Exploring the Antiproliferative Activity of Flavolignans From the Leaves of Casearia arborea (Salicaceae)
Abstract
The Casearia genus (Salicaceae) is well known as a source of natural antitumor prototypes, as described for clerodane diterpenes. In this way, the dichloromethane phase from leaves of Casearia arborea was phytochemically evaluated in liquid chromatography coupled to high‐resolution mass spectrometry, furnishing flavonoid, flavolignan, and phenylpropanoid derivatives, and apocarotenoid terpenoids according to Global Natural Products Social Molecular Networking (GNPS) dereplication. The flavolignans were chemically monitored by liquid chromatography using a UV–Vis detector and purified by a semi‐preparative liquid chromatographic system, furnishing tricin, tricin‐4″‐O‐(threo‐guaiacylglyceryl) ether (salcolin A), and tricin‐4″‐O‐(erythro‐guaiacylglyceryl) ether (salcolin B). Additionally, it was observed that tricin was able to reduce live cells and increase apoptotic cell lines of papillary thyroid cancer and induced only necrosis in this HTH83 cell line, whereas salcolins A and B induced cell death in anaplastic thyroid cancer, increasing cell necrosis. Still, there is no significant induction of apoptotic cell death or necrosis in papillary thyroid cancer cells.
Article type: Research Article
Keywords: cytotoxic activity | flavolignan | molecular networking | thyroid cancer | tricin
Affiliations: Institute of Environmental, Chemical and Pharmaceutical Sciences Federal University of São Paulo São Paulo Brazil; Programa De Pós‐Graduação em Biologia Estrutural e Funcional Federal University of São Paulo São Paulo Brazil; Institute of Chemistry Federal University of Alfenas Alfenas Minas Gerais Brazil; Botany Department, Institute of Biosciences University of São Paulo São Paulo Brazil
License: © 2025 The Author(s). Chemistry & Biodiversity published by Wiley‐VHCA AG. CC BY 4.0 This is an open access article under the terms of the http://creativecommons.org/licenses/by/4.0/ License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Article links: DOI: 10.1002/cbdv.202501489 | PubMed: 40911853 | PMC: PMC12716014
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (2.1 MB)
Introduction
Thyroid cancer incidence is rising, accounting for 3% of all diagnosed cancers [ref. 1, ref. 2]. Papillary thyroid carcinoma (PTC) is the most common type, making up about 80% of cases and with a 5‐year survival rate higher than 90% [ref. 3]. Although anaplastic thyroid cancer (ATC) is rare (1%–2% of cases), its aggressiveness accounts for one third of thyroid cancer deaths, with around 50% of patients having metastatic disease at the first diagnosis, and 25% developing metastasis and rapid disease progression [ref. 4]. ATC patients do not respond to radioiodine or chemotherapy, with a survival of 3–9 months after diagnosis [ref. 5].
Plants serve as sources for discovering new prototypes with antitumor potential because they contain a wide range of bioactive secondary metabolites [ref. 6]. In this context, Brazil has the highest biodiversity of plants in the world, including many medicinal species, and the Atlantic Forest is a conservation hotspot due to its high number of endemic species, making it one of the world’s most important biomes [ref. 7, ref. 8, ref. 9]. The genus Casearia Jacq. (Salicaceae) includes roughly 217–277 species of shrubs and trees [ref. 10, ref. 11, ref. 12] found across the pantropical region, including Africa, Asia, Australia, the Americas, and Pacific islands [ref. 10, ref. 13]. In Brazil, about 50 species of Casearia occur in nearly all biomes, with 24 being endemic [ref. 11, ref. 14, ref. 15]. The pharmacological properties of Casearia are varied, and researchers have been investigating secondary metabolites from 35 different Casearia species [ref. 16, ref. 17, ref. 18, ref. 19, ref. 20, ref. 21]. Regarding chemical composition, clerodane diterpenes are chemical markers of Casearia, demonstrating high cytotoxic activity against various cancer cell lines, as shown by several authors in the review by Xia et al. [ref. 18]. Recently, a variety of flavonoids has been identified in different Casearia species, including C. arborea [ref. 20, ref. 22, ref. 23, ref. 24], C. decandra [ref. 20], C. gossypiosperma [ref. 19], C. grandiflora, C. javitensis, C. lasiophylla [ref. 20], C. sylvestris var. lingua [ref. 25], and C. ulmifolia [ref. 20]. Additionally, the Global Natural Products Social Molecular Networking (GNPS) platform offers a way to organize the metabolic profiles of plant and microorganism extracts. GNPS collects similar spectra obtained from high‐resolution mass spectrometry tandem mass spectrometry (HRMS/MS) of compounds in complex mixtures. It constructs molecular networks based on similar MS/MS spectra, forming spectral families or molecular families due to shared fragmentation patterns. This process also suggests analog compounds and biosynthetic pathways, supporting phytochemical studies [ref. 26, ref. 27], including the investigation of cytotoxic compounds from plant metabolomes [ref. 28].
This work describes the liquid chromatography equipped with diode‐array‐detector (LC‐DAD) guided isolation of flavolignans from C. arborea leaves, including the HRMS/MS dereplication of a polar extract fraction using classical molecular networking (CMN) as a reliable computational tool for annotations in the chemical discovery of plant secondary metabolites. Additionally, this work demonstrates the antiproliferative potential of tricin and flavolignan derivatives, such as reducing metabolic activity, limiting colony growth, and inducing cell death.
Materials and Methods
Plant Materials
Leaves of C. arborea (Rich.) Urb. were collected at a specific farm in Alfenas, MG, Brazil, in February 2013 (coordinates 21°21.747′ S 45°50.417′ W) and were named “Paraíso” (P). Two additional samples were collected in June 2016 in Alfenas, MG, Brazil, from different farms, and labeled “Gaspar Lopes” (G) (21°22′53.8″ S 045°55′46.4″ W) and “Matão” (M) (21°30′09.7″ S 045°53′13.5″ W). The plants were identified by taxonomist Marcelo Pólo. They were deposited in the herbarium of the University of Alfenas (UNIFAL‐MG), with the code “1388” for the 2013 collection (Paraíso). The collections from 2016 were stored in the SPF herbarium at São Paulo University, under the codes “Elias, J.P.C. 01” and “Elias, J.P.C. 02,” for Matão and Gaspar Lopes, respectively.
General Experimental Procedures
1H and 13C‐NMR spectra (including uni‐ and bi‐dimensional) were recorded at 300.13 MHz (1H) and 75.77 MHz (13C) using an Ultrashield 300 Advance III spectrometer (Bruker‐Biospin, Germany). The instrument was equipped with a 5‐mm trinuclear, inverse detection probe with a z‐gradient (TXI, 5.0 mm). The temperature was maintained at 25°C with a BCU 0.5 I accessory. DMSO‐d6 (Sigma‐Aldrich) served as the solvent and internal standard. HRMS/MS data were acquired on a Shimadzu Nexera X2 liquid chromatograph system (Shimadzu, Japan) equipped with an SPD‐M20A Prominence Diode Array detector, coupled to a quadrupole coupled to time‐of‐flight mass analyzer (QTOF) mass spectrometer analyzer (MicroTOF‐QII; Bruker Daltonics, USA) operating with electron spray ionization (ESI) in positive ion mode at 18 000 FWHM for mass resolution. For extracts and compounds, ultra‐performance liquid chromatography coupled to high‐resolution mass spectrometry (UPLC–HRMS) analyses employed a C18 Reverse Phase (RP) Kinetex EVO column (2.6 µm, 100 × 2.1 mm2). All solvents used were AR grade (Merck and J. T. Baker), and formic acid was MS grade (Merck). The dichloromethane phase of the methanolic leaf extract was analyzed on an analytical high‐performance liquid chromatography (HPLC) system with an Agilent 1260 liquid chromatograph equipped with a UV‐DAD detector (Agilent), using a Zorbax Eclipse Plus RP column (3.5 µm, 150 × 4.6 mm2). Compound purification was performed using a semi‐preparative HPLC system on an Agilent 1260 liquid chromatograph with an automatic sample collector (Agilent), equipped with a UV‐DAD detector (Agilent) and a Zorbax Eclipse Plus column (3.5 µm, 150 × 4.6 mm2). All solvents involved in analytical and preparative chromatography were AR grade.
