
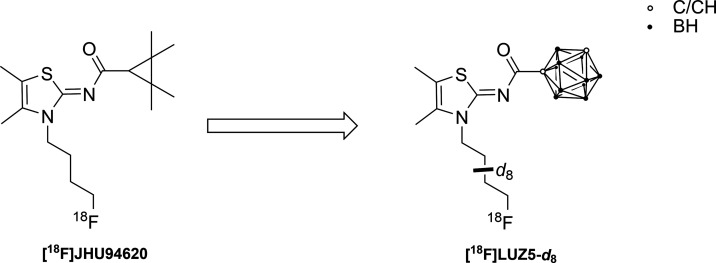
Development of the High-Affinity Carborane-Based Cannabinoid Receptor Type 2 PET Ligand [18F]LUZ5-d8
Abstract
The development of cannabinoid receptor type 2 (CB2R) radioligands for positron emission tomography (PET) imaging was intensively explored. To overcome the low metabolic stability and simultaneously increase the binding affinity of known CB2R radioligands, a carborane moiety was used as a bioisostere. Here we report the synthesis and characterization of carborane-based 1,8-naphthyridinones and thiazoles as novel CB2R ligands. All tested compounds showed low nanomolar CB2R affinity, with (Z)-N-[3-(4-fluorobutyl)-4,5-dimethylthiazole-2(3H)-ylidene]-(1,7-dicarba-closo-dodecaboranyl)-carboxamide (LUZ5) exhibiting the highest affinity (0.8 nM). Compound [18F]LUZ5-d8 was obtained with an automated radiosynthesizer in high radiochemical yield and purity. In vivo evaluation revealed the improved metabolic stability of [18F]LUZ5-d8 compared to that of [18F]JHU94620. PET experiments in rats revealed high uptake in spleen and low uptake in brain. Thus, the introduction of a carborane moiety is an appropriate tool for modifying literature-known CB2R ligands and gaining access to a new class of high-affinity CB2R ligands, while the in vivo pharmacology still needs to be addressed.
Affiliations: †Universität Leipzig, Faculty of Chemistry and Mineralogy, Institute of Inorganic Chemistry, Johannisallee 29, 04103 Leipzig, Germany; ‡Helmholtz-Zentrum Dresden-Rossendorf (HZDR), Institute of Radiopharmaceutical Cancer Research, Department of Neuroradiopharmaceuticals, Research Site Leipzig, 04318 Leipzig, Germany; §Faculty of Chemistry and Food Chemistry, School of Science, TU Dresden, 01069 Dresden, Germany; ∥The Lübeck Institute of Experimental Dermatology, University Medical Center Schleswig-Holstein, 23562 Lübeck, Germany
License: © 2023 The Authors. Published by American Chemical Society CC BY 4.0 Permits the broadest form of re-use including for commercial purposes, provided that author attribution and integrity are maintained (https://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.1021/acs.jmedchem.3c00195 | PubMed: 36944112 | PMC: PMC10782483
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (7.3 MB)
Introduction
The endocannabinoid system (ECS), named after the cannabis plant,1 has become the focus of medicinal research, due to its involvement in the modulation of various physiological and pathological processes.1,2 With the biological investigation of the main active substance in cannabis, (−)-Δ9–trans-tetrahydrocannabinol (THC), cannabinoid receptors type 1 (CB1R, identified in 1988, cloned in 1990) and 2 (CB2R, cloned in 1993) were discovered and studied.2−7 The endocannabinoid system is defined as the entirety of the cannabinoid receptors, the endogenous lipids, and the enzymes responsible for the synthesis and degradation thereof8 and is still being continuously studied. Other receptors like GPR18 and GPR55 have also been shown to be modulated by cannabinoids, despite sharing a low degree of sequence homology with the CB1R and CB2R.9,10
The CB1R and CB2R are part of the family of G protein-coupled receptors (GPCRs) and exhibit a degree of similarity of 44% for amino acids across the entire protein and of 68% regarding the transmembrane domains.4,11 The binding pockets of the CB1R and CB2R differ partly from each other.12 Recently, the antagonist and agonist bonding of the CB2R in the complex with corresponding ligands was revealed through crystal structures.11,12 With the knowledge of similarities and differences between the two receptors, especially the constitution and conformation of their binding pockets, the development of selective and high-affinity ligands is being actively pursued.
While the CB1R is mainly expressed in the central nervous system (CNS), the level of expression of the CB2R in the brain is significantly lower under physiological conditions.13−17 The CB2R is mainly associated with the immune system and predominantly expressed among others in spleen, tonsils, and thymus.18,19 In the brain, the CB2R is overexpressed under pathological conditions like inflammation,20 neurodegenerative diseases, such as Alzheimer’s, Huntington’s, and Parkinson’s21 diseases, or cancer.20 The CB2R is also connected to rheumatoid arthritis or arteriosclerosis.19 The activation of the CB2R can lead to beneficial effects, like causing apoptosis of cancer cells.20 The diagnosis and treatment of the aforementioned pathological conditions could be achieved by the use of selective CB2R ligands. The development of such compounds is ongoing and challenging, because of the strong requirements. The affinity must be high [(sub)-nanomolar range], due to the low expression density of the CB2R, and the selectivity must be high (1000-fold higher than for the CB1R) to avoid the undesired psychedelic effects of the CB1R. Other important characteristics are the metabolic stability, bioavailability, and ability to penetrate membranes or the blood–brain barrier (BBB) (LogP range of 1–3.5).15,22 To date, no selective CB2R ligand is approved for routine clinical use. To gain more insights into the involvement of the CB2R in physiological and pathological processes and to take action for their treatment, the theranostic approach is greatly important. A suitable tool for diagnosis is positron emission tomography (PET), a non-invasive imaging method, with which biochemical features in the body can be monitored.23 In addition to the already mentioned requirements for suitable CB2R ligands, it is necessary that these compounds can be radiolabeled with a suitable radionuclide for PET. The most frequently used radioisotopes for PET are 11C (half-life of 20.4 min) and 18F (half-life of 109.8 min).24 For the usage of PET tracers, it is important that no radiometabolites are present in the brain and that the level of interaction with plasma proteins is as low as possible.15 Evens et al. developed the oxoquinoline-based compound [11C]NE40 ([11C]1)25 (Figure ), a CB2R radiotracer tested in patients with Alzheimer’s disease (AD). Whereas promising results have been obtained in healthy humans, the expected better binding of the tracer was not seen in the AD patients, which could possibly be related to the unfavorable biochemical properties of the radioligand.26 Other scaffolds of interest are naphthyridinones, indoles, indazoles, pyridines, oxadiazoles, carbazoles, imidazoles, thiophenes, or thiazoles.15,27−29 Spinelli et al. published a review in 2017 about structure–affinity and structure–activity relationships of several CB2R scaffolds and identified their influence and the importance of different functional groups within the structures on their suitability as diagnostic and therapeutic CB2R ligands.27 A selection of known radiolabeled CB2R ligands is shown in Figure . The thiazole-based [11C]A-836339 ([11C]2) is an auspicious lead structure. The non-radiolabeled compound was synthesized by Dart et al.,30 further investigated by Yao et al.,31 and radiolabeled by Horti et al.32 Extensive medicinal chemistry studies performed by us [[18F]JHU94620 ([18F]3)]28,33 and Caillé [[18F]FC0(3)24, in the literature as either FC024 or FC0324 ([18F]4)]24,34−36 led to the development of 18F-labeled analogues of [11C]2. While [18F]3 is a low-nanomolar affinity and selective CB2R PET radioligand, the presence of large fractions of radiometabolites in mouse brain limits its application as a PET tracer.28 The preclinical evaluation of [18F]4 in Rhesus monkeys also showed a high in vivo metabolism.35 The pyridine-based scaffold is also in the focus of research toward the development of a PET radioligand for CB2R imaging.27,37,38 Slavik et al. developed [11C]RSR-056 ([11C]5)37 and Haider et al. prepared and preclinically investigated [18F]RoSMA-18-d6 ([18F]6).38 Recently, Modemann et al. reported an indole-based 18F-labeled radioligand ([18F]9).39 The 1,8-naphthyridine-2-one, as another lead structure, was systematically and manifold modified in the past several years.20,27 We recently reported [18F]LU14 ([18F]7),40 a stereochemically pure 1,8-naphthyridine-2-one radioligand, and [18F]LU13 ([18F]8),41 a promising high-affinity radiotracer for targeting CB2R overexpression in neurological disease.
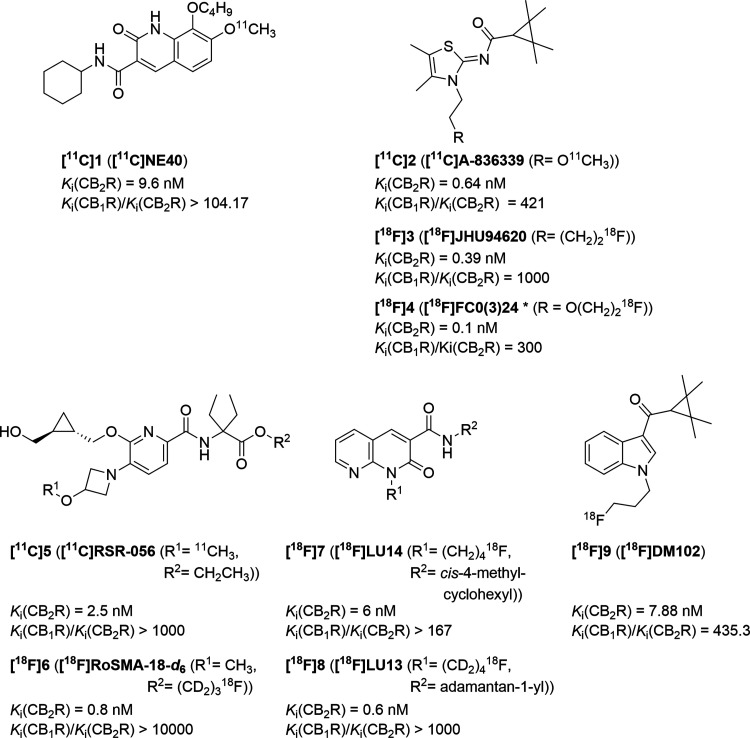
Common drawbacks of the reported CB2R radioligands that hinder the in vivo PET imaging of the neuronal CB2R are the low brain uptake and the low metabolic stability.28,29,35,37,42−45 No CB2R PET tracer is currently available for routine clinical usage. The common structure motifs found in most of the published CB2R ligands are shown in Figure .27
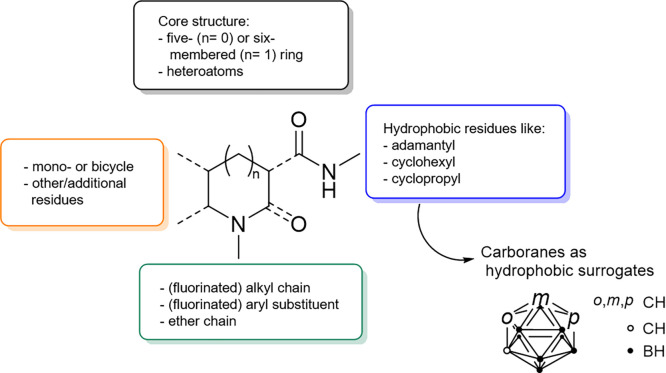
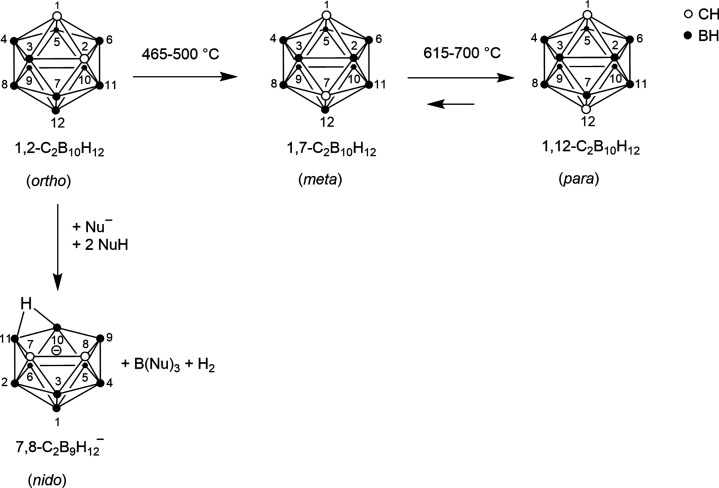
To overcome the fast metabolism and to hone promising candidates, carboranyl moieties can be introduced as substituents to modify organic CB2R ligands.46−50 The icosahedral dicarba-closo-dodecaboranes(12) are cluster compounds consisting of two carbon, 10 boron, and 12 hydrogen atoms and can be divided into ortho (o)-, meta (m)-, and para (p)-carborane (Figure ).51 The three isomers can be transformed into each other (Figure ). They differ not only in size and volume [core volumes of 11.79 Å3 (ortho), 11.72 Å3 (meta), and 11.71 Å3 (para); van der Waals volumes (VvdW) based on crystal structures of 148 Å3 (ortho), 143 Å3 (meta), and 141 Å3 (para)]51 but also in their reactivity. In particular, the reactivity toward Lewis bases is highest for o-carborane and lowest for p-carborane. The o-carborane is most prone to undergo a deboronation reaction that leads to the nido cluster (Figure ).51,52
Due to the delocalization of σ-bonding electrons of the cluster, carboranes are often termed three-dimensional σ-aromatic compounds and compared to benzene (π aromaticity);52 however, the average VvdW of carboranes (144 Å3) is comparable to that of adamantane (136 Å3).51 Considering the chemical difference between the acidic CH units and the hydridic BH units, orthogonal regioselective substitution reactions at both moieties are possible51 and offer a very powerful tool for modifying and adjusting the properties.46 Carborane derivatives are applied in catalysis, in polymers, but are also of particular interest as pharmacophores in medicinal chemistry.46,50−61 Here, the hydrophobicity can facilitate the passage of cellular membranes and the BBB and is therefore especially important for monitoring effects in the brain.46 In addition, due to the inorganic nature of the carborane scaffold, a greater metabolic stability can be expected.46−50 Furthermore, the carborane cluster can interact in a noncovalent way with the chosen target, e.g., by forming dihydrogen bonds.46
In the past several decades, carborane-based drug mimetics have attracted much attention.46 Endo et al. synthesized a carborane-based estradiol analogue62 [BE120 (CB-1) (Figure )]. Scholz et al. reported asborin63 [CB-2 (Figure )], the carborane analogue of aspirin (acetylsalicylic acid). Vázquez et al. published a rimonabant-derived CB1R antagonist64 [CB-3 (Figure )] bearing a carborane moiety. So far, no CB2R ligand bearing a carboranyl moiety has been published, neither as a drug nor as a radiolabeled PET tracer.
