
Structure–Activity Relationships and Evaluation of 2-(Heteroaryl-cycloalkyl)-1H-indoles as Tauopathy Positron Emission Tomography Radiotracers
Abstract
Structure–activity relationship studies were performed on a library of synthesized compounds based on previously identified tau ligands. The top 13 new compounds had Ki values in the range of 5–14 nM in Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD) post-mortem brain tissues. One of the more promising new compounds ([3H]75) bound with high affinity in AD, PSP, and CBD tissues (KD’s = 1–1.5 nM) and Pick’s disease tissue (KD = 3.8 nM). Autoradiography studies with [3H]75 demonstrated specific binding in AD, PSP, and CBD post-mortem tissues. Nonhuman primate brain PET imaging with [18F]75 demonstrated a peak standardized uptake value (SUV) of ∼5 in the cerebellum, ∼4.5 in the cortex, and ∼4 in whole brain with SUV 2-to-90 min ratios of 3.9 in whole brain, 4.9 in cortex, and 4.5 in cerebellum. Compound [18F]75 is a promising candidate for translation to human brain PET imaging studies.
Affiliations: †Department of Radiology, University of Pittsburgh, Pittsburgh, Pennsylvania 15213, United States; ‡Department of Radiology, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, United States; §Department of Psychiatry, University of Pittsburgh, Pittsburgh, Pennsylvania 15213, United States; ∥X-ray Crystallography Laboratory, Department of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15213, United States; ⊥Geriatric Research and Clinical Education, VA Pittsburgh Healthcare System, Pittsburgh, Pennsylvania 15240, United States
License: © 2025 The Authors. Published by American Chemical Society CC BY 4.0 Permits the broadest form of re-use including for commercial purposes, provided that author attribution and integrity are maintained (https://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.1021/acs.jmedchem.4c02988 | PubMed: 40068019 | PMC: PMC11956013
Relevance: Relevant: mentioned in keywords or abstract
Full text: PDF (14.4 MB)
Introduction
Positron emission tomography (PET) imaging of aggregated microtubule-associated protein tau1,2 in the living human Alzheimer’s disease (AD) brain has enabled the assessment of tau burden, disease staging, and monitoring of disease progression.3−6 The commonly used tau PET tracers7 [18F]1 ([18F]T807, [18F]AV-1451, [18F]flortaucipir, Tauvid), [18F]2 ([18F]PI-2620), [18F]3 ([18F]PM–PBB3, [18F]MNI-958, [18F]APN-1607, [18F]florzolotau), and [18F]4 ([18F]MK-6240, [18F]MNI-946, florquinitau) (Figure ) have demonstrated the ability to image AD-tau8,9 but can also suffer from off-target binding10−16 or are less effective in other tauopathies.17−19 Thus, more effective tau PET tracers are needed to successfully image aggregated tau in non-AD tauopathies such as chronic traumatic encephalopathy (CTE), Pick’s disease (PiD), corticobasal degeneration (CBD), and progressive supranuclear palsy (PSP).20−25
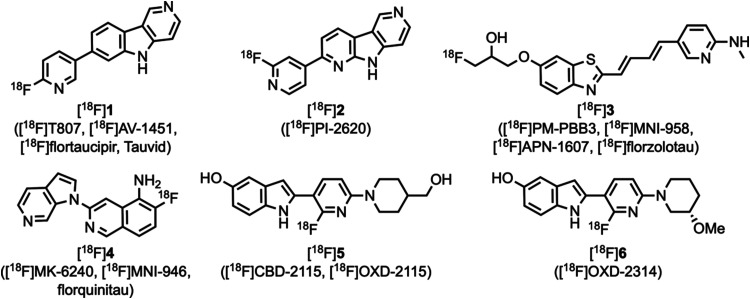
Tau in adult human brain consists of six isoforms resulting from 0, 1, or 2 inserts in the N-terminal domain (0N, 1N, 2N) and either three or four imperfect repeats (3R, 4R) in the microtubule-binding domain.26−29 Aggregates of the different isoforms constitute different tauopathies with AD and CTE filaments being composed primarily of mixed 3R/4R-tau, PiD filaments being composed mainly of 3R-tau, and CBD and PSP filaments being composed predominantly of 4R-tau.30,31 Recent work with cryogenic electron microscopy (cryo-EM) has demonstrated that the different tauopathies can be classified according to the structure formed by the tau filaments.29,32−34 Computer modeling studies of tau fibrils have predicted several potential binding sites for existing PET tracers,35−40 and cryo-EM structures with bound PET ligands have provided insight into possible binding modes of these compounds.41−43 Thus, to image non-AD tauopathies with PET, it may be necessary to develop ligands that bind with high affinity to sites on tau filaments other than those where compounds [18F]1–[18F]4 bind. In an effort to develop a more potent and effective 4R-tau PET tracer, [18F]5 ([18F]CBD-2115, [18F]OXD-2115) (Figure ) was developed, but it suffered from low brain entry as shown by PET studies in mouse, rat, and nonhuman primate.44 Structural modifications of 5 resulted in the identification of [18F]6 ([18F]OXD-2314), which demonstrated improved brain entry in rat and nonhuman primate PET imaging studies and high binding affinity in AD, PSP, CBD, and PiD post-mortem brain tissue homogenate assays.45
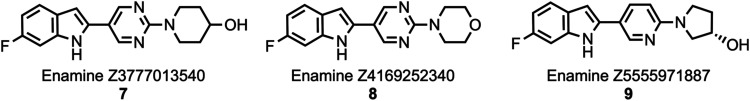
Compounds 7, 8, and 9 (Figure ) were previously identified utilizing an in silico chemical database fingerprint search of the Enamine REAL collection.46 Evaluation of [3H]7 and [3H]8 in post-mortem human AD, PSP, CBD, and PiD brain tissue homogenates demonstrated that these compounds bind with high affinity to aggregated tau, yet 7 and 8 did not compete effectively against [3H]2 or the structurally similar aryl-indole [3H]5 in AD, PSP, and CBD brain tissue homogenate assays. Compounds 7 and 8 did compete against [3H]3 in AD, PSP, and CBD brain tissue homogenate assays, thus demonstrating that 3, 7, and 8 apparently bind to similar locations on both aggregated mixed 3R/4R- and 4R-tau. With the goal of identifying even more potent tau ligands and improved selectivity for 4R-tau over mixed 3R/4R- and 3R-tau species, structure–activity relationship (SAR) studies were performed by synthesizing and evaluating a library of compounds based on 7, 8, and 9 with potential sites for F-18 radiolabeling at the 5- or 6-position of the indole ring,47−51 the center pyrimidine/pyridine ring,52−54 or the cycloalkyl ring.55−60
Results and Discussion
Chemical Synthesis
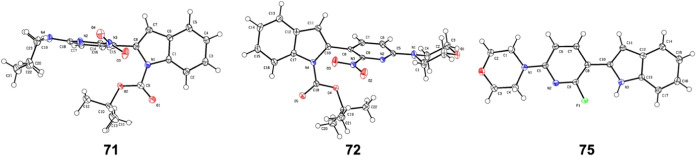
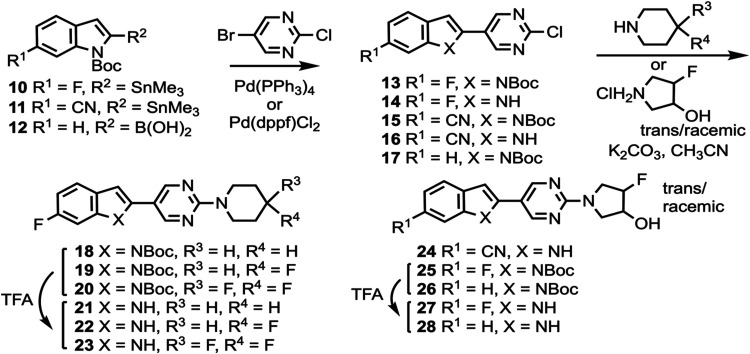
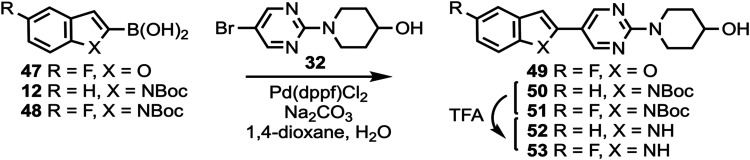
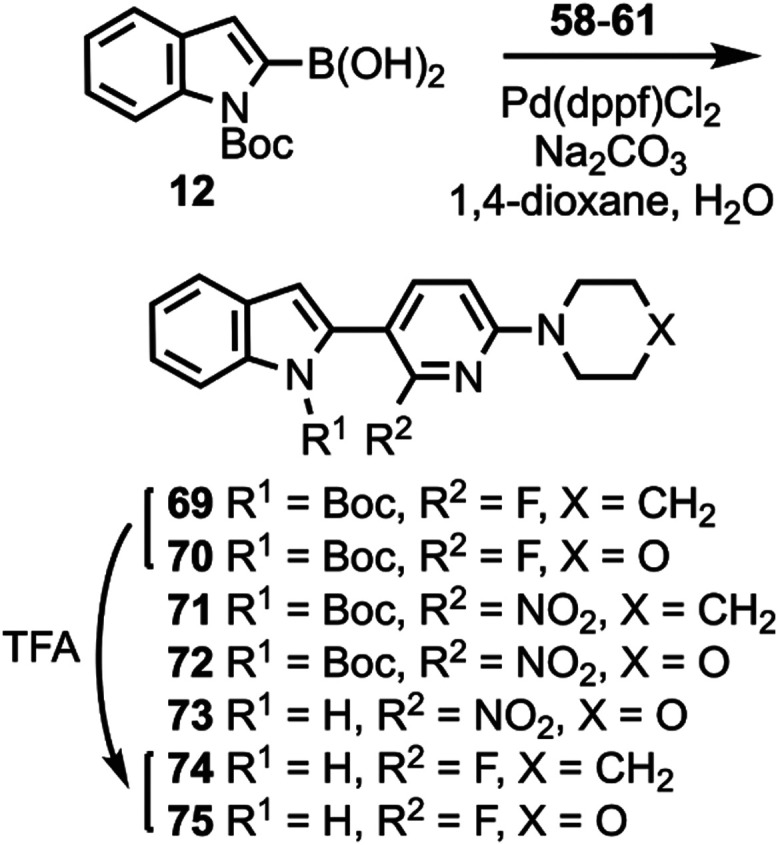
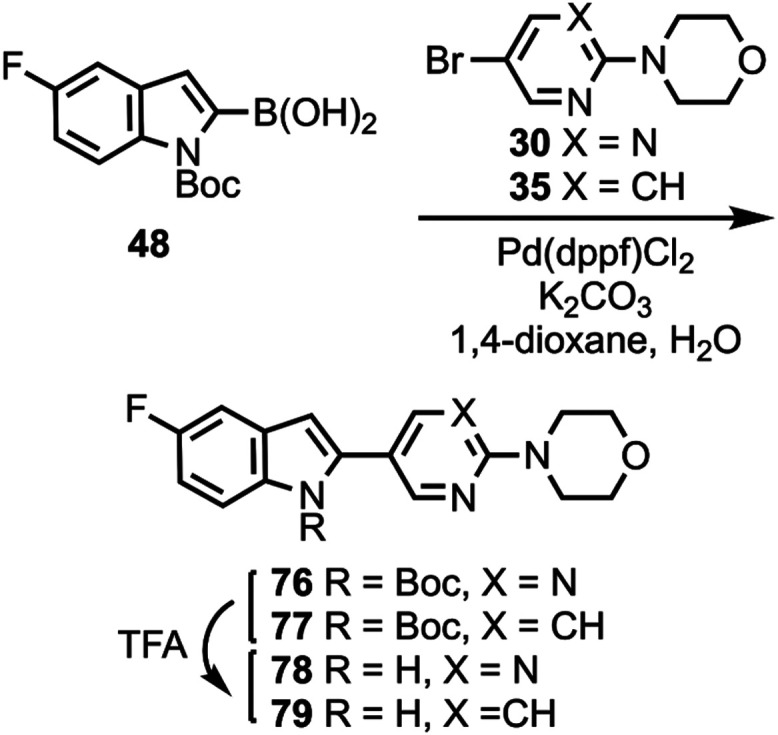
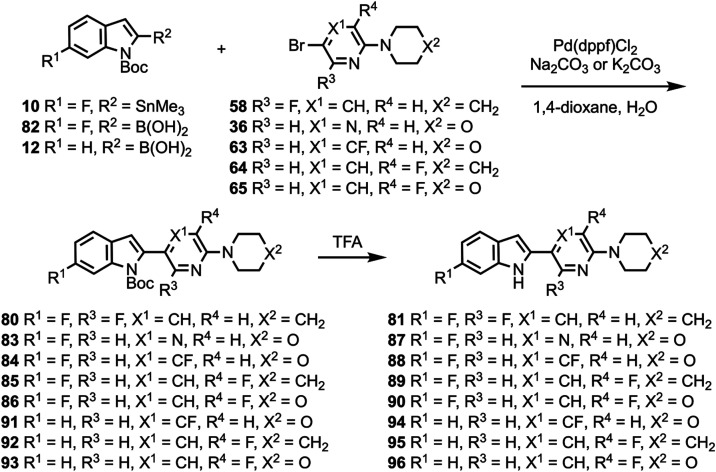
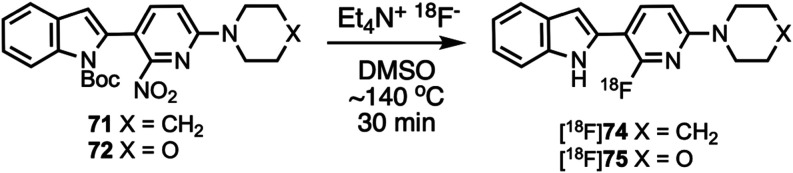
Compounds 10 and 11 (Scheme 1) were prepared according to a previously described procedure;61 compound 12 is commercially available. Compounds 10–12 were coupled to 5-bromo-2-chloropyrimidine to give intermediates 13–17 which were reacted with various cyclic amines to give compounds 18–20 and 24–26. N-Boc deprotection then afforded compounds 21–23, 27, and 28. Substituted heterocyclic intermediates 29–36 (Scheme S1, Supporting Information) were coupled to 10 (Scheme 2) to give intermediates 37–41 which were then deprotected to give compounds 42–46 (the structure of 44 was confirmed by X-ray crystallography, Figure S1, Supporting Information). Commercially available compounds 47, 12, and 48 (Scheme 3) were coupled to 32 to give compounds 49–51, followed by N-Boc deprotection to afford compounds 52 and 53. Substituted pyridine intermediates 54–57 and 62 (Scheme S2, Supporting Information) were brominated with N-bromosuccinimide (NBS) to give compounds 58–61 and 63.62 Substituted pyridines 64–68 (Scheme S3, Supporting Information) were obtained by coupling either piperidine or morpholine63,64 to commercially available pyridines. Compounds 58–61 were coupled to compound 12 (Scheme 4) to give compounds 69–73, followed by deprotection to give compounds 74 and 75. The chemical structures of 71, 72, and 75 were confirmed by X-ray crystallography (Figure ).
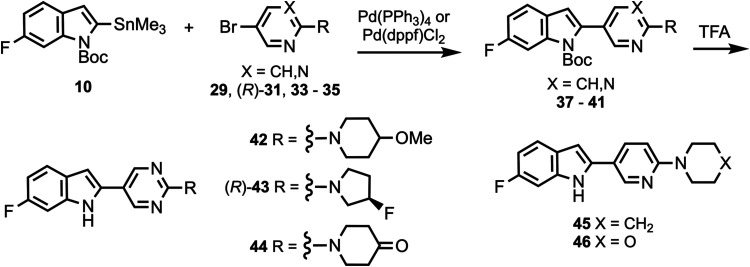
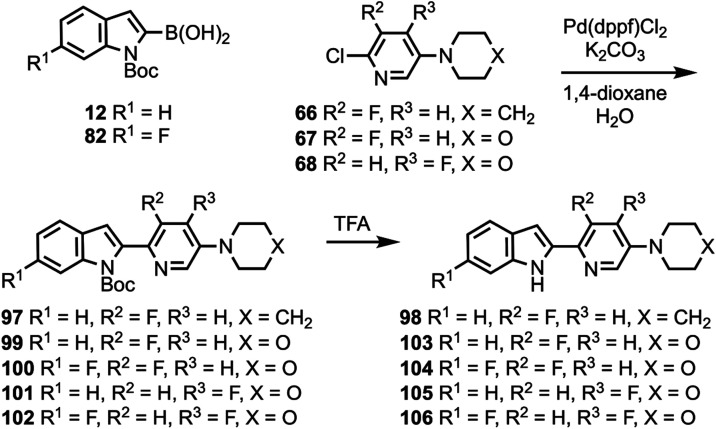
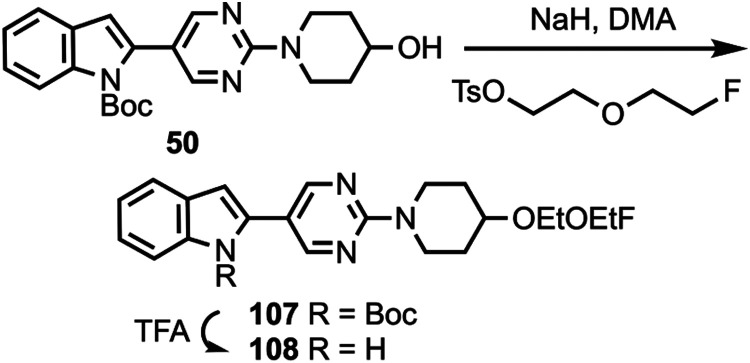
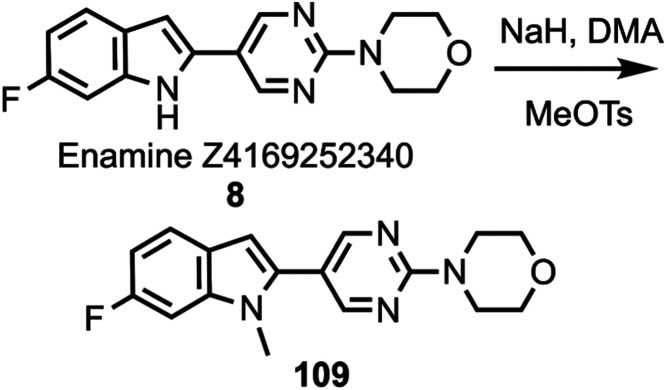
Compound 48 was coupled to compounds 30 and 35 (Scheme 5) to give compounds 76 and 77, followed by deprotection to give compounds 78 and 79. Compounds 10, 82, and 12 were coupled to substituted heterocycles (Scheme 6) to give compounds 80, 83–86, and 91–93 followed by deprotection to afford compounds 81, 87–90, and 94–96. Compounds 12 and 82 were coupled to substituted pyridines (Scheme 7) to give compounds 97 and 99–102, followed by deprotection to afford compounds 98 and 103–106. Compound 50 was O-alkylated (Scheme 8) to give compound 107 followed by deprotection to give compound 108, while compound 8 was N-methylated (Scheme 9) to afford compound 109. A library of compounds was thus synthesized with variable substitution patterns for SAR testing while also maintaining a fluorine atom as a potential site for radiolabeling with F-18. Additionally, compounds 42 and 109 can potentially be radiolabeled with C-11(as O-[11C]CH3 or N-[11C]CH3).
Binding Assays and SAR Studies
All screened compounds were >95% pure as determined by analytical HPLC (Table S8 and Figures S2–S7, Supporting Information). In vitro binding assays in post-mortem brain tissue homogenates were performed as previously described.46,65,66 The inhibition constant (Ki) values of all screened candidate compounds versus [3H]7, [3H]8, and [3H]3 are shown in Table S1 (Supporting Information). Derivatives of 7 were prepared where the 4-hydroxypiperidine ring was held constant (compounds 49, 52, and 53) and where the 4-hydroxy group was O-alkylated (compound 108) (Table S2, Supporting Information). Moving the 6-fluoro group of 7 to the 5-position of the indole ring to give 53, or removing the fluorine atom completely to give 52, did not change the affinity in PSP tissue relative to 7 when competed against [3H]7, while O-alkylation of 52 to give 108 also did not change the affinity in PSP tissue relative to 7 when competed against [3H]7, but did result in a loss of affinity in AD, PSP, and CBD tissue relative to 7 when competed against [3H]8. Changing the indole ring of 53 to benzofuran to give 49 resulted in a loss of affinity in AD and PSP tissue when competed against [3H]7, suggesting that the indole NH group may be necessary as an H-bond donor.
The 6-fluoroindole and pyrimidine rings of 7 were held constant while the 4-substituent of the piperidine ring was varied to give compounds 21, 22, 23, 42, and 44 (Table S3, Supporting Information). O-methylation of the hydroxy group of 7 to give 42 did not significantly alter binding affinity in AD, PSP, or CBD tissue relative to 7 when competed against [3H]7, [3H]8, or [3H]3, indicating O-methylation as a potential strategy to increase brain uptake for PET imaging relative to what was observed for [18F]7.46 Conversion of the hydroxy group of 7 to a carbonyl group to give 44 did not improve affinity when competed against [3H]7 or [3H]8, and resulted in a loss of affinity when competed against [3H]3. Removal of the hydroxy group of 7 to give the unsubstituted piperidine 21 improved the affinity in AD, PSP, and CBD when competed against [3H]7, maintained affinity when competed against [3H]8, and reduced affinity slightly when competed against [3H]3. Replacing the hydroxy group of 7 with fluorine to give 22 did not improve affinity when competed against [3H]7 or [3H]8 and resulted in a loss of affinity when competed against [3H]3, while difluorination to give 23 reduced affinity in AD and PSP tissue when competed against [3H]7 relative to 22.
The piperidine ring of 21 was held constant while the indole and pyrimidine rings were varied to give compounds 45, 74, 81, 89, 95, and 98 (Table S4, Supporting Information). Replacement of the pyrimidine ring of 21 with pyridine to give 45, and movement of the fluorine atom of 45 to the pyridine ring to give 74, generally maintained affinities in AD, PSP, and CBD tissue when competed against [3H]7, [3H]8, and [3H]3. Combining 45 and 74 to give the difluorinated compound 81 reduced affinity in AD, PSP, and CBD tissue when competed against [3H]8, whereas the difluorinated compound 89 maintained affinity in AD, PSP, and CBD tissue relative to 74 when competed against [3H]8. Moving the fluorine atom of 74 across the pyridine ring to give 95 (equivalent to removing the indole fluorine of 89) reduced affinity in AD, PSP, and CBD tissues relative to 74 and 89 when competed against [3H]8. Flipping the fluoropyridine ring of 95 to give 98 did not change the affinities in AD, PSP, and CBD tissues when competed against [3H]8.
SAR studies were performed around the structure of 8 by holding the morpholine ring constant and changing the indole and pyrimidine rings (Table S5, Supporting Information). Replacing the pyrimidine ring of 8 with pyridine to give 46, and then moving the fluorine atom of 46 to the 2-position of the pyridine ring to give 75, generally maintained affinities for both 46 and 75, relative to 8, in AD, PSP, and CBD tissue when competed against [3H]7, [3H]8, and [3H]3. Moving the indole 6-fluoro group of 8 to the indole 5-position to give 78 also maintained binding affinity when competed against [3H]8, while moving the indole 6-fluoro group of 46 to the indole 5-position to give 79 (equivalent to changing the pyrimidine ring of 78 to pyridine) resulted in a slight improvement in affinities in AD, PSP, and CBD tissue, relative to 46 and 78, when competed against [3H]8. Replacing the pyrimidine ring of 8 with pyrazine to give 87 maintained affinities in AD, PSP, and CBD tissue when competed against [3H]8. Moving the fluorine atom of 75 to different positions on the pyridine ring to give 94 and 96 generally maintained affinities, but moving the nitrogen atom to the adjacent position of the ring to give 103 and 105 reduced affinities when competed against [3H]8. Converting 94 to difluorinated 88, and converting 103 to difluorinated 104 both maintained affinities, while converting 96 to difluorinated 90, and converting 105 to difluorinated 106 both resulted in reduced affinities. N-methylation of 8 to give 109 reduced affinities in AD and PSP tissue when competed against [3H]7, further demonstrating the potential H-bonding role of the indole NH group.
SAR studies were also performed around the structure of 9 to give compounds 24, 27, 28, and (R)-43 (Table S6, Supporting Information). Replacing the pyridine ring of 9 with pyrimidine, along with exchanging the OH with F and inverting the chirality to give (R)-43 resulted in a loss of affinity across all tissues and tritiated ligands relative to 9. Changing the pyrrolidine ring substitution of (R)-43 to F/OH-disubstituted to give 27 retained affinities across all tissues and tritiated ligands relative to 9. The pyrrolidine ring of 27 is a racemic mixture of trans-configuration, and an individual enantiomer of 27 may have improved binding affinity and/or improved 4R- over mixed 3R/4R- and 3R-tau selectivity relative to the racemic mixture. Removing the indole fluorine atom of 27 to give 28 or replacing the indole fluorine atom of 27 with cyano to give 24 both resulted in losses of affinity in AD and PSP tissues when competed against [3H]7 and [3H]8.
Compounds 21, 27, 45, 46, 74, and 75 did not compete strongly against [3H]5 (Table S7, Supporting Information) in AD, PSP, or CBD tissues, similarly to what was observed with 7 and 8,46 indicating that these compounds do not bind to the site on aggregated tau where 5 binds. Compounds 21, 45, 46, and 75 did not compete well against [3H]PiB in AD tissue (Table S7, Supporting Information) indicating that these compounds do not bind strongly to the PiB binding site on amyloid-β.65,67
Table 1 ranks the top 13 candidate compounds by PSP Ki values (range: 5–13 nM) when competed against [3H]8. Compound 8 has Ki values (nM) 7.7 ± 0.6 (AD), 8.5 ± 1.2 (PSP), and 11 ± 1.3 (CBD) when competed against [3H]8 (Table S1, Supporting Information). Thus, a series of substituted-indole compounds have been identified with similar Ki values as 8 in AD, PSP, and CBD tissues when competed against [3H]8. All compounds have an indole NH group for hydrogen bonding which was indicated as necessary by the reduction in affinity through N-methylation to give 109 (Table S5, Supporting Information) or conversion to a benzofuran to give 49 (Table S2, Supporting Information). Seven of the compounds in Table 1 have fluoroindole substitution (45, 79, 87, 21, 78, 42, 46), three compounds have fluoropyridine substitution (74, 96, 75), and three compounds are difluoro-substituted (89, 88, 27). Compound 27, as stated above, may have higher affinity as a single enantiomer, and the synthesis of each enantiomer is underway. All of these compounds can potentially be F-18 radiolabeled on the indole ring,47−51 the pyridine ring,52−54 or the pyrrolidine ring55−60 in the case of 27. Compounds 74 and 75, although not the highest affinity compounds in Table 1, represent the most easily accessible F-18 radiolabeled compounds through an [18F]fluoro-denitration mechanism of a 2-nitropyridine ring system,52 as well as a fairly simple radiolabeling precursor synthesis (compounds 71 and 72, Scheme 4). Therefore, compounds 74 and 75 were evaluated with in silico prediction models to gauge whether these compounds have the potential to be successful central nervous system (CNS) PET agents.
Table 1: Top 13 Candidate Ligands Ranked by PSP Ki Values versus [3H]8a
In Silico CNS Exposure Predictions
Compounds 74 and 75 were evaluated with the Pfizer CNS multiparameter optimization (MPO) and PET MPO calculators,68,69 the blood–brain barrier (BBB) Score calculator,70 and the SwissADME BOILED-Egg brain penetration predictor.71,72 The CNS MPO and PET MPO calculators predict the likelihood of success of a CNS-targeting agent by taking into consideration physiochemical properties along with absorption, distribution, metabolism, and excretion (ADME) properties. The BBB Score estimates whether a molecule will be CNS or non-CNS active based on five physiochemical descriptors. The BOILED-Egg predictor calculates properties based on chemical structure and then creates a two-dimensional plot of WLOGP vs tPSA to predict gastrointestinal absorption (not relevant to iv-administered PET tracers) and brain permeation. These models can be used as guides for the selection of compounds with the potential to enter the CNS73−75 although they cannot predict other parameters that could affect the success of a candidate CNS PET tracer such as the extent of nonspecific binding and clearance rates from nontarget brain tissues.
Compound 74 (Figure S8, Supporting Information) has a CNS MPO score of 3.0 (>4 preferred, although there are several known PET tracers with CNS MPO scores <4)73 and a PET MPO score of 2.1 (>3 preferred) which predict a low likelihood of the compound being a successful CNS agent. But compound 74 has a BBB score of 5.01 which predicts a 90% probability that it will penetrate the BBB. Compound 75 (Figure S9, Supporting Information) has a CNS MPO score of 4.1 and a PET MPO score of 3.5 which predict that the compound will be a successful CNS agent. The BBB score for compound 75 is 4.71 which predicts that it will penetrate the BBB, but the probability is less than that of compound 74. The BOILED-Egg plot (Figure S10, Supporting Information) predicts that both 74 and 75 will enter the brain. Thus, both compounds were advanced to radiolabeling development and PET imaging evaluation.
Radiochemistry
The structures of radiolabeling precursors 71 and 72 were confirmed by X-ray crystallography (Figure ). For preliminary radiolabeling development work samples of the crude reaction mixtures were analyzed by analytical HPLC to monitor reaction progress. A sample of the crude reaction mixture of [18F]74 (Figure S11, Supporting Information) demonstrated that after 45 min N-Boc intermediate [18F]69 was not present, but N-deprotected [18F]74 was present. Samples of the crude reaction mixture of [18F]75 were taken at 15, 30, and 45 min (Figures S12–S14, Supporting Information) and showed that [18F]70 was initially formed but then the Boc group was lost over time to give [18F]75, similarly to what was previously reported for [18F]1.76,77 For PET studies with [18F]74 and [18F]75, the radiolabeling reaction (Scheme 10) was performed in dimethyl sulfoxide (DMSO) at ∼140 °C for 30 min, then the crude reaction mixture was purified by semipreparatory HPLC (Figures S15 and S16, Supporting Information). Radiochemical identity of the reformulated radiotracers was confirmed by co-injection with the nonradioactive standards (Figures S17 and S18, Supporting Information).
Lipophilicity
The log D7.4 values of the radiolabeled compounds [18F]74 and [18F]75 were determined by the octanol/aqueous buffer shake-flask method using glass vials and glass pipettes, and only using plastic at the last step when a pipettor with a plastic tip was used to transfer samples to counting tubes. Initial attempts to measure the log D7.4 values by performing the study with plastic 2 mL Eppendorf tubes resulted in variable amounts of activity remaining adhered to the Eppendorf tubes. Using glass vials, the values obtained were [18F]74 log D7.4 = 1.80 ± 0.01 (n = 4) and [18F]75 log D7.4 = 1.96 ± 0.03 (n = 4), which are in the range (log D ∼ 1–5)78 optimal for passive diffusion across the BBB. Each of these values is less than the in silico Clog D predictions used for the MPO and BBB Score calculators (Figures S8 and S9, Supporting Information).
PET Imaging Studies
Dynamic PET brain imaging studies and metabolite analysis in male rhesus macaque monkeys (Macaca mulatta) were performed as previously described.45 Evaluation of candidate tau PET tracers in nonhuman primates (NHP) that do not harbor pathological tau aggregates enables the assessment of normal brain uptake, distribution, and clearance of nonspecific binding, as well as analysis of potential radiometabolites that should more closely resemble the metabolic profile of humans compared to that obtained from rodents. Injection of [18F]74 into a macaque resulted in rapid brain entry with peak standardized uptake value (SUVmax) values of ∼1.5–2.5 (Figure and Table 2). The earlier peak time of cerebellar radioactivity resulted in an initial period of accelerated clearance compared to cortex or whole brain, which peaked later (∼15 min postinjection). Thereafter, all regions exhibited a similar average clearance rate of ∼0.01 SUV/min. This phenomenon is common in PET studies of anesthetized primates and likely results from differential cerebral blood flow rates under isoflurane anesthesia79 and potentially the additive contribution of spill-in of radioactivity signal from multiple adjacent and convergent dural venous sinuses into the cerebellar region of interest. The 2-to-60 min and 2-to-90 min SUV ratios for [18F]74 (Table 2) were whole brain = 1.2 and 1.5, cortex = 1.3 and 1.5, and cerebellum = 2.0 and 2.5. The summed PET images (Figure ) indicate that from 0 to 15 min, [18F]74 distributed uniformly throughout much of the brain, while the images summed from 60 to 90 min demonstrate significant clearance of brain radioactivity. The lack of skull uptake indicates that [18F]74 is stable against [18F]defluorination and suggests that the activity in the spine evident in both early and late summed images is not the result of bone uptake of free [18F]fluoride. Venous metabolite analysis (Figure S19, Supporting Information) indicated that only more polar radiometabolites were formed and that ∼35% intact [18F]74 remained after 10 min and ∼10% intact [18F]74 remained after 30 min.
