
Evaluation of Small-Molecule Candidates as Modulators of M-Type K+ Currents: Impacts on Current Amplitude, Gating, and Voltage-Dependent Hysteresis
Abstract
The core subunits of the KV7.2, KV7.3, and KV7.5 channels, encoded by the KCNQ2, KCNQ3, and KCNQ5 genes, are expressed across various cell types and play a key role in generating the M-type K+ current (IK(M)). This current is characterized by an activation threshold at low voltages and displays slow activation and deactivation kinetics. Variations in the amplitude and gating kinetics of IK(M) can significantly influence membrane excitability. Notably, IK(M) demonstrates distinct voltage-dependent hysteresis when subjected to prolonged isosceles-triangular ramp pulses. In this review, we explore various small-molecule modulators that can either inhibit or enhance the amplitude of IK(M), along with their perturbations on its gating kinetics and voltage-dependent hysteresis. The inhibitors of IK(M) highlighted here include bisoprolol, brivaracetam, cannabidiol, nalbuphine, phenobarbital, and remdesivir. Conversely, compounds such as flupirtine, kynurenic acid, naringenin, QO-58, and solifenacin have been shown to enhance IK(M). These modulators show potential as pharmacological or therapeutic strategies for treating certain disorders linked to gain-of-function or loss-of-function mutations in M-type K+ (KV7x or KCNQx) channels.
Article type: Review Article
Keywords: M-type K, M-type (K, current kinetics, small-molecule modulator
Affiliations: Department of Pharmacy, China Medical University, Taichung 406040, Taiwan; lutl@mail.cmu.edu.tw; Neuroscience Institute, Medical Academy, Lithuanian University of Health Sciences, Eiveniu 2, 50161 Kaunas, Lithuania; rasa.liutkeviciene@lsmuni.lt; Latvian Biomedical Research and Study Centre (BMC), LV-1067 Riga, Latvia; vita.rovita@biomed.lu.lv; Institute of Basic Medical Sciences, College of Medical, National Cheng Kung University, Tainan City 701401, Taiwan; s58131110@gs.ncku.edu.tw; Department of Research and Education, An Nan Hospital, China Medical University, Tainan City 709204, Taiwan; School of Medicine, National Sun Yat-sen University, Kaohsiung 804201, Taiwan
License: © 2025 by the authors. CC BY 4.0 Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Article links: DOI: 10.3390/ijms26041504 | PubMed: 40003973 | PMC: PMC11855363
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (3.9 MB)
1. Introduction
The KCNQ2, KCNQ3, and KCNQ5 genes are known to encode the core subunits of the KV7.2, KV7.3, and KV7.5 K+ channels, respectively. These channels are expressed across a wide range of excitable and non-excitable cells [ref. 1,ref. 2,ref. 3]. Upon activation by appropriate membrane depolarizations, these KV channels generate the macroscopic M-type K+ current (IK(M)), which is characterized by its low voltage activation threshold as well as slow activation and deactivation kinetics [ref. 4,ref. 5,ref. 6]. The low voltage activation threshold allows IK(M) to become active at a membrane potential close to the resting potential of the cell. The designation “M” in IK(M) reflects its modulation by muscarinic receptors, originally identified as being activated by acetylcholine [ref. 5,ref. 7,ref. 8,ref. 9,ref. 10,ref. 11].
Among these channels, KV7.2 and KV7.3 serve as the primary molecular components of IK(M), a key regulator of neuronal excitability, including spike frequency adaptation and synaptic transmission [ref. 1,ref. 2,ref. 6,ref. 12,ref. 13,ref. 14,ref. 15]. However, in specific locations, other subunits may also contribute to M-like currents [ref. 6,ref. 16]. KV7.1 (KCNQ1) is expressed in the heart, particularly in atrial and ventricular myocytes, where it contributes to the slow delayed-rectified K+ current (IKs) [ref. 17]. In contrast, KV7.4 (KCNQ4) is primarily involved in the auditory system, where it plays a key role in maintaining K+ balance within the cochlea [ref. 18]. KV7.5 (KCNQ5) channels play a critical role in regulating cellular electrical excitability and are essential for the proper functioning of the nervous system and other excitable tissues. The dysfunction of KV5 channels has been implicated in various neurological and muscular disorders [ref. 2,ref. 14]. The IK(M) is particularly sensitive to inhibition by linopirdine and can be differentiated from ether-à-go-go-related (erg)-mediated K+ currents [ref. 5,ref. 19,ref. 20,ref. 21]. Recent studies have suggested that the magnitude of IK(M) may regulate the availability of voltage-gated Na+ (NaV) channels during prolonged high-frequency firing of action potentials, thereby affecting reliable presynaptic spikes and synaptic transmission in a quantum-dependent fashion [ref. 1,ref. 14,ref. 22,ref. 23].
Modulation of IK(M) has gained increasing recognition as a potential therapeutic approach for treating a variety of neurological disorders associated with either excessive neuronal activity or dysfunctional autonomic control [ref. 2,ref. 3,ref. 10,ref. 12,ref. 15,ref. 24,ref. 25,ref. 26,ref. 27,ref. 28]. Genetic variants in the KCNQx genes, including both gain-of-function and loss-of-function mutations, have been associated with various conditions such as social impairments, cognitive dysfunction, neuropathic pain, and epilepsy [ref. 24,ref. 25,ref. 26,ref. 27,ref. 29,ref. 30,ref. 31].
2. Biophysical Characteristics of IK(M)
As illustrated in Figure 1, pituitary GH3 cells were exposed to a high-K+, Ca2+-free solution containing 1 μM tetrodotoxin, while the measuring electrode was filled with a K+-enriched solution. Tetrodotoxin was included in the bathing medium to eliminate interference from other ion channels, including NaV channels and Na+-activated K+ channels [ref. 32]. Using a high-K+ bathing solution offers the advantage of conveniently distinguishing between different types of voltage-gated K+ currents, including erg-mediated K+ currents. These erg-mediated currents may partially overlap with IK(M) in various cell types [ref. 19,ref. 33]. During the recordings, the cell was held at a membrane potential of −50 mV, and a 1 − s depolarizing pulse to −10 mV was applied to evoke IK(M). Under these experimental conditions, IK(M) displayed a characteristic slow activation profile in response to sustained depolarization, with activation and deactivating time constants of approximately 100 and 30 ms, respectively. Given that the reversal potential for K+ ions is approximately 0 mV in this context, the resulting IK(M) was an inward K+ current that activated gradually, promoting the influx of K+ ions into the cell [ref. 4,ref. 5,ref. 21,ref. 34]. Moreover, when the voltage returns to the original holding potential (which is −50 mV), a large deactivating tail inward current is also generated (Figure 1).
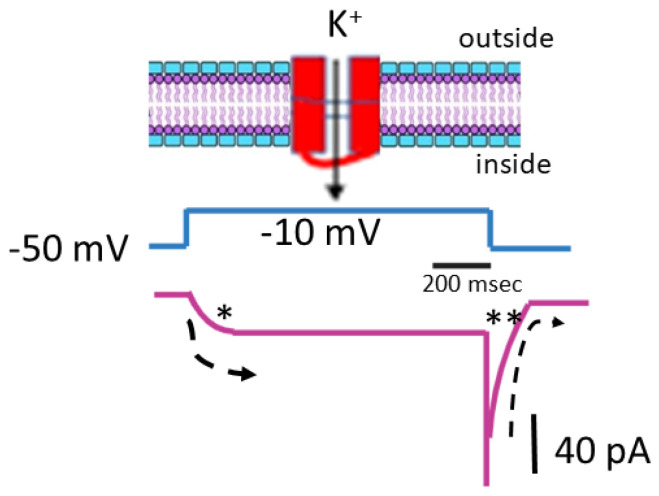
Figure 2 presents the steady-state activation curve of IK(M) in GH3 cells, illustrating its relationship with membrane potential. The amplitude of IK(M) was measured at the end of each 1 − s depolarizing step from a holding potential of −50 mV. Data points recorded at various membrane potentials were fitted using the Boltzmann equation (or Fermi-Dirac distribution) [ref. 35]. The Boltzmann equation is given by
where G represents the ionic conductance of IK(M), calculated as G = I/(V − Erev), (with I and V denoting the current amplitude and membrane potential, respectively, and Erev being the reversal potential for K+ ions), Gmax is the maximal conductance of IK(M), V1/2 is the voltage at which half-maximal activation occurs, q is the apparent gating charge in units of the elementary charge (e), F is Faraday’s constant, R is the universal gas constant, T is the absolute temperature, and F/RT equals 0.04 mV−1.
In this context, the apparent gating charge refers to the amount of electrical charge that moves across the membrane during the activation of ion channels in response to a voltage change [ref. 35,ref. 36,ref. 37]. This “gating” charge corresponds to the movement of charged particles, typically amino acid residues in the ion channel protein, which is necessary for the channel to open [ref. 36]. Under our experimental conditions, the steady-state activation curve of IK(M) (Figure 2) was optimally fitted with an upward nonlinear sigmoidal function, yielding a half-maximal voltage (V1/2) of −23.4 mV and an effective gating charge (q) of 5.1 e.
The difference in free energy associated with the gating of IK(M) at 0 mV (ΔG0) was calculated based on a 2-state gating model, which includes a closed (resting) state and an open state. According to the model, ΔG0 for IK(M) activation at 0 mV can be expressed as q × F × V1/2, where F is Faraday’s constant [ref. 35,ref. 38]. Using the values for q and V1/2, the difference in free energy involved in the gating of IK(M) at 0 mV (ΔG0) was thus estimated to be 11.5 kJ/mol (or 2.75 kcal/mol). The role of phosphosphoinositide metabolism in the gating of IK(M) and its relationship to free energy [ref. 39,ref. 40] warrants further investigation.
Upon exposure to a prolonged upright triangular ramp voltage (Vramp), distinct forward and backward amplitudes of IK(M) were observed, indicating the presence of nonlinear and non-equilibrium voltage-dependent hysteresis (Hys(V)) of IK(M) (Figure 3) [ref. 35,ref. 37,ref. 41]. The Hys(V) property of IK(M) plays a critical role in modulating the overall behaviors of excitable cells, including pituitary GH3 lactotrophs. This phenomenon represents a unique and distinct shift in ion-channel gating, where the voltage sensitivity governing charge movement depends on the prior state of the KM (KV7x or KCNQx) channel involved [ref. 35,ref. 37,ref. 41].
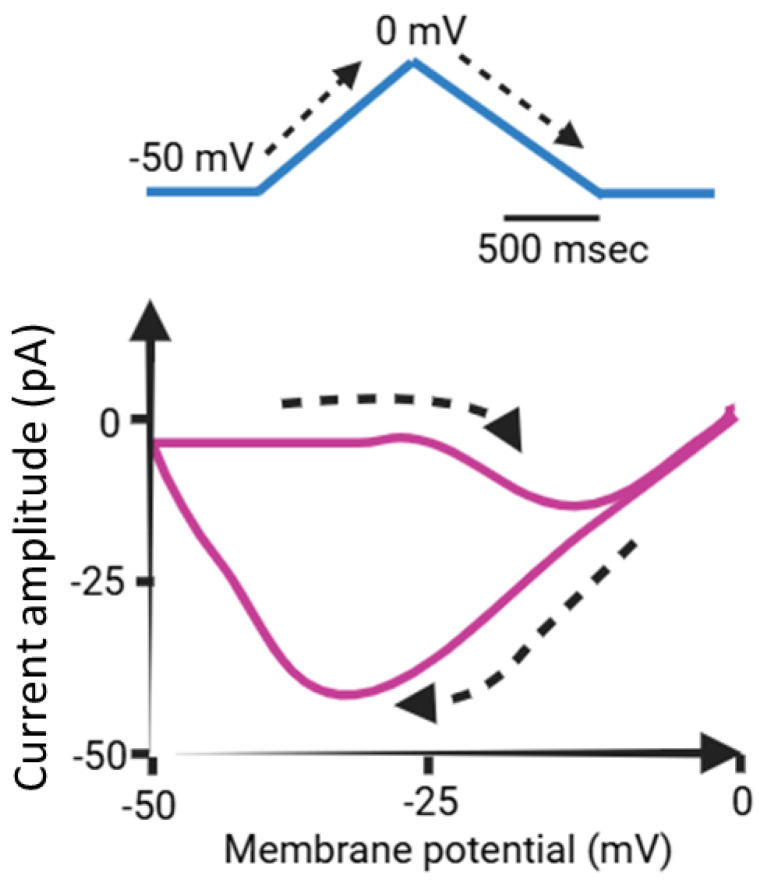
When the membrane potential of an excitable cell undergoes depolarization, specifically along the ascending limb of the upright isosceles-triangular Vramp, the current strength remains relatively small as indicated by the Hys(V) loop (Figure 3). However, during membrane repolarization, namely along the descending limb of this double Vramp, the amplitude of IK(M) increases significantly, resulting in a pronounced alteration in the membrane potential. Consequently, the influence of IK(M) on the excitable membrane is more pronounced during repolarization than during depolarization [ref. 37]. Like perovskite solar cells (PSCs) [ref. 42,ref. 43,ref. 44,ref. 45]), the Hys(V) behaviors may therefore occur across various ionic currents, including IK(M). The Hys(V) behavior is a major challenge in PSCs as it affects the accuracy and stability of efficiency measurements since it is manifested as a discrepancy between the forward (voltage increased from short circuit to open circuit) and the reverse scan (voltage decreased from open circuit to short circuit) [ref. 42,ref. 44,ref. 45,ref. 46,ref. 47]. However, in small cells, the primary effect of IK(M) may be its modulation of the resting potential and the associated input resistance.
