
Structural basis of Δ9-THC analog activity at the Cannabinoid 1 receptor
Abstract
Δ9-tetrahydrocannabinol (THC) is the principal psychoactive compound derived from the cannabis plant Cannabis sativa and approved for emetic conditions, appetite stimulation and sleep apnea relief. THC’s psychoactive actions are mediated primarily by the cannabinoid receptor CB1. Here, we determine the cryo-EM structure of HU210, a THC analog and widely used tool compound, bound to CB1 and its primary transducer, Gi1. We leverage this structure for docking and 1,000 ns molecular dynamics simulations of THC and 10 structural analogs delineating their spatiotemporal interactions at the molecular level. Furthermore, we pharmacologically profile their recruitment of Gi and β-arrestins and reversibility of binding from an active complex. By combining detailed CB1 structural information with molecular models and signaling data we uncover the differential spatiotemporal interactions these ligands make to receptors governing potency, efficacy, bias and kinetics. This may help explain the actions of abused substances, advance fundamental receptor activation studies and design better medicines.
Affiliations: University of Copenhagen; University of Nottingham; Aarhus University; Northeastern University
License: CC BY 4.0 This work is licensed under a Creative Commons Attribution 4.0 International License, which allows reusers to distribute, remix, adapt, and build upon the material in any medium or format, so long as attribution is given to the creator. The license allows for commercial use.
Article links: DOI: 10.21203/rs.3.rs-4277209/v1 | PubMed: 38826401 | PMC: PMC11142349
Relevance: Relevant: mentioned in keywords or abstract
Full text: PDF (4.5 MB)
Introduction
Δ9-tetrahydrocannabinol (THC) is the principal psychoactive compound derived from the cannabis plant, Cannabis sativa. The beneficial physiological effects of THC include analgesia and appetite stimulation1, while harmful effects include e.g., paranoia and anxiety2. As a marketed pharmaceutical Dronabinol, THC is approved as an antiemetic, appetite stimulant and reliever of sleep apnea3. The THC analog Cannabidiol is used for managing anxiety, insomnia, paediatric epilepsy and chronic pain, and these two drugs are combined in Nabiximols, treating neuropathic pain and spasticity in multiple sclerosis. A synthetic THC analog, Nabilone, is also available as an antiemetic and adjunct analgesic for neuropathic pain effective in relieving fibromyalgia4 and multiple sclerosis5. Clinical trials are investigating cannabinoids for treating glioblastoma and a variety of neurological disorders, including Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, amyotrophic lateral sclerosis, traumatic brain injury and stroke6.
The main targets of THC are the cannabinoid receptors, CB1 and CB2. Psychoactive effects are mediated by CB1 in the cerebral cortex, cerebellum, and basal ganglia, whereas CB2 is mainly expressed in cells of the immune system7. CB1 mediates its signaling primarily by Gi/o that inhibits adenylate cyclase which in turn decreases the cellular concentration of cyclic AMP8. In addition, CB1 also couples to Gs, Gq/11 and arrestins9–11. Several CB1 ligands exhibit pathway-biased signaling12,13 i.e., preferential activation of a specific transducer pathway. Biased signaling can lead to functionally selective responses opening for the design of drugs with more beneficial therapeutic profiles and fewer side effects14,15. The multiple pathways, biased ligands and clinical indications make CB1 an appealing system for studying biased signaling.
The structural coverage of complexes of THC analogs and cannabinoid receptors presently spans AM84116,17, CP5594018–20 and AM1154216 bound to CB1 and AM1203317, CP5594021 and HU24321 bound to CB2. The resolution of the structures spans 2.8–3.4 Å and three structures are fully activated cryo-EM complexes with Gi1-2, whereas the AM11542-CB1 complex is a crystal structure with a fusion protein, flavodoxin, that restrains TM6 in an active state. The structure coordinates’ coverage of the overall CB1 protein sequence range between 58 and 62% of residues among all active-state structures of this receptor (Extended Data Table 1) due to the intrinsic flexibility of the N-/C-termini and long third intracellular loop (ICL3).
THC has a tricyclic core consisting of phenyl, pyran and cyclohexene rings that are indexed A, B and C, respectively (Extended Data Fig. 1). Opening of the pyran ring leads to compounds referred to as Cannabidiols that have weaker affinity to CB1–2 and psychoactivity22 than the THC class. Numerous THC analogs have been published, whereof HU210 is among the most potent tool compounds. HU210 is 100–800 times more potent than naturally sourced THC in discrimination tests in animal studies23 and has a binding affinity at human CB1 that is ~700 higher than for THC24. HU243 is a close structural analog of HU210, differing just by the lack of a double bond in C8-C9 of the C ring, sharing its high potency24. Additional potent synthetic THC analogs include the widely used tool compound, CP55940 as well as AM841 and AM11542. In contrast, the phytocannabinoid Cannabinol25 and synthetic ligands L75963326 and JWH13327 have lower affinities and potencies at CB1 and less strong psychoactive effects than THC. Inverse agonism is observed for Tetrahydrocannabivarin (THCv) that has the opposite effect of THC by suppressing appetite and controlling blood glucose levels28, making it a potential treatment for obesity and diabetes.
Structure-activity relationship studies of Δ9-THC and Δ8-THC have focused mainly on the three major pharmacophore features: i) the phenolic hydroxyl group at the C1 position on the A-ring, ii) the methyl group at the C9 position on the C-ring, and iii) the alkyl side chain at the C3 position on the A-ring29–31 (Extended Data Fig. 1). C1 phenolic hydroxyl removal (JWH133) and etherification (L759633) increase selectivity for CB2 over CB1 activation by 153-fold27 and 200-fold26, respectively. Substitution of the C9 methyl with hydroxyl (CP55940) or carbonyl (Nabilone) groups improves affinity, but not selectivity31. A shorter and longer alkyl tail compared to THC decreases and increases binding affinity and potency, respectively,32–34 and five to eight carbons are considered optimal32,35. Elongation with large halogens at the terminal position (as in AM11542) increases binding affinity. Binding affinity can also be enhanced by methyl substitution at the C1’ and C2’ positions31. Here, 1’,2’-substitution gives as good binding affinity but 1’,1’-dimethyl substituted compounds are easier to synthesize. Hitherto, these and other structure-activity studies have predominantly focused on analog substitutions correlated to binding activity or selectivity, and assessment of potency has typically addressed effective dose in vivo29–31. This leaves a need for more molecular mechanistic studies covering ligand-receptor interactions and dissecting additional pharmacological parameters under consistent experimental conditions.
Here, we benchmark the potency and efficacy of THC and 10 analogs for the recruitment of the primary transducer family, Gi/o and b-arrestin. Furthermore, we solve the cryo-EM structure of the HU210/CB1/Gi1 signaling complex and leverage this template to obtain consistent ligand binding modes and spatiotemporal interactions through 1,000 ns molecular dynamics (MD) simulations. Combining these approaches, we expand the structural rationale of THC analogs beyond ligand moieties to dynamic receptor interactions delineating determinants for potency, efficacy, kinetics and biased signaling.
Results
Cryo-EM structure of the HU210/CB1/Gi1 signaling complex
The human CB1 receptor (residues 2–472) and a Gi1 double mutant (G203A, A326S) with increased propensity to form complex with the receptor36,37 were co-expressed in Sf9 insect cells. A stable HU210/CB1/Gi1 complex was formed in cell membranes by binding of the ScFv16 fragment antibody (Fab), and hydrolysis of released GDP nucleotides using Apyrase enzyme. The complex was purified in LMNG detergent micelles, using a M1 anti-Flag affinity resin and size exclusion chromatography. The complex was blotted onto carbon gold grids and imaged using a Titan Krios microscope (Extended Data Fig. 2). The derived HU210/CB1/Gi1 structure (Fig. 1a) has a global resolution of 2.9 Å and large overall similarity to published CB1/Gi1–2 complexes (Fig. 1b and Extended Data Table 2). The HU210-bound structure has the largest coverage of receptor residues among all reported active state CB1 structures16–19,38,39 (Extended Data Table 1). It covers 294 residues, which is four residue backbones and seven sidechains more than the second most complete structure19. The structure reveals more of the N-terminus wherein residues 100–105 form a b-hairpin-like bend with van der Waals contacts to residues F108N-ter, Q261ECL2, C264ECL2 and M371ECL3 (Fig. 1c). Intriguingly, we also observed structural changes in the G protein binding site, far away from the ligand binding site. The HU210 complex (with Gi1) and CP55940 complex19 (with PAM and Gi2) structures demonstrate distinct conformations of the modelled residues in the intracellular loop ICL3 (Fig. 1d). Altogether, the new HU210-bound structure expands our insights into the CB1/Gi1 signaling complex and adds a high-resolution template for THC structure-activity studies.
