
From Symptomatic Therapies to Disease-Modifying Approaches for Neuronal Sodium Channel Disorders
Abstract
Variants in neuronal sodium channel genes are responsible for a spectrum of neurological disorders, including developmental and epileptic encephalopathies (DEEs), with considerable genetic and phenotypic heterogeneity and drug resistance. Gene variants can produce loss-, gain-, or mixed-function effects, resulting in complex genotype-phenotype correlations. Current treatments rely mainly on symptomatic polytherapy with antiseizure medications, with sodium channel blockers contraindicated in loss-of-function cases but beneficial in gain-of-function forms. Existing therapies often provide limited benefit or even no seizure control at all and fail to address developmental impairments, highlighting the need for novel approaches. Emerging strategies include antisense oligonucleotides, gene therapy, and selective small-molecule modulators, which have shown antiseizure potential in preclinical models and in initial clinical studies by modulating SCN gene expression and function. Additionally, pharmacological agents such as fenfluramine, stiripentol, and cannabidiol, although not acting directly on sodium channels, represent recognized therapeutic options for SCN1A-related Dravet syndrome. This review summarizes recent advances in approved and investigational treatments for sodium channel-related neurological disorders, highlighting the transition from symptomatic to precision therapies.
Article type: Review Article
Keywords: sodium channels, Dravet syndrome, zorevunersen, ETX-101, elsunersen, fenfluramine
Affiliations: Department of Pharmacy-Drug Sciences, University of Bari “Aldo Moro”, Via Orabona 4, 70125 Bari, Italy; giorgia.dinoi@uniba.it (G.D.); ileana.canfora@uniba.it (I.C.); brigida.boccanegra@uniba.it (B.B.); elena.conte@uniba.it (E.C.); annamaria.deluca@uniba.it (A.D.L.); antonella.liantonio@uniba.it (A.L.); Children Epilepsy and EEG Center, San Paolo Hospital, ASL Bari, 70132 Bari, Italy; daniela.dagnano@gmail.com (D.D.); vittorio.sciruicchio@asl.bari.it (V.S.)
License: © 2025 by the authors. CC BY 4.0 Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Article links: DOI: 10.3390/ijms27010032 | PubMed: 41515912 | PMC: PMC12786012
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (745 KB)
1. Introduction
Neuronal sodium channel genes, SCN1A, SCN2A and SCN8A, have been extensively involved in the pathophysiology of a broad spectrum of neurological disorders, including developmental and epileptic encephalopathy (DEE) in which epilepsy and neurodevelopmental comorbidities coexist [ref. 1]. Variants in these genes can result in loss-of-function (LOF), gain-of-function (GOF), or mixed LOF/GOF effects, leading to diverse and complex clinical phenotypes. In the absence of approved therapies that specifically target the underlying molecular defects, the current standard of care of sodium channel-related neuronal disorders mainly relies on polytherapy with antiseizure medications (ASMs), often in combination with drugs aimed at managing associated comorbidities. Importantly, sodium channel blockers (SCBs) are contraindicated in individuals carrying LOF variants but can be effective in those carrying GOF variants, and a specific treatment algorithm has been established for Dravet syndrome (DS), the most severe DEE linked to SCN1A mutations [ref. 2]. However, existing treatments have limited efficacy, often carry substantial side effects, and do not always address the neurodevelopmental impairments that extend beyond seizure control especially in SCN-related DEEs. This highlights an urgent need for novel therapeutic strategies [ref. 3]. Actually, the therapeutic management of DEEs is facing a paradigm shift, moving from a symptomatic treatment approach with classical ASMs to disease-modifying therapies aimed at producing a global improvement [ref. 4]. Emerging approaches for treating SCN-related neurologic disorders include antisense oligonucleotides (ASOs), gene therapy, and novel selective small molecules [ref. 5]. ASOs consist of a single-stranded base sequence that can act on specific RNA or DNA molecules, thereby modulating the expression of proteins encoded by the specific transcript. By addressing the underlying genetic cause of the disease, ASOs have the potential to achieve seizure freedom and potentially improve developmental comorbidities. Preclinical evidence suggests that selective modulation of SCN1A, SCN2A and SCN8A expression and function through ASOs targeting human mRNA or gene therapy could offer a promising solution for LOF and GOF variants [ref. 5,ref. 6]. Among other strategies, drug repurposing led to the repositioning of fenfluramine that emerged as a highly effective and well-tolerated treatment for seizures associated with DS, with a dual serotonergic and sigma-1 receptor activity. Clinical trials support fenfluramine’s role not only in seizure control but also in ameliorating the neurodevelopmental trajectory and in the reduction in SUDEP risk in DS [ref. 7,ref. 8]. Additional serotonergic modulators, such as bexicaserin and clemizole, are under investigation and represent potential therapeutic opportunities for patients with DEE. The exploration of novel drug targets and the growing understanding of the interplay between epilepsy and metabolic pathways have opened new avenues for therapies targeting metabolic enzymes, such as stiripentol and soticlestat, specifically for DS [ref. 7]. This narrative review summarizes some of the most recent therapeutic approaches approved or under investigation for the treatment of SCN1A-, SCN2A– and SCN8A-related neurologic disorders (Figure 1; Table 1).
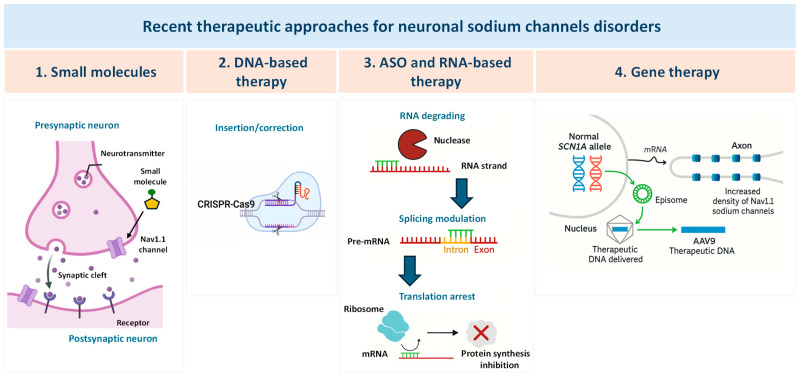
Table 1: Recent therapeutic approaches for neuronal sodium channel disorders.
| Therapeutic Strategy | Drug | Mechanism of Action | Drug Development Process | References |
|---|---|---|---|---|
| Smallmolecules | Stiripentol | GABA modulation and LDH inhibition | Approved for DS | [ref. 9,ref. 10] |
| Fenfluramine | 5-HT and sigma1 receptor modulation | Approved for DS, LGS | [ref. 11,ref. 12] | |
| Cannabidiol | GPR55 antagonism, TRPV1 agonism, adenosine transporter inhibition | Approved for DS, TSC | [ref. 13] | |
| Soticlestat | Cholesterol 24-hydroxylase inhibition | Discontinued | [ref. 14] | |
| Relutrigine (PRAX-562) | Use-dependent and persistent current Nav1.2/Nav1.6 block | Phase 3 EMERALD trial ongoing for DEE regardless of the cause | [ref. 15] | |
| NBI-921352(XEN901) | Use-dependent selective Nav1.6 block | Phase 2 KAYAK trial ongoing for SCN8A-DEE | [ref. 16] | |
| Vormatrigine (PRAX-628) | Use-dependent and persistent current pan-Nav inhibition | Phase 3 RADIANT trial ongoing for adult focal onset epilepsy | [ref. 17,ref. 18] | |
| ASO and RNA/DNA-based therapies | Zorevunersen (STK-001) | ASO, Exon 20N skipping targeting SCN1A | Phase 3 EMPEROR trial initiated for DS | [ref. 19,ref. 20,ref. 21] |
| Elsunersen (PRAX-222) | Gapmer ASO; downregulation of SCN2A | Phase 1/2 EMBRAVE trial ongoing for GOF SCN2A DEE | [ref. 22] | |
| ASO targeting SCN1A-dsAS | Targeting long non-coding downstream RNA and increased expression of SCN1A | Preclinical stage for DS | [ref. 23] | |
| CRISPR/dCas9-based approach | Increased expression of SCN1A | Preclinical stage for DS | [ref. 24] | |
| ASO targeting SCN8A | Downregulation of SCN8A | Preclinical stage for GOF SCN8A-DEE | [ref. 25,ref. 26] | |
| Genetherapy | ETX101 (AAV9-REGABA-eTFSCN1A) | Regulatory gene therapy increasing expression of SCN1A in GABAergic interneurons | Phase 1/2 ENDEAVOR trial ongoing for DS | [ref. 17,ref. 27] |
DS, Dravet syndrome; LGS, Lennox–Gastaut syndrome; TSC, Tuberous Sclerosis Complex; LDH, lactate dehydrogenase; ASO, antisense oligonucleotide.
2. SCN1A-DEE
The SCN1A gene encodes the α-subunit of the voltage-gated sodium channel Nav1.1, which plays a crucial role in the initiation and propagation of action potentials, particularly within fast-spiking GABAergic interneurons of the neocortex, hippocampus, and cerebellum. Mutations in SCN1A can result in LOF, GOF, or mixed functional effects and have been linked to a broad spectrum of neurological disorders with highly variable clinical features and treatment responses [ref. 1,ref. 28]. LOF variants have been classically associated with DS, a severe, drug-resistant DEE, as well as with milder forms such as genetic epilepsy with febrile seizures plus (GEFS+), which are more responsive to antiseizure medications. GOF variants, originally identified in familial hemiplegic migraine type 3 (FHM3), have more recently been linked to neonatal-onset epileptic encephalopathies with or without movement disorders, which often show a positive response to SCBs [ref. 29,ref. 30,ref. 31]. Mixed GOF/LOF variants have been described in early infantile DEEs characterized by neonatal seizures, profound developmental delay, and hyperkinesia [ref. 32]. Studies in mouse models have demonstrated that LOF in Scn1a leads to reduced inhibitory output from GABAergic interneurons, resulting in cortical hyperexcitability and seizures [ref. 33,ref. 34,ref. 35]. This impaired GABAergic signaling, including a delayed GABA switch, is also believed to contribute to the cognitive and behavioral comorbidities observed in patients [ref. 36,ref. 37]. As in many genetic disorders, the genotype-phenotype correlation in SCN1A-related epilepsy is often not straightforward. Clinical variability among individuals carrying the same variant, even within families, suggests a more complex disease mechanism involving genetic background, gene regulation, and neuronal network remodeling, beyond the primary ion channel dysfunction [ref. 38,ref. 39,ref. 40,ref. 41]. Therapeutic response in SCN1A-related epilepsies strongly depends on the functional nature of the causative variant. While, as said, SCBs (e.g., carbamazepine, phenytoin) may be effective in some GOF cases, they are contraindicated in LOF disorders such as DS, where they can exacerbate seizure activity [ref. 2]. Accordingly, the functional characterization of SCN1A variants is critical not only for variant classification but also for guiding treatment decisions. Based on preclinical research in animal models, the most beneficial therapeutic approaches in DS are based on drugs enhancing GABAergic transmission; commonly used medications include valproate, clobazam and stiripentol. More recently, cannabidiol and fenfluramine have been approved as adjunctive therapies in children of >2 years of age [ref. 2]. As mentioned before, given the limitations of current treatments, novel therapeutic strategies are under investigation. These include ASOs and gene therapy designed to upregulate functional SCN1A expression. Additionally, the interplay between epilepsy and metabolism has stimulated interest in therapies targeting also metabolic pathways, such as stiripentol and soticlestat, which offer alternative mechanisms to modulate neuronal excitability.