The activation and deactivation time courses of the IK(M) occur with a time frame of tens to hundreds of milliseconds. When an agonist is applied, the activation of IK(M) takes several seconds, and the recovery of IK(M) following agonist removal can extend beyond a minute [ref. 40]. However, it is important to note that when a prolonged upright isosceles-triangular Vramp, lasting up to 2 s (Figure 3), is applied, the concentration of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2)—which resides almost exclusively in the cytoplasmic leaflet of the plasma membrane—may be altered, potentially influencing Hys(V) strength. This change occurs because the breakdown of PtdIns(4,5)P2 by phospholipase C happens within a few seconds [ref. 39,ref. 40]. However, after the agonist is removed, the resynthesis of PtdIns(4,5)P2 from phosptatidiylinositol (PtdIns) can take anywhere from one to several minutes, depending on the cell type.
Moreover, a recent study reported that compounds containing COOH groups can activate IK(M), along with modifications on the activation curve of this current [ref. 48]. These COOH-containing compounds may activate IK(M) through a lipoelectric mechanism, which involves the lipophilic compounds interacting at the interface between the lipid bilayer and the voltage sensor of the channel [ref. 48]. However, it remains unclear how these lipophilic compounds specifically influence the voltage sensor of the channel and how this interaction affects the strength of Hys(V) in response to a double Vramp stimulus.
3. Small-Molecule Modulators Targeting IK(M)
A variety of both natural and synthetic molecules have been demonstrated to play key roles in regulating IK(M) activity across different cell types, as summarized in Table 1 and Table 2.
Table 1: Two-dimensional chemical structures of drugs and compounds known to inhibit IK(M), as detailed in this study. The data were sourced from PubChem (https://pubchem.ncbi.nlm.nih.gov/) (accessed on 9 February 2025).
| Compound or Drug | Abbreviated Name | PubChem Identifier | Chemical Structure |
|---|---|---|---|
| Bisoprolol | BIS | 2405 | |
| Brivaracetam | BRV | 9837243 | |
| Cannabidiol * | CBD | 644019 | |
| Nalbuphine | NAL | 5311304 | |
| Phenobarbital | PHB | 4763 | |
| Remdesivir | RDV | 121304016 |
* The effect of CBD on IK(M) is still somewhat controversial, with reports suggesting a stimulatory effect on it [23,49]. Therefore, there is still some debate on this matter.
Table 2: Two-dimensional chemical structures of drugs and compounds known to stimulate IK(M), as detailed in this study. The data were sourced from PubChem (https://pubchem.ncbi.nlm.nih.gov/) (accessed on 7 February 2025).
| Compound or Drug | Abbreviated Name | PubChem Identifier | Chemical Structure |
|---|---|---|---|
| Flupirtine | FLU | 53276 | |
| Kynurenic acid | KYNA | 3845 | |
| Naringenin | NGEN | 439246 | |
| QO-58 | – | 51351551 | |
| Solifenacin | SOL | 154059 |
3.1. Small Molecules Known to Inhibit IK(M) (Table 1)
3.1.1. Bisoprolol (BIS, Concor®, Cardicor®, Zebeta®, 1-[4-[(2-Isopropoxyethoxy)methyl]phenoxy]-3-(isopropylamino)propan-2-ol))
BIS is recognized as an oral selective β1 adrenergic receptor blocker commonly used in the treatment of hypertension and heart-related conditions, such as atrial fibrillation, heart failure, and postural tachycardia syndrome [ref. 50,ref. 51]. Because of its lipophilic nature, it facilitates entry into brain tissue to produce regulatory actions on central neurons [ref. 52]. Earlier reports have shown that BIS could bind to β1-adrenergtic receptors inherently existing in brain areas including the pituitary gland and hippocampus [ref. 53,ref. 54]. A previous study also disclosed the effectiveness of BIS in increasing blood prolactin levels [ref. 55].
Notably, previous studies have demonstrated that when GH3 cells were exposed to BIS, the amplitude of IK(M) in response to sustained depolarization was effectively suppressed with an IC50 value of 1.2 μM [ref. 56]. However, the BIS-induced inhibition of IK(M) amplitude in these cells was not affected by the subsequent addition of isoproterenol or ractopamine but was attenuated by flupirtine or ivabradine. Isoproterenol and ractopamine are known to bind to and activate β-adrenergic receptors, while flupirtine and ivabradine can increase the amplitude of IK(M) [ref. 56,ref. 57,ref. 58]. Furthermore, in the cell-attached current recordings, BIS decreased the open probability of KM (K7x or KCNQx) channels, along with a significant reduction in the mean open time of the channel [ref. 56]. The exposure to BIS not only produced a decrease in the maximal open probability of KM channels but also shifted the steady-state activation curve along the voltage axis to depolarized potentials by approximately 7 mV. However, minimal change in the gating charge of such an activation curve was demonstrated in BIS presence. Consequently, as cells were exposed to 1 μM BIS, the difference in free energy (ΔG0) required for activation of the KM channel at 0 mV in GH3 cells was reduced from 1.25 to 0.79 kcal/mol [ref. 56].
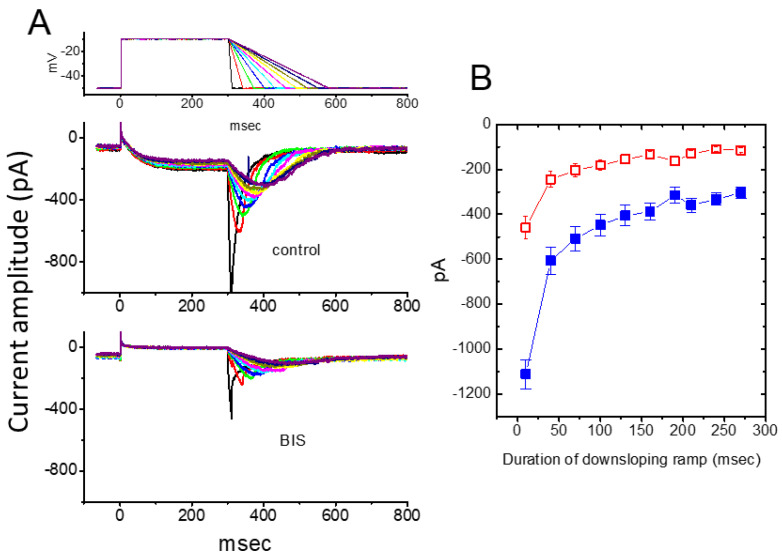
Earlier findings have suggested that the magnitude of IK(M) may influence the falling phase of bursting firing or spike after depolarization [ref. 2,ref. 13,ref. 59]. The falling phase of burst firing refers to the part of the action potential (or series of action potentials) where the membrane potential rapidly repolarizes or returns to a more negative value after the peak of the action potential. More recent research has shown that BIS can significantly affect the deactivation of IK(M) in response to a downsloping Vramp, ranging from −10 to −50 mV, with varying durations [ref. 56]. As depicted in Figure 4, upon returning to −50 mV, a slower ramping rate of Vramp led to a progressive exponential reduction in the peak amplitude of deactivating IK(M), with an estimated time constant of 98 ± 8 ms (n = 11). However, when cells were exposed to 3 μM BIS, the peak amplitude of the current was significantly reduced in an exponential manner, with a time constant of 65 ± 7 ms (n = 11). These results indicate that, as the duration of the duration of the downsloping Vramp is increased, the amplitude of deactivating IK(M) decreases exponentially. Furthermore, the presence of BIS resulted in a time-dependent reduction of IK(M) [ref. 56]. Therefore, the BIS-induced block of IK(M) is not instantaneous but develops gradually over time, when the channels are opened upon rapid membrane depolarization. Additionally, the BIS-induced inhibition of IK(M) in GH3 cells does not seem to be solely dependent on binding to β-adrenergic receptors, although these receptors may exhibit constitutive activity [ref. 54].
3.1.2. Brivaracetam (BRV, (2S)-2-[(4R)-2-Oxo-4-propylpyrrolidin-1-yl]butanamide)
BRV (Brivact®, Brivlera®), a chemical analog of levetiracetam, is an orally or intravenously bioavailable racetam derivative with anticonvulsant (antiepileptic) properties that has appeared in a growing number of research papers [ref. 60,ref. 61]. Notably, it has also been recognized to be efficacious in the treatment of epilepsy and status epilepticus [ref. 60,ref. 61,ref. 62,ref. 63].
BRV has been shown to reduce pain behavior in a murine model of neuropathic pain [ref. 60,ref. 64]. Additionally, this compound has demonstrated anti-neoplastic effects in glioma cells [ref. 65]. It has also been reported that BRV can influence the functional activity of neurons (e.g., hippocampal neurons) or endocrine cells (e.g., pituitary lactotrophs) by binding with high affinity to the synaptic vesicle protein2A (SV2A) [ref. 66,ref. 67,ref. 68]. SV2A is recognized as a critical broad marker for neuroendocrine cells and can be bound to anticonvulsants [ref. 66,ref. 69].
An earlier study has shown that the exposure to BRV in pituitary GH3 cells led to a concentration-dependent inhibition of IK(M) with an IC50 value of 6.5 μM [ref. 70]. The activation time constant of IK(M) was effectively increased during GH3-cell exposure to 10 μM BRV. However, BRV can also suppress the peak amplitude of INa with an IC50 value of 12.2 μM in GH3 cells. A leftward shift in the steady-state inactivation curve of INa was observed in the presence of BRV [ref. 70]. Under the inside-out current recordings, addition of BRV to the intracellular side of the excised patch enhanced the probability of BKCa channels that would be open, without affecting the single-channel conductance of the channel. These observations therefore suggest that, in addition to being a high-affinity ligand for SV2A [ref. 66,ref. 67,ref. 68], BRV is capable of perturbing the amplitude and kinetics of ionic currents, including IK(M). This reveals a potential unintended effect on the functional activities of different excitable cells, such as synaptic transmission [ref. 14].
3.1.3. Cannabidiol (CBD, 2-[(1R,6R)-3-Methyl-6-(prop-1-en-2-yl)cyclohex-2-en-1-yl]-5-pentylbenzeine-1,3-diol)
CBD is a non-psychoactive cannabinoid derived from the Cannabis plant, known for its potential therapeutic effects. CBD was previously reported to modulate the activity of μ- and δ-opioid receptors [ref. 71,ref. 72]. It is among over 100 cannabinoids present in the plant and has been demonstrated to be effective in treating various medical conditions, such as epilepsy, bipolar disorder, inflammation, and cancer [ref. 73,ref. 74,ref. 75].
A recent study by Liu et al. (2023) demonstrated that exposure to CBD led to a concentration-dependent reduction in the amplitude of IK(M) in pituitary GH3 cells, with an IC50 value of 3.6 μM. CBD also caused a rightward shift in the steady-state activation curve of IK(M) without affecting the gating charge of the curve. Notably, the inhibition of IK(M) by CBD was not reversed by the subsequent addition of naloxone, an opioid receptor antagonist. However, the amplitude of IK(M) in GH3 cells was effectively reduced in the presence of either thyrotropin releasing hormone (1 μM) or liraglutide (1 μM) [ref. 4,ref. 76]. The liraglutide-mediated inhibition of IK(M) may result from its interaction with glucagon-like peptide-1 (GLP-1) receptors, which are expressed in pituitary cells, as liraglutide is a synthetic analog of GLP-1 [ref. 77].