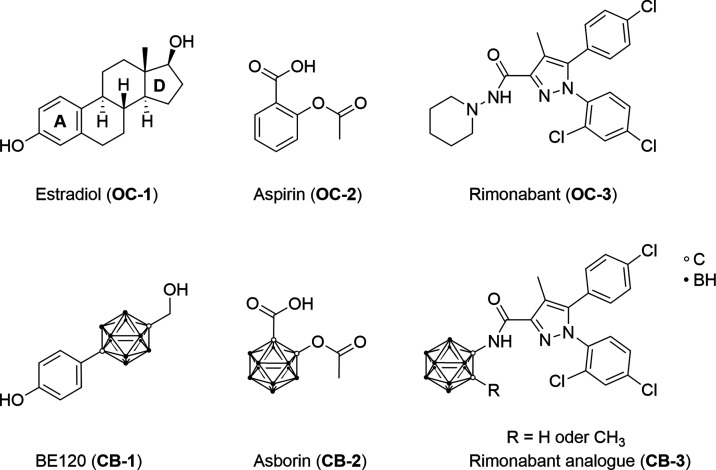
We here report the synthesis and investigation of the first carborane-based CB2R ligands, which were obtained by replacing either the tetramethylcyclopropyl unit in JHU94620(ref. 28) or the adamantyl residue in LU13(ref. 41) with o-, m-, or p-carborane. All new closo-carborane-containing CB2R ligands described herein were evaluated by in vitro metabolism studies using liver microsomes. To gain the first insights into the in vivo behavior of such carborane compounds, we designed a radiosynthesis for our most CB2R potent derivative LUZ5 [compound 16]. While a great number of aliphatic radiofluorinated PET candidates suffer from fast in vivo metabolism yielding free [18F]fluoride,67,68 the use of deuterated alkyls was shown to considerably improve the in vivo stability.69 Thus, a precursor bearing a fully deuterated N-butyl chain was synthesized and radiofluorinated in the presence of K[18F]F-K2.2.2 to give [18F]LUZ5-d8 (Figure ). The in vivo behavior of [18F]LUZ5-d8 was evaluated in metabolic and PET studies in vivo in healthy female CD-1 mice and healthy male Wistar rats, which are the first preliminary steps in the development of a theranostic agent.
Results and Discussion
Synthesis
The synthesis of the starting material 4-fluorobutyl-substituted 1,8-naphthyridin-2-one-3-carboxylic acid 4 was performed in three steps starting from 2-aminopyridine-3-carbaldehyde (1) as described by Lucchesi et al. and Sircar et al.20,70 In brief, aldehyde 1 was reacted in a Knoevenagel condensation with diethylmalonate and piperidine as the base. A nucleophilic substitution at the NH group of ester 2 introduced the 4-fluorobutyl chain followed by a basic ester hydrolysis of 3 with LiOH·H2O to give 4 (Scheme 1).71
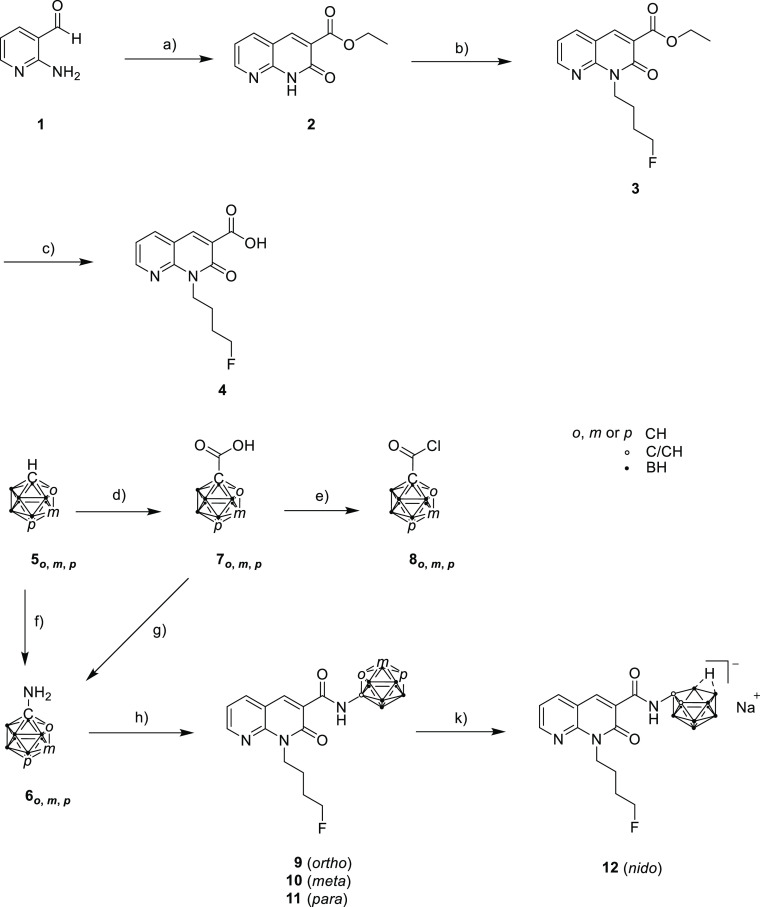
For the next step (Scheme 1, h), the corresponding carboranyl amines were required. 1-Amino-1,2-dicarba-closo-dodecaborane (6o) and 1-amino-1,7-dicarba-closo-dodecaborane (6m) were synthesized as published by Nie et al.72 starting from o– (5o) or m-carborane (5m) (Scheme 1). However, 1-amino-1,12-dicarba-closo-dodecaborane (6p) could not be obtained by the same procedure or by varying the reaction time and temperature. Carboranyl amine 6p could finally be obtained by following the synthetic approach of Tsuji et al.73 via a multistep synthesis starting from the monosubstituted p-carborane-1-carboxylic acid 7p, which is first chlorinated in situ followed by nucleophilic attack of trimethylsilyl azide at the carboxylic acid chloride and a Curtius rearrangement. The resulting isocyanate is reacted with tert-butanol to form the corresponding carbamate. Finally, the tert-butyloxycarbonyl (Boc) group was removed with trifluoroacetic acid (TFA) at room temperature (rt) to give compound 6p in 8% overall yield. Compound 6p was used without further purifications for the next synthetic step.
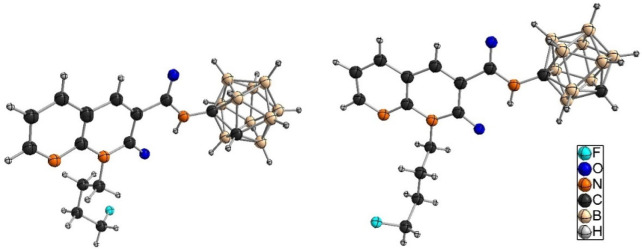
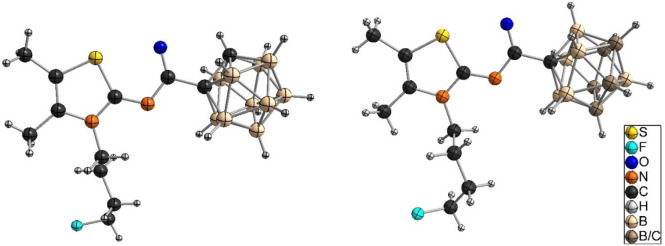
The synthesis of naphthyridine-based target compounds 9–11 (Scheme 1) was adapted from Vázquez et al.64 Carboxylic acid 4 was first converted to the corresponding acid chloride that was further reacted with the respective carboranyl amines 6o, 6m, and 6p in a microwave reaction at 150 °C to give amides 9–11, respectively, which were isolated in 42–50% yield by column chromatography. The three compounds were characterized by one-dimensional (1H, 11B{1H}, and 13C{1H}) and two-dimensional (COSY, HSQC, and HMBC) NMR spectroscopy and ESI-HRMS. Single crystals of o-carborane derivative 9 (Figure , left) and m-carborane analogue 10 (Figure , right) suitable for X-ray structure determination could be obtained from CDCl3 (9) or n-hexane/EtOAc/EtOH (10 and 11) (additional information is available in Table S1; the molecular structure of 11 is shown in Figure S82).
9 was deboronated with NaF in EtOH/H2O [1:1 (v/v)] in a microwave reaction,74,75 yielding racemic nido-carborate(−1) derivative 12 with sodium as the counterion in 79% yield.
The synthesis of compound 14 was performed starting from 2-amino-4,5-dimethylthiazole (13) and 1-bromo-4-fluorobutane (Scheme 2) as described in the literature.28 Target compounds 15, 16 (LUZ5), and 17 (Scheme 2) were obtained by reaction of 14 with carboranyl acid chlorides 8o, 8m, and 8p, respectively, which were obtained following literature procedures (Scheme 1).76−78
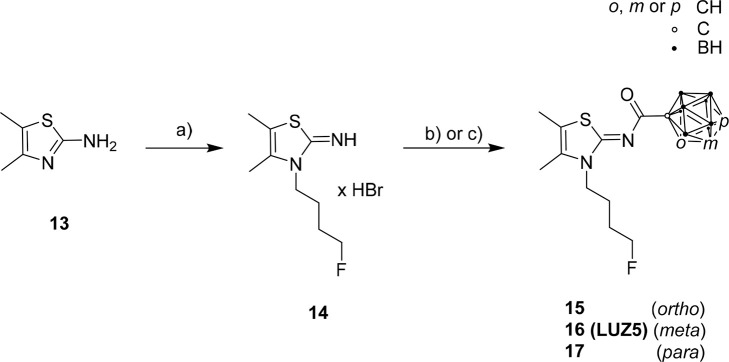
Compound 15 was synthesized in two ways, either in DCM at rt in 48 h (procedure 1), adapted from Moldovan et al.28 (Scheme 2, b), or in CH3CN at 80 °C in 20 h (procedure 2), adapted from Richter et al.79 (Scheme 2, c). The reaction in acetonitrile at increased temperature was also used for the synthesis of LUZ5 (45% yield) and 17 (36% yield). The molecular structures of 15 (Figure , left, crystallized from CDCl3), LUZ5 (Figure , right, crystallized from CDCl3), and 17 (Figure S83, crystallized from n-hexane/DCM) were obtained by X-ray crystallography.
One of the (major) side products in the synthesis of LUZ5 could be identified as the N3-unsubstitued thiazole-carborane acid amide (SP1) (Figures S39, S40, and S58) as shown by 1H and 11B{1H} NMR spectroscopy.
Stability Tests
A high chemical and metabolic stability of the compounds is required for biological evaluation and application in vivo. The method of choice for evaluating the chemical stability was 1H and 11B{1H} NMR spectroscopy, with the latter being particularly suitable for recognizing deboronation. Since the in vitro binding assays are performed in aqueous media, the stability tests should be performed under similar conditions. However, due to the high hydrophobicity of the carborane moieties, target compounds 9–11 and 15–17 were not soluble in aqueous solution without the addition of dimethyl sulfoxide (DMSO). Since a maximum of 1% DMSO can be used in the in vitro binding assay, stability measurements were performed in aqueous DMSO-d6 to see especially if deboronation occurs in the presence of water. Approximately 10 mg of each compound (9–11 and 15–17) was dissolved in the deuterated NMR solvent and monitored via NMR spectroscopy in time intervals of a few minutes (after the addition of the solvent to the respective solid compound) to at least 23 days for the o– (9 and 15) and m-carborane derivatives (10 and LUZ5) and beyond one year for the p-carborane analogues (11 and 17) (Figures S68–S79).
A general stability trend was observed with o-carborane derivatives having the lowest and p-carborane derivatives having the highest stability. This observation was in agreement with our expectations, because o-carboranes have the highest chemical reactivity of the three isomers and are most prone to undergo deboronation reactions, especially in aqueous solutions or in the presence of strong bases.51
Thiazole-based compounds 15 and LUZ5 proved to be more stable than naphthyridine isomers 9 and 10, respectively. For compound 9, additional signals were visible already after 2 h in the 1H or 11B{1H} NMR spectra, indicating that a deboronation to nido-cluster derivative 12 had started. Thus, the stability of 9 is too low considering the time necessary to perform the binding affinity assay, which is 90 min. The other target compounds (10, 11, 15, LUZ5, and 17) showed a stability beyond the binding affinity assay time frame with the following determined stability order: 9 < 15 < 10 < LUZ5 < 11 = 17. p-Carborane derivatives 11 and 17 were stable for more than one year.
For compound LUZ5, additional stability tests were performed with an increased concentration of H2O or D2O in DMSO-d6 [1:5 (v/v), in contrast to aqueous DMSO-d6 for previous measurements]. H2O or D2O was added to the clear solution, which immediately turned turbid indicating the formation of aggregates.80 However, investigation with 11B{1H} NMR spectroscopy was still possible and indicated that compound LUZ5 was stable in the solvent mixture with increased amounts of H2O for at least 19 days and in the mixture with increased amounts of D2O for at least 8 days (Figures S80 and S81).
