Aggregation, Transmission, and Toxicity of the Microtubule-Associated Protein Tau: A Complex Comprehension
Abstract
The microtubule-associated protein tau is an intrinsically disordered protein containing a few short and transient secondary structures. Tau physiologically associates with microtubules (MTs) for its stabilization and detaches from MTs to regulate its dynamics. Under pathological conditions, tau is abnormally modified, detaches from MTs, and forms protein aggregates in neuronal and glial cells. Tau protein aggregates can be found in a number of devastating neurodegenerative diseases known as “tauopathies”, such as Alzheimer’s disease (AD), frontotemporal dementia (FTD), corticobasal degeneration (CBD), etc. However, it is still unclear how the tau protein is compacted into ordered protein aggregates, and the toxicity of the aggregates is still debated. Fortunately, there has been considerable progress in the study of tau in recent years, particularly in the understanding of the intercellular transmission of pathological tau species, the structure of tau aggregates, and the conformational change events in the tau polymerization process. In this review, we summarize the concepts of tau protein aggregation and discuss the views on tau protein transmission and toxicity.
Article type: Review Article
Keywords: tau aggregates, tau toxicity, tau transmission, tauopathy
Affiliations: Key Laboratory of Systems Health Science of Zhejiang Province, School of Life Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China; hujiaxin21@mails.ucas.ac.cn (J.H.); 3018001612@tju.edu.cn (W.S.); shuangshuangyuan@163.com (S.Y.); Key Laboratory of Systems Biology, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Hangzhou 310024, China
License: © 2023 by the authors. CC BY 4.0 Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.3390/ijms241915023 | PubMed: 37834471 | PMC: PMC10573976
Relevance: Relevant: mentioned in keywords or abstract
Full text: PDF (2.4 MB)
1. Introduction
The microtubule-associated protein tau (MAPT; here, tau is used for short), is one of the microtubule-associated proteins (MAPs), and plays a crucial role in regulating the dynamics of microtubules (MTs) through binding and detaching from them. Tau plays a crucial role in axonal transport [ref. 1], synaptic transmission, and connectivity [ref. 2,ref. 3,ref. 4], as well as in maintaining genomic stability and regulating gene expression [ref. 5,ref. 6,ref. 7]. However, in various neurodegenerative diseases, including Alzheimer’s disease (AD), frontotemporal dementia (FTD) with parkinsonism linked to chromosome 17 (FTDP-17), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick’s disease (PiD), etc., tau is abnormally phosphorylated, leading to the formation of protein inclusions such as neurofibrillary tangles (NFTs), which are collectively referred to as “tauopathies” (Table 1) [ref. 8]. Tau is an intrinsically disordered protein lacking a defined structure, and poses challenges in understanding its mechanism of polymerization into oligomers and ordered aggregates, such as paired helical and straight filaments (PHFs and SFs), which ultimately lead to detrimental consequences [ref. 9].
Table 1: Relevance of microtubule-associated protein tau deposition in neurodegenerative disorders.
| Tau Isoforms | Tauopathies | Neuropathological Hallmarks | References |
|---|---|---|---|
| 3R Tau | Pick’s disease (PiD) | Ballooned neurons, gliosis, Pick bodies | [ref. 10,ref. 11] |
| 3R + 4R Tau | Primary age-related tauopathy (PART) | Neurofibrillary tangles (NFTs), absence of amyloid (Aβ) plaques | [ref. 12] |
| Alzheimer’s disease (AD) | NFTs, Aβ plaques, dystrophic neurites Plaques, pretangle neurons, tangle neurons | [ref. 11] | |
| Familial British dementia (FBD) | Cerebral amyloid angiopathy (CAA), parenchymal Aβ plaques, NFTs | [ref. 13] | |
| Familial Danish dementia (FDD) | CAA, NFTs, Aβ plaques, Danish amyloid | [ref. 13] | |
| Chronic traumatic encephalopathy (CTE) | NFTs, astrocytic tangles | [ref. 14] | |
| 4R Tau | Progressive supranuclear paralysis (PSP) | NFTs, tufted tau-positive astrocytes, coiled bodies | [ref. 15] |
| Globular glial tauopathy (GGT) | Globular oligodendrocytic inclusions, globular astrocytic inclusions | [ref. 16] | |
| Corticobasal degeneration (CBD) | Astrocytic plaques, preganglionic neurons, coiled bodies, argyrophilic threads | [ref. 17,ref. 18] | |
| Argyrophilic grain disease (AGD) | Argyrophilic grains, small spindle-shaped lesions, pretangle neurons, oligodendroglial coiled bodies | [ref. 19,ref. 20] | |
| Aging-related tau astrogliopathy (ARTAG) | Thorny or granular/fuzzy astrocytic tau | [ref. 21] |
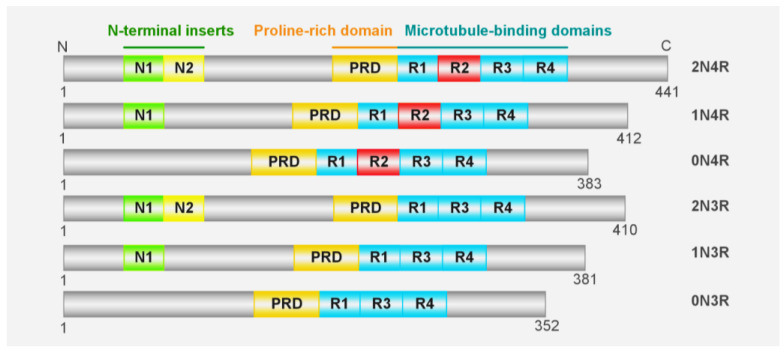
Tau is encoded by the MAPT gene on chromosome 17q21.31, which contains 16 exons that can generate six isoforms due to the alternative splicing of exons 2, 3, and 10 (Figure 1). The longest isoform of tau consists of 441 residues and can be divided into the N-terminus, C-terminus, a proline-rich region, a microtubule binding region (MTBR) encompassing R1-R4 segments, and two acidic N-terminal inserts that contain 29 amino acids each [ref. 22]. The concept that tau is involved in neurodegenerative diseases is supported by the evidence that tau aggregations and mutations correlate with several neurodegenerative diseases, including AD, FTDP17, PSP, etc. Furthermore, the presence of mutations on tau is adequate to elicit neurological pathology in both human and transgenic animal models [ref. 23,ref. 24]. It is interesting to note that, similar to the propagation of tau pathology in patients, the results from animal models also provide strong evidence to support the transmissibility of tau [ref. 25,ref. 26,ref. 27]. However, the underlying mechanism of tau aggregation and toxicity, as well as its intercellular transmission, remains to be elucidated.
2. Tau Aggregates
In vitro and in vivo studies have shown that the conformational change and subsequent exposure of aggregation-prone motifs serve as crucial intermediate events in the formation of oligomeric and fibrillar tau aggregates [ref. 28,ref. 29,ref. 30]. The monomeric form of tau is intrinsically unfolded, lacks a defined tertiary structure, and consists of a large proportion of random coli regions; the absence of ordered secondary and tertiary structures poses challenges in determining its structures [ref. 31]. It was proposed that the tau monomer is compacted into a “paper clip” conformation (Figure 2), in which the C-terminus folds back and interacts with the MTBR, while the N-terminus folds back over the C-terminus, forming a local compacted structure by encircling the core aggregation-prone motifs. The core aggregation-prone motifs encompass the segments “275VQIINK280” and “306VQRVYK311” on R2 and R3, respectively, which adopt a β-strand conformation [ref. 31,ref. 32,ref. 33,ref. 34], and can further facilitate polymerization through intermolecular interactions. However, intramolecular compaction prevents the intermolecular interaction of aggregation-prone motifs, thereby blocking tau aggregation [ref. 35]. Given the significant influence of intermolecular interactions on tau, factors such as heparin, PTMs, and RNA, which are capable of neutralizing and modulating the electrical properties of tau, can facilitate conformational changes and the subsequent exposure of aggregation-prone motifs.
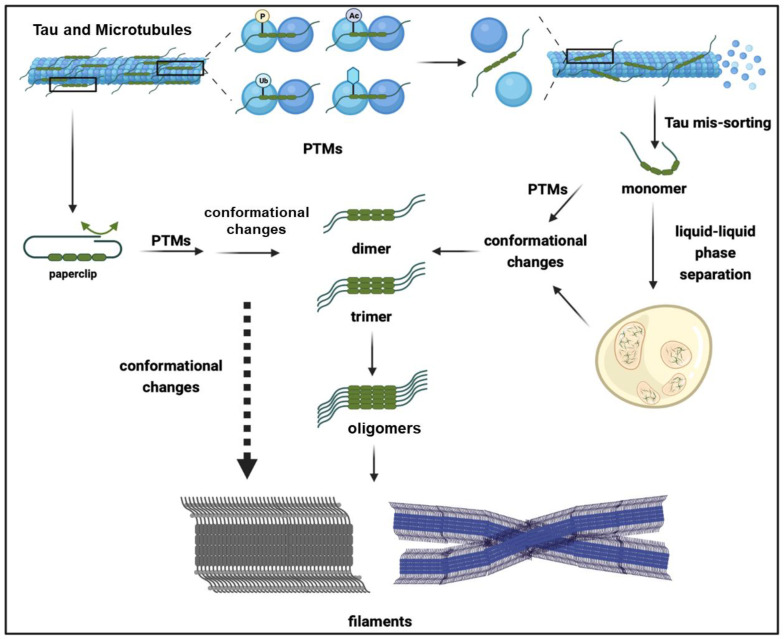
In addition to the “paper clip” model, an intermolecular interaction can also occur through alternative mechanisms. The electricity of the tau protein is distinct across its four segments; the N-terminus of the tau protein, particularly the N1 and N2 regions, exhibits a negative charge with a combined charge of −11.8 when coupled with the proline-rich region. In contrast, the MTBR carries a positive charge of +4.5. Thereby, the N-terminus of tau can interact with the MTBR and mask its core aggregation motifs. A further neutralization of the positive charge of tau via PTMs and heparin can also hinder the intramolecular interactions, leading to the exposure of the aggregation-prone motifs [ref. 36]. Interestingly, tau monomers are predominantly present in normal brains, whereas AD brains contain a substantial proportion of tau oligomers, encompassing dimers, trimers, and high-molecular-weight polymers [ref. 37]. Accordingly, tau oligomers can be classified into two types, sarcosyl-soluble oligomers that are undetectable via AFM, and AFM-detectable oligomers, which mainly consist of sarcosyl-insoluble granular oligomers containing more than 40 mers [ref. 38,ref. 39]. In contrast to tau filaments, the structural determination of tau oligomers poses a significant challenge. The results obtained from MT-associated tau dimers indicate that they are formed through an intermolecular interaction between the N-terminus and the proline-rich region in an antiparallel manner, facilitated by electrostatic complementation involving salt bridges. Subsequently, the N-terminus, C-terminus, and MTBR regions undergo an extension to facilitate the binding of tau towards MTs. The artificial condensation of dimeric tau has been shown to increase its binding affinity towards MTs [ref. 40], particularly with respect to tubulin heterodimers [ref. 41].
The tau dimers and trimers formed under physiological conditions exhibit distinct characteristics from pathological tau oligomers, particularly in terms of their association with different phosphorylation patterns and their status. These findings suggest that normal and pathological tau oligomers possess different properties and undergo distinct biological processing [ref. 42]. Prior to the formation of oligomers, monomeric tau undergoes a series of conformational changes, particularly adopting the β-strand structure. The disulfide-cross-linked intermolecular interaction further facilitates the formation of dimers, and is accompanied by the disulfide-bond-independent intermolecular bridging of the MTBR. These dimers can further polymerize into oligomers with a distinct fate compared to the dimers that are formed physiologically [ref. 36,ref. 43].
Dimerization is likely to serve as a rate-limiting step in the aggregation of the tau protein. The tau dimer acts as the core, and it can be further expanded by the addition of monomers [ref. 44], which undergo polymerization to form multimers, and have been proven in vitro [ref. 45,ref. 46]. Moreover, tau trimers can also serve as the aggregation seed, implying that both dimers and trimers possess the potential to function as fundamental units for aggregation [ref. 37]. In particular, they are abundant in AD brains [ref. 47].
Accordingly, the formation of tau granules precedes the formation of tau filaments, which can be detected in the early stages of AD, even in the absence of AD-related symptoms. In particular, tau granules appear at Braak stages 0 and I, which implies that their genesis is an early event and may relate to the progression of AD [ref. 39]. In contrast, tau filaments can be detected at stage V instead of stages 0, I, and III, which does not exhibit a strong correlation with the early cognitive changes [ref. 48].
Unlike unstructured monomers, tau dimers, trimers, and oligomers tend to gradually adopt the β-sheet conformation. Accordingly, the content of the β-sheet conformation is increased stepwise from soluble oligomers to granular oligomers and filaments, indicating that the structure change is accompanied by tau aggregation and may facilitate the formation of higher-molecular-weight aggregates [ref. 38]. Interestingly, granular oligomers contain more β-sheet structures than randomly coli-enriched monomers, and the proportion of β-sheet structures is continuously increased in the filaments [ref. 48]. The gradual increase in β-sheet conformation is likely critical for tau polymerization [ref. 38].
Although the toxicity of tau filaments is still uncertain and debated, their structures have been investigated by several Cryo-EM studies, since they tend to form defined structures in vitro and in vivo. Unexpectedly, the structures of tau filaments are diverse in different types of tauopathy diseases [ref. 49].
On the basis of the number of protofilaments, the CBD filaments can be divided into types 1 and 2, which correspond to narrow and wide tau filaments, respectively. The ratio of type 1 to type 2 filaments in CBD patients varies from 1:1 to 1:3 [ref. 50]. The narrow tau filament consists of a single protofilament exhibiting a four-layer fold, while the wide filament comprises pairs of identical narrow filaments [ref. 50]. The CBD protofilaments’ core sequence comprises residues K274-E380 and features 11 β-strands linked by turns and arcs, forming a four-layer structure. The antiparallel stacking of the 343KLDFKDR349 motifs from two protofilaments further shapes the type 2 filaments [ref. 50]. In contrast to CBD filaments, the PHFs and SFs observed in AD patients comprise two identical protofilaments encompassing residues 306–378, with the disordered N-terminus and C-terminus forming a fuzzy coat. The filament’s core comprises eight β-sheet structures, including β1–3 of R3, β4–7 of R4, and β8 at the C-terminus. It adopts a C-shaped architecture without R1 and R2, where three β-sheets are arranged in a triangular manner. Additionally, it exhibits two regions with a cross-β architecture, wherein pairs of antiparallel β-sheets are packed together. The cross-β interface is formed by the interaction between β1–2 and β8, which is facilitated by 306VQIVYK311 and residues 373–378 from the opposing β8 sheet. The antiparallel stacking of residues 332PGGGQ336 between two helically symmetric protofilaments further contributes to the formation of PHF aggregates. In contrast, the SF stacking involves the asymmetric packing of 321KCGS324 from protofilament 1 and 313VDLSK317 from protofilament 2 at their interfaces along the helical axis [ref. 49].