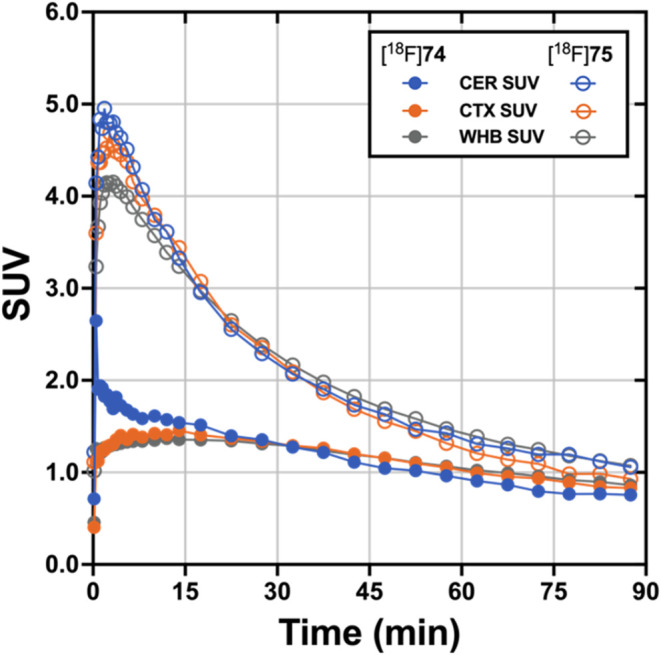
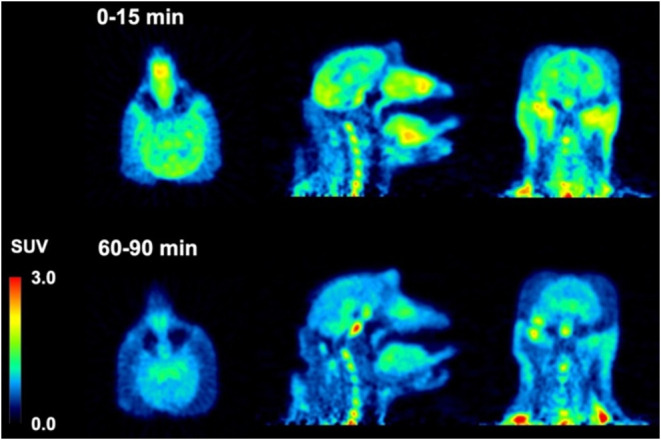
Table 2: Comparison of [18F]74 and [18F]75 SUVmax, 2-to-60 min and 2-to-90 min SUV Ratios In Macaque Brain
| whole Brain | cortex | cerebellum | |||||||
|---|---|---|---|---|---|---|---|---|---|
| SUVmax | 2′:60′ | 2′:90′ | SUVmax | 2′:60′ | 2′:90′ | SUVmax | 2′:60′ | 2′:90′ | |
| [18F]74 | 1.4 | 1.2 | 1.5 | 1.5 | 1.3 | 1.5 | 2.7 | 2.0 | 2.5 |
| [18F]75 | 4.2 | 3.0 | 3.9 | 4.7 | 3.7 | 4.9 | 5.0 | 3.8 | 4.5 |
Injection of [18F]75 into a male macaque resulted in rapid brain entry with SUVmax values of ∼4–5 (Figure and Table 2). Brain radioactivity clearance of [18F]75 was more rapid on average (∼0.04 SUV/min) than we observed for [18F]74, yielding 2-to-60 min and 2-to-90 min SUV ratios (Table 2) of whole brain = 3.0 and 3.9, cortex = 3.7 and 4.9, and cerebellum = 3.8 and 4.5. The summed PET images (Figure ) indicate that from 0 to 15 min, [18F]75 distributed uniformly throughout the cortex, cerebellum, and midbrain, and then largely cleared from the brain by the 60-to-90 min image interval. Primate studies showing high brain uptake, uniform brain distribution, and rapid clearance of nonspecifically bound [18F]75 suggest favorable properties for translational human studies for the visualization of pathologic tau aggregates. The lack of skull uptake indicates that [18F]75 is stable against [18F]defluorination. Venous metabolite analysis (Figure S19, Supporting Information) indicated that only more polar radiometabolites were formed and that ∼35% intact [18F]75 remained after 10 min, ∼20% intact [18F]75 remained after 30 min, and ∼10% intact [18F]75 remained after 90 min.
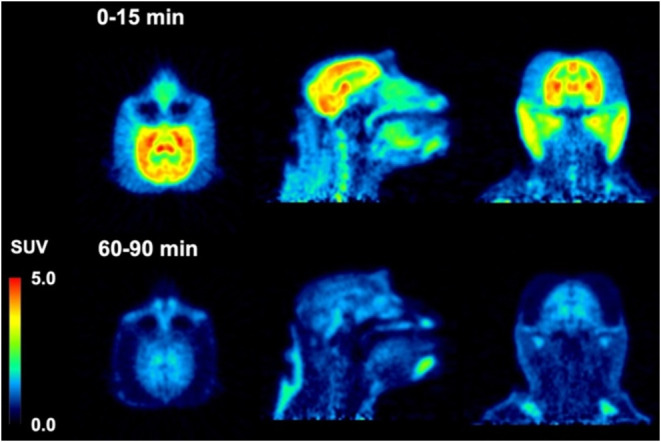
Compounds 74 and 75 differ structurally (Scheme 10) only by the replacement of a CH2 group with an O (piperidine changed to morpholine), and they have similar Ki values in AD, PSP, and CBD tissue samples (Table 1). Both [18F]74 and [18F]75 rapidly entered the brain during a nonhuman primate PET study, but [18F]75 had superior uptake and clearance properties (Figure ) as evidenced by greater 2-to-60 min and 2-to-90 min SUV ratios (Table 2). Thus, 74 was dropped from further consideration and additional studies were performed with 75.
Additional Evaluations of 75 and [3H]75
Compound 75 was screened at a 10-μM concentration against 98 CNS protein targets in the Eurofins CNS SafetyScreen panel (Supporting Information). Compound 75 displayed ≤25.3% inhibition at 96 targets, 44.6% inhibition at human 5-HT2B (vs ± DOI), and 61% inhibition at human monoamine oxidase-B (MAO-B) (vs deprenyl). Off-target binding of some tau PET tracers to MAOs has been previously observed and is a potential concern.12,14,80 The binding of 75 to human recombinant MAO-B was determined by Eurofins (Figure S20, Supporting Information) and was found to be IC50 = 3.0 μM, which is expected to be too weak to result in off-target binding during PET imaging studies. The NHP brain PET study with [18F]75 (Figures and 6) demonstrated clearance of activity with no apparent retention pattern that would indicate significant off-target binding to any constitutive binding site in normal brain tissue.
Compound [3H]75 was tritiated by Novandi Chemistry AB (www.novandi.se) via direct H/T exchange (37 MBq/mL (1.0 mCi/mL), Am = 1.74 TBq/mmol (47 Ci/mmol), radiochemical purity >99%, chemical purity 99%, and stored at a reduced temperature in an EtOH + 0.01% ascorbic acid solution). The KD and Bmax values of [3H]75 were determined by homologous binding assays46,65,66,81 with post-mortem brain tissue samples from AD, PSP, CBD, PiD, young control (CT), elderly CT, Parkinson’s disease (PD), and transactive response DNA-binding protein 43 (TDP-43)82,83 cases with neuropathological confirmation, as well as P301L transgenic mouse brain84,85 (Table 3). Compound [3H]75 binds with high affinity in AD, PSP, CBD, PiD, and P301L brain tissues with KD values of ∼1 nM and Bmax values ranging from 800 to 1800 nM, while only weak binding was observed in young CT, elderly CT, PD, and TDP-43 tissues. Thus, [3H]75 binds avidly to aggregated tau, but is not selective for 4R-tau (PSP, CBD, P301L) over 3R-tau (PiD) or mixed 3R/4R-tau (AD). The weak binding of [3H]75 in PD tissue suggests that off-target binding to α-synuclein copathologies86−88 is not likely to confound tau PET imaging studies of [18F]75 in human subjects, and further suggests the possibility of imaging tau in synucleinopathies.89−92 Additionally, the weak binding in TDP-43 tissue is important due to the possible presence of TDP-43 aggregates in PSP and CBD93 and a recently characterized age-related TDP-43 proteinopathy, termed limbic-predominant age-related TDP-43 encephalopathy (LATE) in subjects of advanced age (>80 y).94
Table 3: Equilibrium Dissociation Constant (KD) and Maximum Binding Density (Bmax) Values of [3H]75 in Post-Mortem Brain Tissue Homogenatesa
| AD | PSP | CBD | P301L | PiD | |
|---|---|---|---|---|---|
| KD (nM) | 1.5 ± 0.0 | 1.0 ± 0.1 | 1.3 ± 0.2 | 2.6 ± 0.2 | 3.8 ± 0.1 |
| Bmax (nM) | 1086 ± 102 | 766 ± 64 | 801 ± 58 | 1782 ± 64 | 1049 ± 55 |
a n = 3, mean ± standard deviation (SD).
The inhibition constants (Ki) of the established tau tracers 1–4, and the experimental tau tracers 5 and 8, were determined versus [3H]75 in post-mortem brain tissue samples from AD, PSP, CBD, and PiD (Table 4). Compounds 1, 2, 4, and 5 competed poorly against [3H]75 in all four tissue types. Computational studies have predicted several potential binding sites on tau fibrils35−40 and the inability of compounds 1, 2, 4, and 5 to potently displace [3H]75 demonstrates that these compounds do not bind to the same location on aggregated tau where [3H]75 binds. Compound 3 had Ki values of ∼100 nM in all four tissue types demonstrating that there may be an overlap of binding sites between 3 and [3H]75 on aggregated tau. Cryo-EM studies41 identified multiple binding sites for 3 on the Alzheimer’s tau fold, and the weak competition (Ki ∼ 86 nM) of 3 against [3H]75 in AD tissue suggests that [3H]75 may bind in proximity to one of the identified sites. Compound 8 had Ki values versus [3H]75 of ∼15 nM in AD, PSP, and CBD tissues, and a Ki value of ∼24 nM in PiD tissue, indicating overlap of binding sites for the two structurally similar compounds in these tissues.
Table 4: Inhibition Constant (Ki) Values of Tau Ligands versus [3H]75 in AD, PSP, CBD, and PiD Brain Tissue Homogenatesa
| AD tissue | PSP tissue | CBD tissue | PiD tissue | |
|---|---|---|---|---|
| competitor | Ki (nM) | Ki (nM) | Ki (nM) | Ki (nM) |
| 1b | 1485 ± 53 | 1860 ± 39 | 2131 ± 58 | 1494 ± 26 |
| 2 | 3023 ± 283 | 10,000 | 10,000 | 1967 ± 96 |
| 3 | 86 ± 5 | 123 ± 7 | 106 ± 6 | 124 ± 3 |
| 4 | 240 ± 6 | 780 ± 32 | 888 ± 32 | 656 ± 36 |
| 5 | 1209 ± 47 | 566 ± 42 | 572 ± 32 | 575 ± 36 |
| 8 | 15 ± 1 | 15 ± 2 | 16 ± 0 | 24 ± 2 |
a n = 3, mean ± SD.
b +10 μM Ro-41-1040 (MAO-A inhibitor) and 10 μM deprenyl (MAO-B inhibitor).
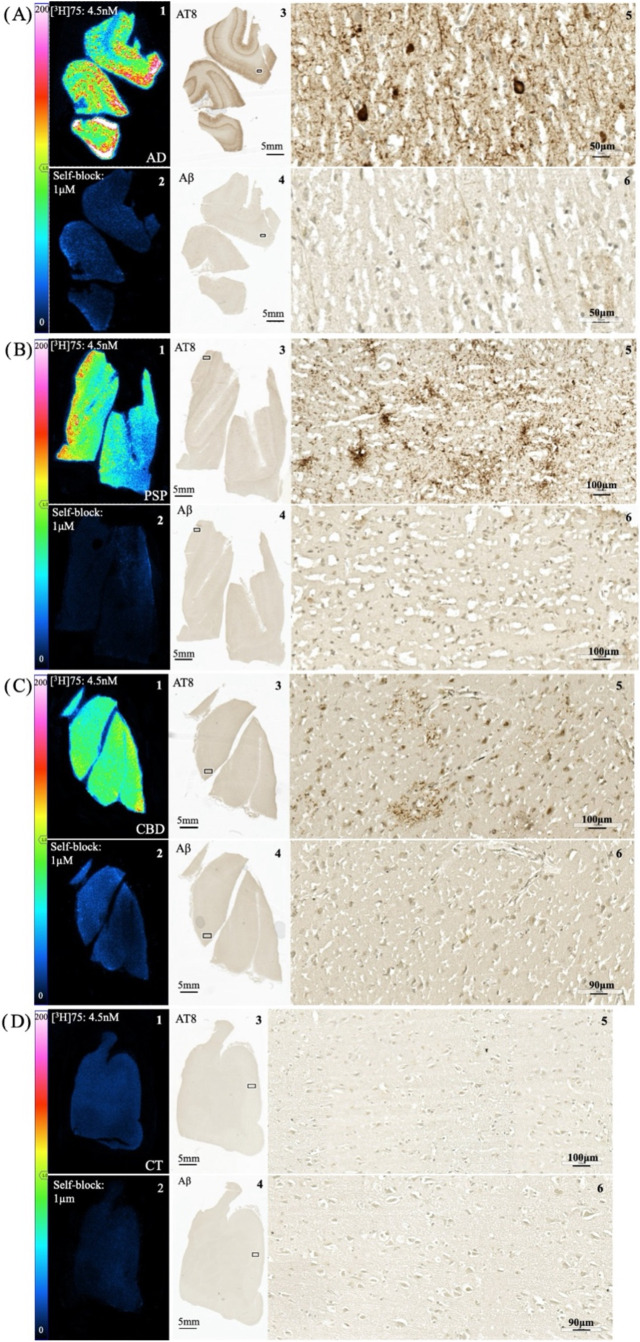
Autoradiography studies with [3H]75 were performed using formalin-fixed, paraffin-embedded (FFPE) post-mortem brain sections from the parietal cortex of AD and PSP subjects, the frontal cortex of a CBD subject, and the cingulate gyrus of an elderly CT subject (Figure ). A clear autoradiographic signal could be observed in AD, PSP, and CBD tissue that corresponded to phospho-tau (AT8) antibody immunohistochemistry (IHC) on adjacent tissue sections. No colocalization was observed with Aβ plaques in AD, PSP, and CBD tissue sections. Specific binding in AD, PSP, and CBD tissue was demonstrated by blocking with 75 (1 μM), while no binding was detected in elderly CT tissue.
Summary and Conclusions
Compound library synthesis and SAR studies based upon the previously identified tau ligands 7, 8, and 9 (Figure ) were performed with the goal of identifying higher affinity tau ligands that also have the potential for radiolabeling with F-18. Thirteen candidate compounds were identified with Ki values vs [3H]8 in the ranges of 5–13 nM in AD and PSP tissues, and 6–14 nM in CBD tissue (Table 1), all 13 of which can potentially be radiolabeled with F-18. The synthesis of precursors for radiolabeling as a 5- or 6-[18F]fluoroindole is more complex due to the need to couple the center aromatic ring to the 2-position of the indole ring without affecting the 5- or 6-position of the indole ring. These syntheses are underway and will be reported in due course. Likewise, the synthesis of individual enantiomers of 27 is underway. Compounds 74 and 75 were thus chosen to be evaluated first due to the ease of radiolabeling precursor synthesis (Scheme 4) and the ease of radiolabeling a 2-[18F]fluoropyridine ring system (Scheme 10). Compounds [18F]74 and [18F]75 were each radiolabeled and evaluated in NHP brain with PET imaging to assess normal brain uptake, clearance, and retention. Both compounds rapidly entered the brain, but [18F]75 showed higher brain uptake and more rapid nonspecific binding clearance (Figure and Table 2). Compound 75 was determined to have low affinity for MAO-B (IC50 = 3.0 μM, Figure S20, Supporting Information), and no other appreciable off-target binding in the Eurofins CNS SafetyScreen panel (Supporting Information). Compound 75 also competed weakly against [3H]PiB in AD tissue (Ki = 61 ± 3 nM, Table S7, Supporting Information) indicating that it does not bind strongly to the PiB binding site on amyloid-β. Homologous binding assays in human post-mortem brain tissue (Table 3) demonstrated that [3H]75 binds with high affinity (KD = 1–1.5 nM) in AD (mixed 3R/4R-tau), PSP (4R-tau), and CBD (4R-tau), and with slightly reduced affinity (KD ∼ 3.8 nM) in PiD (3R-tau). Weak binding was observed in young CT, elderly CT, PD, and TDP-43 cases. Competition binding of existing tau tracers 1–5 against [3H]75 (Table 4) demonstrated that 1, 2, 4, and 5 bind to different locations on tau aggregates than where [3H]75 binds, and that the binding site of 3 has some overlap with the binding site of [3H]75. Autoradiography studies in post-mortem human brain tissue samples (Figure ) demonstrated specific binding of [3H]75 in AD, PSP, and CBD with no significant specific binding in elderly CT tissue. Therefore, [18F]75 is a promising candidate for translation to human brain PET imaging studies with the potential to image tau aggregates not only in 4R-tauopathies, but also in 3R-tauopathies and mixed 3R/4R-tauopathies without the confound of differential sources of off-target binding that are observed with some of the current AD-tau PET radiopharmaceuticals.
Methods
General
Solvents and reagents were used as received. Tetrahydrofuran (THF) was distilled from sodium benzophenone ketyl radical. NMR spectra were obtained on Bruker Avance III spectrometers at the specified frequencies. 1H NMR spectra in CDCl3 are referenced to internal tetramethylsilane (TMS), whereas spectra in DMSO-d6, CD3OD, or acetone-d6 are referenced to solvent residual protons. 13C NMR spectra are referenced to solvent resonances. 19F NMR spectra are unreferenced. Radial chromatography was performed on a Harrison Research Chromatotron using silica rotors from Miles Scientific. Dry silica gel purifications were performed by placing silica gel in a medium-fritted glass filter funnel attached to a vacuum-takeoff adapter (24/40 joint), eluting under vacuum, and collecting fractions in flat-bottom boiling flasks (24/40 joint). Silica gel used was Silicycle SiliFlash P60 40–63 μm (230–400 mesh). Solvent removal was performed on a Buchi Rotary Evaporator. Nonhuman primate PET studies were conducted in accordance with the guidelines set forth by the University of Pittsburgh Institutional Animal Care and Use Committee. In vitro binding assays were performed as previously described.46,65,66 All screened compounds were >95% pure as determined by analytical HPLC (Table S8 and Figures S2–S7, Supporting Information).
Chemistry
tert-Butyl 6-Fluoro-2-(trimethylstannyl)-1H-indole-1-carboxylate (10)
Compound 110 (2.23 g, 9.48 mmol) and Me3SnCl (2.09 g, 10.5 mmol, 1.1 equiv) were flushed with N2(g) for 40 min, then dissolved in freshly distilled THF (100 mL) and cooled in a CH3CN/dry ice bath. LDA solution (2.0 M THF/heptane/ethylbenzene, 6 mL, 12 mmol, 1.3 equiv) was added dropwise over a period of 3 min, the reaction mixture was stirred at CH3CN/dry ice temperature for 5 min, then warmed to ambient temperature and stirred for 4 h. H2O (0.5 mL) was added, the mixture was stirred for 5 min, then concentrated to a dark brown oil. CH2Cl2 and hexane were added and removed to give a dark green/brown syrup/residue that was dissolved in CH2Cl2 and purified by vacuum flash chromatography on silica (15 cm h × 4 cm i.d.): % CH2Cl2/hexane–25% (200 mL), 50% (100 mL) to give a colorless syrup that slowly solidified (3.64 g). The crude product was purified twice by radial chromatography (4 mm silica): hexane (100 mL) to afford 10 (3.52 g, 93%) as a white crystalline solid: 1H NMR (500 MHz, CDCl3) δ 7.64 (dd, 1H, J = 10.5 Hz, J = 2.0 Hz), 7.41 (dd, 1H, J = 8.5 Hz, J = 5.5 Hz), 6.94 (td, 1H, J = 9.0 Hz, J = 2.0 Hz), 6.68 (t, 1H, 3JSnH = 9.0 Hz), 1.70 (s, 9H), 0.30 (t, 9 H, 2JSnH = 28.0 Hz); 13C NMR (100 MHz, CDCl3) δ 160.70 (d, 1JFC = 237.2 Hz), 151.99, 143.73 (d, JFC = 3.9 Hz), 137.71 (d, JFC = 12.4 Hz), 128.72 (t, JSnC = 21.5 Hz), 120.61 (d, JFC = 10.0 Hz), 117.98 (t, JSnC = 21.5 Hz), 110.66 (d, JFC = 24.0 Hz), 102.85 (d, JFC = 28.0 Hz), 84.71, 28.35, −7.01 (t, 1J117SnC = 187.0 Hz, 1J119SnC = 195.5 Hz); HRMS (ESI) [M + H]+ Calcd for C16H23O2NFSn: 400.0729, found: 400.0734.
tert-Butyl 2-(2-Chloropyrimidin-5-yl)-6-fluoro-1H-indole-1-carboxylate (13) and 2-(2-Chloropyrimidin-5-yl)-6-fluoro-1H-indole (14)
Compound 10 (0.380 g, 0.955 mmol), 5-bromo-2-chloropyrimidine (0.250 g, 1.29 mmol, 1.4 equiv), Pd(PPh3)4 (0.080 g, 0.069 mmol, 0.07 equiv), and toluene (25 mL) were stirred at reflux under N2(g) for 15 h, then cooled to ambient temperature and stirred for 3 h. The reaction mixture was poured onto dry silica (55 mm h × 45 mm i.d.) and eluted under vacuum: hexane (50 mL), CH2Cl2 (100 mL), %MeOH/CH2Cl2–1% (100 mL), 2.5% (200 mL), 5% (100 mL), 10% (50 mL) to give an orange/brown residue (0.33 g). Purification by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (100 mL), 50:45:5 (50 mL) afforded 13 (0.057 g, 17%) as an off-white solid, and 14 (0.030 g, 13%) as a light tan solid.
Compound 13
1H NMR (300 MHz, CDCl3) δ 8.68 (s, 2H), 7.94 (dd, 1H, J = 10.5 Hz, J = 2.4 Hz), 7.53 (dd, 1H, J = 8.4 Hz, J = 5.4 Hz), 7.06 (td, 1H, J = 8.7 Hz, J = 2.4 Hz), 6.68 (d, 1H, J = 0.6 Hz), 1.48 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C17H16ClFN3O2: 348.0910, found: 348.0902.
Compound 14
1H NMR (300 MHz, acetone-d6) δ 11.10 (br s, 1H), 9.15 (s, 2H), 7.64 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.19 (m–overlapping resonances, 2H), 6.91 (ddd, 1H, J = 9.9 Hz, J = 8.7 Hz, J = 2.1 Hz); HRMS (ESI) [M + H]+ Calcd for C12H8ClFN3: 248.0385, found: 248.0382.
tert-Butyl 2-(2-Chloropyrimidin-5-yl)-6-cyano-1H-indole-1-carboxylate (15) and 2-(2-Chloropyrimidin-5-yl)-1H-indole-6-carbonitrile (16)
1,4-Dioxane was purged with N2(g) for 40 min. Compound 11 (0.460 g, 1.14 mmol), 5-bromo-2-chloropyrimidine (0.230 g, 1.19 mmol), Pd(dppf)Cl2 (0.078 g, 0.11 mmol, 0.09 equiv), and Na2CO3 (0.140 g, 1.32 mmol, 1.2 equiv) were flushed with N2(g) for 15 min, then 1,4-dioxane (20 mL) was added. The reaction mixture was stirred at reflux under N2(g) for 4 h, cooled, and the solvent was removed to give a brown residue that was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (100 mL), %MeOH/CH2Cl2–1% (100 mL), 2% (100 mL), 3% (200 mL), 4% (100 mL) to give an orange/brown residue (0.160 g, crude 15) and an orange/brown solid (0.062 g, crude 16).
Compound 15
The residue was dissolved in CH2Cl2/MeOH, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: %MeOH/CH2Cl2–1% (100 mL), 2% (100 mL) to give a dark orange residue that was dried under vacuum briefly. The residue was dissolved/suspended in CHCl3 (1 mL), filtered, and the precipitate was rinsed with CHCl3 (1 mL × 2). The filtrate was purified by radial chromatography (1 mm silica): 75:20:5 v/v/v hexane/EtOAc/NEt3 (100 mL) to afford 15 (0.038 g, 9%) as a white solid: 1H NMR (300 MHz, CDCl3) δ 8.71 (s, 2H), 8.58 (d, 1H, J = 0.6 Hz), 7.69 (d, 1H, J = 8.1 Hz), 7.56 (dd, 1H, J = 8.1 Hz, J = 1.5 Hz), 6.77 (s, 1H), 1.49 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C18H16O2N4Cl: 355.0956, found: 355.0958.
Compound 16
The solid was dissolved/suspended in CHCl3 (2.5 mL), filtered, and the precipitate was rinsed with CHCl3 (2.5 mL × 3), then dried under vacuum to afford 16 (0.029 g, 10%) as a yellow/orange solid: 1H NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 9.31 (s, 2H), 7.96 (s, 1H), 7.79 (d, 1H, J = 8.0 Hz), 7.40 (d, 1H, J = 8.0 Hz), 7.36 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 158.62, 156.62, 136.80, 134.87, 131.37, 125.01, 122.21, 121.53, 120.45, 116.71, 103.61, 101.88; HRMS (ESI) [M + H]+ Calcd for C13H8N4Cl: 255.0432, found: 255.0430.
tert-Butyl 2-(2-Chloropyrimidin-5-yl)-1H-indole-1-carboxylate (17)
1-Boc-indole-2-boronic acid (12) (0.250 g, 0.958 mmol), 5-bromo-2-chloropyrimidine (0.180 g, 0.931 mmol), and Pd(dppf)Cl2 (0.052 g, 0.071 mmol, 0.08 equiv) were flushed with N2(g) for 10 min, then 1,4-dioxane (10 mL) was added followed by a solution of K2CO3 in H2O (1.5 mL, 2 M, 3.0 mmol, 3.2 equiv). The reaction mixture was stirred at reflux under N2(g) for 7 h, cooled to ambient temperature, stirred overnight, then filtered through Celite, and the Celite was rinsed with 1,4-dioxane. The filtrate was concentrated to a brown oil, then CHCl3 and hexane were added and removed to give a brown syrup that was dissolved in CH2Cl2, poured onto dry silica (45 mm h × 45 mm i.d.) and eluted under vacuum: CH2Cl2 (200 mL), %MeOH/CH2Cl2–1% (100 mL), 2% (50 mL), 3% (50 mL) to give a tan solid (0.19 g). Purification by radial chromatography (2 mm silica): CH2Cl2 (100 mL) gave an off-white solid (0.11 g) that was again purified by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (35 mL), 85:12:3 (100 mL), 75:20:5 (50 mL) to afford 17 (0.082 g, 27%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.69 (s, 2H), 8.20 (d, 1H, J = 8.4 Hz), 7.60 (d, 1H, J = 7.6 Hz), 7.40 (ddd, 1H, J = 8.4 Hz, J = 7.2 Hz, J = 1.2 Hz), 7.30 (ddd, 1H, J = 7.8 Hz, J = 7.2 Hz, J = 0.8 Hz), 6.71 (s, 1H), 1.49 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 160.12, 158.61, 149.85, 137.70, 131.83, 128.95, 128.04, 125.88, 123.79, 121.23, 116.08, 113.04, 85.33, 28.11; HRMS (ESI) [M + H]+ Calcd for C17H17O2N3Cl: 330.1004, found: 330.1018.
tert-Butyl 6-Fluoro-2-(2-(piperidin-1-yl)pyrimidin-5-yl)-1H-indole-1-carboxylate (18)
Compound 13 (0.042 g, 0.12 mmol), piperidine (0.05 mL, 0.51 mmol, 4.2 equiv), K2CO3 (0.024 g, 0.17 mmol, 1.4 equiv), and CH3CN (10 mL) were stirred at reflux under N2(g) for 75 min, then cooled to ambient temperature. The CH3CN was removed to give a residue that was dissolved in CH2Cl2, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: CH2Cl2 (25 mL), %MeOH/CH2Cl2–1% (50 mL), 2% (75 mL), 3% (50 mL) to afford 18 (0.047 g, 98%) as a light tan solid: 1H NMR (500 MHz, CDCl3) δ 8.33 (s, 2H), 7.93 (dd, 1H, J = 10.5 Hz, J = 2.5 Hz), 7.45 (dd, 1H, J = 8.5 Hz, J = 5.5 Hz), 7.01 (td, 1H, J = 8.5 Hz, J = 2.5 Hz), 6.48 (s, 1H), 3.84 (t, 4H, J = 5.0 Hz), 1.71 (m, 2H), 1.62 (m, 4H), 1.48 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 161.06 (d, 1JFC = 238.8 Hz), 160.86, 157.36, 150.08, 137.63 (d, JFC = 13.8 Hz), 135.95 (d, JFC = 3.8 Hz), 125.64, 121.04 (d, JFC = 10.0 Hz), 116.48, 111.50 (d, JFC = 23.8 Hz), 109.95, 103.42 (d, JFC = 28.8 Hz), 84.63, 45.24, 28.11, 25.96, 25.09; HRMS (ESI) [M + H]+ Calcd for C22H26O2N4F: 397.2034, found: 397.2040.
tert-Butyl 6-Fluoro-2-(2-(4-fluoropiperidin-1-yl)pyrimidin-5-yl)-1H-indole-1-carboxylate (19)
Compound 13 (0.060 g, 0.17 mmol), 4-fluoropiperidine·HCl (0.034 g, 0.24 mmol, 1.4 equiv), K2CO3 (0.088 g, 0.64 mmol, 3.7 equiv), and CH3CN (5 mL) were stirred at reflux under N2(g) for 5 h, then cooled. The solvent was removed to give a residue that was dissolved in CH2Cl2, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum with CH2Cl2 (125 mL) to afford 19 (0.064 g, 89%) as a white foam: 1H NMR (300 MHz, CDCl3) δ 8.35 (s, 2H), 7.93 (dd, 1H, J = 10.8 Hz, J = 2.4 Hz), 7.46 (dd, 1H, J = 8.7 Hz, J = 5.7 Hz), 7.01 (td, 1H, J = 8.7 Hz, J = 2.4 Hz), 6.50 (s, 1H), 4.99 (tt, 0.5 H, J = 6.3 Hz, J = 3.3 Hz) and 4.83 (dt, 0.5 H, J = 9.3 Hz, J = 4.8 Hz) (2JHF = 48.3 Hz), 3.97 (t, 4H, J = 5.7 Hz), 1.93 (m, 4H), 1.48 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H25F2N4O2: 415.1940, found: 415.1944.
tert-Butyl 2-(2-(4,4-Difluoropiperidin-1-yl)pyrimidin-5-yl)-6-fluoro-1H-indole-1-carboxylate (20)
Compound 13 (0.095 g, 0.27 mmol), 4,4-difluoropiperidine·HCl (0.060 g, 0.38 mmol, 1.4 equiv), K2CO3 (0.186 g, 1.35 mmol, 5 equiv), and CH3CN (20 mL) were stirred at reflux under N2(g) for 21 h, then cooled to ambient temperature, filtered, and the precipitate was rinsed with CH3CN. The CH3CN was removed to give an orange residue, then CH2Cl2 and hexane were added and removed to give a light orange solid that was dried under vacuum. The solid was dissolved in CH2Cl2, poured onto dry silica (45 mm h × 45 mm i.d.), and eluted under vacuum with CH2Cl2 (350 mL) to give a colorless residue. Purification by radial chromatography (2 mm silica): %CH2Cl2/hexane–25% (50 mL), 50% (125 mL) gave a white solid (0.046 g) that was purified again by radial chromatography (1 mm silica): %CH2Cl2/hexane–25% (100 mL), 35% (100 mL), 50% (50 mL) to afford 20 (0.032 g, 27%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.37 (s, 2H), 7.93 (dd, 1H, J = 10.8 Hz, J = 2.0 Hz), 7.46 (dd, 1H, J = 8.4 Hz, J = 5.6 Hz), 7.02 (td, 1H, J = 8.8 Hz, J = 2.0 Hz), 6.51 (s, 1H), 4.03 (t, 4H, J = 6.0 Hz), 2.02 (septet, 4H, J = 6.0 Hz), 1.48 (s, 9H) HRMS (ESI) [M + H]+ Calcd for C22H24O2N4F3: 433.1846, found: 433.1836.
6-Fluoro-2-(2-(piperidin-1-yl)pyrimidin-5-yl)-1H-indole (21)
Compound 18 (0.041 g, 0.10 mmol) was dissolved in trifluoroacetic acid (TFA) (1 mL, 13 mmol, 130 equiv), stirred at ambient temperature for 15 min, then poured into a mixture of NaHCO3 (1.23 g, 14.64 mmol, 1.1 equiv TFA), H2O (30 mL), and CH2Cl2 (30 mL). The mixture was stirred, the layers were separated, and the aqueous layer was extracted with CH2Cl2 (10 mL). The combined CH2Cl2 layers were washed with brine (15 mL) and dried over MgSO4. The solution was concentrated, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 90:8:2 (50 mL), 75:20:5 (50 mL), 50:45:5 (75 mL) to afford 21 (0.027 g, 88%) as a light yellow solid: 1H NMR (300 MHz, CDCl3) δ 8.56 (s, 2H), 8.20 (br s, 1H), 7.49 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.08 (dd, 1H, J = 9.6 Hz, J = 2.1 Hz), 6.88 (ddd, 1H, J = 9.6 Hz, J = 8.7 Hz, J = 2.4 Hz), 6.62 (dd, 1H, J = 2.1 Hz, J = 0.9 Hz), 3.84 (t, 4H, J = 5.4 Hz), 1.68 (m, 6 H); 13C NMR (125 MHz, DMSO-d6) δ 160.08, 158.71 (d, 1JFC = 234.4 Hz), 154.49, 136.68 (d, JFC = 13.0 Hz), 134.17 (d, JFC = 3.4 Hz), 125.44, 120.46 (d, JFC = 10.2 Hz), 114.48, 107.73 (d, JFC = 24.1 Hz), 97.05 (d, JFC = 25.7 Hz), 96.91, 44.34, 25.25, 24.27; 19F NMR (470.6 MHz, DMSO-d6) δ −121.61 (m); HRMS (ESI) [M + H]+ Calcd for C17H18FN4: 297.1510, found: 297.1515.