In Vitro Binding Assay
Target compounds 9–11, 15, LUZ5, and 17 were evaluated in a competitive binding affinity assay using hCB2R Chinese hamster ovary (CHO) cells and the agonistic high-affinity CB2R radioligand [3H]WIN55212-2 (Figure S96).
For all target compounds, Ki values in the nanomolar range could be determined (Table 1 and Figures S84–S87). Naphthyridine-based compounds 9–11 showed an increasing affinity of the o– to p-carborane derivatives toward the CB2R. Compound 11 (Ki = 3.9 nM) showed an affinity (∼10-fold) considerably higher than those of the other two compounds. Despite the low nanomolar range, the affinity of compound 11 is lower than that of reference compound LU13 (Figure ). Due to the low stability of 9, the binding affinity determination might be influenced by the presence of small amounts of the nido derivative 12 that is currently under evaluation.
Table 1: Binding Affinities of Target Compounds 9–11, 15, LUZ5, 17, and Reference Compounds to the CB2R
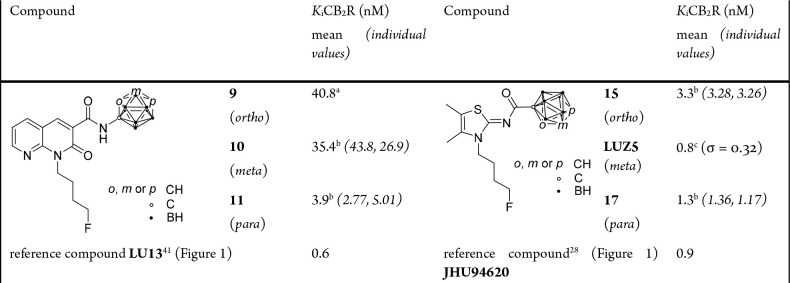
From the thiazole-based compounds, 17 and LUZ5 showed remarkably high affinities of 1.3 and 0.8 nM, respectively, for the CB2R, surpassing the affinity of lead compound JHU94620 (Figure ). Moreover, compound LUZ5 proved to be selective for the CB2R with a CB1R binding affinity of >10 μM (Figure S88). Compounds 9–11, 15, and 17 at concentrations of up to 1 μM did not displace the CB1R-specific radioligand [3H]SR141716A (Figure S96), and thus, we conclude that these compounds bind with only negligible affinity for the CB1R (Figure S89).
The finding that the thiazole-based compounds display a binding affinity higher than those of the respective naphthyridine-based ones is in agreement with the binding energies of the best docked positions (Figure S90). Therefore, for 9 with the lowest binding affinity, the binding energy is the highest (−4.53 kcal/mol) compared to those of 15 (−10.87 kcal/mol) and LUZ5 (−9.68 kcal/mol). Details about the calculation of binding energies can be found in the Supporting Information [Chapter 7: Docking Data of Compounds 9, 15 (Procedure 2), LUZ5].
In Vitro Metabolism Studies
In vitro metabolism studies were performed for target compounds 9–11, 15, LUZ5, and 17. Each of the compounds was incubated with mouse liver microsomes (MLMs) in phosphate-buffered saline (PBS) in the presence of nicotinamide adenine dinucleotide phosphate (NADPH) for 30 and 60 min at 37 °C, according to a literature protocol.81,82 After the addition of cold acetonitrile, the obtained supernatants were analyzed by HPLC-UV-MS (exemplified chromatogram and spectrum in Figures S91 and S92). The correct performance of the experimental setup was confirmed by complete conversion of testosterone under similar conditions. In addition, negative control samples without NADPH or without NADPH and MLM were prepared and analyzed for each target compound.
For each of the compounds investigated, no significant formation of metabolites was detected under the used conditions. This leads to the assumption that cytochrome P450-catalyzed functionalization reactions (phase I), such as oxidation, are not involved in the metabolism of this class of compounds in MLMs. However, the use of human liver microsomes (HLMs) instead of MLMs might provide a different picture, as species differences are described well in the literature83,84 and were already reported for CB2R ligands by Aly et al.85 However, as discussed in the section In Vivo Metabolism, radiometabolites were detected in plasma from rodents after administration of [18F]LUZ5-d8. These findings suggest further in vitro investigations with regard to conjugation reactions (phase II), to understand the complexity of the processes taking place in vivo.
Radiochemistry
On the basis of the binding affinity and stability tests, LUZ5 was selected as the most promising candidate for developing a CB2R radioligand for further biological evaluations.
The synthesis of precursor 19 was performed in two steps starting from 2-amino-4,5-dimethylthiazole (13) and m-carboranyl acid chloride (8m) (Scheme 3). The synthesis of intermediate 18 was analogous to the syntheses of 15, LUZ5, and 17 (Scheme 2).79 For alkylation, the N3 position of 18 was deprotonated with NaH and reacted with 1,4-dibromobutane-d8 to give 19 (Scheme 3).28 The fully deuterated chain was introduced to confer metabolic stability.40,41,69 Brominated precursor 19 was purified by HPLC and then radiofluorinated via nucleophilic substitution under thermal conditions to give [18F]LUZ5-d8.
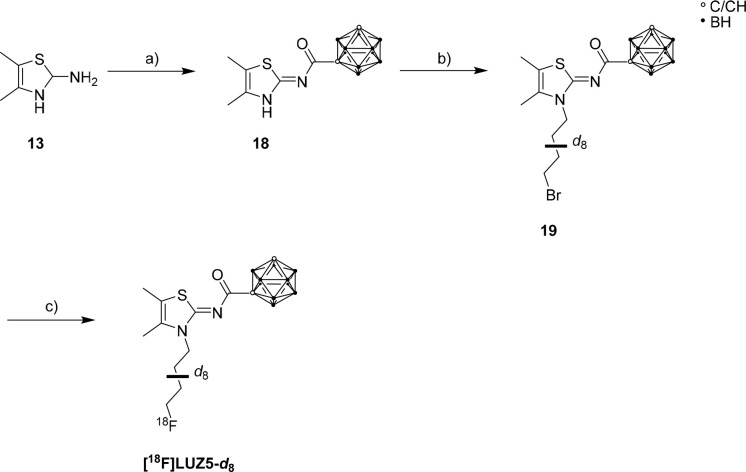
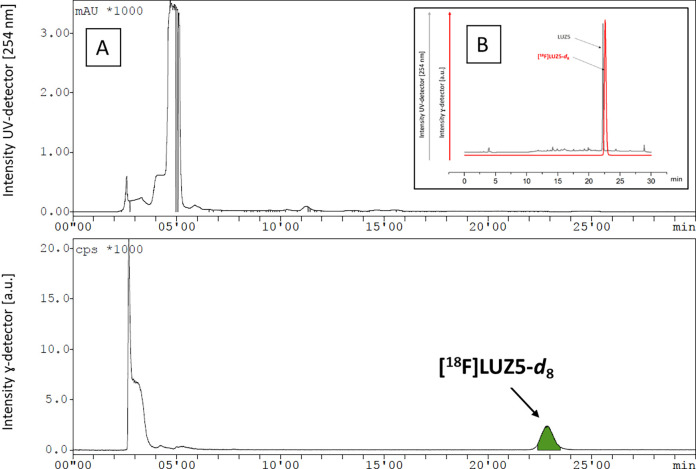
The reaction conditions for the radiosynthesis of [18F]LUZ5-d8 were investigated by using various amounts of 2.2.2-cryptand (K2.2.2) (13–40 μmol), K2CO3 (7.5–15 μmol), and precursor 19 (2.3−4.5 μmol) in CH3CN as the solvent under thermal conditions. Under each of the tested reaction conditions, [18F]LUZ5-d8 was detected in the range between 25% and 50% (radio-HPLC, not isolated). On the basis of these preliminary results, an automated radiosynthesis for [18F]LUZ5-d8 was developed on an Elysia-Raytest radiosynthesizer. The product was purified by HPLC (Figure A) followed by trapping the radiotracer [18F]LUZ5-d8 on a reverse phase (RP) cartridge and elution with EtOH. For biological experiments, the solvent was evaporated under a stream of nitrogen at 70 °C. The final product was formulated in sterile isotonic saline up to a final concentration of <10% EtOH. The overall synthesis time was ∼85 min. [18F]LUZ5-d8 was obtained in 5–8% radiochemical yield (decay-corrected from the end of bombardment), high radiochemical purity [>99% (Figure B)], and high molar activity in the range of 170–190 GBq/μmol at the end of the synthesis (n = 5). The identity of the final product [18F]LUZ5-d8 was confirmed with analytical HPLC by co-injection with the corresponding reference compound [co-injection (Figure B); UV profile of the isolated radiotracer (Figure S93)]. No decomposition of the formulated product was observed within 24 h at room temperature. A logD7.4 of 3.0 was experimentally determined for [18F]LUZ5-d8 by the shake-flask method (n = 3), which is well within the range of 1–3.5 recommended for brain-targeting compounds.22,86
In Vivo Metabolism
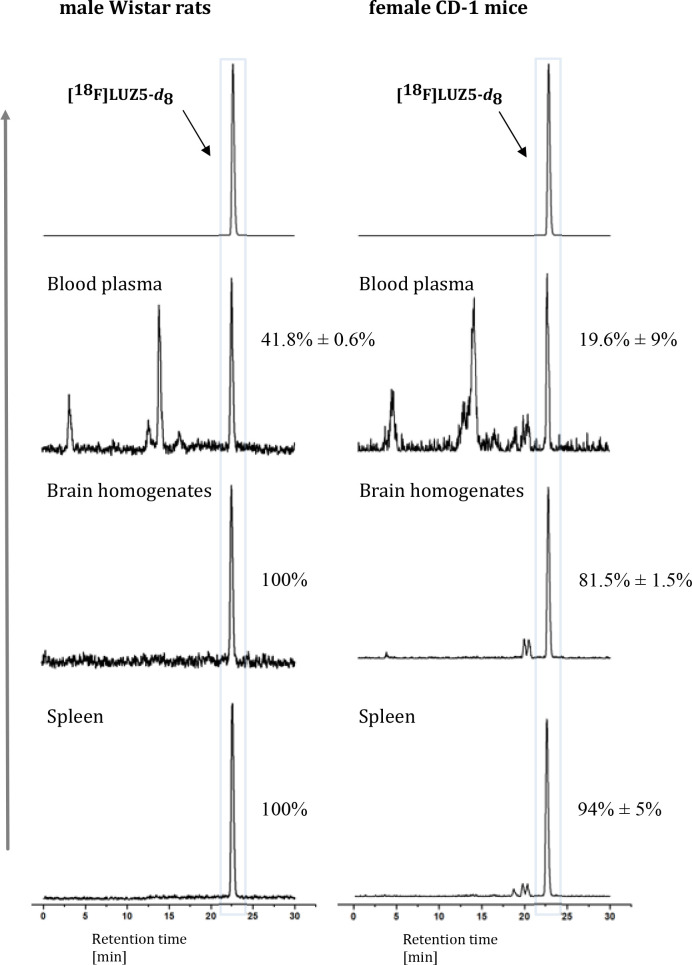
In vivo metabolism studies of [18F]LUZ5-d8 were carried out in healthy female CD-1 mice and healthy male Wistar rats. Samples of blood plasma, brain homogenates, spleen homogenates, and urine were obtained 30 min post-injection (p.i.), and organic solvent denaturation was performed to remove proteins. The extraction was carried out in mice samples by using either MeOH/H2O [9:1 (v/v)] or CH3CN/H2O [9:1 (v/v)], whereby an extraction efficiency of >94% was achieved using the former (n = 3). As a result, these conditions were also used to investigate the metabolic stability of [18F]LUZ5-d8 in rats (n = 2).
In all investigated rat samples, larger amounts of an intact radiotracer were found compared to mice (Figure ). For both mammals, significant amounts of radiometabolites were determined in blood plasma, with mean values of 19.6 ± 9% intact radiotracer in mice and 41.8 ± 0.6% in rats. Compared to the organic analogues [18F]JHU94620 and [18F]FC0(3)24 (Figure ), an average larger amount of intact [18F]LUZ5-d8 could be found in plasma of mice (19.6 ± 9% compared to 7% for [18F]JHU94620)28 and rats [41.8 ± 0.6% compared to 25% for [18F]FC0(3)24],24 respectively. Also in the brain samples, a much larger amount of intact radioligand was found in both mouse (81.5 ± 1.5%) and rat samples (100%) when compared to that of [18F]JHU94620 [36%, data not reported for [18F]FC0(3)24].24,28 In the spleen samples, 94 ± 5% of intact tracer for mice and 100% of intact tracer for rats were found (Figure ). The high metabolic stability can be explained by the presence of the carborane moiety and the implementation of the deuterated substituent at the N1 position of the molecule. Both structural changes might be influencing the stability toward enzymes in vivo and therefore increase the metabolic stability. In urine, no intact radiotracer could be detected.
Assessment of the Biodistribution of [18F]LUZ5-d8 in Rodents
The biodistribution of [18F]LUZ5-d8 over time was evaluated by dynamic PET imaging under control and blocking conditions using CB2R agonist GW405833 (Figure S96) in healthy male Wistar rats (Figure and Table 2) and in a healthy female CD-1 mouse (Figure S95).