Additionally, CBD was found to suppress the density of both activating and deactivating IK(M) in response to pulse-train depolarizing stimuli ranging from −50 to −10 mV, as shown in Figure 5. These results suggest that CBD-induced inhibition of IK(M) remains effective even under conditions of high-frequency pulse-train stimulation [ref. 76]. However, the presence of 10 μM CBD had no effect on INa in GH3 cells. Previous studies have demonstrated that a train of depolarizing pulses can significantly alter the magnitude of INa, a current that decays exponentially over time [ref. 78,ref. 79,ref. 80,ref. 81]. Furthermore, it has been shown that the IK(M) magnitude can regulate the availability of NaV channels during prolonged high-frequency firing [ref. 81]. Taken together, these findings indicate that CBD exposure in GH3 cells reduces the magnitude of IK(M) during pulse-train stimuli. As a result, the availability of NaV channels during sustained high-frequency firing may be significantly diminished, potentially impairing reliable presynaptic spiking and reducing synaptic transmission at elevated frequencies [ref. 1,ref. 14,ref. 22,ref. 23].
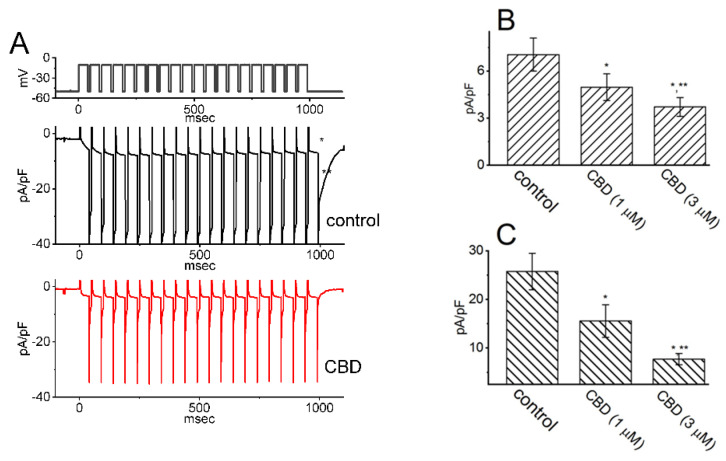
Since naloxone, an opioid receptor antagonist, did not affect the CBD-induced reduction in IK(M), this suggests that CBD’s effect on IK(M) is not mediated through opioid receptor binding. These findings indicate that CBD likely exerts a direct and rapid influence on IK(M), independent of interactions with the cannabinoid or opioid receptor [ref. 75,ref. 76]. If similar effects are observed under culture conditions or in vivo, they could potentially influence the firing behavior of action potentials in cells. However, whether CBD inhibits or stimulates IK(M) remains a topic of debate [ref. 23,ref. 49]. It is possible that its effects vary depending on the specific subtypes of KM (KV7x or KCNQx) channels expressed in different cell types.
3.1.4. Nalbuphine (NAL, Nubain®, 17-[Cyclobutylmethyl]-4,5-epoxymorphinan-3,6,14-triol Hydrochloride)
NAL has been recognized as a moderate-efficacy partial agonist or antagonist of the μ-opioid receptor and as a high-efficacy partial agonist of the κ-opioid receptor with its low affinity for either the δ-opioid receptor or the σ receptor [ref. 82,ref. 83]. It can be used to balance anesthesia, for preoperative and postoperative analgesia, and for obstetrical analgesia [ref. 84,ref. 85].
A previous report has demonstrated the effectiveness of NAL in suppressing the amplitude of IK(M) occurring in mHippoE-14 hippocampal neurons [ref. 86]. This inhibitory effect appears to be direct and independent of its binding to opioid receptors [ref. 85,ref. 87]. The IC50 value required for NAL-mediated inhibition of IK(M) in mHippoE-14 neurons is estimated to be 5.7 μM. These results reflect that NAL exerts a concentration-dependent action on the suppression of IK(M) in these cells.
As mHippoE-14 neurons were exposed to NAL, the amplitude of INa was also effectively suppressed, with an IC50 value of 1.9 μM. The presence of NAL not only inhibited the maximal conductance of peak INa but also shifted the steady-state inactivation curve to hyperpolarized potentials by 12 mV; consequently, the difference in free energy required for NAL-inhibited INa of the curve was altered. In contrast, the gating charge of the curve obtained between the absence and presence of 3 μM NAL did not differ significant. These results indicate that the NAL exposure can inhibit the peak amplitude of INa in a voltage-dependent manner in mHippoE-14 neurons [ref. 86]. In light of these observations, it is conceivable that the inhibitory effect of NAL on multiple ion currents (e.g., INa and IK(M)) tends to be a direct interaction between the drug and the channels themselves. Such an inhibition occurring within a clinically therapeutic range is not linked to agonistic or antagonistic effects on opioid receptors.
3.1.5. Phenobarbital (PHB, Luminal Sodium®, 5-Ethyl-5-phenylbarbituric Acid)
PHB is a medication belonging to a group known as barbiturate. PHB has been recognized to be an anticonvulsant and a hypnotic because it can facilitate synaptic inhibition in the central nervous system by acting on the γ-aminobutyric acid (GABA) type A (GABAA) receptors [ref. 88,ref. 89,ref. 90]. GABAA receptor is a ligand-gated chloride ion channel, which is the most common inhibitory channel in the brain. Pentobarbital also belongs to barbiturates and is similar to PHB. PHB has been disclosed to suppress neurogenic inflammation and exert neuroprotective and even anti-neoplastic activities by modifying membrane ion channels other than chloride channels [ref. 91,ref. 92].
Recently, it has been demonstrated that PHB can regulate the magnitude of multiple types of ionic currents, including IK(M), residing in Neuro-2a neuroblastoma cells [ref. 79]. The magnitude of PHB-induced suppression in IK(M) was not be reversed by the subsequent addition of flumazenil or chlorotoxin. Chlorotoxin, derived originally from scorpion venom, is a blocker of Cl− channels (e.g., CIC-3 channel), while flumazenil is a benzodiazepine receptor antagonist. Therefore, the PHB-mediated inhibition of IK(M) in Neuro-2a cells tends to be acute and may be independent of GABAA receptor-mediated Cl− currents [ref. 88]. Moreover, Neuro-2a-cell exposure to 100 or 300 μM PHB also decreased the amplitudes of activating and deactivating IK(M) during 1 − s pulse train stimulation. These findings showed that a PHB-mediated block of IK(M) remained efficacious upon high pulse-train stimulation. It is therefore conceivable that the availability of NaV channels during high-frequency firing of action potentials in unclamped cells became further retarded during the exposure to PHB, despite its effectiveness in suppressing the INa amplitude directly [ref. 79]. However, since the reported PHB effects range between 100 and 300 μM, it remains to be determined whether its selective effects are directly related to its influence on IK(M).
3.1.6. Remdesivir (RDV, GS-5734, Veklury®, (S)-2-Ethyl-6-methyl-1-(2-propylthazol-4-yl)-4,5-dihydro-1H-pyrrolo[3,4-d]pyrimidin-7(6H)-one)
RDV (GS-5734), a broad-spectrum antiviral agent, is recognized as a mono-phosphoramidate prodrug of an adenosine analog that metabolizes into its active form GD-44524, which is a C-adenosine nucleoside analog [ref. 93]. This compound, a nucleotide-analog inhibitor of RNA-dependent RNA polymerase, is thought to be highly active against coronaviruses (CoVs), including MERS-Cov and SARS-CoV-2 [ref. 93,ref. 94,ref. 95,ref. 96].
It has been recognized as a promising antiviral drug against an array of RNA viruses, predominantly through the targeting of the viral RNA dependent RNA polymerase [ref. 93,ref. 96,ref. 97]. Recent reports have also shown the effectiveness of RDV in treating patients with certain types of panencephalitis [ref. 98,ref. 99,ref. 100].
As shown in Figure 6, previous studies have demonstrated that RDV can inhibit the amplitude of IK(M) in GH3 cells, with an IC50 value of 2.5 μM [ref. 101]. Cell exposure to RDV, using a long-lasting triangular Vramp, noticeably suppressed the strength of Hys(V) for IK(M) elicitation. Therefore, it is likely that RDV exerts a perturbing effect on this non-equilibrium property in KM (KV7x or KCNQx) channels within excitable membranes [ref. 97,ref. 101].
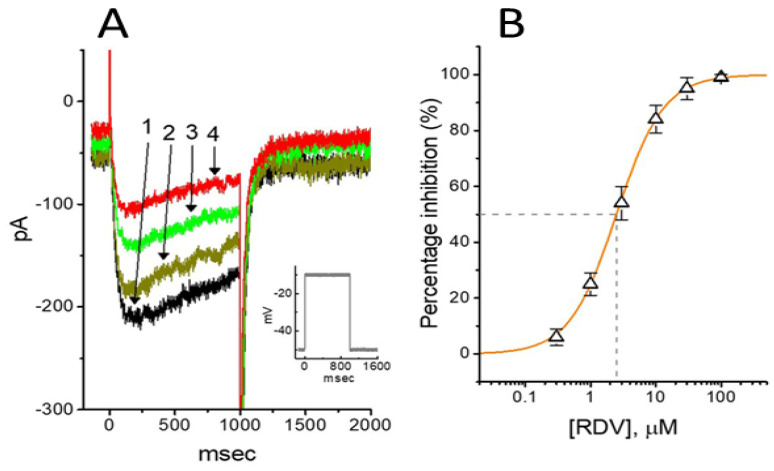
Besides its inhibitory effect on IK(M), RDV can inhibit the amplitude of IK(DR) along with the increased rate of current inactivation during prolonged membrane depolarizations [ref. 101]. The RDV molecules tend to accelerate IK(DR) inactivation in a concentration- and state-dependent fashion, implying that they reach the blocking site of the channel, only when the channel involved resides in the open conformational state. The value of the dissociation constant (KD) required for an RDV-induced block of IK(DR) in GH3 cells was 3.0 μM. The EC50 value of RDV against SAR-CoV-2 residing in Vero E6 cells was reported to be 1.76 μM [ref. 95,ref. 102]. Therefore, RDV-induced inhibition of IK(M), IK(DR) or erg-mediated K+ current is more than likely achieved in vivo [ref. 97,ref. 101,ref. 103]. Therefore, RDV is not classified as a prodrug, and its inhibition of these K+ currents [ref. 101,ref. 102] seems to occur independently of its possible effects on RNA polymerase activity [ref. 94,ref. 103,ref. 104]. It needs to be mentioned that the administrated of RDV in patients with COVID-19 infection may cause cardiac adverse events, possibly due its perturbations on KV channels in heart cells [ref. 105,ref. 106].
3.2. Small Molecules Known to Stimulate IK(M) (Table 2)
3.2.1. Flupirtine (FLU, Katadolon®, 3-(4-Fluorophenyl-2-methylamino-1-pyridin-1-yl-propan-1-one)
FLU belongs to a class of triaminopyridines and is a centrally acting nonopioid analgesic agent with muscle relaxing properties. It has been used for a variety of neurological disorders involving neuronal overexcitability such as epilepsy and neuropathic pain [ref. 15,ref. 107]. In addition to its use in pain management, FLU was reported to display muscle relaxant and anticonvulsant activity. Notably, this compound was demonstrated to be beneficial in treating the patients with human prion diseases [ref. 108,ref. 109,ref. 110].