In patients with PSP, tau polymerizes to form subcortical neurofibrillary tangles and neuropil threads, along with tufted astrocytes and oligodendroglial coiled bodies. Intriguingly, a recent study has revealed that tau exhibits the novel three-layered folded filaments in PSP, which is herein referred to as the PSP fold [ref. 51]. PSP filaments are composed of a single protofilament featuring an ordered core spanning residues 272–381 and adopting the three-layer PSP fold. This fold involves the stacking of R2–R4 and a turn at the “PGGG” motifs located at the end of each repeat. The central layer is formed by R3, with R2 and R4 packing on either side of it. Additionally, the chain undergoes another hairpin turn at the “PGGG” motif within R4, while the C-terminus forms a short fourth layer that covers the end of R2 [ref. 51].
Pick’s disease is classified as a 3R tauopathy. It results in the degeneration of the frontotemporal lobar, which is accompanied by the accumulation of abundant Pick bodies composed of narrow and wide 3R tau filaments. The narrow Pick’s filament consists of a single elongated protofilament, which exhibits structure differences from the PHFs observed in AD. Furthermore, the association of two narrow Pick’s protofilaments results in the formation of wide filaments. The Pick’s filament fold comprises a core consisting of residues Lys254-Phe378, which adopts nine β-strands (β1–β9) organized into four cross-β packing stacks that are connected by turns and arcs. Specifically, β1 and 2 of R1, β3–β5 of R3, and β6–β8 of R4 are tightly packed together to form a hairpin-like structure [ref. 52]. As revealed by the cryo-EM analysis, while R3, R4, and an additional 10–13 residues from the C-terminus of tau contribute to the assembly of the common folded core in tau filaments, distinct types of tau folds are observed in different tauopathy diseases, which can be further classified into three classes and eight subclasses (Table 2, see review [ref. 51]). Disease-associated folding specificity helps to elucidate the underlying mechanisms behind the diverse protein inclusions, subcellular distributions, and cell-type-specific aggregations of tau in various diseases. In particular, it helps to explain why the tau protein can deposit diversely in neuronal cell bodies, synapses, and dendritic compartments, leading to distinct pathological manifestations [ref. 53]. Interestingly, a recently discovered isoform of tau, known as w-tau, exhibits the intriguing ability to mitigate tau aggregation in AD brains [ref. 54]. Moreover, it has been demonstrated that w-tau possesses the capacity to attenuate the aggregation propensity of tau [ref. 55]. Interestingly, w-tau is generated by intron retention, which results in the absence of exon 13 and the addition of the translated intron 12 sequence after exon 12 [ref. 54].
Table 2: Monomeric, oligomeric, and filamentary forms of tau protein.
| Tau Species | Molecular Weight (kDa) | Features | Isoforms or Compositions | Folding Types | References |
|---|---|---|---|---|---|
| Monomer | 55–74 | soluble | tau0N3R, tau1N3R, tau2N3R, tau0N4R, tau1N4R, and tau2N4R | [ref. 56] | |
| Dimer | 180 | cysteine-dependent and reducible | 2 Tau monomers | [ref. 45] | |
| 130 | cysteine-independent and unreducible | [ref. 57] | |||
| Small oligomers | 300–500 | soluble | 6–8 Tau monomers | [ref. 43] | |
| Granular oligomers | 1800 | insoluble, granule | 40 Tau monomers | ||
| Filaments | two-layered | 3R | Narrow Pick filaments | [ref. 51] | |
| Wide Pick filaments | |||||
| three-layered | 4R | PSP fold | |||
| GPT fold | |||||
| GGT type 1, type 2, and type 3 folds | |||||
| four-layered | AGD type 1, type 2 folds | ||||
| CBD fold | |||||
| two-layered | 3R + 4R | AD fold (including FBD and FDD folds) | |||
| CTE fold |
In addition to a structural investigation, despite its inherent challenges, tau aggregation can be investigated by using several methodologies and models (Supplementary Table S1).
3. Factors Facilitating Tau Aggregation
The properties and electrostatics of tau can be influenced by various factors, including post-translational modifications (PTMs) of proteins, polyanions such as RNAs, heavy metals, heat shock proteins, membrane binding, and local condensation of tau (LLPS). PTMs such as phosphorylation, acetylation, truncation, nitration, glycation, glycosylation, and ubiquitination serve as crucial regulators of tau aggregation and toxicity. The primary impact of PTMs is to modify the local charge distribution of tau proteins. For instance, the attachment of a single phosphate group (Pi) to tau can result in an increase in the net charge by −1 on the tau protein; therefore, the enrichment of phosphor sites and phosphor groups can significantly diminish the positive charge of tau [ref. 58,ref. 59,ref. 60].
4. Post-Translational Modifications
The binding of phosphor groups to the MTBR can neutralize its positive charge, thereby inducing its dissociation from the MTs [ref. 61]. For instance, the binding affinity of MTs is reduced by the phosphorylation of the p-epitopes within the MTBR, such as S262/S214, and the epitopes located in the proline-rich domain, like T231 [ref. 62].
Once dissociated from MTs, phosphorylated tau can further undergo pre-aggregation events. Since the charge change, the interaction between the negatively charged C-terminus and MTBR is impeded, leading to the release of the “paper clip” structure and resulting in an enhanced hydrophilic property. The subsequent intermolecular interaction of two tau proteins with the exposed aggregation-prone motifs further facilitates the formation of tau dimers and multimers [ref. 63]. Notably, some in vitro evidence suggests that the phosphorylation of the C-terminal epitopes, including Ser396, Ser404, and Ser422, does not alter the binding affinity of MTs; however, the propensity for conformational changes and aggregation is enhanced [ref. 60,ref. 64,ref. 65]. Interestingly, phosphorylation at Ser396 appears to attenuate the binding affinity of MTs in cell culture models [ref. 66,ref. 67], suggesting potential variations in tau properties across different environments. In addition to reducing the binding affinity of MTs, phosphorylation at AD-related serine (S) and threonine (T) epitopes can increase the propensity for aggregation (Figure 3). The in vitro phosphorylation of tau at AT8, AT100, AT180, and PHF-1 epitopes leads to the self-aggregation of tau, resulting in the spontaneous formation of small amorphous aggregates without the need for aggregation inducers [ref. 68]. Moreover, in vitro phosphorylation at S208 through a phosphoryl-mimetic mutation, S202E/T205E/S208E, induces Tau self-aggregation independently of an aggregation seed [ref. 69], further suggesting that phosphorylation can modulate Tau properties and drive its aggregation. The linking of polyphosphate chains also induces a conformation change in Tau, facilitating the formation of intermolecular cross-linking and aggregation [ref. 70].
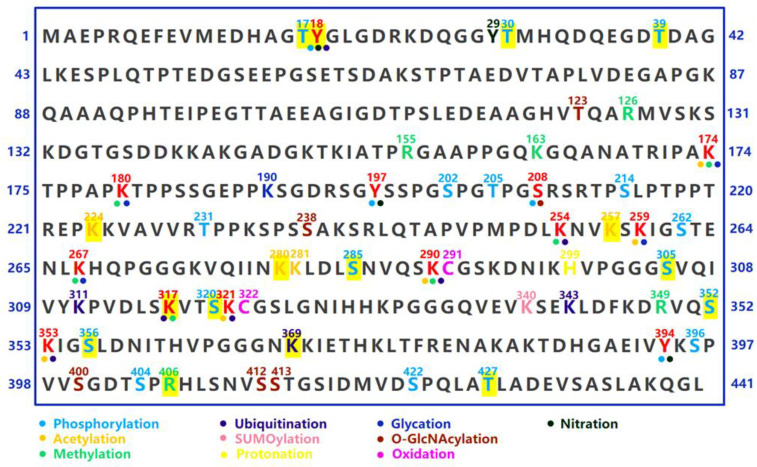
Lysine (K) acetylation, achieved by introducing an acyl group to the -NH side chain, possesses the capacity to neutralize a positive charge and increase the negative charge near the lysine residue, which can also change the properties of tau [ref. 71,ref. 72]. In particular, acetylation at the K280 residue of the MTBR reduces the binding affinity of MTs (Figure 3), increases the proportion of the β-sheet structure, and enhances the self-assembly capability of tau, which can be attributed to the interaction between the hydrophobic chemical groups within the PHF6* motifs [ref. 72,ref. 73].
Similarly, acetylation at K281 also hampers the capacity for MT binding and enhances the propensity for aggregation [ref. 74]. Acetylation at K259, K290, K321, and K353 outside the aggregation-prone motif (PHF6* and PHF6), exhibits a similar impact on reducing the binding affinity of MTs. Surprisingly, mimicking acetylation at K321 and K353 exerts an opposing effect on tau aggregation by influencing the stability of its β-sheet structure [ref. 75]. These findings suggest that PTMs with different effects on tau conformation may differentially modulate tau aggregation [ref. 64].
In addition to acetylation, lysine residues on tau can undergo modifications such as ubiquitination, SUMOylation, methylation, and glycation et al., suggesting the potential for competition and crosstalk among different PTMs (Figure 3) [ref. 60,ref. 76].
The ubiquitination of tau exerts diverse effects. It can facilitate the degradation of tau through the ubiquitin–proteasome and autolysosome systems, while also promoting the aggregation and insoluble inclusions of the tau protein [ref. 77,ref. 78]. The ubiquitination of K18 at the N-terminus of tau disrupts aggregate formation and sequesters tau oligomers into the proteasome for degradation [ref. 79]. The ubiquitination of K63 promotes the autolysosome-mediated degradation of tau. Moreover, the ubiquitination of K48 and K63 also promotes tau aggregation due to the interaction of the enriched ubiquitin chains [ref. 79,ref. 80]. In fact, ubiquitin moieties have been detected on human-brain-derived tau aggregates, including ubiquitin chain-linked K321, K343, K353, and K369 in CBD, as well as K317/K321 in AD [ref. 81].
In particular, CHIP is a predominant ubiquitin ligase for tau. The deletion of CHIP results in an accelerated accumulation of non-aggregated and ubiquitin-negative forms of tau [ref. 82]. The up-regulation of CHIP expression in Cos7 cells results in the formation of ubiquitin-positive tau aggregates, which can be attenuated by Hsp70 [ref. 83]. The AD brain-derived tau conjugated to K63 ubiquitin chains is likely to exhibit a propensity for aggregation and propagation [ref. 84]. The above evidence suggests that ubiquitin conjugation not only influences the degradation of tau, but also modulates its aggregation propensity, since ubiquitin chains can provide additional interactions among tau molecules [ref. 81].
Although the methylation of Tau has been detected in both normal and AD human brains [ref. 85,ref. 86], the functional implications of this epigenetic modification remain to be elucidated. The methylation of both mono- and di-methylated lysine residues occurs along the entire tau protein. However, it does not impact MT polymerization unless the lysine residues of tau are methylated to a high stoichiometry. Surprisingly, even when the methylation levels are elevated, tau still maintains a disordered conformation. Additionally, overall, methylation exerts a primary influence on tau by suppressing its aggregation propensity through impairing the nucleation rate, thereby slowing down the extension of aggregates, and ultimately reducing the stability of tau filaments [ref. 86].
Intriguingly, the methylation of certain residues, such as K317, elicits an opposing effect. K317 methylation decreases tau solubility and facilitates intermolecular interactions and dimerization, which is concomitant with MT destruction [ref. 87]. In addition to lysine residues, methylation can also occur on certain arginine residues (R), including R126, R155, and R349 [ref. 88,ref. 89]. However, the functional implications of arginine methylation remain to be elucidated. Similar to acetylation, the methylation of lysine residues can induce charge neutralization and conformational changes [ref. 58], thereby influencing both intra- and intermolecular interactions [ref. 87]. The additional consequence of lysine methylation is the perturbation of other PTMs. For instance, methylation at K254 blocks Tau ubiquitination and degradation [ref. 89], while methylation at K267 influences the phosphorylation of Ser262, albeit the underlying mechanism remains unknown [ref. 90].
5. Heavy Metal Elements
The disruption of heavy metal homeostasis is implicated in the pathogenesis of several tau-related neurodegenerative diseases, such as AD, which accompanies the accumulation of copper, iron, and zinc. Furthermore, the exposure to certain heavy metals also augments the incidence of AD and other tauopathies [ref. 91]. In particular, heavy metals are postulated to act as cofactors to induce tau aggregation through direct binding [ref. 92,ref. 93,ref. 94]. They possess the capability to induce conformational changes and promote phase separation of tau, thereby enhancing its propensity for aggregation [ref. 95,ref. 96]. However, it is unlikely that they share identical binding sites and kinetics.
The imbalance and accumulation of copper ions have been demonstrated in AD brains [ref. 97]. In vitro studies suggest that copper interacts with the R2 and R3 regions of tau, and that the histidine residues within tau play a vital role in the binding of copper ions. Consequently, copper induces a conformational shift in the aggregation-prone motifs, leading to an increased proportion of alpha-helix and β-sheet structures [ref. 96,ref. 98,ref. 99], facilitating tau self-assembly [ref. 98]. In addition to direct binding, copper can also catalyze the formation of intermolecular disulfide bonds, thereby promoting tau dimerization [ref. 96].
The zinc ion is abundantly accumulated in the AD brain and possesses the capacity to induce the formation of the β-sheet conformation, thus facilitating the formation of tau aggregates. Under reducing conditions, the interaction between tau and zinc is mediated by Cys291 and Cys322, with the additional involvement of certain histidine residues. Interestingly, distinct effects on tau aggregation are observed at low and high levels of zinc. In particular, low zinc levels promote the fibrillization of Tau by promoting the formation of a β-sheet structure, whereas high zinc levels induce the formation of granular aggregation. This phenomenon can be attributed to a binding site switch that occurs with increased zinc concentrations [ref. 95,ref. 100,ref. 101].
In addition, the presence of iron (Fe2+) facilitates the aggregation of tau by inducing a reversible conformational change through an interaction with the threonine residues [ref. 102]. Interestingly, lithium, an alkali metal, exhibits the potential to mitigate brain iron accumulation induced by tau and consequently may reduce iron-induced tau toxicity [ref. 103].
Besides the direct interaction effects, heavy metal elements can modulate the conformation and aggregation of tau by influencing the flux of PTMs; for instance, zinc and copper can impact the activities of tau kinase and phosphatase, thereby augmenting tau phosphorylation and inclusion [ref. 93]. In addition to iron accumulation correction, lithium has the ability to attenuate tau phosphorylation by inhibiting GSK3 activity and modulating the AKT and PKA levels [ref. 104,ref. 105,ref. 106,ref. 107]. Interestingly, it also directly reduces tau expression; however, the underlying mechanism remains to be elucidated [ref. 103].
