
Natural Product-Inspired Dopamine Receptor Ligands
Abstract
Due to their evolutionary bias as ligands for biologically relevant drug targets, natural products offer a unique opportunity as lead compounds in drug discovery. Given the involvement of dopamine receptors in various physiological and behavioral functions, they are linked to numerous diseases and disorders such as Parkinson’s disease, schizophrenia, and substance use disorders. Consequently, ligands targeting dopamine receptors hold considerable therapeutic and investigative promise. As this perspective will highlight, dopamine receptor targeting natural products play a pivotal role as scaffolds with unique and beneficial pharmacological properties, allowing for natural product-inspired drug design and lead optimization. As such, dopamine receptor targeting natural products still have untapped potential to aid in the treatment of disorders and diseases related to central nervous system (CNS) and peripheral nervous system (PNS) dysfunction.
Affiliations: †Department of Chemistry, Hunter College, City University of New York, 695 Park Avenue, New York, New York 10065, United States; ‡Program in Biochemistry, CUNY Graduate Center, 365 Fifth Avenue, New York, New York 10016, United States; §Program in Chemistry, CUNY Graduate Center, 365 Fifth Avenue, New York, New York 10016, United States
License: © 2024 The Authors. Published by American Chemical Society CC BY 4.0 Permits the broadest form of re-use including for commercial purposes, provided that author attribution and integrity are maintained (https://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.1021/acs.jmedchem.4c00537 | PubMed: 39038276 | PMC: PMC11320586
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (3.9 MB)
Introduction
Isolated from natural sources as byproducts of primary or secondary metabolism, natural products are important conduits in drug discovery as lead compounds.1 The significance of natural products in pharmaceuticals cannot be overstated as over 30% of all FDA-approved new molecular entities (NMEs) are derived directly or inspired from microbial and plant species.2 Optimized by natural selection pressures, natural products are refined to have optimal interactions with biological macromolecules, membrane permeability, biomolecular compatibility, and stability, thus sharing key properties of drug likeness.3,4 A recent analysis revealed that over 80% of CNS agents are derived directly or inspired from natural products, and the scaffolds of just 20 natural products yielded more than 400 clinically approved CNS drugs.5
Characteristically defined by their seven transmembrane domains, G protein-coupled receptors (GPCRs) are a superfamily of evolutionarily related integral membrane proteins. GPCRs are canonically coupled to heterotrimeric G proteins, consisting of three associated protein subunits (α, β, and γ). Activation of GPCRs results in the dissociation of the heterotrimeric G proteins into α-subunit and βγ-complex, further transducing the signal to stimulate the effector systems (which are dependent on the specific type of G protein). It should be noted that other signaling mechanisms (e.g., involving ion channels, receptor tyrosine kinases, or β-arrestins) are now known to exist. GPCRs are a critical drug target due to their implication in a wide range of diseases (including cancers, inflammatory diseases, mental disorders, metabolic disorders, cardiovascular diseases, and sensory disorders). In fact, over 30% of FDA-approved drugs target GPCRs.6
Dopamine receptors are GPCRs that recognize the endogenous, catecholamine dopamine (1, Figure ). The five subtypes of dopamine receptors are categorized into two families based on whether they stimulate cAMP production (the D1-like family: D1R and D5R subtypes) via activating Gαs or Gαolf proteins or attenuate cAMP production (the D2-like family: D2R, D3R, and D4R subtypes) via activating Gαi/o proteins (Table 1).7−9 It should be noted that two splice variants of the D2R occur (D2shortR or D2SR and D2longR or D2LR), where D2LR has a 29-mer peptide insertion in the third intracellular loop. These splice variants are predominantly localized to pre- and postsynaptic membranes, respectively.10
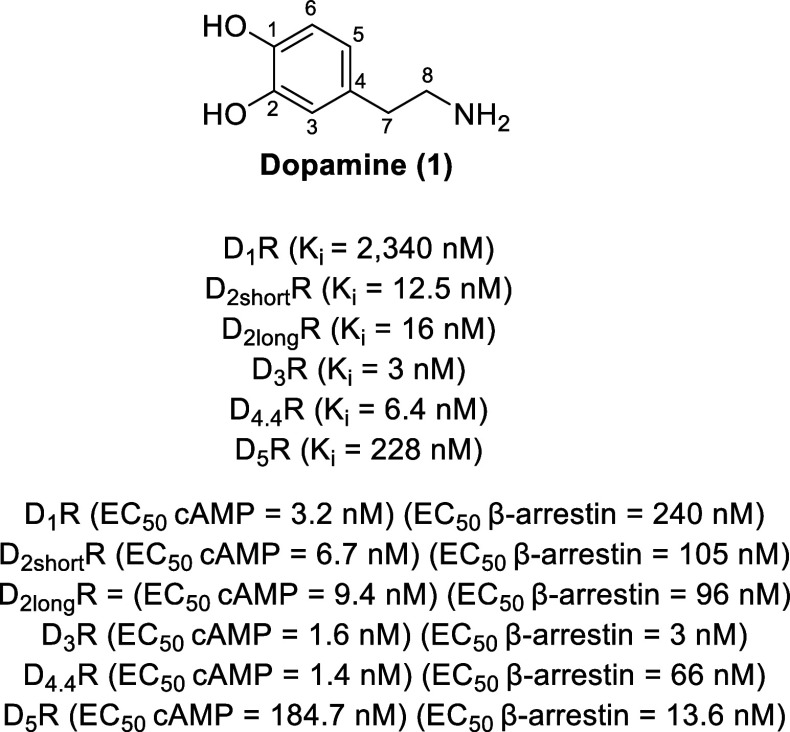
Table 1: Characteristics of Dopamine Receptors
| D1-like family | D2-like family | ||||
|---|---|---|---|---|---|
| D1R | D5R | D2R | D3R | D4R | |
| expression | high levels of expression: mesocortical mesolimbic, and nigrostriatal areas | adrenal glands, blood vessels, cortex, dental gyrus, GI tract, heart, hippocampus, hypothalamus, kidneys, substantia nigra, sympathetic ganglia | high levels of expression: caudate, nucleus accumbens, olfactory bulb putamen, striatum, substantia nigra, tubercle, and ventral tegmental area | hippocampus, hypothalamus, nucleus accumbens, olfactory bulb, striatum, and substantia nigra | adrenal glands, amygdala, blood vessels, frontal cortex, GI tract, heart, hippocampus, hypothalamus, kidneys, mesencephalon, nucleus accumbens, substantia nigra, sympathetic ganglia, and thalamus |
| low levels of expression: cerebellum, hippocampus, hypothalamic areas, kidneys, and thalamus | low levels of expression: adrenal glands, blood vessels, cortex, GI tract, heart, hypothalamus, septum, kidneys, and sympathetic ganglia | ||||
| functions | attention, control of rennin in kidney, impulse control, locomotor activity, regulation of feeding, regulation of memory and learning, reproductive behaviors, reward and reinforcement, sleep | affective functions, endocrine functions, pain process | GI motility, regulation of aldosterone and prolactin secretion, regulation of blood pressure, renal functions, reward and reinforcement, vasodilatations, working memory | cognition, emotions, endocrine functions, regulation of locomotor functions | GI motility, modulations of cognitive functions, regulation of renal functions, vasodilatations, blood pressure, |
| (potential) therapeutic applications | addiction/substance abuse disorder, antihypertensive, obesity, Parkinson’s disease, schizophrenia, Tourette’s syndrome | antipsychotics, depression, Parkinson’s disease, schizophrenia | addiction/substance abuse disorder, Parkinson’s disease, schizophrenia | addiction/substance abuse disorder, ADHD, neuropsychiatric disorders, sexual dysfunction | |
| associated adverse drug reactions | cardiovascular side effects | hyperprolactinemia, metabolic dysfunction | extrapyramidal motor symptoms, hyperprolactinemia, metabolic dysfunction | metabolic dysfunction | |
In addition to canonical signaling via G proteins, dopamine receptors are also known to signal via β-arrestin-based pathways. This (relatively recent) recognition of alternative signal transduction pathways has opened up the possibility to discover ligands that are selective for either G protein or β-arrestin pathways (so-called “functionally selective” or “biased ligands”), leading to different downstream effects.9,11−15 Functional selectivity broadens the conventional definitions of pharmacology to expand the possibility of ligands to act with a mixture of classic characteristics (agonist, antagonist, and/or inverse agonist) depending on the effector pathway.16,17 In other words, functional selectivity describes the ability of a particular ligand to differentially activate distinct subsequent signaling cascades or the selective activation of a specific G protein subtype.18−20 This allows for the potential to mediate divergent processes or the ability to bypass observed adverse effects through the activation of distinct signaling pathways.21−25
As the physiological functions of dopamine are quite extensive (including functions such as reward, sleep regulation, voluntary movement, penile erection, sympathetic regulation, cognitive function, olfaction, and hormonal regulation), dopamine receptors are attractive drug targets.26
At this time, no selective D1R agonist has been commercialized as a CNS-marketed drug. Traditional D1R agonists, such as A-86929 (2, Figure ), dihydrexidine (3), and dinapsoline (4), are characterized by a catechol moiety as typified by dopamine. These ligands have poor CNS penetration and oral bioavailability due to the polarity of the catechol moiety.27,28 Fenoldopam (5), a synthetic benzazepine derivative, is an FDA-approved D1-like agonist, but due to its short duration of action and poor pharmacokinetics, fenoldopam use is limited to emergency medicine as an IV injection in the treatment of hypertensive crisis.29−31 Fenoldopam’s mechanism of action relies on the activation of peripheral D1-like receptors (no blood–brain barrier (BBB) permeability), which results in the reduction of systemic arterial blood pressure through renal vasodilation.32
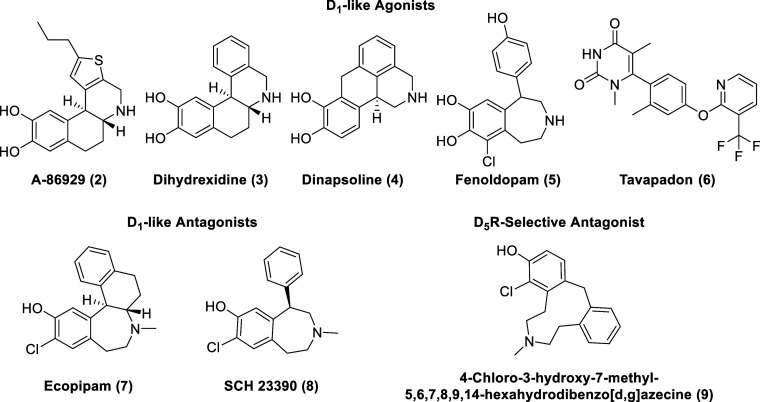
The high degree of structural homology between the ligand binding site of D1R and D5R further exacerbates the difficulties in obtaining selective D1R ligands. This may be of therapeutic relevance since D5R is associated with cardiovascular side effects.33 Nevertheless, D1R is an important target for the treatment of cognitive deficits, particularly those associated with schizophrenia (which are not currently addressed by antipsychotic medications) and Parkinson’s disease. This is because full or partial agonists of D1-like receptors have been shown to reverse working memory in aged monkeys or from deficits induced by ketamine administration.34−36
Relatively recently, researchers at Pfizer identified a new class of potent noncatechol D1-like orthosteric agonists through a high-throughput screening (HTS) campaign of approximately 3 million compounds and subsequent structure–activity relationship (SAR) studies.37,38 The clinical efficacity of these agonists are still being evaluated with the optimized drug candidate, Tavapadon (6, Ki D1R = 8.54 nM), currently under phase 3 clinical trials for Parkinson’s disease in the United States (under development by Cerevel Therapeutics) [NCT04201093, NCT04223193].39,40
D1 antagonism may also hold therapeutic promise as well, as exemplified by Ecopipam (SCH 39166, 7). Ecopipam, a more conformationally restricted benzazepine analog of SCH 23390 (8, the most used D1R reference antagonist), is a selective Dl-like dopamine receptor antagonist (in vitro and in vivo) (Ki at D1R of 1.2 nM, Ki at D5R of 2 nM) with improved selectivity over 5-HT2 and duration of action.41 Ecopipam displays a poor pharmacokinetic profile with extensive N-dealkylation of the N-methyl group and O-glucuronidation of the phenol as well as low oral bioavailability (0.6%).42,43 Ecopipam was initially studied as a treatment for schizophrenia, but it displayed no efficacy (with reported side effects of anxiety, restlessness, sedation, and vomiting).44,45 Clinical studies for cocaine addiction revealed that while Ecopipam is an effective antagonist of the acute euphoric effects of cocaine, repeated administration failed to attenuate the subjective effects of cocaine.46,47 Clinical studies also revealed that Ecopipam is an effective obesity treatment; however, undesirable side effects (depression and reversible anxiety) halted its development as an antiobesity drug.48,49 Ecopipam is currently under phase 3 clinical trials for Tourette’s syndrome in the United States [NCT06021522, NCT05615220].50,51
As one of the most established therapeutic targets for endocrine and neuropsychiatric disorders, the D2-like family of dopamine receptors are promising therapeutic targets. Despite their importance, most investigational and clinically approved drugs are not subtype selective but rather are generally subfamily selective for all three D2-like receptors (D2R, D3R, and D4R) due to the high degree of similarity between the transmembrane (TM) regions.52 For example, antipsychotic drugs such as haloperidol (Haldol, 10, Figure ), paliperidone (Invega, 11), and fluphenazine (Prolixin, 12) not only function as D2-like receptor antagonists but also engage serotonin and adrenergic receptors.53−57
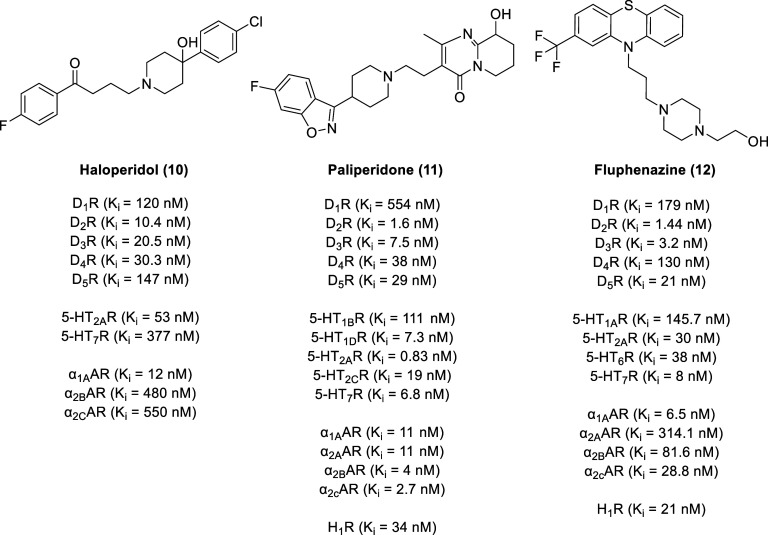
Due to the prevalence of dopamine D2-like signaling in the periphery (mainly in the pancreatic α- and β-cells), D2-like ligands often display metabolic dysfunction (e.g., systemic insulin resistance and dysglycemia).58 Moreover, side effects can result from antagonism of D2R signaling in the nigrostriatal system, causing extrapyramidal motor symptoms, and in the tuberoinfundibular pathway, resulting in elevated prolactin levels.9,59
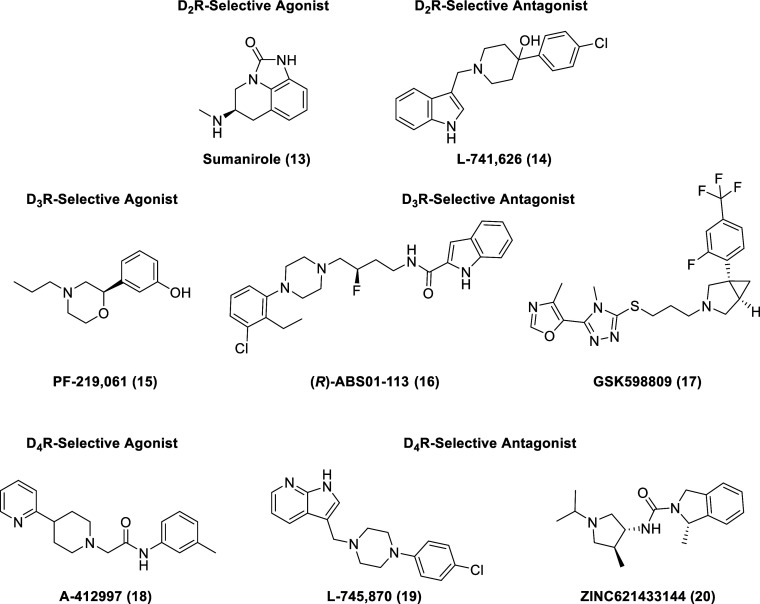
Nevertheless, subtype-selective ligands for each D2-like subtype (representative examples shown, 13–20, in Figure ) are proven to have significant therapeutic applications. All FDA-approved antipsychotics are D2R partial agonists or antagonists. In addition, D2R ligands are used in the treatment of restless legs syndrome, postoperative nausea and vomiting, hyperprolactinemia, and Tourette’s syndrome. D3R ligands are implicated in neurological disorders and are being investigated as potential treatments for substance use disorders.60,61 D4R agonists are proposed therapeutic agents for attention deficit hyperactivity disorder (ADHD) and sexual dysfunction, while D4R antagonists are candidates for the treatment of neuropsychiatric disorders.62,63
Despite their clinical use, conventional dopamine receptor targeting drugs have debilitating side effects mainly due to suboptimal pharmacokinetic and pharmacodynamic properties. The identification of new chemotypes for dopamine receptor ligand discovery and the refinement of existing chemotypes are avenues that may lead to novel and pharmacologically advanced therapeutics. Natural products such as l-DOPA, benzyltetrahydroisoquinolines, aporphine alkaloids, tetrahydroprotoberberines, and ergolines have inspired and will continue to stimulate dopamine receptor ligand discovery efforts in that regard. Herein, we provide an informed perspective on the status of the aforementioned structural classes as a source of ligands for dopamine receptors.