2.1. Stiripentol
Stiripentol is a structurally unique ASM that enhances central GABAergic transmission through multiple mechanisms. It prolongs the opening duration of GABAA receptors and inhibits both GABA reuptake and metabolism, thereby increasing synaptic GABA availability and neuronal inhibition [ref. 42]. In the early 2000s, stiripentol received orphan drug designation and regulatory approval in Europe for the treatment of DS [ref. 43]. Currently, stiripentol is indicated specifically as an adjunctive therapy in combination with valproate and clobazam for patients with DS who are not adequately controlled with the two drugs alone. This combined therapy has demonstrated robust efficacy in reducing seizure frequency, including clinically meaningful (≥50%) and profound (≥75%) reductions in monthly convulsive seizure frequency (MCSF), in multiple clinical trials [ref. 43,ref. 44].
Beyond its well-known GABAergic mechanisms, preclinical studies have shown that stiripentol at a concentration of 500 μM inhibits lactate dehydrogenase (LDH), a key enzyme in the astrocyte-neuron lactate shuttle, responsible for the bidirectional conversion between lactate and pyruvate. Inhibition of LDH disrupts this metabolic coupling, suppressing neuronal hyperexcitability and contributing to reduce seizure susceptibility in a mouse model [ref. 9]. This emerging mechanism suggests a potential role for stiripentol in targeting metabolic pathways contributing to epileptogenesis.
The efficacy of stiripentol has also been explored in other DEEs beyond DS, including Lennox–Gastaut syndrome (LGS) and CDKL5 deficiency disorder. Across these studies, stiripentol was generally well tolerated, with the most common adverse effects being somnolence, anorexia, and weight loss [ref. 45]. Importantly, these adverse events were often reversible upon dose adjustment or discontinuation [ref. 46,ref. 47,ref. 48].
A recent comparative study has evaluated the efficacy and safety of stiripentol along other FDA- and EMA-approved therapies for DS, notably fenfluramine and cannabidiol. Stiripentol and fenfluramine demonstrated comparable efficacy in achieving ≥50% and ≥75% reductions in monthly convulsive seizure frequency (MCSF), with both agents significantly surpassing cannabidiol at all doses [ref. 49]. Importantly, stiripentol was the only agent showing a statistically significant advantage over both fenfluramine and cannabidiol in achieving complete seizure freedom from baseline. Furthermore, while the incidence of serious adverse events was similar within all three therapies, stiripentol exhibited a lower risk of treatment discontinuation due to adverse effects, particularly at its standard dose of 20 mg/kg/day [ref. 10,ref. 49].
These findings support current international consensus guidelines, which prioritize stiripentol and fenfluramine over cannabidiol in the adjunctive management of DS [ref. 2]. Therefore, stiripentol remains a mainstay therapy in DS, with growing interest in its broader applications across the epilepsy spectrum.
2.2. Fenfluramine
In 2020, fenfluramine received FDA and EMA approval as an adjunctive therapy to standard of care for seizures associated with DS and, later, LGS, for individuals aged 2 years and older [ref. 50]. Along with its active metabolite norfenfluramine, this drug acts primarily on the serotonergic system promoting serotonin (5-HT) release and selectively activating 5-HT1D and 5-HT2C receptors. In addition, a modulatory action on the sigma-1 receptor, a chaperone protein known to interact also with ion channels, has been supposed to contribute to its antiseizure effects. Thus, through this dual mechanism of action, fenfluramine potentiates the GABAergic transmission while reducing the glutamatergic one [ref. 51]. This drug was originally developed and approved as an anorectic agent due to its appetite-suppressing effects. However, by the late 1990s, safety concerns about cardiac valvulopathy and pulmonary arterial hypertension led to its withdrawal from the market [ref. 52]. Only recently, a high-throughput drug screening study using scn1Lab−/− mutant zebrafish larvae, a validated model of DS, identified fenfluramine as promising candidates for repurposing in epilepsy [ref. 11,ref. 53]. In this preclinical study, fenfluramine significantly reduced zebrafish locomotor activity, as well as the frequency and duration of epileptiform discharges. Beyond seizure reduction, the anti-neuroinflammation and pro-survival effect of fenfluramine have been recently shown in a mammalian DS model [ref. 54]. Clinical efficacy has been demonstrated in multiple phase 3 randomized, placebo-controlled trials. In clinical studies, fenfluramine significantly reduced MCSF by 54% to 72.9% compared to placebo [ref. 55,ref. 56]. These findings were corroborated by open-label extension studies showing sustained seizure reduction and improvements in quality-of-life metrics. A more recent phase 3 trial further confirmed fenfluramine efficacy in DS, demonstrating a dose-dependent reduction in MCSF. Among participants receiving 0.7 mg/kg/day, 73% achieved a ≥50% reduction in seizures, and 48% achieved a ≥75% reduction, with significantly longer seizure-free intervals than placebo-treated patients [ref. 57]. Fenfluramine was well tolerated, with no reported cases of cardiac valvulopathy or pulmonary hypertension in any trial participant, as confirmed by regular echocardiographic monitoring. Real-world data from European cohorts, including Italy and Germany, have further supported fenfluramine safety and efficacy in DS clinical practice [ref. 10,ref. 12,ref. 58].
Although fenfluramine is approved only for patients aged ≥2 years, a recent prospective study evaluated its use in infants under 2 years with DS. In a median follow-up of 13 months, two of five patients achieved ≥50% seizure reduction, with one achieving seizure freedom for ≥6 months. Additionally, reductions in status epilepticus and seizure clusters were observed. No clinical or echocardiographic signs of valvular disease were detected, and none of the patients discontinued treatment due to adverse events [ref. 8]. Interestingly, long-term responders (≥50% seizure reduction) demonstrated improved executive function compared to non-responders. Neuropsychological assessments showed stable or improved developmental trajectories, with no evidence of regression and mild cognitive gains in 2 out of 5 patients. This encouraging outcome is especially relevant in an early onset syndrome typically associated with progressive developmental delay [ref. 8].
Like fenfluramine, lorcaserin, a selective 5-HT2C receptor agonist, was originally approved by the FDA for weight management and was withdrawn from the market in 2020 following data suggesting a possible increased risk of cancer [ref. 59]. Despite this, interest in lorcaserin potential in treating epilepsy, particularly DEE, has continued. Like fenfluramine, lorcaserin was identified as a potential antiseizure agent in the scn1Lab−/− zebrafish DS model. At concentrations of 10–12.5 µM, lorcaserin significantly reduced both zebrafish locomotor activity and the frequency and duration of epileptiform discharges [ref. 60]. However, a phase 3 clinical trial to determine lorcaserin efficacy and safety as an adjunctive therapy for DS was recently discontinued [ref. 61]. Bexicaserin, another selective 5-HT2C receptor agonist, showed sustained reductions in motor seizure frequency over two years and a favorable safety profile, in a phase 1/2 open label expanded access program involving DEE patients. The ongoing phase 3 trial will further assess bexicaserin’s efficacy and safety in a larger DEE cohort [ref. 4,ref. 7].
2.3. Cannabidiol
In 2018 cannabidiol (CBD) was approved by FDA and in 2019 by EMA for adjunctive treatment of seizures, in conjunction with clobazam, in DS and LGS in patients aged ≥2 years; since then, indications have expanded to include tuberous sclerosis complex. Cannabidiol is a non-psychoactive phytocannabinoid derived from Cannabis sativa exhibiting a broad spectrum of therapeutic effects [ref. 62]. CBD antiseizure potential is mediated by its ability to modulate neuronal excitability and neuroinflammation and grounds on a muti-target mechanism of action. This drug inhibits the GPR55 receptor which is implicated in excitatory neurotransmission and epileptogenesis; activates and subsequently desensitizes TRPV1 channels thus reducing neuronal hyperexcitability; inhibits the equilibrative nucleoside transporter 1 (ENT1) in microglia and astrocytes, thereby increasing extracellular adenosine and enhancing adenosine anticonvulsant effect; potentiates GABAA receptor–mediated currents by interacting with an allosteric site [ref. 62]. Additional studies suggest that CBD modulates different ion channels including Nav1 neuronal sodium channels [ref. 63].
