
Cannabinoid-based drugs targeting CB1 and TRPV1, the sympathetic nervous system, and arthritis
Abstract
Chronic inflammation in rheumatoid arthritis (RA) is accompanied by activation of the sympathetic nervous system, which can support the immune system to perpetuate inflammation. Several animal models of arthritis already demonstrated a profound influence of adrenergic signaling on the course of RA. Peripheral norepinephrine release from sympathetic terminals is controlled by cannabinoid receptor type 1 (CB1), which is activated by two major endocannabinoids (ECs), arachidonylethanolamine (anandamide) and 2-arachidonylglycerol. These ECs also modulate function of transient receptor potential channels (TRPs) located on sensory nerve fibers, which are abundant in arthritic synovial tissue. TRPs not only induce the sensation of pain but also support inflammation via secretion of pro-inflammatory neuropeptides. In addition, many cell types in synovial tissue express CB1 and TRPs. In this review, we focus on CB1 and transient receptor potential vanilloid 1 (TRPV1)-mediated effects on RA since most anti-inflammatory mechanisms induced by cannabinoids are attributed to cannabinoid receptor type 2 (CB2) activation. We demonstrate how CB1 agonism or antagonism can modulate arthritic disease. The concept of functional antagonism with continuous CB1 activation is discussed. Since fatty acid amide hydrolase (FAAH) is a major EC-degrading enzyme, the therapeutic possibility of FAAH inhibition is studied. Finally, the therapeutic potential of ECs is examined since they interact with cannabinoid receptors and TRPs but do not produce central side effects.
Affiliations: Laboratory of Experimental Rheumatology and Neuroendocrine Immunology, University Hospital of Regensburg, D-93053 Regensburg, Germany
License: © Lowin and Straub. 2015 CC BY 4.0 Open AccessThis article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Article links: DOI: 10.1186/s13075-015-0743-x | PubMed: 26343051 | PMC: PMC4561168
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (1.5 MB)
Introduction
Rheumatoid arthritis (RA) is a debilitating disease that affects around 1.3 million people in the US alone [ref. 1]. Important characteristics of RA are inflammation of the joint with subsequent destruction of cartilage, pannus formation and infiltrates of immune cells [ref. 2–ref. 4]. Ongoing inflammation also leads to systemic changes manifesting in co-morbidities like dyslipidemia, depression, fatigue, insulin resistance, activation of the sympathetic nervous system, and cachexia [ref. 5, ref. 6]. Changes in sympathetic activity lead to a metabolic switch, which is in part responsible for the perpetuation of inflammation and the increase in cardiovascular risk in RA patients [ref. 7].
Cannabis has been used since 4000 BC for the treatment of spasms and post-operative pain [ref. 8]. In the 1990s, the two main receptors for cannabinoids (cannabinoid receptors I and II; CB1 and CB2) were identified [ref. 9, ref. 10]. Both receptors are activated by the psychoactive component of cannabis, tetrahydrocannabinol (THC), and several other synthetic and plant-derived cannabinoids [ref. 11]. Two major endogenous cannabinoids (endocannabinoids, ECs), arachidonylethanolamine (anandamide, AEA) and 2-arachidonylglycerol (2-AG), were described shortly after the discovery of CB1 and CB2 [ref. 12, ref. 13]. In recent years, other receptors such as transient receptor potential vanilloid 1 (TRPV1), GPR55, or GPR18 were found to bind cannabinoids, and activation of these receptors is responsible for the off-target effects of several cannabinoids [ref. 14–ref. 18]. Transient receptor potential channel (TRP) modulation by cannabinoids might be explicitly important since these receptors not only influence sensation of pain, but also support inflammation [ref. 19].
This review describes physiological aspects of CB1 receptors, pharmacological roles of ECs and the EC-degrading enzyme fatty acid amid hydrolase (FAAH), functional crosstalk between ECs and TRPV1, the interaction between ECs and the sympathetic nervous system in RA, the influence of ECs on arthritis disease sequelae in mice and humans, and direct immunomodulatory effects of CB1 signaling in the periphery and in the brain. Considering this knowledge we finally try to demonstrate an optimum therapeutic EC approach in RA.
Physiology
CB1 influences cell function by controlling neurotransmitter levels
The classic function of ECs in the nervous system is the regulation of neurotransmitter release via CB1, which is also responsible for the psychotropic effects of cannabis [ref. 20–ref. 23]. CB1 is mainly located on presynaptic nerve terminals, and activation of this receptor reduces the release of neurotransmitter from corresponding neurons in a heteroreceptor-typical way [ref. 24]. Thus, cannabinoids can increase or decrease neuronal excitability depending on neurotransmitter and brain region affected. CB1 receptors are also abundant on peripheral sympathetic nerve terminals, where they modulate adrenergic signaling. This influence on sympathetic nerves can alter lipolysis, cytokine production, ghrelin production, heart rate and bone resorption [ref. 20,ref. 25–ref. 28]. The effects of CB1 activation or inhibition on neurotransmitter release in a given peripheral tissue are depicted in Fig. 1. In addition, CB1 receptors are located on nociceptive nerve fibers. Here, CB1 agonism increases the threshold for the generation of action potentials via modulation of ion channels and TRPs [ref. 29, ref. 30].
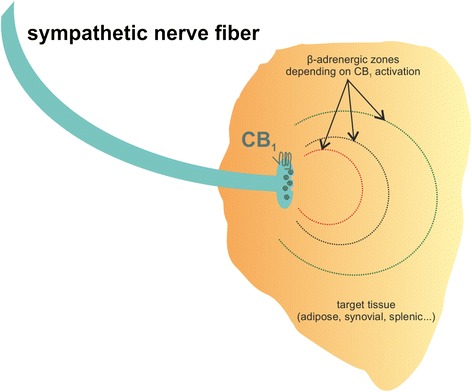
Direct effects of CB1 activation on immune cells have only been scarcely described. Our group but also others demonstrated an influence of cell adhesion in response to CB1 agonism; this effect might also modulate immune function by regulating cell trafficking and tissue extravasation [ref. 31, ref. 32].
CB2 regulates immune cell function directly
While CB1 functions mainly through modulation of central and peripheral neurotransmitter release, activation of CB2 elicits direct anti-inflammatory effects in target cells [ref. 33]. This includes reduction of cytokine and matrix metalloproteinase production, modulation of adhesion and migration but also induction of apoptosis [ref. 33]. The anti-inflammatory potential of CB2 was also confirmed in mouse models of arthritis [ref. 34, ref. 35]. While the impact of CB2 on immune function has already been investigated and reviewed elsewhere [ref. 33, ref. 36], this review focuses on CB1.
Pharmacology
Role of the ECs anandamide and 2-AG
The action of ECs is limited by rapid degradation involving FAAH, which degrades AEA and related N-acylethanolamines, and monoacylglycerol-lipase (MAGL), which degrades 2-AG [ref. 37]. In addition, several enzymes like cyclooxygenase-2, lipoxygenase or cytochrome P450 and others contribute to EC metabolism [ref. 38]. Characteristics of AEA, 2-AG, THC and the CB1 antagonist rimonabant are given in Table 1. Inhibition of FAAH raises the levels of the N-acylethanolamines AEA, palmitoylethanolamine (PEA) and oleoylethanolamine (OEA) [ref. 39]. While AEA is responsible for maintaining basal EC signaling, 2-AG mediates strong and rapid feedback via CB1 receptors [ref. 40]. This is also reflected by the fact that AEA is a partial agonist at CB1, while 2-AG acts as full agonist [ref. 41]. Due to its full agonistic properties, elevation of 2-AG by inhibition of MAGL leads to functional antagonism (discussed below) of CB1, although this might be prevented by reduced dosing [ref. 42, ref. 43]. Furthermore, MAGL inhibition might be detrimental in some situations, since 2-AG is also degraded by cyclooxygenase-2 leading to pro-inflammatory metabolites [ref. 44]. Therefore, this review only covers the consequences of FAAH inhibition.