Dopamine and l-DOPA
The endogenous neurotransmitter dopamine (1, Figure ) functions to activate dopamine receptors with the affinity trend of D3R (Ki = 3 nM) > D4.4R (Ki = 6.4 nM) > D2SR (Ki = 12.5 nM) > D2LR (Ki = 16 nM) ≫ D5R (Ki = 228 nM) ≫ D1R (Ki = 2340 nM) (D4.4R is a human polymorphic variant).64−67 While dopamine cannot be used as a CNS drug, as it cannot cross the protective BBB, it can still be used as a peripheral vasostimulant for the treatment of low heart rate, cardiac arrest, and low blood pressure.68,69 Originally believed to be a biologically inactive amino acid, l-3,4-dihydroxyphenylalanine (l-DOPA, 21, Figure ) was isolated from seedlings of Vicia faba beans in 1913.70,71l-DOPA is transported across the BBB by the large neutral amino acid transporter (LAT1) and is subsequently decarboxylated to dopamine by the endogenous enzyme aromatic l-amino acid decarboxylase. l-DOPA bypasses the rate-limiting step in dopamine synthesis (involving the enzyme tyrosine hydroxylase) and is more quickly converted to dopamine than the biosynthetic starting material for dopamine production, the amino acids l-tyrosine or l-phenylalanine. As such, l-DOPA is able to increase dopamine concentrations and reduce the motoric symptoms of Parkinson’s disease.72 Parkinson’s disease is a long-term CNS degenerative disorder, characterized by the idiopathic loss of dopamine neurons in the caudate-putamen.73 The loss of these neurons causes the manifestation of Parkinson’s disease motor symptoms, characterized by rigidity, slowed movements, difficulty with walking, and tremors.
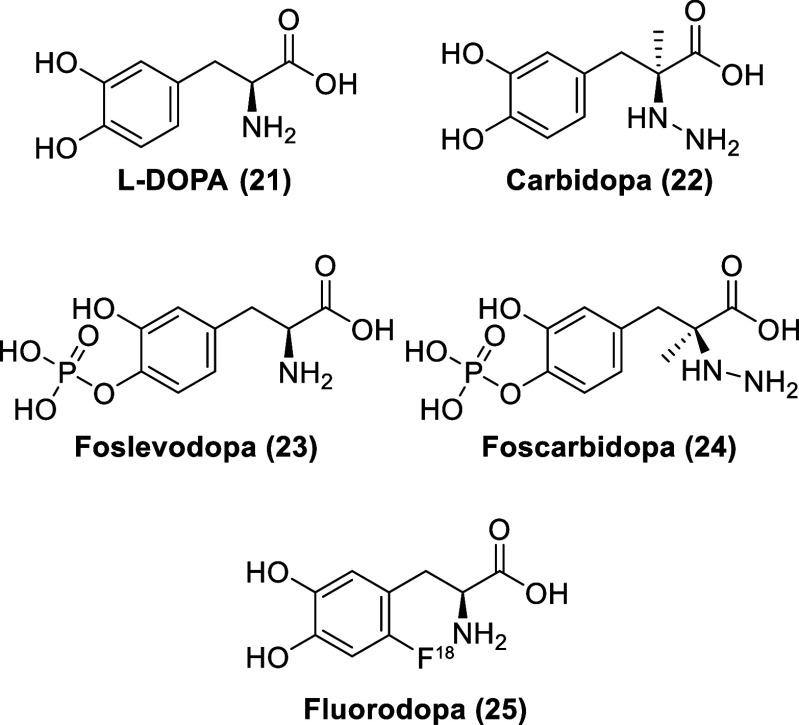
Most patients treated with l-DOPA will eventually develop l-DOPA-induced dyskinesia and motor fluctuations due to dysfunction in dopaminergic pathways.72 Particularly, dyskinesia is believed to be caused by pathological alterations in the nigrostriatal pathway.74 It should be noted that l-DOPA will increase dopamine concentration not just in the CNS but also in the peripheral nervous system (yielding undesirable side effects). As such, it is a standard clinical practice to coadminister a peripheral DOPA decarboxylase inhibitor (DDCI) (e.g., carbidopa, 22) with l-DOPA to prevent the conversion of l-DOPA to dopamine in the periphery.75 But, even with the coadministration of carbidopa, levodopa still has a relatively short half-life in plasma.76 Moreover, gastric motility is typically impaired as Parkinson’s disease progresses, rendering the absorption of oral tablets of levodopa to be unpredictable due to erratic gastric emptying (levodopa is only absorbed in the superior part of the duodenum, the first part of the small intestine). Duopa (AbbVie), a gel formulation of levodopa and carbidopa, allows for the prevention of the peaks and troughs associated with traditional levodopa and carbidopa tablet therapy through continuous dosing of levodopa and carbidopa via a jejunal tube (requiring surgery at the beginning of the therapy).77 As an alternative, phosphonate prodrugs of levodopa (foslevodopa, 23) and carbidopa (foscarbidopa, 24) enable a more effective subcutaneous delivery with significant improvement of aqueous solubility at physiological pH (7.4).78,79
6-[18F]Fluoro-l-DOPA (FDOPA, 25) is a radiolabeled analog of l-DOPA, used in positron emission tomography (PET) imaging of dopaminergic nerve terminals in the striatum in patients with suspected Parkinsonian syndromes.80
Benzylisoquinoline Alkaloids (BIAs)
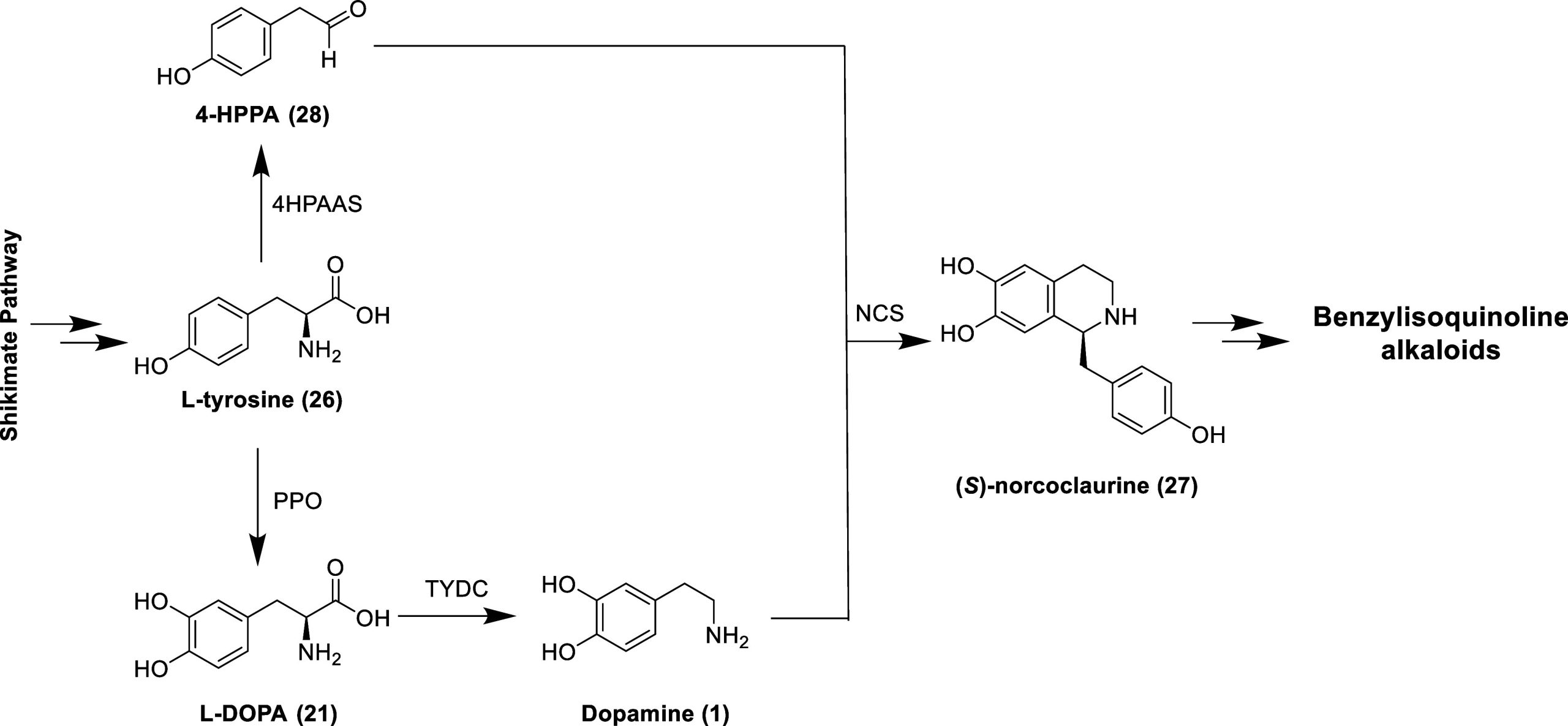
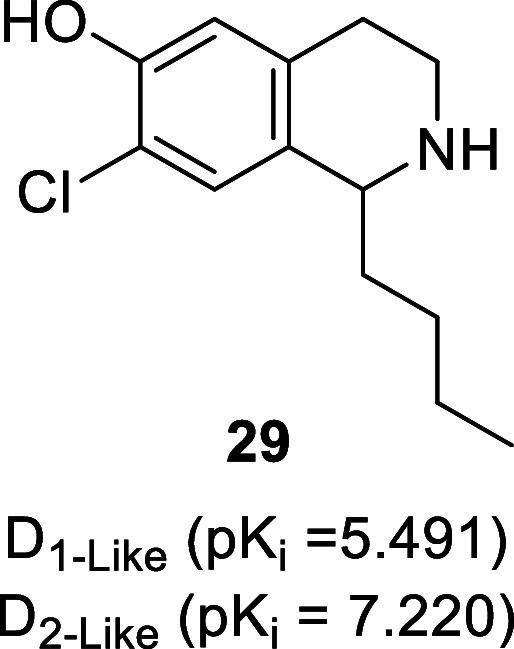
Derived biosynthetically from the amino acid l-tyrosine (Tyr, 26), the benzylisoquinoline alkaloids (BIAs) are a large structural group of plant secondary metabolites found primarily in the order Magnoliales, Laurales, Ranunculales, Berberidales, and Papaverales.81 (S)-Norcoclaurine (27, Figure ) is the common precursor to the benzylisoquinoline derivatives (e.g., aporphine, benzophenanthridine, protoberberine, and pavine alkaloids) and is generated via the enantioselective condensation of dopamine and 4-hydroxyphenylacetaldehyde (4-HPPA, 28) by the enzyme norcoclaurine synthase (NCS).82 4-HPPA is produced directly from Tyr via decarboxylation–oxidative deamination by 4-hydroxyphenylacetaldehyde synthase (4HPAAS), whereas dopamine is formed in a two-step process (with l-DOPA as an intermediate) starting with the hydroxylation of Tyr by polyphenol oxidase (PPO) and subsequential decarboxylation by l-tyrosine decarboxylase (TYDC).83 As the BIAs are biogenetically derived from dopamine, it should not be surprising that several natural and synthetic benzylisoquinoline derivatives have dopaminergic activities as highlighted in the following subsections.84 The presence of the 1-benzyl moiety of BIAs is not essential for dopaminergic activity as seen with 1-butyl-7-chloro-6-hydroxy-tetrahydroisoquinoline (29, Figure , D2-like receptors Ki = 66 nM), which displays antidepressant-like activity in mice.85
Benzyltetrahydroisoquinolines (BTHIQs) and Bis-Benzyltetrahydroisoquinolines (BBTHIQs)
Originally isolated from the roots of Bullock’s heart (Annona reticulata), (S)-reticuline (30, Figure ) is a dopamine receptor ligand with low micromolar or submicromolar affinities to both D1 and D2 rat striatal receptors with IC50 values of 1.8 (D1-like receptors) and 0.47 μM (D2-like receptors).86,87 Ethanolic extracts of stems and roots of the evergreen shrub Cocculus laurifolius revealed (S)-coclaurine (31), a benzyltetrahydroisoquinoline with IC50 values of 0.24 (D1-like receptors) and 0.13 μM (D2-like receptors), based on the displacement of either tritiated raclopride (a D2 dopamine receptor-selective ligand) or SCH23390 (a D1 dopamine receptor-selective ligand) from their specific binding sites in rat striatum.86,88 (R)-(+)-Nor-roefractine (32) is a coclaurine analogue with a 4′-methoxy group (enhancing lipophilicity) and C-6 and C-7 substituents exchanged and displays 6-fold selectivity for D2 receptors although with lower potency compared to other benzyltetrahydroisoquinolines (BTHIQs). The IC50 values for the displacement of tritiated raclopride (D2-like receptors) or SCH23390 (D1-like receptors) by (R)-(+)-nor-roefractine are 5.0 and 32.9 μM, respectively.89
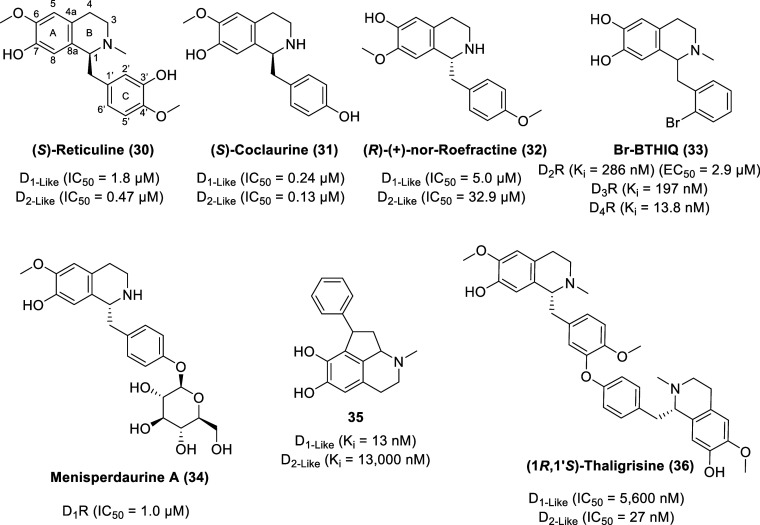
1-(2′-Bromobenzyl)-6,7-dihydroxy-N-methyl-tetrahydroisoquinoline (Br-BTHIQ, 33) displays a partial agonist effect through cAMP signaling at D2R with an EC50 value of 2.9 μM (Emax = 31.9% at 10 μM, Emax = 48.4% at 100 μM), quantified using the homogeneous time-resolved fluorescence (HTRF)-based cAMP kit in Chinese hamster ovary (CHO)-K1 cells stably expressing the cloned human D2SR.90 Br-BTHIQ displays nanomolar affinity to dopamine receptors with the order of D4R (Ki = 13.8 nM) ≫ D3R (Ki = 197 nM) > D2R (Ki = 286 nM).
Menisperdaurine A (34) is a glycosidic benzylisoquinoline alkaloid isolated from the rhizomes of Menispermum dauricum, known as Bian-Fu-Ge-Gen or Bei-Dou-Gen in traditional Chinese medicine with analgesic and antipyretic effects.91 Menisperdaurine A displays D1R antagonistic activity with an IC50 value of 1.0 μM [via fluorometric imaging plate reader (FLIPR) assay, monitoring cellular Ca2+ responses].
