
The Leu/Val6.51 Side Chain of Cannabinoid Receptors Regulates the Binding Mode of the Alkyl Chain of Δ9-Tetrahydrocannabinol
Abstract
(−)-Δ9–trans-tetrahydrocannabinol (THC), which is the principal psychoactive constituent of Cannabis, mediates its action by binding to two members of the G-protein-coupled receptor (GPCR) family: the cannabinoid CB1 (CB1R) and CB2 (CB2R) receptors. Molecular dynamics simulations showed that the pentyl chain of THC could adopts an I-shape conformation, filling an intracellular cavity between Phe3.36 and Trp6.48 for initial agonist-induced receptor activation, in CB1R but not in CB2R. This cavity opens to the five-carbon chain of THC by the conformational change of the γ-branched, flexible, Leu6.51 side chain of CB1R, which is not feasible by the β-branched, mode rigid, Val6.51 side chain of CB2R. In agreement with our computational results, THC could not decrease the forskolin-induced cAMP levels in cells expressing mutant CB1RL6.51V receptor but could activate the mutant CB2RV6.51L receptor as efficiently as wild-type CB1R. Additionally, JWH-133, a full CB2R agonist, contains a branched dimethyl moiety in the ligand chain that bridges Phe3.36 and Val6.51 for receptor activation. In this case, the substitution of Val6.51 to Leu in CB2R makes JWH-133 unable to activate CB2RV6.51L. In conclusion, our combined computational and experimental results have shown that the amino acid at position 6.51 is a key additional player in the initial mechanism of activation of GPCRs that recognize signaling molecules derived from lipid species.
Affiliations: †Laboratory of Computational Medicine, Biostatistics Unit, Faculty of Medicine, Universitat Autònoma Barcelona, 08193 Bellaterra, Barcelona, Spain; ‡Department of Biochemistry and Molecular Biomedicine, School of Biology, University of Barcelona, 08028 Barcelona, Spain; §Centro de Investigación en Red, Enfermedades Neurodegenerativas (CIBERNED), Instituto de Salud Carlos III, 28031 Madrid, Spain; ∥Department of Biochemistry and Physiology, Faculty of Pharmacy and Food Sciences, Universitat de Barcelona, 08028 Barcelona, Spain; ⊥Phytoplant Research S.L.U., 14014 Córdoba, Spain; #Institute of Neuroscience, University of Barcelona (NeuroUB), Av Joan XXIII 27-31, 08028 Barcelona, Spain
License: © 2023 The Authors. Published by American Chemical Society CC BY 4.0 Permits the broadest form of re-use including for commercial purposes, provided that author attribution and integrity are maintained (https://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.1021/acs.jcim.3c01054 | PubMed: 37644761 | PMC: PMC10523433
Relevance: Relevant: mentioned in keywords or abstract
Full text: PDF (4.9 MB)
Introduction
Cannabinoids are naturally occurring compounds found in the Cannabis sativa plant (more commonly known as marijuana). There are over 180 cannabinoids out of the 1600 chemical compounds that have been isolated from Cannabis, with a characteristic oxygen containing C21 aromatic hydrocarbons.1 These exogenous cannabinoids can be further classified into 11 subclasses: cannabichromene (CBC), cannabidiol (CBD), cannabielsoin (CBE), cannabigerol (CBG), cannabicyclol (CBL), cannabinol (CBN), cannabinodiol (CBND), cannabitriol (CBT), (−)-Δ8-trans-tetrahydrocannabinol (Δ8-THC), (−)-Δ9-trans-tetrahydrocannabinol (Δ9-THC), and miscellaneous-type cannabinoids.2 The Δ9-THC subclass contains 25 compounds with common structural features such as a dibenzopyran ring and a hydrophobic alkyl chain. This class includes the most abundant phytocannabinoids: (−)-Δ9–trans-tetrahydrocannabinol (THC), which is the principal psychoactive constituent of Cannabis, and (−)-Δ9-trans-tetrahydrocannabivarin (THCV), which is homologous to THC but has a 3-carbon (propyl chain) instead of a 5-carbon (pentyl chain) in the alkyl chain (Figure ). THCV lacks the psychoactive effects of THC and upregulates energy metabolism, converting it a clinically useful remedy for weight loss, obesity management, and type 2 diabetic patients.3,4 In addition, THCV can produce beneficial antipsychotic effects.5 Endogenous cannabinoids are N-arachidonylethanolamide (anandamide) and 2-arachidonoylglycerol (2-AG) that possess long hydrophobic moieties.6
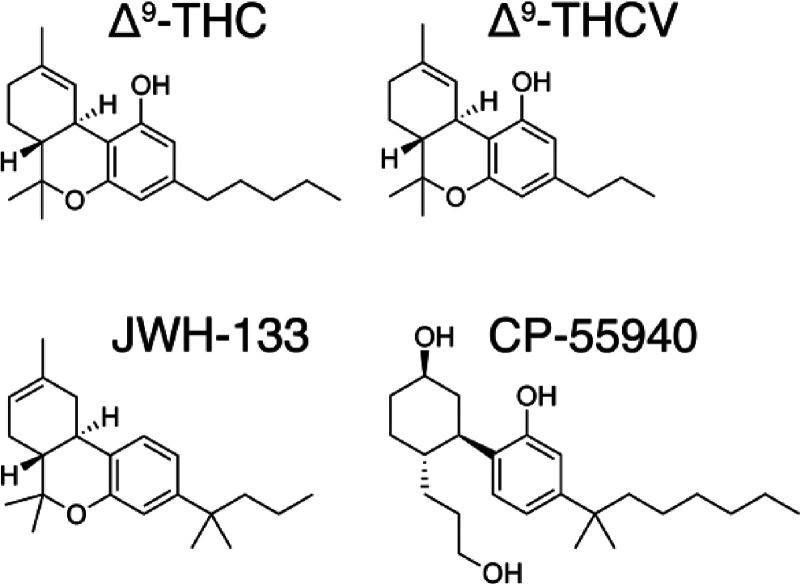
The effects of cannabinoids are primarily mediated through two members of the G-protein-coupled receptor (GPCR) family,7 the cannabinoid CB1 (CB1R) and CB2 (CB2R) receptors. CB1R is one of the most abundant GPCRs in the central nervous system, whereas CB2R is mainly expressed in the immune system.8 However, other molecular targets for certain cannabinoids, aside from CB1R and CB2R, have also been identified.9 Some authors propose that THCV is a CB1R and CB2R antagonist,10 whereas others suggest that THC and THCV are partial agonists at both receptors.11 We have recently shown that THC acts as a partial agonist in CB1R and as an antagonist in CB2R, whereas THCV acts as an antagonist on both receptors.12
Here, we have used the recently released structures of CB1R13−15 and CB2R16−18 in their inactive and active, Gi-bound, conformations to delineate the individual signaling contributions of THC and THCV to modulate both receptors. Phe3.36 and Trp6.48 have been described as conformational toggle or trigger switches involved in the initial agonist-induced receptor activation in CB1R,15,19 CB2R16 and other GPCRs.20−22 However, these amino acids are conserved in CB1R and CB2R (44% sequence identity between receptors), thus they cannot explain the different pharmacological profile of THC and THCV. In this manuscript, a combination of molecular dynamics (MD) simulations and site-directed mutagenesis have permitted to propose residue at position 6.51, which is Leu at CB1R and Val at CB2R, as an additional player capable to selectively recognize the alkyl chain of these ligands, further supporting the yin-yang functional relationship already described for CB1 and CB2 receptors.16 This knowledge could be of great use to facilitate the future design of selective drugs in the endocannabinoid system.