Previous research has shown that FLU enhances the activity of GABAA receptors, which is associated with increased stimulation of KV7 channels [ref. 111]. It is also well established that FLU can bind to and activate KV7.2–7.5 (KCNQ2–5) channels [ref. 6,ref. 112]. However, a study conducted in motor neuronal NSC-34 cells revealed that FLU induces a time-, concentration-, and state-dependent reduction in delayed-rectifier K+ current (IK(DR)) elicited by prolonged depolarizing pulses, without affecting the activation kinetics of the current [ref. 32]. Notably, unlike IK(M), this type of IK(DR), such as KV3.1-encoded current, is characterized by an activation threshold at high voltages, rapid deactivation kinetics, and minimal influence on the resting membrane potential [ref. 101,ref. 113,ref. 114,ref. 115,ref. 116,ref. 117]. Additionally, earlier studies have shown that suppressing KV3.1, a key component of the IK(DR) in high-spiking neurons, can reduce membrane excitability and consequently decrease neuronal firing [ref. 14,ref. 113,ref. 114,ref. 115,ref. 116,ref. 117]. This decrease in excitability is attributed to weakened resurgent K+ currents or repolarizing efficiency, which delay the repolarization of high-frequency action potentials and subsequently slow the recovery of INa [ref. 14,ref. 113,ref. 115,ref. 116]. The resurgent K+ current refers to a unique and relatively less common form of K+ current that can occur in certain excitable cells, including neurons and cardiac myocytes. Similar to the resurgent Na+ current, the resurgent K+ current is characterized by a brief transient outflow of K+ ions during the repolarization phase of an action potential, but the dynamics and mechanisms behind it are distinct.
Importantly, a detailed description of the IK(DR) inactivation time course at varying FLU concentrations, combined with simulations using minimal binding model that yielded a KD value of 8.9 μM, led the researchers to propose that FLU may act as a state-dependent blocker for IK(DR) [ref. 57]. Furthermore, cell treatment with N-methyl-D-aspartate (NMDA), an NMDA receptor agonist, did not alter FLU-induced suppression of IK(DR), suggesting that the inhibitory effect of FLU on IK(DR) is independent of interactions with NMDA receptors. Therefore, the dual effects of IK(M) stimulation and IK(DR) inhibition induced by FLU may synergistically reduce motor neuron activity, particularly during high-frequency action potential firing, assuming similar outcomes occur in vivo [ref. 10,ref. 14,ref. 27,ref. 32,ref. 57,ref. 113,ref. 116].
3.2.2. Kynurenic Acid ((2E)-2-Amino-3-carboxy-5,6,7,8-tetrahydrobenzo[b]pyridine-1 Carboxylic Acid) and Its Aminoalkylated Derivatives
Kynurenic acid (KYNA) is a naturally occurring produce of the normal metabolism of amino acid L-tryptophan that has been reported to inhibit N-methyl-D-aspartate receptor (NMDAR) and neuronal nicotinic α7 receptors [ref. 118,ref. 119]. This compound, together with L-kynurenine, is thought to be an endogenous metabolite of L-tryptophan known to block NMDAR, and it has been frequently demonstrated to exert neuroprotective or anticonvulsant properties in the brain [ref. 119]. Previous studies have disclosed the effectiveness of KYNA in ameliorating the magnitude of KCNQ gene-(KCNQ)- or HCN gene-(HCN)-encoded currents [ref. 120,ref. 121].
It has been recently demonstrated that in GH3 cells, KYNA or KYNA-A4, another aminoalkylated amide derivative, could produce a stimulatory effect on IK(M) in a concentration-, voltage-, and state-dependent fashion [ref. 34]. The EC50 value required for KYNA or KYNA-A4-stimulated IK(M) was yielded to be 18.1 or 6.4 μM, respectively. The presence of KYNA or KYNA-A4 shifted the relationship of normalized IK(M)-conductance and membrane potential to a more depolarized potential with no change in the gating charge of the current. The presence of KYNA was found to increase the probability of KM-channel openings in GH3 cells [ref. 56], with no clear change in single-channel conductance [ref. 34]. Under whole-cell current-clamp potential recordings, KYNA and KYNA-A4 were found to reduce the frequency of spontaneous action potentials in GH3 cells [ref. 34]. This reduction in action potential firing is likely attributed to the activation of IK(M), rather than an effect on either NMDAR activity or aryl hydrocarbon receptors [ref. 56,ref. 120,ref. 122].
3.2.3. Naringenin (NGEN, 4′,5,7-Trihydroxyflavan-4-one)
NGEN is a major dietary flavanone, a type of flavonoid commonly found in citrus fruits, and is known for its potential bioactive effects on human health. Several studies have reported that it and other structurally related compounds could produce anxiolytic and antinociceptive actions [ref. 123,ref. 124].
A previous study provides direct evidence that NGEN has a stimulatory action on IK(M) in NSC motor neuron-like cells [ref. 59]. The IC50 value required for NGEN-mediated stimulation of IK(M) was found to be 9.8 μM, which is significantly lower than the value reported for the inhibition of HERG channels [ref. 125]. When NGEN was added, it shifted the steady-state activation curve of IK(M) conductance to a more hyperpolarized potential in NSC-34 cells [ref. 59]. The lack of effect by NGEN on the gating charge of KM channels in NSC-34 cells led the investigators to propose that the stimulatory effect of NGEN on KM channels in NSC-34 cells is not mediated through a direct interaction on the voltage sensor in the channel.
Furthermore, earlier studies have demonstrated that NGEN directly stimulates BKCa channels [ref. 59,ref. 126]. Consistent with previous findings in vascular myocytes [ref. 126], NGEN-induced enhancement of the IK(Ca) amplitude in HEK293T cells transfected with α-hSlo was associated with an increased probability of BKCa-channel openings [ref. 59]. Thus, it is plausible that NGEN, along with other structurally similar compounds, may interact at specific regions to modulate the activity of both BKCa channels and KV7-encoded channels [ref. 59]. Additionally, in a mathematical model of hippocampal pyramidal neuron [ref. 127], doubling the conductances of both KM– and BKCa-channels to simulate the effect of NGEN (10 μM) led to a significant reduction in the bursting activity of action potentials in the modeled hippocampal neuron [ref. 14,ref. 59].
Because of its lipophilicity, NGEN was reported to transverse the blood–brain barrier and subsequently penetrate into different brain regions [ref. 128], although the brain concentrations of NGEN vary based on local extracellular milieu in and around membranes and synapses. Therefore, NGEN-induced actions on the stimulation of KM and BKCa channels may combine to affect the functional activities, if both ion channels are functionally expressed in central neurons in vivo [ref. 14].
3.2.4. QO-58 (5-(2,6-Dichloro-5-fluoropyridin-3-yl)-3-phenyl-2-(trifluoromethyl)-1H-pyrazolol[1,5-a]pyrimidin-7-one)
QO-58 has been previously demonstrated to be an opener of the KCNQx (KV7x) channel [ref. 129,ref. 130]. It has been reported that this compound could increase the pain threshold of neuropathic pain in a rat model, namely chronic constriction injury of the sciatic nerve [ref. 130]. QO-58 could also exercise anti-nociceptive action on inflammatory pain in rodents [ref. 131,ref. 132]. The ameliorating effects of this compound have been viewed to be closely linked to its activation of KCNQ (KV7) channels [ref. 130,ref. 131]. However, QO-40 (5-(chloromethyl)-3-(nathphalen-1-yl)-2-(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-7(4H)-one), a compound structurally similar to QO-58, has been reported to stimulate the activity of large-conductance Ca2+-activated K+ (BKCa) channels [ref. 133].
It has been shown that the presence of QO-58 can concentration-dependently increase the amplitudes of IK(M) and Ca2+-activated K+ current (IK(Ca)) in pituitary GH3 cells with EC50 values of 3.1 and 4.2 μM, respectively [ref. 134]. Under GH3-cell exposure to QO-58, the steady-state activation curve of IK(M) was shifted along the voltage axis to a hyperpolarized potential with no change in the gating charge, leading to changes in free energy of IK(M) activation. The Hys(V) strength of IK(M) activated by triangular Vramp measurably increased by the QO-58 presence. Furthermore, cell exposure to QO-58 enhanced the probability of BKCa-channel openings as well as shifting the activation curve of the channel at stead state toward the less depolarized potential; however, neither the gating charge nor single-channel conductance of the channel was affected during its exposure.
Alternatively, based on molecular docking predictions, the interactions between the KCa1.1 channel and QO-58, as well as between the KCNQ2 channel and QO-58, were clearly demonstrated [ref. 134]. These findings suggest that QO-58-mediated dual activation of KM (KV7x or KCNQx) and BKCa channels observed in GH3 cells may hold pharmacological or therapeutic significance, provided that similar effects can be confirmed in vivo [ref. 135,ref. 136]. Notably, advances in computational biology have significantly enhanced the ability to predict the druggability of ion-channel gene products, including KM (KV7x or KCNQx) channels [ref. 137,ref. 138,ref. 139].
3.2.5. Solifenacin (SOL, Vesicare®, [(3R)-1-Azabicyclo[2,2,2]octan-3-yl](1S)-1-phenyl-3,4-dihydro-1H-isoquinoline-2-carboxylate)
SOL, a member of isoquinolines, has been viewed as an oral anticholinergic, namely a competitive muscarinic (M1 and M3) receptor antagonist. It is also an antispasmodic agent used to treat the symptoms of overactive bladder, neurogenic detrusor overactivity, or urinary incontinence [ref. 140,ref. 141]. because of muscarinic (M2 and M3) receptor antagonists that have anticholinergic effects such as relaxation of the detrusor muscle in urinary bladder [ref. 140,ref. 141,ref. 142,ref. 143].
An earlier study demonstrated that in GH3 cells, exposure to SOL resulted in a concentration-dependent increase in the amplitude of IK(M) during prolonged membrane depolarization, with a concurrent shortening of the current’s activation time course [ref. 21]. The EC50 or KD value of SOL-stimulated IK(M) was calculated to be 0.34 or 0.55 μM, respectively. Exposure to SOL caused a leftward shift in the steady-state activation curve of IK(M) in GH3 cells. While SOL increased the activity of single KM (or KCNQx) channels, no change in the single-channel conductance was observed [ref. 21]. Additionally, the IK(M) in hippocampal mHippoE-14 neurons was also subject to simulation by SOL. Although the precise ionic mechanism underlying SQL actions on KM (or KCNQx) channels remains unresolved, these findings suggest a novel and non-canonical mechanism by which the SOL molecule interacts with the KM channel to enhance the whole-cell IK(M), ultimately reducing the firing frequency of spontaneous action potentials [ref. 21].
As demonstrated above in Figure 3, the Hys(V) of IK(M) evoked by the long isosceles-triangular Vramp (i.e., the upsloping and downsloping ramp) was revealed in GH3 cells [ref. 101]. The adjustments of such Hys(V) have been noticed to serve a role in fine-tuning the activity of ionic channels (e.g., KM channels) to respond when they are virtually needed [ref. 41,ref. 70,ref. 101,ref. 144]. Moreover, as shown in Figure 7, previous findings have disclosed the effectiveness of SOL in increasing the hysteretic strength of IK(M) associated with the voltage-dependent activation of instantaneous IK(M) in response to isosceles-triangular Vramp [ref. 101]. The SOL-mediated increase in IK(M)’s hysteretic area was suppressed by further addition of linopirdine, an inhibitor of IK(M). Under such scenarios, it is possible that intrinsic changes in the voltage dependence of the voltage-sensing machinery in KM (or KCNQx) channels, namely voltage-sensing domain relaxation [ref. 35,ref. 41], would be instantaneously and dynamically modulated during exposure to SOL. Whether the Hys(V) strength of IK(M) can be linked to the phosphoinositide metabolism in different cell types remains to be fully elucidated.