Synthetic BTHIQ derivatives bearing a cyclopentane motif and a phenyl substituent display high selectivity and affinity toward D2-like receptors as exemplified by 35, affinity values (Ki) of 13 nM at D1-like receptors and 13 000 nM at D2-like receptors (Ki D1/D2 = 1000).92
Bis-benzyltetrahydroisoquinolines (BBTHIQs) are biosynthetically generated by dimerization of trioxygenated benzyltetrahydroisoquinolines via intermolecular oxidative coupling of (S)- and/or (R)-coclaurine and/or its N-methyl derivatives.84 BBTHIQs with one diaryl ether bridge display higher dopaminergic activity (compared to BBTHIQs with two diaryl ether bridges with one diaryl ether bridge and one biphenyl bridge, or seco derivatives) as exemplified by (1R,1′S)-thaligrisine (36) with IC50 values based on the displacement of tritiated raclopride (D2-like receptors) and SCH23390 (D1-like receptors) of 27 and 5600 nM, respectively.93
Aporphine Alkaloids
Comprising one of the largest groups of natural isoquinolines, aporphine alkaloids are widely distributed in flowering plant families, (e.g., Annonaceae, Aristolochiaceae, Berberidaceae, Canellaceae, Eupomatiaceae, Hernandiaceae, Lauraceae, Leguminosae, Magnoliaceae, Menispermaceae, Monimiaceae, Papaveraceae, Piperaceae, Ranunculaceae, Rhamnaceae, Saururaceae, and Symplocaceae families).94−97 The tetracyclic backbone of naturally occurring aporphine alkaloids is typically decorated with substituents such as hydroxyl, methoxy, and methylenedioxy groups on the two aromatic rings. The aporphine alkaloids are a privileged scaffold in drug discovery, as natural aporphine alkaloids are reported to have a myriad of pharmacological activities including antioxidant, antitumor, anticonvulsant, antiplasmodial, antiparkinsonian, antimalarial, antiprotozoal, and cytotoxic effects.15
The acid-catalyzed decomposition of morphine (37, Figure ) led to the discovery of the nonselective dopamine receptor partial agonist, apomorphine (38) with the affinity trend of D4R (Ki = 4.37 nM) > D3R (Ki = 26 nM) > D2R (Ki = 52 nM) ≫ D5R (Ki = 188.9 nM) > D1R (Ki = 484 nM).98−100 Apomorphine is also an antagonist at serotonin receptors (Ki 5-HT2A = 120 nM, Ki 5-HT2B = 132 nM, Ki 5-HT2C = 102 nM) and α-adrenergic receptors (Ki α1D = 64.6 nM, Ki α2A = 141 nM, Ki α2B = 66.1 nM, Ki α2C = 36.3 nM).98−100 Apomorphine was initially explored as an experimental therapeutic for various conditions such as alcoholism, opioid addiction, erectile dysfunction, schizophrenia, and coughing.101,102 It is now FDA approved (marketed under the name Apokyn) for the treatment of acute intermittent hypomobility—off episodes with late-stage Parkinson’s disease patients treated with l-DOPA.103
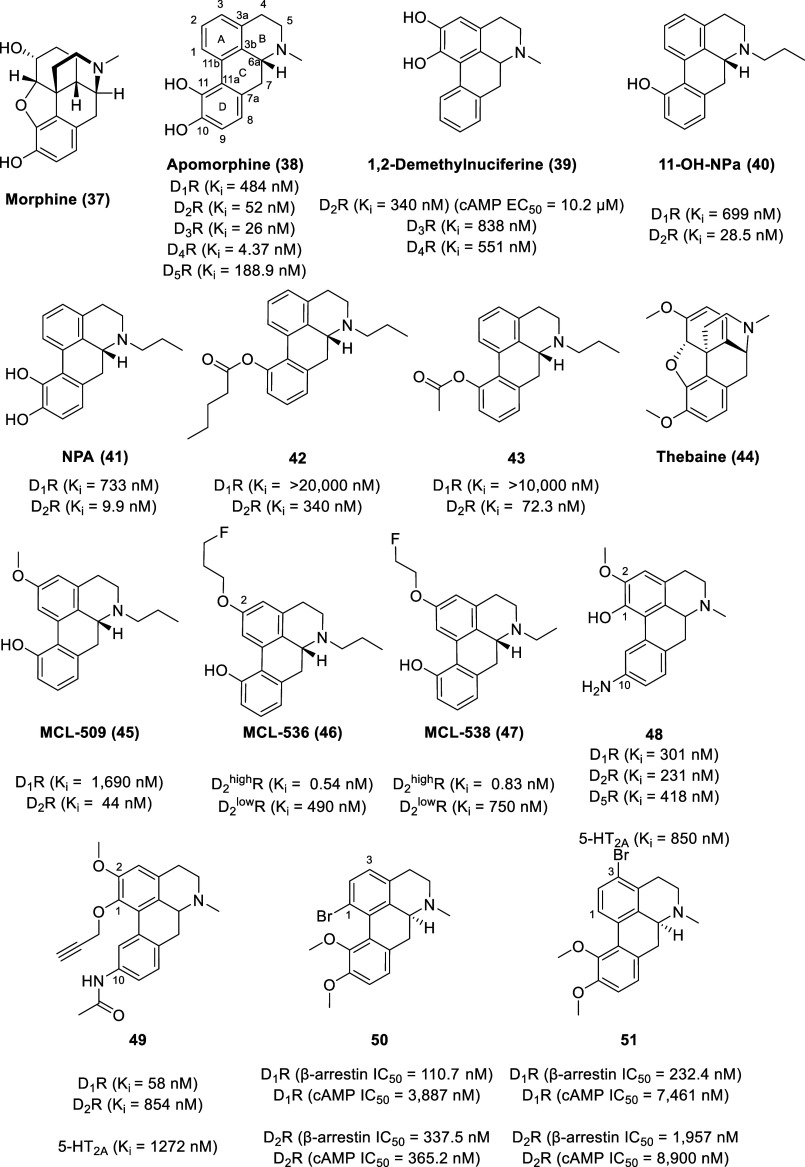
By virtue of the ortho-catechol ring and phenethylamine moiety, apomorphine shares structural similarity to dopamine (a fundamental rationale for its affinity to dopamine receptors).104,105 Due to almost complete first-pass hepatic metabolism (via catechol-O-methylation, sulfation, and glucuronidation), apomorphine has very limited oral bioavailability (<4%) and has a short duration of action.106 However, apomorphine has the ability to cross the blood–brain barrier freely due to its lipophilic tetracyclic structure. In fact, apomorphine appears to concentrate in the brain, with a brain-to-blood concentration ratio of 8:1.107
Apomorphine has undergone extensive SAR studies in efforts to optimize its selectivity, potency, and pharmacokinetic profile. The biphenyl unit, 11-hydroxy substitution, N-alkylation, and C-6α (R) configuration of aporphines are recognized as essential for dopaminergic activities.94,95,108N–n-Propyl substitution improves D2R activity, whereas an N-methyl substituent improves D1R activity.109,110 This propyl effect of improving D2R activity can be applied to other dopaminergic molecules.111 The presence of a catechol group at the 1,2 position of the aporphine scaffold also favors dopamine receptor affinity, as exemplified by 1,2-demethylnuciferine (39).90 Despite 1,2-demethylnuciferine displaying submicromolar affinity with the trend of D2R (Ki = 340 nM) > D4R (Ki = 551 nM) > D3R (Ki = 838 nM), 1,2-demethylnuciferine is a weak D2R agonist through cAMP signaling with an EC50 value of 10.2 μM (Emax = 50.7% at 10 μM, Emax = 92.7% at 100 μM) quantified using the HTRF-based cAMP kit in CHO-K1 cells stably expressing the cloned human D2SR.
A series of N-substituted 11-hydroxynoraporphines and their esters of varying chain lengths was synthesized from morphine and evaluated for binding affinities at dopamine receptor sites in rat caudate-putamen membranes.112 From the series, the N–n-propyl compound 40 (11-OH-NPa) exhibited the highest selectivity and affinity at D2R (Ki D1R = 699 nM, Ki D2R = 28.5 nM); however, its D2R affinity is lower than that of its catechol precursor NPA (41, Ki D1R = 733 nM, Ki D2R = 9.9 nM). The valeryl ester of 11-OH-NPa, compound 42, displayed maximal behavioral potency (via motor activity in normal adult male rats) after systemic injection, whereas the acetate ester of 11-OH-NPa (43) showed maximal behavioral potency (via motor activity in normal adult male rats) after enteric (intragastric) administration. Similarly, a series of N-alkyl-2-methoxy-11-hydroxynoraporphines was synthesized from thebaine (44) and evaluated for binding affinities at dopamine receptors in rat forebrain tissue.113 The most selective 11-monohydroxy aporphine was MCL-509 (45) (Ki D1 = 1,690 nM, Ki D2 = 44 nM), an orally active Parkinson’s disease drug candidate.114
D2 receptors exist in two states, either as a functionally inert low-affinity state (D2lowR) or as a functional high-affinity state (D2highR).115 The high-affinity and low-affinity receptors show a biphasic competition curve and have different binding affinities to various ligands. Evidence increasingly indicates that alterations in the density of D2 receptors in the high-affinity state are more important to pathophysiological processes than alterations in the total receptor density. As such, the development of a selective D2R agonist that is capable of distinguishing between the high- and the low-affinity states is critical as a tool for diagnostics and therapeutics for psychosis and Parkinson’s disease.115 Fluoroalkylation of a hydroxyaporphine substrate led to the development of MCL-536 (46; Ki D2highR = 0.54 nM, Ki D2lowR = 490 nM) and MCL-538 (47; Ki D2highR = 0.83 nM, Ki D2lowR = 750 nM) as novel D2highR-targeted probes for the dopaminergic system.116−118
Due to the high sequence identity (82% in transmembrane regions) between D1R and D5R, the development of subtype-selective D1-like ligands is challenging and limited to only a few compounds.7,119−121 The development of ligands with D1R versus D5R selectivity (and vice versa) remains an area of significant interest in the field. In 2020, our group reported the synthesis and evaluation of two series of aporphine analogs bearing either a sole C-10 nitrogen substituent on the tetracyclic aporphine core or 1,2,10-trisubstituted aporphines (with both groups containing a nitro, aniline, or amide moiety at the C-10 position).122 All compounds except compound 48 lacked D5R affinity. The C-10 nitrogen monosubstituted analogs exhibited a preference for the serotonin 5-HT1AR, while the 1,2,10-trisubstituted analogs generally displayed dopamine receptor selectivity. Out of the two series of compounds, the 1,2,10-trisubstituted aporphine 49 was identified as the ligand with the highest affinity toward D1R (Ki = 58 nM) with low affinity toward 5-HT2AR (Ki = 1272 nM) and D2R (Ki = 854 nM) and no affinity for the other receptors tested (5-HT1AR and D5R). Compound 49 displays higher metabolic stability than apomorphine in human liver microsomes, which is not surprising as it lacks any common conjugation sites (amino, carboxy, hydroxy, or thiol groups) for phase II metabolism. Computational simulations revealed that the D1R versus D5R selectivity of 49 might arise from stronger hydrophobic contacts (primarily with phenylalanine residues) with D1R compared to D5R.
With the potential to bypass observed adverse effects, functional selectivity enables the activation of distinct subsequent signaling cascades or even the selective activation of a particular G protein subtype.22,123 As functional selectivity is still a relatively new paradigm in drug discovery, very few such studies have been performed on aporphines. A structure–functional–selectivity relationship (SFSR) study of the apomorphine scaffold revealed the structural motifs responsible for biased activity at both D1-like and D2-like receptors.124 It was found that alkyl and halogen substituents on the catechol ring generally do not contribute to functional selectivity; rather, these substituents reduce agonist activity at dopamine receptors for both β-arrestin 2 and G protein pathways. β-Arrestin-biased antagonism at both D1 and D2 receptors was observed with bromo substitution at either the C-1 (50) [D1R (β-arrestin IC50 = 110.7 nM, cAMP IC50 = 3,887 nM), D2R (β-arrestin IC50 = 337.5 nM, cAMP IC50 = 365.2 nM)] or the C-3 (51) [D1R (β-arrestin IC50 = 232.4 nM, cAMP IC50 = 7,461 nM), D2R (β-arrestin IC50 = 1,957 nM, cAMP IC50 = 8,900 nM)] position measured by cAMP GloSensor and β-arrestin bioluminiscence resonance energy transfer (BRET) assays. These compounds may be useful for further hit-to-lead optimizations, particularly for the treatment of hyperdopaminergia, which is implicated in psychiatric disorders such as ADHD and schizophrenia.
The natural aporphine, S-(+)-boldine (52, Figure ), well known as a constituent of the bark and leaves of the Chilean tree Boldo (Peumus boldus Molina, Monimiaceae),125−127 has served as a template for the construction of new aporphines. Boldo is used in folk medicine, primarily for the treatment of liver ailments. Although boldine is recognized as an effective antioxidant, it also displays antagonist activity at D1-like and D2-like receptors with IC50 values of 400 and 520 nM, respectively, based on the displacement of [3H]-SCH 23390 or [3H]-raclopride from their specific binding sites in rat striatum.128 Despite the affinity toward dopamine receptors, boldine does not display effective central dopamine antagonist activities in vivo as the high dose of 40 mg/kg boldine i.p. failed to produce any effect on striatal [3H]raclopride binding in rat forebrain while [3H]-SCH 23390 binding decreased by 25%. Furthermore, orally administered boldine is rapidly glucuronidated in the liver with a plasma elimination half-life of 31 min.129 Molecular bromine in acetic acid or N-halosuccinimides in trifluoroacetic acid was used to halogenate boldine to afford 3-haloboldines and 3,8-dihaloboldines.130 Halogenation of boldine at C-3 favors affinity for rat brain D1-like dopamine receptors compared to D2-like dopamine receptors, as exemplified by 3-iodoboldine (53) with IC50 values of 3 (D1-like receptors) and 96 nM (D2-like receptors) based on radioligand displacement studies in rat-brain membranes.
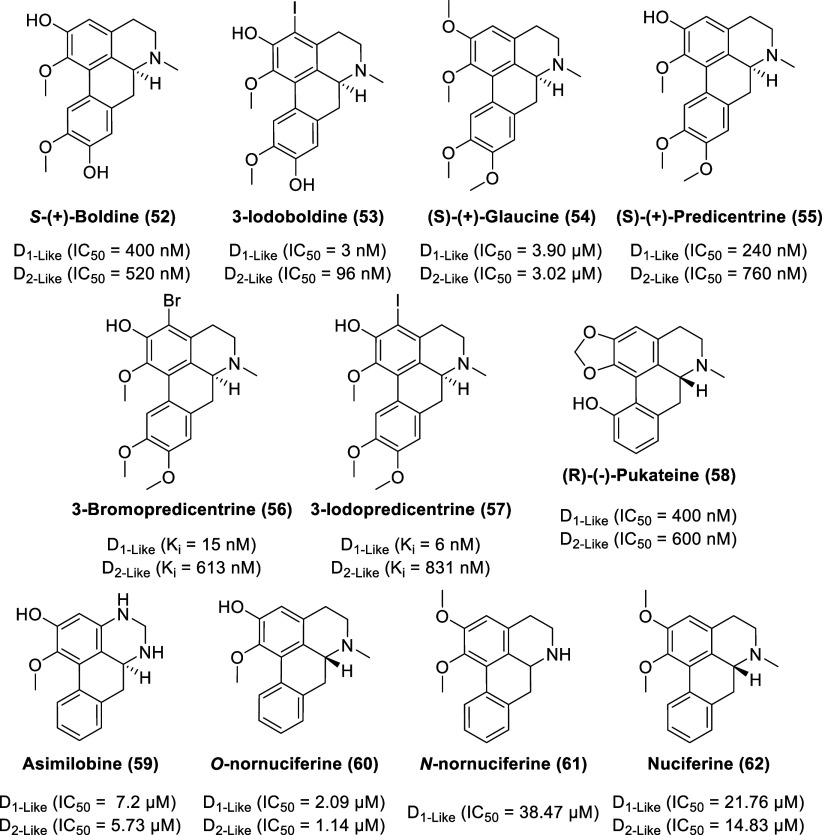
(S)-(+)-Glaucine (54) is a natural derivative of boldine, with no free hydroxyl group, and is used as an antitussive agent (mainly due to Ca2+ channel antagonism). It is commercially isolated from the yellow horned poppy (Glaucium flavum Crantz).131 Glaucine displays a lower affinity toward dopamine receptors than boldine, with IC50 values of 3.90 (D1-like receptors) and 3.02 μM (D2-like receptors), measured by binding studies in rat-striatal membranes using [3H]-SCH 23390 or [3H]-raclopride.128 In vivo studies reveal some antidopaminergic properties of glaucine as the high dose of 40 mg/kg glaucine i.p. elicited a reduction of 50% for both [3H]-SCH 23390 and [3H]-raclopride binding in rat forebrain. Both boldine and glaucine inhibited penile erection, and apomorphine (0.1 mg/kg s.c.) induced yawning in rat models by over 50% at 40 mg/kg (i.p.) without impacting the metabolism of dopamine in rat and mouse forebrain tissues. Similar to boldine and glaucine, (S)-(+)-predicentrine (55) displays moderate activity at both D1-like and D2-like receptors with IC50 values of 240 (D1-like receptors) and 760 nM (D2-like receptors) based on radioligand displacement studies in rat-striatal membranes.132 3-Halogenated and 3,8-dihalogenated derivatives of predicentrine had improved affinity for D1-like receptors, as demonstrated by 3-bromopredicentrine (56; Ki D1-like = 15 nM, Ki D2-like = 613 nM) and 3-iodopredicentrine (57; Ki D1-like = 6 nM, Ki D2-like = 831 nM).132 Halogenated glaucine derivatives failed to exhibit any clear trend toward enhanced selectivity or any improvement with potency, indicating that the D1-like enhanced potency with halogenation at C-3 is conditional with the presence of the 2-hydroxy group on the aporphine skeleton (and it is this moiety that predominantly determines the binding mode that favors D1-like over D2-like receptors).
As an isolate from the bark of the pukatea tree (Laurelia novae-zelandiae), (R)-(−)-pukateine (58) is another aporphine congener with antioxidant and dopaminergic properties.133 The monohydroxyaporphine derivative displays IC50 values of 400 (at D1-like receptors) and 600 nM (at D2-like receptors) based on radioligand binding experiments. Pukateine bears a meta hydroxyphenyl (at C-11) and 6aR configuration, both of which are associated with dopamine receptor activation.94,134 In 6-hydroxydopamine unilaterally denervated rats, a dose of 8 mg/kg but not 4 mg/kg of pukateine elicited a significant contralateral circling, which is a behavior classically associated with the actions of a dopamine agonist. Perfusion of pukateine (at 340 μM) through a microdialysis probe inserted into the striatum induced a significant increase in dopamine levels, indicating an agonist-like interaction with dopamine receptors.
Extracted from the leaves of the sacred lotus Nelumbo nucifera Gaertn., four bioactive aporphine alkaloids [asimilobine (59), O-nornuciferine (60), N-nornuciferine (61), and nuciferine (62)], were identified as antagonists with IC50 values in the mid- to low-micromolar range for dopamine receptors.135O-Nornuciferine exhibited the highest potency at both D1R (IC50 = 2.09 μM) and D2R (IC50 = 1.14 μM), using FLIPR assays in HEK293 cell lines.
Cularine Alkaloids
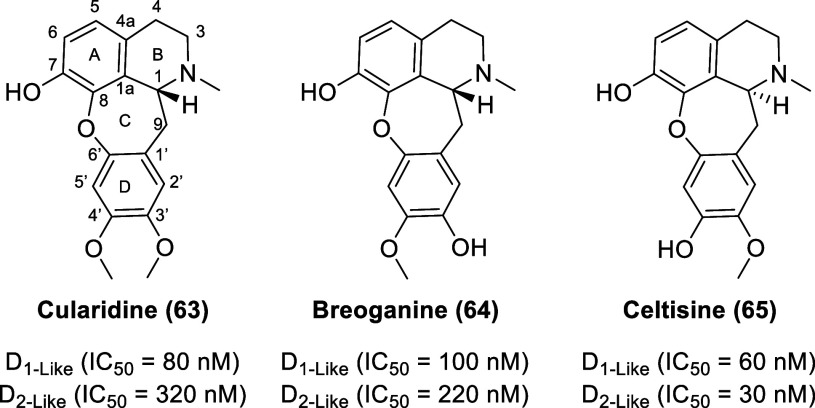
Isolated from the aerial parts of the Spanish shrub Sarcocapnos crassifolia (Fumariaceae), the natural cularines—cularidine (63, Figure ), breoganine (64), and celtisine (65)—display dopamine receptor affinity.136 The cularines bear structural resemblance to aporphines but have a tetracyclic skeleton featuring a 7-membered oxepine system rather than the 6-membered carbocyclic ring C moiety found in aporphines. Cularines and aporphines are biosynthetically derived from similar benzylisoquinoline precursors.137 Cularidine displays IC50 values of 80 (D1-like receptors) and 320 nM (D2-like receptors) (0.25-fold selectivity for DlR versus D2R), whereas breoganine displays IC50 valuesof 100 (D1-like receptors) and 220 nM (D2-like receptors) (0.45-fold selectivity for DlR versus D2R) derived from radioligand displacement studies in rat-striatal membranes. Celtisine displays IC50 values of 60 (D1-like receptors) and 30 nM (D2-like receptors) (2-fold selectivity for DlR/D2R). To date, there have been no follow-up studies to explore the potential of cularine derivatives as dopamine receptor ligands.
