Plectranthus‐derived Abietanes as Protein Kinase C‐δ Activators: In Silico Design, Human Serum Albumin Interaction, and Stability Evaluation
Abstract
Cancer remains a major global health challenge. Among protein kinases (PKCs), PKC‐δ acts as a tumor suppressor in colon cancer and represents a valuable therapeutic target. Human serum albumin (HSA) is gaining attention as an efficient drug carrier, while Plectranthus spp. offers a rich source of bioactive compounds. One such molecule is 7α‐acetoxy‐6β‐hydroxyroyleanone (Roy, 1), a cytotoxic abietane diterpenoid with modifiable hydroxyl groups, making it a promising scaffold for drug development. This study aimed to design a theoretical library of Roy 1 derivatives targeting PKC‐δ. Hydroxyl groups at positions C6 and C12 were modified to explore interactions through molecular docking against the PKC‐δ regulatory domain (1PTR). Compound 16 emerged as the most promising candidate. Additionally, the binding of Roy 1 to HSA was evaluated by steady‐state fluorimetry, revealing moderate affinity near Trp‐214 and enhancing the thermal stability of the complex. Roy 1 exhibits excellent aqueous stability (0.1 mM, pH 7.4, 37°C), with similar results for two benzoylated derivatives (RoyBz and Roy12Bz), and no ester hydrolysis was detected. These findings highlight Roy 1’s potential as a stable, bioactive lead compound for developing PKC‐δ‐targeted therapeutics, with HSA as a suitable delivery vehicle.
Article type: Research Article
Keywords: abietane diterpene, antioxidant activity, HSA, molecular docking, PKC‐δ activation
Affiliations: CBIOS – Universidade Lusófona’s Research Center for Biosciences & Health Technologies Universidade Lusófona Lisboa Portugal; Faculty of Chemistry University of Belgrade Belgrade Serbia; Faculty of Agriculture University of Belgrade Belgrade Serbia; Department of Biotechnology, Chemistry and Pharmacy Università Degli Studi di Siena Siena Italy; Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy Universidade De Lisboa Lisboa Portugal
License: © 2025 The Author(s). Chemistry & Biodiversity published by Wiley‐VHCA AG. CC BY 4.0 This is an open access article under the terms of the http://creativecommons.org/licenses/by/4.0/ License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Article links: DOI: 10.1002/cbdv.202500670 | PubMed: 41061020 | PMC: PMC12716025
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (2.6 MB)
Introduction
Cancer stands as one of the most predominant causes of death worldwide [ref. 1]. The protein kinase (PKC) family plays a pivotal role in tumorigenesis and metastatic dissemination [ref. 2]. Clinical trials involving PKC modulators have generally demonstrated limited success, likely due to the complex and diverse biological functions of PKC isozymes. These functions can be both redundant and opposing, with expression levels varying significantly across different cancer types and isoforms displaying a high degree of structural similarity [ref. 3]. PKC‐δ has been widely characterized as a pro‐apoptotic and anti‐proliferative kinase [ref. 4, ref. 5]. However, PKC‐δ promotes therapy resistance in several tumor models, including lung, pancreatic, and liver cancers [ref. 2].
Human serum albumin (HSA) has emerged as a versatile and invaluable drug carrier with significant therapeutic potential. It is a soluble and monomeric, multi‐domain macromolecule abundantly present in human blood plasma, where it plays a key role in pH buffering and maintaining oncotic pressure [ref. 6]. HSA functions as the primary transporter of a wide range of endogenous and exogenous compounds. Its structural features, including hydrophobic binding pockets, a free thiol group, and surface‐exposed termini, make it particularly well‐suited for the targeted delivery of diverse therapeutic agents. HSA has shown promise as a carrier for drugs used in the diagnosis and treatment of various conditions, including carcinomas, hyperglycemia, infectious diseases, and rheumatoid arthritis. HSA can facilitate drug accumulation in tumors or inflamed tissues through receptor‐mediated transport mechanisms. Furthermore, HSA exhibits notable antioxidant properties that can be leveraged to improve therapeutic efficacy and reduce drug‐induced toxicity [ref. 7, ref. 8]. In addition, HSA plays an essential role in enhancing the circulation and bioavailability of anticancer drugs within the bloodstream [ref. 9].
Plants represent a valuable source of novel biologically active compounds with significant potential for promoting human health and treating various diseases [ref. 10]. The genus Plectranthus L’Hér., commonly referred to as spurflowers, belongs to the Lamiaceae family and comprises more than 300 species, widely distributed across the Old World tropics and subtropical regions [ref. 11]. Many Plectranthus species have been traditionally used in folk medicine to manage a range of conditions, including neurological and blood complaints, infections and fevers, pain, and inflammation [ref. 12]. Plectranthus is particularly rich in bioactive compounds, such as abietane‐type terpenoids. Abietanes have demonstrated a broad spectrum of biological activities, including antibacterial, antitumoral, anti‐inflammatory, antidiabetic, and antioxidant effects. Structurally, Abietane diterpenoids are characterized by a tricyclic abietane carbon skeleton substituted with various functional groups [ref. 13]. The abietane diterpenoid 7α‐acetoxy‐6β‐hydroxyroyleanone (Roy, 1, Figure 1) isolated from P. grandidentatus Gürke has exhibited notable anti‐proliferative activity against several cancer cell lines [ref. 14, ref. 15].
