Interstitial lung disease and pancreatic exocrine insufficiency in CADDS: Phenotypic expansion and literature review
Abstract
Contiguous ABCD1/ DXS1357E deletion syndrome (CADDS) is a rare deletion syndrome involving two contiguous genes on Xq28, ABCD1 and BCAP31 (formerly known as DXS1357E). Only nine individuals with this diagnosis have been reported in the medical literature to date. Intragenic loss‐of‐function variants in BCAP31 cause the deafness, dystonia, and cerebral hypomyelination syndrome (DDCH). Isolated pathogenic intragenic variants in ABCD1 are associated with the most common peroxisomal disorder, X‐linked adrenoleukodystrophy (X‐ALD), a single transporter deficiency, which in its more severe cerebral form is characterised by childhood‐onset neurodegeneration and high levels of very‐long‐chain fatty acids (VLCFA). While increased VLCFA levels also feature in CADDS, the few patients described to date all presented as neonates with a severe phenotype. Here we report a tenth individual with CADDS, a male infant with dysmorphic facial features who was diagnosed through ultra‐rapid whole genome sequencing (WGS) in the setting of persistent cholestatic liver disease, sensorineural hearing loss, hypotonia and growth failure and developmental delay. Biochemical studies showed elevated VLCFA and mildly reduced plasmalogens. He died at 7 months having developed pancreatic exocrine deficiency and interstitial lung disease, two features we propose to be possible extensions to the CADDS phenotype. We also review the genetic, phenotypic, and biochemical features in previously reported individuals with CADDS.
Article type: Case Report
Keywords: BAP31, CADSS, cholestasis, lungs, pancreas, peroxisomal
Affiliations: Department of Metabolic Medicine The Royal Children’s Hospital Melbourne Australia; Victorian Clinical Genetics Services Murdoch Children’s Research Institute Melbourne Australia; Department of Radiology The Royal Children’s Hospital Melbourne Australia; Department of Respiratory Medicine The Royal Children’s Hospital Melbourne Australia; Department of Gastroenterology The Royal Children’s Hospital Melbourne Australia
License: © 2023 The Authors. JIMD Reports published by John Wiley & Sons Ltd on behalf of SSIEM. CC BY 4.0 This is an open access article under the terms of the http://creativecommons.org/licenses/by/4.0/ License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Article links: DOI: 10.1002/jmd2.12390 | PubMed: 37701323 | PMC: PMC10494507
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (1.6 MB)
Synopsis
This paper describes a new patient with CADDS who developed pancreatic exocrine deficiency and interstitial lung disease. We summarise the genetic, phenotypic, and biochemical findings of all previously reported individuals with CADDS.
INTRODUCTION
Contiguous ABCD1/DXS1357E deletion syndrome (CADDS) is a rare deletion syndrome involving two contiguous genes that lie head‐to‐head on chromosome region Xq28, ABCD1 and BCAP31 (formerly known as DXS1357E).