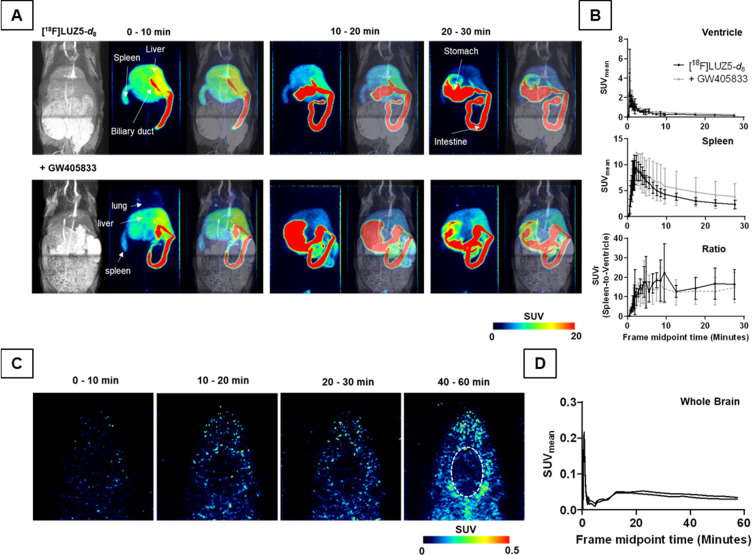
Table 2: Noncompartmental Analysis of PET-Derived Rat Tissue TACs after i.v. Injection of [18F]LUZ5-d8 with or without Pretreatment with GW405833 (n = 3)
| tissue | treatment | time to peak (min) | TAC peak value (SUV) | AUC0–30 min (SUV min) |
|---|---|---|---|---|
| ventricle | vehicle | 0.6 ± 0 | 4.5 ± 2.4 | 10 ± 2 |
| block | 0.6 ± 0 | 3.7 ± 1.3 | 14 ± 1 | |
| p value | 1.000 | 0.6180 | 0.0516 | |
| spleen | vehicle | 2.1 ± 0.3 | 9.5 ± 2.6 | 112 ± 21 |
| block | 2.4 ± 0.8 | 9.5 ± 2.9 | 150 ± 80 | |
| p value | 0.6164 | 0.9958 | 0.4761 | |
| liver | vehicle | 3.4 ± 0.3 | 13 ± 0.5 | 193 ± 16 |
| block | 4.7 ± 0.7 | 14 ± 0.5 | 245 ± 46 | |
| p value | 0.0495 | 0.0962 | 0.1369 | |
| kidney | vehicle | 1.3 ± 0.8 | 1.4 ± 0.1 | 18 ± 2 |
| block | 10 ± 15 | 1.8 ± 0.3 | 30 ± 8 | |
| p value | 0.4325 | 0.1256 | 0.055 | |
| jejunum | vehicle | 12 ± 5 | 42 ± 7 | 702 ± 82 |
| block | 23 ± 9 | 15 ± 8 | 210 ± 128 | |
| p value | 0.1374 | 0.0130 | 0.0050 | |
| lung | vehicle | 0.7 ± 0.1 | 10 ± 6 | 35 ± 29 |
| block | 0.6 ± 0 | 12 ± 9 | 39 ± 30 | |
| p value | 0.1835 | 0.7244 | 0.8793 | |
| stomach | vehicle | 53 ± 9 | 16 ± 23 | 94 ± 149 |
| block | 16 ± 13 | 0.9 ± 0.8 | 15 ± 14 | |
| p value | 0.0144 | 0.3689 | 0.4552 | |
| bone | vehicle | 24 ± 25 | 0.2 ± 0 | 2.8 ± 1.3 |
| block | 14 ± 13 | 0.8 ± 0.9 | 8.7 ± 5.4 | |
| p value | 0.5494 | 0.3598 | 0.1411 | |
| muscle | vehicle | 13 ± 0 | 0.2 ± 0 | 3.9 ± 0.7 |
| block | 16 ± 3 | 0.2 ± 0 | 5.1 ± 0.8 | |
| p value | 0.1835 | 0.1106 | 0.1354 |
In rats, the analysis of the time–activity curves (TACs) of the left ventricle revealed a comparable radioactivity concentration over time in the blood pool AUC0–30 min (area under the curve between 0 and 30 min p.i.) with comparable time-to-peak (TTP) and TAC peak values (TPV) between the control and blocking group. The spleen uptake of [18F]LUZ5-d8 with a TPV of 9.5 ± 2.6, 2.1 ± 0.3 min post-injection, was high and comparable to the uptake after blocking. This was also reflected by the normalized spleen SUV (standardized uptake value) to the blood pool activity with a mean SUVr (SUV ratio of x to reference) between 10 and 22 (control) and 10 and 16 (blocking) from 2 to 30 min p.i. of the radiotracer. Thus, a displaceable uptake of [18F]LUZ5-d8 into the spleen under these conditions could not be shown in vivo. The radiotracer was mainly hepatobiliary excreted as shown by the high AUC0–30 min values of the liver, jejunum, and stomach compared to kidney and the rather low accumulated bladder radioactivity of 0.3 ± 0.1% of the injected activity after 60 min p.i. in the control group (Figure S94). The hepatobiliary excretion was delayed by the blocking compound, which most likely caused the higher renal radioactivity accumulation as shown by the TTP and AUC0–30 min values and the decreased radioactivity concentration in the small intestine. The dynamic PET recordings were limited by the field of view of the device, so altered bladder activity under blocking conditions could unfortunately not be measured, as determination of blood pool activity was prioritized. A negligible radioactivity uptake could be observed in lung, muscle, and bone. Furthermore, the brain uptake of [18F]LUZ5-d8 in the initial distribution phase was rather low and followed by a fast washout in rats as shown in panels C and D of Figure .
Compared to rats, in an exemplary CD-1 mouse (Figure S95) the spleen uptake was lower, with a TPV of 1.2 after 7.5 min p.i. A high hepatobiliary excretion in mouse was found, as well, whereas the activity in bladder after 30 min was 1.9% of the injected radioactivity and therefore 10 times higher than that in rats, which could be in part explained by species-dependent radiometabolite formation. In mouse, the brain uptake as well as uptake in muscle and bone was low and comparable to that in rats.
Taken together, the uptake of [18F]LUZ5-d8 in the CB2R-rich tissue spleen was higher in rats than in mice but could not be blocked by the CB2R agonist GW405833. With the exception of the excretion pathway (mainly hepatobiliary), hints of an increased off-target uptake of the radioligand in tissues with a low CB2R density could not be found in both species. However, the brain uptake is rather low and a slow defluorination of [18F]LUZ5-d8 can be assumed due to the low level of accumulation of radioactivity in the bone.
Conclusions
In this study, a new class consisting of seven carborane-based CB2R ligands has been developed and evaluated. The lead structures of target compounds 9–11, 15, 16 (LUZ5), and 17, based on 1,8-naphthyridinone or thiazole scaffolds, were modified by introducing o-, m-, and p-carborane moieties. The nido-compound 12 was generated via deboronation of 9. Compounds 11, 15, and 17 showed good chemical stability in aqueous DMSO-d6 and low nanomolar affinities (Ki = 3.3, 3.9, and 1.3 nM, respectively). The binding affinity of the thiazole derivatives surpassed that of the naphthyridine derivatives. In this study, we identified LUZ5 with subnanomolar affinity exceeding the reference compound JHU94620. Encouraged by these results, we developed an automated radiofluorination for [18F]LUZ5-d8. The in vivo metabolic stability of [18F]LUZ5-d8 bearing a deuterated butyl chain was investigated in rodents. While in mouse plasma only a small fraction of intact tracer was found 30 min p.i., the percentage of intact radioligand in mouse brain and spleen was high (81.5 ± 1.5% and 94 ± 5%, respectively) and superior to that of [18F]JHU94620. The metabolic stability of [18F]LUZ5-d8 in rats exceeded that in mice. Dynamic PET scans in rats with and without blocking agent GW405833 revealed a fast uptake in spleen and liver, a fast hepatic clearance, and nondisplaceable binding in spleen. The uptake of radiotracer in brain was low. The development of [18F]LUZ5-d8 gave access to a new class of ligands for CB2R bearing a carborane moiety as a bioisostere for an adamantyl or tetramethylcyclopropyl group, which could alter and improve the properties of existing ligands and help to overcome the metabolic stability problems of the known CB2R ligands. For the use of such ligands as PET tracers targeting brain, further structural adjustments have to be considered. The foundation for further research toward diagnostic agents for theranostic application has been laid with this preliminary evaluation of an 18F-labeled radiotracer.
Experimental Section
General Information
All reactions involving carboranes were carried out under a nitrogen atmosphere using the Schlenk technique. Anhydrous DCM, Et2O, n-hexane, and toluene were dried with the MB SPS-800 solvent purification system (MBRAUN, M.Braun Inertgas-Systeme GmbH, Garching, Germany). Acetonitrile and DMF were dried over calcium hydride and distilled. Dry solvents were stored over molecular sieves (4 or 5 Å). Benzyl azide was prepared according to the literature.87 Dicarba-closo-dodecaborane-1-carboxylic acids (7o, 7m, and 7p) can be prepared according to the literature.76,77 Dicarba-closo-dodecaborane-1-carboxylic acid chlorides (8o, 8m, and 8p) can be synthesized as described by Kasar et al.78 1,2-and 1,12-Dicarba-closo-dodecaborane-1-amine 6o and 6p can be prepared as described by Nie et al.72 and Tsuji et al.73 The synthesis of 1,7-dicarba-closo-dodecaborane-1-amine 6m was adapted from Nie et al.72 Compound 4 can be prepared according to a previously published protocol.20,41,70,71 Compound 14 can be synthesized as published by Moldovan et al.28 All other solvents and chemicals were commercially available and used without further purification. Microwave reactions were performed by using an Initiator+ microwave from Biotage (Uppsala, Sweden). Reaction progress and purification of products were monitored by thin-layer chromatography (TLC) using precoated silica gel 60 F254 Alumgram plates (Xtra SIL G) from Macherey-Nagel (Düren, Germany). Parts of TLC plates containing carboranes were stained with a 5–10% PdCl2 solution in methanol. Chromatography was performed in air, with silica gel (60 Å, 0.035–0.070 mm particle diameter) or in an automated fashion with an Isolera-4 and ELSD 1080 (Biotage) with commercially available solvents.
NMR spectra were recorded with Avance III HD 400 and Avance DRX 400 spectrometers from Bruker (Billerica, MA). Measurements were performed at 400.13 MHz (1H), 128.38 MHz (11B), and 100.63 MHz (13C). Chemical shifts (δ) are given in parts per million (ppm). 1H and 13C NMR spectra were referenced to internal deuterated solvent, and 11B{1H} NMR spectra to the Ξ scale.88 Deuterated solvents (CDCl3, DMSO-d6, and acetone-d6) were purchased from Eurisotop (Saint-Aubin, France) with a deuteration rate of 99.80%. High-resolution mass spectrometry (HRMS) was conducted in positive ion mode with an ESI-TOF microTOF instrument from Bruker Daltonik GmbH (Bremen, Germany). The simulation of mass spectra was carried out with the Web-based MS online tool of Scientific Instrument Services (SISweb, Palmer, MA). The analysis of NMR and MS data was done with MestReNova version 14.1.0.89 To determine melting point ranges, the model MPD350.BM2.5 melting point apparatus from Gallenkamp (Cambridge, U.K.) was used. The values are uncorrected. Samples were tested as triplicates, and the mean was calculated. X-ray analysis was performed with single crystals, obtained from CDCl3, EtOH/EtOAc/n-hexane, or n-hexane/DCM at rt by slow evaporation of the solvent or by heating and slowly cooling saturated solutions. The crystals were measured with a Gemini diffractometer (Rigaku Oxford Diffraction) with Mo Kα radiation (λ = 71.073 pm) or Cu-Kα radiation (λ = 154.184 pm) in ω-scan mode. Data reduction was performed with CrysAlis Pro.90 Empirical absorption correction was performed with SCALE3 ABSPACK.91 Structure solution and anisotropic refinement of all atoms except hydrogen atoms and some disordered parts of molecules were performed with SHELXT92 and SHELXL.93 The position of the hydrogen atoms was calculated by locating them on difference Fourier maps calculated at the final stage of the structure refinement excluding disordered fragments. For LUZ5 and for disordered fragments, all hydrogen atoms were calculated on idealized positions. Further details on disordered moieties, fundamental structure parameters and CCDC deposition numbers are given in the Supporting Information (Table S1). The visualizations (figures) were generated with Diamond version 4.94 The purity of all of the compounds evaluated in biological tests was ≥95% as determined by HPLC [Jasco, Pfungstadt, Germany, MD-2010Plus, LG-2080-04S, DG-2080-54, AS-2055Plus, LC-NetII/ADC, λ = 280 nm, column ReproSil-Pur C18-AQ (250 mm × 4.6 mm, 5 μm, Dr. Maisch GmbH, Ammerbuch-Entringen, Germany), gradient of CH3CN/20 mM ammonium acetate, flow rate of 1 mL/min; method: 10% CH3CN/20 mM NH4OAcaq from 0 to 5 min, 10% to 90% CH3CN/20 mM NH4OAcaq from 5 to 20 min, 90% CH3CN/20 mM NH4OAcaq from 20 to 35 min, 90% to 10% CH3CN/20 mM NH4OAcaq from 35 to 36 min, and 10% CH3CN/20 mM NH4OAcaq from 36 to 40 min]. Additionally, for target compound LUZ5, an isocratic HPLC method (66% CH3CN/20 mM NH4OAcaq, flow rate of 1 mL/min) was used.