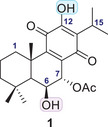
The cytotoxic potential of bioactive compounds can be effectively assessed using in silico approaches. Due to its speed and cost‐effectiveness, molecular docking serves as a valuable alternative for high‐throughput screening of potential drug candidates [ref. 16]. In the present study, molecular docking and molecular dynamics (MD) were employed to investigate the impact of structural modifications on the hydroxyl groups of royleanone 1, with a focus on its ability to activate PKC‐δ. A recent in silico analysis highlighted the potential of Roy 1 to interact with PKC isoforms, suggesting that subtle modifications in its substitution pattern could enhance isoform‐specific binding, particularly toward PKC‐δ [ref. 17]. In light of earlier findings, the derivative 7α‐acetoxy‐6β‐benzoyloxy‐12‐O‐benzoylroyleanone (RoyBz) has emerged as the first selective PKC‐δ activator. RoyBz binds specifically to the PKCδ‐C1‐domain and has been shown to inhibit the proliferation of colon cancer cells by inducing apoptosis [ref. 18]. Additionally, RoyBz shows the ability to inhibit both mitochondrial respiration and glycolysis, thereby reinforcing its potential as an antitumor agent in colon cancer therapy [ref. 19]. Extending this approach, the present work aims to investigate novel royleanone‐based derivatives capable of modulating PKC‐δ for the treatment of colon cancer. Additionally, the interaction between Roy 1 and HSA was evaluated in vitro, alongside an assessment of its aqueous stability, to support future pharmaceutical formulation strategies.
Results and Discussion
Molecular Docking on PKCδ‐C1‐domains
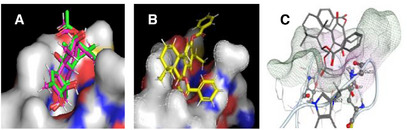
In this study, molecular docking was employed as a key tool to support the discovery of novel royleanone derivatives with potential therapeutic application in colon cancer, specifically through PKC‐δ modulation. Knowledge of the ligand‐binding site prior to docking significantly enhances the efficiency and accuracy of the docking process [ref. 20]. In this case, the binding site was well‐characterized, as the selected crystallographic structure of the PKC‐δ‐C1 domain (PDB ID:1PTR) from the protein data bank [ref. 21] was co‐crystallized with phorbol‐13‐acetate (PA), an endogenous activator of PKC. An initial comparative docking study was conducted using three widely used docking programs: FRED, GOLD, and AutoDock. The accuracy and reliability of each program were assessed by comparing the predicted ligand‐binding poses to two benchmarks: the crystallographic pose of PA [ref. 21] and the reported docking results of the known PKC‐δ activator RoyBzBz [ref. 18].
Among the evaluated tools, FRED software [ref. 22] demonstrated superior performance by accurately reproducing the binding pose of the crystallized PA molecule (Figure 2A), as well as the previously reported docking configuration of RoyBz [ref. 18] (Figure 2B,C). While GOLD and AutoDock are both well‐established molecular docking tools, they were not selected due to their lower accuracy in reproducing the crystallographic pose of PA. GOLD showed deviations in ligand orientation, while AutoDock produced inconsistent results across runs. In contrast, FRED consistently aligned with experimental data. As a result, FRED was selected as the primary docking platform for the subsequent screening studies.
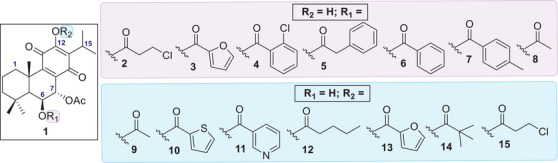
Libraries of theoretical compounds were constructed for docking screening with the dual objectives of increasing structural diversity and exploring the reactive hydroxyl groups of compound 1 (highlighted in Figure 1) for potential esterification. Nineteen substituents (S1–S19, Table 1) were selected to generate novel esters, drawing inspiration from literature‐reported bioactive moieties. The selection of ester types followed a rational design strategy aimed at covering a broad range of physicochemical and steric properties, including lipophilicity, electronic effects (e.g., π‐interactions), flexibility, and steric hindrance. This diverse set of substituents (Table 1) was intended to identify favorable interactions within the PKC‐δ binding site and guide the design of derivatives with enhanced specificity and activity.
TABLE 1: Summary of ester substituents (S1–S19) used for Roy 1 derivatization.
| Code | Substituent type | Representative group | Design rationale | Code | Substituent type | Representative group | Design rationale |
|---|---|---|---|---|---|---|---|
| S1 | Simple acyl | Baseline lipophilicity and minimal steric hindrance | S13‐S14 | Spacer group | Conformational flexibility and extended reach | ||
| S2 | Aliphatic chain | Flexibility and increased chain length | S15 | Bulky aliphatic | High steric hindrance and hydrophobicity | ||
| S3 | Aromatic benzoyl | Enhanced rigidity and π‐stacking potential and aromaticity | S16 | Bulky aromatic | π‐system extension and rigidity | ||
| S4‐S5 | Electron‐donating | Increased electron density and π‐donor properties | S17‐S18 | Heterocyclic | π‐interactions, polar interactions, and hydrogen bonding | ||
| S6 | Electron‐withdrawing | Flexibility, strong lipophilicity, and electron deficiency | S19 | Hetero‐aromatic | Rigid, planar moiety and hydrogen bonding | ||
| S7‐S12 | Electron‐withdrawing | Strongly lipophilic and polar and π‐acceptor properties | |||||
Molecular docking screening revealed several promising candidates with higher predicted binding affinities than the known selective PKC‐δ activator RoyBz. When both hydroxyl positions were derivatized simultaneously, only one compound, the previously reported 7α,6β‐diacetoxy‐12‐O‐acetylroyleanone [ref. 23], displayed favorable results. This finding suggests that maintaining at least one free hydroxyl group may be important for binding affinity. To systematically assess the impact of structural modifications, each hydroxyl position was modified independently, while the other remained unaltered. As shown in Figure 3, theoretical derivative 2 emerged as the most promising compound. Overall, esterification at the C6‐OH position resulted in better docking scores than modifications at C12, suggesting a higher potential for PKC‐δ activation through derivatization at C6. The C6 position appears to tolerate a wide range of substituents, including linear chains, heterocyclic rings, and substituted or unsubstituted benzene rings (compounds 2 ‐ 8, highlighted in purple in Figure 3). In contrast, derivatives at C12 showed a preference for aliphatic chains or aromatic rings with heteroatoms (analogs 9–15, highlighted in blue in Figure 3).