Materials and Methods
Initial CB1R and CB2R Models
The CB1R-AM841-Gi (PDB id 6KPG) and CB2R-AM12033-Gi (6KPF) cryo-EM structures18 were used in docking studies and MD simulations. Missing residues 55–180 of αi in the CB1R-AM841-Gi structure were built from the structure of Gi (6CRK);23 and missing residues 55–181 and 233–239 of αi in the CB2R-AM12033-Gi structure were built from the CB2R-WIN55,212–2-Gi structure (6PT0),17 using AutoModel class24 of MODELER v10.1.25 Protonation states were assigned with the PDB 2PQR tool26 using PROPKA to predict the pKa values of ionizable groups in the proteins at pH 6.5.27 Disulfide bonds between cysteines were built using the tleap module of Ambertools19. Internal water molecules were added to CB1R and CB2R using HomolWat.28 THC and THCV were docked into the orthosteric binding cavity of CB1R and CB2R and JWH-133 into CB2R by using AM841 in 6KPG and AM12033 in 6KPF structures as a reference. Thus, the alkyl chains of THC, THCV, and JWH-133 were initially modeled in the L-shape conformation above Trp2795.43 and Trp1945.43 of CB1R and CB2R, respectively, as observed in the cryo-EM structures. These systems were oriented by the Orientations of Proteins in Membranes (OPM) database,29 and embedded in a lipid bilayer box, constructed using PACKMOL-memgen,30 containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), cholesterol (CHL) (10:1 POPC:CHL ratio), water molecules (TIP3P), and monatomic Na+ and Cl– ions (0.15 M). The resulting systems comprise between 225 and 250k atoms in a box of ∼120 Å × 120 Å × 140 Å (see the Supporting Information in the Zenodo repository for detailed values).
Molecular Dynamics Simulations
MD simulations of these models were performed with GROMACS2018.5.31 The amber14sb-ildn force field was used for the protein, solvent, and ions,32 a GROMACS adaptation of lipid14 for lipids,33 and the general Amber force field (GAFF2) with HF/6-31G*-derived RESP atomic charges for THC, THCV, and JWH-133.34 Molecular systems were subjected to 5000 steps of energy minimization, using the steepest descent algorithm, PME electrostatics, with the Verlet cutoff scheme. This was followed by a 25 ns equilibration protocol consisting of six steps, in which positional restraints are progressively removed, from all heavy atoms to only helix Cα carbons being restricted, meanwhile gradually reducing the applied forces, from 1000 kJ mol–1 nm–2 to 0 kJ mol–1 nm–2. After equilibration, three replicas of a 1 μs unrestrained MD trajectory were generated at a constant temperature of 300 K using separate v-rescale thermostats for the receptor, ligand, lipids, and solvent molecules. Initial velocities were randomly generated for each replica from a Maxwell distribution, using different random seeds. A time step of 2.0 fs was used for the integration of equations of motions using the leapfrog algorithm. Bonds involving hydrogen atoms were kept frozen by using the LINCS algorithm. Lennard-Jones interactions were computed using a cutoff of 1.1 nm under the Verlet cutoff scheme for neighbor searching, and the electrostatic interactions were treated using PME with the same real-space cutoff under periodic boundary conditions. Center of mass motion was removed from all systems. The Berendsen pressure control algorithm was chosen for equilibration and Parrinello–Rahman for production MDs. For complete details, see the Supporting Information in the Zenodo repository.
MD Analysis and Data Visualization
The analysis of the trajectories was performed using MDAnalysis;35 visualization and image rendering were performed with PyMOL36 and VMD,37 and graphical representations were obtained with the Seaborn Package.38
CB1R and CB2R Mutants
Mutations were produced using the QuikChange Site-Directed Mutagenesis Kit. The cDNA for hCB1R and hCB2R, cloned into pcDNA3.1, was amplified using sense and antisense primers harboring the triplets for the desired point mutation (Pfu turbo polymerase was used). The nonmutated DNA template was digested for 1 h with DpnI. PCR products were used to transform XL1-blue supercompetent cells. Finally, positive colonies were tested by sequencing to select those expressing the correct DNA sequence.
cAMP Determination Assays
Determination of cAMP levels in HEK-293T cells transiently expressing CB1R or CB2R (1 μg of cDNA) or the mutant version of the receptor was performed by using the Lance-Ultra cAMP kit (PerkinElmer). Two hours before initiating the experiment, the medium was substituted by a serum-free medium. Then, transfected cells were dispensed in white 384-well microplates at a density of 4000 cells per well and incubated for 15 min at room temperature with compounds, followed by 15 min incubation with forskolin, and 1 h more with homogeneous time-resolved fluorescence (HTRF) assay reagents. Fluorescence at 665 nm was analyzed on a PHERAstar Flagship microplate reader equipped with an HTRF optical module (BMG Labtech). Data analysis was made based on the fluorescence ratio emitted by the labeled cAMP probe (665 nm) over the light emitted by the europium cryptate-labeled anti-cAMP antibody (620 nm). A standard curve was used to calculate cAMP concentration. Forskolin-stimulated cAMP levels were normalized to 100%.