Phenanthrene Alkaloids
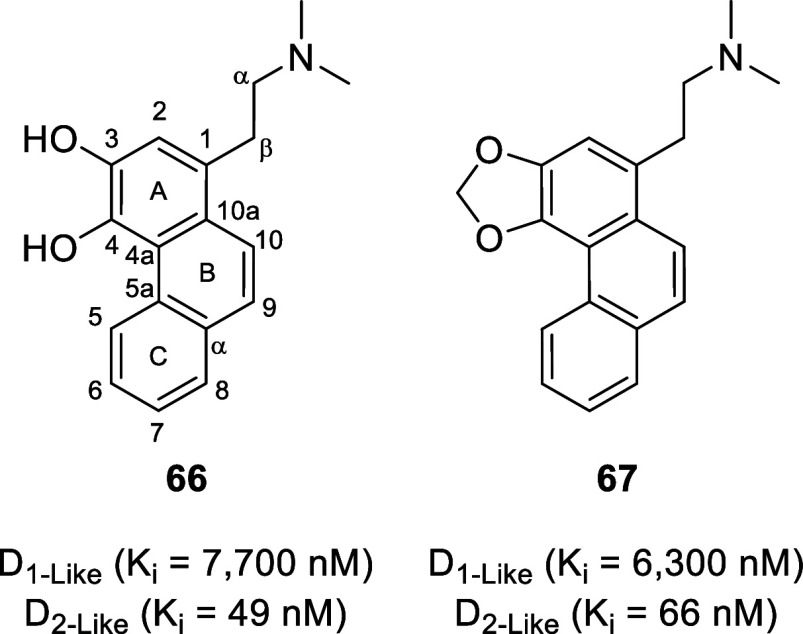
While phenanthrene alkaloids do not contain a nitrogen heterocycle, they are considered alkaloids due to being biogenetically derived from aporphines (thus occurring in the same plant families as aporphines).138 The degradation of aporphines (for the most part) readily yields phenanthrene alkaloids. Hoffmann degradation is the classical (synthetic) method for the transformation of aporphines into phenanthrene alkaloids. The reaction involves the formation of a quaternary ammonium salt by treatment with an alkylating agent (usually methyl iodide or dimethyl sulfate) followed by anion exchange with silver oxide to form a quaternary ammonium hydroxide which can then be thermolyzed to the phenanthrene. 3,4-Dihydroxy- and 3,4-methylenedioxyphenanthrenic derivatives (66 and 67, respectively, Figure ) displayed high selectivity toward D2-like receptors.13966 displays affinity values (Ki) of 7700 nM at D1-like receptors and 49 nM at D2-like receptors (Ki D1/D2 = 157), whereas 67 displays affinity values of 6300 nM at D1-like receptors and 66 nM at D2-like receptors (Ki D1/D2 = 96).
Berberine and Tetrahydroprotoberberines (THPBs)
The bright yellow compound berberine (68, Figure ), a characteristic isolate of most barberries (Berberis plants), of the American goldenseal Hydrastis canadensis, and of the Chinese herb Coptis chinensis, is traditionally used as a textile dye and antiparasitic agent by many cultures in Eurasia and the Americas.140,141 Berberine displays a wide variety of pharmacological properties with implications for cancer, digestive diseases, neurological diseases, cardiovascular diseases, and metabolism disorders. Berberine is a weak dopamine D1-like and D2-like receptor antagonist (D1R IC50 = 15.5 μM, D2LR IC50 = 17.1 μM, D2SR IC50 = 38.6 μM) (using HTRF assay for D1R and [35S]GTPγS binding assay for D2SR and D2LR) and ameliorates the development of experimentally induced colitis in mice by suppressing innate and adaptive immune responses.142
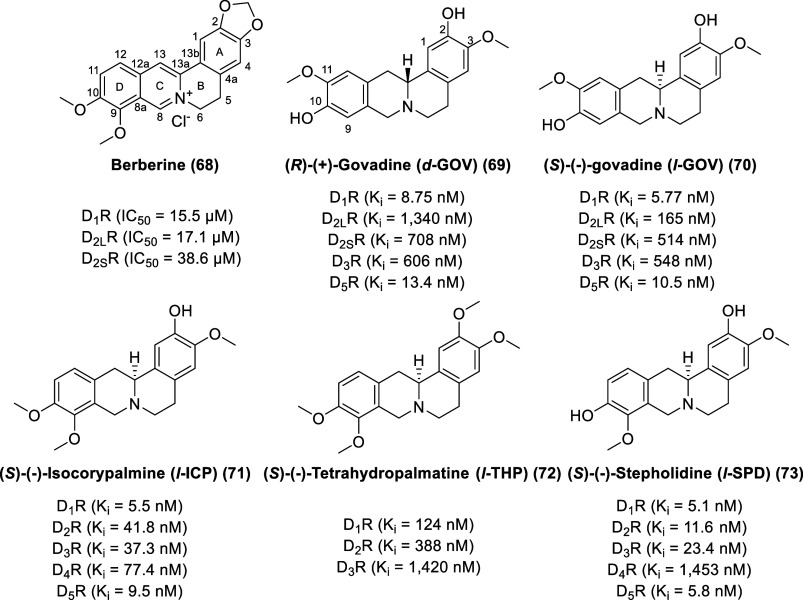
Usually isolated from Corydalis and Stephania species, tetrahydroprotoberberines (THPBs) have been found to display profound effects on the dopaminergic system.143,144 Several THPB natural products and their synthetic derivatives have been identified as dopamine receptor ligands with affinities and activities being primarily reported at D1R, D2R and (to a lesser extent) D3R. In THPB natural products, as in all of the biosynthetically related isoquinoline alkaloids, the aryl rings of the tetracyclic nucleus are usually substituted with hydroxyl or methoxyl groups in the two aryl rings. With a clear consensus, behavioral and biochemical evidence has illustrated that THPBs are antagonists at the D2 family of dopamine receptors.145 The intrinsic activity of THPBs at the D1 family of dopamine receptors is variable, with a majority of compounds being reported as antagonists and comparatively fewer as partial agonists.
Enantiomers of govadine exhibited drastically different behavioral and pharmacological properties.146 (R)-(+)-Govadine (d-GOV, 69) displays dopamine receptor binding affinity with the order of D1R (Ki = 8.75 nM) > D5R (Ki = 13.4 nM) ≫ D3R (Ki = 606 nM) > D2SR (Ki = 708 nM) ≫ D2LR (Ki = 1,340 nM), whereas (S)-(−)-govadine (l-GOV, 70) displays dopamine receptor binding affinity with the trend of D1R (Ki = 5.77 nM) > D5R (Ki = 10.5 nM) ≫ D2LR (Ki = 165 nM) > D2SR (Ki = 514 nM) > D3R (Ki = 548 nM). d-GOV and l-GOV exhibit modest affinity for adrenergic receptors [d-GOV (Ki α1A = 648 nM, Ki α1B = 2,590 nM, Ki α1D = 263 nM, Ki α2A = 304 nM), l-GOV (Ki α1A = 369 nM, Ki α1B = 2480 nM, Ki α1D = 235 nM, Ki α2A = 223 nM)]. Rodent models predictive of antipsychotic efficacy and of positive symptoms of schizophrenia showed that l-GOV (but not d-GOV) had improved these measures. On the other hand, d-GOV (but not l-GOV) may be an effective cognitive enhancer, as it improved impairments in compromised memory function in delayed response tasks.
Isolated from the plant Corydalis yanhusuo, (S)-(−)-isocorypalmine (l-ICP, 71) exhibits strong dopamine receptor binding affinity with the order of D1R (Ki = 5.5 nM) > D5R (Ki = 9.5 nM) > D3R (Ki = 37.3 nM) > D2R (Ki = 41.8 nM) > D4R (Ki = 77.4 nM).147,148l-ICP is a partial agonist at D1-like receptors and antagonist at D2-like receptors.
(S)-(−)-Tetrahydropalmatine (l-THP, 72) displays modest nanomolar to micromolar affinity and antagonist activity toward dopamine receptors with the order of D1R (Ki = 124 nM) > D2R (Ki = 388 nM) ≫ D3R (Ki = 1420 nM).145,149 The levo isomer of THP is responsible for the analgesic, sedative, and neuroleptic properties, whereas the dextro (R) isomer of THP has a distinct toxicologic action.143,150l-THP has been approved for use as an analgesic and sedative in China for over 50 years, originally isolated from Stephania rotunda, under the brand Rotundine or Rotundin.
(S)-(−)-Stepholidine (l-SPD, 73), a constituent of Stephania spp, displays higher dopamine receptor affinity than l-THP, with the trend of D1R (Ki = 5.1 nM) > D5R (Ki = 5.8 nM) > D2R (Ki = 11.6 nM) > D3R (Ki = 23.4 nM) ≫ D4R (Ki = 1453 nM).151l-SPD was originally reported to exhibit sedative and analgesic effects on the CNS and to decrease blood pressure without adverse cardiac effects.152−154 Its antipsychotic potential has also been investigated due to its reported mixed functional profile at D1R and D2R (D1R agonist/D2R antagonist).155−158
Substance use disorder is defined as a compulsive and habitual nonmedical self-administration of drugs despite having severe adverse consequences.159 Addictive drugs elevate extracellular levels of dopamine, rewiring the brain’s dopaminergic mechanisms.160 These alterations in the dopaminergic pathways result in decreased dopamine receptor expression in the brain, reducing interest in all activities but those that arise from habitual rewards. Due to the dopamine receptor polypharmacology of THPBs, these compounds have been proposed to be a novel source of pharmacotherapies for substance use disorders.145,149 In animal models of substance misuse, l-THP demonstrated attenuation of self-administration of cocaine, methamphetamine, heroin, and nicotine.161−167l-ICP was also shown to reduce behavioral sensitization and rewarding effects of cocaine in mice.147 Likewise, l-SPD displayed attenuation of reinstatement of drug and cue-induced self-administration of both opiates and psychostimulants.168−171
The clinical evidence for the efficacy of THPBs in substance use disorders is limited only to a single preliminary clinical trial conducted in China, examining the effects of l-THP on ameliorating heroin craving and increasing the abstinence rate in heroin users via a randomized, double-blinded, and placebo-controlled study of 120 heroin-dependent patients.172 The results suggest that l-THP has presumably therapeutic utility in the treatment of heroin addiction as the drug significantly ameliorated protracted abstinence withdrawal syndrome (PAWS). This pilot study does present several methodological limitations including not separating treatment seekers and nontreatment seekers (as they may have different responses to l-THP treatment), employing only pharmacotherapy without cognitive behavioral treatment (CBT), and utilizing only one low dose of l-THP with a short duration of 1 month to limit the risk of possible hepatic toxicity. Additional clinical evidence revealed that short-term l-THP administration does not exhibit significant differences in vital signs or side effects compared to the placebo group in cocaine users.173
THPBs can be semisynthetically prepared by the reduction of the relatively abundant protoberberines such as berberine, palmatine, and their partially O-demethylated analogs or synthetically prepared via Pictet–Spengler reaction, Bischler–Napieralski reaction, Pomeranz–Fritsch reaction, or Mannich reaction to construct the B-ring and C-ring in sequence.174
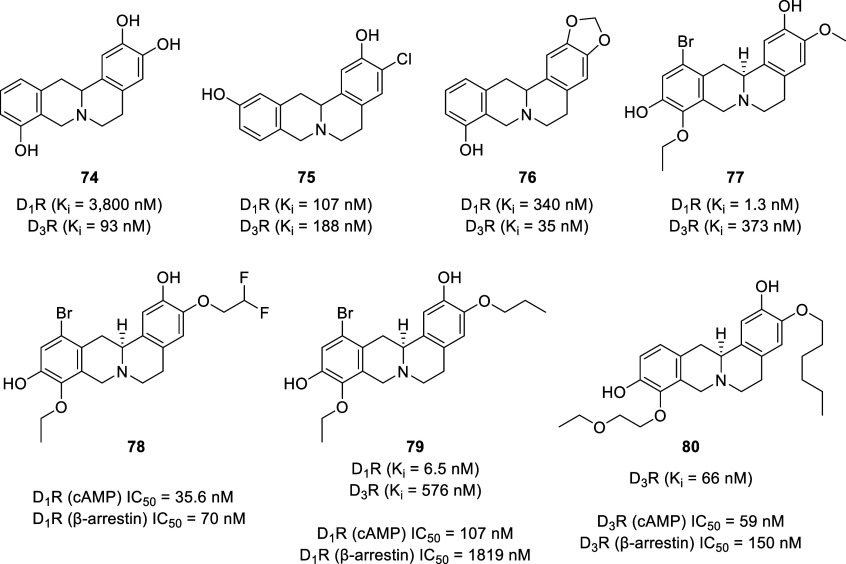
Initial studies on synthetic tetrahydroprotoberberines, focusing on 2,3,9- and 2,3,11-trisubstitution patterns, revealed several ligands with dopaminergic activity.175 The 2,3,9-trihydroxy-THPB (74, Figure ) exhibited the highest selectivity for the D2-like receptors (Ki D1/D2 = 41) with affinity values (Ki) of 3800 nM at D1-like receptors and 93 nM at D2-like receptors. Replacement of the 3-OH with a chlorine atom on the A-ring (3-Cl-2,11-diOH, 75) resulted in increased affinity for D1-like receptors (Ki = 107 nM) but dramatically reduced the selectivity (D2-like receptors, Ki = 188 nM) (Ki D1/D2 = 0.6), which is in contrast to previous studies on BTHIQs where chlorination ortho to the oxygenated group in the A-ring resulted in increased affinity and selectivity toward D2-like receptors.85,176,177 The presence of a methylenedioxy group in the 2,3 position (2,3-OCH2O-9-OH, 76) exerted lower selectivity for the D2-like receptors (Ki D1/D2 = 10) with improved affinity for both dopamine types (D1-like receptors, Ki = 340 nM; D2-like receptors, Ki = 35 nM).
With a lower risk of drug–drug interactions compared to multicomponent drugs (drug cocktails), a designed multiple ligand is a single chemical entity that can modulate multiple targets simultaneously, thereby enhancing efficacy and optimizing dosing and safety.178−180 Dual-acting ligands may be similar to bivalent ligands as they may also combine two pharmacophoric units linked by a spacer, except the spacer is shorter than those of bivalent ligands (with the aim not to simultaneously bind to both protomers but rather to simply deliver both pharmacophoric moieties simultaneously).178,181 Due to their inherent pharmacological properties, bivalent ligands tend to be better suited as pharmacological tools whereas dual-acting ligands may have further application as therapeutic agents.179
In efforts to improve the efficacy and safety of THPBs, our group is actively working on fine tuning the polypharmacology of THPBs as selective D1R partial agonist/D3R antagonist dual-acting ligands.182−185 This targeting strategy is based on the observation that coadministration of a D1R partial agonist (SKF 77434) and a D3R antagonist (NGB 2904) produced robust decreases in cocaine seeking and reward in rats. This indicates a synergistic effect between D1R agonism and D3R antagonism.186 Our initial efforts were focused on SAR studies with l-SPD as a lead molecule. So far, our attempts to identify new selective D1R partial agonist/D3R antagonist dual-acting ligands from THPBs have not been successful, although we have identified several dopamine receptor subtype-specific ligands (e.g., 77–80).182−185,187 Our structure–affinity relationship studies on l-SPD revealed that bromination at the C-12 position improved D1R affinity but generally diminished D3R affinity. Homologation at the C-9 position improved D1R selectivity versus D2R and D3R (often lacking affinity at D2R), whereas alkylation of the C-10 phenolic group with up to 6 carbon atoms in length was tolerated for D1R affinity. Homologation at the C-3 position generally led to retention of the magnitude of D3R affinities with the notable exception of fluoroethylation, which resulted in an approximate 3-fold reduction in D3R affinity compared to l-SPD.