6-Fluoro-2-(2-(4-fluoropiperidin-1-yl)pyrimidin-5-yl)-1H-indole (22)
Compound 19 (0.059 g, 0.14 mmol) was dissolved in TFA (1.5 mL, 19.5 mmol, 139 equiv), stirred at ambient temperature for 20 min, then poured into a mixture of NaHCO3 (1.820 g, 21.67 mmol, 1.1 equiv TFA), H2O (45 mL), and CH2Cl2 (45 mL). The mixture was stirred until the color was gone, then the layers were separated, and the aqueous layer was extracted with CH2Cl2 (10 mL). The combined CH2Cl2 layers were washed with brine (20 mL) and dried over MgSO4. The solution was concentrated, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 90:8:2 (50 mL), 75:20:5 (50 mL), 50:45:5 (100 mL) to afford 22 (0.044 g, 99%) as a light tan solid: 1H NMR (300 MHz, CDCl3) δ 8.58 (s, 2H), 8.21 (br s, 1H), 7.50 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.08 (dd, 1H, J = 9.6 Hz, J = 2.1 Hz), 6.89 (ddd, 1H, J = 9.6 Hz, J = 8.7 Hz, J = 2.1 Hz), 6.64 (dd, 1H, J = 2.1 Hz, J = 0.9 Hz), 5.00 (tt, 0.5 H, J = 6.0 Hz, J = 3.3 Hz) and 4.83 (dt, 0.5 H, J = 9.6 Hz, J = 4.8 Hz) (2JHF = 48.3 Hz), 3.97 (m, 4H), 1.96 (m, 4 H); 1H NMR (300 MHz, DMSO-d6) δ 11.58 (s, 1H), 8.83 (s, 2H), 7.49 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.13 (dd, 1H, J = 9.9 Hz, J = 2.1 Hz), 6.85 (ddd, 1H, J = 9.9 Hz, J = 8.7 Hz, J = 2.1 Hz), 6.82 (d, 1H, J = 1.8 Hz), 4.93 (dtt, 1H, 2JHF = 48.6 Hz, J = 7.2 Hz, J = 3.6 Hz), 3.97 (m, 2H), 3.78 (m, 2H), 1.93 (m, 2H), 1.72 (m, 2H); HRMS (ESI) [M + H]+ Calcd for C17H17F2N4: 315.1416, found: 315.1420.
2-(2-(4,4-Difluoropiperidin-1-yl)pyrimidin-5-yl)-6-fluoro-1H-indole (23)
Compound 20 (0.027 g, 0.062 mmol) was dissolved in TFA (0.6 mL, 7.8 mmol, 126 equiv), stirred at ambient temperature for 20 min, then poured into a mixture of NaHCO3 (0.856 g, 10.2 mmol, 1.3 equiv TFA), H2O (20 mL), and CH2Cl2 (20 mL). The mixture was stirred for 15 min, then the layers were separated, and the aqueous layer was extracted with CH2Cl2 (5 mL × 2). The combined CH2Cl2 layers were washed with brine (15 mL) and dried over MgSO4. The solution was poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: CH2Cl2 (50 mL) %MeOH/CH2Cl2–1% (50 mL), 2% (25 mL), 4% (50 mL) to afford 23 (0.018 g, 87%) as an off-white solid: 1H NMR (300 MHz, CDCl3) δ 8.60 (s, 2H), 8.23 (br s, 1H), 7.51 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.08 (dd, 1H, J = 9.6 Hz, J = 2.1 Hz), 6.90 (ddd, 1H, J = 9.6 Hz, J = 8.7 Hz, J = 2.1 Hz), 6.66 (d, 1H, J = 1.5 Hz), 4.03 (t, 4H, J = 5.7 Hz), 2.04 (tt, 4H, 3JHF = 13.5 Hz, J = 5.7 Hz); HRMS (ESI) [M + H]+ Calcd for C17H16F3N4: 333.1322, found: 333.1314.
2-(2-(3-Fluoro-4-hydroxypyrrolidin-1-yl)pyrimidin-5-yl)-1H-indole-6-carbonitrile (24)
Compound 16 (0.029 g, 0.11 mmol), trans/racemic-4-fluoro-3-hydroxypyrrolidine·HCl (0.033 g, 0.23 mmol, 2 equiv), K2CO3 (0.120 g, 0.868 mmol, 7.9 equiv), and CH3CN (10 mL) were stirred at reflux under N2(g) for 3 h, cooled, and CH3CN was removed to give a dark orange residue. The residue was dissolved/suspended in CH2Cl2/MeOH, filtered, and the precipitate was rinsed with CH2Cl2. The solvent was removed from the filtrate to give an orange residue that was dried under vacuum briefly (0.056 g). The residue was dissolved in CH2Cl2/MeOH, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (100 mL) to afford 24 (0.009 g, 24%) as a light tan solid: 1H NMR (300 MHz, DMSO-d6) δ 12.08 (s, 1H), 8.92 (s, 2H), 7.82 (s, 1H), 7.67 (d, 1H, J = 8.1 Hz), 7.33 (dd, 1H, J = 8.1 Hz, J = 1.5 Hz), 6.98 (d, 1H, J = 1.5 Hz), 5.61 (d, 1H, J = 3.9 Hz), 5.10 (dm, 1H, 2JHF = 49.8 Hz), 4.37 (m, 1H), 3.88 (s, 1H), 3.77 (m, 1H), 3.69 (s, 2H), HRMS (ESI) [M – H]+ Calcd for C17H13FN5O: 322.1099, found: 322.1093.
tert-Butyl 6-Fluoro-2-(2-(3-fluoro-4-hydroxypyrrolidin-1-yl)pyrimidin-5-yl)-1H-indole-1-carboxylate (25)
Compound 13 (0.056 g, 0.16 mmol), trans/racemic-4-fluoro-3-hydroxypyrrolidine·HCl (0.052 g, 0.37 mmol, 2.3 equiv), K2CO3 (0.168 g, 1.22 mmol, 7.5 equiv), and CH3CN (15 mL) were stirred at reflux under N2(g) for 3 h, then cooled to ambient temperature. The CH3CN was removed to give an off-white solid that was dissolved/suspended in CH2Cl2, filtered, and the precipitate was rinsed with CH2Cl2. The CH2Cl2 was removed from the filtrate to give a yellow syrup that was dried under vacuum (89 mg), then dissolved in CH2Cl2/MeOH, poured onto dry silica (45 mm h × 45 mm i.d.), and eluted under vacuum: %MeOH/CH2Cl2–1% (50 mL), 2% (75 mL), 3% (100 mL), 4% (50 mL). The desired fractions were combined and purified by radial chromatography (1 mm silica): %MeOH/CH2Cl2–1% (50 mL), 2% (75 mL), 3% (25 mL) to afford 25 (0.041 g, 61%) as an off-white solid: 1H NMR (300 MHz, CDCl3) δ 8.39 (s, 2H), 7.91 (dd, 1H, J = 10.8 Hz, J = 2.4 Hz), 7.46 (dd, 1H, J = 8.7 Hz, J = 5.7 Hz), 7.01 (td, 1H, J = 8.7 Hz, J = 2.4 Hz), 5.11 (dm, 1H, 2JHF = 51.0 Hz), 4.59 (dm, 1H, J = 6.9 Hz), 4.03 (m, 1H), 3.90 (m, 3H), 1.92 (br d, 1H, J = 2.7 Hz), 1.50 (s, 9H).
tert-Butyl 2-(2-(3-Fluoro-4-hydroxypyrrolidin-1-yl)pyrimidin-5-yl)-1H-indole-1-carboxylate (26)
Compound 17 (0.079 g, 0.24 mmol), trans/racemic-4-fluoro-3-hydroxypyrrolidine·HCl (0.097 g, 0.69 mmol, 2.9 equiv), K2CO3 (0.158 g, 1.14 mmol, 4.7 equiv), and CH3CN (15 mL) were stirred at reflux under N2(g) for 5 h, then cooled to ambient temperature. The CH3CN was removed to give a residue that was dried under vacuum briefly, then dissolved in CH2Cl2, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: CH2Cl2 (25 mL), %MeOH/CH2Cl2–1% (50 mL), 2% (75 mL), 3% (50 mL) to afford 26 (0.085 g, 89%) as an off-white foam: 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 2H), 8.17 (d, 1H, J = 8.4 Hz), 7.55 (d, 1H, J = 7.5 Hz), 7.33 (partially resolved ddd, 1H, J = 7.8 Hz, J = 1.2 Hz), 7.25 (m–partially obscured by CHCl3 resonance, 1H), 6.55 (s, 1H), 5.10 (dm, 1H, 2JHF = 50.7 Hz), 4.59 (m, 1H), 4.03 (m, 1H), 3.91 (m, 3H), 1.89 (d, 1H, J = 3.9 Hz), 1.50 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 161.10 (d, 1JFC = 238.8 Hz), 159.49, 157.47, 150.07, 137.50 (d, JFC = 12.5 Hz), 135.44 (d, JFC = 3.5 Hz), 125.59, 121.21 (d, JFC = 10.0 Hz), 117.49, 111.60 (d, JFC = 25.0 Hz), 110.38, 103.44 (d, JFC = 28.8 Hz), 95.00 (d, 1JFC = 178.8 Hz), 84.94, 73.02 (d, JFC = 27.5 Hz), 52.91, 51.04 (d, JFC = 22.5 Hz), 28.13; HRMS (ESI) [M + H]+ Calcd for C21H23O3N4F2: 417.1733, found: 417.1739.
4-Fluoro-1-(5-(6-fluoro-1H-indol-2-yl)pyrimidin-2-yl)pyrrolidin-3-ol (27)
Compound 25 (0.027 g, 0.065 mmol) was dissolved in TFA (0.75 mL, 9.7 mmol, 149 equiv), stirred at ambient temperature for 15 min, then poured into a mixture of NaHCO3 (0.910 g, 10.8 mmol, 1.1 equiv TFA), H2O (25 mL), and CH2Cl2 (25 mL). The mixture was stirred for 5 min, then the layers were separated, and the aqueous layer was extracted with CH2Cl2 (5 mL × 2). The combined CH2Cl2 layers were dried over MgSO4 and the solvent was removed to give an off-white solid that was dried under vacuum to afford 27 (0.017 g, 83%): 1H NMR (300 MHz, acetone-d6) δ 10.71 (br s, 1H), 8.80 (s, 2H), 7.52 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.11 (dd, 1H, J = 9.9 Hz, J = 2.1 Hz), 6.84 (ddd, 1H, J = 9.9 Hz, J = 8.7 Hz, J = 2.4 Hz), 6.79 (m, 1H), 5.12 (dm, 1H, 2JHF = 51.0 Hz), 4.67 (d, 1H, J = 3.9 Hz), 4.51 (m, 1H), 3.96 (d, 1H, J = 2.1 Hz), 3.85 (m, 1H), 3.79 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 158.74 (d, 1JFC = 234.6 Hz), 159.10, 154,57, 136.75 (d, JFC = 12.6 Hz), 134.13 (d, JFC = 3.5 Hz), 125.43, 120.51, 115.14, 107.76 (d, JFC = 24.3 Hz), 97.09 (d, JFC = 25.5 Hz), 97.07, 95.16 (d, JFC = 177.0 Hz), 71.40 (d, JFC = 27.2 Hz), 52.55, 50.71 (d, JFC = 21.9 Hz); 19F NMR (470.6 MHz, DMSO-d6) δ −121.54 (m), −181.85 (m); HRMS (ESI) [M – H]+ Calcd for C16H13F2N4O: 315.1052, found: 315.1048.
1-(5-(1H-Indol-2-yl)pyrimidin-2-yl)-4-fluoropyrrolidin-3-ol (28)
Compound 26 (0.079 g, 0.20 mmol) was dissolved in CH2Cl2 (5 mL), HCl/1,4-dioxane (4 M, 1 mL, 4 mmol) was added, the mixture was stirred for 5 h, then filtered, and the precipitate was rinsed with EtOEt (2 mL × 3). The solvent was removed from the filtrate to give a light yellow solid that was dried under vacuum briefly, and then dissolved in CH2Cl2 (10 mL). H2O (10 mL) was added followed by conc. NH4OH(aq) (5 drops) and the mixture was stirred for 5 min (pH 11–12). Brine (5 mL) was added, the layers were mixed and separated, and the CH2Cl2 layer was dried over MgSO4. Analysis by TLC (3% MeOH/CH2Cl2) indicated N-Boc protected 26 remained. The solvent was removed to give a white foam that was dried under vacuum (0.050 g). The foam was dissolved in TFA (2 mL, 26 mmol), stirred for 15 min, then poured into a mixture of CH2Cl2 (40 mL), H2O (40 mL), and NaHCO3 (2.420 g, 28.81 mmol, 1.1 equiv TFA). The mixture was stirred for 15 min, then the layers were separated, and the H2O layer was extracted with CH2Cl2 (10 mL). The combined CH2Cl2 layers were washed with brine (25 mL), dried over MgSO4, and the solvent was removed to give an off-white solid (24 mg). Purification by flash column chromatography on silica (3% MeOH/CH2Cl2) afforded 28 (0.021 g, 36%) as an off-white solid: 1H NMR (400 MHz, DMSO-d6) δ 11.47 (s, 1H), 8.86 (s, 2H), 7.50 (d, 1H, J = 8.0 Hz), 7.38 (d, 1H, J = 8.0 Hz), 7.08 (partially resolved ddd, 1H, J = 7.2 Hz, J = 1.2 Hz), 6.99 (partially resolved ddd, 1H, J = 7.2 Hz, J = 1.2 Hz), 6.81 (d 1 H, J = 1.2 Hz), 5.60 (d, 1H, J = 3.6 Hz), 5.09 (dm, 1H, 2JHF = 50.8 Hz), 4.37 (m, 1H), 3.86 (s, 1H), 3.79 (m, 1H), 3.67 (s, 2H); HRMS (ESI) [M – H]+ Calcd for C16H14FN4O: 297.1152, found: 297.1145.
5-Bromo-2-(4-methoxypiperidin-1-yl)pyrimidine (29)
5-Bromo-2-chloropyrimidine (0.880 g, 4.55 mmol), 4-methoxypiperidine (0.65 mL, 5.3 mmol, 1.2 equiv), K2CO3 (0.890 g, 6.44 mmol, 1.4 equiv), and CH3CN (25 mL) were stirred at reflux under N2(g) for 1 h, then cooled to ambient temperature, filtered, and the precipitate was rinsed with CH3CN. The CH3CN was removed to give a faint yellow residue that was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (50 mL), %MeOH/CH2Cl2–1% (100 mL), 2% (100 mL), 3% (100 mL), 4% (100 mL) to afford 29 (1.21 g, 98%) as a white solid: 1H NMR (300 MHz, CDCl3) δ 8.27 (s, 2H), 4.19 (m, 2H), 3.44 (m, 3H), 3.39 (s, 3H), 1.92 (m, 2H), 1.57 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 160.03, 158.08, 105.53, 76.35, 55.92, 41.66, 30.65; HRMS (ESI) [M + H]+ Calcd for C10H15ON3Br: 272.0393, found: 272.0398.
4-(5-Bromopyrimidin-2-yl)morpholine (30)
5-Bromo-2-chloropyrimidine (1.48 g, 7.65 mmol), morpholine (1.0 mL, 12 mmol, 1.6 equiv), K2CO3 (1.30 g, 9.41 mmol, 1.2 equiv), and CH3CN (55 mL) were stirred at reflux under N2(g) for 3 h, then cooled to ambient temperature, filtered, and the precipitate was rinsed with CH3CN. The solvent was removed from the filtrate to give a white solid that was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (100 mL), %MeOH/CH2Cl2–1% (100 mL), 2% (100 mL), 3% (100 mL), 4% (100 mL) to afford 30 (1.74 g, 93%) as a white solid: 1H NMR (300 MHz, CDCl3) δ 8.31 (s, 2H), 3.76 (s, 8H); 13C NMR (125 MHz, CDCl3) δ 160.15, 158.09, 106.39, 66.85, 44.52; HRMS (ESI) [M + H]+ Calcd for C8H11BrN3O: 244.0080, found: 244.0084.
(R)-5-Bromo-2-(3-fluoropyrrolidin-1-yl)pyrimidine ((R)-31)
5-Bromo-2-chloropyrimidine (0.510 g, 2.64 mmol), (R)-3-fluoropyrrolidine·HCl (0.490 g, 3.90 mmol, 1.5 equiv), K2CO3 (1.13 g, 8.18 mmol, 3.1 equiv), and CH3CN (30 mL) were stirred at reflux under N2(g) for 90 min, then cooled to ambient temperature, filtered, and the precipitate was rinsed with EtOAc 3×. The solvent was removed from the filtrate to give a white solid that was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: %CH2Cl2/hexane–50% (50 mL), 75% (100 mL), CH2Cl2 (200 mL), %MeOH/CH2Cl2–1% (100 mL), 2% (100 mL), 3% (100 mL), 5% (100 mL), 10% (50 mL) to afford (R)-31 (0.500 g, 77%) as an off-white solid: 1H NMR (300 MHz, CDCl3) δ 8.32 (s, 2H), 5.36 (dt, 1H, 2JHF = 52.8, J = 3.3 Hz), 3.99–3.57 (m, 4H), 2.45–1.99 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 158.72, 158.18, 106.12, 92.93 (JCF = 174.0 Hz), 53.74 (JCF = 23.0 Hz), 44.69, 32.50 (JCF = 21.0 Hz); HRMS (ESI) [M + H]+ Calcd for C8H10N3BrF: 246.0037, found: 246.0042.
1-(5-Bromopyrimidin-2-yl)piperidin-4-ol (32)
5-Bromo-2-chloropyrimidine (0.970 g, 5.01 mmol), 4-hydroxy-piperidine (0.560 g, 5.54 mmol, 1.1 equiv), K2CO3 (0.920 g, 6.66 mmol, 1.3 equiv), and CH3CN (30 mL) were stirred at reflux under N2(g) for 1 h, then cooled to ambient temperature, filtered, and the precipitate was rinsed with CH3CN. The CH3CN was removed from the filtrate to give a solid that was dissolved/suspended in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: %MeOH/CH2Cl2–1% (100 mL), 2% (100 mL), 3% (200 mL), 5% (150 mL) to afford 32 (1.27 g, 98%) as a white solid: 1H NMR (300 MHz, CDCl3) δ 8.27 (s, 2H), 4.33 (dtd, 2 H, J = 13.8 Hz, J = 4.8 Hz, J = 0.9 Hz), 3.96 (apparent octet, 1H, J = 4.2 Hz), 3.33 (ddd, 2 H, J = 13.5 Hz, J = 9.6 Hz, J = 3.3 Hz), 1.94 (m, 2H), 1.53 (m, 3 H); 13C NMR (75 MHz, CDCl3) δ 160.02, 158.11, 105.66, 68.19, 41.75, 34.22; HRMS (ESI) [M + H]+ Calcd for C9H13ON3Br: 258.0237, found: 258.0242.
1-(5-Bromopyrimidin-2-yl)piperidin-4-one (33)
Compound 32 (0.540 g, 2.09 mmol) was dissolved in CH2Cl2 (25 mL), then H2O (0.05 mL) was added, followed by Dess–Martin periodinane (1.03 g, 2.43 mmol, 1.2 equiv). The reaction mixture was stirred at reflux for 7 h, then cooled to ambient temperature, stirred for 17 h, and poured into a mixture of CH2Cl2 (25 mL), H2O (50 mL), Na2S2O3·5H2O (2.17 g, 8.74 mmol), and NaHCO3 (2.25 g, 26.8 mmol). The mixture was stirred for 2 h, then the layers were separated, and the H2O layer was extracted with CH2Cl2 (10 mL × 2). The combined CH2Cl2 layers were washed with brine (25 mL) and dried over MgSO4. The solution was concentrated, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (200 mL), %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (200 mL) to give an off-white solid (0.55 g). Purification by radial chromatography (4 mm silica): %MeOH/CH2Cl2–1% (100 mL), 2% (100 mL), 4% (50 mL), 5% (100 mL) afforded recovered 32 (0.110 g, 20%) and 33 (0.380 g, 71%) as a white solid: 1H NMR (300 MHz, CDCl3) δ 8.35 (s, 2H), 4.10 (t, 4H, J = 6.3 Hz), 2.50 (t, 4H, J = 6.3 Hz); 13C NMR (125 MHz, CDCl3) δ 208.14, 159.69, 158.42, 107.07, 43.67, 41.04; HRMS (ESI) [M + H]+ Calcd for C9H11BrN3O: 256.0080; found: 256.0092.
5-Bromo-2-(piperidin-1-yl)pyridine (34)
2,5-Dibromopyridine (0.870 g, 3.67 mmol) and piperidine (5.0 mL, 51 mmol, 14 equiv) were stirred under N2(g) in a heated sand bath (105 °C) for 15 h, then cooled to ambient temperature. EtOAc (5 mL) was added, the mixture was filtered, and the precipitate was rinsed with EtOAc (5 mL × 2). The filtrate was concentrated to an oil/residue, then dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane (50 mL), hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (100 mL) to afford 34 (0.860 g, 97%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 8.16 (d, 1H, J = 2.4 Hz), 7.48 (dd, 1H, J = 9.3 Hz, J = 2.4 Hz), 6.54 (d, 1H, J = 9.3 Hz), 3.50 (m, 4H), 1.63 (m, 6 H); 13C NMR (100 MHz, CDCl3) δ 158.35, 148.67, 139.73, 108.61, 106.78, 46.55, 25.59, 24.81; HRMS (ESI) [M + H]+ Calcd for C10H14BrN2: 241.0335, found: 241.0339.
4-(5-Bromopyridin-2-yl)morpholine (35)
2,5-Dibromopyridine (0.870 g, 3.67 mmol) and morpholine (5.0 mL, 58 mmol, 16 equiv) were stirred under N2(g) in a heated sand bath (∼110 °C) for 15 h, then cooled to ambient temperature. EtOAc (5 mL) was added, the mixture was filtered, and the precipitate was rinsed with EtOAc (5 mL × 2). The filtrate was concentrated, diluted with hexane, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (200 mL) to afford 35 (0.860 g, 96%) as an off-white solid: 1H NMR (300 MHz, CDCl3) δ 8.21 (d, 1H, J = 2.4 Hz), 7.56 (dd, 1H, J = 9.0 Hz, J = 2.4 Hz), 6.53 (d, 1H, J = 9.0 Hz), 3.81 (t, 4H, J = 4.8 Hz), 3.47 (t, 4H, J = 4.8 Hz); 13C NMR (125 MHz, CDCl3) δ 158.27, 148.74, 139.97, 108.41, 66.79, 45.71; HRMS (ESI) [M + H]+ Calcd for C9H12ON2Br: 243.0128, found: 243.0139.
4-(5-Bromopyrazin-2-yl)morpholine (36)
Morpholine (0.3 mL, 3.5 mmol) was added to a suspension of NaH (0.087 g, 90%, 3.26 mmol) in THF (6 mL) at 0 °C under N2(g), the mixture was stirred for 15 min, then 2,5-dibromopyrazine (0.806 g, 3.39 mmol) in THF (6 mL) was added, and the reaction mixture was stirred at reflux overnight. The solvent was evaporated, and the residue was dissolved in EtOAc (20 mL), washed with H2O (10 mL), and dried over MgSO4. The solvent was removed and the residue was purified by flash column chromatography (4:1 v/v hexanes/EtOAc) to afford 36 as a white solid (0.858 g, quantitative): 1H NMR (CDCl3, 500 MHz): δ 8.16 (d, 1H, J = 2.0 Hz), 7.86 (d, 1H, J = 2.5 Hz), 3.83 (apparent t, 4H, J = 8.0 Hz), 3.53 (apparent t, 4H, J = 8.0 Hz). HRMS (ESI) [M + H]+ Calcd for C8H11ON3Br: 244.0080, found: 244.0076.
tert-Butyl 6-Fluoro-2-(2-(4-methoxypiperidin-1-yl)pyrimidin-5-yl)-1H-indole-1-carboxylate (37)
Compound 10 (0.650 g, 1.63 mmol), compound 29 (0.490 g, 1.80 mmol, 1.1 equiv), Pd(dppf)Cl2 (0.110 g, 0.150 mmol, 0.09 equiv), and Na2CO3 (0.560 g, 5.28 mmol, 3.2 equiv) were flushed with N2(g) for 10 min, then 1,4-dioxane (45 mL) was added. The reaction mixture was stirred at reflux under N2(g) for 15 h, then cooled to ambient temperature, filtered through Celite, and the Celite was rinsed with 1,4-dioxane. The filtrate was concentrated to a brown syrup that was dried under vacuum, then dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (100 mL), %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (150 mL) to give a brown residue (0.65 g). The residue was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane (25 mL), hexane/EtOAc/NEt3 v/v/v 75:20:5 (100 mL), 50:45:5 (100 mL), 20:75:5 (150 mL) to give a dark orange solid (0.520 g). Purification by radial chromatography (2 mm silica): CH2Cl2 (100 mL), 1% MeOH/CH2Cl2 (50 mL) gave crude 37 (0.300 g, light brown syrup) and crude 42 (0.091 g, light brown residue–further purified below).
Crude 37 was purified by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 95:4:1 (100 mL), 90:8:2 (25 mL) to give a sticky, faint yellow residue that was again purified by radial chromatography (2 mm silica): CHCl3 (50 mL), %EtOH/CHCl3–1% (50 mL), 2% (25 mL) to afford 37 (0.260 g, 37%) as a faint yellow viscous syrup that slowly solidified: 1H NMR (300 MHz, CDCl3) δ 8.34 (s, 2H), 7.93 (dd, 1H, J = 10.8 Hz, J = 2.4 Hz), 7.45 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.01 (td, 1H, J = 8.7 Hz, J = 2.4 Hz), 6.49 (s, 1H), 4.34 (m, 2H), 3.48 (m, 3H), 3.41 (s, 3H), 1.96 (m, 2H), 1.59 (m, 2H), 1.47 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C23H28FN4O3: 427.2140, found: 427.2131.
tert-Butyl (R)-6-Fluoro-2-(2-(3-fluoropyrrolidin-1-yl)pyrimidin-5-yl)-1H-indole-1-carboxylate ((R)-38)
Compound 10 (0.400 g, 1.00 mmol), compound (R)-31 (0.300 g, 1.22 mmol, 1.2 equiv), Pd(dppf)Cl2 (0.076 g, 1.1 mmol, 0.1 equiv), Na2CO3 (0.170 g, 1.60 mmol, 1.6 equiv), and 1,4-dioxane (25 mL) were stirred at reflux under N2(g) for 6 h, then cooled to ambient temperature. The reaction mixture was filtered through Celite, the Celite was rinsed with EtOAC, the filtrate was concentrated to a brown syrup, then CH2Cl2 and hexane were added and removed to give a brown residue that was dried under vacuum. The residue was dissolved in CH2Cl2 (+ a few drops MeOH), poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (100 mL), %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (200 mL) to give a dark orange/brown residue (0.370 g). The residue was dissolved in CH2Cl2/MeOH and purified by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL) to give a light yellow solid (0.170 g) that was purified again by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (100 mL) to give a light yellow solid (0.140 g). A third purification by radial chromatography (2 mm silica): CHCl3 (150 mL), 1% EtOH/CHCl3 (50 mL) afforded (R)-38 (0.130 g, 32%) as an off-white solid: 1H NMR (400 MHz, CDCl3) δ 8.39 (s, 2H), 7.92 (dd, 1H, J = 10.6 Hz, J = 2.4 Hz), 7.46 (dd, 1H, J = 8.8 Hz, J = 5.4 Hz), 7.01 (td, 1H, J = 8.8 Hz, J = 2.4 Hz), 6.51 (s, 1H), 5.40 (dt, 1H, 2JHF = 52.8 Hz, J = 3.2 Hz), 4.03 (ddd, 1H, 3JHF = 25.2 Hz, J = 13.6 Hz, J = 1.6 Hz), 3.93 (t, 1H, J = 10.0 Hz), 3.81 (dd, 0.5 H, J = 13.6 Hz, J = 3.6 Hz), 3.76–3.69 (m–overlapping resonances, 1.5 H), 2.46–2.37 (m, 1H), 2.26–2.06 (m, 1H), 1.49 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C21H23O2N4F2: 401.1784, found: 401.1774.
tert-Butyl 6-Fluoro-2-(2-(4-oxopiperidin-1-yl)pyrimidin-5-yl)-1H-indole-1-carboxylate (39)
1,4-Dioxane was bubbled with N2(g) for 30 min. Compound 10 (0.240 g, 0.603 mmol), compound 33 (0.180 g, 0.703 mmol, 1.2 equiv), Pd(dppf)Cl2 (0.047 g, 0.064 mmol, 0.1 equiv), and Na2CO3 (0.140 g, 1.32 mmol, 2.2 equiv) were flushed with N2(g) for 10 min, then 1,4-dioxane (25 mL) was added, the mixture was stirred at reflux under N2(g) for 5 h, then cooled to ambient temperature. The mixture was filtered through Celite, and the Celite was rinsed with EtOAc. The filtrate was concentrated to a dark orange oil, then CH2Cl2 and hexane were added and removed to give a dark orange residue that was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (100 mL), 50:45:5 (200 mL), 20:75:5 (100 mL) to give a light orange solid (0.134 g). Purification by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (100 mL) gave an off-white solid (0.079 g) that was again purified by radial chromatography (2 mm silica): %MeOH/CH2Cl2–1% (50 mL), 2% (50 mL) to afford 39 (0.070 g, 28%) as an off-white solid: 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 2H), 7.93 (dd, 1H, J = 10.8 Hz, J = 2.4 Hz), 7.47 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.02 (td, 1H, J = 8.7 Hz, J = 2.4 Hz), 6.53 (s, 1H), 4.20 (t, 4H, J = 6.3 Hz), 2.54 (t, 4H, J = 6.3 Hz), 1.49 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H24O3N4F: 411.1827, found: 411.1815.
tert-Butyl 6-Fluoro-2-(6-(piperidin-1-yl)pyridin-3-yl)-1H-indole-1-carboxylate (40)
1,4-Dioxane (30 mL) was purged with N2(g) for 30 min. Compound 10 (0.440 g, 1.11 mmol), compound 34 (0.290 g, 1.20 mmol, 1.1 equiv), Pd(dppf)Cl2 (0.100 g, 0.140 mmol, 0.1 equiv), and Na2CO3 (0.540 g, 5.09 mmol, 4.6 equiv) were flushed with N2(g) for 10 min, then 1,4-dioxane was added. The reaction mixture was stirred at reflux under N2(g) for 5 h, then cooled to ambient temperature, filtered through Celite, and the Celite was rinsed with EtOAc. The solvent was removed from the filtrate to give a brown oil, then CH2Cl2 and hexane were added and removed to give a brown residue that was dried under vacuum. The residue was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: %CH2Cl2/hexane–50% (100 mL), 75% (100 mL), CH2Cl2 (200 mL), %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (100 mL) to give recovered 10 (0.220 g crude, subsequently purified to give 0.094 g, 21% recovery) and a brown residue (0.310 g). The brown residue was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (100 mL), %MeOH/CH2Cl2–1% (100 mL), 2% (100 mL), 3% (200 mL), 5% (200 mL) to give a brown residue (0.270 g) that was purified by radial chromatography (2 mm silica): CHCl3 (100 mL) to give a light brown solid (0.120 g). Purification by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (25 mL) afforded 40 (0.089 g, 20%) as an off-white solid: 1H NMR (300 MHz, CDCl3) δ 8.22 (d, 1H, J = 2.4 Hz), 7.92 (dd, 1H, J = 10.8 Hz, J = 2.4 Hz), 7.45 (m, 2H), 6.99 (td, 1H, J = 8.7 Hz, 2.4 Hz), 6.67 (d, 1H, J = 8.7 Hz), 6.46 (s, 1H), 3.59 (m, 4H), 1.67 (m, 6H), 1.43 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C23H27FN3O2: 396.2082, found: 396.2081.
tert-Butyl 6-Fluoro-2-(6-morpholinopyridin-3-yl)-1H-indole-1-carboxylate (41)
Compound 10 (0.410 g, 1.03 mmol), compound 35 (0.260 g, 1.07 mmol, 1 equiv), Pd(dppf)Cl2 (0.081 g, 0.11 mmol, 0.1 equiv), Na2CO3 (0.320 g, 3.02 mmol, 2.9 equiv) and 1,4-dioxane (25 mL) were stirred at reflux under N2(g) for 6 h, then cooled to ambient temperature. The reaction mixture was filtered through Celite, the Celite was rinsed with EtOAC, the filtrate was concentrated to a brown oil, then CH2Cl2 and hexane were added and removed to give a brown residue that was dried under vacuum. The residue was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 90:8:2 (125 mL), 75:20:5 (100 mL), 50:45: (150 mL), 20:75:5 (200 mL) to give a yellow syrup (0.14 g). Purification by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (125 mL), 75:20:5 (100 mL) afforded 41 (0.066 g, 16%) as a light yellow foam: 1H NMR (400 MHz, CDCl3) δ 8.26 (d, 1H, J = 2.0 Hz), 7.91 (dd, 1H, J = 10.6 Hz, J = 2.0 Hz), 7.54 (dd, 1H, J = 8.8 Hz, J = 2.4 Hz), 7.45 (dd, 1H, J = 8.8 Hz, J = 5.4 Hz), 7.00 (td, 1H, J = 8.8 Hz, J = 2.4 Hz), 6.67 (d, 1H, J = 8.8 Hz), 6.48 (s, 1H), 3.85 (t, 4H, J = 4.8 Hz), 3.56 (t, 4H, J = 4.8 Hz), 1.44 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H25FN3O3: 398.1875, found: 398.1866.
6-Fluoro-2-(2-(4-methoxypiperidin-1-yl)pyrimidin-5-yl)-1H-indole (42)
Crude 42 (0.091 g–from coupling reaction of 37 above) was purified by radial chromatography (1 mm silica): CH2Cl2 (50 mL), %MeOH/CH2Cl2–1% (50 mL), 2% (75 mL) to afford 42 (0.046 g, 9%) as a tan solid: 1H NMR (300 MHz, CDCl3) δ 8.57 (s, 2H), 8.23 (br s, 1H), 7.50 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.08 (dd, 1H, J = 9.6 Hz, J = 2.1 Hz), 6.89 (ddd, 1H, J = 9.7 Hz, J = 8.7 Hz, J = 2.4 Hz), 6.63 (d, 1H, J = 1.5 Hz), 4.31 (m, 2H), 3.50 (m, 3H), 3.41 (s, 3H), 1.97 (m, 2H), 1.62 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 160.04, 158.74 (d, 1JFC = 234.4 Hz), 154.52, 136.70 (d, JFC = 12.7 Hz), 134.06 (d, JFC = 3.5 Hz), 125.42, 120.51 (d, JFC = 10.2 Hz), 114.81, 107.76 (d, JFC = 24.4 Hz), 97.08 (d, JFC = 25.7 Hz), 97.05, 75.49, 54.97, 40.97, 30.20; 19F NMR (470.6 MHz, DMSO-d6) δ −121.53 (m).