Crude Extract Preparation
The leaves were dried for 48 h in an oven at 40°C. The dry leaves were then ground into powder and extracted with methanol (1 L × 5) at room temperature. After filtration and concentration in a rotary evaporator, the methanolic crude extract of the leaves was obtained. The extract was resuspended in methanol:water (2:1, v/v), and liquid–liquid partitioning was performed with solvents of increasing polarity, producing four fractions: n‐hexane, dichloromethane, ethyl acetate, and n‐butanol. Each fraction was concentrated using a rotary evaporator at 40°C. From 40 g of P, approximately 2.1 g of the dichloromethane phase was obtained, labeled PD , and from 10 g of G and M, 0.41 and 0.40 g of the dichloromethane phase were obtained, labeled GD and MD , respectively. The dichloromethane fractions PD , GD , and MD were combined (CarD) for phytochemical analysis, focusing on purifying flavolignan derivatives. The yield of the dichloromethane fractions was around 4%–5%, the lowest among the extracts, but these fractions were rich in target compounds.
UPLC–HRMS Data Acquisition
A sample of CarD was dissolved in methanol, producing 10 mg in 1 mL. The solution was centrifuged at 10 000 rpm for 15 min. The supernatant (100 µL) was transferred to a vial and diluted with 900 µL of a methanol:water (1:1, v/v) mixture to reach a final concentration of 1 mg/mL. It was then analyzed by UPLC–HRMS with a 3 µL injection into the LC system using RP column at 50°C in the column chamber. Separation was achieved with mobile phases (A) water with 0.2% formic acid and (B) acetonitrile (ACN), flowing at 300 µL/min. The gradient method was as follows: 0–1 min, 5% B; 1–4 min, 5%–20% B; 4–8 min, 20% B; 8–12 min, 20%–60% B; 12–14 min, 60%–98% B; and 14–15 min, 98% B. HRMS/MS data were acquired in positive mode ESI over a mass range of m/z 75–1200. The ionization parameters were set as follows: capillary tune voltage at 4500 V, end plate offset at 500 V; nitrogen dry gas flow at 8.0 mL/min; pressure at 4.0 bar; and temperature at 200°C. Auto‐MS/MS selected three precursor ions, and the CID ramp energy was based on literature [ref. 29], with modifications.
HRMS/MS Data Processing and Molecular Networking Workflow
All HRMS/MS data obtained from the analysis of CarD acquired in Bruker microTOF‐QII‐ESI were converted to the “.mzML” extension using the free software MSConvert (Proteowizard) [ref. 30, ref. 31] and conferred in TOOPView (OpenMS) [ref. 32]. The “.mzML” data were then uploaded to the Mass Spectrometry Interactive Virtual Environment (MassIVE) Web server using WinSCP to create the molecular networking using the GNPS platform with the dataset (MSV000089469, doi:10.25345/C59P2W96N), as well as to perform the dereplication for database matches [ref. 26, ref. 29]. To create the molecular networking, the acquired data were treated in the GNPS Data Analysis platform, removing fragments of ±17 Da of precursor m/z. HRMS/MS spectra were filtered, choosing only the six top fragments in the ±50 Da window in all ranges of spectra. The basic options for mass tolerance ions were set to 0.02 Da for precursor and QTOF fragment ions and 0.02 Da. A network was then created using the MS‐cluster algorithm [ref. 33], according to a cosine score above 0.7, more than two matched peaks, and a minimum cluster size of one spectrum. Further, edges between two nodes were kept in the network if and only if each of the nodes appeared in the other’s respective top 10 most similar nodes. The maximum size of a molecular family was set to 100, and the lowest scoring edges were removed from molecular families until the molecular family size was below this threshold. The spectra in the network were then searched against GNPS’ spectral libraries. The library spectra were filtered in the same manner as the input data. All matches kept between network spectra and library spectra were required to have a score above 0.6 and at least three matched peaks. The software Cytoscape was used to visualize and edit the entire molecular networking [ref. 34], as well as on the GNPS website (https://gnps.ucsd.edu/ProteoSAFe/static/gnps‐splash.jsp).
Compound Purification
Tricin (1) was obtained from previous work using PD fraction [ref. 24] and analyzed by UPLC–DAD–HRMS to guide the purification of tricin derivatives. Flavolignan derivatives were identified by DAD–HRMS/MS spectra in the PD , GD , and MD and then collected into CarD for chromatographic purification. The CarD was subsequently fractionated by CC using Sephadex LH‐20 as the stationary phase and methanol as the mobile phase. Fractions with higher concentrations of targeted peaks in the flavonoid UV pattern were monitored using an HPLC–DAD system to prepare for the isolation of flavolignan derivatives 2 and 3. This was performed in a semi‐preparative HPLC–DAD system with mobile phases (A) H2O (0.1% acetic acid) and (B) ACN, flowing at 1.0 mL/min. The gradient method was as follows: 0–5 min, 10%–25% B; 5–10 min, 25%–31% B; 10–20 min, 31%–37.5% B; and 20–26 min, 37.5%–100% B. Under these conditions, three bands/peaks were collected at 356 nm detection.
Structural Identification
Tricin (1)—DMSO‐d61H‐NMR 300 MHz, δH: 6.87 (s, 1H, H‐3), 6.08 (d, 1.5 Hz, 1H, H‐6), 6.41 (d, 1.5 Hz, 1H, H‐8), 7.29 (s, 2H, H‐2′/6′), 3.87 (s, 6H, H‐7′). 13C‐NMR 75 MHz, δC: 165.0 (C‐2), 102.6 (C‐3), 181.2 (C‐4), 163.2 (C‐5), 99.5 (C‐6), 164.6 (C‐7), 94.6 (C‐8), 157.6 (C‐9), 103.2 (C‐10), 120.0 (C‐1′), 104.6 (C‐2′/6′), 148.3 (C‐3′/5′), 132.2 (C‐4), 56.3 (C‐7′) [ref. 35].
Tricin‐4″‐O‐(threo‐guaiacylglyceryl) ether, salcolin A (2)—DMSO‐d61H‐NMR 300 MHz, δH: 7.0 (s, 1H, H‐3), 6.18 (d, 1.4 Hz, 1H, H‐6), 6.52 (d, 1.4 Hz, 1H, H‐8), 7.29 (s, 2H, H‐2′/6′), 3.87 (s, 6H, H‐7′), 6.93 (d, 1.4 Hz, 1H, H‐1″), 6.69 (d, 8.5 Hz, 1H, H‐5″), 6.75 (dd, 8.5, 1.4 Hz, 1H, H‐6″), 4.79 (d, 4.7 Hz, 1H, H‐7″), 4.34 (dd, 8.1, 4.7 Hz, 1H, H‐8″), 3.74 (dd, 12.0, 8.1 Hz, 1H, H‐9″), 3.51 (dd, 12.0, 8.1 Hz, 1H, H‐9″), 3.74 (s, 3H, H‐10″). 13C‐NMR 75 MHz, δC: 165.0 (C‐2), 103.4 (C‐3), 181.7 (C‐4), 161.4 (C‐5), 99.5 (C‐6), 162.9 (C‐7), 94.6 (C‐8), 157.5 (C‐9), 104.2 (C‐10), 119.4 (C‐1′), 104.7 (C‐2′/6′), 147.0 (C‐3′/5′), 133.2 (C‐4), 56.4 (C‐7′), 133.2 (C‐1″), 110.8 (C‐2″), 147.0 (C‐3″), 145.4 (C‐4″), 114.6 (C‐5″), 119.6 (C‐6″), 72.0 (C‐7″), 86.3 (C‐8″), 60.2 (C‐9″), 55.5 (C‐10″) [ref. 36].