ABCD1 encodes the peroxisomal integral membrane ALD protein (ALDP) and belongs to the ATP‐binding cassette (ABC) transporter superfamily.ref. jmd212390-bib-0001 Intragenic loss‐of‐function variants in ABCD1 are associated with X‐linked adrenoleukodystrophy (X‐ALD; MIM#300100), a peroxisomal disorder and single enzyme (transporter) deficiency characterised by reduced ß‐oxidation of VLCFA, white matter demyelination and adrenal cortex atrophy. Elevated plasma VLCFA is present at birth in all affected individuals.ref. jmd212390-bib-0001 Cerebral X‐ALD is the most severe form of the disorder characterised by childhood‐onset neurodegeneration from 3 years of age that, in the absence of bone marrow transplantation very early in the disease course, progresses to severe disability and death.ref. jmd212390-bib-0002
BCAP31 encodes B‐cell‐receptor associated protein 31 (BAP31), a ubiquitously expressed transmembrane protein located in the endoplasmic reticulum (ER), including in mitochondria‐associated membranes (MAMs).ref. jmd212390-bib-0003 BAP31 functions both as a protein chaperone and quality control factor involved in ER export or retention, and ER‐associated degradation. It also acts as a MAM tetherer and regulatory protein, supporting mitochondrial homeostasis and regulating autophagy and apoptosis.ref. jmd212390-bib-0003 Intragenic loss‐of‐function variants in BCAP31 cause the deafness, dystonia and cerebral hypomyelination syndrome (DDCH; MIM#300475).ref. jmd212390-bib-0004
CADDS was first described in 2002 by Corzo et al.,ref. jmd212390-bib-0005 in three male infants with large ABCD1 deletions extending into BCAP31. Since then, a further six patients with a similar phenotype and variable deletions involving these contiguous genes have been reported, including one symptomatic girl.ref. jmd212390-bib-0006, ref. jmd212390-bib-0007, ref. jmd212390-bib-0008, ref. jmd212390-bib-0009 As in DDCH, severe developmental delay, dystonia, deafness, and white matter abnormalities are commonly reported, while the presence of raised VLCFA is like that seen in isolated ABCD1 defects. However, unlike either DDCH or X‐ALD, CADDS affected males have persistent cholestatic liver disease and commonly die in infancy.ref. jmd212390-bib-0005, ref. jmd212390-bib-0006, ref. jmd212390-bib-0007, ref. jmd212390-bib-0008, ref. jmd212390-bib-0009
Here, we describe a severely affected male infant with CADDS who was diagnosed through ultra‐rapid whole genome sequencing (WGS). We summarise the clinical and biochemical findings from all previously published cases of CADDS, while also suggesting that pancreatic insufficiency and interstitial lung disease may represent extensions to the phenotype.
CASE REPORT
The proband was a male infant with hepatic cholestasis, hypotonia, sensorineural hearing loss (SNHL), and failure to thrive referred for metabolic assessment at 4 weeks of age. He was the only child to non‐consanguineous Caucasian healthy parents and was born at term, small for gestational age (birthweight 2.44 kg, Z = −2.2) despite an otherwise uncomplicated pregnancy. He developed jaundice on day 2 with raised total bilirubin of 203 μmol/L (reference range: 0–15 μmol/L) and GGT of 1287 U/L (RR: 0–40 U/L). His stools remained acholic amidst persistent cholestatic liver dysfunction (Table 1). Failure to thrive was compounded by persistent feeding difficulties with poor suck and frequent emesis, together with ongoing malabsorption and steatorrhea due to pancreatic insufficiency (faecal elastase < 15 μg/g; Table 1).
TABLE 1: Proband’s biochemical profile.
| Units | 1 month*☨ | 3 months# | 6 months^ | RR | |
|---|---|---|---|---|---|
| Liver function | |||||
| ALT | U/L | 196 | 199 | 178 | <50 |
| ALP | U/L | 290 | 626 | 418 | 100–350 |
| GGT | U/L | 267 | 585 | 644 | 0–40 |
| INR | Ratio | 1.1 | 2.7 | 1.1 | 0.8–1.2 |
| Total bilirubin (% conjugated) | μmol/L | 65 (66) | 76 (73) | 26 (62) | 0–15 |
| Fat‐soluble vitamins | |||||
| Vitamin A | μmol/L | 1.3 | <0.2 | 0.2 | 0.6–1.8 |
| Vitamin D | nmol/L | 15 | 50–160 | ||
| Vitamin E | μmol/mmol | 2 | 0.7 | 1.3 | 0.9–7.1 |
| Exocrine/endocrine function | |||||
| Faecal elastase | μg/g | <15 | <15 | <15 | >200 |
| AM cortisol | nmol/L | 191 | 268 | 552 | 100–440 |
| ACTH | pmol/L | 15 | 16.2 | <20 | |
| Peroxisomal studies | |||||
| VLCFA | |||||
| C24/C22 | ratio | 1.727 | 0.550–1.050 | ||
| C26/C22 | ratio | 0.144 | 0–0.03 | ||
| Pristanate | μmol/L | 0.01 | 0–2.5 | ||
| Phytanate | μmol/L | 0.9 | 0–20.0 | ||
| RBC plasmalogens | |||||
| Plasmalogen C16:0/Hb | μg/g Hb | 125 | 140–300 | ||
| Plasmalogen C18:0/Hb | μg/g Hb | 184 | 265–475 | ||
| Bile acids | |||||
| Total plasma BA (fasting) | μmol/L | 92 | 316 | 0–36 | |
| Urine taurocholic | +++ | +++ | |||
| Urine THCA | ++ | ++ | |||
Note: Start of supplementation with ursodeoxycholic acid (*), fat‐soluble vitamins (☨), Creon (#) and cholic acids (^).