Chemical Synthesis
1-(4-Fluorobutyl)-2-oxo-N-(dicarba-closo-dodecaboranyl)-1,2-dihydro-1,8-naphthyridine-3-carboxamide (9–11).64 SOCl2 (4.01 equiv) was added to a solution of 4 (0.378 mmol, 1.00 equiv) in dry toluene (10 mL) and stirred for 3 h under reflux. The solvent was removed under reduced pressure after cooling to rt. The acid chloride in dry toluene (1–1.5 mL) was added to a microwave vial of the respective dicarba-closo-dodecaborane-1-amine (6, 0.377 mmol, 0.996–1.01 equiv), dissolved in dry toluene (1 mL). The reaction mixture was stirred for 15 h at rt and kept for 50 min in the microwave at 150 °C for the ortho isomer. For the meta and para isomers, the microwave reaction was carried out for 50 min at 150 °C, and then the mixture was stirred for 16 h at rt and again microwaved for 50 min at 150 °C. The solvent was removed under reduced pressure, and the crude product was purified by column chromatography. 1-(4-Fluorobutyl)-2-oxo-N-(1,2-dicarba-closo-dodecaboranyl)-1,2-dihydro-1,8-naphthyridine-3-carboxamide (9) [n-hexane/EtOAc, 4:1 (v/v) → 1.5:1 (v/v)] was obtained as a white solid: 0.073 g (0.180 mmol, 48%); 1H NMR (400 MHz, CDCl3) δ 1.43–3.64 (br, 10H), 1.87 (m, 4H), 4.54 (dt, 2JHF = 47.5 Hz, 3J = 5.7 Hz, 2H), 4.64 (t, 3J = 7.3 Hz, 2H), 5.13 (s, 1H, CH), 7.34 (dd, 3J = 7.8, 4.6 Hz, 1H), 8.11 (dd, 3J = 7.8, 1.9 Hz, 1H), 8.78 (dd, 3J = 4.6, 1.8 Hz, 1H), 8.81 (s, 1H), 11.67 (s, 1H); 11B{1H} NMR (128 MHz, CDCl3) δ −13.9 (s, 3B), −10.9 (s, 5B), −7.1 (s, 1B), −4.0 (s, 1B); 13C{1H} NMR (101 MHz, CDCl3) δ 24.1 (d, 3JCF = 5.1 Hz), 28.1 (d, 2JCF = 20.1 Hz), 42.0, 60.6, 78.9, 83.8 (d, 1JCF = 165.3 Hz), 114.8, 119.9, 121.0, 139.2, 143.7, 150.0, 153.6, 161.9, 162.7; HRMS (ESI+) m/z for C15H24B10FN3NaO2 [M + Na]+ 428.2764, calcd 428.2754; melting range 177–178 °C.
1-(4-Fluorobutyl)-2-oxo-N-(1,7-dicarba-closo-dodecaboranyl)-1,2-dihydro-1,8-naphthyridine-3-carboxamide (10) [5:1 (v/v) n-hexane/EtOAc → 100% EtOAc] was obtained as a white solid: 0.065 g (0.160 mmol, 42%); 1H NMR (400 MHz, CDCl3) δ 1.40–4.16 (br, 10H), 1.84 (m, 4H), 2.96 (s, 1H), 4.52 (dt, 2JHF = 47.6 Hz, 3J = 5.6 Hz, 2H), 4.61 (t, 3J = 7.2 Hz, 2H), 7.31 (dd, 3J = 7.8, 4.7 Hz, 1H), 8.07 (dd, 3J = 8.3, 1.8 Hz, 1H), 8.74 (dd, 3J = 4.7, 1.7 Hz, 1H), 8.83 (s, 1H), 10.94 (s, 1H); 11B{1H} NMR (128 MHz, CDCl3) δ −15.2 (s, 5B), −12.4 (s, 2B), −10.7 (s, 2B), −3.8 (s, 1B); 13C{1H} NMR (101 MHz, CDCl3) δ 24.1 (d, 3JCF = 5.2 Hz), 28.1 (d, 2JCF = 20.0 Hz), 41.8, 53.2, 79.8, 83.9 (d, 1JCF = 164.9 Hz), 114.9, 119.6, 121.8, 139.0, 143.3, 150.0, 153.1, 161.2, 162.6; HRMS (ESI+) m/z for C15H25B10FN3O2 [M + H]+ 406.2940, calcd 406.2934; melting range 176–178 °C.
1-(4-Fluorobutyl)-2-oxo-N-(1,7-dicarba-closo-dodecaboranyl)-1,2-dihydro-1,8-naphthyridine-3-carboxamide (11) [9:1 (v/v) n-hexane/EtOAc → 100% EtOAc] was obtained as a white solid: 0.077 g (0.190 mmol, 50%); 1H NMR (400 MHz, CDCl3) δ 1.40–3.58 (br, 10H), 1.83 (m, 4H), 2.71 (s, 1H), 4.49 (dt, 2JHF = 47.7 Hz, 3J = 6.0, 5.6 Hz, 2H), 4.59 (t, 3J = 7.1 Hz, 2H), 7.28 (dd, 3J = 8.5, 5.3 Hz, 1H), 8.04 (dd, 3J = 7.9, 1.9 Hz, 1H), 8.72 (dd, 3J = 4.8, 1.8 Hz, 1H), 8.76 (s, 1H), 10.63 (s, 1H); 11B{1H} NMR (128 MHz, CDCl3) δ −16.5 (s, 5B), −12.5 (s, 5B); 13C{1H} NMR (101 MHz, CDCl3) δ 24.0 (d, 3JCF = 5.2 Hz), 28.0 (d, 2JCF = 20.1 Hz), 41.7, 56.3, 83.8 (d, 1JCF = 165.0 Hz), 88.0, 114.8, 119.5, 122.0, 138.8, 143.1, 149.8, 152.9, 160.3, 162.5; HRMS (ESI+) m/z for C15H24B10FN3NaO2 [M + Na]+ 428.2755, calcd 428.2754; melting range 209–213 °C.
Sodium 1-(4-fluorobutyl)-2-oxo-N-(7,8-dicarba-nido-dodeca-hydroundecaborate(−1))-1,2-dihydro-1,8-naphthyridine-3-carboxamide (12, racemate).74,75 A microwave vial was filled with 9 (65.1 mg, 0.160 mmol, 1.00 equiv), NaF (50.0 mg, 1.19 mmol, 7.43 equiv), and a mixture of degassed EtOH/H2O [4.0 mL, 1:1 (v/v)]. The white suspension was stirred for 5 min at 150 °C in the microwave. The solution was decanted, and the solvent was removed under reduced pressure: 1H NMR (400 MHz, acetone-d6) δ −2.03 (d, 3J = 66.9 Hz, 1H), −0.55–2.96 (br, 9H), 1.77 (m, 4H), 3.26 (s, 1H, CH), 4.42 (t, 3J = 5.8 Hz, 1H), 4.54 (m, 3H), 7.44 (dd, 3J = 7.8, 4.6 Hz, 1H), 8.40 (dd, 3J = 7.8, 1.8 Hz, 1H), 8.74 (dd, 3J = 4.7, 1.8 Hz, 1H), 8.91 (s, 1H), 10.12 (s, 1H); 11B{1H} NMR (128 MHz, acetone-d6) δ −37.9 (s, 1B), −33.5 (s, 2B), −24.2 (s, 1B), −19.6 (s, 1B), −18.1 (s, 1B), −14.3 (s, 1B), −10.5 (s, 2B); 13C{1H} NMR (101 MHz, acetone-d6) δ 24.0 (d, 3JCF = 5.3 Hz), 28.1 (d, 2JCF = 19.9 Hz), 41.3, 83.8 (d, 1JCF = 163.2 Hz), 115.2, 119.7, 123.3, 139.4, 142.1, 149.9, 152.5, 162.5, 163.6.
Compound 12 was purified by HPLC using a Reprosil-Pur 120 C18-AQ (250 mm × 20 mm, 10 μm) column with 50% CH3CN/20 mM ammonium acetate with a flow rate of 6 mL/min. The retention time was 16 min. 12 was obtained as a yellow solid (53.0 mg, 0.127 mmol, 79% yield).
(Z)-N-[3-(4-Fluorobutyl)-4,5-dimethylthiazole-2(3H)-ylidene]-(dicarba-closo-dodecaboranyl)-carboxamide (15, LUZ5, and 17). Procedure 1.28(Z)-N-[3-(4-Fluorobutyl)-4,5-dimethylthiazole-2(3H)-ylidene]-(1,2-dicarba-closo-dodecaboranyl)-carboxamide (15). A solution of 8o (0.348 mmol, 0.987 equiv) in dry DCM (3 mL) was added to a solution of 14 (0.353 mmol, 1.00 equiv) in dry DCM (2 mL). Triethylamine (0.361 mmol, 1.02 equiv) was added, and the mixture was stirred for 48 h at rt. The reaction was quenched by the addition of H2O (2 mL), an aqueous saturated NaHCO3 solution (10 mL), and EtOAc (15 mL). The phases were separated, and the aqueous layer was washed with EtOAc (2 × 15 mL). The combined organic layers were dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. The solid was stored under a nitrogen atmosphere and purified by column chromatography [n-hexane/EtOAc, 4:1 (v/v) → 1:1 (v/v)]. Compound 15 was obtained as a white solid (0.032 g, 0.086 mmol, 24% yield): 1H NMR (400 MHz, CDCl3) δ 1.46–3.25 (br, 10H), 1.82 (m, 4H), 2.24 (m, 6H), 4.18 (m, 2H), 4.31 (s, 1H), 4.51 (dt, 2JHF = 47.4 Hz, 3J = 5.6 Hz, 2H); 11B{1H} NMR (128 MHz, CDCl3) δ −14.0 (s, 2B), −11.8 (s, 4B), −9.5 (s, 2B), −3.9 (s, 1B), −3.4 (s, 1B); 13C{1H} NMR (101 MHz, CDCl3) δ 11.2, 11.7, 25.2 (d, 3JCF = 4.0 Hz), 27.6 (d, 2JCF = 20.1 Hz), 46.7, 56.7, 75.4, 83.5 (d, 1JCF = 165.6 Hz), 117.1, 129.4, 166.5, 166.8; HRMS (ESI+) m/z for C12H26B10FN2OS [M + H]+ 373.2761, calcd 373.2753.
Procedure 2.79 The appropriate thiazole was dissolved in dry CH3CN and heated to 80 °C. The respective dicarba-closo-dodecaborane-1-carboxylic acid chloride (8) in dry CH3CN was added, and the reaction mixture was stirred for 2 h under reflux. Triethylamine was added at once, and the mixture was stirred for 16–20 h at 80 °C. The reaction flask was placed in an ice bath, and the precipitate formed was isolated by filtration, suspended in CH3CN, and filtered. The solvent was removed under reduced pressure. The crude product was stored under nitrogen and purified by column chromatography [n-hexane/EtOAc, 4:1 (v/v) → 1:1 (v/v)]. (Z)-N-[3-(4-Fluorobutyl)-4,5-dimethylthiazole-2(3H)-ylidene]-(1,2-dicarba-closo-dodecaboranyl)-carboxamide (15) was synthesized from 14 (0.353 mmol, 1.00 equiv) in dry CH3CN (1.3 mL), 8o (0.348 mmol, 0.987 equiv) in dry CH3CN (1.6 mL), triethylamine (0.361 mmol, 1.02 equiv), suspension in CH3CN (5 mL), yield of compound 15: 0.041 g (0.110 mmol, 31%), white solid; 1H NMR (400 MHz, CDCl3) δ 1.46–3.24 (br, 10H), 1.82 (m, 4H), 2.25 (s, 6H), 4.18 (m, 2H), 4.31 (s, 1H), 4.51 (dt, 2JHF = 47.5 Hz, 3J = 5.5 Hz, 2H); 11B{1H} NMR (128 MHz, CDCl3) δ −14.0 (s, 2B), −11.7 (s, 4B), −9.5 (s, 2B), −4.0 (s, 1B), −3.4 (s, 1B); HRMS (ESI+) m/z for C12H26B10FN2OS [M + H]+ 373.2753, calcd 373.2753; melting range 184–187 °C.
(Z)-N-[3-(4-Fluorobutyl)-4,5-dimethylthiazole-2(3H)-ylidene]-(1,7-dicarba-closo-dodecaboranyl)-carboxamide (LUZ5) was synthesized from 14 (0.551 mmol, 1.00 equiv) in dry CH3CN (2.0 mL), 8m (0.561 mmol, 1.02 equiv) in dry CH3CN (2.6 mL), triethylamine (0.577 mmol, 1.05 equiv) in dry CH3CN (1.0 mL), suspension in CH3CN (15 mL), yield of compound LUZ5: 0.093 g (0.250 mmol, 45%), white solid; 1H NMR (400 MHz, CDCl3) δ 1.45–3.78 (br, 10H), 1.80 (m, 4H), 2.21 (s, 3H), 2.22 (s, 3H), 2.96 (s, 1H), 4.17 (t, 3J = 7.2 Hz, 2H), 4.51 (dt, 2JHF = 47.6 Hz, 3J = 5.6 Hz, 2H); 11B{1H} NMR (128 MHz, CDCl3) δ −15.6 (s, 2B), −13.8 (s, 2B), −11.1 (s, 4B), −8.2 (s, 1B), −4.8 (s, 1B); 13C{1H} NMR (101 MHz, CDCl3) δ 11.3, 11.9, 25.2 (d, 3JCF = 4.1 Hz), 27.8 (d, 2JCF = 20.1 Hz), 46.7, 54.4, 83.6 (d, 1JCF = 165.4 Hz), 116.5, 129.1, 167.0, 168.2; HRMS (ESI+) m/z for C12H26B10FN2OS [M + H]+ 373.2753, calcd 373.2753; melting range 199–202 °C.
(Z)-N-[3-(4-Fluorobutyl)-4,5-dimethylthiazole-2(3H)-ylidene]-(1,12-dicarba-closo-dodecaboranyl)-carboxamide (17) was synthesized from 14 (0.565 mmol, 1.00 equiv) in dry CH3CN (2.0 mL), 8p (0.847 mmol, 1.50 equiv) in dry CH3CN (2.8 mL), triethylamine (0.577 mmol, 1.02 equiv) in dry CH3CN (1.0 mL), suspension in CH3CN (15 mL), yield of compound 17: 0.075 g (0.201 mmol, 36%), white solid; 1H NMR (400 MHz, CDCl3) δ 1.00–3.27 (br, 10H), 1.79 (m, 4H), 2.17 (s, 3H), 2.19 (s, 3H), 2.73 (s, 1H), 4.12 (t, 3J = 7.2 Hz, 2H), 4.51 (dt, 2JHF = 47.7 Hz, 3J = 5.5 Hz, 2H); 11B{1H} NMR (128 MHz, CDCl3) δ −15.6 (s, 5B), −13.2 (s, 5B); 13C{1H} NMR (101 MHz, CDCl3) δ 11.2, 11.8, 25.1 (d, 3JCF = 4.5 Hz), 27.8 (d, 2JCF = 20.1 Hz), 46.6, 60.7, 83.7 (d, 1JCF = 165.4 Hz), 116.2, 128.9, 167.0, 168.9; HRMS (ESI+) m/z for C12H26B10FN2OS [M + H]+ 373.2755, calcd 373.2753; melting range 205–213 °C.