Pure Cannabinoids
Δ9-THC and Δ9-THCV substances were provided by Phytoplant Research S.L.U. Δ9-THC and Δ9-THCV were purified from the Moniek (CPVO/20160114) and Raquel (CPVO/20180114) varieties, respectively, using countercurrent chromatography as previously described.39 The purity of both cannabinoids was set at >95%.
Results
THC Adopts Two Distinct Binding Modes in CB1R But Not in CB2R
To understand the different molecular signatures of THC and THCV, at CB1R and CB2R, we first performed three replicate runs of unbiased 1 μs MD simulations of these compounds bound to the CB1R-Gi and CB2R-Gi complexes (see the Methods section). We have used Gi-bound active states, instead of inactive structures, despite its higher computational cost, because agonists alone are not capable to stabilize the fully active conformation in the absence of the G protein, as shown by NMR experiments.40 Similarly, MD simulations of agonist bound to the inactive state of the receptor are not capable of reaching active-like conformations in the absence of the G protein. Moreover, MD simulations of active, G protein-bound, conformations have permitted to identify additional cavities to accommodate hydrophobic chains of ligands in sphingosine-1-phosphate41 and muscarinic42 receptors, which were not identified in similar simulations of inactive structures.
THC and THCV were docked into these structures with the hydrophobic alkyl tail in the L-shape conformation, as observed in the cryo-EM structures of structurally similar ligands (see the Methods section). Root-mean-square deviations (rmsd) of the ligand heavy atoms show that THC visited during the MD simulations two different poses in the binding pocket of CB1R but not in CB2R (Figure c). In CB1R, THC adopts the initial L-shape conformation, in which the hydrophobic alkyl tail occupies a cavity between TMs 3 and 5 (Figure a), and an I-shape conformation, in which the alkyl tail occupies an intracellular cavity between TMs 3 and 6 (Figure b). The structure–function of the alkyl chain of THC has been reviewed,43 and this dual orientation is consistent with previous studies by others.15,44 The different conformation of the pentyl chain of THC is achieved by a change of a single dihedral angle in the chain, from – anticlinal (dihedral 1–2–3–4 around −90°) in the L-shape to + anticlinal (around 90°) in the I-shape (Figure g). Heatmaps showing when THC adopts the – anticlinal (dihedral <0°) or + anticlinal (>0°) conformation correlates with large (I-shape) and small (L-shape) rmsd values, respectively (Figures c and 2e). Notably, the pentyl chain of THC in CB2R rarely adopts the + anticlinal conformation (Figure e), thus no large rmsd values could be observed from the initial L-shape conformation during the MD simulations (Figure c). The shorter 3-carbon propyl chain of THCV has fewer steric constraints and can visit the + anticlinal conformation in both CB1R and CB2R simulations (Figures f–h).
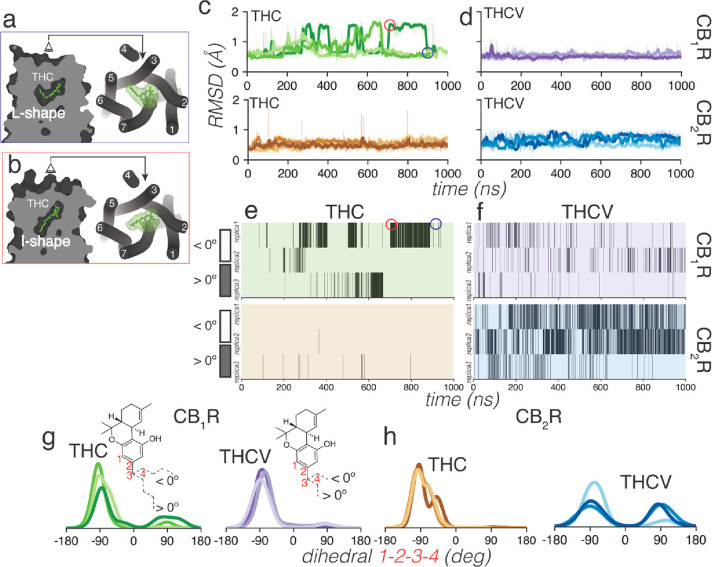
It is not clear why the five-carbon pentyl chain of THC can also adopt the I-shape conformation, in which the alkyl tail occupies an intracellular cavity between TMs 3 and 6, in CB1R but not in CB2R. This intracellular cavity is delineated by the amino acid at position 6.51 (Figure a), which is Leu at CB1R and Val at CB2R (Figure b). The probability to undergo a side chain conformational change in Val is smaller than in Leu,45 due to the β-branched side chain that is shorter than the γ-branched side chain of Leu. Val is generally found with the γ-carbons flanking the small Hα in the trans (t, χ1 = 180°) rotamer conformation, whereas Leu can adopt the more stable trans (t, χ1 = 180°) and less stable gauche+ (g+, χ1 = −60°) rotamer conformations, as observed in the dynameomics rotamer library.46Figure c shows the histogram distributions of the χ1 dihedral angle of Leu6.51 along the MD simulations of CB1R. These panels illustrate that the three-carbon propyl chain of THCV maintains Leu6.51 in the more stable t conformation during the simulation time, whereas the five-carbon pentyl chain of THC in the I-shape conformation triggers or stabilizes the g+ conformation of Leu6.51 in CB1R, opening the access to the intracellular cavity between TMs 3 and 6 (Figure d). In contrast, the conformation of the bulky, β-branched, and more rigid side chain of Val6.51 in CB2R cannot be modified by the pentyl chain of THC (not shown), closing the access to the intracellular cavity.