Table 1: Characteristics of selected cannabinoid receptor ligands
| Ligand | Target receptors | Ki at CB1 in nM | Ki at CB2 in nM | Emax/IC50 at TRPV1 (nM) | Route of degradation |
|---|---|---|---|---|---|
| Anandamide | CB1, CB2, GPR55, TRPV1, TRPA1, TRPM8 (antagonist) | 239.2 ± 61.77 [ref. 158] | 439.5 ± 95.89 [ref. 158] | 458 (Emax) [ref. 159] | FAAH, FAAH-2, NAAA, COX-2, LOX [ref. 160] |
| 2-AG | CB1, CB2, TRPV1, GABAA | 3423.6 ± 3288.24 [ref. 158] | 1193.8 ± 327.71 [ref. 158] | 750 ± 40 (IC50) [ref. 161] | MAGL, COX-2, LOX, ABHD6/12 [ref. 160,ref. 162] |
| Delta9-THC | CB1, CB2, GPR18 | 25.1 ± 5.54 [ref. 158] | 35.2 ± 5.86 [ref. 158] | NA | CYP2C [ref. 163] |
| Rimonabant | CB1, MOR | 1.98 ± 0.36 [ref. 164] | NA | NA | CYP3A [ref. 165] |
Anandamide, 2-arachidonylglycerol (2-AG) and tetrahydrocannabinol (THC) are CB1/CB2 agonists, rimonabant is a CB1/MOR antagonist/inverse agonist. Anandamide and THC are partial CB1/CB2 agonists, 2-AG is a full agonist at both receptors. The main degrading enzyme for each compound is highlighted in bold. ABHD, α/β-hydrolase domain; CB 1 /CB 2, cannabinoid receptor I/II; COX-2, cyclooxygenase-2; CYP, cytochrome P450; Delta9 THC, delta9 tetrahydrocannabinol; Emax, maximal functional response; FAAH, fatty acid amide hydrolase; IC50, half maximal inhibitory concentration; Ki, dissociation constant; LOX, lipoxygenase; MAGL, monoacylglycerol lipase; MOR, μ opoid receptor; NA, not applicable; NAAA, N-acylethanolamine-hydrolyzing acid amidase; TRPA1, transient receptor potential ankyrin I; TRPM8, transient receptor potential melastatin 8; TRPV1, transient receptor potential vanilloid I
The conundrum of functional antagonism at CB1 and TRPV1
Throughout this review, similar effects of CB1 agonists and CB1 antagonists on features of arthritic inflammation are described. This conundrum can be explained by rapid desensitization and downregulation/internalization of CB1 upon agonist exposure [ref. 45–ref. 47]. If desensitization is disturbed due to mutations in crucial CB1 phosphorylation sites, CB1 agonism leads to enhanced acute effects and delayed tolerance [ref. 48]. Consequently, CB1 signaling diminishes in response to repeated agonist exposure [ref. 49]. This feature of CB1 explains functional antagonism: administration of exogenous cannabinoids or elevation of endogenous levels of the full CB1 agonist 2-AG leads to downregulation of CB1. If levels drop low enough, production of ECs is not sufficient to activate CB1 or CB1 signaling pathways. This phenomenon was described with MAGL inhibitors, which increase levels of 2-AG [ref. 42]. Another possibility to achieve antagonistic effects with agonists is the use of CB1 partial agonists like AEA, which lack full activation of CB1 signaling pathways. These partial agonists act as antagonists when full agonists are also present [ref. 50].
TRPs, in particular TRPV1, TRPV2, TRPV3, TRPV4, TRPA1 and TRPM8, serve as ionotropic cannabinoid receptors and they also desensitize upon agonist exposure [ref. 51–ref. 55]. The EC AEA is an agonist at TRPV1 with a binding affinity similar to that of the hot pepper ingredient capsaicin, although it does not activate the receptor like capsaicin [ref. 56]. Therefore, although being an agonist itself, AEA prevents the effects of high efficacy agonists like capsaicin, thus serving as antagonist in this setting. Furthermore, AEA rapidly desensitizes TRPV1, which results in reduced calcium influx [ref. 57]. In addition, the AEA congeners and FAAH substrates PEA and OEA also desensitize TRPV1 [ref. 58, ref. 59]. Although there are no data available regarding the desensitization of other TRPs by N-acylethanolamines, it is likely that this also occurs since there is extensive crosstalk between, for example, TRPV1 and TRPA1 via intracellular calcium [ref. 60]. Moreover, it has been demonstrated that synthetic cannabinoid ligands binding TRPA1 also desensitized target cells to the action of TRPV1 agonists [ref. 61].
FAAH inhibition does not produce central side effects and bridges TRPs and cannabinoid receptors
Central activation of CB1 has psychotropic side effects and this problem is circumvented by the use of FAAH inhibitors [ref. 62]. In contrast to exogenous cannabinoids, AEA does not lead to tolerance at CB1 or psychotropic effects [ref. 63]. Therapeutically, reduction of tolerance to CB1 agonists with FAAH inhibitors can be important since this process leads to a loss of efficacy when repeatedly administered [ref. 63]. In addition, elevation of OEA and PEA also provide anti-inflammatory, neuroprotective effects and they enhance neurogenesis mostly via peroxisome-proliferator activated receptors [ref. 64–ref. 66]. FAAH inhibition has already been demonstrated to be effective in collagen-induced arthritis in mice, although this was attributed to CB2 activation [ref. 34]. Furthermore, FAAH inhibition not only combines anti-inflammatory effects of several N-acylethanolamines but also targets additional receptors such as TRPV1 and peroxisome proliferator activated receptors [ref. 65, ref. 67–ref. 69]. One important receptor for AEA and its congeners OEA and PEA is the TRPV1 cation channel, although other TRPs are similarly activated by AEA [ref. 69–ref. 71].
Besides CB1 and CB2, ECs as well as synthetic and phytocannabinoids bind to members of the TRP family [ref. 54, ref. 61, ref. 72–ref. 74]. Several of these non-selective cation channels integrate external and endogenous stimuli and are sensitized and activated during inflammation [ref. 19, ref. 75]. Pharmacological elevation of AEA in the rat leads to activation but also desensitization of TRPV1, resulting in increased pain thresholds [ref. 69]. In contrast to CB1 activation, TRP activation increases cell excitability leading to increased release of neurotransmitters [ref. 76–ref. 78]. When co-expressed, CB1 agonism decreases TRPV1 channel activity by dephosphorylation, which increases the threshold for agonists [ref. 78]. Although mainly located on sensory Aδ and C-fibers, TRPs are also expressed on peripheral cells such as synoviocytes, and activation results in increased expression of inflammatory mediators [ref. 75, ref. 79, ref. 80]. The best described example of subsequent TRPV1 and CB1 activation is the regulation of blood pressure, where only the CB1/TRPV1 agonist AEA elicited a triphasic response involving both receptors [ref. 81]. First, AEA activates TRPV1 causing hypotension and bradycardia followed by a pressor phase with increased heart rate. In the final phase, prolonged hypotension by AEA is observed and this effect was inhibited by CB1 antagonism. The sequential activation of TRPV1 and CB1 in the context of blood pressure regulation has been reviewed elsewhere [ref. 81].
Clinical relevance
The sympathetic nervous system supports chronic inflammation in arthritis – links to endocannabinoids
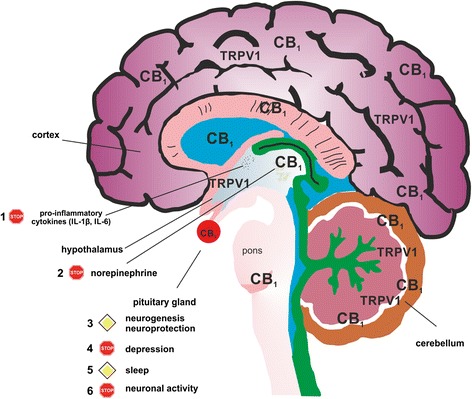
Sympathectomy in arthritic patients has already been performed in the 1920s and follow-up studies showed reduced joint swelling and pain in sympathectomized patients [ref. 82]. The neuroinflammatory component of arthritis has been revealed in studies by Levine and colleagues [ref. 83, ref. 84]. In the mouse model of collagen-induced arthritis it was shown that chemical sympathectomy before or during the time of immunization results in less severe disease [ref. 85]. Late sympathectomy, however, results in exacerbation of experimental arthritis, which might be due to deletion of tyrosine hydroxylase-positive catecholamine-producing cells that appear in synovial tissue during the course of the disease [ref. 86]. The beneficial effects of tyrosine hydroxylase-positive cells on the development of collagen-induced arthritis was demonstrated by our group. In vitro, tyrosine hydroxylase controls cytokine production in mixed synovial cells, whereas in vivo introduction of these cells into arthritic mice reduced arthritic score [ref. 87]. During arthritic inflammation in mice and humans, production of nerve repulsion factors by macrophages leads to the retraction of sympathetic but not sensory fibers from synovial tissue [ref. 88]. As a result, synovial concentration of norepinephrine falls under the threshold for anti-inflammatory β2 receptor activation and this favors pro-inflammatory effects via α-adrenergic signaling [ref. 89, ref. 90]. However, sympathetic signaling is increased in adipose tissue surrounding the synovium, which is responsible for generating energy-rich substrates to support inflammation [ref. 91]. These changes in sympathetic activity during the course of arthritis might be limited or even reversed by altering either EC production or CB1 function, since this receptor controls norepinephrine release. Reduction of EC production by blocking appropriate synthesizing enzymes leads to a functional loss of CB1 since low levels of ECs can no longer activate the receptor. This was already demonstrated in a mouse model of constipation, where inhibition of diacylglycerol lipase α lowered levels of the CB1 agonist 2-AG with concomitant increases in gut motility [ref. 92]. The same effect is achieved by antagonizing CB1 directly [ref. 93]. The loss of sympathetic nerves, altered adrenergic signaling and the possible influence of ECs in the joint is visualized in Fig. 2. In parallel with the disappearance of sympathetic nerve fibers in the joint, hypothalamic norepinephrine, interleukin (IL)-6 and IL-1β increase during the induction phase of experimental arthritis [ref. 94] (Fig. 3). In addition, these changes in cytokine levels and disruption of adrenergic signaling are not accompanied by an adequate response of the hypothalamus-pituitary-adrenal (HPA) axis, resulting in low cortisol levels in relation to inflammation in humans and rodents [ref. 94]. A more detailed description of the influence of the sympathetic nervous system on inflammation has recently been published by our group [ref. 95].