Only a handful of studies have been undertaken to examine the functional selectivity bias of THPBs. Data gathered to date by our group suggest that THPBs function as antagonists at both G protein and arrestin pathways (Figure ) (cAMP functional assays, Promega’s split luciferase-based GloSensor cAMP biosensor technology, or Lance Ultra cAMP IC50 assays; β-arrestin assays, TANGO assays, or DiscoveRx PathHunter technology). l-SPD was found to be a potent pan-dopamine receptor antagonist of both G protein- and β-arrestin-mediated signaling (thus lacking functional selectivity), determined using DiscoveRx PathHunter technology.151
Ergolines
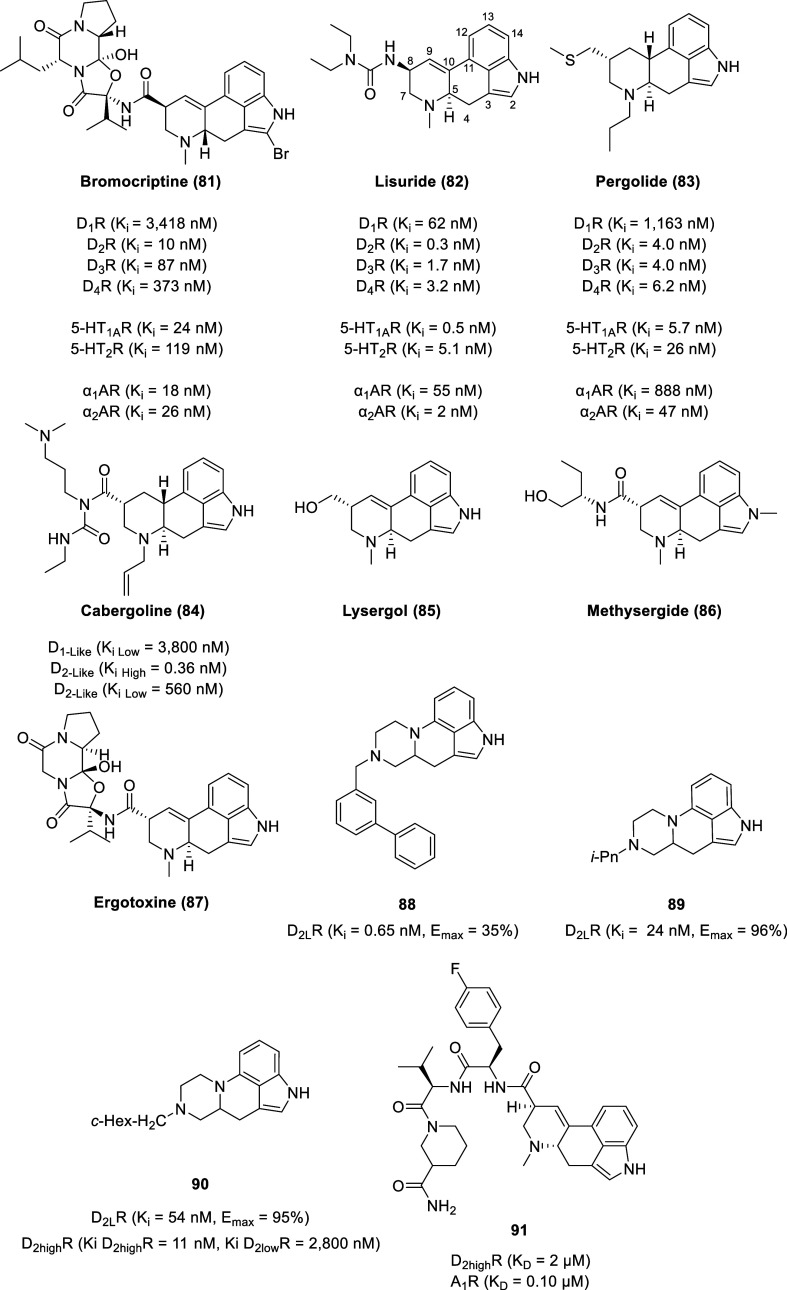
First identified in the dark dense sclerotia of infected grains and grasses by parasitic fungi of the genus Claviceps, ergot alkaloids are also produced by a wide variety of other filamentous fungi, including species in the genus Arthroderma, Aspergillus, Balansia, Epichloe, Neotyphodium, Penicillium, and Periglandula.188−193 Embedded within the tetracyclic structure of the ergolines are the defining features of the monoamine neurotransmitters serotonin, noradrenaline, and dopamine, empowering ergolines to act as potent agonists, partial agonists, or antagonists at these receptors.194 Derivatives of ergolines are categorized into 3 main classes: lysergic acid and its simple amides, ergopeptines, and clavines.195,196 Lysergic acid derivatives contain a carboxylic acid group at position 17 with most lysergic acid derivatives being functionalized by an amide linkage to that position (e.g., ergine and ergonovine). The peptide ergot alkaloids are defined by a tripeptide-derived, cyclol-lactam structure attached as a C-17 amide substituent (e.g., ergocristine, ergotamine, and ergovaline). The clavine alkaloids are classified as the derivatives of 6,8-dimethylergoline or the tricyclic precursors (i.e., agroclavine, elymoclavine, and festuclavine).
While naturally occurring ergolines generally lack selectivity between dopamine receptor subtypes and other biogenic amine receptors, a select number of semisynthetic ergolines display a preference toward dopamine receptors.194 Several ergolines are used as antiparkinsonian drugs [e.g., bromocriptine (81), lisuride (82), and pergolide (83), Figure ] and inhibitors of prolactin release [e.g., 81–83 and cabergoline (84)] due to their agonist activity at dopamine receptors.197,198 The simulation of neostriatal D1 and D2 receptors is associated with the antiparkinsonian effect of ergolines, whereas the simulation of D2 or D4 receptors mediates the suppression of prolactin secretion from the anterior pituitary.199,200
Reduction of the methyl ester of lysergic acid with lithium aluminum hydride yields lysergol (85), a key intermediate in the preparation of semisynthetic ergoline analogs.201 As the hydroxy group of lysergol could undergo nucleophilic substitution upon conversion to sulfonic ester (e.g., mesylate or tosylate), researchers at Lilly were able to introduce a methylthio moiety leading to pergolide.202 Zikán and Semonský at the Research Institute for Pharmacy and Biochemistry at Prague (later SPOFA) developed lisuride via derivation of methysergide (86) in efforts toward the development of an antimigraine agent.203 In 1954, Moses Shelesnyak recognized that ergotoxine (87) could inhibit deciduoma formation that was associated with ovum implantation in rats, attributable to inhibition of prolactin secretion.201,204 This led to researchers at Sandoz developing ergot analogs that selectively inhibited prolactin secretion, leading to the development of bromocriptine.201,205
Distal substituents on an ergoline-derived scaffold by combining two privileged scaffolds, indole and phenylpiperazine (88–90), yielded potent D2R ligands with intrinsic activities ranging from full agonism to partial agonism.206 Alkylation on the nitrogen on the piperazine moiety led to a series of D2R agonists with a wide range of binding affinities with the most potent D2LR ligand bearing an N-(1,1′-biphenyl-3-yl)methyl group (88) (Ki = 0.65 nM, Emax = 35%). An inverse relationship was observed between the size of the amine substituent and the D2R efficacy, where smaller alkyl substituents are full agonists (e.g., 89 and 90). Alkylation of the nitrogen on the indole moiety resulted in ligands with serotonin 5-HT6 receptor affinity.
On both a functional and a molecular level, adenosine and dopamine receptors have interactions with each other as heteromeric complexes.207 Adenosine receptor-selective antagonists display antiparkinsonian activities and exhibit neuroprotective properties (rarely seen with currently used antiparkinsonian drugs). As such, combining adenosine A1 receptor (A1R) and A2AR antagonism with dopamine D1R and D2R agonism into a single multivalent ligand could lead to novel treatments for Parkinson’s disease with improved efficacy and safety.208,209 As ergot alkaloids are privileged structures with an inherent ability to interact with diverse biological receptors, a combinatorial approach based on the ergoline scaffold allowed the identification of compounds with dual adenosine antagonist and dopamine agonist activity as exemplified by 91 (A1R KD = 0.10 μM, D2highR KD = 2 μM).210
Miscellaneous Dopamine Receptor-Targeted Natural Products
Cannabidiol (CBD, 92, Figure ) is a phytocannabinoid found in the Cannabis sativa plant, accounting for up to 40% of the plant’s extract.211 Compared to Δ9-tetrahydrocannabinol (Δ9-THC, 93), CBD lacks psychotomimetic and other psychotropic effects. Δ9-THC displayed binding affinity and functional activity (using a GTPγS functional bioassay) in the nanomolar range for both cannabinoid receptors [(Ki (CB1) = 18 nM, Ki (CB2) = 42 nM)] [(EC50 (CB1) = 269 nM, EC50 (CB2) = 327 nM)].212 With the exception of functional activity at CB2, CBD displays lower binding affinity and functional activity than Δ9-THC at the cannabinoid receptors [(Ki (CB1) = 151 nM, Ki (CB2) = 4582 nM)] [(EC50 (CB1) = 1469 nM, EC50 (CB2) = 104 nM)].
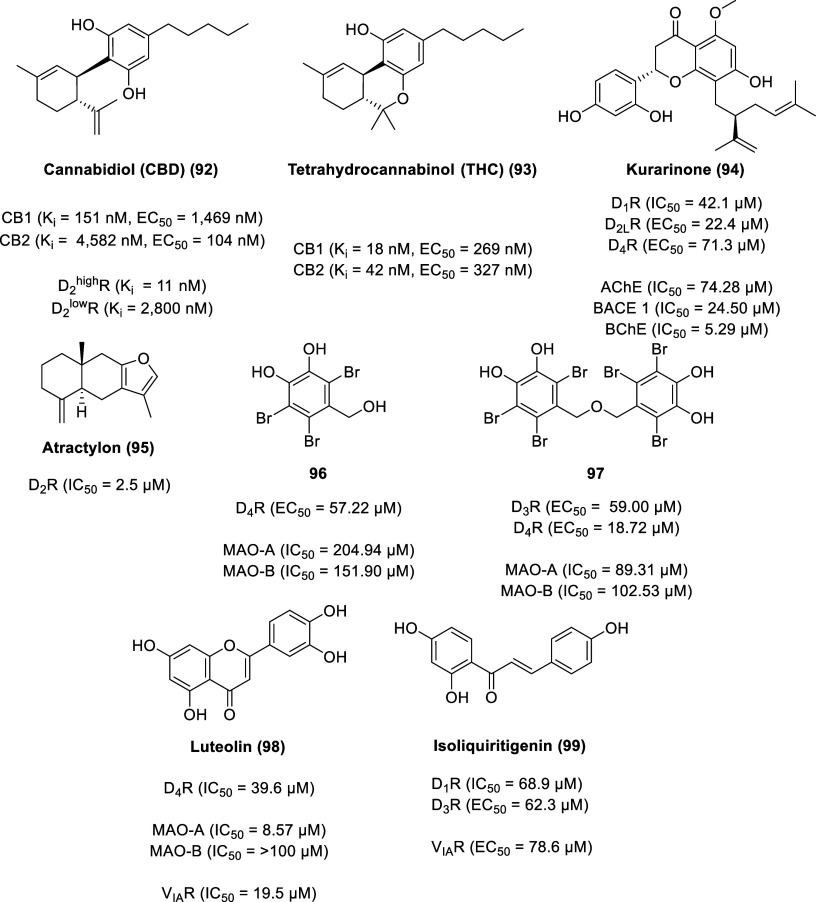
CBD is FDA approved for the treatment of seizures associated with Lennox–Gastaut syndrome, Dravet syndrome, and tuberous sclerosis complex in patients 1 year of age and older (under the brand name Epidiolex). CBD is implicated in other medical illnesses including anxiety, psychosis, and depression, although it is not FDA approved for these conditions.211,213 The clinical antipsychotic effects of CBD correlate with its partial agonist activity at D2highR (Ki D2highR = 11 nM, Ki D2lowR = 2800 nM) in a similar biphasic manner as aripiprazole, a dopamine partial agonist antipsychotic drug.214
Kurarinone (94) is an abundant lavandulylated flavonoid isolated from the perennial shrub Sophora flavescens (Fabaceae).215 Known as Kushen, the dried roots of S. flavescens are used in traditional Chinese medicine for the treatment of cancer and inflammatory diseases.216 Kurarinone displays moderate to potent inhibitory activity on acetylcholinesterase (AChE, IC50 = 74.28 μM), beta-secretase 1 (BACE 1, IC50 = 24.50 μM), and butyrylcholinesterase (BChE, IC50 = 5.29 μM) using either Ellman’s colorimetric method (for AChE and BChE) or with BACE1 FRET assay kit (β-Secretase) kit.217,218 Functional HTRF-based cAMP assays (for D1R and D4R) and Ca2+ flux assays (for D2LR) revealed kurarinone exhibited an antagonist effect on D1R (IC50 = 42.1 μM) and an agonist effect on D2LR (EC50 = 22.4 μM) and D4R (EC50 = 71.3 μM), suggesting its potential to alleviate various neurodegenerative diseases (NDDs).215
Isolated from the perennial herb Atractylodes macrocephala Koidz, atractylon (95) is a furan-containing compound with several pharmacological activities, including anticancer, antiviral, anti-inflammation, and gastroprotective properties.219 Recently, it was demonstrated that atractylon is a D2R agonist in a dose-dependent manner (using a piggyBac-TANGO assay) with an IC50 of 2.5 μM.220 Atractylon attenuated motor deficits and abnormal gait disturbances as well as protected dopaminergic neurons in MPTP-induced PD mice, indicating potential as a therapeutic agent in the treatment of motor symptoms of Parkinson’s disease.
Monoamine oxidase (MAO) is a mammalian flavoenzyme that catalyzes the inactivation pathway of several neurotransmitters (e.g., adrenaline, dopamine, norepinephrine, and serotonin) through oxidative deamination.221 In the biochemical process, oxidative damage can occur as hydrogen peroxide is the major and direct side product of MAO activity. Thus, inhibitors of MAO have neuroprotective properties.222 MAO exists in two isoforms, MAO-A and MAO-B, with different tissue distribution and substrate-inhibitor recognition sites. Bromophenols (96 and 97) from the red alga Symphyocladia latiuscula are good D3R/D4R agonists and are moderate MAO inhibitors and as such are implicated in the management of neurodegenerative diseases.223 Compound 96 displayed inhibition of MAO (MAO-A IC50 = 204.94 μM, MAO-B IC50 = 151.90 μM) and had a potent agonist effect with D4R (EC50 = 57.22 μM), whereas 97 displayed inhibition of MAO (MAO-A IC50 = 89.31 μM, MAO-B IC50 = 102.53 μM) and had an agonist effect with both D4R (EC50 = 18.72 μM) and D3R (EC50 = 59.00 μM) (calculated from a log dose–inhibition curve or concentration-dependent agonist response curve).
Luteolin (98) is another flavonoid, widely distributed in the plant kingdom (e.g., Artemisia montana, Cirsium japonicum, Cynara scolymus, and Taraxacum officinale).224 Luteolin exhibited selective inhibition of MAO-A [MAO-A (IC50 = 8.57 μM), MAO-B (IC50 > 100 μM)] and is a selective antagonist of D4R (IC50 = 39.6 μM) and vasopressin receptor 1A (VIAR) (IC50 = 19.5 μM) calculated from a dose–response curve.
Isoliquiritigenin (99), isolated from Glycyrrhizae rhizoma, is a potent MAO inhibitor (MAO-A IC50 = 0.68 μM, MAO-B IC50 = 0.33 μM), measured by Promega’s MAO-Glo assay system.225 Isoliquiritigenin also displays modulatory functions on dopamine receptors (D1R IC50 = 68.9 μM, D3R EC50 = 62.3 μM) measured by HTRF-based cAMP assay and on VIAR (EC50 = 78.6 μM) measured by a fluorescent Ca2+ influx assay.
Conclusion and Perspectives
Natural products are an invaluable resource as lead compounds in drug discovery, inspiring a countless number of drugs in the treatment of debilitating diseases and illnesses. Despite the prevailing notion highlighting the potential for dopamine receptor-targeted ligands as therapeutic agents for the treatment of numerous diseases and illnesses, currently available ligands have limited success due to inadequacy in selectivity and other pharmacological properties. Subtype-selective dopaminergic drugs are lacking in the market.
Among the natural product scaffolds described, aporphines, THPBs, and ergolines have received the most scientific attention in relation to their dopamine receptor targeting. However, the cularines and miscellaneous natural products described have been relatively underexplored in this regard.
The development of functionally selective dopamine receptor tools is an area where natural product templates can have a significant impact. The identification of β-arrestin-biased D2R antagonists from the aporphine scaffold makes for a promising outlook in that direction.
Compared to the one-target, one-disease paradigm, polypharmacology enables compounds to have enhanced efficacy and safety through synergetic interactions. There is clear evidence that some diseases with complex etiology and/or symptomatology may be remedied optimally by drugs with multiple pharmacological actions. As privileged dopamine receptor-targeted scaffolds, aporphines, THPBs, and ergolines, in particular, offer a unique entry point to develop ligands with multireceptor activities.
While significant progress has been made in the development of dopamine receptor agents, it is clear that further progress is needed as the pharmacological properties can still be improved upon. Dopamine receptor-targeted natural products still have untapped potential in the development of therapeutic and investigative agents, and it is anticipated that they will continue to form a nucleus of inspiration for synthetic studies and drug discovery.
