The Golden Age of Thermally Activated Delayed Fluorescence Materials: Design and Exploitation
Abstract
Since the seminal report by Adachi and co-workers in 2012, there has been a veritable explosion of interest in the design of thermally activated delayed fluorescence (TADF) compounds, particularly as emitters for organic light-emitting diodes (OLEDs). With rapid advancements and innovation in materials design, the efficiencies of TADF OLEDs for each of the primary color points as well as for white devices now rival those of state-of-the-art phosphorescent emitters. Beyond electroluminescent devices, TADF compounds have also found increasing utility and applications in numerous related fields, from photocatalysis, to sensing, to imaging and beyond. Following from our previous review in 2017 (Adv. Mater.2017, 1605444), we here comprehensively document subsequent advances made in TADF materials design and their uses from 2017–2022. Correlations highlighted between structure and properties as well as detailed comparisons and analyses should assist future TADF materials development. The necessarily broadened breadth and scope of this review attests to the bustling activity in this field. We note that the rapidly expanding and accelerating research activity in TADF material development is indicative of a field that has reached adolescence, with an exciting maturity still yet to come.
Affiliations: Organic Semiconductor Centre, EaStCHEM School of Chemistry, 7486University of St Andrews, St Andrews, Fife KY169ST, UK; Department of Physics, 151527Durham University, Durham DH1 3LE, UK; Organic Semiconductor Centre, SUPA School of Physics and Astronomy, University of St Andrews, St Andrews, Fife KY169SS, UK; EaStCHEM School of Chemistry, 151018The University of Edinburgh, Edinburgh, EH9 3FJ, UK; Institute of Organic Chemistry (IOC), 98929Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany; Department of Chemistry, University of Delhi, Delhi 110007, India; Laboratory for Computational Modeling of Functional Materials, Namur Institute of Structured Matter, Université de Namur, Rue de Bruxelles, 61, 5000 Namur, Belgium
License: © 2024 The Authors. Published by American Chemical Society CC BY 4.0 This article is licensed under CC-BY 4.0
Article links: DOI: 10.1021/acs.chemrev.3c00755 | PubMed: 39666979 | PMC: PMC12132800
Relevance: Moderate: mentioned 3+ times in text
Introduction
Being able to control the evolution, energy and spin of excitons in advanced materials underpins technologies ranging from organic light-emitting diodes (OLEDs), to solar cells, to optical sensing and imaging, to photocatalysis and wider technological applications. Many of these applications rely on efficient radiative decay of the generated exciton, that is, the generation of light. Light is not only generated as a result of photoexcitation (photoluminescence) but can be produced following electrical excitation (electroluminescence), chemical reaction (chemiluminescence), biochemical reaction (bioluminescence), application of mechanical force (mechanoluminescence), changes in crystallographic structure (crystalloluminescence), external sound (sonoluminescence), or high-energy ionized particle bombardment (cathodoluminescence, radioluminescence). In particular, the use of OLEDs (applied electroluminescent devices) has exploded over the last decade due to their superior performance in displays and promise for solid-state lighting (SSL) over preceding technologies such as liquid crystalline displays (LCDs), plasma display panels (PDPs), and inorganic light-emitting diodes (LEDs). Unlike now-ubiquitous LCDs, OLED display pixels are self-illuminating and individually addressable, and so do not require a uniform backlight pane. This allows pure black to be produced, resulting in a simpler and more energy-efficient display architecture with deeper achievable visual contrast. Unlike LCD or inorganic LED displays, OLED displays can also be fabricated on a wide range of substrates, offering ultrathin, foldable, flexible and even transparent displays supporting innovative technological applications. Primarily because of their superior picture quality and color gamut (supported by the endless tunability of photophysical properties of the organic materials) OLED displays are now used in the majority of high-end smartphoneref. ref1 and smartwatchref. ref2 screens, and are being increasingly adopted in the large-area television,ref. ref3 monitor, and automotive markets.ref. ref4
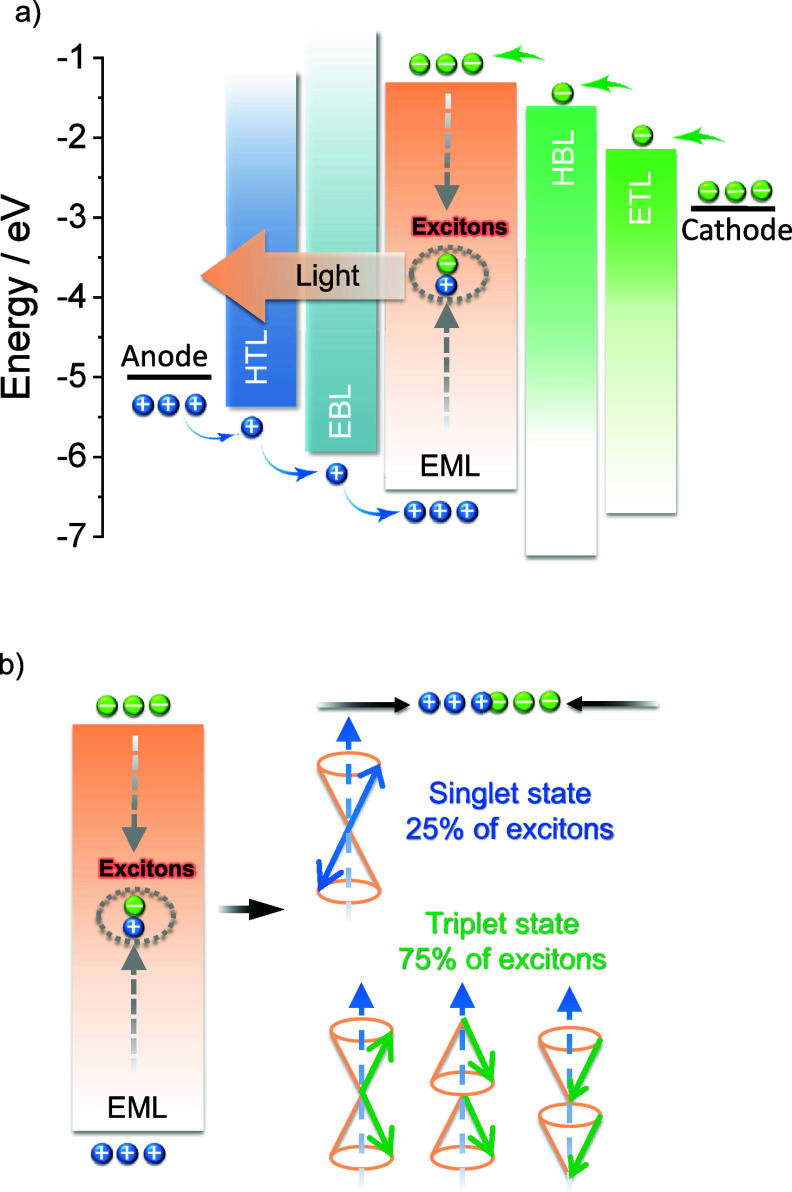
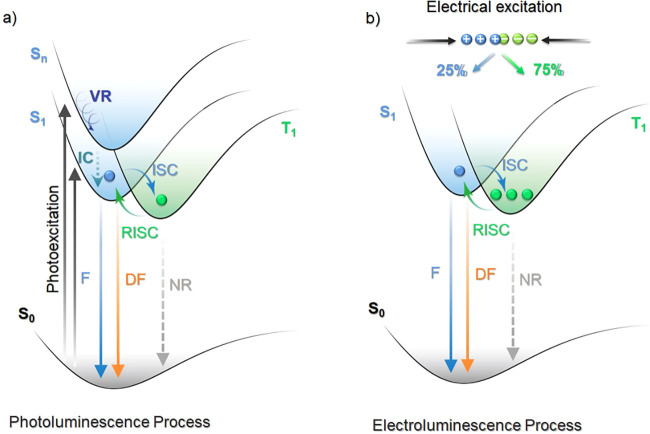
OLEDs consist of a multilayer stack of organic semiconductor materials that are sandwiched between the cathode and anode. These devices produce light upon the application of a voltage, which leads to the injection of charges (holes from the anode and electrons from the cathode) that migrate through the layers of the device, ultimately recombining within the emissive layer (EML) to form excitons (bound electron-hole pairs, Figure a). As both holes and electrons – which correspond directly to molecular radical cations and anions – possess spin 1/2, random recombination and Fermionic spin statistics dictate that the excitons formed will exist in a 1:3 ratio of singlet:triplet excited states (Figure b).ref. ref5 Subsequent radiative decay from the excited states to the ground state produces light emission.
OLED Performance Metrics
OLED performance is assessed primarily in terms of color, operational stability, and efficiency, the latter of which is quantified in terms of its external quantum efficiency (EQE). The EQE, η E QE, of the OLED is defined as the ratio of the number of photons exiting the device to the number of injected charges, and is dependent on the product of four terms according to equation eq1 :ref. ref6
In this expression γ is the Langevin recombination factor of the electron and holes, which is taken to be unity in efficient OLEDs but can be substantially less when charge recombination is not confined to the emissive layer. Φ PL is the photoluminescence quantum yield of the emissive layer or emissive dopant contained therein, which is the ratio of photons emitted to photons absorbed and quantifies the efficiency of light produced upon photoexcitation. β is the fraction of electrically produced excitons that can decay radiatively, which is typically 1 for singlet excitons, 0 for triplet excitons emanating from most organic compounds, and hence 0.25 for a simple 1:3 mixture of singlets and triplets (Figure b). The ability of advanced emitters to harvest otherwise non-emissive triplet excitons can in practice restore β to 1, overcoming this fundamental limit imposed by charge recombination. The combination of these three terms (γβΦ PL) is termed the internal quantum efficiency (IQE) and represents the ratio of photons generated within the OLED compared to charges injected.ref. ref7 The final term, χ out, is the light outcoupling efficiency, which is the fraction of light that escapes the device through a transparent electrode. This term is discussed in greater detail in the context of the orientation of the transition dipole moment (TDM) of the emitter below, although assuming isotropic TDM orientation of the emitter molecules, χ out is around 20–30% for devices fabricated on a flat glass substrate.
In the academic literature, the overall performance of an OLED is frequently judged simply on the maximum achieved value of external quantum efficiency, EQEmax. It is important to note that the EQEmax value typically occurs at very low luminance values, as OLEDs frequently operate most efficiently under minimal current density and corresponding low electrical stress. EQEmax values are consequently often reported at <1 cd m–2, corresponding to an impractically large 1 m2 OLED at this brightness giving off the same total light as a single candle. For applications in displays and lighting, much higher brightnesses on the order of hundreds or thousands cd m–2, respectively, are typically required, and so EQEmax is not a sufficient metric to judge the suitability of the device for most applications.ref. ref8 We therefore quote not only EQEmax, but also EQE at 100 cd m–2 (EQE100) and at 1,000 cd m–2 (EQE1000) wherever possible in this review, and encourage this practice in research articles.
For display applications, the color coordinate of the OLEDs, as defined by the Commission Internationale de l’Éclairage (CIE), is another key parameter which is directly linked to the spectral profile of the electroluminescence, Figure . A subset of all human-visible colors can be demarcated by the standard red-blue-green colur space (sRBG), which assigns ‘pure’ red, blue, and green as (0.64, 0.33), (0.15, 0.06), and (0.30, 0.60), respectively.ref. ref9 All other color points contained within a triangle of the connecting points (Figure , circles and solid white line) can then be generated from mixtures of the red, green, and blue primary colors. Reflecting consumer demand for more vibrant color displays (with access to a wider color gamut), the current industry standard for ultra HD-TVs advancing towards Rec. 2020, redefines the primary colors as (0.13, 0.05), (0.17, 0.80), and (0.71, 0.29) for blue, green and red, respectively (Figure , squares and dotted white line).ref. ref10 Achieving these more deeply saturated color coordinates remains an ongoing challenge for the OLED research community.
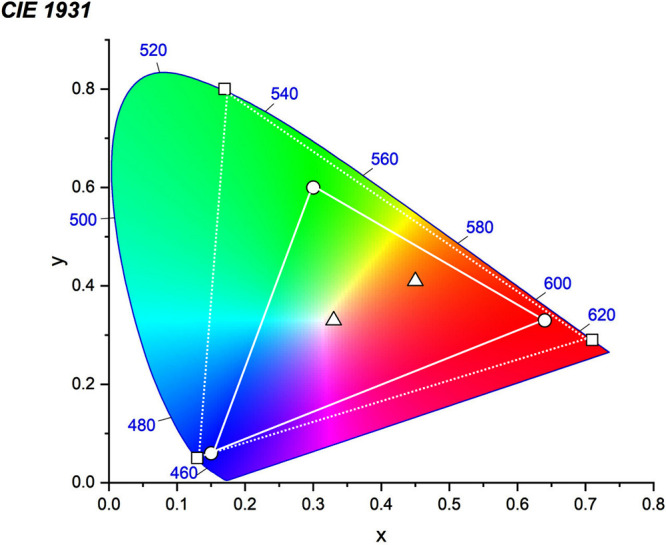
In contrast to multicolor displays, for ‘white’ lighting applications there are two key relevant CIE values. Pure white is defined as having CIE coordinates of (0.33, 0.33) and is similar to outdoor daylight, while warm white has orange-tinted CIE coordinates of (0.45, 0.41).ref. ref11 Warm white is the color associated with an incandescent light bulb, and is the most comfortable for human eyes. Ultimately, the CIE coordinates of an emitter are dependent on its emission peak, but also the spectral width of emission often quoted as the emission full width at half of the emission intensity maximum (FWHM). ‘Narrowband’ emission with small FWHM, frequently discussed in terms of color purity, is particularly prized as it allows emitters to more easily attain the saturated extreme ‘corner’ CIE coordinates required by evolving color standards. Broader emission spectra instead correspond to CIE values closer to white (0.33,0.33), as they contain a larger fraction of the entire visible spectrum. OLED pixels with these broad emission profiles therefore produce color displays limited to unappealing ‘white-ish’ desaturated color.
Beyond the efficiency and color of the device, its operational stability is also central to its performance and commercial applicability. The decrease in EQE with increasing driving voltage and luminance (efficiency roll-off) provides a useful insight into the stability of the OLED, where a stable device shows only a minimal efficiency roll-off and retains high EQE even at high luminances.ref. ref12 Another important and related metric to assess the stability of an OLED is the operational lifetime of the device (LTn), which is defined as the time taken for the device performance under constant driving current to degrade to a certain percentage of its initial brightness (subscript n). There is to date no universally agreed starting luminescence value nor a target percentage decrease used to report device lifetimes in the literature; however, LT90 and LT50 using an initial luminance of 1,000 cd m–2 are the most frequently reported device lifetime metrics. Short operational lifetimes are deeply unappealing for consumer applications of OLEDs, as the brightness of the panel will reduce noticeably through normal use. Different operational lifetimes of the differently colored display subpixels can also lead to a color-shift of the display, as the different colors reduce in achievable brightnesses at different rates, and so industry is most interested in LT90 or LT95.
Device stability is directly associated with the photochemical and electrochemical degradation of OLED materials. Under electrical excitation, high-energy species can form through undesired competing bi-excitonic processes such as triplet-triplet annihilation (TTA), singlet-triplet annihilation (STA), singlet-polaron annihilation (SPA), and triplet-polaron annihilation (TPA), which then initiate unwanted chemical transformations and degradation of OLED materials.ref. ref12 The frequency of these bi-excitonic processes is dependent on the density of the excitons, and become more prevalent at high exciton concentration and higher driving currents. Consequently, device lifetimes are not linear with driving current or starting luminance, as higher driving currents will cause the OLED to operate at lower EQE, with this lower emission efficiency permitting faster degradation within the device and a shorter lifetime. Therefore, the longer-lived triplet excitons in devices are often considered the primary driver of degradation. Rapidly harvesting these triplet excited states to efficiently produce light (or even just quenching them to the inert ground state) is viewed as the key to improving both the device efficiency and stability.
Exciton Harvesting in OLEDs
The first-generation of OLEDs contained simple fluorescent emitters, and thus light was only produced from the radiative decay of the singlet excitons, as radiative triplet exciton decay is a spin-forbidden process and thus in these devices these excitons only decayed non-radiatively (Figure ).ref. ref5 As a result, the β of these devices was 0.25 and the maximum IQE (IQEmax) of early fluorescent OLEDs was capped at 25%. In 1987, Tang and VanSlyke at Kodak were the first to report a functional fluorescent OLED that could operate at modest electric potential, employing Alq3 as the emitter with an EQEmax of ∼1%.ref. ref13 Despite the exploration of a wide range of fluorescent emitters in OLEDs, the limit of 25% IQEmax along with typical outcoupling capped the overall EQEmax to no greater than around 5% for these first-generation OLEDs.
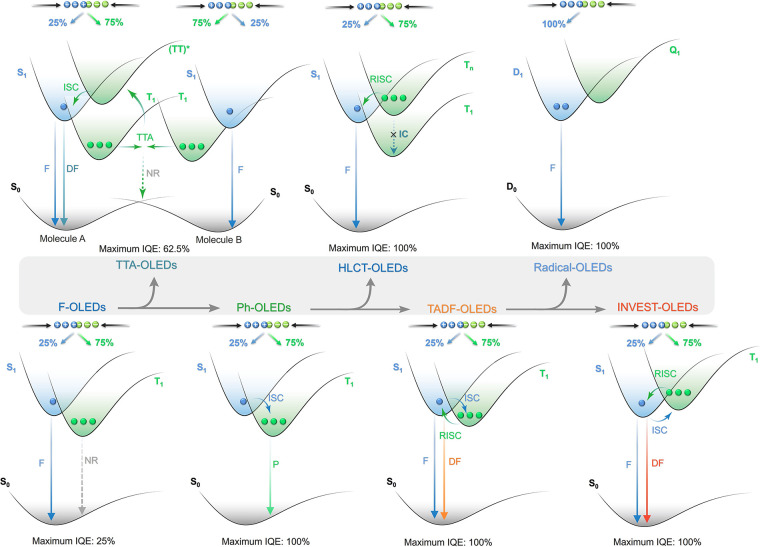
A step-change in efficiency was realized in 1998 when Baldo et al.ref. ref14 produced devices that exceeded the 5% EQEmax limit using phosphorescent emitters materials, developing so-called PhOLEDs. Organometallic phosphorescent emitters can harvest both singlet and triplet excitons to produce light because of the strong spin-orbit coupling (SOC) mediated by a central heavy transition metal ion (e.g., Pt(II), Ir(III)) within the material. The large SOC mediates singlet and triplet spin mixing that enables both intersystem crossing (ISC) of singlet excitons to become triplets, and radiative decay from the triplet excited state in the form of phosphorescence (Figure ). Thus, PhOLEDs can achieve up to 100% IQEmax.ref. ref15 This exciton harvesting strategy has now been widely adopted by industry and in commercialized OLEDs, with both the green and red subpixels of OLED displays typically employing phosphorescent emitters.ref. ref16
However, blue phosphorescent emitters have so far failed to – and may be fundamentally incapable of – delivering the required stability demanded by industry, and so blue subpixels typically contain a fluorescent TTA material.ref. ref17 These TTA or ‘triplet fusion’ materials are highly stable and can still harvest triplet excitons, but require two triplet excitons to generate one singlet, and so have a limiting β of ∼0.63 and maximum achievable IQE of ∼63% (Figure ). There thus remains a search for new emitter materials that (1) address the color and stability deficiencies of blue phosphorescent complexes and (2) can be produced more cheaply than those containing noble metals.ref. ref18 This context also explains the keen focus of the OLED community specifically on new blue emitters (as well as UV and NIR OLEDs),ref19,ref20 with other visible colors largely considered ‘solved’ problems,ref. ref21 with mature, commercialized products.
Beyond phosphorescence, a number of exciton harvesting mechanisms exist that can convert both singlet and triplet excitons into light. These include TTA discussed above, dynamics of excited states with hybridized local and intramolecular charge transfer (HLCT) character, materials with inverted singlet-triplet gap (INVEST), doublet organic radical emitters, and thermally activated delayed fluorescence (TADF). A ‘hot exciton’ or HLCT strategyref22−ref23ref24 involves the conversion of higher-energy triplet states (Tn>1) into singlets via reverse intersystem crossing (RISC), followed by radiative decay from the singlet manifold (Figure ).ref. ref23 Despite an IQEmax of up to 100%, such a RISC process from Tn must compete with typically rapid internal conversion to T1, and the device must also efficiently produce the higher-energy Tn triplet excitons in the device. This Tn recombination process remains poorly understood, and there are thus relatively few reports of devices using this mechanism to date.
Reports of molecules emitting via an INVEST mechanism have recently garnered much excitement in the organic semiconductor community, as this mechanism offers a tantalizingly simple mechanism for converting long-lived triplet excitons into light. Computational studiesref25−ref26ref27 have provided a preliminary framework for materials design, and the first report of an INVEST OLED has recently been published.ref. ref28 The INVEST mechanism involves a fundamental violation of Hund’s rule, where the S1 state is lower in energy than the T1 state, rendering RISC a formally exothermic process that should thus be accelerated (Figure ). The core challenge for INVEST research is to thus fully understand and apply design rules that can deliver materials with this ‘impossible’ ordering of excited states.
Beyond the singlet-triplet picture of excited states, recent work from Ai et al. has highlighted that organic radicals can be used as emitters in OLEDs.ref. ref29 As open shell systems, the excited states have spin multiplicity, as such there are no non-radiative tripletsref. ref30 yet IQEmax can still reach 100% (Figure ). Despite this promise, the chemical space is narrowly explored, based only on donor-decorated tris(trichlorophenyl) radicals, and emission is limited to the red region.ref. ref30
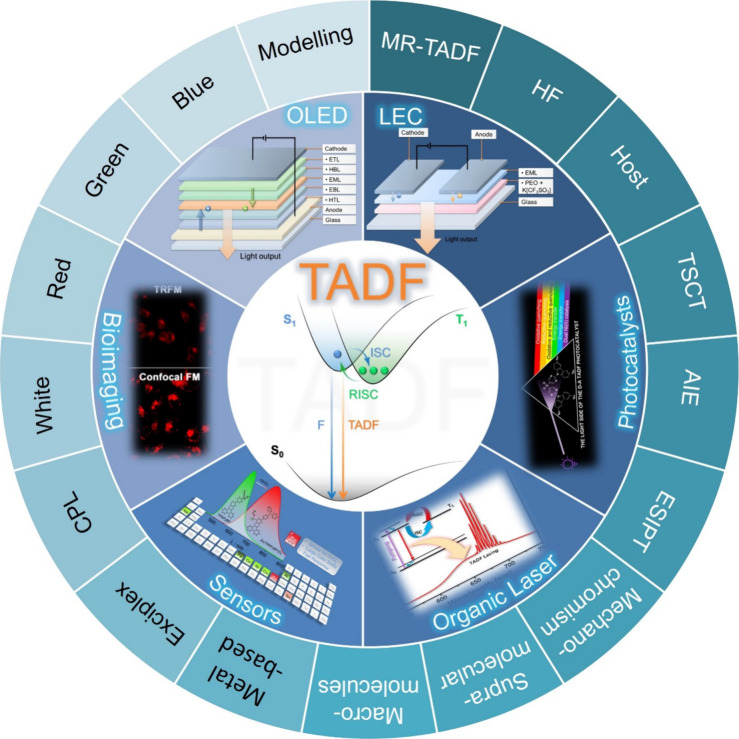
Now an established research theme globally, TADF involves the endothermic upconversion of triplet excitons into singlets followed by radiative decay, ensuring 100% IQEmax is possible (Figure ).ref. ref31 The research and development of TADF-based materials has progressed rapidly since the first report of a TADF material used in an OLED in 2009.ref. ref32 As well as driving progress in state-of-the-art device efficiency, the use of TADF materials has also branched out to include other uses in OLEDs such as host materials,ref. ref33 exciton harvesting materials in hyperfluorescent OLEDs,ref34,ref35 in other electroluminescent devices such as light-emitting electrochemical cells (LECs), as photocatalysts,ref. ref36 bioimaging reagents,ref. ref37 optical components in sensors,ref. ref38 and as materials in photovoltaics and lasing.ref. ref39
Since our last comprehensive review of TADF materials in 2017,ref. ref40 several other reviews have been published, focusing on various facets of TADF materials design and their applications.ref36,ref37,ref41−ref42ref43ref44ref45ref46ref47ref48ref49ref50ref51ref52ref53ref54ref55ref56 Readers are recommended to these reviews to gain an appreciation of the evolution of our understanding of TADF and the materials that operate via this mechanism. In this review we focus on the use of TADF in OLEDs as well as emphasising their wider applications.ref. ref40 We document the diversity of material categories that show TADF, moving beyond organic twisted donor-acceptor (D-A) systems and covering multi-resonant TADF (MR-TADF) materials, exciplexes, macromolecules such as polymers and dendrimers, and metal complexes. We discuss how TADF materials can also exhibit other interesting and valuable photophysical properties such as circularly polarized luminescence (CPL), aggregation induced emission (AIE), mechanochromism, and excited-state intramolecular proton transfer (ESIPT). Beyond their use as emitters in OLEDs, we also discuss examples where TADF materials have been employed as hosts, and as both terminal emitters and as exciton harvesters in hyperfluorescent OLEDs (HF-OLEDs). Finally, we cover their use in applications such as bioimaging, sensors, photocatalysis, supramolecular chemistry, and lasers.
Early History of Thermally Activated Delayed Fluorescence (TADF)
While fluorescence is typically a fast (ns timescale) process, the recognition of ‘slow’ microsecond-to-millisecond TADF is not new, and there are reports of this photophysical process dating back to 1929 (Figure ). Delayed emission was first reported by Perrin while studying Eosin Y,ref. ref57 where it was referred to as “fluorescence with long duration”, distinct from phosphorescence, which was termed “true phosphorescence” in this work.ref. ref58 Subsequent studies by Boudin in 1930, again studying the long-lived emission observed in a solution of Eosin Y, miscategorized the delayed emission as room-temperature phosphorescence (RTP).ref. ref59 Subsequent reports expanded on this initial incorrect assignment (vide infra).ref. ref60 TADF was described qualitatively to occur in fluorescein (Figure ) by Lewis et al. in the 1940s, with measurements made in boric acid glass showing distinct phosphorescence and fluorescence bands.ref. ref58 A temperature-dependent delayed fluorescence was reported as a “thermally activated” process, disappearing below −35 °C and with an approximate activation energy of 8 ± 1 kcal/mol. The putative mechanism was presented in the form of a Jablonski diagram, mimicking the TADF picture that is widely reproduced today, where TADF was called the “alpha process” to distinguish it from phosphorescence, termed the “beta process”. In 1961, studies of Eosin Y in solution undertaken by Parker and Hatchard demonstrated conclusively that the detected photoluminescence (PL) resulted from TADF,ref. ref60 work that directly led from the earlier observations of Boudin.ref. ref59 The measurements performed by Parker and Hatchard were in ethanol and glycol solutions, with the researchers firstly noting a low intensity red-shifted emission peak, missed by Boudin, which they ascribed to phosphorescence, while the main peak was assigned to TADF.ref. ref60 Emission intensity differences as a function of temperature between the two peaks helped confirm the TADF mechanism analogous to the earlier observations of Lewis et al. An in-depth kinetics study revealed an approximate rate constant for ISC (k ISC ∼ 4 × 107 s–1) and one for the “reverse process” (5 × 107 s–1), which we now know as RISC. They concluded that the activation energy should be equal to the energetic difference between singlet and triplet excited states, which we now know to be a crude approximation of the activation energy for RISC (vide infra). The changes in k RISC between ethanol and glycol were ascribed to their differing viscosities, with the greater viscosity of glycol translated to faster k RISC. Subsequent work in the 1960s sought to distinguish the delayed emission in TADF from the newly identified TTA mechanism, with TADF now referred as E-type fluorescence, distinct from P-type fluorescence (TTA), where the E and P monickers referring to Eosin-type and Pyrene-type emission, respectively, the molecules wherein these phenomena were observed.ref61,ref62
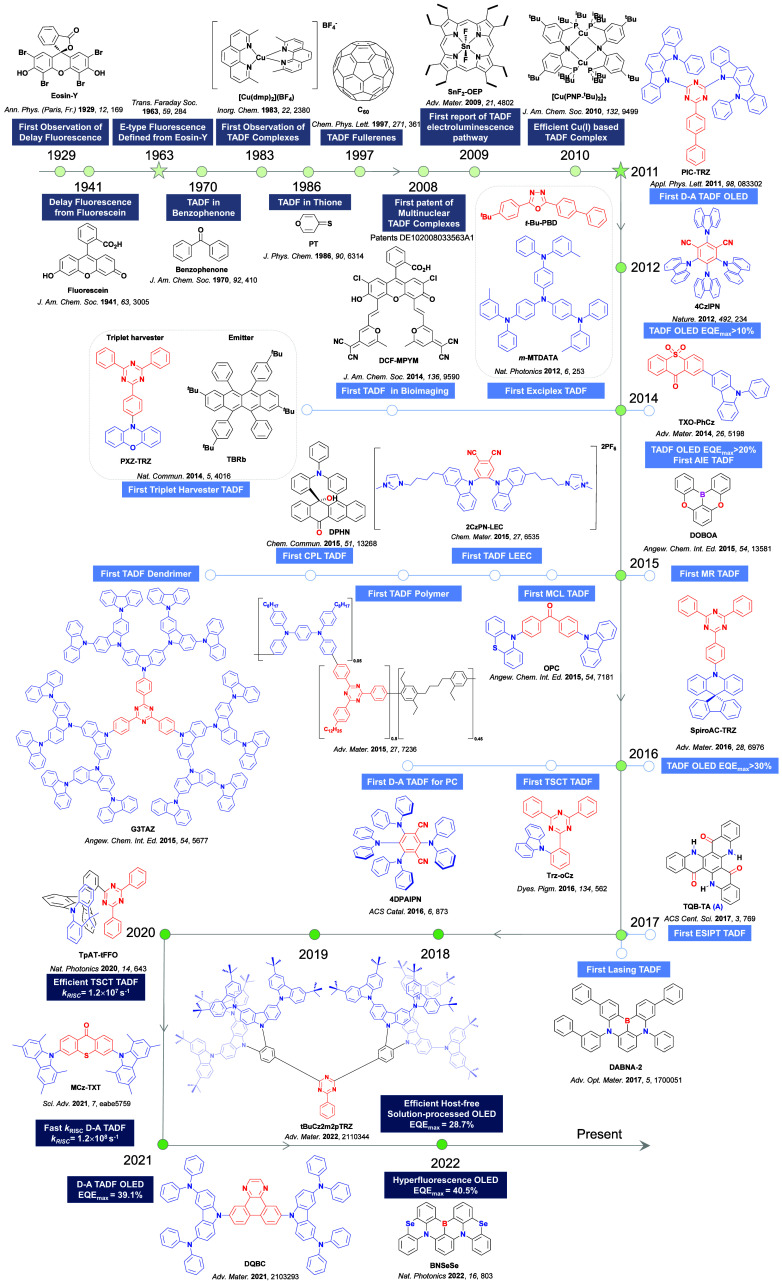
In the 1970s the origin of the delayed emission of benzophenone was probed independently by several groups (Figure ), with TADF initially proposed as the emission mechanism by Saltiel et al.ref. ref63 They observed a high-energy shoulder in the benzophenone emission spectra in carbon tetrachloride, assigned to fluorescence, and noted that the intensity of this band increased with temperature. Time-resolved PL studies by Parks, Brown and Singer, corroborated this assignment where they observed that fluorescence band persisted even after 10 ns in benzene solution and assigned this longer-lived emission as a delayed fluorescence distinct from prompt fluorescence.ref. ref64 Subsequent in-depth analysis by the same group using benzophenone and several derivativesref. ref65 demonstrated that the decay mechanism of benzophenone type materials is complex, with contributions to the PL from prompt fluorescence, TADF, TTA, and RTP. They calculated the triplet to singlet activation energy to be 3.9–5.1 kcal/mol across their series. Work on structurally related thiones undertaken initially by Maciejewski et al.ref. ref66 revealed similar behavior. They studied four structurally distinct thiones, each showing the same phenomenon of a high-energy shoulder of the PL spectra in deoxygenated non-polar solvents. Due to its long PL lifetime, the origin of this shoulder was ascribed to TADF. At temperatures below 220 K this spectral feature disappeared, indicating its appearance to originate from an endothermic process, while both the intensity of the TADF and phosphorescence bands showed a sensitivity to oxygen. Across the series of thiones, as ΔE ST decreased, the amount of TADF increased, with PT (Figure ) having the smallest ΔE ST of the series.
Observation of TADF was also documented in the late 1990s in C60 and C70 by Berberan-Santos and co-workers.ref67,ref68 It was first noted in C70, where the usually weak fluorescence observed was enhanced by two orders of magnitude with increasing temperature in liquid paraffin under deoxygenated conditions thanks to the TADF.ref. ref67 The ΔE ST was measured to be 26 kJ mol–1 (0.26 eV). The study of C60 followed shortly thereafter, with a somewhat larger measured ΔE ST of 35 kJ mol–1 in USP light oil solution.
TADF has also been observed in transition metal complexes, first noted in Cu(I) complexes in the 1980s, though this assignment was initially in dispute.ref69−ref70ref71 McMillin and co-workers first reported TADF in three mononuclear Cu(I) complexes containing different nitrogen heterocyclic ligands, with [Cu(dmp)2]BF4 investigated in detail (Figure ). In degassed DCM solutions a decreased emission intensity was observed with decreasing temperature, which the authors assigned to TADF. A thermal equilibrium between the triplet and singlet excited states was posited to occur due to the modest calculated ΔE ST of 1,800 cm–1 (0.22 eV). This two-state TADF mechanism was disputed by Parker and Crosby, who ascribe the emission decay to occur exclusively from the triplet state in this class of material.ref. ref69 Subsequent temperature-dependent measurements by McMillin and co-worker confirmed the original TADF mechanism.ref. ref71 The first example of a patent protecting the IP surrounding TADF metal complexes was authored by Yersin and Monkowius and had a priority filing in 2008 (published in 2010).ref. ref72 The patent disclosed the use of di- and trinuclear metal complexes that possessed small ΔE ST (50–2,000 cm–1/0.006–0.25 eV) to achieve triplet harvesting following thermal activation. Metals disclosed in the patent included mainly 2nd and 3rd transition row elements. This patent has now been withdrawn.
In 2009, the first example of a non-transition metal TADF emitter for OLEDs was used in terms of a tin(IV) porphyrin-based complex.ref. ref32 Six emitters were investigated photophysically, with an enhancement in emission intensity with increasing temperature confirming their TADF character. Of the family of six emitters studied, all of which were demonstrated to emit TADF from temperature-dependent PL studies, SnF2-OEP was probed in the greatest detail as a 2 wt% doped film in PVCz. Streak camera images showed TADF until 200 K, while overall ΦPL increased from 1.2% at this temperature to approximately 3.0% at 400 K, again consistent with TADF. The ΔE ST extracted from an Arrhenius analysis was 0.24 eV, this moderate gap resulted in inefficient TADF associated with a k RISC of 5 × 101 s–1 at 300 K. Devices were fabricated though no EQEmax was reported, expected to be small given the low ΦPL and inefficient k RISC. Although TADF was not conclusively demonstrated as the electroluminescent pathway given the poor device efficiency, the streak camera showed an enhancement in the electroluminescence intensity at elevated temperatures that is consistent with TADF as the emission mechanism. EQEmax values far surpassing the 5% fluorescence limit were first reported in 2010ref. ref73 in devices with the copper(I) complex, [Cu(PNP-tBu)2]2 where the EQEmax of a green-emitting device was 16.1% (Figure ). Though not explicitly discussed, it is likely that earlier examples of copper(I) based OLEDs likely emit by TADF.ref74,ref75 See Section sec9 for more details surrounding TADF metal complexes.
The first all-organic TADF OLEDs were reported in 2011 by Adachi and co-workers,ref. ref76 who developed the D-A emitter PIC-TRZ (Figure ); it is likely that other organic compounds have been miss-reported as fluorescent or TTA emitters prior to this report. In doped films and solution PIC-TRZ showed an oxygen sensitive delayed emission that is consistent with TADF. Devices showed an EQEmax reaching only 5.3%, this due to the low ΦPL of this compound. Streak camera and time-resolved EL studies confirmed that TADF was operational in the devices; the calculated IQE was 34%, surpassing the theoretical limit imposed on fluorescent systems. The following year the same group disclosed a new family of D-A compounds based on carbazolyl dicyanobenzenes (CDCBs).ref. ref31 In this seminal report, the authors reported sky-blue to red emitters and their use in state-of-the art OLEDs using all-organic emitters. The green-emitting OLED using 4CzIPN performed exceptionally well, with a EQEmax of 19.1%. This work demonstrated conclusively that high EQEmax devices could be fabricated using purely organic compounds as emitters. Since then, thousands of materials based on their initial D-A design have been reported. Since then, TADF emitters have been the subject of numerous studies and applications as represented in Figure .
A Deep Dive into the TADF Mechanism
TADF involves the upconversion of T1 excitons to S1 excitons via a RISC process, evidenced by a biexponential decay profile in the transient PL.ref. ref77 When a TADF compound is excited by light (photoexcitation), singlet excited states are first populated. These singlet excitons typically relax to S1 by rapid internal conversion (IC) and vibrational relaxation (VR) processes, typically following Kasha’s rule.ref. ref78 The generated S1 excitons can either decay radiatively or non-radiatively to the ground state, or be converted to T1 or Tn triplet excitons by ISC owing to the non-trivial SOC and the small singlet-triplet energy gap, ΔE ST, whereby these all rapidly populate T1 by IC and VR processes. The radiative decay from the S1 state is experimentally detected as prompt fluorescence with emission lifetimes, tp, on the order of 10–9 – 10–7 s. The triplet excitons can also decay radiatively as phosphorescence or non-radiatively. In TADF molecules, however, thermal upconversion to the singlet state via RISC can occur. The emission from S1 that results from the eventual radiative decay following RISC (or potentially several ISC/RISC cycles) is observed as delayed fluorescence, with the same emission spectrum associated with a distinct delayed emission lifetime, τd, of 108–102 s–1.ref. ref77 RISC is formally a spin-forbidden process based on the spin selection rules; however, RISC becomes possible once state mixing occurs. As RISC is an endothermic process, an increase in the temperature will result in a faster RISC rate.ref. ref79 This is manifested in an observed increase in the intensity and an acceleration in the decay rate of the delayed fluorescence with temperature, which partially distinguishes this mechanism from TTA.ref. ref77 Under electrical excitation singlet and triplet excitons are formed in a ratio of 1:3, resulting in a significantly larger initial triplet exciton population. The emission in the device results from fluorescence from singlet states, populated simultaneously by direct formation of singlet excitons and by RISC acting on triplet excitons (Figure ). In this process, the RISC is typically the rate-limiting step to delayed emission and a key determinant of OLED performance. Therefore, a deep understanding of the mechanism of RISC, methods to reliably quantify it, and an appreciation of the ratio of ISC:RISC that affects the relative population of singlet and triplet excitons are required to push TADF materials design further.
First-Order State Mixing
The strength of the first-order mixing between singlet and triplet excited states wavefunctions is governed by the first-order mixing coefficient, λ, (equation eq2 );ref. ref31
where H SOC is the SOC between the relevant singlet and triplet states, and ΔE ST is the energy difference between these states. Thus, λ is directly proportional to the magnitude of the SOC and inversely proportional to ΔE ST. The magnitude of the SOC is affected by the nature of the excited states and orbital types as described empirically by El-Sayed,ref. ref80 as well as the atomic mass of the atoms involved in the transitions to these states, known as the heavy atom effect. El-Sayed’s rule effectively states that ISC/RISC become less forbidden (partially allowed) when accompanied by a change in orbital angular momentum, as this ensures that the total angular momentum is conserved.ref81,ref82 In the original paper, this was exemplified by 1ππ* → 3ππ* and 1nπ* → 3nπ* transitions having negligible SOC and small transition rates while 1ππ* → 3nπ* and 1nπ* → 3ππ* transitions have much larger SOC and rates.ref. ref82 In TADF materials, most of the singlet and triplet excited states involves electronic transitions between π orbitals so that El-Sayed’s rule has to be revised in terms of the spatial localization of the molecular orbitals (MOs) involved in the excited state description. We distinguish cases whether the excited states are locally-excited (LE) and charge-transfer (CT) states (Figure ).ref. ref83
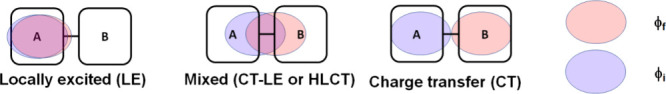
For an LE state the molecular orbitals (MOs) involved in the transition from the ground state are localised on the same part (or evenly throughout) the molecule (Figure ) leading to a strong MO spatial overlap, large ΔE ST and oscillator strength. There is thus a minimal electronic reorganisation upon the transition to the LE excited state, resulting in a very similar transition dipole in both the ground and in the LE excited state.
By contrast, a CT state is described by a transition from an occupied MO to an unoccupied MO that are relatively spatially segregated, and so there is a small exchange integral. Therefore, a large electronic density reorganisation upon transition to the CT excited state is observed, resulting in a large increase of the transition dipole in comparison to the ground state (Figure ).ref. ref84 Intermediate cases, termed mixed CT-LE states (sometimes also referred to as hybrid locally charge transfer – HLCT – states), can also exist where partial overlap between the occupied and the unoccupied MOs exists.ref. ref85 In this picture, there is a relatively larger SOC between a triplet state of LE character and a singlet state of CT character, while the SOC is much smaller when both the singlet and triplet states have CT character.ref. ref83 Accordingly, RISC occurring between a 3LE state and 1CT state would be allowed, while the direct upconversion from a 3CT to 1CT would be formally forbidden, assuming the transition between these excited states involves the exact same MOs. Since the majority of TADF emitters have S1 and T1 states carrying a strong CT character, SOC between these states remains very small and to thus ISC/RISC between these states is inefficient.ref. ref83
It is clear from equation eq2 that for efficient RISC to occur ΔE ST must be minimized. The threshold value of ΔE ST where non-negligible RISC is reported is often presented as <0.2 eV, with thermal energy at room temperature able to overcome the energy barrier between excited states.ref. ref86 When approximating that both T1 and S1 originate from HOMO to LUMO transitions, ΔE ST as well as the energies of the two states (E T and E S) can be framed within Hartree-Fock theory (equation eq3 ):
Since the HOMO-LUMO orbital energy difference, E, as well as the Coulomb repulsion energy, J, are the same for T1 and S1, ΔE ST can be expressed solely in terms of the exchange integral, K. The exchange integral quantifies the interaction between the unpaired electrons in S1 or T1 and S0, where the distribution can be approximated to the LUMO and HOMO, respectively (equation eq4 ):
where ϕ HOMO and ϕ LUMO are the spatial wavefunctions of the HOMO and LUMO with the respective complex conjugates ϕ HOMO * and ϕ LUMO *, e is the electronic charge, ε 0 is the vacuum permittivity, and r 1 and r 2 are the positions of electron 1 and electron 2, respectively. Based on equation eq4 , the simplest strategy to reduce the magnitude of K and thus also ΔE ST is to minimize the overlap of the electron density in the HOMO and LUMO. From a molecular design point of view, the principal manner to localize the HOMO and LUMO on different parts of the emitter is to adopt a twisted D-A architecture (vide infra) to induce a charge transfer character in the S1 and T1 excited states. A negative consequence of segregating the HOMO and LUMO onto different parts of the molecule results in a decrease in the radiative rate constant, k r, owing to reduced wavefunction overlap with the ground state that is quantified in terms of the oscillator strength, f, of the transition.ref31,ref77 The optimal emitter design therefore must carefully balance reducing ΔE ST (to improve the RISC efficiency) while preserving an adequately large f and fast k r, which both contribute to ΦPL.ref. ref87
The value of ΔE ST can be obtained spectroscopically from the measured fluorescence and phosphorescence spectra at low temperature. Either spectral onsets or peak values of these spectra have be used to estimate the energies of their corresponding states (E S and E T), with ΔE ST = E S – E T. Additionally, as the rate of RISC (k RISC) is temperature-dependent it can be approximated using an Arrhenius analysis (equation eq5 ):
where ΔE a is the activation energy, k B is the Boltzmann constant and T is temperature. If RISC were solely dependent on energetics, a direct correlation from E a and ΔE ST to k RISC would be expected.ref. ref88 Indeed, while there is a strong trend of smaller ΔE ST producing faster RISC, this relationship is not always linear, with numerous anomalous examples in the literature where the emitter possesses a relatively large ΔE ST yet unexpectedly fast k RISC as inferred from photophysical data.ref. ref89 Therefore, TADF efficiency cannot be explained only in terms of the first-order mixing of states; spin-vibronic coupling of states may also be important, which implies second-order mixing.
Second-Order State Mixing
Indeed, in recent years it has become widely accepted that the three-state model (S1, T1, and S0, that invokes only first-order mixing between S1 and T1) is too simple to account for the observed photophysics in many organic TADF emitters. In a second-order state mixing picture the Born–Oppenheimer approximation is broken,ref. ref90 and interactions of electronic and vibrational degrees of freedom must also be considered. In this mechanism, upconversion from T1 to S1 occurs through the involvement of higher-lying triplet states (Tn>1), which are accessible via reverse internal conversion (RIC) due to strong vibrational coupling between T1 and Tn>1.ref. ref83 If one of these higher-lying triplet states is of a different orbital nature than that of S1 (which is typically CT), then according to El-Sayed’s rule the SOC will be significantly enhanced, and RISC can then proceed much more readily. The vibrational coupling between T1 and Tn>1 is maximized when the T1 and Tn>1 states are sufficiently close in energy to enable efficient RIC and state-mixing to occur.ref83,ref91 Such a mechanism is frequently invoked to account for an efficient TADF process and the seemingly required involvement of both CT and LE states; however, evidence to support such a mechanism is most usually inferred using computational approaches (vide infra).ref90−ref91ref92ref93
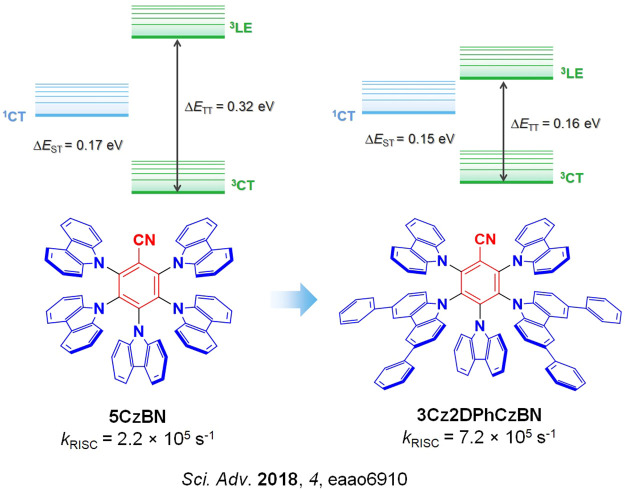
A clear example of second-order mixing was reported by Noda et al.,ref. ref91 who studied a series of structurally related emitters for which k RISC correlates very well with the evolution of the 3CT and 3LE energy gaps (ΔE TT), Figure . Based on the parent emitter, 5CzBN, the energy difference between 3CT and 3LE was calculated to be 0.32 eV (ΔE ST measured to be 0.17 eV) and the corresponding k RISC was 2.2 × 105 s–1. Replacing two of the carbazole donor groups for phenyl-substituted carbazoles introduced 3LE states closer to the lowest-lying 3CT, reducing the calculated ΔE TT to 0.16 eV for 3Cz2DPhCzBN, which translated into a faster k RISC of 7.2 × 105 s–1 (Figure ) while the measured ΔE ST remained similar at 0.15 eV. OLEDs using these two emitters showed strongly contrasting stability, where the LT97 device lifetime improved from 3 hours for the OLED with 5CzBN to 110 hours for the device with 3Cz2DPhCzBN.
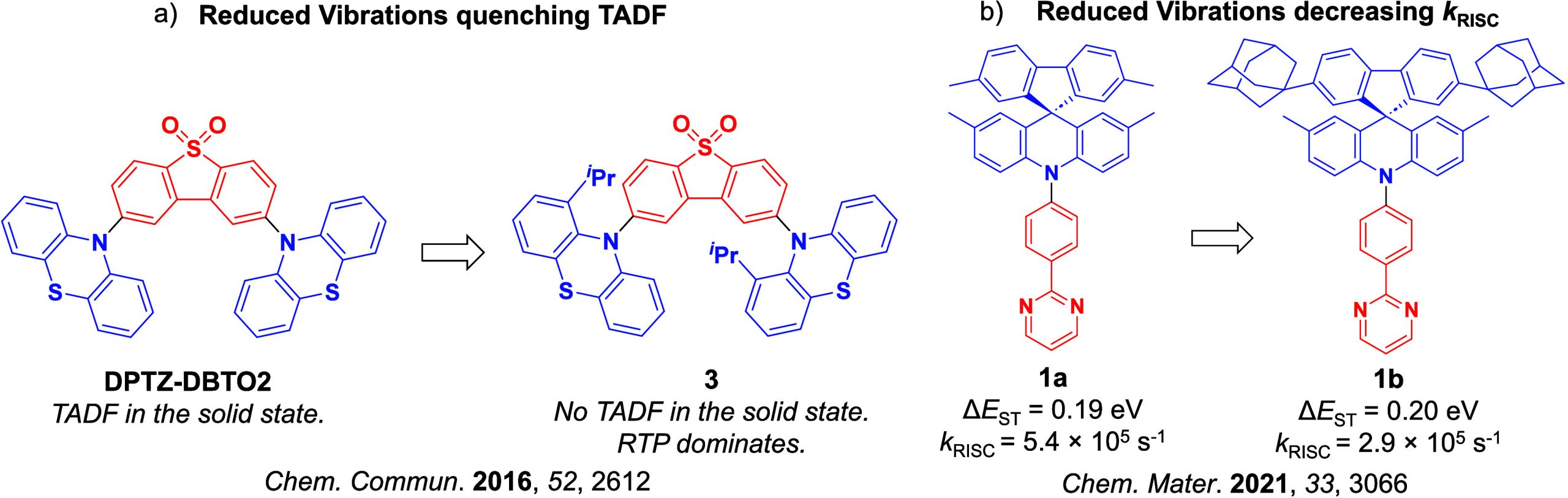
Due to large-scale dipole rearrangement and relaxation, the energy of CT states is dependent on the polarity of the medium surrounding the emitter, while the energy of LE states is largely insensitive to the surroundings. Thus, as the S1 state of TADF emitters is almost always CT in character, altering the polarity of the medium will also affect ΔE ST and k RISC.ref. ref83 External fine-tuning of the energy of the 1CT and 3CT states to closely align with the static 3LE levels was found to be possible with DPTZ-DBTO2 (Figure ), where the fastest k RISC was observed in hosts and solvents that simultaneously minimized the 1CT–3LE and 3CT–3LE gaps. The conformational flexibility inherent in the emitter can also affect the RISC rate.ref94,ref95 The rigidity of the compound can hinder the necessary vibrational motion that is required for coupling to occur between triplet states, ultimately suppressing RISC. Two studies are summarized here to exemplify this effect (Figure ). In the first, Ward et al. incorporated bulky substituents onto a phenothiazine (PTZ) donor of the parent emitter, DPTZ-DBTO2 (Figure a).ref. ref94 Clear TADF was observed for this compound in doped films; however, upon addition of isopropyl groups at the 1-position that reduces conformational flexibility as a result of increased sterics, TADF in material 3 was no longer observable, and instead RTP was the dominant emission mechanism. The second illustrative example was reported by Hempe et al., who showed that despite essentially unchanged singlet and triplet energy levels and similar ΔE ST of 0.19 eV and 0.20 eV for compounds 1a and 1b (Figure b), the addition of the bulky adamantly groups led to a decreased k RISC from 5.4 × 105 s–1 to 2.9 × 105 s–1 in ortho-dichlorobenzene, presumably due to their inertial impact on dampening D-A dihedral vibrations.ref. ref95
TADF Kinetics
Control of the various decay processes for the excited states in TADF compounds is crucial to both understand and account for the efficiency of the TADF molecules.ref. ref89 By analysing the transient PL of the compounds, many of the rate constants shown in equations eq6 –eq11 can be extracted. However, extracting every rate constant remains challenging, and in the case of k RISC is contentious, with several methods suggested across the literature, each using a different set of assumptions to simplify the mathematics. Most of these methods rely on fitting the emission decay to a pair of mono-exponential lifetimes in the prompt (k PF –1) and in the delayed (k DF –1) florescence regime.
Adachi and co-workers first calculated k RISC by assuming that there is no non-radiative decay from the singlet state (k nr S ≈ 0) and no phosphorescence (k r T ≈ 0) as equation eq6 :
where ΦPF is the photoluminescence quantum yield due to only the prompt fluorescence, and ΦDF is photoluminescence quantum yield from the delayed emission that is enabled by RISC.ref. ref96 These components of ΦPL are typically approximated from measurements in the presence (ΦPF) or absence (ΦPF + ΦDF) of atmospheric oxygen, although this has been demonstrated to introduce its own set of issues.ref. ref97
For TADF emitters that show significant delayed fluorescence (ΦDFΦPF –1 > 4) in the transient PL, Dias et al. proposed that k RISC could be approximated by equation eq7 ,ref. ref77 where the authors assumed that there is no non-radiative decay from the triplet state nor any phosphorescence (i.e., ΦRISC ≈ 1):
This model has been further refined by Kaji and co-workers to allow for the extraction of rate constants from samples that do not show a strong DF contribution in the transient PL (equation eq8 ):ref. ref98
To avoid the somewhat subjective and artificial nature of manually identifying and fitting exponential lifetimes to the prompt and delayed emission regimes, Monkman and co-workers have advanced a strategy that relies on simultaneous fitting of the entire transient PL to a three-level kinetic model using equation eq9 , under the assumption that the intensity of the PL is proportional to the singlet population:ref. ref99
To simplify the fit parameters, any non-radiative decay as well as phosphorescence were assumed to contribute negligibly (i.e., ΦPL ≈ 1). Transient absorption spectroscopy was used to independently assess the applicability of the fitting, which simultaneously generates a decay trace of triplet population [T1]. Similar to this approach, kinetics modelling of the transient electroluminescence has also been employed in a device context.ref. ref100 This approach can also be extended with additional kinetics terms, for example with the inclusion of ΦPL measurements to quantify nonradiative rates.ref101,ref102
Nguyen et al. developed a method to determine k RISC from the transient PL in the presence of an exciton quencher using a Stern–Volmer quenching experiments.ref. ref103 The prompt and delayed fluorescence rate constants are extracted for different quencher concentrations, where the prompt and delayed fluorescence rate constants for the pristine film (k PF,0 and k DF,0, respectively) are extrapolated by a fit. The fit of the delayed emission yields k ISC and the RISC rate is calculated according to equation eq10 , assuming no exciton decay from the triplet state. A similar approach was also recently reported for measuring energy transfer rates in hyperfluorescence (HF) blends, although this revealed that distributions of emitter-quencher distances in these films results in time-dependant quenching rates, which can lead to initially misleading trends.ref. ref104
Recently, Tsuchiya et al. presented a full analysis of the three-level system, which does not require any assumptions to be made and permits the extraction of all kinetics parameters from the photophysical experiments.ref. ref102 The RISC rate constant is calculated according to equation eq11 :
where k S = k r S + k nr S + k ISC. To calculate k ISC, the ratio of the delayed emission originating from S1 (i.e., fluorescence) to the delayed emission originating from T1 (phosphorescence) must be determined from an analysis of the spectral shift of the delayed emission over time.
As mentioned previously, most methods for determining k RISC rely on different sets of assumptions. Therefore, analysing the same material and photophysics using different models can lead to a range of values for k RISC.ref99,ref102,ref103 For example, by assuming no non-radiative decay from the singlet state, the value of k RISC is underestimated for materials with a ΦPL of less than 80%.ref. ref102 Recognizing this limitation, Tsuchiya et al. introduced an evaluation of the rate constants obtained with equations eq6 –eq8, revealing a range of k RISC due to over- and underestimations when assuming negligible non-radiative decay from either the singlet or triplet states. Despite this diversity of calculation methods, it is widely recognized that new D-A TADF materials require RISC rates of ∼106 or faster in order to achieve leading device performance.
OLED Fabrication
While relatively quick and convenient photophysical measurements can guide the design of TADF materials, once high-performance candidates are identified their electroluminescence performance must be directly confirmed. Generally, the fabrication of OLEDs occurs using one of two approaches: thermal evaporation under vacuum, which is restricted to low molecular weight-based materials (typically <1000 dalton),ref105,ref106 and solution-processing techniques such as spin-coating, inkjet printing, or doctor blading (which require minimum levels of solubility).ref107−ref108ref109 Solution-processed OLEDs therefore are the only option for high molecular weight materials such as polymersref. ref110 and dendrimers,ref. ref111 which cannot be thermally evaporated; solution-processed OLEDs using low molecular weight emitter is also possible, assuming their solubility and film-forming properties allow formation of homogenous amorphous films. OLEDs based on TADF emitters usually employ a multilayer architecture. Careful choice of the materials in these emissive and transport/injection layers permits optimal charge injection, transport, and exciton recombination kinetics that support high-efficiency devices (Figure and Figure ).