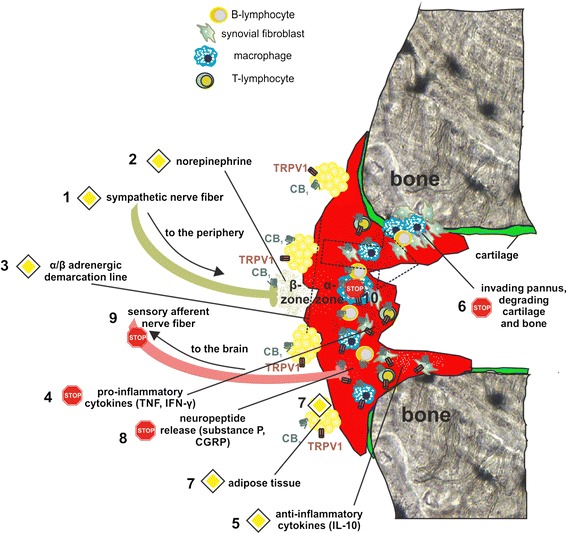
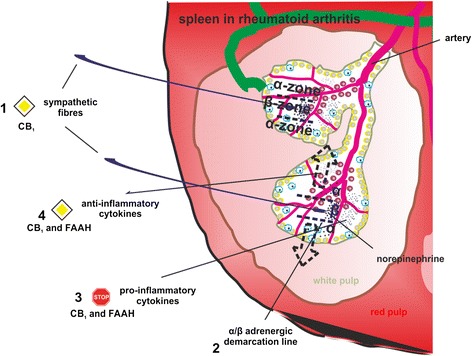
Modulation of adrenergic signaling via CB1 might be beneficial in arthritis
In adjuvant arthritis, immune cells respond to adrenergic β2 receptor stimulation with decreased production of tumor necrosis factor (TNF), an increase in anti-inflammatory IL-10, and a shift to a T-helper type 2 and T-regulatory immune response [ref. 96]. Antagonism of CB1 at splenic sympathetic terminals provides strong anti-inflammatory effects and ameliorates collagen-induced arthritis in mice via reduction of TNF levels, which was inhibited by β2 adrenergic antagonism [ref. 26] (Fig. 4). Furthermore, β2 adrenergic activation on murine B-lymphocytes increases production of the anti-inflammatory cytokine IL-10, which inhibits inflammation [ref. 97]. The time window for anti-arthritic β2 adrenergic effects in mice is crucial since early activation (during the induction phase of experimental arthritis) of sympathetic signaling in the spleen increases interferon (IFN)-γ production [ref. 98]. Sympathetic innervation of the spleen is reduced during the course of experimental arthritis, comparable to the situation in synovial tissue [ref. 99]. This has profound effects on local adrenergic signaling since low concentrations of norepinephrine favor pro-inflammatory α-adrenergic receptor activation [ref. 100, ref. 101] (Fig. 4). Although the beneficial outcome of CB1 receptor antagonism in collagen-induced arthritis in mice was attributed to β2-receptor activation on splenocytes, several other mechanisms might contribute to the therapeutic effects. CB1 antagonism at sympathetic terminals surrounding the synovium might have different outcomes depending on the magnitude of recovery of norepinephrine levels in the joint. If β2 signaling is restored in synovial tissue, local concentrations of IFN-γ and TNF might decline, leading to an overall decrease in joint destruction, synovial inflammation and pain [ref. 102, ref. 103] (Fig. 2). On the other hand, since we demonstrated an increase of sympathetic fibers in human synovial adipose tissue, increased norepinephrine release might further increase lipolysis and thereby fuel inflammation [ref. 91]. Thus, it is imperative to maintain norepinephrine levels over a certain ‘β2 activation threshold’ in the synovium, which might only be achieved with continuous high doses of CB1 antagonists. Consequences of enhanced β2 signaling by CB1 antagonism are depicted in Fig. 2.
Although the above mentioned stimulating effects of CB1 antagonism on adrenergic signaling are evident, CB1 agonists might also prove useful in modulating arthritis. As mentioned earlier, sympathectomy in the early phase ameliorates experimental arthritis in mice [ref. 85]. This indicates a pro-inflammatory influence of adrenergic signaling at the beginning of the disease, which might be counteracted by CB1 agonists decreasing norepinephrine levels [ref. 20]. Arthritis is accompanied by a loss of sympathetic nerve fibers from sites of inflammation and this might also be counteracted by CB1 activation, since neurogenesis is disturbed in CB1 knock-out mice, although we do not know whether this also applies for sympathetic nerve fibers [ref. 104].
The development of comorbidities such as bone resorption, depression and water retention/volume expansion in RA is partly driven by changes in sympathetic activity [ref. 19, ref. 105]. Osteoporosis is a major contributor to RA-associated complications and osteoclasts and osteoblasts respond to cannabinoid receptor activation [ref. 106, ref. 107]. Activation of CB1 results in enhanced osteoblast differentiation, which leads to reduced osteoporosis. Blockade of CB1 disturbs osteoclast function and increases bone mass in the young, but leads to osteoporosis later on due to decreased bone formation [ref. 108].
One major disability associated with RA is the development of depression, which affects around 17 % of patients and is associated with poorer disease outcome [ref. 109]. Depression and CB1 are connected since side effects of rimonabant, a first generation CB1 inverse agonist/antagonist, include depression and anxiety while CB1 agonism has anxiolytic-like and antidepressant-like activities [ref. 110, ref. 111]. The effects of CB1 agonism by FAAH inhibition in the brain are depicted in Fig. 3.
Overactivity of the sympathetic nervous system in RA also leads to water retention via activation of the renin-aldosterone-angiotensin system [ref. 19]. Cannabinoids not only induce diuretic effects but also decrease aldosterone secretion from the adrenal glands by activation of CB1 [ref. 112, ref. 113].
CB1 antagonism activates the HPA axis and reverses insulin resistance
Although modulation of immune cell function via β2-adrenergic receptors is important, CB1 antagonism also supports beneficial systemic changes. One of the hallmarks of RA is an inadequate cortisol secretion in relation to inflammation [ref. 114]. Antagonism at CB1 might counteract this phenomenon, since CB1 knock-out mice had higher levels of adrenocorticotropic hormone and corticosterone under basal but also under stressed conditions [ref. 115]. ECs control glucocorticoid feedback and, therefore, CB1 antagonism increases circulating adrenocorticotropic hormone levels [ref. 116]. Interestingly, high doses of a CB1 agonist also increase the activity of the HPA axis, although this is due to alteration of serotonergic and adrenergic transmission [ref. 117]. The same outcome using CB1 antagonism or agonism on HPA axis activation might also depend on the concentration of CB1 agonists and whether central or peripheral CB1 receptors are targeted. Peripheral agonism at CB1 leads to subsequent activation of α and β adrenoreceptors, which are linked to the antinociceptive effects of CB1 in a rat pain model [ref. 118]. Increases in adrenergic signaling by CB1 agonists might be due to decreased inhibitory gamma-aminobutyric acid (GABA) signaling since release of this neurotransmitter is also controlled by CB1 [ref. 22]. Thus, enhanced GABA signaling reduces sympathetic activity and vice versa [ref. 119]. Central activation of CB1 mediates the rapid effect of glucocorticoid negative feedback and this might explain the necessity for high peripheral doses of the CB1 antagonist rimonabant to increase cortisol levels [ref. 120, ref. 121].