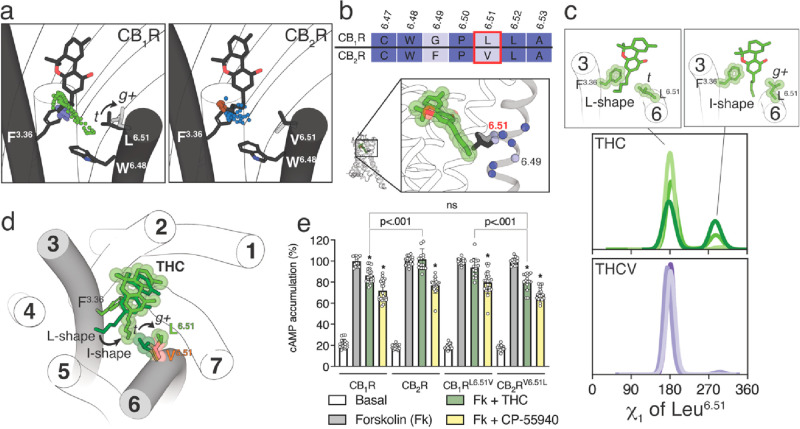
The CB1RL6.51V and CB2RV6.51L Mutations Reverse the Pharmacology of THC
To experimentally validate the proposed different conformations of THC in CB1R and CB2R, we mutated Leu6.51 to Val in CB1R (CB1RL6.51V) and Val6.51 to Leu in CB2R (CB2RV6.51L), and we measured cAMP production in HEK-293T cells (Figure e). The nonselective CP-55940 agonist (100 nM) decreased, as expected for a Gi-coupled receptor, cAMP formation induced by forskolin (500 nM), in a statistically significant manner, in wild-type CB1R and CB2R and mutant CB1RL6.51V and CB2RV6.51L. In contrast, THC (10 μM) can significantly decrease forskolin-induced cAMP accumulation in CB1R but not in CB2R. We used high concentrations of THC to evaluate the greatest attainable response (ceiling effect). These results suggest that, in cAMP measurements, THC acts as a weak partial agonist only in CB1R. Remarkably, the pharmacological profile of THC changes in the mutant receptors. THC can significantly decrease forskolin-induced cAMP accumulation in CB2RV6.51L but not in CB1RL6.51V. Moreover, cAMP accumulation induced by THC is not statistically different between CB1R and CB2RV6.51L. These experimental results, together with computational simulations, suggest that the residue at position 6.51, which is Leu at CB1R and Val at CB2R, is an additional element in the mechanism of receptor activation (see the Discussion section).
JWH-133 Activates CB2R via the Substituted Methyl Groups
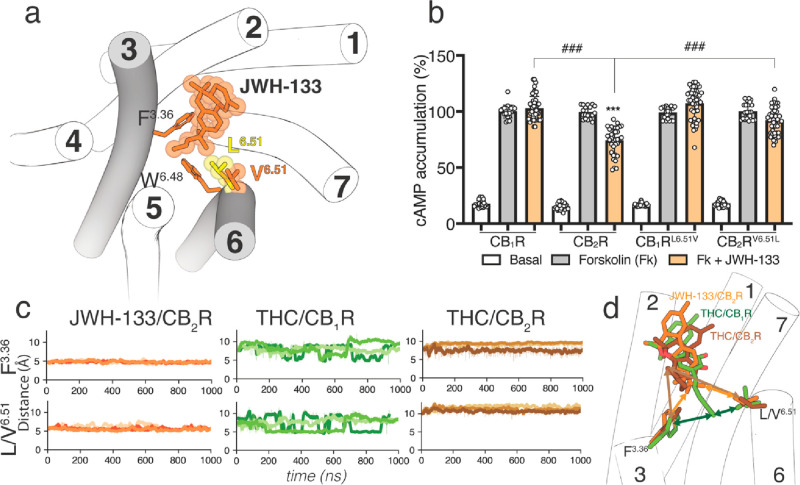
JWH-133 is a potent CB2R agonist, with little affinity for CB1R.47 The structure of JWH-133 is like THC and THCV with a 4-carbon butyl chain, instead of the 3-carbon propyl chain of THCV or the 5-carbon pentyl chain of THC (Figure ). A significant difference between JWH-133 and either THC or THCV is the methyl substitutions on the chain (Figure ). Branching close to the aromatic ring might restrict the dimethylbutyl chain conformation of JWH-133. Thus, it seems reasonable to study the molecular properties of JWH-133, as a full agonist, in complex with CB2R-Gi to challenge our proposed molecular models of THC and THCV (see section 3.1). Consequently, we performed simulations similar to those with THC and THCV to evaluate the binding mode of JWH-133 in CB2R (see the Methods section). The alkyl chain of JWH-133 always adopts the L-shape conformation in the + anticlinal conformation, filling the cavity between TMs 3 and 5 (Figure a). The dimethyl moiety of the dimethylbutyl chain mediates hydrophobic interactions with Phe3.36 and Val6.51, during the simulation time (Figure c). To experimentally validate the key role of Val6.51 in JWH-133-induced CB2R activation, we measured the level of production of cAMP in CB1RL6.51V and CB2RV6.51L mutant receptors expressed in HEK-293T cells (Figure b). JWH-133 (100 nM) was unable to decrease forskolin-induced cAMP formation in CB1R but was statistically significantly lower in CB2R, as expected for a CB2R selective agonist. Substitution of Leu6.51 with Val in CB1R does not facilitate activation of CB1RL6.51V by JWH-133. However, substitution of the single Val6.51 amino acid with Leu in CB2R makes JWH-133 unable to activate CB2RV6.51L. This points to both the bulky, β-branched, and rigid Val6.51 in CB2R and the bulky, branched dimethyl group of the dimethylbutyl chain of JWH-133 as key elements for CB2R activation (see the Discussion section). It was recently shown that the CB2RV6.51L mutation also impeded HU308 and CP-55940, both containing the branched dimethyl group in the alkyl chain, to activate the G protein at CB2R.48
Discussion and Conclusions
Among the ∼350 GPCRs for nonsensory functions, ∼35 are activated by hormone-like signaling molecules derived from lipid species with long hydrophobic chains.6,49 Some of these receptors possess distinctive structural signatures relative to other class A GPCRs such as the N-terminus and ECL-2 folding over the binding site,50,51 which causes the entry of the ligand to the orthosteric site through a tunnel formed between TMs 1 and 7;52,53 or lacking the highly conserved Pro5.50, part of the PIF motif that transmits the signal from the orthosteric ligand binding site to the G protein binding site.54,55 In PIF-containing GPCRs, the interaction of agonists with TM 5 triggers an inward movement of TM 5 at P5.50, a rotation of TM 3 at I3.40, and an outward movement of TM 6 at F6.44.56,57 In GPCRs lacking P5.50, agonists can alter the rotamer of the amino acid at position 3.3620 to trigger the rotation of TM 3 at I3.40 and outward movement of TM 6 at F6.44.41 For instance, in the active crystal structure of S1P3 bound to the endogenous agonist sphingosine-1-phosphate (S1P),58 the long hydrophobic side chain of d18:1 S1P binds in an extended conformation (I-shape) between TMs 4 and 5 (Figure a). S1P triggers conformational changes of Leu1223.36 from t to g+, among others (see quartet core in58), to accommodate the