2016. [DOI | PubMed]
- H-XB Zhang, BP Bean. Cannabidiol Inhibition of Murine Primary Nociceptors: Tight Binding to Slow Inactivated States of Nav18 Channels. J Neurosci, 2021. [DOI | PubMed]
- 255.Zhang HB, Heckman L, Niday Z, Jo S, Fujita A, Shim J, et al (2022) Cannabidiol activates neuronal Kv7 channels. Elife 11 10.7554/eLife.73246