6. Phase Separation
In the protein crowding niche, tau can be concentrated by LLPS, which has been proposed to facilitate conformational change [ref. 68,ref. 108]. Interestingly, even at a physiological concentration of as low as 1–3 µM, tau exhibits the ability to undergo LLPS. In particular, phosphorylation is crucial for tau LLPS, and unphosphorylated tau is unable to undergo LLPS, even in a crowding environment [ref. 108]. Since the phosphorylation of tau can increase its negative charge, it induces conformational changes and enhances intermolecular interactions, thereby facilitating tau LLPS. Overall, the phosphorylation of tau and site-specific phosphorylation, such as phosphorylation at the AT180 and AT8 epitopes, both enhance the propensity for LLPS [ref. 109]. In particular, at high protein concentrations exceeding 50 µM, the phosphorylated tau can undergo LLPS spontaneously, even in the absence of crowding agents. In contrast to phosphorylation, the implications of acetylation on Tau LLPS remain a subject of ongoing debate and necessitate meticulous clarification [ref. 110].
In addition, tau LLPS can be further facilitated by heparin, RNA, and genetic mutations of tau. In particular, even in the absence of crowding circumstances, tau can undergo spontaneous LLPS when carrying pathological mutations such as P301L and delaK280, which have been implicated in inducing β-sheet conformational changes in tau [ref. 108,ref. 111].
Droplets of Tau LLPS exhibit a dynamic exchange with the surrounding environment and undergo rapid gel-like condensation, which can further progress to β-sheet-enriched protein aggregates. These findings suggest that LLPS enhances aggregation propensity [ref. 108,ref. 111]. From this perspective, it is plausible that after mis-sorting from axonal MTs, the subsequent PTMs, such as phosphorylation, are able to promote the LLPS of tau, leading to its condensation (Figure 2). It has been demonstrated that LLPS can enrich the tau concentration by up to ~100-fold [ref. 111], thereby facilitating conformational changes including the extension of the N- and C-terminus, exposing the MTBR, and promoting intermolecular contact for the cluster formation of tau molecules, ultimately leading to tau aggregation and fibrillization [ref. 111,ref. 112]. Surprisingly, although the MTBR is prone to form aggregation, LLPS is predominantly reliant on the N-terminus of tau. Tau clustering in LLPS likely occurs through the interface of its N-terminus, and the removal of the N-terminus impedes LLPS, indicating that the mechanisms of tau LLPS differ from those driving tau polymerization, although LLPS can promote the aggregation of tau [ref. 111].
7. RNAs and RNA-Binding Proteins
Although the precise roles of RNA and RNA-binding proteins in tau aggregation remain elusive, their association with pathological tau aggregates has been demonstrated in vivo [ref. 113,ref. 114]. Tau interacts with RNA molecules through electrostatic interactions. The negatively charged phosphate backbones of RNAs bind to the positively charged segments of tau, facilitating tau condensation, particularly the LLPS [ref. 115], increasing its propensity for aggregation [ref. 114,ref. 116,ref. 117].
Structural studies of tau fibers induced by RNA molecules-reveal the incorporation of two molecular weight RNAs. The intermolecular interaction between two parallel protofilaments, mediated by the Glu391-Ala426 segments, further gives rise to an aggregation core comprising five β-strand structures [ref. 118]. The presence of RNA molecules not only serves as a structural factor that stabilizes tau aggregates, but also acts as a catalyst for tau aggregation by catalyzing the formation of tau filaments. Surprisingly, the removal of RNA molecules can lead to the unexpected dissociation of tau aggregates, indicating their crucial role as molecular glue [ref. 118,ref. 119]. In vitro evidence also suggests that tau has the ability to sequester RNA molecules and subsequently undergo co-polymerization, leading to the formation of amyloid-like fibrils [ref. 120].
However, the properties of tau aggregates induced by RNAs exhibit distinct dissimilarities compared to those induced by heparin. In addition to poly (U), (A), (C), and (G), tRNA, m6A-RNA, snRNAs, and snoRNAs also exhibit the capability of interacting with tau, leading to the LLPS and subsequent polymerization with distinct stoichiometry [ref. 119,ref. 120,ref. 121]. Tau LLPS is also implicated in the indirect interaction between tau and RNAs mediated by RNA-binding proteins such as TIA and HNRNPA2B1 [ref. 122,ref. 123].
8. Interplay between Tau Protein and Membrane Architecture (Membrane Binding)
In the AD brain, tau has been demonstrated to exhibit an association with the cellular membrane [ref. 124]. Furthermore, PHF filaments co-deposit with membrane lipids, including phosphatidylcholine (PC), cholesterol, and sphingolipid [ref. 125], thereby suggesting a link between tau pathology and its interaction with membrane lipids. Furthermore, in vivo and in vitro studies have demonstrated that tau exhibits a propensity to directly associate with membrane lipid species, which is driven by electrostatic interactions. In particular, the hydrophobic hexapeptide motifs PHF6*/PHF6 possess the ability to interact with lipids, leading to the formation of the core structure within the tau-lipid complex, which can further enhance the formation of the β-sheet structure, thereby augmenting the aggregation propensity of tau [ref. 126,ref. 127]. The binding affinity of tau and membrane lipids is predominantly determined by the intensity of the electrostatic interactions and the composition of the phospholipid headgroups [ref. 128]. The specific lipid species capable of binding to tau have been reviewed by Bok et al. [ref. 126]. Interestingly, tau has the capacity to remodel and extract phospholipids from cell membranes, leading to the formation of the tau-lipid complex. The R2 and R3 regions of tau, particularly the PHF6 motif, exhibit a robust capacity for remodeling membrane lipids.
The formation of the tau-lipid complex facilitates a favorable shift in electrical properties [ref. 127], and the presence of a hydrophobic environment may potentially facilitate the enrichment of the tau protein, thereby promoting the intermolecular interfaces. However, it remains to be determined whether lipid binding can facilitate tau LLPS, and the role of membrane lipids in enhancing and stabilizing tau aggregates remains unclear. The binding of tau to lipids also facilitates tau propagation and transmission [ref. 126], since membrane binding enhances tau internalization and exocytosis across the cell membrane. Besides the lipids, membrane-associated proteoglycans and proteins also possess the capacity to interact with tau and contribute to tau pathology. Heparan sulphate proteoglycans (HSPGs), which are ubiquitously distributed on the extracellular matrix and plasma membrane, have the capacity to interact with tau and promote its spreading [ref. 129]. The muscarinic cholinergic receptor can interact with tau, leading to the disruption of calcium homeostasis [ref. 130]. However, the interaction between tau and lipids is predominantly limited to in vitro experiments; therefore, further investigations are warranted to elucidate the binding of tau to the membrane lipids in vivo, as well as the impact of phosphorylation and its relevance to tau aggregation.
9. Propagation and Transmission of Tau
Similar to prions, pathological tau can serve as a nucleating seed for aggregation, inducing the conformational transformation of intrinsically disordered monomers of tau. This process leads to the formation of a conformation that is prone to aggregation and further polymerization, ultimately resulting in the expansion of tau toxicity, which is demonstrated as “tau transmission and propagation” [ref. 131,ref. 132,ref. 133,ref. 134] (Figure 4).
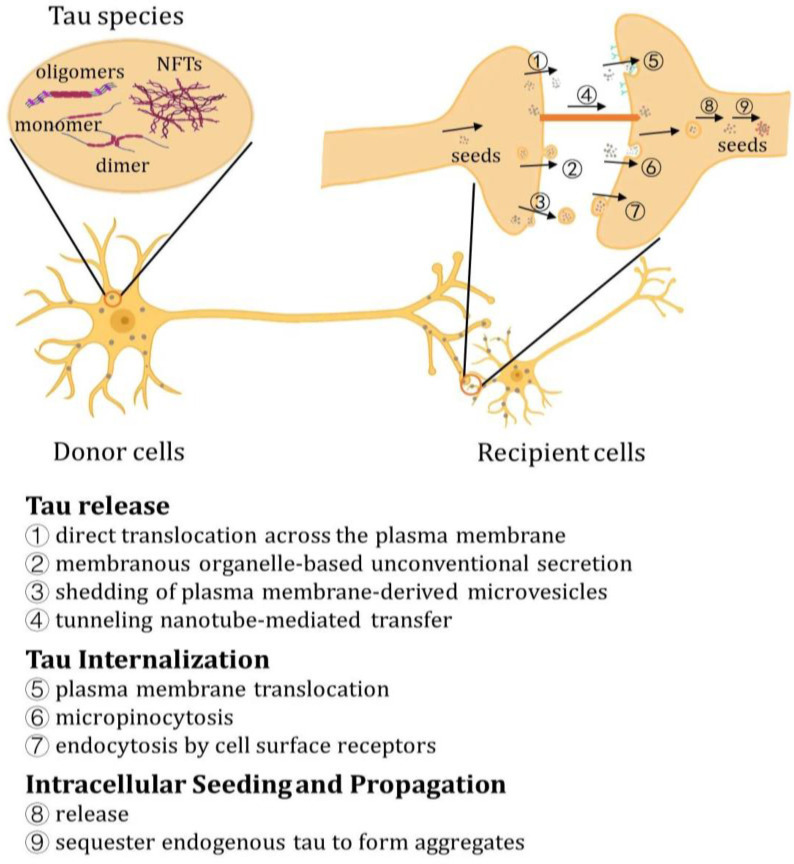
The transmission process involves the formation of pathological tau species, which serve as propagating seeds and can be secreted from donor cells into the extracellular space. Subsequently, they are internalized by recipient cells, where they act as seeds to facilitate the sorting and conversion of normal monomeric tau into aggregation-prone species [ref. 135,ref. 136]. The propagation and transmission of Tau have been unveiled and can be investigated through various models and methodologies (Supplementary Table S2).
10. Release of Tau
In the absence of a canonical secretion signal peptide, tau is likely secreted by the unconventional protein secretory pathway (UPS) rather than the conventional ER-Golgi secretory pathway. According to current knowledge, tau can be secreted through three mechanisms: (1) direct translocation across the plasma membrane; (2) membranous organelle-based unconventional secretion (MOBS); and (3) shedding of plasma membrane-derived microvesicles [ref. 135].
It has been demonstrated that the direct translocation of tau across the plasma membrane is facilitated by its binding to certain lipid species located on the inner leaflet of the plasma membrane, such as PI (4,5)P2. Furthermore, tau is released from the membrane, which occurs through an interaction with HSPGs that are present on the outer leaflet of the plasma membrane [ref. 135], which is similar to the release process of FGF2, and can be enhanced by phosphorylated tau [ref. 137]. In fact, elevated levels of HSPGs such as syndecan 4 and serglycin have been implicated in the presence of amyloid and tau pathology [ref. 138]. The inhibition of HSPGs and their interaction with tau has demonstrated a reduction in in vivo tau propagation [ref. 129], thus highlighting the critical role played by HSPGs in facilitating tau release. It is still not clear whether the pathological species of tau can produce a transmembrane pore similar to FGF2, and additional investigation is required [ref. 139].
Moreover, intracellular tau can be packaged within membranous organelles, such as exosomes, which then fuse with the plasma membrane, thus facilitating the release of organelle-encapsulated tau into the extracellular space. The exosome-mediated secretion of tau is initiated by the engulfment of tau into intraluminal vesicles derived from the inward budding of the late endosome. These vesicles contain hyperphosphorylated species of both monomeric and aggregated forms of tau [ref. 135,ref. 140,ref. 141]. In particular, hyperphosphorylated tau has been detected in the exosome fraction of the cerebrospinal fluid in Alzheimer’s, suggesting that tau is potentially secreted by exosomes [ref. 141], and it has been verified in cell cultures and mouse models [ref. 140,ref. 142].
Although exosome-mediated transmission only contributes to a small fraction of tau propagation, patient-derived exosomes have demonstrated the ability to propagate tau pathology in cell culture and mouse models. Moreover, inhibiting exosome formation can effectively impede tau propagation, providing evidence to suggest that exosome-mediated tau secretion is involved in this process [ref. 135,ref. 143]. In particular, exosome-mediated tau secretion also contributes to the trans-synaptic transmission of tau, a process that is dependent on synaptic connectivity and neuronal transmission activity, although the precise mechanism is not fully understood [ref. 140].
It has been suggested that the disruption of the fusion event between the autophagosome and the lysosome can potentiate autophagosome-mediated cargo release [ref. 135]. Although the underlying mechanism remains elusive, impaired autophagy and the accumulation of autophagic vacuoles have been broadly demonstrated in AD [ref. 144]. Once engulfed by autophagosomes, tau can either be secreted by the fusion of autophagosomes with the plasma membrane [ref. 145], or degraded by the fusion of autophagosomes with lysosomes [ref. 135]. Tau is able to block autophagosome–lysosome fusion by downregulating the expression of the IST1 factor associated with ESCRT-III (IST1). Given that the interaction between IST1 and charged multivesicular body protein 2B\4B (CHMP2B\CHMP4B) plays a crucial role in the assembly of the endosomal sorting complex required for transport complex III (ESCRT-III complex), tau can hinder the formation of the ESCRT-III complex, thereby inhibiting the activity of autophagosome–lysosome-mediated protein degradation. Moreover, tau can disrupt lysosome-mediated protein degradation by enlisting and sequestering FKBP4, resulting in diminished lysosome clustering [ref. 146,ref. 147]. Consequently, tau may enhance autophagosome-mediated secretion by inhibiting the functionality of the autophagosome–lysosome pathway [ref. 148]. In contrast, augmenting autophagic flux by inhibiting p300/CBP activity can conversely attenuate the secretion and propagation of tau [ref. 145,ref. 149].
Unlike exosomes, ectosomes are larger extracellular vesicles that are shed directly from the plasma membrane through budding [ref. 150]. Tau-containing ectosomes have been characterized in the cerebrospinal fluids in Alzheimer’s [ref. 151]; in particular, tau can indeed be secreted by ectosomes in cell culture and rat models [ref. 152]. However, the precise mechanism underlying ectosome-mediated tau secretion remains to be elucidated, particularly regarding its potential involvement in tau propagation in vivo.