(Z)-N-[4,5-Dimethylthiazole-2(3H)-ylidene]-(1,7-dicarba-closo-dodecaboranyl)-carboxamide (18). The preparation of the compound was analogous to procedure 279 given above, synthesized from 13 (1.47 mmol, 1.00 equiv) in dry CH3CN (6 mL), 10m (1.50 mmol, 1.02 equiv) in dry CH3CN (8 mL), and triethylamine (1.51 mmol, 1.03 equiv) in dry CH3CN (4 mL), via column chromatography with 100% n-hexane → 4:1 (v/v) n-hexane/EtOAc → 1:1 (v/v) n-hexane/EtOAc → 100% EtOAc, yielding 0.113 g (0.379 mmol, 26%) of compound 18 as a white solid: 1H NMR (400 MHz, CDCl3) δ 1.45–3.87 (br, 10H), 2.21 (s, 3H), 2.27 (s, 3H), 3.09 (s, 1H), 8.74 (s, 1H); 11B{1H} NMR (128 MHz, CDCl3) δ −15.6 (s, 2B), −13.0 (s, 2B), −11.5 (s, 1B), −10.6 (s, 2B), −6.6 (s, 2B), −5.8 (s, 1B).
(Z)-N-[3-(4-Fluorobutyl-d8)-4,5-dimethylthiazole-2(3H)-ylidene]-(1,7-dicarba-closo-dodecaboranyl)-carboxamide (19).28 NaH (0.027 g, 1.13 mmol, 2.98 equiv) and 1,4-dibromobutane-d8 (0.5 mL, 0.937 g, 4.18 mmol, 11.0 equiv) were added to a solution of 18 (0.113 g, 0.379 mmol, 1.00 equiv) in dry DMF (0.5 mL), and it was stirred for 21 h at rt. Water (2 mL) was added to stop the reaction. NaHCO3 (15 mL) and EtOAc (20 mL) were added, and the phases were separated. After washing with EtOAc (20 mL), the organic phase was dried over MgSO4 and filtered, and the solvents were removed under vacuum. The crude product was purified by column chromatography [n-hexane/EtOAc, 5:1 (v/v) → 2:1 (v/v), yielding 0.034 g (0.078 mmol, 20%) of compound 19 as a white solid: 1H NMR (400 MHz, CDCl3) δ 1.00–3.88 (br, 10H), 2.21 (s, 3H), 2.23 (s, 3H), 2.96 (s, 1H); 11B{1H} NMR (128 MHz, CDCl3) δ −15.5 (s, 2B), −13.7 (s, 2B), −11.1 (s, 4B), −8.1 (s, 1B), −4.7 (s, 1B); 13C{1H} NMR (101 MHz, CDCl3) δ 11.3, 11.8, 26.2 (m), 28.6 (m), 32.3 (m), 45.5 (m), 54.4, 116.5, 129.1, 166.9, 168.1; HRMS (ESI+) m/z for C12H18D8B10BrN2OS [M + H]+ 442.2446, calcd 442.2418.
Compound 19 (10 mg, 0.023 mmol) was purified by HPLC using a Reprosil-Pur 120 C18-AQ (250 mm × 10 mm, 5 μm) column, with 75% aqueous CH3CN with a flow rate of 5 mL/min. The retention time was 18 min.
Stability
Stability tests were performed with 1H and 11B{1H} NMR spectroscopy. Approximately 10 mg of each compound was dissolved in aqueous DMSO-d6 and measured directly thereafter and at increasing time intervals from a few minutes to a couple of days or beyond a year.
The stability of LUZ5 was additionally investigated under the conditions described above, but in the presence of larger amounts of H2O or D2O. For this, LUZ5 was dissolved in DMSO-d6 (0.5 mL) and either H2O (0.1 mL) or D2O (0.1 mL) was added.
Binding Affinity
The in vitro binding affinity assays were performed with membrane homogenates from Chinese hamster ovary (CHO) cells stably transfected with the human CB2R according to a previously published protocol.95 The screening for CB1R binding affinity determination was performed with membrane homogenates from CHO cells stably transfected with the human CB1R as described previously.96
In Vitro Metabolism Studies
For in vitro evaluation of metabolites, the following instruments were used: BioShake iQ (QUANTIFOIL Instruments, Jena, Germany), Centrifuge 5424 (Eppendorf, Hamburg, Germany), UltiMate 3000 UHPLC System (Thermo Scientific, Germering, Germany) including a DAD detector (DAD-3000RS) coupled to an MSQ Plus single quadrupole mass spectrometer (Thermo Scientific, Austin, TX). Compounds 9–11, 15, LUZ5, and 17 were investigated with regard to their metabolism in vitro by incubation with mouse liver microsomes (MLMs) and analyses by HPLC-UV-MS according to an already published protocol.81,82 In brief, each of the test compounds (dissolved in DMSO, final concentration of 10 μM, final DMSO percentage of 1%) and MLMs (final protein concentration of 1 mg/mL) in PBS (pH 7.4) were preincubated in PBS (pH 7.4) for 5 min at 37 °C. Similarly preincubated NADPH (final concentration of 2 mM) was added to start the incubation. The final volume of each incubation mixture was 250 μL, and samples were prepared in duplicate. After the mixture had been gently shaken for 30 or 60 min at 37 °C, 1 mL of ice-cold CH3CN was added. The mixture was shaken vigorously for 30 s, rested on ice for 5 min, shaken for an additional 30 s, and then centrifuged at 14 000 rpm for 10 min. The supernatants were stored at 4 °C until they were measured by HPLC-UV-MS. As a positive control, samples with testosterone (final concentration of 20 μM) as the substrate were prepared like the test compounds, with or without NADPH and incubation for 60 or 90 min. Negative controls without NADPH as well as without both NADPH and MLM were also prepared for each test substance as well as controls without a substrate.
HPLC-UV-MS was performed on a Poroshell 120 EC-C18 column (100 mm × 3 mm, 2.7 μm) (Agilent Technologies, Waldbronn, Germany) at 25 °C (eluent A, H2O and 0.1% formic acid; eluent B, CH3CN and 0.1% formic acid) at a flow rate of 0.7 mL/min and monitoring a wavelength of 323 nm. The gradient elution method was as follows: 20% B from 0 to 1.5 min, 20% to 100% B from 1.5 to 10 min, 100% B from 10 to 12 min, and 20% B from 12 to 15 min. The MSQ Plus single quadrupole mass spectrometer was operated in positive and negative electrospray ionization mode: probe temperature of 550 °C, needle voltage of 3 V, and cone voltage of 75 V. On the basis of the obtained UV chromatograms, the percentage of unchanged compound was calculated from the peak area divided by the sum of peak areas of all relevant signals.
Radiochemistry
Automated Radiosynthesis of [18F]LUZ5-d8
Remote-controlled radiosynthesis was performed using a Synchrom R&D EVO III automated synthesizer (Elysia-Raytest). Briefly, [18F]fluoride (5–12 GBq) was trapped on a Waters QMA cartridge, eluted with a H2O/CH3CN [1 mL, 1:4 (v/v)] solution containing 2.2.2-cryptand (K2.2.2) (11 mg) and K2CO3 (75 μL) into the reaction vessel, and dried via azeotropic distillation. To complete the azeotropic distillation, additional dry CH3CN (1.5 mL) was added. After complete dryness, precursor 19 (1 mg, 22.65 μmol) in CH3CN (1 mL) was added and the reaction mixture was stirred at 90 °C for 10 min. The reaction mixture was diluted with 4 mL of H2O/CH3CN (1:1), and the solution was transferred to the semipreparative HPLC instrument. [18F]LUZ5-d8 was collected via the HPLC collection vial containing 40 mL of H2O and trapped in the Sep-Pak C18 light cartridge. The cartridge was washed with 2 mL of H2O, and [18F]LUZ5-d8 was eluted with 1.3 mL of EtOH. This ethanolic solution was transferred outside of the shielded cell; the solvent was evaporated at 70 °C in a gentle stream of nitrogen for 5–10 min, and [18F]LUZ5-d8 was reconstituted in an isotonic saline solution for further biological characterization. The total synthesis time was ∼85 min.
Quality Control
Radio-thin-layer chromatography was performed on Alugram SIL G/UV254 precoated plates (Macherey-Nagel) with PE:EtOAc [1:1 (v/v)]. The plates were exposed to storage phosphor screens (BAS IP MS 2025 E, GE Healthcare Europe GmbH, Freiburg, Germany), and images were recorded using the Amersham Typhoon RGB Biomolecular Imager (GE Healthcare Life Sciences). Images were quantified with ImageQuant version TL8.1 (GE Healthcare Life Sciences).
Analytical chromatographic separations were performed on a JASCO LC-2000 system, incorporating a PU-2080Plus pump, a model AS-2055Plus auto injector (100 μL sample loop), and a model UV-2070Plus detector coupled with a γ-detector (GABI Star; raytest Isotopenmessgeräte GmbH, Straubenhardt, Germany). Data analysis was performed with the Galaxie chromatography software (Agilent Technologies) using the chromatograms obtained at 254 nm.
The radiochemical yield, radiochemical purity, and analyses of plasma and brain samples were assessed via reverse phase HPLC (RP-HPLC) in gradient mode (10% CH3CN/20 mM NH4OAcaq from 0 to 5 min, 10% → 90% CH3CN/20 mM NH4OAcaq from 5 to 18 min, 90% CH3CN/20 mM NH4OAcaq from 18 to 25 min, 90% → 10% CH3CN/20 mM NH4OAcaq from 25 to 26 min, and 10% CH3CN/20 mM NH4OAcaq from 26 to 30 min).
The molar activity was determined using analytical radio-HPLC with a Reprosil-Pur C18-AQ column (250 mm × 4.6 mm, 5 μm) and 66% CH3CN/20 mM NH4OAcaq as the eluent at a flow rate of 1 mL/min and UV detection at 312 nm.
Determination of Lipophilicity (logD7.4)
The logD7.4 of [18F]LUZ5-d8 was experimentally determined in n-octanol/phosphate-buffered saline (PBS; 0.01 M, pH 7.4) at rt by the shake-flask method. The measurement was performed twice in triplicate.22
Quantification of Radiometabolites
20 to 30 megabecquerels of [18F]LUZ5-d8 dissolved in ∼150 μL of isotonic saline was administered intravenously as a bolus in the tail vein of awake female CD-1 mice weighing ∼33 g (n = 3) and male Wistar rats weighing 240 and 380 g (n = 2). At 30 min p.i., the animals were anesthetized and blood was withdrawn by retrobulbar bleeding using glass capillaries. Immediately afterward, the animals were euthanized by cervical dislocation, and the released urine was sampled. Blood plasma was obtained from the whole blood sample by centrifugation (2 min, 8000 rpm, room temperature). In addition, the brain and spleen were isolated and homogenized in 1 mL of demineralized water on ice (1000 rpm, 10 strokes; glass vessel, PTFE plunger; Potter S, B. Braun Biotech International, Goettingen, Germany).
The samples were further processed for subsequent radio-chromatographic analyses. Two consecutive extractions were performed as duplicates for plasma and brain determinations. Plasma and brain samples were added to an ice-cold mixture of either MeOH and H2O [9:1 (v/v); n = 3] or CH3CN and H2O [9:1 (v/v); n = 1] with extraction efficiencies of ≥94% for the former and ≥89% for the latter. The samples were vortexed for 3 min, incubated on ice for 5 min, vortexed for 3 min, and centrifuged at 10 000 rpm for 5 min. Supernatants were collected; the precipitates were redissolved in 100 μL of extraction solvent, and the extraction procedure was repeated. The activities of supernatants and precipitates were measured in a γ-counter (1480 WIZARD, Fa. PerkinElmer), and the extraction efficiencies were calculated as the ratio of radioactivity in the supernatant to the radioactivity in the original sample (supernatant + precipitate). The supernatants from both extractions were combined, concentrated at 70 °C under argon up to a remaining volume of 100 μL, and subsequently analyzed by analytical radio-HPLC with a gradient system (see Quality Control).
PET Experiments
The in vivo biodistribution of [18F]LUZ5-d8 in male Wistar rats and a female CD-1 mouse was assessed by dynamic small animal PET (Nanoscan, Mediso, Budapest, Hungary), 30 min recordings, followed by T1-weighted (GRE, TR/TE = 15.0/2.4 ms, 252/252, FA = 25°) magnetic resonance (MR) imaging with whole-body coils for anatomical correlation and attenuation correction. Animals were initially anesthetized with 5% isoflurane and placed on a thermostatically heated animal bed where anesthesia was maintained with 2% isoflurane in 60% oxygen/38% room air. The radiotracer was injected into the lateral tail vein (bolus within 5 s) at the start of the PET acquisition.
For the baseline study, 15 ± 3 MBq of the radiotracer was injected into three rats with a body weight of 230 ± 13 g. The blocking studies were conducted by the injection of 1.5 mg of the CB2R agonist GW405833 per kilogram (predissolved in 1:2:7 DMSO/Kolliphor/0.9% NaCl) 10 min prior to the radiotracer (21.6 ± 0.9 MBq) into three rats weighing 228 ± 10 g. Brain uptake studies were performed with two rats (body weights of 213 and 242 g, injected doses of 15.2 and 17.0 MBq, respectively); for the whole body biodistribution study in mouse, one animal was used (body weight of 30.6 g, injected dose of 15.2 MBq).