Tricin‐4″‐O‐(erythro‐guaiacylglyceryl) ether, salcolin B (3)—DMSO‐d61H‐NMR 300 MHz, δH: 6.87 (s, 1H, H‐3), 6.12 (d, 1.0 Hz, 1H, H‐6), 6.46 (d, 1.0 Hz, 1H, H‐8), 7.29 (s, 2H, H‐2′/6′), 3.85 (s, 6H, H‐7′), 6.98 (d, 1.6 Hz, 1H, H‐2″), 6.69 (d, 8.0 Hz, 1H, H‐5″), 6.79 (dd, 8.1, 1.6 Hz, 1H, H‐6″), 4.84 (d, 4.3 Hz, 1H, H‐7″), 4.24 (dd, 9.5, 4.4 Hz, 1H, H‐8″), 3.80 (dd, 12.0, 9.5 Hz, 1H, H‐9″), 3.64 (dd, 12.0, 4.4 Hz, 1H, H‐9″), 3.85 (s, 3H, H‐10″). 13C‐NMR 75 MHz, δC: 165.0 (C‐2), 103.4 (C‐3), 181.7 (C‐4), 161.4 (C‐5), 99.5 (C‐6), 162.9 (C‐7), 94.6 (C‐8), 157.5 (C‐9), 104.2 (C‐10), 119.4 (C‐1′), 104.7 (C‐2′/6′), 147.0 (C‐3′/5′), 133.2 (C‐4), 56.4 (C‐7′), 133.5 (C‐1″), 111.7 (C‐2″), 148.7 (C‐3″), 147.2 (C‐4″), 115.9 (C‐5″), 120.9 (C‐6″), 74.4 (C‐7″), 88.8 (C‐8″), 62.0 (C‐9″), 56.4 (C‐10″) [ref. 36].
Cell Culture
Two thyroid cancer cell lines were used to assess the antitumor activity of the compounds. HTH83 is derived from ATC, whereas TPC‐1 originates from PTC, each harboring different gene mutations. Both were grown in RPMI medium supplemented with 10% fetal bovine serum at 37°C with 5% CO2 [ref. 37]. The medium was changed every 48 h, and when necessary, cells were detached using Trypsin‐EDTA (0.25%) (Gibco) when needed. Prof. Edna Kimura kindly donated the cell lines from the University of São Paulo.
IC50 Determination
The concentration that reduces cell viability to 50% (IC50) was determined after 72 h of treatment using PrestoBlue Cell Viability Reagent (Thermo Fisher Scientific), following the manufacturer’s instructions. Briefly, cell lines were seeded into 96‐well plates at an initial density of 3000 cells and incubated at 37°C with 5% CO2 for 24 h. Cells were then treated with compounds 1–3 in a serial dilution starting at 250 µM. After 72 h of treatment, 10% of PrestoBlue reagent was added to each well. Plates were incubated for 1 h and 30 min at 37°C with 5% CO2, protected from light. Fluorescence (540 nm excitation, 590 nm emission) was measured using a microplate reader M3 (molecular devices). The relative luminescence units of treated cells were normalized to the fluorescence of vehicle‐treated control cells (DMSO) and expressed as a percentage of viable cells. IC50 was determined from a nonlinear regression analysis of the dose–response curve using Prism 5 (GraphPad software).
Clonogenic Assay
To evaluate colony formation after treatment, 250 cells were seeded in a 6‐well plate and incubated for 24 h at 37°C with 5% CO2. Next, the cells were treated with the IC50 of compounds 1–3 for 72 h, then cultured for more than 10–15 days, with media changed twice a week without treatment. Each well was then washed with 1 mL of PBS, fixed with a methanol (33%) and acetic acid (33%) solution for 10 min at 4°C, and stained with 0.5 mL of 1% crystal violet for another 10 min at room temperature. Excess crystal violet was washed off with distilled water, and plates were allowed to dry at room temperature. The number of colonies was counted manually, and the treated groups were normalized against the number of colonies in the DMSO‐treated group.
Annexin‐V Assay
Annexin‐V‐FITC/Propidium Iodide (PI) kit (Thermo Fisher Scientific) was used to investigate which cell death mechanism could be activated after treatments. Briefly, 2 × 105 cells were seeded into a 6‐well plate and incubated for 24 h. Cells were treated with the IC50 of compounds 1–3 for 48 h. Cells were detached with trypsin‐EDTA (0.25%) (Gibco), and 1 × 105 cells were incubated for 30 min, in the dark, at 37°C with Annexin‐V‐FITC (0.2 mg/mL) and PI (0.05 µg). Then, samples were analyzed in a flow cytometer (detection at 488 and 617 nm wavelengths) (Becton Dickinson, FACSCalibur).
Results
Phytochemical Characterization
The dichloromethane phase obtained from the liquid–liquid partition of methanolic extracts (CarD) was analyzed by UPLC–HRMS to determine the chemical profile. The DAD chromatogram of the dichloromethane fraction showed multiple UV spectra indicating the flavone backbone chromophore (λ max 270, 330). The mass spectra confirmed the presence of flavone tricin and its derivatives, based on HRMS/MS fragments of positive ion m/z 331. This flavonoid was previously identified in the dichloromethane phase of C. arborea leaves by our group [ref. 24]. Two tricin derivative ions were detected (m/z 527) in the CarD sample using HR‐ESI positive mode, as well as in negative mode (m/z 525), suggesting the structures of two tricin‐4′‐O‐guaiacylglyceryl ethers according to the literature [ref. 36]. Accordingly, the CarD was subjected to semi‐preparative purification via HPLC–DAD to isolate the flavolignans for cytotoxic assays.
Dereplication
The chemical profile of the CarD was organized using CMN, and dereplication was performed on the basis of ESI‐(+)‐HRMS/MS analysis and GNPS. A total of 39 annotations were organized, including 29 spectral annotations from the GNPS dereplication workflow and 10 spectral annotations based on the CMN spectral organization, through MS/MS analysis of correlated cluster spectra (Figure 1, Table 1).
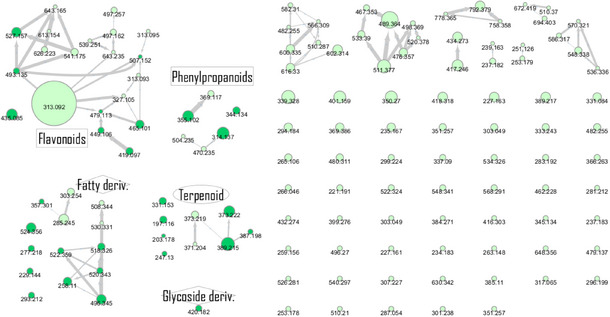
TABLE 1: Chemical profile of dichloromethane fraction from leaves of Casearia arborea.
