
Design of Small Non-Peptidic Ligands That Alter Heteromerization between Cannabinoid CB1 and Serotonin 5HT2A Receptors
Abstract
Activation of cannabinoid CB1 receptors (CB1R) by agonists induces analgesia but also induces cognitive impairment through the heteromer formed between CB1R and the serotonin 5HT2A receptor (5HT2AR). This side effect poses a serious drawback in the therapeutic use of cannabis for pain alleviation. Peptides designed from the transmembrane helices of CB1R, which are predicted to bind 5HT2AR and alter the stability of the CB1R-5HT2AR heteromer, have been shown to avert CB1R agonist-induced cognitive impairment while preserving analgesia. Using these peptides as templates, we have now designed nonpeptidic small molecules that prevent CB1R-5HT2AR heteromerization in bimolecular fluorescence complementation assays and the heteromerization-dependent allosteric modulations in cell signaling experiments. These results provide proof-of-principle for the design of optimized ligand-based disruptors of the CB1R-5HT2AR heteromer, opening new perspectives for in vivo studies.
Affiliations: †Department of Biomedical Engineering, University of West Attica, Ag. Spyridonos, Egaleo 12243, Greece; ‡Laboratori de Medicina Computacional, Unitat de Bioestadística, Facultat de Medicina, Universitat Autónoma de Barcelona, Bellaterra 08193, Spain; §Department of Biochemistry and Molecular Biomedicine, Faculty of Biology, Institute of Biomedicine of the University of Barcelona (IBUB),University of Barcelona, Barcelona 08028, Spain; ∥Department of Medicine and Life Sciences (MELIS-UPF), Universitat Pompeu Fabra, Barcelona 08003, Spain; ⊥Integrative Neurobiology Section, National Institute on Drug Abuse, Intramural Research Program, National Institutes of Health, Baltimore, Maryland 21224, United States
License: © 2024 The Authors. Published by American Chemical Society CC BY 4.0 Permits the broadest form of re-use including for commercial purposes, provided that author attribution and integrity are maintained (https://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.1021/acs.jmedchem.4c01796 | PubMed: 39726149 | PMC: PMC11726681
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (3.7 MB)
Introduction
Cannabinoid agonists have proven their analgesic potential in clinical studies of cancer pain,1,2 neuropathic pain,3 and other types of pain. The effects of cannabinoids are primarily mediated through the cannabinoid CB1 receptor (CB1R) and CB2 receptor (CB2R). CB1R is abundant in the central nervous system, whereas CB2R is mainly expressed in the immune system.4 However, the therapeutic response elicited by cannabinoid agonists such as delta-9-tetrahydrocannabinol (THC), the principal psychoactive constituent of the Cannabis sativa plant (more commonly known as marijuana), is hampered by side effects, namely memory impairment.5 Recent findings linked the cognitive side effects of THC to complexes of CB1R and serotonin 5-HT2A receptor (5HT2AR).6 CB1R and 5HT2AR belong to the G protein-coupled receptor (GPCR) family, which frequently forms oligomers in cells.7 Importantly, the formation of the CB1R-5HT2AR heteromer has been characterized by several techniques, including in situ proximity ligation assays and mice expressing heteromerization-deficient receptors,6 which strongly support their existence in native tissues.8 Moreover, it was also shown that the expression of the CB1R-5HT2AR heteromer was enhanced in olfactory neuroepithelium cells of cannabis users9 and schizophrenia patients.10
The use of synthetic peptides with the sequence of the transmembrane (TM) domains of the receptor fused to the cell-penetrating HIV transactivator of transcription (Tat) peptide (GRKKRRQRRR)11 has been very valuable in predicting contacts among TMs of interacting GPCRs.12−15 Using this approach, we have previously identified that synthetic peptides TM5-Tat and Tat-TM6 (but not TM7-Tat), derived from CB1R, were able to decrease bimolecular fluorescence complementation (BiFC) signals elicited by receptors fused to two complementary halves of YFP (5HT2AR-cYFP and CB1R-nYFP), which suggests that the arrangement of CB1R and 5HT2AR protomers in the heteromer is via TMs 5 and 6 (TM5/6 interface).6 More importantly, interfering TM5-Tat and Tat-TM6, but not TM7-Tat, peptides led to a selective abrogation of memory impairments caused by exposure to THC in vivo.6
It was thus reasonable to assume that altering the formation of the CB1R-5HT2AR heteromer might be an effective strategy for harnessing the therapeutic potential of THC while avoiding its side effects. Accordingly, we recently developed a drug-like 16-residue peptide, displaying 9 key amino acids from the 37-residue TM5-Tat peptide, which could penetrate the blood–brain barrier via a cell-penetrating sequence of 7 amino acids.16 Following up on these results, we now report the design of small nonpeptidic molecules capable of mimicking the interaction of the peptides with 5HT2AR and, thus, capable of disrupting the CB1R-5HT2AR heteromer. As previously proposed, a heteromer-disrupting ligand would allow the use of cannabis to fight pain while avoiding cognitive side effects. This is relevant because it has been suggested that cannabinoid ligands could fill the therapeutic gap between opioids and nonsteroidal anti-inflammatories in multiple moderate pain conditions.17
Results
VYAYMYILW5.63-Tat is the Shortest Peptide That Alters CB1R-5HT2AR Heteromerization as Efficiently as TM5-Tat
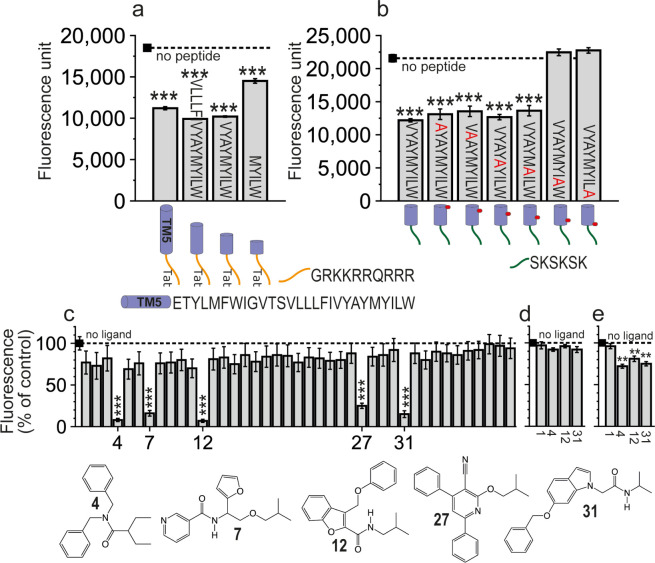
We had previously identified the TM5-Tat peptide, derived from TM 5 of CB1R, that interacts with TMs 5 and 6 of 5HT2AR and alters CB1R-5HT2AR heteromerization.6 Here, we wanted to delineate the minimal region of the 37-residue TM5-Tat peptide (27 amino acids from CB1R plus 10 from Tat) that is capable of interacting with 5HT2AR. Therefore, we tested a variety of shorter peptides, spanning the TM5-Tat peptide, to alter BiFC signals between 5HT2AR-cYFP and CB1R-nYFP in HEK-293T cells (Figure a). We have designed the 5.49VLLLFIVYAYMYILW5.63-Tat, 5.55VYAYMYILW5.63-Tat, and 5.59MYILW5.63-Tat peptides using our previous MD simulations of TM5-Tat in complex with 5HT2AR.16 The residues that form the peptides will be named using the one-letter code of the amino acids and the Ballesteros–Weinstein nomenclature18 of the CB1R sequence as a superscript. The peptides are three, five, and six helical turns shorter, respectively, than the reference TM5-Tat peptide. All caused a clear decrease in BiFC signals, though the shortest one, 5.59MYILW5.63-Tat, being less effective than TM5-Tat or the other two peptides (Figure a).