Outcoupling
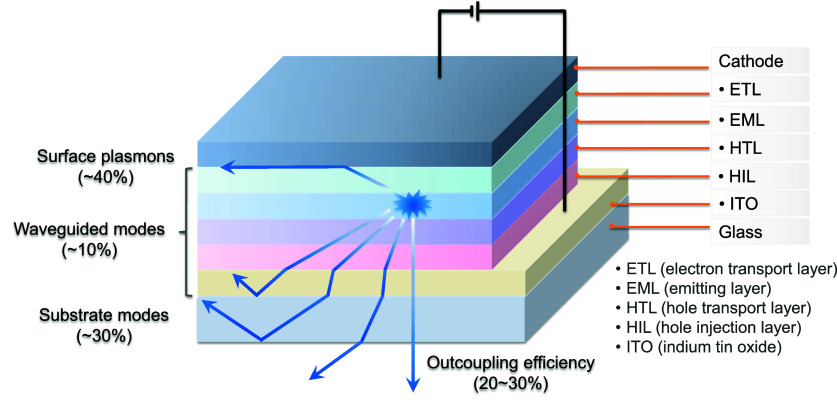
The photons generated inside the emissive layer of an OLED have a number of pathways by which their energy can dissipate, although only those that can escape the device are useful. These pathways include waveguided modes, substrate modes, surface plasmon polaritons (SPPs), (re)absorption, and the aforementioned direct emission (Figure ), which are described in detail in the literature.ref112−ref113ref114ref115 The proportion of light energy that is lost to each of these pathways depends on a number of properties of the device itself, including the thickness and refractive indices of each of the constituent layers, the wavelength of the emitted light and any cavity effects related to the metallic electrode(s), the surface morphology of the glass-air interface, and, most importantly, the angle relative to the surface of the device at which the light is emitted.ref. ref113 At shallow angles, waveguided modes in either the glass substrate or in the organic layers themselves are greatly favoured, as these pathways trap light by total internal reflection at either the organic layer interfaces or the glass/air interface. Likewise, SPPs require very shallow emission angles as they are dependent on near-field coupling of the generated light to the metallic cathode surface. All of these aforementioned modes, which are unproductive and lead to lower efficiency devices, are, however, avoided at high emission angles (i.e., emission normal to the substrate plane), where instead photons can more easily escape the device layers and thus be used productively in the outside world.
Outcoupling Efficiency and Emitter Orientation
The outcoupling efficiency, χ out of a device is the ratio of internally generated photons to externally emitted photons, ideally 100%. χ out is one of the four crucial parameters of an OLED that constitutes its EQE (equation eq1 ) and is reduced by photons that are coupled into waveguided or SPP modes, and thus is dependent on the angle of emission of the photons from within the device. The angle at which light is emitted from an excited molecule is itself not random, but rather is dependent on the orientation of the TDM of the emissive electronic transition. The majority of light emitted will be perpendicular to the TDM vector, as this is the direction in which the interaction between the oscillating molecular electric dipole and propagating light wave is strongest and thus emission is most likely to occur.ref. ref112 Therefore, if a majority of the emitting molecular TDMs are orientated parallel to the plane of the OLED, then a greater proportion of light will leave the device via direct emission, resulting in greater overall efficiency. Unfortunately most molecules are deposited randomly to form isotropic films, and with no preferential orientation of the ensemble of TDMs in the EML, light outcoupling efficiency is typically limited to only 20–30%, meaning that as much as 80% of the generated photons remain trapped within the layers of the OLED. This is the origin of the 20–30% EQEmax limit experienced by many devices.
The overall χ out of a device is influenced not only by any TDM orientation of the EML, but also by other properties of the device such as the emission wavelength and the thickness and refractive indices of the device layers. However, optical simulations have demonstrated that the χ out of a device can be increased by at least as much as 50% by preferentially orientating the TDMs, compared to the isotropic case. In a device with an EQEmax of 20% and with an isotropic arrangement of TDMs, an equivalent device where all TDMs are preferentially orientated would therefore achieve an EQEmax of 30%, and reports have demonstrated that even this limit can be surpassed with further device engineering.ref. ref113 This headroom for significantly improved OLED efficiency has attracted significant attention from the community to design emitters that possess a preferentially (horizontally) orientated TDM in the as-deposited film state. Indeed, as the IQE of modern devices has effectively reached 100% with triplet harvesting strategies, light outcoupling remains as a key factor for advancing higher efficiency devices.
Controlling Orientation
A number of molecular properties are now known to influence the TDM alignment of emitters in a film.ref. ref112 Of these, the most well documented are the length and/or width of the emitter (and/or the aspect ratio of the emitter), the glass-transition temperature of the host, the temperature of the substrate during deposition and, perhaps most importantly, the deposition mechanism by which the film is made. No single one of these parameters can be used in isolation to control or predict the TDM alignment of the emitters in a film, but TDM alignment is typically achieved by using emitters with greater molecular length and/or width (i.e., larger aspect ratio), hosts with a higher glass-transition temperature, substrates with a lower temperature and films synthesised by vacuum deposition (as opposed to solution-processing such as spin-coating). In particular any preferential horizontal alignment of the TDMs exhibited in a vacuum-deposited film is typically lost in an equivalent solution-processed film, as the molecules deposit to form a film all-at-once from a randomised solution state, rather than gradually building up from a molecular beam on a surface.
Despite these advancements, a unifying theory by which complete horizontal TDM alignment can be reliably achieved remains elusive, as the interplay between the different effects influencing TDM alignment is poorly understood.ref. ref112 In addition, new parameters that impact TDM orientation are still being reported, and it is more than likely that yet more await discovery. We recently reviewed this topic in-depth,ref. ref112 and concluded that the following are all parameters that can induce horizontal TDM orientation in TADF emitters: high molecular weight of the emitter; high linearity of the emitter; high molecular weight of the host; small thickness of the emitter; greater length of the emitter relative to the host; and high glass transition temperature of the host. It was additionally found that the relative importance of each of these parameters depends on the exact system under study. For example, for low molecular weight emitters (MW < 600 g mol–1) the most influential parameter is the glass transition temperature of the host, while for heavier emitters the degree of horizontal orientation is better correlated to the molecular weight of the emitter itself. Finally, in the literature, many authors have used arguments relating to the high aspect ratio of the emitter to explain preferential horizontal orientation and the resulting high EQEmax. These arguments are supported by the extensive work by Yokoyama et al.ref. ref116 in demonstrating that molecules with higher aspect ratios tend to preferentially orient horizontally in thin films, thus also aligning the TDM horizontally so long as the TDM is aligned along the plane of the molecule itself. However, it is rare for the aspect ratio of a molecule to be quantified in the literature, making the true strength of this relationship hard to ascertain.ref. ref112 Further, it is unclear whether the aspect ratio of a molecule is a meaningful predictor of TDM orientation in its own right. Instead, it may merely be a proxy for other parameters, such as molecular length and weight, as molecules with higher aspect ratios tend to be longer and therefore heavier. Thus, the challenge of controlling the TDM orientation of the emitters within the EML remains unsolved, and further research is required in order to construct a set of comprehensive design paradigms by which perfectly horizontal TDM orientation can be reliably enforced.
Outlook
Although the mechanism for TADF is much more complex than simple thermal upconversion of T1 to S1 states, in practice the magnitude of ΔE ST largely dictates the feasibility of the process and reducing ΔE ST is almost always a desirable strategy for the design of new TADF materials. According to equation eq3 , reducing the electron density overlap between HOMO and LUMO can effectively reduce the ΔE ST, provided the transition is predominantly HOMO to LUMO. This has been achieved in D-A systems, which can be in the form of twisted intramolecular D-A compounds or molecules that possess pseudo co-planar D and A groups that possess through-space charge transfer (TSCT) states, or in exciplexes where distinct donor and acceptor molecules interact weakly intermolecularly via π-π bonding. In this context a donor is an electron-rich group while an acceptor is an electron-deficient moiety, where the HOMO is situated on the donor and the LUMO on the acceptor.ref. ref86
For twisted D-A compounds, minimization of the exchange integral and thus ΔE ST can be achieved through (1) the use of substituents close to the D-A bond such as addition of methyl groups to confer a highly twisted conformation,ref. ref117 or (2) the inclusion of multiple donors or acceptors, which forces large torsions to mitigate steric congestion between these moieties.ref. ref31 Donors like 9,9-dimethyl-9,10-dihydroacridine (DMAC), phenoxazine (PXZ), and phenothiazine (PTZ) that are linked to acceptors via the nitrogen atom adopt highly twisted conformations owing to their bulky nature.ref. ref117 Although thousands of D-A based TADF emitters have been reported, they are ultimately composed of a relatively limited diversity of D or A units (Figure ).ref40,ref42,ref87 Color tuning in D-A TADF systems is possible by altering the strength of the donor and acceptor groups, which affects both the band gap, ΔE, as well as the energy of the excited states. Increasing the donor strength destabilizes the HOMO, while increasing the acceptor strength stabilizes the LUMO, both of which decrease the energy of the excited states. The emission spectrum in D-A TADF compounds is generally broad, which is due to a large geometric reorganization in the CT excited state, characterized by a large FWHM.ref. ref118 To improve the emission purity of the molecule, incorporation of substituents that not only suppress vibrations but also increase rigidity are needed. Beyond adjusting the structures of the donors and acceptors, the properties are also dependent on their relative regiochemistry.ref119−ref120ref121 Intramolecular interactions can also influence both the emission color and the TADF efficiencies.ref122,ref123 As well, the photophysical properties of compounds are also affected by their environment and intermolecular interactions.ref124−ref125ref126
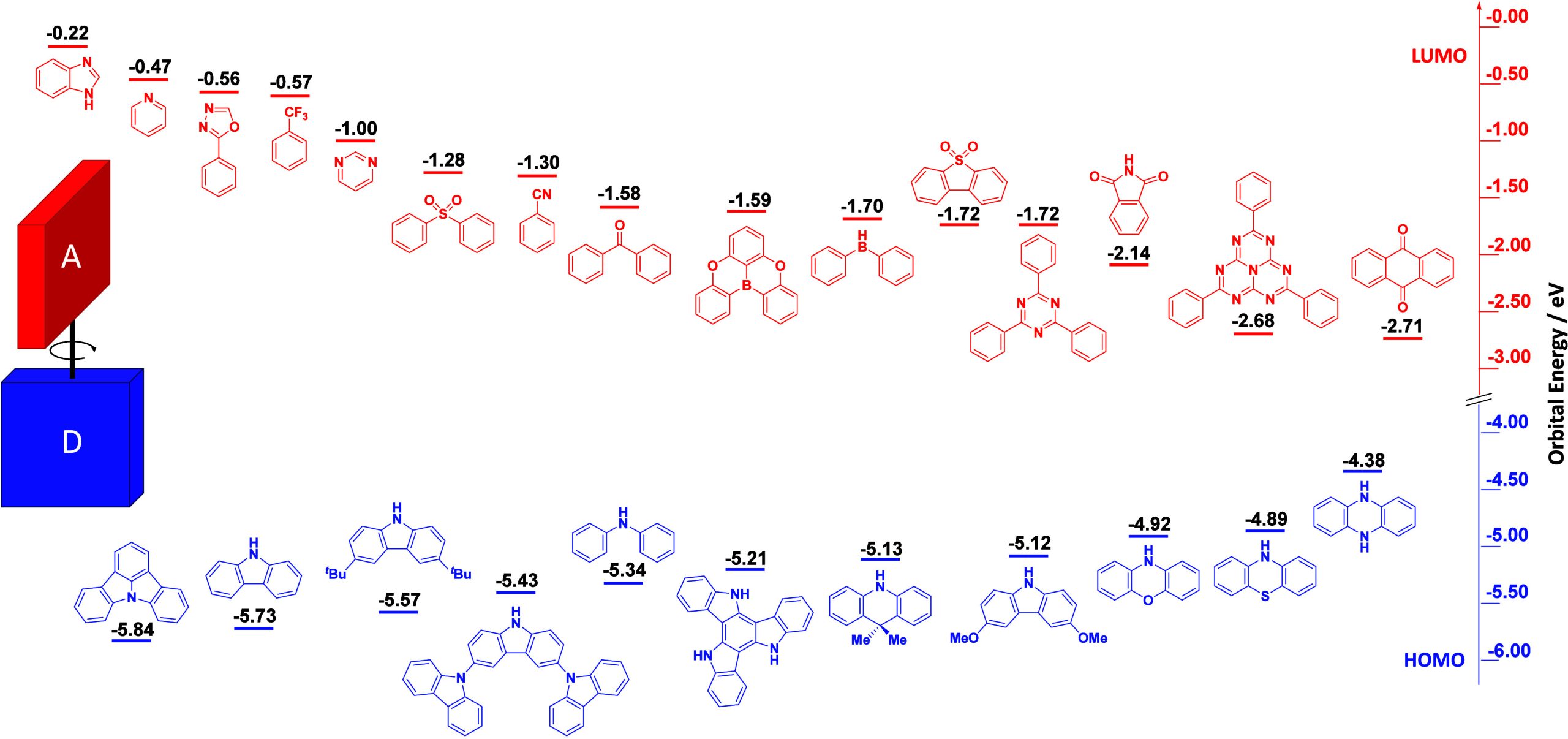
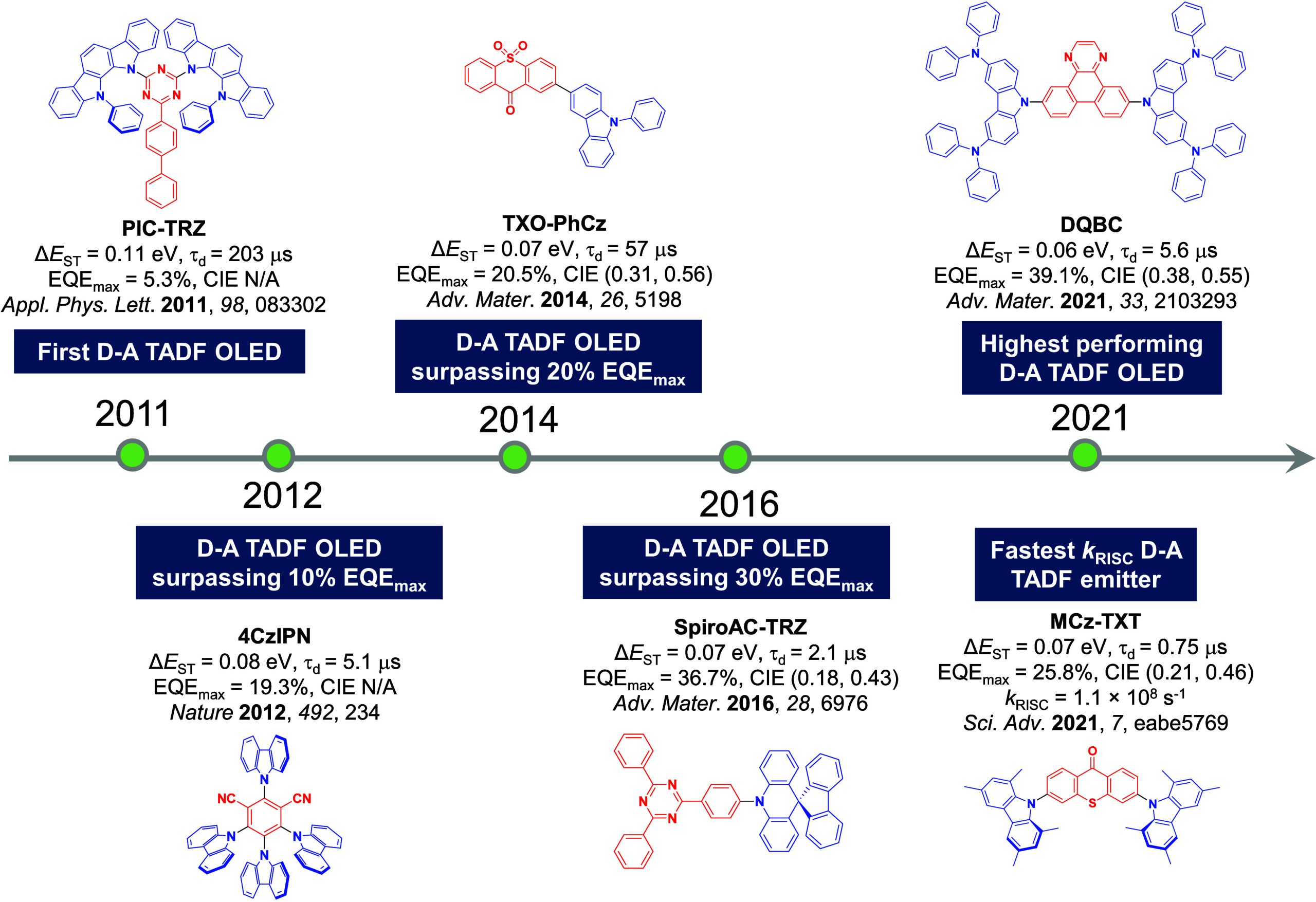
This simple D-A design paradigm is nonetheless the most commonly adopted by the community and has led to an explosion of examples since 2011,ref. ref76 aided by the predictive power of density functional theory calculations (Figure ). A steady increase in overall EQEmax has been driven by a combination of improved emitter and OLED design. It is now much more common, for instance, to witness reports of OLED efficiencies surpassing 30%. Blue, green, and red donor-acceptor designs are surveyed in Sections sec3 , sec4, and sec5, respectively. Each of these sections focuses on trends in properties as a function of common structural motifs. Design rules for other classes of TADF compounds such as TSCT emitters (Section sec12 ), exciplexes (Section sec8 ), metal complexes (Section sec9 ), and MR-TADF materials (Section sec11 ) will be covered separately. Regardless of structure, the impact, interest, and pace of exploration of TADF and the materials that emit via this mechanism have clearly captured the interest and imagination of chemists, physicists, and materials scientists globally.
Molecular Modelling
Introduction
Computational chemistry is now routinely used in the literature as a valuable predictive tool to design and understand new TADF materials. Concurrently, the TADF field has inspired computational chemists and physicists to develop and refine new methodologies to accurately describe the nature, energies, and transition rates between the excited states of existing emitters. These methodologies are essentially divided in two categories: time-dependent density functional theory (TD-DFT) and wave-function-based approaches.ref. ref127 An accurate description of the excited states is key to gaining insight into the mechanistic aspects behind the TADF process, especially when modelling the excited state dynamics.ref128−ref129ref130ref131ref132ref133 This requires identification of key intrinsic features associated with the excited states of TADF materials as well as their interactions, each of which must be accurately modelled. Specifically, an ideal computational protocol must be able to accurately model the orbitals and energies of singlet and triplet excited states, which for organic compounds are either LE, CT, or mixed CT-LE (HLCT).ref. ref134 Compounding this challenge, the effect of the solvent or host environment can play a significant role, even wholly reshuffling the relative energies of both singlet and triplet excited states. Indeed, this new ordering of excited states can be significantly different than in gas-phase modelling which affects both the TADF mechanism and its efficiency.ref. ref135 Furthermore, since electron-phonon coupling is usually large in organic π-conjugated materials, and since molecular vibrations are fundamental to the electronic processes governing TADF, the dynamic nature of the excited-state landscape makes TADF particularly challenge to accurately model. Beyond detailed investigation of excited states for single compounds, large scale high-throughput computational screening protocols have also been implemented to assist in materials design and identification. Here, we will briefly discuss these different computational approaches in view of the recent literature.
Excited-State Energy Level Calculations and the Prediction of Their Nature
Predicting ΔE ST
Although the mechanism behind TADF is frequently more complex than direct RISC from T1 to S1,ref. ref83 ΔE ST remains the key guiding parameter that both experimentalists and theoreticians use to identify emitters as promising targets.ref. ref136 The community has largely used TD-DFT, which is well-adapted for organic D-A (see for instance Sections sec3 –sec5) and carbene metal amide (CMA) (see Section sec9 ) TADF emitters. However, the features of the excited states of MR-TADF emitters (Section sec11 ) makes them incompatible with TD-DFT approaches, as we have recently demonstrated (vide infra).ref137,ref138
In the literature, vertical excitation calculations based on the optimized ground-state geometry are most frequently reported, and the vertical ΔE ST is computed from the difference in vertical excitation energies to S1 and T1 using TD-DFT methods (Figure ).ref. ref139 These calculations are particularly cost-effective as they do not require excited-state geometry optimisations, and essentially mimic an absorption process; however, they are often misguidedly used to then interpret the emission properties of TADF materials. When investigating emission properties, it is instead recommended to commit to optimization of the geometry in the excited states, with S1 and T1 optimizations used to model fluorescence phosphorescence spectra, respectively, since molecular relaxation occurs at much faster timescales (ps and faster) than these radiative processes and thus originating from the relaxed geometry of the excited state. However, this is a more computationally costly approach as each excited state of interest must be reoptimized separately, and as such, this approach is less frequently employed.
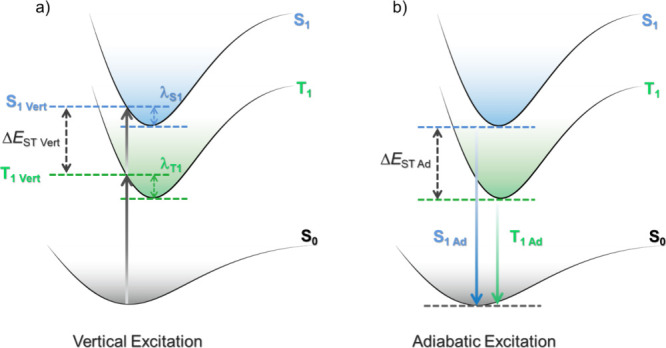
However, even this more careful approach can fail when excited states are close in energy, and start to acquire a multiconfigurational character which cannot be captured by TD-DFT. In these cases, one must rely instead on appropriate wavefunction-based methodologies such as the complete active space self-consistent field (CASSCF).ref. ref140 The adiabatic excitation energy corresponds to the difference in energy between the optimized (relaxed) ground and excited states (Figure ).ref. ref139 Thus, the adiabatic ΔE ST is determined from the difference in energy between the adiabatic S1 and T1 excitation energies. Although the adiabatic ΔE ST is more closely related to the measured ΔE ST, it has been highlighted that the vertical ΔE ST and the adiabatic ΔE ST can provide similar results when both S1 and T1 share a similar electronic configuration, (i.e. the same nature) resulting in similar relaxation energies.ref. ref139 This is often the case for D-A TADF compounds which frequently possess S1 and T1 excited states with a strong CT character, with the modest accuracy and lower cost of vertical ΔE ST explaining its persistent use.
Faced with a plurality of potential computational methods, the preferred choice for characterizing TADF emitters is almost entirely dependent on whether the D-A TADF or MR-TADF D-A TADF materials are capably described using TD-DFT, especially within the Tamm–Dancoff approximation (TDA) that relaxes the triplet instability issue, which results in an over-stabilized T1 with respect to S1.ref141,ref142 The modelling of MR-TADF compounds requires the use of wavefunction-based methods through either single-reference couple-cluster methods or multi-reference protocols such as CASSCF/CASPT2 (Complete active space 2nd order perturbation theory) or CASSCF/NEVTP2 (n-Electron Valence 2nd-order Perturbation Theory) to improve the description of the S1 state with a proper inclusion of (dynamic) electron correlation (which is not as important for T1).
Characterizing the Nature of the Excited States
As highlighted in the introduction, alongside energies, the nature of the excited states and the resulting spin-orbit coupling between them (governed by El-Sayed’s rule) are critical in order to infer the mechanism of a particular TADF process.ref. ref83 Although the nature of these states is still often discussed in terms of the electron density distribution of the HOMO and LUMO (Figure ), this can be misleading, as excited states can display contributions from more than the simple one-electron transition.ref. ref143 More compact pictures of excited states such as natural transition orbitals (NTOs), attachment/detachment frameworks, or the difference in electron density between the ground and excited state are becoming increasingly popular in order to more accurately characterize the nature of the excited states (Figure ).ref. ref127 These methods each portray the spatial distribution or changes of the hole and electron densities for the singlet and triplet excited states, thus accounting for contributions of all relevant orbitals.ref. ref134
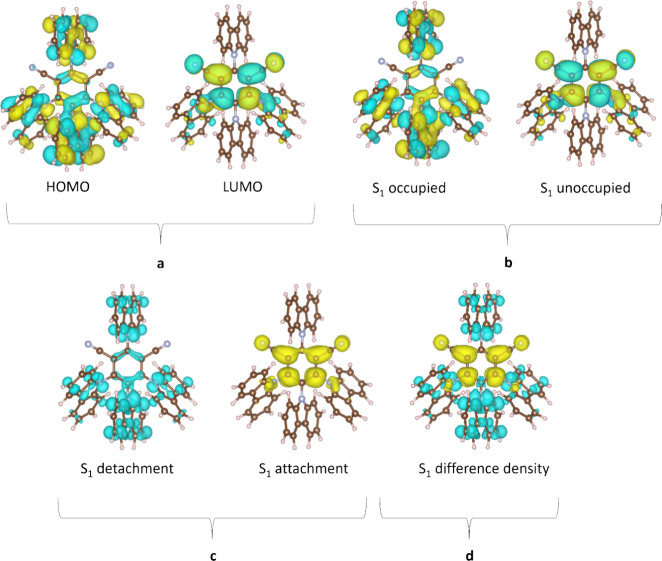
The nature of the excited states can in some cases be qualitatively inferred by inspection of orbital visualisations but more quantitatively by computing metrics relying on the difference in distance between the hole and electron density barycenters, such as DCT.ref. ref144 Alternatively, overlap indices include Λ, which describes the overlap between pairs of (NTO) orbitals,ref145,ref146 or ΦS, which is a measure of the overlap between the hole and the electron densities computed within the attachment/detachment formalism. Typically, CT (or LE) excited states exhibit large (or small) DCT beyond (below) 1.6 Å or small (large) Λ/ΦS of around 0 (1).ref85,ref147 Several studies have highlighted clearly that the nature of the excited state in D-A TADF emitters is never purely LE or CT, but a mixture of both.ref. ref85 In a recent effort, we identified that for D-A TADFs the nature of each of S1, T1, and T2 is well-reproduced when TDA-CAM-B3LYP or TDA-M06-2X functionals are used, when compared to Spin Component Scaling Second order Couple Cluster (SCS-CC2) calculations.ref148,ref149 However, irrespective of the functional, the nature of both T1 and T2 computed at the TDA level does not match as accurately with the SCS-CC2 prediction as it does for the S1 state.
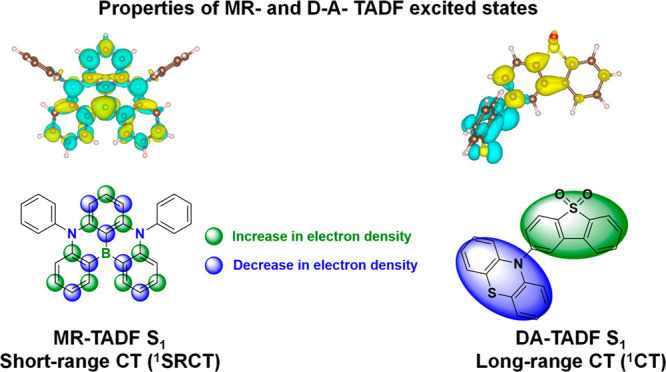
In contrast, for MR-TADF compounds, the lower-lying singlet and triplet excited states consistently exhibit a short-range charge transfer-like (SRCT) character with a difference density pattern showing excess hole and electron densities on adjacent atomic sites (Figure with DABNA-1).ref137,ref138 While this does not exclude the possibility of higher-lying excited states with LE or a long-range CT-like (LRCT) character (for example triggered by the presence of peripheral substituents possessing a significant electron-donating or -withdrawing character)ref. ref150 as mentioned above these SRCT require alternate computational treatment to account for an accurate account of electron correlation. For MR-TADF emitters, difference density plots provide the clearest picture of the alternating hole-electron density pattern present within their excited states.ref. ref138
The relationship between the extent of CT character of the lower-lying singlet and triplet excited states and ΔE ST has been probed using several metrics. Work by Moral et al.ref. ref151 demonstrated, considering six molecules (three hosts and three emitters), that a larger CT character of both S1 and T1 states results in a reduced ΔE ST, in line with equations eq3 and eq4 (see Section 1.4.1). Their analysis was based on the calculation of the Δr metric, namely the distance between the hole and electron density barycenters as computed from NTOs to quantify the extent of CT in the compounds. Lee and Kimref. ref152 investigated the influence of donor substitution and showed that the inclusions of additional donors increase the CT content of T1 while having a minimal effect on S1, which in turn reduce ΔE ST. Along this line, Olivier et al. showed in a series of D-A and D-A-D emitters that the biggest challenge to decreasing ΔE ST consists of increasing the CT character of the T1.ref. ref153 This is exemplified when computing the ΔE ST, the ΦS metric for S1 and T1 and oscillator strength along the torsional profiles of the D-A single bond. Typically, we observed a faster decrease (increase) of the T1 as compared to S1 CT (LE) character. It arises because the T1 state is subject to the exchange interaction (while not the S1 state), which induces a localization of the T1 wavefunction on either the D or A units and significantly increases the LE character of that state. Inducing a larger CT character in T1 results in a larger energy difference between the 3LE and 3CT as compared to the energy difference between the 1LE and 1CT for the corresponding S1 state. A similar study, undertaken by us,ref. ref123 where we performed a torsional screen of four emitters, supports this finding. Here, ΔE ST is smallest when the CT content of S1 and T1 is greatest. By observing the difference in CT character between S1 and T1 ΦS, (ΔΦS), Olivier et al. reported a decreasing ΔE ST when ΔΦS is the smallest, namely when the CT character in both states is the largest.ref. ref85 These studies show that it is easier to induce a larger CT character in S1 state than in T1. However, reaching a large CT character in both states is key to minimizing ΔE ST and it has been the most popular design strategy thus far. In rare exceptions, ΔE ST was made rather small (below 0.1 eV) even though T1 and S1 bore, respectively, an LE and a large CT character, but this requires a careful engineering of the energies of the 3LE and 3CT states.ref. ref154
Benchmarking ΔE ST
D-A TADF Emitters
The variety of DFT functionals are essentially distinguished by the way the exchange-correlation potential is defined, and this also introduces a significant disparity between the excited-state pictures associated with D-A TADF emitters. The most popular functionals employed to model TADF emitters are usually global hybrids such as B3LYP and PBE0, meta-GGA (Generalized Gradient Approximation) such as M06-2X, and long-range corrected functionals such as CAM-B3LYP and LC-ωPBE. Basis set effects can also lead to a further variation of the absolute energies of the excited states. However, no dramatic variation in relative energy has been observed going from Pople-based basis sets such as 6-31G(d,p) to Karlsruhe basis sets such as def2-TZVP.ref. ref153
In the computational chemistry literature, consensus on different methodologies is often approached (although rarely fully achieved) by identifying the most accurate methodologies across a group of known compounds.ref139,ref142,ref155,ref156 These benchmark studies involve comparison between a calculated property of interest at a level of theory under assessment (y i calc) with the corresponding experimental or ‘trusted’ higher-level method property (y i ref). The mean average difference (MAD), the root mean square deviation (RMSD) and the standard deviation (σ) each allow for the determination of the most appropriate methodology based on a statistical analysis.
The accuracy of the selected method(s) with respect to a test data set is assessed by these three metrics, with smaller values corresponding to better performance, although with no consideration for different computational costs.
Moral et al. compared several TD-DFT approaches against experimental data and highlighted that using TDA-DFT compared to TD-DFT produced a more accurate ΔE ST prediction, essentially because the triplet instability issue was better handled.ref. ref142 A study using a larger data set of 17 emitters was undertaken by Sun et al.,ref. ref139 where M06-2X and ω-tuned LC-ωPBE showed excellent agreement between calculated and experimental ΔE ST for both vertical and adiabatic excitations. In ω-tuned LC-ωPBE, the electron repulsion operator is divided into a short-range description at the DFT level and a long-range domain described at the Hartree-Fock level.ref. ref133 The range separation, ω, delimits the two domains and is often optimized to tune the HOMO and LUMO energies to the ionization potential and the electron affinity, respectively. However, this parameter must be optimized for every compound and potentially for each different starting geometry.ref. ref157 Using a similar ω optimisation procedure, other long-range corrected functionals such as LC-B3LYPref. ref158 or LC-ωHPBE have been employed within the literature, differing only in their DFT exchange-correlation potential.
Moving away from TD-DFT, Kunze et al. employed spin-unrestricted (UKS) and restricted open-shell Kohn-Sham (ROKS) SCF calculations to investigate 32 emitters, covering a range of structures.ref. ref155 Their study showed a remarkably small MAD for predicted ΔE ST of 0.025 eV. This impressive accuracy was assigned to an improved CT description owing to the inclusion of orbital relaxation, which other computational schemes based on TD-DFT do not include. However, excited state transition properties are not accessible with ROKS, and TD-DFT should be invoked to access them.
The community primarily still uses hybrid functionals like B3LYP and PBE0, despite the conclusions from these benchmark studies that have highlighted that both produce excessive stabilization of CT states due to their low Hartree-Fock exchange content.ref139,ref155
MR-TADF Emitters
Unlike the modelling of the excited states of D-A TADF compounds, TD-DFT struggles to accurately predict the excited states of MR-TADF emitters,ref137,ref138 where there is a consistent overestimation of the ΔE ST. Despite documenting the inaccurate prediction of the excited-state energies, the community continues to employ TD-DFT methods to model MR-TADF emitters. Recently, we highlighted that coupled cluster calculations can accurately predict the nature and energies of the excited states of MR-TADF compounds as these calculations include a double excitation contribution that current implementation of linear response TD-DFT neglects in the adiabatic approximation.ref25,ref137,ref138 We anticipate that the use of TD-DFT will be rapidly superseded by wavefunction-based methods as new MR-TADF materials continue to be reported at an accelerating pace.
This was initially demonstrated for two emitters for DABNA-1 and TABNA, where excellent agreement was obtained between experimental and calculated ΔE ST values (Figure ).ref. ref137 Using this method, a series of linearly extended systems were also designed, and it was demonstrated that increasing the length produced an increase in f and a reduced ΔE ST. However, this came at the price of a predicted red-shift of the emission. However, suitable substitutions with nitrogen and/or boron atoms enables either a blue- or a red-shift of the emission energy. Ultimately, increased charge transfer character and a reduced CT distance ensured a reduced exchange energy in these compounds, resulting in smaller ΔE ST, and stronger polarizability, leading to a larger f.
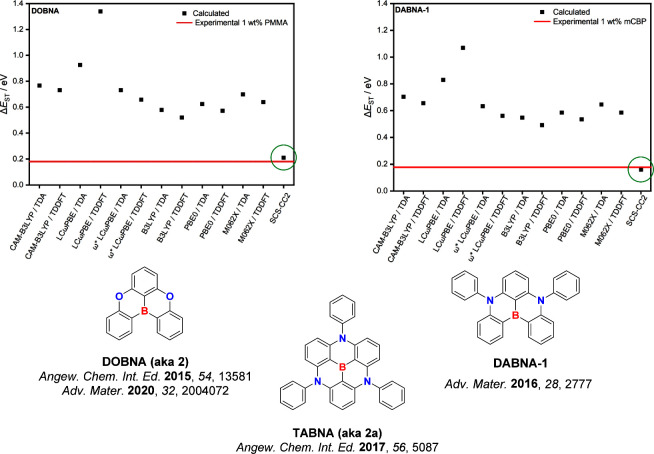
We also recently compared the experimentally and calculated ΔE ST values across 35 literature MR-TADF emitters using SCS-CC2/cc-pVDZ as well as a range of DFT functionals (B3LYP, PBE0, M062X, LC-ωPBE, CAM-B3LYP and ω-tuned LC-ωPBE).ref. ref138 TD(A)-DFT calculations consistently overestimated ΔE ST, with MAD values ranging between 0.29 eV and 0.98 eV. When employing SCS-CC2/cc-pVDZ, the MAD significantly decreases to only 0.04 eV, highlighting the ability of this method to accurately predict the ΔE ST (see Figure ). When considering exclusively boron-acceptor MR-TADF emitters, a strong correlation becomes apparent between SCS-CC2 and the experimental T1 and S1 energies. However, the correlation between calculated and experimental ΔE ST of ketone-acceptor MR-TADF derivatives is not as strong, Figure . This was attributed both to the fact that vertical ΔE ST were considered, thus neglecting excited-state relaxation, and to solvent interactions with the lone pair of the carbonyl functionalities that result in a stabilization of the excited states of this class of MR-TADF emitters. Since the original report, this methodology has enjoyed wide and growing implementation in materials design.ref55,ref150,ref159−ref160ref161ref162ref163ref164ref165ref166ref167ref168ref169ref170ref171ref172ref173ref174ref175ref176ref177ref178ref179ref180
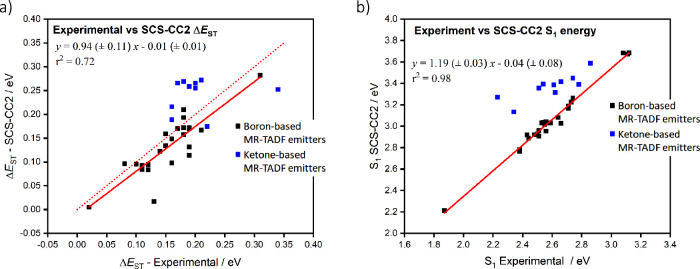
Recently, Sotoyama reported an alternative strategy for the accurate prediction of the ΔE ST of MR-TADF emitters using ΔSCF.ref. ref181 ΔSCF calculations involved two SCF calculations; a first one where an electron with either a spin-up or spin-down is promoted from the occupied to the virtual orbitals. This results in a state halfway between a singlet and a triplet spin configuration, leading to some spin contamination from the triplet state configuration. A second SCF calculation is then performed on the triplet state configuration. The energy of the singlet state is thus obtained as the difference between twice the energy of the first calculation (emulating the singlet state, and doubled to account for the degeneracy of the spin up and spin down electronic configuration) and the energy of the triplet state. The correlation between ΔSCF-calculated versus experimental ΔE ST was investigated across 13 MR-TADF emitters and compared to conventional the results from TD-DFT methodologies. Here, MADs of 0.04 eV using both the B3LYP and the PBE0 functionals using ΔSCF were reported, performing similarly to SCS-CC2 calculationsref. ref138 although at notably reduced computational cost.ref. ref181 The author attributed the accurate prediction of ΔE ST to the orbital relaxation rather than the inclusion of double excitation.
Recently, a benchmark study of both D-A TADF and MR-TADF emitters was performed using the hole-hole (h-h) TDA-DFT method.ref. ref182 This method includes static electronic correlation in a similar fashion to CASSCF, considering electronic transitions within an active space including only single and double excitations while dynamic correlation is introduced through the exchange-correlation functional. Interestingly, these calculations revealed a very good agreement with SCS-CC2 calculations yet at a much cheaper computational cost, with a MAD of 0.04 eV for ΔE ST predictions using the h-h TDA-B3LYP method.
Higher-Lying Triplet States
Understanding the role of higher-lying triplet states is becoming increasingly crucial in explanation otherwise anomalously fast k RISC observed in some TADF materials. Spectroscopically, these states are difficult to observe since they are either a ‘dark state’ or internal conversion to lower-lying excited states outcompetes any radiative decay. Their existence can be detected indirectly using transient absorption spectroscopy (TAS) methods, where their own photoinduced absorption (PIA) features emerge either though T1-Tn or Tn-Tm transitions. However, TAS often requires extensive excited-state calculations to model the triplet absorption spectrum from the optimized T1 or higher Tn states. Therefore, in many of the examples where fast k RISC is reported experimentally, the role of an intermediate upper triplet state has been either asserted speculatively or inferred solely from calculations (Figure ).ref92,ref98,ref183 As an example, among D-A TADF emitters, the fast k RISC of TAT-3DBTO2 was rationalized using TDA-DFT calculations that predicted the existence of 12 higher-lying triplet states within 0.2 eV of T1. This high density of triplet states within a small energy window was proposed to favor fast RISC.ref. ref92 A similar conclusion was obtained also in the study of MCz-TXT, which has one of the fastest reported RISC to date.ref. ref183 TD-DFT was also used to explain the very fast k RISC in TpAT-tFFO, where a subtle alignment of 3CT and 1CT as well as a higher-lying 3LE state was proposed to explain the efficient TADF in this compound.ref. ref98 This is further confirmed by constructing linearly interpolated potential energy profiles (LIPs) between target singlet and triplet states minimum showing potential multiple conical intersections between closely-lying singlet and triplet excited states.ref. ref184 Closely-lying singlet and triplet excited states has often been used to identify compounds emitting from a Hot Exciton mechanism in the device, where close alignment of a higher-lying Tn+1 with either S1 or S2 is implicated to explain the origins of the RISC.ref. ref185
Further establishing useful methods to investigate higher-lying triplet states, a benchmark computational study of 10 different D-A TADF emitters was undertaken by Cardeynaels et al.,ref. ref156 which compared DFT functionals and higher-level CC2 calculations. The study revealed that of the DFT functionals investigated, M06-2X provided the smallest MAD for the absolute energy of T2 with respect to the CC2 calculations. Our recent investigation of 14 chemically diverse D-A TADF emitters supports these previous results,ref. ref147 demonstrating that both M06-2X and CAM-B3LYP perform as well as ω-tuned functionals LCωPBE and LCωHPBE, all relative to SCS-CC2 calculations.
Focussing on MR-TADF emitters, single triangulene core structures usually possess a large energy gap between T1 and T2 as exemplified in DABNA-1 and DiKTa, which appears to hinder upconversion from T1 to T2.ref. ref138 As discussed in the section on MR-TADF Emitters, Section sec11 , expansion of the size of the MR-TADF skeleton is seen as a promising strategy to simultaneously improve f while decreasing ΔE ST. This was confirmed experimentally when comparing DABNA-1 and ν-DABNA (Figure ). In addition, there is a shrinking of the T1-T2 gap from 0.75 eV to 0.12 eV from DABNA-1 to ν-DABNA, computed at SCS-CC2 level based on the ground-state optimized geometry, leading to a significant boost in k RISC from 103 s–1 to 105 s–1.ref. ref173 This behavior is also observed to a lesser extent when comparing ICzMes3 and DiICzMes4 , Figure , where the T1-T2 gap decreases from 0.46 eV to 0.33 eV using the same method.ref. ref161
Spin-Orbit Coupling and RISC
SOC can be considered as the dominant source for spin mixing, driving the triplet-singlet interconversion in D-A TADF and MR-TADF materials and thus crucial for computing k RISC. When considering RISC as an intramolecular process, SOC is often computed to be on the order of a few tenths of meV, while hyperfine interaction (HFI) and spin-spin dipolar coupling are calculated to be much smaller, on the order of a few μeV, and hence can be neglected.ref. ref90 The magnitude of the computed SOC is also very important, as El-Sayed’s rule only qualitatively establishes that SOC vanishes between singlet and triplet excited states of the same nature, while remaining sizeable between excited states of different character.ref81,ref82 The SOC is never exactly zero though, as triplets rarely exhibit perfectly identical nature as their corresponding singlets, as the electronic exchange interaction acts more strongly on triplets than singlets, conferring a higher degree of LE character.
This is not to say that SOC is exclusively the dominant factor in RISC. In bimolecular TADF exciplexes, it is often hypothesized that RISC must be driven by hyperfine interactions as the singlet and triplet excited states are intermolecular states possess a strong CT character between electron-donating and accepting moieties that are not covalently linked and with vanishing hole and electron densities overlap.ref. ref186 Recently, a combination of TAS, transient electron paramagnetic (TrEPR) measurements, and TD-DFT calculations showed that BF2 (Figure ), a curcuminoid derivative, possesses delocalized intermolecular CT states from which intermolecular RISC is driven by a hyperfine intermolecular process.ref. ref187 While this intermolecular RISC mechanism is not the most common spin interconversion mechanism, it should not be neglected outright, especially when devices exhibit high EQE despite the large ΔE ST of the emitter.
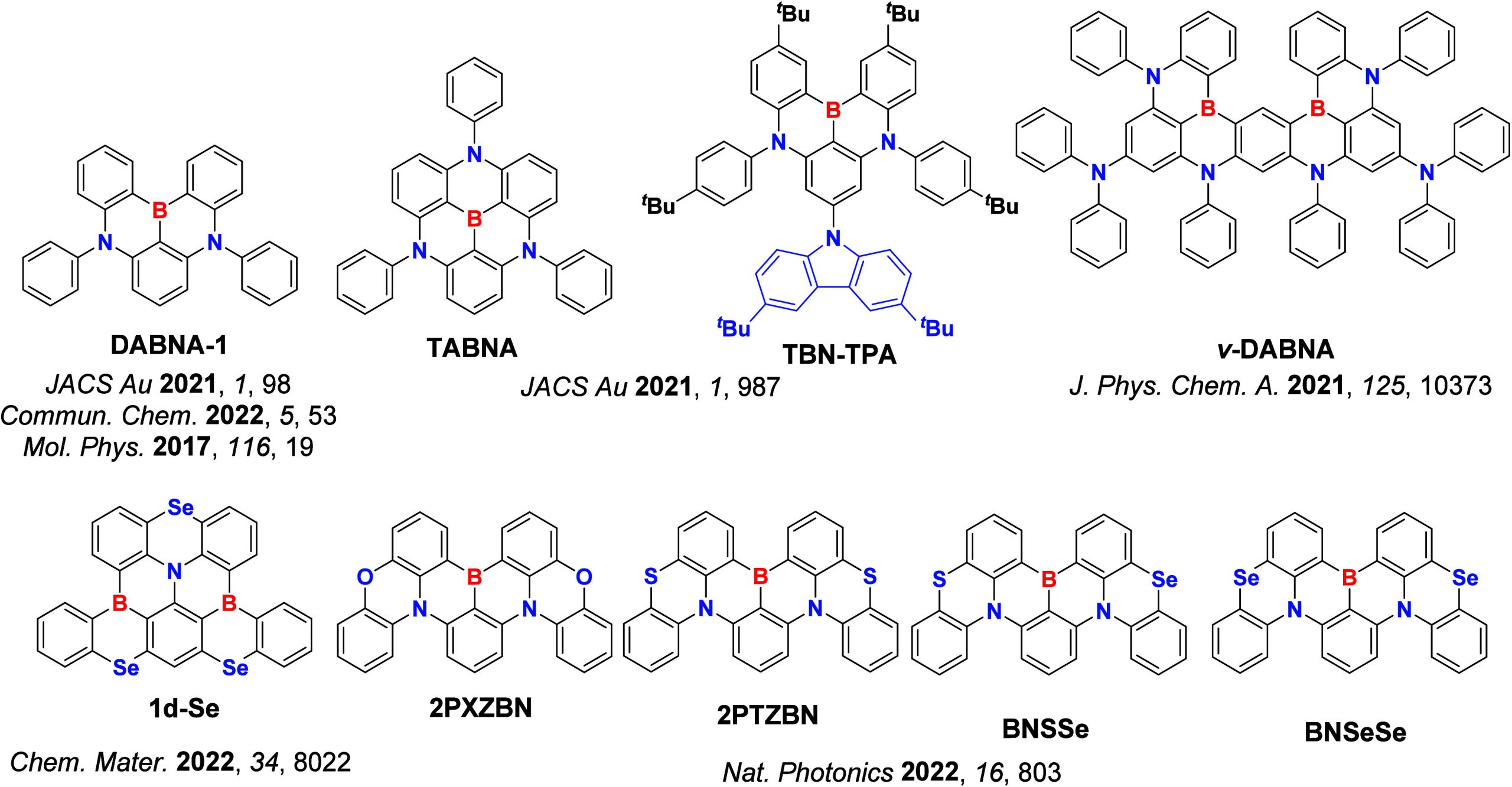
Returning to the intramolecular RISC mechanisms as operant in D-A TADF emitters, computational modelling of 2CzPN and 4CzIPN (Figure ) revealed that the increase of SOC between S1 and T1 occurs at the expense of an increase of ΔE ST.ref. ref85 In line with El-Sayed’s rule, the SOC between S1 and T1 in these compounds increases as the natures of S1 and T1 diverge, and is strictly zero when S1 and T1 are pure CT. The mixed CT-LE nature of S1 and T1 in both 2CzPN and 4CzIPN is dynamically modulated by fluctuations of the torsion angle between the donor and the acceptor, as probed along a molecular dynamics simulation, which impacts the D-A electronic interaction and instantaneous SOC value. This finding was further confirmed by a study of 15 D-A TADF emitters in which Marcus-type rate expressions for k RISC indicated a careful balance is needed between ΔE ST and SOC to ensure fast k RISC.ref. ref154 When the nature of S1 and T1 are significantly different, ΔE ST is very large and RISC is slow regardless of the value of SOC because ΔE ST appears in the exponential factor of the Marcus rate expression. However, while ΔE ST decreases as S1 and T1 take on increasingly similar natures, the reduced contribution from SOC becomes increasingly prominent in the overall k RISC. Although identifying candidate emitters by finding an optimum balance of SOC and ΔE ST seems promising, large variations in both SOC and ΔE ST appear when changing functionals, hence comparison between calculations and experiments must be made with caution.
While the RISC rates for leading D-A TADFs now often exceed 106 s–1, the experimental k RISC values of MR-TADF emitters are significantly slower, on the order of 103–104 s–1 (see Section sec11 for more detail). The direct SOC between T1 and S1 in MR-TADFs is often quite small due to the very similar nature and electronic configuration of both SRCT states. Instead, a recent computational study carried out on DABNA-1, TABNA, and TBN-TPA (Figure ) revealed that a superexchange mechanism should drive RISC involving primarily the much higher-lying T3 states.ref. ref188 Shizu et al. instead reported that RISC in DABNA-1 is actually a two-step process entailing RIC from T1 to T2 followed by efficient RISC from T2 to S1.ref189,ref190 Lin et al. also tried to rationalize the DABNA-1 excited-state dynamics by computing the RISC rate between the first three triplets and S1, and concluded instead that the T1-S1 channel is the most likely route.ref. ref190 These three independent studies, involved three different computational protocols and resulting in very different pictures of the detailed RISC mechanism of the same molecule, certainly needs further clarification.
An extended analogue of DABNA-1, the k RISC mechanism was simulated for v-DABNA (Figure ), achieving good agreement between calculated and experimental values when considering a direct T1 to S1 conversion.ref. ref181 Despite the small SOC between these states, a very small ΔE ST was believed to be sufficient to account for the efficient k RISC reported, while the computed k RISC assuming a T2-mediated RISC mechanism leads to a difference in magnitude compared with experimental findings.
The role of heavy atom effects in modulating k RISC in MR-TADF emitters was recently discussed by Pratik et al. In their two studies, they investigated the role of embedding heavy atoms into a MR-TADF structure using the DLPNOCCSD/def2-TZVP method.ref178,ref191 They focused on chalcogen elements of group 8, probing oxygen, sulfur, and selenium derivatives and the resulting changes in SOC and k RISC. A range of emitters was presented, with SOC and in turn k RISC increasing significantly in selenium-containing materials, compared to oxygen and sulfur congeners; 1d-Se was proposed as a particularly promising emitter wherein k RISC was predicted to outcompete k r (Figure ). Simply considering SOC, Hu et al. have computationally investigated changing k RISC for a similar series of compounds comparing their calculated SOC with the experimental k RISC.ref. ref180 They observed a steady increase in experimental k RISC from 0.04 × 106 s–1 (2PXZBN) and 0.19 × 106 s–1 (2PTZBN) to 0.60 × 106 s–1 (BNSSe) and 2.0 × 106 s–1 (BNSeSe), that is broadly in-line with their calculated T1 – S1 SOC values (Figure ). BNSeSe was calculated to have the most active RISC, which they attributed to a larger SOC; two orders of magnitude larger for S1-T1 in BNSeSe than for 2PXZBN and 2PTZBN. The S1 – T2 SOC was calculated to be largest for BNSeSe, which explains the faster k RISC than for BNSSe, which actually has a larger S1 – T1 SOC. In accordance with these computational studies, and as with D-A TADF systems, introduction of the heavy atom has proved an attractive strategy to increase k RISC in MR-TADF emitters in experimental studies.ref. ref103
Conformational and Vibronic Effects
It is important to note that the approaches mentioned so far primarily involve isolated molecules in either their ground or excited-state optimized geometry. In the OLED, this is clearly not an accurate representation of the emitting layer (EML). The amorphous nature of the EML means that different conformers of the TADF emitters will be present, while simultaneously intramolecular vibrational modes will modulate their geometries over time.
In 2017, Olivier et al. ref. ref153 identified the critical role of torsional vibrational modes around the donor-acceptor single bond in D-A and linear D-A-D TADF emitters, impacting both the energies of the S1 and T1 excited states (and so ΔE ST) and the oscillator strength of the S0-S1 transition. This rather simple approach helped to resolve the apparent contradiction of how a TADF emitter with seemingly negligible S0-S1 oscillator strength (because of the large CT character of the S1 state) may nonetheless possess decent radiative lifetime and so a high ΦPL. The role of vibrations within TADF systems is indeed becoming recognized as ever more important as our understanding develops. In 2016, Marian discussed a mechanism involving an intramolecular vibrational mode of the C=O bond of the xanthen-9-one donor in ACRXTN to justify the promising RISC rate of the emitter (Figure ).ref. ref192 Olivier et al. showed that direct T1-S1 k RISC was significantly boosted due to non-Condon effects on the SOC – i.e. the impact of geometry distortions associated with active vibrational modes on the SOC.ref. ref85 Brédas and co-workers investigated carbazole-based TADF emitters and highlighted that the rotation of the carbazole group in between the two cyano groups in 4CzIPN (Figure ) triggered the RIC from T1 to T2 from which RISC was computed to take place.ref. ref193
Computational chemistry was recently employed to rationalize the exciton harvesting mechanism present in carbene metal amide (CMA) TADF emitters,ref. ref194 notably so as Di et al. had originally invoked an unusual RISC mechanism implicating a negative ΔE ST. Torsional freedom of the carbazolate bonded directly to the coinage metal was believed to be the driving force, but studies by Foller et al. and Taffet et al. contradicted this putative mechanism.ref195,ref196 Initially, it was shown that rotation around the NCz-Metal bond in fact did reduce ΔE ST, but not to the extent that an inverted gap was calculated. The reduced ΔE ST comes at a cost of reduced k f, hence this cannot be responsible for the extremely efficient emitters presented in the original study such as CMA1 (Figure ).ref195,ref196 In a subsequent study, Taffet et al. again highlighted the problems associated with the rotating ligand model, showing that SOC decreases along with ΔE ST, suggesting RISC will be less efficient as in the case of CMA2 (Figure ).ref. ref196 Instead they proposed that the metal-carbene C-N bond deformation and the resulting bond length changes from a bending mode increase both f and SOC, enabling efficient RISC. It was hypothesised in both works that the coplanar geometry is responsible for the TADF observed in these two emitters.ref195,ref196
Role of Solvent and Solid-State Host Matrix: Polarization Effects
Excited states carrying a significant CT character are extremely sensitive to their environment, stabilizing their energy thanks to both electronic (and nuclear polarization) in host/guest systems in the solution state. When modelling emitters in solution, the polarizable continuum model (PCM) is often applied to address this behavior, partly because of its widespread availability within computation software packages and low computational cost. The PCM model approximates the solvent as a continuous medium of fixed dielectric, and therefore also considers that the solvent reorganization around the solute is slow (adiabatic approximation) so that the solute problem is solved for a fixed ‘configuration’ of the solvent molecules. This approach remains largely valid for highly polar solutions for which solvent molecules explore their available (electronic and nuclear) degrees of freedom much more slowly than that of the solute. The SRCT excited states nature typical of MR-TADF emitters are much less sensitive to host environment though, and so these considerations are typically restricted to D-A TADF emitters.
Recently, Painelli and co-workers developed in the frame of Onsager model (point dipole approximation), an anti-adiabatic approach where the electronic degrees of freedom of the solvent are considered to rearrange instantaneously.ref. ref197 This approach is particularly relevant for solvents of low polarity. Interestingly for DPTZ-DBTO2 (Figure ), while Corrected Linear Response-PCM and State Specific-PCM approaches wrongly predict a negative ΔE ST, the anti-adiabatic approach predicts a positive ΔE ST using a dielectric constant corresponding to toluene. The complex photophysics of DMAC-TRZ, Figure , have been modelled by Huu et al. who used a model including four electronic states (S0, one zwitterionic singlet and one zwitterionic triplet state, one LE state) that are coupled to a high frequency and a torsion vibrational modes.ref. ref131 Solvent effects are also included within the frame of the Onsager model considering an anti-adiabatic approach for the electronic degrees of freedom of the solvent while the orientational ones is treated adiabatically. Their model nicely reproduced the absorption (emission) weak (strong) solvatochromism observed experimentally because of the very similar (different) relaxation of the electronic (orientational) part of the solvent response. Moving towards organic matrices, their model attributed the non-exponential behavior in the time-evolution of the S1 population to conformational static disorder and the large spectral shift of the delayed fluorescence (red- and blue-shift) to dielectric disorder.
Along the same line, Gillet et al. performed QM/MM adiabatic molecular dynamics along the S1 and T1 potential energy surfaces of the TXO-TPA D-A TADF emitters in a toluene solvent.ref. ref135 The S1 dynamics revealed that TXO-TPA evolves from a conformation where the D-A torsion is around 40 degrees in the ground state to a completely orthogonal one. The S1 state acquires a stronger CT character resulting in a drastic decrease of ΔEST from 0.3 eV to a nearly vanishing value. The highly twisted conformation is stabilized by the reorganization of the orientation of the toluene solvent molecules around the TADF emitter occurring within few picoseconds which further stabilize the highly CT S1 state. The T1 dynamics shows that in some instances T1, T2 and S1 are close to each other such as RISC becomes active. As a result, when going to vacuum to toluene, the RISC rate is boosted by three orders of magnitude due to the significant decrease of the RISC activation energy arising from both conformational changes and solvent reorganization.