A major problem during the course of RA is the development of insulin resistance with systemic metabolic changes [ref. 122, ref. 123]. Insulin resistance is a direct consequence of enhanced pro-inflammatory cytokine signaling and TNF, IL-6, IL-1β as well as other cytokines are responsible for these changes [ref. 124]. From 2006 to 2008 the CB1 antagonist rimonabant was marketed for use against obesity but was withdrawn due to central side effects [ref. 125]. However, the drug proved to be effective at decreasing important parameters associated with metabolic syndrome. Rimonabant reduces leptin expression, decreases atherosclerosis, and reverses insulin resistance in rodents and humans [ref. 126, ref. 127]. In this respect, CB1 antagonism might also be beneficial in reversing metabolic changes in RA. Insulin resistance is induced by the immune system to divert energy to active immune cells, which are not dependent on insulin for glucose utilization [ref. 105]. Therefore, CB1 antagonism might normalize energy distribution throughout the body and this might deprive the activated immune system of nutrients important for the perpetuation of inflammation. Interestingly, CB1 activation by the phytocannabinoid δ9-tetrahydrocannabinol (THC) corrects hyperlipidemia and hyperglycemia [ref. 128]. This effect might be relevant when using CB1 partial agonists like THC that act as antagonists when full agonists like the EC 2-AG are present. In mice, THC antagonized the effects of the synthetic CB1 agonist AM2389 on hypothermia, although it elicited hypothermic effects by itself [ref. 129]. Furthermore, repeated administration of cannabinoids leads to desensitization and downregulation of CB1, resulting in functional antagonism [ref. 42, ref. 48].
Systemic leptin levels are decreased by CB1 inhibition, and this adipokine is associated with higher IL-6 production and it also initiates production of TH1 cytokines [ref. 130, ref. 131]. In addition, cardiovascular events are a major risk in RA and CB1 antagonists might be effective in decreasing vascular inflammation [ref. 132, ref. 133].
Direct anti-inflammatory effects of CB1 on immune cells
Although most changes associated with CB1 antagonism are mediated via the sympathetic nervous system, direct effects on the immune system are also described. In macrophages from CB1 knock-out mice, TLR4 expression and concomitant pro-inflammatory cytokine production were reduced [ref. 134]. Anti-inflammatory effects of CB1 inhibition were also demonstrated in THP-1 macrophages, where rimonabant decreased TNF and increased IL-10 production [ref. 135]. Furthermore, in a mouse model of sponge-induced angiogenesis, CB1 antagonism reduced leukocyte infiltration and chemokine/cytokine production [ref. 136].
CB1 agonism also has anti-inflammatory effects on immune cells – for example, decreased activation of T lymphocytes by downregulating IL-2 [ref. 137]. However, direct effects of CB1 agonists are most prominent when injected into the brain, where CB1 activation reduces the severity of intestinal inflammation and decreases the activity of microglial cells via reduction of pro-inflammatory cytokines in mice [ref. 138, ref. 139]. Therefore, CB1 activation might alleviate arthritis through central nervous pathways, since neuroinflammation and concomitant increases in brain cytokine levels contribute to the disease [ref. 94, ref. 140].
Central effects of CB1 ligands limit their therapeutic use
Although therapeutically active when administered intrathecally, the use of CB1 agonists or antagonists is limited due to their central adverse events. While CB1 antagonists/inverse agonists like rimonabant induce depression and anxiety in some patients, CB1 agonists like THC have psychotropic properties [ref. 110, ref. 141]. This might derive from reduction of glutamatergic neurotransmission in response to CB1 activation leading to effects similar to NMDA antagonism [ref. 142]. This problem might be circumvented by using peripherally restricted CB1 ligands, which have been generated as second generation cannabinoid therapeutics with proven effects [ref. 143, ref. 144]. Furthermore, neutral antagonists with limited brain penetration and which lack the adverse effects of the inverse agonist rimonabant have been developed [ref. 145]. Neutral antagonists do not influence the constitutive activity of CB1 and therefore do not mediate some of the adverse effects observed with rimonabant therapy [ref. 146]. In contrast to neutral antagonists, inverse agonists like rimonabant not only block CB1 but also stabilize the receptor in an inactive conformation. This diminishes basal signaling and leads to a reciprocal receptor response. In the case of CB1, cAMP is increased by inverse but not by neutral antagonists [ref. 146, ref. 147].
Crosstalk between CB1 and TRPV1 modulates pain and inflammation in arthritis
The importance of TRPV1 in arthritis is emphasized in knock-out animals that show an attenuated disease [ref. 148, ref. 149]. In TRPV1−/− animals, pain thresholds were increased with a concomitant reduction of joint inflammation [ref. 149]. The same protective effect was achieved by oral administration of the TRPV1 agonist SA13353, which reduced TNF production and provided anti-arthritic effects in the rat [ref. 150]. Interestingly, this effect was mediated by TRPV1 located on sensory neurons, emphasizing the neuronal component of arthritis [ref. 150]. This might disrupt a positive feedback loop, since TNF and other pro-inflammatory cytokines sensitize TRPV1 and enhance its activity [ref. 102]. The paradoxical finding that TRPV1 agonists also act in an anti-inflammatory fashion is explained by rapid desensitization of TRPV1 in response to agonist treatment, which depends on the agonist used [ref. 151]. Findings in synovial fibroblasts support this notion, where the TRPV1 agonist capsaicin increases IL-6 production, while AEA, a low efficacy TRPV1 agonist, decreased IL-6 levels under TNF stimulation (T Lowin, unpublished data) [ref. 80].
Since some peripheral effects of TRPV1 are attributed to receptors located on sensory nerve terminals which co-express CB1, crosstalk between both receptors might define the outcome of inflammation [ref. 152]. This can be important in RA, since elevated synovial levels of nerve growth factor sensitize TRPV1 to inflammatory stimuli and CB1 agonism counteracts this response [ref. 153, ref. 154]. In this respect, FAAH inhibition might be superior to selective CB1 agonists since AEA or its metabolites not only activate CB1 but also desensitize TRPV1, leading to analgesia [ref. 69]. Neuronal TRPV1 increases neurotransmitter and pro-inflammatory neuropeptide release via elevation of intracellular calcium levels and the same mechanism often induces the secretion of cytokines from immune cells [ref. 155–ref. 157]. Inhibition of TRPV1 function by concomitant CB1 activation and AEA-induced desensitization (FAAH inhibition) might be a promising strategy to reduce RA disease activity and pain.
Conclusion: is there a perfect cannabinoid-based therapy for the treatment of RA?
The question arises how to modulate the EC system for the treatment of RA. The best treatment option might be a combination of a peripherally restricted CB1 antagonist and a FAAH inhibitor raising systemic levels of N-acylethanolamines. CB1 antagonism has already been shown to result in anti-arthritic effects in mice and this treatment might also increase adrenergic signaling in RA, thereby reducing TNF and IFN-γ and decreasing joint inflammation and cartilage destruction. Potential effects of CB1 antagonism (also of FAAH inhibition) in arthritic synovium and spleen are shown in Figs. 1 and 3, respectively.
Furthermore, CB1 antagonists might reverse metabolic alterations associated with RA: for example, insulin resistance, enhanced leptin expression, depression/fatigue or atherosclerosis. FAAH inhibition on the other hand can counteract the neuroinflammatory component of RA by activating neuronal CB1 and TRPV1 (Fig. 3). Furthermore, the FAAH substrates OEA and PEA can support anti-inflammatory and neurogenic effects of central CB1 activation via peroxisome-proliferator activated receptors. In addition, CB1 activation in the brain lowers sympathetic activity, which can decrease disease-related problems like hypertension. In addition, increases in brain AEA can have antidepressant effects and since many RA patients suffer from mood disorders, FAAH inhibition might help to counteract this central nervous system problem.
In the periphery, FAAH inhibition leads to analgesic and anti-inflammatory effects via desensitization of TRPV1. Moreover, FAAH inhibition has been shown to have high efficacy in arthritic mice through activation of CB2, which might also be beneficial in patients by downregulating cytokine production. In summary, therapeutic intervention in RA with a peripherally restricted CB1 antagonist and a FAAH inhibitor might offer a promising strategy to ameliorate RA.