| Code | RT (s) | Annotation | Parent mass (m/z)Ion formulaAdduct | Cosine (GNPS) | Error (ppm) | HRMS/MS CID |
|---|---|---|---|---|---|---|
| A1 | 52.8 | Choline alfoscerate | 258.110C8H21NO6P+ [M + H]+ | 0.93 | 0 | 104.11184.07125.00 |
| A2 | 204.8 | 4‐O‐β‐d‐glucosyl‐4‐coumaric acid | 344.134C15H22NO8 + [M + NH4]+ | 0.89 | 0 | 147.04165.0585.03127.04 |
| A3 | 224.3 | Chlorogenic acid | 355.102C16H19O9 + [M + H]+ | 0.98 | 0 | 163.04181.05 |
| A4 | 239.5 | Icariside F2 | 420.182C18H30NO10 + [M + NH4]+ | 0.81 | 9.5 | 115.04133.0591.05 |
| A5 | 257.7 | Corchoionoside C | 387.198C19H33O7 + [M + H]+ | 0.66 | 7.7 | 209.15 (207)149.09191.14123.09 (125)135.1195.08 |
| A6 | 285.0 | Icariside B5 | 389.215C19H33O8 + [M + H]+ | 0.81 | 2.6 | 209.15149.10 (151)191.14125.09227.16 |
| A7 | 309.0 | Chlorogenic acid methyl ester | 369.117C17H21O9 + [M + H]+ | — | 2Calcd.369.1186 | 163.04181.05 |
| A8 | 330.1 | Loliolide | 197.116C11H17O3 + [M + H]+ | 0.78 | 5.1 | 133.10 (135)107.08161.09 |
| A9 | 338.3 | Isoquercetin | 465.101C21H21O12 + [M + H]+ | 0.86 | 2.4 | 303.05153.13 |
| A10 | 350.6 | (6R,9S)‐3‐oxo‐α‐ionol‐β‐d‐glucoside (from PubChem) | 373.219C19H31O7 + [M + H]+ | — | 35.0Calcd.371.2064 | 133.10 (135, 137)193.16 (191)211.17 (207, 209)151.04109.10 |
| A11 | 363.1 | Quercetin‐3‐O‐pentoside | 435.085C20H19O11 + [M + H]+ | 0.92 | 0 | 303.05237.18 |
| A12 | 365.0 | Byzantionoside B isomer | 373.219C19H33O7 + [M + H]+ | — | 8.0Calcd.373.2221 | 133.10 (135, 137)191.14209.15151.11109.10 |
| A13 | 377.2 | Byzantionoside B or Blumenol‐C‐glucoside | 373.222C19H33O7 + [M + H]+ | 0.91 | 0 | 211.17193.16135.12109.10175.15119.09 |
| A14 | 408.2 | Benzoic acid derivative | 313.092[M + H]+ | — | — | 151.04 |
| A15 | 419.7 | Cyanidin‐3‐O‐α‐arabinoside | 419.097C20H19O10 + M+ | 0.90 | 0 | 287.05 |
| A16 | 463.0 | Quercetin‐3‐O‐methoxy 3′‐O‐β‐d‐xylopyranose | 449.106C21H21O11 + [M + H]+ | 0.87 | 4.5 | 317.06 |
| A17 | 468.0 | 1,6‐di‐Acetyl‐furannoeremophilone | 331.153C19H23O5 + [M + H − H2O]+ | 0.62 | 0 | 151.07 (149)137.06285.11 (287)255.10227.10189.09 |
| A18 | 479.2 | Abscisic acid | 247.130C15H19O3 + [M + H − H2O]+ | 0.60 | 12.1 | 187.11201.13121.07 (119) |
| A19 | 506.7 | Feruloyltyramine | 314.137C18H20NO4 + [M + H]+ | 0.96 | 6.4 | 177.05121.07145.03151.04 |
| A20 | 550.1 | Isorhamnetin 3‐O‐glucoside | 479.113C22H23O12 + [M + H]+ | 0.89 | 10.4 | 317.06 |
| A21 | 573.5 | Malvidin 3‐O‐galactoside | 493.135C23H25O12 + M+ | 0.84 | 4.1 | 331.08151.04 |
| A22 | 588.1 | Phenylpropanoid derivativeSimilar fragments of feruloyltyramine compound | 626.223[M + H]+ | — | — | 314.14177.05151.04 |
| A23 | 620.2 | Feruloyltyramine derivative | 504.235[M + H]+ | — | — | 177.05151.04251.12328.19 |
| A24 | 622.8 | Traumatic acid | 229.144C12H21O4 + [M + H]+ | 0.72 | 2.1 | 147.12165.13123.12175.11 |
| A25 | 646.4 | β‐Caryophyllene oxide | 203.178C15H23 + [M + H − H2O]+ | 0.73 | 9.8 | 119.0995.09105.07 (107)81.07 |
| A26 | 655.6 | Malvidin derivative | 643.165C31H31O15 + [M + H]+ | — | 1.0Calcd.643.1657 | 331.08151.04 |
| A27 | 656.9 | Peonidin derivative | 613.154C30H29O14 + [M + H]+ | — | 2Calcd.613.1552 | 301.07151.04 |
| A28 | 676.2 | Quercetin‐3‐O‐hexoside‐7,3′,4′‐trimethyl ether | 507.152C24H27O12 + [M + H]+ | 0.76 | 5.9 | 345.10151.04 |
| A29 | 676.4 | Feruloyltyramine derivative(+C6H12N4O) | 470.235C24H32N5O5 + [M + H]+ | — | 11.3Calcd.470.2398 | 177.05294.19 |
| A30 | 716.0 | Salcolin or tritin‐4′‐O‐guaiacyglyceryl ether | 527.157C27H27O11 + [M + H]+ | 0.83 | 1.8 | 331.08 |
| A31 | 747.7 | 9,12,13‐Trihydroxy‐10,15‐octadecadienoic acid(9,12,13‐TriHODE) | 293.212C18H29O3 + [M + H − 2H2O]+ | 0.61 | 3.4 | 107.08147.0281.06151.04 |
| A32 | 761.5 | Tricin‐4′‐O‐veratrylglyceryl ether | 541.175C27H27O11 + [M + H]+ | — | 37.0Calcd.541.1548 | 331.08 |
| A33 | 846.1 | LPC (18:3/0:0) or γ‐Linoleate phosphotidylcholine | 518.326C26H49NO7P+ [M + H]+ | 0.86 | 2.6 | 184.07104.11 |
| A34 | 871.1 | 13‐Keto‐9Z,11E‐octadecadienoic | 277.218C18H29O2 + [M + H − H2O]+ | 0.68 | 10.8 | 149.07135.12121.1093.07 (95) |
| A35 | 875.1 | LPC (18:2/0:0) | 520.343C18H29O2 + [M + H]+ | 0.89 | 5.2 | 184.07104.11 |
| A36 | 896.5 | PC (0:0/16:0) | 496.345C24H51NO7P+ [M + H]+ | 0.89 | 9.5 | 184.07104.11151.04 |
| A37 | 897.5 | Monoolein | 357.301C21H39O4 + [M + H]+ | 0.71 | 2.8 | 95.09 (97)109.10135.1283.08 |
| A38 | 907.5 | 1‐Oleoyl‐sn‐glycero‐3‐phosphocholine | 522.359C26H51NO7P+ [M + H]+ | 0.78 | 11.4 | 184.07104.11283.26 |
| A39 | 950.1 | 1‐Stearoyl‐sn‐glycero‐3‐phosphocholine | 524.356C26H53NO7P+ [M + H]+ | 0.60 | 24.8 | 313.28184.07239.24104.11 |
Note: Spectral annotations of Global Natural Products Social Molecular Networking (GNPS) library hits based on UPLC‐ESI‐(+)‐HRMS‐MS2 results of CarD sample analysis.
The dichloromethane phase CarD showed a polar profile with shikimate derivative compounds, as indicated by spectral annotations of two benzoic compounds (A4 and A14), phenylpropanoids (A2, A3, A7, A19, A22, A23, and A29) (Supporting Information S1), and flavonoids (A9, A11, A15, A16, A20, A21, A26, A27, and A28) or flavolignans (A30 and A32) (Supporting Information S2). Apolar compounds, including terpene derivatives such as one carotenoid (A8), one eremophilane (A17), one sesquiterpenoid (A25), and six megastimane derivatives (A5, A6, A10, A12, A13, and A18), were also identified (Supporting Information S3). Additionally, the apolar compounds in the CarD fraction included fatty acid derivatives, annotated as A1, A24, A31, and A33–A39 (Supporting Information S4).