Moving toward atomistic models for sold films, molecular dynamics (MD) calculations were used by Olivier et al. to simulate the organization of 2CzPN and 4CzIPN as neat films, Figure .ref. ref198 In this study, they performed atomistic microelectrostatic calculations to understand solid-state polarization effects. Upon introducing this polarization treatment both the S1 and the T1 states, which are mixed CT-LE states, are each stabilized by 0.2 to 0.3 eV. This shift in energy is much smaller than the solid-state stabilization observed for charge carriers (holes and electrons), which is on the order of 1 eV for crystalline oligoacenes.ref199,ref200 This contrasting behavior is readily explained by the dipolar character of the CT-like excitation, as opposed to monopolar electrical excitations that are relevant in the case of the studies on oligoacenes. In the case of 2CzPN, solid-state polarization leads to a decrease of ΔE ST, acting through the larger CT character computed of the S1 state compared with the T1 state. However, for 4CzIPN, ΔE ST is almost unaffected since both the S1 and the T1 states have nearly the same nature.
Investigating the role of host-guest interactions over time was performed on the emitter PTZ-DBTO2 (Figure ), considering DPEPO, PY2D and CBP as hosts.ref. ref201 Using a combination of MD and TD-DFT calculations, it was shown that a blue-shift of the delayed emission of D-A TADF emitters in films at the longest time scale does not result from host reorganization, (and thus on specific host-guest interactions) but rather from a distribution of CT states each with different emission energies. The prompt fluorescence is essentially governed by a subset of higher-energy CT states that exhibit the largest hole–electron wave function overlap and therefore have the lowest CT content and thus the larger oscillator strength. As for the delayed fluorescence, the early part of the signal appears to be red-shifted in comparison to the prompt fluorescence because RISC occurs most rapidly in the subset of molecules with lower-energy CT states that have the smallest ΔE ST. The late delayed fluorescence component then occurs through higher-lying but more emissive CT states with slower RISC, thereby rationalizing the subtle transient blue-shift of the emission spectrum. Similarly, as a consequence of distributions of molecular geometries in experimental films, QM/MM simulations, where the QM calculations were performed at the TD-DFT level, revealed that the AIE of DBT-BZ-DMAC, Figure , and the resulting increase in ΦPL,ref. ref202 was a consequence of restricted low energy (< 200 cm–1) torsional motion in the solid state, suppressing non-radiative decay to the ground state.
Emission Spectra Prediction
Prediction of the emission spectrum of TADF emitters is key to identifying whether or not the emission of a candidate material fulfils desired criteria in terms of color coordinate and purity. In this context, both the energy of the emission peak and the FWHM of the emission spectrum are relevant parameters to predict the color coordinate. At the molecular scale, calculations can also provide information as to which vibrational modes can contribute to the broadness of the emission spectrum, potentially driving molecular design rules to dampen their impact and reduce FWHM.ref. ref203
Prediction of the emission spectrum of D-A emitters is quite a complex task because of the flexibility around the single bond connecting the donor and acceptor moieties. This broadness is usually emulated by reasonably assuming a Gaussian broadening, centred around the computed emission energy at the optimized geometry of the excited state.ref. ref131 Another approach involves TD(A)-DFT calculations carried out on selected configurations extracted from a simulated MD trajectory. The broadening of the spectrum naturally arises from the fluctuations in the S1 excitation energy due to changes in conformation along the trajectory, and corresponds more directly to the experimental scenario.ref. ref85 The difficulty with computational treatment of flexible vibrational modes primarily arises from their high anharmonicity, which requires treatment with more advanced methodologies such as adiabatic molecular dynamics. We refer the reader to a recent review for further information on these methods.ref. ref204
Prediction of the emission spectrum of MR-TADF compounds has also been carried out, taking into consideration appropriate vibronic models.ref173,ref205,ref206 Due to the rigidity of the compounds, the most popular approaches reported in the literature assume that the potential energy surfaces of the ground and the excited states are described within an harmonic approximation. The transition dipole moment is approximated as a Taylor expansion up to the first order. The 0th order term is the transition dipole moment at the S1 equilibrium geometry and corresponds to the Franck-Condon contribution, while the 1st order term is the Herzberg-–eller contribution and accounts for the variation of the transition dipole moment along the 3N-6 ground-state normal modes of a given compound. The emission cross-section is obtained by thermal averaging over the vibrational manifold, usually performed in the time-domain. This approach allows for the determination of the temperature dependence of the population of the different vibrational levels, and thus of the emission spectrum. The undistorted harmonic oscillator approach is also commonly employed. This approximation assumes that the normal modes of vibration and their frequencies are identical between the ground and the excited state, and that the wavefunction of the vibrational modes are in their ground state (i.e. no thermal excitation of the vibrational modes).ref207,ref208 In most MR-TADF simulations the Herzberg–Teller contribution is neglected and only the Franck-Condon contribution is retained, because of the usually high 0th order component of the transition dipole moment.
Demonstrating the utility of these approaches, a recent study of four DiKTa derivatives, QA-PF, QA-PCN, QA-PMO, and QA-PCZ (Figure ), presented a normal mode analysis that identified the broadening of the emission spectra as arising due to two specific low-frequency vibrational modes below 130 cm–1. A higher-frequency mode was associated with the twisting of the DiKTa core, and a lower-frequency torsional mode involved the phenyl ring substituents.ref. ref203 The addition of the phenyl substituents slightly reduces the Huang–Rhys factor of the higher-energy mode that primarily contributes to the width of the emission in DiKTa, resulting in comparably narrow emission spectra for these modified derivatives. Pei et al. studied two molecules, m-Cz-BNCz and p-Cz-BNCz (Figure ), displaying significantly different emission spectra. The meta-substituted compound has a broad emission spectrum that is red-shifted with respect to the parent BNCz compound, while the para-substituted compound has a very similar and narrow emission spectrum to the parent compound. The difference in emission is attributed to the differing nature of the excited states where p-Cz-BNCz exhibits a typical SRCT-like emission spectrum while m-Cz-BNCz possesses an excited state with stronger LRCT character.ref. ref209 TD-DFT calculations using a ω-tuned LRC-ωPBE/6-311G(d,p) method simulated the emission spectrum within the Franck-Condon approximation. The red-shifted emission in m-Cz-BNCz compared to p-Cz-BNCz was properly predicted, attributed to antibonding mixing of the HOMOs of the Cz and BNCz units, which allows for the spread of the HOMO electron density of m-Cz-BNCz onto the Cz substituent but not for for p-Cz-BNCz. Due to this anti-bonding interaction, the orbital localization of the HOMO is highly sensitive to variation in the torsion between the MR-TADF core and the Cz substituent, resulting in a modulation of the CT character upon variation of the torsion angle. This results in a broadened simulated spectrum in this MR-TADF material akin to those of D-A TADF emitters, which was also observed experimentally.ref. ref210
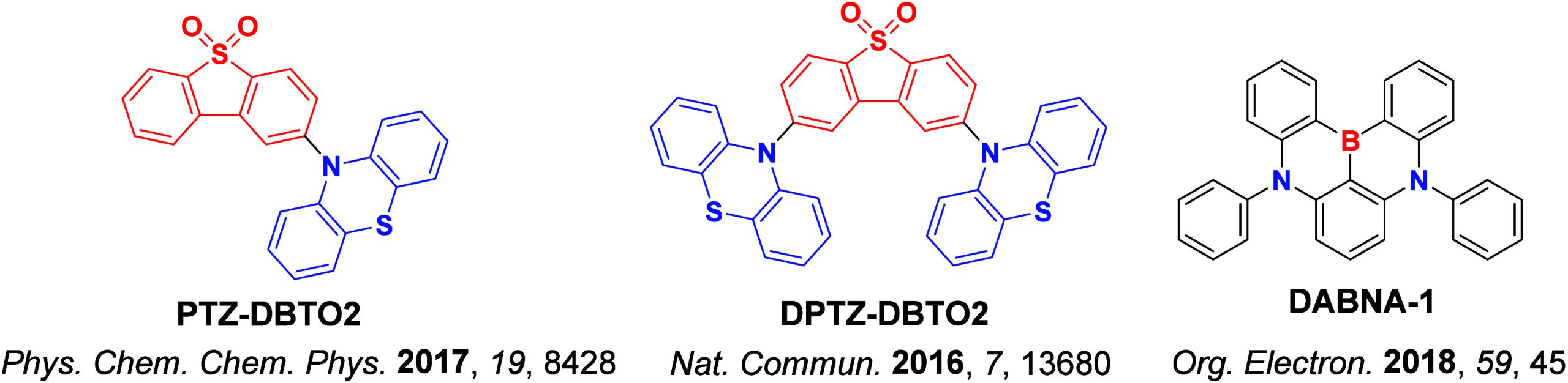
Again highlighting the importance of considering of vibronic effects, we recently performed distorted harmonic oscillator modelling of the emission spectrum of a deep-blue nonacene emitter, NOBNacene (Figure ).ref. ref211 These simulations revealed that the main peak is broadened due to two lower-frequency vibrational modes at around 180 and 650 cm–1 involving out-of-plane distortion of the conjugated core, while the side band is dominated by a high-frequency stretching mode at around 1600 cm–1. These findings support the narrowband emission reported for this compound, with FHWM of 40 nm (0.29 eV) in THF.
Excited-State Dynamics
The excited-state dynamics of TADF emitters are typically investigated using either a rate approach or quantum dynamics simulations. k RISC and radiative k r rates as well as non-radiative processes such as k IC and k ISC have been computed both in a fully quantum Fermi Golden rule treatment, or using the derived semi-classical Marcus rate expressionref. ref189 that was largely invoked in Section sec4 .ref189,ref190 Of these, quantum dynamics has the advantage of accounting equally for nuclear and electronic degrees of freedom, allowing for the study of the excited-state dynamics without making any assumptions about the interconversion mechanism.ref. ref81 Following this strategy, Gibson et al. highlighted early on the importance of vibronic coupling in the upconversion of triplet excitons into singlets.ref83,ref212 Specifically, investigating PTZ-DBTO2 and DPTZ-DPTO2 (Figure ), they showed using Multi Configuration Time-Dependent Hartree quantum dynamics simulations that RISC takes place through an intermediate LE triplet state and that RISC is strongly coupled to torsional vibrational modes. They also concluded that ΔE ST is not the sole consideration when discussing RISC, but also identified the importance of the magnitude of the S1-T2 gap. Using a similar method, Northey et al. later showed that the accurate prediction of the size of the S1-T2 gap was also crucial to reproduce the timescale of the RISC process in DABNA-1 (Figure ).ref. ref213
Machine Learning Screening of TADF Emitters
Considering the large volume of new TADF materials reported annually and the vast potential chemical space for their design, it is unsurprising that machine learning (ML) and the high-throughput computational screening of molecules have emerged as strategies to identify and assess promising candidate materials. The first study in 2015 used a tree-based genetic algorithm looking for compounds exhibiting a balance between small ΔE ST and a large S1 transition dipole moment.ref. ref214 A sea of 1.26 × 106 fragments were used as the building blocks and 4000 potential targets were identified using the genetic algorithm, although none of the proposed emitters were synthesised in this study. Aspuru–Guzik and co-workers screened approximately a million candidate molecules using a combination of DFT ground-state optimization and TD-DFT excitation energies. ML techniques allowed selection of compounds offering the best balance between a small ΔE ST, a large f and a fast k TADF.ref. ref136 Within this study and following these calculations, experts (mostly synthetic organic chemists) rated the promising emitters (over a thousand) based on their predicted properties, their novelty, and their synthetic accessibility – identifying a set of four compounds to be synthesized and incorporated into OLEDs. A promising EQEmax of 22% was achieved for a device using one of the compounds, J1 (Figure ).
Along the same lines of computer-aided design, a study was carried out to identify the best combination of host materials and green TADF emitters.ref. ref215 This was achieved by training an ML model that considered the ΦPL of the guest materials in a host matrix, the frontier orbital energy differences between the host and the guest materials, and the ΔE ST obtained experimentally in order to predict the EQEmax of the OLED device. This model appears to be quite reliable and allows identification of the best combination of host and green TADF emitters without the need for preparing the OLED. A similar study was carried out by Shi et al. to predict the EQE of TADF OLEDs based on the properties of the TADF emitters and the host in the EML, the transport layers, and the interfaces.ref. ref216 Among the four algorithms employed, the neural network provides the greatest accuracy in predicting the EQE of the TADF OLEDs. Andrienko and co-workers reported a virtual screening study of TADF emitters for single-layer OLEDs.ref217,ref218 The requirements of the proposed emitters included a small ΔE ST, ambipolar transport (guaranteed by an ionization potential larger than 6.5 eV and an electron affinity below −2.5 eV), and small energetic disorder (supported by a small electric dipole). These three criteria were used to identify devices with a high expected EQE, supported by efficient triplet upconversion and charge recombination occurring far from the electrodes to avoid exciton quenching. After selection of potential candidates, amorphous phase molecular dynamics simulations were carried out to characterize the width of the energetic disorder associated with hole σh and electron transport σe. Using this approach, 37bdt1-ant2 (Figure ) was identified as a promising candidate due to its small σh and σe.
Troisi and co-workers performed a high-throughput screening study on a series of compounds found in the Cambridge Structural Database without imposing a D-A type of structure.ref. ref219 Interestingly, they found a category of compound not based on the typical D-A scaffold and subsequently designed new compounds based on these hits to optimize ΔE ST together with the oscillator strength. A promising candidate, ZERJEL02 (Figure ), represents how compounds far removed from conventional D-A design rules can be identified using ML models, and shows reasonable calculated ΔE ST of 0.35 eV and f of 0.06. The authors then refined the structure to improve TADF performance, with ZERJEL02-Mod (Figure ) having a smaller calculated ΔE ST of 0.19 eV and a comparable f of 0.03.
Zhu et al. reported the high-throughput screening of D–A–D triads designed to emit via a “Hot Exciton” (HCLT) mechanism.ref. ref220 The strategy consisted of first establishing the threshold for large triplet–triplet splitting and a small singlet–triplet gap with the higher-lying triplet, then filtering combinations through rate comparison of competitive crossing pathways, and finally confirming RISC predictions with a more expensive evaluation of the magnitude of the spin-orbit coupling. Based on a dataset of 234 compounds, this protocol identified 31 candidate “hot exciton” emitters, four of which were indeed reported in the literature (DMF-DPP-DMF is one of the promising candidates, Figure ). Remarkably, while most of the promising systems show prominent HLCT character, several candidates did not fulfil this condition, indicating that unidentified design principles exist to afford efficient OLED materials.
Tan et al. trained an ML model based on set of D-A TADF emitters that was used in combination with an adversarial autoencoder to generate new chemical structures of emitters.ref. ref221 Among the large set of compounds generated, the ones with a ΔE ST smaller than 0.4 eV and a f value larger than 0.02 were taken on for subsequent vertical excitation TD-DFT calculations. The set of compounds was further refined by computing ΔE ST and f for the relaxed excited states geometry. Besides some known electron-donating and electron-accepting groups, the authors uncovered some new electro-active groups (such as mol_10, Figure ). In the end, they identified 19 compounds with ΔE ST smaller than 0.2 eV and S1-T1 SOC of tenths of meV.
Outlook
As summarised in this section, computational chemistry has proven to be an essential tool for generating mechanistic insight, and increasingly in recent years for actively guiding TADF emitter design. Computational chemistry is now arguably one of the key drivers of new TADF emitter development, allowing for quick and accurate screening of candidate structures. Supporting this utility, over the past 10 years there has been significant refinement in the computational methodologies used to calculate ΔE ST to a high degree of accuracy; however, holistic prediction of efficient emitters beyond this parameter remains challenging. Aside from ΔE ST the oscillator strength is a crucial indicator of ΦPL, but computing accurately the relevant rates of non-radiative processes remains non-trivial. Studies aiming at determining computationally the emission FWHM and k RISC are also becoming more common, which while computationally demanding are welcome additional lenses for assessing TADF materials designs. For MR-TADF compounds specifically, although we can now accurately predict their ΔE ST using wavefunction-based approaches, large-scale accurate screening has not yet been demonstrated due to their increased computational cost beyond the DFT approaches suitable for D-A TADF emitters. For both categories of TADF emitters finding a workable balance between computational cost and accuracy remains elusive, which is a key prerequisite before chemical space can be reliably explored and rapidly charted purely in silico.
In this context we note that while computational approaches towards assessing individual molecules are now reasonably mature (commonly performed in the gas phase or employing polarizable continuum solvent approximations), for yet deeper understanding and predictive power future efforts must increasingly focus on large multimolecular systems. The ability to simulate a molecule (or group of molecules) in an environment closely corresponding to real-world applications – namely by modelling atomistic solid-state host-guest morphologies – will give the most direct insight into real-world systems. These morphologies are what should then be used for subsequent excited-state calculations, rather than the more accessible relaxed geometries of isolated molecules typically used at present. Such calculations should also account for host-guest polarization effects, which are experimentally known to influence the ordering and spacings of excited states within the singlet and triplet manifolds, and therefore radically change the mechanistic picture of the whole TADF process. With the aforementioned effects already representing a challenging scale of simulation, for supreme accuracy these systems must also incorporate dynamic effects, as vibrational motion is also known experimentally to play a significant role in both the radiative and non-radiative processes governing TADF. Further, the involvement of higher-lying triplet states in RISC is already experimentally established, and computational approaches are uniquely suitable for probing these upper-state interactions towards building a more complete understanding of the structural features that support efficient TADF.
Aside from direct computations for specific molecular structures, recently we have also witnessed an increase in large-scale molecule screenings using machine learning tools. These strategies are welcome, likely speeding emitter design and pointing towards molecular structures far outside typical human intuition or imagination. However, the true impact of this approach is yet to be realised, with the optimal predicted emitters often being either synthetically very challenging (a difficult feature to quantify for model training), or not particularly novel (likely reflective of a limited training dataset of structures). We note that the utility of any machine learning model is intimately tied to the quality and size of its training data as well as identifying the desirable molecular features to be optimized. Moreover, the current academic research/cultural practices deprive the field of knowledge of TADF materials that do not reach “publishable” thresholds of performance or novelty. It is unclear how the research community’s decentralised and nebulous understanding of what doesn’t work – equally precious to the data scientist as what does work – could be made more accessible to support these data-driven efforts in materials design.
Blue TADF Emitters λEL < 490 nm
Introduction
Blue electroluminescence (EL) is uniquely challenging because blue photons are the highest in energy needed for human color vision (for a discussion of green and red EL see Sections sec4 and sec5, respectively). To produce these high photon energies large band gap emitters are required, with S1 energies typically > 2.7 eV. As most OLEDs contain an EML with an emitter doped into a host, these hosts must also be stable towards charge carriers and excitons of such high energy, significantly limiting the choice of usable chemical groups. Furthermore, in order to support high device efficiencies unlocked by triplet-harvesting in the emissive dopants, the host must also possess a higher triplet energy than that of the emitter. While these considerations are also applicable to red and green OLEDs, at the high energies associated with blue emission these requirements become especially limiting, with energies coming close or even exceeding bond dissociation energies of some of the organic materials. The relatively weak metal-ligand bonds associated with phosphorescent complexes are thought to be a main reason why blue PhOLEDs have not developed as rapidly as other colors, and these material stability issues are also a main factor that contributes to the severe efficiency roll-off in blue OLEDs.
To obtain blue emission, the bandgap of the emitter can be increased by stabilizing the HOMO and/or destabilizing the LUMO energy levels. In D-A TADF compounds this is typically achieved by combining weak electron donors (stabilized HOMO) with weak electron acceptors (destabilized LUMO), or by connecting multiple weak donors to a moderate acceptor. However, the energy of the emissive CT state is sensitive to the polarity of the environment, which leads to undesired emission red-shifting and broadening in the solid state. Together with the large FWHM typical of emissive CT states, this makes it very challenging to obtain deep blue emission that meets the Rec. 2020 standard.ref. ref222 The emergence of MR-TADF emitters has helped to address the color purity of blue TADF OLEDs (see Section sec11 ); however, their RISC rates are often far lower and to preserve this blue color most MR-TADF emitters must be doped into a host at a very low doping concentrations to avoid aggregation. These restrictions in applications lead to poorer exciton harvesting within the EML and a sub-optimally situated recombination zone. D-A TADF emitters, on the other hand, can be doped at higher concentrations or even used neat, and in many cases also contribute to charge balance in the devices.
In this context there has therefore been a particularly focused effort to develop efficient blue TADF emitters across the years 2017–2022.ref40,ref223−ref224ref225ref226ref227ref228 Many design strategies have been reported, almost always with the aims of achieving deep-blue emission while maintaining rapid RISC and high ΦPL. Some investigated facets of molecular design include dihedral angle control (increasing the D-A dihedral angle, and restricting D-A rotation), positional tuning between the donor and acceptor, heteroatom or heavy atom doping, rigidification of the emitter structure, and varying the number of donors and acceptors to optimize the D-A interaction. In this section we compare and analyze these blue D-A TADF emitters in terms of their photophysical properties and OLED performance, considering those having λEL < 490 nm and where the device showed an EQEmax > 10%, while the criterion for deep-blue emitters included the OLEDs having a CIEy coordinate < 0.10. For the sake of clarity, the summarized examples are divided into subsections according to the electron-acceptor and their properties are summarized in Table S1.
History and Context
In their seminal 2012 paper Uoyama et al. reported 2CzPN (Figure ), a blue TADF emitter based on a phthalonitrile acceptor and carbazole donors.ref. ref31 The design uses a moderate electron acceptor phthalonitrile coupled with multiple (in this case two) weak carbazole donors to give blue emission. The two donors are ortho to each other, which also helps to restrict the D-A rotation. This compound hence became one of the early benchmark emitters, with sky-blue emission (λPL = 473 nm, ΦPL = 47%) and a delayed lifetime, τd, of 166 μs in toluene (no ΔE ST was measured). OLEDs with 5 wt% 2CzPN in PPT host showed an EQEmax of 8%. Subsequent reports documented optimization of 2CzPN-based OLEDs and 5% of 2CzPN in a mixed cohost system of mCP:PPT (named PO15 in that work) showed the highest EQEmax of 21.8% at λEL = 480 nm [CIE coordinates of (0.17, 0.27)]. However, the efficiency roll-off remained severe with an EQE1000 of only 2.8%.ref. ref229 Such a large drop in efficiency was attributed to the relatively slow RISC rate in 2CzPN.
Zhang et al. reported the first deep-blue TADF OLED containing an emitter based on carbazole and diphenylsulfone, DTCz-DPS (originally named 3 in that work, Figure ).ref. ref230 Diphenylsulfone is a weak acceptor and two tert-butylcarbazoles served as moderate donors that yielded a deep-blue TADF emitter (λPL = 423 nm and ΦPL = 80% in 10 wt% doped films in DPEPO), albeit with a long τd of 2.6 ms and a large ΔE ST of 0.32 eV. OLEDs with DTCz-DPS showed an EQEmax of 9.9% at λEL = 423 nm [CIE coordinates of (0.15, 0.07)], but the efficiency roll-off was expectedly high considering the long τd. Since this first report, the sulfone acceptor has been employed widely within blue TADF emitters.
Another benchmark TADF emitter, DMAC-TRZ (Figure ),ref. ref231 despite being a sky-blue emitter, provided a good starting point for further fine-tuning and enhancing of emission properties. Many subsequent blue TADF emitters are based on similar structures, and the 9,10-dihydro-9,9-dimethylacridine (DMAC) donor has become extremely popular for its moderate donating strength and near orthogonal conformation adopted when it is N-bound to an acceptor (or bridge). The acceptor in DMAC-TRZ is 2,4,6-triphenyl-1,3,5-triazine (TRZ), which has also become popular in both blue and green TADF materials. DMAC-TRZ shows efficient sky-blue emission (λPL = 495 nm and ΦPL = 90% in 8 wt% doped mCPCN films) with a fast τd of 1.9 μs (Table S1). Such fast TADF is a consequence of the very small ΔE ST of 0.046 eV. Efficient triplet harvesting was evident in the device, with EQEmax of 26.5% at λEL of 495 nm, and showing a negligible efficiency roll-off with EQE100 of 25.1%.
While in this section we do not review the multi-resonant TADF (MR-TADF) emitters (see Section sec11 ), MR-TADF compounds have infiltrated the D-A world too; most notably employing MR-TADF moieties as acceptor, exemplified by DOBNA (Figure ).ref. ref232 Its intrinsically high triplet energy of 2.97 eV, its high ΦPL of 72% and the ease of chemical functionalization of this molecule has made DOBNA a very component of blue D-A TADF emitters since its first report in 2015.
Triazine-Containing Emitters
The TRZ moiety is one of the most widely employed acceptors used in blue TADF emitter design, and is the subject of a detailed review previously published by our group.ref. ref51 The popularity of TRZ stems from a reasonably shallow calculated LUMO energy of −1.72 eV suitable for blue emission, thermal stability and rigidity, and the ease of chemical substitution at the 2,4,6-carbon atoms. The chemical structures of recent TRZ-based blue TADF emitters summarized here are shown in Figure –Figure . One of the simplest recent examples of a blue D-A emitter containing TRZ is Cz-Ph-TRZ ref. ref233 (also reported as pCzTPTZ or CzTRZ,ref234,ref235 Figure ). Doped at 10 wt% in DPEPO, this compound emits at λPL of 438 nm and has a ΦPL of 71%, however the large ΔE ST of 0.36 eV prohibits TADF and Cz-Ph-TRZ is classed as a purely fluorescent blue emitter – often used as a reference or control material in the development of other new TADF emitters. OLEDs with Cz-Ph-TRZ showed no indications of triplet harvesting, with the EQEmax reached only 4.1% (or 5.8% in a separate reportref. ref236) at λEL = 446 nm [CIE coordinates of (0.14, 0.12)].
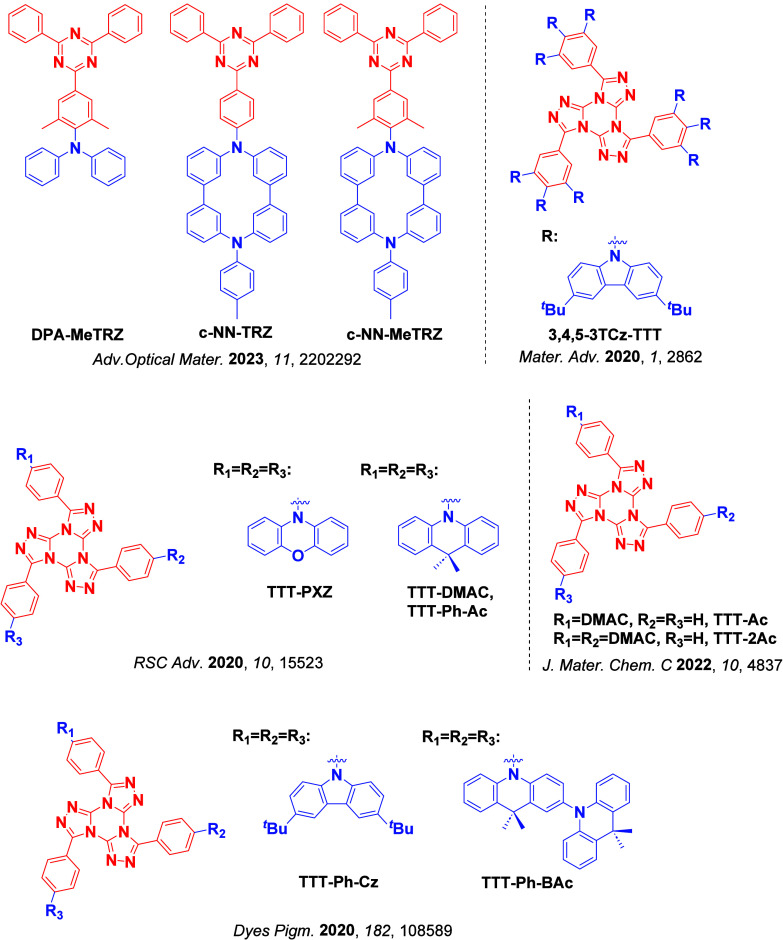
In tandem with TRZ, carbazole is one of the most commonly used donors for the design of TADF emitters, with its rigid structure and relatively weak calculated electron-donating strength (HOMO = −5.73 eV) supporting blue and deep-blue emission. However, unlike other larger donors such as DMAC, PXZ, or PTZ, carbazole has a smaller five-membered central ring which leads to it adopting less twisted conformations when linked to an acceptor (or bridge) via the nitrogen atom. This increased planarity can lead to a larger HOMO/LUMO overlap that translates to larger ΔE ST and poor TADF performance, for example in the aforementioned Cz-Ph-TRZ. The ΔE ST can, however, be reduced by increasing the donor strength of the carbazole by adding electron-donating substituents. The simplest example of such a modification was reported by Liu et al., who introduced tert-butyl substituents at the 3- and 6-positions of carbazole in the compound BuCz-TRZ (Figure ).ref. ref237 The increased donor strength was enough to turn on TADF in this emitter, and BuCz-TRZ doped at 6 wt% in DPEPO film emits at λPL = 439 nm, has a ΦPL of 83%, and a τd of 68.1 μs. The presence of TADF can be rationalized by the smaller ΔE ST to 0.29 eV, compared to 0.36 eV in Cz-Ph-TRZ. The OLED with BuCz-TRZ showed an EQEmax of 9.3% at λEL of 459 nm [CIE coordinates (0.15, 0.15)], although severe efficiency roll-off of 69% at 100 cd m–2 was reported (Table S1). Employing the same strategy, bulkier analogue DPFCz-TRZ emits at λPL = 429 nm, has a ΦPL of 88%, and a surprisingly short τd of 0.64 μs in toluene despite its moderately large ΔE ST of 0.21 eV.ref. ref238 The devices with 40 wt% DPFCz-TRZ doped in DPEPO showed an EQEmax of 15.5% and deep-blue emission at λEL = 445 nm [CIE coordinates of (0.15, 0.10)], although again with severe efficiency roll-off of 47% at 1000 cd m–2.
The electronics of carbazole as a donor can also be modified by fusing additional rings to it, with these extended donors also impacting the conformation of the emitter. Two isomeric hybrids based on indolocarbazole (ICz), IndCzpTr-1 and IndCzpTr-2 (Figure ), exemplify this strategy.ref. ref239 Both isomers showed comparable photophysics in neat films (λPL = 492 and 510 nm, ΦPL = 75 and 71%, τd = 35 and 34 μs, and ΔE ST = 0.13 and 0.11 eV, all respectively), with the less sterically crowded IndCzpTr-2 also having preferential horizontal dipole orientation (Table S1). This resulted in a twofold increase of EQEmax in the OLED with IndCzpTr-2 (30%) compared to that with IndCzpTr-1 (14.5%). However, the indolo[3,2-b]carbazole of IndCzpTr-2 is a stronger electron donor than indolo[2,3-a]carbazole of IndCzpTr-1, which led to a red-shift in the emission of the former as was also observed in film photoluminescence. Similarly, fused carbazolyl donors incorporating spiro-fluorenyl fragments are another well-studied category of extended donors. A family of four donors featuring differently substituted fluorenyl groups with a spirofluorene (InCz23FTz, λPL = 470 nm), diphenyl groups (InCz23DPhTz with λPL = 471 nm, and InCz34DPhTrz with λPL = 475 nm) and a dimethyl analogue (InCz23DMeTz, λPL = 488 nm) were reported.ref. ref240 Each of the emitters displayed high ΦPL values between 86 and 98% in 10 wt% doped films in DPEPO; however, in each case the delayed emission contribution was low (between 11 and 17%), suggesting inefficient triplet harvesting. Long τd between 70 and 98 ms support this conclusion, which was surprising given the relatively small ΔE ST values between 0.11 to 0.19 eV. The devices with InCz23FTz (λEL = 468 nm), InCz23DPhTz (λEL = 472 nm), In23DMeTz (λEL = 480 nm), and InCz34PhTz (λEL = 472 nm) showed EQEmax of 17.2, 17.9, 22.8, and 25.9%, respectively, although with significant efficiency roll-off of 63, 55, 43, and 45% at 100 cd m–2, and 82, 83, 75, and 80% at 1000 cd m–2.
Fusing a carbazole donor with a benzofuran group and attachment of an additional secondary carbazole unit gave the D-A emitter Trz-BFCzCz (Figure ).ref. ref241 The compound doped at 20 wt% in DPEPO emits at λPL of 460 nm, has a ΦPL of 75%, a τd of 37 μs, and a moderate ΔE ST of 0.13 eV in frozen toluene. Compared to reference emitter Trz-CzCz containing only bicarbazole and no fused furan group, the ΔE ST was decreased by 100 meV and the τd was shortened by 70 μs with only a minor sacrifice in ΦPL (ΦPL of Trz-CzCz is 89%). The sky-blue OLED [CIE coordinates of (0.18, 0.32)] with Trz-BFCzCz showed an EQEmax of 23.3%, but the EQE1000 dropped considerably to 13.1%. Despite the efficiency roll-off being high, the Trz-BFCzCz OLED showed an improvement of four percentage points in the EQE1000 compared to device with Trz-CzCz (EQEmax = 23.8%, EQE1000 = 8.9%).
The electron-donating strength of carbazole derivatives can be further tuned by incorporating heteroaromatic substituents at the 3- and 6-positions. Examples include the use of dibenzothiophene (DBTCz-Trz) and dibenzofuran (DBFCz-Trz and BDBFCz-Trz, Figure ).ref. ref242 These compounds showed moderate ΔE ST values of 0.20–0.23 eV and high ΦPL of >89% in 15 wt% doped films in DPEPO. The OLEDs with DBTCz-Trz, DBFCz-Trz, and BDBFCz-Trz showed EQEmax of 21.7, 21.6, and 21.5% at λEL of 472, 472, and 488 nm, all respectively (Table S1). However, the efficiency roll-off was very high (88, 85, and 80%, respectively at 1000 cd m–2), which was attributed to singlet polaron quenching resulting from charge imbalance when using DPEPO host. Wang et al. reported a similar triazine-carbazole hybrid named PPCTRZ that featured phenanthroimidazole substitution on the carbazole and showed deep-blue emission, with a λPL of 411 nm and ΦPL of 38% in 10 wt% doped films in CBP.ref. ref243 The reference compound DCBTRZ contained a second carbazole instead of the phenanthroimidazole, and also showed similar photophysics with λPL of 435 nm and ΦPL of 39% in doped films. Both compounds have large ΔE ST of 0.39 and 0.38 eV for DCBTRZ and PPCTRZ, respectively, and transient PL decay measurements of 10 wt% doped CBP films showed multiexponential decay kinetics with only short nanosecond lifetimes. Analysis of the variable-temperature data did not reveal any notable TADF behavior, despite the claims by the authors that the compounds are TADF-active. Deep-blue OLEDs with DCBTRZ and PPCTRZ in CBP host showed λEL of 440 and 442 nm (CIEy of 0.059 and 0.063), which was very red-shifted compared to the film PL yet exhibited rather lower EQEmax values of 6.6 and 6.5% – likely illustrating the lack of triplet harvesting in the devices.
Frequently unmodified carbazole in D-A compounds adopts a relatively less twisted conformation with the acceptor, which can lead to large HOMO/LUMO overlap that is detrimental to overall TADF performance. Significant efforts have therefore been devoted to modifying carbazole to increase the steric congestion close to the nitrogen atom and tune the D-A dihedral angles. This is often achieved by either introducing substituents on a π-bridge connecting D-A moieties or attaching directly on the donor itself at the 1- and 8-positions. For example, cyano groups ortho-disposed to the donor act not only as a functional steric control units but can also tune the electronics of the resulting compound.ref119,ref120 This strategy was demonstrated in TrzCNBFCz and Trz2CNBFCz containing one or two CN groups, which were compared to reference material TrzBFCz without such modification (Figure ).ref. ref244 TrzCNBFCz and Trz2CNBFCz in THF at 77 K have smaller ΔE ST of 0.13 and 0.10 eV (respectively) compared to 0.27 eV for TrzBFCz. As well as providing steric control, the CN groups also act to stabilize both the HOMO and LUMO levels of the emitters as determined by CV, where the HOMO of TrzCNBFCz was stabilized by ca. 100 meV and the LUMO by 200 meV in comparison to TrzBFCz. In Trz2CNBFCz the influence of the CN groups on the orbital energies is even more dramatic, with the HOMO stabilized by 200 meV and LUMO by 670 meV. Consequently, the reduced HOMO-LUMO gap in Trz2CNBFCz resulted in a red-shifted emission (λPL = 432 nm) while the λPL is 407 nm for TrzCNBFCz. TrzCNBFCz has 100% ΦPL in 10 wt% doped films in DPEPO, while the ΦPL of Trz2CNBFCz is lower at 62% and short τd were registered for both compounds (τd = 9.4 μs for TrzCNBFCz and 3.1 μs for Trz2CNBFCz) (Table S1). Deep-blue OLEDs with TrzBFCz [CIE coordinates of (0.15, 0.10)] showed an EQEmax of 18% which decreased by 55% at 1000 cd m–2. The OLED with TrzCNBFCz showed sky-blue emission [CIE coordinates of (0.17, 0.31)], an improved EQEmax of 20.9%, and a reduced efficiency roll-off of 37% at 1000 cd m–2. The OLED with Trz2CNBFCz showed CIE coordinates of (0.27, 0.52) and EQEmax of 15%, a consequence of the considerably lower ΦPL.
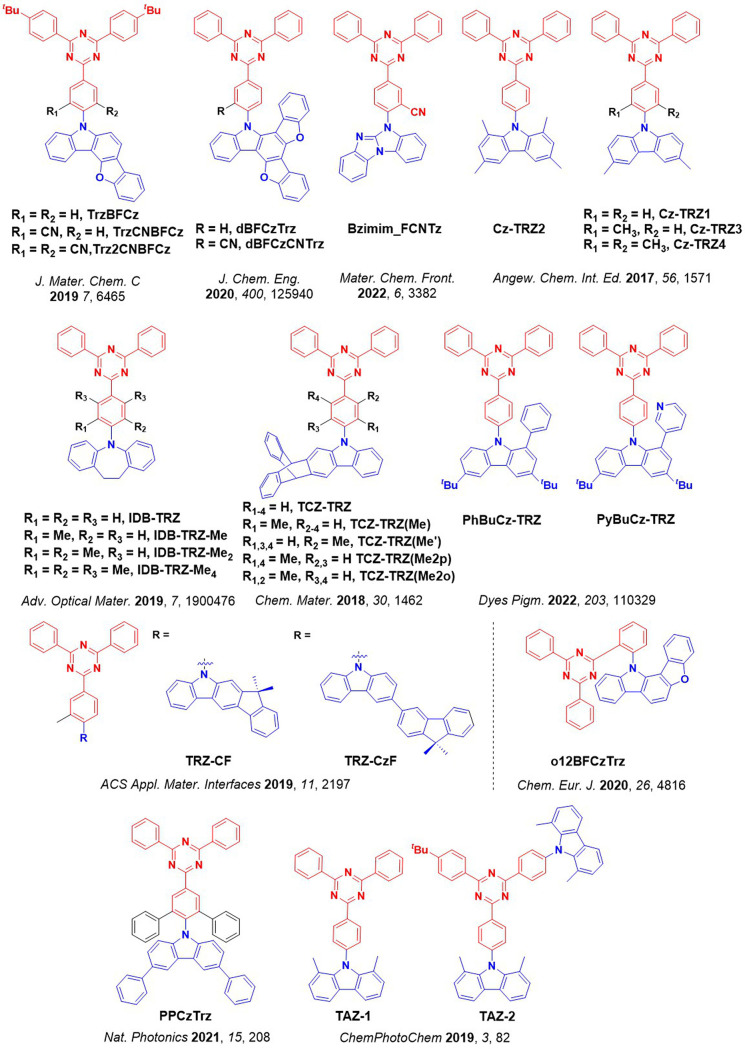
Demonstrating a similar impact of CN substitution on the π-bridge connecting D-A emitters, dBFCzTrz and dBFCzCNTrz containing a dioxoazatruxene type donor were developed (Figure ).ref. ref245 As expected the bulky donor, both compounds adopt a strongly twisted geometry, with triplet energies found to be 2.88 and 3.02 eV for dBFCzCNTrz and dBFCzTrz, respectively. While both molecules have near-unity ΦPL, dBFCzTrz has a longer τd of 30 μs compared to 4.9 μs for dBFCzCNTrz, each at 20 wt% in DPEPO (Table S1). The OLED with dBFCzTrz is bluer [CIE coordinates (0.16, 0.27)] although showed a lower EQEmax of 22.6% (efficiency roll-off of 46% at 1000 cd m–2) compared to its counterpart with dBFCzCNTrz ([CIE coordinates (0.22, 0.47)] and EQEmax of 27.5%, and efficiency roll-off of 12% at 1000 cd m–2). The same research group also investigated the effect of changing the donor position in dBFCzTrz, where three benzofurocarbazole isomers were ortho-connected to the triazine moiety.ref. ref246 While all three of the newly reported compounds showed similar ΦPL (82–88%), τd (3–4 μs), and triplet energy values (2.9–3.1 eV), the device with o12BFCzTrz showed the bluest emission with λEL = 478 nm [CIE coordinates of (0.16, 0.29)], an EQEmax of 19.2%, and a surprisingly low efficiency roll-off of 11% at 1000 cd m–2.
A fused imidazole-carbazole based donor in conjunction with a CN-substituted π-bridge was similarly explored for blue TADF emission in Bzimim_FCNTz (Figure ).ref. ref247 Bzimim_FCNTz emits at λPL of 461 nm with moderate ΔE ST of 0.17 eV in toluene, and has unity ΦPL with short τd of 7.9 μs in 20 wt% doped films in DPEPO. OLEDs with 20 wt% Bzimim_FCNTz in DPEPO demonstrated the best performance with an EQEmax of 22.6% [CIE coordinates of (0.17, 0.27)], and a low efficiency roll-off of only 11% at 100 cd m–2 (Table S1).
Replacing electron-withdrawing cyano groups on phenylene bridges with donating methyl groups should achieve similar steric control while also supporting larger HOMO-LUMO gaps and blue-shifted emission. Cui et al. reported a series of carbazole-triazine compounds featuring such methyl substituents, used to fine-tune the blue emission color and TADF performance (Figure ).ref. ref248 DFT calculations showed that the dihedral angle between the donor and acceptor planes was tuned from 49.8° in reference emitter Cz-TRZ1 (no methyl group between D-π-A) to 71.3° in Cz-TRZ3 (1 methyl group on the π-bridge) to 86.7° in Cz-TRZ2 (2 methyl groups on the donor) and 82.3° in Cz-TRZ4 (2 methyl groups on the π-bridge). Incorporation of methyl groups on the π-bridge resulted in a modest red-shift in the emission in Cz-TRZ3 and Cz-TRZ4, with λPL of 435 and 432 nm respectively. Cz-TRZ2 bears two additional methyl groups on the donor, which increases the electron-donating strength and leads to a larger red-shift of the emission to a λPL of 465 nm in toluene. As with previous examples, the steric control of the dihedral angle between donor and acceptor has a significant impact on ΔE ST. Cz-TRZ1 has a ΔE ST of 0.43 eV in toluene, while introduction of methyl groups decreases ΔE ST to 0.07, 0.17, and 0.15 eV in Cz-TRZ2, Cz-TRZ3, and Cz-TRZ4, respectively. This steric control has a negative impact on ΦPL though, which falls from 72% for Cz-TRZ1 to 35% for Cz-TRZ4 in toluene. Notably, in the 6 wt% doped films in DPEPO the ΦPL for Cz-TRZ1–4 increases from 87 to 98, 92, and 85%, respectively. The impacts of steric control on the emitter conformation is also reflected in τd, with unsubstituted Cz-TRZ1 having the longest τd of 29 μs, while the τd of Cz-TRZ2–4 are 3.5, 13, and 10 μs, respectively. Deep-blue OLEDs with Cz-TRZ3 and Cz-TRZ4 both showed CIE coordinates of (0.15, 0.10) and respective EQEmax of 19.2 and 18.3%, as well as efficiency roll-off of approximately 19 and 23% at 100 cd m–2. The OLED with Cz-TRZ2 showed an EQEmax of 22.0% and sky-blue electroluminescence due to its stronger donor, with CIE coordinates (0.16, 0.24).
Triptycene can also direct steric interactions to enhance frontier molecular orbital spatial separation, which was shown in a series of triptycene-modified carbazole-triazine materials. The presence of triptycene fused to carbazole enables TADF in TCZ-TRZ (λPL = 432 nm, ΦPL = 77%, ΔE ST = 0.27 eV, τd = 38 μs in toluene) while the reference carbazole-triazine compound Cz-Ph-TRZ is only fluorescent.ref. ref249 Introduction of a methyl moiety ortho to the carbazole in TCZ-TRZ(Me) or ortho to the TRZ in TCZ-TRZ(Me′) results in further reduction in ΔE ST for both methylated derivatives, while the specific position of the methyl group significantly affects the ΦPL. TCZ-TRZ(Me) and TCZ-TRZ(Me′) emit at λPL = 431 and 429 nm, have ΦPL of 60 and 80%, ΔE ST of 0.16 and 0.12 eV, and τd of 51 and 58 μs in toluene, all respectively. Additional emitters with two methyl groups decorating the bridging moiety have reduced ΦPL although an improvement in RISC efficiency, as seen in TCZ-TRZ(Me2p) and TCZ-TRZ(Me2o) with λPL for both at 427 nm, ΦPL of 46 and 47%, ΔE ST of 0.14 and 0.18 eV, and τd of 39 and 37 μs in toluene, all respectively (Table S1). The devices with TCZ-TRZ and TCZ-TRZ(Me) showed EQEmax of 10.4% [CIE coordinates of (0.16, 0.14)] and 11.1% [CIE coordinates of (0.17, 0.18)], respectively. Unfortunately, the devices suffered from a severe efficiency roll-off with EQE50 being only 3.4 and 2%, which was attributed to the long excited state lifetimes of the emitters.
The impact of methyl substitution on the linking phenylene bridge was also studied in a family of emitters containing an iminodibenzyl donor. The reported molecules were IDB-TRZ (unsubstituted π-bridge), IDB-TRZ-Me (1 methyl group adjacent to the donor), IDB-TRZ-Me2 (2 methyl groups adjacent to the donor), and IDB-TRZ-Me4 (4 methyl groups, Figure ).ref. ref250 While the donor itself is quite flexible due to the ethyl bridge, it became locked in a highly twisted geometry in IDB-TRZ-Me2 which resulted in decreased non-radiative decay, leading to a high ΦPL of 98% in 20 wt% doped films in PPF. The steric control of the donor conformation also impacted the ΔE ST, which decreased progressively from 0.182 eV in IDB-TRZ to 0.093 eV in IDB-TRZ-Me, 0.077 eV in IDB-TRZ-Me2 , and ∼0 eV in IDB-TRZ-Me4 . The negligible ΔE ST in IDB-TRZ-Me4 supported a two-order magnitude accelerated k RISC of 120 × 104 s–1 compared to the other materials in the family (k RISC = 1.7, 4.3, and 6.4 × 104 s–1 for IDB-TRZ, IDB-TRZ-Me, and IDB-TRZ-Me2 , respectively). However, this faster RISC came at a cost of a relatively low ΦPL of 37%. The devices employing the IDB-TRZ derivatives showed sky-blue emission with λEL ranging from 474 to 496 nm, and with device efficiency reflecting the underlying photophysics of the emitter. The device with IDB-TRZ-Me2 showed the highest EQEmax at 28.3%, followed by IDB-TRZ-Me4 (16.4%), IDB-TRZ-Me (12.3%), and IDB-TRZ (6.8%). A relatively low efficiency roll-off of ∼14% at 100 cd m–2 was additionally reported for the device with IDB-TRZ-Me2 .
Using a previously discussed fused carbazole-fluorene donor, emitter TRZ-CF (Figure ) also contained a methyl group on the phenylene bridge adjacent to the donor. Comparator emitter TRZ-CzF contained the same modified acceptor with a carbazole donor instead featuring a pendant (rather than fused) fluorenyl group at the 2-position.ref. ref251 TRZ-CF and TRZCzF have long τd of 7.3 and 11 ms in 20 wt% DPEPO films, with moderately large ΔE ST of 0.22 and 0.31 eV in 2-MeTHF glass, all respectively. TRZ-CF with more conjugated fused donor emits at λPL = 474 nm, which is red-shifted compared to TRZCzF (λPL = 458 nm). Consistent with the respective ΦPL values of 86 and 69%, the devices with TRZ-CF and TRZ-CzF showed EQEmax of 20 and 13.3% at λEL of 476 and 460 nm (Table S1). The magnitudes of the efficiency roll-off at 1000 cd m–2 were 43 and 72% respectively, which correlated with the magnitude of the delayed lifetime.
Bulky electron-withdrawing groups positioned at the C-1 position of carbazole were introduced to increase the torsion between triazine and the donor.ref. ref237 The properties of five molecules containing phenyl, (PhBuCz-TRZ), pyridinyl (PyBuCz-TRZ, PyBuCz-MeTRZ), and cyano groups (CNBuCz-TRZ, Figure ) were compared to reference compound BuCz-TRZ (Figure ). The compounds showed near-UV to deep-blue emission with λPL ranging from 398–440 nm in toluene and high ΦPL ranging from 77–87% in 6 wt.% doped films in DPEPO. Introduction of bulky group on the donor positively affected the delayed lifetimes, with the τd of 68.1 μs for the unsubstituted BuCz-TRZ dropping to 44.1 μs for phenyl-substituted PhBuCz-TRZ, and then to 35.8 μs for pyridinyl-substituted PyBuCz-TRZ, and to 30.1 and 23.6 μs for methyl-substituted BuCz-MeTRZ and PyBuCz-MeTRZ, respectively. Surprisingly, donor modifications had only a minor effect on the excited state energies with ΔE ST ranging narrowly between 0.27–0.30 eV for BuCz-TRZ, PhBuCz-TRZ, and PyBuCz-TRZ. Introduction of the methyl group, however, resulted in a further reduction of the ΔE ST to 0.25 eV and 0.24 eV for BuCz-MeTRZ and PyBuCz-MeTRZ, respectively. The reference OLED with BuCz-TRZ showed an EQEmax of 9.3% with λEL of 459 nm [CIE coordinates (0.15, 0.15)], but the devices with PhBuCz-TRZ and PyBuCz-TRZ showed higher EQEmax of 12.1 and 15.3%, respectively (Table S1). These OLEDs were also bluer, with respective λEL of 458 and 455 nm [CIE coordinates of (0.15, 0.16) and (0.15, 0.13)]. On the other hand, OLEDs containing methyl and cyano-substituted derivatives showed significantly different performance metrics. The OLEDs with BuCz-MeTRZ and PyBuCz-MeTRZ showed triplet harvesting with EQEmax of 15.5 and 11.7%, respectively, while the EQEmax of CNBuCz-TRZ OLED was only 4.1%. The devices suffered from severe efficiency roll-off though, with EQE dropping to 50% of the maximum values at 100 cd m–2 and no data provided at 1000 cd m–2.
The conformation of carbazole donors can also be twisted through the introduction of methyl substituents at both the 1- and 8-positions, as in TAZ-1 and TAZ-2 (Figure ).ref. ref252 Steric locking of carbazole not only yielded smaller ΔE ST values (0.15 and 0.10 eV for TAZ-1 and TAZ-2, respectively), but also improved ΦPL of 88 and 100% at λPL = 468 or 476 nm in 20 wt% doped films in PPF, respectively (Table S1). The OLEDs with TAZ-1 and TAZ-2 showed EQEmax of 17.7 and 21.2%, and emitted at λEL of 478 and 479 nm [CIE coordinates of (0.16, 0.25) and (0.16, 0.27)], all respectively. Both OLEDs showed an efficiency roll-off of ∼40% at 1000 cd m–2. Yeon et al. instead explored the use of phenyl substituents on the phenylene bridged, ortho to the donor in PPCzTrz.ref. ref253 The compound emits at λPL of 444 nm, has ΦPL of 93%, τd of 25 μs, and ΔE ST of 0.16 eV in 20 wt% doped films in DPEPO (Table S1). The OLEDs showed high EQEmax of 34% [CIE coordinates of (0.13, 0.20)] and a moderate efficiency roll-off of 24% at 1000 cd m–2. When a mixed co-host system of oCBP:CNmCBPCN was used the EQEmax dropped to 10%, but the device lifetime (LT50) was improved significantly from 1 to 24 h running at 1000 cd m–2.
DMAC-TRZ (Figure ) was designed relatively early in the current boom of TADF research, and has become a popular reference compound due to its high solid-state ΦPL (90%), remarkably short τd of 1.9 μs, and negligible ΔE ST of just 46 meV in 8 wt% doped mCPCN films.ref. ref231 However, the compound is a sky-blue-emitter with λPL of 495 nm, and often green-emissive in other solvents and hosts. Significant effort has been devoted to derivatizing this model structure in order to retain the efficient TADF properties and tune the emission color deeper into the blue. For example, the methyl moieties in DMAC were substituted for an adamantyl group in a-DMAc-TRZ.ref. ref254 Dual fluorescence was observed as a result of quasi-equatorial (QEC) and quasi-axial (QAC) excited state conformers. At 1 wt% doping in DPEPO the λPL of 430 nm was attributed to locally-excited fluorescence from QAC, exhibiting only a prompt lifetime of 15.45 ns and a large ΔE ST of 0.31 eV (Table S1). Increasing the doping concentration to 20 wt% caused a red-shift in the emission to λPL = 490 nm along with activating efficient TADF with τd = 4.1 μs, ΦPL = 86%, and a reduced ΔE ST of 0.20 eV attributed to dominant QEC (Table S1). The OLED showed a high EQEmax of 28.9% at CIE coordinates of (0.18, 0.35), however the device showed a rather severe efficiency roll-off at 100 cd m–2 of 56%.
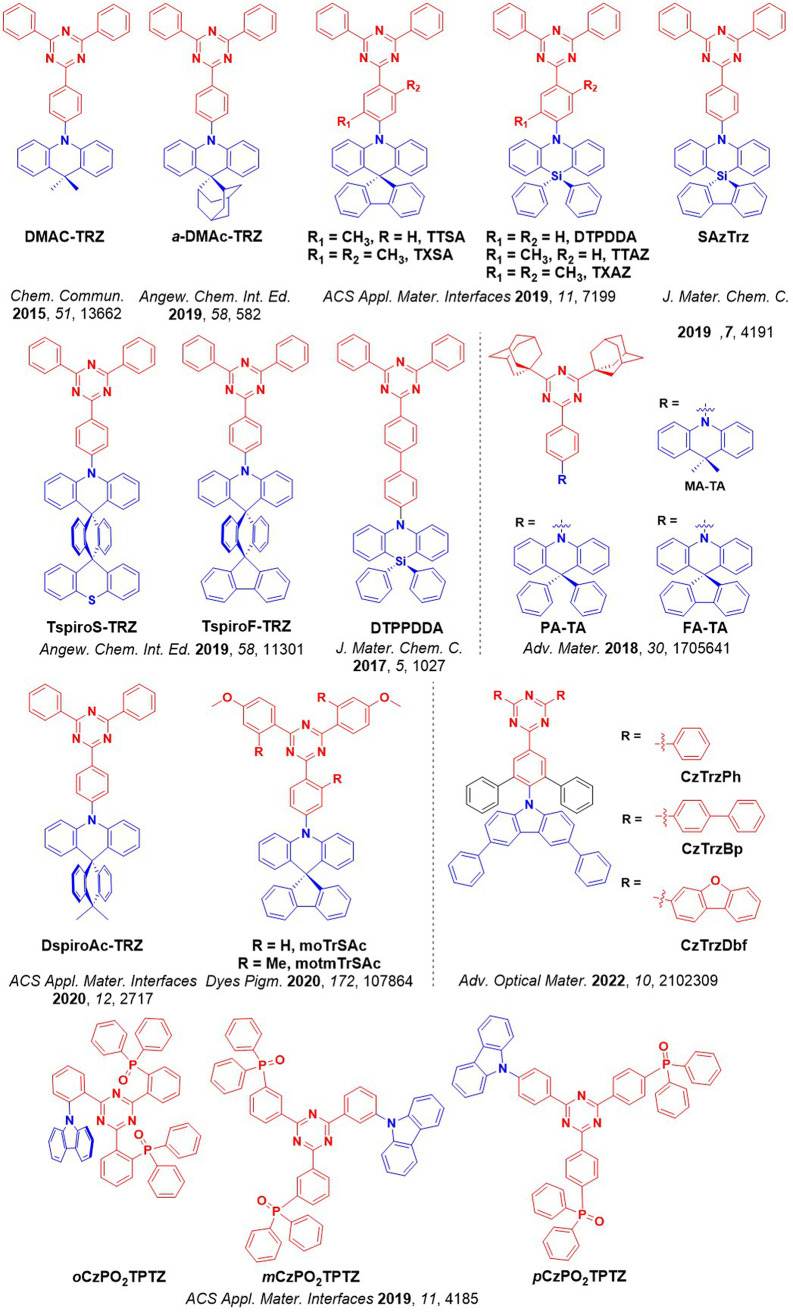
Similar to the carbazole-containing examples discussed further above, methyl groups can be installed into the linking phenylene bridge to influence the acridine torsional angle.ref. ref255 TTSA (λPL = 481 nm in 10 wt% mCP:TSPO1) and TTAZ (λPL = 465 nm) feature only one methyl group ortho to the donor (Figure ). TXSA (λPL = 475 nm in 10 wt% DPEPO) and TXAZ (λPL = 458 nm) have two methyl groups to further twist the Ph-acceptor torsion, resulting in a blue-shifted emission due to the reduced conjugation. The increased twisting of the bridging phenylene had only a minor effect on ΦPL and ΔE ST, with TTSA, TXSA showing near-unity ΦPL values and ΔE ST as small as 60 meV. Phenazasiline derivatives TTAZ and TXAZ showed much lower ΦPL of 68 and 50% and much larger ΔE ST of 0.16 and 0.18 eV, respectively (Table S1). These values were found to be comparable to the ones of the previously reported unsubstituted phenazasiline-triazine hybrid, DTPDDA (ΦPL = 74% and ΔE ST of 0.14 eV in 16 wt% doped films in mCP:TSPO1).ref. ref256 The EQEmax for the devices with TTSA and TXSA were 27.9 and 20.7% at λEL of 480 and 476 nm respectively, with reasonable efficiency roll-offs of 19 and 22% at 100 cd m–2. The devices with phenazasiline emitters TTAZ and TXAZ showed EQEmax values of 23.7 and 16.0% at λEL = 464 and 456 nm, with efficiency roll-off of 39 and 47% at 100 cd m–2.