Abbreviations: ACTH, adrenocorticotropic hormone; ALP, alkaline phosphatase; ALT, alanine transaminase; BA, bile acids; Hb, haemoglobin; INR, international normalised ratio; RBC, red blood cell; RR, reference range; THCA, tauro tetrahydroxycholestanoic acid; VLCFA, very long chain fatty acids.
At 4 weeks of age, he appeared jaundiced and cachectic, his weight was 2.65 kg (Z = − 3.88) and head circumference 34.5 cm (Z = −2.20). He had triangular faces with a broad forehead and a pointed chin, and an occipital haemangioma that had remained unchanged since birth. His anterior fontanelle was not enlarged. He had central hypotonia with prominent head lag, but otherwise normal reflexes and anti‐gravity movements. There was no organomegaly. His vision was intact and auditory brainstem response audiometry confirmed bilateral moderate SNHL. Brain MRI at 4 weeks showed normal myelination and no evidence of cortical dysplasia, ischemia, or haemorrhage.
Tyrosinemia, galactosemia and Nieman Pick disease type C, together with more common causes of intra‐and extra‐hepatic cholestasis were excluded following initial investigations. Other monogenic differential diagnoses subsequently considered included peroxisomal disorders, disorders of bile acid metabolism, congenital disorders of glycosylation (e.g., ATP6AP1‐CDG), mitochondrial disorders and JAG1 or NOTCH2 defects.
METHODS
Biochemical and Fibroscan analyses
Pre‐treatment blood, urine and CSF samples were collected and processed for routine biochemistry, amino acids, organic acids, bile acids, VLCFA and plasmalogens according to standard protocols. Other urine metabolites were measured by flow‐injection tandem mass spectrometry using targeted multiple reaction monitoring as previously described.ref. jmd212390-bib-0010 FibroScan™ was undertaken with the Echosens machine, S1 probe and 10 successful measurements.
Next‐generation sequencing analysis
Trio whole genome sequencing (WGS) was performed on DNA isolated from blood using massively parallel sequencing (Nextera™ DNA Flex Library Prep kit, Illumina Sequencers) with a mean target coverage of 30x and a minimum of 90% of bases sequenced to at least 10× for nuclear DNA (nDNA) and a minimum of 800× mean coverage for mitochondrial DNA (mtDNA). Data was processed, including read alignment to the reference genome (GRCh38) and to the revised Cambridge Reference Sequence (rCRS) mitochondrial genome (NC_012920.1). Variant calling was carried out using Cpipe or Mutect2 for nDNA and mtDNA, respectively.ref. jmd212390-bib-0011 For nDNA, variant analysis and interpretation within the target region (RefSeq genes ±1 kb) was performed using Agilent Alissa Interpret and reported in accordance with HGVS nomenclature. Copy number variants were screened for using an internal detection tool, CxGo.ref. jmd212390-bib-0012 Curation of nDNA variants was phenotype‐driven with custom gene and pre‐curated gene lists (https://panelapp.agha.umccr.org/) used for variant prioritisation. Classification of nDNA and mtDNA variants was based on ACMG guidelines.ref. jmd212390-bib-0013
RESULTS
Ultra‐rapid, five‐day turnaround WGS identified a 67 kb de novo hemizygous deletion involving ABCD1 and part of BCAP31 on Xq28 (chrX: 153714363–153 781 647), that was orthogonally validated with chromosomal microarray. He had markedly abnormal peroxisomal studies, with raised very long chain fatty acids and reduced plasmalogens (Table 1). He was nutritionally supported with a medium chain triglyceride‐based formula. Pancreatic exocrine insufficiency was managed with Creon and fat‐soluble vitamin supplementation. Cholestatic treatment with ursodeoxycholic acid (10 mg/kg BD) was commenced from 4 weeks of age with limited effect.