To address a major limitation of this compound, its poor water solubility [ref. 24, ref. 25], a new strategy involving amino acids was explored. These organic molecules, which feature both an amino group (‐NH2) and a carboxylic acid group (‐COOH), along with distinct side chains, offer varying degrees of hydrophilicity. For instance, amino acids such as lysine and arginine have positively charged side chains, while serine contains a hydroxyl group, all of which contribute to enhanced water solubility [ref. 26, ref. 27]. Previous studies have demonstrated that conjugating bioactive molecules with amino acids can improve both their pharmacological properties and solubility [ref. 26, ref. 28]. Likewise, the introduction of an amino acid was investigated to improve water solubility. Based on this rationale, a new library of theoretical derivatives was developed using the top‐scoring compound (theoretical derivative 2) as the template. In this new series, amino acids were introduced at position C12, including proline, glycine, alanine, tryptophan, and aspartate. Among these, the glycine‐containing derivative (compound 16, Figure 4a) showed the most favorable interactions, forming three hydrogen bonds with PKC‐δ. These involved the acetoxy group at C‐17 and carbonyl at C‐14 interacting with Gln257 and the carbonyl of the propionic side chain (C‐21) forming a bond with Gly253 (Figure 4b).
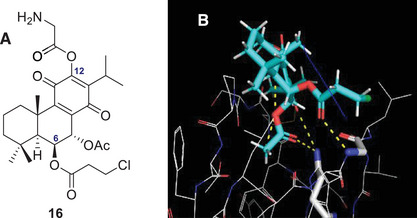
These three hydrogen bonds are also present in other derivatives, including compound 2. However, the glycine derivative (compound 16) forms an additional hydrogen bond through its amine group with Met238, potentially enhancing its binding affinity. Notably, compound 16 appears to exhibit a stronger predicted affinity for PKC‐δ than the other tested derivatives, although its isoform selectivity has not yet been determined. These findings provide important insights that can guide the selection of promising candidates for future synthesis and biological evaluation.
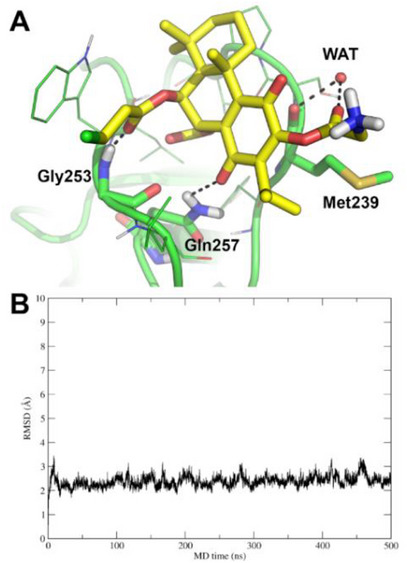
Molecular Dynamics Simulations of Compound 16
To confirm the docking pose of 16, 500 ns of unrestrained MD simulations were run in explicit solvent. The MD trajectory was then analysed for the binding mode of 16 through frames cluster analysis and calculation of the root‐mean square deviation (RMSD). Results unequivocally confirm that the binding mode of 16, such as predicted by molecular docking, is highly stable in the MD simulation. Indeed, the most populated cluster of frames is composed of 94.9% of MD frames, whose representative structure is shown in Figure 5A. Overall, 16 interacts with PKC‐δ with a pose that is highly similar to that of the crystallographic ligand PA [ref. 29] by direct H‐bonds to Gly253 and Gln257, as well as a water‐bridged H‐bond to the backbone of Met239. In addition to cluster analysis, the conformational stability of the PKC‐δ/16 complex was assessed by RMSD calculation (Figure 5B).
Fluorimetric Analysis of the HSA/Roy Interactions
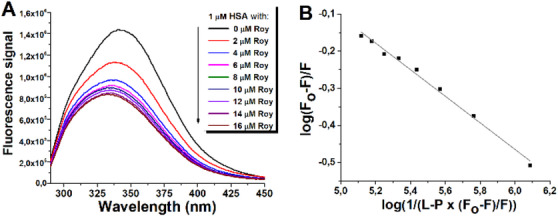
Given the importance of HSA for the delivery of anticancer drugs through the bloodstream [ref. 9], interactions between 1 and this protein were studied in vitro under simulated physiological conditions. Ligand binding often induces quenching of intrinsic protein fluorescence. HSA has only one Trp residue (Trp‐214 at subdomain IIA) and 18 Tyr residues [ref. 30]. The interactions between HSA and compound 1 caused the concentration‐dependent reduction of the intrinsic fluorescence of protein, followed by a blue shift of the emission maximum from 342 to 333 nm (Figure 6A). Using the quenching data (Figure 6B), a binding constant for the HSA/1 complex was determined to be 6.5 × 104 M−1 at 37°C. The resulting association constant was of moderate strength, similar to most anticancer drugs that have been shown to bind to HSA, mainly to subdomain II [ref. 31].
Synchronous fluorescence is a valuable tool for separating the contribution of Trp and Tyr residues to total protein intrinsic fluorescence. The fluorescence of HSA with Δλ (Δλ = λem—λex) of 60 and 15 nm is characteristic of Try and Tyr, respectively [ref. 32]. From Figures 6A and 6B, it was evident that the binding of compound 1 induced greater Trp residue fluorescence quenching compared to the decrease in fluorescence arising from Tyr residues, with no significant shifts of emission maxima, indicating that bound Roy 1 was situated closer to Trp residues. Therefore, the observed blue shift seen in the HSA fluorescence spectrum (Figure 7A) is due to preferential quenching of the Trp‐214 residues by royleanone 1 binding. In contrast, the relative contribution of Tyr residues to protein fluorescence increases upon ligand binding (Figure 7B).