In preclinical models of DS, CBD significantly reduced seizure burden and improved autistic-like social deficits [ref. 13,ref. 64]. Following, CBD has undergone extensive evaluation in randomized controlled trials for DS. In a breakthrough phase 3, double-blind, placebo-controlled study involving 120 children and young adults with DS, adjunctive CBD (20 mg/kg/day) reduced MCSF from 12.4 to 5.9, compared with a smaller reduction in the placebo group (14.9 to 14.1). Although the ≥50% responder rate favored CBD (43% vs. 27%), this effect did not reach statistical significance in the primary analysis [ref. 65]. However, subsequent trials and open-label extensions confirmed significant and durable seizure reductions. A long-term extension study (median treatment duration of about 80 weeks) demonstrated a median 50% reduction in major motor seizures and 44% in total monthly seizures after 12 weeks, these effects being sustained for up to 96 weeks [ref. 66]. Among patients with DS or LGS, 53% showed ≥50% seizure reduction, 23% achieved ≥75%, and 6% became seizure-free for major motor seizures; respective rates for total seizures reduction were 46%, 26%, and 5%. Recent real-world studies have further supported the efficacy and safety of CBD in drug-resistant epilepsies, including DS, although responses vary and appear to be modulated by concomitant medications such as clobazam and valproate [ref. 67,ref. 68]. Common adverse events include somnolence (30%), diarrhea (24%), decreased appetite, and fatigue. Serious adverse events reported in long-term studies include convulsions (14%), status epilepticus (9%), pneumonia (5%), and fever (4%) [ref. 68]. Importantly, no new safety signals have emerged in post-marketing studies, and the rate of SUDEP remained consistent with background rates for severe epilepsies, with two cases reported in a large cohort over the observation period [ref. 68].
CBD is known to interact with multiple antiseizure drugs necessitating careful monitoring [ref. 69]. Due to inhibition of CYP2C19, co-administration of CBD with clobazam increases serum levels of the active metabolite nor-desmethylclobazam (norclobazam) principally metabolized by CYP2C19, which likely contributes to increased sedation. The combination of CBD and valproate has been associated with elevated liver transaminases and hyperammonemia, requiring routine monitoring of hepatic function and ammonia levels during co-therapy [ref. 70].
Despite other newer therapies like stiripentol or fenfluramine having demonstrated greater efficacy in terms of seizure reduction, CBD remains an important therapeutic option, particularly for drug-resistant patients [ref. 10].
2.4. Soticlestat
Soticlestat is a selective inhibitor of cholesterol 24-hydroxylase (CH24H or CYP46A1), an enzyme expressed predominantly in the brain that catalyzes the conversion of cholesterol to 24S-hydroxycholesterol (24S-HC). By reducing the amount of 24S-HC, soticlestat reduces glutamatergic excitatory transmission and attenuates neuroinflammation and glial activation, producing an anticonvulsant effect [ref. 71,ref. 72]. Inhibition of CH24H by soticlestat reduces central 24S-HC levels, a potent positive allosteric modulator of NMDA receptors, implicated in the pathogenesis of excitotoxicity and epileptogenesis [ref. 14,ref. 72]. Excess CH24H in astrocytes in epilepsy, aging, Alzheimer disease disrupts lipid rafts containing EEAT2, thus impairing glutamate uptake and increasing glutamate neurotoxicity. In addition, 24HC, via the integrin pathway, increases TNF-α and IL6. TNF-α enhances glutamate release and inhibits glutamate uptake, hence contributing to neural overexcitation. This novel mechanism of action was expected to provide seizure control with a distinct approach from current ASMs [ref. 17].
In preclinical studies, including Scn1a+/− mouse models of DS, soticlestat significantly reduced seizure frequency, neural hyperexcitability, and SUDEP. Notably, it also conferred protection against hyperthermia-induced seizures, a hallmark of DS [ref. 14].
Early-phase clinical evaluation in patients with DEE initially provided evidence for efficacy. In a phase 1b/2a study, soticlestat adjunctive to standard of care resulted in 36.4% median reduction in seizure frequency, with an acceptable safety profile [ref. 73]. The subsequent phase 2 ELEKTRA trial, a randomized, double-blind, placebo-controlled study, assessed soticlestat as adjunctive therapy to standard of care in children with either DS or LGS. Among DS participants, soticlestat produced a statistically significant reduction in convulsive seizure frequency compared with placebo, whereas LGS patients experienced only a non-significant numerical decline in drop seizures, despite comparable reductions in 24S-HC levels across groups. In both cohorts, treatment was associated with improvements in global clinical functioning as assessed by caregiver and clinician impression scales. Common adverse events included lethargy and constipation, though tolerability was overall favorable [ref. 74,ref. 75]. The SKYLINE and SKYWAY multicenter, randomized, double-blind Phase 3 studies evaluated soticlestat plus standard of care versus placebo plus standard of care in patients with refractory DS and LGS, respectively. Soticlestat hardly missed the primary endpoint of reduction from baseline in convulsive seizure frequency and reduction from baseline in major motor drop seizure frequency as compared with placebo. Following these results in 2024, Takeda announced the discontinuation of the soticlestat development program in January 2025.
2.5. Zorevunersen (STK-001)
Zorevunersen (STK-001) is an ASO developed by Stoke Therapeutics using the TANGO (Targeted Augmentation of Nuclear Gene Output) platform. It has been designed to exclude an alternative poison exon (termed exon 20N), which introduces an in-frame premature stop codon into a subset of transcripts, resulting in non-productive mRNA and reduced Nav1.1 channel expression. Indeed, by sterically blocking exon 20N inclusion, zorevunersen reduces non-functional transcripts while enhancing productive SCN1A mRNA and restoring Nav1.1 protein levels from the wild-type allele [ref. 19,ref. 76]. In vitro, zorevunersen decreased the proportion of poison exon–containing transcripts from approximately 60% to under 5%. In wild-type neonatal mice (P2), intracerebroventricular (ICV) administration increased full-length SCN1A mRNA threefold along corresponding rise in Nav1.1 protein. Interestingly, in Scn1a-haploinsufficient mice, zorevunersen prevented the typical ~50% mortality by week 4, delayed seizure onset, decreased seizure frequency, reduced SUDEP, and improved interneuron excitability [ref. 19,ref. 20,ref. 21].
The Phase 1/2a open-label trials (MONARCH in USA, and ADMIRAL in UK) enrolled drug-resistant children and adolescents (ages 2–18) with genetically confirmed SCN1A variants, most receiving ≥3 antiseizure medications, with ~54% on ≥4 medications and half on fenfluramine. In dose cohorts receiving two or three 70 mg injections of zorevunersen, median convulsive seizure reduction was 85% at 3 months and 74% at 6 months post-last dose; lower reductions (~43–57%) were observed after a single 70 mg dose.
Open-label extension studies (SWALLOWTAIL and LONGWING) involving lower maintenance doses (30–45 mg every 4 months) demonstrated durable reductions in seizures over 12 months and improvements in cognition and behavior.
Approximately 30% of participants experienced treatment-emergent adverse events related to zorevunersen, primarily elevated cerebrospinal fluid protein and procedural vomiting; serious adverse events occurred in ~22%, though most were considered unrelated to the drug. In extension studies, cerebrospinal fluid protein elevations were more common (~74–79%), but did not exhibit clinical manifestations.
Zorevunersen has been granted breakthrough therapy designation by the FDA, as well as orphan drug designations from both the FDA and EMA. A global, randomized, double-blind, sham-controlled phase 3 trial (EMPEROR) was planned to commence mid-2025 to evaluate the efficacy, safety and tolerability of zorevunersen in children aged 2 to <18 with DS with a confirmed LOF variant in SCN1A. The trial is expected to enroll participants across the US, Japan, UK and EU. Participants will be randomized 1:1 to receive either zorevunersen via intrathecal administration or a sham comparator for a 52-week treatment period following an 8-week baseline period. Following the completion of the study, eligible participants will be offered ongoing treatment with zorevunersen as part of an open-label extension study. The primary endpoint of the study is percent change from baseline in major motor seizure frequency at week 28 in patients receiving zorevunersen as compared to sham. The key secondary endpoints are the durability of effect on major motor seizure frequency and improvements in behavior and cognition as measured by Vineland-3 subdomains, including expressive communication, receptive communication, interpersonal relationships, coping skills and personal skills. Additional endpoints include safety, Clinician Global Impression of Change, Caregiver Global Impression of Change, and the Bayley Scales of Infant Development [ref. 76].
As said before, the therapeutic strategy is applicable to DS patients with SCN1A LOF variants (e.g., truncating, nonsense, frameshift, or deletions) whose mutant alleles are subject to nonsense-mediated decay. It is contraindicated in patients with missense variants displaying GOF or dominant-negative effects, as increasing Nav1.1 expression may exacerbate disease pathology [ref. 77].
2.6. ETX101 (AAV9-REGABA-eTFSCN1A)
ETX101 (AAV9-REGABA-eTFSCN1A) is a gene regulatory therapy that delivers a transgene coding for an engineered SCN1A-specific transcription factor (eTFSCN1A) to upregulate the expression of the endogenous SCN1A gene. eTF binds to a conserved regulatory region upstream of the SCN1A transcription start site, enhancing SCN1A expression and protein translation. Expression of the transgene is controlled by a GABAergic inhibitory neuron-selective regulatory element (REGABA). This approach increases the production of NaV1.1 at endogenous levels preferentially in GABAergic neurons, thereby restoring inhibitory function and minimizing potential off-target effects in excitatory cells [ref. 27]. ETX101 uses a clinically validated adeno-associated virus (AAV9) capsid and is delivered via ICV infusion in a one-time administration [ref. 17]. AAV-based vectors are considered safe as they cannot replicate or integrate into the genome. Local ICV administration of AAV-mediated gene therapy enhances delivery to the CNS, thereby increasing the potential for efficacy [ref. 5]. Preclinical data for ETX101 demonstrated, indeed, broad distribution into relevant CNS structures, including the cortex and hippocampus, in non-human primates (juvenile cynomolgus macaque), as well as long-term survival and reduction in seizure frequency and SUDEP in DS mouse models. No relevant adverse clinical or histopathological findings were reported [ref. 5,ref. 27].
POLARIS is the principal program designed to optimize and accelerate clinical development of ETX101 for the treatment of people with DS. This program includes phase 1/2 clinical studies to assess the safety and efficacy of ETX101 in infants and young children (ENDEAVOR, WAYFINDER, EXPEDITION). This investigational program builds on several initiatives, including Dravet ENGAGE study, which explored the needs and experiences of DS patients and families; ELUCIDATE, an ongoing biomarker discovery project aimed at identifying novel measures of disease progression; and ENVISION, the largest longitudinal natural history study of DS conducted to date, which provided insights into the disease course in infants and young children. ETX101 is currently in Phase 1/2 trials for infants and children with DS.