At 6 months, he was hospitalised for nutritional and respiratory assessments. His weight had stagnated (weight 3.55 kg, Z = −7.20), necessitating a transition to continuous nasojejunal feeds because of recurrent vomiting. He had global developmental delay with profound hypotonia and poor head control. He had stridor and increased work of breathing and lung CT showed unexplained diffuse interstitial opacities (Figure 1C), without specific features of aspiration pneumonitis and unremarkable infective indices. Liver stiffness measurement measured by Fibroscan was 12.9 kPa. This would be consistent with significant fibrosis; however, interpretation is limited due to the size of the patient and the lack of well‐established normal values in infants under 5 kg. Hepatic ultrasound at 1 month old showed normal echogenicity. Due to ongoing cholestasis a trial of oral cholic acid (15 mg/kg/day) was commenced, but he died at 7 months, before the effect of this medication could be ascertained.
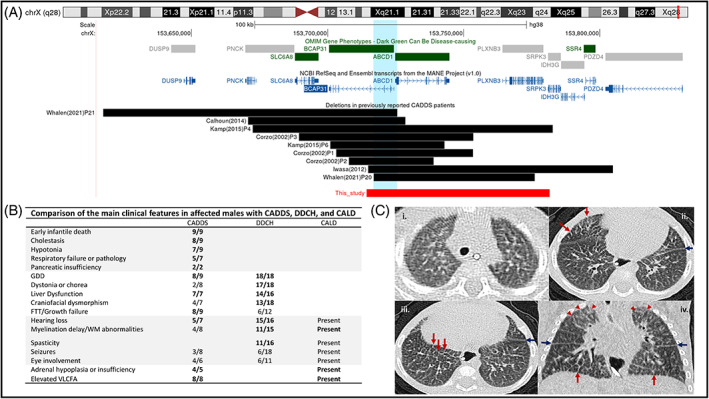
DISCUSSION
Contiguous ABCD1/DXS1357E deletion syndrome (CADDS) is an ultra‐rare condition with only 10 individuals reported, including this patient. Their genetic, phenotypic, and biochemical characteristics are summarised in Table 2.
TABLE 2: Clinical, biochemical, and molecular features of CADDS.
| Reference | Whalen P21 | Kamp P4 | Calhoun | Corzo P3 | Kamp P6 | Corzo P1 | Corzo P2 | Iwasa | Whalen P20 | This report | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular | |||||||||||
| Deletion size | 108 kb | 110 kb | 58 kb | 64 kb | 34–42 kb | 50 kb | 31 kb | 90 kb | 60 kb | 67 kb | |
| Involved genes | DUSP9, PNCK, SLC6A8, BCAP31, ABCD1 | PNCK, SLC6A8, BCAP31, ABCD1, PLXNB3, SRPK3 | SLC6A8, BCAP31, ABCD1 | SLC6A8, BCAP31, ABCD1 | BCAP31, ABCD1 | BCAP31, ABCD1 | BCAP31, ABCD1 | BCAP31, ABCD1, PLXNB3, SRPK3, IDH3G, SSR4, PDZ4 | BCAP31, ABCD1, PLXNB3 | BCAP31, ABCD1, PLXNB3, SRPK3 | |
| Inheritance | nd | nd | Maternal | De novo | nd | Maternal | Maternal | De novo | De novo | De novo | |
| Clinical | |||||||||||
| Sex | F | M | M | M | M | M | M | M | M | M | 9M:1F |
| Age | 3 year | Died <5 month | Died 11 month | Died 4 month | Died 8 month | Died 11 month | Died 4 month | Died 8 month | Died 16 month | Died 7 month | Died: 9 |
| Cause of death | nd | LF | LF, RF | nd | LF, GI bleeding | RF, GI bleeding | Pneumonia, sepsis | RF, lung infiltrates | RF, lung infiltrates | ||
| Growth | |||||||||||
| IUGR/SGA | − | nd | + | + | − | − | − | + | + | + | 5/9 |
| FTT | − | nd | + | + | + | + | + | + | + | + | 8/9 |
| Developmental | |||||||||||
| GDD | + | nd | + | + | + | + | + | + | + | + | 9/9 |
| GI | |||||||||||
| Cholestasis | + | + | + | + | + | + | + | + | − | + | 9/10 |
| Liver dysfunction | nd | nd | + | + | nd | + | + | + | + | + | 7/7 |
| Pancr. insufficiency | − | nd | nd | nd | nd | nd | nd | nd | + | + | 2/3 |