The exocytosis of tau can also be facilitated by the secretion of late endosomes/lysosomes [ref. 153]. In particular, the up-regulation of the late endosome/lysosome-associated small GTPase Rab7 has been observed in post-mortem AD brains, suggesting a potential increase in Rab7-mediated late endosome/lysosome secretion that may contribute to disease progression, which has been verified in cell culture models. The reduction in Rab7 expression leads to a decrease in tau secretion, while the up-regulation of Rab7 expression exerts the opposite effect [ref. 154,ref. 155,ref. 156]. Furthermore, the secretion of tau through late endosome/lysosome-mediated pathways is also modulated by Hsc70 and its partner DnaJC5, which has been termed as misfolding-associated protein secretion (MAPS) [ref. 153,ref. 157,ref. 158]. The MAPS pathway is initiated by the recognition of tau by the ER-bound chaperone USP19, followed by the processing of tau by Hsc70 and DNAJ5C. This leads to the translocation of tau into the lumen of ER-associated late endosomes, which are then secreted into the extracellular space via the SNAP23- and syntaxin-4-mediated plasma membrane fusion of tau-containing late endosomes/lysosomes [ref. 153,ref. 159], leading to the free distribution of tau in the extracellular milieu, thereby facilitating its propagation [ref. 157]. The MAPS pathway is likely to function in parallel with the proteasome-mediated protein degradation system, serving as an additional mechanism for maintaining protein quality control (PQC). The initiation of the proteasome pathway is also facilitated by the USP19-mediated sorting of protein clients, and its functional disruption indeed has the potential to augment MAPS [ref. 160]. It is worth noting that the accumulation of tau in AD may impede proteasome function, potentially leading to the pathological promotion of MAPS-mediated tau secretion as a compensatory mechanism for impaired PQC, thereby reducing the intracellular levels of misfolded tau [ref. 161,ref. 162,ref. 163].
Nanotubes represent cellular protrusions that facilitate the direct intercellular delivery of cargos such as prion proteins, viruses, and organelles. F-actin-rich tunneling nanotubes are capable of facilitating the intercellular transportation of tau, thereby facilitating tau transmission [ref. 134,ref. 164]. Interestingly, nanotubes possess the capacity to facilitate the transportation of tau fibrils between neuronal cells with the aid of Actin [ref. 165,ref. 166]. In particular, the formation of nanotubes can be promoted via extracellular tau, which is likely involved in the formation of nanotubes [ref. 165]. Cellular stress, such as the accumulation of misfolded proteins like alpha-synuclein and tau, along with lysosomal and proteasomal dysfunction, may drive the formation of nanotubes to alleviate protein entrapment and aggregation-related stress [ref. 136,ref. 167]. Furthermore, the involvement of nanotubes in the propagation of tau in animal models and patients remains unclear.
11. Tau Internalization
The internalization of extracellular tau and tau-containing organelles is predominantly reliant on endocytosis. The internalization of tau fibrils is mediated by micropinocytosis through the invagination of the plasma membrane, which is a process modulated by Actin. In particular, the binding and internalization of tau fibrils are modulated by HSPGs on the cell surface, and inhibiting the binding of tau and HSPGs can prevent the uptake and propagation of tau [ref. 129]. In contrast to fibrils, the internalization of low-molecular-weight species such as tau oligomers is mediated via bulk endocytosis, which is akin to the fluid-phase endocytosis of dextran, and is mediated by dynamin. Relevantly, bulk endocytosis is thereby able to enrich tau within endosomes [ref. 168,ref. 169]. In particular, monomeric tau species are also able to be slowly internalized by actin-dependent micropinocytosis and rapidly internalized via dynamin-dependent endocytosis [ref. 135]. In addition, the endocytosis of free tau species, including monomeric tau and tau oligomers, is also modulated by cell surface receptors such as LRP1. The inhibition of LRP1 significantly impedes the propagation of tau in mice, suggesting that interfering with its internalization can potentially reduce tau pathology. However, it should be noted that the inhibition of LRP1 only partially reverses the uptake of sonicated Tau fibrils [ref. 129,ref. 170].
The uptake of tau exosomes is modulated via filopodia-mediated internalization, involving filopodial surfing, grabbing, and pulling motions. This dynamic filopodia extends to endocytic hot spots to facilitate the delivery of exosomes into the endosomes [ref. 171].
12. Intracellular Seeding and Propagation of Tau
Once internalized, tau can be released from endosomes or macropinosomes by permeabilizing through vesicular membranes, although the precise mechanism underlying this process remains to be elucidated. Internalized tau aggregates can further induce damage to the membrane of endosomes [ref. 172]. Importantly, tau has the ability to interact with vesicular membranes by sequestering phospholipids, thereby leading to membrane rupture [ref. 127]. In fact, other protein aggregates like alpha-synuclein can also cause a similar rupture of endosome membranes [ref. 173].
Following its release, tau can induce the formation and aggregation of endogenous tau by adopting a conformation that is prone to aggregation, resembling the behavior exhibited by intracellular tau seeds [ref. 36,ref. 120,ref. 132,ref. 174,ref. 175]. Remarkably, monomeric tau also has the ability to sequester endogenous tau and form the nucleation core, thus facilitating tau polymerization in a prion-like manner [ref. 36,ref. 176]. The internalized monomeric tau can also activate calpain, thereby facilitating the cleavage of endogenous tau [ref. 177]. In accordance with the propagation of tau, tau pathology exhibits gradual progression and spread patterns, which initially manifest in the transentorhinal cortex, and subsequently spread to the entorhinal cortex, hippocampus, deeper cortical layers, and subcortical nuclei [ref. 178,ref. 179,ref. 180].
13. Revisiting the Pathogenic Mechanisms of Tau Toxicity
Given its pivotal role in regulating microtubule dynamics, aberrant PTMs and other factors can disrupt the normal physiological function of tau by altering its properties, subcellular localization, and promoting the formation of insoluble tau aggregates, thereby leading to at least two detrimental toxic effects (Figure 5): (1) the loss of normal physiological function and (2) the gain of toxic effects resulting from tau aggregates.
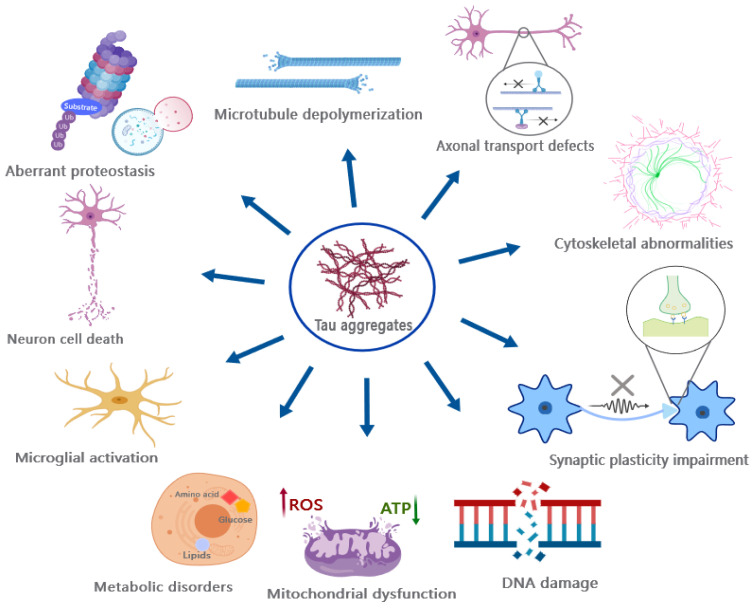
Physiologically, tau binds to MTs and stabilizes MTs by acting as a spacer between tubulin dimers [ref. 181,ref. 182]. However, hyperphosphorylation of tau abolishes its ability to bind MTs, resulting in their dissociation [ref. 183,ref. 184,ref. 185] and subsequent destabilization. Since MTs serve as the structural skeleton and corridor for efficient axonal transport, they facilitate the movement of cargo between the neuronal cell body and their axon terminals, such as nutrients, signaling molecules, and organelles [ref. 186,ref. 187]. Therefore, the dissociation of MTs leads to the disruption of axonal transport and synaptic dysfunction [ref. 188,ref. 189], manifesting as cognitive decline, memory loss, and other associated symptoms [ref. 190,ref. 191].
The toxicological effects of tau aggregates, particularly the oligomers, exhibit a complex and diverse nature (Figure 5) [ref. 192]. For instance, tau oligomers have been shown to exert detrimental effects on mitochondria, disrupt fast axonal transport, impair genomic stability, interfere with synaptic transmission and function, destabilize MTs and the cytoskeleton including the Actin network, disrupt protein degradation systems, as well as induce cell death such as apoptosis [ref. 192,ref. 193,ref. 194,ref. 195].
The toxicity of NFTs and filaments remains debated despite their association with tauopathy as one of the pathological features. However, it is important to note that neuronal dysfunction and cell death precede the formation of NFTs in both human patients and animal models, suggesting that NFTs may not be the primary causative factor responsible for exerting tau toxicity [ref. 193,ref. 196,ref. 197], although several lines of evidence suggest that there is a correlation between neuronal apoptosis and the formation of NFTs [ref. 24], and the neuronal dysfunction and behavioral changes in tauopathy animals coincide with the occurrence of NFTs [ref. 198]. A precise study by Santacruz et al. demonstrates that tau toxicity is independent of the accumulation of NFTs, which is revealed by expressing a repressible human tau variant in mice. The suppression of tau expression significantly attenuates tau toxicity in mice, while the formation of NFTs continues [ref. 199]. The assessment of the toxicity of different tau species in vivo is challenging since monomeric and oligomeric tau species also serve as precursors of NFTs. Thereby, animals with NFTs exhibit the coexistence of monomeric and oligomeric tau species, implicating that the toxic effects associated with NFTs may also be caused by the intermediate oligomeric tau and the hyperphosphorylated monomeric tau.
It is noteworthy that tau toxicity extends beyond synapses, MTs, and axons as mentioned [ref. 192]. In addition, tau can interfere with various biological processes, and induce genomic instability, neuroinflammation, metabolic disorders, and membrane remodeling [ref. 192]. Neuroinflammation has been suggested as an important consequence of pathological tau accumulation in various neurodegenerative diseases, such as FTD, Pick’s disease, and PSP, which can result in synaptic clearance and neuronal damage [ref. 200,ref. 201,ref. 202,ref. 203]. Additional evidence from mouse models has demonstrated that tau possesses the capacity to elicit hyperactivated inflammation, encompassing the excessive activation of microglia and T-cells [ref. 204,ref. 205]. Interestingly, the inhibition of microglial and T-cell activation has been shown to effectively block tau pathology [ref. 205], thereby suggesting a significant contribution of inflammatory responses in tau pathology.
A new perspective regarding tau toxicity is that it has the potential to disrupt cellular metabolism [ref. 206,ref. 207]. Tau deposition is correlated with impaired metabolisms of glucose, norepinephrine, and purine [ref. 208,ref. 209], reduced glucose utilization and oxidative phosphorylation activity [ref. 209,ref. 210], as well as the interference of the arginine, ornithine, and methionine metabolisms [ref. 211]. Although the metabolic decline in AD can be attributed to the synergistic effect of Aβ and tau [ref. 207], it is plausible that tau itself exerts a significant influence on inducing metabolic disturbances. The expression of only the tauP301L mutant in a transgenic mouse model can eventually result in metabolic disturbance [ref. 212,ref. 213], including the impaired metabolism of glucose and glutamate, which is attributed to the reduced expression of metabolic enzymes [ref. 214]. Additionally, it has been demonstrated that induced neurons (iNs) derived from Alzheimer’s fibroblasts undergo a metabolic switch to facilitate aerobic glycolysis as a result of a pathological isoform transition of pyruvate kinase M [ref. 215]. Furthermore, tau can disrupt energy biogenesis by interfering with the activity of mitochondrial complex-I [ref. 214]. The interaction between tau and mitochondrial proteins like NDUFS5, 6, NDUFA8, etc., can also impact the mitochondrial proteomes [ref. 216]. However, the diverse metabolic perturbing effects of tau, the link between metabolic disturbance and tau toxicity, and the underlying mechanisms remain poorly understood.
14. Discussion
The properties and structures of tau have been shown to be diverse in various tauopathy diseases, which highlights the complexity of tau polymerization. The mechanism by which tau adopts the aggregation process in vivo remains incompletely understood despite the initial electrical change and subsequent gradual conformation shift model that have been demonstrated. The diversity of tau aggregation courses may hinder the efficacy of targeted compounds and antibodies, necessitating careful consideration in future therapeutic approaches for tauopathies.
To propagate its pathology, tau undergoes a series of transmission events involving the release of transmissible seeds and subsequent internalization by recipient cells. It is noteworthy that the modulation of the tau transmission process, such as perturbing HSPGs and endocytosis, has the potential to slow down tau propagation in animal models, thereby offering a promising avenue for endeavors aimed at attenuating the progression of tau pathology in vivo.
Importantly, the multifaceted downstream consequences of tau dysfunction and aggregation necessitate a comprehensive approach, and targeting a single consequence may yield limited efficacy. It is worth noting that prioritizing the upstream driving forces and the tau protein itself holds greater potential for mitigating tau toxicity.