References
- CG Helmick, DT Felson, RC Lawrence, S Gabriel, R Hirsch, CK Kwoh. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I Arthritis Rheum, 2008. [PubMed]
- IB McInnes, G Schett. The pathogenesis of rheumatoid arthritis. N Engl J Med, 2011. [PubMed]
- N Bottini, GS Firestein. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol, 2013. [PubMed]
- TJ Smeets, RJ Dolhain, FC Breedveld, PP Tak. Analysis of the cellular infiltrates and expression of cytokines in synovial tissue from patients with rheumatoid arthritis and reactive arthritis. J Pathol, 1998. [PubMed]
- RD Situnayake, G Kitas. Dyslipidemia and rheumatoid arthritis. Ann Rheum Dis, 1997. [PubMed]
- PH Dessein, BI Joffe, AE Stanwix. Inflammation, insulin resistance, and aberrant lipid metabolism as cardiovascular risk factors in rheumatoid arthritis. J Rheumatol, 2003. [PubMed]
- RH Straub, JW Bijlsma, A Masi, M Cutolo. Role of neuroendocrine and neuroimmune mechanisms in chronic inflammatory rheumatic diseases – the 10-year update. Semin Arthritis Rheum, 2013. [PubMed]
- H-L Li. An archaeological and historical account of cannabis in China. Economic Bot, 1974
- M Herkenham, AB Lynn, MD Little, MR Johnson, LS Melvin, BR de Costa. Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A, 1990. [PubMed]
- S Munro, KL Thomas, M Abu-Shaar. Molecular characterization of a peripheral receptor for cannabinoids. Nature, 1993. [PubMed]
- RG Pertwee. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther, 1997. [PubMed]
- WA Devane, L Hanus, A Breuer, RG Pertwee, LA Stevenson, G Griffin. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science, 1992. [PubMed]
- R Mechoulam, S Ben-Shabat, L Hanus, M Ligumsky, NE Kaminski, AR Schatz. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol, 1995. [PubMed]
- RA Ross. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol, 2003. [PubMed]
- E Ryberg, N Larsson, S Sjogren, S Hjorth, NO Hermansson, J Leonova. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol, 2007. [PubMed]
- CM Henstridge. Off-target cannabinoid effects mediated by GPR55. Pharmacology, 2012. [PubMed]
- D McHugh, J Page, E Dunn, HB Bradshaw. Delta(9)-Tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells. Br J Pharmacol, 2012. [PubMed]
- JP Buchweitz, PW Karmaus, KJ Williams, JR Harkema, NE Kaminski. Targeted deletion of cannabinoid receptors CB1 and CB2 produced enhanced inflammatory responses to influenza A/PR/8/34 in the absence and presence of Delta9-tetrahydrocannabinol. J Leukoc Biol, 2008. [PubMed]
- RH Straub. TRPV1, TRPA1, and TRPM8 channels in inflammation, energy redirection, and water retention: role in chronic inflammatory diseases with an evolutionary perspective. J Mol Med (Berl), 2014. [PubMed]
- J Tam, V Trembovler, MV Di, S Petrosino, G Leo, A Alexandrovich. The cannabinoid CB1 receptor regulates bone formation by modulating adrenergic signaling. FASEB J, 2008. [PubMed]
- A Polissidis, O Chouliara, A Galanopoulos, G Naxakis, D Papahatjis, Z Papadopoulou-Daifoti. Cannabinoids negatively modulate striatal glutamate and dopamine release and behavioural output of acute D-amphetamine. Behav Brain Res, 2014. [PubMed]
- GG Szabo, N Lenkey, N Holderith, T Andrasi, Z Nusser, N Hajos. Presynaptic calcium channel inhibition underlies CB(1) cannabinoid receptor-mediated suppression of GABA release. J Neurosci, 2014. [PubMed]
- D Cota, MA Steiner, G Marsicano, C Cervino, JP Herman, Y Grubler. Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology, 2007. [PubMed]
- MR Carey, MH Myoga, KR McDaniels, G Marsicano, B Lutz, K Mackie. Presynaptic CB1 receptors regulate synaptic plasticity at cerebellar parallel fiber synapses. J Neurophysiol, 2011. [PubMed]
- B Kola, I Farkas, M Christ-Crain, G Wittmann, F Lolli, F Amin. The orexigenic effect of ghrelin is mediated through central activation of the endogenous cannabinoid system. PLoS One, 2008
- SJ Mnich, RR Hiebsch, RM Huff, S Muthian. Anti-inflammatory properties of CB1-receptor antagonist involves beta2 adrenoceptors. J Pharmacol Exp Ther, 2010. [PubMed]
- MR Boon, S Kooijman, AD van Dam, LR Pelgrom, JF Berbee, CA Visseren. Peripheral cannabinoid 1 receptor blockade activates brown adipose tissue and diminishes dyslipidemia and obesity. FASEB J, 2014. [PubMed]
- T Pfitzer, N Niederhoffer, B Szabo. Search for an endogenous cannabinoid-mediated effect in the sympathetic nervous system. Naunyn Schmiedebergs Arch Pharmacol, 2005. [PubMed]
- I Devesa, A Ferrer-Montiel. Neurotrophins, endocannabinoids and thermo-transient receptor potential: a threesome in pain signalling. Eur J Neurosci, 2014. [PubMed]
- M Kress, R Kuner. Mode of action of cannabinoids on nociceptive nerve endings. Exp Brain Res, 2009. [PubMed]
- T Lowin, W Zhu, K Dettmer-Wilde, RH Straub. Cortisol-mediated adhesion of synovial fibroblasts is dependent on the degradation of anandamide and activation of the endocannabinoid system. Arthritis Rheum, 2012. [PubMed]
- B Rossi, E Zenaro, S Angiari, L Ottoboni, S Bach, L Piccio. Inverse agonism of cannabinoid CB1 receptor blocks the adhesion of encephalitogenic T cells in inflamed brain venules by a protein kinase A-dependent mechanism. J Neuroimmunol, 2011. [PubMed]
- AM Malfitano, S Basu, K Maresz, M Bifulco, BN Dittel. What we know and do not know about the cannabinoid receptor 2 (CB2). Semin Immunol, 2014. [PubMed]
- SG Kinsey, PS Naidu, BF Cravatt, DT Dudley, AH Lichtman. Fatty acid amide hydrolase blockade attenuates the development of collagen-induced arthritis and related thermal hyperalgesia in mice. Pharmacol Biochem Behav, 2011. [PubMed]
- S Fukuda, H Kohsaka, A Takayasu, W Yokoyama, C Miyabe, Y Miyabe. Cannabinoid receptor 2 as a potential therapeutic target in rheumatoid arthritis. BMC Musculoskelet Disord, 2014. [PubMed]
- JP Pereira, J An, Y Xu, Y Huang, JG Cyster. Cannabinoid receptor 2 mediates the retention of immature B cells in bone marrow sinusoids. Nat Immunol, 2009. [PubMed]
- RA Kohnz, DK Nomura. Chemical approaches to therapeutically target the metabolism and signaling of the endocannabinoid 2-AG and eicosanoids. Chem Soc Rev, 2014. [PubMed]
- S Zelasko, WR Arnold, A Das. Endocannabinoid metabolism by cytochrome P450 monooxygenases. Prostaglandins Other Lipid Mediat, 2015
- T Bisogno, PL De, M Di. V. Fatty acid amide hydrolase, an enzyme with many bioactive substrates. Possible therapeutic implications. Curr Pharm Des, 2002. [PubMed]
- T Ohno-Shosaku, M Kano. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr Opin Neurobiol, 2014. [PubMed]
- CJ Hillard. Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat, 2000. [PubMed]
- JE Schlosburg, JL Blankman, JZ Long, DK Nomura, B Pan, SG Kinsey. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci, 2010. [PubMed]
- SG Kinsey, LE Wise, D Ramesh, R Abdullah, DE Selley, BF Cravatt. Repeated low-dose administration of the monoacylglycerol lipase inhibitor JZL184 retains cannabinoid receptor type 1-mediated antinociceptive and gastroprotective effects. J Pharmacol Exp Ther, 2013. [PubMed]
- S Valdeolivas, MR Pazos, T Bisogno, F Piscitelli, FA Iannotti, M Allara. The inhibition of 2-arachidonoyl-glycerol (2-AG) biosynthesis, rather than enhancing striatal damage, protects striatal neurons from malonate-induced death: a potential role of cyclooxygenase-2-dependent metabolism of 2-AG. Cell Death Dis, 2013
- T Luk, W Jin, A Zvonok, D Lu, XZ Lin, C Chavkin. Identification of a potent and highly efficacious, yet slowly desensitizing CB1 cannabinoid receptor agonist. Br J Pharmacol, 2004. [PubMed]
- W Jin, S Brown, JP Roche, C Hsieh, JP Celver, A Kovoor. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J Neurosci, 1999. [PubMed]