LW5.63 Are the Residues Contributing More Significantly to 5HT2AR Binding
In protein–protein interactions, “hot spots” are amino acids that contribute more significantly to the binding affinity. To experimentally probe the key molecular interactions of the minimal VYAYMYILW5.63-Tat peptide with 5HT2AR, we mutated the amino acids predicted to interact with 5HT2AR to Ala (Figure b). Since the 6-residue SKSKSK19 cell-penetrating sequence can replace Tat with similar effects,16 the experiments were performed with SKSKSK-extended peptides (Figure b). Both the L5.62 (VYAYMYIAW5.63-SKSKSK) and W5.63 (VYAYMYILA5.63-SKSKSK) mutants were unable to decrease fluorescence; hence, residues LW5.63 were deemed key for the interaction with 5HT2AR.
A 3-Point Pharmacophore Model for the Interaction with 5HT2AR
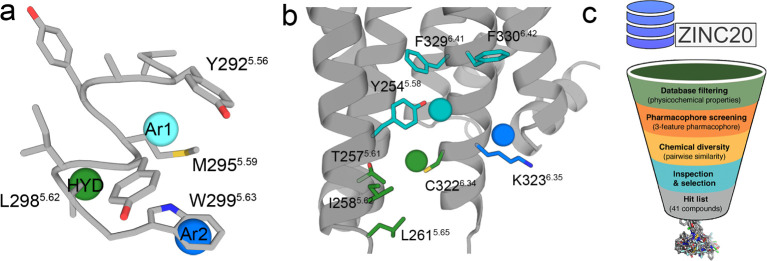
We aimed to develop a model of a pharmacophore that embodies the structural features necessary for 5HT2AR recognition. We inferred 3-point pharmacophoric features (Figure a) based on the above experimental results with different peptides (Figure a,b) and the previously reported computational model between TM5-Tat and 5HT2AR (Figure b).16 The model includes first an aromatic feature (Ar1) that mimics the key W5.63 side chain identified in the Ala scan (Figures b, 2a) predicted to interact with Lys3236.35 and Phe3306.42 of 5HT2AR (Figure b) (the 5HT2AR residues will be named using the three-letter amino acid code to distinguish them from the peptide residues). Second, a hydrophobic feature (HYD) mimicking the L5.62 side chain of the peptide (Figure a) that would be in a small membrane-facing site between TMs 5 and 6 of 5HT2AR, flanked by Thr2575.61, Ile2585.62, Leu2615.65, and Cys3216.34 of 5HT2AR (Figure b). Third, an additional aromatic feature (Ar2) would interact with the Tyr2545.58, Phe3296.41, and Phe3306.42 cluster of aromatic rings of 5HT2AR (Figure b). The rationale for adding this aromatic feature, placed approximately between Y5.56 and M5.59 of the peptide (Figure a), is that the removal of a helical turn (VYAY) in the 5.55VYAYMYILW5.63-Tat peptide makes the resulting 5.59MYILW5.63-Tat peptide clearly less efficient in decreasing BiFC signals than TM5-Tat (Figure a).
A Pharmacophore-Based Virtual Screening Identified 41 Small Nonpeptidic Ligands That Potentially Bind 5HT2AR and Alter CB1R-5HT2AR Heteromerization
Approximately 12 M compounds of the Clean Drug-like subset of the ZINC database20 were filtered to obtain the screening subset. Since the binding site is in the hydrophobic membrane environment, the criteria for filtering the library were Lipinski’s rules (maximum 5 hydrogen bond donors, 10 hydrogen bond acceptors, molecular weight of 500 Da, and log P of 5)21 slightly modified for this specific task. Explicitly, the molecular weight limits were set at 250–500 Da, the maximum hydrogen bond donors (<4) and acceptors (<8) were decreased to reduce polarity, log P was increased to the 2–7 range to favor hydrophobicity, and the number of aromatic groups in the ligand was set to ≥2 to meet the pharmacophore requirements. The initial database was filtered using Openbabel,22 leading to a subset of ∼4 M compounds (see Methods). Diverse conformations (250 per compound) of these ∼4 M compounds were rigid body-fitted (no additional conformational changes in the ligand were allowed) to the 3-point pharmacophore model. These were ranked from best to worst fitting (see Methods), and ∼300 k putative disruptors were mapped again onto the pharmacophore model using a flexible fit (additional conformational changes in the ligand are allowed). Finally, top scoring ∼4 k compounds (see Methods) were visually inspected for chemical complementation between the ligand and the predicted 5HT2AR cavity. These procedures (Figure c) finally yielded 41 diverse compounds (Table S1) that were selected for purchasing and experimental validation.