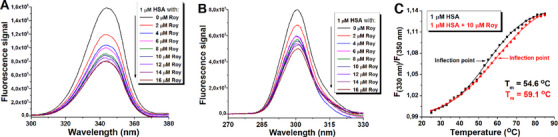
Experiments comparing HSA and HSA/1 complex properties have shown a difference in their thermal stability. The calculated melting point was higher (on average 4.5°C or 8.2%) in the complex than in the protein alone (Figure 7C). This result indicated that Roy 1 binding stabilized HSA.
Taken together, these results point to moderate affinity binding of compound 1 to human serum albumin (HSA) at subdomain IIA (Sudlow site I), increasing the protein’s thermal stability. This suggests that HSA could be a suitable carrier protein for transporting Roy 1 through systemic circulation.
In Vitro Antioxidant Capacity
The antioxidant capacity of Roy 1 was evaluated alongside antioxidant standards, Vitamin C, and Trolox, using six different assays: 2,2‐diphenyl‐1‐picrylhydrazyl (DPPH), 2,2′‐azino‐bis(3‐ethylbenzothiazoline‐6‐sulfonic acid) (ABTS), nitric oxide (NO), total antioxidant capacity assay (TAOC), oxygen radical absorbance capacity (ORAC), and hydroxyl radical antioxidant capacity (HORAC). DPPH, ABTS, and NO are spectrophotometric assays that measure the ability of compounds to quench DPPH stable radical, in situ formed ABTS cation radical, and ●NO radical, respectively. TAOC is a spectrophotometric redox‐based assay that assesses the total antioxidant capacity through metal ion reduction. For consistency, antioxidant activity in these assays is expressed as half‐maximal inhibitory concentration (IC50) values for DPPH, ABTS, and NO assays and as EC50 values for the TAOC test. HORAC and ORAC are fluorometric assays that evaluate the capacity of compounds to scavenge hydroxyl and peroxyl radicals, respectively, with results expressed as Trolox equivalents (TEs). As shown in Table 2, Roy 1 exhibited low antioxidant capacity (about 7–10 times less than selected standard antioxidants), showing modest activity only in the ●NO scavenging assay compared to Vitamin C.
TABLE 2: In vitro antioxidant capacity of Roy 1, Vitamin C, and Trolox.
| Parameter (Units) | IC50 (mM) | EC50 (mM) | (TE) | (TE) | ||
|---|---|---|---|---|---|---|
| Test | DPPH | ABTS | NO | TAOC | ORAC | HORAC |
| Roy 1 | >1.5 | ∼1.3 | 0.800 | >2.0 | 0.178 | 0.142 |
| Vitamin Ccbdv70537-tbl2-note-0001 | 0.275 | 0.240 | 0.702 | 0.234 | 0.895 | 0.911 |
| Troloxcbdv70537-tbl2-note-0001 | 0.211 | 0.143 | 0.160 | >1.25 | 1 | 1 |
In DMSO (as for all other samples), numerical data represent means from two independent experiments performed in duplicate (standard deviations were below 7%).
Evaluation of Roy 1 Stability in Aqueous Medium
To support future pharmaceutical formulation studies using Roy 1 as a lead compound, its stability in aqueous media was investigated. Stability testing was conducted in phosphate‐buffered saline (PBS, pH 7.4) at 37°C, a physiologically relevant environment [ref. 33, ref. 34]. As previously reported for other royleanones [ref. 25, ref. 35], Roy 1 has poor water solubility and thus limited solubility in PBS.[ref. 24] To address this, co‐solvents were employed. Acetonitrile (ACN, 17%) [ref. 36] and dimethyl sulfoxide (DMSO, 10%) [ref. 34, ref. 37] were used to enhance solubility. Stock solutions of Roy 1 were prepared at 0.1 mM in PBS/ACN (17%), 0.4 mM in PBS/ACN (17%), and 0.4 mM in PBS/DMSO (10%). These were incubated at 37°C for periods ranging from 10 to 40 days and monitored over time. The concentration of Roy 1 was determined by high‐performance liquid chromatography‐diode array detector (HPLC‐DAD) at a detection wavelength of 270 nm using a calibration curve. Regression analysis showed good linearity (r2 = 0.9960). The retention time (Rt), regression equation, correlation coefficient (r2), limit of detection (LOD), and limit of quantification (LOQ) are summarized in Table 3.
TABLE 3: Parameters of calibration curve for compound 1.
| Rt (min) | Regression equation | Correlation coefficient (r2) | LOD (µg/mL) | LOQ (µg/mL) |
|---|---|---|---|---|
| 5.6 | y = 25793x + 668.36 | 0.9960 | 6.6 | 19.9 |
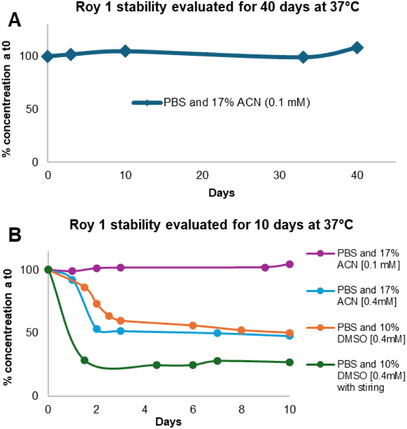
The results indicate that compound 1 remains fully stable in aqueous medium at a concentration of 0.1 mM, pH 7.4, and 37°C, with no degradation detected over a 40‐day period (Figure 8A). However, at a higher concentration (0.4 mM), a noticeable reduction in the peak corresponding to compound 1 was observed as early as day 2 of the experiment (Figure 8B). This effect occurred in both solvent systems (PBS/ACN and PBS/DMSO) but was more pronounced in PBS/DMSO, particularly under stirring conditions. Importantly, no additional peaks were detected in any chromatograms, suggesting compound 1 underwent precipitation rather than degradation. This precipitation tendency, especially at elevated concentrations, is consistent with the compound’s low water solubility. At 0.1 mM in PBS/ACN, approximately 16% of Roy 1 precipitated, with minimal variation over time. In contrast, at 0.4 mM in PBS/ACN, precipitation increased from 35% on day 1% to 66% by day 2. The collected precipitate was reinjected into the HPLC, confirming the exclusive presence of compound 1. These findings support the conclusion that Roy 1 is chemically stable under the tested conditions and that its apparent concentration loss is due to precipitation, not decomposition, across the tested range (0.1–0.4 mM).