Structural Identification of Compounds 1–3 by NMR and HRMS/MS
Compound 1 was identified as tricin by HR‐ESI‐(+) with m/z 331.0800 and NMR data. Additionally, the structure of tricin in flavolignans 2 (CarD‐9/2) and 3 (CarD‐9/3) was confirmed by 1H and 13C‐NMR according to the literature [ref. 36]. Compounds 2 and 3 showed m/z 527.1572 and 527.1581, respectively, consistent with the [M + H]+ adduct and the molecular formula C27H26O11 (exact mass 527.1553, Supporting Information S5). The negative mode further verified the tricin derivative, with [M–H]− matching m/z 525.1439 (same molecular formula, C27H26O11). HR‐ESI‐(−)‐MS/MS analysis revealed the guaiacyl fragment, based on the proposed fragmentation mechanism (Supporting Information S6). The fragment at m/z 329.0653 in the negative mode spectrum corresponds to the tricin anion, indicating a guaiacyl structure (m 196), confirmed by the negatively charged fragment at m/z 195.0657.
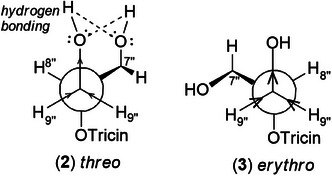
To confirm the flavolignan structures, compounds 2 and 3 were analyzed using 1H and 13C‐NMR spectra, showing tricin chemical shifts along with signals assigned to the coniferyl acid derivative through HSQC spectra. The aromatic region includes δ H‐2″ 6.9/δ C‐2″ 110.8, δ H‐5″ 6.7/δ C‐5″ 114.6, and δ H‐6″ 6.75/δ C‐6″ 119.2. Carbinolic carbons appear at δ H‐7″ 4.8/δ C‐7″ 72.0, δ H‐8″ 4.3/δ C‐8″ 86.3, and δ H‐9″ 3.5 and 3.7/δ C‐9″ 60, along with one aromatic methoxyl group at δ H‐10″ 3.7/δ C‐10″ 55.3. The 4′‐O‐linkage was determined via NOESY spectra (Supporting Information S7), where proton couplings between methoxyl hydrogen of tricin (H‐7′) and guaiacyl (H‐7″, H‐8″, H‐9″) were observed at δ H‐7′ 4.0/δ H‐7″ 5.0, δ H‐7′ 4.0/δ H‐8″ 3.9, and δ H‐7′ 4.0/δ H‐9″ 3.9. Compound 3 (erythro) displayed a slightly deshielded H‐9″ compared to 2 (threo), according to the literature [ref. 36]. Furthermore, the Newman projection of both diastereoisomers helps visualize the relative positions of hydroxyl groups in each configuration. In compound 2 (threo), the hydroxyl groups are in the same spatial orientation, enabling hydrogen bonding that minimizes the inductive effect on H‐9″ and H‐7″. Conversely, compound 3 (erythro) has the 7″‐OH near H‐9″, but no hydrogen bond occurs with 9″‐OH. The higher inductive effect of 9″‐OH, due to the absence of hydrogen bonding with 7″‐OH, results in deshielding of H‐9″ (Figure 2).
Cytotoxic Potential for Compounds 1–3
Tricin (1), salcolin A (2), and salcolin B (3) were tested against thyroid cancer cell lines (Table 2) and shown to decrease cell viability (Figure 3). The IC50 values for each compound in each cell line are listed in Table 3. Tricin (1) had similar IC50 values in HTH83 and TPC1 (177.5 and 187.0 µM, respectively); however, lower concentrations of compounds 2 and 3 were needed to reduce cell viability by 50% in both cell lines (66.69 and 84.98 µM for HTH83, respectively; 56.12 and 56.5 µM for TPC1, respectively).
TABLE 2: Cell lines used in the study and the harboring mutations.
| Cell line | Type | Mutations |
|---|---|---|
| HTH83 | ATC | AR p.G456_G457insG; HRASQ61R; TERT c.228C>T; TP53 p.P154Afs*28 |
| TPC1 | PTC | RET/PTC1; CDKN2A p.A68fs; STAG2Q1089X; TERT c.228C>T |
Abbreviations: ATC, anaplastic thyroid cancer; PTC, papillary thyroid cancer.
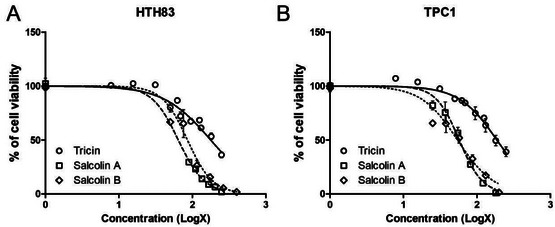
TABLE 3: IC50 (µM) determined for HTH83 (anaplastic thyroid carcinoma cells—ATC) and TPC‐1 (papillary cancer cells—PTC).
| Compounds | HTH‐83 | TPC‐1 |
|---|---|---|
| Tricin (1) | 177.5 | 187.0 |
| Salcolin A (2) | 66.69 | 56.12 |
| Salcolin B (3) | 84.98 | 56.5 |
Note: The IC50 value was calculated using non‐linear regression in GraphPad Prism 5.0.
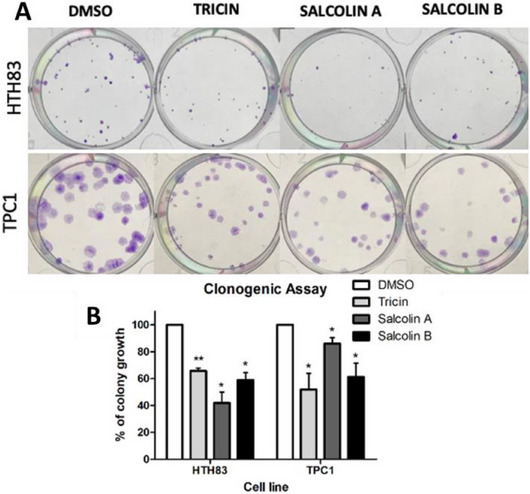
To confirm the reduction in cell viability, clonogenic assays were performed using the IC50 concentration of each compound to treat both cell lines (Figure 4). Compound 1 decreased colony growth by 34.58% in HTH83 and 48.04% in TPC1 (p < 0.01 and p < 0.05). Compound 2 reduced colony growth by 58.05% in HTH83 and 13.97% in TPC1 (p < 0.05), whereas Compound 3 decreased it by 41.09% in HTH83 and 38.68% in TPC1 (p < 0.05). Similar results were reported for tricin in other cancer histology samples. A reduction in proliferation, colony growth, and cell cycle arrest was observed in a breast cancer cell line [ref. 38]. In a colon cancer cell line, treatment with tricin lowered the number of colonies [ref. 39].
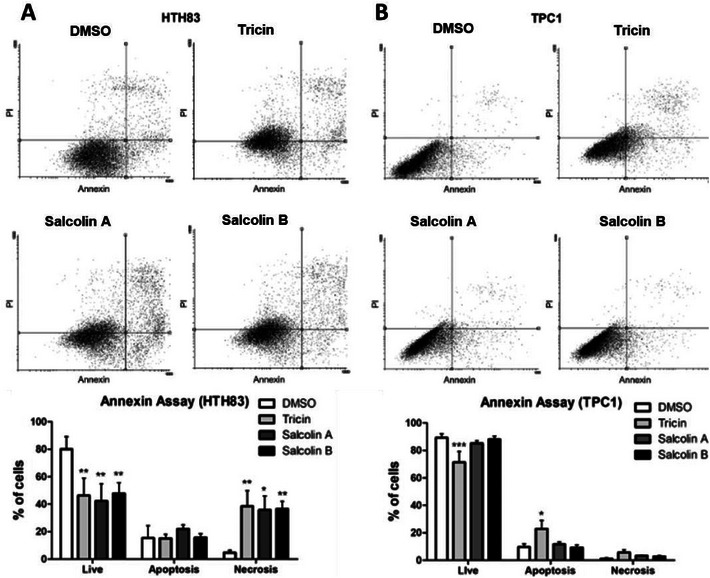
To better understand that cell death mechanism might be activated after flavone treatments, Annexin‐V assays were conducted. Treatment with compounds 1–3 reduced the percentage of live cells (cells annexin−/PI−) by 33.73%, 37.67%, and 32.23%, respectively, in HTH83 (p < 0.01) (Figure 5A), whereas the percentage of necrotic cells (cells annexin−/PI+) increased to 31.83%, 31.09%, and 33.89%, respectively (p < 0.01, p < 0.05, and p < 0.01) (Figure 5A). Compound 1 was the only compound that reduced the percentage of live cells in TPC1 (17.81%, p < 0.001), and it increased the percentage of apoptotic cells (cells annexin+/PI− and annexin+/PI+) by 13.29% (p < 0.05) (Figure 5B).