Five Compounds Alter CB1R-5HT2AR Heteromerization in BiFC Assays
The 41 small nonpeptidic compounds were tested in BiFC assays (Figure c) for their ability to alter CB1R-5HT2AR heteromerization (see Methods). Five of the 41 compounds (12% hit rate) caused a significant decrease of BiFC signals, like the reported peptides. All five structures (Figure c) contain at least two aromatic rings, phenyl and/or aromatic heterocycles, and a branched hydrophobic group. Among these, compounds 4, 12, and 31, causing the largest decrease in fluorescence, were selected for further analysis. Inactive compound 1 (negative control) and active compounds 4, 12, and 31 were also tested to alter BiFC signals between CB1R-cYFP and CB1R-nYFP (Figure d) and between 5HT2AR-cYFP and 5HT2AR-nYFP (Figure e). These experiments show that compounds 4, 12, and 31, but not compound 1, bind to 5HT2AR, but not to CB1R, and influence the homodimerization of 5HT2AR.
Compounds 4, 12, and 31 Block Cross-Antagonism and Negative Crosstalk Biochemical Signatures of the CB1R-5HT2AR Heteromer
We have shown that the formation of the CB1R-5HT2AR heteromer alters the signaling response compared to receptors expressed individually on the cell surface.6,9,10,16,23 Heteromer formation triggers a switch in the G-protein coupling for 5HT2AR from Gq to Gi proteins. Antagonist binding to one of the receptors blocks signaling of the interacting receptor (cross-antagonism), and costimulation with both agonists does not produce an additive effect (negative crosstalk) (see (ref. 24) for definition of these effects). Aiming to ascertain the biochemical signatures of the CB1R-5HT2AR heteromer in the presence of 4, 12, and 31, we monitored receptor activation by measuring cAMP levels [decrease in forskolin (FK)-induced cAMP] or the increase in pERK in the ERK1/2 phosphorylation pathway. Cells stimulated with FK and treated with the CB1R agonist WIN 55,212-2 (WIN) or the 5HT2AR agonist 2,5-dimethoxy-4-iodoamphetamine (DOI) showed reduced levels of cAMP production in the absence and presence of compounds 4, 12, and 31 (Figure a). Hence, none of these compounds, neither the full-length TM5-Tat or Tat-TM6 peptides6 nor their shortened versions,16 influenced G protein coupling preferences (5HT2AR remained Gi-coupled). In the CB1R-5HT2AR heteromer, decreased cAMP or increased pERK caused by 5HT2AR agonist DOI is similar (no statistical differences) in the absence and presence of compounds 4, 12, and 31 (Figure ). Thus, these compounds do not allosterically influence the activity of the orthosteric agonist DOI in 5HT2AR activation. Moreover, coadministration of WIN and DOI agonists does not lead to a further statistically significant decrease in cAMP or an increase in pERK (negative crosstalk) in the absence of the ligands. In contrast, in the presence of 4, 12, and 31, simultaneous addition of both agonists causes a statistically significant cAMP decrease or pERK increase (absence of negative crosstalk), relative to single agonist administration (Figure ).
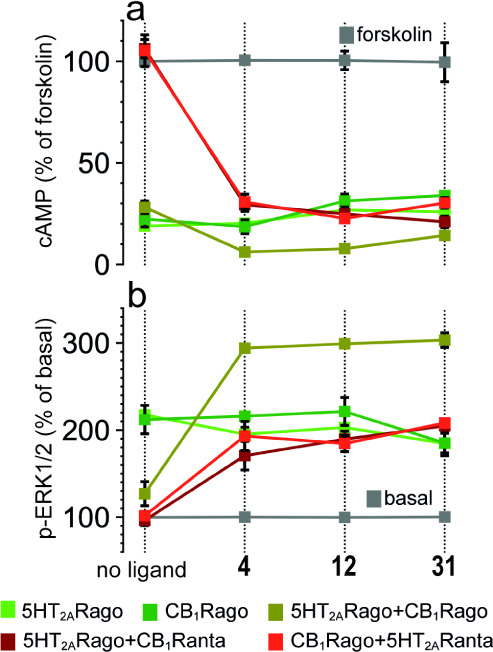
The CB1R antagonist rimonabant (RIM) blocked the decrease in FK-induced cAMP or the increase in pERK triggered by the 5HT2AR agonist DOI, and the 5HT2AR antagonist MDL 100,907 (MDL) also blocked the decrease in cAMP and increase in pERK induced by the CB1R agonist WIN (bidirectional cross-antagonism). Cross-antagonism is due to the formation of a high surface complementarity between TMs 5 and 6 of the two protomers, via a four-helix bundle,25 which blocks the opening of the intracellular cavity for G protein binding at the other protomer.7 Notably, compounds 4, 12, and 31 eliminated this bidirectional cross-antagonism (Figure ). Thus, in the presence of 4, 12, and 31, the CB1R agonist WIN plus the 5HT2AR antagonist MDL and the 5HT2AR agonist DOI plus the CB1R antagonist (RIM) can decrease cAMP or increase pERK, as efficiently as the CB1R agonist WIN and the 5HT2AR agonist DOI alone, respectively. Because orthosteric antagonists do not signal on their own, cross-antagonism requires direct protein–protein interaction at the TM level. The fact that these compounds cancel the bidirectional cross-antagonism suggests that they change these pharmacological properties of the CB1R-5HT2AR heteromer by altering the heteromeric interface.
In summary, alteration of the heteromeric interface of the CB1R-5HT2AR heteromer with ligands did not block the coupling of 5HT2AR to Gi but abolished the reduced signaling upon simultaneous activation of both receptors (negative crosstalk) and the bidirectional cross-antagonism. This suggests that ligands that interfere with the heteromeric interface can remove the heteromer-dependent structural restrictions for receptor activation without disrupting heteromerization and its G protein-coupling preferences.