- M Rinaldi-Carmona, DA Le, D Oustric, F Barth, M Bouaboula, P Carayon. Modulation of CB1 cannabinoid receptor functions after a long-term exposure to agonist or inverse agonist in the Chinese hamster ovary cell expression system. J Pharmacol Exp Ther, 1998. [PubMed]
- DJ Morgan, BJ Davis, CS Kearn, D Marcus, AJ Cook, J Wager-Miller. Mutation of putative GRK phosphorylation sites in the cannabinoid receptor 1 (CB1R) confers resistance to cannabinoid tolerance and hypersensitivity to cannabinoids in mice. J Neurosci, 2014. [PubMed]
- TL Daigle, CS Kearn, K Mackie. Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacology, 2008. [PubMed]
- W Gonsiorek, C Lunn, X Fan, S Narula, D Lundell, RW Hipkin. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: antagonism by anandamide. Mol Pharmacol, 2000. [PubMed]
- RG Pertwee, AC Howlett, ME Abood, SP Alexander. Di M, V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB(1) and CB(2). Pharmacol Rev, 2010. [PubMed]
- J Joseph, S Wang, J Lee, JY Ro, MK Chung. Carboxyl-terminal domain of transient receptor potential vanilloid 1 contains distinct segments differentially involved in capsaicin- and heat-induced desensitization. J Biol Chem, 2013. [PubMed]
- PL De, A Ligresti, AS Moriello, M Allara, T Bisogno, S Petrosino. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol, 2011. [PubMed]
- PL De, P Orlando, AS Moriello, G Aviello, C Stott, AA Izzo. Cannabinoid actions at TRPV channels: effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol (Oxf), 2012. [PubMed]
- M Nabissi, MB Morelli, C Amantini, S Liberati, M Santoni, L Ricci-Vitiani. Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int J Cancer, 2015
- RA Ross, TM Gibson, HC Brockie, M Leslie, G Pashmi, SJ Craib. Structure-activity relationship for the endogenous cannabinoid, anandamide, and certain of its analogues at vanilloid receptors in transfected cells and vas deferens. Br J Pharmacol, 2001. [PubMed]
- E Lizanecz, Z Bagi, ET Pasztor, Z Papp, I Edes, N Kedei. Phosphorylation-dependent desensitization by anandamide of vanilloid receptor-1 (TRPV1) function in rat skeletal muscle arterioles and in Chinese hamster ovary cells expressing TRPV1. Mol Pharmacol, 2006. [PubMed]
- WS Ho, DA Barrett, MD Randall. ‘Entourage’ effects of N-palmitoylethanolamide and N-oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br J Pharmacol, 2008. [PubMed]
- P Ambrosino, MV Soldovieri, C Russo, M Taglialatela. Activation and desensitization of TRPV1 channels in sensory neurons by the PPARalpha agonist palmitoylethanolamide. Br J Pharmacol, 2013. [PubMed]
- MJ Patil, NA Jeske, AN Akopian. Transient receptor potential V1 regulates activation and modulation of transient receptor potential A1 by Ca2+. Neuroscience, 2010. [PubMed]
- AN Akopian, NB Ruparel, A Patwardhan, KM Hargreaves. Cannabinoids desensitize capsaicin and mustard oil responses in sensory neurons via TRPA1 activation. J Neurosci, 2008. [PubMed]
- Z Justinova, RA Mangieri, M Bortolato, SI Chefer, AG Mukhin, JR Clapper. Fatty acid amide hydrolase inhibition heightens anandamide signaling without producing reinforcing effects in primates. Biol Psychiatry, 2008. [PubMed]
- KW Falenski, AJ Thorpe, JE Schlosburg, BF Cravatt, RA Abdullah, TH Smith. FAAH−/− mice display differential tolerance, dependence, and cannabinoid receptor adaptation after delta 9-tetrahydrocannabinol and anandamide administration. Neuropsychopharmacology, 2010. [PubMed]
- BK Ormerod, SJ Hanft, A Asokan, U Haditsch, SW Lee, TD Palmer. PPARgamma activation prevents impairments in spatial memory and neurogenesis following transient illness. Brain Behav Immun, 2013. [PubMed]
- M Fidaleo, F Fanelli, MP Ceru, S Moreno. Neuroprotective properties of peroxisome proliferator-activated receptor alpha (PPARalpha) and its lipid ligands. Curr Med Chem, 2014. [PubMed]
- T Bandiera, S Ponzano, D Piomelli. Advances in the discovery of N-acylethanolamine acid amidase inhibitors. Pharmacol Res, 2014. [PubMed]
- F Borrelli, B Romano, S Petrosino, E Pagano, R Capasso, D Coppola. Palmitoylethanolamide, a naturally occurring lipid, is an orally effective intestinal anti-inflammatory agent. Br J Pharmacol, 2015. [PubMed]
- M Alhouayek, P Bottemanne, KV Subramanian, DM Lambert, A Makriyannis, PD Cani. N-Acylethanolamine-hydrolyzing acid amidase inhibition increases colon N-palmitoylethanolamine levels and counteracts murine colitis. FASEB J, 2014. [PubMed]
- K Starowicz, W Makuch, M Korostynski, N Malek, M Slezak, M Zychowska. Full inhibition of spinal FAAH leads to TRPV1-mediated analgesic effects in neuropathic rats and possible lipoxygenase-mediated remodeling of anandamide metabolism. PLoS One, 2013
- H Watanabe, J Vriens, J Prenen, G Droogmans, T Voets, B Nilius. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature, 2003. [PubMed]
- WJ Redmond, L Gu, M Camo, P McIntyre, M Connor. Ligand determinants of fatty acid activation of the pronociceptive ion channel TRPA1. PeerJ, 2014
- M Patil, A Patwardhan, MM Salas, KM Hargreaves, AN Akopian. Cannabinoid receptor antagonists AM251 and AM630 activate TRPA1 in sensory neurons. Neuropharmacology, 2011. [PubMed]
- A Toth, PM Blumberg, J Boczan. Anandamide and the vanilloid receptor (TRPV1). Vitam Horm, 2009. [PubMed]
- NA Jeske, AM Patwardhan, N Gamper, TJ Price, AN Akopian, KM Hargreaves. Cannabinoid WIN 55,212-2 regulates TRPV1 phosphorylation in sensory neurons. J Biol Chem, 2006. [PubMed]
- N Hatano, Y Itoh, H Suzuki, Y Muraki, H Hayashi, K Onozaki. Hypoxia-inducible factor-1alpha (HIF1alpha) switches on transient receptor potential ankyrin repeat 1 (TRPA1) gene expression via a hypoxia response element-like motif to modulate cytokine release. J Biol Chem, 2012. [PubMed]
- MA Engel, I Izydorczyk, SM Mueller-Tribbensee, C Becker, MF Neurath, PW Reeh. Inhibitory CB1 and activating/desensitizing TRPV1-mediated cannabinoid actions on CGRP release in rodent skin. Neuropeptides, 2011. [PubMed]
- K Weller, PW Reeh, SK Sauer. TRPV1, TRPA1, and CB1 in the isolated vagus nerve – axonal chemosensitivity and control of neuropeptide release. Neuropeptides, 2011. [PubMed]
- Y Yang, H Yang, Z Wang, K Varadaraj, SS Kumari, S Mergler. Cannabinoid receptor 1 suppresses transient receptor potential vanilloid 1-induced inflammatory responses to corneal injury. Cell Signal, 2013. [PubMed]
- MY Kochukov, TA McNearney, Y Fu, KN Westlund. Thermosensitive TRP ion channels mediate cytosolic calcium response in human synoviocytes. Am J Physiol Cell Physiol, 2006
- A Engler, A Aeschlimann, BR Simmen, BA Michel, RE Gay, S Gay. Expression of transient receptor potential vanilloid 1 (TRPV1) in synovial fibroblasts from patients with osteoarthritis and rheumatoid arthritis. Biochem Biophys Res Commun, 2007. [PubMed]
- B Malinowska, M Baranowska-Kuczko, E Schlicker. Triphasic blood pressure responses to cannabinoids: do we understand the mechanism?. Br J Pharmacol, 2012. [PubMed]
- BL Kidd, S Cruwys, PI Mapp, DR Blake. Role of the sympathetic nervous system in chronic joint pain and inflammation. Ann Rheum Dis, 1992. [PubMed]
- JD Levine, MA Moskowitz, AI Basbaum. The contribution of neurogenic inflammation in experimental arthritis. J Immunol, 1985. [PubMed]
- JD Levine, SJ Dardick, MF Roizen, C Helms, AI Basbaum. Contribution of sensory afferents and sympathetic efferents to joint injury in experimental arthritis. J Neurosci, 1986. [PubMed]
- P Härle, G Pongratz, J Albrecht, IH Tarner, RH Straub. An early sympathetic nervous system influence exacerbates collagen-induced arthritis via CD4 + CD25+ cells. Arthritis Rheum, 2008. [PubMed]
- S Capellino, K Weber, M Gelder, P Härle, RH Straub. First appearance and location of catecholaminergic cells during experimental arthritis and elimination by chemical sympathectomy. Arthritis Rheum, 2012. [PubMed]
- Z Jenei-Lanzl, S Capellino, F Kees, M Fleck, T Lowin, RH Straub. Anti-inflammatory effects of cell-based therapy with tyrosine hydroxylase-positive catecholaminergic cells in experimental arthritis. Ann Rheum Dis, 2015. [PubMed]