alkyl chain. Similar results were observed in S1P bound to S1P159 and S1P260 (Figure a). In the cryo-EM structure of active LPA1 bound to the lysophosphatidic acid (LPA),61 the alkyl chain of LPA cannot extend to the cleft between TMs 4 and 5 as S1P, due to the presence of the bulky Trp2105.43 in LPA1 (S1P1–3 possess the less bulky Cys2065.43, Val1945.43, or Cys2005.43), blocking the access. LPA adopts an U-shaped conformation bending backward and triggering the g+ conformation of Leu1323.36 (Figure b). The long acyl chain of the ONO-0740556 agonist, a more rigid and potent LPA analog, binds LPA1 in a different bent conformation than LPA62 (Figure c). In this case, the aromatic ring of ONO-0740556 triggers the g+ conformation of Leu1323.36. The lack of side chain in Gly2746.51 (LPA1–3 possess Gly at this key position) permits LPA1 to have a small pocket in front of Leu1323.36 encaging the terminus of LPA or the phenyl ring of ONO-0740556.61,62
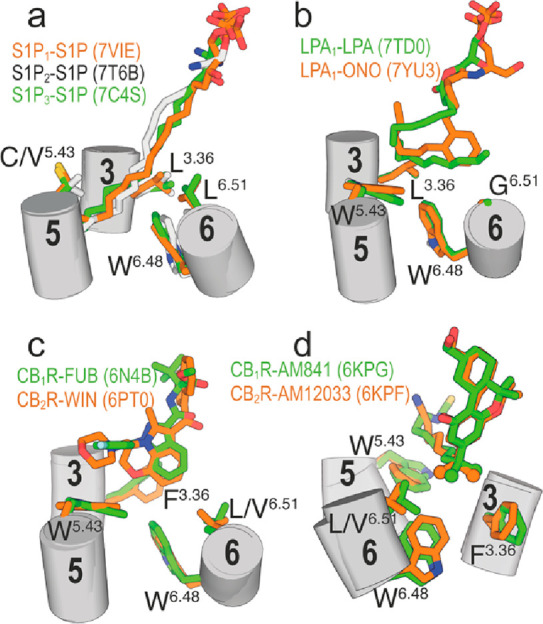
CB1R and CB2R possess Trp2795.43 and Trp1945.43, respectively, thus blocking the TMs 4 and 5 cleft created in S1P binding to S1P1–3; and contain Phe2003.36 and Phe1173.36, respectively, as conformational toggle or trigger switch involved in the initial agonist-induced receptor activation. Notably, the indazole ring of MDMB-Fubinaca triggers the active g+ conformation of Phe2003.36 in CB1R by an aromatic–aromatic interaction (Figure c),15 and the aromatic core of WIN 55,212–2 also forms aromatic–aromatic interactions with Phe1173.36 in g+ of CB2R (Figure c).17 In other known structures of active CB1R and CB2R bound to agonists, the bulky and branched dimethyl groups of the alkyl chain of AM841 and AM12033 bridge Phe2003.36 in g+ and Leu3596.51 of CB1R and Phe1173.36 in g+ and Val2616.51 of CB2R (Figure d), respectively.18 The conformational change in Phe3.36, from pointing toward TM 6 in t to pointing toward TM 7 in g+, permits Trp6.48 to move toward TM 5 for receptor activation.
These data indicate that the hydrophobic alkyl chain of the signaling molecule is key in the process of ligand-induced receptor activation. Thus, in this paper, we have studied the conformation of the alkyl chain of THC, THCV, and JWH-133 bound to CB1R and CB2R and calculated their distances to Phe3.36 along the MD simulations. Among them, the distances between the terminal methyl group of the alkyl chain of THC and the centroid of the aromatic ring of Phe3.36 and either the δ- or γ- carbon of Leu/Val6.51 are important to highlight (see Figures c and 4d). They fluctuated from >5 Å to <5 Å in CB1R and are always >5 Å in CB2R, indicating a dual orientation of the alkyl chain in CB1R, which either occupies a cavity above Trp5.43 (between TMs 3 and 5) in an L-shape conformation, or an intracellular cavity between Phe3.36 and Trp6.48 (TMs 3 and 6) in an I-shape conformation (Figure d). The main achievement of this work is the discovery that THC in CB1R, but not in CB2R, can adopt this I-shape conformation. The intracellular cavity between Phe3.36 and Trp6.48 is also delineated by the amino acid at position 6.51, which is the γ-branched, flexible Leu side chain in CB1R and the β-branched, model rigid Val side chain in CB2R (Figure b). We have shown that the five-carbon pentyl chain of THC can trigger the conformational change of Leu6.51 from t, blocking the access of the chain to the intracellular cavity, to g+, opening the access (Figures c and 3d). This opening of the chain access to the intracellular cavity is not feasible with the rigid Val6.51 side chain of CB2R. The binding mode of THC in the I-shape conformation positions the alkyl chain between Phe3.36 in the active g+ conformation and Trp6.48 that is involved in the initial mechanism of agonist-induced receptor activation. Thus, these computational results are compatible with our experiments, showing that THC acts as a partial agonist in CB1R and as an antagonist in CB2R (Figure e).12 In agreement with our computational results, THC could not activate the mutant CB1RL6.51V receptor and activated the mutant CB2RV6.51L receptor as efficiently as wild type CB1R (Figure e). We have recently shown that the alkyl chain of cannabidiol, in the allosteric binding mode, also expands toward the intracellular cavity modulating the conformation of Phe3.36.63
The predicted binding mode of the dimethylbutyl chain conformation of the full agonist JWH-133 at CB2R is always in the L-shape conformation, filling the cavity between TMs 3 and 5 (Figure a). However, the branched dimethyl moiety of the ligand chain mediates hydrophobic interactions with Phe3.36 in the active g+ conformation and Val6.51 (Figure c). Notably, substitution of Val6.51 for Leu in CB2R makes JWH-133 unable to activate CB2RV6.51L (Figure b). This supports the concept that the branched dimethylbutyl chain conformation of JWH-133 needs a foothold on the rigid Val6.51 to move Phe3.36 to the active g+ conformation for receptor activation.