References
- J. W. Li, J. C. Vederas. Drug discovery and natural products: end of an era or an endless frontier?.. Science, 2009. [DOI | PubMed]
- E. Patridge, P. Gareiss, M. S. Kinch, D. Hoyer. An analysis of FDA-approved drugs: natural products and their derivatives.. Drug Discovery Today, 2016. [DOI | PubMed]
- R. A. Shenvi. Natural Product Synthesis in the 21st Century: Beyond the Mountain Top.. ACS Cent. Sci., 2024. [DOI | PubMed]
- P. Ertl, S. Roggo, A. Schuffenhauer. Natural product-likeness score and its application for prioritization of compound libraries.. J. Chem. Inf. Model., 2008. [DOI | PubMed]
- S. S. Bharate, S. Mignani, R. A. Vishwakarma. Why Are the Majority of Active Compounds in the CNS Domain Natural Products? A Critical Analysis.. J. Med. Chem., 2018. [DOI | PubMed]
- K. Sriram, P. A. Insel. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs?.. Mol. Pharmacol., 2018. [DOI | PubMed]
- K. A. Neve. The dopamine receptors;, 2010
- Z. U. Khan, L. Mrzljak, A. Gutierrez, A. de la Calle, P. S. Goldman-Rakic. Prominence of the dopamine D2 short isoform in dopaminergic pathways.. Proc. Natl. Acad. Sci. U S A, 1998. [DOI | PubMed]
- D. L. Gray, J. A. Allen, S. Mente, R. E. O’Connor, G. J. DeMarco, I. Efremov, P. Tierney, D. Volfson, J. Davoren, E. Guilmette. Impaired beta-arrestin recruitment and reduced desensitization by non-catechol agonists of the D1 dopamine receptor.. Nat. Commun., 2018. [DOI | PubMed]
- Z. U. Khan, L. Mrzljak, A. Gutierrez, A. de la Calle, P. S. Goldman-Rakic. Prominence of the dopamine D2 short isoform in dopaminergic pathways.. Proc. Natl. Acad. Sci. U S A, 1998. [DOI | PubMed]
- D. L. Gray, J. A. Allen, S. Mente, R. E. O’Connor, G. J. DeMarco, I. Efremov, P. Tierney, D. Volfson, J. Davoren, E. Guilmette. Impaired beta-arrestin recruitment and reduced desensitization by non-catechol agonists of the D1 dopamine receptor.. Nat. Commun., 2018. [DOI | PubMed]
- E. Von Moo, K. Harpsøe, A. S. Hauser, I. Masuho, H. Bräuner-Osborne, D. E. Gloriam, K. A. Martemyanov. Ligand-directed bias of G protein signaling at the dopamine D2 receptor.. Cell Chemical Biology, 2022. [DOI | PubMed]
- Y. Yang. Functional Selectivity of Dopamine D(1) Receptor Signaling: Retrospect and Prospect.. Int. J. Mol. Sci., 2021. [DOI | PubMed]
- J. D. Violin, A. L. Crombie, D. G. Soergel, M. W. Lark. Biased ligands at G-protein-coupled receptors: promise and progress.. Trends Pharmacol. Sci., 2014. [DOI | PubMed]
- T. Kenakin. Biased Receptor Signaling in Drug Discovery.. Pharmacol. Rev., 2019. [DOI | PubMed]
- T. Kenakin. Functional selectivity and biased receptor signaling.. J. Pharmacol. Exp. Ther., 2011. [DOI | PubMed]
- S. D. Chang, M. R. Bruchas. Functional selectivity at GPCRs: new opportunities in psychiatric drug discovery.. Neuropsychopharmacology, 2014. [DOI | PubMed]
- M. L. Martini, J. Liu, C. Ray, X. Yu, X. P. Huang, A. Urs, N. Urs, J. D. McCorvy, M. G. Caron, B. L. Roth. Defining Structure-Functional Selectivity Relationships (SFSR) for a Class of Non-Catechol Dopamine D1 Receptor Agonists.. J. Med. Chem., 2019. [DOI | PubMed]
- H. Yano, N. S. Cai, M. Xu, R. K. Verma, W. Rea, A. F. Hoffman, L. Shi, J. A. Javitch, A. Bonci, S. Ferre. Gs- versus Golf-dependent functional selectivity mediated by the dopamine D1 receptor.. Nat. Commun., 2018. [DOI | PubMed]
- L. M. Luttrell, F. L. Roudabush, E. W. Choy, W. E. Miller, M. E. Field, K. L. Pierce, R. J. Lefkowitz. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds.. Proc. Natl. Acad. Sci. U S A, 2001. [DOI | PubMed]
- N. M. Urs, S. Bido, S. M. Peterson, T. L. Daigle, C. E. Bass, R. R. Gainetdinov, E. Bezard, M. G. Caron. Targeting beta-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease.. Proc. Natl. Acad. Sci. U S A, 2015. [DOI | PubMed]
- L. Tan, W. Yan, J. D. McCorvy, J. Cheng. Biased Ligands of G Protein-Coupled Receptors (GPCRs): Structure-Functional Selectivity Relationships (SFSRs) and Therapeutic Potential.. J. Med. Chem., 2018. [DOI | PubMed]
- J. D. Violin, A. L. Crombie, D. G. Soergel, M. W. Lark. Biased ligands at G-protein-coupled receptors: promise and progress.. Trends Pharmacol. Sci., 2014. [DOI | PubMed]
- C. L. Schmid, N. M. Kennedy, N. C. Ross, K. M. Lovell, Z. Yue, J. Morgenweck, M. D. Cameron, T. D. Bannister, L. M. Bohn. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics.. Cell, 2017. [DOI | PubMed]
- K. L. White, J. E. Robinson, H. Zhu, J. F. DiBerto, P. R. Polepally, J. K. Zjawiony, D. E. Nichols, C. J. Malanga, B. L. Roth. The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo.. J. Pharmacol. Exp. Ther., 2015. [DOI | PubMed]
- J. M. Beaulieu, S. Espinoza, R. R. Gainetdinov. Dopamine receptors – IUPHAR Review 13.. Br. J. Pharmacol., 2015. [DOI | PubMed]
- A. Antonini, E. Tolosa. Apomorphine and levodopa infusion therapies for advanced Parkinson’s disease: selection criteria and patient management.. Expert Rev. Neurother., 2009. [DOI | PubMed]
- M. Wood, V. Dubois, D. Scheller, M. Gillard. Rotigotine is a potent agonist at dopamine D1 receptors as well as at dopamine D2 and D3 receptors.. Br. J. Pharmacol., 2015. [DOI | PubMed]
- R. N. Brogden, A. Markham. Fenoldopam: a review of its pharmacodynamic and pharmacokinetic properties and intravenous clinical potential in the management of hypertensive urgencies and emergencies.. Drugs, 1997. [DOI | PubMed]
- A. J. Nichols, R. R. Ruffolo, D. P. Brooks. The pharmacology of fenoldopam.. Am. J. Hypertens., 1990. [DOI | PubMed]
- A. Grenader, D. P. Healy. Fenoldopam is a partial agonist at dopamine-1 (DA1) receptors in LLC-PK1 cells.. J. Pharmacol. Exp. Ther., 1991. [PubMed]
- C. E. McCoy, F. L. Douglas, L. I. Goldberg. Selective antagonism of the hypotensive effects of dopamine agonists in spontaneously hypertensive rats.. Hypertension, 1986. [DOI | PubMed]
- C. Zeng, M. Zhang, L. D. Asico, G. M. Eisner, P. A. Jose. The dopaminergic system in hypertension.. Clin. Sci. (Lond), 2007. [DOI | PubMed]
- T. Nakako, T. Murai, M. Ikejiri, T. Ishiyama, M. Taiji, K. Ikeda. Effects of a dopamine D1 agonist on ketamine-induced spatial working memory dysfunction in common marmosets.. Behav. Brain Res., 2013. [DOI | PubMed]
- J. X. Cai, A. F. Arnsten. Dose-dependent effects of the dopamine D1 receptor agonists A77636 or SKF81297 on spatial working memory in aged monkeys.. J. Pharmacol. Exp. Ther., 1997. [PubMed]
- S. A. Castner, P. S. Goldman-Rakic. Enhancement of working memory in aged monkeys by a sensitizing regimen of dopamine D1 receptor stimulation.. J. Neurosci., 2004. [DOI | PubMed]
- S. A. Castner, P. S. Goldman-Rakic. Enhancement of working memory in aged monkeys by a sensitizing regimen of dopamine D1 receptor stimulation.. J. Neurosci., 2004. [DOI | PubMed]
- A. Hall, L. Provins, A. Valade. Novel Strategies To Activate the Dopamine D(1) Receptor: Recent Advances in Orthosteric Agonism and Positive Allosteric Modulation.. J. Med. Chem., 2019. [DOI | PubMed]
- Fixed-Dose Trial in Early Parkinson’s Disease (PD) (TEMPO-1); https://clinicaltrials.gov/study/NCT04201093?term=Tavapadon&aggFilters=status:act&rank=2 (accessed June 30, 2024).
- Flexible-Dose Trial in Early Parkinson’s Disease (PD) (TEMPO-2); https://clinicaltrials.gov/study/NCT04223193?term=Tavapadon&aggFilters=status:act&rank=1 (accessed June 30, 2024).
- R. E. Chipkin, L. C. Iorio, V. L. Coffin, R. D. McQuade, J. G. Berger, A. Barnett. Pharmacological profile of SCH39166: a dopamine D1 selective benzonaphthazepine with potential antipsychotic activity.. J. Pharmacol. Exp. Ther., 1988. [PubMed]
- C. E. Tedford, V. L. Coffin, V. Ruperto, M. Cohen, R. D. McQuade, R. Johnson, H. K. Kim, C. C. Lin. Determination of plasma and brain concentrations of SCH 39166 and their correlation to conditioned avoidance behavior in rats.. Psychopharmacology (Berl), 1993. [DOI | PubMed]
- C. E. Tedford, V. B. Ruperto, A. Barnett. Characterization of a rat liver glucuronosyltransferase that glucuronidates the selective D1 antagonist, SCH 23390, and other benzazepines.. Drug Metab. Dispos., 1991. [PubMed]
- P. Karlsson, L. Smith, L. Farde, C. Harnryd, G. Sedvall, F. A. Wiesel. Lack of apparent antipsychotic effect of the D1-dopamine receptor antagonist SCH39166 in acutely ill schizophrenic patients.. Psychopharmacology (Berl), 1995. [DOI | PubMed]
- J. A. Den Boer, H. J. van Megen, W. W. Fleischhacker, J. W. Louwerens, B. R. Slaap, H. G. Westenberg, G. D. Burrows, O. N. Srivastava. Differential effects of the D1-DA receptor antagonist SCH39166 on positive and negative symptoms of schizophrenia.. Psychopharmacology (Berl), 1995. [DOI | PubMed]
- M. Haney, A. S. Ward, R. W. Foltin, M. W. Fischman. Effects of ecopipam, a selective dopamine D1 antagonist, on smoked cocaine self-administration by humans.. Psychopharmacology (Berl), 2001. [DOI | PubMed]
- E. Nann-Vernotica, E. C. Donny, G. E. Bigelow, S. L. Walsh. Repeated administration of the D1/5 antagonist ecopipam fails to attenuate the subjective effects of cocaine.. Psychopharmacology (Berl), 2001. [DOI | PubMed]
- A. Astrup, F. L. Greenway, W. Ling, L. Pedicone, J. Lachowicz, C. D. Strader, R. Kwan, G. Ecopipam Obesity Study. Randomized controlled trials of the D1/D5 antagonist ecopipam for weight loss in obese subjects.. Obesity (Silver Spring), 2007. [DOI | PubMed]
- A. A. Coulter, C. J. Rebello, F. L. Greenway. Centrally Acting Agents for Obesity: Past, Present, and Future.. Drugs, 2018. [DOI | PubMed]
- A Study to Evaluate Long-term Safety of Ecopipam Tablets in Children, Adolescents and Adults With Tourette’s Disorder; https://classic.clinicaltrials.gov/ct2/show/NCT06021522?term=Ecopipam&phase=23&draw=2&rank=1 (accessed. June 26, 2024).
- Ecopipam Tablets to Study Tourette’s Disorder in Children, Adolescents and Adults (D1AMOND); https://classic.clinicaltrials.gov/ct2/show/NCT05615220?term=Ecopipam&phase=23&draw=2&rank=3 (accessed. June 26, 2024).
- D. R. Sibley, F. J. Monsma. Molecular biology of dopamine receptors.. Trends Pharmacol. Sci., 1992. [DOI | PubMed]
- B. A. Bricker, K. Peprah, H. J. Kang, S. Y. Ablordeppey. Evaluation of SYA16263 as a new potential antipsychotic agent without catalepsy.. Pharmacol., Biochem. Behav., 2019. [DOI | PubMed]
- W. K. Kroeze, S. J. Hufeisen, B. A. Popadak, S. M. Renock, S. Steinberg, P. Ernsberger, K. Jayathilake, H. Y. Meltzer, B. L. Roth. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs.. Neuropsychopharmacology, 2003. [DOI | PubMed]
- M. Corena-McLeod. Comparative Pharmacology of Risperidone and Paliperidone.. Drugs R D, 2015. [DOI | PubMed]
- K. Yonemura, K. Miyanaga, Y. Machiyama. Profiles of the affinity of antipsychotic drugs for neurotransmitter receptors and their clinical implication.. Kitakanto Medical Journal, 1998. [DOI]
- Y. von Coburg, T. Kottke, L. Weizel, X. Ligneau, H. Stark. Potential utility of histamine H3 receptor antagonist pharmacophore in antipsychotics.. Bioorg. Med. Chem. Lett., 2009. [DOI | PubMed]
- D. Aslanoglou, S. Bertera, M. Sanchez-Soto, R. Benjamin Free, J. Lee, W. Zong, X. Xue, S. Shrestha, M. Brissova, R. W. Logan. Dopamine regulates pancreatic glucagon and insulin secretion via adrenergic and dopaminergic receptors.. Transl. Psychiatry, 2021. [DOI | PubMed]
- P. Stepnicki, M. Kondej, A. A. Kaczor. Current Concepts and Treatments of Schizophrenia.. Molecules, 2018. [DOI | PubMed]
- A. H. Newman, Z. X. Xi, C. Heidbreder. Current Perspectives on Selective Dopamine D(3) Receptor Antagonists/Partial Agonists as Pharmacotherapeutics for Opioid and Psychostimulant Use Disorders.. Curr. Top. Behav. Neurosci., 2022. [DOI]
- E. M. Pich, G. Collo. Pharmacological targeting of dopamine D3 receptors: Possible clinical applications of selective drugs.. Eur. Neuropsychopharmacol., 2015. [DOI | PubMed]
- S. Ferre, A. M. Belcher, J. Bonaventura, C. Quiroz, M. Sanchez-Soto, V. Casado-Anguera, N. S. Cai, E. Moreno, C. A. Boateng, T. M. Keck. Functional and pharmacological role of the dopamine D(4) receptor and its polymorphic variants.. Front Endocrinol (Lausanne), 2022. [DOI | PubMed]
- L. Botticelli, E. Micioni Di Bonaventura, F. Del Bello, G. Giorgioni, A. Piergentili, A. Romano, W. Quaglia, C. Cifani, M. V. Micioni Di Bonaventura. Underlying Susceptibility to Eating Disorders and Drug Abuse: Genetic and Pharmacological Aspects of Dopamine D4 Receptors.. Nutrients, 2020. [DOI | PubMed]
- M. Dorfler, N. Tschammer, K. Hamperl, H. Hubner, P. Gmeiner. Novel D3 selective dopaminergics incorporating enyne units as nonaromatic catechol bioisosteres: synthesis, bioactivity, and mutagenesis studies.. J. Med. Chem., 2008. [DOI | PubMed]
- R. K. Sunahara, H. C. Guan, B. F. O’Dowd, P. Seeman, L. G. Laurier, G. Ng, S. R. George, J. Torchia, H. H. Van Tol, H. B. Niznik. Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1.. Nature, 1991. [DOI | PubMed]
- M. Sanchez-Soto, A. Bonifazi, N. S. Cai, M. P. Ellenberger, A. H. Newman, S. Ferre, H. Yano. Evidence for Noncanonical Neurotransmitter Activation: Norepinephrine as a Dopamine D2-Like Receptor Agonist.. Mol. Pharmacol., 2016. [DOI | PubMed]
- J. L. Conroy, R. B. Free, D. R. Sibley. Identification of G Protein-Biased Agonists That Fail To Recruit β-Arrestin or Promote Internalization of the D1 Dopamine Receptor.. ACS Chem. Neurosci., 2015. [DOI | PubMed]
- J. E. Hardebo, C. Owman. Barrier mechanisms for neurotransmitter monoamines and their precursors at the blood-brain interface.. Ann. Neurol., 1980. [DOI | PubMed]
- J. Sonne, A. Goyal, W. Lopez-Ojeda. Dopamine.. StatPearls [Internet];, 2023
- O. Hornykiewicz. L-DOPA: from a biologically inactive amino acid to a successful therapeutic agent.. Amino Acids, 2002. [DOI | PubMed]
- K. J. Tang, Y. Zhao, X. Tao, J. Li, Y. Chen, D. C. Holland, T. Y. Jin, A. Y. Wang, L. Xiang. Catecholamine Derivatives: Natural Occurrence, Structural Diversity, and Biological Activity.. J. Nat. Prod., 2023. [DOI | PubMed]
- M. A. Cenci, M. Lundblad. Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia.. J. Neurochem., 2006. [DOI | PubMed]
- H. Ehringer, O. Hornykiewicz. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system.. Parkinsonism Relat. Disord., 1998. [DOI | PubMed]
- M. A. Cenci. Presynaptic Mechanisms of l-DOPA-Induced Dyskinesia: The Findings, the Debate, and the Therapeutic Implications.. Front. Neurol., 2014. [DOI | PubMed]
- O. S. Gershanik. Improving L-dopa therapy: the development of enzyme inhibitors.. Mov. Disord., 2015. [DOI | PubMed]
- D. R. Hill, A. D. Huters, T. B. Towne, R. E. Reddy, J. L. Fogle, E. A. Voight, P. R. Kym. Parkinson’s Disease: Advances in Treatment and the Syntheses of Various Classes of Pharmaceutical Drug Substances.. Chem. Rev., 2023. [DOI | PubMed]
- D. Nyholm. Duodopa(R) treatment for advanced Parkinson’s disease: a review of efficacy and safety.. Parkinsonism Relat. Disord., 2012. [DOI | PubMed]
- M. Rosebraugh, E. A. Voight, E. M. Moussa, F. Jameel, X. Lou, G. G. Z. Zhang, P. T. Mayer, D. Stolarik, R. A. Carr, B. P. Enright. Foslevodopa/Foscarbidopa: A New Subcutaneous Treatment for Parkinson’s Disease.. Ann. Neurol., 2021. [DOI | PubMed]