2.7. Additional ASO and Gene Editing Approaches
Recent advances in precision medicine for DS have identified a long antisense non-coding RNA, SCN1A-dsAS (downstream antisense), that negatively regulates SCN1A expression in human brain tissue, likely via transcriptional inhibition. Elevated levels of this antisense transcript correlate with reduced Nav1.1, contributing to the haploinsufficiency characteristic of DS [ref. 5]. To counter this, AntagoNAT ASOs targeting SCN1A-dsAS have been developed. In human DS patient fibroblasts, mouse models, and non-human primates, these ASOs upregulated SCN1A expression, restored Nav1.1 levels in fast-spiking inhibitory interneurons, normalized interneuron activity, and reduced seizure burden [ref. 7,ref. 23]. Further preclinical characterization of one of these ASOs, ASO3 (CO-3527), demonstrated efficacy in neuroblastoma-derived SK-N-AS cells [ref. 78]. ASO3 significantly downregulated SCN1A-dsAS transcripts and increased SCN1A mRNA levels, outdoing another ASO (Cur-1740-11), likely due to enhanced chemical stability and higher binding affinity [ref. 5]. These findings highlight the regulatory role of SCN1A-dsAS in SCN1A transcription and validate antisense-mediated modulation as a viable therapeutic strategy. By suppressing the inhibitory long non-coding RNAs, AntagoNATs can restore Nav1.1 expression and function, offering a novel precision approach for addressing the genetic alteration underlying DS.
Strategies based on genome editing have also been employed to upregulate SCN1A expression. The CRISPR/Cas9 system employs the Cas9 endonuclease, a molecular scissor, to introduce targeted double-strand breaks in DNA, enabling precise genome editing in animal, human, and plant cells [ref. 79]. A modified version of this system, in which the nuclease activity of Cas9 is inactivated (dCas9) and fused to transcriptional activator domains, can be directed to specific gene promoters to enhance gene expression. Targeting dCas9 fused to a transcriptional activator (Scn1a-dCas9A) to the SCN1A promoter has been shown to upregulate SCN1A transcription and Nav1.1 protein expression in primary mouse neurons isolated from Scn1a+/R1407X DS pups [ref. 24]. In addition, DS mouse pups administered with the Scn1a-dCas9 system delivered ICV using a dual AAV9-based system showed increased firing rates in GABAergic interneuron and reduced hyperthermia-induced seizures, with an increased threshold for seizure induction. These results supported the therapeutic potential of this transcriptional activation strategy [ref. 24].
3. SCN8A-DEE
The SCN8A gene encodes the Nav1.6 voltage-gated sodium channel, a key determinant of neuronal excitability [ref. 1,ref. 80]. Nav1.6 is widely expressed throughout the brain, predominantly in excitatory neurons and, to a lesser extent, in inhibitory neurons, localizing primarily to the axon initial segment and nodes of Ranvier of myelinated axons. Like SCN1A, SCN8A expression is lowest during prenatal development and increases progressively toward adulthood [ref. 1]. Experimental studies in mice have demonstrated that selective deletion of Nav1.6 in cerebellar Purkinje neurons results in pronounced cognitive and motor deficits, highlighting the essential role of this channel in cerebellar and higher-order brain functions [ref. 81,ref. 82].
Clinically, SCN8A-related disorders exhibit a wide phenotypic spectrum, with disease manifestations classified into five groups based on age of onset and electroclinical biomarkers [ref. 83]. Epileptic phenotypes range from early-onset severe to moderate DEE, with severity influenced by the extent of developmental impairment and seizure refractoriness. Other phenotypes include Self-limiting familial infantile epilepsy (SeLFIE), also referred to as benign familial infantile epilepsy (BFIE), characterized by normal cognition and early-onset seizures that respond well to ASMs. Later-onset and no seizure phenotypes include Neurodevelopmental delay with generalized epilepsy (NDDwGE) and Neurodevelopmental delay without epilepsy (NDDwoE), which typically presents with mild to moderate intellectual disability (severe in ~10% of cases). Additional features may include hypotonia and movement disorders such as dystonia, ataxia, and choreoathetosis [ref. 7]. Importantly, SCN8A mutations are also associated with an increased risk of SUDEP. SCN8A variants can disrupt the normal function of Nav1.6 channels, leading to either GOF or LOF effects [ref. 1]. Most severe early-onset epilepsy have been associated with missense variants causing GOF and may respond to high-dose SCBs. GOF mutations often result in increased neuronal excitability due to altered channel kinetics, such as channels opening more readily, remaining open longer, or inactivating more slowly. Some mutations also generate persistent sodium currents between action potentials, further contributing to neuronal hyperexcitability. In contrast, LOF variants, presenting in individuals with intellectual disability without seizures, impair Nav1.6 function by reducing the number of channels or altering gating properties [ref. 1]. This leads to impaired action potential initiation and propagation and, consequently, disrupted synaptic communication, primarily during development. This early change might cause a persistent alteration in neuronal activity, leading to the autism spectrum disorder phenotype [ref. 81,ref. 82]. According to global consensus on the diagnosis and treatment of SCN8A-related disorders, SCBs, such as oxcarbazepine or carbamazepine, are the first-line treatment for GOF variants, severe DEE, mild/moderate DEE, and SeLFIE, whereas levetiracetam may worsen seizures and contribute to developmental regression in GOF patients. First-line treatment for NDDwGE is valproate, ethosuximide, or lamotrigine; SCBs are relatively contraindicated in LOF patients [ref. 83].
3.1. Relutrigine (PRAX 562)
Relutrigine is an orally active Nav1 blocker under development for the treatment of DEEs regardless of the etiology. This small molecule showed a preferential inhibition of persistent sodium current, a key driver of seizure symptoms in early-onset SCN2A-DEE and SCN8A-DEE and in other severe DEEs [ref. 15].
Relutrigine showed not only a 60-fold preference for persistent sodium current over peak sodium current but also a remarkable and unexpected 30-fold preference for use-dependent block over tonic block on Nav channels expressed in HEK-293 cell lines. Compared with available SCBs, such as lacosamide and carbamazepine, relutrigine showed an improved efficacy profile and improved tolerability, both in cell lines and animal models [ref. 4,ref. 84]. In vivo studies of relutrigine have demonstrated dose-dependent inhibition of seizures up to complete control in SCN2A, SCN8A and other DEE mouse models. The anticonvulsant activity of relutrigine was evaluated preclinically also in the MES (maximal electroshock seizure) animal model where it was again compared with standard SCBs carbamazepine and lamotrigine. Unlike carbamazepine and lamotrigine, the maximum effective dose of relutrigine (10 mg/kg) prevented seizures without affecting locomotor function. Regarding the safety profile, in preclinical MES models and spontaneous locomotor activity (sLMA) models, relutrigine showed a 3-fold higher protective index than carbamazepine and lamotrigine, although further studies are required to investigate the preclinical protective index of relutrigine after chronic treatment, which would reflect the planned clinical use.
Relutrigine has been generally well-tolerated in three phase 1 studies and has demonstrated biomarker changes indicative of Nav channel modulation. The EMBOLD study is a multicenter, randomized phase 2 clinical trial designed to explore the safety, tolerability, efficacy and pharmacokinetics of relutrigine in pediatric (aged 2–18 years) patients with early-onset SCN2A-DEE and SCN8A-DEE. EMBOLD cohort 1 study showed a placebo-adjusted monthly motor seizure reduction of 46% during the double-blind period and seizure freedom in over 30% of patients while on relutrigine. Additionally, significant improvements were observed in alertness, communication and seizure severity as noted by both clinicians and caregivers and 77% reduction in median seizure rate seen for patients in the long-term extension. At 11-month open-label extension period of the trial, an average of approximately 90% seizure reduction was observed in patients, and a sustained and continuous improvement in seizure-free periods was observed, with a mean of 67 days without seizures compared to 3 days in the baseline period. There were no new safety signals, drug related serious adverse events or dose reductions needed.
Based on the results of this study, the EMBOLD registrational cohort 2 is currently ongoing and continues to enroll, with initial results expected to come in the first half of 2026, followed by a potential new drug application filing [ref. 85].
Relutrigine has received orphan drug designation and rare pediatric disease designation from the FDA for the treatment of SCN2A-DEE, SCN8A-DEE and DS, as well as breakthrough therapy designation, and orphan drug designation from the EMA for the treatment of SCN2A-DEE and SCN8A-DEE. The breakthrough therapy designation from FDA was granted based on the highly compelling results from the phase 2 EMBOLD trial in SCN2A– and SCN8A-DEEs. Praxis has also recently initiated the EMERALD study, a registrational, 16-week, placebo-controlled trial evaluating relutrigine effect in seizure reduction in patients diagnosed with DEEs, regardless of etiology [ref. 86].
Thus, relutrigine demonstrated robust anticonvulsant activity in vivo, with good tolerability compared to other SCBs, suggesting potential for an improved clinical therapeutic window especially in patients where standard SCBs have limited efficacy due to poor tolerability. Given the role of persistent sodium current in modulating excitability, relutrigine has the potential to be a widely effective and well-tolerated ASM for both genetic and non-genetic epilepsies.
3.2. NBI-921352 (XEN901)
NBI-921352 is a selective inhibitor of Nav1.6 channels (IC50 ~ 0.051 µM) with high affinity towards the inactivated state of the channel and preferentially binding the voltage sensor domain of DIV. In a SCN8A GOF mouse model, NBI-921352 inhibited action potential generation in glutamatergic excitatory pyramidal neurons, bypassing GABAergic interneurons where Nav1.1 is dominant, and prevented electrically induced seizures [ref. 16]. Efficacy appeared superior to standard SCBs. Phase I clinical studies demonstrated that NBI-921352 was well tolerated at plasma concentrations above those required for efficacy in preclinical rodent studies, without signs of ataxia and motor symptoms. NBI-921352 is under development for both SCN8A-DEE epilepsy and adult focal seizures for oral administration. A phase II trial (KAYAK study) evaluating NBI-921352 in children and young adults with SCN8A-DEE is ongoing, with an estimated completion date in December 2025.