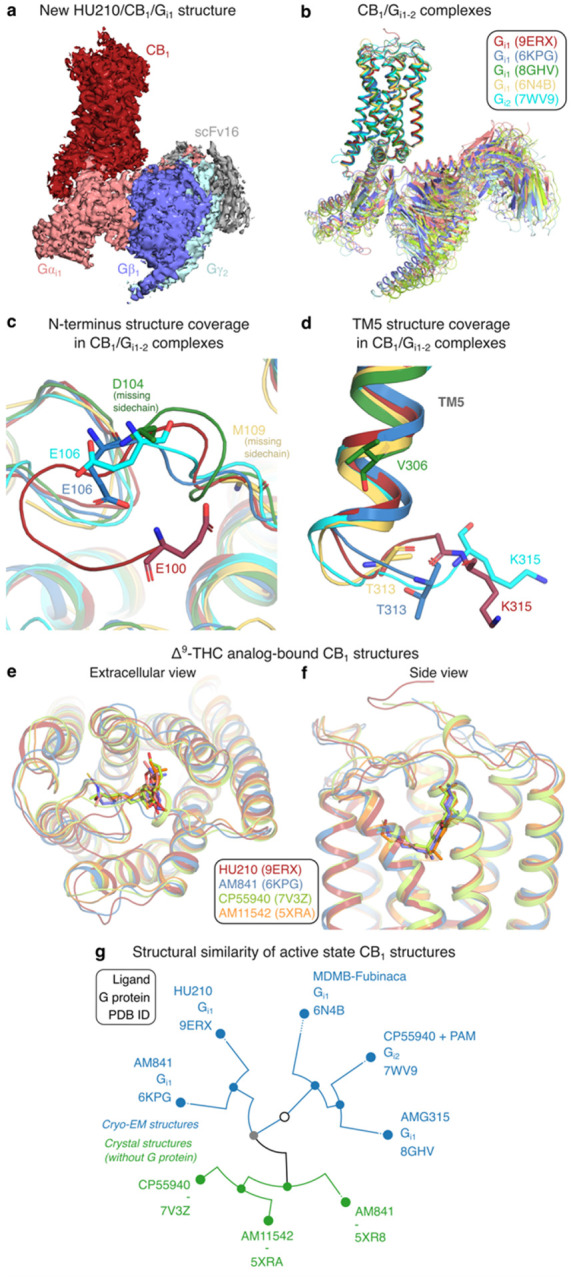
HU210 shares binding site with the THC analogs AM84116 (Extended Data Fig. 3), CP5594018,19 (cryo-EM Gi1-2 complexes), AM1154216 (crystal structure with a fusion protein, flavodoxin in ICL3) (Fig. 1e–f) bound to CB1 and AM12033-bound CB2ref. 17 (Gi1 cryo-EM structure). Conformational clustering of the transmembrane helices of all active-state CB1 structures shows that ligands have a large impact on the structural similarity of receptors (Fig. 1g). Whereas the receptors first separate into crystal structures with fusion proteins (inducing an active-like conformation) and cryo-EM structures with G proteins, HU210-bound CB1 then groups closest to the cryo-EM complex with the closest analog of HU210, AM841 (Extended Data Table 1). These two ligands have a Tanimoto coefficient of 0.82 and their respective CB1/Gi1 structure complexes have an RMSD value of 0.81 Å. Furthermore, the crystal and cryo-EM structures grouping the closest contain the same ligand, AM841 (Fig. 1g). This demonstrates a notably high conformational impact of the relatively small ligand on the overall CB1/Gi structure. Taken together, we extend the number of THC analogs with a CB1–2 structure to five and find that these bind with similar conformations in the same site and have a clear impact on the receptor conformation. These structures provide a strong support for structure-activity relationship analysis aiming to delineate common and unique receptor interactions (below).
THC analogs exhibit diverse pharmacological activity
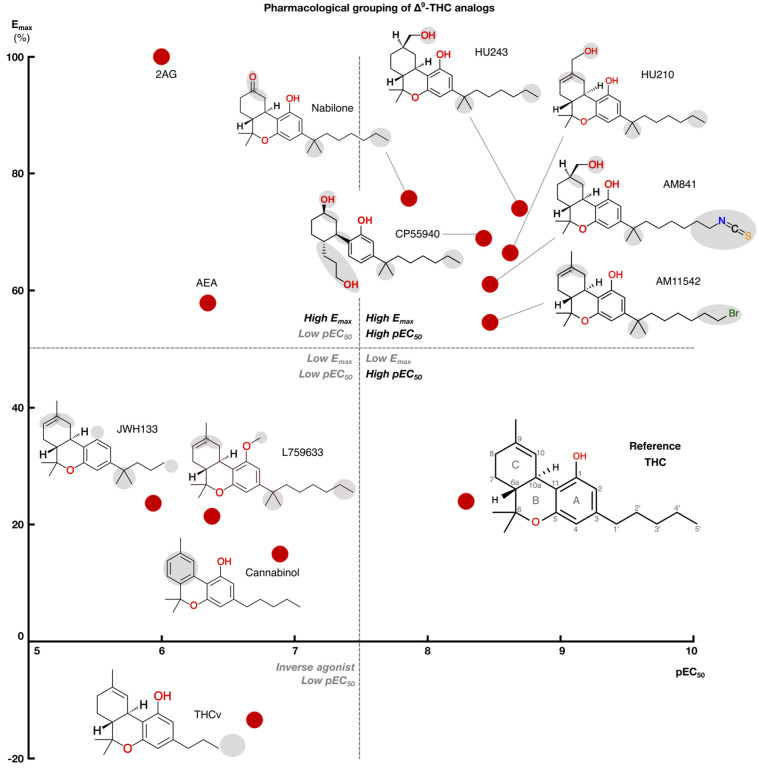
We selected 10 structurally diverse analogs of THC for investigation by docking, molecular dynamics and pharmacological assaying (Extended Data Fig. 1). This includes C1 phenolic hydroxyl removal (JWH133) and etherification (L759633), and C9 methyl replacement with carbonyl (Nabilone), hydroxy (CP55940) or hydroxymethyl (AM841, HU210 and HU243) groups. For the C3 alkyl tail, the analogs span propyl (THCv), butyl (JWH133), pentyl (THC and Cannabinol), heptyl (L759633, Nabilone, CP55940, HU243 and HU210) as well as heptyl substituted with isothiocyanate (AM841) or bromo (AM11542) groups. Of these 11 ligands, eight have an initial branching (1’,1’-dimethyl substitution) of the alkyl chain whereas THC, THCv and cannabinol lack this feature. Altogether, the selected 11 ligands allow for informative and efficient characterization of structure-function covering the major determinants29–31.
We profiled the recruitment of the primary transducer, Gi/o to CB1 for THC, the 10 analogs and the endocannabinoids 2-Arachidonoylglycerol (2AG) and N-arachidonoylethanolamine (AEA, Fig. 2 and Extended Data Fig. 1). We find that the tested ligands fall into five major groups by their activity: i) low-potency/high-efficacy (2AG and AEA), ii) high-potency/high-efficacy (AM841, AM11542, CP55940, HU210, HU243 and Nabilone), iii) low-potency/low-efficacy (Cannabinol, JWH133, L759633), iv) high-potency/low-efficacy (THC) and v) inverse agonist/antagonist (THCv) (Fig. 3). The endocannabinoid 2AG has the highest efficacy and was selected as the reference ligand for comparisons of efficacy (Emax=100%).