In conclusion, our findings have shown that, in cannabinoid receptors and probably other receptors that recognize signaling molecules derived from lipid species with long hydrophobic chains, the amino acid at position 6.51 defines the size and shape of the cavity near Phe3.36 and Trp6.48 and is a key additional player in the mechanism of activation of this type of GPCRs.
References
- M. N. Tahir, F. Shahbazi, S. Rondeau-Gagne, J. F. Trant. The biosynthesis of the cannabinoids.. J. Cannabis Res., 2021. [DOI | PubMed]
- M. M. Radwan, S. Chandra, S. Gul, M. A. ElSohly. Cannabinoids, Phenolics, Terpenes and Alkaloids of Cannabis.. Molecules, 2021. [DOI | PubMed]
- K. A. Jadoon, S. H. Ratcliffe, D. A. Barrett, E. L. Thomas, C. Stott, J. D. Bell, S. E. O’Sullivan, G. D. Tan. Efficacy and Safety of Cannabidiol and Tetrahydrocannabivarin on Glycemic and Lipid Parameters in Patients With Type 2 Diabetes: A Randomized, Double-Blind, Placebo-Controlled, Parallel Group Pilot Study.. Diabetes Care, 2016. [DOI | PubMed]
- A. Abioye, O. Ayodele, A. Marinkovic, R. Patidar, A. Akinwekomi, A. Sanyaolu. Delta9-Tetrahydrocannabivarin (THCV): a commentary on potential therapeutic benefit for the management of obesity and diabetes.. J. Cannabis Res., 2020. [DOI | PubMed]
- M. G. Cascio, E. Zamberletti, P. Marini, D. Parolaro, R. G. Pertwee. The phytocannabinoid, Delta(9)-tetrahydrocannabivarin, can act through 5-HT(1)A receptors to produce antipsychotic effects.. Br. J. Pharmacol., 2015. [DOI | PubMed]
- R. N. V. Krishna Deepak, R. K. Verma, Y. D. Hartono, W. S. Yew, H. Fan. Recent Advances in Structure, Function, and Pharmacology of Class A Lipid GPCRs: Opportunities and Challenges for Drug Discovery.. Pharmaceuticals (Basel), 2022. [DOI]
- S. Ferre, F. Ciruela, C. W. Dessauer, J. Gonzalez-Maeso, T. E. Hebert, R. Jockers, D. E. Logothetis, L. Pardo. G protein-coupled receptor-effector macromolecular membrane assemblies (GEMMAs).. Pharmacol. Ther., 2022. [DOI | PubMed]
- L. H. Parsons, Y. L. Hurd. Endocannabinoid signalling in reward and addiction.. Nat. Rev. Neurosci., 2015. [DOI | PubMed]
- P. Morales, D. P. Hurst, P. H. Reggio. Molecular Targets of the Phytocannabinoids: A Complex Picture.. Prog. Chem. Org. Nat. Prod., 2017. [DOI | PubMed]
- A. Thomas, L. A. Stevenson, K. N. Wease, M. R. Price, G. Baillie, R. A. Ross, R. G. Pertwee. Evidence that the plant cannabinoid Delta9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist.. Br. J. Pharmacol., 2005. [DOI | PubMed]
- A. Zagzoog, K. A. Mohamed, H. J. J. Kim, E. D. Kim, C. S. Frank, T. Black, P. D. Jadhav, L. A. Holbrook, R. B. Laprairie. In vitro and in vivo pharmacological activity of minor cannabinoids isolated from Cannabis sativa.. Sci. Rep., 2020. [DOI | PubMed]
- I. Raich, R. Rivas-Santisteban, A. Lillo, J. Lillo, I. Reyes-Resina, X. Nadal, C. Ferreiro-Vera, V. S. de Medina, M. Majellaro, E. Sotelo, G. Navarro, R. Franco. Similarities and differences upon binding of naturally occurring Delta(9)-tetrahydrocannabinol-derivatives to cannabinoid CB1 and CB2 receptors.. Pharmacol. Res., 2021. [DOI | PubMed]
- Z. Shao, J. Yin, K. Chapman, M. Grzemska, L. Clark, J. Wang, D. M. Rosenbaum. High-resolution crystal structure of the human CB1 cannabinoid receptor.. Nature, 2016. [DOI | PubMed]
- T. Hua, K. Vemuri, M. Pu, L. Qu, G. W. Han, Y. Wu, S. Zhao, W. Shui, S. Li, A. Korde, R. B. Laprairie, E. L. Stahl, J. H. Ho, N. Zvonok, H. Zhou, I. Kufareva, B. Wu, Q. Zhao, M. A. Hanson, L. M. Bohn, A. Makriyannis, R. C. Stevens, Z. J. Liu. Crystal Structure of the Human Cannabinoid Receptor CB1.. Cell, 2016. [DOI | PubMed]
- K. Krishna Kumar, M. Shalev-Benami, M. J. Robertson, H. Hu, S. D. Banister, S. A. Hollingsworth, N. R. Latorraca, H. E. Kato, D. Hilger, S. Maeda, W. I. Weis, D. L. Farrens, R. O. Dror, S. V. Malhotra, B. K. Kobilka, G. Skiniotis. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex.. Cell, 2019. [DOI | PubMed]
- X. Li, T. Hua, K. Vemuri, J. H. Ho, Y. Wu, L. Wu, P. Popov, O. Benchama, N. Zvonok, K. Locke, L. Qu, G. W. Han, M. R. Iyer, R. Cinar, N. J. Coffey, J. Wang, M. Wu, V. Katritch, S. Zhao, G. Kunos, L. M. Bohn, A. Makriyannis, R. C. Stevens, Z. J. Liu. Crystal Structure of the Human Cannabinoid Receptor CB2.. Cell, 2019. [DOI | PubMed]