List-mode PET data were binned as a series of attenuation-corrected sinogram frames and reconstructed by ordered subset expectation maximization (OSEM3D) with four iterations, six subsets, and a voxel size of 0.4 mm3 (Nucline version 2.01, Mediso). The analysis of reconstructed data sets was performed with PMOD version 4.103 (PMOD Technologies LLC, Zurich, Switzerland). Non-parametrical analysis of achieved time-activity curves (TACs) was performed with Microsoft Excel to determine the time to peak, the TAC peak value, and the area under the curve (AUC):
where c(radioactivity) is expressed as the standardized uptake value normalized to the body weight in grams (SUV).
Data are shown as means ± the standard deviation. Group differences were tested by a Student’s t test, with p < 0.05 designated as significant. Graphs were generated with GraphPad Prism version 9.3.1.
References
- B. E. Alger. Getting high on the endocannabinoid system.. Cerebrum, 2013. [PubMed]
- J. Guindon, A. G. Hohmann. The endocannabinoid system and pain.. CNS Neurol. Disord. Drug Targets, 2009. [DOI | PubMed]
- L. A. Matsuda, S. J. Lolait, M. J. Brownstein, A. C. Young, T. I. Bonner. Structure of a cannabinoid receptor and functional expression of the cloned cDNA.. Nature, 1990. [DOI | PubMed]
- S. Munro, K. L. Thomas, M. Abu-Shaar. Molecular characterization of a peripheral receptor for cannabinoids.. Nature, 1993. [DOI | PubMed]
- E. Moreno, M. Cavic, A. Krivokuca, V. Casadó, E. Canela. The Endocannabinoid System as a Target in Cancer Diseases:Are We There Yet?.. Front. Pharmacol., 2019. [DOI | PubMed]
- W. A. Devane, F. A. Dysarz, M. R. Johnson, L. S. Melvin, A. C. Howlett. Determination and characterization of a cannabinoid receptor in rat brain.. Mol. Pharmacol., 1988. [PubMed]
- A. C. Howlett, M. E. Abood. CB1 and CB2 Receptor Pharmacology.. Adv. Pharmacol., 2017. [DOI | PubMed]
- H.-C. Lu, K. Mackie. Review of the Endocannabinoid System.. Biol. Psychiatry Cogn. Neurosci. Neuroimaging, 2021. [DOI | PubMed]
- P. Morales, A. Lago-Fernandez, D. P. Hurst, N. Sotudeh, E. Brailoiu, P. H. Reggio, M. E. Abood, N. Jagerovic. Therapeutic Exploitation of GPR18: Beyond the Cannabinoids?.. J. Med. Chem., 2020. [DOI | PubMed]
- E. Tudurí, M. Imbernon, R. J. Hernández-Bautista, M. Tojo, J. Fernø, C. Diéguez, R. Nogueiras. GPR55: a new promising target for metabolism?.. J. Mol. Endocrinol., 2017. [DOI | PubMed]
- F. Shahbazi, V. Grandi, A. Banerjee, J. F. Trant. Cannabinoids and Cannabinoid Receptors:The Story so Far.. iScience, 2020. [DOI | PubMed]
- X. Li, T. Hua, K. Vemuri, J.-H. Ho, Y. Wu, L. Wu, P. Popov, O. Benchama, N. Zvonok, K. Locke, L. Qu, G. W. Han, M. R. Iyer, R. Cinar, N. J. Coffey, J. Wang, M. Wu, V. Katritch, S. Zhao, G. Kunos, L. M. Bohn, A. Makriyannis, R. C. Stevens, Z.-J. Liu. Crystal Structure of the Human Cannabinoid Receptor CB2.. Cell, 2019. [DOI | PubMed]
- R. G. Biringer. Endocannabinoid signaling pathways: beyond CB1R and CB2R.. J. Cell Commun. Signal., 2021. [DOI | PubMed]
- G. F. Mangiatordi, F. Intranuovo, P. Delre, F. S. Abatematteo, C. Abate, M. Niso, T. M. Creanza, N. Ancona, A. Stefanachi, M. Contino. Cannabinoid Receptor Subtype 2 (CB2R) in a Multitarget Approach: Perspective of an Innovative Strategy in Cancer and Neurodegeneration.. J. Med. Chem., 2020. [DOI | PubMed]
- R. Ni, L. Mu, S. Ametamey. Positron emission tomography of type 2 cannabinoid receptors for detecting inflammation in the central nervous system.. Acta Pharmacol. Sin., 2019. [DOI | PubMed]
- D.-J. Chen, M. Gao, F.-F. Gao, Q.-X. Su, J. Wu. Brain cannabinoid receptor 2: expression, function and modulation.. Acta Pharmacol. Sin., 2017. [DOI | PubMed]
- P. Jain, A. M. Chaney, M. L. Carlson, I. M. Jackson, A. Rao, M. L. James. Neuroinflammation PET Imaging: Current Opinion and Future Directions.. J. Nucl. Med., 2020. [DOI | PubMed]
- M. Argenziano, C. Tortora, G. Bellini, A. Di Paola, F. Punzo, F. Rossi. The Endocannabinoid System in Pediatric Inflammatory and Immune Diseases.. Int. J. Mol. Sci., 2019. [DOI | PubMed]
- C. Turcotte, M.-R. Blanchet, M. Laviolette, N. Flamand. The CB2 receptor and its role as a regulator of inflammation.. Cell. Mol. Life Sci., 2016. [DOI | PubMed]
- V. Lucchesi, D. P. Hurst, D. M. Shore, S. Bertini, B. M. Ehrmann, M. Allarà, L. Lawrence, A. Ligresti, F. Minutolo, G. Saccomanni, H. Sharir, M. Macchia, V. Di Marzo, M. E. Abood, P. H. Reggio, C. Manera. CB2-selective cannabinoid receptor ligands:Synthesis, pharmacological evaluation, and molecular modeling investigation of 1,8-Naphthyridin-2(1H)-one-3-carboxamides.. J. Med. Chem., 2014. [DOI | PubMed]
- G. Navarro, P. Morales, C. Rodríguez-Cueto, J. Fernández-Ruiz, N. Jagerovic, R. Franco. Targeting Cannabinoid CB2 Receptors in the Central Nervous System. Medicinal Chemistry Approaches with Focus on Neurodegenerative Disorders.. Front. Neurosci., 2016. [DOI | PubMed]
- R. N. Waterhouse. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents.. Mol. Imaging Biol., 2003. [DOI | PubMed]
- P. Brust, J. van den Hoff, J. Steinbach. Development of (18)F-labeled radiotracers for neuroreceptor imaging with positron emission tomography.. Neurosci. Bull., 2014. [DOI | PubMed]
- F. Caillé, F. Cacheux, M.-A. Peyronneau, B. Jego, E. Jaumain, G. Pottier, C. Ullmer, U. Grether, A. Winkeler, F. Dollé, A. Damont, B. Kuhnast. From Structure-Activity Relationships on Thiazole Derivatives to the In Vivo Evaluation of a New Radiotracer for Cannabinoid Subtype 2 PET Imaging.. Mol. Pharmaceutics, 2017. [DOI]
- N. Evens, G. G. Muccioli, N. Houbrechts, D. M. Lambert, A. M. Verbruggen, K. van Laere, G. M. Bormans. Synthesis and biological evaluation of carbon-11- and fluorine-18-labeled 2-oxoquinoline derivatives for type 2 cannabinoid receptor positron emission tomography imaging.. Nucl. Med. Biol., 2009. [DOI | PubMed]
- R. Ahmad, A. Postnov, G. Bormans, J. Versijpt, M. Vandenbulcke, K. van Laere. Decreased in vivo availability of the cannabinoid type 2 receptor in Alzheimer’s disease.. Eur. J. Nucl. Med. Mol. Imaging, 2016. [DOI | PubMed]
- F. Spinelli, E. Capparelli, C. Abate, N. A. Colabufo, M. Contino. Perspectives of Cannabinoid Type 2 Receptor (CB2R) Ligands in Neurodegenerative Disorders:Structure-Affinity Relationship (SAfiR) and Structure-Activity Relationship (SAR) Studies.. J. Med. Chem., 2017. [DOI | PubMed]
- R.-P. Moldovan, R. Teodoro, Y. Gao, W. Deuther-Conrad, M. Kranz, Y. Wang, H. Kuwabara, M. Nakano, H. Valentine, S. Fischer, M. G. Pomper, D. F. Wong, R. F. Dannals, P. Brust, A. G. Horti. Development of a High-Affinity PET Radioligand for Imaging Cannabinoid Subtype 2 Receptor.. J. Med. Chem., 2016. [DOI | PubMed]
- F. Basagni, M. Rosini, M. Decker. Functionalized Cannabinoid Subtype 2 Receptor Ligands:Fluorescent, PET, Photochromic and Covalent Molecular Probes.. ChemMedChem., 2020. [DOI | PubMed]
- M. J. Dart, W. A. Carroll, A. S. Florjancic, J. M. Frost, M. E. Gallagher, T. Li, D. W. Nelson, M. V. Patel, S. Peddi, M. A. Perez, K. B. Ryther, K. R. Tietje, T. Kolasa. Novel Compounds as Cannabinoid Receptor Ligands and Uses Thereof. PCT US. 2007
- B. B. Yao, G. Hsieh, A. V. Daza, Y. Fan, G. K. Grayson, T. R. Garrison, O. El Kouhen, B. A. Hooker, M. Pai, E. J. Wensink, A. K. Salyers, P. Chandran, C. Z. Zhu, C. Zhong, K. Ryther, M. E. Gallagher, C.-L. Chin, A. E. Tovcimak, V. P. Hradil, G. B. Fox, M. J. Dart, P. Honore, M. D. Meyer. Characterization of a cannabinoid CB2 receptor-selective agonist, A-836339 2,2,3,3-tetramethyl-cyclopropanecarboxylic acid 3-(2-methoxy-ethyl)-4,5-dimethyl-3H-thiazol-(2Z)-ylidene-amide, using in vitro pharmacological assays, in vivo pain models, and pharmacological magnetic resonance imaging.. J. Pharmacol. Exp. Ther., 2009. [DOI | PubMed]
- A. G. Horti, Y. Gao, H. T. Ravert, P. Finley, H. Valentine, D. F. Wong, C. J. Endres, A. V. Savonenko, R. F. Dannals. Synthesis and biodistribution of 11CA-836339, a new potential radioligand for PET imaging of cannabinoid type 2 receptors (CB2).. Bioorg. Med. Chem., 2010. [DOI | PubMed]
- R.-P. Moldovan, W. Deuther-Conrad, R. Teodoro, Y. Wang, S. Fischer, M. Pomper, D. Wong, R. Dannals, P. Brust, A. Horti. 18F-JHU94620, a high affinity PET radioligand for imaging of cannabinoid subtype 2 receptors (CB2R).. J. Nucl. Med., 2015. [PubMed]
- Poster Presentations.. J. Label Compd. Radiopharm., 2019. [DOI]
- S. Auvity, F. Caillé, S. Demphel, B. Kuhnast, M.-A. Peyronneau. Preclinical evaluation of [18F]FC0324, a new CB2R PET radiolidand in non-human primates.. J. Nucl. Med., 2019
- F. Caillé, S. Auvity, B. Attili, G. Bormans, M. A. Peyronneau, B. Kuhnast. Développement translationnel d’un traceur TEP pour l’imagerie des récepteurs cannabinoïdes de type 2.. Med. Nucleaire, 2018. [DOI]
- R. Slavik, U. Grether, A. Müller Herde, L. Gobbi, J. Fingerle, C. Ullmer, S. D. Krämer, R. Schibli, L. Mu, S. M. Ametamey. Discovery of a high affinity and selective pyridine analog as a potential positron emission tomography imaging agent for cannabinoid type 2 receptor.. J. Med. Chem., 2015. [DOI | PubMed]
- A. Haider, J. Kretz, L. Gobbi, H. Ahmed, K. Atz, M. Bürkler, C. Bartelmus, J. Fingerle, W. Guba, C. Ullmer, M. Honer, I. Knuesel, M. Weber, A. Brink, A. M. Herde, C. Keller, R. Schibli, L. Mu, U. Grether, S. M. Ametamey. Structure-Activity Relationship Studies of Pyridine-Based Ligands and Identification of a Fluorinated Derivative for Positron Emission Tomography Imaging of Cannabinoid Type 2 Receptors.. J. Med. Chem., 2019. [DOI | PubMed]
- D. J. Modemann, A. B. Mahardhika, S. Yamoune, A.-K. Kreyenschmidt, F. Maaß, S. Kremers, C. Breunig, C.-O. Sahlmann, J. Bucerius, D. Stalke, J. Wiltfang, Y. Bouter, C. E. Müller, C. Bouter, B. Meller. Development of high-affinity fluorinated ligands for cannabinoid subtype 2 receptor, and in vitro evaluation of a radioactive tracer for imaging.. Eur. J. Med. Chem., 2022. [DOI | PubMed]
- R. Teodoro, D. Gündel, W. Deuther-Conrad, L. Ueberham, M. Toussaint, G. Bormans, P. Brust, R.-P. Moldovan. Development of 18FLU14 for PET Imaging of Cannabinoid Receptor Type 2 in the Brain.. Int. J. Mol. Sci., 2021. [DOI | PubMed]
- D. Gündel, W. Deuther-Conrad, L. Ueberham, S. Kaur, E. Otikova, R. Teodoro, M. Toussaint, T. H. Lai, O. Clauß, M. Scheunemann, G. Bormans, M. Bachmann, K. Kopka, P. Brust, R.-P. Moldovan. Structure-Based Design, Optimization, and Development of 18FLU13: A Novel Radioligand for Cannabinoid Receptor Type 2 Imaging in the Brain with PET.. J. Med. Chem., 2022. [DOI | PubMed]
- L. Hou, J. Rong, A. Haider, D. Ogasawara, C. Varlow, M. A. Schafroth, L. Mu, J. Gan, H. Xu, C. J. Fowler, M.-R. Zhang, N. Vasdev, S. Ametamey, B. F. Cravatt, L. Wang, S. H. Liang. Positron Emission Tomography Imaging of the Endocannabinoid System: Opportunities and Challenges in Radiotracer Development.. J. Med. Chem., 2021. [DOI | PubMed]
- D. Heimann, F. Börgel, H. de Vries, M. Patberg, E. Jan-Smith, B. Frehland, D. Schepmann, L. H. Heitman, B. Wünsch. Optimization of the metabolic stability of a fluorinated cannabinoid receptor subtype 2 (CB2) ligand designed for PET studies.. Eur. J. Med. Chem., 2018. [DOI | PubMed]
- R. Slavik, A. Müller Herde, A. Haider, S. D. Krämer, M. Weber, R. Schibli, S. M. Ametamey, L. Mu. Discovery of a fluorinated 4-oxo-quinoline derivative as a potential positron emission tomography radiotracer for imaging cannabinoid receptor type 2.. J. Neurochem., 2016. [DOI | PubMed]
- G. Pottier, V. Gómez-Vallejo, D. Padro, R. Boisgard, F. Dollé, J. Llop, A. Winkeler, A. Martín. PET imaging of cannabinoid type 2 receptors with 11CA-836339 did not evidence changes following neuroinflammation in rats.. J. Cereb. Blood Flow Metab., 2017. [DOI | PubMed]
- P. Stockmann, M. Gozzi, R. Kuhnert, M. B. Sárosi, E. Hey-Hawkins. New keys for old locks:Carborane-containing drugs as platforms for mechanism-based therapies.. Chem. Soc. Rev., 2019. [DOI | PubMed]
- P. Hoppenz, S. Els-Heindl, M. Kellert, R. Kuhnert, S. Saretz, H.-G. Lerchen, J. Köbberling, B. Riedl, E. Hey-Hawkins, A. G. Beck-Sickinger. A Selective Carborane-Functionalized Gastrin-Releasing Peptide Receptor Agonist as Boron Delivery Agent for Boron Neutron Capture Therapy.. J. Org. Chem., 2020. [DOI | PubMed]
- B. Grüner, J. Brynda, V. Das, V. Šícha, J. Štěpánková, J. Nekvinda, J. Holub, K. Pospíšilová, M. Fábry, P. Pachl, V. Král, M. Kugler, V. Mašek, M. Medvedíková, S. Matějková, A. Nová, B. Lišková, S. Gurská, P. Džubák, M. Hajdúch, P. Řezáčová. Metallacarborane Sulfamides: Unconventional, Specific, and Highly Selective Inhibitors of Carbonic Anhydrase IX.. J. Med. Chem., 2019. [DOI | PubMed]
- M. Couto, M. F. García, C. Alamón, M. Cabrera, P. Cabral, A. Merlino, F. Teixidor, H. Cerecetto, C. Viñas. Discovery of Potent EGFR Inhibitors through the Incorporation of a 3D-Aromatic-Boron-Rich-Cluster into the 4-Anilinoquinazoline Scaffold: Potential Drugs for Glioma Treatment.. Chem. – Eur. J., 2018. [DOI | PubMed]
- A. Marfavi, P. Kavianpour, L. M. Rendina. Carboranes in drug discovery, chemical biology and molecular imaging.. Nat. Rev. Chem., 2022. [DOI | PubMed]
- M. Scholz, E. Hey-Hawkins. Carbaboranes as pharmacophores:Properties, synthesis, and application strategies.. Chem. Rev., 2011. [DOI | PubMed]
- R. N. Grimes. Carboranes,, 2016
- Boron science: New technologies and applications; CRC Press, 2012.