References
- B. Esmaeli-Azad, J.H. McCarty, S.C. Feinstein. Sense and antisense transfection analysis of tau function: Tau influences net microtubule assembly, neurite outgrowth and neuritic stability. J. Cell Sci., 1994. [DOI | PubMed]
- G. Siano, M. Varisco, M.C. Caiazza, V. Quercioli, M. Mainardi, C. Ippolito, A. Cattaneo, C. Di Primio. Tau Modulates VGluT1 Expression. J. Mol. Biol., 2019. [DOI | PubMed]
- F. Biundo, D. Del Prete, H. Zhang, O. Arancio, L. D’Adamio. A role for tau in learning, memory and synaptic plasticity. Sci. Rep., 2018. [PubMed]
- S.A. Kent, T.L. Spires-Jones, C.S. Durrant. Durrant, The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol., 2020. [PubMed]
- M.K. Sjoberg, E. Shestakova, Z. Mansuroglu, R.B. Maccioni, E. Bonnefoy. Tau protein binds to pericentromeric DNA: A putative role for nuclear tau in nucleolar organization. J. Cell Sci., 2006. [DOI | PubMed]
- H. Qi, F.X. Cantrelle, H. Benhelli-Mokrani, C. Smet-Nocca, L. Buee, G. Lippens, E. Bonnefoy, M.C. Galas, I. Landrieu. Nuclear magnetic resonance spectroscopy characterization of interaction of Tau with DNA and its regulation by phosphorylation. Biochemistry, 2015. [DOI | PubMed]
- Z. Mansuroglu, H. Benhelli-Mokrani, V. Marcato, A. Sultan, M. Violet, A. Chauderlier, L. Delattre, A. Loyens, S. Talahari, S. Begard. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep., 2016. [DOI | PubMed]
- V.M. Lee, M. Goedert, J.Q. Trojanowski. Neurodegenerative tauopathies. Annu. Rev. Neurosci., 2001. [DOI | PubMed]
- P. Gómez-Ramos, M.A. Morán. Ultrastructural aspects of neurofibrillary tangle formation in aging and Alzheimer’s disease. Microsc. Res. Tech., 1998. [DOI | PubMed]
- S. Murayama, H. Mori, Y. Ihara, M. Tomonaga. Immunocytochemical and ultrastructural studies of Pick’s disease. Ann. Neurol., 1990. [DOI | PubMed]
- I. Ferrer, I. López-González, M. Carmona, L. Arregui, E. Dalfó, B. Torrejón-Escribano, R. Diehl, G.G. Kovacs. Glial and neuronal tau pathology in tauopathies: Characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol., 2014. [DOI | PubMed]
- J.F. Crary, J.Q. Trojanowski, J.A. Schneider, J.F. Abisambra, E.L. Abner, I. Alafuzoff, S.E. Arnold, J. Attems, T.G. Beach, E.H. Bigio. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol., 2014. [DOI | PubMed]
- H.J. Garringer, J. Murrell, L. D’Adamio, B. Ghetti, R. Vidal. Modeling familial British and Danish dementia. Brain Struct. Funct., 2010. [DOI | PubMed]
- T.B. VanItallie. Traumatic brain injury (TBI) in collision sports: Possible mechanisms of transformation into chronic traumatic encephalopathy (CTE). Metabolism, 2019. [DOI | PubMed]
- T. Nishimura, K. Ikeda, H. Akiyama, H. Kondo, M. Kato, F. Li, E. Iseki, K. Kosaka. Immunohistochemical investigation of tau-positive structures in the cerebral cortex of patients with progressive supranuclear palsy. Neurosci. Lett., 1995. [DOI | PubMed]
- E.H. Bigio, A.M. Lipton, S.H. Yen, M.L. Hutton, M. Baker, P. Nacharaju, C.L. White, P. Davies, W. Lin, D.W. Dickson. Frontal lobe dementia with novel tauopathy: Sporadic multiple system tauopathy with dementia. J. Neuropathol. Exp. Neurol., 2001. [DOI | PubMed]
- T. Komori, N. Arai, M. Oda, H. Nakayama, H. Mori, S. Yagishita, T. Takahashi, N. Amano, S. Murayama, S. Murakami. Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol., 1998. [DOI | PubMed]
- D.W. Dickson. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J. Neurol., 1999. [DOI | PubMed]
- K. Ikeda, H. Akiyama, H. Kondo, C. Haga. A study of dementia with argyrophilic grains. Possible cytoskeletal abnormality in dendrospinal portion of neurons and oligodendroglia. Acta Neuropathol., 1995. [DOI | PubMed]
- M. Tolnay, M.G. Spillantini, M. Goedert, J. Ulrich, D. Langui, A. Probst. Argyrophilic grain disease: Widespread hyperphosphorylation of tau protein in limbic neurons. Acta Neuropathol., 1997. [DOI | PubMed]
- H. McCann, B. Durand, C.E. Shepherd. Aging-Related Tau Astrogliopathy in Aging and Neurodegeneration. Brain Sci., 2021. [DOI | PubMed]
- A. Didonna. Tau at the interface between neurodegeneration and neuroinflammation. Genes Immun., 2020. [DOI | PubMed]
- M. Goedert. Tau gene mutations and their effects. Mov. Disord., 2005. [DOI]
- Y. Tatebayashi, T. Miyasaka, D.H. Chui, T. Akagi, K. Mishima, K. Iwasaki, M. Fujiwara, K. Tanemura, M. Murayama, K. Ishiguro. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc. Natl. Acad. Sci. USA, 2002. [DOI | PubMed]
- M. Hosokawa, M. Masuda-Suzukake, H. Shitara, A. Shimozawa, G. Suzuki, H. Kondo, T. Nonaka, W. Campbell, T. Arai, M. Hasegawa. Development of a novel tau propagation mouse model endogenously expressing 3 and 4 repeat tau isoforms. Brain, 2022. [DOI | PubMed]
- S. Wegmann, R.E. Bennett, A.S. Amaral, B.T. Hyman. Studying tau protein propagation and pathology in the mouse brain using adeno-associated viruses. Methods Cell Biol., 2017. [DOI | PubMed]
- M. Colom-Cadena, C. Davies, S. Sirisi, J.E. Lee, E.M. Simzer, M. Tzioras, M. Querol-Vilaseca, É. Sánchez-Aced, Y.Y. Chang, K. Holt. Synaptic oligomeric tau in Alzheimer’s disease—A potential culprit in the spread of tau pathology through the brain. Neuron, 2023. [DOI | PubMed]
- N.L. Zabik, M.M. Imhof, S. Martic-Milne. Structural evaluations of tau protein conformation: Methodologies and approaches. Biochem. Cell Biol., 2017. [DOI | PubMed]
- N.A. Eschmann, E.R. Georgieva, P. Ganguly, P.P. Borbat, M.D. Rappaport, Y. Akdogan, J.H. Freed, J.E. Shea, S. Han. Signature of an aggregation-prone conformation of tau. Sci. Rep., 2017. [DOI | PubMed]
- D. Chen, K.W. Drombosky, Z. Hou, L. Sari, O.M. Kashmer, B.D. Ryder, V.A. Perez, D.R. Woodard, M.M. Lin, M.I. Diamond. Tau local structure shields an amyloid-forming motif and controls aggregation propensity. Nat. Commun., 2019. [DOI | PubMed]
- J. Avila, J.S. Jiménez, C.L. Sayas, M. Bolós, J.C. Zabala, G. Rivas, F. Hernández. Tau Structures. Front. Aging Neurosci., 2016. [DOI | PubMed]
- Y. Zeng, J. Yang, B. Zhang, M. Gao, Z. Su, Y. Huang. The structure and phase of tau: From monomer to amyloid filament. Cell Mol. Life Sci., 2021. [DOI | PubMed]
- S. Jeganathan, M. von Bergen, H. Brutlach, H.J. Steinhoff, E. Mandelkow. Global hairpin folding of tau in solution. Biochemistry, 2006. [DOI | PubMed]
- M.D. Mukrasch, S. Bibow, J. Korukottu, S. Jeganathan, J. Biernat, C. Griesinger, E. Mandelkow, M. Zweckstetter. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol., 2009. [DOI | PubMed]
- H. Mirbaha, D. Chen, O.A. Morazova, K.M. Ruff, A.M. Sharma, X. Liu, M. Goodarzi, R.V. Pappu, D.W. Colby, H. Mirzaei. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife, 2018. [DOI | PubMed]
- Z. Hou, D. Chen, B.D. Ryder, L.A. Joachimiak. Biophysical properties of a tau seed. Sci. Rep., 2021. [DOI | PubMed]
- H. Mirbaha, B.B. Holmes, D.W. Sanders, J. Bieschke, M.I. Diamond. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J. Biol. Chem., 2015. [DOI | PubMed]
- A. Takashima. Tauopathies and tau oligomers. J. Alzheimers Dis., 2013. [DOI | PubMed]
- S. Maeda, N. Sahara, Y. Saito, S. Murayama, A. Ikai, A. Takashima. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci. Res., 2006. [DOI | PubMed]
- X. Zhang, M. Vigers, J. McCarty, J.N. Rauch, G.H. Fredrickson, M.Z. Wilson, J.E. Shea, S. Han, K.S. Kosik. The proline-rich domain promotes Tau liquid-liquid phase separation in cells. J. Cell Biol., 2020. [DOI | PubMed]
- H. Kadavath, R.V. Hofele, J. Biernat, S. Kumar, K. Tepper, H. Urlaub, E. Mandelkow, M. Zweckstetter. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA, 2015. [DOI | PubMed]
- M.T. Gyparaki, A. Arab, E.M. Sorokina, A.N. Santiago-Ruiz, C.H. Bohrer, J. Xiao, M. Lakadamyali. Tau forms oligomeric complexes on microtubules that are distinct from tau aggregates. Proc. Natl. Acad. Sci. USA, 2021. [DOI | PubMed]
- N. Sahara, S. Maeda, M. Murayama, T. Suzuki, N. Dohmae, S.H. Yen, A. Takashima. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci., 2007. [DOI | PubMed]
- E.E. Congdon, S. Kim, J. Bonchak, T. Songrug, A. Matzavinos, J. Kuret. Nucleation-dependent tau filament formation: The importance of dimerization and an estimation of elementary rate constants. J. Biol. Chem., 2008. [DOI | PubMed]
- K.R. Patterson, C. Remmers, Y. Fu, S. Brooker, N.M. Kanaan, L. Vana, S. Ward, J.F. Reyes, K. Philibert, M.J. Glucksman. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem., 2011. [DOI | PubMed]
- S. Maeda, A. Takashima. Tau Oligomers. Adv. Exp. Med. Biol., 2019. [DOI | PubMed]
- U. Sengupta, E. Portelius, O. Hansson, K. Farmer, D. Castillo-Carranza, R. Woltjer, H. Zetterberg, D. Galasko, K. Blennow, R. Kayed. Tau oligomers in cerebrospinal fluid in Alzheimer’s disease. Ann. Clin. Transl. Neurol., 2017. [DOI | PubMed]
- S. Maeda, N. Sahara, Y. Saito, M. Murayama, Y. Yoshiike, H. Kim, T. Miyasaka, S. Murayama, A. Ikai, A. Takashima. Granular tau oligomers as intermediates of tau filaments. Biochemistry, 2007. [DOI | PubMed]
- A.W.P. Fitzpatrick, B. Falcon, S. He, A.G. Murzin, G. Murshudov, H.J. Garringer, R.A. Crowther, B. Ghetti, M. Goedert, S.H.W. Scheres. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature, 2017. [DOI | PubMed]
- W. Zhang, A. Tarutani, K.L. Newell, A.G. Murzin, T. Matsubara, B. Falcon, R. Vidal, H.J. Garringer, Y. Shi, T. Ikeuchi. Novel tau filament fold in corticobasal degeneration. Nature, 2020. [DOI | PubMed]
- Y. Shi, W. Zhang, Y. Yang, A.G. Murzin, B. Falcon, A. Kotecha, M. van Beers, A. Tarutani, F. Kametani, H.J. Garringer. Structure-based classification of tauopathies. Nature, 2021. [DOI | PubMed]
- B. Falcon, W. Zhang, A.G. Murzin, G. Murshudov, H.J. Garringer, R. Vidal, R.A. Crowther, B. Ghetti, S.H.W. Scheres, M. Goedert. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature, 2018. [DOI | PubMed]
- H. Zempel, E. Mandelkow. Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci., 2014. [DOI | PubMed]
- V. García-Escudero, D. Ruiz-Gabarre, R. Gargini, M. Pérez, E. García, R. Cuadros, I.H. Hernández, J.R. Cabrera, R. García-Escudero, J.J. Lucas. A new non-aggregative splicing isoform of human Tau is decreased in Alzheimer’s disease. Acta Neuropathol., 2021. [DOI | PubMed]
- F. Hernández, I. Ferrer, M. Pérez, J.C. Zabala, J.A. Del Rio, J. Avila. Tau Aggregation. Neuroscience, 2023. [DOI | PubMed]
- C.M. Cowan, A. Mudher. Are tau aggregates toxic or protective in tauopathies?. Front. Neurol., 2013. [DOI | PubMed]
- V. Makrides, T.E. Shen, R. Bhatia, B.L. Smith, J. Thimm, R. Lal, S.C. Feinstein. Microtubule-dependent oligomerization of tau. Implications for physiological tau function and tauopathies. J. Biol. Chem., 2003. [DOI | PubMed]
- S. Wegmann, J. Biernat, E. Mandelkow. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol., 2021. [DOI | PubMed]
- M. Pevalova, P. Filipcik, M. Novak, J. Avila, K. Iqbal. Post-translational modifications of tau protein. Bratisl. Lekárske Listy, 2006
- C. Alquezar, S. Arya, A.W. Kao. Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol., 2020. [DOI | PubMed]
- E. Sontag, V. Nunbhakdi-Craig, G. Lee, G.S. Bloom, M.C. Mumby. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron, 1996. [DOI | PubMed]
- J.-J. Pei, C. Björkdahl, H. Zhang, X. Zhou, B. Winblad. p70 S6 kinase and tau in Alzheimer’s disease. J. Alzheimers Dis., 2008. [DOI | PubMed]
- Y. Chen, X. Chen, Z. Yao, Y. Shi, J. Xiong, J. Zhou, Z. Su, Y. Huang. 14-3-3/Tau Interaction and Tau Amyloidogenesis. J. Mol. Neurosci., 2019. [DOI | PubMed]
- C. Haase, J.T. Stieler, T. Arendt, M. Holzer. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem., 2004. [DOI | PubMed]
- F. Liu, B. Li, E.J. Tung, I. Grundke-Iqbal, K. Iqbal, C.X. Gong. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci., 2007. [DOI | PubMed]
- G.T. Bramblett, M. Goedert, R. Jakes, S.E. Merrick, J.Q. Trojanowski, V.M. Lee. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron, 1993. [DOI | PubMed]
- S. Sato, Y. Tatebayashi, T. Akagi, D.H. Chui, M. Murayama, T. Miyasaka, E. Planel, K. Tanemura, X. Sun, T. Hashikawa. Aberrant tau phosphorylation by glycogen synthase kinase-3β and JNK3 induces oligomeric tau fibrils in COS-7 cells. J. Biol. Chem., 2002. [DOI | PubMed]