3.3. Additional ASO and RNA-Based Approaches
Additional strategies to treat SCN8A-DEE, still at the preclinical stage, include ASO and RNA-based therapies. Both repeated ASO therapy and a single AAV10-shRNA dose after seizure onset prevented recurrent seizures and extended survival over 12 months of follow-up in SCN8A mouse models. Indeed, in a conditional mouse model with Cre-dependent expression of the pathogenic SCN8A GOF mutation R1872W, an ASO targeting Scn8a downregulated Scn8a expression, extended lifespan and delayed seizure onset by 25 to 50% when administered after birth. This model exhibits early onset seizures, rapid progression, and 100% penetrance and survives for only 2 weeks [ref. 25]. Repeated ASO administration initiated after seizure onset, a timing more relevant to clinical treatment, provided sustained seizure control and significantly prolonged survival throughout the 12-month observation period. Similarly, a single neonatal treatment on postnatal day 1 with an adeno-associated virus serotype 10 (AAV10) vector expressing a short hairpin RNA (shRNA) targeting Scn8a (AAV10-shRNA) conferred long-term seizure protection and extended survival for up to one year [ref. 26]. These results support the disease-modifying potential of these approaches as a post-onset, long-term treatment for SCN8A-DEE.
3.4. Vormatrigine (PRAX 628)
Vormatrigine (PRAX-628) is a novel small molecule designed to reduce pathological neuronal hyperexcitability by targeting sodium channels in the brain, and it is currently under development as a once-daily, oral treatment for adult focal onset seizures and generalized epilepsy [ref. 18]. Vormatrigine exhibits a dual mechanism of action: selective modulation of persistent sodium current and enhanced use-dependent inhibition of peak sodium current. These properties enable precise targeting of hyperexcitable neuronal states while sparing normal activity. Pharmacological evaluations on different human Nav1 isoforms (Nav1.1, 1.2, 1.5, 1.6, 1.7, 1.8, and 1.9) revealed pan-Nav strong inhibitory activity. Even if vormatrigine has not been specifically developed for SCN8A-DEE, this drug induced significant suppression of Nav1.6 persistent sodium current (IC50 ~ 128 nM) and strong use-dependent block of peak sodium current, effects occurring at concentrations below those causing undesired tonic block. Vormatrigine was more potent compared with standard SCBs (e.g., carbamazepine, lamotrigine). These findings position this drug as a promising therapeutic candidate with a potentially superior safety and efficacy profile compared to existing SCBs [ref. 87,ref. 88]. Preclinical in vivo studies demonstrated the antiseizure efficacy of vormatrigine in the MES (maximal electroshock seizure) model, a translational model of focal epilepsy, at lower doses than standard-of-care ASMs. The in vivo anticonvulsant efficacy of vormatrigine (ED50 ~ 0.42 mg/kg) was approximately ten times higher than that of carbamazepine, cenobamate, lamotrigine and of the Kv7 opener XEN1101 (3.8–5.4 mg/kg). Data from the first cohort of patients in the RADIANT study demonstrated a robust seizure reduction and good safety profile [ref. 87,ref. 88]. The POWER1 study is enrolling.
4. SCN2A-DEE
The SCN2A gene encodes for the Nav1.2 voltage-gated sodium channel isoform widely expressed in developing and mature brain, especially in excitatory neurons [ref. 5]. As seen for other neuronal isoforms, variants in SCN2A have been associated with a broad spectrum of neurodevelopmental disorders, with or without epilepsy. Non-seizure phenotypes are characterized by severe encephalopathy, with developmental delays, movement disorders, schizophrenia, and various other conditions. SCN2A is also a candidate risk gene for autism spectrum disorder and nonsyndromic intellectual disability [ref. 1,ref. 28]. Seizure-related phenotypes include self-limited familial (inherited) neonatal–infantile epilepsy (SeLFNIE), previously called benign familial neonatal–infantile seizures (BFNIS), and early-infantile DEE such as Ohtahara syndrome, West syndrome, and LGS. SCN2A variants associated with neonatal and early infantile epilepsies generally produce increased Nav1 activity (GOF) and typically respond well to SCBs. Conversely, variants found in later-onset epilepsies and autism and intellectual disability without seizures cause LOF and show little to no response to SCBs. However, LOF SCN2A truncation can also occur in severe epilepsies including West syndrome and LGS. Thus, variant classification is not straightforward [ref. 28]. Different molecular consequences are associated with SCN2A LOF variants including reduced sodium current density, reduced protein expression, increased protein degradation, defects in trafficking, shifts in the voltage-dependent activation towards depolarized potentials, reduced probability of channel opening, and accelerated inactivation. GOF variants instead cause a shift in the voltage-dependent activation towards more hyperpolarized potentials, increased probability of channel opening, slowed inactivation, and persistent sodium current, leading to increased Nav1.2 channel activity [ref. 1].
In the absence of approved therapies for SCN2A-related disorders, clinical management is based on combination of ASMs and often drugs for managing debilitating comorbidities with several limitations [ref. 5,ref. 7]. However, a novel therapeutic approach based on ASO targeting SCN2A has been developed.
Elsunersen (PRAX 222)
Elsunersen (PRAX-222) is an ASO designed to selectively reduce SCN2A gene expression, offering potential therapeutic benefit to patients with GOF SCN2A variants associated with early-onset SCN2A-DEE [ref. 5]. In vitro studies have demonstrated that elsunersen effectively decreases both SCN2A mRNA and protein levels. Preclinical in vivo studies using mouse models showed dose-dependent reductions in seizure frequency, improved behavioral and locomotor function, and increased survival rates. These data suggest the potential of elsunersen as the first disease-modifying treatment for SCN2A-DEE linked to GOF variants. The ongoing phase 1/2 EMBRAVE study represents the first-in-human clinical trial evaluating elsunersen safety and efficacy in pediatric patients (aged 2–18 years) with early-onset SCN2A-DEE [ref. 89]. Early clinical data of elsunersen in combination with SCBs in 5 patients indicated safety and seizure reduction, including cessation of previous refractory status epilepticus. In a single patient, the combination of intrathecal administration of elsunersen and relutrigine induced a significant reduction in seizures and status epilepticus highlighting the potential for complementary precision sodium channel modulation in early onset SCN2A-DEE [ref. 22,ref. 90]. Elsunersen has received orphan drug designation from both the FDA and EMA, as well as rare pediatric disease designation from the FDA for SCN2A-DEE treatment.
5. Challenges and Future Directions
The therapeutic management of DEEs is moving from a traditional, symptomatic therapy using combinations of conventional ASMs to disease-modifying approaches that either directly target the underlying etiology or exploits alternative pathways and aim for more thorough, sustained clinical improvement [ref. 4,ref. 28] (Figure 1; Table 1).
As multiple genetic and repurposed treatments become available, new clinical, methodological, and ethical challenges are expected to emerge [ref. 3].
The complex relationship between genotype and phenotype in many neurodevelopmental disorders requires precise variant interpretation to guide appropriate treatment and predict clinical outcomes. Both GOF and LOF variants within the same sodium channel gene have been identified, which can lead to markedly different phenotypes requiring different therapeutic strategies [ref. 29]. For instance, in SCN-related DEEs, GOF variants may necessitate gene-silencing strategies or specific SCBs, whereas LOF variants often result in haploinsufficiency phenotypes demanding increased protein expression [ref. 31]. Consequently, variant classification according to the functional defect is essential to inform therapeutic approaches aimed at upregulating, restoring, or suppressing gene or protein activity [ref. 91,ref. 92,ref. 93,ref. 94].
Understanding the natural history of the diseases and developing clinical biomarkers for deep phenotyping are fundamental to recognizing the populations most likely to benefit from specific therapies and to defining outcome measures for future clinical trials (e.g., ELUCIDATE and ENVISION studies for DS). Robust longitudinal and real-world evidence studies are essential to evaluate the long-term effectiveness and safety of both approved and investigational drugs in defined patient subgroups and to assess their impact on neurodevelopmental outcomes and other comorbidities [ref. 3]. In this regard, stiripentol and fenfluramine have shown the strongest seizure reductions in RCTs and network meta-analyses whereas cannabidiol is effective but, on average, less potent than the other two especially towards neurodevelopmental impairment [ref. 12,ref. 46,ref. 58]. Among novel small molecules, relutrigine and vormatrigine are next-generation Nav modulators designed to selectively inhibit persistent sodium current, a mechanism intended to improve both efficacy and tolerability compared with traditional SCBs. Relutrigine has shown promising clinical activity in SCN2A– and SCN8A-DEEs, with meaningful seizure reductions in some patients and better tolerability compared to older SCBs, despite RCT data remaining limited.
In addition, the definition of the optimal therapeutic window for intervention becomes relevant for maximizing clinical benefit. Many genetic epilepsies and neurodevelopmental disorders have early-onset and progressive courses, implying that treatment may be the most effective during defined developmental windows. Interventions initiated after these critical periods may have limited capacity to modify the overall trajectory of the disease. The attempt to administer fenfluramine off-label to children younger than 2 years highlights the need for timely intervention in DS [ref. 8,ref. 28].
Although ASO and gene therapy hold great promise for treating DEE and other neurodevelopmental disorders, several limitations must be overcome to fully exploit their potential [ref. 6,ref. 77]. Currently, ASO formulations may not readily cross the blood–brain barrier, requiring intrathecal or intracerebroventricular administration to achieve therapeutic CNS concentrations. While these delivery routes warrant targeted exposure, they are invasive and may carry procedural risks. Emerging strategies, such as nanoparticle encapsulation, receptor-mediated transcytosis, or chemical modification to enhance blood–brain barrier permeability, may enable less invasive and broader delivery. In addition, ASO pharmacokinetics pose challenges: their limited in vivo half-life necessitates repeated dosing, often multiple times per year, to maintain efficacy. The development of long-acting formulations could reduce treatment troubles and improve patient compliance. Similarly, AAV vectors face several key limitations. Their small packaging capacity (~4.7 kb) restricts use to smaller genes. Tissue-specific targeting and, conversely, off-target effects remain a technical challenge, limiting wide clinical translation [ref. 95]. In addition, immune and dose-related toxicities may raise safety concerns. To overcome these difficulties, emerging strategies include engineered capsids with improved trophism and immune evasion, novel promoters for tissue-specific delivery (e.g., AAV9 in ETX101) and cost-effective enhanced production platforms [ref. 95]. Despite these challenges, early-phase clinical data show promising seizures reduction and significant changes in cognition/behavioral impairment for some participants, consistent with disease-modifying intent of ASO and gene therapy approaches. These results have supported progression toward larger clinical trials.