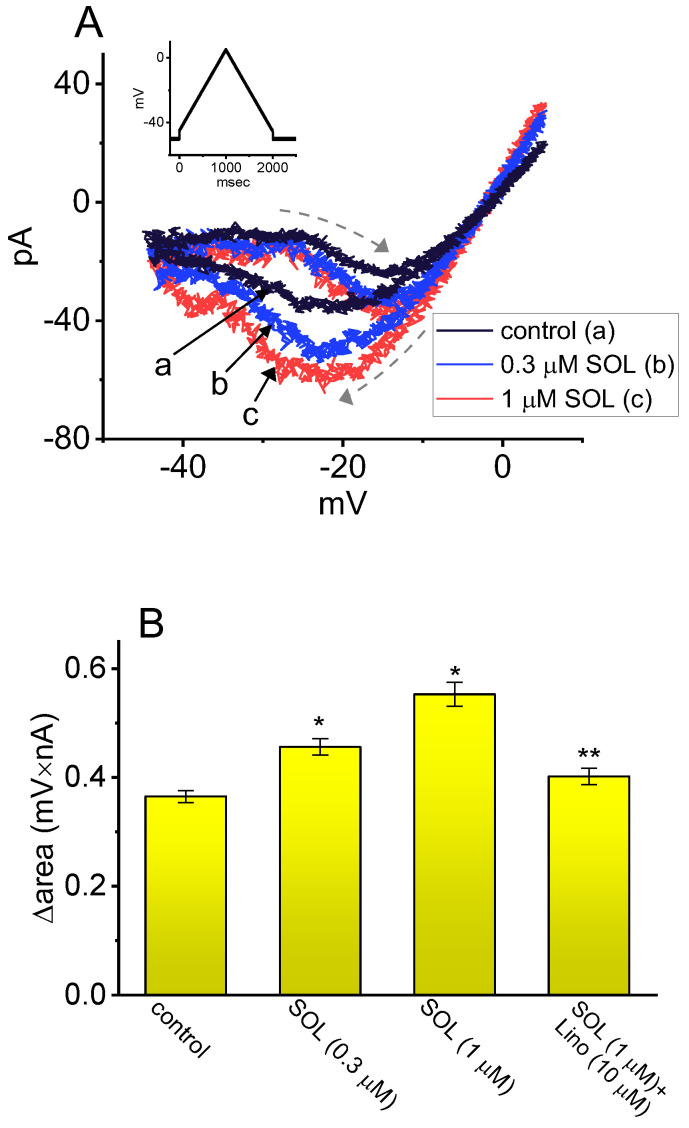
Several studies have shown that many compounds or drugs—such as cibenzoline, doxorubicin, and quinidine—exert their effects not by directly targeting cardiac muscarinic receptors but rather by modulating G protein-regulated inwardly rectifying K+ (GIRK) channels themselves [ref. 145,ref. 146,ref. 147]. It has been also reported that, during visceral contraction or glandular over-secretion, atropine, an antagonist of muscarinic receptors, does not demonstrate significant reversal effects on its own [ref. 148]. Therefore, whether certain compounds with anticholinergic activity, such as SOL, act indirectly on muscarinic receptors or on the activity of KM (KV7x, KCNQx) channels themselves remains a notable subject that requires further in-depth investigation and research in the future.
Recent ECG tracings have highlighted variability in the RR interval, a key indicator of heart variability [ref. 149]. This variability is closely linked to the function of the autonomic nervous system, particularly the balance of sympathetic and parasympathetic nerve activity. Heart rate variability testing combined with posture change maneuvers (such as supine to standing transitions) allows for the assessment of autonomic nervous system variations and the effects of small molecular modulators targeting IK(M) on autonomic function [ref. 150]. Therefore, investigating the regulation of the IK(M) and its influence on autonomic functions as demonstrated previously [ref. 9,ref. 10,ref. 11,ref. 12,ref. 30,ref. 136,ref. 151], including heart rate variability, offers a compelling opportunity for in-depth analysis and research.
4. Conclusions
Experimental studies have demonstrated that a range of compounds or drugs can directly and dynamically influence the magnitude and gating properties of IK(M), either through inhibition or stimulation. Table 1 and Table 2 provide the two-dimensional chemical structures of the compounds that inhibit or stimulate IK(M), respectively. These alterations in IK(M) can significantly affect cellular excitability, modifying firing frequencies and patterns across various cell types [ref. 6]. The slow activation and deactivation kinetics of IK(M), with time constants of approximately 100 and 30 ms, respectively, are believed to play a crucial role in synaptic delays. This is especially evident during slow synaptic processes in chemical transmission, such as long-term facilitation and depression [ref. 2,ref. 6,ref. 14,ref. 15,ref. 81]. Moreover, the subcellular and molecular basis for both the slow deactivation and the residual activation of IK(M) [ref. 39,ref. 40] are needed to be further explored.
It has been demonstrated that magnetoelectrical nanoparticles can modulate the firing activity, including the generation and propagation action potentials [ref. 139,ref. 152,ref. 153,ref. 154]. The hysteretic behavior induced by these nanoparticles under direct-current magnetic field highlights a potential mechanism for aberrant electrical activity. This effect may stem from phenomena such as magneto-Coulomb interactions or electroporation-induced currents, which could subsequently alter neural network structures and function [ref. 2,ref. 38,ref. 43,ref. 152,ref. 154,ref. 155,ref. 156,ref. 157,ref. 158,ref. 159,ref. 160].
However, it remains unclear whether magnetic fields combined with superferromagnetic nanoparticles, as described previously [ref. 161], influence the Hys(V) behavior of IK(M), the gating of other ionic currents, or currents generated by membrane electroporation in various cell types [ref. 38,ref. 43,ref. 152,ref. 157,ref. 160]. Addressing these uncertainties thus requires further research. Similarly, the combined application of transcranial magnetic stimulation and intravenously administered magnetic nanoparticles are important and warrant investigation for its potential impact on the Hys(V) behavior of ionic channels, electroporation-induced currents, or both. Despite these open questions, this non-invasive therapeutic technique shows promise for treating psychiatric conditions such as major depressive disorder, obsessive-compulsive disorder, and autism [ref. 162].
Mutations in KCNQx genes, leading to either gain-of-function or loss-of-function, can disrupt the electrical activity of excitable cells, assuming these genes are functionally and bioactively expressed [ref. 25,ref. 30,ref. 151]. This study provides proof-of-concept evidence that small molecules can effectively modulate the amplitude, gating kinetics, and instantaneous Hys(V) behavior of IK(M) in electrically excitable membranes. Because these proteins are predominantly located on the cell surface membrane, rather than sequestered in the cytoplasm or nucleus, they are more accessible to externally administered compounds. Furthermore, KCNQx-encoded proteins are strongly implicated in various diseases, such as epileptic disorders [ref. 25]. Their significant druggability, including the potential for subunit-specific modulation, makes them attractive targets for pharmaceutical development [ref. 16,ref. 81,ref. 137,ref. 138]. While techniques like polymerase chain reaction (PCR) testing and Western blotting are reliable for detecting gene and protein expression, additional experimental validation using electrophysiological methods across various cell types is crucial to uncover their functional and bioactive effects, fully confirming their therapeutic potential.
References
- F. Miceli, M.V. Soldovieri, M. Martire, M. Taglialatela. Molecular pharmacology and therapeutic potential of neuronal Kv7-modulating drugs. Curr. Opin. Pharmacol., 2008. [DOI | PubMed]
- E. Dossi, L. Zonca, H. Pivonkova, G. Milior, J. Moulard, L. Vargova, O. Chever, D. Holcman, N. Rouach. Astroglial gap junctions strengthen hippocampal network activity by sustaining afterhyperpolarization via KCNQ channels. Cell Rep., 2024. [DOI | PubMed]
- B. Graziano, L. Wang, O.R. White, D.H. Kaplan, J. Fernandez-Abascal, L. Bianchi. Glial KCNQ K+ channels control neuronal output by regulating GABA release from glia in C. elegans. Neuron, 2024. [DOI | PubMed]
- S. Sankaranarayanan, S.M. Simasko. Characterization of an M-like current modulated by thyrotropin-releasing hormone in normal rat lactotrophs. J. Neurosci., 1996. [DOI | PubMed]
- A.A. Selyanko, D.A. Brown. M-channel gating and simulation. Biophys. J., 1999. [DOI | PubMed]
- D.A. Brown, G.M. Passmore. Neural KCNQ (Kv7) channels. Br. J. Pharmacol., 2009. [DOI | PubMed]
- D.A. Brown, P.R. Adams. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature, 1980. [DOI | PubMed]
- P.R. Adams, D.A. Brown, A. Constanti. Pharmacological inhibition of the M-current. J. Physiol., 1982. [DOI | PubMed]
- H. Chen, P.A. Smith. M-currents in frog sympathetic ganglion cells: Manipulation of membrane phosphorylation. Br. J. Pharmacol., 1992. [DOI | PubMed]
- P.A. Smith, H. Chen, D.E. Kurenny, A.A. Selyanko, J.A. Zidichouski. Regulation of the M current: Transduction mechanism and role in ganglionic transmission. Canadian Journal of Physiology and Pharmacology, 1992. [DOI | PubMed]
- D.E. Kurenny, H. Chen, P.A. Smith. Effects of muscarine on K+-channel currents in the C-cells of bullfrog sympathetic ganglion. Brain Res., 1994. [DOI | PubMed]
- T. Mochidome, H. Ishibashi, K. Takahama. Bradykinin activates airway parasympathetic ganglion neurons by inhibiting M-currents. Neuroscience, 2001. [DOI | PubMed]
- A. Caspi, F. Benninger, Y. Yaari. KV7/M channels mediate osmotic modulation of intrinsic neuronal excitability. J. Neurosci., 2009. [DOI | PubMed]
- Z.W. Wang, L.O. Trussell, K. Vedantham. Regulation of Neurotransmitter Release by K+ Channels. Adv. Neurobiol., 2023. [PubMed]
- S. Jiang, B. Liu, K. Lin, L. Li, R. Li, S. Tan, X. Zhang, L. Jiang, H. Ni, Y. Wang. Impacted spike frequency adaptation associated with reduction of KCNQ2/3 exacerbates seizure activity in temporal lobe epilepsy. Hippocampus, 2024. [DOI | PubMed]
- J. Lemke, M. Gollasch, D. Tsvetkov, L. Schulig. Advances in the design and development of chemical modulators of the voltage-gated potassium channels K(V)7.4 and K(V)7.5. Expert. Opin. Drug Discov., 2025. [DOI | PubMed]
- R. Schimpf, C. Wolpert, F. Gaita, C. Giustetto, M. Borggrefe. Short QT syndrome. Cardiovasc. Res., 2005. [DOI | PubMed]
- C. Kubisch, B.C. Schroeder, T. Friedrich, B. Lütjohann, A. El-Amraoui, S. Marlin, C. Petit, T.J. Jentsch. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell, 1999. [DOI | PubMed]
- H. Meves, J.R. Schwarz, I. Wulfsen. Separation of M-like current and ERG current in NG108-15 cells. Br. J. Pharmacol., 1999. [DOI | PubMed]
- A.A. Selyanko, P. Delmas, J.K. Hadley, L. Tatulian, I.C. Wood, M. Mistry, B. London, D.A. Brown. Dominant-negative subunits reveal potassium channel families that contribute to M-like potassium currents. J. Neurosci., 2002. [DOI | PubMed]
- H.-Y. Cho, T.-H. Chuang, S.-N. Wu. The Effectiveness in Activating M-Type K+ Current Produced by Solifenacin ([(3R)-1-azabicyclo[2.2.2]octan-3-yl] (1S)-1-phenyl-3,4-dihydro-1H-isoquinoline-2-carboxylate): Independent of Its Antimuscarinic Action. Int. J. Mol. Sci., 2021. [DOI | PubMed]
- A.D. Wei, J.M. Ramirez. Presynaptic Mechanisms and KCNQ Potassium Channels Modulate Opioid Depression of Respiratory Drive. Front. Physiol., 2019. [DOI | PubMed]
- X. Zhan, C. Drummond-Main, D. Greening, J. Yao, S.W.R. Chen, J.P. Appendino, P.Y.B. Au, R.W. Turner. Cannabidiol counters the effects of a dominant-negative pathogenic Kv7.2 variant. iScience, 2022. [DOI | PubMed]