- J.X. Meng, Y. Zhang, D. Saman, A.M. Haider, S. De, J.C. Sang, K. Brown, K. Jiang, J. Humphrey, L. Julian. Hyperphosphorylated tau self-assembles into amorphous aggregates eliciting TLR4-dependent responses. Nat. Commun., 2022. [DOI | PubMed]
- Y. Xia, S. Prokop, K.M. Gorion, J.D. Kim, Z.A. Sorrentino, B.M. Bell, A.N. Manaois, P. Chakrabarty, P. Davies, B.I. Giasson. Tau Ser208 phosphorylation promotes aggregation and reveals neuropathologic diversity in Alzheimer’s disease and other tauopathies. Acta Neuropathol. Commun., 2020. [DOI | PubMed]
- S.P. Wickramasinghe, J. Lempart, H.E. Merens, J. Murphy, P. Huettemann, U. Jakob, E. Rhoades. Polyphosphate Initiates Tau Aggregation through Intra- and Intermolecular Scaffolding. Biophys. J., 2019. [DOI | PubMed]
- A. Drazic, L.M. Myklebust, R. Ree, T. Arnesen. The world of protein acetylation. Biochim. Biophys. Acta, 2016. [DOI | PubMed]
- T.J. Cohen, J.L. Guo, D.E. Hurtado, L.K. Kwong, I.P. Mills, J.Q. Trojanowski, V.M. Lee. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun., 2011. [DOI | PubMed]
- Y. Zou, L. Guan. Unraveling the Influence of K280 Acetylation on the Conformational Features of Tau Core Fragment: A Molecular Dynamics Simulation Study. Front. Mol. Biosci., 2021. [DOI | PubMed]
- H. Trzeciakiewicz, J.H. Tseng, C.M. Wander, V. Madden, A. Tripathy, C.X. Yuan, T.J. Cohen. A Dual Pathogenic Mechanism Links Tau Acetylation to Sporadic Tauopathy. Sci. Rep., 2017. [DOI | PubMed]
- Y. Xia, B.M. Bell, B.I. Giasson. Tau K321/K353 pseudoacetylation within KXGS motifs regulates tau-microtubule interactions and inhibits aggregation. Sci. Rep., 2021. [DOI | PubMed]
- X.J. Yang, E. Seto. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol. Cell, 2008. [DOI | PubMed]
- L. Li, Y. Jiang, J.Z. Wang, R. Liu, X. Wang. Tau Ubiquitination in Alzheimer’s Disease. Front. Neurol., 2021. [DOI | PubMed]
- F. Munari, C.G. Barracchia, C. Franchin, F. Parolini, S. Capaldi, A. Romeo, L. Bubacco, M. Assfalg, G. Arrigoni, M. D’Onofrio. Semisynthetic and Enzyme-Mediated Conjugate Preparations Illuminate the Ubiquitination-Dependent Aggregation of Tau Protein. Angew. Chem. Int. Ed. Engl., 2020. [DOI | PubMed]
- Y. Ye, D. Klenerman, D. Finley. N-Terminal Ubiquitination of Amyloidogenic Proteins Triggers Removal of Their Oligomers by the Proteasome Holoenzyme. J. Mol. Biol., 2020. [DOI | PubMed]
- J.M. Tan, E.S. Wong, D.S. Kirkpatrick, O. Pletnikova, H.S. Ko, S.-P. Tay, M.W. Ho, J. Troncoso, S.P. Gygi, M.K. Lee. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet., 2008. [DOI | PubMed]
- T. Arakhamia, C.E. Lee, Y. Carlomagno, M. Kumar, D.M. Duong, H. Wesseling, S.R. Kundinger, K. Wang, D. Williams, M. DeTure. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell, 2021. [DOI | PubMed]
- C.A. Dickey, M. Yue, W.L. Lin, D.W. Dickson, J.H. Dunmore, W.C. Lee, C. Zehr, G. West, S. Cao, A.M. Clark. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J. Neurosci., 2006. [DOI | PubMed]
- L. Petrucelli, D. Dickson, K. Kehoe, J. Taylor, H. Snyder, A. Grover, M. De Lucia, E. McGowan, J. Lewis, G. Prihar. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet., 2004. [DOI | PubMed]
- N. Puangmalai, U. Sengupta, N. Bhatt, S. Gaikwad, M. Montalbano, A. Bhuyan, S. Garcia, S. McAllen, M. Sonawane, C. Jerez. Lysine 63-linked ubiquitination of tau oligomers contributes to the pathogenesis of Alzheimer’s disease. J. Biol. Chem., 2022. [DOI | PubMed]
- C.J. Huseby, C.N. Hoffman, G.L. Cooper, J.C. Cocuron, A.P. Alonso, S.N. Thomas, A.J. Yang, J. Kuret. Quantification of Tau Protein Lysine Methylation in Aging and Alzheimer’s Disease. J. Alzheimers Dis., 2019. [DOI | PubMed]
- K.E. Funk, S.N. Thomas, K.N. Schafer, G.L. Cooper, Z. Liao, D.J. Clark, A.J. Yang, J. Kuret. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem. J., 2014. [DOI | PubMed]
- H. Shams, A. Matsunaga, Q. Ma, M.R.K. Mofrad, A. Didonna. Methylation at a conserved lysine residue modulates tau assembly and cellular functions. Mol. Cell Neurosci., 2022. [DOI | PubMed]
- M. Morris, G.M. Knudsen, S. Maeda, J.C. Trinidad, A. Ioanoviciu, A.L. Burlingame, L. Mucke. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci., 2015. [DOI | PubMed]
- C. Kontaxi, P. Piccardo, A.C. Gill. Lysine-Directed Post-translational Modifications of Tau Protein in Alzheimer’s Disease and Related Tauopathies. Front. Mol. Biosci., 2017. [DOI | PubMed]
- S.N. Thomas, K.E. Funk, Y. Wan, Z. Liao, P. Davies, J. Kuret, A.J. Yang. Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: A mass spectrometry approach. Acta Neuropathol., 2012. [DOI | PubMed]
- F. Islam, S. Shohag, S. Akhter, M.R. Islam, S. Sultana, S. Mitra, D. Chandran, M.U. Khandaker, G.M. Ashraf, A.M. Idris. Exposure of metal toxicity in Alzheimer’s disease: An extensive review. Front. Pharmacol., 2022. [DOI | PubMed]
- L. Wang, Y.L. Yin, X.Z. Liu, P. Shen, Y.G. Zheng, X.R. Lan, C.B. Lu, J.Z. Wang. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener., 2020. [DOI | PubMed]
- A.C. Kim, S. Lim, Y.K. Kim. Metal Ion Effects on Aβ and Tau Aggregation. Int. J. Mol. Sci., 2018. [DOI | PubMed]
- R. La Rocca, P.O. Tsvetkov, A.V. Golovin, D. Allegro, P. Barbier, S. Malesinski, F. Guerlesquin, F. Devred. Identification of the three zinc-binding sites on tau protein. Int. J. Biol. Macromol., 2022. [DOI | PubMed]
- Y. Huang, Z. Wu, Y. Cao, M. Lang, B. Lu, B. Zhou. Zinc binding directly regulates tau toxicity independent of tau hyperphosphorylation. Cell Rep., 2014. [DOI | PubMed]
- K. Zubčić, P.R. Hof, G. Šimić, M. Jazvinšćak Jembrek. The Role of Copper in Tau-Related Pathology in Alzheimer’s Disease. Front. Mol. Neurosci., 2020. [DOI | PubMed]
- R. Squitti, C.C. Quattrocchi, C. Salustri, P.M. Rossini. Ceruloplasmin fragmentation is implicated in ‘free’ copper deregulation of Alzheimer’s disease. Prion, 2008. [DOI | PubMed]
- Q.F. Ma, Y.M. Li, J.T. Du, K. Kanazawa, T. Nemoto, H. Nakanishi, Y.F. Zhao. Binding of copper (II) ion to an Alzheimer’s tau peptide as revealed by MALDI-TOF MS, CD, and NMR. Biopolymers, 2005. [DOI | PubMed]
- M. Sadqi, F. Hernández, U. Pan, M. Pérez, M.D. Schaeberle, J. Avila, V. Muñoz. Alpha-helix structure in Alzheimer’s disease aggregates of tau-protein. Biochemistry, 2002. [DOI | PubMed]
- Z.Y. Mo, Y.Z. Zhu, H.L. Zhu, J.B. Fan, J. Chen, Y. Liang. Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J. Biol. Chem., 2009. [DOI | PubMed]
- A.Y. Roman, F. Devred, D. Byrne, R. La Rocca, N.N. Ninkina, V. Peyrot, P.O. Tsvetkov. Zinc Induces Temperature-Dependent Reversible Self-Assembly of Tau. J. Mol. Biol., 2019. [DOI | PubMed]
- S. Ahmadi, I.I. Ebralidze, Z. She, H.-B. Kraatz. Electrochemical studies of tau protein-iron interactions—Potential implications for Alzheimer’s Disease. Electrochim. Acta, 2017. [DOI]
- A. Rametti, F. Esclaire, C. Yardin, N. Cogné, F. Terro. Lithium down-regulates tau in cultured cortical neurons: A possible mechanism of neuroprotection. Neurosci. Lett., 2008. [DOI | PubMed]
- M. Hong, D.C. Chen, P.S. Klein, V.M. Lee. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J. Biol. Chem., 1997. [DOI | PubMed]
- V.J. De-Paula, O.V. Forlenza. Lithium modulates multiple tau kinases with distinct effects in cortical and hippocampal neurons according to concentration ranges. Naunyn-Schmiedeberg’s Arch. Pharmacol., 2022. [DOI | PubMed]
- K.M. Brown, D.K. Tracy. Lithium: The pharmacodynamic actions of the amazing ion. Ther. Adv. Psychopharmacol., 2013. [DOI | PubMed]
- R.H. Lenox, C.G. Hahn. Overview of the mechanism of action of lithium in the brain: Fifty-year update. J. Clin. Psychiatry, 2000
- S. Wegmann, B. Eftekharzadeh, K. Tepper, K.M. Zoltowska, R.E. Bennett, S. Dujardin, P.R. Laskowski, D. MacKenzie, T. Kamath, C. Commins. Tau protein liquid-liquid phase separation can initiate tau aggregation. Embo J., 2018. [DOI | PubMed]
- S.K. Rai, A. Savastano, P. Singh, S. Mukhopadhyay, M. Zweckstetter. Liquid-liquid phase separation of tau: From molecular biophysics to physiology and disease. Protein Sci., 2021. [DOI | PubMed]
- J.S. Rane, A. Kumari, D. Panda. The Acetyl Mimicking Mutation, K274Q in Tau, Enhances the Metal Binding Affinity of Tau and Reduces the Ability of Tau to Protect DNA. ACS Chem. Neurosci., 2020. [DOI | PubMed]
- N.M. Kanaan, C. Hamel, T. Grabinski, B. Combs. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat. Commun., 2020. [DOI | PubMed]
- J. Wen, L. Hong, G. Krainer, Q.Q. Yao, T.P.J. Knowles, S. Wu, S. Perrett. Conformational Expansion of Tau in Condensates Promotes Irreversible Aggregation. J. Am. Chem. Soc., 2021. [DOI | PubMed]
- E. Lester, F.K. Ooi, N. Bakkar, J. Ayers, A.L. Woerman, J. Wheeler, R. Bowser, G.A. Carlson, S.B. Prusiner, R. Parker. Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron, 2021. [DOI | PubMed]
- P.J. McMillan, S.J. Benbow, R. Uhrich, A. Saxton, M. Baum, T. Strovas, J.M. Wheeler, J. Baker, N.F. Liachko, C.D. Keene. Tau-RNA complexes inhibit microtubule polymerization and drive disease-relevant conformation change. Brain, 2023. [DOI | PubMed]
- G. Lee, N. Cowan, M. Kirschner. The primary structure and heterogeneity of tau protein from mouse brain. Science, 1988. [DOI | PubMed]
- T. Kampers, P. Friedhoff, J. Biernat, E.M. Mandelkow, E. Mandelkow. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett., 1996. [DOI | PubMed]
- S. Ambadipudi, J. Biernat, D. Riedel, E. Mandelkow, M. Zweckstetter. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun., 2017. [DOI | PubMed]
- R. Abskharon, M.R. Sawaya, D.R. Boyer, Q. Cao, B.A. Nguyen, D. Cascio, D.S. Eisenberg. Cryo-EM structure of RNA-induced tau fibrils reveals a small C-terminal core that may nucleate fibril formation. Proc. Natl. Acad. Sci. USA, 2022. [DOI | PubMed]
- A.N. Zwierzchowski-Zarate, A. Mendoza-Oliva, O.M. Kashmer, J.E. Collazo-Lopez, C.L. White, M.I. Diamond. RNA induces unique tau strains and stabilizes Alzheimer’s disease seeds. J. Biol. Chem., 2022. [DOI | PubMed]
- P. Chakraborty, G. Rivière, S. Liu, A.I. de Opakua, R. Dervişoğlu, A. Hebestreit, L.B. Andreas, I.M. Vorberg, M. Zweckstetter. Co-factor-free aggregation of tau into seeding-competent RNA-sequestering amyloid fibrils. Nat. Commun., 2021. [DOI | PubMed]
- X. Zhang, Y. Lin, N.A. Eschmann, H. Zhou, J.N. Rauch, I. Hernandez, E. Guzman, K.S. Kosik, S. Han. RNA stores tau reversibly in complex coacervates. PLoS Biol., 2017. [DOI | PubMed]
- P.E.A. Ash, S. Lei, J. Shattuck, S. Boudeau, Y. Carlomagno, M. Medalla, B.L. Mashimo, G. Socorro, L.F.A. Al-Mohanna, L. Jiang. TIA1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc. Natl. Acad. Sci. USA, 2021. [DOI | PubMed]
- L. Jiang, W. Lin, C. Zhang, P.E.A. Ash, M. Verma, J. Kwan, E. van Vliet, Z. Yang, A.L. Cruz, S. Boudeau. Interaction of tau with HNRNPA2B1 and N6-methyladenosine RNA mediates the progression of tauopathy. Mol Cell, 2021. [DOI | PubMed]
- E.G. Gray, M. Paula-Barbosa, A. Roher. Alzheimer’s disease: Paired helical filaments and cytomembranes. Neuropathol. Appl. Neurobiol., 1987. [DOI | PubMed]
- G.P. Gellermann, T.R. Appel, P. Davies, S. Diekmann. Paired helical filaments contain small amounts of cholesterol, phosphatidylcholine and sphingolipids. Biol. Chem., 2006. [DOI | PubMed]
- E. Bok, E. Leem, B.R. Lee, J.M. Lee, C.J. Yoo, E.M. Lee, J. Kim. Role of the Lipid Membrane and Membrane Proteins in Tau Pathology. Front. Cell Dev. Biol., 2021. [DOI | PubMed]
- N. Ait-Bouziad, G. Lv, A.L. Mahul-Mellier, S. Xiao, G. Zorludemir, D. Eliezer, T. Walz, H.A. Lashuel. Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes. Nat. Commun., 2017. [DOI | PubMed]
- C.A. Sallaberry, B.J. Voss, J. Majewski, J. Biernat, E. Mandelkow, E.Y. Chi, C.M. Vander Zanden. Tau and Membranes: Interactions That Promote Folding and Condensation. Front. Cell Dev. Biol., 2021. [DOI | PubMed]
- B.B. Holmes, S.L. DeVos, N. Kfoury, M. Li, R. Jacks, K. Yanamandra, M.O. Ouidja, F.M. Brodsky, J. Marasa, D.P. Bagchi. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA, 2013. [DOI | PubMed]