Pires et al. highlighted that ester groups are generally prone to hydrolysis under various conditions [ref. 38]. Therefore, the stability of two bioactive ester derivatives, RoyBz and the 12‐benzoyl derivative 7α‐acetoxy‐6β‐hydroxy‐12‐O‐benzoylroyleanone (Roy12Bz), [ref. 39, ref. 40] was evaluated as representative examples. As previously demonstrated, Roy 1 showed poor solubility in PBS solutions, even with the use of co‐solvents. Similarly, RoyBz and Roy12Bz were insoluble in water and only moderately soluble in DMSO.
Stock solutions of each compound were prepared at 0.1 mM in DMSO/PBS. RoyBz formed a yellow solution with needle‐like crystals, while Roy12Bz resulted in a suspension. Both solutions were incubated at pH 7.4 and 37°C for 10 days. The resulting precipitates were collected, redissolved in methanol, and analyzed by HPLC. Each compound was identified by its retention time and UV spectrum. The chromatograms showed a single peak for each derivative, corresponding to the intact RoyBz and Roy12Bz compounds. These results confirm their stability under the tested conditions. Notably, Roy12Bz contains an ester group at C12, and RoyBz has esters at both C6 and C12, positions typically prone to hydrolysis. However, no ester cleavage was detected in either case. These findings support the feasibility of synthesizing new ester derivatives based on Roy 1 for therapeutic development.
Conclusions
This study explored the potential of novel theoretical royleanone derivatives as modulators of PKC‐δ, with a focus on applications in colon cancer therapy. Roy 1 served as a lead compound for structure‐based design, with modifications targeting the hydroxyl groups at positions C12 and C6. Docking studies revealed that aliphatic substitutions at C12 are favorable for PKC‐δ activation, while C6 can accommodate a broader range of functional groups. Among the tested derivatives, compound 16 showed the most promising binding profile. Overall, in silico screening identified several candidates worthy of further synthesis.
The interaction between Roy 1 and HSA was evaluated through steady‐state fluorimetric analysis. Results showed moderate binding affinity, with the main binding site located near the Trp‐214 residue in subdomain IIA (Sudlow site I). Furthermore, thermal denaturation studies revealed a significant increase in the melting temperature of the HSA/Roy complex compared to HSA alone, suggesting that Roy binding stabilizes the protein structure. These findings support the potential of HSA as a carrier for Roy 1 in systemic circulation.
Regarding compound stability, Roy 1 demonstrated full stability in aqueous medium at 0.1 mM, pH 7.4, and 37°C over a 40‐day period. Its stability appeared largely independent of concentration (0.1–0.4 mM). Similarly, the derivatives RoyBz and Roy12Bz remained intact, with no signs of ester cleavage under the same conditions. These findings reinforce the potential of Roy 1 as a robust lead molecule for the development of novel royleanone‐based therapeutics with enhanced biological activity.
Experimental
General
Essentially fatty acid‐free HSA, type A1887, was purchased from Sigma‐Aldrich (USA) and used without further purification. HSA concentration was determined using an extinction coefficient of 35 700 M−1 cm−1 at 280 nm. Roy 1 was dissolved in DMSO. All HSA‐related measurements were duplicated in 50 mM phosphate buffer, pH 7.4. Final concentrations of DMSO in HSA‐1 mixtures did not exceed 1% (v/v).
Plant Materials
The plant material, P. grandidentatus Gürke, was cultivated in Parque Botânico da Tapada da Ajuda (Instituto Superior Agrário, Lisbon, Portugal) from cuttings obtained from the Kirstenbosch National Botanical Garden (Cape Town, South Africa). Voucher specimens (572/2008) were deposited in Herbarium João de Carvalho e Vasconcellos (ISA). The plant name has been checked using https://wfoplantlist.org/ [ref. 41].
Extraction and Isolation
The extraction and isolation process was adapted from Isca, V.M.S. et al. [ref. 23]. The leaves and stems of P. grandidentatus were air‐dried, ground into powder, and extracted with acetone in ultrasound equipment (Sonorex Super RK 510 H; Bandelin, Berlin, Germany). The isolation was performed by dry flash chromatography, using silica gel (Merck 9385) as the stationary phase and mixtures of Hex: AcOEt and AcOEt: MeOH as eluents. Roy1 was obtained from recrystallization from Hex.
Molecular Docking of Theoretical Derivatives on PKC‐δ
The crystallographic structure of protein kinase delta CYS2 complexed with PA (1PTR.pdb from the protein data bank [ref. 21]) was used to perform the redocking of the known activator PA. The possible binding mode of RoyBz, reported as a PKC‐δ selective activator [ref. 18] was also assessed by molecular docking. FRED, Gold, and AutoDock programs were tested. FRED software [ref. 22] was selected to perform the docking studies. The binding pocket was defined according to Bessa et al. [ref. 18] Software Vida [ref. 42] was used to draw the structures’ databases. Docking poses were visually inspected using Pymol software [ref. 43].