Compounds 4, 12, and 31 Bind in a Membrane-Facing Site between TMs 5 and 6 of 5HT2AR
Our previously reported 5HT2BR-based homology model of 5HT2AR bound to the TM5-Tat peptide16 predicted that short peptides would bind in a small membrane-facing site between TMs 5 and 6 of 5HT2AR. Here, we have used the recently released structures of 5HT2AR26,27 to identify the binding mode of compounds 4, 12, and 31. Among the multiple docking models (see Figure S1 and Methods), we have first selected poses in which the terminal phenyl ring of the ligands is placed between the group of aromatic rings of 5HT2AR formed by Tyr2545.58, Phe3296.41, and Phe3306.42. Second, we evaluated the stability of the binding mode using unbiased MD simulations (see Methods). Figure a-c shows root-mean-square deviations (RMSDs) of the heavy atoms of the ligands and the C atoms of 5HT2AR for simulations of docking poses that are stable and reproduced repeatedly. As expected, the RMSD values of the ligands (<4 Å) are higher than those of the protein (<2 Å) because, unlike orthosteric binding sites, the cavities of membrane-facing sites are formed in part by the surrounding, flexible lipids. Hence, compounds 4, 12, and 31 have 33%, 38%, and 32%, respectively, of the total solvent-accessible area (SASA of 511 Å2, 522 Å2, and 523 Å2, respectively) exposed to the membrane (MESA of 171 Å2, 197 Å2, and 167 Å2, respectively), calculated from three replicates of 500 ns of unbiased MD simulations. Moreover, the distance between the center of mass (COM) of compounds 4, 12, and 31 and the residues in the binding site, along the trajectories (Figure a-c), shows that all three ligands remained stable at the predicted binding site. Inactive compound 40, used as a negative control, is less stable in the binding site than these active compounds 4, 12, and 31 in analogous MD simulations (Figure S2).
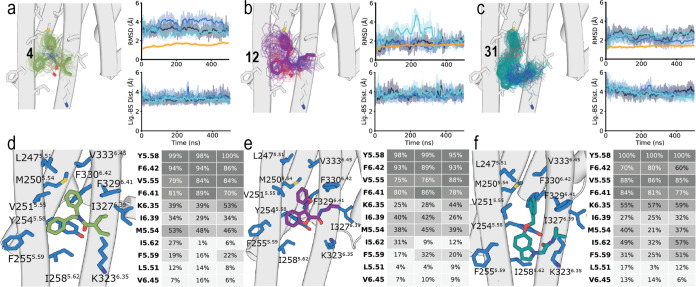
Figure d–f shows heatmaps of the predicted interactions of compounds 4, 12, and 31 with amino acids of the membrane-facing pocket between TMs 5 and 6 of 5HT2AR. The three ligands have very similar binding modes, interacting with the same side chains of 5HT2AR. First, the common terminal phenyl ring of the ligands maintains the initially modeled aromatic interactions with the group of aromatic rings of 5HT2AR formed by Tyr2545.58, Phe3296.41, and Phe3306.42. Additional interactions between this aromatic ring of the ligand and Met2505.54 and Val2515.55 are observed. Notably, the highly polarizable sulfur atom of Met can form stronger interactions, than aromatic or hydrophobic side chains, with aromatic groups.28 On the other side of the molecule, the branched hydrophobic group of the ligands interacts with Lys3236.35, Ile3276.39, and Phe3306.42. The central aromatic heterocycle of 12 and 31, or the phenyl ring of 4, interacts with Tyr2545.58, Phe2555.59, and Ile2585.62. Finally, the carbonyl groups of all three ligands form a hydrogen bond interaction with Tyr2545.58. This lipid-facing site between TMs 5 and 6 was labeled Orphan Site 5 (OS5) in the recent analysis of the pocketome of GPCRs.29
Discussion and Conclusions
The analysis of the pocketome of GPCRs has shown multiple nonconserved pockets in addition to the orthosteric pocket, where endogenous ligands bind.29 Many of these sites had already been identified in structures of allosteric ligands with GPCRs. In contrast, other sites remained untargeted (orphan) sites. One of these orphan sites (OS5) is located at the membrane-facing part of 5HT2AR, between TMs 5 and 6, where compounds 4, 12, and 31 are predicted to bind. However, these compounds do not allosterically influence the activity of the orthosteric agonist DOI in 5HT2AR activation, so we cannot define this site as allosteric or these compounds as allosteric modulators. An alternative role of these membrane-facing cavities is to modulate the stability of the GPCR oligomers. The TM5/6 interface is particularly relevant because it is frequently predicted in GPCR heteromers6,15,30 and directly participates in the allosteric modulation of the partner receptor.7
The design of small molecules targeting protein–protein interfaces, and GPCR interfaces in particular, is specially challenging as they have large and flat contact areas without well-defined pockets, which would be “undruggable” according to the classical definition of druggable sites.31 Therefore, most ligands binding these pockets have a complex structure and are very lipophilic, as part of their binding cavity is promoted by the lipidic membrane.32 This makes their optimization to drug candidates more challenging.33 However, the field of designing molecular inhibitors34 or molecular glues35 to modulate protein–protein interactions with small molecules is rapidly growing.
We developed an approach to design molecular inhibitors of GPCR interactions. First, after characterizing a GPCR heteromer of interest,6 we designed synthetic peptides with the sequence of TM domains of one of the receptors fused to cell-penetrating peptides such as the Tat11 or SKSKSK19 sequence. The ability of these peptides to disrupt BiFC signals of the two receptors separately fused to the complementary cYFP and nYFP predicts contacts among TM domains of interacting GPCRs.36 The 37-residue-long TM5-Tat and 36-residue-long Tat-TM6 peptides, derived from the CB1R sequence, were able to disrupt BiFC signals between 5HT2AR-cYFP and CB1R-nYFP, suggesting a TM5/6 heteromeric interface.6 Second, by downsizing the length of the 37-residue TM5-Tat interfering peptide to only include amino acids essential for receptor interaction, we designed a fully druggable 16-mer that alters CB1R-5HT2AR heteromerization as efficiently.16 Third, Ala scanning mutagenesis identified “hot spots”, i.e., side chains that are more relevant than others for peptide-receptor interaction. And fourth, we designed small nonpeptidic molecules capable of mimicking the “hot spot” recognition elements with 5HT2AR. We accomplished this by performing pharmacophore-guided virtual screening, a well-established successful computational tool for rational drug design.37
The candidate compounds, selected in silico from chemical databases, required experimental validation. Typically, for orthosteric ligands, pharmacological assays are performed to measure affinity or/and efficacy. However, when targeting orphan sites, with no known ligands binding to this site and without allosteric effects on orthosteric agonists, alternative assays must be used. We tested the ability of these compounds to alter CB1R-5HT2AR heteromerization in BiFC assays, in which five of the 41 compounds (12% hit rate) caused a significant disruptive effect. More importantly, we also tested whether these compounds counteract the heteromerization-dependent allosteric effects that CB1R ligands exert on 5HT2AR ligands and vice versa.7,24 In agreement, compounds 4, 12, and 31 blocked part of the allosteric effects within the CB1R-5HT2AR heteromer. In fact, the presence of these compounds counteracted the previously described CB1R-5HT2AR heteromer-dependent negative crosstalk and cross-antagonism.6 However, these ligands did not influence G protein coupling preferences, as 5HT2AR remained Gi-coupled. This suggests that other domains of CB1R (intracellular loops or/and C-terminal) might contribute to the Gq to Gi switch in 5-HT2AR. We favor the hypothesis that dissociation of the CB1R-5HT2AR heteromer depends, in addition to the heteromeric interface, on the existence of additional direct interactions between intracellular receptor domains, as previously described for CB1R with adenosine and dopamine receptors.38,39 Those interactions might still remain after alteration of the TM heteromeric interface, and Gi coupling to 5-HT2AR remained unaltered.