- A. Wysocka, E. Palasz, M. Steczkowska, G. Niewiadomska. Dangerous Liaisons: Tau Interaction with Muscarinic Receptors. Curr. Alzheimer. Res., 2020. [DOI | PubMed]
- P. Brundin, R. Melki, R. Kopito. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol, 2010. [DOI | PubMed]
- J.L. Guo, V.M. Lee. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem., 2011. [DOI | PubMed]
- J.L. Guo, V.M. Lee. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med., 2014. [DOI | PubMed]
- S. Dujardin, B.T. Hyman. Tau Prion-Like Propagation: State of the Art and Current Challenges. Adv. Exp. Med. Biol., 2019. [DOI | PubMed]
- M. Merezhko, R.L. Uronen, H.J. Huttunen. The Cell Biology of Tau Secretion. Front. Mol. Neurosci., 2020. [DOI | PubMed]
- N. Annadurai, J.B. De Sanctis, M. Hajdúch, V. Das. Tau secretion and propagation: Perspectives for potential preventive interventions in Alzheimer’s disease and other tauopathies. Exp. Neurol., 2021. [DOI | PubMed]
- T. Katsinelos, M. Zeitler, E. Dimou, A. Karakatsani, H.M. Müller, E. Nachman, J.P. Steringer, C. Ruiz de Almodovar, W. Nickel, T.R. Jahn. Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep., 2018. [DOI | PubMed]
- L. Lorente-Gea, B. García, C. Martín, H. Ordiales, O. García-Suárez, K.M. Piña-Batista, J. Merayo-Lloves, L.M. Quirós, I. Fernández-Vega. Heparan Sulfate Proteoglycans Undergo Differential Expression Alterations in Alzheimer Disease Brains. J. Neuropathol. Exp. Neurol., 2020. [DOI | PubMed]
- J.P. Steringer, S. Bleicken, H. Andreas, S. Zacherl, M. Laussmann, K. Temmerman, F.X. Contreras, T.A. Bharat, J. Lechner, H.M. Müller. Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2)-dependent oligomerization of fibroblast growth factor 2 (FGF2) triggers the formation of a lipidic membrane pore implicated in unconventional secretion. J. Biol. Chem., 2012. [DOI | PubMed]
- Y. Wang, V. Balaji, S. Kaniyappan, L. Krüger, S. Irsen, K. Tepper, R. Chandupatla, W. Maetzler, A. Schneider, E. Mandelkow. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener., 2017. [DOI | PubMed]
- S. Saman, W. Kim, M. Raya, Y. Visnick, S. Miro, S. Saman, B. Jackson, A.C. McKee, V.E. Alvarez, N.C. Lee. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem., 2012. [DOI | PubMed]
- J.C. Polanco, B.J. Scicluna, A.F. Hill, J. Götz. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J. Biol. Chem., 2016. [DOI | PubMed]
- H. Asai, S. Ikezu, S. Tsunoda, M. Medalla, J. Luebke, T. Haydar, B. Wolozin, O. Butovsky, S. Kügler, T. Ikezu. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci., 2015. [DOI | PubMed]
- T. Hamano, S. Enomoto, N. Shirafuji, M. Ikawa, O. Yamamura, S.H. Yen, Y. Nakamoto. Autophagy and Tau Protein. Int. J. Mol. Sci., 2021. [DOI | PubMed]
- X. Chen, Y. Li, C. Wang, Y. Tang, S.-A. Mok, R.M. Tsai, J.C. Rojas, A. Karydas, B.L. Miller, A.L. Boxer. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol. Neurodegener., 2020. [DOI | PubMed]
- Q. Feng, Y. Luo, X.N. Zhang, X.F. Yang, X.Y. Hong, D.S. Sun, X.C. Li, Y. Hu, X.G. Li, J.F. Zhang. MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: A vicious cycle in Alzheimer neurodegeneration. Autophagy, 2020. [DOI | PubMed]
- B. Chambraud, C. Daguinot, K. Guillemeau, M. Genet, O. Dounane, G. Meduri, C. Poüs, E.E. Baulieu, J. Giustiniani. Decrease of neuronal FKBP4/FKBP52 modulates perinuclear lysosomal positioning and MAPT/Tau behavior during MAPT/Tau-induced proteotoxic stress. Autophagy, 2021. [DOI | PubMed]
- N.V. Mohamed, V. Plouffe, G. Rémillard-Labrosse, E. Planel, N. Leclerc. Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci. Rep., 2014. [DOI | PubMed]
- I.H. Lee, T. Finkel. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem., 2009. [DOI | PubMed]
- E. Cocucci, J. Meldolesi. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol., 2015. [DOI | PubMed]
- P. Spitzer, L.M. Mulzer, T.J. Oberstein, L.E. Munoz, P. Lewczuk, J. Kornhuber, M. Herrmann, J.M. Maler. Microvesicles from cerebrospinal fluid of patients with Alzheimer’s disease display reduced concentrations of tau and APP protein. Sci. Rep., 2019. [DOI | PubMed]
- S. Dujardin, S. Bégard, R. Caillierez, C. Lachaud, L. Delattre, S. Carrier, A. Loyens, M.C. Galas, L. Bousset, R. Melki. Ectosomes: A new mechanism for non-exosomal secretion of tau protein. PLoS ONE, 2014. [DOI | PubMed]
- M. Yan, T. Zheng. Role of the endolysosomal pathway and exosome release in tau propagation. Neurochem. Int., 2021. [DOI | PubMed]
- L. Rodriguez, N.V. Mohamed, A. Desjardins, R. Lippé, E.A. Fon, N. Leclerc. Rab7A regulates tau secretion. J. Neurochem., 2017. [DOI | PubMed]
- S.D. Ginsberg, E.J. Mufson, S.E. Counts, J. Wuu, M.J. Alldred, R.A. Nixon, S. Che. Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis., 2010. [DOI | PubMed]
- A. Armstrong, N. Mattsson, H. Appelqvist, C. Janefjord, L. Sandin, L. Agholme, B. Olsson, S. Svensson, K. Blennow, H. Zetterberg. Lysosomal network proteins as potential novel CSF biomarkers for Alzheimer’s disease. Neuromolecular. Med., 2014. [DOI | PubMed]
- S.N. Fontaine, D. Zheng, J.J. Sabbagh, M.D. Martin, D. Chaput, A. Darling, J.H. Trotter, A.R. Stothert, B.A. Nordhues, A. Lussier. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J., 2016. [DOI | PubMed]
- M.E. Cheetham, B.H. Anderton, A.P. Jackson. Inhibition of hsc70-catalysed clathrin uncoating by HSJ1 proteins. Biochem. J., 1996. [DOI | PubMed]
- Y. Xu, L. Cui, A. Dibello, L. Wang, J. Lee, L. Saidi, J.G. Lee, Y. Ye. DNAJC5 facilitates USP19-dependent unconventional secretion of misfolded cytosolic proteins. Cell Discov., 2018. [DOI | PubMed]
- J.G. Lee, S. Takahama, G. Zhang, S.I. Tomarev, Y. Ye. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol., 2016. [DOI | PubMed]
- H.C. Tai, A. Serrano-Pozo, T. Hashimoto, M.P. Frosch, T.L. Spires-Jones, B.T. Hyman. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol., 2012. [DOI | PubMed]
- S. Keck, R. Nitsch, T. Grune, O. Ullrich. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem., 2003. [DOI | PubMed]
- L. Alvarez-Erviti, Y. Seow, A.H. Schapira, C. Gardiner, I.L. Sargent, M.J. Wood, J.M. Cooper. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis., 2011. [DOI | PubMed]
- S. Gurke, J.F. Barroso, E. Hodneland, N.V. Bukoreshtliev, O. Schlicker, H.H. Gerdes. Tunneling nanotube (TNT)-like structures facilitate a constitutive, actomyosin-dependent exchange of endocytic organelles between normal rat kidney cells. Exp. Cell Res., 2008. [DOI | PubMed]
- M. Tardivel, S. Bégard, L. Bousset, S. Dujardin, A. Coens, R. Melki, L. Buée, M. Colin. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun., 2016. [DOI | PubMed]
- S. Abounit, J.W. Wu, K. Duff, G.S. Victoria, C. Zurzolo. Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion, 2016. [DOI | PubMed]
- G.S. Victoria, C. Zurzolo. The spread of prion-like proteins by lysosomes and tunneling nanotubes: Implications for neurodegenerative diseases. J. Cell Biol., 2017. [DOI | PubMed]
- J.W. Wu, M. Herman, L. Liu, S. Simoes, C.M. Acker, H. Figueroa, J.I. Steinberg, M. Margittai, R. Kayed, C. Zurzolo. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem., 2013. [DOI | PubMed]
- Y. Imoto, S. Raychaudhuri, Y. Ma, P. Fenske, E. Sandoval, K. Itoh, E.M. Blumrich, H.T. Matsubayashi, L. Mamer, F. Zarebidaki. Dynamin is primed at endocytic sites for ultrafast endocytosis. Neuron, 2022. [DOI | PubMed]
- J.N. Rauch, G. Luna, E. Guzman, M. Audouard, C. Challis, Y.E. Sibih, C. Leshuk, I. Hernandez, S. Wegmann, B.T. Hyman. LRP1 is a master regulator of tau uptake and spread. Nature, 2020. [DOI | PubMed]
- W. Heusermann, J. Hean, D. Trojer, E. Steib, S. von Bueren, A. Graff-Meyer, C. Genoud, K. Martin, N. Pizzato, J. Voshol. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell Biol., 2016. [DOI | PubMed]
- S. Calafate, W. Flavin, P. Verstreken, D. Moechars. Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep., 2016. [DOI | PubMed]
- D. Freeman, R. Cedillos, S. Choyke, Z. Lukic, K. McGuire, S. Marvin, A.M. Burrage, S. Sudholt, A. Rana, C. O’Connor. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS ONE, 2013. [DOI | PubMed]
- J.H. Tseng, A. Ajit, Z. Tabassum, N. Patel, X. Tian, Y. Chen, A.W. Prevatte, K. Ling, F. Rigo, R.B. Meeker. Tau seeds are subject to aberrant modifications resulting in distinct signatures. Cell Rep., 2021. [DOI | PubMed]
- A.D. Alonso, I. Grundke-Iqbal, H.S. Barra, K. Iqbal. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA, 1997. [DOI | PubMed]
- K.H. Strang, C.L. Croft, Z.A. Sorrentino, P. Chakrabarty, T.E. Golde, B.I. Giasson. Distinct differences in prion-like seeding and aggregation between Tau protein variants provide mechanistic insights into tauopathies. J. Biol. Chem., 2018. [DOI | PubMed]
- G. Manassero, M. Guglielmotto, D. Monteleone, V. Vasciaveo, O. Butenko, E. Tamagno, O. Arancio, M. Tabaton. Dual Mechanism of Toxicity for Extracellular Injection of Tau Oligomers versus Monomers in Human Tau Mice. J. Alzheimers Dis., 2017. [DOI | PubMed]
- H. Braak, E. Braak. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol., 1991. [DOI | PubMed]
- H. Braak, D.R. Thal, E. Ghebremedhin, K. Del Tredici. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol., 2011. [DOI | PubMed]
- D.P. Hanger, D.H. Lau, E.C. Phillips, M.K. Bondulich, T. Guo, B.W. Woodward, A.M. Pooler, W. Noble. Intracellular and extracellular roles for tau in neurodegenerative disease. J. Alzheimers Dis., 2014. [DOI | PubMed]
- K.J. Rosenberg, J.L. Ross, H.E. Feinstein, S.C. Feinstein, J. Israelachvili. Complementary dimerization of microtubule-associated tau protein: Implications for microtubule bundling and tau-mediated pathogenesis. Proc. Natl. Acad. Sci. USA, 2008. [DOI | PubMed]
- X. Dong, S. Bera, Q. Qiao, Y. Tang, Z. Lao, Y. Luo, E. Gazit, G. Wei. Liquid–liquid phase separation of tau protein is encoded at the monomeric level. J. Phys. Chem. Lett., 2021. [DOI | PubMed]
- K. Iqbal, F. Liu, C.-X. Gong, I. Grundke-Iqbal. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res., 2010. [DOI | PubMed]
- C.M. Cowan, T. Bossing, A. Page, D. Shepherd, A. Mudher. Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol., 2010. [DOI | PubMed]
- V.H. Man, X. He, J. Gao, J. Wang. Phosphorylation of Tau R2 Repeat Destabilizes Its Binding to Microtubules: A Molecular Dynamics Simulation Study. ACS Chem. Neurosci., 2023. [DOI | PubMed]
- G.B. Stokin, L.S. Goldstein. Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem., 2006. [DOI | PubMed]
- H. Sherizadeh. What is axoplasmic transport? Considering the role of exercise training: A mini review. J. Exerc. Organ Cross Talk, 2022
- A. Venkatramani, D. Panda. Regulation of neuronal microtubule dynamics by tau: Implications for tauopathies. Int. J. Biol. Macromol., 2019. [DOI | PubMed]
- R. Prior, L. Van Helleputte, V. Benoy, L. Van Den Bosch. Defective axonal transport: A common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol. Dis., 2017. [DOI | PubMed]
- E. Kesidou, P. Theotokis, O. Damianidou, M. Boziki, N. Konstantinidou, C. Taloumtzis, S.-A. Sintila, P. Grigoriadis, M.E. Evangelopoulos, C. Bakirtzis. CNS Ageing in Health and Neurodegenerative Disorders. J. Clin. Med., 2023. [DOI | PubMed]
- R.J. Castellani, R.K. Rolston, M.A. Smith. Alzheimer disease. Disease-a-Month DM, 2010. [DOI | PubMed]
- G. Niewiadomska, W. Niewiadomski, M. Steczkowska, A. Gasiorowska. Tau Oligomers Neurotoxicity. Life, 2021. [DOI | PubMed]
- S.S. Shafiei, M.J. Guerrero-Muñoz, D.L. Castillo-Carranza. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci., 2017. [DOI | PubMed]
- S.M. Ward, D.S. Himmelstein, J.K. Lancia, L.I. Binder. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem. Soc. Trans., 2012. [DOI | PubMed]
- J.E. Gerson, R. Kayed. Formation and propagation of tau oligomeric seeds. Front. Neurol., 2013. [DOI | PubMed]
- Y. Yoshiyama, M. Higuchi, B. Zhang, S.M. Huang, N. Iwata, T.C. Saido, J. Maeda, T. Suhara, J.Q. Trojanowski, V.M. Lee. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron, 2007. [DOI | PubMed]