| Hepatomegaly | − | nd | + | nd | nd | nd | nd | + | − | − | 2/5 |
| Splenomegaly | − | nd | + | nd | nd | nd | nd | − | − | − | 1/5 |
| Neurological | |||||||||||
| Hypotonia | − | − | + | + | + | + | + | + | − | + | 7/10 |
| Seizures | − | nd | + | + | − | − | + | − | − | − | 3/9 |
| Dystonia/chorea | − | nd | − | − | − | + | − | − | + | − | 2/9 |
| Myelination delay | + | + | − | + | − | nd | + | + | − | − | 5/9 |
| SNHL | + | + | − | nd | nd | + | + | + | − | + | 6/8 |
| Ophthalmological | + | nd | + | nd | − | + | nd | + | + | − | 5/7 |
| Endocrine | |||||||||||
| Adrenal hypoplasia/insufficiency | nd | nd | + | + | nd | nd | nd | + | + | − | 4/5 |
| Craniofacial/skin | |||||||||||
| Dysmorphism | − | nd | + | − | + | − | − | + | nd | + | 4/8 |
| Haemangiomas | − | − | + | − | − | − | − | − | − | + | 2/10 |
| Biochemical | |||||||||||
| VLCFA | nd | Elevated | Elevated | Elevated | Elevated | Elevated | Elevated | Elevated | nd | Elevated | |
| Plasmalogens | nd | nd | nd | Normal | nd | Normal | Normal | Normal | nd | Low | |
| Phytanic acid | nd | nd | nd | Normal | nd | Normal | Normal | Normal | nd | Normal | |
| Plasma L‐pipecolic | nd | nd | nd | Normal | nd | Normal | Normal | nd | nd | nd | |
| Urine organic acids | nd | nd | nd | Normal | Normal | nd | nd | Dicarboxylic p‐OH‐phenylacetate | nd | Mild dicarboxylic aciduria | |
| Bile acids | |||||||||||
| Plasma | nd | nd | nd | Elevated | nd | nd | Elevated | nd | nd | Elevated | |
| Urine | nd | nd | nd | − | nd | nd | − | nd | nd | Elevated taurocholic | |
Note: Patient order is based on the location of their deletion (centromeric to telomeric).
Abbreviations: +, presence of feature; −, absence of feature; F, female; FTT, failure to thrive; GDD, global developmental delay; GI, gastrointestinal; IUGR, intrauterine growth restriction; LF; liver failure; M, male; nd, no data; Pancr, pancreatic; RF, respiratory failure; SGA, small for gestational age; SNHL, sensorineural hearing loss; VLCFA, very long chain fatty acids.
In the current genomic era, the increased clinical availability of NGS platforms has greatly contributed to the discovery of rare diseases, also shortening diagnostic odysseys, and allowing earlier implementation of relevant management and/or surveillance measures. The CADDS diagnosis in our patient was established in an intensive care setting through access to ultra‐rapid turnaround trio WGS. This avoided the need for a liver biopsy and other invasive procedures during the patient’s diagnostic workup, reflecting the clinical utility of rapid WGS in decreasing infant morbidity.ref. jmd212390-bib-0014, ref. jmd212390-bib-0015 We acknowledge that this deletion would have been identified by microarray, albeit at a slower turnaround time, and that in general, performing microarray before genomic sequencing should be the norm unless there is particular urgency to go straight to WES/WGS.