- C. Xing, Y. Zhuang, T. H. Xu, Z. Feng, X. E. Zhou, M. Chen, L. Wang, X. Meng, Y. Xue, J. Wang, H. Liu, T. F. McGuire, G. Zhao, K. Melcher, C. Zhang, H. E. Xu, X. Q. Xie. Cryo-EM Structure of the Human Cannabinoid Receptor CB2-Gi Signaling Complex.. Cell, 2020. [DOI | PubMed]
- T. Hua, X. Li, L. Wu, C. Iliopoulos-Tsoutsouvas, Y. Wang, M. Wu, L. Shen, C. A. Johnston, S. P. Nikas, F. Song. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures.. Cell, 2020. [DOI | PubMed]
- S. D. McAllister, D. P. Hurst, J. Barnett-Norris, D. Lynch, P. H. Reggio, M. E. Abood. Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: the importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation.. J. Biol. Chem., 2004. [DOI | PubMed]
- L. P. Pellissier, J. Sallander, M. Campillo, F. Gaven, E. Queffeulou, M. Pillot, A. Dumuis, S. Claeysen, J. Bockaert, L. Pardo. Conformational toggle switches implicated in basal constitutive and agonist-induced activated states of 5-hydroxytryptamine-4 receptors.. Mol. Pharmacol., 2009. [DOI | PubMed]
- G. Navarro, A. Gonzalez, S. Campanacci, R. Rivas-Santisteban, I. Reyes-Resina, N. Casajuana-Martin, A. Cordomi, L. Pardo, R. Franco. Experimental and computational analysis of biased agonism on full-length and a C-terminally truncated adenosine A2A receptor.. Comput. Struct. Biotechnol. J., 2020. [DOI | PubMed]
- B. A. Ersoy, L. Pardo, S. Zhang, D. A. Thompson, G. Millhauser, C. Govaerts, C. Vaisse. Mechanism of N-terminal modulation of activity at the melanocortin-4 receptor GPCR.. Nat. Chem. Biol., 2012. [DOI | PubMed]
- S. Maeda, A. Koehl, H. Matile, H. Hu, D. Hilger, G. F. X. Schertler, A. Manglik, G. Skiniotis, R. J. P. Dawson, B. K. Kobilka. Development of an antibody fragment that stabilizes GPCR/G-protein complexes.. Nat. Commun., 2018. [DOI | PubMed]
- B. Webb, A. Sali. Comparative Protein Structure Modeling Using MODELLER.. Curr. Protoc. Bioinformatics, 2016. [DOI]
- M. A. Marti-Renom, A. C. Stuart, A. Fiser, R. Sanchez, F. Melo, A. Sali. Comparative protein structure modeling of genes and genomes.. Annu. Rev. Biophys. Biomol. Struct., 2000. [DOI | PubMed]
- T. J. Dolinsky, J. E. Nielsen, J. A. McCammon, N. A. Baker. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations.. Nucleic Acids Res., 2004. [DOI | PubMed]
- C. R. Sondergaard, M. H. Olsson, M. Rostkowski, J. H. Jensen. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values.. J. Chem. Theory Comput., 2011. [DOI | PubMed]
- E. Mayol, A. Garcia-Recio, J. K. S. Tiemann, P. W. Hildebrand, R. Guixa-Gonzalez, M. Olivella, A. Cordomi. HomolWat: a web server tool to incorporate ‘homologous’ water molecules into GPCR structures.. Nucleic Acids Res., 2020. [DOI | PubMed]
- M. A. Lomize, I. D. Pogozheva, H. Joo, H. I. Mosberg, A. L. Lomize. OPM database and PPM web server: resources for positioning of proteins in membranes.. Nucleic Acids Res., 2012. [DOI | PubMed]
- S. Schott-Verdugo, H. Gohlke. PACKMOL-Memgen: A Simple-To-Use, Generalized Workflow for Membrane-Protein-Lipid-Bilayer System Building.. J. Chem. Inf. Model., 2019. [DOI | PubMed]
- M. J. Abraham, T. Murtola, R. Schulz, S. Pall, J. C. Smith, B. Hess, E. Lindahl. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers.. SoftwareX, 2015. [DOI]
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser, C. Simmerling. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB.. J. Chem. Theory Comput., 2015. [DOI | PubMed]
- C. J. Dickson, B. D. Madej, A. A. Skjevik, R. M. Betz, K. Teigen, I. R. Gould, R. C. Walker. Lipid14: The Amber Lipid Force Field.. J. Chem. Theory Comput., 2014. [DOI | PubMed]
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman, D. A. Case. Development and testing of a general amber force field.. J. Comput. Chem., 2004. [DOI | PubMed]
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf, O. Beckstein. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations.. J. Comput. Chem., 2011. [DOI | PubMed]
- The PyMOL Molecular Graphics System, Version 1.3r1; Schrodinger, LLC, 2010.