Discussion
The chemical diversity of the dichloromethane fraction from leaves of C. arborea is similar to C. sylvestris var. lingua [ref. 25], presenting flavonoid‐3‐O‐glycosides, phenylpropanoids, and flavolignans. Our group has also identified flavone‐3‐O‐glycosides in the ethyl acetate fraction from C. arborea [ref. 23]. Here, we report the chemical profile of the dichloromethane phase from leaves of C. arborea methanolic extracts, including the presence of tricin and rare flavolignans presenting 4′‐O‐8″‐guaiacylglyceryl derivatives. The free occurrence of tricin may be linked to enzymatic hydrolysis of tricin derivatives during extraction [ref. 40], as seen in the extraction of tricin from bamboo, palms, sugarcane, grains, seeds [ref. 41], and plant juice [ref. 42]. The first natural tricin flavolignan derivative was reported from Aegilops ovata, named aegicin (4′‐O‐p‐coumaroylglyceryl ether), which inhibits plant germination [ref. 43]. Two flavolignans, salcolins A and B, were isolated from Salsola collina, with tricin detected in large amounts. These compounds were obtained with threo and erythro configurations of tricin‐4′‐O‐guaiacylglyceryl ether [ref. 44]. Tricin and its derivatives are common among many monocotyledonous plants, such as Gramineae [ref. 45] and Cyperaceae [ref. 46, ref. 47]. Tricin has biological activities related to plant protection. Its accumulation boosts wheat seedlings treated with herbicide safener [ref. 48], and it has demonstrated antibacterial, antifungal, and insecticidal properties [ref. 49]. Tricin and its derivatives also exhibit antioxidant, anti‐plant‐hopper, anti‐weed, and anti‐herbicide effects, act as gene inducers, protect against biotic and abiotic stresses, and show allelopathic activity [ref. 50, ref. 51].
Tricin and its derivatives have pharmaceutical potential due to their antioxidant activity, ability to act as anti‐inflammatory agents, inhibit exocytosis from antigen‐stimulated rat leukemia basophils and hepatitis B, serve as chemopreventive agents against intestinal carcinogenesis, and inhibit breast cancer cells, among other applications [ref. 50]. They inhibit P‐glycoprotein activity in adriamycin‐resistant human breast cancer cells, delay spontaneous mammary tumor development, and suppress oxidative stress‐induced apoptosis [ref. 52]. Tricin exhibits anti‐clonogenic activity in vitro and has been reported for use in treating human‐derived breast (MDA‐MB‐468) and colon carcinoma (SW‐480) [ref. 39], including antineoplastic properties in vivo against P388 leukemia [ref. 53] and promyelocytic leukemia cells (HL‐60) [ref. 54]. It was shown to promote cell cycle arrest in the MDA‐MB‐486 cell line without inducing apoptosis [ref. 38]. In glioma cells (C6), tricin inhibited proliferation and invasion [ref. 55]. In Lewis lung carcinoma, tricin inhibited proliferation and migration and induced apoptosis [ref. 56]. Tricin improved docetaxel efficacy in prostate cancer cells (PCSs) [ref. 57].
Our results show that tricin inhibits the growth of thyroid cancer cell lines. Different results were observed in the two studied cell lines. In the ATC cell line, there is an increased necrotic effect, whereas in the PTC cell line, there is evidence of apoptotic cell death. PTC is the most common type of thyroid cancer and generally has a good prognosis [ref. 58]. Approximately 70% of PTCs have activation of the MAPK pathway due to BRAF and RAS mutations, as well as RET fusions, which promote cancer progression [ref. 59]. TPC1, a PTC cell line, contains the RET/PTC1 rearrangement, which affects several transcription factors involved in cell proliferation, differentiation, and apoptosis [ref. 60]. Cancer cells with RET/PTC1 rearrangements rely more on the RAF‐1 pathway for survival and growth than on the BRAF‐ERK pathway, which is typically needed in cells with RAS or BRAF mutations [ref. 61]. ATC is rare and the most aggressive form of thyroid cancer (accounting for 1%–2% of cases), with a survival time ranging from 0.5 to 3 years [ref. 62]. HTH83 is a cell line derived from ATC that contains the H‐Ras Q61R mutation, which is associated with the MEK‐ERK and PI3K/AKT pathways, both involved in cancer progression [ref. 63]. We hypothesize that the different mutation profiles of HTH83 and TPC1 influence their responses to tricin in this study. Tricin successfully reduced the number of live cells and increased apoptotic cells in TPC1, BRAF‐mutated cells. However, it did not induce significant apoptosis in HTH83, instead causing only necrosis in this cell line harboring the H‐RAS mutation. These findings prompt us to consider whether flavolignans induce cell death or simply decrease cell viability in TPC1.
In an in vivo experiment, Cai et al. [ref. 64] reported a controlled diet, including tricin, in mice. The consumption of food supplemented with tricin reduced the number of intestinal adenomas and the mechanisms by which tricin inhibits or downregulates the COX‐2 enzyme and decreases PGE2 production. The potential of flavonoids for prevention and chemoprevention is well known [ref. 65, ref. 66, ref. 67, ref. 68]. Seki et al. [ref. 69] suggest tricin as a therapeutic prototype for hepatic fibrogenesis in in vivo assays. The flavone inhibits PDGF‐induced proliferation of HSCs in vitro by blocking PDGF‐R phosphorylation and downstream signaling pathways rather than by competing for PDGF receptor binding. An acylated tricin glucoside demonstrated antioxidant and antiproliferative activities against breast (MCF‐7, NCI‐ADR), prostate (PC3), ovary (OVCAR‐03), non‐small cell lung (NCI460), and colon cancer cell lines (HT‐29). In melanoma (UACC‐62) and kidney cell lines (786‐0), activity was only observed at high concentrations [ref. 70]. The compound 5,7,3′,4′,5′‐pentamethoxyflavone (PMF) contains methoxy groups, which appear to enhance metabolic stability, resulting in slower removal compared to tricin [ref. 71].
Mohanlal et al. [ref. 72] first reported the cytotoxicity of salcolins A and B in vitro against HCT 116 (colon), SKOV3 (ovary), and MCF‐7 (breast) human cancer cell lines. Apoptosis was observed and confirmed by the decreased mitochondrial membrane potential. Another activity of salcolin B was a dose‐dependent inhibition of nitric oxide production in macrophage cell lines without evidence of cytotoxicity [ref. 73], along with higher anti‐inflammatory and anti‐allergy activities of tricin derivatives, including salcolins A and B, compared to tricin [ref. 74], and neuroprotective effects [ref. 75]. Our results show that for ATC, both salcolins A and B induced necrotic cell death at the same levels as tricin but at lower concentrations. In the PTC cell line, salcolins A and B did not significantly induce apoptosis or necrosis, unlike tricin. These findings highlight the influence of cell genotype on the biological activity of the flavolignan derivatives.