In this framework, reliable cell and animal models of disease together with clinically oriented preclinical studies are essential to enhance the translatability of safety and efficacy data about new therapies to patients and de-risk clinical research [ref. 96,ref. 97]. In addition, improved knowledge of the mechanisms underlying SCN-related disorders, at the molecular, cellular and network levels in disease models, may allow for the identification of novel druggable pathways and/or facilitate drug repurposing [ref. 38,ref. 39]. For example, delayed GABA switch has emerged as a novel mechanism contributing to neurodevelopmental deficits in DS and a potential drug target [ref. 98].
Finally, the complexity involved in developing biological and innovative drugs presents significant challenges related to cost, time, long-term sustainability and equitable access, particularly for patients in low- and middle-income countries, that must be addressed in the future.
References
- A. Brunklaus, D. Lal. Sodium channel epilepsies and neurodevelopmental disorders: From disease mechanisms to clinical application. Dev. Med. Child Neurol., 2020. [DOI | PubMed]
- E.C. Wirrell, V. Hood, K.G. Knupp, M.A. Meskis, R. Nabbout, I.E. Scheffer, J. Wilmshurst, J. Sullivan. International consensus on diagnosis and management of Dravet syndrome. Epilepsia, 2022. [DOI | PubMed]
- S. Balestrini, D. Mei, S.M. Sisodiya, R. Guerrini. Steps to Improve Precision Medicine in Epilepsy. Mol. Diagn. Ther., 2023. [DOI | PubMed]
- P. Klein, R.M. Kaminski, M. Koepp, W. Löscher. New epilepsy therapies in development. Nat. Rev. Drug Discov., 2024. [DOI | PubMed]
- R. Lersch, R. Jannadi, L. Grosse, M. Wagner, M.F. Schneider, C. von Stülpnagel, F. Heinen, H. Potschka, I. Borggraefe. Targeted Molecular Strategies for Genetic Neurodevelopmental Disorders: Emerging Lessons from Dravet Syndrome. Neuroscientist, 2023. [DOI | PubMed]
- S.F. Hill, M.H. Meisler. Antisense Oligonucleotide Therapy for Neurodevelopmental Disorders. Dev. Neurosci., 2021. [DOI | PubMed]
- D. Samanta, S. Bhatia, S.E. Hunter, C.K. Rao, K. Xiong, C. Karakas, P.C. Reeders, G. Erdemir, S. Sattar, E. Axeen. Current and Emerging Precision Therapies for Developmental and Epileptic Encephalopathies. Pediatr. Neurol., 2025. [DOI | PubMed]
- N. Pietrafusa, M. Trivisano, S. Casellato, C. Correale, S. Cappelletti, P. De Liso, I. Onida, S. Sotgiu, A. Butera, N. Specchio. Fenfluramine below the age of 2 years in Dravet syndrome: What about safety and efficacy?. Epilepsia, 2024. [DOI | PubMed]
- N. Sada, S. Lee, T. Katsu, T. Otsuki, T. Inoue. Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science, 2015. [DOI | PubMed]
- S. Balestrini, I. Galli, M.L. Ricci, E. Parrini, D. Mei, M. Mastrangelo, F. Pisani, C. Filippi, L. Giordano, E. Cesaroni. Clinical and Genetic Landscape of Epilepsies with Absence Seizures and Single-Gene Etiology. Epilepsia, 2025. [DOI]
- J. Sourbron, I. Smolders, P. de Witte, L. Lagae. Pharmacological Analysis of the Anti-epileptic Mechanisms of Fenfluramine in scn1a Mutant Zebrafish. Front. Pharmacol., 2017. [DOI | PubMed]
- A. Boncristiano, S. Balestrini, V. Doccini, N. Specchio, N. Pietrafusa, M. Trivisano, F. Darra, A. Cossu, D. Battaglia, M. Quintiliani. Fenfluramine treatment for Dravet syndrome: Long term real-world analysis demonstrates safety and reduced health care burden. Epilepsia, 2025. [DOI | PubMed]
- J.S. Kaplan, N. Stella, W.A. Catterall, R.E. Westenbroek. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA, 2017. [DOI | PubMed]
- N.A. Hawkins, M. Jurado, T.T. Thaxton, S.E. Duarte, L. Barse, T. Tatsukawa, K. Yamakawa, T. Nishi, S. Kondo, M. Miyamoto. Soticlestat, a novel cholesterol 24-hydroxylase inhibitor, reduces seizures and premature death in Dravet syndrome mice. Epilepsia, 2021. [DOI | PubMed]
- K.M. Kahlig, L. Scott, R.J. Hatch, A. Griffin, G. Martinez Botella, Z.A. Hughes, M. Wittmann. The novel persistent sodium current inhibitor PRAX-562 has potent anticonvulsant activity with improved protective index relative to standard of care sodium channel blockers. Epilepsia, 2022. [DOI | PubMed]
- J.P. Johnson, T. Focken, K. Khakh, P.K. Tari, C. Dube, S.J. Goodchild, J.C. Andrez, G. Bankar, D. Bogucki, K. Burford. NBI-921352, a first-in-class, Na[V]1.6 selective, sodium channel inhibitor that prevents seizures in Scn8a gain-of-function mice, and wild-type mice and rats. Elife, 2022. [DOI | PubMed]
- M. Bialer, S.I. Johannessen, M.J. Koepp, E. Perucca, P. Perucca, T. Tomson, H.S. White. Progress report on new medications for seizures and epilepsy: A summary of the 17th Eilat Conference on New Antiepileptic Drugs and Devices [EILAT XVII]. I. Drugs in preclinical and early clinical development. Epilepsia, 2024. [DOI | PubMed]
- K. Kahlig, M. Chapman, S. Petrou. PRAX-628: A Novel Sodium Channel Blocker with Greater Potency and Activity Dependence Compared to Standard of Care. Proceedings of the American Epilepsy Society Congress
- K.H. Lim, Z. Han, H.Y. Jeon, J. Kach, E. Jing, S. Weyn-Vanhentenryck, M. Downs, A. Corrionero, R. Oh, J. Scharner. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun., 2020. [DOI | PubMed]
- Y. Yuan, L. Lopez-Santiago, N. Denomme, C. Chen, H.A. O’Malley, S.L. Hodges, S. Ji, Z. Han, A. Christiansen, L.L. Isom. Antisense oligonucleotides restore excitability, GABA signalling and sodium current density in a Dravet syndrome model. Brain, 2024. [DOI | PubMed]
- Z. Han, C. Chen, A. Christiansen, S. Ji, Q. Lin, C. Anumonwo, C. Liu, S.C. Leiser, M. Meena, I. Aznarez. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med., 2020. [DOI | PubMed]
- M. Wagner, G. Berecki, W. Fazeli, C. Nussbaum, A.W. Flemmer, S. Frizzo, F. Heer, F. Heinen, R. Horton, H. Jacotin. Antisense oligonucleotide treatment in a preterm infant with early-onset SCN2A developmental and epileptic encephalopathy. Nat. Med., 2025. [DOI | PubMed]
- J. Hsiao, T.Y. Yuan, M.S. Tsai, C.Y. Lu, Y.C. Lin, M.L. Lee, S.W. Lin, F.C. Chang, H. Liu Pimentel, C. Olive. Upregulation of haploinsufficient gene expression in the brain by targeting a long non-coding RNA improves seizure phenotype in a model of Dravet syndrome. EBioMedicine, 2016. [DOI | PubMed]
- G. Colasante, G. Lignani, S. Brusco, C. Di Berardino, J. Carpenter, S. Giannelli, N. Valassina, S. Bido, R. Ricci, V. Castoldi. dCas9-Based Scn1a Gene Activation Restores Inhibitory Interneuron Excitability and Attenuates Seizures in Dravet Syndrome Mice. Mol. Ther., 2020. [DOI | PubMed]
- G.M. Lenk, P. Jafar-Nejad, S.F. Hill, L.D. Huffman, C.E. Smolen, J.L. Wagnon, H. Petit, W. Yu, J. Ziobro, K. Bhatia. Scn8a Antisense Oligonucleotide Is Protective in Mouse Models of SCN8A Encephalopathy and Dravet Syndrome. Ann. Neurol., 2020. [DOI | PubMed]
- S.F. Hill, W. Yu, J. Ziobro, S. Chalasani, F. Reger, M.H. Meisler. Long-Term Downregulation of the Sodium Channel Gene Scn8a Is Therapeutic in Mouse Models of SCN8A Epilepsy. Ann. Neurol., 2024. [DOI | PubMed]
- A. Tanenhaus, T. Stowe, A. Young, J. McLaughlin, R. Aeran, I.W. Lin, J. Li, R. Hosur, M. Chen, J. Leedy. Cell-Selective Adeno-Associated Virus-Mediated SCN1A Gene Regulation Therapy Rescues Mortality and Seizure Phenotypes in a Dravet Syndrome Mouse Model and Is Well Tolerated in Nonhuman Primates. Hum. Gene Ther., 2022. [DOI | PubMed]
- R. Guerrini, V. Conti, M. Mantegazza, S. Balestrini, A.S. Galanopoulou, F. Benfenati. Developmental and epileptic encephalopathies: From genetic heterogeneity to phenotypic continuum. Physiol. Rev., 2023. [DOI | PubMed]
- A. Brunklaus, T. Brünger, T. Feng, C. Fons, A. Lehikoinen, E. Panagiotakaki, M.A. Vintan, J. Symonds, J. Andrew, A. Arzimanoglou. The gain of function SCN1A disorder spectrum: Novel epilepsy phenotypes and therapeutic implications. Brain, 2022. [DOI | PubMed]
- J. Clatot, S. Parthasarathy, S. Cohen, J.L. McKee, S. Massey, A. Somarowthu, E.M. Goldberg, I. Helbig. SCN1A gain-of-function mutation causing an early onset epileptic encephalopathy. Epilepsia, 2023. [DOI | PubMed]
- Y. Kobayashi Takahashi, K. Tabata, S. Baba, E. Takeshita, N. Sumitomo, Y. Shimizu-Motohashi, T. Saito, E. Nakagawa, A. Ishii, S. Hirose. SCN1A gain of function effects in Dravet syndrome: Insights into clinical phenotypes and therapeutic implications. Epilepsia Open, 2025. [DOI | PubMed]