It is important to note that there are similarities and differences between the predicted binding of the 3-point pharmacophore model, derived from the 5.55VYAYMYILW5.63 peptide, to the previously reported 5HT2BR-based homology model of 5HT2AR6,16 and the predicted binding mode of compounds 4, 12, and 31 to the recently released structure of 5HT2AR obtained by MD simulations. In both cases, the aromatic Ar2 element and the terminal phenyl ring of the ligands are placed in similar positions to interact with the Tyr2545.58, Phe3296.41, and Phe3306.42 cluster of aromatic rings. What remains different is the branched hydrophobic group of the ligands, which occupies the position of the aromatic Ar1 pharmacophoric element (W5.63 of the peptide), and the central aromatic heterocycle of 12 and 31, or the phenyl ring of 4, occupy the position of the hydrophobic HYD (L5.62 of the peptide). Moreover, the proposed hydrogen bond interaction of these ligands with Tyr2545.58 was not included in the pharmacophore model.
Tyr5.58 and the NPxxY and DRY motifs of class A GPCRs are responsible for opening the intracellular cavity, through the outward movement of TMs 5 and 6, for G protein binding.40,41 The binding mode of compounds 4, 12, and 31 was studied in the inactive conformations of TMs 5 and 6 of 5HT2AR, so the effect of the active conformations of these helices on their binding mode has not been evaluated. However, measurements of cAMP and pERK indicate that the binding of compounds 4, 12, and 31 to 5HT2AR does not affect receptor activation.
In conclusion, we believe compounds 4, 12, and 31 provide proof-of-principle for the design of optimized ligand-based disruptors of the CB1R-5HT2AR heteromer, opening new perspectives for in vivo studies.
Experimental Section
Peptide Synthesis, Analysis, and Purification
Peptides were assembled in a Prelude instrument (Protein Technologies, Tucson, AZ) running optimized Fmoc synthesis protocols as described before.42 Final deprotection and cleavage were performed with a CF3COOH/H2O/3,6-dioxa-1,8-octanedithiol/triisopropylsilane (94:2.5:2.5:1 v/v) cocktail for 90 min. Peptide analysis and purification were performed as previously detailed.42 Fractions of satisfactory purity (>90%) by analytical high-performance liquid chromatography (HPLC) were pooled, lyophilized, and analyzed for identity by HPLC–MS.
Expression Vectors, HEK-293T Cell Culture, and Transient Transfection
Sequences encoding YFP Venus protein amino acid residues 1–155 and 156–238 were subcloned into the pcDNA3.1 vector to obtain the YFP Venus hemitruncated proteins. The human cDNAs for 5HT2AR and CB1R, cloned into the pcDNA3.1 were amplified and subcloned as described6 to give 5HT2AR-cYFP and CB1R-nYFP. Human embryonic kidney (HEK-293T) cells obtained from ATCC were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) supplemented with 2 mM l-glutamine, 100 μg/mL sodium pyruvate, 100 U/mL penicillin/streptomycin, MEM nonessential amino acid solution (1/100), and 5% (v/v) heat-inactivated fetal bovine serum (FBS) (all supplements from Invitrogen, Paisley, Scotland, UK). Cells were maintained at 37 °C under an atmosphere of 5% CO2. HEK-293T cells were transfected with the corresponding fusion protein cDNA by the polyethylenimine (Sigma) method, as described before.6,16
BiFC Assay
HEK-293T cells, after 48 h transient cotransfection with the cDNA encoding for 5HT2AR-cYFP and CB1R-nYFP (4 μg of cDNA for each construct), were treated with vehicle or 4 μM peptide or 4 μM ligand for 4 h at 37 °C. To quantify protein-reconstituted YFP Venus expression, cells (20 μg protein) were distributed in 96-well microplates (black plates with a transparent bottom, Porvair, King’s Lynn, UK), and emission fluorescence at 530 nm was read in a Fluo Star Optima fluorimeter (BMG Labtechnologies, Offenburg, Germany) equipped with a high-energy Xe flash lamp, using a 10 nm bandwidth excitation filter at 400 nm reading. Protein fluorescence expression was determined as the fluorescence of the sample minus the fluorescence of cells not expressing the fusion proteins (basal). Cells expressing 5HT2AR-cVenus and nVenus or CB1R-nVenus and cVenus showed similar fluorescence levels to nontransfected cells.
cAMP Production and ERK-1/2 Phosphorylation Assays
For cAMP production, homogeneous time-resolved fluorescence energy transfer (HTRF) assays were performed as previously described.6,16 Cells (1000 cells/well) growing in medium containing 50 μM zardeverine were pretreated with the CB1R antagonist rimonabant (1 μM, RIM), and the 5-HT2AR antagonist MDL 100,907 (300 nM, MDL), or the corresponding vehicle in white ProxiPlate 384-well microplates (PerkinElmer, Waltham, MA, US) at 25 °C for 20 min and stimulated with the CB1R agonist WIN 55,212-2 (100 nM, WIN) and the 5HT2AR agonist DOI (100 nM, DOI) for 15 min before adding 0.5 μM FK or vehicle and incubated for an additional 15 min period. Fluorescence at 665 nm was analyzed on a PHERAstar Flagship microplate reader equipped with an HTRF optical module (BMG Lab Technologies, Offenburg, Germany). For ERK-1/2 phosphorylation assays, HEK-293 cells (30,000 cells/well) seeded in 96-well poly d-lysine-coated plates (Sigma-Aldrich, Madrid, Spain) were pretreated at 25 °C for 15 min with RIM (1 μM) and MDL (300 nM) or the corresponding vehicle and stimulated for an additional 7 min with WIN (100 nM) and DOI (100 nM). Phosphorylation was determined in white ProxiPlate 384-well microplates (PerkinElmer Life Sciences) by the α-screen bead-based technology using the amplified luminescent proximity homogeneous assay kit (PerkinElmer Life Sciences) and by using the Enspire multimode plate reader (PerkinElmer Life Sciences). Phosphorylation is expressed in arbitrary units, ALPHAcounts, as measured by light emission at 520–620 nm of the acceptor beads.