- A. de Calignon, L.M. Fox, R. Pitstick, G.A. Carlson, B.J. Bacskai, T.L. Spires-Jones, B.T. Hyman. Caspase activation precedes and leads to tangles. Nature, 2010. [DOI | PubMed]
- T. Ishihara, M. Hong, B. Zhang, Y. Nakagawa, M.K. Lee, J.Q. Trojanowski, V.M. Lee. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron, 1999. [DOI | PubMed]
- K. Santacruz, J. Lewis, T. Spires, J. Paulson, L. Kotilinek, M. Ingelsson, A. Guimaraes, M. DeTure, M. Ramsden, E. McGowan. Tau suppression in a neurodegenerative mouse model improves memory function. Science, 2005. [DOI | PubMed]
- C. Laurent, L. Buée, D. Blum. Tau and neuroinflammation: What impact for Alzheimer’s Disease and Tauopathies?. Biomed. J., 2018. [DOI | PubMed]
- A. Bellucci, O. Bugiani, B. Ghetti, M.G. Spillantini. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener. Dis., 2011. [DOI | PubMed]
- E. Schofield, C. Kersaitis, C.E. Shepherd, J.J. Kril, G.M. Halliday. Severity of gliosis in Pick’s disease and frontotemporal lobar degeneration: Tau-positive glia differentiate these disorders. Brain, 2003. [DOI | PubMed]
- R. Fernández-Botrán, Z. Ahmed, F.A. Crespo, C. Gatenbee, J. Gonzalez, D.W. Dickson, I. Litvan. Cytokine expression and microglial activation in progressive supranuclear palsy. Parkinsonism. Relat. Disord., 2011. [DOI | PubMed]
- M. Jin, H. Shiwaku, H. Tanaka, T. Obita, S. Ohuchi, Y. Yoshioka, X. Jin, K. Kondo, K. Fujita, H. Homma. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun., 2021. [DOI | PubMed]
- X. Chen, M. Firulyova, M. Manis, J. Herz, I. Smirnov, E. Aladyeva, C. Wang, X. Bao, M.B. Finn, H. Hu. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature, 2023. [DOI | PubMed]
- V. Kalia, M.M. Niedzwiecki, J.M. Bradner, F.K. Lau, F.L. Anderson, M.L. Bucher, K.E. Manz, A.P. Schlotter, Z.C. Fuentes, K.D. Pennell. Cross-species metabolomic analysis of tau- and DDT-related toxicity. PNAS Nexus, 2022. [DOI | PubMed]
- T.A. Pascoal, S. Mathotaarachchi, S. Mohades, A.L. Benedet, C.O. Chung, M. Shin, S. Wang, T. Beaudry, M.S. Kang, J.P. Soucy. Amyloid-β and hyperphosphorylated tau synergy drives metabolic decline in preclinical Alzheimer’s disease. Mol. Psychiatry, 2017. [DOI | PubMed]
- J.N. Adams, S.N. Lockhart, L. Li, W.J. Jagust. Relationships Between Tau and Glucose Metabolism Reflect Alzheimer’s Disease Pathology in Cognitively Normal Older Adults. Cereb. Cortex, 2019. [DOI | PubMed]
- R. Kaddurah-Daouk, H. Zhu, S. Sharma, M. Bogdanov, S.G. Rozen, W. Matson, N.O. Oki, A.A. Motsinger-Reif, E. Churchill, Z. Lei. Alterations in metabolic pathways and networks in Alzheimer’s disease. Transl. Psychiatry, 2013. [DOI | PubMed]
- A. Strom, L. Iaccarino, L. Edwards, O.H. Lesman-Segev, D.N. Soleimani-Meigooni, J. Pham, S.L. Baker, S.M. Landau, W.J. Jagust, B.L. Miller. Cortical hypometabolism reflects local atrophy and tau pathology in symptomatic Alzheimer’s disease. Brain, 2022. [DOI | PubMed]
- A. York, A. Everhart, M.P. Vitek, K.W. Gottschalk, C.A. Colton. Metabolism-Based Gene Differences in Neurons Expressing Hyperphosphorylated AT8- Positive (AT8+) Tau in Alzheimer’s Disease. ASN Neuro., 2021. [DOI | PubMed]
- A. Joly-Amado, K.S. Serraneau, M. Brownlow, C. Marín de Evsikova, J.R. Speakman, M.N. Gordon, D. Morgan. Metabolic changes over the course of aging in a mouse model of tau deposition. Neurobiol. Aging, 2016. [DOI | PubMed]
- D.J. Koss, L. Robinson, B.D. Drever, K. Plucińska, S. Stoppelkamp, P. Veselcic, G. Riedel, B. Platt. Mutant Tau knock-in mice display frontotemporal dementia relevant behaviour and histopathology. Neurobiol. Dis., 2016. [DOI | PubMed]
- L.H. Nilsen, C. Rae, L.M. Ittner, J. Götz, U. Sonnewald. Glutamate metabolism is impaired in transgenic mice with tau hyperphosphorylation. J. Cereb. Blood Flow Metab., 2013. [DOI | PubMed]
- L. Traxler, J.R. Herdy, D. Stefanoni, S. Eichhorner, S. Pelucchi, A. Szücs, A. Santagostino, Y. Kim, R.K. Agarwal, J.C.M. Schlachetzki. Warburg-like metabolic transformation underlies neuronal degeneration in sporadic Alzheimer’s disease. Cell Metab., 2022. [DOI | PubMed]
- T.E. Tracy, J. Madero-Pérez, D.L. Swaney, T.S. Chang, M. Moritz, C. Konrad, M.E. Ward, E. Stevenson, R. Hüttenhain, G. Kauwe. Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell, 2022. [DOI | PubMed]
- R. Crespo, W. Koudstaal, A. Apetri. In Vitro Assay for Studying the Aggregation of Tau Protein and Drug Screening. J. Vis. Exp., 2018. [DOI]
- B. Aulston, Q. Liu, P. Reilly, S.H. Yuan. An In Vitro Model for Studying Tau Aggregation Using Lentiviral-mediated Transduction of Human Neurons. J. Vis. Exp., 2019. [DOI]
- K. Kitoka, R. Skrabana, N. Gasparik, J. Hritz, K. Jaudzems. NMR Studies of Tau Protein in Tauopathies. Front. Mol. Biosci., 2021. [DOI | PubMed]
- S. Lim, M.M. Haque, D. Kim, D.J. Kim, Y.K. Kim. Cell-based Models To Investigate Tau Aggregation. Comput. Struct. Biotechnol. J., 2014. [DOI | PubMed]
- G. Limorenko, M. Tatli, R. Kolla, S. Nazarov, M.-T. Weil, D.C. Schöndorf, D. Geist, P. Reinhardt, D.E. Ehrnhoefer, H. Stahlberg. Fully co-factor-free ClearTau platform produces seeding-competent Tau fibrils for reconstructing pathological Tau aggregates. Nat. Commun., 2023. [DOI | PubMed]
- J.D. Manos, C.N. Preiss, N. Venkat, J. Tamm, P. Reinhardt, T. Kwon, J. Wu, A.D. Winter, T.R. Jahn, K. Yanamandra. Uncovering specificity of endogenous TAU aggregation in a human iPSC-neuron TAU seeding model. iScience, 2022. [DOI | PubMed]
- H. Shimada, Y. Sato, T. Sasaki, A. Shimozawa, K. Imaizumi, T. Shindo, S. Miyao, K. Kiyama, T. Kondo, S. Shibata. A next-generation iPSC-derived forebrain organoid model of tauopathy with tau fibrils by AAV-mediated gene transfer. Cell Rep. Methods, 2022. [DOI | PubMed]
- M.F. Gutknecht, H. Kaku, T.L. Rothstein. Microparticle immunocapture assay for quantitation of protein multimer amount and size. Cell Rep. Methods, 2022. [DOI | PubMed]
- S.A. Levy, G. Zuniga, E.M. Gonzalez, D. Butler, S. Temple, B. Frost. TauLUM, an in vivo Drosophila sensor of tau multimerization, identifies neuroprotective interventions in tauopathy. Cell Rep. Methods, 2022. [DOI | PubMed]
- Y. Jiang, Y. Lin, S. Krishnaswamy, R. Pan, Q. Wu, L.A. Sandusky-Beltran, M. Liu, M.-H. Kuo, X.-P. Kong, E.E. Congdon. Single-domain antibody–based noninvasive in vivo imaging of α-synuclein or tau pathology. Sci. Adv., 2023. [DOI | PubMed]
- J. Zhao, L. Jiang, A. Matlock, Y. Xu, J. Zhu, H. Zhu, L. Tian, B. Wolozin, J.-X. Cheng. Mid-infrared chemical imaging of intracellular tau fibrils using fluorescence-guided computational photothermal microscopy. Light Sci. Appl., 2023. [DOI | PubMed]
- T. Kimura, M. Ono, C. Seki, K. Sampei, M. Shimojo, K. Kawamura, M.-R. Zhang, N. Sahara, Y. Takado, M. Higuchi. A quantitative in vivo imaging platform for tracking pathological tau depositions and resultant neuronal death in a mouse model. Eur. J. Nucl. Med. Mol. Imaging, 2022. [DOI | PubMed]
- V.L. Villemagne, M.T. Fodero-Tavoletti, C.L. Masters, C.C. Rowe. Tau imaging: Early progress and future directions. Lancet Neurol., 2015. [DOI | PubMed]
- H. Watanabe, M. Ono, H. Saji. Novel PET/SPECT Probes for Imaging of Tau in Alzheimer’s Disease. Sci. World J., 2015. [DOI]
- C. Chen, R.R. Kumbhar, H. Wang, X. Yang, K. Gadhave, C. Rastegar, Y. Kimura, A. Behensky, S. Katakam, D. Jeong. Pathological Tau transmission initiated by binding lymphocyte-activation gene 3. bioRxiv, 2023. [DOI]
- R. Zhu, K.M. Makwana, Y. Zhang, B. Rajewski, J. Del Valle, Y. Wang. Blocking Tau Transmission by Biomimetic Graphene Nanoparticles. J. Mater. Chem. B, 2023. [DOI | PubMed]
- N. Puangmalai, N. Bhatt, M. Montalbano, U. Sengupta, S. Gaikwad, F. Ventura, S. McAllen, A. Ellsworth, S. Garcia, R. Kayed. Internalization mechanisms of brain-derived tau oligomers from patients with Alzheimer’s disease, progressive supranuclear palsy and dementia with Lewy bodies. Cell Death Dis., 2020. [DOI | PubMed]
- T. Mothes, B. Portal, E. Konstantinidis, K. Eltom, S. Libard, L. Streubel-Gallasch, M. Ingelsson, J. Rostami, M. Lindskog, A. Erlandsson. Astrocytic uptake of neuronal corpses promotes cell-to-cell spreading of tau pathology. Acta Neuropathol. Commun., 2023. [DOI | PubMed]
- K.L. Batenburg, C. Sestito, P. Cornelissen-Steijger, J.R. van Weering, L.S. Price, V.M. Heine, W. Scheper. A 3D human co-culture to model neuron-astrocyte interactions in tauopathies. Biol. Proced. Online, 2023. [DOI | PubMed]
- S. Narasimhan, L. Changolkar, D.M. Riddle, A. Kats, A. Stieber, S.A. Weitzman, B. Zhang, Z. Li, E.D. Roberson, J.Q. Trojanowski. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J. Exp. Med., 2020. [DOI | PubMed]
- T. Bouillet, M. Ciba, C.L. Alves, F.A. Rodrigues, C. Thielemann, M. Colin, L. Buée, S. Halliez. Revisiting the involvement of tau in complex neural network remodeling: Analysis of the extracellular neuronal activity in organotypic brain slice co-cultures. J. Neural Eng., 2022. [DOI]
- A. Mewes, H. Franke, D. Singer. Organotypic brain slice cultures of adult transgenic P301S mice—A model for tauopathy studies. PLoS ONE, 2012. [DOI | PubMed]
- C.L. Croft, M.A. Wade, K. Kurbatskaya, P. Mastrandreas, M.M. Hughes, E.C. Phillips, A.M. Pooler, M.S. Perkinton, D.P. Hanger, W. Noble. Membrane association and release of wild-type and pathological tau from organotypic brain slice cultures. Cell Death Dis., 2017. [DOI | PubMed]
- A. Katsikoudi, E. Ficulle, A. Cavallini, G. Sharman, A. Guyot, M. Zagnoni, B.J. Eastwood, M. Hutton, S. Bose. Quantitative propagation of assembled human Tau from Alzheimer’s disease brain in microfluidic neuronal cultures. J. Biol. Chem., 2020. [DOI | PubMed]
- S. Calafate, A. Buist, K. Miskiewicz, V. Vijayan, G. Daneels, B. De Strooper, J. de Wit, P. Verstreken, D. Moechars. Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep., 2015. [DOI | PubMed]
- M. Usenovic, S. Niroomand, R.E. Drolet, L. Yao, R.C. Gaspar, N.G. Hatcher, J. Schachter, J.J. Renger, S. Parmentier-Batteur. Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J. Neurosci., 2015. [DOI | PubMed]
- C.N. Winston, B. Aulston, E.M. Rockenstein, A. Adame, O. Prikhodko, K.N. Dave, P. Mishra, R.A. Rissman, S.H. Yuan. Neuronal exosome-derived human tau is toxic to recipient mouse neurons in vivo. J. Alzheimers Dis., 2019. [DOI | PubMed]
- F. Clavaguera, T. Bolmont, R.A. Crowther, D. Abramowski, S. Frank, A. Probst, G. Fraser, A.K. Stalder, M. Beibel, M. Staufenbiel. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol., 2009. [DOI | PubMed]
- E. Peeraer, A. Bottelbergs, K. Van Kolen, I.-C. Stancu, B. Vasconcelos, M. Mahieu, H. Duytschaever, L. Ver Donck, A. Torremans, E. Sluydts. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol. Dis., 2015. [DOI | PubMed]
- M. Darricau, T. Katsinelos, F. Raschella, T. Milekovic, L. Crochemore, Q. Li, G. Courtine, W.A. McEwan, B. Dehay, E. Bezard. Tau seeds from patients induce progressive supranuclear palsy pathology and symptoms in primates. Brain, 2023. [DOI | PubMed]
- A.P.T. Nguyen, G. Daniel, P. Valdés, M.S. Islam, B.L. Schneider, D.J. Moore. G2019S LRRK2 enhances the neuronal transmission of tau in the mouse brain. Hum. Mol. Genet., 2018. [DOI | PubMed]
- Y. Kim, H. Park, Y. Kim, S.-H. Kim, J.H. Lee, H. Yang, S.J. Kim, C.M. Li, H. Lee, D.-H. Na. Pathogenic role of RAGE in tau transmission and memory deficits. Biol. Psychiatry, 2023. [DOI | PubMed]
- Z. He, J.D. McBride, H. Xu, L. Changolkar, S.-j. Kim, B. Zhang, S. Narasimhan, G.S. Gibbons, J.L. Guo, M. Kozak. Transmission of tauopathy strains is independent of their isoform composition. Nat. Commun., 2020. [DOI | PubMed]
- S. Wegmann, E.A. Maury, M.J. Kirk, L. Saqran, A. Roe, S.L. DeVos, S. Nicholls, Z. Fan, S. Takeda, O. Cagsal-Getkin. Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J., 2015. [DOI | PubMed]
- J.W. Wu, S.A. Hussaini, I.M. Bastille, G.A. Rodriguez, A. Mrejeru, K. Rilett, D.W. Sanders, C. Cook, H. Fu, R.A. Boonen. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci., 2016. [DOI | PubMed]