- V.K. Gribkoff. The therapeutic potential of neuronal K V 7 (KCNQ) channel modulators: An update. Expert. Opin. Ther. Targets, 2008. [DOI | PubMed]
- X. Chen, Y. Zhang, X. Ren, Q. Su, Y. Liu, X. Dang, Y. Qin, X. Yang, Z. Xing, Y. Shen. The SUMO-specific protease SENP2 plays an essential role in the regulation of Kv7.2 and Kv7.3 potassium channels. J. Biol. Chem., 2021. [DOI | PubMed]
- V.K. Gribkoff, R.J. Winquist. Potassium channelopathies associated with epilepsy-related syndromes and directions for therapeutic intervention. Biochem. Pharmacol., 2023. [DOI | PubMed]
- H. Oh, S. Lee, Y. Oh, S. Kim, Y.S. Kim, Y. Yang, W. Choi, Y.-E. Yoo, H. Cho, S. Lee. Kv7/KCNQ potassium channels in cortical hyperexcitability and juvenile seizure-related death in Ank2-mutant mice. Nat. Commun., 2023. [DOI | PubMed]
- B.C. Baculis, J. Zhang, H.J. Chung. The Role of Kv7 Channels in Neural Plasticity and Behavior. Front. Physiol., 2020. [DOI | PubMed]
- S. Maljevic, T.V. Wuttke, G. Seebohm, H. Lerche. KV7 channelopathies. Pflugers Arch., 2010. [DOI | PubMed]
- S.L. Vanhoof-Villalba, N.M. Gautier, V. Mishra, E. Glasscock. Pharmacogenetics of KCNQ channel activation in 2 potassium channelopathy mouse models of epilepsy. Epilepsia, 2018. [DOI | PubMed]
- P. Nappi, F. Miceli, M.V. Soldovieri, P. Ambrosino, V. Barrese, M. Taglialatela. Epileptic channelopathies caused by neuronal Kv7 (KCNQ) channel dysfunction. Pflugers Arch., 2020. [DOI | PubMed]
- S.N. Wu, M.C. Hsu, Y.K. Liao, F.T. Wu, Y.J. Jong, Y.C. Lo. Evidence for inhibitory effects of flupirtine, a centrally acting analgesic, on delayed rectifier k+ currents in motor neuron-like cells. Evid. Based Complement. Alternat Med., 2012. [DOI | PubMed]
- A.A. Selyanko, J.K. Hadley, I.C. Wood, F.C. Abogadie, P. Delmas, N.J. Buckley, B. London, D.A. Brown. Two types of K+ channel subunit, Erg1 and KCNQ2/3, contribute to the M-like current in a mammalian neuronal cell. J. Neurosci., 1999. [DOI | PubMed]
- Y.C. Lo, C.L. Lin, W.Y. Fang, B. Lőrinczi, I. Szatmári, W.H. Chang, F. Fülöp, S.N. Wu. Effective Activation by Kynurenic Acid and Its Aminoalkylated Derivatives on M-Type K+ Current. Int. J. Mol. Sci., 2021. [DOI | PubMed]
- C.A. Villalba-Galea, A.T. Chiem. Hysteretic Behavior in Voltage-Gated Channels. Front. Pharmacol., 2020. [DOI | PubMed]
- S. Chowdhury, B. Chanda. Estimating the voltage-dependent free energy change of ion channels using the median voltage for activation. J. Gen. Physiol., 2012. [DOI | PubMed]
- S.N. Wu, C.L. Wu, H.Y. Cho, C.W. Chiang. Effective Perturbations by Small-Molecule Modulators on Voltage-Dependent Hysteresis of Transmembrane Ionic Currents. Int. J. Mol. Sci., 2022. [DOI | PubMed]
- S.N. Wu, C.C. Yeh, H.C. Huang, W.H. Yang. Cholesterol depletion with (2-hydroxypropyl)-β-cyclodextrin modifies the gating of membrane electroporation-induced inward current in pituitary tumor GH3 cells: Experimental and analytical studies. Cell. Physiol. Biochem., 2011. [DOI | PubMed]
- B. Hille, E.J. Dickson, M. Kruse, O. Vivas, B.C. Suh. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta, 2015. [DOI | PubMed]
- J.B. Jensen, B.H. Falkenburger, E.J. Dickson, L. de la Cruz, G. Dai, J. Myeong, S.R. Jung, M. Kruse, O. Vivas, B.C. Suh. Biophysical physiology of phosphoinositide rapid dynamics and regulation in living cells. J. Gen. Physiol., 2022. [DOI | PubMed]
- A. Suma, D. Sigg, S. Gallagher, G. Gonnella, V. Carnevale. Ion Channels in Critical Membranes: Clustering, Cooperativity, and Memory Effects. PRX Life, 2024. [DOI]
- H.E. Balaguera, J. Bisquert. Accelerating the Assessment of Hysteresis in Perovskite Solar Cells. ACS Energy Lett., 2024. [DOI | PubMed]
- J. Bisquert. Hysteresis, Impedance, and Transients Effects in Halide Perovskite Solar Cells and Memory Devices Analysis by Neuron-Style Models. Adv. Energy Mater., 2024. [DOI]
- W.M. Postiglione, G. Yu, V. Chaturvedi, H. Zhou, K. Heltemes, A. Jacobson, M. Greven, C. Leighton. Mechanisms of Hysteresis and Reversibility across the Voltage-Driven Perovskite-Brownmillerite Transformation in Electrolyte-Gated Ultrathin La0.5Sr0.5CoO3-δ. ACS Appl. Mater. Interfaces, 2024. [DOI | PubMed]
- S. Tammireddy, M.N. Lintangpradipto, O. Telschow, M.H. Futscher, B. Ehrler, O.M. Bakr, Y. Vaynzof, C. Deibel. Hysteresis and Its Correlation to Ionic Defects in Perovskite Solar Cells. J. Phys. Chem. Lett., 2024. [DOI | PubMed]
- Z.S. Wang, Y. An, X. Ren, H. Zhang, Z. Huang, H.-L. Yip, Z. Huang, W.C.H. Choy. Device deficiency and degradation diagnosis model of Perovskite solar cells through hysteresis analysis. Nat. Commun., 2024. [DOI | PubMed]
- Z. Zou, H. Qiu, Z. Shao. Unveiling heterogeneity of hysteresis in perovskite thin films. Discov. Nano, 2024. [DOI | PubMed]
- O. Rönnelid, F. Elinder. Carboxyl-group compounds activate voltage-gated potassium channels via a distinct mechanism. J. Gen. Physiol., 2024. [DOI | PubMed]
- P.S. Cogan. A cautionary tale of paradox and false positives in cannabidiol research. Expert. Opin. Drug Discov., 2025. [DOI | PubMed]
- E. Agabiti Rosei, D. Rizzoni. Metabolic profile of nebivolol, a beta-adrenoceptor antagonist with unique characteristics. Drugs, 2007. [DOI | PubMed]
- T. Yndigegn, B. Lindahl, K. Mars, J. Alfredsson, J. Benatar, L. Brandin, D. Erlinge, O. Hallen, C. Held, P. Hjalmarsson. Beta-Blockers after Myocardial Infarction and Preserved Ejection Fraction. N. Engl. J. Med., 2024. [DOI | PubMed]
- A. Sigaroudi, M. Kinzig, O. Wahl, C. Stelzer, M. Schroeter, U. Fuhr, U. Holzgrabe, F. Sörgel. Quantification of Bisoprolol and Metoprolol in Simultaneous Human Serum and Cerebrospinal Fluid Samples. Pharmacology, 2018. [DOI | PubMed]
- D.V. Soloviev, M. Matarrese, R.M. Moresco, S. Todde, T.A. Bonasera, F. Sudati, P. Simonelli, F. Magni, D. Colombo, A. Carpinelli. Asymmetric synthesis and preliminary evaluation of (R)- and (S)-[11C]bisoprolol, a putative beta1-selective adrenoceptor radioligand. Neurochem. Int., 2001. [DOI | PubMed]
- K. Janssens, O. Krylyshkina, N. Hersmus, H. Vankelecom, C. Denef. Beta1-adrenoceptor expression in rat anterior pituitary gonadotrophs and in mouse alphaT3-1 and LbetaT2 gonadotrophic cell lines. Endocrinology, 2008. [DOI | PubMed]
- G.S. Nurmamedova, N.B. Gumbatov, I.I. Mustafaev. Level of hormones of pituitary-gonadal axis, penile blood flow and sexual function in men with arterial hypertension during monotherapy with bisoprolol and nebivolol. Kardiologiia, 2007. [PubMed]
- E.C. So, N.P. Foo, S.Y. Ko, S.N. Wu. Bisoprolol, Known to Be a Selective β1-Receptor Antagonist, Differentially but Directly Suppresses I(K(M)) and I(K(erg)) in Pituitary Cells and Hippocampal Neurons. Int. J. Mol. Sci., 2019. [DOI | PubMed]
- S.N. Wu, C.C. Yeh, H.C. Huang, E.C. So, Y.C. Lo. Electrophysiological characterization of sodium-activated potassium channels in NG108-15 and NSC-34 motor neuron-like cells. Acta Physiol., 2012. [DOI]
- H.T. Hsiao, Y.C. Liu, P.Y. Liu, S.N. Wu. Concerted suppression of I(h) and activation of I(K(M)) by ivabradine, an HCN-channel inhibitor, in pituitary cells and hippocampal neurons. Brain Res. Bull., 2019. [DOI | PubMed]
- H.T. Hsu, Y.T. Tseng, Y.C. Lo, S.N. Wu. Ability of naringenin, a bioflavonoid, to activate M-type potassium current in motor neuron-like cells and to increase BKCa-channel activity in HEK293T cells transfected with α-hSlo subunit. BMC Neurosci., 2014. [DOI | PubMed]
- B. Malawska, K. Kulig. Brivaracetam: A new drug in development for epilepsy and neuropathic pain. Expert. Opin. Investig. Drugs, 2008. [DOI]
- A. Strzelczyk, L. Kay, S. Bauer, I. Immisch, K.M. Klein, S. Knake, A. Kowski, R. Kunz, G. Kurlemann, L. Langenbruch. Use of brivaracetam in genetic generalized epilepsies and for acute, intravenous treatment of absence status epilepticus. Epilepsia, 2018. [DOI | PubMed]
- S.M. Hoy. Brivaracetam: A Review in Partial-Onset (Focal) Seizures in Patients with Epilepsy. CNS Drugs, 2016. [DOI | PubMed]
- P. Klein, J. Schiemann, M.R. Sperling, J. Whitesides, W. Liang, T. Stalvey, C. Brandt, P. Kwan. A randomized, double-blind, placebo-controlled, multicenter, parallel-group study to evaluate the efficacy and safety of adjunctive brivaracetam in adult patients with uncontrolled partial-onset seizures. Epilepsia, 2015. [DOI | PubMed]
- S. Tsymbalyuk, M. Smith, C. Gore, O. Tsymbalyuk, S. Ivanova, C. Sansur, V. Gerzanich, J.M. Simard. Brivaracetam attenuates pain behaviors in a murine model of neuropathic pain. Mol. Pain., 2019. [DOI | PubMed]
- A. Rizzo, S. Donzelli, V. Girgenti, A. Sacconi, C. Vasco, A. Salmaggi, G. Blandino, M. Maschio, E. Ciusani. In vitro antineoplastic effects of brivaracetam and lacosamide on human glioma cells. J. Exp. Clin. Cancer Res., 2017. [DOI | PubMed]
- G.M. Portela-Gomes, A. Lukinius, L. Grimelius. Synaptic vesicle protein 2, A new neuroendocrine cell marker. Am. J. Pathol., 2000. [DOI | PubMed]
- X. Yang, J. Bognar, T. He, M. Mohammed, I. Niespodziany, C. Wolff, M. Esguerra, S.M. Rothman, J.M. Dubinsky. Brivaracetam augments short-term depression and slows vesicle recycling. Epilepsia, 2015. [DOI | PubMed]
- J.M. Nicolas, J. Hannestad, D. Holden, S. Kervyn, N. Nabulsi, D. Tytgat, Y. Huang, H. Chanteux, L. Staelens, A. Matagne. Brivaracetam, a selective high-affinity synaptic vesicle protein 2A (SV2A) ligand with preclinical evidence of high brain permeability and fast onset of action. Epilepsia, 2016. [DOI | PubMed]
- A. Mittal, M.F. Martin, E.J. Levin, C. Adams, M. Yang, L. Provins, A. Hall, M. Procter, M. Ledecq, A. Hillisch. Structures of synaptic vesicle protein 2A and 2B bound to anticonvulsants. Nat. Struct. Mol. Biol., 2024. [DOI | PubMed]