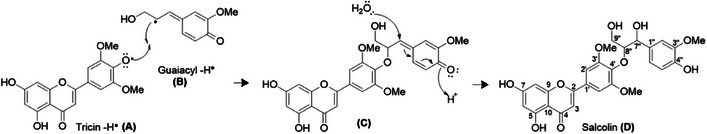
Regarding the structural identification of flavolignan tricin derivatives, Yan et al. [ref. 76] reported the identification of lycopodone, a rare compound with no activity against A549 and K562 tumor cell lines. Still, tricin showed moderate cytotoxic activity against K562. Yan et al. used HMBC projection to conclude that the 2D‐NMR flavolignan coupling is only visible in ROESY or NOESY experiments [ref. 36, ref. 77]. We also used NOESY to observe the correlation of the 4′‐O‐linkage, defined in proton couplings between methoxyl hydrogen of the B ring of tricin (H‐7′) and guaiacyl (H‐7″, H‐8″, H‐9″), to confirm the tricin‐4′‐O‐8″‐guaiacylglyceryl linkage in the salcolin diastereoisomers, which is different from the lycopodone reported by Yan et al.
The tricin‐4′‐O‐7″‐guaiacylglyceryl linkage in lycopodone initially appears incorrect because the preferred biogenesis of the tricin‐4′‐O‐guaiacylglyceryl linkage occurs at C‐8″ of guaiacol glyceryl (Figure 6), unless the radical is absent at C‐7″, as confirmed by the resonance structures of the delocalized electrons in the guaiacyl ℼ system.
Conclusions
In conclusion, we demonstrated that tricin, salcolin A, and salcolin B induce cell death in ATC cell lines by increasing necrosis, and their activity may depend on the origin of the thyroid cell lines. The phytochemical analysis of the dichloromethane phase revealed a chemical profile similar to other species of Casearia, suggesting that the shikimate biosynthesis pathway is prominently present.
Author Contributions
Augusto L. Santos: conceptualization, data curation, formal analysis, investigation, methodology, software, writing original draft. Mariana T. Rodrigues: conceptualization, data curation, formal analysis, investigation, methodology, software. Ana Paula Michelli: data curation, formal analysis, investigation, methodology, software. Rodrigo E. Tamura: formal analysis, methodology, writing original draft. Ileana G. S. de Rubió: formal analysis, methodology, conceptualization. Marisi G. Soares: data curation. Marcelo J. P. Ferreira: formal analysis, methodology, funding acquisition, review and editing. Patricia Sartorelli: conceptualization, funding acquisition, project administration, visualization, resources, review and editing, supervision.
Conflicts of Interest
The authors declare no conflicts of interest.
Supplementary Materials
References
- Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries,”. CA: A Cancer Journal for Clinicians, 2018. [DOI | PubMed]
- Estimating the Global Cancer Incidence and Mortality in 2018: GLOBOCAN Sources and Methods,”. International Journal of Cancer, 2019. [DOI | PubMed]
- 2015 American Thyroid Association Management Guidelines for Adult Patients With Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer,”. Thyroid: Official Journal of the American Thyroid Association, 2015. [DOI]
- Anaplastic Carcinoma of the Thyroid: A 24‐Year Experience,”. Head & Neck, 1995. [DOI | PubMed]
- Evaluation of Overall Survival in Patients with Anaplastic Thyroid Carcinoma, 2000–2019,”. JAMA Oncology, 2020. [DOI | PubMed]
- Natural Products as Sources of New Drugs Over the Nearly Four Decades From 01/1981 to 09/2019,”. Journal of Natural Products, 2020. [DOI | PubMed]
- O Brasil no Contexto de Controle de Qualidade de Plantas Medicinais,”. Revista Brasileira De Farmacognosia, 2010. [DOI]
- Biodiversity Hotspots for Conservation Priorities,”. Nature, 2000. [DOI | PubMed]
- A Hot‐Point Within a Hot‐Spot: A High Diversity Site in Brazil’s Atlantic Forest,”. Biodiversity and Conservation, 2007. [DOI]
- GBIF Backbone Taxonomy 2023 (. 2023. [DOI]
- Species in Casearia (. 2013
- 12 D. J. Mabberley , Mabberley’s Plant‐Book, 4th ed. (Cambridge University Press, 2017), 10.1017/9781316335581.
- 13 H. O. Sleumer , Flora Neotropica Monograph Number 22 ‐ Flacourtiaceae (The New York Botanic Garden, 1980).
- 14 H. Lorenzi , Árvores Brasileiras: Manual de Identificação e Cultivo de Plantas Arbóreas Nativas do Brasil, 2nd ed. (Platarum, 1998).
- 15 R. Marquete and E. Medeiros , Salicaceae in Flora e Funga do Brasil (Jardim Botânico do Rio de Janeiro, 2022).
- Cytotoxic Clerodane Diterpenoids From the Leaves and Twigs of Casearia balansae ,”. Journal of Natural Products, 2013. [DOI | PubMed]
- Discovery of Tricyclic Clerodane Diterpenes as Sarco/Endoplasmic Reticulum Ca2+‐ATPase Inhibitors and Structure–Activity Relationships,”. Journal of Natural Products, 2015. [DOI | PubMed]
- The Genus Casearia: A Phytochemical and Pharmacological Overview,”. Phytochemistry Reviews, 2015. [DOI]
- Chemical Constituents From Casearia spp. (Flacourtiaceae/Salicaceae sensu lato),”. Revista Brasileira De Farmacognosia‐Brazilian Journal of Pharmacognosy, 2017. [DOI]
- Non‐Polar and Polar Chemical Profiling of Six Casearia Species (Salicaceae),”. Biochemical Systematics and Ecology, 2019. [DOI]
- Cytotoxic Clerodane Diterpenoids From the Leaves of Casearia kurzii ,”. Bioorganic Chemistry, 2019. [DOI | PubMed]
- Phytochemistry Investigation of Casearia arborea (Rich.) Urb. (Salicaceae) and Antimicrobial Analysis of Its Diterpene,”. Quimica Nova, 2018. [DOI]
- Identification of Flavonoid‐3‐O‐Glycosides From Leaves of Casearia arborea (Salicaceae) by UHPLC‐DAD‐ESI‐HRMS/MS Combined With Molecular Networking and NMR,”. Phytochemical Analysis, 2021. [DOI | PubMed]
- Antileishmanial Activity and Immunomodulatory Effects of Tricin Isolated From Leaves of Casearia arborea (Salicaceae),”. Chemistry and Biodiversity, 2017. [DOI]
- Flavonoids From Casearia sylvestris Swartz Variety Lingua (Salicaceae),”. Biochemical Systematics and Ecology, 2016. [DOI]
- Sharing and Community Curation of Mass Spectrometry Data With Global Natural Products Social Molecular Networking,”. Nature Biotechnology, 2016. [DOI]
- Metabolômica de Plantas: Métodos E Desafios,”. Química Nova, 2021. [DOI]
- A Molecular Networking Strategy: High‐Throughput Screening and Chemical Analysis of Brazilian Cerrado Plant Extracts Against Cancer Cells,”. Cells, 2021. [DOI | PubMed]
- Reproducible Molecular Networking of Untargeted Mass Spectrometry Data Using GNPS,”. Nature Protocols, 2020. [DOI | PubMed]
- ProteoWizard: Open Source Software for Rapid Proteomics Tools Development,”. Bioinformatics (Oxford, England), 2008. [DOI | PubMed]
- A Cross‐Platform Toolkit for Mass Spectrometry and Proteomics,”. Nature Biotechnology, 2012. [DOI]
- OpenMS: A Flexible Open‐Source Software Platform for Mass Spectrometry Data Analysis,”. Nature Methods, 2016. [DOI | PubMed]
- Clustering Millions of Tandem Mass Spectra,”. Journal of Proteome Research, 2008. [DOI | PubMed]
- Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks,”. Genome Research, 2003. [DOI | PubMed]
- Quiquelignan A–H, Eight New Lignoids From the Rattan Palm Calamus quiquesetinervius and Their Antiradical, Anti‐Inflammatory and Antiplatelet Aggregation Activities,”. Bioorganic & Medicinal Chemistry, 2010. [DOI | PubMed]
- Flavonolignans From Hyparrhenia hirta ,”. Phytochemistry, 2002. [DOI | PubMed]
- MicroRNA Deregulation in Anaplastic Thyroid Cancer Biology,”. International Journal of Endocrinology, 2014. [DOI]
- Growth‐Inhibitory and Cell Cycle‐Arresting Properties of the Rice Bran Constituent Tricin in Human‐Derived Breast Cancer Cells In Vitro and in Nude Mice In Vivo ,”. British Journal of Cancer, 2004. [DOI | PubMed]
- Characterization of Potentially Chemopreventive Phenols in Extracts of Brown Rice That Inhibit the Growth of Human Breast and Colon Cancer Cells,”. Cancer Epidemiology and Prevention Biomarkers, 2000
- 40 T. W. Goodwin , Chemistry and Biochemistry of Plant Pigments (Academic Press, 1965).