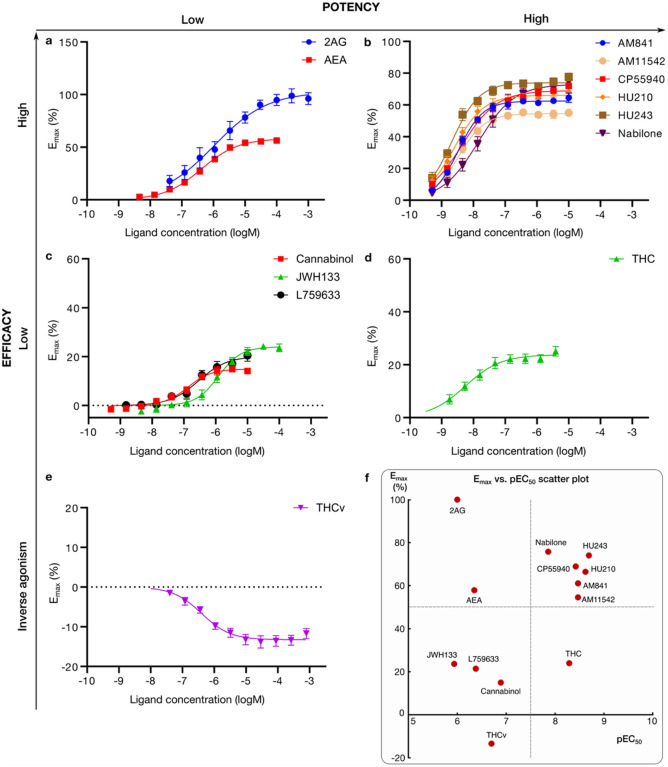
SAR of high-potency/high-efficacy THC analogs
All high-potency/high-efficacy THC analogs (AM841, AM11542, CP55940, HU210, HU243 and Nabilone) have an alkyl tail consisting of seven carbons – two more than the reference ligand THC. Through molecular dynamics (MD) simulations, we find that the high activity of the long alkyl chains may be attributed to favorable contacts to T1973×33, I271ECL2, Y2755×39 and W27 95×43 that are 19, 22, 40 and 21% more frequent, respectively relative to THC (Fig. 4c). Conversely, a comparison with AM841, shows that a further extension of HU243 with an isothiocyanate moiety lowers ligand efficacy from 74% to 61% showing that a too long, or polar alkyl chain is not favorable. In MD simulations, AM841 positions the isothiocyanate substituent in an angled conformation between TM3 and TM4 where it induces a rotamer shift of Y2755×39 indicating a strained and/or higher-energy binding conformation (Fig. 5a). Like AM841, AM11542 also has a further elongated alkyl tail, being bromo-substituted, and its efficacy is lower than that of AM841.

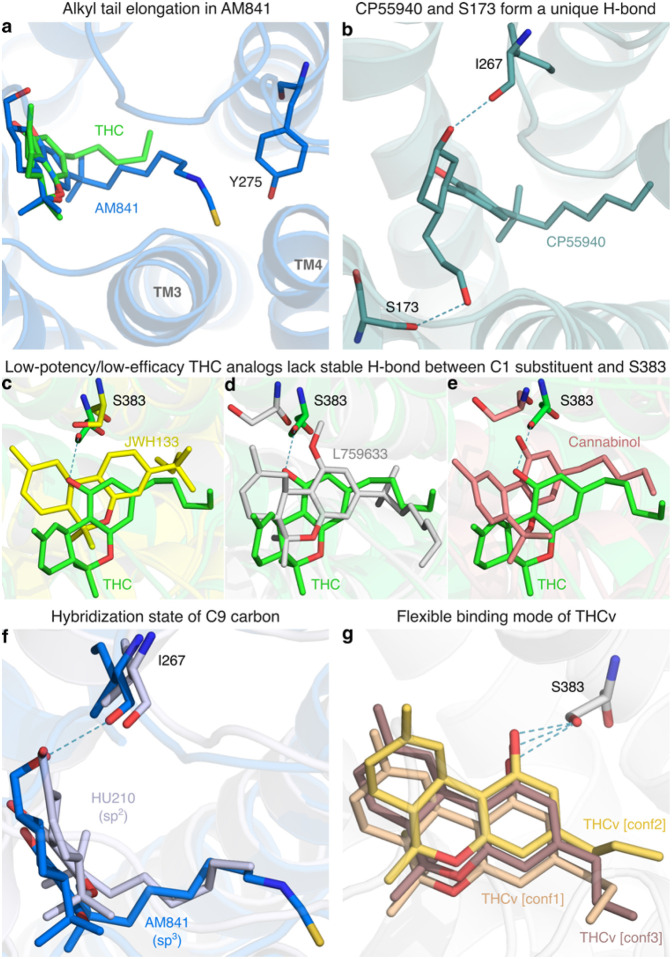
The low efficacy of AM11542, compared to other analogs in this group, could also be attributed to the lack of a hydrogen bonding substituent at C9 at the opposite end of the ligand. Furthermore, Nabilone, which has the lowest potency in this group, has a carbonyl at C9 indicating that hydrogen bond donor capacity is essential for high activity. Consistently, the three agonists with the highest efficacy and potency have a hydroxyl (CP55940) or hydroxymethyl (AM841, HU210 and HU243) that can be hydrogen bond donors. In our MD simulations, CP55940’s C9 hydroxyl forms a very stable hydrogen bond to the backbone carbonyl of I267ECL2 (Fig. 5b). The hydroxymethyl-containing ligands share the hydrogen bond to I267ECL2 and have additional van der Waals contacts with H1782×65. Another difference at the C9 carbon is the hybridization state, which is sp3 for AM841, CP55940 and HU243, and sp2 for AM11542, HU210 and Nabilone. Our MD simulations show that ligands with sp3-hybridized C9 carbons, especially CP55940 and HU243, have a more stable hydrogen bond to the backbone carbonyl of I267ECL2 than observed for sp2-hybridized agonists (Fig. 5f). Nabilone instead has a unique, although low frequent, hydrogen bond between its C9 carbonyl and H1782×65.
All the ligands in this group have a 1’,1’-dimethyl substitution, which is important for activity. Uniquely, CP55940 lacks the B-ring and instead has a rotatable bond between the C10a and C11 atoms. As CP55940 groups close to the middle of the five other analogs it is possible that this flexibility does not affect activity. Alternatively, its effect could be masked by favorable interactions of the unique 6a hydroxypropyl. MD simulations show that this hydroxypropyl forms a very stable hydrogen bond to S1732×60 (Fig. 5b). Given this observation, and the major increase in conformational freedom at the center of the tricyclic core, the alternative explanation of a masked effect seems most plausible. Taken together, this presents a dynamic structural basis for high potency and efficacy that extends and agrees with previous structure-activities studies demonstrating the benefit of the seven-carbon alkyl chain32,35 and hydrogen bonding functionality (ideally donor) in position C931.
SAR of low-potency/low-efficacy THC analogs
Cannabinol is the single analog that groups closest to THC and it differs only by a single feature – an aromatic benzene in the C-ring. However, this difference alone leads to a 25-fold potency reduction (Extended Data Table 3). JWH133 has the lowest potency of all THC analogs and, like the inverse agonist THCv, has a shorter alkyl tail (one and two carbons fewer, respectively) than THC. JWH133 and L759633 both lack the 1-hydroxyl present in THC and the high-potency/high-efficacy analogs. JWH133, which has no 1-substituent, lacks an electron-donating group on the A-ring – weakening its aromatic interactions – and the hydrogen bond to S3837×38 in MD (Fig. 4c and Fig. 5c). Furthermore, in L759633, which has a 1-methoxy substituent, this hydrogen bond is 79% less frequent than in THC (Fig. 5d). Notably, although this hydroxyl group is preserved in Cannabinol this ligand also has an infrequent hydrogen bond – due to the conformational change induced by the planar C-ring (Fig. 4b and Fig. 5e). Hence, the 1-hydroxyl functionality and S3837×38 interaction are major contributors to potency. L759633 is structurally similar to HU210 differing only by the 1-substituent (methoxy vs. hydroxyl) and 9-substituent (methyl vs. hydroxymethyl). When considering the structure-activity relationships of all analogs, these two structural differences explain the low potency and efficacy, respectively of L759633.
SAR of the inverse agonist THCv
Strikingly, THCv only differs from THC by a two-carbon reduction of the alkyl chain but has the opposite modality, inverse agonism and 39-fold lower potency. The large effect on potency is remarkable, given that the high-potency/high-efficacy analogs also differ by addition of two-carbons relative THC (albeit addition at a different site) but have similar potencies. The MD simulations show that the short propyl tail of THCv renders a part of the binding pocket vacant. Compared to THC, this leads to a loss of interactions in MD simulations to the residues Y2755×39, L27 65×40 and I271ECL2. However, the MD analysis shows that a more profound characteristic of THCv is a much more dynamic overall binding mode (Fig. 5g). Throughout the simulations, THCv is anchored by a stable hydrogen bond between the central C1 hydroxyl and S3837×38 but its tricyclic ring moiety has less stable interactions with F268ECL2 and L1933×29 and the two distal ends are considerably more mobile than for THC. Altogether, the inverse agonism of THCv may be attributed in part to lost interactions of the tail but predominantly to the reduced stability of many more interactions across the scaffold.
Low-potency/low-efficacy THC analogs exhibit G protein recruitment bias
To assess if THC analogs display different activity also in terms of biased signaling, we complemented the Gi1 recruitment with arrestin recruitment experiments. By generating bias plots (Extended Data Fig. 4), we find that all ligands show a stronger recruitment of mGi than arrestin. This indicates a general difference in the sensitivity of the two systems, whereas ligand bias can only be defined relative to a reference ligand40. We therefore calculated recruitment bias of tested ligands relative to two reference ligands, the endogenous ligand 2AG and THC. Interestingly, 2AG has low potency/high efficacy and THC has high potency/low efficacy (Fig. 3), but these ligands are nearly unbiased relative to each other because their opposite efficacy/potency relationships equal out when calculating log(Emax/EC50) values. Specifically, their relative bias factor (fold-difference in log(Emax/EC50) across ligands and pathways) is a moderate 1.3 in both cases, for arrestin (THC) or Gi1 (2AG).