The use of a phenazasiline donor coupled to an extended triazine yields deep-blue TADF emitter DTPPDDA (Figure ).ref. ref257 When doped at 8 wt% in a cohost of mCP:TSPO1, DTPPDDA emits at λPL of 439 nm and has a ΦPL of 38%. Despite the small ΔE ST of 0.04 eV, no delayed emission was reported (Table S1). Deep-blue OLEDs with CIE coordinates (0.15, 0.09) nonetheless showed a moderate EQEmax of 4.7%, exceeding the theoretical limiting EQEmax of 4.4% for fluorescence and demonstrating at least some triplet harvesting activity.ref. ref257 The absence of effective triplet harvesting resulted in a large efficiency roll-off of 50% at 100 cd m–2 and a low maximum luminance of 281 cd m–2. Similar silicon-containing analogues DTPDDA and SAzTrz, were also reported.ref. ref258 SAzTrz doped at 10 wt% in mCP:TSPO1 co-host emits at λPL of 465 nm and has a ΦPL of 65%, however a moderately large ΔE ST of 0.25 eV resulted in slow TADF with τd of 173 μs (Table S1). The device employing the same co-host system emitted at CIE coordinates of (0.15, 0.18) and showed an EQEmax of 20.6%, but again severe efficiency roll-off of 64% at 100 cd m–2 was reported with a maximum luminance of 440 cd m–2.
A so-called tri-spiro donor strategy was shown to be effective in reducing ACQ, as well as in increasing the horizontal orientation of the TDMs in TspiroS-TRZ and TspiroF-TRZ (Figure ).ref. ref259 Perpendicular chromophore orientation ensured sufficient frontier orbital separation, which resulted in ΔE ST values as small as 0.05 and 0.08 eV for TspiroS-TRZ and TspiroF-TRZ respectively. Both materials showed sky-blue emission in 30 wt% doped films in DPEPO (λPL = 470 and 479 nm) with τd of 3.0 and 4.5 μs, and ΦPL of 75 and 82%, all respectively (Table S1). Such outstanding photophysics was reflected in the device performance, with EQEmax values of 33.3 and 28.1% for the OLEDs with TspiroS-TRZ and TspiroF-TRZ at λEL = 481 and 493 nm. These devices also displayed moderate efficiency roll-off of 29 and 18% at 100 cd m–2. Remarkably, a non-doped device containing TspiroS-TRZ demonstrated an EQEmax of 20%, which at the time was one of the most efficient sky-blue non-doped OLEDs. The same authors subsequently reported a slightly modified emitter structure, DspiroAc-TRZ.ref. ref260 Studying intermolecular interactions in the crystalline state, the authors discovered that the intermolecular distances were sufficiently long to decrease the HOMO-LUMO interactions of dimers while still allowing for horizontal orientation of their TDM. This afforded high ΦPL in crystalline and amorphous non-doped films of 78.5% at λPL = 496 nm and 83.7% at λPL = 482 nm, respectively. The ΔE ST, determined in frozen toluene, was 0.04 eV and the τd of the neat film was τd = 3.2 μs. The non-doped sky-blue device consequently outperformed the parent device with TspiroS-TRZ, with an EQEmax of 25.7% and an efficiency roll-off of 36% at 1000 cd m–2.
In addition to donor modification, modulating of the degree of conjugation in TRZ has been probed as a strategy to blue-shift the emission. Compounds moTrSAc and motmTrSAc (Figure )ref. ref261 contain a spiro-DMAC donor linked to a modified TRZ acceptor. Twisting of the TRZ phenylenes by means of ortho-methyl groups resulted in destabilization of both the singlet and triplet energies by ca. 100 meV in motmTrSAc compared to the unsubstituted parent moTrSAc, with very low ΔE ST of 0.01 eV for both compounds. Short τd of 3.4 and 3.0 μs and ΦPL of 70% at λPL = 482 nm or 51% at λPL = 469 nm were reported for moTrSAc and motmTrSAc in respective 10 wt% doped films in DPEPO (Table S1). The bluest device incorporating motmTrSAc showed an EQEmax of 19.5% at CIE coordinates of (0.16, 0.22).
Starting from the basis of the previously discussed PPCzTrz (Figure ), Kang et al. modified the TRZ acceptor moiety in the hope of localizing the triplet excitons far from the weak D-A C-N bond.ref. ref262 A series of triazine-carbazole compounds was designed with a focus on expanding conjugation in the TRZ moiety through introduction of biphenyl (CzTrzBp) or dibenzofuranyl fragments (CzTrzDbf). SCS-ADC(2) calculations revealed migration of the 3LE state, from localization on the π-spacer with slight extension into the donor in reference molecule CzTrzPh, to localization mainly on the distal arms of the acceptor in CzTrzBp and CzTrzDbf, thereby reducing excited state electron density near the vulnerable C-N bond. All three compounds showed deep-blue emission with λPL ranging from 444–451 nm in toluene and have moderate ΦPL of 34, 35, and 49% for CzTrzPh, CzTrzBp, and CzTrzDbf respectively in 15 wt% doped films in CNmCBPCN. The extension of the π-conjugation on the acceptor however increased the ΔE ST values from 0.16 to 0.26 and 0.31 eV, which translated into lengthening of the τd from 15.3 to 26.3 and 30.3 μs for CzTrzPh, CzTrzDbf, and CzTrzBp, all respectively. Blue OLEDs with CzTrzDbf and CzTrzBp showed EQEmax of 12.4 and 9.2% at CIE coordinates of (0.16, 0.19) and (0.16, 0.14), respectively. In terms of initial performance, the CzTrzPh containing OLED showed almost identical values to the device with CzTrzDbf; however, the LT80 of the former was only 17.2 hours compared to 40.3 hours for the latter, both running at an initial 500 cd m–2. The CzTrzBp OLED also showed an improved device lifetime with LT80 of 30.5 hours.
Replacing the peripheral rings of TRZ with adamantyl groups not only improved solubility, allowing solution-processed devices to be fabricated, but also resulted in a destabilized LUMO and a blue-shifted emission.ref. ref263 Wada et al. employed this acceptor in combination with DMAC donors in three blue TADF emitters, MA-TA, FA-TA, and PA-TA (Figure ).ref. ref263 MA-TA, FA-TA, and PA-TA doped at 10 wt% in CzSi have ΦPL of 83% [CIE coordinates of (0.15, 0.19)], 76% [CIE coordinates of (0.15, 0.13)], and 70% [CIE coordinates of (0.15, 0.10)], respectively. The solution-processed devices showed respective EQEmax of 22.1, 11.2, and 6.7% at the same CIE coordinates as the photoluminescence, with efficiency roll-off of 37% at 100 cd m–2 noted for the device with MA-TA. Luminance of 100 cd m–2 could not be reached for the devices using the other two emitters.
Using a carbazole donor and a TRZ acceptor functionalized with phosphine oxide groups, excellent TADF efficiency was observed in oCzPO2TPTZ, mCzPO2TPTZ, and pCzPO2TPTZ (Figure ).ref. ref264 These compounds emit at λPL of 470–485 nm and showed a clear trend in ΦPL across the o/m/p isomers of 25, 53 and 75%, respectively. The trends in ΦPL were then reflected in the OLED performance: an EQEmax of 20.9% was reported for the device with pCzPO2TPTZ, which decreased to 11.6% for the device with mCzPO2TPTZ and to 6.7% for the device with oCzPO2TPTZ (Table S1). Compared to the non-phosphine oxide parent, pCzTPTZ (Figure ), the presence of the secondary acceptor led to a smaller ΔE ST, higher ΦPL, and faster k RISC (ΔE ST = 0.01 and 0.17 eV, ΦPL = 73 and 11%, and k RISC = 7.1 and 0.6 × 104 s–1 for pCzPO2TPTZ and pCzTPTZ, respectively).
Blue TADF can alternatively be enabled by incorporating multiple weak donors about a central triazine acceptor. Oh et al. explored the impact on blue TADF devices of changing the relative position of a pair of carbazole groups about the same phenylene bridge attached to a triazine acceptor.ref. ref265 A blue-shift was observed in moving from ortho-meta substitution [23CT, CIE coordinates of (0.17, 0.33)] to ortho-para [24CT, CIE coordinates of (0.15, 0.26)] and finally to meta-para substitution [34CT, CIE coordinates of (0.15, 0., 0.17)], which is in line with the decreasing D-A dihedral angles that result in the later compounds having excited states with more of LE character (Figure ). This blue-shift is accompanied by a negative impact on the TADF properties though, with ΔE ST = ∼0, 0.11, and 0.29 eV for 23CT, 24CT, and 34CT, respectively (Table S1). The devices showed EQEmax of 21.8% [CIE coordinates of (0.17, 0.33)], 22.4% [CIE coordinates of (0.15, 0.26)], and 13.3% [CIE coordinates of (0.15, 0.17)], respectively, with extraordinary efficiency roll-off of only 5% at 1000 cd m–2 for the device with 23CT. The efficiency roll-off for the devices with 24CT (32%) and 34CT (58%) was considerably larger. The improved device performance for 23CT was attributed to efficient RISC as a result of its well-aligned 1CT and 3LE states. The same authors also explored the benefit of having three carbazole donors similarly substituted about the same phenylene at different positions.ref. ref266 While the greenest compound from the series 234CzTrz has the shortest τd of 4.1 μs and a ΦPL of 90%, the other two compounds, 235CzTrz and 245CzTrz, have longer τd of 8.4 and 9.7 μs respectively and also almost unity ΦPL values in 30 wt% doped films in DPEPO. The bluest device using 245CzTrz [CIE coordinates of (0.17, 0.39)] showed an EQEmax of 22% as well as an efficiency roll-off of 37% at 1000 cd m–2. Comparing the device performance of emitters with 2 vs 3 carbazoles, i.e., OLEDs with 245CzTrz vs 23CT, the latter outperformed the former in terms of color purity and efficiency roll-off despite having almost identical EQEmax. In separate report, excellent efficiency roll-off of 5% at 1000 cd m–2 with CIE coordinates of (0.15, 0.22) was achieved using trisCz-TRZ, an emitter containing three carbazoles symmetrically ortho-substituted to a triphenyltriazine core.ref. ref267 This performance was supported by its short τd of 5.0 μs and small ΔE ST of 0.03 eV, although the EQEmax was only 16.5%.
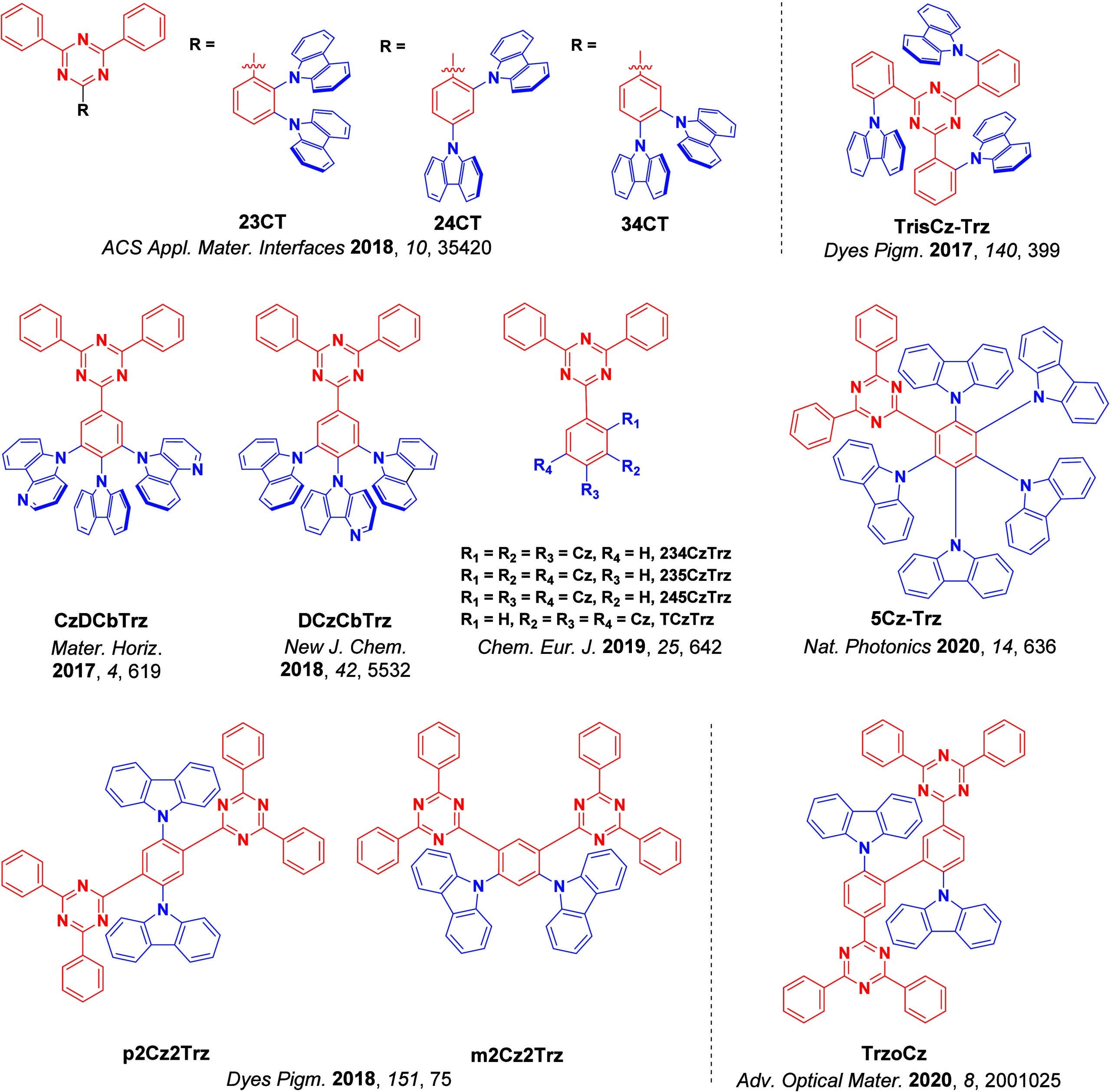
Another multi-donor-substituted example is CzDCbTrz, whereby two δ-carbolines are attached at meta positions to TRZ with a carbazole at the para position (Figure ). The EQEmax of the device with CzDCbTrz was 22.0%, which is an improvement from the 19.0% reported for tris-carbazole containing reference emitter TCzTrz.ref. ref268 A shorter τd of 7.5 μs (compared to 9.2 μs TCzTrz) likely contributes to the improved device performance while a slight blue-shift was also reported for the CzDCbTrz λEL, shifting from 476 to 471 nm on the inclusion of carbolines. Similar to CzDCbTrz, DCzCbTrz instead contains one para-connected δ-carboline and two meta-substituted carbazole donors around the bridging phenylene.ref. ref269 The OLED with DCzCbTrz showed a similar EQEmax of 22.1% and a similar efficiency roll-off at 1000 cd m–2 (54% for the device with DCzCbTrz and 57% for the device with CzDCbTrz), which was again an improvement from carboline-free TCzTrz. Other emitters featuring α- or δ- carbolines paired with benzonitrile acceptors are discussed further below.
Increasing further the number of carbazole donors, compound 5Cz-Trz (Figure ) is an example of how multiple donor units can form “charge-resonance-type hybrid triplet states” leading to large spin–orbit coupling and a dense manifold of triplet states energetically close to the singlets.ref. ref93 5Cz-Trz emits at λPL of 486 nm and has almost unity ΦPL, a short τd of 2.1 μs, and a negligible ΔE ST of 0.02 eV (Table S1). The device with 5Cz-Trz showed very high EQEmax of 29.3% at λEL of 486 nm, along with negligible efficiency roll-off. Moreover, the sky-blue device showed very high operational stability, with LT90 at 1,000 cd m–2 of ca. 600 h.
Multiple carbazoles can also be combined with multiple triazines to produce blue TADF emitters. A pair of rigid bistriazine and biscarbazole isomers, p2Cz2Trz and m2Cz2Trz, were studied by Lee et al. (Figure ).ref. ref270 Each of the two TRZ acceptors and two carbazoles are disposed para to each other in p2Cz2Trz, such that each donor is ortho to one acceptor and meta to the other. In m2Cz2Trz each donor is arranged in para and ortho dispositions to the two acceptors, resulting in different orbital separation among the D-A pairs. Both molecules have deep-blue emission in 1 wt% doped films in PMMA, with the singlet state energies estimated at S1 = 2.88 and 3.02 eV, with ΦPL values of 82 and 91% in air, and τd of 16.6 and 12.2 μs for p2Cz2Trz and m2Cz2Trz, all respectively (Table S1). The device with p2Cz2Trz displayed green emission and showed an EQEmax of 12.5% [CIE coordinates of (0.39, 0.58)], while that with m2Cz2Trz remained sky-blue [CIE coordinates of (0.20, 0.47)] with an EQEmax of 18.5% and low efficiency roll-off of 12% at 1000 cd m–2. In another similar structure, substituents ortho– to the carbazole moiety restrain molecular motion as well as increase the D-A dihedral angle in TrzoCz (Figure ). This has the potential of boosting the solid-state ΦPL while yielding a smaller ΔE ST, and indeed TrzoCz doped at 10 wt% in DPEPO emits at λPL = 450 nm, has close-to-unity ΦPL, and a τd of 20 μs.ref. ref271 The TrzoCz devices showed an EQEmax of 28% at λEL of 484 nm [(CIE of (0.15, 0.32)] although suffered from a severe efficiency roll-off of 60% at 1000 cd m–2.
An unconventional macrocyclic triphenylamine donor in conjunction with TRZ was recently explored for the construction of blue TADF emitters. Lin et al. designed c-NN-TRZ and c-NN-MeTrz either with or without methyl groups ortho to the donor to help to maintain the strongly twisted conformation, with DPA-MeTRZ serving as an uncyclized reference compound (Figure ).ref. ref272 DPA-MeTrz, c-NN-TRZ, and c-NN-MeTrz emit at λPL of 466, 476, and 467 nm respectively, and each have unity ΦPL. Moderately large ΔE ST values of between 0.24–0.32 eV led to τd on the millisecond timescale though. OLEDs with 12 wt% of c-NN-TRZ or DPA-MeTRZ doped in mCPCN film showed EQEmax of 26.3 and 19.1%, respectively (Table S1), while the device incorporating c-NN-MeTRZ showed the highest EQEmax of 32.2% which was attributed to the more horizontally oriented TDMs. However, all the devices suffered from severe efficiency roll-off of between 59–70% at 500 cd m–2, likely a consequence of the long τd.
In 2020 three research groups simultaneously reported tristriazolotriazine (TTT) TADF derivatives, combined with various donors.ref273−ref274ref275 TTT is an extended and planar 1,3,5-triazine derivative, and hence was originally used for the design of discotic liquid crystals.ref. ref276 The first reported TTT-based TADF compounds were triply substituted with either carbazole (TTT-Ph-Cz), DMAC (TTT-DMAC), PXZ (TTT-PXZ), or biacridine (TTT-Ph-Bac) moieties, or instead substituted with 9 carbazoles (3,4,5-3TCz-TTT) (Figure ).ref273,ref275 The highest solid-state ΦPL were reported for TTT-DMAC and 3,4,5-3TCz-TTT, reaching values of 79 and 80% respectively, while phenoxazine-based green TTT-PXZ has a more moderate ΦPL of 39% and the triply substituted carbazole derivative TTT-Ph-Cz has a ΦPL of only 42%. TTT-DMAC in 5 wt% doped films in CzSi films has a τd of 4.6 μs (ΔE ST = 0.20 eV), while in 3 wt% DPEPO it was separately reported to have much longer τd of 142 μs (ΔE ST = 0.24 eV), and even longer in 15 wt% CzSi at 4.7 ms (ΔE ST = 0.27 eV) (Table S1). The inclusion of additional Cz donors in 3,4,5-3TCz-TTT resulted in a much smaller ΔE ST of 0.21 eV, compared to 0.43 eV for TTT-Ph-Cz, which did not show any delayed fluorescence in its transient PL. 3,4,5-3TCz-TTT instead showed a τd of 3.1 ms in 15 wt% doped film in CzSi. TTT-Ph-Bac demonstrated only a moderate ΦPL at 32%, but also the smallest ΔE ST of 0.09 eV among the TTT derivatives reported in 2020. Solution-processed OLEDs with green TTT-PXZ showed a moderate EQEmax of 6.2%, while the device with TTT-DMAC achieved only an EQEmax of 1.9% at λEL of 480 nm. The TTT-DMAC device was substantially improved by adding a PVK layer, which possibly helped to better confine the excitons and raised the EQEmax to 11%. The device with 3,4,5-3TCz-TTT showed an EQEmax of 5.8%, while the device with purely fluorescent derivative TTT-Ph-Cz showed an even lower EQEmax of 3.3%.
Recently Fang et al. reported asymmetrical singly or double substituted TTT derivatives, TTT-Ac and TTT-2Ac (Figure ).ref. ref277 The two compounds emit at λPL of 468 and 471 nm and have moderate ΦPL of 63 and 47%, respectively. Introduction of the extra donor in TTT-2Ac results in a much smaller ΔE ST (0.19 eV) compared to TTT-Ac (0.35 eV), with the smaller ΔE ST translating to a shorter τd of 20 μs compared to TTT-Ac (27 μs) (Table S1). Solution-processed OLEDs with TTT-Ac and TTT-2Ac emitting at λEL of 470 and 474 nm [CIE coordinates of (0.16, 0.21) and (0.17, 0.26)] showed EQEmax of 9.2 and 8.1%, respectively.
In summary, the nature of the donor can have a strong influence on the photophysical and device properties of triazine-based TADF emitters, as is evident in the comparison of previously discussed TspiroS-TRZ and Tris-Cz-TRZ (Figure ). Rigid and sterically twisted D-A structures lead to small ΔE ST and efficient TADF with short excited-state lifetimes, that often translate into high-efficiency OLEDs. However, OLEDs employing these emitters are frequently sky-blue at best, with CIEy coordinates far from Rec. 2020 standard for blue. A handful of examples do show deep-blue emission with CIEy coordinates of < 0.1, however OLEDs utilizing such emitters as DCBTRZ struggle to achieve efficiencies exceeding 10%, and the efficiency roll-off remains high.
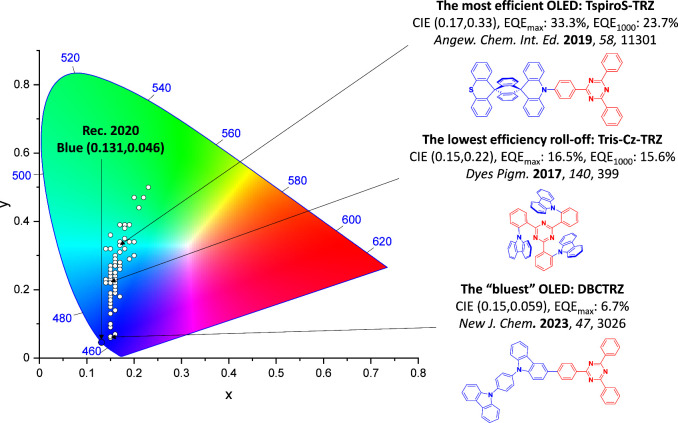
Other Nitrogen Heterocycles: Pyrazine- and Pyrimidine-Containing Emitters
Similar to TRZ, a variety of blue TADF emitters have been designed using pyrimidine and pyrazine as acceptor moieties. These possess shallower LUMOs than TRZ, hence are weaker electron acceptors and thus compatible with a wider range of (stronger) donors while maintaining blue emission.ref. ref278 The 2,4,6-positions of the pyrimidine ring and 2,3,5,6-positions of the pyrazine ring are also easily functionalized, which adds to the attractiveness of these heterocycles in the construction of blue TADF emitters. The pyrimidine and pyrazine-based blue TADF emitters discussed here are shown in Figure .
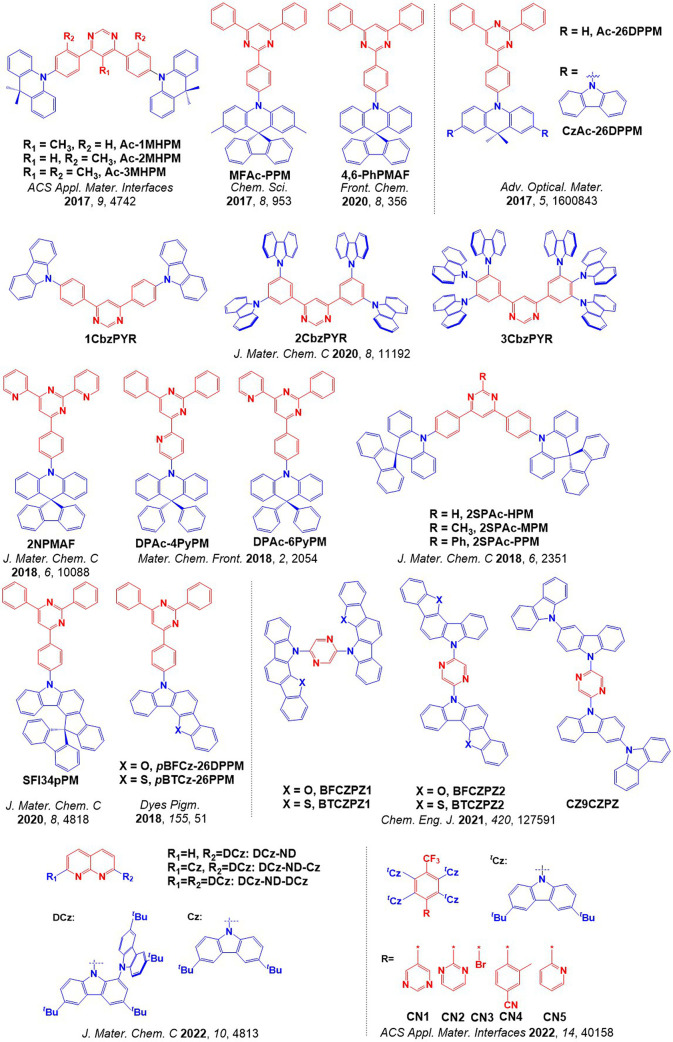
The introduction of methyl groups around a pyrimidine acceptor unit leads to high torsions between the pyrimidine and adjacent phenylenes, causing an increase in the excited-state energies and producing deep-blue emission in combination with DMAC donors.ref. ref279 Moving from one central (Ac-1MHPM) to two symmetric methyl groups (Ac-2MHPM, Figure ) resulted in limited changes to the photophysics (λPL = 481 and 477 nm, with ΦPL = 75 and 71%, respectively). The conjugation was dramatically reduced with the introduction of three methyl groups though (Ac-3MHPM), affording a more twisted structure with the λPL blue-shifted to 454 nm and the ΦPL decreased to 47% (Table S1). However, the number of methyl groups had surprisingly little impact on the TADF properties, with τd ranging between 44 and 50 ms and ΔE ST between 0.22–0.24 eV for the three compounds. The OLEDs with Ac-1MHPM, Ac-2MHPM, and Ac-3MHPM showed EQEmax values of 24.0, 19.8, and 17.8%, respectively, and the EL reflected in the λPL values with the Ac-3MHPM device having CIE coordinates of (0.15, 0.16) compared to (0.15, 0.27) and (0.15, 0.28) for the devices with Ac-1MHPM and Ac-2MHPM, respectively. A large efficiency roll-off was observed for each of the devices, with the bluest device using Ac-3MHPM unable to reach 1000 cd m–2.
When using the less sterically bulky donor carbazole, both the position and number of donors must be carefully optimized to achieve efficient TADF. Serevičius et al. employed a symmetric bis(phenyl)pyrimidine acceptor in this way, and when carbazole groups were substituted at the para positions the resulting 1CbzPYR (Figure ) possessed a large ΔE ST of 0.48 eV and showed no TADF in 1 wt% doped films in PMMA.ref. ref280 When carbazole groups were instead substituted at the meta positions as in 2CbzPYR, a smaller ΔE ST of 0.27 eV was achieved but with a low ΦPL of 22% (Table S1). Compound 3CbzPYR featuring a full set of meta and para substituted carbazoles has a much higher ΦPL of 81%, benefitting from both reduced non-radiative decay and enhanced radiative decay. The OLEDs with 3CbzPYR emitted at λEL of 464 nm [CIE coordinates of (0.16, 0.23)] and showed EQEmax of 19.7%. Although the maximum luminance approached 10,000 cd m–2, severe efficiency roll-off of 55% at 100 cd m–2 was observed due to the large ΔE ST of 0.32 eV.
Li et al. reported three D-A-D structures using various pyrimidine acceptors coupled to spiro-acridine donors, which exhibited good performance in OLED devices.ref. ref281 The pyrimidine acceptors were either unsubstituted (2SPAc-HPM), methyl substituted (2SAPAc-MPM), phenyl substituted (2SPAc-PPM, Figure ), and showed moderate ΔE ST between 0.15 and 0.19 eV with high ΦPL ranging from 82–97%. The devices consequently showed high EQEmax values of 25.6, 24.3, and 31.5%, respectively; however, severe efficiency roll-off of 26, 34, and 43% was reported at 100 cd m–2 arising from the long τd (52–57 ms). Another spiro-acridine donor with methyl groups at the 2,7-positions was instead coupled between the heteroatoms of diphenyl pyrimidine to give MFAc-PPM. This emitter has a moderately high ΔE ST of 0.25 eV and of τd of 78 μs in 18 wt% doped films in PPF, yet was nonetheless able to produce sky blue OLEDs with CIE coordinates of (0.16, 0.23), an EQEmax of 20.4%, and reasonable efficiency roll-off of 24% at 100 cd m–2 (Table S1).ref. ref282 A corresponding analogue without methyl groups on the donor, 4,6-PhPMAF, gave deep-blue emission in a device using 22 wt% doping in DPEPO, with λEL of 458 nm and CIE coordinates of (0.15, 0.11).ref. ref283 The device however showed an EQEmax of only 3% due to the low ΦPL of 17%, while the efficiency roll-off associated with the relatively large ΔE ST of 0.27 eV and long τd of 0.3 ms was so severe that even 200 cd m–2 was not achieved. Ac-26DPPM and CzAc-26DPPM feature asymmetric substitution of the pyrimidine acceptor with acridine-based donors, and in 10 wt% doped films in DPEPO exhibited sky-blue emission with λPL of 476 and 496 nm, respectively. Ac-26DPPM and CzAc-26DPPM both have ΦPL of 81%, and τd of 87 and 55 μs in the same DPEPO. The OLEDs with Ac-26DPPM and CzAc-26DPPM showed sky-blue emission at CIE of (0.18, 0.32) and (0.21, 0.37), EQEmax of 19.3 and 23.7%, and efficiency roll-offs of 67 and 60% at 1000 cd m–2, all respectively.ref. ref284
Decorating pyrimidine with pyridines in conjunction with spiro-acridine donors afforded efficient sky-blue TADF emitters 2NPMAF, DPAc-4PyPM, and DPAc-6PyPM (Figure ). Higher ΦPL (>80%) and faster k RISC (∼105 s–1) were observed for DPAc-4PyPM and DPAc-6PyPM in comparison to reference emitter DPAc-TPPM (ΦPL = 70%, k RISC = 6.9 × 104 s–1) without pyridines.ref. ref285 Intramolecular H-bonding between the pyridine units and the pyrimidine core was suggested as responsible, however its significance in relation to the TADF mechanism was not apparent. The devices with 2NPMAF, DPAc-4PyPM, and DPAc-6PyPM showed EQEmax of 23.6, 24.3, and 22.4% at λEL of 481, 484, and 472 nm, respectively (Table S1).ref. ref286
Again, featuring pyrimidine, SFI34pPM (Figure ) features a sterically hindered spiro-fluorene-fused carbazole derivative as the electron donor, leading to internal rigidity, excellent thermal stability, and a ΦPL of 74% in 10 wt% doped films in DPEPO. A deep-blue device with SFI34pPM showed CIEy of 0.09 and an EQEmax of 8.2%.ref. ref287 Another family of emitters containing asymmetric pyrimidine acceptors coupled to functionalized carbazoles also produced deep-blue OLEDs.ref. ref288 Benzofuro- and benzothieno- carbazoles were used as donors in pBFcz-2,6DPPM and pBTCz-2,6DPPM, which emit similarly at λPL = 437 and 435 nm with ΦPL of 71 and 75%, respectively. However, long τd of 200 and 383 μs were measured due to large ΔE ST of 0.27 and 0.34 eV, also respectively (Table S1). The OLEDs using 10 wt% doping in DPEPO host retained identical deep-blue emission with CIE coordinates of (0.15, 0.05), and EQEmax of 6.2 and 5.4% respectively. The efficiency roll-off was severe though, with neither emitter able to achieve 1000 cd m–2 and only pBFCz-2,6DPPM able to reach 100 cd m–2 (with efficiency roll-off of ∼29% at that brightness). Non-doped devices with pBFcz-2,6DPPM and pBTCz-2,6DPPM showed a slight red-shift in the emission (CIEy shifting to 0.07), with EQEmax of 5.8 and 5.4% and both achieving brightness of over 3000 cd m–2.
The effect of heteroatoms on spin-orbit coupling (SOC) between S1 and T1 states was investigated within a series of pyrazine-based TADF emitters bearing donors of benzofuran fused carbazole (BFCZPZ1 and BFCZPZ2), benzothiophene carbazole (BTCZPZ1 and BTCZPZ2), or a 9-bicarbazole (CZ9CZPZ, Figure ).ref. ref289 BTCZPZ1 possesses the smallest ΔE ST (0.24 eV), while the others have ΔE ST values range between 0.31–0.37 eV (Table S1). The ΦPL of BFCZPZ1 is 68%, while for the other emitters it is above 91%, all in 7 wt% doped films in PPF. TD-DFT calculation showed that in emitters with shorter distances between the donor heteroatoms and the acceptor moiety the SOC between the S1 and T1 states was enhanced. Accordingly, the spin-orbital coupling matrix elements between S1 and T1 of BFCZPZ1 and BTCZPZ1 were 0.311 and 0.980 cm–1, compared to 0.122, 0.149, and 0.252 cm–1 for BFCZPZ2, BTCZPZ2, and CZ9CZPZ, all respectively. As a result, BTCZPZ1 showed the shortest τd of 90 μs and fastest kRISC of 8.5 × 104 s–1 of this series. The device with BFCZPZ1 showed the bluest emission with λEL of 436 nm and CIE coordinates of (0.15, 0.06), while the λEL for the devices with BFCZPZ2, BTCZPZ1, BTCZPZ2, and CZ9CZPZ, were shifted to 464, 472, 468, and 468 nm, respectively. The EQEmax of the device with BFCZPZ1 was 6.5% due to the lower ΦPL, while the EQEmax for BFCZPZ2, BTCZPZ1, BTCZPZ2, and CZ9CZPZ, were much improved at 21.3, 21.1, 19.7, and 20.0%, respectively. The devices with BFCZPZ1 and BTCZPZ1 showed the best efficiency roll-off of 20 and 26% at 100 cd m–2, while for the other three devices the efficiency roll-off was around 50% at the same brightness level.
Banevičius et al. reported a series of naphthyridine-carbazole hybrids with particular focus on asymmetric derivative DCz-ND-Cz (Figure ). This emitter which showed a shortened τd (4.4 μs), faster k RISC, and a high ΦPL of 74% compared to the singly substituted analogue DCz-ND and symmetric congener DCz-ND-DCz (τd of 6 and 7μs, ΦPL of 46 and 72%, respectively), all in 20 wt% doped films in DPEPO (Table S1).ref. ref290 The OLEDs showed comparable performance with EQEmax ranging between 18.1–20.8% at λEL of 464–469 nm, however the device with DCz-ND-Cz demonstrated the smallest efficiency roll-off of 53% at 1000 cd m–2 compared to 67% for DCz-ND-DCz and 70% for DCz-ND.
Mahmoudi et al. reported a series of multicarbazole derivatives in the structural template of 4CzTPN featuring various electron-acceptor moieties and a common trifluoromethyl substitutent.ref. ref291 The bluest compounds were CN1 and CN4 (Figure ), which in neat films emitted sky-blue at λPL of 482 and 490 nm, and have ΦPL 76 and 27%, respectively. Short τd were reported for these emitters (2.4 and 1.8 μs), accompanied by small ΔE ST values of 0.03 and 0.04 eV. Non-doped devices achieved sky-blue emission with λEL of 481 and 476 nm [CIE coordinates (0.16, 0.27) and (0.17, 0.24)] and EQEmax of 8.4 and 5.5% respectively. These EQEs nearly doubled when a doped device architecture (20 wt% in mCBP) was used.
Of these examples, the best non-triazine nitrogen-heterocycle blue TADF emitters all feature pyrimidine (Figure ). Sky-blue-emitting OLEDs achieved efficiencies as high as 31% with 2SPAc-PPM, while the OLEDs with the lowest efficiency roll-off employed 2NPMAF featuring the same spiro-donor. In terms of color, the OLED with pBFCz-2,6DPPM most closely approaches the target CIE coordinates of Rec. 2020 standard, however poor efficiency is still unavoidable in this color region.
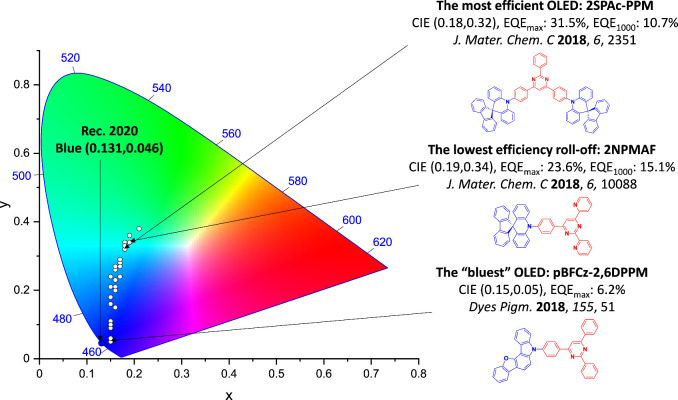
Boron-Containing Emitters
The use of boron as an acceptor has been widely reported in the literature.ref226,ref292 Generally tri- or tetra- substituted boron acceptors are decorated with donors to form D-A TADF emitters, although recent boron-containing MR-TADF emittersref118,ref293 are discussed separately in Section sec11 . The configuration of tri-substituted boron acceptors can be classified as either fully fused (a boron atom directly attached to three contiguous aryl units) or unfused (a boron atom directly connected to at least one isolated aryl group). Examples of tetra-substituted boron acceptors are typically composed of a BF2 group linked to aryl units.
As an early and structurally simple example, the ortho regiochemistry in CzoB (Figure ) resulted in the Cz donor adopting a twisted conformation that produces a significantly smaller ΔE ST than the equivalent para-congener, with ΔE ST of 0.15 and 0.39 eV for CzoB and CzpB, respectively, in toluene. CzoB emits at λPL of 463 nm in toluene and has ΦPL of 84% in 20 wt% doped films in DPEPO. The corresponding OLED showed an EQEmax of 22.6% with CIE coordinates of (0.14, 0.15), although the long τd of 56.3 μs resulted in large efficiency roll-off of 19 and 77% at 100 and 1000 cd m–2.ref. ref294
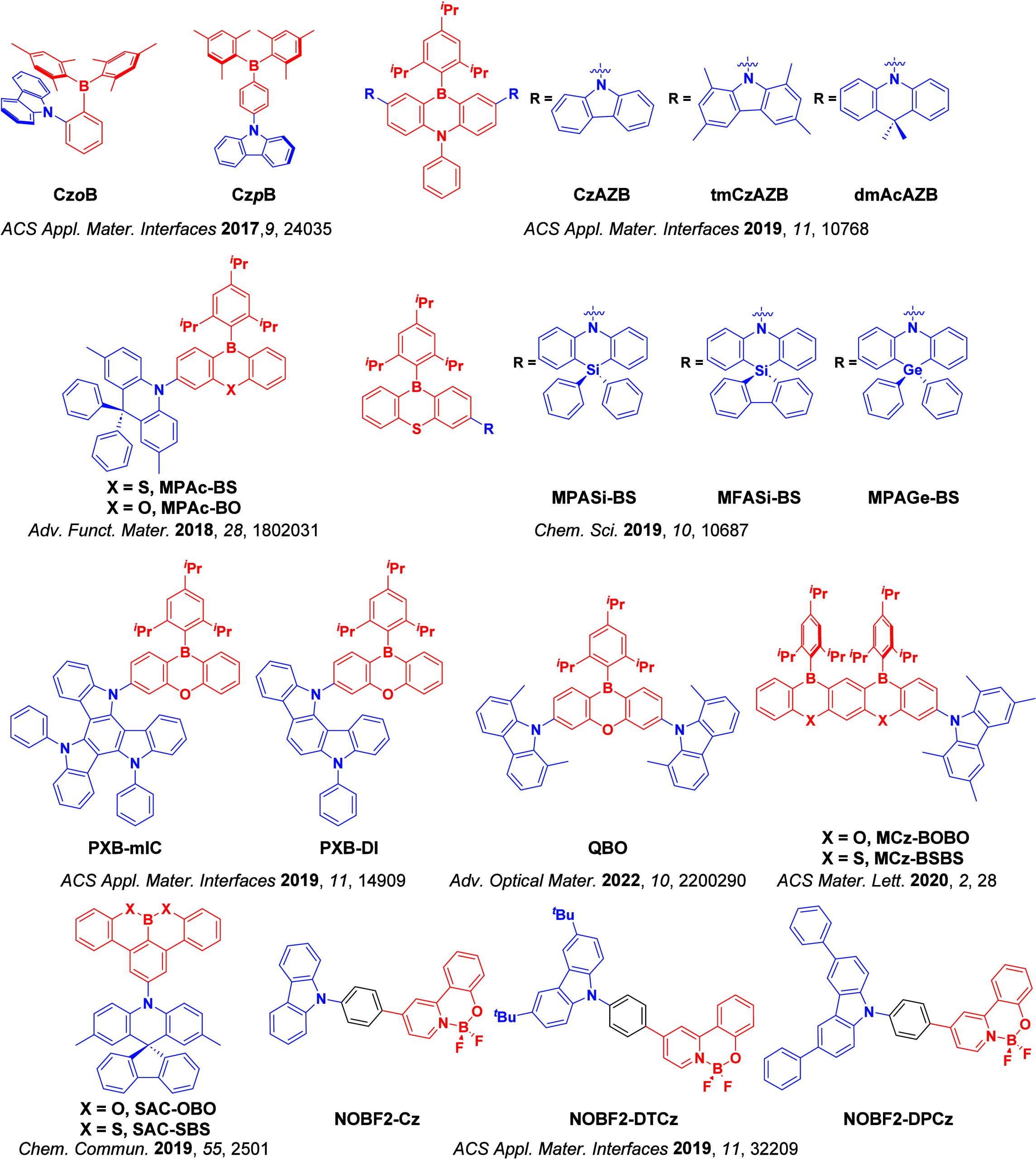
Using unfused boron acceptor dibenzo-1,4-azaborine, a series of three emitters were designed bearing DMAC (dmAcAZB), tetramethylcarbazole (tmCzAZB), and carbazole donors (CzAZB, Figure ).ref. ref117 Forcing a near-orthogonal D-A conformation was key to promoting RISC and TADF, and indeed CzAZB displayed no delayed fluorescence due to its more planarized structure and large ΔE ST of 0.31 eV compared to 0.26 and 0.11 eV for tmCzAZB and dmAcAZB, respectively. CzAZB, tmCzAZB, and dmAcAB emit at λPL of 452, 451, and 469 nm, and although CzAZB exhibited an excellent ΦPL of 99% its lack of TADF resulted in poor device EQEmax of 5.5%. tmCzAZB and dmAcAB have τd of over 150 μs, but were nonetheless able to achieve EQEmax of 12.4 and 20.8% in 10 wt% doped mCP host. The emission of these latter two was at λEL of 464 and 469 nm with CIE coordinates of (0.14, 0.15) and (0.14, 0.19), but significant efficiency roll-off at 100 cd m–2 of 56 and 38% was reported, all respectively.
MPAc-BS and MPAc-BO (Figure ) contain bulky dibenzoheteraborin acceptors containing either sulfur or oxygen atoms, connected to a dimethyldiphenylacridine (MPAc) donor.ref. ref295 MPAc-BS and MPAc-BO emit at λPL 481 and 466 nm and have high ΦPL values of 99 and 98% respectively, as neat films. Along with suppression of ACQ to support these ΦPL, the neat films also have short τd of 1.7 and 2.4 μs and fast k RISC of 3.5 and 1.0 × 106 s–1, all respectively. The sulfur atom in MPAc-BS was proposed to enhance the SOC, resulting in faster k RISC than in MPAc-BO. Non-doped devices with MPAc-BS and MPAc-BO showed EQEmax of 22.8 and 21.3%, emitting at λEL of 487 and 474 nm [CIE coordinates of (0.15, 0.36) and (0.14, 0.23)], respectively. Additionally, the devices showed low efficiency roll-off of 1 and 14% at 100 cd m–2, respectively. The rigid nature of these sterically crowded emitters helps to explain both the high ΦPL, resistance to ACQ, and narrow emission FWHM of 63 and 59 nm.
Matsuo et al. have also explored incorporating heavy atoms in an effort to boost SOC and thus k RISC. A family of blue emitters containing phenothiaborin (BS) as the acceptor was developed using acridan-analogue donors, whereby the bridging carbon atom of the donor is substituted by silicon or germanium atoms. Compounds MPASi-BS, MFASi-BS, and MPAGe-BS (Figure ) emit at λPL of 479, 483, and 468 nm, respectively, in 50 wt% doped films in PPF.ref. ref296 The closely lying 1CT, 3CT, and 3LE states along with heavy atom effects combined to improve the SOC between the singlet and triplet states, and thus accelerate RISC. The emitters involving a donor heavy atom (MPASi-BS, MFASi-BS, and MPAGe-BS) all exhibited k RISC above 1 × 107 s–1 in doped PPF film, much faster than the green-emissive reference material MPAc-BS ref. ref297 (3.5 × 106 s–1). In addition to the fast k RISC, the high ΦPL (close to 100%), and moderate ΔE ST (<0.11 eV) of MPASi-BS, MFASi-BS, and MPAGe-BS allowed them to support strong performance in OLEDs. The devices with MPASi-BS and MFASi-BS showed sky blue emission with λEL of 478 and 484 nm, CIE coordinates of (0.14, 0.26) and (0.14, 0.32), and EQEmax of 27.6 and 23.9% with only 5 and 8% efficiency roll-off at 1000 cd m–2, all respectively. Interestingly, for MPAGe-BS the excitons were initially generated on high energy quasi-axial (QA) conformers, requiring subsequent energy transfer to the lower energy emissive quasi-equatorial (QE) conformer. As a result, the device EQEmax was diminished to 15.7% (16% efficiency roll-off at 1000 cd m–2).
Combining rigid diindolocarbazole or indolocarbazole donors with a dibenzooxaborin acceptor afforded the efficient blue TADF emitters PXB-DI and PXB-mIC (Figure ).ref. ref298 As 20 wt% doped films in PPBI, PXB-DI has a higher ΦPL of 79%, faster k RISC of 1.17 × 106 s–1 and a red-shifted emission of λPL = 470 nm compared to PXB-mIC (ΦPL: 51%, k RISC: 5.22 × 105 s–1, and λPL = 425 nm). The ΔE ST values are 0.09 and 0.19 eV for PXB-DI and PXB-mIC, respectively. The enhanced TADF properties of PXB-DI than PXB-mIC were attributed by the authors to the stronger donor strength and extended rigid structure of diindolocarbazole. The OLEDs with PXB-mIC showed an EQEmax of 12.5% at CIE coordinates of (0.15, 0.08), although the efficiency roll-off (58% at 1000 cd m–2) was rather severe. The devices with PXB-DI instead showed sky-blue emission at CIE coordinates of (0.16, 0.34) and very high EQEmax of 37.4%, with only a 15% roll-off of the efficiency at 1000 cd m–2.
Quadrupolar D-A-D blue TADF emitter QBO (Figure ) was designed using the same phenoxaborin acceptor and 1,8-dimethylcarbazole donors.ref. ref299 The key to this design strategy was to generate doubly degenerate CT excited states associated with the two separate donor units, which would enhance the density of excited states and SOC – and indeed a high SOCME of 0.41 cm–1 was calculated for QBO. The λPL, ΔE ST, ΦPL, τ d, and k RISC values are 455 nm, 0.01 eV, 83%, 0.65 μs, and 19×105 s–1 in 20 wt% doped films in PPF. The resulting OLEDs emitted at λEL at 460 nm with CIE coordinates of (0.14, 0.12) and EQEmax/EQE1000 of 20.5 and 17.7%. Similar derivatives MCz-BOBO and MCz-BSBS featured an acceptor extended with additional boron-oxygen/boron-sulfur moieties.ref. ref300 These blue emitters, employing a ladder-shaped heteraborin acceptor and tetramethyl carbazole as the donor, achieved sufficiently separated HOMO/LUMO for small ΔE ST. Indeed, MCz-BSBS and MCz-BOBO emit at λPL of 483 and 476 nm and have ΔE ST of 0.17 and 0.01 eV, along with ΦPL of 93 and 100% and τd of 2.7 and 0.78 μs in 20 wt% doped films in PPF, all respectively. TD-DFT calculations revealed similar excited states topologies for the two emitters, with the closely lying S1 and T1 states showing similar CT character while the slightly higher T2 states exhibited LE character. The SOCME value between El-Sayeed-allowed T2 and S1 in the sulfur-containing MCz-BSBS (2.93 cm–1) was more than 30 times higher than the value for oxygen-containing MCz-BOBO (0.09 cm–1), demonstrating the impact of heavy-atom effects in this context. These calculations aligned well with the faster experimental k RISC of MCz-BSBS (8.8 and 2.5 × 106 s–1). The OLEDs with MCz-BSBS and MCz-BOBO emitted with CIE coordinates of (0.14, 0.33) and (0.13, 0.20), and showed EQEmax of 25.9 and 20.1% with low efficiency roll-offs of 25 and 12% at 1000 cd m–2, all respectively.
An unusual boron-containing acceptor with thioether linking groups produced efficient sky-blue emitter SAC-SBS (Figure ).ref. ref301 In 20 wt% doped films in PPF SAC-SBS showed promising photophysical properties with λPL of 491 nm, ΦPL of 81%, ΔE ST of 0.12 eV, τd of 22 μs, and k RISC of 32×105 s–1 compared to its ether-linked analogue SAC-OBO (λPL of 470 nm, ΦPL of 28%, ΔE ST of 0.30 eV, τd of 140 μs, and k RISC of 3.5 × 105 s–1) (Table S1). Devices with SAC-SBS emitted at λEL of 489 nm [CIE coordinates of (0.17, 0.39)] and showed EQEmax of 20.9%. The efficiency roll-off was moderate, at 16% at 100 cd m–2. SAC-OBO was found to deactivate the boron center too much, producing a worse triplet harvester albeit accompanied by a blue-shift in the emission. The OLED with SAC-OBO consequently showed an EQEmax of only 5.2% at λEL of 471 nm and CIE coordinates of (0.16, 0.22).
Contrasting to the previous examples, a tetra-coordinated boron acceptor was used in conjunction with carbazole-based donors in NOBF2-Cz, NOBF2-DTCz, and NOBF2-DPCz (Figure ).ref. ref302 These emitters use boron difluoride (BF2) in the chelating acceptor 4-phenylpyridin-2-yl)phenol (PPyPOH) moiety, which increased the overall acceptor strength enough to enable TADF in these materials. Compounds NOBF2-Cz, NOBF2-DTCz, and NOBF2-DPCz emit at respective λPL of 449, 473, and 471 nm in toluene. In 10 wt% doped films in DPEPO they show high ΦPL values (70 to 99%), moderately large ΔE ST values (0.20 to 0.22 eV) and long τ d (110 to 132 μs). The blue OLEDs with NOBF2-Cz, NOBF2-DTCz, and NOBF2-DPCz nonetheless showed EQEmax values of 11.0, 12.7, and 15.8% at CIE coordinates of (0.14, 0.16), (0.14, 0.21), and (0.14, 0.28), all respectively. This study hence demonstrated the utility of tetra-coordinated boron in the acceptors of D-A TADF emitters.
The fully fused triaryl-boron acceptor 5,9-dioxa-13b-boranaphtho[3,2,1-de]anthracene (DBA) has a triangulene shape, with Hirai et al. first developing the material for use in OLEDs.ref. ref303 Later, a tert-butyl modified DBA as acceptor (TDBA) was joined with DMAC or diindolocarbazole as donors, giving blue-emitting materials TDBA-Ac and TDBA-DI (Figure ).ref. ref304 These compounds emit at λPL of 458 and 456 nm, have ΔE ST of 0.06 and 0.11 eV in toluene, and high ΦPL of 93 and 99% in 20 wt% doped films in DBFPA, all respectively. The device with TDBA-Ac in PPBI showed an EQEmax of 21.5% at CIE coordinates of (0.15, 0.06), with the rigid structure conferring a reasonably narrow emission FWHM of 48 nm. In contrast, the devices with TDBA-DI in either PPBI or DBFPO emitted with CIE coordinates of (0.14, 0.15) and (0.15, 0.28), achieving very high EQEmax values of 32.2 and 38.2% and small efficiency roll-off of 17 and 10% at 1000 cd m–2, respectively. The observed spectral shift to sky-blue in DBFPO host was attributed to its more polar nature compared to PPBI. The outstanding EL performance of TDBA-DI is supported by its almost unity ΦPL, fast k RISC (1.1 × 106 s–1 in DBFPO) and high horizontal TDM orientation (89% in DBFPO).ref. ref304 Subsequently reported DBA-DI is a close analogue of TDBA-DI without tert-butyl groups on the DBA moiety. This compound showed improved electrochemical stability with higher bond dissociation energies, an important trait for device stability.ref. ref305 DBA-DI emits at λPL of 467 nm in toluene (456 nm for TDBA-DI) and maintains a high ΦPL (95.3% in mCBP-CN), small ΔE ST (0.03 eV in toluene), short τd (1.25 μs in mCBP-CN), and fast k RISC of 6.2 × 106 s–1 (in mCBP-CN). The device with DBA-DI in mCBP-CN exhibited sky-blue emission with CIE coordinates of (0.16, 0.39). The device also achieved a high EQEmax of 28.1%, with only 1% of efficiency roll-off at 1000 cd m–2 and a maximum luminance as high as 126 200 cd m–2. The device lifetime (LT50) also reached 329 hours, running at an initial 1000 cd m–2. Longer device lifetime (540 h) was achieved when a mixed host of mCBP-CN:DDBFT was adopted, however the emission color was slightly red-shifted with CIE coordinates of (0.17, 0.40).
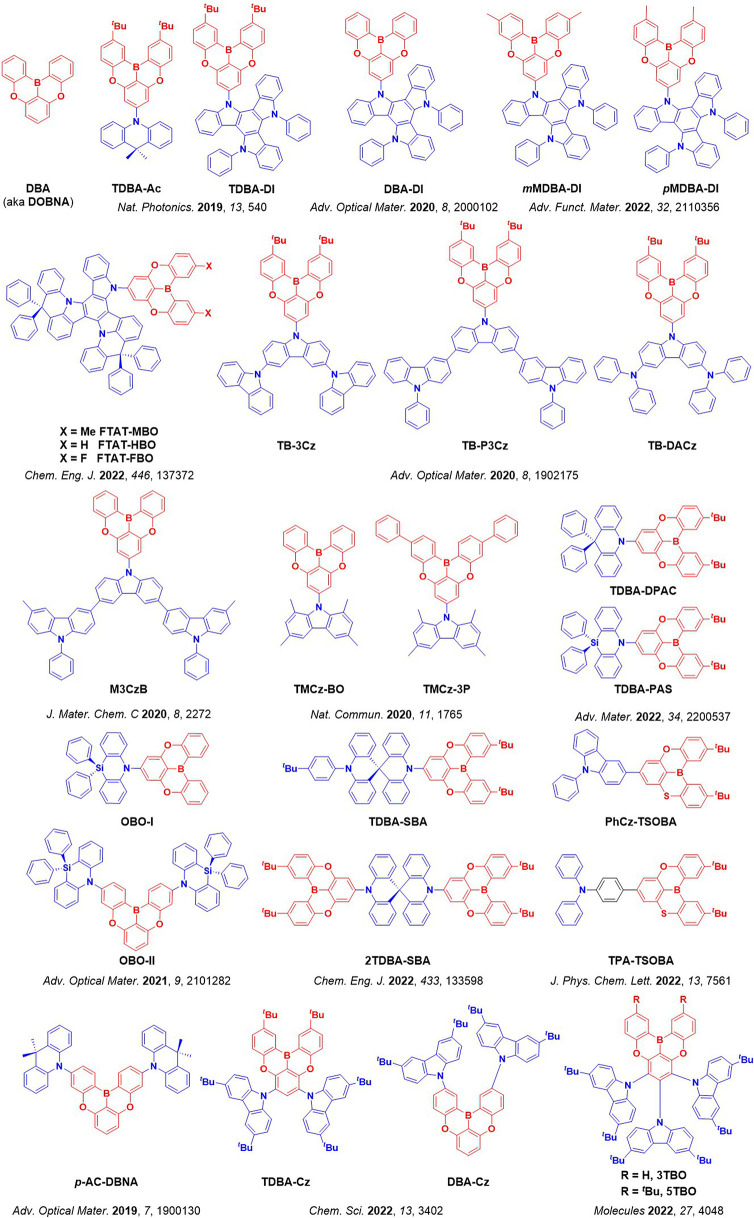
In a subsequent study, the acceptor strength and CT character of DBA-DI ref. ref305 was manipulated by incorporating methyl groups para or meta to the oxygen atoms to afford pMDBA-DI and mMDBA-DI (Figure ).ref. ref306 This design strategy resulted in blue-shifted emission with λPL = 451 nm for mMDBA-DI and 460 nm for pMDBA-DI, while retaining moderately small ΔE ST and short τd at 0.12 eV and 1.90 μs for mMDBA-DI and 0.07 eV and 1.60 μs for pMDBA-DI in toluene. Furthermore, near unity ΦPL of 97.3 and 97.8% were recorded, respectively, for mMDBA-DI and pMDBA-DI in 30 wt% doped DBFPO films. The respective OLEDs with pMDBA-DI and mMDBA-DI showed EQEmax of 33.1 and 32.8% at λEL of 483 and 474 nm [CIE coordinates of (0.15, 0.31) and (0.14, 0.23)], with low efficiency roll-off of 2.4 and 13.4% at 1000 cd m–2. Additional blue TADF emitters FTAT-MBO, FTAT-HBO, and FTAT-FBO containing an intramolecular-locked triazatruxene (FTAT) donor moiety were designed, featuring DBA acceptors substituted with either methyl, hydrogen, or fluorine.ref. ref307 The ΔE ST values are 0.24, 0.21, 0.09 eV in toluene, with associated τd of 3.6, 3.5, 1.8 μs and ΦPL of 55, 66, 90% in 20 wt% doped films in mCP, all respectively. The solution-processed OLEDs with FTAT-FBO emitted at λEL = 473 nm and CIE coordinates of (0.15, 0.25), and showed the best performance with an EQEmax of 17.5% which decreased only slightly to 17.3% at 1000 cd m–2.