- T.Y. Hung, S.N. Wu, C.W. Huang. The Integrated Effects of Brivaracetam, a Selective Analog of Levetiracetam, on Ionic Currents and Neuronal Excitability. Biomedicines, 2021. [DOI | PubMed]
- M. Kathmann, K. Flau, A. Redmer, C. Tränkle, E. Schlicker. Cannabidiol is an allosteric modulator at mu- and delta-opioid receptors. Naunyn Schmiedebergs Arch. Pharmacol., 2006. [DOI | PubMed]
- S.C. Britch, S. Babalonis, S.L. Walsh. Cannabidiol: Pharmacology and therapeutic targets. Psychopharmacology, 2021. [DOI | PubMed]
- A.I. Fraguas-Sánchez, A.I. Torres-Suárez. Medical Use of Cannabinoids. Drugs, 2018. [DOI | PubMed]
- L.D. Schurman, D. Lu, D.A. Kendall, A.C. Howlett, A.H. Lichtman. Molecular Mechanism and Cannabinoid Pharmacology. Handb. Exp. Pharmacol., 2020. [PubMed]
- A. Sideris, L.V. Doan. An Overview of Cannabidiol. Anesth. Analg., 2024. [DOI | PubMed]
- Y.C. Liu, E.C. So, S.N. Wu. Cannabidiol Modulates M-Type K+ and Hyperpolarization-Activated Cation Currents. Biomedicines, 2023. [DOI | PubMed]
- R.L. Dods, D. Donnelly. The peptide agonist-binding site of the glucagon-like peptide-1 (GLP-1) receptor based on site-directed mutagenesis and knowledge-based modelling. Biosci. Rep., 2015. [DOI | PubMed]
- A. Taddese, B.P. Bean. Subthreshold Sodium Current from Rapidly Inactivating Sodium Channels Drives Spontaneous Firing of Tuberomammillary Neurons. Neuron, 2002. [DOI | PubMed]
- P.M. Wu, P.C. Lai, H.Y. Cho, T.H. Chuang, S.N. Wu, Y.F. Tu. Effective Perturbations by Phenobarbital on I(Na), I(K(erg)), I(K(M)) and I(K(DR)) during Pulse Train Stimulation in Neuroblastoma Neuro-2a Cells. Biomedicines, 2022. [DOI | PubMed]
- P.M. Wu, Y.C. Lin, C.W. Chiang, H.Y. Cho, T.H. Chuang, M.C. Yu, S.N. Wu, Y.F. Tu. Effective Modulation by Lacosamide on Cumulative Inhibition of I(Na) during High-Frequency Stimulation and Recovery of I(Na) Block during Conditioning Pulse Train. Int. J. Mol. Sci., 2022. [DOI | PubMed]
- Y. Zhang, D. Li, Y. Darwish, X. Fu, L.O. Trussell, H. Huang. KCNQ Channels Enable Reliable Presynaptic Spiking and Synaptic Transmission at High Frequency. J. Neurosci., 2022. [DOI | PubMed]
- R.J. Bodnar. Endogenous Opiates and Behavior: 2016. Peptides, 2018. [PubMed]
- R. Raghav, R. Jain, A. Dhawan, T.S. Roy, P. Kumar. Chronic co-administration of nalbuphine attenuates the development of opioid dependence. Pharmacol. Biochem. Behav., 2018. [DOI | PubMed]
- T.K. Bindra, P. Kumar, G. Jindal. Postoperative Analgesia with Intrathecal Nalbuphine versus Intrathecal Fentanyl in Cesarean Section: A Double-Blind Randomized Comparative Study. Anesth. Essays Res., 2018. [DOI | PubMed]
- S. Singh, V. Sri Krishna, G. Cherian Ambooken, D.K. Peter. Nalbuphine: An underrecognized battlefield analgesic and its utilization in combat care and peripheral areas. Med. J. Armed Forces India, 2024. [DOI | PubMed]
- Y.Y. Liu, H.T. Hsiao, J.C. Wang, Y.C. Liu, S.N. Wu. Effectiveness of nalbuphine, a κ-opioid receptor agonist and μ-opioid receptor antagonist, in the inhibition of I(Na), I(K(M)), and I(K(erg)) unlinked to interaction with opioid receptors. Drug Dev. Res., 2019. [DOI | PubMed]
- L. Moravčíková, J. Královičová, Ľ. Lacinová. SNC80 and naltrindole modulate voltage-dependent sodium, potassium and calcium channels via a putatively delta opioid receptor-independent mechanism. Gen. Physiol. Biophys., 2018. [DOI | PubMed]
- A. Zeller, M. Arras, R. Jurd, U. Rudolph. Identification of a molecular target mediating the general anesthetic actions of pentobarbital. Mol. Pharmacol., 2007. [DOI | PubMed]
- A. Asgari, S. Semnanian, N. Atapour, A. Shojaei, H. Moradi-Chameh, S. Ghafouri, V. Sheibani, J. Mirnajafi-Zadeh. Low-frequency electrical stimulation enhances the effectiveness of phenobarbital on GABAergic currents in hippocampal slices of kindled rats. Neuroscience, 2016. [DOI | PubMed]
- C. Chu, N. Li, R. Zhong, D. Zhao, W. Lin. Efficacy of Phenobarbital and Prognosis Predictors in Women With Epilepsy From Rural Northeast China: A 10-Year Follow-Up Study. Front. Neurol., 2022. [DOI | PubMed]
- C. Onizuka, M. Irifune, A. Mukai, Y. Shimizu, M. Doi, K. Oue, M. Yoshida, T. Kochi, E. Imado, T. Kanematsu. Pentobarbital may protect against neurogenic inflammation after surgery via inhibition of substance P release from peripheral nerves of rats. Neurosci. Lett., 2022. [DOI | PubMed]
- Q. Wang, X. Liu, B. Li, X. Yang, W. Lu, A. Li, H. Li, X. Zhang, J. Han. Sodium Pentobarbital Suppresses Breast Cancer Cell Growth Partly via Normalizing Microcirculatory Hemodynamics and Oxygenation in Tumors. J. Pharmacol. Exp. Ther., 2022. [DOI | PubMed]
- M.K. Lo, R. Jordan, A. Arvey, J. Sudhamsu, P. Shrivastava-Ranjan, A.L. Hotard, M. Flint, L.K. McMullan, D. Siegel, M.O. Clarke. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses. Sci. Rep., 2017. [DOI | PubMed]
- C.J. Gordon, E.P. Tchesnokov, J.Y. Feng, D.P. Porter, M. Götte. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem., 2020. [DOI | PubMed]
- M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi, Z. Hu, W. Zhong, G. Xiao. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res., 2020. [DOI | PubMed]
- H.S. Andrews, J.D. Herman, R.T. Gandhi. Treatments for COVID-19. Annu. Rev. Med., 2024. [DOI | PubMed]
- G.-B. Lin, C.-L. Shih, R. Liutkevičienė, V. Rovite, E.C. So, C.-L. Wu, S.-N. Wu. Assessing the Impact of Agents with Antiviral Activities on Transmembrane Ionic Currents: Exploring Possible Unintended Actions. Biophysica, 2024. [DOI]
- N. Nallathambi, S.P. Naidu, S. Yogesh, P. Balamanikandan, C. Adithyan, S. Navvin, H. Seshadri, N. Mohanapriya, S. Prakash. A Rare Case of Coronavirus Disease 2019 Encephalitis Mimicking Creutzfeldt-Jacob Disease in an Immunocompromised Patient: A Case Report. Case Rep. Neurol., 2024. [DOI | PubMed]
- K.S. Schmitz, K. Handrejk, L. Liepina, L. Bauer, G.D. Haas, F. van Puijfelik, E.J.B. Veldhuis Kroeze, M. Riekstina, J. Strautmanis, H. Cao. Functional properties of measles virus proteins derived from a subacute sclerosing panencephalitis patient who received repeated remdesivir treatments. J. Virol., 2024. [DOI | PubMed]
- W. Trender, P.J. Hellyer, B. Killingley, M. Kalinova, A.J. Mann, A.P. Catchpole, D. Menon, E. Needham, R. Thwaites, C. Chiu. Changes in memory and cognition during the SARS-CoV-2 human challenge study. eClinicalMedicine, 2024. [DOI | PubMed]
- W.T. Chang, P.Y. Liu, Z.H. Gao, S.W. Lee, W.K. Lee, S.N. Wu. Evidence for the Effectiveness of Remdesivir (GS-5734), a Nucleoside-Analog Antiviral Drug in the Inhibition of I (K(M)) or I (K(DR)) and in the Stimulation of I (MEP). Front. Pharmacol., 2020. [DOI | PubMed]
- E. Amarh, J.E. Tisdale, B.R. Overholser. Prolonged Exposure to Remdesivir Inhibits the Human Ether-A-Go-Go-Related Gene Potassium Current. J. Cardiovasc. Pharmacol., 2023. [DOI | PubMed]
- F. Han, Y. Liu, M. Mo, J. Chen, C. Wang, Y. Yang, J. Wu. Current treatment strategies for COVID-19 (Review). Mol. Med. Rep., 2021. [DOI | PubMed]
- W.-M. Ng, S.-N. Wu, B.-M. Huang, E.C. So. Investigating the influence of XAV-939, a tankyrase inhibitor, on the density and gating of erg-mediated K+ currents in mouse MA-10 Leydig tumor cells. Eur. J. Pharmacol., 2024. [DOI | PubMed]
- F. Abedipour, H.H. Mirzaei, H. Ansari, N. Ehsanzadeh, A. Rashki, M.M. Vahedi, A. Rashki. Remdesivir-Related Cardiac Adverse Effects in COVID-19 Patients: A Case-Control Study. Drug Res., 2024. [DOI | PubMed]
- V. Terzić, J. Miantezila Basilua, N. Billard, L. de Gastines, D. Belhadi, C. Fougerou-Leurent, N. Peiffer-Smadja, N. Mercier, C. Delmas, A. Ferrane. Cardiac Adverse Events and Remdesivir in Hospitalized Patients With COVID-19: A Post Hoc Safety Analysis of the Randomized DisCoVeRy Trial. Clin. Infect. Dis., 2024. [DOI | PubMed]
- S. Harish, K. Bhuvana, G.M. Bengalorkar, T. Kumar. Flupirtine: Clinical pharmacology. J. Anaesthesiol. Clin. Pharmacol., 2012. [DOI | PubMed]
- M. Otto, L. Cepek, P. Ratzka, S. Doehlinger, I. Boekhoff, J. Wiltfang, E. Irle, G. Pergande, B. Ellers-Lenz, O. Windl. Efficacy of flupirtine on cognitive function in patients with CJD: A double-blind study. Neurology, 2004. [DOI | PubMed]
- B.S. Appleby, C.G. Lyketsos. Rapidly progressive dementias and the treatment of human prion diseases. Expert. Opin. Pharmacother., 2011. [DOI | PubMed]
- L.H.L. Miranda, A. Oliveira, D.M. Carvalho, G.M.F. Souza, J.G.M. Magalhães, J.A.C. Júnior, P. Lima, R.M.A. Júnior, S.P.L. Filho, H.M.A. Melo. Systematic review of pharmacological management in Creutzfeldt-Jakob disease: No options so far?. Arq. Neuropsiquiatr., 2022. [DOI | PubMed]
- F. Klinger, P. Geier, M.M. Dorostkar, G.K. Chandaka, A. Yousuf, I. Salzer, H. Kubista, S. Boehm. Concomitant facilitation of GABAA receptors and KV7 channels by the non-opioid analgesic flupirtine. Br. J. Pharmacol., 2012. [DOI | PubMed]
- Q. Xiong, Z. Gao, W. Wang, M. Li. Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol. Sci., 2008. [DOI | PubMed]
- G. Baranauskas, T. Tkatch, K. Nagata, J.Z. Yeh, D.J. Surmeier. Kv3.4 subunits enhance the repolarizing efficiency of Kv3.1 channels in fast-spiking neurons. Nat. Neurosci., 2003. [DOI | PubMed]
- G. Baranauskas. Ionic channel function in action potential generation: Current perspective. Mol. Neurobiol., 2007. [DOI | PubMed]
- C.-W. Huang, J.-J. Tsai, C.C. Huang, S.-N. Wu. Experimental and simulation studies on the mechanisms of levetiracetam-mediated inhibition of delayed-rectifier potassium current (KV3.1): Contribution to the firing of action potentials. J. Physiol. Pharmacol., 2009
- A.J. Labro, M.F. Priest, J.J. Lacroix, D.J. Snyders, F. Bezanilla. Kv3.1 uses a timely resurgent K+ current to secure action potential repolarization. Nat. Commun., 2015. [DOI | PubMed]