The group of low-potency/low-efficacy THC analogs (Fig. 2c) show either very weak or no arrestin recruitment. For example, in the case of JWH133 its arrestin Emax was estimated to be 9.1 % of the 2AG response, while the response to L759633 was too low to fit to a curve and Cannabinol did not show any arrestin recruitment. Our observation that only low-potency/low-efficacy THC analogs display recruitment bias indicates that more chemical diversity would be required to achieve strong bias, while maintaining potency and efficacy.
Structure-kinetics relationship
There have been contradictory results describing the binding of AM841 to CB1. Early studies based on washout41,42 experiments suggested that AM841 binding to CB141 and CB242 is irreversible and that this is due to the formation of a dithiocarbamate group between its isothiocyanate moiety and C3556×47 43. Additionally, irreversible AM841-CB2 binding was also suggested by a mass spectrometry study43. However, recent structures of AM841-bound CB1 provided evidence against this covalent interaction, as the ligand tail was not near C3 556×47 in TM6 but instead occupied a hydrophobic pocket between TM3-TM516,17. Furthermore, new washout experiments conducted with AM841 and analogs lacking the isothiocyanate moiety showed that although binding is “tight”44 or “wash-resistant”16, it is not irreversible or covalent. Notably, a study on functional effects in rhesus monkeys showed that THC does not share the pseudo-irreversible stimulation exhibited by HU21045. Altogether, these data point to different experimental effects with THC and its analogs. Therefore, we conducted a series of kinetics experiments designed to benchmark the apparent koff values of THC and five analogs differing structurally in their tricyclic ring, alkyl branch and alkyl tail (Fig. 6, Extended Data Fig. 4 and Extended Data Table 4).
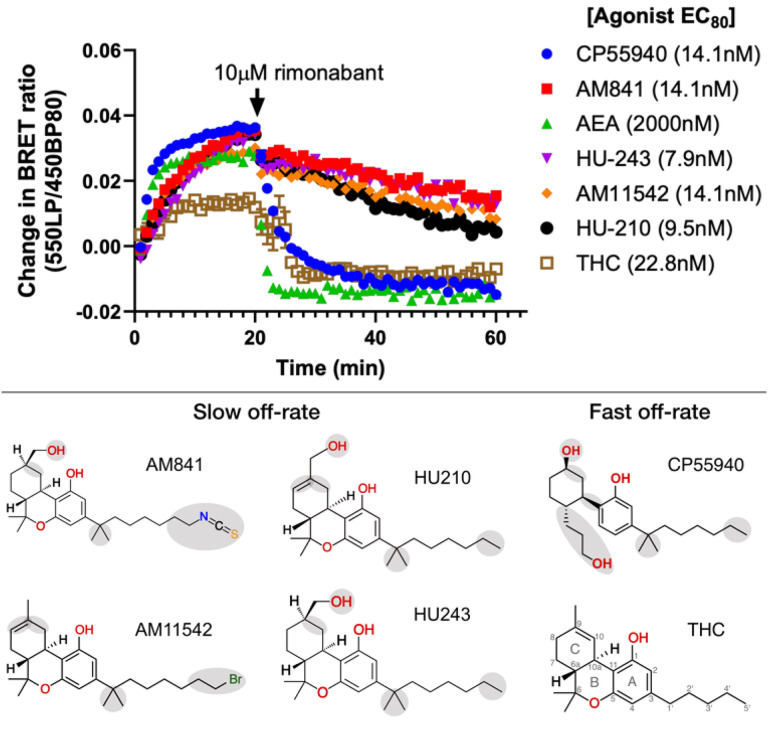
These experiments show the relative differences in ligand dissociation on mGi binding following application of a saturating concentration of the CB1R specific inverse agonist rimonabant. We find that AM841, AM11542, HU210 and HU243 exhibit slow off-rates from the activated CB1 whereas THC, CP55940 and the endogenous CB1R agonist AEA instead show relatively fast off-rates. This is in agreement with previous studies demonstrating that the slow off-rate of these ligands is independent of an isothiocyanate moiety and covalent bond16,44, and that some ligands in this series have fast off-rates45. All four ligands with slow dissociation share the tricyclic ring, alkyl branch and alkyl tail with seven carbons. Given that CP55940 is the only ligand lacking the B-ring its difference in kinetics is likely caused by an increased conformational freedom or reduced bulk at the core scaffold. In MD simulations, CP55940 has an increased flexibility in this region destabilizing key hydrophobic interactions. For THC, the fast off-rate can instead be attributed to the unique shorter, five-carbon alkyl tail. As THC has a lower efficacy than its studied analogs it displays a smaller total drop in BRET ratio, whereas the steepest drop in binding is observed for the endocannabinoid AEA. Although its long aliphatic chain is similar to the C3 alkyl tail of THC analogs, AEA lacks the tricyclic scaffold fitting tightly into the binding pocket and making many hydrophobic interactions. Altogether, the kinetics experiments demonstrated that the tricyclic moiety, alkyl branch (1’,1’-dimethyl) and seven-carbon alkyl tail are essential features for THC analogs to bind to CB1 in a slow off-rate, “wash-resistant” manner.
Discussion
Structural biology on GPCRs is approaching a throughput which allows comparison of complexes of close ligand analogs, especially when, like here they are complemented with manual docking or ligand substitution. Furthermore, high-performance computing has transformed molecular dynamics simulations, enabling the exploration of complex interactions involving multiple ligands bound to fully activated receptors, which in turn are bound to their cognate G proteins e.g.,46,47. A combination of these techniques allowed us to delineate detailed structure-activity relationships on the level of spatiotemporal ligand-receptor interactions. Moving beyond a solely static ligand-based analyses to one which encompasses receptor-effector complex explorations unlocks the potential for identifying and exploiting previously unexplored proximal binding sites, thus broadening the scope of therapeutic intervention and drug design strategies. Furthermore, it provides a deeper, target-complementarity rationale for favorable pharmacophore elements informing the optimal bioisosteric replacements. Exciting new prospects are currently opening for such rational, structure-based drug design from recent breakthroughs demonstrating highly accurate modeling of ligand-protein complexes using AlphaFold48 or RoseTTAFold All-Atom49. Finally, to attain the structural basis for design of better drugs through biased signaling, it would be highly desirable to determine complexes of differentially biased ligands bound to the same receptor and their respective preferred cognate transducer protein.
Our structure-activity relationship study covers diverse structural analogs of THC with modifications in key pharmacophore features and consistent pharmacological evaluation of potency, efficacy and signaling bias for Gi/o and b-arrestin. Our analysis adds to previous studies by plotting ligands by potency and efficacy (Fig. 3), allowing separation of ligands and determinants by both pharmacological properties. This is also the first comprehensive analysis of ligand-receptor binding modes. This took advantage of the new, high-resolution/-coverage CB1/Gi1 structure providing a consistent receptor, while we could also preserve ligand conformations from recent experimental structures, where available16–18. The molecular dynamics simulations could explain differential activities not readily understood from ligand structures or static binding modes alone. For example, THCv only differs from THC by a two-carbon alkyl tail reduction but displays dramatic inverse agonism and low potency. Here, MD revealed a highly “wobbly” binding causing the receptor interactions to become unstable and infrequent. Another point in case for MD is that although preserving a C1 hydroxyl, Cannabinol – like the non-hydroxyl analogs JWH133 and L759633 – loses the hydrogen bond to S3837×38. This observation provides a structural basis contributing to Cannabinol’s relatively low affinity and potency at CB1 and to JWH133’s and L759633’s CB2 selectivity.
Our study is the first to report the relative rates of THC analog dissociation from an active CB1 receptor complex under consistent experimental conditions. These studied compounds vary in alkyl tail length (shorter in THC) and modifications (isothiocyanate in AM841 and bromine in AM11542), B-ring (open in CP55940) and C1’ alkyl branch (missing in THC). Furthermore, we included the endocannabinoid AEA as an additional, non-THC-analog reference ligand. Our results demonstrate that the length of the alkyl tail and completeness of the tricyclic ring moiety are structural determinants for dissociation kinetics from the active form of the receptor. This corroborates a recent study that performed washout experiments of 14 compounds similar to AM84144 – all of which had a heptyl or octyl tail and displayed wash-resistant binding. The tight binding which results in slow dissociation, is likely due to the ligands’ fit in the binding pocket and molecular interactions, the majority of which are hydrophobic contributing to the free energy of binding. The early belief that AM841 binds irreversibly due to a covalent bond was based on the observation that mutations of C3 556×47 to serine, alanine and leucine significantly increased radioligand binding relative to wildtype CB1 in the presence of AM84141. This has now been disproven by AM841-CB1 structure complexes16,17 and recent washout studies16,44. Of note, C3 556×47 is part of a highly conserved C3 556×47WxP motif in TM6 involved in ligand binding and activation of class A GPCRs50. Hence, the most plausible explanation of the mutagenesis effects is through weakening of ligand binding and receptor activation.