Compound 42 from N-Boc Deprotection of 37
Compound 37 (0.065 g, 0.15 mmol) and TFA (1.5 mL, 20 mmol, 129 equiv) were stirred at ambient temperature for 15 min, then poured into a mixture of NaHCO3 (1.84 g, 21.9 mmol, 1.1 equiv TFA), H2O (45 mL), and CH2Cl2 (45 mL). The mixture was stirred, then the layers were separated, and the H2O layer was extracted with CH2Cl2 (10 mL). The combined CH2Cl2 layers were washed with brine (20 mL) and dried over MgSO4. The solution was concentrated, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 90:8:2 (50 mL), 75:20:5 (50 mL), 50:45:5 (100 mL) to afford 42 (0.030 g, 61%) as a light yellow solid: 1H NMR (300 MHz, CDCl3) δ 8.57 (s, 2H), 8.24 (br s, 1H), 7.49 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.07 (dd, 1H, J = 9.3 Hz, J = 2.1 Hz), 6.89 (ddd, 1H, J = 9.6 Hz, J = 8.7 Hz, J = 2.4 Hz), 6.62 (partially resolved dd, 1H, J = 2.1 Hz, J = 0.6 Hz), 4.31 (m, 2H), 3.50 (m, 3H), 3.41 (s, 3H), 1.97 (m, 2H), 1.62 (m, 2H); HRMS (ESI) [M + H]+ Calcd for C18H20FN4O: 327.1616, found: 327.1620.
(R)-6-Fluoro-2-(2-(3-fluoropyrrolidin-1-yl)pyrimidin-5-yl)-1H-indole ((R)-43)
Compound (R)-38 (0.120 g, 0.300 mmol) was dissolved in TFA (3 mL, 39 mmol, 130 equiv), stirred at ambient temperature for 20 min, then poured into a mixture of NaHCO3 (3.95 g, 47.0 mmol, 1.2 equiv TFA), H2O (75 mL), and CH2Cl2 (75 mL). The mixture was stirred for 20 min, then the layers were separated, and the aqueous layer was extracted with CH2Cl2 (25 mL × 2). The combined CH2Cl2 layers were washed with brine (25 mL), dried over MgSO4, and concentrated (precipitate formed). MeOH was added to redissolve the precipitate, then the solution was poured onto dry silica (55 mm h × 45 mm i.d.) and eluted under vacuum: %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (200 mL), 10% (50 mL) to give a yellow solid (85 mg). The solid was placed in a medium-fritted filter, rinsed with 1:1 v/v CH2Cl2/hexane (1 mL × 7), and dried under vacuum to afford (R)-43 (0.067 g, 74%) as a light yellow solid: 1H NMR (300 MHz, acetone-d6) δ 10.71 (br s, 1H), 8.80 (s, 2H), 7.52 (dd, 1H, J = 8.4 Hz, J = 5.4 Hz), 7.10 (dd, 1H, J = 9.9 Hz, J = 2.1 Hz), 6.84 (ddd, 1H, J = 9.9 Hz, J = 8.7 Hz, J = 2.4 Hz), 6.79 (s, 1H), 5.45 (dm, 1H, 2JHF = 52.8 Hz), 4.01–3.58 (m, 4H), 2.37–2.12 (m, 2H); 13C NMR (MHz, CDCl3) δ 158.76 (d, 1JFC = 233.8 Hz), 158.93, 154.54, 136.80 (d, J = 12.5 Hz), 134.17 (d, J = 3.8 Hz), 125.45, 120.53 (d, J = 10.0 Hz), 115.03, 107.79 (d, J = 23.8 Hz), 97.10 (d, J = 26.3 Hz), 97.07, 93.10 (d, J = 171.3 Hz), 53.24 (d, J = 22.5 Hz), 44.28, 31.50 (d, J = 21.3 Hz); HRMS (ESI) [M + H]+ Calcd for C16H15N4F2: 301.1259, found: 301.1253.
1-(5-(6-Fluoro-1H-indol-2-yl)pyrimidin-2-yl)piperidin-4-one (44)
Compound 39 (0.065 g, 0.16 mmol) was dissolved in TFA (1.6 mL, 21 mmol, 131 equiv), stirred at ambient temperature for 20 min, then poured into a mixture of NaHCO3 (2.13 g, 25.4 mmol, 1.2 equiv TFA), H2O (40 mL), and CH2Cl2 (40 mL). The mixture was stirred for 10 min, then the layers were separated, and the aqueous layer was extracted with CH2Cl2 (15 mL × 2). The combined CH2Cl2 layers were washed with brine (25 mL), dried over MgSO4, concentrated, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (200 mL), 10% (50 mL) to give an off-white solid (43 mg). Purification by radial chromatography (1 mm silica): %MeOH/CH2Cl2–1% (50 mL), 2.5% (25 mL) afforded 44 (0.038 g, 77%) as a light tan solid: 1H NMR (300 MHz, CDCl3) δ 8.65 (s, 2H), 8.27 (br s, 1H), 7.52 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.10 (dd, 1H, J = 9.3 Hz, J = 2.1 Hz), 6.91 (ddd, 1H, J = 9.6 Hz, J = 8.7 Hz, J = 2.1 Hz), 6.68 (dd, 1H, J = 2.1 Hz, J = 0.6 Hz), 4.20 (t, 4H, J = 6.0 Hz), 2.55 (t, 4H, J = 6.0 Hz); HRMS (ESI) [M + H]+ Calcd for C17H16FN4O: 311.1303, found: 311.1296. X-ray quality crystals were grown by slow evaporation of acetone.
6-Fluoro-2-(6-(piperidin-1-yl)pyridin-3-yl)-1H-indole (45)
Compound 40 (0.082 g, 0.21 mmol) was dissolved in TFA (2 mL, 26 mmol, 126 equiv), stirred at ambient temperature for 20 min, then poured into a mixture of Na2CO3 (2.43 g, 28.9 mmol, 1.1 equiv TFA), H2O (60 mL), and CH2Cl2 (60 mL). The mixture was stirred for 10 min, then the layers were separated and the aqueous layer was extracted with CH2Cl2 (10 mL × 3). The combined CH2Cl2 layers were washed with brine (30 mL), dried over MgSO4, then poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: %MeOH/CH2Cl2–1% (50 mL), 2% (75 mL), 3% (50 mL) to give an off-white solid (0.058 g). Purification by radial chromatography (1 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (40 mL), 75:20:5 (50 mL) afforded 45 (0.050 g, 82%) as an off-white solid: 1H NMR (300 MHz, CDCl3) δ 8.46 (d, 1H, J = 2.4 Hz), 8.28 (br s, 1H), 7.68 (dd, 1H, J = 9.0 Hz, J = 2.4 Hz), 7.48 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.06 (dd, 1H, J = 9.6 Hz, J = 1.8 Hz), 6.86 (ddd, 1H, J = 9.6 Hz, J = 8.7 Hz, J = 2.1 Hz), 6.70 (d, 1H, J = 8.7 Hz), 6.61 (d, 1H, J = 1.5 Hz), 3.59 (m, 4H), 1.67 (m, 6 H); 13C NMR (125 MHz, DMSO-d6) δ 158.57 (d, 1JFC = 233.9 Hz), 157.98, 144.42, 136.86 (d, JFC = 3.5 Hz), 136.66 (d, JFC = 12.6 Hz), 134.13, 125.61, 120.25 (d, JFC = 9.9 Hz), 116.64, 107.49 (d, JFC = 24.0 Hz), 106.98, 96.95 (d, JFC = 25.5 Hz), 96.51, 45.50, 24.99, 24.29; 19F NMR (470.6 MHz, DMSO-d6) δ −122.04 (m); HRMS (ESI) [M + H]+ Calcd for C18H19FN3: 296.1558, found: 296.1568.
4-(5-(6-Fluoro-1H-indol-2-yl)pyridin-2-yl)morpholine (46)
Compound 41 (0.061 g, 0.15 mmol) was dissolved in TFA (1.5 mL, 20 mmol, 127 equiv), stirred at ambient temperature for 20 min, then poured into a mixture of Na2CO3 (1.82 g, 21.7 mmol, 1.1 equiv TFA), H2O (40 mL), and CH2Cl2 (40 mL). The mixture was stirred for 15 min, then the layers were separated, and the aqueous layer was extracted with CH2Cl2 (15 mL × 2). The combined CH2Cl2 layers were washed with brine (25 mL), dried over MgSO4, then poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: %MeOH/CH2Cl2–1% (50 mL), 2% (75 mL), 3% (50 mL) to afford 46 (0.042 g, 92%) as a light tan solid: 1H NMR (300 MHz, CDCl3) δ 8.50 (d, 1H, J = 2.1 Hz), 8.28 (br s, 1H), 7.75 (dd, 1H, J = 8.7 Hz, J = 2.4 Hz), 7.49 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.07 (dd, 1H, J = 9.3 Hz, J = 2.1 Hz), 6.88 (ddd, 1H, J = 9.6 Hz, J = 8.7 Hz, J = 2.1 Hz), 6.71 (d, 1H, J = 8.7 Hz), 6.65 (dd, 1H, J = 2.1 Hz, J = 0.6 Hz), 3.85 (t, 4H, J = 4.8 Hz), 3.57 (t, 4H, J = 4.8 Hz); 13C NMR (125 MHz, DMSO-d6) δ 158.66 (d, 1JFC = 234.3 Hz), 158.19, 144.28, 136.72 (d, JFC = 12.7 Hz), 136.54 (d, JFC = 3.4 Hz), 134.19, 125.55, 120.41 (d, JFC = 10.1 Hz), 117.95, 107.60 (d, JFC = 24.3 Hz), 106.92, 97.00 (d, JFC = 25.5 Hz), 96.99, 65.91, 45.04; 19F NMR (470.6 MHz, DMSO-d6) δ −121.79; HRMS (ESI) [M + H]+ Calcd for C17H17FN3O: 298.1350, found: 298.1345.
1-(5-(5-Fluorobenzofuran-2-yl)pyrimidin-2-yl)piperidin-4-ol (49)
1,4-Dioxane (30 mL) and H2O (6 mL) were combined and purged with N2(g) for 45 min. 5-Fluorobenzofuran-2-boronic acid (47) (0.250 g, 1.39 mmol), compound 32 (0.420 g, 1.63 mmol, 1.2 equiv), Pd(dppf)Cl2·CH2Cl2 (0.097 g, 0.12 mmol, 0.09 equiv), and Na2CO3 (0.710 g, 6.70 mmol, 4.8 equiv) were flushed with N2(g) for 10 min, then the H2O/1,4-dioxane mixture (17 mL) was added. The reaction mixture was stirred under N2(g) in a heated sand bath (∼95 °C) for 14 h, then cooled to ambient temperature, filtered through Celite, and the Celite was rinsed with EtOAc. The filtrate was concentrated, then EtOAc was added and removed to give a brown residue that was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 75:20:5 (100 mL), 50:45:5 (150 mL), 20:75:5 (250 mL), EtOAc (50 mL). The desired fractions were combined and purified by radial chromatography (2 mm silica): %EtOH/CHCl3–1% (50 mL), 2% (75 mL), 3% (100 mL) to give a tan solid (77 mg). The solid was suspended in CHCl3 (2 mL), filtered, rinsed with CHCl3 (1.5 mL × 2) and dried under vacuum to afford 49 (0.047 g, 11%) as a white solid: 1H NMR (300 MHz, DMSO-d6) δ 8.85 (s, 2H), 7.60 (dd, 1H, J = 9.0 Hz, J = 4.2 Hz), 7.43 (dd, 1H, J = 9.0 Hz, J = 2.4 Hz), 7.26 (s, 1H), 7.10 (td, 1H, J = 9.0 Hz, J = 2.4 Hz), 4.77 (d, 1H, J = 4.2 Hz), 4.30 (partially resolved dddd, 2 H, J = 13.5 Hz, J = 4.5 Hz), 3.77 (apparent octet, 1H, J = 4.2 Hz), 3.39 (ddd–partially obscured by H2O resonance, 2 H, J = 9.6 Hz, J = 3.3 Hz), 1.80 (m, 2H), 1.36 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 160.37, 158.80 (d, 1JFC = 235.0 Hz), 154.72, 154.02, 150.27, 129.89 (d, J = 11.3 Hz), 112.27, 111.90 (d, J = 10.0 Hz), 111.31 (d, J = 26.3 Hz), 106.16 (d, J = 25.0 Hz), 100.00 (d, J = 3.8 Hz), 65.86, 41.17, 33.91; HRMS (ESI) [M + H]+ Calcd for C17H17FN3O2: 314.1299, found: 314.1296.
tert-Butyl 2-(2-(4-Hydroxypiperidin-1-yl)pyrimidin-5-yl)-1H-indole-1-carboxylate (50)
1,4-Dioxane (10 mL) and H2O (2 mL) were combined and purged with N2(g) for 10 min. Compound 32 (0.240 g, 0.930 mmol), compound 12 (0.260 g, 0.996 mmol, 1.1 equiv), Pd(dppf)Cl2 (0.094 g, 0.12 mmol, 0.1 equiv), and Na2CO3 (0.450 g, 4.25 mmol, 4.6 equiv) were flushed with N2(g) for 15 min, then the H2O/1,4-dioxane mixture was added. The reaction mixture was stirred under N2(g) in a heated sand bath (85 °C) for 16 h, then cooled to ambient temperature, filtered through Celite, and the Celite was rinsed with EtOAc. The solvent was removed to give a black syrup that was dissolved in CH2Cl2, poured onto dry silica (45 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (100 mL), %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (100 mL). The desired fractions were combined, concentrated, poured onto dry silica (45 mm h × 45 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 90:8:2 (50 mL), 75:20:5 (100 mL), 50:45:5 (100 mL), 20:75:5 (200 mL) to give an orange/tan foam (0.25 g). Purification by radial chromatography (2 mm silica): CHCl3 (50 mL), %EtOH/CHCl3–1% (50 mL), 2% (75 mL), 3% (50 mL) afforded 50 (0.230 g, 63%) as a light yellow foam: 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 2H), 8.19 (d, 1H, J = 8.4 Hz), 7.54 (d, 1H, J = 6.0 Hz), 7.32 (apparent t, 1H, J = 7.6 Hz), 7.25 (apparent t–partially obscured by CHCl3 resonance, 1H, J = 7.6 Hz), 6.53 (s, 1H), 4.46 (partially resolved dddd, 2 H, J = 13.6 Hz, J = 4.4 Hz), 3.99 (br s, 1H), 3.40 (partially resolved ddd, 2 H, J = 11.4 Hz, J = 2.8 Hz), 1.98 (m, 2H), 1.57 (m, 2H), 1.48 (s, 10 H); 13C NMR (125 MHz, CDCl3) δ 160.70, 157.39, 150.29, 137.39, 135.32, 129.34, 124.63, 123.28, 120.56, 117.25, 115.87, 110.52, 84.25, 68.31, 41.75, 34.32, 28.13; HRMS (ESI) [M + H]+ Calcd for C22H27O3N4: 395.2078, found: 395.2073.
tert-Butyl 5-Fluoro-2-(2-(4-hydroxypiperidin-1-yl)pyrimidin-5-yl)-1H-indole-1-carboxylate (51)
1,4-Dioxane (30 mL) and H2O (6 mL) were combined and purged with N2(g) for 45 min. Compound 48 (0.250 g, 0.896 mmol), compound 32 (0.250 g, 0.969 mmol, 1.1 equiv), Pd(dppf)Cl2·CH2Cl2 (0.079 g, 0.097 mmol, 0.1 equiv), and Na2CO3 (0.470 g, 4.43 mmol, 5 equiv) were flushed with N2(g) for 10 min, then the H2O/1,4-dioxane mixture (16 mL) was added. The reaction mixture was stirred under N2(g) in a heated sand bath (∼95 °C) for 14 h, then cooled to ambient temperature, filtered through Celite, and the Celite was rinsed with EtOAc. The filtrate was concentrated to an oil, then EtOAc was added and removed to give a brown residue that was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 75:20:5 (100 mL), 50:45:5 (150 mL), 20:75:5 (200 mL) to give a tan foam (0.230 g). Purification by radial chromatography (2 mm silica): %EtOH/CHCl3–1% (50 mL), 2% (75 mL) afforded 51 (0.170 g, 46%) as an off-white foam: 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 2H), 8.14 (dd, 1H, J = 8.8 Hz, J = 4.8 Hz), 7.19 (dd, 1H, J = 8.8 Hz, J = 2.4 Hz), 7.04 (td, 1H, J = 9.2 Hz, J = 2.4 Hz), 6.49 (s, 1H), 4.46 (partially resolved dddd, 2 H, J = 13.6 Hz, J = 4.4 Hz), 4.00 (apparent octet, 1H, J = 4.4 Hz), 3.40 (ddd, 1H, J = 10.0 Hz, J = 3.2 Hz), 1.98 (m, 2H), 1.56 (m, 3H), 1.47 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 160.81, 159.58 (d, 1JFC = 238.0 Hz), 157.42, 150.10, 137.01, 133.80, 130.17 (d, JFC = 9.9 Hz), 116.99 (d, JFC = 4.9 Hz), 116.94, 112.32 (d, JFC = 24.6 Hz), 110.12 (d, JFC = 3.9 Hz), 105.91 (d, JFC = 23.5 Hz), 84.56, 68.36, 41.75, 34.36, 28.14; HRMS (ESI) [M + H]+ Calcd for C22H26O3N4F: 413.1984, found: 413.1987.
1-(5-(1H-Indol-2-yl)pyrimidin-2-yl)piperidin-4-ol (52)
Compound 50 (0.083 g, 0.21 mmol) was dissolved in TFA (2.0 mL, 26 mmol, 124 equiv), stirred at ambient temperature for 20 min, then poured into a mixture of NaHCO3 (2.40 g, 28.6 mmol, 1.1 equiv TFA), H2O (60 mL), and CH2Cl2 (60 mL). The mixture was stirred until gas evolution ceased, then the layers were separated, and the aqueous layer was extracted with CH2Cl2 (10 mL). The combined CH2Cl2 layers were washed with brine (25 mL), dried over MgSO4, and concentrated which produced a precipitate. MeOH was added (several drops) until the precipitate dissolved, then the solution was poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (150 mL), 10% (50 mL) to give an off-white solid. The solid was dissolved/suspended in CHCl3 (∼4 mL), then MeOH (∼0.5 mL) was added to get the remaining solid to dissolve. The solution was purified by radial chromatography (2 mm silica): CHCl3 (25 mL), %EtOH/CHCl3–1% (50 mL), 2% (75 mL), 3% (50 mL), 4% (50 mL), 10% (60 mL) to afford 52 (0.058 g, 94%) as an off-white solid: 1H NMR (400 MHz, acetone-d6) δ 10.59 (br s, 1H), 8.80 (s, 2H), 7.54 (d, 1H, J = 7.6 Hz), 7.37 (d, 1H, J = 8.4 Hz), 7.08 (td, 1H, J = 7.6 Hz, J = 1.2 Hz), 7.01 (td, 1H, J = 7.6 Hz, J = 1.2), 6.78 (d, 1H, J = 1.2 Hz), 4.42 (partially resolved dddd, 2 H, J = 13.6 Hz, J = 4.4 Hz), 3.91 (apparent octet, 1H, J = 4.4 Hz), 3.84 (d, 1H, J = 4.4 Hz), 3.42 (ddd, 2 H, J = 13.4 Hz, J = 9.8 Hz, J = 3.2 Hz), 1.90 (m, 2H), 1.48 (m, 2H); HRMS (ESI) [M + H]+ Calcd for C17H19N4O: 295.1553, found: 295.1553.
1-(5-(5-Fluoro-1H-indol-2-yl)pyrimidin-2-yl)piperidin-4-ol (53)
Compound 51 (0.090 g, 0.22 mmol) was dissolved in TFA (2 mL, 26 mmol, 119 equiv), stirred at ambient temperature for 20 min, then poured into a mixture of NaHCO3 (2.45 g, 29.2 mmol, 1.1 equiv TFA), H2O (60 mL), and CH2Cl2 (60 mL). The mixture was stirred for 20 min, the layers were separated, and the aqueous layer was extracted with CH2Cl2 (10 mL). The combined CH2Cl2 layers were washed with brine (25 mL) and dried over MgSO4. The solution was concentrated, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (50 mL), %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (100 mL), 10% (100 mL) to give a faint yellow solid (54 mg). Purification by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 75:20:5 (100 mL), 50:45:5 (100 mL), 20:75:5 (250 mL) afforded 53 (0.047 g, 69%) as a light tan solid: 1H NMR (300 MHz, acetone-d6) δ 10.67 (br s, 1H), 8.79 (s, 2H), 7.36 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.23 (dd, 1H, J = 9.6 Hz, J = 2.7 Hz), 6.87 (ddd, 1H, J = 9.6 Hz, J = 8.7 Hz, J = 2.7 Hz), 6.77 (m, 1H), 4.41 (partially resolved dddd, 2 H, J = 13.5 Hz, J = 4.5 Hz), 3.92 (apparent octet, 1H, J = 4.2 Hz), 3.83 (d, 1H, J = 4.2 Hz), 3.43 (ddd, 2 H, J = 13.2 Hz, J = 9.6 Hz, J = 3.3 Hz), 1.90 (m, 2H), 1.48 (dddd, 2 H, J = 13.2 Hz, J = 9.0 Hz, J = 4.2 Hz); 13C NMR (125 MHz, DMSO-d6) δ 160.16, 157.19 (d, 1JFC = 230.0 Hz), 154.77, 135.39, 133.50, 128.96 (d, J = 10.0 Hz), 114.52, 111.83 (d, J = 10.0 Hz), 109.11 (d, J = 25.0 Hz), 104.15 (d, J = 23.8 Hz), 97.18 (d, J = 5.0 Hz), 66.04, 41.23, 33.92; HRMS (ESI) [M + H]+ Calcd for C17H18FN4O: 313.1459, found: 313.1456.
2-Fluoro-6-(piperidin-1-yl)pyridine (54)
2,6-Difluoropyridine (1.340 g, 11.64 mmol), piperidine (1.2 mL, 12 mmol, 1 equiv), i-Pr2NEt (3.2 mL, 18 mmol, 1.6 equiv), and 1,4-dioxane (10 mL) were stirred at reflux under N2(g) for 90 min, then cooled to ambient temperature. The reaction mixture was concentrated to a yellow oil, CH2Cl2 and hexane were added, the solution was poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane (100 mL), % CH2Cl2/hexane–10% (100 mL), 25% (200 mL), 50% (100 mL), 75% (100 mL), CH2Cl2 (100 mL) to afford 54 (1.500 g, 71%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.48 (dd, 1H, JHF = 16.7 Hz, J = 8.1 Hz), 6.40 (dd, 1H, J = 8.1 Hz, J = 2.7 Hz), 6.09 (dd, 1H, J = 7.8 Hz, J = 3.0 Hz), 3.51 (m, 4H), 1.64 (m, 6 H); 19F NMR (282.5 MHz, CDCl3) δ −68.60 (d, J = 6.5 Hz); 13C NMR (125 MHz, CDCl3) δ 162.96 (d, 1JCF = 233.0 Hz), 158.58 (d, 3JCF = 15.9 Hz), 141.73 (d, 3JCF = 8.4 Hz), 102.69 (d, 4JCF = 4.0 Hz), 94.86 (d, 2JCF = 37.6 Hz), 46.12, 25.46, 24.70; HRMS (ESI) [M + H]+ Calcd for C10H14FN2: 181.1136, found: 181.1133.
4-(6-Fluoropyridin-2-yl)morpholine (55)
2,6-Difluoropyridine (1.340 g, 11.64 mmol), morpholine (1.0 mL, 12 mmol, 1 equiv), i-Pr2NEt (3 mL, 17 mmol, 1.5 equiv), and 1,4-dioxane (10 mL) were stirred at reflux under N2(g) for 90 min, cooled to ambient temperature, and concentrated to an oil that was dried under vacuum to give an off-white solid. The solid was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane (50 mL), % CH2Cl2/hexane–25% (50 mL), 50% (75 mL), 75% (100 mL), CH2Cl2 (500 mL), %MeOH/CH2Cl2–2% (75 mL), 5% (50 mL), 10% (100 mL) to afford 55 (0.720 g, 34%) as a colorless oil that slowly became a white solid: 1H NMR (300 MHz, CDCl3) δ 7.55 (dd, 1H, JCF = 16.5 Hz, J = 8.1 Hz), 6.41 (dd, 1H, J = 8.1 Hz, J = 2.7 Hz), 6.21 (dd, 1H, J = 7.8 Hz, J = 2.7 Hz), 3.80 (t, 4H, J = 4.8 Hz), 3.50 (t, 4H, J = 4.8 Hz); 19F NMR (282.5 MHz, CDCl3) δ −68.47 (d, J = 7.1 Hz); 13C NMR (125 MHz, CDCl3) δ 162.87 (d, 1JCF = 234.6 Hz), 158.64 (d, 3JCF = 15.5 Hz), 142.07 (d, 3JCF = 8.3 Hz), 102.74 (d, 4JCF = 4.0 Hz), 96.74 (d, 2JCF = 37.1 Hz), 66.65, 45.40; HRMS (ESI) [M + H]+ Calcd for C9H12FN2O: 183.0928, found: 183.0926.
2-Nitro-6-(piperidin-1-yl)pyridine (56)
2-Chloro-6-nitropyridine (0.280 g, 1.77 mmol), piperidine (0.2 mL, 2 mmol, 1.1 equiv), i-Pr2NEt (0.5 mL, 3 mmol, 1.6 equiv), and 1,4-dioxane (5 mL) were stirred at reflux under N2(g) for 2 h, then cooled to ambient temperature. The reaction mixture was poured onto dry silica (55 mm h × 45 mm i.d.) and eluted under vacuum: hexane (50 mL), hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (150 mL) to give a yellow/orange syrup (0.270 g). Purification by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (25 mL) afforded 56 (0.160 g, 44%) as an orange syrup: 1H NMR (300 MHz, CDCl3) δ 7.64 (dd, 1H, J = 8.4 Hz, J = 7.5 Hz), 7.37 (d, 1H, J = 7.5 Hz), 6.89 (d, 1H, J = 8.4 Hz), 3.63 (m, 4H), 1.67 (m, 6 H); 13C NMR (125 MHz, CDCl3) δ 157.98, 156.15, 140.02, 111.88, 104.61, 46.03, 25.55, 24.62; HRMS (ESI) [M + H]+ Calcd for C10H14O2N3: 208.1081, found: 208.1083.
4-(6-Nitropyridin-2-yl)morpholine (57)
2-Chloro-6-nitropyridine (0.280 g, 1.77 mmol), morpholine (0.2 mL, 2 mmol, 1.3 equiv), i-Pr2NEt (0.5 mL, 3 mmol, 1.6 equiv), and 1,4-dioxane (5 mL) were stirred at reflux under N2(g) for 4 h, then cooled to ambient temperature, filtered, and the precipitate was rinsed with EtOAc. The solvent was removed from the filtrate to give an orange residue that was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (200 mL), %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (150 mL) to give a yellow/orange solid. Purification by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (75 mL) afforded 57 (0.140 g, 38%) as a yellow/orange solid: 1H NMR (300 MHz, CDCl3) δ 7.73 (dd, 1H, J = 8.4 Hz, J = 7.5 Hz), 7.49 (d, 1H, J = 7.5 Hz), 6.90 (d, 1H, J = 8.4 Hz), 3.83 (t, 4H, J = 5.1 Hz), J = 3.63 (t, 4H, J = 5.1 Hz); 13C NMR (125 MHz, CDCl3) δ 158.17, 156.06, 140.49, 111.79, 106.21, 66.67, 45.27; HRMS (ESI) [M + H]+ Calcd for C9H12N3O3: 210.0873, found: 210.0871.
3-Bromo-2-fluoro-6-(piperidin-1-yl)pyridine (58)
Compound 54 (0.770 g, 4.27 mmol) was dissolved in CH3CN (25 mL) under N2(g) and cooled to 0 °C. NBS (0.860 g, 4.83 mmol, 1.1 equiv) was added, the reaction mixture was stirred at 0 °C for 5 min, then warmed to ambient temperature and stirred for 6 h. The CH3CN was removed to give a light tan residue that was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum with CH2Cl2 (150 mL) to give a colorless oil (1.26 g). Purification by radial chromatography (2 mm silica): %CH2Cl2/hexane–10% (100 mL), 25% (50 mL) afforded 58 (0.780 g, 70%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.57 (apparent t, 1H, J = 8.7 Hz), 6.33 (dd, 1H, J = 8.7 Hz, J = 1.5 Hz), 3.49 (m, 4H), 1.62 (m, 6 H); 19F NMR (470.6 MHz, CDCl3) δ −66.52 (d, J = 8.5 Hz); 13C NMR (125 MHz, CD3OD) δ 159.43 (d, 1JCF = 229.3 Hz), 158.53 (d, 3JCF = 14.9 Hz), 145.54 (d, 3JCF = 2.4 Hz), 106.08 (d, 4JCF = 4.4 Hz), 87.26 (d, 2JCF = 38.4 Hz), 47.17, 26.48, 25.66; HRMS (ESI) [M + H]+ Calcd for C10H1379BrFN2: 259.0241, found: 259.0238.
4-(5-Bromo-6-fluoropyridin-2-yl)morpholine (59)
Compound 55 (0.360 g, 1.98 mmol) was dissolved in CH3CN (12 mL) under N2(g) and cooled to 0 °C. NBS (0.390 g, 2.19 mmol, 1.1 equiv) was added, the reaction mixture was stirred at 0 °C for 5 min, then warmed to ambient temperature and stirred for 6 h. The CH3CN was removed to give a red/brown syrup that was dissolved in CH2Cl2, poured onto dry silica (45 mm h × 45 mm i.d.), and eluted under vacuum with CH2Cl2 (300 mL), %MeOH/CH2Cl2–1% (100 mL), 2% (100 mL) to give a white solid (0.54 g). Purification by radial chromatography (2 mm silica): %CH2Cl2/hexane–25% (100 mL), 50% (100 mL), 75% (100 mL) afforded 59 (0.280 g, 54%) as a white solid: 1H NMR (300 MHz, CDCl3) δ 7.65 (apparent t, 1H, J = 8.7 Hz), 6.34 (dd, 1H, J = 8.7 Hz, J = 1.5 Hz), 3.79 (t, 4H, J = 4.8 Hz), 3.48 (t, 4H, J = 4.8 Hz); 19F NMR (470.6 MHz, CDCl3) δ −66.22 (d, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 158.35 (d, 1JCF = 241.5 Hz), 157.37 (d, 3JCF = 23.8 Hz), 144.64 (d, 3JCF = 2.6 Hz), 104.63 (d, 4JCF = 4.5 Hz), 89.09 (d, 2JCF = 38.5 Hz), 66.60, 45.44; HRMS (ESI) [M + H]+ Calcd for C9H1179BrFN2O: 261.0033, found: 261.0063.
3-Bromo-2-nitro-6-(piperidin-1-yl)pyridine (60)
Compound 56 (0.160 g, 0.772 mmol) was flushed with N2(g), then dissolved in CH3CN (10 mL) and cooled to 0 °C. NBS (0.140 g, 0.787 mmol) was added, the reaction mixture was warmed to ambient temperature, and stirred under N2(g) for 5.5 h. The CH3CN was removed to give an orange syrup, then CH2Cl2 and hexane were added and removed to give an orange residue that was dissolved in CH2Cl2, poured onto dry silica (45 mm h × 45 mm i.d.), and eluted under vacuum: hexane (50 mL), %CH2Cl2/hexane–25% (100 mL), 50% (100 mL), CH2Cl2 (150 mL) to give a dark yellow syrup. Purification by radial chromatography (2 mm silica): %CH2Cl2/hexane–25% (100 mL), 50% (50 mL) afforded 60 (0.150 g, 68%) as a yellow syrup: 1H NMR (500 MHz, CDCl3) δ 7.64 (d, 1H, J = 9.0 Hz), 6.66 (d, 1H, J = 9.0 Hz), 3.54 (t, 4H, J = 5.5 Hz), 1.66 (m, 2H), 1.62 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 156.45, 156.14, 144.12, 111.10, 92.85, 46.17, 25.49, 24.52; HRMS (ESI) [M + H]+ Calcd for C10H1379BrN3O2: 286.0186, found: 286.0172.
4-(5-Bromo-6-nitropyridin-2-yl)morpholine (61)
Compound 57 (0.120 g, 0.574 mmol) was dissolved in CH3CN (10 mL), cooled to 0 °C, and NBS (0.150 g, 0.843 mmol, 1.5 equiv) was added. The reaction mixture was stirred in a capped flask at ambient temperature for 6 h, concentrated to an orange oil, then CH2Cl2 and hexane were added and removed 2× to give an orange solid that was dried under vacuum. The solid was dissolved in CH2Cl2, poured onto dry silica (45 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (400 mL), 1% MeOH/CH2Cl2 (100 mL) to give a yellow solid (0.130 g). Purification by radial chromatography (2 mm silica) with CHCl3 (100 mL) afforded 61 (0.082 g, 50%) as a yellow solid: 1H NMR (400 MHz, CDCl3) δ 7.76 (d, 1H, J = 9.2 Hz), 6.68 (d, 1H, J = 9.2 Hz), 3.80 (t, 4H, J = 4.8 Hz), 3.54 (t, 4H, J = 4.8 Hz); 13C NMR (125 MHz, CDCl3) δ 156.65, 156.07, 144.61, 110.99, 94.88, 66.51, 45.21; HRMS (ESI) [M + H]+ Calcd for C9H1179BrN3O3: 287.9978, found: 287.9981.