Employing peripheral carbazole or diphenylamine substituents on the main carbazole donor leads to deep-blue to sky-blue solution-processable emitters TB-3Cz, TB-P3Cz, and TB-DACz (Figure ).ref. ref308 The emission of TB-DACz (λPL of 493 nm) is red-shifted compared to TB-3Cz (413 nm) and TB-P3Cz (433 nm) in toluene, which was attributed to the diphenylamine axillary donor strengthening the D-A interactions. The sufficiently separated HOMO/LUMO nonetheless enables small ΔE ST values of 0.06, 0.11, and 0.07 eV respectively for TB-3Cz, TB-P3Cz, and TB-DACz. All the three emitters showed AIE behavior and high ΦPL (>90%) in non-doped films, while the τd values were <10 μs. Solution-processed devices with TB-3Cz and TB-P3Cz showed deep-blue emission at λEL of 424 and 428 nm and [CIE coordinates of (0.17, 0.07) and (0.15, 0.08)], while the λEL of the TB-DACz-based device was red-shifted to 492 nm. The EQEmax for the solution-processed devices with TB-3Cz and TB-3PCz were as high as 9.9 and 6.1% respectively, but unfortunately the devices suffered from severe efficiency roll-off of ∼93 and 43% at 1000 cd m–2. Vacuum-deposited devices with TB-P3Cz showed a much higher EQEmax of 29.1% albeit still with 54% efficiency roll-off at 1000 cd m–2, along with LT50 of 60 h and blue emission at CIE coordinates (0.14, 0.19). By removing the tert-butyl groups from the acceptor unit of TB-P3Cz and adding methyl groups to the same donor moiety, an optimized structure M3CzB was developed and separately reported. The device with M3CzB in DBFPO showed an even higher EQEmax of 30.7% and with a lower efficiency roll-off of 30% at 1000 cd m–2, albeit with red-shifted emission [λEL = 470 nm, CIE coordinates (0.14, 0.26)]. A device fabricated with 20 wt% M3CzB in mCBP-CN as the EML for improved stability exhibited an LT50 of 81 h at an initial 400 cd m–2.ref. ref309
Kim et al. used a tetramethylcarbazole donor with the DBA acceptor in blue TADF emitters TMCz-BO and TMCz-3P (Figure ). TMCz-BO and TMCz-3P emit at λPL of 446 and 455 nm respectively in toluene, with TMCz-BO having degenerate 1CT, 3CT, and 3LE states which resulted in small ΔE ST of 0.02 eV, short τd of 0.75 μs, and fast k RISC of 1.9 × 106 s–1 in 30 wt% doped films in PPF. In TMCz-3P the geometry and orbital character of the 3LE and 3CT states deviate more from 1CT, resulting in a larger ΔE ST of 0.13 eV, longer τd of 14.5 μs, and reduced k RISC of 0.03 × 106 s–1. As a result the device with TMCz-BO showed an EQEmax of 20.7% and efficiency roll-off of 16% at 1000 cd m–2 [λEL of 471 nm and CIE coordinates of (0.14, 0.18)], outperforming the OLED with TMCz-3P with a similar EQEmax of 20.4% but more severe efficiency roll-off of 37% at 1000 cd m–2, and with a somewhat red-shifted λEL of 479 nm at CIE coordinates of (0.14, 0.26).ref. ref310
The emitters TDBA-PAS and TDBA-DPAC contain phenazasiline and diphenylacridine donor moieties coupled to the DBA acceptor (Figure ), in which the larger Si center weakens the electron donating strength.ref. ref311 As a result the emission of the former in toluene is blue-shifted (λPL= 427 nm) compared to the latter (λPL = 444 nm), and the emission of TDBA-DPAC is itself blue-shifted compared to previously discussed TDBA-Ac (λPL = 458 nm).ref. ref304 TDBA-PAS also shows dual emission at low dopant concentrations, which was attributed to the presence of both quasi-axial (QA) and quasi-equatorial (QE) conformers associated with the higher and lower energy emission respectively, and enabled by the increased flexibility of the Si linking center. After increasing the dopant concentration from 10 to 30 wt% in DPEPO film to minimize the impact of the QA conformer, the emission of TDBA-PAS narrowed to a FWHM of 54 nm and the ΦPL was enhanced from 80.7 to 92.1%. In contrast, for TDBA-DPAC the FWHM increased from 57 to 66 nm and the ΦPL decreased from 85.3 to 76.8%. The increase in ΦPL for TDBA-PAS was attributed to improved energy transfer from the high energy QA conformer to low energy QE conformer, while the decrease of ΦPL in TDBA-DPAC was attributed to concentration quenching. Both emitters nonetheless showed small ΔE ST < 0.06 eV, fast k RISC > 12.3×105 s–1, and short τd < 3.14 μs. The OLEDs with TDBA-PAS and TDBA-DPAC showed respective EQEmax of 22.4 and 24.6% at CIE coordinates of (0.16, 0.04) and (0.15, 0.09). Further enhanced TADF properties were achieved by removal of the tert-butyl group from the DBA acceptor in OBO-I, which was reported alongside a symmetric D-A-D emitter OBO-II.ref. ref312 These structural changes afforded emitters with significantly smaller ΔE ST of 0.007 and 0.013 eV in 20 wt% doped films in PPF, resulting in much shorter τd of 1.60 and 1.70 μs, higher ΦPL of 81 and 98%, and faster k RISC of 11 and 9.2 × 10–5 s–1 for OBO-I and OBO-II, respectively. The OLEDs with OBO-I and OBO-II showed EQEmax of 21.7 and 31.7% at CIE coordinates of (0.14, 0.10) and (0.14, 0.13), and non-doped devices showed EQEmax of 8.7 and 23.1% at CIE coordinates of (0.15, 0.17) and (0.15, 0.20), all respectively. The non-doped devices with OBO-I and OBO-II also showed reduced efficiency roll-off with EQE decreasing from maximum values by 6.9/48.3% and 7.8/43.7% at 100/1000 cd m–2, respectively.
Another strategy to increase the efficiency of OLEDs is to enhance the emitter molecular anisotropy to boost optical outcoupling efficiency. Linearly shaped emitters TDBA-SBA and 2TDBA-SBA (Figure ) containing spiro-bisacridine donors exhibited this desired preferential horizontal orientation of their TDMs, reported at 86 and 88% respectively in 20 wt% doped flims in DBFPO.ref. ref313 Both compounds have small ΔE ST of 0.01 eV leading to similar τd of 1.47 and 1.38 μs alongside ΦPL of 89 and 87%. The blue OLEDs with TDBA-SBA and 2TDBA-SBA showed EQEmax values (with CIE coordinates) of 29.3% (0.13, 0.15) and 18.3% (0.13, 0.21), respectively, supported in part by the horizontal TDMs. The lower efficiency of 2TDBA-SBA was attributed to its higher k nr and slower k RISC compared to TDBA-SBA.
The effect of donor position has also been investigated by incorporating DMAC moieties in meta-, para-, or meta′-positions relative to the boron atom in the DBA acceptor. The para-substituted compound (p-AC-DBNA, Figure ) exhibited sky-blue emission with λPL of 496 nm, higher ΦPL of 96%, smaller ΔE ST of 0.09 eV, shorter τd of 1.5 μs, and faster k RISC of 1.1×106 s–1 in 5 wt% doped BCPO films. The OLEDs with p-AC-DBNA emitted at λEL of 488 nm, and showed an EQEmax of 20.5% and low efficiency roll-off of 8% at 100 cd m–2 and 20% at 1000 cd m–2 (Table S1).ref. ref314 Similarly, two isomeric carbazole-substituted emitters with DBA acceptors, TDBA-Cz and DBA-Cz (Figure ), exhibited narrowband emission at λPL of 461 and 447 nm (FWHM of 43 and 38 nm) and ΔE ST of 0.14 and 0.03 eV, all respectively, in toluene.ref. ref315 The ΦPL in 10 wt% doped films in DPEPO are unity and 90%, while the ΦPL remained relatively high at 88 and 52% in neat films. The devices with TDBA-Cz and DBA-Cz hence showed high respective EQEmax of 31.1 and 30.3% at CIE coordinates of (0.13, 0.13) and (0.14, 0.15), while the non-doped device with TDBA-Cz showed an EQEmax of 21.4% at CIE of (0.14, 0.16). Incorporation of an additional carbazole unit and subsequent removal of the acceptor tert-butyl groups afforded new TADF emitters 5TBO and 3TBO.ref. ref316 5TBO showed a blue-shifted emission (λPL = 457 nm) while retaining a FWHM of 44 nm, while 3BTO showed a slightly red-shifted emission (λPL = 462 nm) and a narrower FWHM of 39 nm. 3BTO and 5BTO have similar ΔE ST values of ∼0.15 eV, yet differ significantly in their ΦPL of 78.4 and 96.7% in respective 30 wt% doped mCBP films. These differences were attributed to smaller conformational freedom in 5BTO, resulting in slower k nr and the larger ΦPL. The device with 5TBO consequently showed a higher EQEmax of 26.2% than the device with 3BTO (17.3%), with both embittering at λEL of 484 nm with similar CIE coordinates of (0.13, 0.28) and (0.12, 0.29), respectively.
Compounds PhCz-TSOBA and TPA-TSOBA replace an oxygen atom in the DBA core with a sulfur atom (Figure ).ref. ref317 PhCz-TSOBA and TPA-TSOBA emit at 444 and 447 nm, respectively, in toluene and show narrowband emission (FWHM of 32 and 34 nm). However, their ΔE ST are large at 0.23 and 0.36 eV in toluene, their ΦPL are moderate at 60.8 and 61.8%, and their k RISC are slow at 4.08 and 1.91×10–4 s–1 in 10 wt% doped films in 2,6-DczPPy, all respectively. The OLEDs with PhCz-TSOBA and TPA-TSOBA showed the same EQEmax of 16.7% at λEL of 456 nm, with FWHM of 57 and 55 nm [CIE coordinates of (0.14, 0.15) and (0.14, 0.12)], respectively.
Summarizing these reported results using boron-containing acceptors, the fully fused DBA-based materials frequently exhibit superior TADF properties and device performances than unfused boron. The more rigid and bulky nature of the DBA acceptor provides an optimal dihedral angle with many types of donor to produce emitters that have well separated frontier orbitals, enhanced ΦPL, and often also horizontally oriented TDM. The most efficient OLED with a boron acceptor (TDBA-DI) showed an EQEmax of 38.2%, and this class of emitters is able to reach rather deep into the blue region of CIE space (Figure ).ref. ref304 Despite these high achievable EQE, common to all current blue TADF OLEDs, these devices with boron-based acceptors still frequently suffer from poor device roll-off and lifetime.
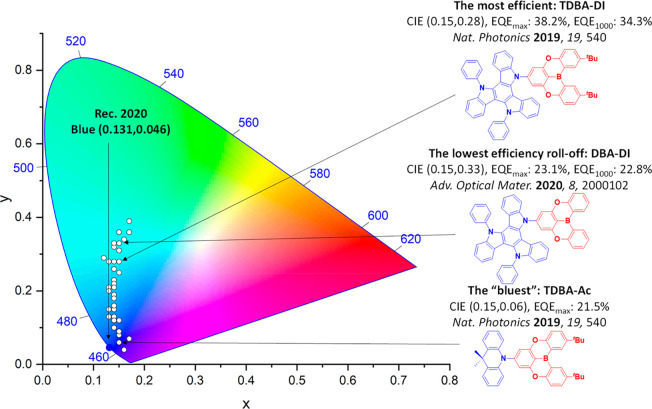
Nitrile-Containing Emitters
Likely due to its compact size and simple chemical structure, many of the early reported organic TADF materials contained nitrile acceptors. This includes the first sky-blue emitter 2CzPN, and materials development using this acceptor has only accelerated in the intervening years. For example, the replacement of Cz in 2CzPN ref. ref31 with a δ-carboline led to the formation of sky-blue emitter δ-2CbPN (Figure ). In toluene this compound has a smaller ΔE ST of 0.13 eV, higher ΦPL of 93%, shorter τd of 180 μs, and blue-shifted λPL of 453 nm compared to 2CzPN (ΔE ST of 0.21 eV, ΦPL of 89%, τd of 270 μs, and λPL of 473 nm). Thus, the corresponding OLED with δ-2CbPN showed an improved EQEmax of 22.5% and blue-shifted λEL= 486 nm compared to 19.2% and 491 nm for 2CzPN, both using the same device structure. The device containing α-2CzPN instead showed a very low EQEmax of 4.2% at λEL of 473 nm, owing to the moderate ΦPL of 37%. Furthermore, the ΔE ST of α-2CzPN in 20 wt% doped mCP films (0.28 eV) is more than double that of δ-2CzPN (0.13 eV). As a result, there is less efficient RISC in α-2CzPN, with the weaker donating properties of the α-carboline directly leading to lower charge-transfer exciton character and thus an increased ΔE ST.
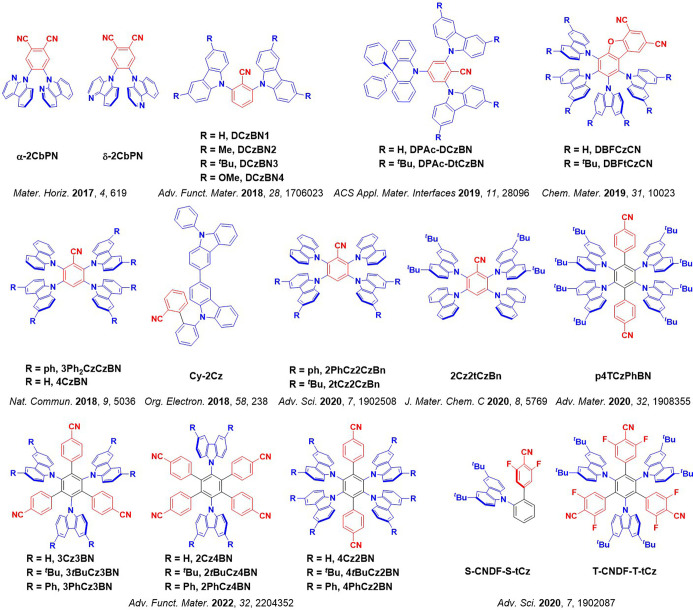
A benzonitrile acceptor was functionalized with pairs of ortho carbazole donor derivatives to produce deep-blue TADF emitters that showed CIEy coordinates < 0.08 in devices: DCzBN1 (0.15, 0.05), DCzBN2 (0.15, 0.07), and DCzBN3 (0.16, 0.06) (Figure ).ref. ref318 The devices showed EQEmax values of 2.5, 7.7, and 10.3%, respectively, with the latter representing one of the highest performing deep-blue TADF OLEDs at that time. An EQEmax of 18.0% was achieved for the device with related structure DCzBN4; however, a red-shift in the EL was observed with CIE coordinates of (0.16, 0.23). Unfortunately, all the devices displayed severe efficiency roll-off, with brightness of 100 cd m–2 not achieved for DCzBN1–3, while the efficiency roll-off for DCzBN4 was 42% at 100 cd m–2. The longer τd for DCzBN1–3 (11.2 to 18.0 μs) compared to DCzBN4 (5.5 μs) is consistent with the difference in efficiency roll-off behavior. Additionally, the high T1 energies (∼3.0 eV) of all the emitters and the use of mCP (T1 ≈ 2.9 eV) as a blocking layer may have contributed to inefficient exciton confinement. Introducing a spiro-acridan donor at the para position of benzonitrile in the DCzBN framework resulted in improved device efficiency, although this was also accompanied by a red-shift in the emission. The stronger ICT between the spiro-acridine and the benzonitrile moieties in DPAc-DCzBN and DPAc-DtCzBN led to higher ΦPL (71 and 64%, respectively) compared to DCzBN1–3 all with a ΦPL below 35%, but broader and red-shifted emission in the doped DPEPO films. The devices with DPAc-DCzBN and DPAc-DtCzBN showed EQEmax of 23% for both devices yet suffered around 50% efficiency roll-off at 1000 cd m–2; the CIE coordinates were (0.17, 0.25) and (0.16, 0.15), respectively.ref. ref319
Substituting a dibenzofuran core with four carbazole donors and two nitrile acceptors was highly effective in achieving efficient TADF in the emitters DBFCzCN and DBFtCzCN (Figure ).ref. ref320 DBFCzCN and DBFtCzCN emit with λPL, ΔE ST, and τd of 451 and 489 nm, 0.28 and 0.18 eV, and 89.0 and 29.1 μs respectively in THF. They also have high ΦPL of 100 and 92% in 20 wt% doped films in DPEPO. The device with DBFCzCN showed an EQEmax of 25.2% with CIE coordinates of (0.15, 0.29), although with a large roll-off (93%) at 1000 cd m–2. The device with DBFtCzCN showed an EQEmax and CIE of 17.4% and (0.19, 0.49), with the green EL arising from the relatively stronger tBuCz donors.
Decoration of carbazole donors with peripheral phenyl groups in 3Ph2CzCzBN (Figure ) led to improved TADF behavior and blue emission in toluene, with λPL, ΔE ST, ΦPL, and τd of 464 nm, 0.19 eV, 95%, and 10 μs. These properties compared favorably to 4CzBN (443 nm, 0.23 eV, 63%, and 50 μs), which contains unsubstituted Cz donors. The device with 3Ph2CzCzBN (15 wt% in mCBP) showed EQEmax and CIE coordinates of 15.9% and (0.17, 0.37), albeit with a red-shifted emission at 482 nm compared to the reference device with 4CzBN (EQEmax of 9.7% and CIE coordinates (0.19, 0.32) at λEL of 471 nm). Further, the device performance of 3Ph2CzCzBN was further improved in an EML consisting of 20 wt% emitter in mCBP, with EQEmax, CIE, and λEL of 17.9%, (0.18, 0.39) and 486 nm, and reduced efficiency roll-off of 1.7% at 1000 cd m–2.ref. ref321
Zou et al. employed a mixture of substituted and unsubstituted carbazole donors in 2tCz2CzBn and 2PhCz2CzBn (Figure ),ref. ref322 both of which showed similar λPL of 455 and 458 nm in toluene, ΔE ST values of 0.15 and 0.17 eV in 2Me-THF, and ΦPL of 87 and 86% in doped mCBP films (20 and 30 wt%), all respectively. The OLED with 2tCz2CzBn showed an EQEmax of 23.8% at λEL of 464 nm with CIE coordinates of (0.15, 0.19), while the OLED based on 2PhCz2CzBn showed an improved EQEmax of 26.6% with an almost identical EL spectrum. In another report, non-doped solution-processed devices with 2tCz2CzBn also exhibited high efficiency, with the EQEmax reaching 24.5% albeit with a red-shifted EL [λEL = 472 nm and CIE coordinates (0.16, 0.24)] compared to an EQEmax of 25.8% [λEL = 488 nm and CIE coordinates (0.21, 0.42)] for the device with isomer 2Cz2tCzBn. The high EQEmax of the devices with 2tCz2CzBn and 2Cz2tCzBn are supported by the AIE properties of these materials as neat films, however both devices showed severe efficiency roll-off of more than 75% at 1000 cd m–2.ref. ref323
To further blue-shift the emission of multi-carbazole emitters, Zhang et al. adopted a weaker cyanophenyl acceptor in lieu of the stronger (directly attached) cyano acceptor. In toluene p4TCzPhBN (Figure ) has λPL, ΔE ST, ΦPL, τd, and k RISC of 452 nm, 0.1 eV, 93.4%, 6.3 μs, and 2.36 × 106 s–1, respectively. The OLED showed an EQEmax of 22.8% at λEL of 456 nm and CIE coordinates of (0.15, 0.10).ref. ref324 Later, Madama et al. developed a series of TADF emitters using the cyanophenyl group as a weak acceptor and different numbers and types of carbazole derivatives (Cz, tBuCz, or PhCz) in the structural template 2-4(D)(2-4)BN (Figure ).ref. ref325 Their preliminary photophysical results revealed that emitters containing carbazole as the donor showed either no (3Cz3BN and 2Cz4BN) or weak (4Cz2BN) TADF character. Likewise, 3tBuCz3BN, 2tBuCz4BN, 3PhCz3BN, and 2PhCz4BN also showed weak TADF character with ΔE ST > 0.18 eV and low ΦPL < 19% in toluene. Only compounds 4tBuCz2BN and 4PhCz2BN exhibited efficient TADF with ΔE ST of 0.07 and 0.03 eV, τd of 29.0 and 5.0 μs, and ΦPL of 45 and 87%, respectively. OLEDs were fabricated with 4PhCz2BN and the optimized devices showed an EQEmax of 18.2% at CIE coordinates of (0.16, 0.28). Non-doped devices also performed well with EQEmax of 16.4% and efficiency roll-off of only 13% at 1000 cd m–2, albeit with a red-shifted λEL = 498 nm and CIE coordinates (0.21, 0.45).
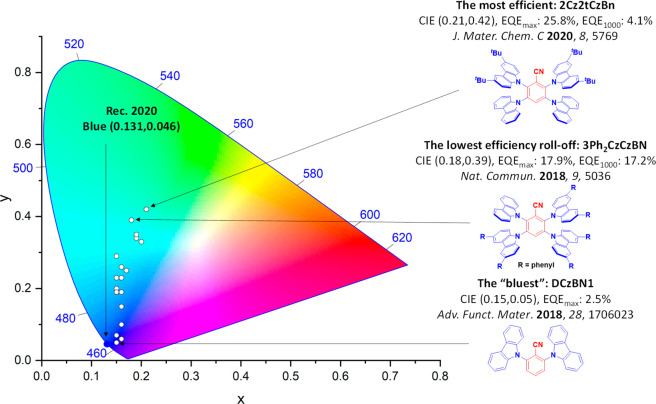
The deep-blue emitter Cy-2Cz contains a benzonitrile acceptor with an ortho-substituted bisN-phenylcarbazole donor (Figure ), and has ΔE ST of 0.18 eV in toluene. The OLED with Cy-2Cz showed deep blue emission with CIE coordinates of (0.16, 0.10) and an EQEmax of 11.9%. The relatively low efficiency roll-off of ∼21% at 1000 cd m–2 results from the short τd of 9.4 μs, indicated the promise of this emitter design strategy.ref. ref326 Through-space charge transfer (TSCT) and through-bond charge transfer (TBCT) contributions to the excited state character were simultaneously incorporated into an emitter with three alternating difluorocyanobenzene acceptor units and three di-tert-butyl carbazole donor groups, connected to a central benzene ring. TD-DFT calculation showed that T-CNDF-T-tCz (Figure ) has multiple degenerate excited singlet and triplet states, leading to a low energy barrier for RISC. T-CNDF-T-tCz emits at λPL of 477 nm, has a small ΔE ST of 0.03 eV, a high ΦPL of 76%, and a τd of 7.79 μs as a neat film (Table S1). The non-doped solution-processed OLED exhibited sky-blue emission with λEL of 484 nm at CIE coordinates of (0.19, 0.35), and showed an EQEmax of 21.0% – nine times higher than the device with reference mono D-A emitter S-CNDF-S-tCz (EQEmax of 2.6%).ref. ref327
The most efficient devices containing benzonitrile-based blue TADF emitters are highlighted in Figure . These devices largely struggle to achieve high efficiency due to their low ΦPL. Further, these devices generally show suboptimal blue color coordinates due to their broad emission. Thus, this molecular design seems to be less promising than those based on, for instance, triazine or boron-containing acceptors.
Oxadiazole-Containing Emitters
The inherently electronegative nature of the heteroatoms in oxadiazole has long been deployed in the design of electron-transporting materials for OLEDs, such as PBD and OXD-7.ref328−ref329ref330 The very shallow LUMO level (−0.55 eV) of oxadiazoleref. ref331 also makes 1,3,4-oxadiazole and its derivatives potentially attractive as acceptors in the design of deep-blue TADF emitters. The oxadiazole functionality can be easily obtained from an existing nitrile precursor, and thus a wide variety of donor-acceptor compounds can be readily accessed. Selected oxadiazole based emitters discussed here are shown in Figure . For example, replacement of the nitrile groups in 2CzPN with weaker electron-accepting oxadiazoles resulted in blue-shifted emission, which could be fine-tuned with the distal aryl groups. The respective λPL of oxadiazole-based 2CzdOXDME, 2CzdOXDPh, and 2CzdOXD4MeOPh (Figure ) are 453, 466, and 459 nm, whereas 2CzdOXD4CF3Ph has a red-shifted λPL of 487 nm due to the auxiliary trifluoromethyl acceptor unit. Further, the oxadiazole-based emitters 2CzdOXDME, 2CzdOXDPh, 2CzdOXD4CF3Ph, and 2CzdOXD4MeOPh have high ΦPL of 28.7, 38.3, 39.1, and 47% respectively, superior to 2CzPN (28.1%).ref. ref332 However, 2CzdOXDME, 2CzdOXDPh, 2CzdOXD4CF3Ph, and 2CzdOXD4MeOPh have long τd of 24.6, 58.6, 31.5, and 64.9 ms in 10 wt% doped films in PMMA and large ΔE ST of 0.31, 0.31, 0.32, and 0.44 eV in 10 wt% doped films in DPEPO, all respectively (Table S1).ref. ref333 The OLEDs with 2CzdOXDMe, 2CzdOXDPh, 2CzdOXD4CF3Ph, and 2CzdOXD4MeOPh showed EQEmax of 11.8, 7.0, 12.3, and 4.8% with λEL of 452, 460, 472, and 452 nm at CIE of (0.17, 0.17), (0.15, 0.16), (0.18, 0.28), and (0.17, 0.17), all respectively. Unfortunately, the large ΔE ST and longer τd led to severe efficiency roll-off (89, 81, 79, and 73% respectively at 100 cd m–2). Therefore, although blue-shifted emission had been achieved, it came at the cost of efficiency and overall performance.ref. ref332
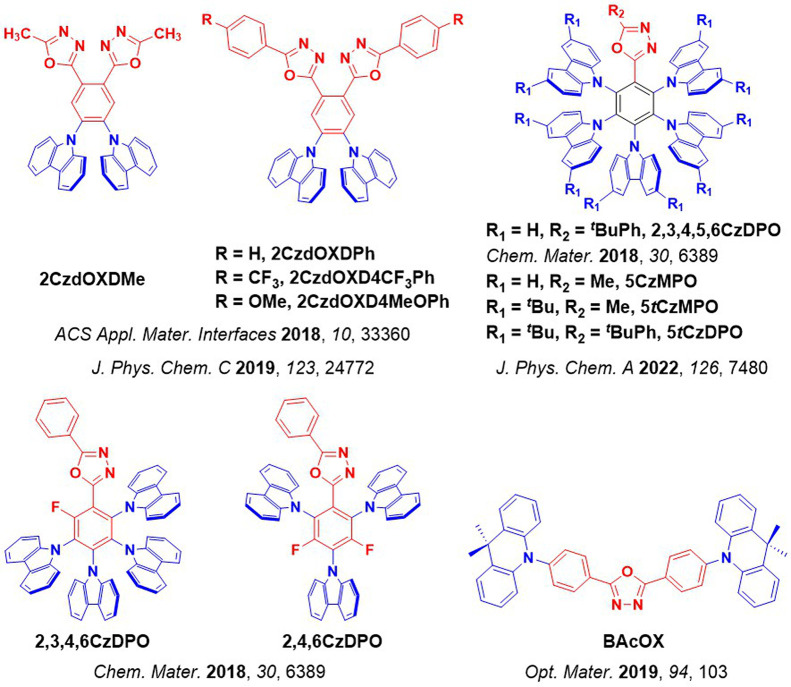
Using a similar oxadiazole acceptor the effect of changing the number of carbazole donors was investigated along with the use of secondary fluorine acceptor moieties connected to the central benzene.ref. ref334 Although six emitters were investigated, of these only 2,4,6CzDPO, 2,3,4,6CzDPO, and 2,3,4,5,6CzDPO (Figure ) displayed TADF, emitting at λPL of 490, 483, and 482 nm respectively in toluene. Increasing the number of donors altered the S1 energy, with substitution at the 3- and 5-positions particularly destabilizing the S1 energy relative to 2,4,6CzDPO. The devices with 2,4,6CzDPO, 2,3,4,6CzDPO, and 2,3,4,5,6CzDPO showed respective EQEmax values of 6.1, 17.8, and 24.4% at CIE coordinates of (0.17, 0.30), (0.18, 0.36) and (0.16, 0.29), far from target blue color coordinates. Relatively high efficiencies roll-off ranging from 23 to 54% at 100 cd m–2 also occurred, due to long τd of between 75 and 308 and 75 μs. Later the same group studied the effect of adding methyl and phenyl substituents on the oxadiazole acceptor of the most promising emitter previously identified (2,3,4,5,6CzDPO) and designed additional compounds 5tCzDPO, 5CzMPO, and 5tCzMPO. The methyl-modified 5CzMPO showed a blue-shifted emission (λPL = 466 nm) while the emission of 5tCzDPO is red-shifted (λPL = 496 nm) compared to 2,3,4,5,6CzDPO (λPL = 482 nm), all in toluene. These three emitters showed smaller ΔE ST in the range of 0.01 to 0.12 eV and higher ΦPL (20 to 30%) compared to 2,3,4,5,6CzDPO (ΔE ST = 0.12 eV and ΦPL = 13%), attributed to the increased donor strength of the tCz donor. The OLED with 5tCzDPO showed a higher EQEmax of 29% but red-shifted CIE coordinates of (0.18, 0.36) compared to the reference device with 2,3,4,5,6CzDPO [EQEmax = 24.4%, CIE coordinates of (0.16, 0.29)] (Table S1).ref. ref335
A simple D-A-D design using DMAC as the donor produced highly efficient sky-blue TADF emitter BAcOX (Figure ).ref. ref336 BAcOX emits at λEL of 461 nm and has a ΔE ST of 0.26 eV in toluene, while the ΦPL and τd are 93% and 84.1 μs in 10 wt% doped films in DPEPO. The OLED showed an EQEmax of 22.3% at λEL of 475 nm and CIE coordinates of (0.16, 0.24), however the device suffered from severe efficiency roll-off and a low maximum luminance of 200 cd m–2 was attributed to TTA and STA quenching permitted by the relatively long τd of 84 μs. This quenching was further exacerbated by unbalanced transport of carriers and inferior exciton confinement in the emission layer. Using DPPOC as the host instead of DPEPO improved the efficiency roll-off, but at a cost to the EQEmax which reached only 16.7%.
To date, only a few oxadiazole-based blue TADF materials have been developed. The best performing devices in terms of efficiency, roll-off, and color are shown in Figure . Though the devices with oxadiazole-based emitters showed high EQE, they exhibit higher CIEy (> 0.05) coordinates compared to the Rec. 2020 blue standard. This is because of the too strong electron-withdrawing nature of the oxadiazole. This results in red-shifted and broadened emission. The generally poor thermal and photochemical stability of this moiety and the sub-optimal performance of these emitters in OLEDs are contributing factors to the lack of popularity of oxadiazole based TADF materials.
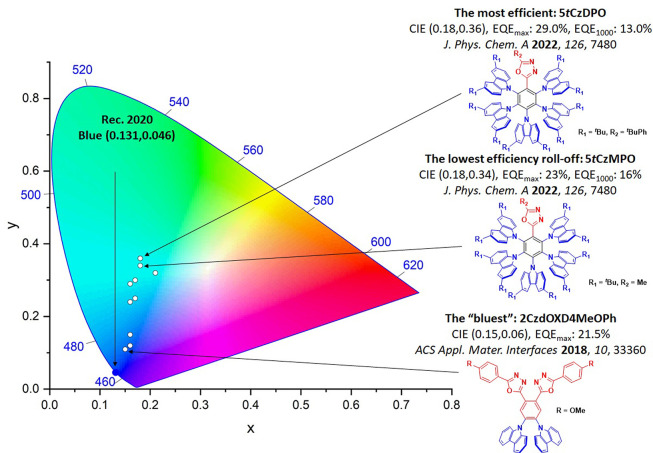
Sulfone-Containing Emitters
Diphenylsulfone (DPS) is a versatile electron-accepting group, with moderate electron-withdrawing ability (LUMO = −1.81 eV)ref. ref337 making it a suitable acceptor for blue TADF emitter design. The very first deep-blue TADF emitter tCz-DPS featured a D-A-D structure with tCz donors and was reported by Adachi and co-workers in 2012.ref. ref230 tCz-DPS exhibited deep blue emission with λPL of 423 nm and ΦPL of 80% in 10 wt% doped films in DPEPO.ref. ref230 However, the ΔE ST is large at 0.32 eV, accompanied by a long τd of 8.2 ms. The OLEDs showed deep-blue emission with λEL of 420 nm and an EQEmax of 9.9% but with very severe efficiency roll-off.ref. ref230 The same group soon after reported DMAC-DPS (Figure ), where the tCz donors were replaced with stronger and bulkier DMAC donors. DMAC-DPS exhibited blue emission with λPL of 464 nm (ΦPL = 80%), and more importantly the ΔE ST decreased to 0.08 eV with τd shortened to 3.1 μs in 10 wt% doped films in mCP.ref. ref338 The device with DMAC-DPS emitted at λEL = 465 nm [CIE coordinates of (0.16, 0.20)], and showed what was a record-high EQEmax of 19.5%. These two benchmark TADF emitters illustrate the potential of DPS in achieving highly efficient blue TADF and have inspired a large number of related emitter designs that contain the sulfone motif in the subsequent years.
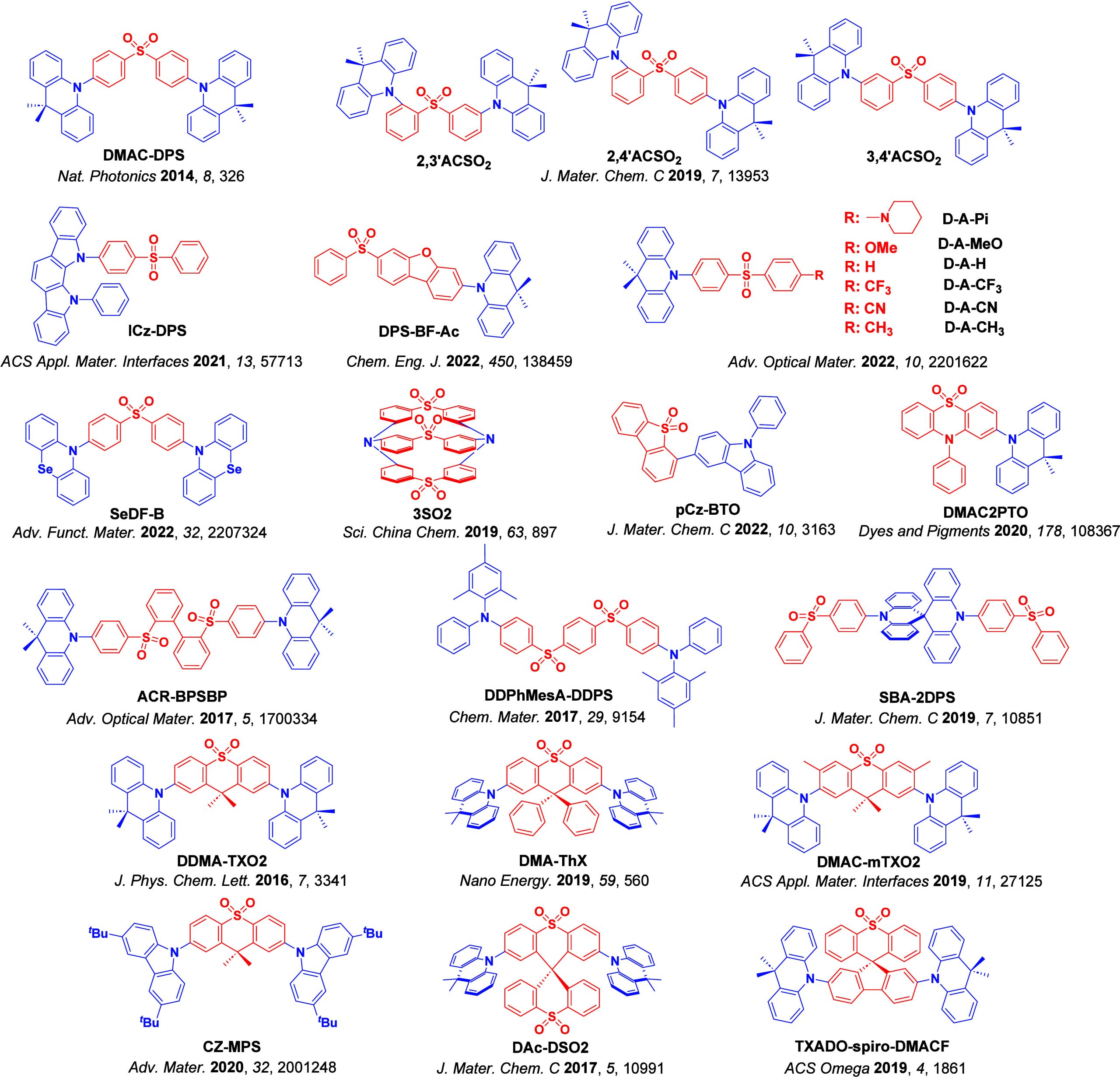
Three isomers of DMAC-DPS were designed to investigate the effect of the donor’s position on the photophysical properties of the emitters.ref. ref339 The molecules 2,3′ACSO2 and 2,4′ACSO2 (Figure ) both have one ortho-substituted acridine donor, with the second acridine placed either in the meta or para position on the opposite side of the central DPS. These two compounds exhibited a red-shift in the emission (λPL of 502 and 499 nm, respectively) and a decrease in their ΦPL (59 and 66%) in 10 wt% doped films in DPEPO compared to DMAC-DPS (λPL of 464 nm and ΦPL of 90% in 10 wt% doped films in mCP). The OLEDs of the two emitters in 10 wt% DPEPO emitted in the green and showed only moderate EQEmax of around 12%. Compound 3,4′ACSO2, containing one meta and one para acridine group, instead emits in the sky-blue at λPL of 476 nm, has a ΦPL of 77%, and τd of 5.4 μs in 10 wt% doped films in DPEPO. The OLED with 3,4′ACSO2 doped in 10 wt% DPEPO displayed sky-blue emission with CIE coordinates (0.17, 0.29) and showed an EQEmax of 20.5% with efficiency roll-off of around 35% at 1000 cd m–2.ref. ref339
To investigate the role of higher-lying LE triplet states (3LE) in the TADF mechanism, Ryoo et al. replaced one of the donors of DMAC-DPS with weakly conjugated electron-donating groups of piperidine (D-A-Pi), -OMe (D-A-MeO), -CH3 (D-A-CH3 ), or -H (D-A-H), or with electron-withdrawing groups of -CF3 (D-A-CF3 ) or -CN (D-A-CN, Figure ).ref. ref340 These six emitters exhibited blue emission in 10 wt% doped films in DPEPO, with λPL ranging from 448 to 488 nm and ΦPL all above 80% apart from D-A-Pi (62%). All six emitters possess S1 and T1 states of CT character, with ΔE ST values all smaller than 0.10 eV. The gaps between the LE T2 state (3.19 eV, T1 from DMAC) the S1 state were calculated to be −0.04, 0.02, 0.05, 0.10, 0.23, and 0.28 eV for D-A-Pi, D-A-MeO, D-A-CH3 , D-A-H, D-A-CF3 , and D-A-CN, respectively. D-A-CF3 and D-A-CN possessing degenerate S1 and T1 states have shorter τd of 3.6 and 2.1 μs and faster k RISC of 1.9 and 2.1×106 s –1 respectively, compared to τd > 5 μs and k RISC < 1.5 ×106 s –1 for D-A-Pi, D-A-MeO, D-A-CH3 each containing electron-donating groups, and also D-A-H. Devices with each of the six emitters exhibited EQEmax values between 17.2 and 23.9% at λEL between 447 and 489 nm. Among these devices those with D-A-CF3 and D-A-CN showed small respective efficiency roll-off of 9.0% (EQEmax = 21.3%) and 14.2% (EQEmax = 20.5%) at a luminance of 100 cd m–2. Zhu et al. also explored D-A molecular design and extended the connecting DPS phenyl ring into a bulky benzofuran group, affording DPS-BF-Ac. This linking unit adopts an almost perpendicular geometry between the donor and acceptor moieties, leading to a small ΔE ST of 0.03 eV in 10 wt% doped films in PMMA. These films emit at λPL of 477 nm, having a ΦPL of 87% and a τd of 14.5 μs.ref. ref341 The solution-processed device using DPS-BF-Ac showed an EQEmax of 24.7%, and sky-blue emission with λEL of 482 nm.
Xi et al. used a bulky and weakly electron-donating syn-indolocarbazole in ICz-DPS (Figure ), which emits at 438 nm and has a ΦPL of 72% with a small ΔE ST of 0.03 eV in 10 wt% doped films in DPEPO.ref. ref342 ICz-DPS exhibited a very short τd of 0.6 μs and thus a fast k RISC of 3.3 × 106 s–1. The device with ICz-DPS showed deep-blue emission with an λEL of 435 nm at CIE coordinates of (0.15, 0.08), and the EQEmax reached 11.6% with an efficiency roll-off of only 6% at 1000 cd m–2.
Sharif et al. explored the effect of enhancing SOC and minimizing ΔE ST by incorporating the heavy atom selenium within the donor in SeDF-B (Figure ).ref. ref343 Theoretical calculations predicted two low-energy conformers, axial and equatorial, with only the latter showing the potential to be TADF-active. SeDF-B emits at λPL of 490 nm but has a very low ΦPL of 3% in 10 wt% doped solution-processed mCBP films. Despite the moderately long τd of 18.5 μs, the k RISC reached 0.6×106 s –1, and the vacuum-processed device with SeDF-B showed an EQEmax of 25.6% at CIE coordinates of (0.17, 0.14) and very small efficiency roll-off (∼10%) below 1000 cd m–2. The operational lifetime (LT80) of the OLED also reached 29 hours. The abnormally high EQEmax considering the low ΦPL was ascribed to the different populations of axial/equatorial conformers during the thermal evaporation process, with the more efficient equatorial conformer being the dominant species in the evaporated films.
A D-A-A-D structure using two sulfone groups coupled with DMAC donors produced the blue emitter ACR-BPSBP (Figure ), with λPL = 460 nm, ΦPL = 82%, and τd = 5.0 μs.ref. ref344 The OLED showed an EQEmax of 24.6% at CIE coordinates of (0.16, 0.21), however the efficiency roll-off at 200 cd m–2 was high (∼47%), and a luminance of 1000 cd m–2 could not be reached. This was attributed to slow RISC resulting from poor alignment of the LE/CT excited states, with long exciton lifetimes allowing increased TTA and SPA in the device.ref. ref344 A deep-blue solution-processed device with an EQEmax of 8.5% and low efficiency roll-off of ∼9% at 1000 cd m–2 at CIE coordinates of (0.16, 0.08) was produced using a similar emitter DDPhMesA-DDPS (named 3b in that work) containing mesityl-diphenylamine donors with the same bis-DPS acceptor.ref. ref345
Sky-blue emitter DAc-DSO2 (Figure ) contains a central acceptor comprised of two spiro-linked DPS groups with two acridine donors, and emits at λPL of ∼465 nm, with a small ΔE ST of 0.01 eV. The device with DAc-DSO2 showed sky-blue emission with CIE coordinates of (0.18, 0.33) and an EQEmax of 25.4%, with very low efficiency roll-off of 13% at 1000 cd m–2 supported by the tailored device structure and small ΔE ST.ref. ref346 A similar DPS-spiro-fluorene acceptor was coupled to acridines affording the blue emitter TXADO-spiro-DMACF. The corresponding non-doped device showed an EQEmax of 5.3% with a CIEy coordinate of 0.09, but suffered from severe efficiency roll-off of 43% at 200 cd m–2 associated with its long τd of 101 μs.ref. ref347 The elongated emitter SBA-2DPS instead contains a central spiro-bis-acridine donor, decorated on each side with terminal DPS groups and resulting in a small ΔE ST of 0.09 eV and fast τd of 4.3 μs.ref. ref348 Owing to its molecular weight and linear shape SBA-2DPS shows preferential horizontal TDM orientation (87%), and the device with SBA-2DPS showed EQEmax of 25.5% linked to its improved light-outcoupling efficiency. Emitting at λEL of 467 nm and CIE coordinates of (0.15, 0.20), the device exhibited modest efficiency roll-off of 10 and 39% at 100 and 1000 cd m–2, respectively.
Linking the phenyl rings of DPS gives the structurally related acceptor dimethylthioxanthene-S,S-dioxide (TXO2), which has also been used in blue TADF emitters as exemplified in DDMA-TXO2 (Figure ).ref. ref349 Like DMAC-DPS, DDMA-TXO2 has a high ΦPL of 95% and shows blue emission (λPL ∼ 460 nm) with a small ΔEST of 0.01 eV, and τd of 44 μs in 13 wt% doped films in DPEPO. The device showed an EQEmax of 22.4% at λEL of 465 nm and CIE coordinates of (0.16, 0.24).ref. ref349 Replacing the methyl groups on the acceptor with phenyl groups in DMA-ThX produced a device with blue-shifted CIE coordinates of (0.14, 0.10) despite the similar λEL of 459 nm, which may be more a function of the change of host to mCBP rather than the intrinsic photophysics of the emitter. Indeed, the balance between color point and efficiency was brought into stark relief when comparing performance for the device in mCBP (EQEmax of 2.9% and Lummax of 848 cd m–2) with that in DPEPO [EQEmax up to 18.4%, λEL of 462 nm, CIE coordinates of (0.14, 0.15), and Lummax of 1460 cd m–2].ref. ref350 Adding additional methyl groups meta to the sulfone in DDMA-TXO2 decreases the acceptor strength and forces the D–A torsion angle to be more twisted, leading to a blue-shift the emission as reported in DMAC-mTXO2.ref. ref351 DMAC-mTXO2 exhibited blue emission with λPL of around 450 nm and ΦPL of 88% in 35 wt% doped DPEPO films. Moreover, the twisted structure resulted in a very small ΔE ST of 0.05 eV and a short τd of 3 μs. The device with DMAC-mTXO2 displayed blue-shifted emission with CIE coordinates of (0.15, 0.18), compared to (0.16, 0.25) for DDMA-TXO2 using the same device stack. The EQEmax of DMAC-mTXO2 device was slightly improved to 22.6% compared to 20.0% for the device with DDMA-TXO2, tracking with the higher ΦPL (88 and 80%, respectively). The device with DMAC-mTXO2 also showed a very small efficiency roll-off of 0.5 and 12% at 100 cd m–2 and 1000 cd m–2 respectively, which were attributed to the fast k RISC of 2.8 × 106 s–1.
Replacing the acridine groups in DDMA-TXO2 with weaker dtCz donors afforded the deep-blue emitter CZ-MPS (Figure ).ref. ref352 The ΔE ST of CZ-MPS in toluene is large at 0.49 eV, which seems to be too large to support effectively RISC. TD-DFT calculations, however, predicted the presence of intermediate T2 and T3 triplet states, with LE character and with large SOCME values of 0.16 and 0.19 cm–1 respectively, that could alternatively contribute to RISC. CZ-MPS emits in the ultraviolet with λPL of 384 nm, ΦPL of 47%, and long τd of 4.8 ms in 10 wt% doped films in PMMA. The device with CZ-MPS in tCzSi showed ultraviolet emission with an λEL of 389 nm, CIEy coordinate of 0.06, and an EQEmax of 9.3% that was the highest reported for a UV-emitting OLED at that time. Considering the ΦPL of the 10 wt% CZ-MPS doped tCzSi film is only 46%, such a high EQEmax indicated that triplet harvesting nonetheless occurs, most likely through TADF.
The TADF emitter DMAC2PTO (Figure ) employs a phenylamine-linked DPS as a stronger acceptor in conjunction with a DMAC donor. The ΔE ST for this compound is small at 0.03 eV in 2-MeTHF glass.ref. ref353 DMAC2PTO emits at λPL of 448 nm with ΦPL of 62% and short τd of 4.2 μs in 15 wt% doped films in DPEPO. The optimized device with DMAC2PTO emitted at λEL of 448 nm with CIE coordinates of (0.15, 0.11) and showed an EQEmax value of 15.2%. The severe efficiency roll-off documented is likely due in part to the use of the notably unstable DPEPO host, along with TTA and STA quenching.ref. ref353
Wang et al. used dibenzo[b,d]thiophene-5,5-dioxide as the acceptor unit and 9-phenyl-9H-carbazole (pCz) as the donor in the AIE compound pCz-BTO (Figure ).ref. ref354 This compound emits at λPL of 438 nm, has a ΦPL of 59%, and a ΔE ST of 0.18 eV. The EQEmax of the non-doped device reached 7.1% with CIE coordinates of (0.15, 0.10), while the device with 10 wt% emitter in DPEPO had EQEmax slightly higher at 9.5% with almost identical CIE coordinates of (0.15, 0.09).
A creatively designed and highly soluble organic cage consisting of three DPS units connected by two donating nitrogen bridges (3SO2, Figure ) shows promising TADF properties.ref. ref355 Due to the cage structure intramolecular conformational flexibility was restricted, and 3SO3 showed narrowed deep blue emission with λPL of 414 nm and FWHM of 34 nm, along with ΔE ST of 0.18 eV, all in toluene. 3SO3 has a low ΦPL of 14% and τd of 8.6 μs in 5 wt% doped films in 26DCzPPY, and the OLED showed narrowband emission at λEL of 413 nm (FWHM of 35 nm) with CIE coordinates of (0.15, 0.04). However, the EQEmax was only 2.6% which was attributed to the low ΦPL, also to challenges selecting an appropriate device structure due to the high S1/T1 energies and shallow LUMO of 3SO2.
Thanks to its relatively weak electron-withdrawing ability, the DPS moiety and its related cyclic structures have become popular in the design of blue TADF emitters, particularly when paired with acridine donors (Figure ). The deepest blue OLED is based on 3SO2 and emits at λEL of 413 nm and has CIE coordinates of (0.14, 0.04). SeDF-B, where DPS moiety is used as acceptor and phenoselenazine is used as donor, exhibited the highest EQEmax of 25.6% among the blue sulfone-based emitters and also exhibited a small efficiency roll-off with EQE1000 of 23.0%. The lowest efficiency roll-off was achieved in the device with ICz-DPS where the EQEmax of 11.6% decreased only to 10.9% at 1000 cd m–2 at CIE coordinates of (0.15, 0.08). Overall, the examples of sulfone-based blue TADF emitters demonstrate a capacity to approach the Rec. 2020 blue emission CIE coordinates; however, the devices based on these materials suffer from severe efficiency roll-off and poor device stability, which are likely unfortunately due to the intrinsically poor photochemical stability of the sulfone group.
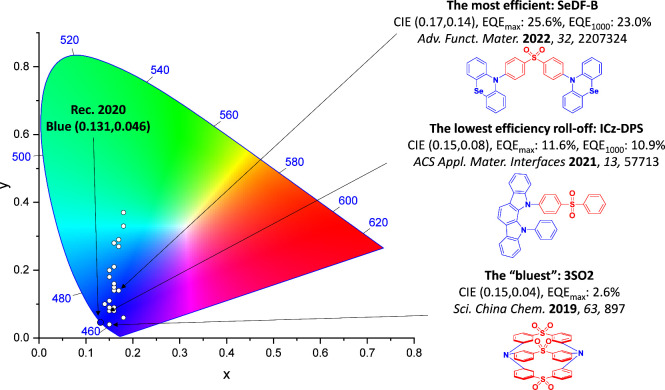
Ketone-Containing Emitters
The ketone moiety was first introduced in TADF emitter design in the form of the benzophenone acceptor in the compound Cz2BP, where the device emitted at λEL of 446 nm [CIE coordinates of (0.16, 0.14)] and showed an EQEmax of 8.1% (Figure ).ref. ref356 More recently, a series of D-A emitters constructed with isobenzofurine (MXAc-BF) or chromone (MXAc-CM and XAc-CM) coupled to a xanthene-spiro-acridine donor unit showed sky-blue to blue TADF emission.ref. ref357 The three compounds emit with λPL ranging from 461–482 nm, with small ΔEST (0.08–0.11 eV) and short delayed lifetimes (τd = 2.8–4.3 μs) in 50 wt% doped films in PPF. Although their EQEmax were moderate (16.2, 15.0, and 12.1% at λEL 478, 478, and 462 nm, respectively), the efficiency roll-off at 100 cd m–2 was very low at only 3–4%. Moreover, the devices with MXAc-BF and MXAc-CM exhibited low efficiency roll-off of 26% at 1000 cd m–2, attributed to their relatively fast k RISC of 4.9 and 7.5 × 105 s–1 respectively.
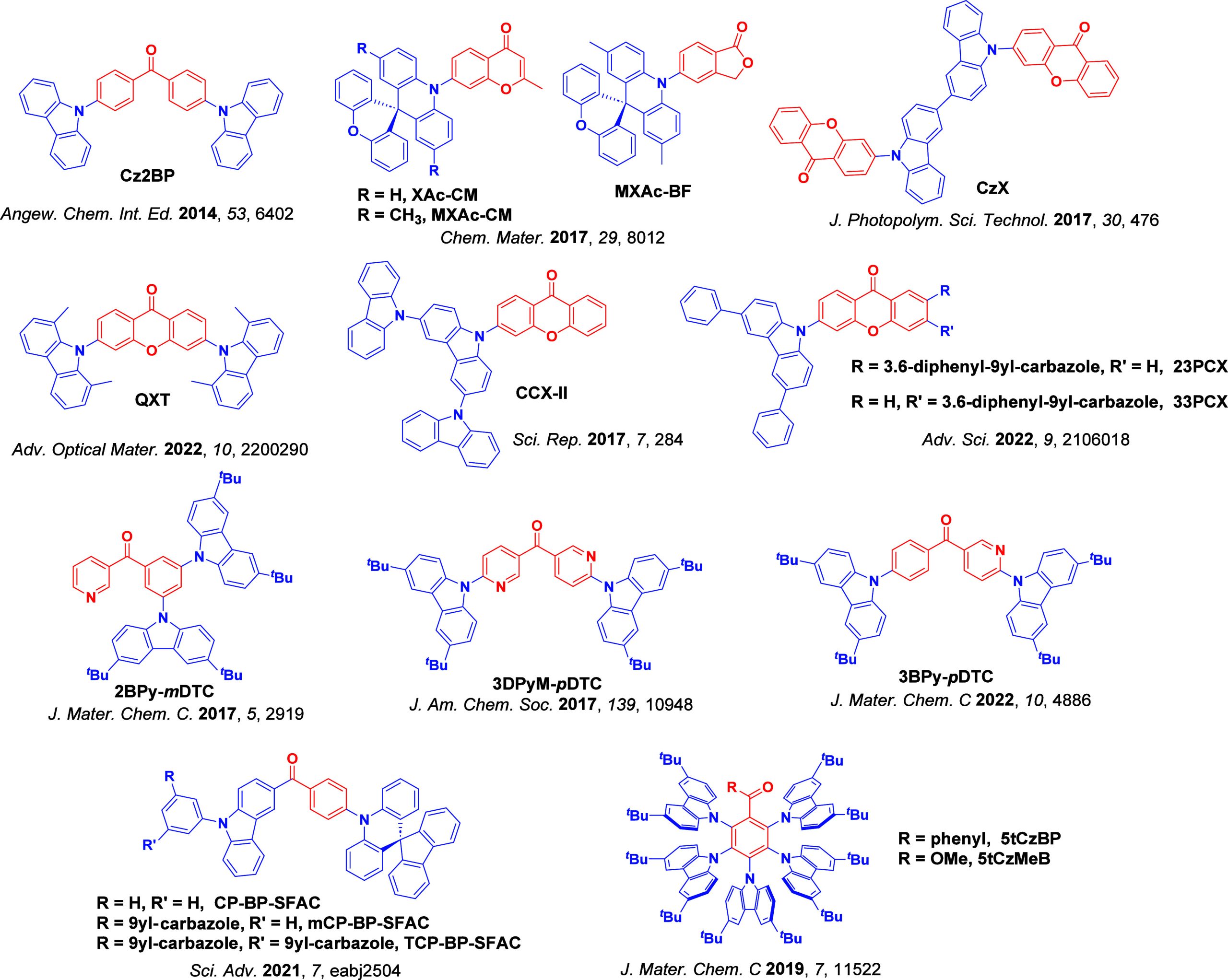
CzX (Figure ) possesses an A-D-A design consisting of a central dicarbazole donor coupled to two terminal xanthone acceptors.ref. ref358 This emitter produced a blue emitting device with λEL 482 nm and EQEmax 19.9%, along with a reasonable efficiency roll-off of ∼25% at 100 cd m–2. No lifetime or ΔE ST were reported, although a modest ΦPL of 54% in toluene suggests very efficient triplet conversion must have occurred. Another xanthone acceptor coupled to a tercarbazole donor dendron yielded the blue emitter CCX-II, which was demonstrated to have a high ΦPL of 97% along with preferentially horizontally orientated TDM in 6 wt% doped films in PPF.ref. ref359 The impressive EQEmax of 25.9% at CIE coordinates of (0.15, 0.22) was further enhanced to 33.3% with the use of an external outcoupling sheet. An extremely small ΔE ST of 0.03 eV helped to support the efficient RISC that led to small efficiency roll-off values of 13 and 34% at 100 and 1000 cd m–2, respectively.