The THC analogs HU210 and CP55940 have been previously reported to be biased towards the Gi/o family (cAMP inhibition) over phosphorylation of extracellular signal-regulated kinase 1/2 (pERK1/2)51. However, pERK1/2 is a downstream protein with activity that is dependent on both G proteins and arrestins. Therefore, its bias cannot be delineated directly at the level of the receptor-binding transducers. Our experiments separate these pathways and show that only low-potency/low-efficacy THC analogs have a strong bias towards G protein over arrestin recruitment, which is weak or abrogated (Extended Data Fig. 4 and Extended Data Table 3). This indicates that that more chemical diversity would be required to achieve strong bias, while maintaining potency and efficacy. Several compound classes activating CB1 have already been found to exhibit biased signaling, including endocannabinoids, phytocannabinoids, synthetic cannabinoids, indoles and biphenylureas13. Furthermore, in our experiments, THC displayed a slight preference (bias factor 1.3) for arrestin over Gi1 compared to the endogenous agonist 2AG. All biased signaling experiments are heavily system-dependent and these experiments, performed on transducers directly binding to the receptor, often do not translate to in vivo settings40. For example, both THC and 2AG have been shown to be arrestin-selective over Gi/o in a cell model of striatal neurons52. Together, this warrants many more biased signaling studies covering different CB1 ligand scaffolds across the transducer, downstream pathway and in vivo levels. For example, to investigate if biased ligands can be identified that induce therapeutic effects, such as analgesia, without common side effects e.g., hypothermia, catalepsy, and hypolocomotion53.
Taken together, the HU210/CB1/Gi1 cryo-EM structure, pharmacological profiling, ligand-receptor docking and molecular dynamics simulations could link functional groups of THC ligands to spatiotemporal interactions determining efficacy, potency, biased signaling and kinetics. This may advance our understanding of fundamental receptor function and support the further development of THC and its analogs as therapeutics.
Methods
Protein expression
Human cannabinoid receptor 1 (CB1) residue 2–472 was fused with a N-terminal hemagglutinin (HA) signal peptide followed by a Flag tag and a short Gly-Ser linker. At its C-terminus, the receptor was fused to a 10-histidine purification tag. The DNA sequence was codon optimized for expression in insect cells and inserted into the pFastBac1 vector using custom DNA synthesis (Genscript) for Bac-to-Bac virus expression. A dominant negative mutant of human GNAi1 (G203A/A326S)37,54 was also inserted into PfastBacl. Viruses co-expressing human GNB1 and GNG2 together, as well as Ric8A were kindly provided as a gift by Daniel Hilger. The CB1 receptor Gi protein complex was formed by co-expression in Spodoptera frugiperda (Sf9) insect cells. Cells were infected at 2 mio/ml cells with vius at a ratio between receptor, GNAil, GNB1/GNG2, and Ric8A at (8:5:2:1). Cells were harvested by centrifugation after 48 hours and frozen before subsequent purification.
A Fab fragment (scFv16) construct essentially identical to the construct reported by 55 was expressed using pFastBacl in BTI-Tn-5B1–4 (High Five/Hi5) cells. Cells were infected at 2 mio/ml cells and harvested after 48 hours. Cells were removed by centrifugation and the supernatant containing the secreted scFv16 protein was frozen with 10% glycerol.
ScFv16 purification
ScFv16 was purified using HisPur™ Ni-NTA resin (Thermofisher sci. Cat. No. 88221). In short, initially impurities were removed by precipitation by adding 30 mM Tris pH 8.0, 1 mM NiSO4 and 5 mM CaCl2 for 1 hour at room temperature followed by centrifugation at 18,000 rpm for 20 min. The supernatant was filtered through a 0.22 mM filter and loaded onto Ni-NTA resin over night at room temperature. The resin was then washed in wash buffers: Wash buffer 1: 20 mM Hepes pH 7.4, 500 mM NaCl and 10 mM imidazole, wash buffer 2: 20 mM Hepes pH 7.4, 100 mM NaCl and 10 mM imidazole and wash buffer 3: 20 mM Hepes pH 7.4, 100 mM NaCl and 30 mM imidazole. The protein was eluted with a buffer containing 20 mM Hepes pH 7.4, 100 mM NaCl and 300 mM imidazole. The histidine tag of the protein was removed by digesting with 3C protease for 3 days at 4 °C. ScFv16 was concentrated to ~1.5 mg/ml and frozen with 20% glycerol.
Complex formation and purification
The cell pellet from 1 L of media containing the CB1 Gi protein complex was lysed at room temperature for 1 hour with stirring. The lysis buffer consisted of: 20 mM Hepes pH 7.4, 50 mM NaCl, 4 mM MgCl2, 2 mM HU-210 (Tocris Cat. No. 0966), ~10 mg/ml ScFv16, 0.025 units/ml Apyrase (NEB Cat. No. M0398S) and Roche EDTA free protease inhibitors (Sigma Cat. No. REF05056489001). The lysed cells were harvested by centrifugation for 20 min at 11,000 rpm. The complex was then solubilized using a 40 ml dounce tissue grinder with 30 strokes and a solubilization buffer containing 20 mM Hepes pH 7.4, 100 mM NaCl, 4 mM MgCl2, 1 mM HU-210, ~20 mg/ml ScFv16, 0.05 units/ml Apyrase (NEB Cat. No. M0398S), 15% glycerol, 0.5% LMNG (Anatrace Cat. No. NG310), 0.03% Cholesteryl hemisuccinate (Sigma Cat. No. C6512–25G) and Roche EDTA free protease inhibitors. The sample was mixed for 2 hours at 4 °C. Non solubilized material was removed by centrifugation for 40 min at 14.000 rpm. 5 mM CaCl2 and 0.75 ml M1 anti flag resin added to the supernatant. The slurry was gently mixed for 1 hour at 4 °C and the beads with bound complex were collected by centrifugation for 5 min at 1000 g and transferred to a 1.5 ml filter column (Bio-Rad Cat. No. #7311550EDU). The resin was washed with purification buffer containing 20 mM Hepes pH 7.4, 100 mM NaCI, 5 mM CaCl2, 2 mM MgCl2, 500 nM HU-210, 0.01% LMNG and 0.006% Cholesteryl hemisuccinate . The protein was eluted in purification buffer supplemented with 10 mM EDTA and 200 mg/ml Flag peptide (Sigma Cat. F3290–4MG). The eluted protein was supplemented with an additional 1 mM HU-210 and concentrated to ~50 ml using 100 kDa spin concentrators (Merck cat. nr. Z614092) at 1,000 g. Subsequently, the complex was frozen in liquid nitrogen. Immediately before preparation of grids for cryo-EM, the sample was run on a Superose 6 Increase 3.2/300 column on a Akta system using purification buffer for elution at 0.05 ml/min.
Cryo-EM
Prior to sample application the cryo-EM Quantifoil R 1/1 C 300 grids were activated using a Gloqube Plus glow discharger at 15 mA for 45 sec. All grids were prepared using a Leica EM2 plunge freezer. 3 ml of complex at ~0.85 mg/ml was loaded using a total blotting time of 6–8 sec.
Cryo-EM data collection
Data collection was performed on a Titan Krios G3i (Thermo Fisher Scientific) with a K3/BioQuantuum detector/energy filter setup (Gatan). Magnification was set at 130kx, resulting in a physical pixel size of 0.647 Å/px. Automated data collection was done using EPU (Thermo Fisher Scientific) set to collect in super resolution with 2x binning to the physical pixel size, with gain correction on the fly. AFIS was used for faster data collection speed with 1 exposure per hole for 11260 movies total. Defocus targets were set from −0.6 to −1.8 μm in steps of 0.2 μm, with autofocus after distance at 6 μm. A 50 μm C2 aperture and no objective aperture was used. The energy filter was tuned and set to a slit width of 20 eV with auto centering of the ZLP every hour. An exposure time of 1.50 s in 56 frames for 60.6 e/Å2 total dose was employed.