4-(4-Fluoropyridin-2-yl)morpholine (62)
2-Bromo-4-fluoropyridine (1.00 g, 5.68 mmol), morpholine (0.500 g, 5.74 mmol), sodium tert-butoxide (0.580 g, 6.04 mmol), toluene (50 mL), Pd2(dba)3 (0.073 g, 0.080 mmol) and XantPhos (0.140 g, 0.242 mmol) were flushed with N2(g) for 10 min, then heated at 100 °C overnight. The reaction mixture was cooled to ambient temperature, filtered through a short silica gel pad, and washed with EtOAc. The filtrate was evaporated to dryness and the residue was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 80:20) to afford 62 (0.880 g, 85%) as white solid: 1H NMR (CDCl3, 300 MHz): δ 8.13 (dd, 1H, J = 9.4 Hz, J = 5.4 Hz), 6.41 (ddd, 1H, J = 8.1 Hz, J = 5.4 Hz, J = 2.1 Hz), 6.28 (dd, 1H, J = 12.3 Hz, J = 2.1 Hz), 3.81 (apparent t, 4H, J = 4.8 Hz), 3.49 (apparent t, 4H, J = 4.8 Hz); HRMS (ESI) [M + H]+ Calcd for C9H12ON2F: 183.0928, found: 183.0931.
4-(5-Bromo-4-fluoropyridin-2-yl)morpholine (63)
Compound 62 (0.870 g, 4.78 mmol) was dissolved in CH3CN (50 mL), cooled to 0 °C, then NBS (0.850 g, 4.78 mmol) was added. The reaction mixture was stirred at 0 °C for 90 min, the solvent was removed, and the residue was purified by flash column chromatography (4:1 v/v hexane/EtOAc) to afford 63 (1.20 g, 96%) as an off-white solid: 1H NMR (300 MHz, CDCl3) δ 8.21 (d, 1H, J = 9.9 Hz), 6.36 (d, 1H, J = 11.5 Hz), 3.80 (apparent t, 4H, J = 4.8 Hz), 3.48 (apparent t, 4H, J = 4.8 Hz). HRMS (ESI) [M + H]+ Calcd for C9H11ON2BrF: 261.0033, found: 261.0029.
5-Bromo-3-fluoro-2-(piperidin-1-yl)pyridine (64)
2,5-Dibromo-3-fluoropyridine (0.270 g, 1.06 mmol), piperidine (0.085 g, 1.0 mmol), sodium tert-butoxide (0.144 g, 1.50 mmol), toluene (20 mL), Pd2(dba)3 (0.018 g, 0.020 mmol), and XantPhos (0.035 g, 0.061 mmol) were flushed with N2(g) for 5 min, then heated at 100 °C overnight. The reaction mixture was cooled to ambient temperature, filtered through a short silica gel pad, and washed with EtOAc. The filtrate was evaporated to dryness and the residue was purified by radial chromatography (7:3 v/v hexane/EtOAc) to afford 64 (0.150 g, 58%) as a white solid: 1H NMR (300 MHz, CDCl3): δ 8.02 (dd, 1H, J = 2.1 Hz, J = 0.9 Hz), 7.33 (dd, 1H, J = 12.0 Hz, J = 2.1 Hz), 3.41 (m, 4H), 1.65 (m, 6 H); HRMS (ESI) [M + H]+ Calcd for C10H13N2BrF: 259.0241, found: 259.0237.
4-(5-Bromo-3-fluoropyridin-2-yl)morpholine (65)
2,5-Dibromo-3-fluoropyridine (0.270 g, 1.06 mmol), morpholine (0.087 mg, 1.0 mmol), sodium tert-butoxide (0.144 g, 1.50 mmol), toluene (20 mL), Pd2(dba)3 (0.018 g, 0.020 mmol), and XantPhos (0.035 g, 0.061 mmol) were flushed with N2(g) for 5 min, then heated at 100 °C overnight. The reaction mixture was cooled to ambient temperature, filtered through a short silica gel pad, and washed with EtOAc (10 mL × 4). The filtrate was evaporated to dryness and the residue was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 80:20) to afford 65 (0.160 g, 61%) as white solid: 1H NMR (300 MHz, CDCl3) δ 8.05 (dd, 1H, J = 1.8 Hz, J = 0.9 Hz), 7.39 (dd, 1H, J = 12.0 Hz, J = 1.8 Hz), 3.82 (apparent t, 4H, J = 4.8 Hz), 3.46 (apparent t, 4H, J = 4.8 Hz); HRMS (ESI) [M + H]+ Calcd for C9H11ON2BrF: 261.0033, found: 261.0044.
2-Chloro-3-fluoro-5-(piperidin-1-yl)pyridine (66)
5-Bromo-2-chloro-3-fluoropyridine (0.270 g, 1.28 mmol), piperidine (0.088 g, 1.03 mmol), sodium tert-butoxide (0.144 g, 1.50 mmol), toluene (20 mL), Pd2(dba)3 (0.018 g, 0.020 mmol), and XantPhos (0.035 g, 0.061 mmol) were flushed with N2(g) for 5 min, then heated at 100 °C overnight. The reaction mixture was cooled to ambient temperature, filtered through a short silica gel pad, and washed with EtOAc. The filtrate was evaporated to dryness and the residue was purified by radial chromatography (7:3 v/v hexane/EtOAc) to afford 66 (0.220 g, 99%) as white solid: 1H NMR (300 MHz, CDCl3) δ 7.84 (d, 1H, J = 2.7 Hz), 6.95 (dd, 1H, J = 11.4 Hz, J = 2.7 Hz), 3.20 (m, 4H), 1.67 (m, 6 H); HRMS (ESI) [M + H]+ Calcd for C10H13N2ClF: 215.0746, found: 215.0743.
4-(6-Chloro-5-fluoropyridin-3-yl)morpholine (67)
5-Bromo-2-chloro-3-fluoropyridine (0.270 g, 1.28 mmol), morpholine (0.110 g, 1.26 mmol), sodium tert-butoxide (0.144 g, 1.50 mmol), toluene (20 mL), Pd2(dba)3 (0.018 g, 0.020 mmol), and XantPhos (0.035 g, 0.061 mmol) were flushed with N2(g) for 5 min, then heated at 100 °C overnight. The reaction mixture was cooled to ambient temperature, filtered through a short silica gel pad, and washed with EtOAc. The filtrate was evaporated to dryness and the residue was purified by radial chromatography (7:3 v/v hexane/EtOAc) to afford 67 (0.250 g, 92%) as white solid: 1H NMR (300 MHz, CDCl3) δ 7.85 (d, 1H, J = 2.7 Hz), 6.97 (dd, 1H, J = 10.8 Hz, J = 2.7 Hz), 3.87 (m–AA’XX’, 4H, J = 4.8 Hz, J = 1.8 Hz), 3.19 (m–AA’XX’, 4H, J = 4.8 Hz, J = 1.8 Hz); HRMS (ESI) [M + H]+ Calcd for C9H11ON2ClF: 217.0539, found: 217.0536.
4-(6-Chloro-4-fluoropyridin-3-yl)morpholine (68)
2-Chloro-4-fluoro-5-bromopyridine (0.270 g, 1.28 mmol), morpholine (0.087 g, 1.0 mmol), sodium tert-butoxide (0.144 g, 1.50 mmol), toluene (20 mL), Pd2(dba)3 (0.018 g, 0.020 mmol), and XantPhos (0.035 g, 0.061 mmol) were flushed with N2(g) for 5 min, then heated at 100 °C overnight. The reaction mixture was cooled to ambient temperature, filtered through a short silica gel pad, and washed with EtOAc. The filtrate was evaporated to dryness and the residue was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 80:20) to afford 68 (0.097 g, 45%) as white solid: 1H NMR (300 MHz, CDCl3) δ 7.98 (d, 1H, J = 10.5 Hz), 7.04 (d, 1H, J = 11.1 Hz), 3.86 (m–AA’XX’, 4H, J = 4.8 Hz, J = 1.8 Hz), 3.12 (m–AA’XX’, 4H, J = 4.8 Hz, J = 1.8 Hz); HRMS (ESI) [M + H]+ Calcd for C9H11ON2ClF: 217.0539, found: 217.0536.
tert-Butyl 2-(2-Fluoro-6-(piperidin-1-yl)pyridin-3-yl)-1H-indole-1-carboxylate (69)
1,4-Dioxane (30 mL) and H2O (6 mL) were combined and purged with N2(g) for 30 min. Compound 12 (0.250 g, 0.958 mmol), compound 58 (0.310 g, 1.20 mmol, 1.2 equiv), Pd(dppf)Cl2 (0.095 g, 0.13 mmol, 0.14 equiv), and Na2CO3 (0.470 g, 4.43 mmol, 4.6 equiv) were flushed with N2(g) for 10 min, then the H2O/1,4-dioxane mixture (15 mL) was added. The reaction mixture was stirred at reflux under N2(g) for 4 h, then cooled to ambient temperature, filtered through Celite, and the Celite was rinsed with 1,4-dioxane, then EtOAc. The solvent was removed from the filtrate to give a brown oil that was dried under vacuum to give a brown residue. The residue was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum with CH2Cl2 (250 mL) to give a faint tan syrup (0.19 g). Purification by radial chromatography (2 mm silica) with 90:8:2 v/v/v hexane/EtOAc/NEt3 (100 mL) afforded 69 (0.087 g, 23%) as a white foam: 1H NMR (300 MHz, CDCl3) δ 8.20 (d, 1H, J = 8.4 Hz), 7.55 (m, 2H), 7.31 (ddd, 1H, J = 8.4 Hz, J = 7.2 Hz, J = 1.2 Hz), 7.22 (td, 1H, J = 7.2 Hz, J = 0.9 Hz), 6.53 (s, 1H), 6.49 (dd, 1H, J = 8.4 Hz, J = 1.8 Hz), 3.58 (m, 4H), 1.66 (m, 6H), 1.45 (s, 9H); 19F NMR (282.5 MHz, CDCl3) δ −67.96 (d, J = 9.6 Hz); HRMS (ESI) [M + H]+ Calcd for C23H27FN3O2: 396.2082, found: 396.2074.
tert-Butyl 2-(2-Fluoro-6-morpholinopyridin-3-yl)-1H-indole-1-carboxylate (70)
1,4-Dioxane (30 mL) and H2O (6 mL) were combined and purged with N2(g) for 30 min. Compound 12 (0.240 g, 0.919 mmol), compound 59 (0.250 g, 0.958 mmol), Pd(dppf)Cl2 (0.110 g, 0.150 mmol, 0.16 equiv), and Na2CO3 (0.300 g, 2.83 mmol, 3.1 equiv) were flushed with N2(g) for 15 min, then the H2O/1,4-dioxane mixture (15 mL) was added. The reaction mixture was stirred at reflux under N2(g) for 4 h, then cooled to ambient temperature, filtered through Celite, and the Celite was rinsed with 1,4-dioxane, then EtOAc. The solvent was removed from the filtrate to give a brown oil that was dried under vacuum to give a brown residue. The residue was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (100 mL), %MeOH/CH2Cl2–1% (100 mL), 2.5% (100 mL), 5% (150 mL). The desired fractions were combined, concentrated, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 75:20:5 (300 mL), 50:45:5 (100 mL) to give a brown residue (0.23 g). Purification by radial chromatography (2 mm silica) hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (100 mL) afforded 70 (0.200 g, 55%) as a white foam: 1H NMR (300 MHz, CDCl3) δ 8.20 (d, 1H, J = 8.1 Hz), 7.62 (dd, 1H, J = 9.6 Hz, J = 8.1 Hz), 7.54 (d, 1H, J = 7.2 Hz), 7.32 (ddd, 1H, J = 8.4 Hz, J = 7.2 Hz, J = 1.2 Hz), 7.23 (td, 1H, J = 7.5 Hz, J = 0.9 Hz), 6.55 (s, 1H), 6.48 (dd, 1H, J = 8.1 Hz, J = 1.8 Hz), 3.82 (t, 4H, J = 4.8 Hz), 3.54 (t, 4H, J = 4.8 Hz), 1.46 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 159.39 (d, 1JFC = 237.7 Hz), 158.10 (d, JFC = 15.6 Hz), 150.26, 142.03 (d, JFC = 4.8 Hz), 137.27, 133.70 (d, JFC = 4.3 Hz), 129.22, 124.62, 123.01, 120.57, 115.69, 110.61, 105.72 (d, JFC = 31.2 Hz), 102.33 (d, JFC = 3.8 Hz), 83.81, 66.73, 45.64, 28.01; 19F NMR (282.5 MHz, CDCl3) δ −67.85 (d, J = 9.9 Hz); HRMS (ESI) [M + H]+ Calcd for C22H25FN3O3: 398.1875, found: 398.1867.
tert-Butyl 2-(2-Nitro-6-(piperidin-1-yl)pyridin-3-yl)-1H-indole-1-carboxylate (71)
1,4-Dioxane (15 mL) and H2O (3 mL) were combined and purged with N2(g) for 30 min. Compound 12 (0.092 g, 0.35 mmol), compound 60 (0.110 g, 0.384 mmol), Pd(dppf)Cl2 (0.032 g, 0.044 mmol, 0.1 equiv), and Na2CO3 (0.196 g, 1.85 mmol, 5.2 equiv) were flushed with N2(g) for 10 min, then the H2O/1,4-dioxane mixture (10 mL) was added. The reaction mixture was stirred at reflux under N2(g) for 5 h, then cooled to ambient temperature, filtered through Celite, and the Celite was rinsed with EtOAc. The solvent was removed from the filtrate to give a dark green/black residue that was dissolved in CH2Cl2, poured onto dry silica (45 mm h × 45 mm i.d.), and eluted under vacuum with 75:20:5 v/v/v hexane/EtOAc/NEt3 (200 mL) to give a dark orange syrup (0.140 g). Purification by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (100 mL) gave a yellow solid (0.065 g) that was again purified by radial chromatography (2 mm silica) with CHCl3 (50 mL) to give an orange foam (0.056 g). Purification by radial chromatography (1 mm silica): %CH2Cl2/hexane–25% (25 mL), 50% (50 mL), CH2Cl2 (25 mL) afforded 71 (0.048 g, 32%) as a yellow/orange foam: 1H NMR (400 MHz, CDCl3) δ 8.20 (d, 1H, J = 8.4 Hz), 7.58 (d, 1H, J = 8.8 Hz), 7.53 (d, 1H, J = 8.0 Hz), 7.33 (apparent t, 1H, J = 7.6 Hz), 7.24 (apparent t – partially obscured by CHCl3 resonance, 1H, J = 7.2 Hz), 6.86 (d, 1H, J = 8.8 Hz), 6.50 (s, 1H), 3.66 (m, 4H), 1.67 (m, 6H), 1.41 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 157.10, 154.90, 150.07, 142.88, 136.97, 134.21, 129.21, 124.83, 123.08, 120.72, 116.06, 111.29, 110.65, 109.53, 83.90, 46.24, 27.97, 25.60, 24.74; HRMS (ESI) [M + H]+ Calcd for C23H27N4O4: 423.2027, found: 423.2030. X-ray quality crystals were grown by slow evaporation of CH2Cl2/hexane.
tert-Butyl 2-(6-Morpholino-2-nitropyridin-3-yl)-1H-indole-1-carboxylate (72)
1,4-Dioxane (15 mL) and H2O (3 mL) were combined and purged with N2(g) for 30 min. Compound 12 (0.060 g, 0.23 mmol), compound 61 (0.066 g, 0.23 mmol), Pd(dppf)Cl2 (0.019 g, 0.026 mmol, 0.1 equiv), and Na2CO3 (0.117 g, 1.10 mmol, 4.8 equiv) were flushed with N2(g), then the H2O/1,4-dioxane mixture (10 mL) was added. The reaction mixture was stirred at reflux under N2(g) for 4 h, then cooled to ambient temperature, filtered through Celite, and the Celite was rinsed with EtOAc. The filtrate was concentrated, then EtOAc was added and removed to give a dark green/brown oil. CH2Cl2 and hexane were added and removed to give a residue that was dissolved in CH2Cl2, poured onto dry silica (45 mm h × 45 mm i.d.), and eluted under vacuum: CH2Cl2 (200 mL), %MeOH/CH2Cl2–1% (100 mL), 2% (150 mL) to give a dark yellow/brown residue (69 mg). Purification by radial chromatography (1 mm silica): CH2Cl2 (100 mL) afforded 72 (0.036 g, 37%) as a yellow/orange foam: 1H NMR (400 MHz, CDCl3) δ 8.18 (d, 1H, J = 8.1 Hz), 7.67 (d, 1H, J = 8.1 Hz), 7.54 (d, 1H, J = 7.6 Hz), 7.34 (ddd, 1H, J = 8.4 Hz, J = 7.2 Hz, J = 1.2 Hz), 7.25 (m – partially obscured by CHCl3 resonance, 1H), 6.88 (d, 1H, J = 8.4 Hz), 6.52 (s, 1H), 3.84 (t, 4H, J = 4.8 Hz), 3.65 (t, 4H, J = 4.8 Hz), 1.43 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 157.25, 154.70, 150.06, 143.25, 136.89, 133.83, 129.17, 125.00, 123.17, 120.83, 116.09, 113.03, 110.87, 109.56, 84.12, 66.67, 45.35, 28.01; HRMS (ESI) [M + H]+ Calcd for C22H25N4O5: 425.1820, found: 425.1815. X-ray quality crystals were grown by slow evaporation of CH2Cl2/hexane.
4-(5-(1H-Indol-2-yl)-6-nitropyridin-2-yl)morpholine (73)
Isolated as a side-product from the reaction of 72: 1H NMR (400 MHz, acetone-d6) δ 10.53 (br s, 1H), 8.09 (d, 1H, J = 8.8 Hz), 7.56 (d, 1H, J = 7.6 Hz), 7.41 (d, 1H, J = 8.4 Hz), 7.20 (d, 1H, J = 8.8 Hz), 7.13 (t, 1H, J = 7.6 Hz), 7.04 (t, 1H, J = 7.6 Hz), 6.49 (s, 1H), 3.77 (t, 4H, J = 4.8 Hz), 3.62 (t, 4H, J = 4.8 Hz); 13C NMR (125 MHz, DMSO-d6) δ 156.31, 154.79, 140.95, 136.77, 130.23, 128.21, 121.94, 120.14, 119.51, 111.30, 110.32, 107.21, 100.09, 65.66, 44.65; HRMS (ESI) [M + H]+ Calcd for C17H17N4O3: 325.1295, found: 325.1295.
2-(2-Fluoro-6-(piperidin-1-yl)pyridin-3-yl)-1H-indole (74)
Compound 69 (0.071 g, 0.18 mmol) and TFA (1.8 mL, 23 mmol, 130 equiv) were stirred for 20 min, then poured into a mixture of NaHCO3 (2.290 g, 27.26 mmol, 1.2 equiv TFA), H2O (45 mL), and CH2Cl2 (45 mL). The mixture was stirred for 10 min, the layers were separated, and the aqueous layer was extracted with CH2Cl2 (20 mL). The combined CH2Cl2 layers were washed with brine (25 mL), dried over MgSO4, concentrated, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 90:8:2 (25 mL), 75:20:5 (200 mL), 50:45:5 (50 mL) to afford 74 (0.025 g, 47%) as an off-white solid: 1H NMR (400 MHz, CDCl3) δ 8.77 (br s, 1H), 7.92 (dd, 1H, J = 10.6 Hz, J = 8.4 Hz), 7.59 (d, 1H, J = 8.0 Hz), 7.39 (d, 1H, J = 7.6 Hz), 7.17 (td, 1H, J = 7.6 Hz, J = 0.8 Hz), 7.10 (td, 1H, J = 7.6 Hz, J = 0.8 Hz), 6.72 (d, 1H, J = 1.6 Hz), 6.54 (dd, 1H, J = 8.4 Hz, J = 2.4 Hz), 3.59 (m, 4H), 1.67 (m, 6 H); 13C NMR (125 MHz, DMSO-d6) δ 158.00 (d, 1JFC = 236.7 Hz), 156.04 (d, JFC = 16.5 Hz), 139.16 (d, JFC = 4.4 Hz), 136.42, 131.47 (d, JFC = 7.3 Hz), 128.57, 121.11, 119.60, 119.21, 111.00, 103.91 (d, JFC = 3.5 Hz), 100.65 (d, JFC = 28.0 Hz), 99.81 (d, JFC = 8.3 Hz), 45.44, 24.93, 24.09; 19F NMR (470.6 MHz, DMSO-d6) δ −67.52; 19F NMR (376.5 MHz, CDCl3) δ −70.32; HRMS (ESI) [M + H]+ Calcd for C18H19FN3: 296.1558, found: 296.1604.
4-(6-Fluoro-5-(1H-indol-2-yl)pyridin-2-yl)morpholine (75, JSS20–183A)
Compound 70 (0.076 g, 0.19 mmol) and TFA (1.8 mL, 23 mmol, 123 equiv) were stirred for 20 min, then poured into a mixture of NaHCO3 (2.220 g, 26.43 mmol, 1.1 equiv TFA), H2O (45 mL), and CH2Cl2 (45 mL). The mixture was stirred for 10 min, the layers were separated, and the aqueous layer was extracted with CH2Cl2 (20 mL). The combined CH2Cl2 layers were washed with brine (25 mL), dried over MgSO4, concentrated, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: hexane/EtOAc/NEt3 v/v/v 75:20:5 (100 mL), 50:45:5 (200 mL), 20:75:5 (75 mL) to afford 75 (0.054 g, 95%) as a light tan solid: 1H NMR (400 MHz, CDCl3) δ 8.78 (br s, 1H), 7.99 (dd, 1H, J = 10.4 Hz, J = 8.8 Hz), 7.60 (d, 1H, J = 7.6 Hz), 7.40 (d, 1H, J = 8.0 Hz), 7.18 (td, 1H, J = 7.6 Hz, J = 0.8 Hz), 7.11 (td, 1H, J = 7.6 Hz, J = 0.8 Hz), 6.77 (d, 1H, J = 1.2 Hz), 6.55 (dd, 1H, J = 8.4 Hz, J = 2.0 Hz), 3.82 (t, 4H, J = 4.8 Hz), 3.57 (t, 4H, J = 4.8 Hz); 1H NMR (500 MHz, DMSO-d6) δ 11.31 (s, 1H), 8.18 (dd, 1H, J = 10.5 Hz, J = 8.5 Hz), 7.51 (d, 1H, J = 8.0 Hz), 7.40 (d, 1H, J = 8.0 Hz), 7.08 (t, 1H, J = 7.5 Hz), 6.99 (t, 1H, J = 7.5 Hz), 6.88 (d, 1H, J = 8.5 Hz), 6.73 (s, 1H), 3.71 (t, 4H, J = 5.0 Hz), 3.51 (t, 4H, J = 5.0 Hz); 1H NMR (500 MHz, acetone-d6) δ 10.45 (br s, 1H), 8.16 (dd, 1H, J = 10.5 Hz, J = 9.0 Hz), 7.54 (d, 1H, J = 8.0 Hz), 7.42 (d, 1H, J = 8.0 Hz), 7.09 (t, 1H, J = 7.5 Hz), 7.01 (t, 1H, J = 7.5 Hz), 6.79 (overlapping resonances, 2H), 3.76 (t, 4H, J = 5.0 Hz), 3.56 (t, 4H, J = 5.0 Hz); 13C NMR (125 MHz, DMSO-d6) δ 157.86 (d, 1JFC = 237.2 Hz), 156.31 (d, JFC = 16.1 Hz), 139.26 (d, JFC = 4.4 Hz), 136.47, 131.13 (d, JFC = 7.2 Hz), 128.50, 121.30, 119.71, 119.27, 111.06, 104.14 (d, JFC = 3.6 Hz), 102.11 (d, JFC = 27.5 Hz), 100.23 (d, JFC = 8.6 Hz), 65.71, 44.83; 19F NMR (376.5 MHz, CDCl3) δ −70.37; HRMS (ESI) [M + H]+ Calcd for C17H17FN3O: 298.1350, found: 298.1396. X-ray quality crystals were grown by slow evaporation of CDCl3.
tert-Butyl 5-Fluoro-2-(2-morpholinopyrimidin-5-yl)-1H-indole-1-carboxylate (76)
Compound 48 (0.210 g, 0.752 mmol), compound 30 (0.100 g, 0.410 mmol), and Pd(dppf)Cl2 (0.020 g, 0.027 mmol) were dissolved in 1,4-dioxane (10 mL), then K2CO3(aq) (2 M, 0.5 mL) was added. The reaction mixture was flushed with N2(g) for 5 min, then heated at 100 °C under N2(g) overnight. The reaction mixture was cooled to ambient temperature, evaporated to dryness, the residue was dissolved in EtOAc (30 mL), washed with H2O (10 mL), dried over MgSO4, and filtered. The filtrate was passed through a silica gel pad, the silica was washed with EtOAc, and the filtrate was concentrated to dryness. The residue was purified by radial chromatography (hexane/EtOAc v/v 90:10 to 80:20) to afford 76 (0.150 g, 92%) as a solid: 1H NMR (300 MHz, CDCl3) δ 8.38 (s, 2H), 8.13 (dd, 1H, J = 9.0 Hz, J = 4.5 Hz), 7.20 (dd, 1H, J = 8.7 Hz, J = 2.7 Hz), 7.05 (td, 1H, J = 9.0 Hz, J = 2.7 Hz), 6.50 (d, 1H, J = 0.3 Hz), 3.88 (m, 4H), 3.79 (m, 4H), 1.49 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C21H24O3N4F: 399.1827, found: 399.1837.
tert-Butyl 5-Fluoro-2-(6-morpholinopyridin-3-yl)-1H-indole-1-carboxylate (77)
Compound 48 (0.210 g, 0.752 mmol), compound 35 (0.100 g, 0.411 mmol), Pd(dppf)Cl2 (0.020 g, 0.027 mmol), 1,4-dioxane (10 mL), and K2CO3(aq) (2 M, 0.5 mL) were flushed with N2(g) for 10 min, then stirred at 100 °C under N2(g) overnight. The reaction mixture was cooled to ambient temperature, evaporated to dryness, the residue was dissolved in EtOAc (30 mL), washed with H2O (10 mL), dried over MgSO4, and filtered. The filtrate was passed through a silica gel pad, the silica was washed with EtOAc, and the filtrate was concentrated to dryness. The residue was purified by radial chromatography (hexane/EtOAc v/v 90:10 to 80:20) to afford 77 (0.120 g, 73%) as a solid: 1H NMR (300 MHz, CDCl3) δ 8.26 (d, 1H, J = 2.1 Hz), 8.12 (dd, 1H, J = 9.0 Hz, J = 4.5 Hz), 7.55 (dd, 1H, J = 8.7 Hz, J = 1.8 Hz), 7.19 (dd, 1H, J = 8.7 Hz, J = 2.4 Hz), 7.03 (td, 1H, J = 9.0 Hz, J = 2.4 Hz), 6.68 (d, 1H, J = 8.7 Hz), 6.48 (s, 1H), 3.85 (t, 4H, J = 4.8 Hz), 3.57 (partially resolved t, 4H, J = 4.8 Hz), 1.43 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H25O3N3F: 398.1875, found: 398.1887.
4-(5-(5-Fluoro-1H-indol-2-yl)pyrimidin-2-yl)morpholine (78)
Compound 76 (0.140 g, 0.351 mmol) was dissolved in TFA (2 mL) and stirred at ambient temperature for 2 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid that was washed with cold CH2Cl2 to afford 78 (0.098 g, 93%) as an off-white solid: 1H NMR (300 MHz, acetone-d6) δ 10.80 (br s, 1H), 8.83 (s, 2H), 7.37 (dd, 1H, J = 8.7 Hz, J = 4.5 Hz), 7.23 (dd, 1H, J = 9.6 Hz, J = 2.4 Hz), 6.88 (dd, 1H, J = 9.6 Hz, J = 8.7 Hz, J = 2.4 Hz), 6.79 (m, 1H), 3.82 (m, 4H), 3.71 (m, 4 H); 13C NMR (125 MHz, DMSO-d6): 160.33, 157.23 (d, 1JFC = 231.4 Hz), 154.69, 135.15, 133.59, 128.93 (d, JFC = 10.3 Hz), 115.41, 111.92 (d, JFC = 9.9 Hz), 109.27 (d, JFC = 25.9 Hz), 104.31, 104.12, 97.50 (d, JFC = 4.7 Hz), 65.95, 44.01; HRMS (ESI) [M + H]+ Calcd for C16H16ON4F: 299.1303, found: 299.1301.
4-(5-(5-Fluoro-1H-indol-2-yl)pyridin-2-yl)morpholine (79)
Compound 77 (0.120 g, 0.302 mmol) was dissolved in TFA (2 mL) and stirred at ambient temperature for 2 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid that was placed in a filter and rinsed with a small amount of acetone to afford 79 (0.066 g, 74%) as an off-white solid: 1H NMR (acetone-d6, 300 MHz): δ 10.62 (s, 1H), 8.65 (dd, 1H, J = 2.5 Hz, J = 0.7 Hz), 7.98 (dd, 1H, J = 8.9 Hz, J = 2.6 Hz), 7.34 (dd, 1H, J = 8.8 Hz, J = 4.5 Hz), 7.21 (dd, 1H, J = 9.9 Hz, J = 2.5 Hz), 6.85 (m, 2H), 6.74 (s, 1H), 3.74 (m, 4H), 3.56 (m, 4 H); 13C NMR (125 MHz, DMSO-d6): 158.30, 157.17 (d, 1JFC = 231.5 Hz), 144.57, 137.81, 134.38, 133.54, 129.07 (d, JFC = 10.6 Hz), 117.78, 111.75 (d, JFC = 9.9 Hz), 108.88 (d, JFC = 26.0 Hz), 106.86, 104.05 (d, JFC = 23.1 Hz), 97.18 (d, JFC = 4.5 Hz), 65.90, 45.01; HRMS (ESI) [M + H]+ Calcd for C17H17ON3F: 298.1350, found: 298.1348.
tert-Butyl 6-Fluoro-2-(2-fluoro-6-(piperidin-1-yl)pyridin-3-yl)-1H-indole-1-carboxylate (80)
1,4-Dioxane (25 mL) was purged with N2(g) for 1 h. Compound 58 (0.250 g, 0.965 mmol), compound 10 (0.410 g, 1.03 mmol, 1.1 equiv), Na2CO3 (0.510 g, 4.81 mmol, 5 equiv), and Pd(dppf)Cl2 (0.089 g, 0.12 mmol, 0.1 equiv) were flushed with N2(g) for 10 min, then the 1,4-dioxane was added. The reaction mixture was stirred at reflux under N2(g) for 3 h, then cooled to ambient temperature, filtered through Celite, and the Celite was rinsed with EtOAc. The filtrate was concentrated to a brown oil, then hexane was added and removed to give a brown residue that was dissolved in CH2Cl2, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane (50 mL), hexane/EtOAc/NEt3 v/v/v 90:8:2 (100 mL), 75:20:5 (100 mL), 50:45:5 (50 mL) to give an orange syrup (0.43 g). Purification by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 95:4:1 (100 mL), 90:8:2 (50 mL) gave an off-white foam (0.16 g) that was again purified by radial chromatography (2 mm silica): %CH2Cl2/hexane–25% (50 mL), 50% (75 mL), 75% (50 mL), CH2Cl2 (25 mL) to give an off-white solid (0.14 g). A final purification by radial chromatography (2 mm silica): hexane/EtOAc/NEt3 v/v/v 90:8:2 (125 mL), 75:20:5 (25 mL) afforded 80 (0.130 g, 33%) as a white solid: 1H NMR (300 MHz, CDCl3) δ 7.95 (dd, 1H, J = 10.8 Hz, J = 2.1 Hz), 7.54 (dd, 1H, J = 9.6 Hz, J = 8.4 Hz), 7.44 (dd, 1H, J = 8.4 Hz, J = 5.4 Hz), 6.99 (td, 1H, J = 8.7 Hz, J = 2.4 Hz), 6.54 (d, 1H, J = 7.5 Hz), 6.49 (s, 1H), 3.57 (m, 4H), 1.67 (s, 6H), 1.44 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 161.06 (d, JFC = 238.0 Hz), 159.46 (d, JFC = 234.5 Hz), 158.04 (d, JFC = 16.2 Hz), 150.10, 141.80 (d, JFC = 4.8 Hz), 137.51 (d, JFC = 12.8 Hz), 134.52 (t, JFC = 4.3 Hz), 125.55 (d, JFC = 1.1 Hz), 120.95 (d, JFC = 9.8 Hz), 111.16 (d, JFC = 24.1 Hz), 109.84, 103.46 (d, JFC = 31.3 Hz), 103.11 (d, JFC = 28.6 Hz), 102.25 (d, JFC = 3.8 Hz), 84.08, 46.45, 27.93, 25.55, 24.84; HRMS (ESI) [M + H]+ Calcd for C23H26F2N3O2: 414.1988, found: 414.1989.
6-Fluoro-2-(2-fluoro-6-(piperidin-1-yl)pyridin-3-yl)-1H-indole (81)
Compound 80 (0.092 g, 0.22 mmol) was dissolved in TFA (2.2 mL, 29 mmol, 128 equiv), stirred at ambient temperature for 20 min, then poured into a mixture of NaHCO3 (2.68 g, 31.9 mmol, 1.1 equiv TFA), H2O (55 mL), and CH2Cl2 (55 mL). The mixture was stirred for 10 min, then the layers were separated, and the H2O layer was extracted with CH2Cl2 (25 mL). The combined CH2Cl2 layers were washed with H2O (50 mL), brine (50 mL), and dried over MgSO4. The solution was concentrated, poured onto dry silica (55 mm h × 45 mm i.d.), and eluted under vacuum: hexane (50 mL), hexane/EtOAc/NEt3 v/v/v 75:20:5 (200 mL), 50:45:5 (150 mL) to give an off-white/tan solid (68 mg). The solid was suspended in CHCl3 (1 mL), filtered, rinsed with CHCl3 (1 mL × 3), then hexane (5 mL), and dried under vacuum to afford 81 (0.049 g, 70%) as a white solid: 1H NMR (300 MHz, DMSO-d6) δ 11.36 (s, 1H), 8.08 (dd, 1H, J = 10.8 Hz, J = 8.7 Hz), 7.49 (dd, 1H, J = 8.7 Hz, J = 5.7 Hz), 7.13 (dd, 1H, J = 9.9 Hz, J = 1.8 Hz), 6.84 (m, 2H), 6.69 (s, 1H), 3.56 (m, 4H), 1.59 (m, 6 H); 13C NMR (125 MHz, DMSO-d6) δ 158.71 (d, 1JFC = 232.9 Hz), 157.84 (d, 1JFC = 234.8 Hz), 157.05 (d, JFC = 16.3 Hz), 139.06 (d, JFC = 4.4 Hz), 136.33 (d, JFC = 12.9 Hz), 132.23 (dd, JFC = 7.3 Hz, JFC = 3.6 Hz), 125.34, 120.53 (d, JFC = 10.0 Hz), 107.61 (d, JFC = 24.3 Hz), 103.91 (d, JFC = 3.5 Hz), 100.40 (d, JFC = 27.9 Hz), 99.70 (d, JFC = 8.0 Hz), 97.03 (d, JFC = 25.8 Hz), 45.41, 24.92, 24.06; HRMS (ESI) [M + H]+ Calcd for C18H18F2N3: 314.1463, found: 314.1454.