- M. Rosebraugh, P. Kym, W. Liu, M. Facheris, J. Benesh. A Novel Levodopa/Carbidopa Prodrug (ABBV-951) 24-h Continuous Subcutaneous Infusion Treatment for Parkinson’s Disease (P3.8–037).. Neurology, 2019. [DOI]
- K. Leung. L-3,4-Dihydroxy-6-[(18)F]fluorophenylalanine.. Molecular Imaging and Contrast Agent Database (MICAD),, 2004
- I. M. Menendez-Perdomo, P. J. Facchini. Benzylisoquinoline Alkaloids Biosynthesis in Sacred Lotus.. Molecules, 2018. [DOI | PubMed]
- R. Stadler, T. M. Kutchan, M. H. Zenk. (S)-norcoclaurine is the central intermediate in benzylisoquinoline alkaloid biosynthesis.. Phytochemistry, 1989. [DOI]
- J. J. Xu, X. Fang, C. Y. Li, L. Yang, X. Y. Chen. General and specialized tyrosine metabolism pathways in plants.. aBIOTECH, 2020. [DOI | PubMed]
- N. Cabedo, I. Berenguer, B. Figadere, D. Cortes. An overview on benzylisoquinoline derivatives with dopaminergic and serotonergic activities.. Curr. Med. Chem., 2009. [DOI | PubMed]
- I. Berenguer, N. E. Aouad, S. Andujar, V. Romero, F. Suvire, T. Freret, A. Bermejo, M. D. Ivorra, R. D. Enriz, M. Boulouard. Tetrahydroisoquinolines as dopaminergic ligands: 1-Butyl-7-chloro-6-hydroxy-tetrahydroisoquinoline, a new compound with antidepressant-like activity in mice.. Bioorg. Med. Chem., 2009. [DOI | PubMed]
- A. Bermejo, P. Protais, M. A. Blazquez, K. S. Rao, M. C. Zafra-polo, D. Cortes. Dopaminergic Isoquinoline Alkaloids from Roots of Xylopia papuana.. Natural Product Letters, 1995. [DOI]
- P. G. Jamkhande, A. S. Wattamwar. Annona reticulata Linn. (Bullock’s heart): Plant profile, phytochemistry and pharmacological properties.. J. Tradit. Complement. Med., 2015. [DOI | PubMed]
- D. S. Bhakuni, S. Jain. Alkaloids of cocculus laurifolius D.C.. Tetrahedron, 1980. [DOI]
- N. Cabedo, P. Protais, B. K. Cassels, D. Cortes. Synthesis and dopamine receptor selectivity of the benzyltetrahydroisoquinoline, (R)-(+)-nor-roefractine.. J. Nat. Prod., 1998. [DOI | PubMed]
- A. G. Silva, L. Vila, P. Marques, L. Moreno, M. Loza, M. J. Sanz, D. Cortes, M. Castro, N. Cabedo. 1-(2’-Bromobenzyl)-6,7-dihydroxy-N-methyl-tetrahydroisoquinoline and 1,2-Demethyl-nuciferine as Agonists in Human D(2) Dopamine Receptors.. J. Nat. Prod., 2020. [DOI | PubMed]
- H. L. Wei, Y. P. Zhao, J. X. Wang, Y. Han, H. Li, H. Zhou, T. Hou, C. J. Wang, Y. M. Yao, X. L. Zhang. Menisperdaurines A-W, structurally diverse isoquinoline alkaloids from Menispermum dauricum and their dopamine D1 receptor activities.. Bioorg. Chem., 2022. [DOI | PubMed]
- J. Párraga, A. Galán, M. J. Sanz, N. Cabedo, D. Cortes. Synthesis of hexahydrocyclopenta[ij]isoquinolines as a new class of dopaminergic agents.. Eur. J. Med. Chem., 2015. [DOI | PubMed]
- D. Cortes, B. Figadere, J. Saez, P. Protais. Displacement Activity of Bisbenzylisoquinoline Alkaloids at Striatal 3H-SCH 23390 and 3H-Raclopride Binding Sites.. J. Nat. Prod., 1992. [DOI | PubMed]
- A. Zhang, Y. Zhang, A. R. Branfman, R. J. Baldessarini, J. L. Neumeyer. Advances in development of dopaminergic aporphinoids.. J. Med. Chem., 2007. [DOI | PubMed]
- R. Zhu, G. Jiang, W. Tang, X. Zhao, F. Chen, X. Zhang, N. Ye. Aporphines: A privileged scaffold in CNS drug discovery.. Eur. J. Med. Chem., 2023. [DOI | PubMed]
- J. Sun, X. Zhan, W. Wang, X. Yang, Y. Liu, H. Yang, J. Deng, H. Yang. Natural aporphine alkaloids: A comprehensive review of phytochemistry, pharmacokinetics, anticancer activities, and clinical application.. J. Adv. Res., 2023. [DOI]
- M. Shamma, H. Guinaudeau. Aporphinoid alkaloids.. Nat. Prod. Rep., 1986. [DOI | PubMed]
- M. J. Millan, L. Maiofiss, D. Cussac, V. Audinot, J. A. Boutin, A. Newman-Tancredi. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes.. J. Pharmacol. Exp. Ther., 2002. [DOI | PubMed]
- A. Newman-Tancredi, D. Cussac, V. Audinot, J. P. Nicolas, F. De Ceuninck, J. A. Boutin, M. J. Millan. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. II. Agonist and antagonist properties at subtypes of dopamine D(2)-like receptor and alpha(1)/alpha(2)-adrenoceptor.. J. Pharmacol Exp Ther, 2002. [DOI | PubMed]
- A. Newman-Tancredi, D. Cussac, Y. Quentric, M. Touzard, L. Verriele, N. Carpentier, M. J. Millan. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. III. Agonist and antagonist properties at serotonin, 5-HT(1) and 5-HT(2), receptor subtypes.. J. Pharmacol. Exp. Ther., 2002. [DOI | PubMed]
- I. Ul Haq, P. A Lewitt, H. H Fernandez. Apomorphine therapy in Parkinson’s disease: a review.. Expert Opin. Pharmacother., 2007. [DOI | PubMed]
- J. A. Subramony. Apomorphine in dopaminergic therapy.. Mol. Pharmaceutics, 2006. [DOI]
- J. J. Chen, C. Obering. A review of intermittent subcutaneous apomorphine injections for the rescue management of motor fluctuations associated with advanced Parkinson’s disease.. Clin. Ther., 2005. [DOI | PubMed]
- M. Auffret, S. Drapier, M. Verin. Pharmacological Insights into the Use of Apomorphine in Parkinson’s Disease: Clinical Relevance.. Clin. Drug Investig., 2018. [DOI]
- F. Carbone, A. Djamshidian, K. Seppi, W. Poewe. Apomorphine for Parkinson’s Disease: Efficacy and Safety of Current and New Formulations.. CNS Drugs, 2019. [DOI | PubMed]
- S. T. Gancher, J. G. Nutt, W. R. Woodward. Absorption of apomorphine by various routes in parkinsonism.. Mov Disord, 1991. [DOI | PubMed]
- D. L. Corboy, M. L. Wagner, J. I. Sage. Apomorphine for motor fluctuations and freezing in Parkinson’s disease.. Ann. Pharmacother., 1995. [DOI | PubMed]
- N. Ye, J. L. Neumeyer, R. J. Baldessarini, X. Zhen, A. Zhang. Update 1 of: Recent progress in development of dopamine receptor subtype-selective agents: potential therapeutics for neurological and psychiatric disorders.. Chem. Rev., 2013. [DOI | PubMed]
- M. V. Koch, J. G. Cannon, A. M. Burkman. Centrally acting emetics. II. Norapomorphine and derivatives.. J. Med. Chem., 1968. [DOI | PubMed]
- M. K. Menon, W. G. Clark, J. L. Neumeyer. Comparison of the dopaminergic effects of apomorphine and (−)-N-n-propylnorapomorphine.. Eur. J. Pharmacol., 1978. [DOI | PubMed]
- J. N. Jacob, D. E. Nichols, J. D. Kohli, D. Glock. Dopamine agonist properties of N-alkyl-4-(3,4-dihydroxyphenyl)-1,2,3,4-tetrahydroisoquinolines.. J. Med. Chem., 1981. [DOI | PubMed]
- C. Csutoras, A. Zhang, K. Zhang, N. S. Kula, R. J. Baldessarini, J. L. Neumeyer. Synthesis and neuropharmacological evaluation of R(−)-N-alkyl-11-hydroxynoraporphines and their esters.. Bioorg. Med. Chem., 2004. [DOI | PubMed]
- Y. G. Si, M. P. Gardner, F. I. Tarazi, R. J. Baldessarini, J. L. Neumeyer. Synthesis and dopamine receptor affinities of N-alkyl-11-hydroxy-2-methoxynoraporphines: N-alkyl substituents determine D1 versus D2 receptor selectivity.. J. Med. Chem., 2008. [DOI | PubMed]
- E. Y. Melikhova, N. Derrien, N. Fleary-Roberts, S. M. Forsyth, S. Papadouli, A. M. Ruda, V. G. Thangavadivale, S. N. G. Tyler, J. D. Moseley. Early Process Development and Scale-Up of Orally Active Apomorphine Drug Candidates.. Org. Process Res. Dev., 2023. [DOI]
- A. Graff-Guerrero, R. Mizrahi, O. Agid, H. Marcon, P. Barsoum, P. Rusjan, A. A. Wilson, R. Zipursky, S. Kapur. The dopamine D2 receptors in high-affinity state and D3 receptors in schizophrenia: a clinical [11C]-(+)-PHNO PET study.. Neuropsychopharmacology, 2009. [DOI | PubMed]
- A. W. Sromek, Y. G. Si, T. Zhang, S. R. George, P. Seeman, J. L. Neumeyer. Synthesis and Evaluation of Fluorinated Aporphines: Potential Positron Emission Tomography Ligands for D2 Receptors.. ACS Med. Chem. Lett., 2011. [DOI | PubMed]
- S. Subburaju, A. W. Sromek, P. Seeman, J. L. Neumeyer. New Dopamine D2 Receptor Agonist, [(3)H]MCL-536, for Detecting Dopamine D2high Receptors in Vivo.. ACS Chem. Neurosci., 2018. [DOI | PubMed]
- S. Subburaju, A. W. Sromek, P. Seeman, J. L. Neumeyer. The High Affinity Dopamine D(2) Receptor Agonist MCL-536: A New Tool for Studying Dopaminergic Contribution to Neurological Disorders.. ACS Chem. Neurosci., 2021. [DOI | PubMed]
- A. Hall, L. Provins, A. Valade. Novel Strategies To Activate the Dopamine D1 Receptor: Recent Advances in Orthosteric Agonism and Positive Allosteric Modulation.. J. Med. Chem., 2019. [DOI | PubMed]
- J. L. Neumeyer, N. S. Kula, J. Bergman, R. J. Baldessarini. Receptor affinities of dopamine D1 receptor-selective novel phenylbenzazepines.. Eur. J. Pharmacol., 2003. [DOI | PubMed]
- A. Sidhu. Coupling of D1 and D5 dopamine receptors to multiple G proteins: Implications for understanding the diversity in receptor-G protein coupling.. Mol. Neurobiol., 1998. [DOI | PubMed]
- A. Karki, R. Juarez, H. K. Namballa, I. Alberts, W. W. Harding. Identification of C10 nitrogen-containing aporphines with dopamine D(1) versus D(5) receptor selectivity.. Bioorg. Med. Chem. Lett., 2020. [DOI | PubMed]
- E. Lorenzen, E. Ceraudo, Y. A. Berchiche, C. A. Rico, A. Furstenberg, T. P. Sakmar, T. Huber. G protein subtype-specific signaling bias in a series of CCR5 chemokine analogs.. Sci. Signal, 2018. [DOI | PubMed]
- H. Park, A. N. Urs, J. Zimmerman, C. Liu, Q. Wang, N. M. Urs. Structure-Functional-Selectivity Relationship Studies of Novel Apomorphine Analogs to Develop D1R/D2R Biased Ligands.. ACS Med. Chem. Lett., 2020. [DOI | PubMed]
- H. Speisky, B. K. Cassels. Boldo and boldine: an emerging case of natural drug development.. Pharmacol. Res., 1994. [DOI | PubMed]
- H. K. Namballa, S. Madapa, D. K. Sigalapalli, W. W. Harding. Semisynthetic Transformations on (+)-Boldine Reveal a 5-HT(2A/2C)R Antagonist.. J. Nat. Prod., 2022. [DOI | PubMed]
- S. Madapa, W. W. Harding. Semisynthetic Studies on and Biological Evaluation of N-Methyllaurotetanine Analogues as Ligands for 5-HT Receptors.. J. Nat. Prod., 2015. [DOI | PubMed]
- M. Asencio, B. Delaquerriere, B. K. Cassels, H. Speisky, E. Comoy, P. Protais. Biochemical and behavioral effects of boldine and glaucine on dopamine systems.. Pharmacol., Biochem. Behav., 1999. [DOI | PubMed]
- I. Jimenez, H. Speisky. Biological disposition of boldine: in vitro and in vivo studies.. Phytother. Res., 2000. [DOI | PubMed]
- E. M. Sobarzo-Sanchez, J. Arbaoui, P. Protais, B. K. Cassels. Halogenated boldine derivatives with enhanced monoamine receptor selectivity.. J. Nat. Prod., 2000. [DOI | PubMed]
- J. Cortijo, V. Villagrasa, R. Pons, L. Berto, M. Marti-Cabrera, M. Martinez-Losa, T. Domenech, J. Beleta, E. J. Morcillo. Bronchodilator and anti-inflammatory activities of glaucine: In vitro studies in human airway smooth muscle and polymorphonuclear leukocytes.. Br. J. Pharmacol., 1999. [DOI | PubMed]
- M. Asencio, C. Hurtado-Guzman, J. J. Lopez, B. K. Cassels, P. Protais, A. Chagraoui. Structure-affinity relationships of halogenated predicentrine and glaucine derivatives at D1 and D2 dopaminergic receptors: halogenation and D1 receptor selectivity.. Bioorg. Med. Chem., 2005. [DOI | PubMed]
- F. A. Dajas-Bailador, M. Asencio, C. Bonilla, M. C. Scorza, C. Echeverry, M. Reyes-Parada, R. Silveira, P. Protais, G. Russell, B. K. Cassels. Dopaminergic pharmacology and antioxidant properties of pukateine, a natural product lead for the design of agents increasing dopamine neurotransmission.. Gen. Pharmacol., 1999. [DOI | PubMed]
- J. L. Neumeyer, M. Froimowitz, R. J. Baldessarini, A. Campbell, Y. G. Gao. Neuropharmacology and stereochemistry of dopamine receptor agonist and antagonist enantiomeric pairs.. J. Recept. Res., 1988. [DOI | PubMed]
- H. Zhou, T. Hou, Z. Gao, X. Guo, C. Wang, J. Wang, Y. Liu, X. Liang. Discovery of eight alkaloids with D1 and D2 antagonist activity in leaves of Nelumbo nucifera Gaertn. Using FLIPR assays.. J. Ethnopharmacol., 2021. [DOI | PubMed]
- P. Protais, D. Cortes, J. -L. Pons, S. Lopez, M. C. Villaverde, L. Castedo. Displacement activity of some natural cularine alkaloids at striatal 3H-SCH 23 390 and 3H-raclopride binding sites.. Experientia, 1992. [DOI]
- M. H. Zenk, M. Rueffer, M. Amann, B. Deus-Neumann, N. Nagakura. Benzylisoquinoline biosynthesis by cultivated plant cells and isolated enzymes.. J. Nat. Prod., 1985. [DOI]
- L. Castedo, G. Tojo, A. Brossi. Chapter 3 Phenanthrene Alkaloids. In. The Alkaloids: Chemistry and Pharmacology,, 1990
- L. Moreno, N. Cabedo, M. D. Ivorra, M. J. Sanz, A. L. Castel, M. Carmen Alvarez, D. Cortes. 3,4-Dihydroxy- and 3,4-methylenedioxy- phenanthrene-type alkaloids with high selectivity for D2 dopamine receptor.. Bioorg. Med. Chem. Lett., 2013. [DOI | PubMed]
- D. Song, J. Hao, D. Fan. Biological properties and clinical applications of berberine.. Front. Med., 2020. [DOI | PubMed]
- J. J. Inbaraj, B. M. Kukielczak, P. Bilski, S. L. Sandvik, C. F. Chignell. Photochemistry and photocytotoxicity of alkaloids from Goldenseal (Hydrastis canadensis L.) 1. Berberine.. Chem. Res. Toxicol., 2001. [DOI | PubMed]
- M. Kawano, R. Takagi, A. Kaneko, S. Matsushita. Berberine is a dopamine D1- and D2-like receptor antagonist and ameliorates experimentally induced colitis by suppressing innate and adaptive immune responses.. J. Neuroimmunol., 2015. [DOI | PubMed]
- H. Chu, G. Jin, E. Friedman, X. Zhen. Recent development in studies of tetrahydroprotoberberines: mechanism in antinociception and drug addiction.. Cell Mol. Neurobiol., 2008. [DOI | PubMed]
- S. X. Xu, L. P. Yu, Y. R. Han, Y. Chen, G. Z. Jin. Effects of tetrahydroprotoberberines on dopamine receptor subtypes in brain.. Zhongguo Yao Li Xue Bao, 1989. [PubMed]
- M. O. Nesbit, A. G. Phillips. Tetrahydroprotoberberines: A Novel Source of Pharmacotherapies for Substance Use Disorders?.. Trends Pharmacol. Sci., 2020. [DOI | PubMed]
- C. C. Lapish, K. C. Ahn, R. A. Chambers, D. M. Ashby, S. Ahn, A. G. Phillips. Selective effects of D- and L-govadine in preclinical tests of positive, negative, and cognitive symptoms of schizophrenia.. Neuropsychopharmacology, 2014. [DOI | PubMed]
- W. Xu, Y. Wang, Z. Ma, Y. T. Chiu, P. Huang, K. Rasakham, E. Unterwald, D. Y. Lee, L. Y. Liu-Chen. L-isocorypalmine reduces behavioral sensitization and rewarding effects of cocaine in mice by acting on dopamine receptors.. Drug Alcohol Depend., 2013. [DOI | PubMed]
- Z. Z. Ma, W. Xu, N. H. Jensen, B. L. Roth, L. Y. Liu-Chen, D. Y. Lee. Isoquinoline alkaloids isolated from Corydalis yanhusuo and their binding affinities at the dopamine D1 receptor.. Molecules, 2008. [DOI | PubMed]
- J. B. Wang, J. R. Mantsch. l-tetrahydropalamatine: a potential new medication for the treatment of cocaine addiction.. Future Med. Chem., 2012. [DOI | PubMed]
- S. X. Xu, G. Z. Jin, L. P. Yu, G. X. Liu, W. W. Lu, S. D. Fang. [Brain dopamine depleted by d-tetrahydropalmatine].. Zhongguo Yao Li Xue Bao, 1987. [PubMed]
- J. A. Meade, R. B. Free, N. R. Miller, L. S. Chun, T. B. Doyle, A. E. Moritz, J. L. Conroy, V. J. Watts, D. R. Sibley. (−)-Stepholidine is a potent pan-dopamine receptor antagonist of both G protein- and beta-arrestin-mediated signaling.. Psychopharmacology (Berl), 2015. [DOI | PubMed]