- W. Humphrey, A. Dalke, K. Schulten. VMD: visual molecular dynamics.. J. Mol. Graph., 1996. [DOI | PubMed]
- M. L. Waskom. seaborn: statistical data visualization.. J. Open Source Softw., 2021. [DOI]
- X. Nadal. Methods of purifying cannabinoids using liquid:liquid chromatography.. 2019
- R. Nygaard, Y. Zou, R. O. Dror, T. J. Mildorf, D. H. Arlow, A. Manglik, A. C. Pan, C. W. Liu, J. J. Fung, M. P. Bokoch, F. S. Thian, T. S. Kobilka, D. E. Shaw, L. Mueller, R. S. Prosser, B. K. Kobilka. The Dynamic Process of beta(2)-Adrenergic Receptor Activation.. Cell, 2013. [DOI | PubMed]
- A. Troupiotis-Tsailaki, J. Zachmann, I. Gonzalez-Gil, A. Gonzalez, S. Ortega-Gutierrez, M. L. Lopez-Rodriguez, L. Pardo, C. Govaerts. Ligand chain length drives activation of lipid G protein-coupled receptors.. Sci. Rep., 2017. [DOI | PubMed]
- A. S. Powers, V. Pham, W. A. C. Burger, G. Thompson, Y. Laloudakis, N. W. Barnes, P. M. Sexton, S. M. Paul, A. Christopoulos, D. M. Thal, C. C. Felder, C. Valant, R. O. Dror. Structural basis of efficacy-driven ligand selectivity at GPCRs.. Nat. Chem. Biol., 2023. [DOI | PubMed]
- E. W. Bow, J. M. Rimoldi. The Structure-Function Relationships of Classical Cannabinoids: CB1/CB2Modulation.. Perspect. Medicin. Chem., 2016. [DOI | PubMed]
- S. W. Jung, A. E. Cho, W. Yu. Exploring the Ligand Efficacy of Cannabinoid Receptor 1 (CB1) using Molecular Dynamics Simulations.. Sci. Rep., 2018. [DOI | PubMed]
- F. Gaudreault, M. Chartier, R. Najmanovich. Side-chain rotamer changes upon ligand binding: common, crucial, correlate with entropy and rearrange hydrogen bonding.. Bioinformatics, 2012. [DOI | PubMed]
- A. D. Scouras, V. Daggett. The Dynameomics rotamer library: amino acid side chain conformations and dynamics from comprehensive molecular dynamics simulations in water.. Protein Sci., 2011. [DOI | PubMed]
- J. W. Huffman, J. Liddle, S. Yu, M. M. Aung, M. E. Abood, J. L. Wiley, B. R. Martin. 3-(1′,1′-Dimethylbutyl)-1-deoxy-delta8-THC and related compounds: synthesis of selective ligands for the CB2 receptor.. Bioorg. Med. Chem. Lett., 1999. [DOI]
- X. Li, H. Chang, J. Bouma, L. V. de Paus, P. Mukhopadhyay, J. Paloczi, M. Mustafa, C. van der Horst, S. S. Kumar, L. Wu, Y. Yu, R. van den Berg, A. P. A. Janssen, A. Lichtman, Z. J. Liu, P. Pacher, M. van der Stelt, L. H. Heitman, T. Hua. Structural basis of selective cannabinoid CB(2) receptor activation.. Nat. Commun., 2023. [DOI | PubMed]
- S. P. Alexander, A. Christopoulos, A. P. Davenport, E. Kelly, N. V. Marrion, J. A. Peters, E. Faccenda, S. D. Harding, A. J. Pawson, J. L. Sharman, C. Southan, J. A. Davies. the Concise Guide to Pharmacology 2017/18: G protein-coupled receptors.. Br. J. Pharmacol., 2017. [DOI | PubMed]
- A. Gonzalez, A. Cordomí, G. Caltabiano, L. Pardo. Impact of helix irregularities on sequence alignment and homology modelling of G protein-coupled receptors.. Chembiochem, 2012. [DOI | PubMed]
- M. Audet, R. C. Stevens. Emerging structural biology of lipid G protein-coupled receptors.. Protein Sci., 2019. [DOI | PubMed]
- N. Stanley, L. Pardo, G. D. Fabritiis. The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor.. Sci. Rep., 2016. [DOI | PubMed]
- N. Casajuana-Martin, G. Navarro, A. Gonzalez, C. Llinas Del Torrent, M. Gomez-Autet, A. Quintana Garcia, R. Franco, L. Pardo. A Single Point Mutation Blocks the Entrance of Ligands to the Cannabinoid CB(2) Receptor via the Lipid Bilayer.. J. Chem. Inf. Model., 2022. [DOI | PubMed]
- W. I. Weis, B. K. Kobilka. The Molecular Basis of G Protein-Coupled Receptor Activation.. Annu. Rev. Biochem., 2018. [DOI | PubMed]
- Q. Zhou, D. Yang, M. Wu, Y. Guo, W. Guo, L. Zhong, X. Cai, A. Dai, W. Jang, E. I. Shakhnovich. Common activation mechanism of class A GPCRs.. Elife, 2019. [DOI]
- K. Sansuk, X. Deupi, I. R. Torrecillas, A. Jongejan, S. Nijmeijer, R. A. Bakker, L. Pardo, R. Leurs. A Structural Insight into the Reorientation of Transmembrane Domains 3 and 5 during Family A G Protein-Coupled Receptor Activation.. Mol. Pharmacol., 2011. [DOI | PubMed]
- S. G. Rasmussen, H. J. Choi, J. J. Fung, E. Pardon, P. Casarosa, P. S. Chae, B. T. Devree, D. M. Rosenbaum, F. S. Thian, T. S. Kobilka, A. Schnapp, I. Konetzki, R. K. Sunahara, S. H. Gellman, A. Pautsch, J. Steyaert, W. I. Weis, B. K. Kobilka. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor.. Nature, 2011. [DOI | PubMed]
- S. Maeda, Y. Shiimura, H. Asada, K. Hirata, F. Luo, E. Nango, N. Tanaka, M. Toyomoto, A. Inoue, J. Aoki, S. Iwata, M. Hagiwara. Endogenous agonist-bound S1PR3 structure reveals determinants of G protein-subtype bias.. Sci. Adv., 2021. [DOI]
- L. Yu, L. He, B. Gan, R. Ti, Q. Xiao, X. Yang, H. Hu, L. Zhu, S. Wang, R. Ren. Structural insights into sphingosine-1-phosphate receptor activation.. Proc. Natl. Acad. Sci. U.S.A., 2022. [DOI | PubMed]
- H. Chen, K. Chen, W. Huang, L. M. Staudt, J. G. Cyster, X. Li. Structure of S1PR2-heterotrimeric G(13) signaling complex.. Sci. Adv., 2022. [DOI | PubMed]
- S. Liu, N. Paknejad, L. Zhu, Y. Kihara, M. Ray, J. Chun, W. Liu, R. K. Hite, X. Y. Huang. Differential activation mechanisms of lipid GPCRs by lysophosphatidic acid and sphingosine 1-phosphate.. Nat. Commun., 2022. [DOI | PubMed]
- H. Akasaka, T. Tanaka, F. K. Sano, Y. Matsuzaki, W. Shihoya, O. Nureki. Structure of the active G(i)-coupled human lysophosphatidic acid receptor 1 complexed with a potent agonist.. Nat. Commun., 2022. [DOI | PubMed]
- G. Navarro, A. Gonzalez, A. Sanchez-Morales, N. Casajuana-Martin, M. Gomez-Ventura, A. Cordomi, F. Busque, R. Alibes, L. Pardo, R. Franco. Design of Negative and Positive Allosteric Modulators of the Cannabinoid CB2 Receptor Derived from the Natural Product Cannabidiol.. J. Med. Chem., 2021. [DOI | PubMed]