(1-(tert-Butoxycarbonyl)-6-fluoro-1H-indol-2-yl)boronic Acid (82)
Compound 110 (2.350 g, 9.989 mmol) was dissolved in THF (100 mL) under N2(g), cooled to 0 °C, then triisopropyl borate (2.820 g, 14.99 mmol) was added. LDA (2 M THF/heptane/ethylbenzene, 6.5 mL, 13 mmol) was added dropwise over a period of 10 min, the reaction mixture was stirred at 0 °C for 2 h, then quenched with 1 M HCl(aq). The reaction mixture was extracted with EtOAc (100 mL × 3), and the combined extracts were washed with H2O (50 mL × 2), dried over MgSO4, filtered, and evaporated to dryness. The residue was recrystallized from 1:2 v/v hexane/EtOAc to afford 82 (1.780 g, 64%) as a white solid: 1H NMR (300 MHz, CDCl3): δ 7.72 (dd, 1H, J = 11.1 Hz, J = 2.4 Hz), 7.52 (dd, 1H, J = 8.4 Hz, J = 5.7 Hz), 7.45 (d, 1H, J = 0.6 Hz), 7.02 (td, 1H, J = 8.7 Hz, J = 2.4 Hz) 6.88 (br s, 2H), 1.75 (s, 9H). Compound 82 decomposes in the mass spectrometer under both positive ion mode and negative ion mode.
tert-Butyl 6-Fluoro-2-(5-morpholinopyrazin-2-yl)-1H-indole-1-carboxylate (83)
Compound 82 (0.210 g, 0.752 mmol), compound 36 (0.100 g, 0.410 mmol), Pd(dppf)Cl2 (0.020 g, 0.027 mmol), 1,4-dioxane (10 mL), and K2CO3(aq) (2 M, 0.5 mL) were flushed with N2(g) for 10 min, then stirred under N2(g) at 100 °C overnight. The reaction mixture was cooled to ambient temperature, evaporated to dryness, the residue was dissolved in EtOAc (30 mL), washed with H2O (10 mL), dried over MgSO4, and filtered. The filtrate was passed through a silica gel pad, the silica was washed with EtOAc, and the filtrate was concentrated to dryness. The residue was purified by radial chromatography (hexane/EtOAc v/v 90:10 to 80:20) to afford 83 (0.078 g, 48%) as a solid: 1H NMR (300 MHz, CDCl3) δ 8.27 (d, 1H, J = 1.2 Hz), 8.17 (d, 1H, J = 1.5 Hz), 7.88 (dd, 1H, J = 10.8 Hz, J = 2.4 Hz), 7.48 (dd, 1H, J = 8.7 Hz, J = 5.7 Hz), 7.01 (td, 1H, J = 9.0 Hz, J = 2.4 Hz), 6.69 (s, 1H), 3.86 (apparent t, 4H, J = 4.8 Hz), 3.62 (apparent t, 4H, J = 4.8 Hz), 1.44 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C21H24O3N4F: 399.1827, found: 399.1829.
tert-Butyl 6-Fluoro-2-(4-fluoro-6-morpholinopyridin-3-yl)-1H-indole-1-carboxylate (84)
Compound 82 (0.340 g, 1.22 mmol), compound 63 (0.150 g, 0.575 mmol), Pd(dppf)Cl2 (0.028 g, 0.038 mmol), 1,4-dioxane (30 mL), and K2CO3(aq) (2 M, 0.7 mL) were flushed with N2(g) for 5 min, then stirred under N2(g) at 100 °C overnight (TLC indicated unreacted 63 remaining). Additional 82 (0.150 g, 0.537 mmol) was added and the reaction mixture was stirred under N2(g) at 100 °C overnight, then cooled to ambient temperature and evaporated to dryness. The residue was dissolved in H2O, extracted with EtOAc (10 mL × 3), and the combined extracts were dried over MgSO4, and filtered. The filtrate was passed through a silica gel pad, the silica was washed with EtOAc, and the solvent was removed to give a residue that was purified by radial chromatography (hexane/EtOAc v/v 99:1 to 80:20) to afford 84 (0.150 g, 63%) as a solid: 1H NMR (500 MHz, CDCl3) δ 8.20 (d, 1H, J = 10.5 Hz), 7.92 (dd, 1H, J = 10.5 Hz, J = 1.5 Hz), 7.47 (dd, 1H, J = 9.0 Hz, J = 5.5 Hz), 7.01 (td, 1H, J = 9.0 Hz, J = 2.5 Hz), 6.53 (s, 1H), 6.36 (d, 1H, J = 12.5 Hz), 3.83 (t, 4H, J = 5.0 Hz), 3.56 (partially resolved m, 4H), 1.47 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H24O3N3F2: 416.1780, found: 416.1793.
tert-Butyl 6-Fluoro-2-(5-fluoro-6-(piperidin-1-yl)pyridin-3-yl)-1H-indole-1-carboxylate (85)
Compound 82 (0.060 g, 0.21 mmol), compound 64 (0.053 g, 0.21 mmol), Pd(dppf)Cl2 (0.010 g, 0.014 mmol), 1,4-dioxane (6 mL), and K2CO3(aq) (2 M, 0.3 mL) were flushed with N2(g) for 5 min, then stirred under N2(g) at 100 °C overnight (TLC indicated unreacted 64 remaining). Additional 82 (0.064 g, 0.23 mmol) was added and the reaction mixture was stirred under N2(g) at 100 °C overnight, then cooled to ambient temperature and evaporated to dryness. The residue was dissolved in H2O, extracted with EtOAc (10 mL × 3), and the combined extracts were dried over MgSO4, and filtered. The filtrate was passed through a silica gel pad, the silica was washed with EtOAc, and the solvent was removed to give a residue that was purified by radial chromatography (hexane/EtOAc v/v 99:1 to 80:20) to afford 85 (0.059 g, 70%) as a solid: 1H NMR (300 MHz, CDCl3) δ 8.00 (m, 1H), 7.89 (dd, 1H, J = 10.8 Hz, J = 1.8 Hz), 7.41 (m, 1H), 7.21 (dd, 1H, J = 13.8 Hz, J = 1.8 Hz), 6.96 (tt, 1H, J = 9.0 Hz, J = 2.1 Hz), 6.47 (s, 1H), 3.45 (m, 4H), 1.64 (m, 6H), 1.39 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C23H26O2N3F2: 414.1988, found: 414.1988.
tert-Butyl 6-Fluoro-2-(5-fluoro-6-morpholinopyridin-3-yl)-1H-indole-1-carboxylate (86)
Compound 82 (0.190 g, 0.681 mmol), compound 65 (0.077 g, 0.295 mmol), Pd(dppf)Cl2 (0.012 g, 0.016 mmol), 1,4-dioxane (10 mL), and K2CO3(aq) (2 M, 0.5 mL) were flushed with N2(g) for 10 min, then stirred under N2(g) at 100 °C overnight. The reaction mixture was cooled to ambient temperature, evaporated to dryness, the residue was dissolved in H2O, and extracted with EtOAc (10 mL × 3). The combined extracts were dried over MgSO4 and filtered, the filtrate was passed through a silica gel pad, and the silica was washed with EtOAc. The solvent was removed to give a residue that was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 80:20) to afford 86 (0.086 g, 70%) as a solid: 1H NMR (400 MHz, CDCl3): δ 8.07 (s, 1H), 7.92 (d, 1H, J = 10.8 Hz), 7.46 (dd, 1H, J = 8.8 Hz, J = 5.6 Hz), 7.30 (d, 1H, J = 13.6 Hz), 7.02 (unresolved apparent td, 1H, J = 8.8 Hz), 6.53 (s, 1H), 3.86 (t, 4H, J = 4.4 Hz), 3.54 (t, 4H, J = 4.4 Hz), 1.45 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H24O3N3F2: 416.1780, found: 416.1781.
4-(5-(6-Fluoro-1H-indol-2-yl)pyrazin-2-yl)morpholine (87)
Compound 83 (0.078 g, 0.20 mmol) was dissolved in TFA (2 mL), stirred at ambient temperature for 2 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid that was washed with cold CH2Cl2 to afford 87 (0.058 g, 99%) as an off-white solid: 1H NMR (500 MHz, acetone-d6) δ 10.75 (br s, 1H), 8.72 (d, 1H, J = 1.0 Hz), 8.26 (d, 1H, J = 1.5 Hz), 7.55 (dd, 1H, J = 9.0 Hz, J = 5.5 Hz), 7.23 (dd, 1H, J = 10.0 Hz, J = 2.0 Hz), 6.84 (ddd, 1H, J = 10.0 Hz, J = 8.5 Hz, J = 2.0 Hz), 3.79 (t, 4H, J = 5.0 Hz), 3.62 (t, 4H, J = 5.0 Hz); 13C NMR (125 MHz, DMSO-d6): 159.06 (d, 1JFC = 234.3 Hz), 153.41, 138.40, 136.93 (d, JFC = 13.2 Hz), 136.29, 135.17, 129.82, 125.49, 121.08 (d, JFC = 9.6 Hz), 107.89 (d, JFC = 24.9 Hz), 98.09, 97.51 (d, JFC = 26.0 Hz), 65.81, 44.54; HRMS (ESI) [M + H]+ Calcd for C16H16ON4F: 299.1303, found: 299.1302.
4-(4-Fluoro-5-(6-fluoro-1H-indol-2-yl)pyridin-2-yl)morpholine (88)
Compound 84 (0.150 g, 0.361 mmol) was dissolved in TFA (2 mL), stirred at ambient temperature for 1.5 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid that was washed with cold CH2Cl2 to afford 88 (0.110 g, 97%) as an off-white solid: 1H NMR (300 MHz, CDCl3) δ 8.71 (br s, 1H), 8.61 (d, 1H, J = 11.1 Hz), 7.52 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.08 (dd, 1H, J = 9.6 Hz, J = 2.1 Hz), 6.89 (ddd, 1H, J = 9.9 Hz, J = 8.7 Hz, J = 2.1 Hz), 6.80 (dd, 1H, J = 1.2 Hz), 6.40 (d, 1H, J = 14.7 Hz), 3.85 (apparent t, 4H, J = 4.8 Hz), 3.57 (apparent t, 4H, J = 4.8 Hz); 13C NMR (125 MHz, DMSO-d6): 166.26 (d, 1JFC = 257.9 Hz), 159.88 (d, JFC = 4.5 Hz), 158.99 (d, JFC = 241.6 Hz), 147.42 (d, JFC = 4.9 Hz), 136.47 (d, JFC = 12.7 Hz), 130.55, 125.23, 120.82 (d, JFC = 10.1 Hz), 107.83 (d, JFC = 24.4 Hz), 107.36 (d, JFC = 10.9 Hz), 100.68 (d, JFC = 7.0 Hz), 97.18 (d, JFC = 25.7 Hz), 93.74 (d, JFC = 22.6 Hz), 65.83, 45.03; HRMS (ESI) [M + H]+ Calcd for C17H16ON3F2: 316.1256, found: 316.1246.
6-Fluoro-2-(5-fluoro-6-(piperidin-1-yl)pyridin-3-yl)-1H-indole (89)
Compound 85 (0.059 g, 0.14 mmol) was dissolved in TFA (1 mL), stirred at ambient temperature for 2.5 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid that was purified by radial chromatography (hexanes/EtOAc v/v 95:5 to 80:20), then purified again by radial chromatography (CHCl3) to afford 89 (0.008 g, 18%) as an off-white solid: 1H NMR (300 MHz, acetone-d6) δ 10.74 (br s, 1H), 8.47 (t, 1H, J = 1.8 Hz), 7.78 (dd, 1H, J = 14.7 Hz, J = 2.1 Hz), 7.53 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.10 (dd, 1H, J = 9.9 Hz, J = 2.4 Hz), 6.86 (m, 1H), 6.84 (ddd, 1H, J = 9.9 Hz, J = 8.4 Hz, J = 2.4 Hz), 3.51 (m, 4H), 1.67 (m, 6 H); 13C NMR (125 MHz, DMSO-d6): 159.12 (d, JFC = 235.1 Hz), 149.17 (d, JFC = 255.3 Hz), 148.54 (d, JFC = 6.4 Hz), 139.22 (d, JFC = 4.5 Hz), 137.06 (d, JFC = 12.7 Hz), 135.17, 125.53, 120.91 (d, JFC = 10.1 Hz), 120.73, 119.83 (d, JFC = 20.8 Hz), 107.98 (d, JFC = 24.4 Hz), 98.56, 97.22 (d, JFC = 25.5 Hz), 48.37 (d, JFC = 5.7 Hz), 25.49, 24.31; HRMS (ESI) [M + H]+ Calcd for C18H18N3F2: 314.1463, found: 314.1455.
4-(3-Fluoro-5-(6-fluoro-1H-indol-2-yl)pyridin-2-yl)morpholine (90)
Compound 86 (0.080 g, 0.19 mmol) was dissolved in TFA (2 mL), stirred at ambient temperature for 1.5 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid that was washed with cold CH2Cl2 to afford 90 (0.060 g, 98%) as an off-white solid: 1H NMR (300 MHz, acetone-d6) δ 10.79 (br s, 1H), 8.51 (apparent t, 1H, J = 1.8 Hz), 7.84 (dd, 1H, J = 14.4 Hz, J = 1.8 Hz), 7.54 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.11 (apparent dd, 1H, J = 9.9 Hz, J = 2.4 Hz), 6.89 (m, 1H), 6.85 (ddd, 1H, J = 9.9 Hz, J = 8.7 Hz, J = 2.4 Hz), 3.78 (apparent t, 4H, J = 4.8 Hz), 3.50 (apparent t, 4H, J = 4.8 Hz); 13C NMR (125 MHz, DMSO-d6): 159.01 (d, JFC = 235.2 Hz), 149.21 (d, JFC = 255.4 Hz), 147.86 (d, JFC = 6.8 Hz), 139.13 (d, JFC = 4.7 Hz), 136.94 (d, JFC = 12.7 Hz), 134.79 (d, JFC = 3.2 Hz), 125.33, 121.48 (d, JFC = 3.0 Hz), 120.90 (d, JFC = 10.1 Hz), 119.98 (d, JFC = 20.6 Hz), 107.98 (d, JFC = 24.4 Hz), 98.82, 97.17 (d, JFC = 25.5 Hz), 65.97, 47.67 (d, JFC = 5.4 Hz); HRMS (ESI) [M + H]+ Calcd for C17H16ON3F2: 316.1256, found: 316.1253.
tert-Butyl 2-(4-Fluoro-6-morpholinopyridin-3-yl)-1H-indole-1-carboxylate (91)
Compound 12 (0.300 g, 1.15 mmol), compound 63 (0.150 g, 0.575 mmol), Pd(dppf)Cl2 (0.028 g, 0.038 mmol), 1,4-dioxane (30 mL), and K2CO3(aq) (2 M, 0.7 mL) were flushed with N2(g) for 10 min, stirred under N2(g) at 100 °C overnight, then cooled to ambient temperature, and evaporated to dryness. The residue was dissolved in H2O and extracted with EtOAc (10 mL × 3), and the extracts were combined, dried over MgSO4, and filtered. The filtrate was passed through a silica gel pad, and the silica was washed with EtOAc. The solvent was removed to give a residue that was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 80:20) to afford 91 (0.160 g, 70%) as a solid: 1H NMR (300 MHz, CDCl3) δ 8.22 (d, 1H, J = 10.5 Hz), 8.19 (d, 1H, J = 7.5 Hz), 7.56 (dm, 1H, J = 7.5 Hz), 7.33 (ddd, 1H, J = 8.4 Hz, J = 7.2 Hz, J = 1.5 Hz), 7.24 (td, 1H, J = 7.5 Hz, J = 1.2 Hz), 6.57 (s, 1H), 6.36 (d, 1H, J = 12.9 Hz), 3.83 (apparent t, 4H, J = 4.8 Hz), 3.56 (apparent t, 4H, J = 4.8 Hz), 1.48 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H25O3N3F: 398.1875, found: 398.1887.
tert-Butyl 2-(5-Fluoro-6-(piperidin-1-yl)pyridin-3-yl)-1H-indole-1-carboxylate (92)
Compound 12 (0.090 g, 0.34 mmol), compound 64 (0.090 g, 0.35 mmol), Pd(dppf)Cl2 (0.019 g, 0.026 mmol), 1,4-dioxane (10 mL), and K2CO3(aq) (2 M, 0.5 mL) were flushed with N2(g) for 5 min, then stirred under N2(g) at 100 °C overnight (TLC indicated unreacted 64 remaining). Additional 12 (0.085 g, 0.33 mmol) was added, and the reaction mixture was stirred under N2(g) at 100 °C for 5 h, then cooled to ambient temperature and evaporated to dryness. The residue was dissolved in H2O and extracted with EtOAc (10 mL × 3), and the extracts were combined, dried over MgSO4, and filtered. The filtrate was passed through a silica gel pad, and the silica was washed with EtOAc. The solvent was removed to give a residue that was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 80:20) to afford 92 (0.120 g, 87%) as a solid:
1H NMR (300 MHz, CDCl3) δ 8.19 (d, 1H, J = 8.4 Hz), 8.07 (t, 1H, J = 1.5 Hz), 7.54 (unresolved apparent dd, 1H, J = 7.8 Hz), 7.33 (partially resolved ddd, 1H, J = 8.1 Hz, J = 7.2 Hz, J = 1.2 Hz), 7.26 (m–obscured by CHCl3 resonance, 3 H) 6.55 (s, 1H), 3.50 (m, 4H), 1.69 (m, 6H), 1.44 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C23H27O2N3F: 396.2082, found: 396.2082.
tert-Butyl 2-(5-Fluoro-6-morpholinopyridin-3-yl)-1H-indole-1-carboxylate (93)
Compound 12 (0.170 g, 0.651 mmol), compound 65 (0.075 g, 0.29 mmol), Pd(dppf)Cl2 (0.011 g, 0.015 mmol), 1,4-dioxane (10 mL), and K2CO3(aq) (2 M, 0.5 mL) were flushed with N2(g) for 5 min, then stirred under N2(g) at 100 °C overnight. The reaction mixture was cooled to ambient temperature and evaporated to dryness. The residue was dissolved in H2O and extracted with EtOAc (10 mL × 3), and the extracts were dried over MgSO4 and filtered. The filtrate was passed through a silica gel pad, and the silica was washed with EtOAc. The solvent was removed to give a residue that was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 80:20) to afford 93 (0.092 g, 80%) as an off-white solid: 1H NMR (400 MHz, CDCl3) δ 8.18 (d, 1H, J = 8.4 Hz), 8.09 (s, 1H), 7.55 (d, 1H, J = 7.6 Hz), 7.33 (m, 2H), 7.26 (m–obscured by CHCl3 resonance, 1H), 6.57 (s, 1H), 3.86 (t, 4H, J = 4.4 Hz), 3.54 (t, 4H, J = 4.4 Hz), 1.45 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H25O3N3F: 398.1875, found: 398.1863.
4-(4-Fluoro-5-(1H-indol-2-yl)pyridin-2-yl)morpholine (94)
Compound 91 (0.054 g, 0.14 mmol) was dissolved in TFA (2 mL), stirred at ambient temperature for 2 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 solution to give a solid that was washed with cold CH2Cl2, then purified by radial chromatography (hexane/EtOAc v/v 90:10 to 80:20) to afford 94 (0.030 g, 74%) as an off-white solid: 1H NMR (300 MHz, acetone-d6) δ 10.52 (br s, 1H), 8.66 (d, 1H, J = 11.4 Hz), 7.56 (d, 1H, J = 8.1 Hz), 7.42 (d, 1H, J = 8.1 Hz), 7.10 (partially resolved ddd, 1H, J = 8.1 Hz, J = 6.9 Hz, J = 1.2 Hz), 7.02 (partially resolved ddd, 1H, J = 7.8 Hz, J = 7.2 Hz, J = 1.2 Hz), 6.81 (m, 1H), 6.71 (d, 1H, J = 14.7 Hz), 3.76 (apparent t, 4H, J = 4.8 Hz), 3.59 (apparent t, 4H, J = 4.8 Hz); 13C NMR (125 MHz, DMSO-d6) δ 166.33 (d, JFC = 257.8 Hz), 159.93 (d, JFC = 11.4 Hz), 147.61 (d, JFC = 3.8 Hz), 136.49, 129.80, 128.42, 121.48, 119.78, 119.32, 111.12, 107.52 (d, JFC = 10.1 Hz), 100.69 (d, JFC = 6.3 Hz), 93.66 (d, JFC = 22.5 Hz), 65.81, 45.00; HRMS (ESI) [M + H]+ Calcd for C17H17ON3F: 298.1350, found: 298.1343.
2-(5-Fluoro-6-(piperidin-1-yl)pyridin-3-yl)-1H-indole (95)
Compound 92 (0.110 g, 0.278 mmol) was dissolved in TFA (2 mL), stirred at ambient temperature for 2 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid which was washed with cold CH2Cl2, then purified by radial chromatography (hexane/EtOAc v/v 90:10) to afford 95 (0.066 g, 80%) as an off-white solid: 1H NMR (300 MHz, acetone-d6) δ 10.61 (br s, 1H), 8.50 (t, 1H, J = 1.8 Hz), 7.80 (dd, 1H, J = 14.7 Hz, J = 1.8 Hz), 7.54 (apparent d, 1H, J = 8.1 Hz), 7.38 (dd, 1H, J = 8.7 Hz, J = 0.6 Hz), 7.10 (ddd, 1H, J = 8.1 Hz, J = 6.9 Hz, J = 1.2 Hz), 7.02 (ddd, 1H, J = 8.1 Hz, J = 6.9 Hz, J = 1.2 Hz), 6.85 (m, 1H), 3.50 (m, 4H), 1.67 (m, 6 H); 13C NMR (125 MHz, DMSO-d6): 149.03 (d, JFC = 255.2 Hz), 148.34 (d, JFC = 6.4 Hz), 139.27 (d, JFC = 4.6 Hz), 137.00, 134.25, 128.59, 121.49, 120.81 (d, JFC = 2.8 Hz), 119.82 (d, JFC = 20.8 Hz), 119.80, 119.43, 111.08, 98.44, 48.25 (d, JFC = 5.8 Hz), 25.37, 24.19; HRMS (ESI) [M + H]+ Calcd for C18H19N3F: 296.1558, found: 296.1550.
4-(3-Fluoro-5-(1H-indol-2-yl)pyridin-2-yl)morpholine (96)
Compound 93 (0.090 g, 0.23 mmol) was dissolved in TFA (2 mL), stirred at ambient temperature for 1 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid that was purified by radial chromatography twice (hexane/EtOAc v/v 95:5 to 90:10) to afford 96 (0.025 g, 37%) as a solid: 1H NMR (300 MHz, acetone-d6) δ 10.65 (br s, 1H), 8.54 (apparent t, 1H, J = 1.8 Hz), 7.86 (dd, 1H, J = 14.4 Hz, J = 1.8 Hz), 7.55 (d, 1H, J = 7.8 Hz), 7.39 (partially resolved dd, 1H, J = 8.1 Hz, J = 0.9 Hz), 7.11 (ddd, 1H, J = 8.7 Hz, J = 6.9 Hz, J = 1.2 Hz), 7.02 (partially resolved ddd, 1H, J = 8.1 Hz, J = 7.8 Hz, J = 1.2 Hz), 6.88 (m, 1H), 3.78 (apparent t, 4H, J = 4.8 Hz), 3.50 (apparent t, 4H, J = 4.8 Hz); 13C NMR (125 MHz, DMSO-d6): 149.29 (d, JFC = 255.4 Hz), 147.87 (d, JFC = 6.9 Hz), 139.24 (d, JFC = 4.8 Hz), 137.09, 136.93, 133.83 (d, JFC = 10.2 Hz), 128.49 (d, JFC = 10.4 Hz), 121.65, 120.08 (d, JFC = 20.5 Hz), 119.88, 119.49, 111.10, 98.78, 66.01, 47.73 (d, JFC = 5.4 Hz); HRMS (ESI) [M + H]+ Calcd for C17H17ON3F: 298.1350, found: 298.1348.
tert-Butyl 2-(3-Fluoro-5-(piperidin-1-yl)pyridin-2-yl)-1H-indole-1-carboxylate (97)
Compound 12 (0.220 g, 0.843 mmol), compound 66 (0.090 g, 0.42 mmol), Pd(dppf)Cl2 (0.019 g, 0.026 mmol), 1,4-dioxane (10 mL), and K2CO3(aq) (2 M, 0.5 mL) were flushed with N2(g) for 5 min, then stirred under N2(g) at 100 °C overnight (TLC indicated unreacted 66 remaining). Additional 12 (0.090 g, 0.35 mmol) was added and stirring under N2(g) at 100 °C was continued overnight, then the reaction mixture was cooled to ambient temperature and evaporated to dryness. The residue was dissolved in H2O and extracted with EtOAc (10 mL × 3), and the extracts were dried over MgSO4 and filtered. The filtrate was passed through a silica gel pad, and the silica was washed with EtOAc. The solvent was removed to give a residue that was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 80:20) to afford 97 (0.075 g, 45%) as a solid: 1H NMR (300 MHz, CDCl3) δ 8.19 (dd, 1H, J = 8.4 Hz, J = 0.6 Hz), 8.15 (dd, 1H, J = 2.7 Hz, J = 1.5 Hz), 7.57 (d, 1H, J = 7.5 Hz), 7.33 (ddd, 1H, J = 8.4 Hz, J = 7.2 Hz, J = 1.2 Hz), 7.23 (apparent td–partially obscured by CHCl3 resonance, 1H, J = 7.5 Hz, J = 0.9 Hz), 6.91 (dd, 1H, J = 12.6 Hz, J = 2.4 Hz), 6.78 (s, 1H), 3.28 (m, 4H), 1.70 (m, 6H), 1.42 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C23H27O2N3F: 396.2082, found: 396.2082.
2-(3-Fluoro-5-(piperidin-1-yl)pyridin-2-yl)-1H-indole (98)
Compound 97 (0.075 g, 0.19 mmol) was dissolved in TFA (1 mL), stirred at ambient temperature for 1 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid which was washed with cold CH2Cl2 to afford 98 (0.044 g, 78%) as an off-white solid: 1H NMR (300 MHz, acetone-d6) δ 10.60 (br s, 1H), 8.20 (t, 1H, J = 2.4 Hz), 7.58 (partially resolved dd, 1H, J = 8.1 Hz), 7.55 (partially resolved dd, 1H, J = 8.1 Hz, J = 0.9 Hz), 7.19 (dd, 1H, J = 14.7 Hz, J = 2.4 Hz), 7.12 (ddd, 1H, J = 8.1 Hz, J = 7.2 Hz, J = 1.2 Hz), 7.01 (ddd, 1H, J = 7.8 Hz, J = 7.2 Hz, J = 1.2 Hz), 6.94 (m, 1H), 3.37 (m, 4H), 1.70 (m, 6 H); 13C NMR (125 MHz, DMSO-d6): 157.07 (d, JFC = 258.7 Hz), 147.40 (d, JFC = 5.4 Hz), 136.20, 132.75 (d, JFC = 7.2 Hz), 132.42, 128.70, 127.30 (d, JFC = 12.2 Hz), 121.70, 120.22, 119.15, 111.69, 108.77 (d, JFC = 22.6 Hz), 100.67 (d, JFC = 11.9 Hz), 48.18, 24.72, 23.64; HRMS (ESI) [M + H]+ Calcd for C18H19N3F: 296.1558, found: 296.1562.
tert-Butyl 2-(3-Fluoro-5-morpholinopyridin-2-yl)-1H-indole-1-carboxylate (99)
Compound 12 (0.170 g, 0.651 mmol), compound 67 (0.078 g, 0.36 mmol), Pd(dppf)Cl2 (0.018 g, 0.025 mmol), 1,4-dioxane (10 mL), and K2CO3(aq) (2 M, 0.5 mL) were flushed with N2(g) for 5 min, then stirred under N2(g) at 100 °C overnight. The reaction mixture was cooled to ambient temperature and evaporated to dryness. The residue was dissolved in H2O, extracted with EtOAc (10 mL × 3), and the extracts were dried over MgSO4 and filtered. The filtrate was passed through a silica gel pad, and the silica was washed with EtOAc. The solvent was removed to give a residue that was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 80:2) to afford recovered 67 (0.058 g, 74%) and 99 (0.029 g, 20%) as a solid: 1H NMR (400 MHz, CDCl3) δ 8.18 (d, 1H, J = 8.4 Hz), 8.15 (s, 1H), 7.58 (d, 1H, J = 7.6 Hz), 7.34 (apparent t, 1H, J = 7.6 Hz), 7.24 (apparent t, 1H, J = 7.2 Hz), 6.92 (dd, 1H, J = 12.0 Hz, J = 2.0 Hz), 6.81 (s, 1H), 3.90 (t, 4H, J = 4.8 Hz), 3.26 (t, 4H, J = 4.8 Hz), 1.43 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H25O3N3F: 398.1875, found: 398.1865.
tert-Butyl 6-Fluoro-2-(3-fluoro-5-morpholinopyridin-2-yl)-1H-indole-1-carboxylate (100)
Compound 82 (0.190 g, 0.681 mmol), compound 67 (0.087 g, 0.40 mmol), Pd(dppf)Cl2 (0.019 g, 0.026 mmol), 1,4-dioxane (10 mL), and K2CO3(aq) (2 M, 0.5 mL) were flushed with N2(g) for 5 min, then stirred under N2(g) at 100 °C overnight (TLC indicated unreacted 67 remaining). Additional 82 (0.093 g, 0.33 mmol) was added, and the reaction mixture was stirred under N2(g) at 100 °C for 5 h, then cooled to ambient temperature and evaporated to dryness. The residue was dissolved in H2O and extracted with EtOAc (10 mL × 3), and the extracts were combined, dried over MgSO4, and filtered. The filtrate was passed through a silica gel pad, and the silica was washed with EtOAc. The solvent was removed to give a residue that was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 80:20) to afford recovered 67 (0.023 g, 26%) and 100 (0.096 g, 57%) as a solid: 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 7.91 (partially resolved dd, 1H, J = 10.8 Hz, J = 1.2 Hz), 7.49 (dd, 1H, J = 8.4 Hz, J = 5.6 Hz), 7.00 (apparent td, 1H, J = 9.6 Hz, J = 1.6 Hz), 6.92 (apparent dd, 1H, J = 12.4 Hz, J = 1.6 Hz), 6.76 (s, 1H), 3.90 (t, 4H, J = 4.8 Hz), 3.26 (t, 4H, J = 4.8 Hz), 1.43 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H24O3N3F2: 416.1780, found: 416.1767.
tert-Butyl 2-(4-Fluoro-5-morpholinopyridin-2-yl)-1H-indole-1-carboxylate (101)
Compound 12 (0.085 g, 0.33 mmol), compound 68 (0.078 g, 0.36 mmol), Pd(dppf)Cl2 (0.018 g, 0.025 mmol), 1,4-dioxane (5 mL), and K2CO3(aq) (2 M, 0.5 mL) were stirred at 100 °C under N2(g) overnight. The reaction mixture was cooled to ambient temperature, evaporated to dryness, and the residue was dissolved in H2O and extracted with EtOAc (10 mL × 3). The combined extracts were dried over MgSO4, filtered, and the filtrate was passed through a silica gel pad and washed with EtOAc. The solution was concentrated to dryness and the residue was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 70:30) to give recovered 68 (0.032 g, 41%) and compound 101 (0.045 g, 35%): 1H NMR (300 MHz, CDCl3) δ 8.28 (d, 1H, J = 11.1 Hz), 8.15 (d, 1H, J = 8.1 Hz), 7.57 (d, 1H, J = 7.8 Hz), 7.35 (partially resolved ddd, 1H, J = 8.4 Hz, J = 7.2 Hz, J = 1.2 Hz), 7.25 (m–partially obscured by CHCl3 resonance, 1H), 7.19 (d, 1H, J = 12.9 Hz), 6.75 (s, 1H), 3.90 (apparent t, 4H, J = 4.5 Hz), 3.21 (apparent t, 4H, J = 4.5 Hz), 1.42 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H25O3N3F: 398.1875, found: 398.1865.
tert-Butyl 6-Fluoro-2-(4-fluoro-5-morpholinopyridin-2-yl)-1H-indole-1-carboxylate (102)
Compound 82 (0.230 g, 0.824 mmol), compound 68 (0.097 g, 0.45 mmol), Pd(dppf)Cl2 (0.018 g, 0.025 mmol), 1,4-dioxane (10 mL), and K2CO3(aq) (2 M, 0.5 mL) were flushed with N2(g) for 5 min, then stirred under N2(g) and heated at 100 °C overnight. The reaction mixture was cooled to ambient temperature, evaporated to dryness, the residue was dissolved in H2O and extracted with EtOAc (10 mL × 3). The combined extracts were dried over MgSO4, filtered, and the filtrate was passed through a silica gel pad and washed with EtOAc. The solution was concentrated to dryness and the residue was purified by radial chromatography (hexane/EtOAc v/v 95:5 to 80:20) to afford recovered 68 (0.068 g, 70%) and 102 (0.029 g, 16%) as a solid: 1H NMR (300 MHz, CDCl3) δ 8.27 (d, 1H, J = 10.8 Hz), 7.88 (dd, 1H, J = 10.5 Hz, J = 2.4 Hz), 7.48 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.18 (d, 1H, J = 12.9 Hz), 7.01 (td, 1H, J = 9.0 Hz, J = 2.4 Hz), 6.70 (s, 1H), 3.90 (m–AA’XX’, 4H, J = 4.8 Hz, J = 1.8 Hz), 3.21 (m–AA’XX’, 4H, J = 4.8 Hz, J = 1.8 Hz), 1.41 (s, 9H); HRMS (ESI) [M + H]+ Calcd for C22H24O3N3F2: 416.1780, found: 416.1767.