Molecular Dynamics Simulations
MD simulations were run with AMBER18, using the ff14SB force field for the protein, and the general Amber force field (GAFF) for the small molecule 16 [ref. 44, ref. 45, ref. 46]. The docking pose was solvated in a rectilinear box of TIP3P‐type explicit water molecules, buffering 15 Å from the macromolecular system, for a total of 6177 water molecules. The total charge of the system was neutralized by Cl‐ ions. The time step was 2 fs. According to a well‐established MD protocol [ref. 47, ref. 48, ref. 49, ref. 50, ref. 51], the initial system was first submitted to an energy minimization of the solvent only, while keeping the solute frozen, for 1000 steps with the steepest descent algorithm (SD), followed by 4000 steps with the conjugate gradient algorithm (CG). Then, the solvated solute was energy minimized for 1000 steps with the SD, followed by 9000 steps with the CG, before being heated to 300 K for 2 ns at constant volume using the Langevin thermostat. Density was equilibrated for 2 ns at constant pressure using the Berendsen barostat. A preliminary MD of 50 ns was run before the final production of MD trajectories lasting 500 ns at constant pressure. In all the steps above, no restraints were used. MD trajectory analysis was carried out with the CPPTRAJ software [ref. 52].
Fluorescence Spectroscopy Measurements
All fluorescence measurements were done on a FluoroMax‐4 spectrofluorometer (HORIBA Scientific, Japan) under temperature‐controlled conditions (Peltier control system), with the width of the excitation and emission slit both adjusted to 5 nm and with 1 cm path length cells. The fluorescence spectra were recorded under thermostated conditions (37°C). HSA concentration was kept constant (1 µM) while the Roy 1 concentration varied from 0–14 µM.
For intrinsic fluorescence experiments, the excitation wavelength was 280 nm, and emission spectra were recorded between 290 and 450 nm. The change in fluorescence emission intensity was measured within 1 min of adding each aliquot of ligand to the protein solution. The emission of Roy 1 solutions without HSA was subtracted to correct background fluorescence. Fluorescence intensities were corrected for inner filter effects according to the equation [ref. 53]:
where Fc is corrected fluorescence, F0 is measured fluorescence, and Aex and Aem are the absorbances of Roy at excitation and peak emission wavelength, respectively.
The association (binding) constant for the HSA‐Roy complex was calculated using the equation [ref. 54]:
where [P] and [L] are the total concentrations of protein (HSA) and ligand (1), respectively, K a is the binding constant, and n is the Hill coefficient (indication of the number of binding sites on the protein).
Synchronous fluorescence spectra of the HSA‐1 complex, with emissions in the range of 290–400 nm, were obtained at two different scanning intervals: Δλ = 15 nm, tyrosine (Tyr) residues excitation, and Δλ = 60 nm, tryptophan (Trp) residues excitation, where Δλ = Δλem—Δλex.
The thermal stability of HSA alone (1 µM) or in the presence of Roy 1 (10 µM) was studied in the temperature range from 25 to 85°C by measuring protein intrinsic fluorescence reduction. The rate of temperature increase was 2°C/min, with the equilibration time set to 1 min. The results were expressed as the change of the (fluorescence) ratio F330/F350 with the temperature and fitted with a sigmoidal function. The inflection point in the plot was taken as the melting point (Tm) of the protein [ref. 55].
Antioxidant Capacity
The antioxidant capacity of Roy 1 was evaluated in six antioxidant assays: DPPH, ABTS, NO, TAOC, ORAC, and HORAC (Table 2) and compared with two standard antioxidants, Trolox (6‐hydroxy‐2,5,7,8‐tetramethylchroman‐2‐carboxylic acid) and Vitamin C (ascorbic acid). ABTS, DPPH, NO, and TAOC are spectrophotometric, while HORAC and ORAC are fluorometric assays. The percentage inhibition in ABTS, DPPH, and NO assays was calculated using the following equation:
where Ai is the initial concentration of the chromophore and As is the absorbance of the remaining concentration of chromophore in the presence of analyzed compounds. To compare the radical scavenging activity of Roy 1 with Trolox and Vitamin C in ABTS, DPPH, and NO assay, IC50 values were calculated from the graph of scavenging activity against the concentrations (from 50 µM to 2 mM) of the investigated compounds. The IC50 value is the sample concentration required for a 50% free radical scavenging inhibition activity.
In the TAOC test, antioxidant capacity was expressed as the EC50 value, which represents the concentration of the investigated compound that results in an absorbance of 0.500. An increase in absorbance is measured after mixing the solution of the studied compound with a mixture of (NH4)6Mo7O24, Na3PO4, and H2SO4.
The results of HORAC and ORAC assays are expressed as Trolox equivalents (TE). TE values are obtained from the equation: AUCS/AUCT, where AUCs = AUC(sample)—AUC(buffer blank) and AUCT = AUC(Trolox)—AUC(buffer blank) (AUC means the net area under the curve). In the case of both assays, reaction mixtures containing standards and fluorescein were used as substrates.
The principles of these assays (Table 4) may be found in the review book chapter written by Stengel et al. [ref. 56], while detailed experimental procedures are given in our recent papers [ref. 57, ref. 58].
TABLE 4: Antioxidant assays used in this study.