Cryo-EM data processing
CryoSPARC Live (Structura Biotechnology, 56) was used to monitor data quality during the data collection and provided an initial 3D volume. The data was pre-processed in Relion 3.157 followed by particle picking using crYOLO58 resulting in 603k particles. The particle stack was cleaned by iterating between jobs of 3D Classification and 3D Auto-refine using Sidesplitter59, ending with a polished stack of 187k particles. The stack was imported to CryoSPARC and subjected to Non-uniform 3D refinement, followed by a Local Refine job with a mask around the TM region resulting in a 2.9 Å overall resolution.
HU210/CB1/Gi1 structure model building
The published structure of CB1 bound to the ligand AM841 (PDB: 6KPG)17 was used as the starting model for our refinement. The starting model for the alpha helical domain of the Gi1 subunit was retrieved from the crystal structure of the scFv16 bound Gi1 heterotrimer (PDB: 6CRK)60. Model building and refinement were done using the Coot61 and Phenix62 software packages.
THC analog – CB1 structure/model preparation
The HU210/CB1/Gi1 structure was prepared for THC analog docking and molecular dynamics simulations with THC and analogs. We removed ScFv16, reverted the two Gai1 mutations (G203A and A326S) to wildtype and modeled-in the missing ICL3 (residues 316–334) of CB1 using MODELLER63. The structure was prepared using the Protein Preparation Wizard tool64 and OPLS3e force field65 in Schrodinger66. For, AM84117, AM1154216 and CP5594018 conformations were taken from their respective CB1 structure complexes (the latter two lacking a G protein) and superposed to HU210 to generate CB1/Gi1 complexes. For the remaining THC analogs, CB1/Gi1 complexes were constructed by using the Maestro 3D Builder Tool66 to edit HU210 in our CB1/Gi1 structure and perform energy minimization.
Molecular dynamics simulations
Protonation states of ionizable residues were assigned using Epik66,67 using a pH value of 7.0 as reference. Restrained minimization was performed using the OPLS3e force field65. A membrane was built and positioned around the receptor with guidance from the PPM 2.0 Web Server of the Orientations of Proteins in Membranes (OPM) database68. An orthorhombic box shape with 11 Å buffer distance in the x, y and z directions was used to build a solvent box around the membrane-bound agonist-receptor-transducer complexes. All atom 1-palmitoyl-2-oleoylphosphatidylcholine bilayer (POPC) and TIP3P water were used for the lipid and water models, respectively. A salt concentration of 0.15 M was set, and all the systems were neutralised using sodium and chloride ions.
All MD simulations were performed using the Desmond Molecular Dynamics System69 with the OPLS3e force field65 and full particle mesh Ewald electrostatics70. The systems were gradually heated to 300 K in the NVT ensemble and allowed to equilibrate for 50 ns. Production MD simulations at constant temperature (300 K) and pressure (1 atm) were performed for 1000 ns for each system. This resulted in a total simulation time of 11 μs from all the simulations. Analysis of MD trajectories to calculate frequencies of receptor-ligand, intra-receptor, and receptor-transducer contacts was done using the GetContacts MD analysis package71.
Cell culture and molecular biology reagents
The HEKT-REx™-293 cell line (Cat. No. R71007) was purchased from Invitrogen (CA, USA). T75 mammalian cell culture flasks were purchased from Fisher Scientific (Loughborough, UK). Cell culture reagents from Sigma Aldrich (St. Louis, MO, USA) include Dulbecco’s Modified Eagle’s Medium (DMEM) – high glucose (Cat. No. D6429), Dulbecco’s Phosphate Buffered Saline (D-PBS, Cat. No. D8537), Hank’s Buffered Saline Solution (HBSS, Cat. No. H8264), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) sodium salt (HEPES, Cat. No. RDD035–100G), bovine serum albumin (FBS, Cat. No. F7524), trypsin/EDTA solution 100mL (Cat. No. R001100). Reagents purchased from Gibco™ (MA, USA) included Blasticidin™ Selection Reagent HCl 10mg/mL (Cat. No. 12172530), Zeocin™ Selection Reagent 100mg/mL (cat. No. R25005), Geneticin™ Selective Antibiotic (G-418 Sulfate) 50mg/mL (Cat. No. 10131035). Reagents from Corning® (Corning, NY, USA) include Corning® 100mL Cellstripper™, liquid (Cat. No. 25–056-Cl). Polyethylenimine (PEI, Cat. No. 23966–1) was obtained from Polysciences Inc (PA, USA). 96-well cell culture plates (Cat. No. 655098) were purchased from Greiner Bio-One (Stonehouse, UK). The expression vectors pcDNA™4/TO was obtained from ThermoFisher Scientific and pcDNA™ 3.1 from Invitrogen™.
Compounds
Reference ligands were obtained from Bio-Techne® Tocris (Abingdon, Oxfordshire, UK) include HU210 or ((6aR)-frans-3-(1,1-Dimethylheptyl)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-methanol) (Cat. No. 0966), anandamide (AEA) or (N-(2-Hydroxyethyl)-5Z,8Z,11Z,14Z-eicosatetraenamide (Cat. No. 1339), 2-Arachidoylglycerol (2AG) or (5Z,8Z,11Z,14Z)-5,8,11,14-Eicosatetraenoic acid, 2-hydroxy-1-(hydroxymethyl)ethyl ester (Cat. No. 1298), and Rimonabant (SR-141716A) or (N-(Piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride) (Cat. No. 0923), (−) Cannabinol or 2-[(1R,6R)-3-Methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-5-pentyl-1,3-benzenediol (Cat. No. 1570), JWH133 or (6aR,10aR)-3-(1,1-Dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran (Cat. No. 1343/10). CP55940 or (5-(1,1-Dimethylheptyl)-2-[5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]phenol) (Cat. No. C1112), Nabilone (Cat. No. N3785), Tetrahydrocannabivarin (THCv) (Cat. No. T-094) and Δ9-Tetrahydrocannabinol solution (THC, Cat. No. T2386) was obtained from Merck. L759633 (Cat. No. CAY10009280–1 mg) was obtained from Cambridge Biosciences.
Cell culture
Cultured cells were maintained in a humidified incubator at 37 °C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich) containing 10% foetal bovine serum (FBS). Stable cell lines expressing both the CB1 plus venus-mGsi, and the CB1 plus venus-β-arrestin2, were created in T75 flasks, using a 3:1 ratio of polyethylenimine (PEI): DNA. 24 hours post transfection cells were split into T175 flasks and maintained as stable cell lines. Antibiotics including blasticidin (5 μg/mL) and Zeocin™ (20 Mg/mL) were introduced as selection agents to create stable cell lines expressing the pcDNA™4/TO a mammalian expression vector which encodes the appropriate CB1 sequence (ThermoFisher Scientific, UK). Whilst Geneticin® (G-418 sulphate, 200 μg/mL) was used to select for cells containing the pcDNA™ 3.1 mammalian expression vector (Invitrogen™), encoding venus-mGsi and venus-β-arrestin2. A mixed population of stable cells was eventually produced with cells having resistance to the selection agents employed.
Mini-G protein and β-arrestin recruitment assays in CB1-expressing cells.
CB1 coupling to G proteins was assessed using a fluorescent G protein surrogate, venus-mini-Gsi1 (vmGsi) protein72. The venus-mGsi subunit is essentially a chimeric protein consisting of C-terminal Gi1 residues grafted onto venus-mGs, and originally engineered from the native Gsa protein. The venus-mGsi is ideal for studying receptor activation of the CB1, as unlike its wild type Gai1 counterpart, the resulting active-receptor complex formed is stable and resistant to nucleotide exchange meaning that active state signalling is maintained if the agonist is present.
HEK293TR-CB1-nLuc cells expressing fluorescently labelled miniG or β-arrestin protein were maintained in a humidified environment at 37 °C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) containing blasticidin (5 mg/ml), Zeocin (20 mg/ml) and G418 (0.2mg/mL) and used to assess compound stimulated β-arrestin recruitment to the human CB1. Cultured cells were harvested upon reaching 70% confluency and plated at a seeding density of 50,000 cells per well in poly-D-lysine (5 mg/mL) coated clear-bottomed 96-well cell culture plates. The cells were grown for 48h until they reached confluency and then stimulated with (1 μg/mL tetracycline) for a further 48h. Media was aspirated, and the cells were washed in 100 μL/well PBS, then 90 μL/well assay buffer (HBSS, 0.5% BSA, 5 mM HEPES) with 10 μM furimazine was applied to each well. A white back seal was applied to plates, which was then incubated for 15 minutes at 37°C to allow the furimazine to enter the cells. Assay plates were then transferred to the PHERAstar FSX set to a temperature of 37°C, and three BRET cycles were run to collect an initial baseline reading, after which 10 μL of compounds diluted in assay buffer was added to the plate, and the plate was read at 1-minute intervals for 30 minutes. Compounds were serially diluted in DMSO, before a 1/10 dilution in assay buffer and a further 1/10 dilution on addition to the assay plate. Buffer containing 10% DMSO (1% final) served as the vehicle control with all responses normalised to the maximal response produced by 2AG. mini-G protein reversal experiments were performed essentially as described above, by applying a concentration of agonist producing 80% of its own maximum response (EC80) to cells in a 96-well cell culture plate, followed by the addition of an excess of rimonabant (10 mM) or vehicle, with the resulting BRET signals monitored for up to 90min.