Virtual Screening
A three-point feature pharmacophore was built using Discovery Studio 3.5 (Accelrys Software Inc., Discovery Studio Environment, Release 3.5, San Diego), using the reported experimental results obtained with different peptides and the previously reported computational model between TM5-Tat and a 5HT2BR-based homology model of 5HT2AR.6,16 Excluded space features were added by using the 5HT2AR homology model. Diverse conformations (250 per compound) of a subset of ∼4 M compounds, split according to heavy atom count in bins of 20–23, 24–26, 27–29, and 30–36, with molecular weight in the 250–500 Da range, A log P in the 2–7 range, and with <8 hydrogen bond acceptors, <4 hydrogen bond donors, and ≥2 aromatic groups, were rigidly fitted to the pharmacophore model. These solutions were scored from best to worst fitting (total score 0–3, with 3 being optimal) with the fit_value scoring function of Discovery Studio, which measures the spatial complementarity of the center of each group to the center of the feature for aliphatic (score 0–1) and combined spatial complementarity and directional for aromatic ring placement (score 0–1) in the two aromatic features. The top 300,000 compounds were further rescored using a flexible fit, in which slight flexibility is allowed for each conformation. Top-scoring 1000 compounds from each molecular size bin were clustered by similarity using DataWarrior.43 The process involves the calculation of a similarity matrix for each bin using fragment descriptors. The most similar compounds form the first cluster, and their similarity values are replaced by the mean similarity between the cluster center and the other compounds. This process of merging the most similar compounds or clusters, updating similarity values as weighted means, continues until the user-defined similarity threshold, which was set at 0.7, is reached. The highest scored compounds of each cluster were visually inspected when fitted in the pharmacophore model, while located in the defined pocket of the 5-HT2AR model, to finally select 41 diverse compounds. All compounds were purchased from different vendors with >95% purity.
MD Simulations of Selected Compounds in Complex with 5HT2AR
The GPCRdb refined structure44 of inactive 5HT2AR (PDB id 7WC4)27 was used. The fusion protein was removed. Compounds 4, 12, and 31 were docked in a membrane-facing binding cavity between TMs 5 and 6 of 5HT2AR using rDock.45 Docking poses were ranked according to Stotal, and the poses in which the terminal phenyl ring of the ligands was placed between the group of aromatic rings of 5HT2AR formed by Tyr2545.58, Phe3296.41, and Phe3306.42 were initially selected (Figure S1). MD simulations of these complexes were performed using GROMACS,46 embedded in a rectangular lipid bilayer box, constructed using PACKMOL-memgen,47 containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and cholesterol (ratio 10:6), water molecules, and a 0.15 M concentration of monatomic Na+ and Cl– ions, using a previously described protocol.48 In particular, these initial systems were energy-minimized and subsequently subjected to various steps of MD equilibration (25 ns in total), where positional restraints on protein coordinates were progressively unreleased to accommodate the lipids and solvent around the protein. MD trajectories were produced at a constant temperature of 300 K by using separate v-rescale thermostats for the protein, ligand, lipids, and solvent molecules. In these simulations, a time step of 2 fs was used for the integration of the equations of motion. All bonds and angles were kept frozen by using the LINCS algorithm. Lennard-Jones interactions were computed by using a cutoff of 10 Å, and the electrostatic interactions were treated by using the particle mesh Ewald method. The amber14sb-ildn force field was used for the protein, solvent, and ions,49 lipid14 for lipids,50 and the general Amber force field (GAFF2) with HF/6-31G*-derived RESP atomic charges for the ligands.51 The analysis of the trajectories was performed using the Python package MDAnalysis,52 and solvent (SASA) and membrane-exposed (MESA) surface areas were calculated using FreeSASA.53
Statistical Analysis
Experimental data were managed and analyzed with GraphPad Prism software, version 10 (San Diego, CA, USA) or IBM SPSS Statistics version 27.0 (IBM Corp., NY, USA). P-values lower than 0.05 were considered statistically significant.