Min et al. designed symmetric D-A-D material QXT (Figure ), featuring 1,8-dimethylcarbazole donors and a xanthone acceptor.ref. ref360 The compound showed fast k RISC of 2.4 ×106 s –1 in 20 wt% doped PPF films, and the OLED emitted at λEL of 480 nm with an EQEmax of 24.9%, along with a small efficiency roll-off of ∼13% at 1000 cd m–2. Zhang et al. instead used 3,6-diphenylcarbazole as the donors and connected them at different positions on a xanthone acceptor in 23PCX and 33PCX.ref. ref361 23PCX and 33PCX in 20 wt% doped films in PPF emit at λPL of 485 and 472 nm, with respective ΦPL of 88 and 92%, and ΔE ST below 0.05 eV for both, compared to λPL of 489 nm, ΦPL of 96% and ΔE ST of 0.02 eV for QXT in the same medium.ref. ref360 The OLEDs with 23PCX and 33PCX emitted at λEL of 484 and 469 nm with CIE coordinates of (0.17, 0.36), and (0.16, 0.25) and EQEmax of 25.5 and 27.5%, all respectively. The efficiency roll-off of at 1000 cd m–2 was moderately large at 38 and 33% respectively.
A dipyridyl-ketone acceptor with tert-butylcarbazole donors (3DPyM-pDTC, Figure ) exhibited blue emission with λPL of 464 nm, a high ΦPL of 98%, a small ΔE ST of 0.02 eV, and a short τd of 10 μs in 7 wt% doped mCBP films.ref. ref362 Moreover, the TDM of 3DPyM-pDTC in films adopts near-perfect horizontal orientation, leading to enhanced light outcoupling and supporting an EQEmax of 31.9%. The device CIE coordinates were (0.14, 0.18), but it also showed moderate efficiency roll-off of 18 and 49% at 100 and 1000 cd m–2. Replacing the one of the pyridines with a phenyl ring in 3BPy-pDTC leads to blue-shifted emission at λPL of 453 nm in 7 wt% doped films in mCBP, compared to 475 nm for 3DPyM-pDTC. This comes but at the cost of ΔE ST increasing from 0.02 eV (for 3DPyM-pDTC) to 0.19 eV though.ref. ref363 The OLED with 3BPy-pDTC showed an EQEmax of 25% at λEL of 458 nm with CIE coordinates of (0.14, 0.13) (Table S1). 2BPy-mDTC is a related compound containing a similar pyridyl ketone acceptor coupled to tert-butylcarbazole donors.ref. ref364 This compound emits at λPL of 476 nm, has a high ΦPL of 92%, a small ΔE ST of 0.05 eV, and τd of 8.3 μs in 7 wt% doped mCBP films. The OLEDs with 2BPy-mDTC showed an EQEmax of 24.6% at CIE coordinates of (0.15, 0.28), along with modest efficiency roll-off at 100 cd m–2 of ∼13%.
Converting the phenyl-ketone in 5tCzBP (Figure ) to a methyl ester in 5tCzMeB blue-shifts the emission, but also suppresses triplet non-radiative decay (3 × 106 s–1 in 5tCzBP to 0.3 × 106 s–1 of 5tCzMeB) while conserving k RISC at around 4 × 106 s–1 in toluene.ref. ref365 The 20 wt% in DPEPO doped and non-doped OLEDs with 5tCzMeB showed divergent performance at similar emission color, with EQEmax of 24.6% [λEL of 481 nm and CIE coordinates of (0.19, 0.32)] and 13.4% [λEL of 488 nm and CIE coordinates of (0.20, 0.36)], and both shared high efficiency roll-off of 33 and 44% at 100 cd m–2, all respectively. By contrast, the emission of the OLEDs with 5tCzBP were shifted to the green, with λEL of 497 nm and a moderate EQEmax ≈ 10%.
Fu et al. constructed D-A emitters CP-BP-SFAC, mCP-BP-SFAC, and TCP-BP-SFAC (Figure ) employing spiro[acridine-9,9′-fluorene] (SFAC) as electron donor and incorporated increasing numbers of terminal carbazoles in structures typically associated with host materials on the opposite side of a benzophenone acceptor.ref. ref366 All three compounds showed AIE with ΦPL above 80% in neat films, and generally similar photophysical properties with λPL at around 480 nm and ΔE ST around 0.10 eV in either 20 wt% doped DPEPO or non-doped films. The non-doped devices with each of the three emitters exhibited sky-blue emission with λEL at around 489 nm and EQEmax values ranging from 22.5 to 26.1%, while for the 20 wt% doped DPEPO devices the EQEmax of each reached above 36.6% at λEL of around 480 nm. The extremely high EQEmax was ascribed to the high ΦPL (∼100%) and preferential horizontal TDM orientation (Θ// > 72%) in the DPEPO films.
The summary of ketone-containing blue TADF emitters is shown in Figure . Compound 3BPy-pDTC exhibited the “bluest” emission with CIE coordinates of (0.14, 0.13), λEL of 458 nm, and EQEmax of 25.3%. The highest EQEmax of 38.6% was achieved in the device with TCP-BP-SFAC, which has CIE coordinates of (0.16, 0.28), whereas the device with QXT exhibited the smallest efficiency roll-off with EQEmax of 24.9% and EQE1000 of 21.7 at CIE coordinates of (0.16, 0.30). A large portion of the emitters based on ketone are classified as sky-blue. Thus, future research should be devoted to modulating the acceptor strength to promote a blue-shift of the emission.
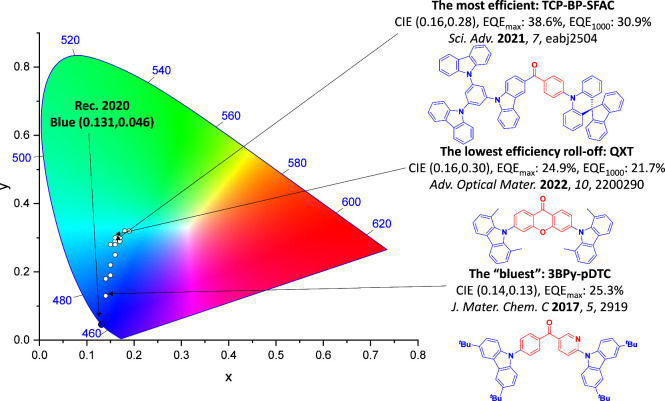
Other Emitters
Beyond the commonly used acceptor groups described above, there are a number of emitters that contain alternative or less frequently used acceptor groups. For example, OLED host materials featuring phosphine oxide groups are also sources of potential inspiration as weak acceptor units in the design of blue TADF emitters. The electron-withdrawing effect of the phosphine oxide (PO) groups adjusts excitonic ICT character for blue emission,ref. ref367 while the sp3-hybridized phosphorus atom enhances molecular distortion of the frontier molecular orbitals, separating them to establish small ΔE ST.ref. ref368
An acceptor with two PO groups at meta positions relative to carbazole donors around a central phenyl linker gave blue emitter m2tBCzPO (Figure ). The corresponding device showed an EQEmax of 21.0% at CIE coordinates (0.16, 0.17) and good efficiency roll-off at 100 and 1000 cd m–2 of 7 and 26%, respectively. The higher efficiency of the device with m2tBCzPO compared to other derivatives where the PO groups are located at the ortho or para positions was ascribed to the better balance between electronic and steric effects that contributed to efficient RISC.ref. ref369 A follow-up study of D–A–D variant 4tBCzDPDPO2A utilized a phosphine oxide homo-conjugated acceptor to bridge four di-tert-butyl-carbazolyl groups.ref. ref370 In comparison to 4tBCzPPOPO and 4tBCzPPODPO, which both adopted a non-conjugated D-A-A-D structure, the through-space conjugation effect in 4tBCzDPDPO2A leads simultaneously to small ΔE ST and improved oscillator strength, evidenced by doubling of ΦPL and a quadrupling of k RISC. The device with 4tBCzDPDPO2A showed an EQEmax of 23.7% with only 6 and 22% efficiency roll-off at 100 and 1000 cd m–2, respectively, with sky-blue emission at λEL of 470 nm and CIE coordinates of (0.18, 0.30). The low efficiency roll-off was attributed to the high RISC efficiency (94%) and fast radiative decay of 3.2 ×107 s–1 of this emitter. The devices with 4tBCzPPOPO and 4tBCzPPODPO showed blue-shifted emission with λEL = 460 nm for both and CIE coordinates of (0.18, 0.23) and (0.19, 0.25), respectively. However, the low ΦPL (∼30%) led to poor device efficiency with EQEmax reaching only 3.6 and 4.0%.
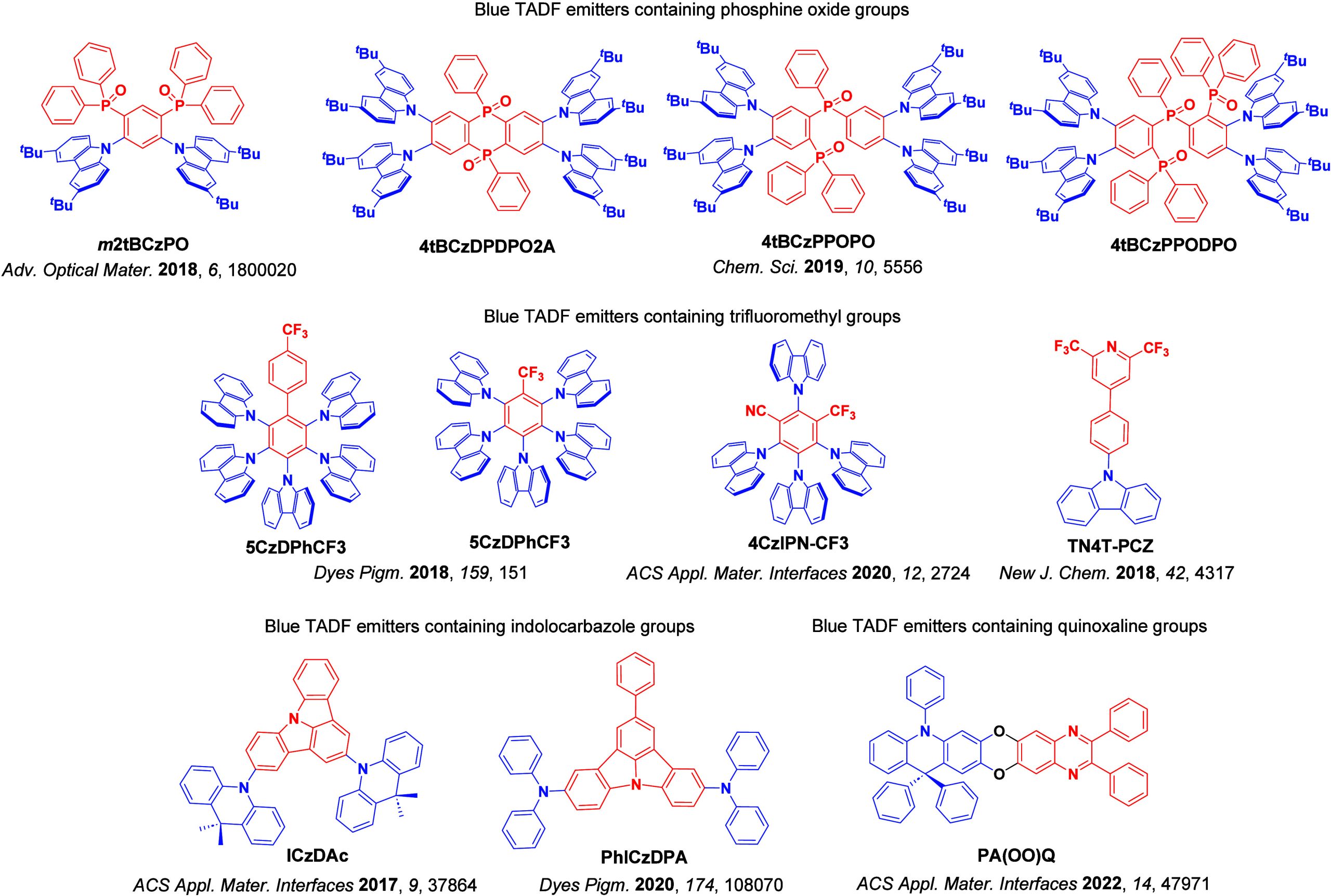
Trifluoromethyl (CF3) is of potential interest for producing deep-blue emission due to its weak electron-withdrawing ability [LUMO for (trifluoromethyl)benzene: −1.71 eV].ref. ref371 CIE coordinates of (0.16, 0.07) were achieved in a device with 5CzDPhCF3 , which pairs this acceptor with carbazoles (Figure ).ref. ref372 A poor EQEmax of 2.0% and high efficiency roll-off of 75% at 100 cd m–2 were noted though, likely due to the low ΦPL of 27% and the long τd of 0.14 ms (Table S1). The additional phenylene spacer in 5CzDPhCF3 proved essential for deep-blue emission, as the CIE coordinates shifted to (0.18, 0.33) for the device with 5CzDPhCF3 . Similarly, replacing one of the cyano groups in 4CzIPN for CF3 in 4CzIPN-CF3 tuned the emission from green to sky-blue.ref. ref373 A device with 4CzIPN-CF3 exhibited sky-blue emission at λEL of 487 nm, benefiting from the high emitter ΦPL (77%) and an exciplex host system to acheive the EQEmax of 23.1% with only 10% efficiency roll-off at 1000 cd m–2. Compound TN4T-PCZ is another carbazole-containing blue emitter coupled with a trifluoromethyl-substituted pyridine as the acceptor.ref. ref374 TN4T-PCZ emits at λPL of 411 nm, has a ΦPL of 87%, and a small ΔE ST of just 100 meV. The device with TN4T-PCZ showed deep-blue emission with λEL of 415 nm, CIE coordinates of (0.16, 0.03), and an EQEmax of 20.4%, making it one of the most efficient deep-blue D-A TADF OLEDs to date.
Coupling iCz with acridine donors produced the blue TADF emitter ICzDAc (Figure ) with ΔE ST of 0.17 eV and 98% ΦPL in 10 wt% doped DPEPO films.ref. ref375 The device with ICzDAc showed an EQEmax of 19.7% although with large 50% efficiency roll-off at 1000 cd m–2, with blue emission at CIE coordinates of (0.15, 0.16). A follow-up study instead used diphenylamines as donor groups in PhICzDPA, which maintained the high ΦPL of 94% and a reduced ΔE ST (0.12 eV) in 10 wt% doped DPEPO films. The device with PhICzDPA exhibited a high EQEmax of 30.4% and sky-blue emission with CIE coordinates of (0.13, 0.32).ref. ref376 However, this device also showed severe efficiency roll-off (43% at 100 cd m–2) and failed to reach 1000 cd m–2. This behavior can be rationalized by the long-lived excitons (τd = 249 μs) that are prone to quenching by multi-excitonic non-radiative decay processes under electrical excitation.
Wang et al. designed unique D–A emitter PA(OO)Q (Figure ), containing a fused ring structure where coplanar acridine donor and quinoxaline acceptor were connected by two oxygen-bridges within a six-membered ring.ref. ref377 Although DFT calculations predicted a small HOMO/LUMO overlap, the emitter with a more planar geometry still has a large ΔE ST of 0.35 eV and a long τd of 1.5 ms in 5 wt% doped films in mCBP. The OLED exhibited sky-blue emission with λEL of 488 nm, CIE coordinates of (0.19, 0.37), and EQEmax of 19.5%. However, the device showed severe efficiency roll-off of 73% at 100 cd m–2 due to the slow RISC process (k RISC = 1.1×103 s –1), likely resulting from its unusual structure.
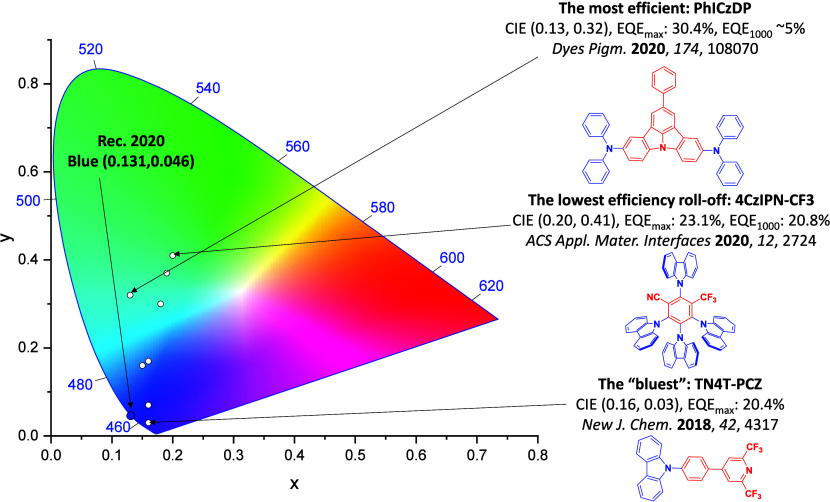
Although the acceptors discussed in this section are under-explored compared to other classes of acceptors discussed above, these studies nonetheless illustrate their great potentials in terms of approaching the standard blue CIE coordinates and exhibiting high device performance (Figure ). Thus, many of these “exotic” acceptors deserve greater attention in the design of blue TADF emitters.
Outlook
The period between 2017–2022 has witnessed an intense search for an ideal blue emitter, which to this day remains elusive. Tremendous efforts have translated into numerous examples of blue TADF OLEDs achieving EQEmax greater than 20%, yet only a handful of examples achieved the desired deep-blue emission (CIEy < 0.1) while maintaining this high efficiency (Figure ). Most of these deep-blue devices also suffer from unacceptable efficiency roll-off at practical brightnesses, and there also remains a lack of concerted effort to quantify device lifetimes necessary to correlate emitter structure to device stability.
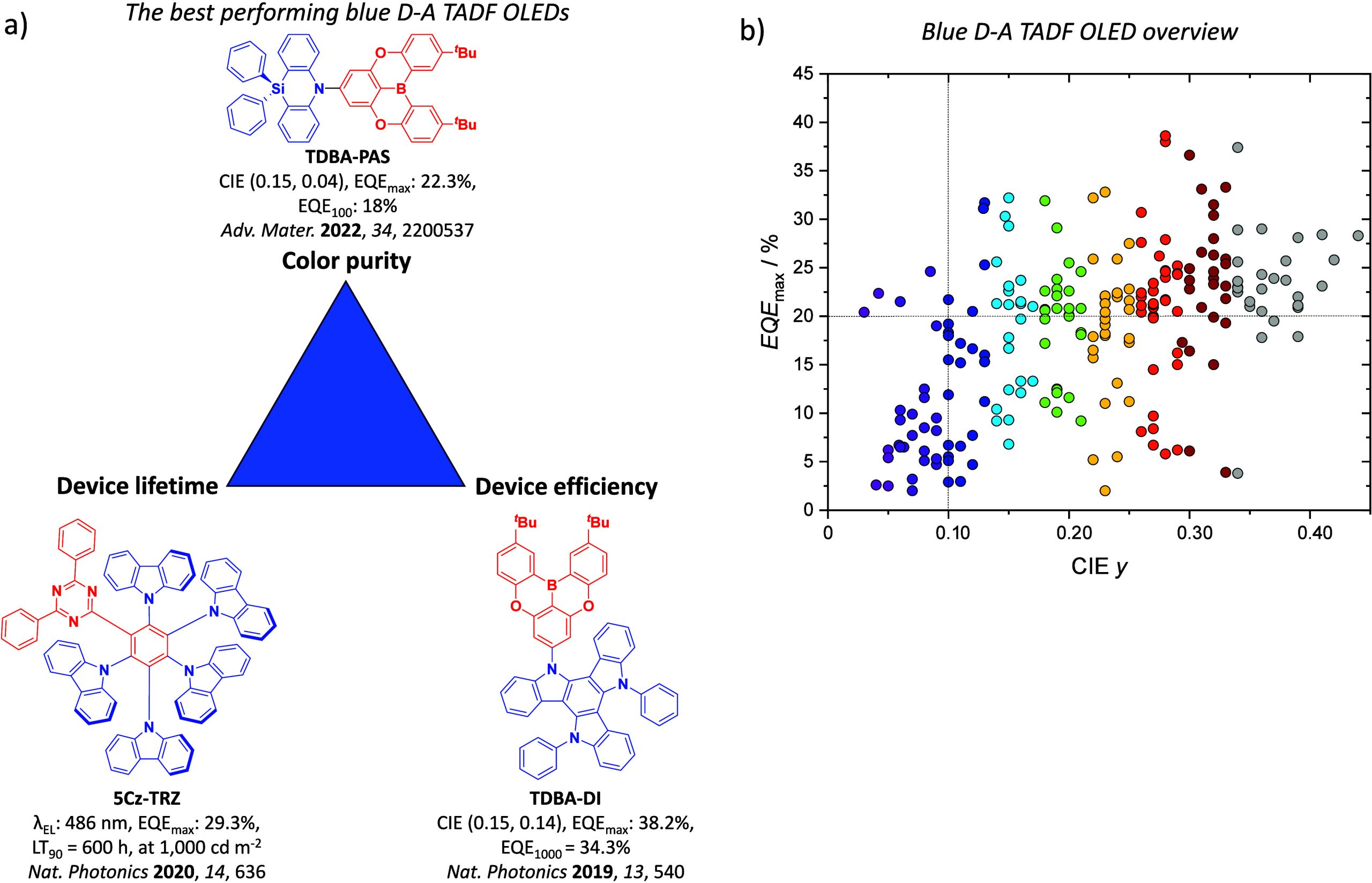
Triazine by far remains the most popular acceptor in the design of blue emitters. Deep blue emission can be achieved when triazine is combined with weak donors (carbazoles, carbolines, imidazoles), as exemplified by DCBTRZ (EQEmax of 6.6%) where the device achieves CIE coordinates of (0.15, 0.056).ref. ref243 There are a large number of highly efficient devices with reported EQEmax exceeding 30%, most notably for TspiroS-TRZ, which achieved EQEmax of 33.3%.ref. ref259 The molecular shape of TspiroS-TRZ helps to promote horizontal emitting dipole orientation, which supports this impressive device performance.
While device lifetime is clearly a central concern for commercial applications, only some studies report device lifetime information. For example, the device with PPCzTrz demonstrated LT50 of 24 h at an initial 1000 cd m–2,ref. ref253 which was later improved to an LT80 of 30.5 hours using a phenylated derivative CzTrzBp.ref. ref262 By far the longest reported blue OLED lifetime belongs to 5Cz-Trz, with a LT90 of ca. 600 h at inital 1,000 cd m–2,ref. ref93 although this device with λEL of 486 nm, actually is sky-blue.
Boron-containing D-A emitters have also emerged as a subclass with generally attractive photophysical properties for blue emitters. Examples of OLEDs based on rigid DBA emitters frequently surpass EQEmax >30%. Of these, the bluest OLED incorporated TDBA-Ac (CIE coordinates of 0.15, 0.05)ref. ref304 and the most efficient and stable OLEDs were fabricated using emitters with a rigid triazatruxene donor: TDBA-DI (EQEmax = 38.2%)ref. ref304 and DBA-DI (EQEmax = 28.1%, with efficiency roll-off of 1% at 1000 cd m–2).ref. ref305 The device with the longest lifetime was one employing DBA-DI, with LT50 of 329 h at an initial 1000 cd m–2, albeit again with sky blue emission (λEL ∼ 470 nm). As Figure depicts, two out of three the best-performing emitters summarized here feature the DBA acceptor. However, device lifetime studies remain limited and the most stable devices to date are still based on early triazine-carbazole hybrids.
Aside from boron or triazine-based acceptors, while deep blue emission is readily achieved using pyrimidine-based emitters, these OLEDs typically struggle to achieve CIEy < 0.1. Relatively stronger electron-accepting nitrile, oxadiazole, and ketone acceptors are less preferred chromophores for the design of deep-blue emitters, but feature heavily in green TADF emitter design (Section sec4 ). The same color-tuning considerations also apply to derivatives that contain multiple donors. Numerous examples of OLEDs with sulfone-containing emitters achieve CIEy < 0.1; however, these devices also typically show a significant efficiency roll-off. This may be due to suspected photochemical instability of the diphenylsulfone-type acceptor, although equally may be due to similar suspected instability of DPEPO and other phosphine oxide materials that are currently the only suitable hosts for such high T1 emitters.
Evidently, the triazine and boron acceptor-based emitters are the most promising designs for highly efficient deep-blue D-A TADF emitters with CIEy < 0.10. In particular, the devices with boron acceptor-based emitters showed excellent color purity in the deep-blue region and had the highest device efficiency but possessed poor device stability, whereas the triazine acceptor-based emitters showed excellent device stability. In analyzing and aggregating the optoelectronic and device data presented in this section there are some important trends that inform the design of efficient deep-blue D-A TADF emitters; 1) a large dihedral angle between appropriately chosen donor and acceptor is essential for spatial HOMO and LUMO separation to attain sufficiently small ΔE ST while maintaining the oscillator strength to the S1 state, which is a very challenging task; 2) a rigid molecular structure is desirable to help avoiding non-radiative decay and to maintain high ΦPL, although this is often counteracted by larger dihedral angles; 3) a large planar (e.g., diindolocarbazole) or linear difunctionalized donor (e.g., spiro-acridine) moiety seems to facilitate the horizontal alignment of the TDM in the emissive layer, leading to enhanced light outcoupling efficiencies. Thus, though much progress has been made and many highly efficient, deeply blue emissive, or highly stable emitters have been designed or discovered, the search for a single material simultaneously possessing all these traits continues as a central research focus of the global organic electronics research community.
Green TADF Emitters λEL 490–580 nm
Introduction
Green emitters, which we define as those having λEL between 490 and 580 nm, have emerged as the largest class of TADF emitters, and ones that lead to OLEDs with some of the highest reported efficiencies. The ground-breaking paper by Adachi and co-workers indeed featured a green device using 4CzIPN (Figure ), with efficiencies nearing 20% that were unprecedented for an OLED using an organic emitter.ref. ref378 Unburdened by molecular instability and restricted choice of hosts faced by blue emitters (Section sec3 ), while still being sufficiently high in energy to avoid the energy gap law that hinders the efficiency of red emitters (Section sec5 ), the reported efficiencies of green TADF devices and the number of reported green-emissive TADF compounds have steadily increased year on year.
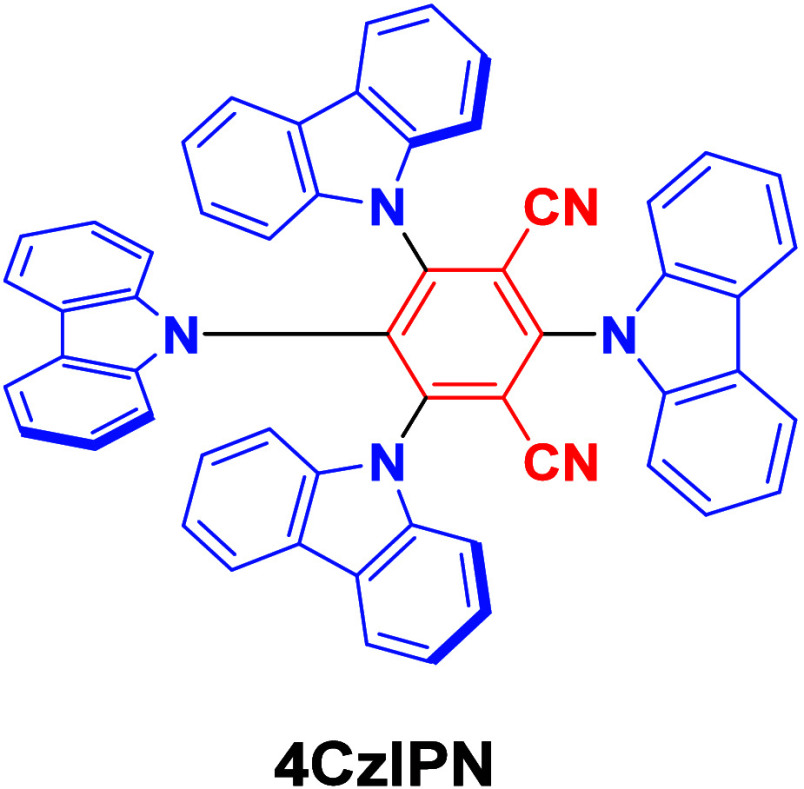
Due to the expansiveness of the green TADF emitter literature, here we restrict our scope to purely organic D-A emitters reported since 2017 where the OLED showed an EQEmax > 20% and/or exhibited notably low efficiency roll-off and high brightness. Similar to the organization in Section sec3 , the emitters in Section sec4 are classified in subsections according to the acceptors and their key photophysical and device properties are summarized in Table S2. Green TADF emitters with other molecular designs (through-space TADF (Section sec12 ), MR-TADF (Section sec11 ), and metal-based TADF (Section sec9 )) or green emitters designed with alternate applications in mind (chiral TADF, assistant dopants, and others) are summarized in other relevant sections of this review.
Nitrile-Based Acceptors
As a relatively simple and synthetically accessible withdrawing group, nitrile acceptors featured heavily in the seminal Nature paper authored by Adachi and co-workers describing TADF from a series of carbozolyl dicyanobenzene compounds.ref. ref378 Many TADF emitters have since been reported using one or more cyano groups within the acceptor moiety, with various donor groups either directly connected to the same aryl ring as the cyano group, or via a bridging aryl group. The number, type, and positions of these substituents impact both the emission energy and the efficiency of the TADF process.ref119−ref120ref121,ref379 For example, by altering the positions of additional benzonitrile substituents in phenoxazine-cyanobenzene compounds from the meta (mPTBC) to the ortho (oPTBC) position, different emission colors were observed (λPL of 518 and 561 nm, respectively, in toluene).ref. ref380 These two compounds have comparable ΦPL and ΔE ST with 58.4% and 0.006 eV for mPTBC and 57.6% and 0.007 eV for oPTBC (Figure ). This in turn resulted in similar device performance with EQEmax of 18.1% (λEL = 516 nm) and 17.8% (λEL = 540 nm) (Table S2). Both devices showed good efficiency roll-off with efficiency at 1000 cd m–2 declining by only 13 and 18% for mPTBC and oPTBC, respectively.
The same group subsequently investigated the impact of restricting molecular motions upon photophysical properties in related structures oAcTBC and mAcTBC, employing an acridine donor instead of phenoxazine (Figure ).ref. ref381 Steric restriction was increased by once again altering the positions of the auxiliary benzonitrile substituents. Replacing DMAC for a spirofluorene derivative afforded oSpTBC and mSpTBC. The emitters oAcTBC, oSpTBC, mAcTBC, and mSpTBC have ΦPL values of 84, 93, 77, and 65% respectively, doped at between 10–27 wt% in mCP. The increase in ΦPL from oAcTBC to oSpTBC was expected since the more rigid spiro-based donor suppresses k nr. A decrease in ΦPL was observed in the meta derivatives, which may be due to the larger dihedral angle between donor and acceptors. The four compounds all show small ΔE ST of between 0.01 and 0.03 eV, along with similar τd between 13.3 and 17.4 μs. EQEmax of 20.9 (λEL = 512 nm), 26.8 (λEL = 508 nm), 19.2 (λEL = 496 nm), and 18.9% (λEL = 492 nm) were obtained for the devices with oAcTBC (10 wt% in mCP), oSpTBC (16 wt% in mCP), mAcTBC (27 wt% in mCP), and mSpTBC (24 wt% in mCP), all respectively, with the device efficiencies correlating with the respective ΦPL (Table S2). Reduced efficiency roll-off at 1000 cd m–2 was observed for the device with oAcTBC (17%) compared to oSpTBC (29%), which was attributed to the higher prompt fluorescence contribution (34% compared to 25%). Efficiency roll-off of 37 and 26% was noted for the devices with mAcTBC and mSpTBC, respectively.
Wang et al. reported the emitter 4tBuCzPN (Figure ) that contains two ortho-bonded benzonitrile groups as the acceptor.ref. ref382 Developing from the simpler D-A-D structure of 2tBuCzPN, the dual-core and axially chiral 4tBuCzPN is more conformationally rigid and also adopts a more twisted structure. The ΦPL of 4tBuCzPN (74%) is much higher than in 2tBuCzPN (29%) while the td decreases from 14.1 to 4.0 μs (Table S2). The resulting OLED performance improve dramatically, with EQEmax of 5.3 and 20.8% for the devices with 2tBuCzPN and 4tBuCzPN, respectively. The chiroptical properties of 4tBuCzPN are discussed in Section sec7 .
A strategy to control the relative energies of 3CT and 3LE states was proposed by Noda et al., whereby the 3LE level was brought close to the 3CT state by the addition of a second type of donor unit.ref. ref91 The structure of the original 5CzBN emitter was altered to phenyl-substitute two of the carbazoles in 3Cz2DPhCzBN (Figure ). The compound emits at λPL of 495 nm and has a ΦPL of 80%, compared to 24% at 520 nm for 5CzBN, both in 20 wt% doped mCBP films. Additionally, 3Cz2DPhCzBN has an improved k RISC of 9.9 × 105 s–1 compared to 3.6 × 105 s–1 for the parent emitter (Table S2). The devices with 5CzBN and 3Cz2DPhCzBN showed similar EQEmax of 18.0 and 20.9%, respectively, and thanks to faster RISC the device with 3Cz2DPhCzBN showed markedly reduced efficiency roll-off (11% at 5000 cd m–2, compared to 23% for 5CzBN). Furthermore, better operational stability was demonstrated for the device with 3Cz2DPhCzBN with an LT97 of 110 hours at 1000 cd m–2, compared to just 3 hours for the device with 5CzBN.
Similar structural modification of 5CzBN was reported by Balijapalli et al., in which phenyl, pyridyl, and trifluoromethyl groups were substituted onto the carbazole donors of 5CzBN.ref. ref383 Of the family of compounds, the one with the most attractive set of emission properties was PyPhBN (Figure ), which possesses three unsubstituted carbazole donors, one carbazole extended with two phenyl units, and another decorated with two pyridine units. These modifications led to a ΦPL of 92% and a ΔE ST of 0.13 eV, which translated into a device with improved EQEmax of 20.6% at 501 nm. Woo et al. also reported a modified version of 5CzBN in compound 4mCzBN-BP, containing four dimethylcarbazole donors about a benzonitrile acceptor core as well as an ortho-biphenyl substituent para to the nitrile.ref. ref384 The ortho-biphenyl enforces a large steric hindrance and larger D-A dihedral angles between the carbazoles and the acceptor core, while the 3LE of the biphenyl group can couple with 3CT of 4mCzBN-BP to accelerate RISC compared to the parent molecule. 4mCzBN-BP emits at λPL of 491 nm, has a ΦPL 95%, and a k RISC of 2.28 × 106 s–1 in 10 wt% doped mCP films (Table S2). The device showed an EQEmax of 23.1% at λEL of 496 and CIE coordinates of (0.20, 0.45), and showed moderate efficiency roll-off of 26% at 400 cd m–2. Zhang et al. reported the emitter 5PCzCN, which has five dimethylcarbazole donors and emits at λPL of 489 nm with a high ΦPL of 96.5% and a small ΔE ST of 0.028 eV, in 10% doped mCP films.ref. ref385 The OLED with 5PCzCN showed green emission at 504 nm [CIE coordinates of (0.21, 0.49)] with an excellent EQEmax of 32.1% and efficiency roll-off of 9.3% at 1000 cd m–2. This increase in device performance compared to 5CzBN highlights the crucial importance of balancing donor strength to achieve efficient RISC, while the peripheral methyl substituents likely also help to suppress concentration quenching. The OLED also displayed high stability with LT50 of 95.5 h at 1000 cd m–2.
Unlike vacuum-deposited OLEDs, which can show enhanced light-outcoupling when the TDM of the emitters are preferentially aligned, solution-processed devices with the same emitter typically exhibit no improved light-outcoupling as the processing technique results in isotropic orientation of the TDMs. However, Zhao et al. demonstrated that by attaching flexible alkyl chains terminated with spirobifluorene groups to 5CzBN, these groups helped not only to improve the solubility for solution-processed OLEDs, aided carrier mobility, and likely assisted in preventing aggregation quenching, but crucially also supported spontaneous horizontal orientation of the emitter TDM. Measurements of Θ// gave values of 72–73% for 5CzBN, compared to 67% (i.e., isotropic alignment) for 5CzBN-Hex in solution-processed neat films.ref. ref386 5CzBN-ESF, containing the shortest alkyl chain of the series, emits at λPL of 480 nm and achieved the highest ΦPL of 80%, with a small ΔE ST of 0.06 eV and a short τd of 1.82 μs in toluene (Table S2). OLEDs with 5CzBN-ESF showed high EQEmax of 30.6% and λEL of 508 nm [CIE coordinates of (0.27, 0.55)], with efficiency roll-off of 33% at 1000 cd m–2.
Boron-Containing Acceptors
Many acceptors have been developed using the inherent electron-withdrawing ability of the lowest-lying vacant p-orbital of boron. Two emitters, ACBM and SACBM (Figure ), containing a simple N-borylated acceptor unit coupled to various acridine-based donors were reported.ref. ref387 ACBM and SACBM emit at λPL of 527 and 518 nm and have ΦPL of 76 and 99%, respectively, in 8 wt% and 4 wt% doped films in 2,6-DCzppy (Table S2). Moderate ΔE ST of 0.11 eV for both ACBM and SACBM along with short τd of 3.0 and 2.6 μs, respectively, resulted in low efficiency roll-off in the devices. The OLEDs with ACBM and SACBM showed EQEmax of 11.2 and 19.1% at CIE coordinates of (0.33, 0.56) and (0.22, 0.59), and efficiency roll-offs of 9 and 2% at 100 cd m–2, all respectively. Significant research effort has since followed in the use of boron as part of acceptor systems for D-A TADF materials.
In another report, D-A-D derivatives using the same acceptor and similar donors attached meta to the acceptor were designed. B-2DMAC (Figure , λPL of 505 nm, ΔE ST of 0.03 eV, ΦPL of 46.8%, τd of 3.4 μs, and k RISC of 0.8 × 105 s–1) gave the highest performance green device, with λEL = 507 nm, EQEmax = 19.3% [CIE coordinates of (0.25, 0.53)], and efficiency roll-off of 6% at 100 cd m–2 and 21% at 1000 cd m–2.ref. ref388 Devices with the phenothiazine (B-2PTZ) and phenoxazine (B-2PXZ) analogues emitted at λEL of 556 and 544 nm [CIE coordinates of (0.43, 0.54) and (0.40, 0.56)] with similar efficiency roll-off but lower EQEmax (7.6 and 10.1%) resulting from the lower ΦPL (18 and 26%). The same acceptor was again used in two similar emitters that have only a single donor group, either with (PXZPBM) or without a phenylene linker (PXZBM).ref. ref389 The steric hindrance between the phenoxazine and dimesitylboryl in PXZBM caused a puckered conformation of the phenoxazine in the ground state, leading to elongation of the N-B bond and reduced π-conjugation in the excited state. The introduction of the spacer in PXZPBM permitted the phenoxazine to adopt a planar conformation (itself perpendicular to the acceptor) and emit from a charge transfer state. The presence of the spacer also improved ΦPL from 36 to 80%, reduced ΔE ST from 0.13 to 0.08 eV, and a shortened τd from 3.6 to 2.2 μs, all respectively in toluene (Table S2). In turn, contrasting device performances were observed with EQEmax of 10.9% compared to 22.6%, at λEL = 567 [CIE coordinates of (0.45, 0.51)] and 505 nm [CIE coordinates of (0.25, 0.54)] for the OLEDs with PXZBM and PXZPBM respectively. Both compounds showed low efficiency roll-off with the EQE100 decreasing by either 10 or 1% compared to their respective maximum values. The EQE1000 remained as high as 20.8% for the device with PXZPBM, representing an 8% efficiency roll-off; by contrast, due to the larger ΔE ST and longer τd of PXZBM there was a significantly larger efficiency roll-off of 63% at 1000 cd m–2 for this device.
The same dimesitylborane acceptor was again used in another report by Qu et al. to produce D-A-A’ emitters PX-TRZ-B and PX-SF-B (Figure ).ref. ref390 Small ΔE ST values 0.037 and 0.013 eV for PX-TRZ-B and PX-SF-B in toluene, respectively, are achieved due to large HOMO-LUMO spatial separation, caused by the expanded LUMO distribution over the tandem acceptor. PX-SF-B has the higher ΦPL of 84% (compared to 65%) in 5 wt% doped CBP films. This translated into devices with higher EQEmax of 24.8% (10 wt% in CBP) at λEL of 535 nm [CIE coordinates of (0.37, 0.55)] for the device with PX-SF-B, compared to PX-TRZ-B with an EQEmax of 18.6% (10 wt% in CBP) at λEL of 557 nm [CIE coordinates of (0.43, 0.54)]; a small increase in EQEmax (19.2%) was observed at lower device loading (5 wt% in CBP). The efficiency roll-off for both materials was impressive with almost no loss in performance up to 1000 cd m–2, likely supported by the additional charge transporting properties of either the triazine or diphenylsulfone groups.
The addition of perfluoroalkyl (CF3 and C3F7) and perfluoroaryl (4-CF3C6F4) units to a reference boron-based emitter, CzoB (Figure ), allowed Kumar et al. to develop a series of compounds that reached ΦPL of up to 100% in toluene.ref. ref391 Amongst this family of emitters, CzCF3oB, BuCzCF3oB, and BuCzTF7oB were identified as the most promising. The CzCF3oB device showed an EQEmax of 22.9% at λEL of 517 nm [CIE coordinates of (0.24, 0.57)] and with an efficiency roll-off of 2 and 23% at 100 and 1000 cd m–2 (Table S2). The device with BuCzTF7oB showed an EQEmax of 21.9% with almost no efficiency roll-off at 100 cd m–2 and a 16% decrease at 1000 cd m–2. This device also showed the most red-shifted emission [λEL = 550 nm, CIE coordinates of (0.33, 0.60)], demonstrating the color tuning utility of these perfluorinated substituents.
A series of emitters using a dibenzo[b,e][1,4]heteraborin acceptor with an acridine donor were reported by Park et al.ref. ref297 Of these, MPAc-BS (Figure ) has the most noteworthy properties and emits at λPL of 497 nm, has ΦPL of 100%, a small ΔE ST of 0.023 eV, and a τd of 1.3 μs in 50 wt% doped films in PPF. The OLED with MPAc-BS showed an EQEmax of 25.3% at λEL of 503 nm [CIE coordinates of (0.20, 0.51)] and showed a very mild efficiency roll-off of only 1.2 and 6.3% at 100 and 1000 cd m–2, respectively. CzDBA and tBuCzDBA contain a similar diboroanthracene acceptor unit in combination carbazole donors and have similarly small ΔE ST of 0.03 and 0.02 eV in 10% doped film in CBP, along with very fast τd of 3.2 and 2.1 μs, and high ΦPL of 100 and 86%, all respectively (Table S2).ref. ref392 The devices showed EQEmax of 37.8 and 32.4% at λEL of 528 [CIE coordinates of (0.31, 0.61)] and 542 nm [CIE coordinates of (0.37, 0.60)], respectively, as well as excellent efficiency roll-off at 1000 cd m–2 of 0.3 and 3%, representing some of the highest-performance green-emissive devices to date. The high EQEmax was attributed to both the high ΦPL and the preferentially horizontally oriented TDMs arising from the rod-like molecular design.
Ouyang et al. reported a derivative of CzoB that contains an additional carbazole donor at the ortho position of triarylborane, resulting in D-A-D materials oB-2Cz and oB-2tCz (Figure ).ref. ref393 The large D-A dihedral angles enforced by the double ortho substitution and the highly rigid structure gave rise to small ΔE ST values of 0.06 and 0.03 eV, high ΦPL of 93 and 96%, and fast k RISC of 5.17 and 17.06 ×105 s–1 for oB-2Cz and oB-2tCz, all respectively (Table S2). The devices with oB-2Cz and oB-2tCz showed EQEmax of 28.1% (efficiency roll-off of 51% at 1000 cd m–2) and 27.5% (efficiency roll-off of 44% at 1000 cd m–2) at λEL of 486 [CIE coordinates of (0.18, 0.37)] and 498 nm [CIE coordinates of (0.22, 0.49)], again respectively.
A rigid and planar hybrid boron-carbonyl group was used by Lee et al. as an acceptor in TMCzBCO and DMACBCO (Figure ).ref. ref394 TMCzBCO and DMACBCO are efficient green emitters, emitting at λPL of 526 and 520 nm and with ΦPL of 94 and 93%, and have small ΔE ST of 0.007 and 0.011 eV and fast k RISC of 5.38 and 6.42 × 106 s–1, all respectively (Table S2). Devices with these two emitters showed EQEmax of 24.7 and 28.4% at λEL of 532 [CIE coordinates of (0.33, 0.59)] and 556 nm [CIE coordinates of (0.43, 0.54)], respectively. Remarkably, the efficiency remains as high as 20.3 and 21.5% respectively at 5000 cd m–2.
To explore the influence of different bulky groups on the horizontal dipole alignment in films, Wu et al. reported a new emitter iCzDBA ref. ref395 and compared it with the previously reported emitters CzDBA and tBuCzDBA ref. ref392 (Figure ). The 10 wt% iCzDBA doped film in CBP showed a small ΔE ST of 0.03 eV. Due to increased steric hindrance of the tert-butyl or isopropyl groups, aggregation induced quenching was alleviated in the films of tBuCzDBA and iCzDBA, evidenced by the high ΦPL of 84 and 88% and short delayed lifetimes of 1.2 and 1.4 μs, all respectively (Table S2). The neat film of tBuCzDBA exhibits a higher Θ// of 92% compared to iCzDBA (Θ// 77%), ascribed to the bulky groups on the terminal ends of tBuCzDBA extending the long axis of the compound. The OLEDs with tBuCzDBA and iCzDBA showed better performance with EQEmax of 26.9% [CIE coordinates of (0.44, 0.55), λEL 558 nm] and 18.7% [CIE coordinates of (0.38, 0.59), λEL 540 nm], respectively, while the device with unsubstituted CzDBA showed an EQEmax of 13.5% [CIE coordinates of (0.44, 0.55, λEL 557 nm]. In addition to this, efficiency remain high with roll-off of 1.1 and 0.5% at 1000 cd m–2 for the devices with tBuCzDBA and iCzDBA, respectively.
All boron-containing acceptors summarized so far have featured 3-coordinate boron centers. A smaller additional class of boron-containing materials also feature 4-coordinate boron. Separating donors and acceptors with a 4-coordinate boron bridge isolates the HOMO and LUMO from one another, resulting in small ΔE ST and allowing TADF to occur. Shiu et al. reported the two such compounds fppyBTPA and dfppyBTPA (Figure ), which emit, respectively, at λPL of 494 and 508 nm, and have ΦPL, ΔE ST, and τd of 72%, ≈ 0 eV, and 2.0 μs (for fppyBTPA in 8 wt% doped film in mCPCN) and 100%, ≈ 0 eV and 2.4 μs (for dfppyBTPA in 25 wt% doped film in mCPCN) (Table S2).ref. ref396 The devices with fppyBTPA and dfppyBTPA showed EQEmax of 20.2 and 26.6% at CIE coordinates of (0.27, 0.54) and (0.26, 0.58), however the OLED with fppyBTPA suffered from large efficiency roll-off of 23% at 100 cd m–2, while the device with dfppyBTPA showed a much smaller efficiency roll-off of just 5% at 100 cd m–2.
Sulfone-Containing Acceptors
Sulfones have been thoroughly explored as acceptors in D-A TADF emitter design and are almost as popular and established as cyano-based acceptors in the context of blue and green emitters (Figure ). In a recent example, a diphenylsulfone acceptor coupled to two acridine-based donor dendrons containing peripheral diphenylamines gave the emitter DDA-DP.ref. ref397 This compound emits at 549 nm as a neat film and has a ΦPL of 12.4% in toluene (Table S2). The ΦPL increases to 45% in 15 wt% doped films, in mCP and with a ΔE ST of 0.04 eV. The solution-processed OLED showed an EQEmax of 8.1% at λEL of 550 nm [CIE coordinates of (0.36, 0.56)]. Notably, the efficiency roll-off at 1000 cd m–2 was only 1%, which was attributed to the very fast τd of 0.45 μs, limiting the accumulation of triplet excitons and associated triplet quenching processes in the device.
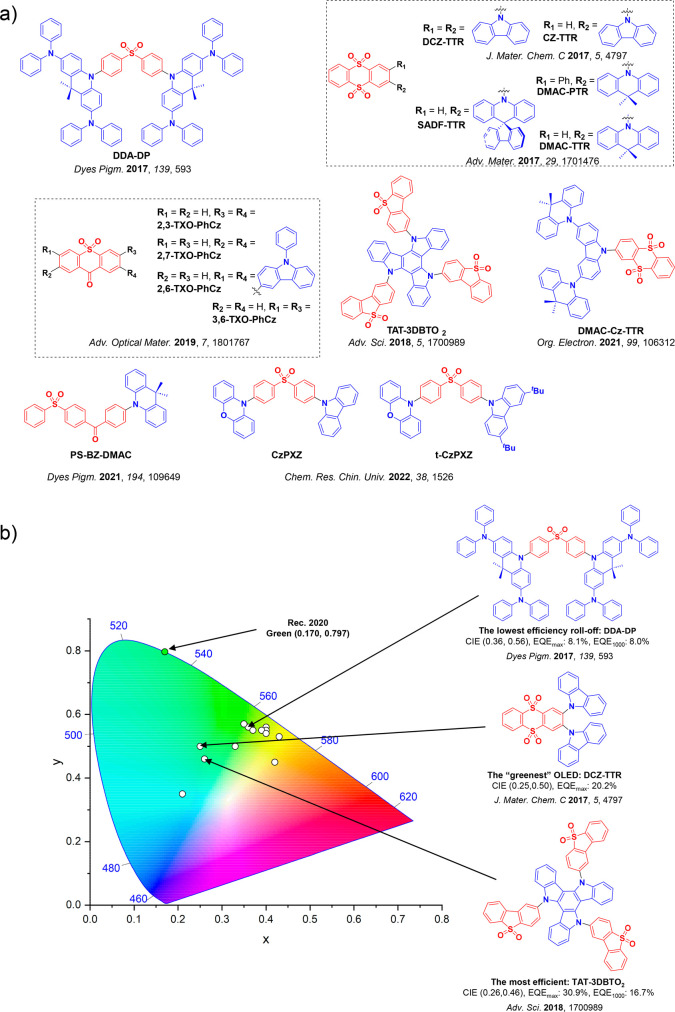
Wang et al. employed a related thianthrene tetraoxide acceptor in combination with carbazole donors in the emitters DCz-TTR and Cz-TTR (Figure ).ref. ref398 With only one carbazole, Cz-TTR emits at 487 nm with a higher ΦPL of 56.5%, although also a larger ΔE ST of 0.10 eV in 6.5 wt% doped films (Table S2). in mCP. The dicarbazole congener DCz-TTR in contrast has λPL of 502 nm, ΦPL of 47.1%, and ΔE ST of 0.03 eV. OLEDs with DCz-TTR showed an EQEmax of 20.1% at λEL of 512 nm (efficiency roll-off of 40% at 1000 cd m–2), while the device with Cz-TTR performed worse, showing an EQEmax of 14.4% (λEL of 492 nm), which decreased by ca. 83% at 1000 cd m–2). Using the same thianthrene tetraoxide acceptor three additional D-A emitters were reported using acridine donor derivatives: DMAC-TTR, DMAC-PTR, and SADF-TTR.ref. ref399 Similar to the aforementioned carbazole analogues, these compounds emit at 555, 572, and 530 nm, have small ΔE ST (0.01, 0.02, and 0.03 eV), fast τd (5.2, 3.4, and 5.2 μs) and moderate ΦPL (43, 59, and 52%) all in 10 wt% doped films in mCP. The OLEDs with DMAC-TTR, DMAC-PTR, and SADF-TTR showed EQEmax values of 13.9, 18.2, and 20.2% at CIE coordinates of (0.33, 0.50), (0.40, 0.56), and (0.35, 0.57), respectively. The device efficiency roll-off was low, reflective in the EQE100 of ∼12, ∼14, and ∼17%, respectively. The thianthrene tetraoxide acceptor was later also combined with a acridine-decorated carbazole donor dendron in the D′-D-A compound DMAC-CZ-TTR.ref. ref400 The acridines act as secondary electron donating group (D′) to fortify the donating strength of the primary carbazole donor. The doped film of 10 wt% DMAC-CZ-TTR in CBP emits at 550 nm and has a ΦPL of 69.5%, a small ΔE ST of 0.066 eV, a τd of 14.7 ms, and k RISC of 7.67×105 s–1. The solution-processed and vacuum-deposited devices showed almost the same EQEmax of 20.6% [λEL 568 nm, CIE coordinates of (0.45, 0.51)] and 21.2% [λEL 550 nm, CIE coordinates of (0.40, 0.54)], respectively.
A related acceptor was generated by replacing one of the SO2 groups in the thianthrene with a ketone.ref. ref401 Coupling this acceptor to N-phenyl carbazole donors resulted in highly efficient emitters in 5 wt% doped films in CBP: 2,3-TXO-PhCz (λPL = 540 nm; ΦPL = 62%), 2,6-TXO-PhCz (λPL = 526 nm; ΦPL = 84%), 2,7-TXO-PhCz (λPL = 530 nm; ΦPL = 89%), and 3,6-TXO-PhCz (λPL = 544 nm; ΦPL = 85%) (Figure ). The OLEDs prepared with 2,6-TXO-PhCz, 2,7-TXO-PhCz, and 3,6-TXO-PhCz showed EQEmax of 23.2, 24.4, and 18.1% respectively, although the τd of 77, 63, and 74 μs proved detrimental to device performance with efficiency roll-off at 100 cd m–2 of ∼59, ∼51, and ∼45% (Table S2). Considerably worse device performance was exhibited for the OLED with 2,3-TXO-PhCz (EQEmax of 11.9%), which was qualitatively in trend with the lower ΦPL of 62.1% and larger ΔE ST of 0.24 eV of this emitter.
Employing a triazatruxene donor coupled to three dibenzothiophene-5,5-dioxide acceptors, dos Santos et al. demonstrated that the D-A3 compound TAT-3DBTO2 (Figure ) showed very efficient TADF owing to the large density of triplet states resulting from the multiple conformers present.ref. ref92 This was reflected in the multiple fitted τd components, which were ascribed by the authors to the different conformers. An average τd of 11.7 μs and very fast k RISC of 1.5×107 s–1 was reported for the fastest delayed emission component (τ1 = 103.9 ns, supported by ΔE ST of 0.03 eV).ref. ref83 The green OLEDs showed very high EQEmax of 30.9% at CIE coordinates of (0.26, 0.46). The efficient TADF was also reflected in the efficiency roll-off, where the EQE100 was maintained at 29%, although the EQE1000 dropped to 16.5%.
Exploring asymmetric D-A-D emitters, PS-BZ-DMAC (Figure ) incorporates an unusual D-A-A’ structure with a sulfone terminal acceptor bridged to the acridine donor by a benzophenone group.ref. ref402 This compound emits at 574 nm, has a ΦPL of 76%, and a τd of 2.83 μs in 5 wt% doped films in CBP. The OLEDs with PS-BZ-DMAC emitted at λEL of 537 nm [CIE coordinates of (0.37, 0.55)] and showed an EQEmax of 20.6%. Gao et al. reported asymmetric TADF materials CzPXZ and t-CzPXZ that also showed AIE (Figure ).ref. ref403 The neat films of CzPXZ and t-CzPXZ emit at 533 and 528 nm, have τd of 3.6 and 1.4 μs, small ΔE ST of 0.03 and 0.04 eV, and ΦPL of 79 and 77% in respective neat films. Non-doped OLEDs showed EQEmax of 21.8% (λEL of 520 nm) and 17.4% (λEL of 514 nm), respectively.