- S. Matricardi, S. Cestèle, M. Trivisano, B. Kassabian, N. Leroudier, R. Vittorini, M. Nosadini, E. Cesaroni, S. Siliquini, C. Marinaccio. Gain of function SCN1A disease-causing variants: Expanding the phenotypic spectrum and functional studies guiding the choice of effective antiseizure medication. Epilepsia, 2023. [DOI | PubMed]
- F.H. Yu, M. Mantegazza, R.E. Westenbroek, C.A. Robbins, F. Kalume, K.A. Burton, W.J. Spain, G.S. McKnight, T. Scheuer, W.A. Catterall. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci., 2006. [DOI | PubMed]
- H. Ogiwara, N. Miyamoto, N. Morita, N. Atapour, E. Mazaki, I. Inoue, T. Takeuchi, S. Itohara, Y. Yanagawa, K. Obata. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci., 2007. [DOI | PubMed]
- M.S. Martin, K. Dutt, L.A. Papale, C.M. Dubé, S.B. Dutton, G. de Haan, A. Shankar, S. Tufik, M.H. Meisler, T.Z. Baram. Altered function of the SCN1A voltage-gated sodium channel leads to gamma aminobutyric acid-ergic [GABAergic] interneuron abnormalities. J. Biol. Chem., 2010. [DOI | PubMed]
- A.C. Bender, R.P. Morse, R.C. Scott, G.L. Holmes, P.P. Lenck-Santini. SCN1A mutations in Dravet syndrome: Impact of interneuron dysfunction on neural networks and cognitive outcome. Epilepsy Behav., 2012. [DOI | PubMed]
- S. Han, C. Tai, R.E. Westenbroek, F.H. Yu, C.S. Cheah, G.B. Potter, J.L. Rubenstein, T. Scheuer, H.O. de la Iglesia, W.A. Catterall. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature, 2012. [DOI | PubMed]
- G. Berecki, A. Bryson, T. Polster, S. Petrou. Biophysical characterization and modelling of SCN1A gain-of-function predicts interneuron hyperexcitability and a predisposition to network instability through homeostatic plasticity. Neurobiol. Dis., 2023. [DOI | PubMed]
- H. Martins Custodio, L.M. Clayton, R. Bellampalli, S. Pagni, K. Silvennoinen, R. Caswell, A. Brunklaus, R. Guerrini, B.P.C. Koeleman. Widespread genomic influences on phenotype in Dravet syndrome, a ‘monogenic’ condition. Brain, 2023. [DOI | PubMed]
- A. Bryson, S. Petrou. SCN1A channelopathies: Navigating from genotype to neural circuit dysfunction. Front. Neurol., 2023. [DOI | PubMed]
- N.A. Koch, L. Sonnenberg, U.B.S. Hedrich, S. Lauxmann, J. Benda. Loss or gain of function? Effects of ion channel mutations on neuronal firing depend on the neuron type. Front. Neurol., 2023. [DOI | PubMed]
- A. Bacq, A. Depaulis, V. Castagné, M.E. Le Guern, E.C. Wirrell, M. Verleye. An Update on Stiripentol Mechanisms of Action: A Narrative Review. Adv. Ther., 2024. [DOI | PubMed]
- C. Chiron, M.C. Marchand, A. Tran, E. Rey, P. d’Athis, J. Vincent, O. Dulac, G. Pons. Stiripentol in severe myoclonic epilepsy in infancy: A randomised placebo-controlled syndrome-dedicated trial. Lancet, 2000. [DOI | PubMed]
- A. Rosati, A. Boncristiano, V. Doccini, A. Pugi, T. Pisano, M. Lenge, S. De Masi, R. Guerrini. Long-term efficacy of add-on stiripentol treatment in children, adolescents, and young adults with refractory epilepsies: A single center prospective observational study. Epilepsia, 2019. [DOI | PubMed]
- N. Specchio, S. Auvin, A. Strzelczyk, F. Brigo, V. Villanueva, E. Trinka. Efficacy and safety of stiripentol in the prevention and cessation of status epilepticus: A systematic review. Epilepsia Open, 2024. [DOI | PubMed]
- S. Balestrini, V. Doccini, A. Boncristiano, M. Lenge, S. De Masi, R. Guerrini. Efficacy and Safety of Long-Term Treatment with Stiripentol in Children and Adults with Drug-Resistant Epilepsies: A Retrospective Cohort Study of 196 Patients. Drugs Real World Outcomes, 2022. [DOI | PubMed]
- A. Gil-Nagel, A. Aledo-Serrano, Á. Beltrán-Corbellini, L. Martínez-Vicente, A. Jimenez-Huete, R. Toledano-Delgado, I. Gacía-Morales, A. Valls-Carbó. Efficacy and tolerability of add-on stiripentol in real-world clinical practice: An observational study in Dravet syndrome and non-Dravet developmental and epileptic encephalopathies. Epilepsia Open, 2024. [DOI | PubMed]
- V. Soto-Insuga, E. González-Alguacil, M. Ballarà-Petitbò, N. Lamagrande-Casanova, A. Duat-Rodríguez, C. Benítez-Provedo, E. Cardenal-Muñoz, J.J. García-Peñas. Efficacy of Stiripentol Beyond Dravet Syndrome: A Retrospective Medical Record Review of Patients with Drug-Resistant Epilepsies. Neurol. Ther., 2025. [DOI | PubMed]
- R. Guerrini, C. Chiron, D. Vandame, W. Linley, T. Toward. Comparative efficacy and safety of stiripentol, cannabidiol and fenfluramine as first-line add-on therapies for seizures in Dravet syndrome: A network meta-analysis. Epilepsia Open, 2024. [DOI | PubMed]
- R. Odi, R.W. Invernizzi, T. Gallily, M. Bialer, E. Perucca. Fenfluramine repurposing from weight loss to epilepsy: What we do and do not know. Pharmacol. Ther., 2021. [DOI | PubMed]
- P. Martin, T. Reeder, J. Sourbron, P.A.M. de Witte, A.R. Gammaitoni, B.S. Galer. An Emerging Role for Sigma-1 Receptors in the Treatment of Developmental and Epileptic Encephalopathies. Int. J. Mol. Sci., 2021. [DOI | PubMed]
- L. Abenhaim, Y. Moride, F. Brenot, S. Rich, J. Benichou, X. Kurz, T. Higenbottam, C. Oakley, E. Wouters, M. Aubier. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N. Engl. J. Med., 1996. [DOI | PubMed]
- M.T. Dinday, S.C. Baraban. Large-Scale Phenotype-Based Antiepileptic Drug Screening in a Zebrafish Model of Dravet Syndrome. eNeuro, 2015. [DOI]
- J. Cha, G. Filatov, S.J. Smith, A.R. Gammaitoni, A. Lothe, T. Reeder. Fenfluramine increases survival and reduces markers of neurodegeneration in a mouse model of Dravet syndrome. Epilepsia Open, 2024. [DOI | PubMed]
- L. Lagae, J. Sullivan, K. Knupp, L. Laux, T. Polster, M. Nikanorova, O. Devinsky, J.H. Cross, R. Guerrini, D. Talwar. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: A randomised, double-blind, placebo-controlled trial. Lancet, 2019. [DOI | PubMed]
- R. Nabbout, A. Mistry, S. Zuberi, N. Villeneuve, A. Gil-Nagel, R. Sanchez-Carpintero, U. Stephani, L. Laux, E. Wirrell, K. Knupp. Fenfluramine for Treatment-Resistant Seizures in Patients with Dravet Syndrome Receiving Stiripentol-Inclusive Regimens: A Randomized Clinical Trial. JAMA Neurol., 2020. [DOI | PubMed]
- J. Sullivan, L. Lagae, J.H. Cross, O. Devinsky, R. Guerrini, K.G. Knupp, L. Laux, M. Nikanorova, T. Polster, D. Talwar. Fenfluramine in the treatment of Dravet syndrome: Results of a third randomized, placebo-controlled clinical trial. Epilepsia, 2023. [DOI | PubMed]
- I.E. Scheffer, R. Nabbout, L. Lagae, O. Devinsky, S. Auvin, E.A. Thiele, E.C. Wirrell, T. Polster, N. Specchio, M. Pringsheim. Long-term safety and effectiveness of fenfluramine in children and adults with Dravet syndrome. Epilepsia, 2025. [DOI | PubMed]
- L. de Andrade Mesquita, G. Fagundes Piccoli, G. Richter da Natividade, B. Frison Spiazzi, V. Colpani, F. Gerchman. Is lorcaserin really associated with increased risk of cancer? A systematic review and meta-analysis. Obes. Rev., 2021. [DOI | PubMed]
- A. Griffin, K.R. Hamling, K. Knupp, S. Hong, L.P. Lee, S.C. Baraban. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain, 2017. [DOI | PubMed]
- M. Bialer, E. Perucca. Lorcaserin for Dravet Syndrome: A Potential Advance Over Fenfluramine?. CNS Drugs, 2022. [DOI | PubMed]
- D.S. Reddy. Therapeutic and clinical foundations of cannabidiol therapy for difficult-to-treat seizures in children and adults with refractory epilepsies. Exp. Neurol., 2023. [DOI | PubMed]
- C.J. Milligan, L.L. Anderson, M.T. Bowen, S.D. Banister, I.S. McGregor, J.C. Arnold, S. Petrou. A nutraceutical product, extracted from Cannabis sativa, modulates voltage-gated sodium channel function. J. Cannabis Res., 2022. [DOI | PubMed]
- E.C. Rosenberg, S. Chamberland, M. Bazelot, E.R. Nebet, X. Wang, S. McKenzie, S. Jain, S. Greenhill, M. Wilson, N. Marley. Cannabidiol modulates excitatory-inhibitory ratio to counter hippocampal hyperactivity. Neuron, 2023. [DOI | PubMed]
- O. Devinsky, J.H. Cross, L. Laux, E. Marsh, I. Miller, R. Nabbout, I.E. Scheffer, E.A. Thiele, S. Wright. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. N. Engl. J. Med., 2017. [DOI | PubMed]