- A Fassold, W Falk, S Anders, T Hirsch, VM Mirsky, RH Straub. Soluble neuropilin-2, a nerve repellent receptor, is increased in rheumatoid arthritis synovium and aggravates sympathetic fiber repulsion and arthritis. Arthritis Rheum, 2009. [PubMed]
- RN Spengler, RM Allen, DG Remick, RM Strieter, SL Kunkel. Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J Immunol, 1990. [PubMed]
- G Pongratz, RH Straub. Role of peripheral nerve fibres in acute and chronic inflammation in arthritis. Nat Rev Rheumatol, 2013. [PubMed]
- RH Straub, T Lowin, S Klatt, C Wolff, L Rauch. Increased density of sympathetic nerve fibers in metabolically activated fat tissue surrounding human synovium and mouse lymph nodes in arthritis. Arthritis Rheum, 2011. [PubMed]
- M Bashashati, Y Nasser, CM Keenan, W Ho, F Piscitelli, M Nalli. Inhibiting endocannabinoid biosynthesis: a novel approach to the treatment of constipation. Br J Pharmacol, 2015. [PubMed]
- MA Storr, M Bashashati, C Hirota, VK Vemuri, CM Keenan, M Duncan. Differential effects of CB(1) neutral antagonists and inverse agonists on gastrointestinal motility in mice. Neurogastroenterol Motil, 2010. [PubMed]
- A Rey, C Wolff, J Wildmann, A Randolf, A Hahnel, HO Besedovsky. Disrupted brain-immune system-joint communication during experimental arthritis. Arthritis Rheum, 2008. [PubMed]
- G Pongratz, RH Straub. The sympathetic nervous system in inflammation. Arthritis Res Ther, 2014. [PubMed]
- CL Lubahn, D Lorton, JA Schaller, SJ Sweeney, DL Bellinger. Targeting alpha- and beta-adrenergic receptors differentially shifts Th1, Th2, and inflammatory cytokine profiles in immune organs to attenuate adjuvant arthritis. Front Immunol, 2014. [PubMed]
- G Pongratz, M Melzer, RH Straub. The sympathetic nervous system stimulates anti-inflammatory B cells in collagen-type II-induced arthritis. Ann Rheum Dis, 2012. [PubMed]
- RH Straub, L Rauch, A Fassold, T Lowin, G Pongratz. Neuronally released sympathetic neurotransmitters stimulate splenic interferon-gamma secretion from T cells in early type II collagen-induced arthritis. Arthritis Rheum, 2008. [PubMed]
- D Lorton, C Lubahn, S Sweeney, A Major, CA Lindquist, J Schaller. Differences in the injury/sprouting response of splenic noradrenergic nerves in Lewis rats with adjuvant-induced arthritis compared with rats treated with 6-hydroxydopamine. Brain Behav Immun, 2009. [PubMed]
- RH Straub, L Rauch, L Rauh, G Pongratz. Sympathetic inhibition of IL-6, IFN-gamma, and KC/CXCL1 and sympathetic stimulation of TGF-beta in spleen of early arthritic mice. Brain Behav Immun, 2011. [PubMed]
- T Meinel, G Pongratz, L Rauch, RH Straub. Neuronal alpha1/2-adrenergic stimulation of IFN-gamma, IL-6, and CXCL-1 in murine spleen in late experimental arthritis. Brain Behav Immun, 2013. [PubMed]
- FA Russell, ES Fernandes, JP Courade, JE Keeble, SD Brain. Tumour necrosis factor alpha mediates transient receptor potential vanilloid 1-dependent bilateral thermal hyperalgesia with distinct peripheral roles of interleukin-1beta, protein kinase C and cyclooxygenase-2 signalling. Pain, 2009. [PubMed]
- HK Takahashi, S Mori, K Liu, H Wake, J Zhang, R Liu. Beta2-adrenoceptor stimulation inhibits advanced glycation end products-induced adhesion molecule expression and cytokine production in human peripheral blood mononuclear cells. Eur J Pharmacol, 2010. [PubMed]
- K Jin, L Xie, SH Kim, S Parmentier-Batteur, Y Sun, XO Mao. Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol Pharmacol, 2004. [PubMed]
- RH Straub, M Cutolo, F Buttgereit, G Pongratz. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med, 2010. [PubMed]
- R Sakthiswary, S Das. The effects of TNF alpha antagonist therapy on bone metabolism in rheumatoid arthritis: a systematic review. Curr Drug Targets, 2013. [PubMed]
- AI Idris, SH Ralston. Cannabinoids and bone: friend or foe?. Calcif Tissue Int, 2010. [PubMed]
- AI Idris, A Sophocleous, E Landao-Bassonga, M Canals, G Milligan, D Baker. Cannabinoid receptor type 1 protects against age-related osteoporosis by regulating osteoblast and adipocyte differentiation in marrow stromal cells. Cell Metab, 2009. [PubMed]
- F Matcham, L Rayner, S Steer, M Hotopf. The prevalence of depression in rheumatoid arthritis: a systematic review and meta-analysis. Rheumatology (Oxford), 2013. [PubMed]
- FA Moreira, JA Crippa. The psychiatric side-effects of rimonabant. Rev Bras Psiquiatr, 2009. [PubMed]
- CH Ashton, PB Moore. Endocannabinoid system dysfunction in mood and related disorders. Acta Psychiatr Scand, 2011. [PubMed]
- CG Ziegler, C Mohn, V Lamounier-Zepter, V Rettori, SR Bornstein, AW Krug. Expression and function of endocannabinoid receptors in the human adrenal cortex. Horm Metab Res, 2010. [PubMed]
- GR Chopda, VK Vemuri, R Sharma, GA Thakur, A Makriyannis, CA Paronis. Diuretic effects of cannabinoid agonists in mice. Eur J Pharmacol, 2013. [PubMed]
- RH Straub, L Paimela, R Peltomaa, J Scholmerich, M Leirisalo-Repo. Inadequately low serum levels of steroid hormones in relation to interleukin-6 and tumor necrosis factor in untreated patients with early rheumatoid arthritis and reactive arthritis. Arthritis Rheum, 2002. [PubMed]
- I Barna, D Zelena, AC Arszovszki, C Ledent. The role of endogenous cannabinoids in the hypothalamo-pituitary-adrenal axis regulation: in vivo and in vitro studies in CB1 receptor knockout mice. Life Sci, 2004. [PubMed]
- AB Ginsberg, NC Pecoraro, JP Warne, HF Horneman, MF Dallman. Rapid alteration of stress-induced hypothalamic-pituitary-adrenal hormone secretion in the rat: a comparison of glucocorticoids and cannabinoids. Stress, 2010. [PubMed]
- RJ McLaughlin, MN Hill, BB Gorzalka. Monoaminergic neurotransmission contributes to cannabinoid-induced activation of the hypothalamic-pituitary-adrenal axis. Eur J Pharmacol, 2009. [PubMed]
- TR Romero, LC Resende, LS Guzzo, ID Duarte. CB1 and CB2 cannabinoid receptor agonists induce peripheral antinociception by activation of the endogenous noradrenergic system. Anesth Analg, 2013. [PubMed]
- J Francis, SM MohanKumar, PS MohanKumar. Leptin inhibits norepinephrine efflux from the hypothalamus in vitro: role of gamma aminobutyric acid. Brain Res, 2004. [PubMed]
- MN Hill, JG Tasker. Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience, 2012. [PubMed]
- RS Goodwin, MH Baumann, DA Gorelick, E Schwilke, DM Schwope, WD Darwin. CB1 – cannabinoid receptor antagonist effects on cortisol in cannabis-dependent men. Am J Drug Alcohol Abuse, 2012. [PubMed]
- MA Gonzalez-Gay, C Gonzalez-Juanatey, TR Vazquez-Rodriguez, JA Miranda-Filloy, J Llorca. Insulin resistance in rheumatoid arthritis: the impact of the anti-TNF-alpha therapy. Ann N Y Acad Sci, 2010. [PubMed]
- VR da Cunha, CV Brenol, JC Brenol, RM Xavier. Rheumatoid arthritis and metabolic syndrome. Rev Bras Reumatol, 2011. [PubMed]
- H Tilg, AR Moschen. Inflammatory mechanisms in the regulation of insulin resistance. Mol Med, 2008. [PubMed]
- A Samat, B Tomlinson, S Taheri, GN Thomas. Rimonabant for the treatment of obesity. Recent Pat Cardiovasc Drug Discov, 2008. [PubMed]
- Z Pang, NN Wu, W Zhao, DC Chain, E Schaffer, X Zhang. The central cannabinoid CB1 receptor is required for diet-induced obesity and rimonabant’s antiobesity effects in mice. Obesity (Silver Spring), 2011. [PubMed]
- J Tam, G Godlewski, BJ Earley, L Zhou, T Jourdan, G Szanda. Role of adiponectin in the metabolic effects of cannabinoid type 1 receptor blockade in mice with diet-induced obesity. Am J Physiol Endocrinol Metab, 2014. [PubMed]
- 128.Coskun ZM, Bolkent S. Biochemical and immunohistochemical changes in delta-9-tetrahydrocannabinol-treated type 2 diabetic rats. Acta Histochem. 2014;116:112–6.