References
- B. Hinz, R. Ramer. Cannabinoids as anticancer drugs: current status of preclinical research.. Br. J. Cancer, 2022. [DOI | PubMed]
- H. R. Wardill, L. T. Wooley, O. M. Bellas, K. Cao, C. B. Cross, M. van Dyk, G. Kichenadasse, J. M. Bowen, A. C. W. Zannettino, S. Shakib. Supporting gut health with medicinal cannabis in people with advanced cancer: potential benefits and challenges.. Br. J. Cancer, 2024. [DOI | PubMed]
- J. S. Hansen, S. Gustavsen, H. Roshanisefat, M. Kant, F. Biering-Sorensen, C. Andersen, A. Olsson, H. H. Chow, N. Asgari, J. R. Hansen. Cannabis-Based Medicine for Neuropathic Pain and Spasticity-A Multicenter, Randomized, Double-Blinded, Placebo-Controlled Trial.. Pharmaceuticals, 2023. [DOI | PubMed]
- L. H. Parsons, Y. L. Hurd. Endocannabinoid signalling in reward and addiction.. Nat. Rev. Neurosci., 2015. [DOI | PubMed]
- D. S. Hasin. US Epidemiology of Cannabis Use and Associated Problems.. Neuropsychopharmacology, 2018. [DOI | PubMed]
- X. Vinals, E. Moreno, L. Lanfumey, A. Cordomi, A. Pastor, R. de La Torre, P. Gasperini, G. Navarro, L. A. Howell, L. Pardo. Cognitive Impairment Induced by Delta9-tetrahydrocannabinol Occurs through Heteromers between Cannabinoid CB1 and Serotonin 5-HT2A Receptors.. PloS Biol., 2015. [DOI | PubMed]
- S. Ferre, F. Ciruela, C. W. Dessauer, J. Gonzalez-Maeso, T. E. Hebert, R. Jockers, D. E. Logothetis, L. Pardo. G protein-coupled receptor-effector macromolecular membrane assemblies (GEMMAs).. Pharmacol. Ther., 2022. [DOI | PubMed]
- I. Gomes, M. A. Ayoub, W. Fujita, W. C. Jaeger, K. D. Pfleger, L. A. Devi. G Protein-Coupled Receptor Heteromers.. Annu. Rev. Pharmacol. Toxicol., 2016. [DOI | PubMed]
- L. Galindo, E. Moreno, F. Lopez-Armenta, D. Guinart, A. Cuenca-Royo, M. Izquierdo-Serra, L. Xicota, C. Fernandez, E. Menoyo, J. M. Fernandez-Fernandez. Cannabis Users Show Enhanced Expression of CB(1)-5HT(2A) Receptor Heteromers in Olfactory Neuroepithelium Cells.. Mol. Neurobiol., 2018. [DOI | PubMed]
- D. Guinart, E. Moreno, L. Galindo, A. Cuenca-Royo, M. Barrera-Conde, E. J. Perez, C. Fernandez-Aviles, C. U. Correll, E. I. Canela, V. Casado. Altered Signaling in CB1R-5-HT2AR Heteromers in Olfactory Neuroepithelium Cells of Schizophrenia Patients is Modulated by Cannabis Use.. Schizophr. Bull., 2020. [DOI | PubMed]
- S. R. Schwarze, A. Ho, A. Vocero-Akbani, S. F. Dowdy. In vivo protein transduction: delivery of a biologically active protein into the mouse.. Science, 1999. [DOI | PubMed]
- S. Q. He, Z. N. Zhang, J. S. Guan, H. R. Liu, B. Zhao, H. B. Wang, Q. Li, H. Yang, J. Luo, Z. Y. Li. Facilitation of mu-opioid receptor activity by preventing delta-opioid receptor-mediated codegradation.. Neuron, 2011. [DOI | PubMed]
- G. Navarro, A. Cordomi, M. Brugarolas, E. Moreno, D. Aguinaga, L. Perez-Benito, S. Ferre, A. Cortes, V. Casado, J. Mallol. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain.. BMC Biol., 2018. [DOI | PubMed]
- G. Navarro, A. Cordomi, V. Casado-Anguera, E. Moreno, N. S. Cai, A. Cortes, E. I. Canela, C. W. Dessauer, V. Casado, L. Pardo. Evidence for functional pre-coupled complexes of receptor heteromers and adenylyl cyclase.. Nat. Commun., 2018. [DOI | PubMed]
- P. A. De Oliveira, E. Moreno, N. Casajuana-Martin, V. Casado-Anguera, N. S. Cai, G. A. Camacho-Hernandez, H. Zhu, A. Bonifazi, M. D. Hall, D. Weinshenker. Preferential Gs protein coupling of the galanin Gal1 receptor in the micro-opioid-Gal1 receptor heterotetramer.. Pharmacol. Res., 2022. [DOI | PubMed]
- M. Gallo, E. Moreno, S. Defaus, A. Ortega-Alvaro, A. Gonzalez, P. Robledo, M. Cavaco, V. Neves, M. Castanho, V. Casado. Orally Active Peptide Vector Allows Using Cannabis to Fight Pain While Avoiding Side Effects.. J. Med. Chem., 2021. [DOI | PubMed]
- P. F. Whiting, R. F. Wolff, S. Deshpande, M. Di Nisio, S. Duffy, A. V. Hernandez, J. C. Keurentjes, S. Lang, K. Misso, S. Ryder. Cannabinoids for Medical Use: A Systematic Review and Meta-analysis.. JAMA, 2015. [DOI | PubMed]
- J. A. Ballesteros, H. Weinstein. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors.. Methods Neurosci., 1995. [DOI]
- B. Jastrzebska, Y. Chen, T. Orban, H. Jin, L. Hofmann, K. Palczewski. Disruption of Rhodopsin Dimerization with Synthetic Peptides Targeting an Interaction Interface.. J. Biol. Chem., 2015. [DOI | PubMed]
- J. J. Irwin, K. G. Tang, J. Young, C. Dandarchuluun, B. R. Wong, M. Khurelbaatar, Y. S. Moroz, J. Mayfield, R. A. Sayle. ZINC20-A Free Ultralarge-Scale Chemical Database for Ligand Discovery.. J. Chem. Inf. Model., 2020. [DOI | PubMed]
- M. D. Shultz. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs.. J. Med. Chem., 2019. [DOI | PubMed]
- N. M. O’Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch, G. R. Hutchison. Open Babel: An open chemical toolbox.. J. Cheminf., 2011. [DOI]
- J. Botta, L. Bibic, P. Killoran, P. J. McCormick, L. A. Howell. Design and development of stapled transmembrane peptides that disrupt the activity of G-protein-coupled receptor oligomers.. J. Biol. Chem., 2019. [DOI | PubMed]
- C. L. Del Torrent, I. Raich, A. Gonzalez, J. Lillo, N. Casajuana-Martin, R. Franco, L. Pardo, G. Navarro. Allosterism in the adenosine A(2A) and cannabinoid CB(2) heteromer.. Br. J. Pharmacol., 2024. [DOI]
- A. Manglik, A. C. Kruse, T. S. Kobilka, F. S. Thian, J. M. Mathiesen, R. K. Sunahara, L. Pardo, W. I. Weis, B. K. Kobilka, S. Granier. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist.. Nature, 2012. [DOI | PubMed]