- H.T. Hsiao, J.C. Wang, S.N. Wu. Inhibitory Effectiveness in Delayed-Rectifier Potassium Current Caused by Vortioxetine, Known to Be a Novel Antidepressant. Biomedicines, 2022. [DOI | PubMed]
- C. Hilmas, E.F. Pereira, M. Alkondon, A. Rassoulpour, R. Schwarcz, E.X. Albuquerque. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci., 2001. [DOI | PubMed]
- S. Erhardt, S.K. Olsson, G. Engberg. Pharmacological manipulation of kynurenic acid: Potential in the treatment of psychiatric disorders. CNS Drugs, 2009. [DOI | PubMed]
- K. Sakakibara, G.G. Feng, J. Li, T. Akahori, Y. Yasuda, E. Nakamura, N. Hatakeyama, Y. Fujiwara, H. Kinoshita. Kynurenine causes vasodilation and hypotension induced by activation of KCNQ-encoded voltage-dependent K+ channels. J. Pharmacol. Sci., 2015. [DOI | PubMed]
- F. Resta, A. Masi, M. Sili, A. Laurino, F. Moroni, G. Mannaioni. Kynurenic acid and zaprinast induce analgesia by modulating HCN channels through GPR35 activation. Neuropharmacology, 2016. [DOI | PubMed]
- A.Y. Grishanova, M.L. Perepechaeva. Kynurenic Acid/AhR Signaling at the Junction of Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci., 2024. [DOI | PubMed]
- A.C. Paladini, M. Marder, H. Viola, C. Wolfman, C. Wasowski, J.H. Medina. Flavonoids and the central nervous system: From forgotten factors to potent anxiolytic compounds. J. Pharm. Pharmacol., 1999. [DOI | PubMed]
- A.G. Guimarães, S.V. Gomes, V.R. Moraes, P.C. Nogueira, A.G. Ferreira, A.F. Blank, A.D. Santos, M.D. Viana, G.H. Silva, L.J. Quintans Júnior. Phytochemical characterization and antinociceptive effect of Lippia gracilis Schauer. J. Nat. Med., 2012. [DOI | PubMed]
- E.P. Scholz, E. Zitron, C. Kiesecker, S. Lück, D. Thomas, S. Kathöfer, V.A. Kreye, H.A. Katus, J. Kiehn, W. Schoels. Inhibition of cardiac HERG channels by grapefruit flavonoid naringenin: Implications for the influence of dietary compounds on cardiac repolarisation. Naunyn Schmiedebergs Arch. Pharmacol., 2005. [DOI | PubMed]
- S. Saponara, L. Testai, D. Iozzi, E. Martinotti, A. Martelli, S. Chericoni, G. Sgaragli, F. Fusi, V. Calderone. (+/−)-Naringenin as large conductance Ca2+-activated K+ (BKCa) channel opener in vascular smooth muscle cells. Br. J. Pharmacol., 2006. [DOI | PubMed]
- J. Xu, C.E. Clancy. Ionic mechanisms of endogenous bursting in CA3 hippocampal pyramidal neurons: A model study. PLoS ONE, 2008. [DOI | PubMed]
- K.A. Youdim, M.Z. Qaiser, D.J. Begley, C.A. Rice-Evans, N.J. Abbott. Flavonoid permeability across an in situ model of the blood-brain barrier. Free Radic. Biol. Med., 2004. [DOI | PubMed]
- J. Qi, F. Zhang, Y. Mi, Y. Fu, W. Xu, D. Zhang, Y. Wu, X. Du, Q. Jia, K. Wang. Design, synthesis and biological activity of pyrazolo[1,5-a]pyrimidin-7(4H)-ones as novel Kv7/KCNQ potassium channel activators. Eur. J. Med. Chem., 2011. [DOI | PubMed]
- F. Zhang, Y. Mi, J.L. Qi, J.W. Li, M. Si, B.C. Guan, X.N. Du, H.L. An, H.L. Zhang. Modulation of K(v)7 potassium channels by a novel opener pyrazolo[1,5-a]pyrimidin-7(4H)-one compound QO-58. Br. J. Pharmacol., 2013. [DOI | PubMed]
- B.C. Teng, Y. Song, F. Zhang, T.Y. Ma, J.L. Qi, H.L. Zhang, G. Li, K. Wang. Activation of neuronal Kv7/KCNQ/M-channels by the opener QO58-lysine and its anti-nociceptive effects on inflammatory pain in rodents. Acta Pharmacol. Sin., 2016. [DOI | PubMed]
- M.Z. Du, R. Duan, D.C. Shao, X.Y. Zhang, F. Zhang, H. Li. [Antinociceptive efficacy of QO-58 in the monosodium lodoacetate rat model for osteoarthritis pain]. Zhonghua Yi Xue Za Zhi, 2017. [PubMed]
- W.-T. Chang, S.-N. Wu. Effective Activation of BKCa Channels by QO-40 (5-(Chloromethyl)-3-(Naphthalen-1-yl)-2-(Trifluoromethyl)Pyrazolo [1,5-a]pyrimidin-7(4H)-one), Known to Be an Opener of KCNQ2/Q3 Channels. Pharmaceuticals, 2021. [DOI | PubMed]
- C.-L. Wu, P. Fu, H.-Y. Cho, T.-H. Chuang, S.-N. Wu. Evidence for Dual Activation of IK(M) and IK(Ca) Caused by QO-58 (5-(2,6-Dichloro-5-fluoropyridin-3-yl)-3-phenyl-2-(trifluoromethyl)-1H-pyrazolol[1,5-a]pyrimidin-7-one). Int. J. Mol. Sci., 2022. [DOI | PubMed]
- J. Zhang, Q. Jia, J. Qi, H. Zhang, Y. Wu, X. Shi. Exploring in vivo metabolism and excretion of QO-58L using ultra-high-performance liquid chromatography coupled with tandem mass spectrometry. Eur. J. Pharm. Sci., 2018. [DOI | PubMed]
- Z.F. Sheng, H. Zhang, P. Zheng, S. Chen, Z. Gu, J.J. Zhou, J.G. Phaup, H.M. Chang, E.T.H. Yeh, H.L. Pan. Impaired Kv7 channel activity in the central amygdala contributes to elevated sympathetic outflow in hypertension. Cardiovasc. Res., 2022. [DOI | PubMed]
- C. Stagno, F. Mancuso, T. Ciaglia, C. Ostacolo, A. Piperno, N. Iraci, N. Micale. In Silico Methods for the Discovery of Kv7.2/7.3 Channels Modulators: A Comprehensive Review. Molecules, 2024. [DOI | PubMed]
- R. Taujale, N. Gravel, Z. Zhou, W. Yeung, K. Kochut, N. Kannan. Informatic challenges and advances in illuminating the druggable proteome. Drug Discov. Today, 2024. [DOI | PubMed]
- S. Zhang, D. Ma, K. Wang, Y. Li, Z. Yang, X. Li, J. Li, J. He, L. Mei, Y. Ye. A small-molecule activation mechanism that directly opens the KCNQ2 channel. Nat. Chem. Biol., 2024. [DOI | PubMed]
- D. Simpson, A.J. Wagstaff. Solifenacin in overactive bladder syndrome. Drugs Aging, 2005. [DOI | PubMed]
- C.K. Payne. Solifenacin in overactive bladder syndrome. Drugs, 2006. [DOI | PubMed]
- P. Abrams, K.E. Andersson. Muscarinic receptor antagonists for overactive bladder. BJU Int., 2007. [DOI | PubMed]
- D. Luo, L. Liu, P. Han, Q. Wei, H. Shen. Solifenacin for overactive bladder: A systematic review and meta-analysis. Int. Urogynecol. J., 2012. [DOI | PubMed]
- S.M. Rappaport, O. Teijido, D.P. Hoogerheide, T.K. Rostovtseva, A.M. Berezhkovskii, S.M. Bezrukov. Conductance hysteresis in the voltage-dependent anion channel. Eur. Biophys. J., 2015. [DOI | PubMed]
- T. Nakajima, Y. Kurachi, H. Ito, R. Takikawa, T. Sugimoto. Anti-cholinergic effects of quinidine, disopyramide, and procainamide in isolated atrial myocytes: Mediation by different molecular mechanisms. Circ. Res., 1989. [DOI | PubMed]
- S.N. Wu, T. Nakajima, T. Yamashita, E. Hamada, H. Hazama, K. Iwasawa, M. Omata, Y. Kurachi. Molecular mechanism of cibenzoline-induced anticholinergic action in single atrial myocytes: Comparison with effect of disopyramide. J. Cardiovasc. Pharmacol., 1994. [DOI | PubMed]
- Y. Hara, K. Temma, Z. Sekiya, A. Chugun, H. Kondo. Molecular mechanism of doxorubicin-induced anticholinergic effect in guinea-pig atria. Can. J. Physiol. Pharmacol., 2000. [DOI | PubMed]
- C.J. Fowler. Intravesical treatment of overactive bladder. Urology, 2000. [DOI | PubMed]
- K.J. Heilman, S.W. Porges. Accuracy of the LifeShirt (Vivometrics) in the detection of cardiac rhythms. Biol. Psychol., 2007. [DOI | PubMed]
- C.J.G. da Cruz, L.G.G. Porto, P. da Silva Rolim, D. de Souza Pires, G.L. Garcia, G.E. Molina. Impact of heart rate on reproducibility of heart rate variability analysis in the supine and standing positions in healthy men. Clinics, 2019. [DOI | PubMed]
- T. Berg. M-currents (Kv7.2-7.3/KCNQ2-KCNQ3) Are Responsible for Dysfunctional Autonomic Control in Hypertensive Rats. Front. Physiol., 2016. [DOI | PubMed]
- Y.C. Liu, P.C. Wu, D.B. Shieh, S.N. Wu. The effects of magnetite (Fe3O4) nanoparticles on electroporation-induced inward currents in pituitary tumor (GH3) cells and in RAW 264.7 macrophages. Int. J. Nanomed., 2012. [DOI | PubMed]
- A.K. Biswas, J. Atulasimha, S. Bandyopadhyay. The straintronic spin-neuron. Nanotechnology, 2015. [DOI | PubMed]
- S. Fiocchi, E. Chiaramello, A. Marrella, G. Suarato, M. Bonato, M. Parazzini, P. Ravazzani. Correction: Modeling of core-shell magneto-electric nanoparticles for biomedical applications: Effect of composition, dimension, and magnetic field features on magnetoelectric response. PLoS ONE, 2024. [DOI | PubMed]
- C. Möller, N. Arai, J. Lücke, U. Ziemann. Hysteresis effects on the input-output curve of motor evoked potentials. Clin. Neurophysiol., 2009. [DOI | PubMed]
- S. Datta, L. Marty, J.P. Cleuziou, C. Tilmaciu, B. Soula, E. Flahaut, W. Wernsdorfer. Magneto-Coulomb Effect in Carbon Nanotube Quantum Dots Filled with Magnetic Nanoparticles. Phys. Rev. Lett., 2011. [DOI | PubMed]
- E.C. So, K.L. Tsai, F.T. Wu, M.C. Hsu, K.C. Wu, S.N. Wu. Identification of minuscule inward currents as precursors to membrane electroporation-induced currents: Real-time prediction of pore appearance. Cell. Physiol. Biochem., 2013. [DOI | PubMed]
- S. Licciardi, G. Ala, E. Francomano, F. Viola, M. Lo Giudice, A. Salvini, F. Sargeni, V. Bertolini, A. Di Schino, A. Faba. Neural Network Architectures and Magnetic Hysteresis: Overview and Comparisons. Mathematics, 2024. [DOI]
- T. Sarkar, S. Biswas, S. Kakkar, A.V. Raghu, S.D. Kaushik, C. Bera, V.B. Kamble. Coexistence of multiple magnetic interactions in oxygen-deficient V2O5 nanoparticles. J. Phys. Condens. Matter, 2024. [DOI | PubMed]
- P. Ye, L. Huang, K. Zhao. Bidirectional Modulation on Electroporation Induced by Membrane Tension Under the Electric Field. ACS Omega, 2024. [DOI | PubMed]
- Z.W. Tay, S. Savliwala, D.W. Hensley, K.L.B. Fung, C. Colson, B.D. Fellows, X. Zhou, Q. Huynh, Y. Lu, B. Zheng. Superferromagnetic Nanoparticles Enable Order-of-Magnitude Resolution & Sensitivity Gain in Magnetic Particle Imaging. Small Methods, 2021. [PubMed]
- R. Li, J. Wang, X. Yu, P. Xu, S. Zhang, J. Xu, Y. Bai, Z. Dai, Y. Sun, R. Ye. Enhancing the effects of transcranial magnetic stimulation with intravenously injected magnetic nanoparticles. Biomater. Sci., 2019. [DOI | PubMed]