Signal detection and data analysis
Raw experimental mGi and β-arrestin2 recruitment data was collected at 1 min intervals on the BMG PHERAstar FSX (BMG Labtech, Offenburg, Germany), and processed using MARS data analysis software (BMG Labtech), as the ratio of BRET 1 (535–30LP/475–30BP). This data was then exported in Microsoft Excel and transferred to GraphPad PRISM 9.2 (GraphPad Software, San Diego, U.S.A.). A kinetic analysis of compound induced response measured over time was completed by plotting the resulting reported BRET ratios. Characterisation of agonist CBR responses was achieved by selecting the concentration-response data at a fixed time point and one producing the maximal observable responses to the ligands under test. Concentration response data was then normalized to the reference ligand 2AG. Individual concentration response data were fitted to sigmoidal (variable slope) curves using a “four parameter logistic equation”:
Where Bottom and Top are the plateaus of the agonist and inverse agonist concentration response curves. LogEC50 is the concentration of agonist/inverse agonist that gives a half-maximal effect, and the Hillslope is the unitless slope factor. Individual agonist EC50 and Emax values are reported as the Mean ± SEM, from the number (n) of individual experiment indicated.
All normalized data of the individual concentration points from each individual experiments were pooled, and bias plots were constructed by means of a ‘centered second-order polynomial’ fitting of the normalized and pooled data obtained for the individual concentration points of the mini-Gi (x-coordinate) and b-arrestin2 (y-coordinate) assay formats.
The rates of mini-Gi protein reversal were estimated for each agonist in Prism 9.0 using the following equation which describes a ‘one phase exponential decay’:
Individual agonist koff values are reported in tables as the Mean ± SEM, from the number (n) of individual experiment indicated.
Compound synthesis
(6aR,9R,10aR)-9-(hydroxymethyl)-3-(8-isothiocyanato-2-methyloctan-2-yl)-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-1-ol (AM841). To a stirred solution of (6aR,9R,10aR)-3-(8-azido-2-methyloctan-2-yl)-9-(hydroxymethyl)-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-1-ol (56 mg, 0.13 mmol) in anhydrous tetrahydrofuran (2.6 mL) at room temperature under an argon atmosphere were added triphenylphosphine (170 mg, 0.65 mmol) followed by carbon disulfide (230 uL, 3.9 mmol) and the mixture was stirred at that temperature for 24 hours. Upon completion, the volatiles were evaporated under reduced pressure and the residue was purified by flash column chromatography (silica gel; 25% ethyl acetate in hexanes as eluent) to afford 49 mg of AM841. Physical, analytical, and spectroscopic data were identical to those we reported earlier16.
(6aR,10aR)-3-(8-bromo-2-methyloctan-2-yl)-6,6,9-trimethyl-6a,7,10,10a-tetrahydro-6H-benzo[c]chromen-1-ol (AM11542). To a mixture of 5-(8-bromo-2-methyloctan-2-yl)benzene-1,3-diol (124 mg, 0.39 mmol) and p-toluenesulfonic acid (14 mg, 0.08 mmol) in anhydrous methylene chloride (3 mL) at 0°C under an argon atmosphere was added a solution of (4R)-1-methyl-4-(prop-1-en-2-yl)cyclohex-2-en-1-ol (71 mg, 47 mmol) in anhydrous methylene chloride (1 mL) and the resulting mixture was stirred at that temperature for 40 minutes. Upon completion, the reaction was quenched by saturated aqueous sodium bicarbonate and the organic layer was dried over sodium sulphate and evaporated. The residue was purified by flash column chromatography (silica gel; 5% diethyl ether in hexanes) to afford 127 mg of (1’R,2’R)-4-(8-bromo-2-methyloctan-2-yl)-5’-methyl-2’-(prop-1-en-2-yl)-1’,2’,3’,6’-tetrahydro-[1,1’-biphenyl]-2,6-diol. The product (122 mg, 0.27 mmol) was dissolved in anhydrous methylene chloride (8 mL) under an argon atmosphere and cooled to 0°C. Boron trifluoride etherate (160 uL, 1.3 mmol) was added and the mixture was stirred for 30 minutes at 0°C followed by 6 hours at ambient temperature. The reaction was quenched by saturated aqueous sodium bicarbonate and the organic layer was dried over sodium sulphate and evaporated. The residue was purified by flash column chromatography (silica gel; 5% ethyl acetate in hexanes) to afford 105 mg of AM11542. Physical, analytical, and spectroscopic data were identical to those we reported earlier73.
References
- M. Maccarrone. Endocannabinoid signaling at the periphery: 50 years after THC.. Trends in pharmacological sciences, 2015. [DOI | PubMed]
- M. Maccarrone. Goods and Bads of the Endocannabinoid System as a Therapeutic Target: Lessons Learned after 30 Years.. Pharmacol. Rev., 2023. [DOI | PubMed]
- S. G. Schutz, A. Dunn, T. J. Braley, B. Pitt, A. V. Shelgikar. New frontiers in pharmacologic obstructive sleep apnea treatment: A narrative review.. Sleep Med. Rev., 2021. [DOI | PubMed]
- P. G. Fine, M. J. Rosenfeld. The endocannabinoid system, cannabinoids, and pain.. Rambam Maimonides Med, 2013. [DOI]
- S. Nielsen. The Use of Cannabis and Cannabinoids in Treating Symptoms of Multiple Sclerosis: a Systematic Review of Reviews.. Curr. Neurol. Neurosci. Rep., 2018. [DOI | PubMed]
- L. Cristino, T. Bisogno, V. Di Marzo. Cannabinoids and the expanded endocannabinoid system in neurological disorders.. Nat. Rev. Neurol., 2020. [DOI | PubMed]
- R. G. Pertwee. The pharmacology of cannabinoid receptors and their ligands: an overview.. Int. J. Obes. (Lond.), 2006. [DOI | PubMed]
- M. R. Elphick, M. Egertova. The neurobiology and evolution of cannabinoid signalling.. Philos. Trans. R. Soc. Lond. B Biol. Sci., 2001. [DOI | PubMed]
- M. Glass, C. C. Felder. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor.. J Neurosci, 1997. [PubMed]
- J. E. Lauckner, B. Hille, K. Mackie. The cannabinoid agonist WIN55,212–2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins.. Proceedings of the National Academy of Sciences of the United States of America, 2005. [DOI | PubMed]
- R. B. Laprairie, E. L. Stahl, L. M. Bohn. Approaches to Assess Biased Signaling at the CB1R Receptor.. Methods Enzymol, 2017. [DOI | PubMed]
- J. Caroli. A community Biased Signaling Atlas.. Nat. Chem. Biol., 2023. [DOI | PubMed]
- R. Al-Zoubi, P. Morales, P. H. Reggio. Structural Insights into CB1 Receptor Biased Signaling.. International journal of molecular sciences, 2019. [DOI]
- J. S. Smith, R. J. Lefkowitz, S. Rajagopal. Biased signalling: from simple switches to allosteric microprocessors.. Nat. Rev. Drug Discov., 2018. [DOI | PubMed]
- T. Kenakin, A. Christopoulos. Signalling bias in new drug discovery: detection, quantification and therapeutic impact.. Nat. Rev. Drug Discov., 2013. [DOI | PubMed]
- T. Hua. Crystal structures of agonist-bound human cannabinoid receptor CB(1).. Nature, 2017. [DOI | PubMed]
- T. Hua. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-G(i) Complex Structures.. Cell, 2020. [DOI | PubMed]
- X. Wang. A Genetically Encoded F-19 NMR Probe Reveals the Allosteric Modulation Mechanism of Cannabinoid Receptor 1.. J. Am. Chem. Soc., 2021. [DOI | PubMed]
- X. Yang. Molecular mechanism of allosteric modulation for the cannabinoid receptor CB1.. Nat. Chem. Biol., 2022. [DOI | PubMed]
- Z. Shao. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor.. Nat. Chem. Biol., 2019. [DOI | PubMed]
- X. Li. Structural basis of selective cannabinoid CB(2) receptor activation.. Nat Commun, 2023. [DOI | PubMed]
- A. Thomas. 6”-Azidohex-2”-yne-cannabidiol: a potential neutral, competitive cannabinoid CB1 receptor antagonist.. European journal of pharmacology, 2004. [DOI | PubMed]