- Plant Polyphenols—XII,”. Phytochemistry, 1964. [DOI]
- Nature of a Muscle‐Inhibiting Compound in Lucerne and Its Possible Connexion With Bloat in Cattle,”. Nature, 1949. [DOI]
- A New Flavolignan of Biogenetic Interest From Aegilops ovata L.—Part I,”. Israel Journal of Chemistry, 1977. [DOI]
- Flavonolignans of Salsola collina ,”. Chemistry of Natural Compounds, 1992. [DOI]
- Flavonoid Patterns in Leaves of the Gramineae,”. Biochemical Systematics and Ecology, 1976. [DOI]
- Flavonoids in Leaves and Inflorescences of Australian Cyperaceae,”. Phytochemistry, 1985. [DOI]
- Leaf Flavonoids of Cyperus Species in Egypt,”. Biochemical Systematics and Ecology, 1989. [DOI]
- Selective Disruption of Wheat Secondary Metabolism by Herbicide Safeners,”. Phytochemistry, 2006. [DOI | PubMed]
- Bioactive Flavonoids From Endophyte‐Infected Blue Grass (Poa ampla),”. Journal of Agricultural and Food Chemistry, 1998. [DOI]
- The Occurrence of Tricin and Its Derivatives in Plants,”. Green Chemistry, 2016. [DOI]
- Tricin—A Potential Multifunctional Nutraceutical,”. Phytochemistry Reviews, 2010. [DOI]
- P‐Glycoprotein Inhibitory Activity of Two Phenolic Compounds, (−)‐Syringaresinol and Tricin From Sasa borealis ,”. Chemistry and Biodiversity, 2007. [DOI | PubMed]
- Antitumor Agents. 49. Tricin, Kaempferol‐3‐O‐β‐d‐Glucopyranoside and (+)‐Nortrachelogenin, Antileukemic Principles From Wikstroemia indica,”. Journal of Natural Products, 1981. [DOI | PubMed]
- Flavonolignans and Other Constituents From Lepidium meyenii With Activities in Anti‐Inflammation and Human Cancer Cell Lines,”. Journal of Agricultural and Food Chemistry, 2015. [DOI | PubMed]
- Inhibition of the Proliferation and Invasion of C6 Glioma Cells by Tricin via the Upregulation of Focal‐Adhesion‐Kinase‐Targeting microRNA‐7,”. Journal of Agricultural and Food Chemistry, 2018. [DOI | PubMed]
- Metabolomics and Integrated Network Pharmacology Analysis Reveal Tricin as the Active Anti‐Cancer Component of Weijing Decoction by Suppression of PRKCA and Sphingolipid Signaling,”. Pharmacological Research, 2021. [DOI | PubMed]
- Tricin Isolated From Allium atroviolaceum Potentiated the Effect of Docetaxel on PC3 Cell Proliferation: Role of miR‐21,”. Natural Product Research, 2019. [DOI | PubMed]
- Papillary Thyroid Carcinoma Managed at the Mayo Clinic During Six Decades (1940–1999): Temporal Trends in Initial Therapy and Long‐Term Outcome in 2444 Consecutively Treated Patients,”. World Journal of Surgery, 2002. [DOI | PubMed]
- Biologic and Clinical Perspectives On Thyroid Cancer,”. New England Journal of Medicine, 2016. [DOI | PubMed]
- RET/PTC Rearrangement in Thyroid Tumors,”. Endocrine Pathology, 2002. [DOI | PubMed]
- Synthesis of Analogues of Thyroid Hormones: Nuclear Receptor Modulators,”. Orbital, 2015. [DOI]
- Anaplastic Thyroid Cancer: Molecular Pathogenesis and Emerging Therapies,”. Endocrine‐Related Cancer, 2009. [DOI | PubMed]
- A Historic Perspective and Overview of h‐Ras Structure, Oncogenicity, and Targeting,”. Molecular Cancer Therapeutics, 2020. [DOI | PubMed]
- The Rice Bran Constituent Tricin Potently Inhibits Cyclooxygenase Enzymes and Interferes With Intestinal Carcinogenesis in ApcMin Mice,”. Molecular Cancer Therapeutics, 2005. [DOI | PubMed]
- Intake of Specific Carotenoids and Flavonoids and the Risk of Gastric Cancer in Spain,”. Cancer Causes & Control, 1999. [DOI | PubMed]
- Vegetables, Fruit, and Cancer Prevention,”. Journal of the American Dietetic Association, 1996. [DOI | PubMed]
- Differential Modulation of Cyclooxygenase‐Mediated Prostaglandin Production by the Putative Cancer Chemopreventive Flavonoids Tricin, Apigenin and Quercetin,”. Cancer Chemotherapy and Pharmacology, 2006. [DOI | PubMed]
- Winter Wheat Hull (husk) Is a Valuable Source for Tricin, a Potential Selective Cytotoxic Agent,”. Food Chemistry, 2013. [DOI | PubMed]
- Tricin Inhibits Proliferation of Human Hepatic Stellate Cells In Vitro by Blocking Tyrosine Phosphorylation of PDGF Receptor and Its Signaling Pathways,”. Journal of Cellular Biochemistry, 2012. [DOI | PubMed]
- Antiproliferative and Antioxidant Activities of a Tricin Acylated Glycoside From Sugarcane (Saccharum officinarum) Juice,”. Phytochemistry, 2007. [DOI | PubMed]
- Pharmacokinetics in Mice and Metabolism in Murine and Human Liver Fractions of the Putative Cancer Chemopreventive Agents 3′,4′,5′,5,7‐Pentamethoxyflavone and Tricin (4′,5,7‐trihydroxy‐3′,5′‐Dimethoxyflavone),”. Cancer Chemotherapy and Pharmacology, 2011. [DOI | PubMed]
- Tricin 4′‐O‐(Erythro‐β‐Guaiacylglyceryl) Ether and Tricin 4′‐O‐(Threo‐β‐Guaiacylglyceryl) Ether Isolated From Njavara (Oryza sativa L. var. Njavara), Induce Apoptosis in Multiple Tumor Cells by Mitochondrial Pathway,”. Journal of Natural Medicines, 2013. [DOI | PubMed]
- A New Flavonolignan From the Aerial Parts of Oryza sativa L. Inhibits Nitric Oxide Production in RAW 264.7 Macrophage Cells,”. Journal of Applied Biological Chemistry, 2011. [DOI]
- Tricin Derivatives as Anti‐Inflammatory and Anti‐Allergic Constituents From the Aerial Part of Zizania latifolia ,”. Bioscience, Biotechnology, and Biochemistry, 2015. [DOI | PubMed]
- Lignan and Flavonoids From the Stems of Zea mays and Their Anti‐Inflammatory and Neuroprotective Activities,”. Archives of Pharmacal Research, 2015. [DOI | PubMed]
- A New Flavone From Lycopodium japonicum ,”. Heterocycles, 2005. [DOI]
- Applying Excitation Sculpting to Construct Singly and Doubly Selective 1D NMR Experiments,”. Journal of Magnetic Resonance, 1997. [DOI]