Triazine-Containing Acceptors
Due to its moderate electron-withdrawing ability, triazine has become a widely used acceptor in TADF emitter design. A prototypical green-emissive compound DMAC-TRZ (Figure )ref. ref231 (λPL of 495 nm, ΦPL of 90%, ΔE ST of 0.046 eV, and τd of 1.9 μs in 8 wt% doped film in mCPCN) has since inspired many derivative molecular designs, much like how 4CzIPN has been the starting point for derivatization of benzonitrile-based TADF materials. For example, Gan et al. used a spiro-acridine-based donor coupled to triazine in the two derivatives TRZ-p-ACRSA and TRZ-m-ACRSA (Figure ).ref. ref404 Both intramolecular and through-space CT interactions were proposed to occur between the donor and acceptor, which also allowed various intermediate 3LE states to be close in energy to 1CT and mediate efficient RISC, reflected in the τd of 4.7 and 5.7 μs, respectively. The two compounds emit at around λPL of 500 nm and have ΦPL of 97 and 70% in 20 wt% doped films in DPEPO. The devices with TRZ-p-ACRSA and TRZ-m-ACRSA showed EQEmax of 28.0% [CIE coordinates of (0.19, 0.42)] and 17.7% [CIE coordinates of (0.22, 0.45)], respectively. At 100 cd m–2 the efficiency roll-off was as low as 1% for the device with TRZ-p-ACRSA and 3% for the device with TRZ-m-ACRSA, while at 1000 cd m–2, the efficiency roll-off was 21 and 24%, respectively.
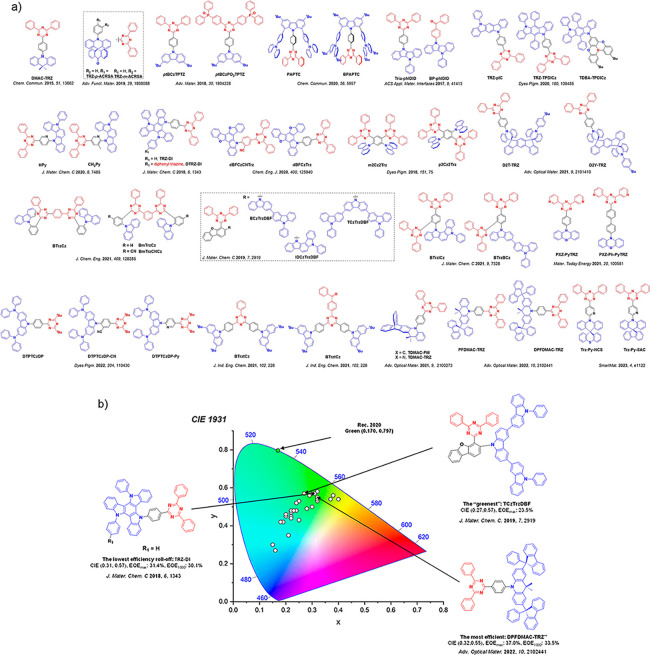
Coupling phosphine oxide auxiliary acceptors to an existing D-A structure produced a high performing green/blue emitter, ptBCzPO2TPTZ (Figure ). Compared to the control material ptBCzTPTZ (λPL of 446 nm, ΦPL of 25%, ΔE ST of 0.24 eV), this compound has a very high ΦPL of 96%, small ΔE ST of 0.01 eV, and good charge balance in 10 wt% doped film in DPEPO, supporting exciton utilization efficiency of 96%. The OLED performance was thus vastly improved, with an EQEmax of 28.9% (λEL 492 nm), increased from just 4.4% for the device with ptBCzTPTZ.ref. ref405 The efficiency roll-off of the device with ptBCzPO2TPTZ at 100 cd m–2 was only 10%, however the performance degraded considerably beyond this point with an efficiency roll-off of 43% at 1000 cd m–2.
Constraining the conformational landscape and balancing of through-space and through-bond CT interactions were used by Li et al. within the design of PAPTC and BPAPTC (Figure ).ref. ref406 The reference emitter TC consists only of triazine and tBu-carbazole that are directly coupled to have a TBCT state. PAPTC and BPAPTC instead feature suppressed rotation of the triazine acceptor about the 9-position of the tBu-carbazole donor resulting from the donor-acceptor-donor “sandwich” structure, which also enabled through-space charge transfer interactions (see Section sec12 ). PAPTC and BPAPTC have much smaller ΔE ST in toluene of 0.07 and 0.06 eV, respectively, compared to 0.28 eV for TC. These two compounds emit at λPL of 509 and 519 nm and have ΦPL of 78 and 90%. The solution-processed devices with PAPTC and BPAPTC both emitted at λEL of 520 nm, showed EQEmax of 17.4 and 24.3%, and efficiency roll-off of 17 and 7% at 1000 cd m–2, all respectively.
Ryoo et al. introduced a novel fused bicarbazole donor that was coupled with either benzophenone in BP-phIDID, or triazine in Tria-phIDID (Figure ).ref. ref407 The two compounds in toluene emit at λPL of 520 and 526 nm and have ΔE ST of 0.20 and 0.12 eV. In 6 wt% doped films in PMMA the τd are 22.8 and 18.4 μs, and in 8 wt% doped films in CBP the ΦPL are 56.9 and 69.8%, all respectively. Other acceptors were also screened using this emitter design, (benzonitrile, benzosulfone, and nitrobenzene) though none of the resulting emitters showed TADF. Devices with BP-phIDID and Tria-phIDID showed EQEmax of 13.9 and 20.8% at λEL of 497 nm [CIE coordinates of (0.25, 0.43)] and 504 nm [CIE coordinates of (0.28, 0.49)], but suffered from severe efficiency roll-off of 56 and 49% at 100 cd m–2, all respectively.
Maeng et al. investigated the impact of phenyl substitution on indolocarbazole donors in emitters TRZ-TPDICz and TDBA-TPDICz (Figure ).ref. ref408 These two emitters were compared with the reference emitter TRZ-pIC, with unsubstituted indolocarbazole. TRZ-TPDICz has nearly the same λPL and ΔE ST in toluene (λPL of 479 nm, ΔE ST of 0.26 eV) as TRZ-pIC (λPL of 478 nm, ΔE ST of 0.29 eV), and possesses the same ΦPL of 86% in 20 wt% doped films in DBFPO (Table S2). TDBA-TPDICz (λPL of 447 nm, ΔE ST of 0.41 eV in toluene) has a higher ΦPL of 96% in the same films than the reference emitter, but at the expense of a much larger ΔE ST. TDBA-TPDICz is bluer due to the weaker DOBNA-based TDBA acceptor. Phenyl-substituted TPDICz turns out to be a stronger donor than pIC evidenced by the shallower HOMO level (-5.37 eV for TRZ-TPDICz vs −5.66 eV for TRZ-pIC), while the steric impact of the modified donor helped to maintain an orthogonal conformation between the donor and acceptor segments. The device with TRZ-TPDICz showed a superior EQEmax of 30.3% at λEL of 509 nm and CIE coordinates of (0.25, 0.53). The device with the reference emitter TRZ-pIC showed an EQEmax of 26.8% [λEL of 507 nm, CIE coordinates of (0.27, 0.53)], while the device with TDBA-TPDICz showed comparatively poorer device performance of the series with EQEmax of 16.9% at λEL of 462 nm and CIE coordinates of (0.14, 0.14) due to its large ΔE ST.
Yoon et al. modified TRZ-pIC by replacing the phenylene bridge between the triazine unit and a different indolocarbazole with pyridine, or by additionally incorporating a methyl group on the pyridyl bridge in HPy and CH3Py (Figure ).ref. ref409 HPy and CH3Py both emit at around 500 nm, and the 10% doped films of these emitters in DPEPO have high ΦPL of 94% (ΔE ST of 0.22 eV, τd of 5.08 ms) and 88.7% (ΔE ST of 0.10 eV, τd of 3.42 ms), respectively (Table S2). The devices with HPy and CH3Py showed EQEmax of 23.6% [CIE coordinates of (0.22, 0.44)], and 24.6% [CIE coordinates of (0.22, 0.44)], respectively. The device with HPy showed high efficiency roll-off of 63% at 1000 cd m–2, whereas the equivalent efficiency roll-off of the device with CH3Py was smaller at only 28%.
Kim et al. explored a similar design strategy to that of TAT-3DBTO2 , ref. ref92 using a triazatruxene donor but with triazines as the acceptorref. ref410 in D-A (TRZ-DI) and D-A2 (DTRZ-DI) structures (Figure ). TRZ-DI and DTRZ-DI both emit at λPL = 521 nm, have high ΦPL of 87 and 83%, small ΔE ST of 0.02 and 0.03 eV, and τd of 1.32 and 1.47 μs, all respectively. This translated to excellent devices with EQEmax of 31.4 and 26.2% reported using TRZ-DI (λEL = 526 nm) and DTRZ-DI (λEL = 526 nm), while the efficiency roll-off at 10, 000 cd m–2 was found to be 19 and 26% respectively, representing two of the best performing green TADF devices to date. The minor difference in device performance was attributed to the differing ΦPL, although it is remarkable that different levels of acceptor functionalization occurred without any change in the emission spectrum itself. A furan modified triazatruxene donor was incorporated within emitters dBFCzCNTrz and dBFCzTrz (Figure ).ref. ref245 The insertion of a nitrile group ortho to the donor in dBFCzCNTrz (λPL of ca. 490 nm) produced a stronger acceptor leading to a red-shifted emission compared to dBFCzTrz (λPL of ca. 455 nm). dBFCzCNTrz and dBFCzTrz have high ΦPL of 80.7 and 89.4%, ΔE ST of 0.09 and 0.13 eV, and τd of 4.9 and 30.4 μs in 20 wt% doped films in DPEPO, all respectively (Table S2). The device with dBFCzCNTrz emitted at λEL of 497 nm with CIE coordinates of (0.22, 0.47), and showed an EQEmax of 27.5% which decreased to 24.3% at 1000 cd m–2. The device with dBFCzTrz emitted at λEL of 470 nm with CIE coordinates of (0.15, 0.18) and showed a lower EQEmax of 22.6% that also suffered more severe efficiency roll-off (EQE1000 of 12.3%).
A pair of isomeric emitters incorporating two triazine acceptors and two carbazoles donors, m2Cz2TRZ and p2Cz2TRZ (Figure ), emit at 465 and 502 nm, have ΔE ST of 0.09 and 0.18 eV (1 wt% doped films in PMMA) and have ΦPL of 96 and 86% with τd of 12.2 and 16.6 μs (10 wt% doped films in DPEPO), all respectively (Table S2).ref. ref270 Due to the higher ΦPL of the emitter, the device with m2Cz2TRZ showed a higher EQEmax of 18.5% (λEL = 493 nm) compared to 12.5% (λEL = 534 nm) for the device with p2Cz2TRZ, and also had lower efficiency roll-off of 13% compared to 26% at 1000 cd m–2, all respectively.
Rather than a typical phenylene linker, BCzTrzDBF, TCzTrzDBF, and IDCzTrzDBF each instead contained a benzofuran unit (Figure ).ref. ref411 The computed ground- and excited-state energies suggested little change arising from the replacement of the phenylene with a benzofuran linker. BCzTrzDBF, TCzTrzDBF, and IDCzTrzDBF have ΦPL of 82, 86, and 85% in 5 wt% doped films in mCBPTrz, respectively. Their small ΔE ST (0.06, 0.01, and 0.05 eV) and short τd (5.4, 4.4, and 2.8 μs) ensured large kRISC of 3.9, 6.0, and 8.1 × 105 s–1, all respectively (Table S2). The OLEDs with BCzTrzDBF, TCzTrzDBF, and IDCzTrzDBF showed EQEmax of 20.1% [λEL = 503 nm, CIE coordinates of (0.24, 0.52)], 23.5% [λEL = 511 nm, CIE coordinates of (0.27, 0.57)] and 12.2% [λEL = 500 nm, CIE coordinates of (0.22, 0.48)]. The devices with BCzTrzDBF, TCzTrzDBF, and IDCzTrzDBF showed efficiency roll-offs of 33, 24, and 13%, respectively, at 3000 cd m–2, which were inversely proportional to the kRISC of the emitters. The TDMs of BCzTrzDBF and TCzTrzDBF were also found to be preferentially horizontally aligned, resulting in enhanced light outcoupling in these devices.
The effect of doping concentration was studied by Liu et al. in emitters D2T-TRZ and D2Y-TRZ, possessing similar donor and acceptor subunits but contrasting molecular shapes (Figure ).ref. ref412 D2T-TRZ and D2Y-TRZ in 10 wt% doped films in DPEPO emit at 489 and 491 nm, have ΦPL of 97 and 71%, and ΔE ST of 0.10 and 0.41 eV respectively (Table S2). The small ΔE ST in D2T-TRZ translated to excellent electroluminescence properties at doping levels below 70 wt%. The highest EQEmax of 27.1% [CIE coordinates of (0.20, 0.45)] was observed for the device with an EML comprising 20 wt% D2T-TRZ in DPEPO. An equivalent device with 30 wt% D2Y-TRZ in DPEPO showed an EQEmax of 16.4% [CIE coordinates of (0.22, 0.47)]. The lower EQEmax in the device with D2Y-TRZ was correlated to both the lower ΦPL and slower kRISC of that emitter. Similar to the device with D2T-TRZ, the EQEmax of the device with D2Y-TRZ was significantly negatively impacted when the doping concentration increased beyond 40 wt%.
Dual-emissive BmTrzCz and BmTrzCNCz (Figure ) are based on the previously reported dual-emissive TADF material BTrzCz.ref413,ref414 The extended conjugation between the para-linked triazines in BTrzCz resulted in a larger ΔE ST of 0.14 eV compared to BmTrzCz (ΔE ST of 0.07 eV), where the two triazine units are meta-disposed to each other. A secondary cyano acceptor present in BmTrzCNCz further reduced the ΔE ST to 0.05 eV (Table S2). BmTrzCz and BmTrzCNCZ have comparable ΦPL of 85 and 84%, and k RISC of 4.95 and 3.70 × 105 s–1, respectively. The devices with BmTrzCz and BmTrzCNCz showed higher EQEmax of 20.3 and 21.9% compared to the OLED with the parent BTrzCz at only 13.5%. The two devices also showed low efficiency roll-off of 18.0 and 19.3% at 1000 cd m–2, respectively. Structurally related compounds BTrzICz and BTrzBCz also have two triazine acceptor units connected meta to each other through a central phenyl linker, with one donor carbazole that is connected ortho to one of the triazine acceptors and para to the other.ref. ref415 Films of BTrzICz and BTrzBCz (5 wt% in CzTrz) emit at 490 and 500 nm, have ΦPL of 97 and 92%, ΔE ST of 0.00 and 0.04 eV, and τd of 3.7 and 7.2 μs, all respectively. In line with their τd the k RISC of BTrzICz is 8.07×105 s–1, 4 times faster than BTrzBCz (k RISC of 2.12 × 105 s–1). The devices with BTrzICz and BTrzBCz showed EQEmax of 20.7 and 20.5% at CIE coordinates of (0.29, 0.56) and (0.30, 0.57), with low efficiency roll-off corresponding to EQE3000 of 20.1 and 18.7%, all respectively.
Zhang et al. replaced two of the peripheral phenyl groups of triazine with electron-withdrawing pyridine in PXZ-PyTRZ, and also obtained PXZ-Ph-PyTRZ by inserting an additional phenylene spacer between the donor and acceptor moieties (Figure ).ref. ref416 PXZ-PyTRZ and PXZ-Ph-PyTRZ emit at λPL of 572 and 538 nm, have ΔE ST of 0.01 and 0.09 eV, and ΦPL of 65 and 76% in 10 wt% doped films in CBP (Table S2). The EQEmax increased from 18.5% at λEL 560 nm and CIE coordinates of (0.44, 0.54) in the device with PXZ-PyTRZ, to 22.2% at λEL 540 nm, CIE coordinates of (0.38, 0.56) in the device with PXZ-Ph-PyTRZ. The efficiency roll-off was milder in the device with PXZ-PyTRZ (6.5% at 1000 cd m–2) than the device with PXZ-Ph-PyTRZ (18.9% at 1000 cd m–2).
BTrztCz (Figure ) contains a benzoylphenyltriazine acceptor and carbazole donors. It has a ΦPL of 70% and a k RISC of 8.70×104 s–1 in 10 wt% doped films in DPEPO (Table S2).ref. ref417 The additional benzoyl unit of BTrztCz compared to TrztCz (Figure ) strengthened the acceptor and produced longer wavelength emission (red-shifted from 466 to 496 nm) in 10 wt% doped films in DPEPO. The OLEDs with BTrztCz showed an EQEmax of 21.4% at λEL of 496 nm.
TDMAC-TRZ and TDMAC-PM (Figure ) were developed by Zhan et al. using a triptycene-fused acridine donor with triazine or pyrimidine acceptors.ref. ref418 TDMAC-TRZ and TDMAC-PM emit at λPL of 525 and 505 nm, have ΔE ST of 0.045 and 0.048 eV, ΦPL of 82 and 77%, and τd of 1.7 and 5.0 μs in 10 wt% doped films in DPEPO (Table S2). OLEDs with TDMAC-TRZ and TDMAC-PM both displayed EQEmax of 24.2% but with diverging respective efficiency roll-off of 45.5 and 78.5% at 1000 cd m–2. These devices emitted at λEL of 525 and 505 nm and CIE coordinates of (0.32, 0.54) and (0.32, 0.53), respectively. The performance of non-doped devices remained high with EQEmax of 23% at λEL 529 nm [CIE coordinates of (0.35, 0.56)] and 18% at λEL 503 nm [CIE coordinates of (0.34, 0.54)], respectively. Both non-doped devices showed severe efficiency roll-off though, of 48.2 and 48.9%, respectively at 1000 cd m–2.
Shi et al. reported three 2,4-di-tert-butyl-1,3,5-triazine based emitters, DTPTCzDP, DTPTCzDP-CN, and DTPTCzDP-Py (Figure ) where substitution of phenyl groups in the typical triphenyltriazine for tert-butyl groups effectively weakened the acceptor.ref. ref419 The π-bridge between the donors and acceptors was also varied from phenylene to pyridyl or benzonitrile to modulate the conformation of the emitter and to further tune the acceptor strength. DTPTCzDP and DTPTCzDP-Py emit at λPL of 508 and 532 nm and have similar ΦPL of 68 and 70%, ΔE ST of 0.14 and 0.08 eV, and τd of 2.78 and 1.76 μs in 7 wt% doped films in DPEPO, all respectively (Table S2). DTPTCzDP-CN instead emits at 545 nm in 7 wt% doped CBP films with a ΦPL of 62%, a somewhat smaller ΔE ST of 0.03 eV, and shorter τd of 1.47 μs. The OLEDs with each of the three emitters at 10 wt% in CBP showed greater than 16% EQEmax: the device with DTPTCzDP emitted at λEL of 510 nm with CIE coordinates of (0.30, 0.50), and had the leading EQEmax of 20.1%. The devices with DTPTCzDP-CN and DTPTCzDP-Py emitted at λEL of 548 and 538 nm with CIE coordinates of (0.40, 0.54) and (0.37, 0.54), and showed EQEmax of 17.8 and 16.9%, all respectively.
PFDMAC-TRZ and DPFDMAC-TRZ (Figure ), inspired by DMAC-TRZ but featuring spirofluorene-substituted acridine donors, were reported by Feng et al.ref. ref420 PFDMAC-TRZ and DPFDMAC-TRZ showed high ΦPL of 93 and 97%, the same ΔE ST of 0.16 eV, and short τd of 1.6 and 1.3 μs in 30 wt% doped films in DPEPO, all respectively (Table S2). These emitters also showed comparably horizontal oriented TDMs of Θ// from 78 and 81%. The device with DPFDMAC-TRZ showed a very high EQEmax of 37.0% [CIE coordinates of (0.32, 0.55)] and excellent efficiency roll-off (EQE1000 of 33.5%) at λEL of 524 nm. The device with PFDMAC-TRZ showed a similar EQEmax of 35.1% [CIE coordinates of (0.32, 0.55)], but with comparatively poorer efficiency roll-off (EQE1000 of 24.9%) at λEL of 521 nm.
In a bid to enhance SOC, Fan et al. reported the compounds Trz-Py-NCS and Trz-Py-SAC (Figure ), which contain a heavy sulfur atom within the spiro-linked acridine donor in the former and pyridine bridges in both.ref. ref421 However, the sulfur atom in Trz-Py-SAC (λPL of ca. 510 nm) imparted no significant changes in the photophysical properties compared to Trz-Py-NCS (λPL of ca. 505 nm). Remarkably, the ΦPL is 100% in the neat films of both emitters, with additional small ΔE ST values of 0.059 and 0.058 eV giving short τd of 1.2 and 1.3 μs and high k RISC of 1.8 and 1.6 × 106 s–1, all respectively (Table S2). The non-doped OLEDs with Trz-Py-NCS and Trz-Py-SAC showed EQEmax of 30.8 and 30.3% and impressive EQE1000 of 29.1 and 28.1%, also respectively (λEL = 520 and 524 nm).
Pyrimidine-Based Acceptors
Beyond triazine, other N-heterocycles have also been used as acceptors or linking groups in green-emissive TADF materials.ref. ref122 Exploiting the heavy-atom effect, Xiang et al. modified the structure of PXZPM with halogens to improve the ΦPL and shorten the τd.ref. ref422 ClPPM and BrPPM (Figure ) have higher ΦPL of 93 and 91% compared to 88% for PXZPM, shorter τd of 1.4 and 1.3 μs (2.6 ms for PXZPM), and smaller ΔE ST of 0.06 and 0.07 eV (0.08 eV for PXZPM) in 1.5 wt% doped films in CBP, all respectively (Table S2). k RISC thus improved from 2.71×105 to ∼106 s–1 for both of the halogenated analogues. The more efficient TADF in these two compounds translated into higher performing devices, with EQEmax of 25.3 and 23.6% (19.9% for the device with PXZPM). Moreover, the EQE1000 of the devices with ClPPM and BrPPM remain as high as 22.2 and 19.8%, respectively as compared to 14.2% for the device with PXZPM. To achieve improved horizontal orientation of the emitter TDM, the same group also elongated the acceptor of PXZPM in PXZPyPM and PXZTAZPM in 6.0 wt% doped films in mCPCN.ref. ref423 These emitters also have 100% ΦPL for PXZPM and PXZPyPM, and 93% for PXZTAZPM. The ΔE ST of all the materials are also very small at 0.04, 0.07 and 0.05 eV respectively. These excellent optical properties then translated into devices which showed respective EQEmax of 29.5, 33.9, and 30.1%, all at λEL of 528 nm.
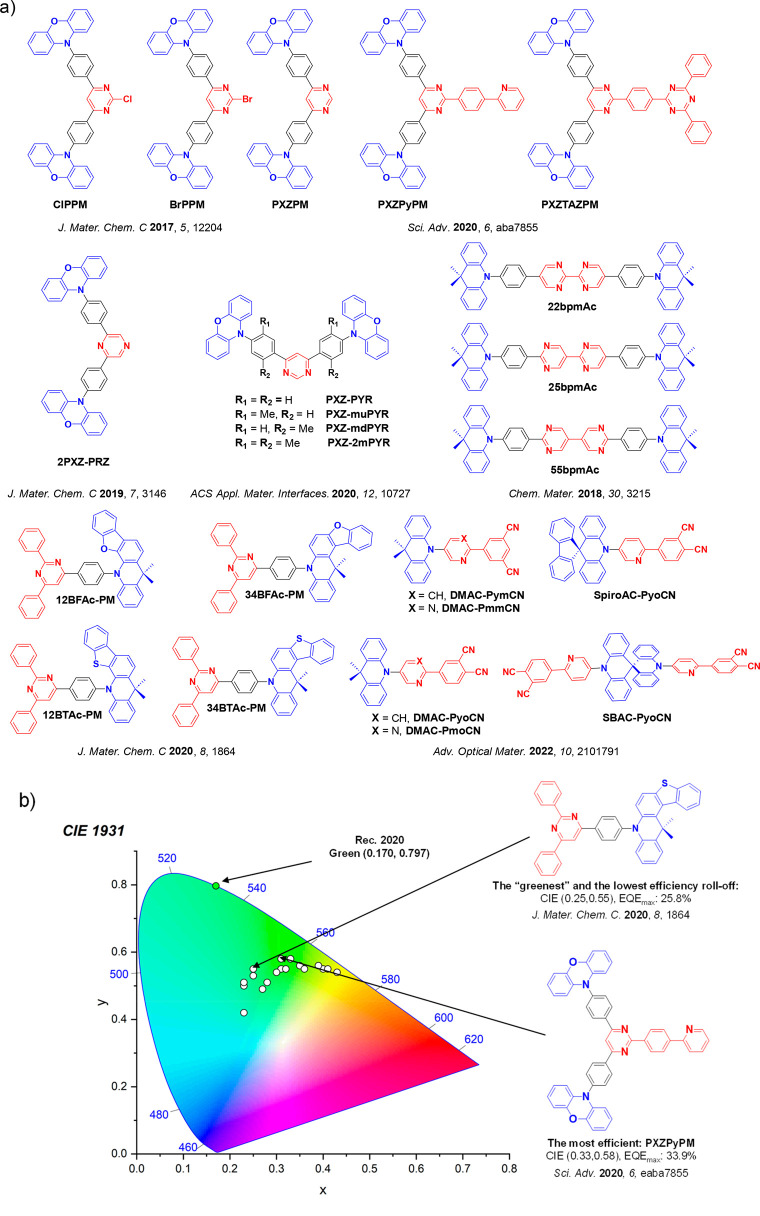
Kato et al. introduced the use of a pyrazine acceptor in an otherwise identical structure to PZXPM, producing the emitter 2PXZ-PRZ (Figure ).ref. ref424 Despite the promising ΦPL of 65%, a relatively long τd of 54 μs and quite a large ΔE ST of 0.21 eV suggested the devices would suffer from significant efficiency roll-off (Table S2). The devices showed an EQEmax of 21.4% at CIE coordinates of (0.31, 0.55), but indeed the device efficiency at 100 and 1000 cd m–2 dropped by 19 and 59%. These differences in device performance compared to the previous examples highlight how a modest structural change, in this case pyrimidine to pyrazine as the acceptor, can lead to significant differences in the optoelectronic properties and device performance.
Phenoxazine was used alongside a central pyrimidine acceptor by Serevičius et al. in the PYR series of emitters.ref. ref425 The reference material PXZ-PYR (identical to the aforementioned PXZPM) emits at λPL of 543 nm, has a ΦPL of 42%, and a τd of 1.6 μs in toluene (Table S2). The device showed an EQEmax of 27.9% at λEL of 536 nm and CIE coordinates of (0.35, 0.56). This reference structure was then modified through the addition of methyl groups at different positions relative to the donors to give PXZ-muPYR, PXZ-mdPYR, and PXZ-2dPYR (Figure ), impacting both electronic and conformational properties. The λPL of PXZ-muPYR, PXZ-mdPYR, and PXZ-2mPYR are all blue-shifted relative to PXZ-PYR (λPL of 530, 528, and 519 nm), have ΦPL of 52, 38, and 53%, τd of 4.2, 1.7, and 4.3 μs in toluene, and ΔE ST of 0.07, 0.15, and 0.13 eV in 1 wt% doped films in PMMA, all respectively. The corresponding devices showed EQEmax of 29.1, 27.5, and 26.3% at λEL of 529, 514, 502 nm with CIE coordinates of (0.32, 0.55), (0.27, 0.49), and (0.23, 0.42), all respectively.
Benzofuran and benzothiophene were fused to acridine donors in different geometries to generate four emitters, each with a pyrimidine acceptor: 12BFAc-PM, 12BTAc-PM, 34BFAc-PM, and 34BTAc-PM (Figure ).ref. ref426 The 30 wt% doped films in DPEPO of 12BFAc-PM emits at λPL of 475 nm while 12BTAc-PM, 34BFAc-PM, and 34BTAc-PM emit in the green region at λPL of 509, 519, and 521 nm. The device with 12BFAc-PM showed a relatively low EQEmax of 12.9% at 482 nm [CIE coordinates of (0.16, 0, 0.29)], due to its large ΔE ST of 0.37 eV and moderate ΦPL of 69%. The related structure 12BTAc-PM has a much smaller ΔE ST of 0.17 eV with high ΦPL of 87%, and the device with this emitter consequently performed better, emitting at λEL of 503 nm and having an EQEmax of 25.6% [CIE coordinates of (0.23, 0, 0.50)]. Compounds 34BFAc-PM and 34BTAc-PM have much smaller ΔE ST of 0.08 eV and much higher ΦPL of 95 and 92%, leading to devices with EQEmax of 27.7 and 25.8% at λEL of 503 and 509 nm [CIE coordinates of (0.25, 0.55) and (0.23, 0.51)], all respectively. The efficiency roll-off of the devices with 12BTAc-PM, 34BFAc-PM, and 34BTAc-PM were also relatively small, with EQE1000 of 22.0, 24.6, and 25.3%, respectively.
The role of intramolecular hydrogen bonding was investigated by Park et al. in a series of emitters containing bi(pyrimidine) acceptors.ref. ref427 Two compounds, 25bpmAc and 55bpmAC (Figure ), adopt a planarized acceptor conformation, while the hydrogen bonding is absent in 22bpmAc which adopts a more twisted conformation. The differing conjugation resulting from this change in conformation is manifested in ΦPL of 75, 98, and 99% for 22bpmAc (λPL of 471 nm), 25bpmAc (λPL of 472 nm), and 55bpmAc (λPL of 466 nm), all respectively in 1 wt% doped films in PS. The ΔE ST of these emitters range narrowly between 0.24 to 0.29 eV and their τd range from 17.1 to 37.5 μs (Table S2). 25bpmAc and 55bpmAc also have narrower emission spectra, with FWHM of 87 nm (25bpmAc) and 82 nm (55bpmAc), compared to 96 nm for 22bpmAc. The corresponding devices showed EQEmax of 20.5, 24.9, and 15.7% for 25bpmAc (λEL = 524 nm), 55bpmAc (λEL = 512 nm), and 22bpmAc (λEL = 517 nm), all respectively. Significant efficiency roll-off was observed at 100 cd m–2 and 1000 cd m–2 though, ranging between 24–49% and 63–83%.
DMAC-PymCN, DMAC-PmmCN, DMAC-PyoCN and DMAC-PmoCN (Figure ) feature combinations of pyridine/pyrimidine and phthalonitrile acceptors.ref. ref428 All four emitters bear highly twisted geometries, giving rise to small ΔE ST values of 0.20, 0.14, 0.13, and 0.11 eV. Among the four emitters DMAC-PyoCN has the highest ΦPL of 91% (8 wt% doped in mCPCN) and slowest k nr of 2.8 × 106 s–1, despite having a moderate ΔE ST of 0.13 eV (Table S2). The device with DMAC-PyoCN showed a EQEmax of 25.9% [CIE coordinates of (0.41, 0.55)], with the devices of the other three emitters having EQEmax no higher than 22.3%. PyoCN was therefore demonstrated to be the best choice of acceptor amongst those studied, and to optimize the emitter design the authors also employed spiro-acridine and spiro-bisacridine donors in SpiroAC-PyoCN (λPL of 518 nm) and SBAC-PyoCN (λPL of 525 nm), both of which have ΦPL of 100% in 8 wt% doped films in mCPCN. The devices with SpiroAC-PyoCN and SBAC-PyoCN showed excellent EQEmax of 33.7% [CIE coordinates of (0.31, 0.58)] and 36.1% [CIE coordinates of (0.31, 0.58)], with moderate efficiency roll-offs of 15.7 and 13.1% at 1000 cd m–2 due to an unremarkable k RISC of 8.3 and 7.7 × 104 s–1, all respectively.
Other N-Heterocycle Acceptors
Extending from small N-heterocycles like pyridine, pyrazine, and pyrimidine, larger or more elaborate π-systems have also been explored in green-emissive D-A TADF emitter design. For example, two pyridine units were fused together to create a napthylpyridine acceptor and coupled with phenoxazine or phenothiazine donors in NyDPO and NyDPt (Figure ).ref. ref429 The most interesting feature of these two linear emitters is the high degree of horizontal TDM orientation they exhibit in 5 wt% doped films in mCP, with Θ// of 81 and 84% respectively. NyDPO has a ΦPL of 79%, which combined with the preferential horizontal dipole orientation led to an EQEmax of 29.9% (Table S2). NyDPt on the other hand has a much lower ΦPL of 45% due to the presence of a non-TADF quasi-axial conformer. The impact of the two conformers could be seen from the ΔE ST measurements, where the ΔE ST NyDPO is small at 0.09 eV, while there are two ΔE ST values for NyDPT from the quasi-axial and quasi-equatorial conformers, at 0.59 eV (too large for TADF to occur) and 0.016 eV, respectively. The OLEDs with NyDPT nonetheless showed an EQEmax of 25.8%. Both materials unfortunately presented long τd which led to severe efficiency roll-off, and especially for the devices with NyDPt where the EQE1000 was only 6.5%. The same group later reported the related structure NyDPAc, composed of the same napthylpyridine acceptor but coupled to DMAC.ref. ref430 NyDPAc emits at λPL of 510 nm, has a ΦPL of 57%, τd of 451 μs, and a large ΔEST of 0.29 eV in 10 wt% doped films in DPEPO. The device with NyDPAc showed an EQEmax of 20.9% [λEL = 516 nm, CIE coordinates of (0.28, 0.53)], which was lower than those with NyDPO and NyDPt, though the preferentially horizontally oriented TDM of NyDPAc compensated somewhat for its lower ΦPL.
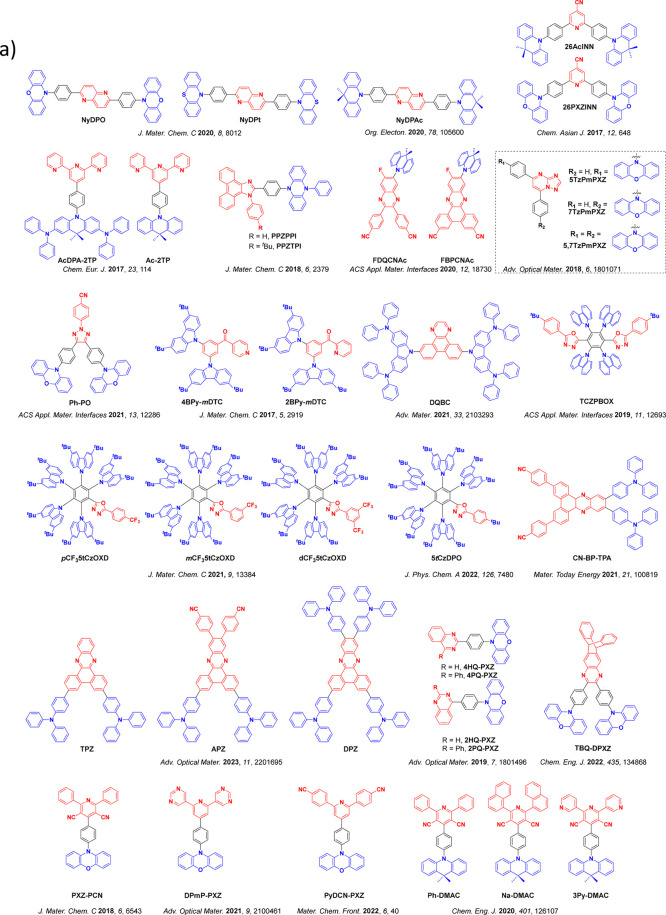
Two D-A-D emitters with a 4-cyanopyridine acceptor, 26AcINN and 26PXZINN (Figure ), were developed by Sasabe et al.ref. ref431 26AcINN and 26PXZINN emit at λPL of 495 and 522 nm, have the same ΦPL of 79%, but divergent τd of 117 and 27 μs, all respectively, in 10 wt% doped films in CBP (Table S2). The devices with 26AcINN and 26PXZINN showed EQEmax of 21.6% [λEL = 501 nm, CIE coordinates of (0.22, 0.45)] and 22.7% [λEL = 527 nm, CIE coordinates of (0.34, 0.58)]. The device with 26AcINN showed considerable efficiency roll-off, with efficiency dropping by 36% at 100 cd m–2 and by 66% at 1000 cd m–2, while the device 26PXZINN showed a much smaller efficiency roll-off of 2% at 100 cd m–2 and 26% at 1000 cd m–2. Because of the use of the stronger PXZ donor a smaller ΔE ST of 0.06 eV and faster τd of 27 μs was achieved for 26PXZINN compared to 26AcINN (ΔE ST = 0.28 eV; τd = 117 μs), which explains the starkly contrasting device efficiency roll-off behavior. The same group also reported a family of emitters containing a new terpyridine acceptor.ref. ref432 An acridine donor either with or without peripheral diphenylamine units was coupled to this terpyridine to give emitters AcDPA-2TP and Ac-2TP. By employing a donor dendron, AcDPA-2TP has a higher ΦPL of 62% compared to Ac-2TP (ΦPL of 53%) while the ΔE ST was reduced from 0.38 eV in Ac-2TP to 0.03 eV in AcDPA-2TP, consequently improving the TADF characteristics as reflected in the much shorter τd of 15 versus 319 ms. The devices with AcDPA-2TP showed a much-improved EQEmax of 23.7% and superior efficiency roll-off (EQE1000 remaining at 21.9%) compared to the devices with Ac-2TP (EQEmax of 9.2% and EQE1000 of 1.1%).
Huang et al. used the strong donor phenazine coupled to imidazole-based acceptors to produce emissive TADF compounds PPZTPI and PPZPPI (Figure ).ref. ref433 With ΦPL of 73% for PPZTPI (λPL of 527 nm) and 99% for PPZPPI (λPL of 533 nm), both emitters also showed comparable ΔE ST of 0.11 and 0.12 eV along with fairly long τd of 127 and 118 μs, all respectively (Table S2). The devices with PPZTPI (λEL of 528 nm) and PPZPPI (λEL of 528 nm) showed EQEmax of 20.5 and 21.1%, however, the long-lived excitons proved damaging to the efficiency roll-off (∼33 and ∼21% at 100 cd m–2, all respectively). Kothavale et al. also employed phenazine as the donor to give FDQCNAc (λPL of 549 nm) and also employed a fused phenanthrene to give red emitter FBPCNAc (λPL of 607 nm) (Figure ).ref. ref434 FDQCNAc and FBPCNAc have ΦPL of 87 and 79% with τd of 24.0 and 11.1 μs, respectively. Introduction of a fluorine atom in the emitter helped to achieve small ΔE ST of 0.08 and 0.04 eV in 1 wt% doped PS films at 300 K. These modifications led to highly efficient devices using 1 wt% emitter doped in PBICT, having EQEmax of 27.6% [λEL of 554 nm, CIE coordinates of (0.42, 0.55),] for the device with FDQCNAc and 23.8% [λEL of 597 nm, CIE coordinates of (0.55, 0.44)] for the device with FBPCNAc.
Singly substituted D-A systems 5TzPmPXZ (λPL = 527 nm) and 7TzPmPXZ (λPL = 532 nm, Figure ) containing an unusual [1,2,4]triazolo[1,5-a]pyrimidine (TzPm) have moderate ΦPL of 64 and 49% and small ΔE ST of 0.10 and 0.07 eV along with fast τd of 2.9 and 2.8 μs in 0.7 wt% doped films in CBP, all respectively (Table S2).ref. ref435 The corresponding D-A-D emitter, 5,7TxPmPXZ (λPL = 543 nm), showed slightly improved TADF behavior and slightly red-shifted emission, with similar ΦPL (66%), ΔE ST (0.06 eV), and τd (2.6 μs). Solution-processed devices showed EQEmax of 9.3% with both D-A emitters (λEL 542 and 552 nm for the devices with 5TzPmPXZ and 7TzPmPXZ, respectively), and 14.3% for the OLED with 5,7TxPmPXZ. Low efficiency roll-off of just 2% at 1000 cd m–2 for the device with 7TzPmPXZ and 13% for the device with 5,7TzPmPXZ were noted while the efficiency dropped by 22% for the device with 5TzPmPXZ.
1,2,4-Triazole was also recently used as an acceptor by Kim et al. in a series of six emitters.ref. ref436 The best performing OLED used the phenoxazine derivative Ph-PO (Figure ), which showed an EQEmax of 20.8% at λEL of 524 nm – this being strongly red-shifted compared to the devices with the other emitters due the use of stronger electron-donating phenoxazine. The high device EQEmax is the result of a confluence of a ΦPL of 78% (λPL of 525 nm), a small ΔE ST 0.14 eV, and a τd of 5.36 μs in 20 wt% doped films in DPEPO (Table S2), which matches the composition of the EML.
Two blue-green OLEDs with λEL at 490 nm were prepared using pyridyl-ketone acceptors coupled to tert-butylcarbazole donors in 4BPy-mDTC and 2Bpy-mDTC (Figure ).ref. ref364 The devices with 4BPy-mDTC and 2Bpy-mDTC showed EQEmax of 28.1 and 28.0% with CIE coordinates of (0.17, 0.37) and (0.16, 0.37), although the efficiency roll-off was significant at ∼58 and ∼53% at 1000 cd m–2, all respectively (Table S2). The very high efficiencies were due to the near unity ΦPL of 97 and 96%, resulting from the pyridyl nitrogen atom restricting conformational changes in the excited state, along with small ΔEST of 0.01 and 0.02 eV.
Chen et al., reported one of the highest EQEmax for green TADF OLEDs using the emitter DQBC (Figure ). DQBC emits at λPL of 551 nm, and the D-A-D structure having an extended π-conjugation helped to achieve a small ΔE ST of 0.06 eV as well as a high ΦPL of 95% with fast kRISC of 1.16 × 106 s–1 and τd of 5.5 μs (Table S2).ref. ref437 Furthermore, the TDM of DQBC is strongly horizontally oriented and this coupled with its high ΦPL explain the outstanding EQEmax of 39.1% (λEL of 534 nm), with an efficiency roll-off of 25.6% at 1000 cd m–2 when doped at 20 wt% in the mCPBC host. When increasing the concentration from 10 wt% (EQEmax 29.3%) to 30 wt% (EQEmax 32.2%), a red-shift of the emission from 528 to 538 nm was observed. The low EQE at other doping ratios was attributed to poor charge transport at low doping and severe aggregation-caused quenching at high doping.
TCZPBOX (Figure ) is an emitter composed of oxadiazole acceptors coupled with carbazole donors.ref. ref438 Only a slight decrease in ΦPL was observed moving from the 40 wt% doped PYD2 films (89%, λPL = 527 nm) to neat films (71%, λPL = 546 nm), with the red-shift suggests some aggregation in the neat film. Both doped and neat films showed TADF characteristics with ΔE ST 0.03 eV for both. A shorter biexponential decay with τd of 4 and 30 μs was reported for the 40% doped PYD2 films, which is nonetheless very similar to the τd of 4 and 26 μs of neat films. The doped and non-doped devices showed EQEmax of 27.9 and 20.2%, respectively, and the efficiency decreased by only 5% at 100 cd m–2 for both and by either 13 or 14% at 1000 cd m–2. This report therefore contained some of the first high-performance non-doped TADF OLEDs. Peripheral substituents in similar carbazole/oxadiazole-based TADF emitters was studied by Hu et al.ref. ref439 In pCF35tCzOXD, mCF35tCzOXD, and dCF35tCzOXD, tert-butyl and CF3 groups were attached to the periphery of both the carbazole and oxadiazole. The purpose of the tert-butyl group was to decrease intermolecular interactions, while electronic tuning was mediated by the electron-withdrawing CF3 groups. pCF35tCzOXD (λPL = 535 nm in CH2Cl2) and mCF35tCzOXD (λPL = 532 nm in CH2Cl2) have ΦPL of 66.7 and 66.2%, small ΔE ST values of 0.12 and 0.005 eV, τd of 2.16 and 1.90 μs, and kRISC values of 2.3 and 2.1 × 106 s–1 in 10 wt% doped films in o-CzOXD, all respectively (Table S2). Devices with pCF35tCzOXD and mCF35tCzOXD doped in 26DCzPPy showed EQEmax of 20.3% (λEL = 494 nm) and 22.1% (λEL = 494 nm). The device with dCF35tCzOXD, which has two CF3 substituents, showed an increased EQEmax of 23.3% (λEL = 496 nm) due to its superior ΦPL of 87.8% and kRISC of 4.6 × 106 s–1. Cooper et al. similarly used oxadiazole as the acceptor in the emitter 5tCzDPO, which contained five tert-butylcarbazole donors.ref. ref335 5tCzDPO emits at λPL of 496 nm in toluene while the emission is blue-shifted to 474 nm in the 12.5 wt% doped films in DPEPO. In DPEPO the ΦPL is 79%, the ΔE ST is 0.01 eV, and the τd is 6.8 μs. The OLED with 5tCzDPO emitted at around λEL of 490 nm and exhibited an EQEmax of 29.0% at CIE coordinates of (0.18, 0.36), however the efficiency roll-off was significant at 55.2% at 1000 cd m–2.
Zhou et al. used dibenzo[a,c]phenazine as an electron acceptor in CN-BP-TPA (Figure ). This compound emits at λPL of 578 nm, has a ΦPL of almost 100% in 10 wt% doped film in CBP and a moderate ΔE ST of 0.19 eV (Table S2).ref. ref440 The device showed an EQEmax of 26.0% at λEL of 580 nm and CIE coordinates of (0.51, 0.47). Strong π-π interactions between the π-conjugated phenazine acceptors in neighboring molecules had an adverse effect on the neat film ΦPL, reflected in the much lower EQEmax of the non-doped device of 5.0% and the strongly red-shifted emission at 607 nm. Liu et al. used similar acceptors with the electron donors introduced at different positions in DPZ, TPZ, and APZ.ref. ref441 These three compounds showed yellow-green emission at λPL = 539, 564, and 577 nm with ΦPL of 54, 67, and 86%, respectively in 10 wt% doped films in CBP. Due to their large ΔE ST values of 0.29, 0.34, and 0.20 eV, the emitters have long τd of 284, 240 and 298 μs, respectively. However, the horizontally oriented TDMs in the 10 wt% doped films were measured to be 84, 92 and 88%, respectively. Thus, the best-performing device in the study with APZ achieved an EQEmax of 27.5% at λEL of 562 nm and CIE coordinates of (0.44, 0.55), although the efficiency roll-off was very poor with the EQE decreasing by 86% at 1000 cd m–2.
Another nitrogen-rich acceptor quinazoline was coupled to phenoxazine to produce a series of emitters 4HQ-PXZ, 4PQ-PXZ, 2HQ-PXZ, and 2PQ-PXZ (Figure ).ref. ref442 Smaller ΔE ST of 0.10 and 0.09 eV were reported for 2HQ-PXZ and 2PQ-PXZ compared to 0.19 and 0.22 eV for 4HQ-PXZ and 4PQ-PXZ, all in 6 wt% doped films in CBP. This contrast was due to more twisted geometries adopted by 2HQ-PXZ and 2PQ-PXZ, associated with substitution of the donor at the 4-position of the quinazoline. This also resulted in faster τd of 35.9 and 28.3 μs along with improved ΦPL of 81.0 and 73.9% for 2HQ-PXZ and 2PQ-PXZ, compared to 48.3 and 40.1 μs and 66.9 and 67.5% for 4HQ-PXZ and 4PQ-PXZ, all respectively. Devices with 2HQ-PXZ and 2PQ-PXZ showed EQEmax of 16.0% [λEL = 538 nm, CIE coordinates of (0.36, 0.57)] and 17.1% [λEL = 538 nm, CIE coordinates of (0.36, 0.56)], while those with 4HQ-PXZ and 4PQ-PXZ showed higher EQEmax of 20.2% [λEL = 511 nm, CIE coordinates of (0.25, 0.54)] and 20.5% [λEL = 518 nm, CIE coordinates of (0.28, 0.57)] (Table S2). The trend in ΦPL is opposite to that of the EQEmax, which was ascribed to different populations of conformers present in the devices for the different materials, resulting from the two PXZ units (crooked and planar form). The OLEDs with 4HQ-PXZ, 4PQ-PXZ, 2HQ-PXZ, and 2PQ-PXZ all showed similar efficiency roll-off at 100 cd m–2 of 22, 37, 14, and 27%, respectively.
Ji et al. reported emitters TBP-DPXZ (Figure ) (a red emitter) and TBQ-DPXZ (Figure ) consisting of phenoxazine donors attached to either locked (triptycene-fused) dibenzophenazine or unlocked (triptycene-fused) 2,3-diphenylquinoxaline acceptors.ref. ref443 Due to the weaker electron-withdrawing ability of the unlocked acceptor and the small HOMO-LUMO overlap, TBQ-DPXZ emits at 537 nm with a ΦPL of 91%, ΔE ST of 0.07 eV, and τd of 3.6 μs in 20 wt% doped films in BCPO (Table S2). TBP-DPXZ with the locked phenanthrene acceptor emits at 586 nm, has a ΦPL of 50.3%, a ΔE ST of 0.03 eV, and a τd of 4.8 μs. The green OLED with TBQ-DPXZ showed an EQEmax of 25.1% at λEL of 533 nm and CIE coordinates of (0.35, 0.55), but the efficiency roll-off was strong with the EQE dropping by 53% at 1000 cd m–2].
Chen et al. reported green emitter PXZ-PCN that contains a PXZ donor and a dicyanopyridine acceptor (Figure ).ref. ref444 This compound emits at λPL of 565 nm and has a ΦPL of 57%, a small ΔE ST of 0.01 eV, and a τd of 1.58 μs in 10 wt% doped films in CBP. The device showed an EQEmax of 15.1% [λEL = 568 nm, CIE coordinates of (0.48, 0.51)], and efficiency roll-off of 21% at 1000 cd m–2. A similar structure with 2,6-di(pyrimidin-5-yl) pyridine as the acceptor (DPmP-PXZ) was used by Shi et al. to develop a highly efficient non-doped OLED.ref. ref445 A putative hydrogen-bonding network present in the neat films of DPmP-PXZ controls the conformation of the emitter and the orientation of the TDM, suppressing exciton annihilation and improving charge mobility in the non-doped device. The non-doped device showed an EQEmax of 21.8% (λEL = 560 nm), while a 60 wt% doped device in mCP host showed a modestly higher EQEmax of 23.6% (Table S2). PyDCN-PXZ is another emitter with a similar acceptor and a phenoxazine donor that emits at λPL of 532 nm in toluene, has a ΦPL of 89.6%, and a small ΔE ST of 0.06 eV in 15 wt% doped CBP film. The devices showed an EQEmax of 26.9% at λEL of 519 nm.ref. ref446
Liu et al. reported three emitters based on differently substituted dicyanopyridine, Ph-DMAC, Na-DMAC, and 3Py-DMAC (Figure ).ref. ref447 The 10 wt% doped films in CBP of these emit at λPL of 532, 531 and 538 nm, respectively. Ph-DMAC has a higher ΦPL of 89% (ΔE ST of 0.06 eV) compared to those of Na-DMAC and 3Py-DMAC with ΦPL of 56 (ΔE ST of 0.15 eV) and 60% (ΔE ST of 0.04 eV). The devices with the best performance employed Ph-DMAC as the emitter and showed an EQEmax of 29.1% at 539 nm, while the OLEDs with Na-DMAC and 3Py-DMAC with stronger acceptors showed red-shifted emission at 554 and 567 nm and lower EQEmax of 21.2 and 21.5%. OLEDs with the three emitters presented quite different efficiency roll-off behavior: the devices with Ph-DMAC and 3Py-DMAC showed EQE100 and EQE1000 of 21.7/18.5% and 18.9/17.3% respectively (Table S2), with the smaller efficiency roll-off of the latter correlated to its shorter τd (2.5 μs for Ph-DMAC and 1.5 μs for 3Py-DMAC) and faster RISC (kRISC of 0.96 × 106 s–1 for Ph-DMAC and 1.83 × 106 s–1 for 3Py-DMAC). By contrast, the device with Na-DMAC (τd of 0.68 μs and kRISC of 1.6 × 106 s–1) showed the most severe efficiency roll-off, with the EQE1000 dropping significantly to 8.3%. Me-DMAC is an emitter that contains a similar dimethyldicyanopyridine acceptor.ref. ref448 Its twisted geometry ensured separation of the HOMO and LUMO, leading to a ΔEST of 0.12 eV, and emitting at λPL of 542 nm in toluene. Photophysical investigations of 10 wt% doped CBP film showed a high ΦPL of 96%, a short τd of 2.7 μs, and a fast kRISC of 1.7 × 106 s–1. The OLED showed an EQEmax of 25.8% [CIE coordinates of (0.28, 0.51)], which decreased to 18.2% at 1000 cd m–2. TPAPPC, TPAmPPC, and tTPAmPPC are three additional emitters that also employ pyridine-carbonitrile acceptors.ref. ref449 Due to the presence of the 3,5-dicyano groups on the pyridine ring alongside 2,6-dimethyl groups on the linking phenyl ring these emitters adopt a strongly twisted confirmation, leading to small ΔE ST of 0.027 and 0.020 eV in TPAmPPC and tTPAmPPC, yet retaining high ΦPL of 100 and 79%, all respectively. The dihedral angle between the D/A planes of TPAPPC, which does not have the methyl groups, is much smaller (38.2°) resulting in comparatively large ΔE ST of 0.21 eV, yet a ΦPL of 100%. The OLEDs with TPAmPPC showed a record-breaking EQEmax of 39.8% at λEL of 537 nm with CIE coordinates of (0.35, 0.57). The device with TPAPPC (λEL = 520 nm) showed an EQEmax of 37.5% [CIE coordinates of (0.28, 0.56)], while the device with tTPAmPPC (λEL = 556 nm) showed a relatively lower EQEmax of 29.8% [CIE coordinates of (0.42, 0.55)] due to its lower ΦPL (79%). A non-doped device was also fabricated using TPAmPPC, showing an EQEmax of 22.2%. Xie et al. reported the emitters 3CPDA-MPC and 9CPDA-MPC, which use a similar dicyanopyridine-based acceptor linked to carbazolyl acridine donor dendrons.ref. ref450 Neat films of 3CPDA-MPC and 9CPDA-MPC emit at λPL of 522 and 509 nm, have ΦPL of 89 and 92%, ΔEST of 0.11 and 0.13 eV, and τd of 1.65 and 2.05 μs as well as showing preferential horizontal TDM orientation (Θ// of 73 and 78%) in neat films, all respectively. Due to the higher ΦPL, enhanced horizontal orientation ratio, and charge carrier mobility, the non-doped OLED with 9CPDA-MPC demonstrated better performance at 510 nm with EQEmax of 29.6%.
The compound BTPDIDCz (Figure ) containing a benzothienopyrimidine acceptor with triazatruxene as the donor moiety was reported by Lee et al.ref. ref451 It emits at λPL of 520 nm and has a ΦPL of 83%, a small ΔE ST of 0.01 eV, and a τd of 4.1 μs. The device with BTPDIDCz showed an EQEmax of 24.5% at CIE coordinates of (0.38, 0.57) and had a very low efficiency roll-off, with EQE3000 of 23.2%. The same group also coupled a 5H-benzofuro[3,2-c]carbazole donor ortho to the benzothienopyrimidine acceptor in BTPBFCz. Three derivatives of this reference emitter, BTPBFCz-D1, BTPBFCz-D2 and BTPBFCz-D3, contain one of the three secondary donors, 5H-benzofuro[3,2-c]carbazole, 12H-benzofuro[3,2-a]carbazole, or 5-phenyl-5,12-dihydroindolo[3,2-a]carbazole, all emit around 480 nm in toluene. These structural modifications result in increased ΦPL of 84, 85, and 92% in 20 wt% doped films in DPEPO, respectively, compared to 74% for BTPBFCz (Table S2).ref. ref452 While the ΔE ST of BTPBFCz is 0.09 eV, those of BTPBFCz-D1, BTPBFCz-D2, and BTPBFCz-D3 are larger at 0.10, 0.23, and 0.12 eV, respectively. The τd of all of the emitters range narrowly from 15.6 to 22.0 μs. The devices with BTPBFCz-D1, BTPBFCz-D2, BTPBFCz-D3 showed comparable EQEmax of 20.7, 20.0, and 22.7% at λEL ranging from 491 to 497 nm and with CIE coordinates of (0.18, 0.39), (0.18, 0.37), and (0.19, 0.41), all respectively. This represents an improvement of more than 40% over the EQEmax of the device with BTPBFCz [EQEmax of 15.8%, CIE coordinates of (0.20, 0.43)].
Carbonyl Containing Acceptors
Similar to sulfones, ketones and other carbonyl-based acceptors are popular in TADF materials design, with the low-lying n-π* transition of the carbonyl able to facilitate ISC/RISC.ref. ref453 For example, an imide acceptor was coupled to two carbazole donors to give two bright emitters AI-Cz and AI-TBCz (Figure ).ref. ref454 They emit at λPL of 510 and 545 nm, have ΦPL of 84 and 72%, ΔE ST of 0.09 and 0.08 eV, and rather long τd of 81 and 64 μs, all respectively (Table S2). OLEDs with AI-Cz and AI-TBCz showed EQEmax of 23.2% (λEL = 510 nm) and 21.1% (λEL = 540 nm) but showed significant efficiency roll-off (EQE100 of 15.2 and 11.5% and EQE1000 of 7 and 5.5%, all respectively).