- J.P. Szaflarski, E.M. Bebin, A.M. Comi, A.D. Patel, C. Joshi, D. Checketts, J.C. Beal, L.C. Laux, L.M. De Boer, M.H. Wong. Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: Expanded access program results. Epilepsia, 2018. [DOI | PubMed]
- S. Lattanzi, G. Zaccara, E. Russo, A. La Neve, M.A.M. Lodi, P. Striano. Practical use of pharmaceutically purified oral cannabidiol in Dravet syndrome and Lennox-Gastaut syndrome. Expert Rev. Neurother., 2021. [DOI | PubMed]
- A.D. Patel, J.P. Szaflarski, P.D. Lyons, M. Boffa, T. Greco, T.B. Saurer, K. Rajasekaran, K.C. Simontacchi, E.A. Thiele. Long-term efficacy and safety of cannabidiol in patients with treatment-resistant focal epilepsies treated in the Expanded Access Program. Epilepsia, 2025. [DOI]
- M. De Bellis, G. d’Orsi, E.M. Rubino, C. Arigliano, M. Carella, V. Sciruicchio, A. Liantonio, A. De Luca, P. Imbrici. Adverse effects of antiseizure medications: A review of the impact of pharmacogenetics and drug interactions in clinical practice. Front. Pharmacol., 2025. [DOI | PubMed]
- G. Morrison, J. Crockett, G. Blakey, K. Sommerville. A phase 1, open-label, pharmacokinetic trial to investigate possible drug-drug interactions between clobazam, stiripentol, valproate, and cannabidiol in healthy subjects. Clin. Pharmacol. Drug Dev., 2019. [DOI | PubMed]
- S. Kondo, V. Murthy, M. Asgharnejad, A. Benitez, K. Nakashima, N. Hawkins, H.S. White. A review of the putative antiseizure and antiepileptogenic mechanisms of action for soticlestat. Epilepsia, 2025. [DOI | PubMed]
- T. Nishi, S. Kondo, M. Miyamoto, S. Watanabe, S. Hasegawa, S. Kondo, J. Yano, E. Watanabe, T. Ishi, M. Yoshikawa. Soticlestat, a novel cholesterol 24-hydroxylase inhibitor shows a therapeutic potential for neural hyperexcitation in mice. Sci. Rep., 2020. [DOI | PubMed]
- J.J. Halford, M.R. Sperling, D. Arkilo, M. Asgharnejad, C. Zinger, R. Xu, M. During, J.A. French. A phase 1b/2a study of soticlestat as adjunctive therapy in participants with developmental and/or epileptic encephalopathies. Epilepsy Res., 2021. [DOI | PubMed]
- C.D. Hahn, Y. Jiang, V. Villanueva, M. Zolnowska, D. Arkilo, S. Hsiao, M. Asgharnejad, D. Dlugos. A phase 2, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of soticlestat as adjunctive therapy in pediatric patients with Dravet syndrome or Lennox-Gastaut syndrome [ELEKTRA]. Epilepsia, 2022. [DOI | PubMed]
- Z. Lanlan, J. Nana, W. Chengzhong. Efficacy, safety, and tolerability of soticlestat [TAK-935] as adjunctive therapy in pediatric patients with Dravet syndrome and Lennox-Gastaut syndrome: A meta-analysis of 3 randomized controlled trials. Front. Pharmacol., 2025. [DOI | PubMed]
- L.L. Isom, K.G. Knupp. Targeted Augmentation of Nuclear Gene Output [TANGO]. Jasper’s Basic Mechanisms of the Epilepsies, 2024
- L.L. Isom, K.G. Knupp. Dravet Syndrome: Novel Approaches for the Most Common Genetic Epilepsy. Neurotherapeutics, 2021. [DOI | PubMed]
- M.F. Schneider, M. Vogt, J. Scheuermann, V. Müller, A.H.L. Fischer-Hentrich, T. Kremer, S. Lugert, F. Metzger, M. Kudernatsch, G. Kluger. Brain expression profiles of two SCN1A antisense RNAs in children and adolescents with epilepsy. Transl. Neurosci., 2024. [DOI | PubMed]
- M. Asmamaw, B. Zawdie. Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing. Biologics, 2021. [DOI | PubMed]
- J.H. Caldwell, K.L. Schaller, R.S. Lasher, E. Peles, S.R. Levinson. Sodium channel Na[v]1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc. Natl. Acad. Sci. USA, 2000. [DOI | PubMed]
- S.I. Levin, Z.M. Khaliq, T.K. Aman, T.M. Grieco, J.A. Kearney, I.M. Raman, M.H. Meisler. Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a [NaV1.6] in cerebellar Purkinje neurons and granule cells. J. Neurophysiol., 2006. [DOI | PubMed]
- X. Yang, H. Yin, X. Wang, Y. Sun, X. Bian, G. Zhang, A. Li, A. Cao, B. Li, D. Ebrahimi-Fakhari. Social Deficits and Cerebellar Degeneration in Purkinje Cell Scn8a Knockout Mice. Front. Mol. Neurosci., 2022. [DOI | PubMed]
- G. Conecker, M.Y. Xia, J. Hecker, C. Achkar, C. Cukiert, S. Devries, E. Donner, M.P. Fitzgerald, E. Gardella, M. Hammer. Global modified Delphi consensus on diagnosis, phenotypes, and treatment of SCN8A-related epilepsy and/or neurodevelopmental disorders. Epilepsia, 2024. [DOI | PubMed]
- L. Anderson, K.M. Kahlig, M. Souza, S. Petrou. Preclinical Findings of Relutrigine, a Precision Sodium Channel Modulator, Point to Anticonvulsant Potential in Dravet Syndrome. Proceedings of the 36th International Epilepsy Congress
- Frizzo. Relutrigine Demonstrates Robust Seizure Reduction and Seizure Freedom in DEEs: Results from the EMBOLD Study. Proceedings of the 36th International Epilepsy Congress
- S. Kamireddy. EMERALD: A Phase 3, Randomized, Multi-Center, Double-Blind, Placebo-Controlled Clinical Trial to Evaluate the Efficacy, Safety, Tolerability, and Pharmacokinetics of Relutrigine in Participants with Developmental and Epileptic Encephalopathies. Proceedings of the 36th International Epilepsy Congress
- K. Hansen, S. Frizzo, H. Jacotin, D. Patel, N. Epstein, A. Patelk, H. Sun, S. Petrou, M. Souza. Updates from the First-in-human Phase 1 Clinical Trial Evaluating the Safety, Tolerability, Pharmacokinetics and Food Effect of Vormatrigine in Healthy Participants. Proceedings of the 36th International Epilepsy Congress
- K. Hansen. RADIANT–An Open Label Phase 2 Clinical Trial to Evaluate the Efficacy and Safety of Vormatrigine in Adult Patients with Focal Onset or Primary Generalized Tonic-Clonic Seizures. Proceedings of the 36th International Epilepsy Congress
- B. Spar, K. Dalby, S. Frizzo, D. Patel, H. Jacotin, M. Souza, S. Petrou. EMBRAVE: A Clinical Trial of PRAX-222, a Novel Antisense Oligonucleotide, in Pediatric Participants with Early Onset SCN2A Developmental and Epileptic Encephalopathy. Proceedings of the 36th International Epilepsy Congress
- B. Spar. Emergency Use Case of Relutrigine, a Functional State Sodium Channel Modulator, in an Infant with SCN2A-DEE and Refractory Status Epilepticus. Proceedings of the 36th International Epilepsy Congress
- G. Dinoi, E. Conte, O. Palumbo, M. Benvenuto, M.A. Coppola, P. Palumbo, P. Lastella, B. Boccanegra, E. Di Muro, M. Castori. The Biallelic Inheritance of Two Novel SCN1A Variants Results in Developmental and Epileptic Encephalopathy Responsive to Levetiracetam. Biomedicines, 2024. [DOI | PubMed]
- G. Dinoi, M. Morin, E. Conte, H. Mor Shaked, M.A. Coppola, M.C. D’Adamo, O. Elpeleg, A. Liantonio, I. Hartmann, A. De Luca. Clinical and Functional Study of a De Novo Variant in the PVP Motif of Kv1.1 Channel Associated with Epilepsy, Developmental Delay and Ataxia. Int. J. Mol. Sci., 2022. [DOI | PubMed]
- P. Imbrici, E. Conte, R. Blunck, F. Stregapede, A. Liantonio, M. Tosi, M.C. D’Adamo, A. De Luca, V. Brankovic, G. Zanni. A Novel KCNA2 Variant in a Patient with Non-Progressive Congenital Ataxia and Epilepsy: Functional Characterization and Sensitivity to 4-Aminopyridine. Int. J. Mol. Sci., 2021. [DOI | PubMed]
- M. De Bellis, B. Boccanegra, A.G. Cerchiara, P. Imbrici, A. De Luca. Blockers of Skeletal Muscle Nav1.4 Channels: From Therapy of Myotonic Syndrome to Molecular Determinants of Pharmacological Action and Back. Int. J. Mol. Sci., 2023. [DOI | PubMed]
- Z. Słyk, N. Stachowiak, M. Małecki. Recombinant adeno-associated virus vectors for gene therapy of the central nervous system: Delivery routes and clinical aspects. Biomedicines, 2024. [DOI | PubMed]
- B. Boccanegra, P. Mantuano, E. Conte, A.G. Cerchiara, L. Tulimiero, R. Quarta, E. Caputo, F. Sanarica, M. Forino, V. Spadotto. LKB1 Signaling Is Altered in Skeletal Muscle of a Duchenne Muscular Dystrophy Mouse Model. Dis. Model. Mech., 2023. [DOI | PubMed]
- R. Willmann, J. Lee, C. Turner, K. Nagaraju, A. Aartsma-Rus, D.J. Wells, K.R. Wagner, C. Csimma, V. Straub, M.D. Grounds. Improving Translatability of Preclinical Studies for Neuromuscular Disorders: Lessons from the TREAT-NMD Advisory Committee for Therapeutics (TACT). Dis. Model. Mech., 2020. [DOI | PubMed]
- L. Pizzamiglio, F. Capitano, E. Rusina, G. Fossati, E. Menna, I. Léna, F. Antonucci, M. Mantegazza. Neurodevelopmental defects in Dravet syndrome Scn1a+/− mice: Targeting GABA-switch rescues behavioral dysfunctions but not seizures and mortality. Neurobiol. Dis., 2025. [DOI | PubMed]