The Xq28 deletions reported in CADDS vary in length between 31 and 110 kb (Figure 1A). The critical region common to all deletions will become better delineated as more affected individuals are identified. The deletions reported to date all span an 11.7 kb area that includes the promoter regions of both genes, the first exon of ABCD1 and exons 1–3 of BCAP31 (Figure 1A). RNA analyses and immunocytochemical studies on cultured fibroblasts from affected CADDS patients have previously confirmed the absence of BCAP31 transcripts and ALDP (the protein encoded by ABCD1), respectively, in keeping with a loss‐of‐function effect.ref. jmd212390-bib-0005, ref. jmd212390-bib-0008
Our proband’s phenotype is consistent with previous reports, where most individuals presented in early infancy with hepatic cholestasis, severe growth failure, profound hypotonia, developmental delay and elevated VLCFAs (Table 2).ref. jmd212390-bib-0005, ref. jmd212390-bib-0006, ref. jmd212390-bib-0007, ref. jmd212390-bib-0008, ref. jmd212390-bib-0009 SNHL occurred in 6/8, while cerebral hypomyelination and white matter changes were described in 5/9. Early death in hemizygous males (3–16 months) is common, although a symptomatic 3‐year‐old girl with CADDS is the longest surviving individual reported.ref. jmd212390-bib-0008 The milder phenotype in this female is likely due to her having an unaffected X chromosome.
The presence of additional genes encompassed by separate deletions makes comparison between patients with CADDS challenging, given the possible contributions of each gene to the overall phenotype. Four affected individuals had deletions involving the creatine transporter gene SLC6A8. Delayed myelination (3/4 vs. 2/5) and seizures (2/3 vs. 1/6) were more frequently reported in this group compared to those whose deletions did not span SLC6A8 (Table 2), reminiscent of features also seen in cerebral creatine deficiency syndrome 1 (CCDS1; MIM#300352). The deletion in our proband includes two other genes currently without gene‐disease associations. PLXNB3 encodes plexin B3 which functions as a receptor for the semaphorin‐5A molecule and plays a role in axon guidance, invasive growth, and cell migration.ref. jmd212390-bib-0016 There has been recent speculation about its involvement in cardiogenesis and neurodevelopment through Notch signalling regulation.ref. jmd212390-bib-0017 However, congenital heart disease did not feature in any of the CADDS patients with PLXNB3 involvement.ref. jmd212390-bib-0006, ref. jmd212390-bib-0008, ref. jmd212390-bib-0009 SRPK3 encodes a protein member of the serine/arginine‐rich specific kinase family that is known to be involved in mRNA processing, and synaptic vesicle and neurotransmitter release.ref. jmd212390-bib-0018 A zebrafish knockout study in which Srpk3 −/− fish were raised to adulthood has recently revealed SRPK3 to be a compelling candidate gene for X‐linked intellectual disability.ref. jmd212390-bib-0019 The impact of its deletion in our patient and two others is unclear, however, due to the early mortality reported in CADDS.ref. jmd212390-bib-0006, ref. jmd212390-bib-0008
The separate contributions of BCAP31 and ABCD1 to the CADDS phenotype can only be partially gleaned from disorders caused by isolated deficiencies of these genes, DDCH and X‐ALD, respectively (Figure 1B). Spasticity, dystonia, chorea, deafness and cerebral hypomyelination appear to feature more prominently in individuals with intragenic loss‐of‐function variants in BCAP31. Neither DDCH nor X‐ALD, can account for the neonatal cholestasis and early infantile death underpinning the severity in CADDS. Rather, the combined loss of BCAP31 and ABCD1 has been suggested to exert a synergistic deleterious effect on bile acid synthesis,ref. jmd212390-bib-0009 possibly accounting for the hepatic cholestasis in this condition.ref. jmd212390-bib-0007, ref. jmd212390-bib-0008 Similar synergistic effects are implicated in the lethality of CADDS.ref. jmd212390-bib-0007 Our patient died at 7 months of age having developed pancreatic insufficiency and unexplained lung infiltrates (Figure 1C). Similar features were reported in another male infant with a 60 kb deletion including BCAP31, ABCD1, and PLXNB3 who died of respiratory failure at 16 months,ref. jmd212390-bib-0009 and overall, death from respiratory causes is reported in 50% of males with CADDS.ref. jmd212390-bib-0005, ref. jmd212390-bib-0006 Pancreatic insufficiency and interstitial lung disease are therefore possible extensions to the CADDS phenotype (Figure 1B).