- CA Paronis, SP Nikas, VG Shukla, A Makriyannis. Delta(9)-Tetrahydrocannabinol acts as a partial agonist/antagonist in mice. Behav Pharmacol, 2012. [PubMed]
- P Harle, RH Straub. Leptin is a link between adipose tissue and inflammation. Ann N Y Acad Sci, 2006. [PubMed]
- S Muraoka, N Kusunoki, H Takahashi, K Tsuchiya, S Kawai. Leptin stimulates interleukin-6 production via janus kinase 2/signal transducer and activator of transcription 3 in rheumatoid synovial fibroblasts. Clin Exp Rheumatol, 2013. [PubMed]
- E Choy, K Ganeshalingam, AG Semb, Z Szekanecz, M Nurmohamed. Cardiovascular risk in rheumatoid arthritis: recent advances in the understanding of the pivotal role of inflammation, risk predictors and the impact of treatment. Rheumatology (Oxford), 2014. [PubMed]
- K Sugamura, S Sugiyama, Y Fujiwara, J Matsubara, E Akiyama, H Maeda. Cannabinoid 1 receptor blockade reduces atherosclerosis with enhances reverse cholesterol transport. J Atheroscler Thromb, 2010. [PubMed]
- M Duncan, MA Galic, A Wang, AP Chambers, DM McCafferty, DM McKay. Cannabinoid 1 receptors are critical for the innate immune response to TLR4 stimulation. Am J Physiol Regul Integr Comp Physiol, 2013. [PubMed]
- A Miranville, AW Herling, G Biemer-Daub, MD Voss. Reversal of inflammation-induced impairment of glucose uptake in adipocytes by direct effect of CB1 antagonism on adipose tissue macrophages. Obesity (Silver Spring), 2010. [PubMed]
- R Guabiraba, RC Russo, AM Coelho, MA Ferreira, GA Lopes, AK Gomes. Blockade of cannabinoid receptors reduces inflammation, leukocyte accumulation and neovascularization in a model of sponge-induced inflammatory angiogenesis. Inflamm Res, 2013. [PubMed]
- J Kraus. Expression and functions of mu-opioid receptors and cannabinoid receptors type 1 in T lymphocytes. Ann N Y Acad Sci, 2012. [PubMed]
- J Fichna, M Bawa, GA Thakur, R Tichkule, A Makriyannis, DM McCafferty. Cannabinoids alleviate experimentally induced intestinal inflammation by acting at central and peripheral receptors. PLoS One, 2014
- R Ribeiro, F Yu, J Wen, A Vana, Y Zhang. Therapeutic potential of a novel cannabinoid agent CB52 in the mouse model of experimental autoimmune encephalomyelitis. Neuroscience, 2013. [PubMed]
- NR Fuggle, FA Howe, RL Allen, N Sofat. New insights into the impact of neuro-inflammation in rheumatoid arthritis. Front Neurosci, 2014. [PubMed]
- GA Cabral, M Jamerson. Marijuana use and brain immune mechanisms. Int Rev Neurobiol, 2014. [PubMed]
- P Sanchez-Blazquez, M Rodriguez-Munoz, J Garzon. The cannabinoid receptor 1 associates with NMDA receptors to produce glutamatergic hypofunction: implications in psychosis and schizophrenia. Front Pharmacol, 2014. [PubMed]
- C Silvestri, V Di Marzo. Second generation CB1 receptor blockers and other inhibitors of peripheral endocannabinoid overactivity and the rationale of their use against metabolic disorders. Expert Opin Investig Drugs, 2012
- XH Yu, CQ Cao, G Martino, C Puma, A Morinville, S St-Onge. A peripherally restricted cannabinoid receptor agonist produces robust anti-nociceptive effects in rodent models of inflammatory and neuropathic pain. Pain, 2010. [PubMed]
- JD Salamone, PJ McLaughlin, K Sink, A Makriyannis, LA Parker. Cannabinoid CB1 receptor inverse agonists and neutral antagonists: effects on food intake, food-reinforced behavior and food aversions. Physiol Behav, 2007. [PubMed]
- J Bergman, MS Delatte, CA Paronis, K Vemuri, GA Thakur, A Makriyannis. Some effects of CB1 antagonists with inverse agonist and neutral biochemical properties. Physiol Behav, 2008. [PubMed]
- M Bouaboula, S Perrachon, L Milligan, X Canat, M Rinaldi-Carmona, M Portier. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. J Biol Chem, 1997. [PubMed]
- A Szabo, Z Helyes, K Sandor, A Bite, E Pinter, J Nemeth. Role of transient receptor potential vanilloid 1 receptors in adjuvant-induced chronic arthritis: in vivo study using gene-deficient mice. J Pharmacol Exp Ther, 2005. [PubMed]
- NJ Barton, DS McQueen, D Thomson, SD Gauldie, AW Wilson, DM Salter. Attenuation of experimental arthritis in TRPV1R knockout mice. Exp Mol Pathol, 2006. [PubMed]
- M Murai, F Tsuji, M Nose, I Seki, K Oki, C Setoguchi. SA13353 (1-[2-(1-Adamantyl)ethyl]-1-pentyl-3-[3-(4-pyridyl)propyl]urea) inhibits TNF-alpha production through the activation of capsaicin-sensitive afferent neurons mediated via transient receptor potential vanilloid 1 in vivo. Eur J Pharmacol, 2008. [PubMed]
- D Ursu, K Knopp, RE Beattie, B Liu, E Sher. Pungency of TRPV1 agonists is directly correlated with kinetics of receptor activation and lipophilicity. Eur J Pharmacol, 2010. [PubMed]
- JS Walczak, F Cervero. Local activation of cannabinoid CB(1) receptors in the urinary bladder reduces the inflammation-induced sensitization of bladder afferents. Mol Pain, 2011. [PubMed]
- SP Raychaudhuri, SK Raychaudhuri, KR Atkuri, LA Herzenberg, LA Herzenberg. Nerve growth factor: a key local regulator in the pathogenesis of inflammatory arthritis. Arthritis Rheum, 2011. [PubMed]
- ZY Wang, T McDowell, P Wang, R Alvarez, T Gomez, DE Bjorling. Activation of CB1 inhibits NGF-induced sensitization of TRPV1 in adult mouse afferent neurons. Neuroscience, 2014. [PubMed]
- A Boillat, O Alijevic, S Kellenberger. Calcium entry via TRPV1 but not ASICs induces neuropeptide release from sensory neurons. Mol Cell Neurosci, 2014. [PubMed]
- K Shoudai, JH Peters, SJ McDougall, JA Fawley, MC Andresen. Thermally active TRPV1 tonically drives central spontaneous glutamate release. J Neurosci, 2010. [PubMed]
- X Jia, H Zhang, X Cao, Y Yin, B Zhang. Activation of TRPV1 mediates thymic stromal lymphopoietin release via the Ca2+/NFAT pathway in airway epithelial cells. FEBS Lett, 2014. [PubMed]
- JM McPartland, M Glass, RG Pertwee. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol, 2007. [PubMed]
- PM Lam, J McDonald, DG Lambert. Characterization and comparison of recombinant human and rat TRPV1 receptors: effects of exo- and endocannabinoids. Br J Anaesth, 2005. [PubMed]
- P Urquhart, A Nicolaou, DF Woodward. Endocannabinoids and their oxygenation by cyclo-oxygenases, lipoxygenases and other oxygenases. Biochim Biophys Acta, 1851
- S Petrosino, MA Schiano, S Cerrato, M Fusco, A Puigdemont, L De Petrocellis. The anti-inflammatory mediator palmitoylethanolamide enhances the levels of 2-arachidonoyl-glycerol and potentiates its actions at TRPV1 cation channels. Br J Pharmacol, 2015
- D Navia-Paldanius, JR Savinainen, JT Laitinen. Biochemical and pharmacological characterization of human alpha/beta-hydrolase domain containing 6 (ABHD6) and 12 (ABHD12). J Lipid Res, 2012. [PubMed]
- TM Bland, RL Haining, TS Tracy, PS Callery. CYP2C-catalyzed delta9-tetrahydrocannabinol metabolism: kinetics, pharmacogenetics and interaction with phenytoin. Biochem Pharmacol, 2005. [PubMed]
- M Rinaldi-Carmona, F Barth, M Heaulme, D Shire, B Calandra, C Congy. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett, 1994. [PubMed]
- MA Bergstrom, EM Isin, N Castagnoli, CE Milne. Bioactivation pathways of the cannabinoid receptor 1 antagonist rimonabant. Drug Metab Dispos, 2011. [PubMed]