4-(5-Fluoro-6-(1H-indol-2-yl)pyridin-3-yl)morpholine (103)
Compound 99 (0.029 g, 0.073 mmol) was dissolved in TFA (1 mL), stirred at ambient temperature for 2 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid that was washed with cold CH2Cl2 to afford 103 (0.017 g, 78%) as an off-white solid: 1H NMR (400 MHz, acetone-d6) δ 10.65 (br s, 1H), 8.22 (t, 1H, J = 2.0 Hz), 7.60 (unresolved dq, 1H, J = 8.0 Hz), 7.56 (partially resolved dq, 1H, J = 8.0 Hz, J = 0.8 Hz), 7.24 (dd, 1H, J = 14.4 Hz, J = 2.4 Hz), 7.14 (ddd, 1H, J = 8.8 Hz, J = 7.2 Hz, J = 1.2 Hz), 7.02 (ddd, 1H, J = 7.8 Hz, J = 7.2 Hz, J = 1.2 Hz), 6.98 (dm, 1H, J = 4.4 Hz), 3.81 (apparent t, 4H, J = 4.8 Hz), 3.32 (partially resolved m–AA’XX’, 4H, J = 4.8 Hz); 13C NMR (125 MHz, DMSO-d6): 156.94 (d, JFC = 258.6 Hz), 147.20 (d, JFC = 4.3 Hz), 136.24, 132.53 (d, JFC = 6.4 Hz), 132.14, 128.65, 128.46 (d, JFC = 10.9 Hz), 121.88, 120.33, 119.22, 111.74, 108.95 (d, JFC = 22.1 Hz), 101.06 (d, JFC = 11.9 Hz), 65.69, 47.23; HRMS (ESI) [M + H]+ Calcd for C17H17ON3F: 298.1350, found: 298.1353.
4-(5-Fluoro-6-(6-fluoro-1H-indol-2-yl)pyridin-3-yl)morpholine (104)
Compound 100 (0.031 g, 0.075 mmol) was dissolved in TFA (1 mL), stirred at ambient temperature for 1 h, and then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid that was washed with cold CH2Cl2 to afford 104 (0.023 g, 98%) as an off-white solid: 1H NMR (300 MHz, acetone-d6) δ 10.77 (br s, 1H), 8.21 (t, 1H, J = 1.8 Hz), 7.58 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.30 (dd, 1H, J = 10.2 Hz, J = 2.4 Hz), 7.23 (dd, 1H, J = 14.4 Hz, J = 2.4 Hz), 6.96 (dm, 1H, J = 4.2 Hz), 6.85 (ddd, 1H, J = 9.9 Hz, J = 8.7 Hz, J = 2.4 Hz), 3.81 (apparent t, 4H, J = 4.8 Hz), 3.32 (apparent t, 4H, J = 4.8 Hz); 13C NMR (125 MHz, DMSO-d6): 159.18 (d, JFC = 235.3 Hz), 156.82 (d, JFC = 258.8 Hz), 147.26 (d, JFC = 5.5 Hz), 136.24 (d, JFC = 13.2 Hz), 133.33 (unresolved m), 132.13 (d, JFC = 2.8 Hz), 128.15 (d, JFC = 11.9 Hz), 125.49, 121.46 (d, JFC = 10.2 Hz), 108.93 (d, JFC = 22.8 Hz), 107.83 (d, JFC = 24.5 Hz), 101.06 (d, JFC = 12.1 Hz), 97.51 (d, JFC = 25.7 Hz), 65.69, 47.21; HRMS (ESI) [M + H]+ Calcd for C17H16ON3F2: 316.1256, found: 316.1258.
4-(4-Fluoro-6-(1H-indol-2-yl)pyridin-3-yl)morpholine (105)
Compound 101 (0.040 g, 0.10 mmol) was dissolved in TFA (1.5 mL), stirred at ambient temperature for 1.5 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid that was washed with hexanes and cold CH2Cl2 to afford 105 (0.029 g, 97%) as an off-white solid: 1H NMR (300 MHz, acetone-d6) δ 10.73 (br s, 1H), 8.28 (d, 1H, J = 11.1 Hz), 7.72 (d, 1H, J = 13.8 Hz), 7.58 (d, 1H, J = 8.1 Hz), 7.53 (d, 1H, J = 8.1 Hz, J = 0.9 Hz), 7.14 (ddd, 1H, J = 8.1 Hz, J = 7.2 Hz, J = 1.2 Hz), 7.07 (m, 1H), 7.02 (ddd, 1H, J = 7.8 Hz, J = 7.2 Hz, J = 0.9 Hz), 3.82 (m–AA’XX’, 4H, J = 4.8 Hz, J = 1.8 Hz), 3.19 (m–AA’XX’, 4H, J = 4.8 Hz, J = 1.8 Hz); 13C NMR (125 MHz, DMSO-d6): 160.55 (d, JFC = 258.7 Hz), 146.18 (d, JFC = 8.0 Hz), 140.75 (unresolved d), 137.10, 136.37 (unresolved d), 134.62 (d, JFC = 5.2 Hz), 128.33, 122.15, 120.43, 119.43, 111.87, 107.55 (d, JFC = 18.4 Hz), 100.24, 65.99, 50.08; HRMS (ESI) [M + H]+ Calcd for C17H17ON3F: 298.1350, found: 298.1347.
4-(4-Fluoro-6-(6-fluoro-1H-indol-2-yl)pyridin-3-yl)morpholine (106)
Compound 102 (0.029 g, 0.070 mmol) was dissolved in TFA (1 mL), stirred at ambient temperature for 2 h, then the TFA was removed with N2(g) flow. The residue was neutralized with aqueous NaHCO3 to give a solid which was washed with cold CH2Cl2 to afford 106 (0.021 g, 95%) as an off-white solid: 1H NMR (300 MHz, acetone-d6) δ 10.86 (br s, 1H), 8.27 (d, 1H, J = 11.1 Hz), 7.71 (d, 1H, J = 13.8 Hz), 7.57 (dd, 1H, J = 8.7 Hz, J = 5.4 Hz), 7.26 (dd, 1H, J = 9.9 Hz, J = 2.4 Hz), 7.08 (m, 1H), 6.85 (ddd, 1H, J = 9.9 Hz, J = 8.7 Hz, J = 2.4 Hz), 3.82 (m–AA’XX’, 4H, J = 4.8 Hz, J = 1.8 Hz), 3.19 (m–AA’XX’, 4H, J = 4.8 Hz, J = 1.8 Hz); 13C NMR (125 MHz, DMSO-d6): 160.56 (d, JFC = 258.8 Hz), 159.30 (d, JFC = 235.7 Hz), 145.85 (d, JFC = 8.3 Hz), 140.71 (d, JFC = 3.0 Hz), 137.16 (dd, JFC = 3.6 Hz), 137.03 (d, JFC = 13.2 Hz), 134.69 (d, JFC = 5.8 Hz), 125.17, 121.59 (d, JFC = 10.2 Hz), 108.11 (d, JFC = 24.5 Hz), 107.46 (d, JFC = 18.5 Hz), 100.26, 97.63 (d, JFC = 25.8 Hz), 65.99, 50.07 (d, JFC = 2.6 Hz); HRMS (ESI) [M + H]+ Calcd for C17H16ON3F2: 316.1256, found: 316.1245.
tert-Butyl 2-(2-(4-(2-(2-Fluoroethoxy)ethoxy)piperidin-1-yl)pyrimidin-5-yl)-1H-indole-1-carboxylate (107)
Compound 50 (0.079 g, 0.20 mmol) was flushed with N2(g), dissolved in N,N-dimethylacetamide (DMA) (1 mL), and cooled to 0 °C. NaH (60%, 0.026 g, 0.65 mmol, 3.2 equiv) was added, the mixture was stirred at 0 °C under N2(g) for 5 min, then a solution of 2-(2-fluoroethoxy)ethyl 4-methylbenzenesulfonate (0.110 g, 0.419 mmol, 2.1 equiv) in DMA (0.5 mL) was added. The reaction mixture was stirred at ambient temperature under N2(g) for 2 h, then H2O (5 drops) was added, the mixture was stirred for 10 min, then poured into a mixture of H2O (20 mL), EtOAc (20 mL), hexane (10 mL). The layers were mixed and separated, and the organic layer was washed with H2O (10 mL × 2) and brine (10 mL) and dried over MgSO4. The solvent was removed to give a yellow residue (0.138 g) that was dissolved in CH2Cl2, poured onto dry silica (55 cm h × 45 cm i.d.), and eluted under vacuum: CH2Cl2 (100 mL), %MeOH/CH2Cl2–1% (100 mL), 2% (100 mL), 3% (100 mL), 5% (100 mL) to give a faint brown syrup (0.126 g). Purification by radial chromatography (1 mm silica): CH2Cl2 (50 mL), %MeOH/CH2Cl2–1% (50 mL), 2% (25 mL) afforded 107 (0.032 g, 33%) as a colorless residue: 1H NMR (300 MHz, CDCl3) δ 8.48 (s, 2H), 7.62 (d, 1H, J = 7.8 Hz), 7.41 (d, 1H, J = 8.1 Hz), 7.24 (td, 1H, J = 8.1 Hz, J = 0.9 Hz), 7.15 (partially resolved td, 1H, J = 7.8 Hz, J = 0.9 Hz), 6.50 (s, 1H), 4.86 (apparent septet, 1H, J = 4.2 Hz), 4.52 (apparent t, 1H, J = 4.2 Hz), 4.39 (m, 3H), 4.30 (t, 2 H, J = 6.0 Hz), 3.80 (t, 2 H, J = 6.0 Hz), 3.55 (m, 4H), 2.06 (m, 2H), 1.77 (m, 2H), 1.52 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 160.98, 158.41, 153.18, 137.63, 136.29, 128.37, 122.06, 120.70, 120.35, 115.32, 110.13, 102.87, 83.83, 82.43 (d, JFC = 13.8 Hz), 73.40, 70.64 (d, JFC = 18.8 Hz), 70.01, 44.09, 41.48, 30.83, 28.02; HRMS (ESI) [M + H]+ Calcd for C26H34O4N4F: 485.2559, found: 485.2556.
2-(2-(4-(2-(2-Fluoroethoxy)ethoxy)piperidin-1-yl)pyrimidin-5-yl)-1H-indole (108)
Compound 107 (0.031 g, 0.064 mmol) and TFA (0.65 mL, 8.4 mmol, 132 equiv) were stirred for 20 min, then poured into a mixture of NaHCO3 (0.790 g, 9.40 mmol, 1.1 equiv TFA), H2O (25 mL), and CH2Cl2 (25 mL). The mixture was stirred for 5 min, the layers were separated, and the aqueous layer was extracted with CH2Cl2 (5 mL × 2). The combined CH2Cl2 layers were washed with brine (20 mL), dried over MgSO4, concentrated, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: %MeOH/CH2Cl2–1% (50 mL), 2% (75 mL), 3% (100 mL) to afford 108 (0.016 g, 65%) as an off-white solid: 1H NMR (MHz, CDCl3) δ 8.48 (s, 2H), 7.62 (d, 1H, J = 7.6 Hz), 7.41 (d, 1H, J = 8.0 Hz), 7.24 (td, 1H, J = 8.0 Hz, J = 0.8 Hz), 7.15 (partially resolved td, 1H, J = 7.6 Hz, J = 0.8 Hz), 6.50 (s, 1H), 4.48 (dt, 2 H, J = 13.4 Hz, J = 4.4 Hz), 4.44 (dt, 2 H, 2JHF = 48.0 Hz, J = 4.4 Hz), 4.30 (t, 2 H, J = 6.0 Hz), 4.01 (apparent septet, 1H, J = 4.4 Hz), 3.80 (t, 2 H, J = 6.0 Hz), 3.55 (dt, 2 H, 3JHF = 29.2 Hz, J = 4.4 Hz), 3.42 (ddd, 2 H, J = 13.4 Hz, J = 10.0 Hz, J = 3.6 Hz), 2.01 (m, 2H), 1.58 (m, 2H); HRMS (ESI) [M + H]+ Calcd for C21H26FN4O2: 385.2034, found: 385.2041.
4-(5-(6-Fluoro-1-methyl-1H-indol-2-yl)pyrimidin-2-yl)morpholine (109)
Compound 8 (0.021 g, 0.070 mmol) was flushed with N2(g), then dissolved in DMA (1 mL), and cooled to 0 °C. NaH (60%, 0.010 g, 0.24 mmol, 3.4 equiv) was added, the reaction mixture was stirred at 0 °C under N2(g) for 18 min, then MeOTs (0.027 g, 0.14 mmol, 2 equiv) was added. The reaction mixture was warmed to ambient temperature, stirred for 2 h, then H2O (1 drop) was added. The reaction mixture was combined with EtOAc (15 mL), hexane (5 mL), and H2O (10 mL), the layers were mixed and separated, and the organic layer was washed with H2O (10 mL × 2) and brine (10 mL) and dried over MgSO4. The solvent was removed to give an off-white solid that was dissolved in CH2Cl2, poured onto dry silica (33 mm h × 33 mm i.d.), and eluted under vacuum: %MeOH/CH2Cl2–1% (100 mL), 2% (50 mL), 3% (50 mL) to afford 109 (0.019 g, 86%) as an off-white solid: 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 2H), 7.52 (dd, 1H, J = 8.4 Hz, J = 5.4 Hz), 7.02 (dd, 1H, J = 10.0 Hz, J = 2.0 Hz), 6.91 (td, 1H, J = 9.0 Hz, J = 2.0 Hz), 6.49 (s, 1H), 3.88 (m, 4H), 3.81 (m, 4H), 3.67 (s, 3H); HRMS (ESI) [M + H]+ Calcd for C17H18FN4O: 313.1459, found: 313.1455.
tert-Butyl 6-Fluoro-1H-indole-1-carboxylate (110)
6-Fluoroindole (3.28 g, 24.3 mmol), Boc2O (6.59 g, 30.2 mmol, 1.2 equiv), i-Pr2NEt (7.0 mL, 40 mmol, 1.7 equiv), DMAP (0.310 g, 2.54 mmol, 0.1 equiv), and CH2Cl2 (210 mL) were stirred at ambient temperature under N2(g) for 16 h. The reaction mixture was concentrated to a yellow/orange oil, hexane was added, and the mixture was purified by vacuum flash chromatography on silica (16 cm h × 4 cm i.d.): hexane (100 mL), %CH2Cl2/hexane–10% (200 mL), 25% (400 mL) to afford 110 (5.67 g, 99%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.86 (br d, 1H, J = 10.2 Hz), 7.56 (d, 1H, J = 3.6 Hz), 7.46 (dd, 1H, J = 8.4 Hz, J = 5.4 Hz), 6.98 (td, 1H, J = 9.0 Hz, J = 2.4 Hz), 6.53 (d, 1H, J = 3.6 Hz), 1.67 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 161.05 (d, 1JFC = 238.8 Hz), 149.71, 135.55 (d, JFC = 12.5 Hz), 126.95, 126.35 (d, JFC = 2.5 Hz), 121.62 (d, JFC = 10.0 Hz), 111.10 (d, JFC = 23.8 Hz), 107.19, 102.74 (d, JFC = 28.8 Hz), 84.25, 28.35; HRMS (ESI) [M + H]+ Calcd for C13H15O2NF: 236.1081, found: 236.1092.
2-(2-[18F]Fluoro-6-(piperidin-1-yl)pyridin-3-yl)-1H-indole ([18F]74)
Et4NHCO3 (70 mg) was dissolved in Milli-Q H2O (10 mL) to give a 7 mg/mL solution. A Waters Sep-Pak Plus Light QMA (WAT023525) was rinsed with Et4NHCO3(aq) solution (8 mL, 7 mg/mL), then Milli-Q H2O (10 mL) and then air (25 mL). A Waters C18 Sep-Pak (WAT020515) was rinsed with EtOH (10 mL) and then Milli-Q H2O (10 mL). No-carrier added [18F]F– was produced via the 18O(p,n)18F reaction (Siemens Eclipse HP cyclotron) and transferred in [18O]H2O to a lead-shielded hot cell (radiolabeling was then performed remotely with the aid of remote manipulators). [18F]F– (26.6 GBq (719 mCi)) was trapped on the QMA and eluted with Et4NHCO3(aq) solution (1 mL, 7 mg/mL), then CH3CN (1 mL) (451.4 MBq (12.2 mCi) remained on the QMA), and collected in a 5 mL V-vial. The solvents were evaporated under argon flow at 110 °C, then the Et4N18F was azeotropically dried with CH3CN aliquots (1 mL × 3) at 110 °C under argon flow. Compound 71 (3.8 mg) was dissolved in DMSO (0.6 mL) and added to the dried Et4N18F, the V-vial was capped and mixed gently for ∼10 s until the solution turned faint pink, then heated at ∼140 °C for 30 min. The V-vial was cooled for ∼1 min, the solution was diluted with HPLC solvent (0.5 mL, 55:45 v/v CH3CN/0.1 M NH4HCO2 pH 4.2), loaded onto the HPLC loop, the V-vial was rinsed with HPLC solvent (0.5 mL), the solvent rinse was loaded onto the HPLC loop, and the sample was purified by prep-HPLC (Phenomenex Gemini, 250 × 10 mm + guard; 5 mL/min for 6 min, then 10 mL/min). The HPLC radioactive peak at ∼24–25 min was collected, diluted with Milli-Q H2O (50 mL), passed through the C18 Sep-Pak, and the Sep-Pak was rinsed with Milli-Q H2O (10 mL). The radiotracer was eluted from the Sep-Pak with EtOH (1 mL), then 0.9% sterile saline (9 mL), passed through a 0.2 μm filter, and collected in a sterile vial to afford [18F]74 (1.2 GBq (31.9 mCi); 4.4% radiochemical yield (not decay-corrected); 99.4% radiochemical purity (Phenomenex Gemini, 5 μm, NX-C18 110 Å, 100 × 4.6 mm 60:40 v/v CH3CN/0.1 M NH4HCO2 pH 4.2; 1 mL/min; 254 nm); apparent95,96Am = 129.5 MBq/nmol (3.5 mCi/nmol); pH 5). Radiochemical identity was confirmed by coinjection with 74. The endotoxin level of the final formulation was <2.00 EU/mL (Charles Rivers Endosafe).
4-(6-[18F]Fluoro-5-(1H-indol-2-yl)pyridin-2-yl)morpholine ([18F]75, [18F]JSS20–183A)
Et4NHCO3 (70 mg) was dissolved in Milli-Q H2O (10 mL) to give a 7 mg/mL solution. A Waters Sep-Pak Plus Light QMA (WAT023525) was rinsed with Et4NHCO3(aq) solution (8 mL, 7 mg/mL), then Milli-Q H2O (10 mL), then air (25 mL). A Waters C18 Sep-Pak (WAT020515) was rinsed with EtOH (10 mL) and then Milli-Q H2O (10 mL). No-carrier added [18F]F– was produced via the 18O(p,n)18F reaction (Siemens Eclipse HP cyclotron) and transferred in [18O]H2O to a lead-shielded hot cell (radiolabeling was then performed remotely with the aid of remote manipulators). [18F]F– (30.3 GBq (818 mCi)) was trapped on the QMA and eluted with Et4NHCO3(aq) solution (1 mL, 7 mg/mL), then CH3CN (1 mL), then air (1 mL) (296 MBq (8.0 mCi) remained on the QMA), and collected in a 5 mL V-vial. The solvents were evaporated under argon flow at 110 °C, then the Et4N18F was azeotropically dried with CH3CN aliquots (1 mL × 3) at 110 °C under argon flow. Compound 72 (2.5 mg) was dissolved in DMSO (0.5 mL), added to the dried Et4N18F (the solution turned light purple), the V-vial was capped, and heated at ∼140 °C for 30 min. The V-vial was cooled for ∼1 min, the solution was diluted with HPLC solvent (0.5 mL, 45:55 v/v CH3CN/0.1 M NH4HCO2 pH 4.2), loaded onto the HPLC loop, the V-vial was rinsed with HPLC solvent (0.5 mL), the solvent rinse was loaded onto the HPLC loop, and the sample was purified by prep-HPLC (Phenomenex Gemini, 250 mm × 10 mm + guard; 5 mL/min for 6 min, then 8 mL/min). The HPLC radioactive peak at ca. 20.4–22 min was collected, diluted with Milli-Q H2O (50 mL), passed through the C18 Sep-Pak, and the Sep-Pak was rinsed with Milli-Q H2O (10 mL). The radiotracer was eluted from the Sep-Pak with EtOH (1 mL), then 0.9% sterile saline (9 mL), passed through a 0.2 μm filter, and collected in a sterile vial to afford [18F]75 (3.5 GBq (94.3 mCi)); 11.5% radiochemical yield (not decay-corrected); 98.9% radiochemical purity (Phenomenex Gemini, 5 μm, NX-C18 110 Å, 100 mm × 4.6 mm; 45:55 v/v CH3CN/0.1 M NH4HCO2 pH 4.2; 1 mL/min; 254 nm); apparent95,96Am = 51.8 MBq/nmol (1.4 mCi/nmol); pH 5. Radiochemical identity was confirmed by co-injection with 75. The endotoxin level of the final formulation was <2.00 EU/mL (Charles Rivers Endosafe).
Log D7.4 Determination of [18F]74
To four 8 mL glass vials with plastic screw caps (pulp/vinyl liner) were added octanol (2 mL) and 20 mM NaH2PO4(aq) solution pH 7.4 (2 mL), and the vials were vortexed for 15 s. To each vial was added radiotracer solution (50 μL, ∼170 kBq (∼4.6 μCi)), then each vial was vortexed for 30 s and let stand for 30 min to allow the layers to separate. From each of the four glass vials, samples of the octanol layer (0.5 mL) were removed using a pipettor with a plastic tip and placed in four counting tubes. Glass pipettes were then used to transfer samples of the aqueous layer (∼1.5 mL) from the four glass vials to glass test tubes. A sample from each aqueous layer (0.5 mL) was removed using a pipettor with a plastic tip and placed in a counting tube. The tubes were counted, and the log(octanol counts/aqueous counts) value was calculated for each pair of tubes and then averaged (log D7.4 = 1.80 ± 0.01 (n = 4)).
Log D7.4 Determination of [18F]75
To four 8 mL glass vials with plastic screw caps (pulp/vinyl liner) was added octanol (2 mL) and 20 mM NaH2PO4(aq) solution pH 7.4 (2 mL), and the vials were vortexed for 15 s. To each vial was added radiotracer solution (20 μL, ∼11.1 kBq (∼0.3 μCi)) that had been diluted with NaH2PO4(aq) solution, then each vial was vortexed for 30 s and let stand for 30 min to allow the layers to separate. From each of the four glass vials samples of the octanol layer (0.5 mL) were removed using a pipettor with a plastic tip and placed in four counting tubes. Glass pipettes were then used to transfer samples of the aqueous layer (∼1.5 mL) from the four glass vials to glass test tubes. A sample from each aqueous layer (0.5 mL) was removed using a pipettor with a plastic tip and placed in counting tubes. The tubes were counted, and the log(octanol counts/aqueous counts) value was calculated for each pair of tubes and then averaged (log D7.4 = 1.96 ± 0.03 (n = 4)).
PET/CT Imaging of [18F]74 and [18F]75
Nonhuman primate (NHP) imaging studies in male macaque (m. mulatta) monkeys (12 and 13 kg) were carried out in accordance with the guidelines set forth by the Institutional Animal Care and Use Committee (IACUC) at the University of Pittsburgh under an IACUC-approved protocol. PET/CT imaging was performed with a Siemens Biograph mCT Flow 64–4 R PET/CT scanner (22.1 mm axial FOV, maximum intrinsic spatial resolution of 4.3 mm fwhm). NHP were initially sedated with ketamine (15 mg/kg, i.m.) and glycopyrrolate (0.01 mg/kg) prior to intubation and induction of isoflurane anesthesia (0.5–2.0%). A venous catheter was placed in the saphenous vein for intravenous injection of [18F]74 or [18F]75 and venous blood sampling. NHPs were positioned using a scout planar radiograph to place the brain in the center of the field of view, after which a low-dose (19 mrem) helical CT scan was collected for attenuation correction of PET emission data. List-mode acquisition of PET emission data commenced with the start of slow (20 s) bolus injections of [18F]74 (101.8 MBq, molar activity 110.6 MBq/nmol) or [18F]75 (96.0 MBq, molar activity 88.4 MBq/nmol) and continued for 90 min. Emission data were binned into a dynamic series of 32 frames of increasing duration (20 s to 10 min) and reconstructed using filtered back projection with Fourier rebinning with standard corrections applied for attenuation, scatter, electronics dead-time, and physical decay.
Radiometabolite Analysis of [18F]74 and [18F]75
Venous blood samples (3 mL) were collected at 2, 10, 30, 45, and 90 min after radiotracer administration. Whole blood samples were transferred from the sample syringe into centrifuge tubes and centrifuged (Eppendorf MiniSpin 5452) for 45 s at 11,300g. The plasma supernatant (∼0.5 mL) was transferred to a centrifuge tube and diluted with an equal volume of acetonitrile. The resultant solution was vortexed for 1 min and then centrifuged for 45 s at 12,500 rpm. The resulting supernatant was analyzed by reverse-phase HPLC (Phenomenex Gemini, 5 μm, NX-C18 110 Å, 100 mm × 4.6 mm; 45:55 v/v CH3CN/0.1 M NH4HCO2 pH 4.2; 1 mL/min) and an inline 3″ NaI(Tl) scintillation detector (GABI Star, Elysia-Raytest, GmBH) enveloping a large-volume (2.0 mL) flow cell for measuring radioactivity in the eluent. Analysis of radiochromatograms was performed using GINA Star software v. 4.07 (Elysia-Raytest, GmBH) to determine the fraction of unmetabolized [18F]74 or [18F]75 at each time point. System suitability of the analytical system was evaluated by injection of the nonradioactive standard using the same analytical system described above using a Waters UV detector (Model 481, 254 nm).
Post-Mortem Tissues
Post-mortem human brain tissues were collected by brain banks at the University of Pittsburgh Alzheimer’s Disease Research Center (ADRC), the University of Pittsburgh Brain Tissue Donation Program, the Center for Neurodegenerative Disease Research (CNDR) at the University of Pennsylvania, the Neurodegenerative Disease Brain Bank at the University of California, San Francisco, and Dr. Thomas Beach at Banner/Sun Health AZ (through the Michael J. Fox Foundation) following informed consent of the donors and utilized at the University of Pittsburgh under the approval of the Committee for Oversight of Research and Clinical Training Involving Decedents (CORID no. 295). The binding assays utilized fresh frozen post-mortem human tissue from autopsy-confirmed cases. Brain tissue blocks (1 cm3) contained only frequent mixed 3R/4R-tau neurofibrillary tangles and amyloid-β plaque aggregates (AD middle frontal gyrus), or only 4R-tau aggregates (PSP superior frontal gyrus and CBD middle frontal gyrus), or only 3R-tau aggregates (PiD middle frontal gyrus) and no detectable amyloid-β or TDP-43 aggregates. Fresh frozen, autopsy-confirmed PD anterior cingulate cortex tissue blocks (1 cm3) contained frequent α-synuclein aggregates and no detectable amyloid-β or TDP-43 aggregates. Fresh frozen brain tissue blocks (1 cm3) of midfrontal gyrus from an autopsy-confirmed FTLD-TDP case contained TDP-43 pathology (score 5), but no amyloid-β, tau, or α-synuclein pathology. Fresh frozen brain tissue blocks (1 cm3) of cortical gray matter (prefrontal, parietal, occipital, and temporal pole) from a young autopsy-confirmed healthy control contained no neuropathological changes. Fresh frozen brain tissue blocks (1 cm3) of midfrontal gyrus from an autopsy-confirmed healthy elderly control contained no amyloid-β, tau, or α-synuclein pathology. The frozen tissue blocks were separately thawed and homogenized in ice-cold pH 7.0 phosphate-buffered saline (PBS) at 300 mg/mL on ice using a glass homogenizer, diluted 30-fold with PBS to 10 mg/mL and homogenized a second time with a Brinkmann Polytron homogenizer before storage at −80 °C. At the time of binding assays, frozen brain tissue homogenates were thawed to room temperature and diluted 10-fold in Tris buffer (pH 7.2) to a concentration of 1 mg/mL.
Fresh frozen whole brains were obtained from three 9-month-old female P301L-JNPL3 mice from the laboratory of Dr. Peter Davies (Feinstein Institute for Medical Research, North Shore-LIJ Health System, Long Island). These mice carry a transgene encoding human tau with four microtubule-binding repeat domains and no N-terminal inserts (4R/0N). The transgene contains the P301L mutation and expression is driven by the mouse prion promoter. Tau inclusions develop in an age- and gene-dose-dependent manner as early as 4.5 months. At 9 months of age, the mice have widespread and abundant tau pathology in the brain, deep cerebellar nuclei, medulla, and spinal cord.84 For the assays, brain tissue homogenates were pooled together from all three mice and samples were frozen until processed for the binding assays as described above.
In Vitro Competition (Ki) Assays
The unlabeled competitor compound equilibrium inhibition constant (Ki) values were determined versus tritium-labeled radioligands according to previously published procedures46,65,66 with minor modifications. A solution of each competitor compound was made in DMSO-d6 containing dimethyl sulfone (1 mM) as internal standard, and the concentration of the competitor compound was then determined by quantitative NMR.97 The solution was then diluted with DMSO to give a stock solution (400 μM) of the competitor compound. Various concentrations of the competitor compound were then made by diluting with Tris buffer. The appropriate concentrations (0.1 nM–1 μM) of unlabeled competitor (400 μL) were combined with solutions of tritium-labeled radioligand in Tris buffer (500 μL, ∼1 nM final concentration of radioligand). Brain tissue homogenate solution (100 μL) in Tris buffer (1 mg/mL) was then added (final concentration = 100 μg brain tissue/mL, 0.25% DMSO). The binding assay solution was incubated at ambient temperature for 60 min, then filtered (Whatman GF/B glass filter) using a Brandel M-24R cell harvester, and rapidly washed with Tris buffer (3 mL × 3). The filter circles were combined with CytoScint-ES, vortexed thoroughly, and counted using a liquid scintillation counter. Complete inhibition (100%) of radioligand-specific binding was defined as the number of counts displaced by a solution (1 μM) of the unlabeled version of the radioligand. All assays were performed in triplicate at each concentration.
Binding Affinity (KD) Assays
Compound [3H]75 homologous81 binding affinity (KD) assays using AD, PSP, CBD, PiD, young CT, elderly CT, PD, TDP-43, and P301L transgenic mouse brain homogenates were performed according to previously published procedures46,65,66 with minor modifications. A solution of 75 in DMSO (400 μM) was prepared as described above, diluted with Tris buffer to give a 20-μM solution (5% DMSO/Tris), then diluted serially with 5% DMSO/Tris. The solutions of 75 (50 μL) were each combined with a solution of [3H]75 (50 μL), Tris buffer (800 μL), and brain tissue homogenate solution (100 μL) (final assay solution: 100 μg brain tissue/mL, ∼1 nM [3H]75, 0.2–1000 nM 75, 0.25% DMSO/Tris buffer). Incubation, filtration, and counting were performed as described above. The concentration of bound [3H]75 was determined from the radioactivity retained on the filter (corrected for the nondisplaceable radioactivity—defined as that remaining with ∼1 μM 75) and the molar activity of [3H]75 after dilution with varying concentrations of 75. A Scatchard plot of the bound/free versus bound radioligand values at the different ligand concentrations was used to determine the KD value (slope = −1/ KD). The Bmax value was determined by the x-axis intercept of the bound/free versus bound line. All assays were performed at least in triplicate.
In Vitro Real-Time Autoradiography and Immunohistochemistry (IHC)
Formalin-fixed paraffin-embedded human brain tissues from AD, PSP, CBD, and elderly CT cases were acquired from the Center for Neurodegenerative Disease Research (CNDR) at the University of Pennsylvania. Clinical demographic data is shown in Table S9 (Supporting Information).
In vitro autoradiography was performed using 6-μm-thick deparaffinized sections derived from AD, PSP, CBD, and elderly CT brains. Brain sections underwent deparaffinization and antigen retrieval step using Citrate buffer (0.5 M, pH 6.0) for 1 h at 70 °C in a preheated water bath. All slides were subsequently equilibrated for 30 min in 1× PBS (Dulbecco’s phosphate-buffered saline) and then incubated for 90 min at ambient temperature with 4.5 nM [3H]75. The sections were rinsed two times in cold buffer 1× PBS + 20% EtOH for 5 min, followed by a quick dip in cold distilled water. Nonspecific binding was determined using 1 μM of unlabeled 75. Slides were then allowed to air-dry before being exposed and scanned in a real-time autoradiography system (BeaQuant instrument, ai4R) for 24 h. ROI delineation and quantification of signal were performed by using the image analysis software Beamage (ai4R). Specific binding was determined by subtracting the nonspecific signal (NSB) from the total signal and expressed as counts/min/mm2.
Immunohistochemistry was performed on deparaffinized sections adjacent to those used for autoradiography. Sections were permeabilized with 0.1% Triton X-100 for 10 min followed by 3 × 5 min washes in PBS Tween 20 (PBST) buffer. Sections underwent hydrogen peroxide blocking for 15 min and PBS + 10% goat serum +1% BSA + 0.1% Tween 20 blocking for 1 h at ambient temperature. Sections were immunostained using Phospho-Tau (Ser202, Thr205) Monoclonal Antibody (AT8) and Antibeta Amyloid antibody (mOC23) (Table S10, Supporting Information) used at 1:500 dilution overnight at 4 °C. After a series of thorough washes with PBST buffer, the slides were incubated with the secondary antibody Goat Anti-Mouse IgG H&L (HRP) and Goat antirabbit IgG H&L (HRP) (Table S11, Supporting Information) at 1:10,000 for 1 h at ambient temperature. The sections were washed again with PBST buffer, 3 × 5 min, treated with DAB substrate for 10 min, and counterstained with Meyer’s hematoxylin dye. Subsequently, the sections were dehydrated sequentially into 50, 75, 95, and 100% ethanol and xylene baths for 5 min each, then mounted with Limonene mounting media and coverslipped for microscopy. Images were captured with a Leica Aperio slide scanner (RRID: SCR_022420) at 40× magnification.