Although the underlying pathophysiology in CADDS remains unclear, further exploration into the role of BAP31 in driving the severity of the phenotype is warranted, given the pleiotropic function of this protein. Concomitant loss of BCAP31 has been suggested to exacerbate the clinical phenotype of SLC6A8 deficiency, with patients exhibiting more pronounced hypotonia and developmental delays than expected, together with severe failure to thrive, dystonia and choreoathetosis.ref. jmd212390-bib-0008, ref. jmd212390-bib-0020, ref. jmd212390-bib-0021 Similar effects are possibly at play in contiguous ABCD1 and BCAP31 deletions. Interestingly, BAP31 has been postulated to be involved in the cystic fibrosis disease process, as it targets CFTRdelta508 for ER associated degradation despite any residual transport activity that the partially misfolded CFTR protein may retain.ref. jmd212390-bib-0003 The cholestatic liver disease and pancreatic insufficiency in our proband are reminiscent of features also seen in cystic fibrosis. Those similarities, however, do not extend to the respiratory findings identified on CT, which showed a diffuse interstitial process rather than bronchiectasis and air‐trapping. BAP31 also interacts with the bile salt export pump BSEP,ref. jmd212390-bib-0003 and its involvement in mitochondrial homeostasis offer alternative avenues of exploration into its possible roles in the aetiology of cholestatic liver disease in CADDS. Unfortunately, we were not able to explore these hypotheses further, as our patient died before an adequate sweat test could be collected or fibroblast cultures established.
Oral cholic acid is an efficacious therapy in bile acid synthesis disorders and adjunctive treatment of peroxisomal disorders in patients who exhibit manifestations of liver disease, steatorrhea, or complications from decreased fat‐soluble vitamin absorption without liver fibrosis.ref. jmd212390-bib-0022 Unfortunately, our patient died before the effects of this medication could be ascertained. His plasmalogens were mildly reduced and urine bile acid intermediates such as tetrahydroxycholestanoic (THCA) remained raised in normal proportion to other markers of liver dysfunction (Table 1), making an underlying peroxisomal biogenesis disorder unlikely. Previous studies, have reported normal plasmalogen levels in blood and cultured fibroblasts, with normal peroxisomal size, number and assembly demonstrated in liver tissue by electron microscopy and immunochemistry.ref. jmd212390-bib-0005, ref. jmd212390-bib-0006 BCAP31 deficiency potentially exacerbates the peroxisomal phenotype in CADDS, although the mechanisms remain unclear.ref. jmd212390-bib-0006
CONCLUSION
This report adds to the limited body of knowledge that currently exists about CADDS, although many questions remain regarding the underlying pathophysiology of this condition. More investigations are needed to understand the synergistic effect of combined ABCD1 and BCAP31 loss of function. Pancreatic insufficiency and interstitial lung disease are possible extensions to the CADDS phenotype, while further biochemical characterisation of affected patients will be important to elucidate the extent to which peroxisome metabolism is compromised in this condition. Finally, ultra‐rapid genomic sequencing can help shorten patients’ diagnostic odysseys, potentially precluding the need for invasive procedures.
AUTHOR CONTRIBUTIONS
Oliver Heath, Dinusha Pandithan, Laura Raiti, Moya Vandeleur, Martin B. Delatycki, Joy Yaplito‐Lee, Winita Hardikar, Rebecca Halligan: Clinical patient care and diagnosis. Oliver Heath, Dinusha Pandithan, Laura Raiti, James Pitt, Elena Savva, Jenny Bracken, Moya Vandeleur, Martin B. Delatycki, Joy Yaplito‐Lee, Winita Hardikar, Rebecca Halligan: Evaluation of metabolic, genetic, and radiologic results. Oliver Heath, Rebecca Halligan: Planning of the manuscript. Oliver Heath: Drafting of the manuscript. Oliver Heath, Dinusha Pandithan, Laura Raiti, James Pitt, Elena Savva, Jenny Bracken, Moya Vandeleur, Martin B. Delatycki, Joy Yaplito‐Lee, Winita Hardikar, Rebecca Halligan: Revision of the manuscript.