- E. Hey-Hawkins, C. Viñas Teixidor. Boron-Based Compounds: Potential and Emerging Applications in Medicine;, 2018
- M. Gozzi, B. Schwarze, E. Hey-Hawkins. Half- and mixed-sandwich metallacarboranes for potential applications in medicine.. Pure Appl. Chem., 2019. [DOI]
- Handbook of boron science with applications in organometallics, catalysis, materials and medicine; World Scientific, 2019.
- N. Murphy, E. McCarthy, R. Dwyer, P. Farràs. Boron clusters as breast cancer therapeutics.. J. Inorg. Biochem., 2021. [DOI | PubMed]
- M. Kugler, J. Nekvinda, J. Holub, S. El Anwar, V. Das, V. Šícha, K. Pospíšilová, M. Fábry, V. Král, J. Brynda, V. Kašička, M. Hajdúch, P. Řezáčová, B. Grüner. Inhibitors of CA IX Enzyme Based on Polyhedral Boron Compounds.. ChemBioChem., 2021. [DOI | PubMed]
- Z. J. Leśnikowski. Challenges and Opportunities for the Application of Boron Clusters in Drug Design.. J. Med. Chem., 2016. [DOI | PubMed]
- K. Messner, B. Vuong, G. K. Tranmer. The Boron Advantage: The Evolution and Diversification of Boron’s Applications in Medicinal Chemistry.. Pharmaceuticals, 2022. [DOI | PubMed]
- B. C. Das, N. K. Nandwana, S. Das, V. Nandwana, M. A. Shareef, Y. Das, M. Saito, L. M. Weiss, F. Almaguel, N. S. Hosmane, T. Evans. Boron Chemicals in Drug Discovery and Development: Synthesis and Medicinal Perspective.. Molecules, 2022. [DOI | PubMed]
- Y. Endo, T. Yoshimi, C. Miyaura. Boron clusters for medicinal drug design: Selective estrogen receptor modulators bearing carborane.. Pure Appl. Chem., 2003. [DOI]
- M. Scholz, K. Bensdorf, R. Gust, E. Hey-Hawkins. Asborin:The carbaborane analogue of aspirin.. ChemMedChem., 2009. [DOI | PubMed]
- N. Vázquez, V. Gómez-Vallejo, J. Llop. Synthesis of novel pyrazole derivatives incorporating one dicarba-closo-dodecaborane unit.. Tetrahedron Lett., 2012. [DOI]
- D. Grafstein, J. Dvorak. Neocarboranes, a New Family of Stable Organoboranes Isomeric with the Carboranes.. Inorg. Chem., 1963. [DOI]
- G. M. Edvenson, D. F. Gaines. Thermal isomerization of regiospecifically boron-10-labeled icosahedral carboranes.. Inorg. Chem., 1990. [DOI]
- R.-P. Moldovan, B. Wenzel, R. Teodoro, W. Neumann, S. Dukic-Stefanovic, W. Kraus, P. Rong, W. Deuther-Conrad, E. Hey-Hawkins, U. Krügel, P. Brust. Studies towards the development of a PET radiotracer for imaging of the P2Y1 receptors in the brain: synthesis, 18F-labeling and preliminary biological evaluation.. Eur. J. Med. Chem., 2019. [DOI | PubMed]
- C. J. R. Fookes, T. Q. Pham, F. Mattner, I. Greguric, C. Loc’h, X. Liu, P. Berghofer, R. Shepherd, M.-C. Gregoire, A. Katsifis. Synthesis and biological evaluation of substituted 18Fimidazo1,2-apyridines and 18Fpyrazolo1,5-apyrimidines for the study of the peripheral benzodiazepine receptor using positron emission tomography.. J. Med. Chem., 2008. [DOI | PubMed]
- M. A. Klenner, G. Pascali, B. H. Fraser, T. A. Darwish. Kinetic isotope effects and synthetic strategies for deuterated carbon-11 and fluorine-18 labelled PET radiopharmaceuticals.. Nucl. Med. Biol., 2021. [DOI]
- J. Sircar, S. K. C. Kumar, W. Ying. Substituted naphthyridine derivatives as inhibitors of macrophage migration inhibitory factor and their use in the treatment of human diseases. PCT US. 2005
- L. Fader, O. Lepage, M. Bailey, P. L. Beaulieu, F. Bilodeau, R. Carson, A. Giroux, C. Godbout, B. Moureau, J. Naud, M. Parisien, M. Poirier, M. Poirier, S. Suprenant, C. Thibeault. Derivates as inhibitors of cytomegalovirus DNA polymerase. PCT US. 2013
- Y. Nie, Y. Wang, J. Miao, Y. Li, Z. Zhang. Synthesis and characterization of carboranyl Schiff base compounds from 1-amino- o -carborane.. J. Organomet. Chem., 2015. [DOI]
- M. Tsuji. On attempts at generation of carboranyl carbocation.. J. Org. Chem., 2003. [DOI | PubMed]
- A. S. Louie, N. Vasdev, J. F. Valliant. Preparation, characterization, and screening of a high affinity organometallic probe for α-adrenergic receptors.. J. Med. Chem., 2011. [DOI | PubMed]
- T. J. Gullon. The Preparation of Metallocarborane and Iodinated Carborane Amino Acid Analogues for Molecular Imaging and Therapy. M.S. Thesis,. 2010
- S. Choi, Y. Byun. Synthesis of sterically-hindered 1,7-dicarba-closo-dodecarborane thiourea analogs.. J. Organomet. Chem., 2013. [DOI]
- M. Scholz, A. L. Blobaum, L. J. Marnett, E. Hey-Hawkins. Ortho-carbaborane derivatives of indomethacin as cyclooxygenase (COX)-2 selective inhibitors.. Bioorg. Med. Chem., 2012. [DOI | PubMed]
- R. A. Kasar, G. M. Knudsen, S. B. Kahl. Synthesis of 3-Amino-1-carboxy-o-carborane and an Improved, General Method for the Synthesis of All Three C-Amino-C-carboxycarboranes.. Inorg. Chem., 1999. [DOI | PubMed]
- R. Richter, B. Tucker, H. Ulrich. Synthesis and reactions of some N-acylated and N-sulfonylated N,N’-dialkylureas.. J. Org. Chem., 1978. [DOI]
- M. Gozzi, B. Schwarze, E. Hey-Hawkins. Preparing (Metalla)carboranes for Nanomedicine.. ChemMedChem., 2021. [DOI | PubMed]
- F.-A. Ludwig, R. Smits, S. Fischer, C. K. Donat, A. Hoepping, P. Brust, J. Steinbach. LC-MS Supported Studies on the in Vitro Metabolism of both Enantiomers of Flubatine and the in Vivo Metabolism of (+)-(18)FFlubatine-A Positron Emission Tomography Radioligand for Imaging α4β2 Nicotinic Acetylcholine Receptors.. Molecules, 2016. [DOI | PubMed]
- R. Ritawidya, F.-A. Ludwig, D. Briel, P. Brust, M. Scheunemann. Synthesis and In Vitro Evaluation of 8-Pyridinyl-Substituted Benzoeimidazo2,1-c1,2,4triazines as Phosphodiesterase 2A Inhibitors.. Molecules, 2019. [DOI | PubMed]
- D. Schneider, D. Bier, M. Holschbach, A. Bauer, B. Neumaier. Species Differences in Microsomal Metabolism of Xanthine-Derived A1 Adenosine Receptor Ligands.. Pharmaceuticals, 2021. [DOI | PubMed]
- I. Y. Bae, M. S. Choi, Y. S. Ji, S.-K. Yoo, K. Kim, H. H. Yoo. Species Differences in Stereoselective Pharmacokinetics of HSG4112, A New Anti-Obesity Agent.. Pharmaceutics, 2020. [DOI | PubMed]
- M. W. Aly, F.-A. Ludwig, W. Deuther-Conrad, P. Brust, A. H. Abadi, R.-P. Moldovan, N. A. Osman. Development of fluorinated and methoxylated benzothiazole derivatives as highly potent and selective cannabinoid CB2 receptor ligands.. Bioorg. Chem., 2021. [DOI | PubMed]
- S. S. Zoghbi, K. B. Anderson, K. J. Jenko, D. A. Luckenbaugh, R. B. Innis, V. W. Pike. On quantitative relationships between drug-like compound lipophilicity and plasma free fraction in monkey and human.. J. Pharm. Sci., 2012. [DOI | PubMed]
- W. Song, N. Zheng, M. Li, J. He, J. Li, K. Dong, K. Ullah, Y. Zheng. Rhodium(I)-Catalyzed Regioselective Azide-internal Alkynyl Trifluoromethyl Sulfide Cycloaddition and Azide-internal Thioalkyne Cycloaddition under Mild Conditions.. Adv. Synth. Catal., 2019. [DOI]
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow, P. Granger. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts(IUPAC Recommendations 2001).. Pure Appl. Chem., 2001. [DOI]
- MestReNova; Mestrelab Research S.L., 2019.
- CrysAlis Pro: Data Collection and Data Reduction Software Package; Rigaku Oxford Diffraction.
- SCALE3 ABSPACK: Empirical Absorption Correction using Spherical Harmonics.
- G. M. Sheldrick. SHELXT – integrated space-group and crystal-structure determination.. Acta Cryst. A, 2015. [DOI]
- G. M. Sheldrick. Crystal structure refinement with SHELXL.. Acta Cryst. C, 2015. [DOI]
- Diamond: Crystal Impact; 2014.
- R.-P. Moldovan, W. Deuther-Conrad, A. G. Horti, P. Brust. Synthesis and Preliminary Biological Evaluation of Indol-3-yl-oxoacetamides as Potent Cannabinoid Receptor Type 2 Ligands.. Molecules, 2017. [DOI | PubMed]
- T. Rühl, W. Deuther-Conrad, S. Fischer, R. Günther, L. Hennig, H. Krautscheid, P. Brust. Cannabinoid receptor type 2 (CB2)-selective N-aryl-oxadiazolyl-propionamides:Synthesis, radiolabelling, molecular modelling and biological evaluation.. Org. Med. Chem. Lett., 2012. [DOI | PubMed]