- Z. L. Xiong, Z. Sun, G. Z. Jin, Y. Chen. [Influence of l-stepholidine on blood pressure and its relation to alpha-adrenoceptors].. Zhongguo Yao Li Xue Bao, 1987. [PubMed]
- Z. D. Zhang, G. Z. Jin, S. X. Xu, L. P. Yu, Y. Chen, F. Y. Jiang, Y. R. Zhang, Z. Sun, Y. L. Ding, C. F. Bian. [Effects of l-stepholidine on the central nervous and cardiovascular systems].. Zhongguo Yao Li Xue Bao, 1986. [PubMed]
- C. F. Bian, S. M. Duan, S. H. Xing, Y. M. Yu, W. Qin, G. Z. Jin, Y. Chen. [Interaction of analgesics and l-stepholidine].. Zhongguo Yao Li Xue Bao, 1986. [PubMed]
- B. A. Ellenbroek, X. X. Zhang, G. Z. Jin. Effects of (−)stepholidine in animal models for schizophrenia.. Acta Pharmacol. Sin., 2006. [DOI | PubMed]
- Y. Guo, H. Zhang, X. Chen, W. Cai, J. Cheng, Y. Yang, G. Jin, X. Zhen. Evaluation of the antipsychotic effect of bi-acetylated l-stepholidine (l-SPD-A), a novel dopamine and serotonin receptor dual ligand.. Schizophr. Res., 2009. [DOI | PubMed]
- G. Z. Jin, Z. T. Zhu, Y. Fu. (−)-Stepholidine: a potential novel antipsychotic drug with dual D1 receptor agonist and D2 receptor antagonist actions.. Trends Pharmacol. Sci., 2002. [DOI | PubMed]
- S. Natesan, G. E. Reckless, K. B. Barlow, J. Odontiadis, J. N. Nobrega, G. B. Baker, S. R. George, D. Mamo, S. Kapur. The antipsychotic potential of l-stepholidine-a naturally occurring dopamine receptor D1 agonist and D2 antagonist.. Psychopharmacology (Berl), 2008. [DOI | PubMed]
- R. A. Wise, M. A. Robble. Dopamine and Addiction.. Annu. Rev. Psychol., 2020. [DOI | PubMed]
- M. Diana. The dopamine hypothesis of drug addiction and its potential therapeutic value.. Front. Psychiatry, 2011. [DOI | PubMed]
- J. R. Mantsch, S. J. Li, R. Risinger, S. Awad, E. Katz, D. A. Baker, Z. Yang. Levo-tetrahydropalmatine attenuates cocaine self-administration and cocaine-induced reinstatement in rats.. Psychopharmacology (Berl), 2007. [DOI | PubMed]
- Z. X. Xi, Z. Yang, S. J. Li, X. Li, C. Dillon, X. Q. Peng, K. Spiller, E. L. Gardner. Levo-tetrahydropalmatine inhibits cocaine’s rewarding effects: experiments with self-administration and brain-stimulation reward in rats.. Neuropharmacology, 2007. [DOI | PubMed]
- J. R. Mantsch, S. Wisniewski, O. Vranjkovic, C. Peters, A. Becker, A. Valentine, S. J. Li, D. A. Baker, Z. Yang. Levo-tetrahydropalmatine attenuates cocaine self-administration under a progressive-ratio schedule and cocaine discrimination in rats.. Pharmacol., Biochem. Behav., 2010. [DOI | PubMed]
- Y. Figueroa-Guzman, C. Mueller, O. Vranjkovic, S. Wisniewski, Z. Yang, S. J. Li, C. Bohr, E. N. Graf, D. A. Baker, J. R. Mantsch. Oral administration of levo-tetrahydropalmatine attenuates reinstatement of extinguished cocaine seeking by cocaine, stress or drug-associated cues in rats.. Drug Alcohol Depend., 2011. [DOI | PubMed]
- K. Yue, B. Ma, Q. Ru, L. Chen, Y. Gan, D. Wang, G. Jin, C. Li. The dopamine receptor antagonist levo-tetrahydropalmatine attenuates heroin self-administration and heroin-induced reinstatement in rats.. Pharmacol., Biochem. Behav., 2012. [DOI | PubMed]
- X. Gong, K. Yue, B. Ma, J. Xing, Y. Gan, D. Wang, G. Jin, C. Li. Levo-tetrahydropalmatine, a natural, mixed dopamine receptor antagonist, inhibits methamphetamine self-administration and methamphetamine-induced reinstatement.. Pharmacol., Biochem. Behav., 2016. [DOI | PubMed]
- S. L. Faison, C. W. Schindler, S. R. Goldberg, J. B. Wang. l-tetrahydropalmatine reduces nicotine self-administration and reinstatement in rats.. BMC Pharmacol. Toxicol., 2016. [DOI | PubMed]
- M. Manuszak, W. Harding, S. Gadhiya, R. Ranaldi. (−)-Stepholidine reduces cue-induced reinstatement of cocaine seeking and cocaine self-administration in rats.. Drug Alcohol Depend., 2018. [DOI | PubMed]
- C. Hicks, P. Huang, L. Ramos, S. U. Nayak, Y. Caro, A. B. Reitz, G. R. Smith, D. Y. Lee, S. M. Rawls, L. Y. Liu-Chen. Dopamine D1-Like Receptor Agonist and D2-Like Receptor Antagonist (−)-Stepholidine Reduces Reinstatement of Drug-Seeking Behavior for 3,4-Methylenedioxypyrovalerone (MDPV) in Rats.. ACS Chem. Neurosci., 2018. [DOI | PubMed]
- W. Wang, Y. Zhou, J. Sun, L. Pan, L. Kang, Z. Dai, R. Yu, G. Jin, L. Ma. The effect of L-stepholidine, a novel extract of Chinese herb, on the acquisition, expression, maintenance, and re-acquisition of morphine conditioned place preference in rats.. Neuropharmacology, 2007. [DOI | PubMed]
- B. Ma, K. Yue, L. Chen, X. Tian, Q. Ru, Y. Gan, D. Wang, G. Jin, C. Li. L-stepholidine, a natural dopamine receptor D1 agonist and D2 antagonist, inhibits heroin-induced reinstatement.. Neurosci. Lett., 2014. [DOI | PubMed]
- Z. Yang, Y. C. Shao, S. J. Li, J. L. Qi, M. J. Zhang, W. Hao, G. Z. Jin. Medication of l-tetrahydropalmatine significantly ameliorates opiate craving and increases the abstinence rate in heroin users: a pilot study.. Acta Pharmacol Sin, 2008. [DOI | PubMed]
- H. E. Hassan, D. Kelly, M. Honick, S. Shukla, A. Ibrahim, D. A. Gorelick, M. Glassman, R. P. McMahon, H. J. Wehring, A. M. Kearns. Pharmacokinetics and Safety Assessment of l-Tetrahydropalmatine in Cocaine Users: A Randomized, Double-Blind, Placebo-Controlled Study.. J. Clin. Pharmacol., 2017. [DOI | PubMed]
- Z. X. Niu, Y. T. Wang, J. F. Wang. Recent advances in total synthesis of protoberberine and chiral tetrahydroberberine alkaloids.. Nat. Prod. Rep., 2024. [DOI]
- J. Párraga, N. Cabedo, S. Andujar, L. Piqueras, L. Moreno, A. Galán, E. Angelina, R. D. Enriz, M. D. Ivorra, M. J. Sanz. 2,3,9- and 2,3,11-Trisubstituted tetrahydroprotoberberines as D2 dopaminergic ligands.. Eur. J. Med. Chem., 2013. [DOI | PubMed]
- N. El Aouad, I. Berenguer, V. Romero, P. Marin, A. Serrano, S. Andujar, F. Suvire, A. Bermejo, M. D. Ivorra, R. D. Enriz. Structure-activity relationship of dopaminergic halogenated 1-benzyl-tetrahydroisoquinoline derivatives.. Eur. J. Med. Chem., 2009. [DOI | PubMed]
- S. A. Andujar, R. D. Tosso, F. D. Suvire, E. Angelina, N. Peruchena, N. Cabedo, D. Cortes, R. D. Enriz. Searching the “Biologically Relevant”Conformation of Dopamine: A Computational Approach.. J. Chem. Inf. Model., 2012. [DOI | PubMed]
- R. Morphy, Z. Rankovic. Designed multiple ligands. An emerging drug discovery paradigm.. J. Med. Chem., 2005. [DOI | PubMed]
- R. Morphy, C. Kay, Z. Rankovic. From magic bullets to designed multiple ligands.. Drug Discovery Today, 2004. [DOI | PubMed]
- I. R. Edwards, J. K. Aronson. Adverse drug reactions: definitions, diagnosis, and management.. Lancet, 2000. [DOI | PubMed]
- M. Decker. Design of hybrid molecules for drug development;, 2017
- S. Madapa, S. Gadhiya, T. Kurtzman, I. L. Alberts, S. Ramsey, M. Reith, W. W. Harding. Synthesis and evaluation of C9 alkoxy analogues of (−)-stepholidine as dopamine receptor ligands.. Eur. J. Med. Chem., 2017. [DOI | PubMed]
- H. K. Namballa, M. Dorogan, A. R. Gudipally, S. Okafor, S. Gadhiya, W. W. Harding. Discovery of Selective Dopamine Receptor Ligands Derived from (−)-Stepholidine via C-3 Alkoxylation and C-3/C-9 Dialkoxylation.. J. Med. Chem., 2023. [DOI | PubMed]
- H. K. Namballa, A. M. Decker, M. Dorogan, A. Gudipally, J. Goclon, W. W. Harding. Fluoroalkoxylated C-3 and C-9 (S)-12-bromostepholidine analogues with D1R antagonist activity.. Bioorg. Chem., 2023. [DOI | PubMed]
- T. Cai, L. Xie, S. Zhang, M. Chen, D. He, A. Badkul, Y. Liu, H. K. Namballa, M. Dorogan, W. W. Harding. End-to-end sequence-structure-function meta-learning predicts genome-wide chemical-protein interactions for dark proteins.. PLoS Comput. Biol., 2023. [DOI | PubMed]
- E. Galaj, W. Harding, R. Ranaldi. Dopamine D1 and D3 receptor interactions in cocaine reward and seeking in rats.. Psychopharmacology (Berl), 2016. [DOI | PubMed]
- S. Gadhiya, S. Madapa, T. Kurtzman, I. L. Alberts, S. Ramsey, N. K. Pillarsetty, T. Kalidindi, W. W. Harding. Tetrahydroprotoberberine alkaloids with dopamine and sigma receptor affinity.. Bioorg. Med. Chem., 2016. [DOI | PubMed]
- D. Jakubczyk, J. Z. Cheng, S. E. O’Connor. Biosynthesis of the ergot alkaloids.. Nat. Prod. Rep., 2014. [DOI | PubMed]
- D. G. Panaccione, K. L. Ryan, C. L. Schardl, S. Florea. Analysis and modification of ergot alkaloid profiles in fungi.. Methods Enzymol., 2012. [DOI | PubMed]
- C. L. Schardl, S. Florea, J. Pan, P. Nagabhyru, S. Bec, P. J. Calie. The epichloae: alkaloid diversity and roles in symbiosis with grasses.. Curr. Opin. Plant Biol., 2013. [DOI | PubMed]
- C. Wallwey, S. M. Li. Ergot alkaloids: structure diversity, biosynthetic gene clusters and functional proof of biosynthetic genes.. Nat. Prod. Rep., 2011. [DOI | PubMed]
- C. L. Schardl, C. A. Young, U. Hesse, S. G. Amyotte, K. Andreeva, P. J. Calie, D. J. Fleetwood, D. C. Haws, N. Moore, B. Oeser. Plant-symbiotic fungi as chemical engineers: multi-genome analysis of the clavicipitaceae reveals dynamics of alkaloid loci.. PLoS Genet., 2013. [DOI | PubMed]
- U. Steiner, S. Leibner, C. L. Schardl, A. Leuchtmann, E. Leistner. Periglandula, a new fungal genus within the Clavicipitaceae and its association with Convolvulaceae.. Mycologia, 2011. [DOI | PubMed]
- V. r. Křen, L. Cvak. Ergot: the genus Claviceps;, 1999
- C. L. Schardl, D. G. Panaccione, P. Tudzynski. Ergot alkaloids-biology and molecular biology.. Alkaloids Chem. Biol., 2006. [DOI | PubMed]
- H. Liu, Y. Jia. Ergot alkaloids: synthetic approaches to lysergic acid and clavine alkaloids.. Nat. Prod. Rep., 2017. [DOI | PubMed]
- P. Seeman. Antiparkinson therapeutic potencies correlate with their affinities at dopamine D2(High) receptors.. Synapse, 2007. [DOI | PubMed]
- M. F. Piercey, W. E. Hoffmann, M. W. Smith, D. K. Hyslop. Inhibition of dopamine neuron firing by pramipexole, a dopamine D3 receptor-preferring agonist: comparison to other dopamine receptor agonists.. Eur. J. Pharmacol., 1996. [DOI | PubMed]
- P. G. Strange. New insights into dopamine receptors in the central nervous system.. Neurochem. Int., 1993. [DOI | PubMed]
- J. Harris, P. M. Stanford, S. R. Oakes, C. J. Ormandy. Prolactin and the prolactin receptor: new targets of an old hormone.. Ann. Med., 2004. [DOI | PubMed]
- W. Sneader. Drug discovery: a history;, 2005
- T. T. Yen, N. B. Stamm, J. A. Clemens. Pergolide: a potent dopaminergic antihypertensive.. Life Sci., 1979. [DOI | PubMed]
- R. Horowski, P. A. Loschmann. Classical dopamine agonists.. J. Neural Transm. (Vienna), 2019. [DOI | PubMed]
- M. C. Shelesnyak. Ergotoxine inhibition of deciduoma formation and its reversal by progesterone.. Am. J. Physiol., 1954. [DOI | PubMed]
- E. Fluckiger, H. R. Wagner. [2-Br-alpha-ergokryptin: influence on fertility and lactation in the rat].. Experientia, 1968. [DOI | PubMed]
- N. Krogsgaard-Larsen, A. A. Jensen, T. J. Schroder, C. T. Christoffersen, J. Kehler. Novel aza-analogous ergoline derived scaffolds as potent serotonin 5-HT(6) and dopamine D(2) receptor ligands.. J. Med. Chem., 2014. [DOI | PubMed]
- K. Fuxe, D. Marcellino, D. O. Borroto-Escuela, M. Guescini, V. Fernandez-Duenas, S. Tanganelli, A. Rivera, F. Ciruela, L. F. Agnati. Adenosine-dopamine interactions in the pathophysiology and treatment of CNS disorders.. CNS Neurosci. Ther., 2010. [DOI | PubMed]
- S. Aoyama, H. Kase, E. Borrelli. Rescue of locomotor impairment in dopamine D2 receptor-deficient mice by an adenosine A2A receptor antagonist.. J. Neurosci., 2000. [DOI | PubMed]
- J. Hockemeyer, J. C. Burbiel, C. E. Muller. Multigram-scale syntheses, stability, and photoreactions of A2A adenosine receptor antagonists with 8-styrylxanthine structure: potential drugs for Parkinson’s disease.. J. Org. Chem., 2004. [DOI | PubMed]
- M. Vendrell, E. Angulo, V. Casado, C. Lluis, R. Franco, F. Albericio, M. Royo. Novel ergopeptides as dual ligands for adenosine and dopamine receptors.. J. Med. Chem., 2007. [DOI | PubMed]
- A. C. Campos, F. A. Moreira, F. V. Gomes, E. A. Del Bel, F. S. Guimaraes. Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders.. Philos. Trans. R Soc. London B Biol. Sci., 2012. [DOI | PubMed]
- A. S. Husni, C. R. McCurdy, M. M. Radwan, S. A. Ahmed, D. Slade, S. A. Ross, M. A. ElSohly, S. J. Cutler. Evaluation of Phytocannabinoids from High Potency Cannabis sativa using In Vitro Bioassays to Determine Structure-Activity Relationships for Cannabinoid Receptor 1 and Cannabinoid Receptor 2.. Med. Chem. Res., 2014. [DOI | PubMed]
- S. C. Britch, S. Babalonis, S. L. Walsh. Cannabidiol: pharmacology and therapeutic targets.. Psychopharmacology (Berl), 2021. [DOI | PubMed]
- P. Seeman. Cannabidiol is a partial agonist at dopamine D2High receptors, predicting its antipsychotic clinical dose.. Transl. Psychiatry, 2016. [DOI | PubMed]
- R. Prajapati, S. H. Seong, P. Paudel, S. E. Park, H. A. Jung, J. S. Choi. In Vitro and In Silico Characterization of Kurarinone as a Dopamine D(1A) Receptor Antagonist and D(2L) and D(4) Receptor Agonist.. ACS Omega, 2021. [DOI | PubMed]
- W. Wang, R. L. You, W. J. Qin, L. N. Hai, M. J. Fang, G. H. Huang, R. X. Kang, M. H. Li, Y. F. Qiao, J. W. Li. Anti-tumor activities of active ingredients in Compound Kushen Injection.. Acta Pharmacol. Sin., 2015. [DOI | PubMed]
- H. A. Jung, T. Yokozawa, B. W. Kim, J. H. Jung, J. S. Choi. Selective inhibition of prenylated flavonoids from Sophora flavescens against BACE1 and cholinesterases.. Am. J. Chin. Med., 2010. [DOI | PubMed]
- G. L. Ellman, K. D. Courtney, V. Andres, R. M. Feather-Stone. A new and rapid colorimetric determination of acetylcholinesterase activity.. Biochem. Pharmacol., 1961. [DOI | PubMed]
- H. Yan, Y. Sun, Y. Ma, B. Ji, X. Hou, Z. Yu, Y. Zhao. Determination of atractylon in rat plasma by a GC-MS method and its application to a pharmacokinetic study.. J. Pharm. Anal., 2015. [DOI | PubMed]
- H. Li, F. Wang, Z. Zhou, X. Jiang, F. Li, Y. Feng, C. Liu, Y. Zhang, S. Fan, X. Wu. Atractylon, a novel dopamine 2 receptor agonist, ameliorates Parkinsonian like motor dysfunctions in MPTP-induced mice.. Neurotoxicology, 2022. [DOI | PubMed]
- S. Manzoor, N. Hoda. A comprehensive review of monoamine oxidase inhibitors as Anti-Alzheimer’s disease agents: A review.. Eur. J. Med. Chem., 2020. [DOI | PubMed]
- M. B. Youdim, Y. S. Bakhle. Monoamine oxidase: isoforms and inhibitors in Parkinson’s disease and depressive illness.. Br. J. Pharmacol., 2006. [DOI | PubMed]
- P. Paudel, S. E. Park, S. H. Seong, H. A. Jung, J. S. Choi. Bromophenols from Symphyocladia latiuscula Target Human Monoamine Oxidase and Dopaminergic Receptors for the Management of Neurodegenerative Diseases.. J. Agric. Food Chem., 2020. [DOI | PubMed]
- S. E. Park, P. Paudel, A. Wagle, S. H. Seong, H. R. Kim, F. M. Fauzi, H. A. Jung, J. S. Choi. Luteolin, a Potent Human Monoamine Oxidase-A Inhibitor and Dopamine D(4) and Vasopressin V(1A) Receptor Antagonist.. J. Agric. Food Chem., 2020. [DOI | PubMed]
- R. Prajapati, S. H. Seong, S. E. Park, P. Paudel, H. A. Jung, J. S. Choi. Isoliquiritigenin, a potent human monoamine oxidase inhibitor, modulates dopamine D(1), D(3), and vasopressin V(1A) receptors.. Sci. Rep., 2021. [DOI | PubMed]