CONFLICT OF INTEREST STATEMENT
Oliver Heath, Dinusha Pandithan, James Pitt, Elena Savva, Laura Raiti, Martin B Delatycki, Jenny Bracken, Moya Vandeleur, Joy Yaplito‐Lee, Winita Hardikar and Rebecca Halligan have approved the manuscript and declare that they have no conflict of interest. They did not receive reimbursements/fees/funds/salaries from an organization that may in any way gain or lose financially from the results reported in the reviewed manuscript in the last 5 years and have no other competing financial or non‐financial interests, as outlined in the JIMD Conflict of Interest form.
AKNOWLEDGEMENTS
The authors thank the patient’s family for their participation.
INFORMED CONSENT
All procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 2000. Written informed consent was obtained from the patient’s parents for collection of samples and publication of medical data.
ETHICAL APPROVAL
Not required.
References
- Putative X‐linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters.. Nature., 1993. [DOI | PubMed]
- 2 Moser HW , Smith KD , Watkins PA , Powers J , Moser AB . In: Scriver CR , Beaudet AL , Sly WS , Valle D , eds. The Metabolic and Molecular Bases of Inherited Diseases. 8th ed. McGraw‐Hill; 2001:3251‐3301.
- BAP31: physiological functions and roles in disease.. Biochimie., 2021. [DOI | PubMed]
- Mutations in BCAP31 cause a severe X‐linked phenotype with deafness, dystonia, and central hypomyelination and disorganize the Golgi apparatus.. Am J Hum Genet., 2013. [DOI | PubMed]
- Contiguous deletion of the X‐linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: a novel neonatal phenotype similar to peroxisomal biogenesis disorders.. Am J Hum Genet., 2002. [DOI | PubMed]
- Contiguous ABCD1 DXS1357E deletion syndrome: report of an autopsy case.. Neuropathology., 2013. [DOI | PubMed]
- Distal Xq28 microdeletions: clarification of the spectrum of contiguous gene deletions involving ABCD1, BCAP31, and SLC6A8 with a new case and review of the literature.. Am J Med Genet A., 2014. [DOI | PubMed]
- Genotype‐phenotype correlation of contiguous gene deletions of SLC6A8, BCAP31 and ABCD1.. Clin Genet., 2015. [DOI | PubMed]
- Further delineation of BCAP31‐linked intellectual disability: description of 17 new families with LoF and missense variants.. Eur J Hum Genet., 2021. [DOI | PubMed]
- Comprehensive screening of urine samples for inborn errors of metabolism by electrospray tandem mass spectrometry.. Clin Chem., 2002. [PubMed]
- Cpipe: a shared variant detection pipeline designed for diagnostic settings.. Genome Med., 2015. [DOI | PubMed]
- Ximmer: a system for improving accuracy and consistency of CNV calling from exome data.. Gigascience., 2018. [DOI | PubMed]
- Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.. Genet Med., 2015. [DOI | PubMed]
- Rapid whole‐genome sequencing decreases infant morbidity and cost of hospitalization.. NPJ Genom Med., 2018. [DOI | PubMed]
- Rapid whole genome sequencing has clinical utility in children in the PICU.. Pediatr Crit Care Med., 2019. [DOI | PubMed]
- Plexin B3 promotes neurite outgrowth, interacts homophilically, and interacts with Rin.. BMC Neurosci., 2005. [DOI | PubMed]
- An X‐linked PLXNB3 mutation identified in patients with congenital heart disease with neurodevelopmental disabilities.. Transl Pediatr., 2022. [DOI | PubMed]
- A presynaptic phosphosignaling hub for lasting homeostatic plasticity.. Cell Rep., 2022. [DOI | PubMed]
- Eye movement defects in KO zebrafish reveals SRPK3 as a causative gene for an X‐linked intellectual disability.. Res Sq., 2023. [DOI]
- X‐linked creatine transporter defect: a report on two unrelated boys with a severe clinical phenotype.. J Inherit Metab Dis., 2006. [DOI | PubMed]
- Contiguous deletion of SLC6A8 and BAP31 in a patient with severe dystonia and sensorineural deafness.. Mol Genet Metab., 2012. [DOI | PubMed]
- Cholbam(R) and Zellweger spectrum disorders: treatment implementation and management.. Orphanet J Rare Dis., 2021. [DOI | PubMed]