| Assay abbreviation | Full name of the antioxidant assay | Radical | Radical generation method/Mechanism of action |
|---|---|---|---|
| DPPH | (2,2‐diphenyl‐1‐picrylhydrazyl/DPPH) radical scavenging assay | DPPH● | — |
| ABTS | (2,2′‐azino‐bis[3‐ethylbenzothiazoline‐6‐sulfonic acid]/ABTS) radical scavenging assay | ABTS+● | The reaction of ABTS with K2S2O8 in water |
| NO | Nitric oxide scavenging | ●NO | Incubation of Na2[Fe(CN)6] solution in light |
| HORAC | Hydroxyl radical antioxidant capacity | ●OH | H2O2/CoF2/picolinic acid |
| ORAC | Oxygen radical absorbance capacity | ●OOH | Thermal homolysis of 2,2’‐azobis (2‐amidino‐propane) dihydrochloride (AAPH) |
| TAOC | Total antioxidant capacity assay | − | The reduction of Mo(VI) to Mo(V), with the subsequent formation of a stable blue‐green phosphate Mo(V)complex at acidic pH. |
Evaluation of Roy Stability in Aqueous Medium
Stock solutions of Roy 1 (0.4 mM) were prepared in PBS (pH 7.4) containing 10% of DMSO (9 mL PBS to 1 mL DMSO), adapted from Mak et al., 2017 [ref. 37]. Solutions were incubated at 37°C for 10 days. One of the stock solutions (PBS/DMSO) was kept under constant stirring during the assay. Stock solutions of compound 1 (0.1 mM and 0.4 mM) were prepared in PBS buffer (pH 7.4), containing 17% acetonitrile (ACN) (20 mL PBS to 4 mL ACN) [ref. 59]. Solutions were incubated at 37°C for 40 and 10 days, respectively. At different times, several aliquots of the reaction mixtures were taken and analyzed by HPLC‐DAD at 270 nm wavelength. The compounds RoyBz and the 12‐benzoyl derivative Roy12Bz were previously synthesized [ref. 39, ref. 40]. Stock solutions of RoyBz and Roy12Bz (0.1 mM) were also tested in PBS (pH 7.4) and 10% of DMSO at 37°C for 10 days. The quantification was carried out in an Agilent Technologies 1200 Infinity Series system with a diode array detector (DAD; Agilent, Santa Clara, CA, USA) equipped with an Eclipse XDB‐C18 (250 x 4.0 mm i.d., 5 µm) column, from Merck and ChemStation Software (Hewlett‐Packard, Palo Alto, CA, USA). Each sample was analyzed (after 20 µL injection), and a gradient elution mixture composed of solution A (methanol), solution B (acetonitrile), and solution C (0.3% trifluoroacetic acid in water) was used as follows: 0 min, 15% A, 5% B, and 80% C; 2 min, 70% A, 30% B, and 0% C; 10 min, 70% A, 30% B, and 0% C; and 15 min, 15% A, 5% B, and 80% C. The flow rate was set at 1 mL.min−1. The column was maintained at 29°C. The analysis time was 15 min, including the stabilization of the RP‐18 column. A calibration curve with acceptable validation parameters was constructed. All samples were filtered before HPLC analysis. Assay conducted in triplicate.
Author Contributions
Patricia Rijo and Mattia Mori conceived the experiments; Vera M. S. Isca and Milan Nikolić conducted the experiments; and Vera M. S. Isca and Nenad Filipović analyzed the results. All authors reviewed the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1 F. Bray , J. Ferlay , I. Soerjomataram , R. L. Siegel , L. A. Torre , and A. Jemal CA: A Cancer Journal for Clinicians 68 (2018): 394–424.30207593
- 2 D. Matias , C. Bessa , M. Fátima Simões , C. P. Reis , L. Saraiva , and P. Rijo , “Natural Products as Lead Protein Kinase C Modulators for Cancer Therapy,” in Studies in Natural Products Chemistry, ed. A. Rahma (Elsevier, 2016): 45–79.
- Wealth of Opportunity—The C1 Domain as a Target for Drug Development,”. Current Drug Targets, 2008. [PubMed]
- Protein Kinase C and Cancer: What We Know and What We Do Not,”. Oncogene, 2014. [PubMed]
- Protein Kinase C (PKC) Isozymes and Cancer,”. New Journal of Science, 2014
- 6 T. Peters , Genetics and Medical Applications (Academic Press, 1996).
- Albumin: An Emerging Opportunity in Drug Delivery,”. Biotechnology and Bioprocess Engineering, 2020
- Unraveling the Versatility of Human Serum Albumin—A Comprehensive Review of Its Biological Significance and Therapeutic Potential,”. Current Research in Structural Biology, 2023. [PubMed]
- Understanding the Interaction between Human Serum Albumin and Anti‐Bacterial/ Anti‐Cancer Compounds,”. Current Pharmaceutical Design, 2015. [PubMed]
- A Comprehensive Review on the Biological, Agricultural and Pharmaceutical Properties of Secondary Metabolites Based‐Plant Origin,”. International Journal of Molecular Sciences, 2023. [DOI | PubMed]
- Plectranthus: A Plant for the Future?”. South African Journal of Botany, 2011
- Plectranthus: A Review of Ethnobotanical Uses,”. Journal of Ethnopharmacology, 2006. [PubMed]
- Reactivity of Diterpenoid Quinones: Royleanones,”. Current Pharmaceutical Design, 2016. [PubMed]
- Insight the Biological Activities of Selected Abietane Diterpenes Isolated From Plectranthus spp,”. Biomolecules, 2020. [DOI | PubMed]
- Transgenesis as a Tool for the Efficient Production of Selected Secondary Metabolites from Plant in Vitro Cultures,”. Pharmaceuticals, 2020
- 16 D. V. S. Green and M. Segall , “Chemoinformatics in Lead Optimization,” in: Chemoinformatics for Drug Discovery, ed. J. Bajorath (John Wiley & Sons, 2013), 149–178.
- Activity to Breast Cancer Cell Lines of Different Malignancy and Predicted Interaction With Protein Kinase C Isoforms of Royleanones,”. International Journal of Molecular Sciences, 2020. [PubMed]
- Discovery of a Small‐molecule Protein Kinase Cδ‐selective Activator With Promising Application in Colon Cancer Therapy,”. Cell Death & Disease, 2018. [DOI]
- Counteracting Colon Cancer by Inhibiting Mitochondrial Respiration and Glycolysis With a Selective PKCδ Activator,”. International Journal of Molecular Sciences, 2023. [DOI | PubMed]
- Molecular Docking: A Powerful Approach for Structure‐Based Drug Discovery,”. Current Computer‐Aided Drug Design, 2011. [PubMed]
- Protein Data Bank ,. 2023
- FRED: Protein‐Ligand Docking . OpenEye Scientific Software.