- W. A. Devane. A novel probe for the cannabinoid receptor.. Journal of medicinal chemistry, 1992. [DOI | PubMed]
- E. Stern, D. M. Lambert. Medicinal chemistry endeavors around the phytocannabinoids.. Chemistry & biodiversity A,, 2007. [DOI]
- M. H. Rhee. Cannabinol derivatives: binding to cannabinoid receptors and inhibition of adenylylcyclase.. Journal of medicinal chemistry, 1997. [DOI | PubMed]
- R. A. Ross. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630.. Br JPharmacol, 1999. [DOI | PubMed]
- M. Soethoudt. Cannabinoid CB2 receptor ligand profiling reveals biased signalling and off-target activity.. Nature Communications, 2017. [DOI]
- A. Abioye. Delta9-Tetrahydrocannabivarin (THCV): a commentary on potential therapeutic benefit for the management of obesity and diabetes.. J Cannabis Res, 2020. [DOI | PubMed]
- A. D. Khanolkar, S. L. Palmer, A. Makriyannis. Molecular probes for the cannabinoid receptors.. Chem Phys Lipids, 2000. [DOI | PubMed]
- C. Prandi, M. Blangetti, D. Namdar, H. Koltai. Structure-Activity Relationship of Cannabis Derived Compounds for the Treatment of Neuronal Activity-Related Diseases.. Molecules, 2018. [DOI]
- E. W. Bow, J. M. Rimoldi. The Structure-Function Relationships of Classical Cannabinoids: CB1/CB2 Modulation.. Perspect. Medicin. Chem., 2016. [DOI | PubMed]
- B. R. Martin. Manipulation of the tetrahydrocannabinol side chain delineates agonists, partial agonists, and antagonists.. The Journal of pharmacology and experimental therapeutics, 1999. [PubMed]
- D. A. Andersson. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ(9)-tetrahydrocannabiorcol.. Nat Commun, 2011. [DOI | PubMed]
- R. K. Razdan. Structure-activity relationships in cannabinoids.. Pharmacol. Rev., 1986. [PubMed]
- A. Thomas. Evidence that the plant cannabinoid Delta9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist.. Br J Pharmacol, 2005. [DOI | PubMed]
- P. Liu. The structural basis of the dominant negative phenotype of the Gai1β1Y2 G203A/A326S heterotrimer.. Acta Pharmacol. Sin., 2016. [DOI | PubMed]
- Y. Kang. Cryo-EM structure of human rhodopsin bound to an inhibitory G protein.. Nature, 2018. [DOI | PubMed]
- K. Krishna Kumar. Structural basis for activation of CB1 by an endocannabinoid analog.. Nat Commun, 2023. [DOI | PubMed]
- K. Krishna Kumar. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex.. Cell, 2019. [DOI | PubMed]
- P. Kolb. Community guidelines for GPCR ligand bias: IUPHAR review 32.. Br J Pharmacol, 2022. [DOI | PubMed]
- R. P. Picone. (−)-7’-Isothiocyanato-11-hydroxy-1’,1’-dimethylheptylhexahydrocannabinol (AM841), a high-affinity electrophilic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor.. Molecular pharmacology, 2005. [DOI | PubMed]
- Y. Pei. Ligand-binding architecture of human CB2 cannabinoid receptor: evidence for receptor subtype-specific binding motif and modeling GPCR activation.. Chem Biol, 2008. [DOI | PubMed]
- D. W. Szymanski. Mass spectrometry-based proteomics of human cannabinoid receptor 2: covalent cysteine 6.47(257)-ligand interaction affording megagonist receptor activation.. J. Proteome Res., 2011. [DOI | PubMed]
- S. Jiang. Novel Functionalized Cannabinoid Receptor Probes: Development of Exceptionally Potent Agonists.. Journal of medicinal chemistry, 2021. [DOI | PubMed]
- L. Hruba, L. R. McMahon. The cannabinoid agonist HU-210: Pseudo-irreversible discriminative stimulus effects in rhesus monkeys.. Eur. J. Pharmacol., 2014. [DOI | PubMed]
- I. Rodriguez-Espigares. GPCRmd uncovers the dynamics of the 3D-GPCRome.. Nat. Methods, 2020. [DOI | PubMed]
- A. S. Powers. Structural basis of efficacy-driven ligand selectivity at GPCRs.. Nat. Chem. Biol., 2023. [DOI | PubMed]
- 48.AlphaFold (Google DeepMind, 2023).
- R. Krishna. Generalized Biomolecular Modeling and Design with RoseTTAFold All-Atom.. bioRxiv,, 2023. [DOI]
- A. S. Hauser. GPCR activation mechanisms across classes and macro/microscales.. Nat. Struct. Mol. Biol., 2021. [DOI | PubMed]
- E. Khajehali. Biased Agonism and Biased Allosteric Modulation at the CB1 Cannabinoid Receptor.. Molecular pharmacology, 2015. [DOI | PubMed]
- R. B. Laprairie, A. M. Bagher, M. E. M. Kelly, D. J. Dupre, E. M. Denovan-Wright. Type 1 Cannabinoid Receptor Ligands Display Functional Selectivity in a Cell Culture Model of Striatal Medium Spiny Projection Neurons*.. J. Biol. Chem., 2014. [DOI | PubMed]
- B. R. Martin. Behavioral, biochemical, and molecular modeling evaluations of cannabinoid analogs.. PharmacolBiochem Behav, 1991. [DOI]
- P. Liu. The structural basis of the dominant negative phenotype of the Galphai1beta1gamma2 G203A/A326S heterotrimer.. Acta Pharmacol Sin, 2016. [DOI | PubMed]
- S. Maeda. Development of an antibody fragment that stabilizes GPCR/G-protein complexes.. Nat Commun, 2018. [DOI | PubMed]
- A. Punjani, J. L. Rubinstein, D. J. Fleet, M. A. Brubaker. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination.. Nat. MethodsH,, 2017. [DOI]
- J. Zivanov. New tools for automated high-resolution cryo-EM structure determination in RELION-3.. Elife, 2018. [DOI]
- T. Wagner. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM.. Commun Biol, 2019. [DOI | PubMed]
- K. Ramlaul, C. M. Palmer, T. Nakane, C. H. S. Aylett. Mitigating local over-fitting during single particle reconstruction with SIDESPLITTER.. J. Struct. Biol., 2020. [DOI | PubMed]
- S. Maeda. Development of an antibody fragment that stabilizes GPCR/G-protein complexes.. Nature Communications, 2018. [DOI]
- P. Emsley, B. Lohkamp, W. G. Scott, K. Cowtan. Features and development of Coot.. Acta Crystallogr. D Biol. Crystallogr., 2010. [DOI | PubMed]
- D. Liebschner. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix.. Acta Crystallogr D Struct Biol, 2019. [DOI | PubMed]
- B. Webb, A. Sali. Protein Structure Modeling with MODELLER.. Methods Mol. Biol., 2021. [DOI | PubMed]
- G. Madhavi Sastry, M. Adzhigirey, T. Day, R. Annabhimoju, W. Sherman. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments.. Journal of computer-aided molecular design, 2013. [DOI | PubMed]
- K. Roos. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules.. J Chem. Theory Comput., 2019. [DOI | PubMed]
- 66.Small Molecule Drug Discovery Suite (Schrodinger LLC, 2023).
- R. C. Johnston. Epik: pKa and Protonation State Prediction through Machine Learning.. J Chem. Theory Comput., 2023. [DOI | PubMed]
- M. A. Lomize, I. D. Pogozheva, H. Joo, H. I. Mosberg, A. L. Lomize. OPM database and PPM web server: resources for positioning of proteins in membranes.. Nucleic Acids Res., 2012. [DOI | PubMed]
- 69.Maestro-Desmond Interoperability Tools (D. E. Shaw Research & Schrodinger, New York, NY, 2023).
- T. Darden, D. York, L. Pedersen. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems.. The Journal of Chemical Physics, 1993. [DOI]
- A. J. Venkatakrishnan. Uncovering patterns of atomic interactions in static and dynamic structures of proteins.. bioRxiv,, 2019. [DOI]
- Q. Wan. Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells.. J Biol. Chem., 2018. [DOI | PubMed]
- S. P. Nikas. The role of halogen substitution in classical cannabinoids: a CB1 pharmacophore model.. The AAPSjournal6,, 2004. [DOI]
- A. J. Kooistra, C. Munk, A. S. Hauser, D. E. Gloriam. An online GPCR structure analysis platform.. Nat. Struct. Mol. Biol., 2021. [DOI | PubMed]
- V. Isberg. Generic GPCR residue numbers – aligning topology maps while minding the gaps.. Trends in pharmacological sciences, 2015. [DOI | PubMed]