- K. Kim, T. Che, O. Panova, J. F. DiBerto, J. Lyu, B. E. Krumm, D. Wacker, M. J. Robertson, A. B. Seven, D. E. Nichols. Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor.. Cell, 2020. [DOI | PubMed]
- D. Cao, J. Yu, H. Wang, Z. Luo, X. Liu, L. He, J. Qi, L. Fan, L. Tang, Z. Chen. Structure-based discovery of nonhallucinogenic psychedelic analogs.. Science, 2022. [DOI | PubMed]
- J. C. Gomez-Tamayo, A. Cordomi, M. Olivella, E. Mayol, D. Fourmy, L. Pardo. Analysis of the interactions of sulfur-containing amino acids in membrane proteins.. Protein Sci., 2016. [DOI | PubMed]
- J. B. Hedderich, M. Persechino, K. Becker, F. M. Heydenreich, T. Gutermuth, M. Bouvier, M. Bunemann, P. Kolb. The pocketome of G-protein-coupled receptors reveals previously untargeted allosteric sites.. Nat. Commun., 2022. [DOI | PubMed]
- T. Gnad, G. Navarro, M. Lahesmaa, L. Reverte-Salisa, F. Copperi, A. Cordomi, J. Naumann, A. Hochhauser, S. Haufs-Brusberg, D. Wenzel. Adenosine/A2B Receptor Signaling Ameliorates the Effects of Aging and Counteracts Obesity.. Cell Metab., 2020. [DOI | PubMed]
- X. Barril. Druggability predictions: methods, limitations, and applications.. Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013. [DOI]
- P. Obi, S. Natesan. Membrane Lipids Are an Integral Part of Transmembrane Allosteric Sites in GPCRs: A Case Study of Cannabinoid CB1 Receptor Bound to a Negative Allosteric Modulator, ORG27569, and Analogs.. J. Med. Chem., 2022. [DOI | PubMed]
- J. Payandeh, M. Volgraf. Ligand binding at the protein-lipid interface: strategic considerations for drug design.. Nat. Rev. Drug Discovery, 2021. [DOI | PubMed]
- M. R. Arkin, Y. Tang, J. A. Wells. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality.. Chem. Biol., 2014. [DOI | PubMed]
- M. Konstantinidou, M. R. Arkin. Molecular glues for protein-protein interactions: Progressing toward a new dream.. Cell Chem. Biol., 2024. [DOI | PubMed]
- R. Franco, A. Cordomi, C. Llinas Del Torrent, A. Lillo, J. Serrano-Marin, G. Navarro, L. Pardo. Structure and function of adenosine receptor heteromers.. Cell. Mol. Life Sci., 2021. [DOI | PubMed]
- H. Li, X. Sun, W. Cui, M. Xu, J. Dong, B. E. Ekundayo, D. Ni, Z. Rao, L. Guo, H. Stahlberg. Computational drug development for membrane protein targets.. Nat. Biotechnol., 2024. [DOI | PubMed]
- G. Navarro, S. Ferre, A. Cordomi, E. Moreno, J. Mallol, V. Casado, A. Cortes, H. Hoffmann, J. Ortiz, E. I. Canela. Interactions between intracellular domains as key determinants of the quaternary structure and function of receptor heteromers.. J. Biol. Chem., 2010. [DOI | PubMed]
- A. Kofalvi, E. Moreno, A. Cordomi, N. S. Cai, V. Fernandez-Duenas, S. G. Ferreira, R. Guixa-Gonzalez, M. Sanchez-Soto, H. Yano, V. Casado-Anguera. Control of glutamate release by complexes of adenosine and cannabinoid receptors.. BMC Biol., 2020. [DOI | PubMed]
- W. I. Weis, B. K. Kobilka. The Molecular Basis of G Protein-Coupled Receptor Activation.. Annu. Rev. Biochem., 2018. [DOI | PubMed]
- Q. Zhou, D. Yang, M. Wu, Y. Guo, W. Guo, L. Zhong, X. Cai, A. Dai, W. Jang, E. I. Shakhnovich. Common activation mechanism of class A GPCRs.. Elife, 2019. [DOI | PubMed]
- M. Gallo, G. Navarro, R. Franco, D. Andreu. A(2A) Receptor Homodimer-Disrupting Sequence Efficiently Delivered by a Protease-Resistant, Cyclic CPP Vector.. Int. J. Mol. Sci., 2019. [DOI | PubMed]
- T. Sander, J. Freyss, M. von Korff, C. Rufener. DataWarrior: an open-source program for chemistry aware data visualization and analysis.. J. Chem. Inf. Model., 2015. [DOI | PubMed]
- G. Pandy-Szekeres, J. Caroli, A. Mamyrbekov, A. A. Kermani, G. M. Keseru, A. J. Kooistra, D. E. Gloriam. GPCRdb in 2023: state-specific structure models using AlphaFold2 and new ligand resources.. Nucleic Acids Res., 2023. [DOI | PubMed]
- S. Ruiz-Carmona, D. Alvarez-Garcia, N. Foloppe, A. B. Garmendia-Doval, S. Juhos, P. Schmidtke, X. Barril, R. E. Hubbard, S. D. Morley. rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids.. PloS Comput. Biol., 2014. [DOI | PubMed]
- M. J. Abraham, T. Murtola, R. Schulz, S. Pall, J. C. Smith, B. Hess, E. Lindahl. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers.. SoftwareX, 2015. [DOI]
- S. Schott-Verdugo, H. Gohlke. PACKMOL-Memgen: A Simple-To-Use, Generalized Workflow for Membrane-Protein-Lipid-Bilayer System Building.. J. Chem. Inf. Model., 2019. [DOI | PubMed]
- C. Llinas Del Torrent, I. Raich, A. Gonzalez, N. Casajuana-Martin, J. Lillo, J. B. Rebassa, C. Ferreiro-Vera, V. Sanchez de Medina, R. Franco, G. Navarro. The Leu/Val(6.51) Side Chain of Cannabinoid Receptors Regulates the Binding Mode of the Alkyl Chain of Delta(9)-Tetrahydrocannabinol.. J. Chem. Inf. Model., 2023. [DOI | PubMed]
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser, C. Simmerling. ff14 SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB.. J. Chem. Theory Comput., 2015. [DOI | PubMed]
- C. J. Dickson, B. D. Madej, A. A. Skjevik, R. M. Betz, K. Teigen, I. R. Gould, R. C. Walker. Lipid14: The Amber Lipid Force Field.. J. Chem. Theory Comput., 2014. [DOI | PubMed]
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman, D. A. Case. Development and testing of a general amber force field.. J. Comput. Chem., 2004. [DOI | PubMed]
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf, O. Beckstein. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations.. J. Comput. Chem., 2011. [DOI | PubMed]
- S. Mitternacht. FreeSASA: An open source C library for solvent accessible surface area calculations.. F1000Research, 2016. [DOI | PubMed]