- Extraction Optimization and Reactivity of 7α‐acetoxy‐6β‐hydroxyroyleanone and Ability of Its Derivatives to Modulate PKC Isoforms,”. Scientific Reports, 2024. [PubMed]
- Self‐Assembly Nanoparticles of Natural Bioactive Abietane Diterpenes,”. International Journal of Molecular Sciences, 2021. [DOI | PubMed]
- Dehydroroyleanone as a Building Block for a Drug Delivery Platform Based on Self‐Assembled Nanoparticles: Structural Studies and Chemical Modification,”. ACS Omega, 2022. [PubMed]
- Application of Amino Acids in the Structural Modification of Natural Products: A Review,”. Frontiers in Chemistry, 2021. [PubMed]
- Chemical and Enzymatic Stability of Amino Acid Prodrugs Containing Methoxy, Ethoxy and Propylene Glycol Linkers,”. Molecular Pharmaceutics, 2009. [PubMed]
- Amino Acids in the Development of Prodrugs,”. Molecules, 2018. [DOI | PubMed]
- Crystal Structure of the Cys2 Activator‐binding Domain of Protein Kinase Cδ in Complex With Phorbol Ester,”. Cell, 1995. [PubMed]
- Atomic Structure and Chemistry of human Serum Albumin,”. Nature, 1992. [PubMed]
- Binding Sites of Anticancer Drugs on Human Serum Albumin (HSA): A Review,”. Protein & Peptide Letters, 2022. [PubMed]
- Recent advances in molecular luminescence analysis,” in:. 1979
- Flavonols With a Catechol or Pyrogallol Substitution Pattern on Ring B Readily Form Stable Dimers in Phosphate Buffered Saline at Four Degrees Celsius,”. Food Chemistry, 2020. [PubMed]
- β‐Casein Nanoparticle‐based Oral Drug Delivery System for Potential Treatment of Gastric Carcinoma: Stability, Target‐activated Release and Cytotoxicity,”. European Journal of Pharmaceutics and Biopharmaceutics, 2012. [PubMed]
- Isolation, Synthesis and Nanoencapsulation of Cytotoxic Compounds from Plectranthus Spp , (. 2019
- 36 M. F. Simões , E. Valente , M. J. R. Gómez , E. Anes , and L. Constantino European Journal of Pharmaceutical Sciences 37 (2009): 257–263.19491013
- Stabilizing Short‐lived Schiff Base Derivatives of 5‐aminouracils That Activate Mucosal‐associated Invariant T Cells,”. Nature Communications, 2017
- Benzoic Acid Derivatives as Prodrugs for the Treatment of Tuberculosis,”. Pharmaceuticals, 2022. [DOI]
- Molecular Docking Studies of Royleanone Diterpenoids From Plectranthus spp. As P‐Glycoprotein Inhibitors,”. ACS Medicinal Chemistry Letters, 2020. [PubMed]
- Royleanone Derivatives From Plectranthus spp. as a Novel Class of P‐glycoprotein Inhibitors,”. Frontiers in Pharmacology, 2020
- WFO Plant List (. 2023
- Scientific Software.
- 2019‐1; Schrödinger.
- An Overview of the Amber Biomolecular Simulation Package,”. WIREs Computational Molecular Science, 2013
- Development and Testing of a General Amber Force Field,”. Journal of Computational Chemistry, 2004. [PubMed]
- ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters From ff99SB,”. Journal of Chemical Theory and Computation, 2015. [PubMed]
- Experimental and Computational Druggability Exploration of the 14‐3‐3ζ/SOS1pS 1161 PPI Interface,”. Journal of Chemical Information and Modeling, 2020. [PubMed]
- Identification of Small Molecular Chaperones Binding P23H Mutant Opsin Through an in Silico Structure‐Based Approach,”. Journal of Chemical Information and Modeling, 2022. [PubMed]
- The Antiepileptic Drug Levetiracetam Inhibits Carbonic Anhydrase: In Vitro and in Silico Studies on Catalytically Active Human Isoforms,”. ACS Medicinal Chemistry Letters, 2024. [PubMed]
- Fighting Antimicrobial Resistance: Insights on How the Staphylococcus aureus NorA Efflux Pump Recognizes 2‐Phenylquinoline Inhibitors by Supervised Molecular Dynamics (SuMD) and Molecular Docking Simulations,”. Journal of Chemical Information and Modeling, 2023. [PubMed]
- Fragment‐Based Interrogation of the 14–3–3/TAZ Protein–Protein Interaction,”. Biochemistry, 2024. [PubMed]
- PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data,”. Journal of Chemical Theory and Computation, 2013. [PubMed]
- 53 J. R. Lakowicz , Principles of Fluorescence Spectroscopy (University of Maryland School of Medicine, 2006).
- Investigation of the Interaction Between Flavonoids and Human Serum Albumin,”. Journal of Molecular Structure, 2004
- Spectroscopic Study of Secondary Structure and Thermal Denaturation of Recombinant Human Factor XIII in Aqueous Solution,”. Archives of Biochemistry and Biophysics, 1997. [PubMed]
- 56 D. B. Stengel and S. Connan , Marine Algae: A Source of Biomass for Biotechnological Applications, in: Methods in Molecular Biology, ed. J. M. Walker (Humana Press, 2015), 1–37.
- Structural, Antioxidant, Antiproliferative and In‐silico Study of Pyridine‐based Hydrazonyl‐1,3‐selenazoles and Their Sulfur Isosteres,”. Journal of Molecular Structure, 2021
- Selenazolyl‐hydrazones as Novel Selective MAO Inhibitors with Antiproliferative and Antioxidant Activities: Experimental and in‐silico Studies,”. Frontiers of Chemistry, 2018
- Sulfur Analogues of Tyrosine in the Development of Triazene Hybrid Compounds: A New Strategy Against Melanoma,”. ACS Medicinal Chemistry Letters, 2021. [PubMed]