References
- E. H. Kellogg, N. M. A. Hejab, S. Poepsel, K. H. Downing, F. DiMaio, E. Nogales. Near-atomic model of microtubule-tau interactions.. Science, 2018. [DOI | PubMed]
- M. D. Weingarten, A. H. Lockwood, S. Y. Hwo, M. W. Kirschner. A protein factor essential for microtubule assembly.. Proc. Natl. Acad. Sci. U.S.A., 1975. [DOI | PubMed]
- V. J. Lowe, E. S. Lundt, S. M. Albertson, H.-K. Min, P. Fang, S. A. Przybelski, M. L. Senjem, C. G. Schwarz, K. Kantarci, B. Boeve, D. T. Jones, R. R. Reichard, J. F. Tranovich, F. S. Hanna Al-Shaikh, D. S. Knopman, C. R. Jack, D. W. Dickson, R. C. Petersen, M. E. Murray. Tau-positron emission tomography correlates with neuropathology findings.. Alzheimer’s Dementia, 2020. [DOI]
- N. Franzmeier, M. Brendel, L. Beyer, L. Slemann, G. G. Kovacs, T. Arzberger, C. Kurz, G. Respondek, M. J. Lukic, D. Biel, A. Rubinski, L. Frontzkowski, S. Hummel, A. Müller, A. Finze, C. Palleis, E. Joseph, E. Weidinger, S. Katzdobler, M. Song, G. Biechele, M. Kern, M. Scheifele, B.-S. Rauchmann, R. Perneczky, M. Rullman, M. Patt, A. Schildan, H. Barthel, O. Sabri, J. J. Rumpf, M. L. Schroeter, J. Classen, V. Villemagne, J. Seibyl, A. W. Stephens, E. B. Lee, D. G. Coughlin, A. Giese, M. Grossman, C. T. McMillan, E. Gelpi, L. Molina-Porcel, Y. Compta, J. C. van Swieten, L. D. Laat, C. Troakes, S. Al-Sarraj, J. L. Robinson, S. X. Xie, D. J. Irwin, S. Roeber, J. Herms, M. Simons, P. Bartenstein, V. M. Lee, J. Q. Trojanowski, J. Levin, G. Höglinger, M. Ewers. Tau deposition patterns are associated with functional connectivity in primary tauopathies.. Nat. Commun., 2022. [DOI | PubMed]
- A. C. Macedo, C. Tissot, J. Therriault, S. Servaes, Y.-T. Wang, J. Fernandez-Arias, N. Rahmouni, F. Z. Lussier, M. Vermeiren, G. Bezgin, P. Vitali, K. P. Ng, E. R. Zimmer, M.-C. Guiot, T. A. Pascoal, S. Gauthier, P. Rosa-Neto. The Use of Tau PET to Stage Alzheimer Disease According to the Braak Staging Framework.. J. Nucl. Med., 2023. [DOI | PubMed]
- C. G. Robinson, J. Lee, P. H. Min, S. A. Przybelski, K. A. Josephs, D. T. Jones, J. Graff-Radford, B. F. Boeve, D. S. Knopman, C. R. Jack, R. C. Petersen, M. M. Machulda, J. A. Fields, V. J. Lowe. Significance of a positive tau PET scan with a negative amyloid PET scan.. Alzheimer’s Dementia, 2024. [DOI]
- H. Wongso, R. Harada, S. Furumoto. Current Progress and Future Directions in Non-Alzheimer’s Disease Tau PET Tracers.. ACS Chem. Neurosci., 2025. [DOI | PubMed]
- Y. T. Wang, P. Edison. Tau Imaging in Neurodegenerative Diseases Using Positron Emission Tomography.. Curr. Neurol. Neurosci. Rep., 2019. [DOI | PubMed]
- Z. Mohammadi, H. Alizadeh, J. Marton, P. Cumming. The Sensitivity of Tau Tracers for the Discrimination of Alzheimer’s Disease Patients and Healthy Controls by PET.. Biomolecules, 2023. [DOI | PubMed]
- M. Marquié, E. E. Verwer, A. C. Meltzer, S. J. W. Kim, C. Agüero, J. Gonzalez, S. J. Makaretz, M. Siao Tick Chong, P. Ramanan, A. C. Amaral, M. D. Normandin, C. R. Vanderburg, S. N. Gomperts, K. A. Johnson, M. P. Frosch, T. Gómez-Isla. Lessons learned about [F-18]-AV-1451 off-target binding from an autopsy-confirmed Parkinson’s case.. Acta Neuropathol. Commun., 2017. [DOI | PubMed]
- J. Y. Choi, H. Cho, S. J. Ahn, J. H. Lee, Y. H. Ryu, M. S. Lee, C. H. Lyoo. Off-Target 18F-AV-1451 Binding in the Basal Ganglia Correlates with Age-Related Iron Accumulation.. J. Nucl. Med., 2018. [DOI | PubMed]
- C. Vermeiren, P. Motte, D. Viot, G. Mairet-Coello, J.-P. Courade, M. Citron, J. Mercier, J. Hannestad, M. Gillard. The tau positron-emission tomography tracer AV-1451 binds with similar affinities to tau fibrils and monoamine oxidases.. Mov. Disord., 2018. [DOI | PubMed]
- S. L. Baker, T. M. Harrison, A. Maass, R. La Joie, W. J. Jagust. Effect of Off-Target Binding on 18F-Flortaucipir Variability in Healthy Controls Across the Life Span.. J. Nucl. Med., 2019. [DOI | PubMed]
- L. R. Drake, J. M. Pham, T. J. Desmond, A. V. Mossine, S. J. Lee, M. R. Kilbourn, R. A. Koeppe, A. F. Brooks, P. J. H. Scott. Identification of AV-1451 as a Weak, Nonselective Inhibitor of Monoamine Oxidase.. ACS Chem. Neurosci., 2019. [DOI | PubMed]
- A. Gogola, D. S. Minhas, V. L. Villemagne, A. D. Cohen, J. M. Mountz, T. A. Pascoal, C. M. Laymon, N. S. Mason, M. D. Ikonomovic, C. A. Mathis, B. E. Snitz, O. L. Lopez, W. E. Klunk, B. J. Lopresti. Direct Comparison of the Tau PET Tracers 18F-Flortaucipir and 18F-MK-6240 in Human Subjects.. J. Nucl. Med., 2022. [DOI | PubMed]
- C. Tissot, S. Servaes, F. Z. Lussier, J. P. Ferrari-Souza, J. Therriault, P. C. L. Ferreira, G. Bezgin, B. Bellaver, D. T. Leffa, S. S. Mathotaarachchi, M. Chamoun, J. Stevenson, N. Rahmouni, M. S. Kang, V. Pallen, N. Margherita-Poltronetti, Y.-T. Wang, J. Fernandez-Arias, A. L. Benedet, E. R. Zimmer, J.-P. Soucy, D. L. Tudorascu, A. D. Cohen, M. Sharp, S. Gauthier, G. Massarweh, B. Lopresti, W. E. Klunk, S. L. Baker, V. L. Villemagne, P. Rosa-Neto, T. A. Pascoal. The Association of Age-Related and Off-Target Retention with Longitudinal Quantification of [18F]MK6240 Tau PET in Target Regions.. J. Nucl. Med., 2023. [DOI | PubMed]
- C. Groot, S. Villeneuve, R. Smith, O. Hansson, R. Ossenkoppele. Tau PET Imaging in Neurodegenerative Disorders.. J. Nucl. Med., 2022. [DOI | PubMed]
- G. Blazhenets, D. N. Soleimani-Meigooni, W. Thomas, N. Mundada, M. Brendel, S. Vento, L. VandeVrede, H. W. Heuer, P. Ljubenkov, J. C. Rojas, M. K. Chen, A. N. Amuiri, Z. Miller, M. L. Gorno-Tempini, B. L. Miller, H. J. Rosen, I. Litvan, M. Grossman, B. Boeve, A. Pantelyat, M. C. Tartaglia, D. J. Irwin, B. C. Dickerson, S. L. Baker, A. L. Boxer, G. D. Rabinovici, R. La Joie. [18F]PI-2620 Binding Patterns in Patients with Suspected Alzheimer Disease and Frontotemporal Lobar Degeneration.. J. Nucl. Med., 2023. [DOI | PubMed]
- M. Malpetti, S. S. Kaalund, K. A. Tsvetanov, T. Rittman, M. Briggs, K. S. J. Allinson, L. Passamonti, N. Holland, P. S. Jones, T. D. Fryer, Y. T. Hong, A. Kouli, W. R. Bevan-Jones, E. Mak, G. Savulich, M. G. Spillantini, F. I. Aigbirhio, C. H. Williams-Gray, J. T. O’Brien, J. B. Rowe. In Vivo 18F-Flortaucipir PET Does Not Accurately Support the Staging of Progressive Supranuclear Palsy.. J. Nucl. Med., 2022. [DOI | PubMed]
- T. W. Rösler, A. Tayaranian Marvian, M. Brendel, N.-P. Nykänen, M. Höllerhage, S. C. Schwarz, F. Hopfner, T. Koeglsperger, G. Respondek, K. Schweyer, J. Levin, V. L. Villemagne, H. Barthel, O. Sabri, U. Müller, W. G. Meissner, G. G. Kovacs, G. U. Höglinger. Four-repeat tauopathies.. Prog. Neurobiol., 2019. [DOI | PubMed]
- M. Stamelou, G. Respondek, N. Giagkou, J. L. Whitwell, G. G. Kovacs, G. U. Höglinger. Evolving concepts in progressive supranuclear palsy and other 4-repeat tauopathies.. Nat. Rev. Neurol., 2021. [DOI | PubMed]
- D. G. Coughlin, I. Litvan. Progressive supranuclear palsy: Advances in diagnosis and management.. Parkinsonism Relat. Disord., 2020. [DOI | PubMed]
- D.-e. C. Chung, S. Roemer, L. Petrucelli, D. W. Dickson. Cellular and pathological heterogeneity of primary tauopathies.. Mol. Neurodegener., 2021. [DOI | PubMed]
- S. Koga, K. A. Josephs, I. Aiba, M. Yoshida, D. W. Dickson. Neuropathology and emerging biomarkers in corticobasal syndrome.. J. Neurol., Neurosurg. Psychiatry, 2022. [DOI | PubMed]
- M. Mimuro, M. Yoshida. Chameleons and mimics: Progressive supranuclear palsy and corticobasal degeneration.. Neuropathology, 2020. [DOI | PubMed]
- M. Goedert, M. G. Spillantini, R. Jakes, D. Rutherford, R. A. Crowther. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease.. Neuron, 1989. [DOI | PubMed]
- M. Goedert, D. S. Eisenberg, R. A. Crowther. Propagation of Tau Aggregates and Neurodegeneration.. Annu. Rev. Neurosci., 2017. [DOI | PubMed]
- M. Goedert. The ordered assembly of tau is the gain-of-toxic function that causes human tauopathies.. Alzheimer’s Dementia, 2016. [DOI]
- T. Arakhamia, C. E. Lee, Y. Carlomagno, M. Kumar, D. M. Duong, H. Wesseling, S. R. Kundinger, K. Wang, D. Williams, M. DeTure, D. W. Dickson, C. N. Cook, N. T. Seyfried, L. Petrucelli, J. A. Steen, A. W. P. Fitzpatrick. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains.. Cell, 2020. [DOI | PubMed]
- M. Goedert. Tau filaments in neurodegenerative diseases.. FEBS Lett., 2018. [DOI | PubMed]
- M. Hasegawa, A. Takashima, B. Wolozin, L. Buee. Structure of NFT: Biochemical Approach. In. Tau Biology;, 2019
- Y. Shi, W. Zhang, Y. Yang, A. G. Murzin, B. Falcon, A. Kotecha, M. van Beers, A. Tarutani, F. Kametani, H. J. Garringer, R. Vidal, G. I. Hallinan, T. Lashley, Y. Saito, S. Murayama, M. Yoshida, H. Tanaka, A. Kakita, T. Ikeuchi, A. C. Robinson, D. M. A. Mann, G. G. Kovacs, T. Revesz, B. Ghetti, M. Hasegawa, M. Goedert, S. H. W. Scheres. Structure-based classification of tauopathies.. Nature, 2021. [DOI | PubMed]
- M. Goedert. Cryo-EM structures of τ filaments from human brain.. Essays Biochem., 2021. [DOI | PubMed]
- B. Falcon, W. Zhang, A. G. Murzin, G. Murshudov, H. J. Garringer, R. Vidal, R. A. Crowther, B. Ghetti, S. H. W. Scheres, M. Goedert. Structures of filaments from Pick’s disease reveal a novel tau protein fold.. Nature, 2018. [DOI | PubMed]
- N. A. Murugan, A. Nordberg, H. Ågren. Different Positron Emission Tomography Tau Tracers Bind to Multiple Binding Sites on the Tau Fibril: Insight from Computational Modeling.. ACS Chem. Neurosci., 2018. [DOI | PubMed]
- G. Kuang, N. A. Murugan, Y. Zhou, A. Nordberg, H. Ågren. Computational Insight into the Binding Profile of the Second-Generation PET Tracer PI2620 with Tau Fibrils.. ACS Chem. Neurosci., 2020. [DOI | PubMed]
- N. A. Murugan, A. Nordberg, H. Ågren. Cryptic Sites in Tau Fibrils Explain the Preferential Binding of the AV-1451 PET Tracer toward Alzheimer’s Tauopathy.. ACS Chem. Neurosci., 2021. [DOI | PubMed]
- Y. Zhou, J. Li, A. Nordberg, H. Ågren. Dissecting the Binding Profile of PET Tracers to Corticobasal Degeneration Tau Fibrils.. ACS Chem. Neurosci., 2021. [DOI | PubMed]
- G. Künze, R. Kümpfel, M. Rullmann, H. Barthel, M. Brendel, M. Patt, O. Sabri. Molecular Simulations Reveal Distinct Energetic and Kinetic Binding Properties of [18F]PI-2620 on Tau Filaments from 3R/4R and 4R Tauopathies.. ACS Chem. Neurosci., 2022. [DOI | PubMed]
- J. Li, A. Kumar, B. Långström, A. Nordberg, H. Ågren. Insight into the Binding of First- and Second-Generation PET Tracers to 4R and 3R/4R Tau Protofibrils.. ACS Chem. Neurosci., 2023. [DOI | PubMed]
- Y. Shi, A. G. Murzin, B. Falcon, A. Epstein, J. Machin, P. Tempest, K. L. Newell, R. Vidal, H. J. Garringer, N. Sahara, M. Higuchi, B. Ghetti, M.-K. Jang, S. H. W. Scheres, M. Goedert. Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607.. Acta Neuropathol., 2021. [DOI | PubMed]
- G. E. Merz, M. J. Chalkley, S. K. Tan, E. Tse, J. Lee, S. B. Prusiner, N. A. Paras, W. F. DeGrado, D. R. Southworth. Stacked binding of a PET ligand to Alzheimer’s tau paired helical filaments.. Nat. Commun., 2023. [DOI | PubMed]
- Y. Shi, B. Ghetti, M. Goedert, S. H. W. Scheres. Cryo-EM Structures of Chronic Traumatic Encephalopathy Tau Filaments with PET Ligand Flortaucipir.. J. Mol. Biol., 2023. [DOI | PubMed]
- A. Lindberg, A. C. Knight, D. Sohn, L. Rakos, J. Tong, A. Radelet, N. S. Mason, J. S. Stehouwer, B. J. Lopresti, W. E. Klunk, J. Sandell, A. Sandberg, P. Hammarström, S. Svensson, C. A. Mathis, N. Vasdev. Radiosynthesis, In Vitro and In Vivo Evaluation of [18F]CBD-2115 as a First-in-Class Radiotracer for Imaging 4R-Tauopathies.. ACS Chem. Neurosci., 2021. [DOI | PubMed]
- A. Lindberg, E. Murrell, J. Tong, N. S. Mason, D. Sohn, J. Sandell, P. Ström, J. S. Stehouwer, B. J. Lopresti, J. Viklund, S. Svensson, C. A. Mathis, N. Vasdev. Ligand-based design of [18F]OXD-2314 for PET imaging in non-Alzheimer’s disease tauopathies.. Nat. Commun., 2024. [DOI | PubMed]
- T. J. A. Graham, A. Lindberg, J. Tong, J. S. Stehouwer, N. Vasdev, R. H. Mach, C. A. Mathis. In Silico Discovery and Subsequent Characterization of Potent 4R-Tauopathy Positron Emission Tomography Radiotracers.. J. Med. Chem., 2023. [DOI | PubMed]
- D. Schäfer, P. Weiß, J. Ermert, J. Castillo Meleán, F. Zarrad, B. Neumaier. Preparation of No-Carrier-Added 6-[18F]Fluoro-l-tryptophan via Cu-Mediated Radiofluorination.. Eur. J. Org. Chem., 2016. [DOI]
- Z. Qiao, K. Mardon, D. H. R. Stimson, M.-a. Migotto, D. C. Reutens, R. Bhalla. Synthesis and evaluation of 6-[18F]fluoro-3-(pyridin-3-yl)-1H-indole as potential PET tracer for targeting tryptophane 2, 3-dioxygenase (TDO).. Nucl. Med. Biol., 2020. [DOI]
- A. Zak, L. Lemaire, S. Chalon, G. Chicheri, H. Marzag, S. Bodard, S. Sérrière, S. Routier, F. Buron, J. Vercouillie. [18F]-labeled positron emission tomography ligand for the histamine H4 receptor.. J. Labelled Compd. Radiopharm., 2021. [DOI]
- R. Teodoro, D. Gündel, W. Deuther-Conrad, A. Kazimir, M. Toussaint, B. Wenzel, G. Bormans, E. Hey-Hawkins, K. Kopka, P. Brust, R.-P. Moldovan. Synthesis, Structure–Activity Relationships, Radiofluorination, and Biological Evaluation of [18F]RM365, a Novel Radioligand for Imaging the Human Cannabinoid Receptor Type 2 (CB2R) in the Brain with PET.. J. Med. Chem., 2023. [DOI | PubMed]
- J. Zischler, N. Kolks, D. Modemann, B. Neumaier, B. D. Zlatopolskiy. Alcohol-Enhanced Cu-Mediated Radiofluorination.. Chem. – Eur. J., 2017. [DOI | PubMed]
- F. Dollé. Fluorine-18-Labelled Fluoropyridines: Advances in Radiopharmaceutical Design.. Curr. Pharm. Des., 2005. [DOI | PubMed]
- T. Wang, Q. Lin, Y. Zhang, Z. Xu, D. Shi, Y. Cheng, Z. Fu, H. Tan, D. Cheng, H. Shi. Synthesis and biological evaluation of novel PET tracers [18F]AG120 & [18F]AG135 for imaging mutant isocitrate dehydrogenase 1 expression.. Bioorg. Med. Chem., 2022. [DOI | PubMed]
- C. Warnier, C. Lemaire, G. Becker, G. Zaragoza, F. Giacomelli, J. Aerts, M. Otabashi, M. A. Bahri, J. Mercier, A. Plenevaux, A. Luxen. Enabling Efficient Positron Emission Tomography (PET) Imaging of Synaptic Vesicle Glycoprotein 2A (SV2A) with a Robust and One-Step Radiosynthesis of a Highly Potent 18F-Labeled Ligand ([18F]UCB-H).. J. Med. Chem., 2016. [DOI | PubMed]
- T. E. F. Morgan, L. M. Riley, A. A. S. Tavares, A. Sutherland. Automated Radiosynthesis of cis- and trans-4-[18F]Fluoro-l-proline Using [18F]Fluoride.. J. Org. Chem., 2021. [DOI | PubMed]
- T. C. Pickel, G. Pashikanti, R. J. Voll, W. Yu, Z. Zhang, J. A. Nye, J. Bacsa, J. J. Olson, L. S. Liebeskind, M. M. Goodman. Synthesis, Radiolabeling, and Biological Evaluation of the trans-Stereoisomers of 1-Amino-3-(fluoro-18F)-4-fluorocyclopentane-1-carboxylic Acid as PET Imaging Agents.. ACS Pharmacol. Transl. Sci., 2021. [DOI | PubMed]
- F. Buckingham, V. Gouverneur. Asymmetric 18F-fluorination for applications in positron emission tomography.. Chem. Sci., 2016. [DOI | PubMed]
- R. Smith, F. Capotosti, M. Schain, T. Ohlsson, E. Vokali, J. Molette, T. Touilloux, V. Hliva, I. K. Dimitrakopoulos, A. Puschmann, J. Jögi, P. Svenningsson, M. Andréasson, C. Sandiego, D. S. Russell, P. Miranda-Azpiazu, C. Halldin, E. Stomrud, S. Hall, K. Bratteby, E. Tampio L’Estrade, R. Luthi-Carter, A. Pfeifer, M. Kosco-Vilbois, J. Streffer, O. Hansson. The α-synuclein PET tracer [18F] ACI-12589 distinguishes multiple system atrophy from other neurodegenerative diseases.. Nat. Commun., 2023. [DOI | PubMed]
- R. Koudih, G. Gilbert, M. Dhilly, A. Abbas, L. Barré, D. Debruyne, F. Sobrio. Synthesis and in vitro characterization of trans- and cis-[18F]-4-methylbenzyl 4-[(pyrimidin-2-ylamino)methyl]-3-fluoropiperidine-1-carboxylates as new potential PET radiotracer candidates for the NR2B subtype N-methyl-d-aspartate receptor.. Eur. J. Med. Chem., 2012. [DOI | PubMed]
- A. Pees, J. Tong, S. Birudaraju, Y. S. Munot, S. H. Liang, D. Saturnino Guarino, R. H. Mach, C. A. Mathis, N. Vasdev. Development of Pyridothiophene Compounds for PET Imaging of α-Synuclein.. Chem. – Eur. J., 2024. [DOI | PubMed]
- A. Kumar, M. Say, D. W. Boykin. Synthesis of New Substituted 2-(Trimethylstannyl)indoles.. Synthesis, 2008. [DOI]
- A. C. Smith, D. W. Kung, A. Shavnya, T. A. Brandt, P. D. Dent, N. E. Genung, S. Cabral, J. Panteleev, M. Herr, K. N. Yip, G. E. Aspnes, E. L. Conn, M. S. Dowling, D. J. Edmonds, I. D. Edmonds, D. P. Fernando, P. M. Herrinton, N. F. Keene, S. Y. Lavergne, Q. Li, J. Polivkova, C. R. Rose, B. A. Thuma, M. G. Vetelino, G. Wang, J. D. Weaver, D. W. Widlicka, K. E. Price Wiglesworth, J. Xiao, T. Zahn, Y. Zhang. Evolution of the Synthesis of AMPK Activators for the Treatment of Diabetic Nephropathy: From Three Preclinical Candidates to the Investigational New Drug PF-06409577.. Org. Process Res. Dev., 2018. [DOI]
- J. Ji, T. Li, W. H. Bunnelle. Selective Amination of Polyhalopyridines Catalyzed by a Palladium–Xantphos Complex.. Org. Lett., 2003. [DOI | PubMed]
- B. W. Stroup, P. V. Szklennik, C. J. Forster, M. H. Serrano-Wu. Chemoselective Amination of 5-Bromo-2-chloro-3-fluoropyridine.. Org. Lett., 2007. [DOI | PubMed]
- W. E. Klunk, Y. Wang, G.-f. Huang, M. L. Debnath, D. P. Holt, L. Shao, R. L. Hamilton, M. D. Ikonomovic, S. T. DeKosky, C. A. Mathis. The Binding of 2-(4′-Methylaminophenyl)Benzothiazole to Postmortem Brain Homogenates Is Dominated by the Amyloid Component.. J. Neurosci., 2003. [DOI | PubMed]
- W. E. Klunk, Y. Wang, G.-f. Huang, M. L. Debnath, D. P. Holt, C. A. Mathis. Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain.. Life Sci., 2001. [DOI | PubMed]
- J. C. Price, W. E. Klunk, B. J. Lopresti, X. Lu, J. A. Hoge, S. K. Ziolko, D. P. Holt, C. C. Meltzer, S. T. DeKosky, C. A. Mathis. Kinetic Modeling of Amyloid Binding in Humans using PET Imaging and Pittsburgh Compound-B.. J. Cereb. Blood Flow Metabol., 2005. [DOI]
- T. T. Wager, X. Hou, P. R. Verhoest, A. Villalobos. Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties.. ACS Chem. Neurosci., 2010. [DOI | PubMed]
- L. Zhang, A. Villalobos, E. M. Beck, T. Bocan, T. A. Chappie, L. Chen, S. Grimwood, S. D. Heck, C. J. Helal, X. Hou, J. M. Humphrey, J. Lu, M. B. Skaddan, T. J. McCarthy, P. R. Verhoest, T. T. Wager, K. Zasadny. Design and Selection Parameters to Accelerate the Discovery of Novel Central Nervous System Positron Emission Tomography (PET) Ligands and Their Application in the Development of a Novel Phosphodiesterase 2A PET Ligand.. J. Med. Chem., 2013. [DOI | PubMed]
- M. Gupta, H. J. Lee, C. J. Barden, D. F. Weaver. The Blood–Brain Barrier (BBB) Score.. J. Med. Chem., 2019. [DOI | PubMed]
- A. Daina, V. Zoete. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules.. ChemMedChem, 2016. [DOI | PubMed]
- A. Daina, O. Michielin, V. Zoete. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules.. Sci. Rep., 2017. [DOI | PubMed]
- E. M. Joshi, A. Need, J. Schaus, Z. Chen, D. Benesh, C. Mitch, S. Morton, T. J. Raub, L. Phebus, V. Barth. Efficiency Gains in Tracer Identification for Nuclear Imaging: Can In Vivo LC-MS/MS Evaluation of Small Molecules Screen for Successful PET Tracers?.. ACS Chem. Neurosci., 2014. [DOI | PubMed]
- E. J. L. Stéen, D. J. Vugts, A. D. Windhorst. The Application of in silico Methods for Prediction of Blood-Brain Barrier Permeability of Small Molecule PET Tracers.. Front. Nucl. Med., 2022. [DOI | PubMed]
- C.-J. Hsieh, S. Giannakoulias, E. J. Petersson, R. H. Mach. Computational Chemistry for the Identification of Lead Compounds for Radiotracer Development.. Pharmaceuticals, 2023. [DOI | PubMed]
- T. M. Shoup, D. L. Yokell, P. A. Rice, R. N. Jackson, E. Livni, K. A. Johnson, T. J. Brady, N. Vasdev. A concise radiosynthesis of the tau radiopharmaceutical, [18F]T807.. J. Labelled Compd. Radiopharm., 2013. [DOI]
- A. V. Mossine, A. F. Brooks, B. D. Henderson, B. G. Hockley, K. A. Frey, P. J. H. Scott. An updated radiosynthesis of [18F]AV1451 for tau PET imaging.. EJNMMI Radiopharm. Chem., 2017. [DOI | PubMed]
- V. W. Pike. Considerations in the Development of Reversibly Binding PET Radioligands for Brain Imaging.. Curr. Med. Chem., 2016. [DOI | PubMed]
- P. Reinstrup, E. Ryding, L. Algotsson, K. Messeter, B. Asgeirsson, T. Uski. Distribution of Cerebral Blood Flow during Anesthesia with Isoflurane or Halothane in Humans.. Anesthesiology, 1995. [DOI | PubMed]
- K. P. Ng, T. A. Pascoal, S. Mathotaarachchi, J. Therriault, M. S. Kang, M. Shin, M.-C. Guiot, Q. Guo, R. Harada, R. A. Comley, G. Massarweh, J.-P. Soucy, N. Okamura, S. Gauthier, P. Rosa-Neto. Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain.. Alzheimer’s Res. Ther., 2017. [DOI | PubMed]
- H. J. Motulsky, R. R. Neubig. Analyzing Binding Data.. Curr. Protoc. Neurosci., 2010. [DOI]
- A. Meneses, S. Koga, J. O’Leary, D. W. Dickson, G. Bu, N. Zhao. TDP-43 Pathology in Alzheimer’s Disease.. Mol. Neurodegener., 2021. [DOI | PubMed]
- A. F. Carlos, K. A. Josephs. Frontotemporal lobar degeneration with TAR DNA-binding protein 43 (TDP-43): its journey of more than 100 years.. J. Neurol., 2022. [DOI | PubMed]
- J. Lewis, E. McGowan, J. Rockwood, H. Melrose, P. Nacharaju, M. Van Slegtenhorst, K. Gwinn-Hardy, M. P. Murphy, M. Baker, X. Yu, K. Duff, J. Hardy, A. Corral, W.-L. Lin, S.-H. Yen, D. W. Dickson, P. Davies, M. Hutton. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein.. Nat. Genet., 2000. [DOI | PubMed]
- M. Ramsden, K. Linda, F. Colleen, P. Jennifer, M. Eileen, S. Karen, G. Aaron, Y. Mei, L. Jada, C. George, H. Michael, H. A. Karen. Age-Dependent Neurofibrillary Tangle Formation, Neuron Loss, and Memory Impairment in a Mouse Model of Human Tauopathy (P301L).. J. Neurosci., 2005. [DOI | PubMed]
- R. L. Hamilton. Lewy Bodies in Alzheimer’s Disease: A Neuropathological Review of 145 Cases Using α-Synuclein Immunohistochemistry.. Brain Pathol., 2000. [DOI | PubMed]
- W. Marui, E. Iseki, K. Uéda, K. Kosaka. Occurrence of human α-synuclein immunoreactive neurons with neurofibrillary tangle formation in the limbic areas of patients with Alzheimer’s disease.. J. Neurol. Sci., 2000. [DOI | PubMed]
- Y. Arai, M. Yamazaki, O. Mori, H. Muramatsu, G. Asano, Y. Katayama. α-Synuclein-positive structures in cases with sporadic Alzheimer’s disease: morphology and its relationship to tau aggregation.. Brain Res., 2001. [DOI | PubMed]
- U. Sengupta, M. J. Guerrero-Muñoz, D. L. Castillo-Carranza, C. A. Lasagna-Reeves, J. E. Gerson, A. A. Paulucci-Holthauzen, S. Krishnamurthy, M. Farhed, G. R. Jackson, R. Kayed. Pathological Interface Between Oligomeric Alpha-Synuclein and Tau in Synucleinopathies.. Biol. Psychiatry, 2015. [DOI | PubMed]
- M. Delgado-Alvarado, B. Gago, A. Gorostidi, H. Jiménez-Urbieta, R. Dacosta-Aguayo, I. Navalpotro-Gómez, J. Ruiz-Martínez, A. Bergareche, J. F. Martí-Massó, P. Martínez-Lage, A. Izagirre, M. C. Rodríguez-Oroz. Tau/α-synuclein ratio and inflammatory proteins in Parkinson’s disease: An exploratory study.. Mov. Disord., 2017. [DOI | PubMed]
- M. Meloni, C. Agliardi, F. R. Guerini, M. Zanzottera, E. Bolognesi, S. Picciolini, M. Marano, A. Magliozzi, A. Di Fonzo, A. Arighi, C. Fenoglio, G. Franco, F. Arienti, F. L. Saibene, J. Navarro, M. Clerici. Oligomeric α-synuclein and tau aggregates in NDEVs differentiate Parkinson’s disease from atypical parkinsonisms.. Neurobiol. Dis., 2023. [DOI | PubMed]
- Y. Chu, W. D. Hirst, H. J. Federoff, A. S. Harms, A. J. Stoessl, J. H. Kordower. Nigrostriatal tau pathology in parkinsonism and Parkinson’s disease.. Brain, 2024. [DOI | PubMed]
- Y. Riku, Y. Iwasaki, S. Ishigaki, A. Akagi, M. Hasegawa, K. Nishioka, Y. Li, M. Riku, T. Ikeuchi, Y. Fujioka, H. Miyahara, J. Sone, N. Hattori, M. Yoshida, M. Katsuno, G. Sobue. Motor neuron TDP-43 proteinopathy in progressive supranuclear palsy and corticobasal degeneration.. Brain, 2022. [DOI | PubMed]
- P. T. Nelson, D. W. Dickson, J. Q. Trojanowski, C. R. Jack, P. A. Boyle, K. Arfanakis, R. Rademakers, I. Alafuzoff, J. Attems, C. Brayne, I. T. S. Coyle-Gilchrist, H. C. Chui, D. W. Fardo, M. E. Flanagan, G. Halliday, S. R. K. Hokkanen, S. Hunter, G. A. Jicha, Y. Katsumata, C. H. Kawas, C. D. Keene, G. G. Kovacs, W. A. Kukull, A. I. Levey, N. Makkinejad, T. J. Montine, S. Murayama, M. E. Murray, S. Nag, R. A. Rissman, W. W. Seeley, R. A. Sperling, C. L. White Iii, L. Yu, J. A. Schneider. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report.. Brain, 2019. [DOI | PubMed]
- H. H. Coenen, A. D. Gee, M. Adam, G. Antoni, C. S. Cutler, Y. Fujibayashi, J. M. Jeong, R. H. Mach, T. L. Mindt, V. W. Pike, A. D. Windhorst. Consensus nomenclature rules for radiopharmaceutical chemistry — Setting the record straight.. Nucl. Med. Biol., 2017. [DOI | PubMed]
- G. Luurtsema, V. Pichler, S. Bongarzone, Y. Seimbille, P. Elsinga, A. Gee, J. Vercouillie. EANM guideline for harmonisation on molar activity or specific activity of radiopharmaceuticals: impact on safety and imaging quality.. EJNMMI Radiopharm. Chem., 2021. [DOI | PubMed]
- S. K. Bharti, R. Roy. Quantitative 1H NMR spectroscopy.. TrAC, Trends Anal. Chem., 2012. [DOI]