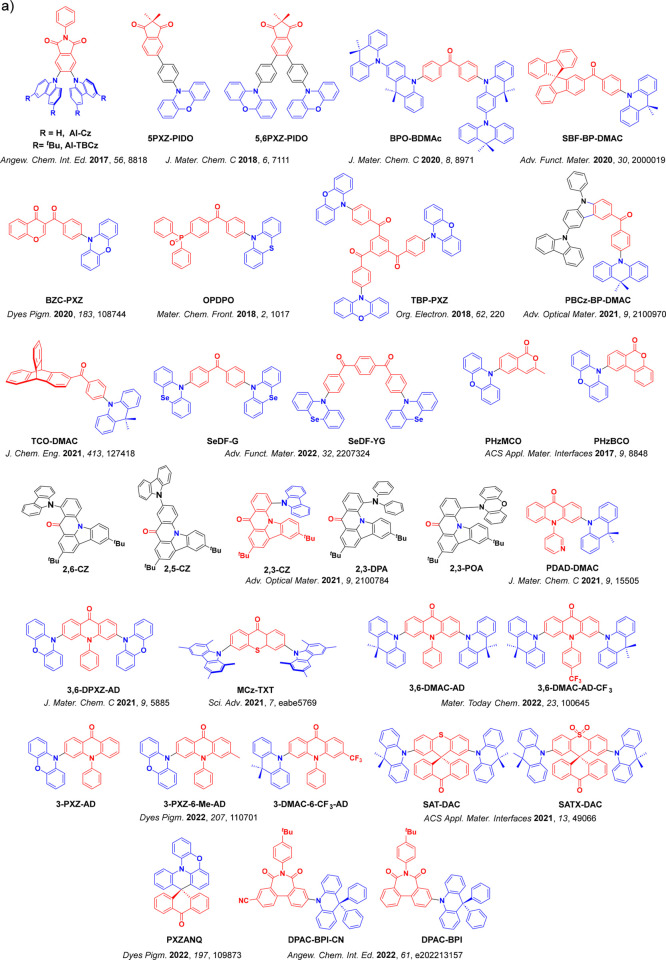
Xiang et al. introduced D-A (5PXZ-PIDO) and D-A-D (5,6PXZ-PIDO) emitters (Figure ) containing a diketone acceptor coupled to PXZ donors.ref. ref455 The two compounds emit at λPL of 535 and 544 nm and have ΦPL of 72 and 76%, respectively, in 1.5 wt% doped films in CBP (Table S2). As a result of the small ΔE ST of 0.11 and 0.06 eV and short τd of 2.37 and 1.98 ms, 5PXZ-PIDO and 5,6PXZ-PIDO have fast k RISC of 4.06 and 5.89 × 105 s–1. Although the OLEDs with 5PXZ-PIDO and 5,6PXZ-PIDO showed only modest EQEmax of 14.4% [CIE coordinates of (0.39, 0.54)] and 16.9% [CIE coordinates of (0.42, 0.53)], the fast RISC ensured relatively low efficiency roll-off of just 25 and 16% at 1000 cd m–2, all respectively.
Liu et al. combined benzophenone with the donor dendron BDMAc (9,9,9′,9′-tetramethyl-9,9′,10,10′-tetrahydro-2,10′-biacridine) to give BPO-BDMAc (Figure ).ref. ref456 BPO-BDMAc emits at λPL of 516 nm, has a ΦPL of 89.1%, a ΔE ST of 0.03 eV, and a τd of 3 μs in 25 wt% doped films in mCPCN. Solution-processed OLEDs emitted at λEL of 522 nm and showed an EQEmax of 22.5% (Table S2).
Benzophenones with ancillary functional substituents alongside the donors are a recurring theme in emitter design. A ketone-based emitter featuring an acridine donor and a spirobifluorene, SBF-BP-DMAC (Figure ), was developed by Zheng et al.ref. ref457 With a high ΦPL of 72.1% as a neat film this compound was employed in both non-doped and doped devices, giving EQEmax of 20.1 and 24.5%, respectively (Table S2). Wang et al. developed emitter BZC-PXZ which similarly features a ketone acceptor unit with a chromone moiety and a phenoxazine donor unit, which emits at λPL of 561 nm, has a small ΔE ST of 0.02 eV, and a high ΦPL of 93% in 5 wt% doped films in mCP.ref. ref458 The OLEDs with BZC-PXZ emitted at λEL of 544 nm and showed an EQEmax of 22.0% with a small efficiency roll off of 7.3% at 1000 cd m–2. In a similar manner asymmetric phosphine oxide-substituted benzophenone-based emitter OPDPO was reported by Chen et al.ref. ref459 The compound emits at λPL of 589 nm and has a small ΔE ST of 0.02 eV as a neat film. OLEDs with OPDPO doped in CBP (10 wt%) showed an EQEmax of 26.7% at λEL 552 nm, as well as a low efficiency roll-off of 18% at 1000 cd m–2. The same emitter was used to prepare a non-doped device that displayed red-shifted emission with λEL of 588 nm, a lower EQEmax of 16.6%, and a higher efficiency roll-off of 29% at 1000 cd m–2. The efficiency roll-off could be improved to 9% by increasing the emitter layer thickness from 7 to 10 nm, however the EQEmax decreased to 12.8%.
Bai et al. introduced an emitter composed of a triketone acceptor coupled to phenoxazine donors, TBP-PXZ (Figure ).ref. ref460 The 10 wt% doped CBP film emits at λPL of 592 nm and has a ΦPL of 68%, a τd of 11.9 μs, and a ΔE ST of 0.02 eV. The relatively short τd was postulated to be due to the presence of multiple conformers, some of which facilitate efficient RISC. The OLEDs with TBP-PXZ emitted at λEL of 564 nm and showed an EQEmax of 17.7% [CIE coordinates of (0.45, 0.53)], which decreased only slightly to 16.0% at 1000 cd m–2.
A self-hosting AIE-based TADF material, PBCz-BP-DMAC (Figure ), was reported by Dong et al.ref. ref461 PBCz, a common host moiety, was coupled to BP-DMAC resulting in an enhancement of the charge transporting properties of the emitter. The 10 wt% doped film of PBCz-BP-DMAC in PPF emits at λPL of 488 nm, has a high ΦPL of 92.3%, a ΔE ST of 0.02 eV, and a τd of 9.2 μs (Table S2). Both doped (10 wt% in PPF, λEL = 492 nm) and non-doped PBCz-BP-DMAC devices (λEL = 494 nm) emitted effectively and give comparable EQEmax of 27.5% [CIE coordinates of (0.21, 0.42)] and 23% [CIE coordinates of (0.21, 0.43)], respectively. Very low efficiency roll-off was also noted for both the doped and non-doped devices, falling by just 8 or 6% at 1000 cd m–2.
Jing et al. designed TCO-DMAC (Figure ), where a triptycene-fused benzophenone serves as an acceptor and is coupled to a dimethylacridine as the donor.ref. ref462 TCO-DMAC emits at λPL of 499 nm and has a high ΦPL of 92%, a small ΔE ST of 0.04 eV, and fast k RISC of 1.33 × 106 s–1 in 20 wt% doped films in BCPO (Table S2). The OLEDs showed an EQEmax of 21.2% at λEL of 499 nm and CIE coordinates of (0.23, 0.45). The efficiency roll-off was also low at 4% at 100 cd m–2 and 17% at 1000 cd m–2. The non-doped OLEDs showed a somewhat lower EQEmax of 15.6% [λEL 501 nm, CIE coordinates of (0.25, 0.48)] but had comparable efficiency roll-off of 4% at 100 cd m–2 and 13% at 1000 cd m–2.
Sharif et al. employed phenoselenazine as the donor, which was coupled to benzophenone or 1,4-phenylenebis(phenylmethanone) acceptors in SeDF-G and SeDF-YG (Figure ).ref. ref343 Due to the strong heavy atom effect of the selenium, enhanced spin-orbit couplings (Hso) of 110 and 52 cm–1 between S1 and T1 and very fast k RISC ≈ 1012 s–1 were calculated using DFT for the quasi-equatorial conformers of SeDF-G and SeDF-YG. Experimentally the ΔE ST are 0.15 eV for both compounds, the τd are 3.9 and 4.6 μs, and the k RISC are 5.7 and 10.6 × 106 s–1 in 10 wt% doped films in mCBP, all respectively (Table S2). OLEDs with SeDF-G and SeDF-YG showed respective EQEmax of 30.8 and 18.8% at CIE coordinates of (0.31, 0.53) and (0.33, 0.48). However, very low ΦPL of 7.6 and 8.5% were measured in the corresponding solution-processed 10 wt% doped films in mCBP, which was suggested to arise from the evaporated films having a different distribution of axial/equatorial conformers compared to solution-processed films. The higher ΦPL and narrower emitting conformer was postulated to be dominant in the evaporated films relevant to the OLEDs.
Two emitters with phenoxazine coupled to coumarin-based acceptors, PHzMCO and PHzBCO, were reported by Chen et al. (Figure ).ref. ref463 Similar emission properties with λPL = 510 and 524 nm, ΦPL = 47 and 52%, and τd = 17.9 and 9.3 μs were observed, all respectively, in 8 wt% doped films in mCP. The very small ΔE ST of 0.018 eV for PHzMCO and 0.006 eV for PHzBCO ensured efficient RISC, and OLEDs with PHzMCO and PHzBCO showed relatively high EQEmax of 17.8% [CIE coordinates of (0.26, 0.50)] and 19.6% [CIE coordinates of (0.32, 0.50)] – surprisingly high considering the low ΦPL of the emitters. The OLEDs also exhibited low efficiency roll-off, with EQE1000 of 15.3 and 17% and EQE10000 of 10.3 and 12.9%, respectively.
A series of five emitters 2,6-CZ, 2,5-CZ, 2,3-CZ, 2,3-DPA, and 2,3-POA contained the same fused carbonyl-carbazole acceptor coupled with different donors featuring differing regiochemistry (Figure ).ref. ref464 Only 2,3-POA showed CT emission due to the use of the strongly electron-donating phenoxazine, with the others being classified as MR-TADF (See Section sec11 ). 2,3-POA emits at λPL of 547 nm (toluene) and has a ΦPL of 82.5%, a ΔE ST of 0.01 eV, and a τd of 6.2 ms in 3.5 wt% doped films in mCBP. The OLEDs with 2,3-POA showed an EQEmax of 21.7% at λEL of 528 nm, with CIE coordinates of (0.30, 0.62).
PDAD-DMAC (Figure ) is an AIE TADF emitter with pyridine-substituted acridone acceptor and an acridine donor.ref. ref465 The 20 wt% doped PPF film of this emitter has a high ΦPL of 94%, a small ΔE ST of 0.029 eV, and short τd of 4.8 μs at λPL of 502 nm. The device showed an EQEmax of 24.1% at a λEL of 492 nm. Using very similar acceptor but with two PXZ donors, Mei et al. reported 3,6-DPXZ-AD, which emits at λPL of 563 nm in toluene. In 7 wt% doped films in CBP, the emitter has a high ΦPL of 94.9% and a k RISC of 1.1 ×106 s–1, arising from the quasi-equatorial conformation of the molecule.ref. ref466 The OLEDs with 3,6-DPXZ-AD emitted at λEL of 552 nm, and showed a high EQEmax of 30.6% and low efficiency roll-off (6% at 100 cd m–2 and 27% at 1000 cd m–2). Mei et al. also incorporated methyl or trifluoromethyl groups at the 6-position of the acridone to tune the energy levels of the 1CT, 3CT, and 3LE states.ref. ref467 3-DMAC-6-CF3-AD, 3-PXZ-AD, and 3-PXZ-6-Me-AD have ΔE ST of near 0 eV in 2-MeTHF, leading to short τd of 3.5 μs for 3-DMAC-6-CF3-AD in 7 wt% doped films in DPEPO, and 2.2 and 2.3 μs for 3-PXZ-AD and 3-PXZ-6-Me-AD in 7 wt% doped films in CBP, respectively. 3-DMAC-6-CF3-AD, 3-PXZ-AD, and 3-PXZ-6-Me-AD emit at λPL of 514, 555, and 533 nm and have high ΦPL of 85% (7 wt% 3-DMAC-6-CF3-AD in DPEPO), 86%, and 91% (7 wt% in CBP) in the doped films, all respectively. The OLEDs with 3-DMAC-6-CF3-AD, 3-PXZ-AD, and 3-PXZ-6-Me-AD emitted at λEL of 512, 519, and 505 nm with CIE coordinates of (0.27, 0.55), (0.32, 0.57), and (0.27, 0.53), and showed comparable EQEmax of 21.7, 21.1, and 23.3%, all respectively (Table S2). The same research group also reported analogous D-A-D emitters 3,6-DMAC-AD and 3,6-DMAC-AD-CF3 , each containing two DMAC donors.ref. ref468 Almost isoenergetic 3LE-3CT states and small ΔE ST (3LE-1CT and 3CT-1CT) in 2-MeTHF resulted in strong SOC, short τd of 3.4 and 2.2 μs, and fast RISC (k RISC = 2.6 and 4.2 × 106 s–1), along with ΦPL of 81.1 and 74.4%, all respectively in 7 wt% doped films in DPEPO. OLEDs with 3,6-DMAC-AD and 3,6-DMAC-AD-CF3 showed EQEmax of 23.2 and 21.6% and had low efficiency roll-off of 20 and 5% at 1000 cd m–2.
Aizawa et al. employed a thioxanthone acceptor in the emitter MCz-TXT (Figure ).ref. ref183 The 10 wt% doped film in mCBP emits at λPL of 490 nm and has a high ΦPL of 92%. The sulfur atom serves to enhance the SOC between S1 and T2, thus accelerating RISC which is reflected in the very short τd of 750 ns and outstanding k RISC of 1.1×108 s–1. The OLED with MCz-TXT showed an EQEmax of 25.8% and excellent efficiency roll-off (5% at 100 cd m–2 and 16% at 1000 cd m–2) (Table S2). Inspired by this field-leading RISC rate, other groups have since studied the use of heavy-atoms in similar acceptors, extending even to polonium derivatives, albeit only computationally.ref. ref469
Wang et al. designed two TADF emitters containing spiro-linked dual acceptors, SAT-DAC and SATX-DAC (Figure ).ref. ref470 These two compounds emit at λPL of 510 and 517 nm, have ΔE ST of 0 and 0.05 eV as neat films, and have ΦPL of 76.8 and 68.1% in 30 wt% doped films in DPEPO, all respectively. OLEDs with SAT-DAC and SATX-DAC emitted at λEL of 520 and 524 nm, and showed EQEmax of 22.6 and 20.9%. The spiro-D-σ-A architecture was proposed to enhance through-space charge transfer and reduce efficiency roll-off (21 and 19% at 1000 cd m–2 , respectively). The added bulk of the spiro design in this emitter design also likely helped alleviate concentration quenching. A similarly spiro-linked TADF emitter with anthracenone acceptor, PXZANQ, was reported by Yang et al.ref. ref471 The rigidly orthogonal arrangement between the donor and acceptor fragments led to a ΦPL of 71%, a small ΔE ST of 0.03 eV, and a τd of 10.2 μs in 10 wt% doped films in DPEPO (Table S2), similar to the previously studied ACRSA.ref453,ref472−ref473ref474ref475 The OLED with PXZANQ emitted at 528 nm with CIE coordinates of (0.33, 0.54), and showed an EQEmax of 22.1% – much higher than the ∼16% EQEmax previously reported for the device with ACRSA.
Huang et al. used an electron-deficient heptagonal diimide to conformationally lock a biphenyl-based acceptor in emitters DPAC-BPI-CN and DPAC-BPI (Figure ).ref. ref476 The stronger electron-withdrawing ability of the cyano-substituted acceptor endowed DPAC-BPI-CN with a smaller ΔE ST of 0.07 eV in toluene (0.15 eV for DPAC-BPI) and a red-shifted emission at 525 nm in neat film (472 nm for DPAC-BPI). The modestly flexible heptagonal geometry suppressed intermolecular interactions, reflected in the high ΦPL of 90.1% of the neat film. The DPAC-BPI-CN neat films also showed a high Θ// of 83%, resulting in device EQEmax of 26.2% at 531 nm.
Other Acceptors
While trifluoromethyl groups have been used as auxiliary electron-withdrawing groupsref. ref477 and investigated as acceptors computationally,ref. ref478 one of the few experimental examples using it directly as an acceptor group is 7CzFDCF3DPh (Figure ).ref. ref479 This emitter is composed of two phenyl rings; one substituted by four carbazoles and a trifluoromethyl group para to the other phenyl, and the other decorated with three carbazoles, one trifluoromethyl at the para position, and a fluorine at the ortho position. This design ensured a strongly twisted conformation between the two halves of the emitter, resulting in a small ΔE ST of 0.05 eV. 7CzFDCF3DPh emits at 555 nm, has a ΦPL of 55%, and a k RISC of 9.5 × 105 s–1 as a neat film. The corresponding OLEDs emitted at CIE coordinates of (0.36, 0.56) and showed an EQEmax of 20.8%, with mild efficiency roll-off (EQE100 and EQE1000 of 18.5 and 16.8% respectively).
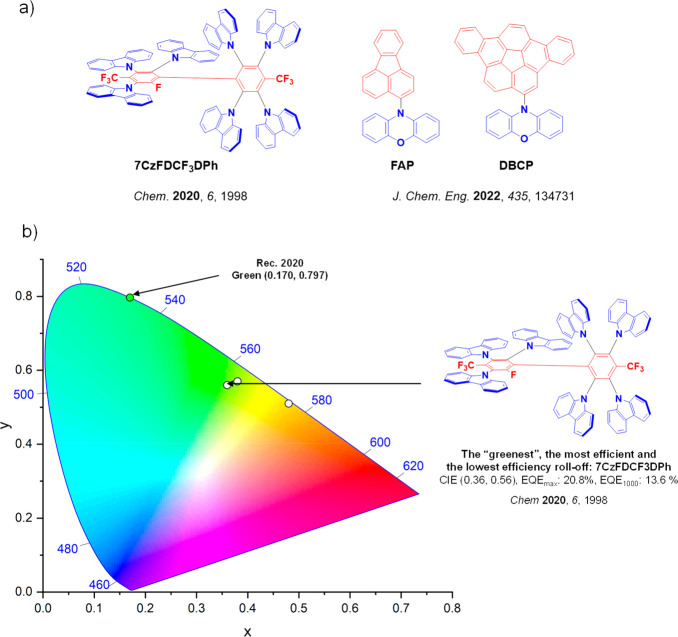
Chen et al. reported the emitters DBCP and FAP that used heteroatom-free polyaromatic hydrocarbons as acceptors (Figure ).ref. ref480 The planar geometry of the fluoranthene showed more π-delocalization than the bowl-like dibenzocorannulene, which lowered the energy of the lowest-lying 3LE state in FAP (5% doped films in mCP) and thus increased the ΔE ST to 0.32 eV compared to 0.10 eV for DBCP. DBCP has a higher ΦPL of 89% and much shorter τd of 30 μs compared to FAP (ΦPL of 54% and τd of 489.4 μs). The OLEDs with DBCP and FAP showed EQEmax of 20.2 and 12.8% at λEL of 544 and 568 nm, respectively.
Outlook
This section provides a detailed overview of green-emitting TADF materials. Numerous green TADF emitters have been reported between 2017 and 2022. These emitters have icorporated a range of acceptor types, including nitrile, boron, sulfone, N-heterocycles, and carbonyl-based acceptors, that permit the fine tuning, and even the enhancement, of the performance of green D-A TADF emitters. This substantial progress has led to the achievement of remarkable efficiencies in green TADF OLEDs, with EQEmax values now routinely exceeding 30%. This level of efficiency was unattainable at the beginning of this period.
By leveraging a preferential horizontal TDM alignment of the emitter, the performance of numerous devices has stood out by achieving EQEmax values approaching 40% in single-stack configurations. Among these, the OLED featuring TPAmPPC, an emitter containing a pyridine-carbonitrile as the acceptor, reached the pinnacle with an EQEmax of 39.8% and CIE coordinates of (0.35, 0.57). CzDBA, with diboroanthracene as the acceptor, is another exemplar emitter used in high-efficiency green-emitting devices. This OLED not only showed a remarkable EQEmax of 38%, but demonstrated a negligible efficiency roll-off of 0.3% at 1000 cd m–2 at CIE coordinates of (0.31, 0.61), in close proximity to the BT.709 green coordinates of (0.300, 0.600).
However, it remains a challenge for D-A TADF devices to meet the demanding Rec. 2020 green coordinates of (0.170, 0.797) due to their generally too broad and unstructured emission spectra that is a consequence of the CT nature of the excited state and inherent conformational flexibility of the emitters. Narrowband MR-TADF emitters (see Section sec11 ) hold promise as candidates to address this design flaw. Furthermore, most reported green-emitting D-A and MR-TADF OLEDs still suffer from a too severe efficiency roll-off. Therefore, ongoing efforts are thus still necessary towards simultaneously reducing the efficiency roll-off while maintaining high device efficiencies, which will eventually pave the way for highly stable and efficient green-emitting OLEDs.
Emitters with smaller ΔE ST and strong SOC are indispensable for facilitating a rapid RISC rate needed to alleviate TTA and STA processes that occur in the device. New OLED fabrication strategies are also promising. For example, hyperfluorescence OLEDs (see Section sec17 ) decouple exciton harvesting from emission by employing separate materials. Rapid FRET from the assistant dopant to the terminal emitter in the device effectively reduces the triplet exciton population and thus minimizes the chance of multiexcitonic quenching. Beyond advancements in the design of the green emitters themselves, we also suggest that much of these future gains will be achieved through the development of new transporting materials and host materials for better charge balance and optimal pairing with TADF emitter.
Red and NIR TADF Emitters λEL > 580 nm
Compared to blue (Section sec3 ) and green (Section sec4 ) emitters, red emitters represent an underdeveloped area of TADF research owing to fundamental difficulties in engineering high ΦPL in the red color region. This is primarily a consequence of the energy gap law, which states that as the energy gap (Eg) decreases between the excited and ground states, the density of vibronic states in both the ground and excited states will increase.ref. ref481 Such an increased density of states (and smaller energy gaps between the S1 and S0 sublevels) leads to increased internal conversion rates for S1 to S0, and accelerated non-radiative decay. Furthermore, the rate of radiative decay is proportional to the cube of the frequency of the transition, with a decreased S1–S0 energy gap therefore leading to a decrease in k r. Thus it becomes fundamentally more difficult to engineer high ΦPL in materials that emit at longer wavelengths, and particularly so for deep red (DR) and near-infrared (NIR) emitting materials. As a separate but additional factor, the low-energy S1 states associated with red emission typically require significantly expanded π-conjugation systems, making π–π stacking interactions more likely and resulting in significant aggregation-caused quenching (ACQ) for red emitters.ref482,ref483 As a result, efforts to design efficient red TADF materials (and red emitters in general) have not progressed as rapidly as for blue and green counterparts.
Following the design rules discussed in previous sections, red TADF emitters typically incorporate strong electron donors (D) and acceptors (A) linked in a strongly twisted D-A geometry. This choice of molecular fragments affords a shallow HOMO for D and a deep LUMO for A, which together induces a narrow bandgap and therefore a low 1CT emission energy.ref. ref484 Examples of chemical structures used as acceptors sorted by their acceptor strength (informed by experimentally inferred LUMO energies) and by the extent of π-conjugation are shown in Figure . To help suppress non-radiative decay pathways, rigid and/or planar fused donors or acceptors are favored, resulting in simultaneously higher ΦPL and a narrowing of the emission spectrum.ref. ref485 This can in turn increase the ΦPL of these emitters and ultimately device EQEmax, reaching above 30% for some vacuum-deposited OLEDs despite intrinsic challenges for this color.ref. ref486 However, increased the planarity of the emitter often increases the likelihood of π–π stacking, worsening aggregation and potentially leading to increased ACQ. Therefore, rationally controlling molecular packing with appropriate intra- and inter-molecular interactions is important for the control of TADF-activity, ΦPL, and effective carrier transport.ref. ref487
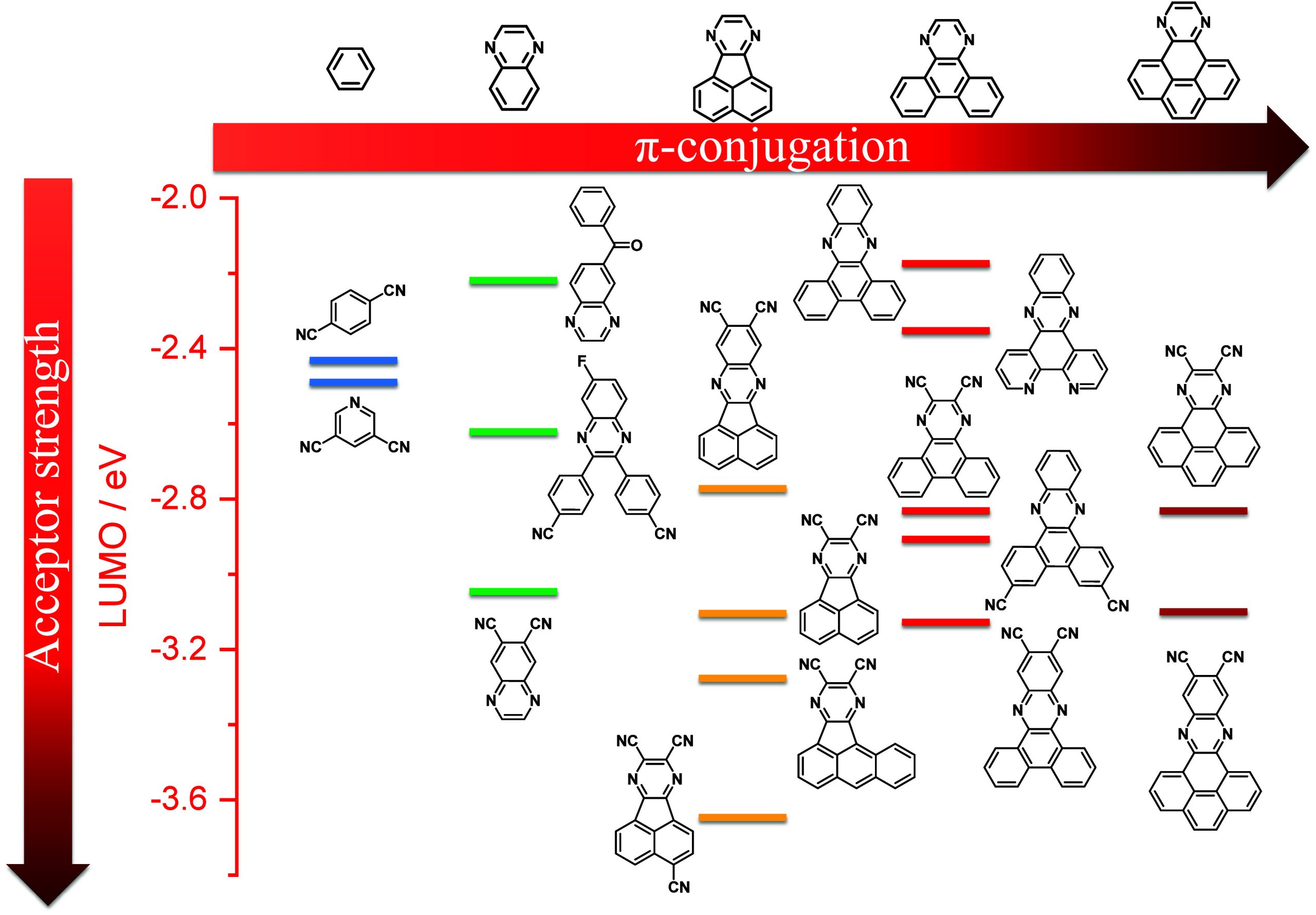
Here we discuss some of the best red and NIR emitters reported between 2017–2022, and summarize their reported photophysical and device performances in Table S3. Despite fewer reports than for the other colors, substantial progress has been made to increase the red OLED efficiencies.ref488,ref489 Red OLEDs using MR-TADF emitters can also reach very high EQEmax and are covered in Section sec11 . For the purpose of this review, we focus only on red/NIR emitters where the device emits at λEL > 580 nm and/or has an EQEmax greater than 9%.
Pyridine-3,5-dicarbonitrile Acceptors
Prior to the timeline of this review, the first red TADF emitter, 1,4-dicyano-2,3,5,6-tetrakis(3,6-diphenylcarbazol-9-yl)benzene (4CzTPN-Ph), was reported by Adachi and co-workers group in 2012 (Figure ).ref. ref31 Composed of a strongly electron-deficient terphthalonitrile acceptor unit and four carbazole derivative donors, 4CzTPN-Ph has a small τd of 1.1 μs and a ΦPL of 26.3% in toluene, emitting at λPL = 577 nm. The device showed an EQEmax of 11.2% at λEL ≈ 580 nm, corresponding to CIE coordinates of (0.52, 0.45) and had efficiency roll-off of ∼20% at 100 cd m–2 and ∼70% at 1000 cd m–2.
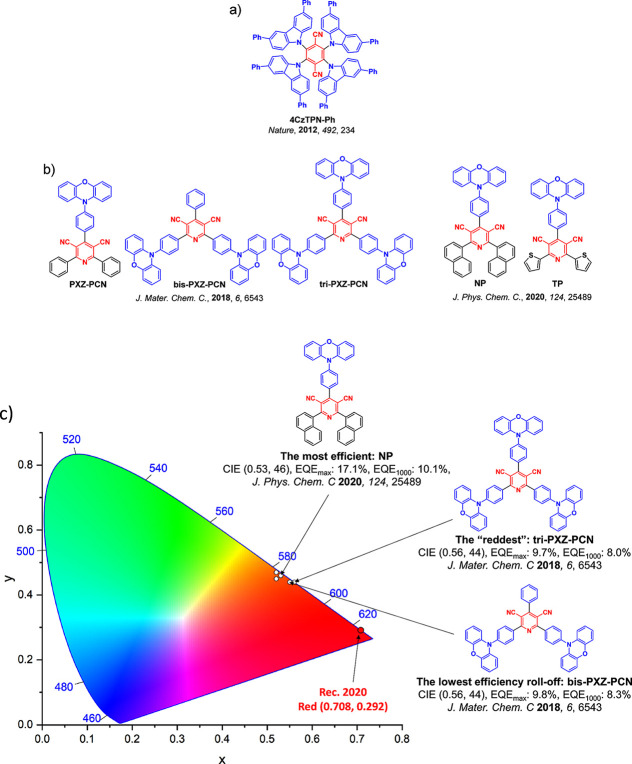
In 2018, Chen et al. reported three emitters PXZ-PCN, bis-PXZ-PCN, and tri-PXZ-PCN (Figure ) that contain one to three PXZ donors with a pyridine-3,5-dicarbonitrile (PCN) acceptor.ref. ref444 The emission of bis– and tri-PXZ-PCN peaks narrowly between λPL of 601 and 606 nm in 10 wt% doped CBP films while PXZ-PCN emits at 565 nm. Bis-PXZ-PCN and tri-PXZ-PCN have low ΦPL of 36 and 34%, yet short τd of 1.40 and 1.48 μs and fast k RISC of 9.8 and 8.8 × 105 s–1, all respectively (Table S3). Whilst the devices with bis-PXZ-PCN and tri-PXZ-PCN showed EQEmax of only 9.8 (λEL = 600 nm) and 9.7% (λEL = 608 nm), their EQE1000 remained at 8.3 and 8.0%, representing a low efficiency roll-off. Liu et al. subsequently reported red-emitters NP and TP (Figure ), with the same PCN acceptor substituted with either naphthyl or thienyl donor groups.ref. ref490 NP and TP emit at 622 and 619 nm in toluene, and at 560 and 555 nm in 10 wt% doped CBP films. They have ΦPL of 50 and 40% and ΔE ST of 0.14 and 0.15 eV, and very short τd of 0.65 and 0.80 μs in the same films, all respectively. The devices with TP showed an EQEmax of 12.4% with λEL at 591 nm while NP showed an EQEmax of 17.1% with λEL at 590 nm.
Quinoxaline Acceptors
Quinoxalines are another example of strong electron-acceptor that have been used in red TADF emitter design. Li et al. reported an asymmetric D-A emitter, TPA-QCN (Figure )ref. ref491 for which varying the concentration in doped TPA-QCN:TPBi films from 15 to 30 wt% shifted the λPL from 649 to 700 nm, with the neat film emitting at λPL = 733 nm. The ΦPL remained high in the doped films (47–70%), although dropped considerably in the neat film (ΦPL = 21%). TPA-QCN has a rather large ΔE ST of 0.23 eV in toluene at 77 K and a long τd of 943 μs. The OLEDs showed an EQEmax of 14.5% at 644 nm (15 wt% doped in TPBi), however this was accompanied by a large efficiency roll-off of ∼72% at 100 cd m–2 (Table S3). A much lower EQEmax of 3.9% was obtained for a non-doped device with NIR emission (λEL = 728 nm).
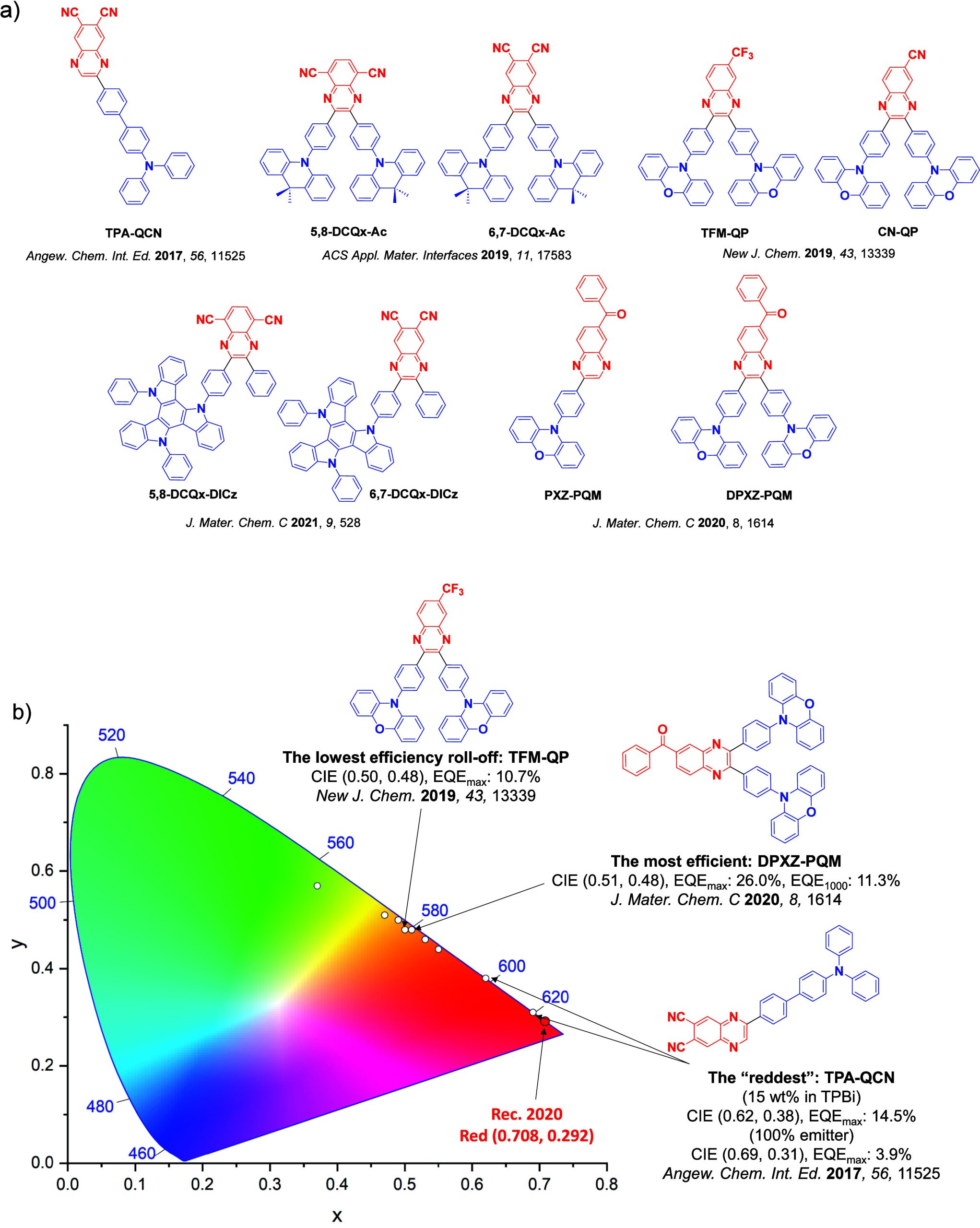
Using a bis-cyano substituted quinoxaline substituted with two DMAC donors, Kothavale et al. reported the emitters 5,8-DCQx-Ac and 6,7-DCQx-Ac (Figure ).ref. ref492 5,8-DCQx-Ac displayed a much deeper LUMO, leading to a red-shift in emission from 620 nm (6,7-DCQx-Ac) to 663 nm (5,8-DCQx-Ac) in toluene. 5,8-DCQx-Ac has a moderate ΦPL of 72%, a ΔE ST of 0.11 eV, and short τd of 3.12 μs (Table S3). The OLEDs with this emitter doped in bipolar host (1 wt% in mCP-PFP) showed an EQEmax of 16.4% at λEL of 602 nm and CIE coordinates of (0.55, 0.44). Despite the 6,7-DCQx-Ac device showing an EQEmax of 21.1%, the λEL was 578 nm. In a subsequent paper by the same group, analogous emitters 6,7-DCQx-DICz and 5,8-DCQx-DICz (Figure ) were reported with a triazatruxene donor in lieu of an acridan.ref. ref493 Changing the position of the two cyano groups from the ortho to the meta positions with respect to the pyrazine ring resulted in a significant increase in ΦPL from 40 (5,8-DCQx-DICz) to 73% (6,7-DCNQx-DICz). However, this improvement is accompanied by a blue-shift of almost 50 nm in the emission λPL, from 651 (5,8-DCQx-DICz) to 603 nm (6,7,DCQx-DICz). The device with 6,7-DCQx-DICz (1 wt% doped in PBICT) showed a higher EQEmax of 23.9% at λEL of 578 nm than the device with 6,7-DCQx-Ac at 21.1% at λEL of 578 nm. The 5,8-DCQx-DICz device, on the other hand, showed an EQEmax of 12.5% at λEL of 603 nm.
The strong donor phenoxazine was combined with similar acceptors 6-(trifluoromethyl)quinoxaline or 6-(cyano)quinoxaline to form compounds TFM-QP and CN-QP (Figure ).ref. ref494 TFM-QP and CN-QP emit at λPL of 613 and 611 nm in toluene, and both have ΦPL of 61% in 5 wt% doped CBP films (Table S3). The compounds showed delayed fluorescence with rather long τd of 5.0 ms (TFM-QP) and 1.6 ms (CN-QP) despite their small ΔE ST of 0.04 and 0.03 eV, all respectively. The yellow OLEDs fabricated with TFM-QP and CN-QP exhibited EQEmax of 14.1 and 9.7%, both with λEL of 584 nm, illustrating that the red emission achieved in solution measurements is not always straightforward to translate into devices.
PXZ-PQM and DPXZ-PQM (Figure ) similarly combine benzoyl and quinoxaline units, with differing numbers of phenoxazine donor units.ref. ref495 The PL spectra of PXZ-PQM (ΔE ST of 0.03 eV) and DPXZ-PQM (ΔE ST of 0.02 eV) in 5 wt% doped films in DCzDPy gave broad orange-to-red emission at λPL of 588 and 586 nm, demonstrating little impact of the number of donor groups in this case. The device with DPXZ-PQM exhibited the best EL performance with an EQEmax of 26.0%, and orange-red emission at λEL of 590 nm corresponding to CIE coordinates of (0.51, 0.48) (Table S3). This performance was attributed to the high ΦPL (88%), relatively small ΔE ST (0.02 eV), and fast RISC rate (k RISC = 2.05 × 105 s–1). The OLED with PXZ-PQM showed a somewhat lower EQEmax of 20.4%, attributed to the ΦPL of 70% of the emitter. The efficiency roll-off at 100 and 1000 cd m–2 were 14 and 45% for the PXZ-PQM-based device, and 23 and 47% for the DPXZ-PQM-based device.
Acenaphtho[1,2-b]pyrazine Acceptors
Acenaphtho[1,2-b]pyrazine-8,9-dicarbonitrile (APDC), a stronger acceptor than quinoxaline, was used in conjunction with two TPA donors to produce the deep-red emitter APDC-DTPA (Figure ).ref. ref496 Doped at 10 wt% in TPBi, APDC-DTPA emits at λPL of 687 nm with a ΦPL of 63% and a ΔE ST of 0.14 eV (Table S3). As neat films the emission is red-shifted to λPL = 756 nm, which is accompanied by a drop in ΦPL to 17% due to ACQ. The OLEDs with APDC-DTPA showed an EQEmax of 10.2% at λEL of 693 nm. Non-doped OLEDs produced NIR λEL of 777 nm although with a lower EQEmax of 2.2%. While both devices represent some of the deepest red TADF OLEDs reported to date, they also suffer from severe efficiency roll-off, with the EQE100 for the doped device being ∼0.8%, while the non-doped device did not reach this level of luminance. This low efficiency was attributed to triplet–triplet or singlet–triplet annihilation arising from the relatively long triplet lifetime, along with low ΦPL in the solid state.ref. ref497
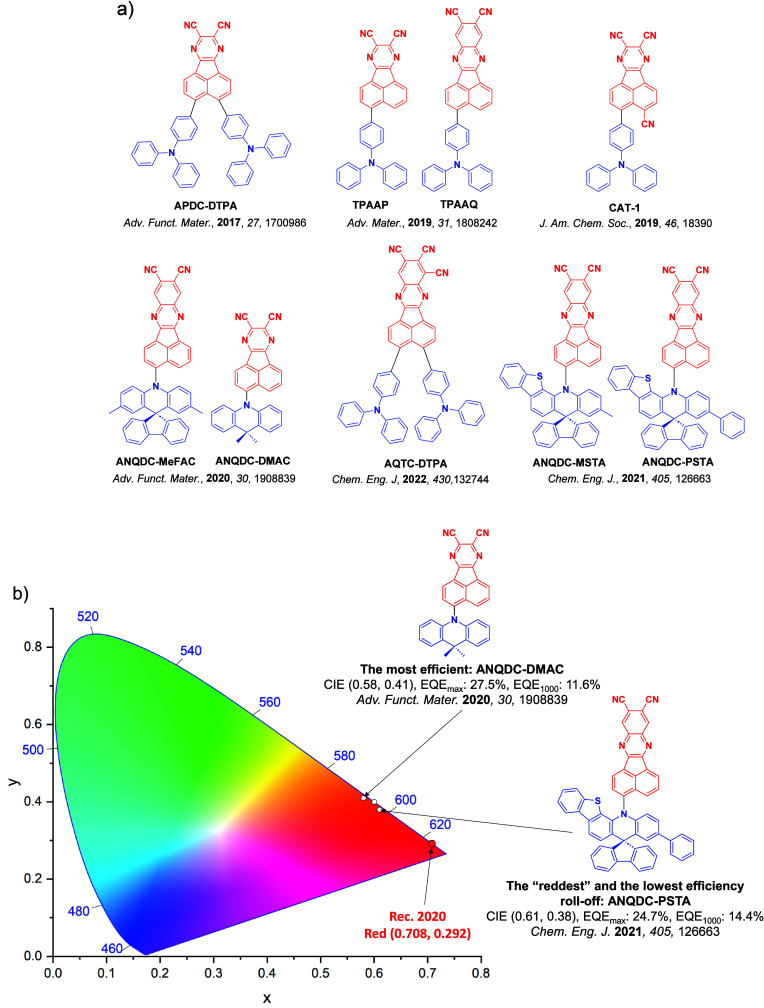
Xue et al. reported two D-A TADF emitters TPAAP and TPAAQ (Figure ), containing the strong electron-drawing acceptors acenaphtho[1,2-b]pyrazine-8,9-dicarbonitrile (AP) and acenaphtho[1,2-b]quinoxaline-8,9-dicarbonitrile (AQ).ref. ref498 Compared to APDC-DTPA both compounds contain only one TPA moiety. In toluene TPAAP emits at λPL of 609 nm, has a high ΦPL of 97% and a ΔE ST of 0.19 eV, while TPAAQ has ΦPL of 93% but with a larger ΔE ST of 0.33 eV (Table S3). Notably, both molecules exhibited significantly red-shifted PL spectra in their aggregated forms, falling into the NIR region with λPL at 777 nm for TPAAP and 716 nm for TPAAQ due to the formation of J-aggregates that possess strong intermolecular CT excited states. OLEDs with 5 or 10 wt% TPAAP in TPBi showed EQEmax of 15.8 and 14.1% with λEL of 630 and 657 nm, all respectively. The non-doped devices with TPAAQ (ΦPL = 16.3%) and TPAAP (ΦPL = 20.3%) exhibited NIR emission with EQEmax of 3.5% at 711 nm and 5.1% at 765 nm, respectively.
Congrave et al. reported a NIR TADF emitter, CAT-1 (3-triphenylamine-4-cyano-acenaphtho[1,2-b]-pyrazine-8,9-dicarbonitrile), which is structurally similar to TPAAP. CAT-1 incorporates triphenylamine as the donor and acenaphtho[1,2-b]-pyrazine as the acceptor, enabling NIR TADF emission (Figure ).ref. ref20 Compared to APDC-DTPA (neat, λPL = 756 nm), CAT-1 in 10 wt% doped CBP films has a red-shifted emission (λPL = 763 nm) at modest ΦPL of 8.8% and a rather long τ d of 80 μs considering its ΔE ST of ca. 0.04 eV (Table S3). Increasing the doping ratio of CAT-1 in the evaporated films led to significant red-shift of the emission and a corresponding decrease in ΦPL; for example, the 40 wt% doped CBP film emission was recorded at λPL = 820 nm with low ΦPL of 2%. Evaporated neat films of CAT-1 emit at λPL of 887 nm, while neat films dropcasted from chlorobenzene solution were further red-shifted to λPL = 950 nm. The non-doped OLEDs showed an EQEmax of 0.019% at an λEL at 904 nm. Computational studies of this material later revealed the potential for intramolecular hydrogen bonding (CH-CN) between the TPA donor and CN acceptor group as assisting in the overall performance compared to TPAAP.ref. ref499
Gong et al. reported the emitters ANQDC-DMAC and ANQDC-MeFAC (Figure ), using the same acceptor as TPAAQ but coupled with either DMAC or MeFAC as the donor unit.ref. ref500 This combination of donor and acceptor resulted in λPL at 596 and 604 nm in 1.5 wt% CBP:TPBi co-host and high ΦPL of 95 and 77%, along with a small ΔE ST values of 0.06 and 0.05 eV, all respectively (Table S3). Both compounds displayed preferential horizontal TD alignments of ∼80%, attributed to the linear and planar acceptor motif and rod-like molecular configuration. The OLEDs with ANQDC-DMAC and ANQDC-MeFAC achieved EQEmax of 27.5 and 26.3% at λEL at 615 and 614 nm respectively, corresponding to CIE coordinates of (0.58, 0.41) and (0.60, 0.40).
Cheng et al. reported a NIR TADF emitter containing an auxiliary electron-withdrawing group attached to the AQ acceptor, AQTC-DTPA (Figure ).ref. ref501 AQTC-DTPA in 10 wt% doped films in CBP emits at λPL of 718 nm and has a ΦPL of 19.1%, while in neat films this compound emits at λPL of 878 nm and has a ΦPL of 1.1%. A large red-shift (82 nm) was observed in the emission spectrum of AQTC-DTPA in the 10 wt% doped film relative in toluene (636 nm). The EL spectra also showed a significant bathochromic shift from 694 to 894 nm when the doping ratio increased in CBP film from 10 to 100 wt% (neat). The red-shifting of the emission was attributed to strong intermolecular interactions in the emissive layer, growing in strength as the doping concentration increased. The large and coplanar AQ unit in AQTC-DTPA favoured π-π stacking interactions in thin films, which was supported by single-crystal X-ray analysis. The solid-state structure of amorphous AQTC-DTPA obtained by cluster analysis indeed showed a tight packing pattern, aggregated in a head-to-head mode with π-π distances below 3.6 Å. A device with AQTC-DTPA (10 wt% doped in CBP) showed an EQEmax of 9.3% at λEL of 694 nm, with the EQEmax decreasing significantly to 0.51/0.41/0.30/0.23% as the doping concentration increased from 60/70/80/100 wt% at λEL of 810/828/852/894 nm, all respectively (Table S3).
Recently, Gong et al. reported red emitters ANQDC-MSTA and ANQDC-PSTA (Figure ) using rigid, linear, and planar ANQDC as the acceptor coupled with thienocarbazole-fused acridine donors MSTA and PSTA.ref. ref502 The compounds emit at λPL of 623 and 618 nm and have ΦPL of 65 and 72% respectively, in 1.5 wt% doped films in a 1:1 CBP:TPBi co-host (Table S3). The ANQDC-MSTA-based OLED (1.5 wt% emitter doped in CBP:TPBi co-host) exhibited an EQEmax of 21.8%, while the ANQDC-PSTA-based OLEDs displaying slightly higher EQEmax of 24.7%. Both OLEDs displayed λEL of 622 nm and CIE coordinates of (0.61, 0.38).
Pyrazino- or Quinoxalino-Expanded Phenanthrene Acceptors
Pyrazine-fused phenanthrene is a large and rigid π-conjugated structure that has been widely used as an acceptor in red TADF emitters. For example, Wang et al. employed dicyano-substituted pyrazino-phenanthrene (DCPP) as the acceptor and DPA or DMAC as the donor in a series red and deep-red TADF emitters.ref. ref503 DMAC derivatives DMAC-DCPP and DMAC-Ph-DCPP (Figure ) emit at 618 and 594 nm, have ΦPL of 33 and 65%, small ΔE ST of 0.08 and 0.05 eV, and short τd of 2.4 and 3.2 μs, all respectively (Table S3). The OLEDs with DMAC-DCPP and DMAC-Ph-DCPP showed EQEmax of 10.1 and 16.5%, with CIE coordinates of (0.60, 0.40) and (0.53, 0.46), respectively. However, large efficiency roll-off were observed with EQE500 of just 4.2% for the device with DMAC-DCPP and 6.3% for the device with DMAC-Ph-DCPP. Replacing DMAC with DPA resulted in red-shifted emission at λPL = 606 and 628 nm for DPA-DCPP and DPA-Ph-DCPP, respectively, along with ΦPL of 64 and 65%. The devices with these two emitters also experienced severe efficiency roll-off (EQEmax of 10.4 and 15.1%, EQE500 of 0.9 and 1.6%), due to their larger ΔE ST (0.28 and 0.10 eV) and much longer delayed lifetimes (τd = 579 and 82 μs).
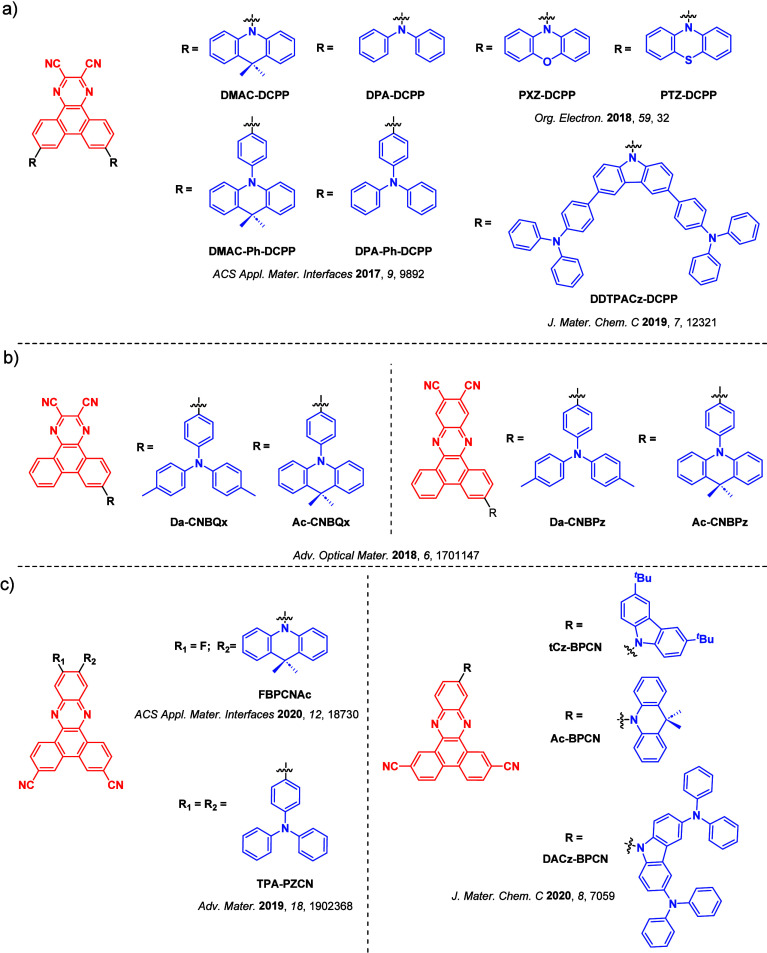
The same DCPP acceptor unit was also reported by Wang et al. coupled with stronger phenoxazine and phenothiazine donors (Figure ).ref. ref504 In toluene, PXZ-DCPP and PTZ-DCPP emit at 564 and 580 nm, have ΦPL of 11.9 and 17.4% and ΔE ST of 0.09 and 0.18 eV, respectively (Table S3). The devices with PXZ-DCPP and PTZ-DCPP showed EQEmax of 17.4 and 12.3% at λEL of 608 and 640 nm with associated CIE coordinates of (0.56, 0.43) and (0.62, 0.36), all respectively. Importantly, these devices exhibited only modest efficiency roll-off with the EQE1000 of 12.9% and 6.1%. A similar red emitter with carbazole donors that are additionally decorated with two TPA units was also reported by Wang et al..ref. ref505 DDTPACz-DCPP (Figure ) has a ΦPL of 53% and emits at λPL of 663 nm in 10 wt% doped films in CBP, along with having a ΔE ST of 0.16 eV and a τd of 9.7 μs. The solution-processed devices with DDTPACz-DCPP showed an EQEmax of 13.6% at λEL of 646 nm and CIE coordinates of (0.63, 0.37).
Furue et al. reported two asymmetric D−π–A emitters Da-CNBPz and Ac-CNBPz (Figure ), consisting of 11,12-dicyanodibenzo[a,c]phenazine (CNBPz) as a strong acceptor unit.ref. ref506 These were compared with D−π–A TADF analogues Da-CNBQx and Ac-CNBQx, containing 2,3-dicyanodibenzo[f,h]quinoxaline (CNBQx) as a less π-conjugated and weaker acceptor unit (Figure ). Comparing Da-CNBQx and Da-CNBPz, the emission red-shifted from 633 to 688 nm and there is a modest decrease in ΦPL from 85 to 72% (Table S3). The same behavior was observed for Ac-CNBQx and Ac-CNBPz, with λPL red-shifted from 561 to 615 nm and ΦPL decreasing from 75 to 67%, respectively. Calculated non-radiative rate constants (k nr) for the emitters showed an increase in non-radiative decay upon extending the π-conjugation of the acceptor, with values of 1.6 × 107 s–1 for Da-CNBQx and 2.4 × 107 s–1 for Da-CNBPz; the same trends were observed for Ac-CNBQx and Ac-CNBPz, where an increase of non-radiative decay from 0.16 to 0.25 × 107 s–1 was seen, in line with the energy gap law. Devices with Da-CNBQx and Da-CNBPz showed EQEmax of 15.0% at λEL of 670 nm and 20.0% at λEL of 617 nm, respectively, representing some of the highest efficiency red TADF OLEDs to date. However, both of these devices suffered severe efficiency roll-off with EQE100 dropping to 3.8 and 7.5%, respectively. Although the devices with Ac-CNBQx and Ac-CNBPz show lower EQEmax (and blue-shifted emission) of 16.2% (λEL = 630 nm) and 14.0% (λEL of 685 nm), their EQE100 were superior at 14.5 and 13.9%, all respectively. This change is due to smaller ΔE ST values when using Ac as the donor of 0.03 and 0.10 eV for Ac-CNBPz and Ac-CNBQx compared to 0.11 and 0.18 eV for Da-CNBPz and Da-CNBQx, respectively, all in 6 wt% doped films in CBP.
In a similar approach to the previous example, cyano groups were added to the 3- and 6- positions of a phenazine core to increase the electron-accepting strength, while two TPA groups were employed as the donors.ref. ref507 TPA–PZCN (Figure ) emits at 610 nm and has a very high ΦPL of 97%, a ΔE ST of 0.13 eV, and a τd of 133 μs. The devices with TPA–PZCN showed an EQEmax of 27.4% at λEL at 628 nm and CIE coordinates of (0.65, 0.35), which represents the best result with a peak wavelength longer than 600 nm among the reported red TADF devices. In a subsequent report, Kothavale et al. functionalized the same acceptor with a fluorine atom and used DMAC as the donor in FBPCNAc (Figure ).ref. ref434 The fluorine substituent was attached ortho to the DMAC, which strengthened the electron-acceptor. FBPCNAc emits at 607 nm, has a ΦPL of 79%, a small ΔE ST of 0.05 eV, and a τd of 11.1 μs. The OLEDs with FBPCNAc realized an EQEmax of 23.8% at λEL of 597 nm and CIE coordinates of (0.55, 0.44). This blue-shift of the emission compared to the previous examples is likely due to there being only one donor unit in this emitter design compared to two for the others. Kothavale et al. also reported two related emitters, Ac-BPCN and DACz-BPCN (Figure ), which differ in the substitution position of the CN group on the BPCN acceptor unit.ref. ref508 Ac-BPCN and DACz-BPCN emit at 618 and 654 nm, have ΦPL of 66 and 47%, ΔE ST of 0.13 and 0.07 eV, and τd of 11.1 and 7.2 μs, all respectively. The OLEDs with Ac-BPCN and DACz-BPCN in the bipolar host PBICT showed EQEmax of 20.7% (λEL = 597 nm) and 11% (λEL = 631 nm) at CIE coordinates of (0.54, 0.45) and (0.60, 0.39), respectively.
Moving away from CN-substituted π-conjugated acceptors, Xie et al. developed three TADF molecules xDMAC-BP (x = 1, 2, 3) containing a rigid planar phenazine acceptor core and different numbers of DMAC donors at the 3-/6-/11-positions (Figure ).ref. ref509 The emission color of the xDMAC-BP series could be tuned from green to orange-red by changing the number of the DMAC units. The reddest emitting analogue 3DMAC-BP emits at λPL of 590 nm, has a high ΦPL of 89%, a small ΔE ST of 0.05 eV, and a short τd of 2.9 μs in 20 wt% doped films in mCBP. The OLEDs with 3DMAC-BP showed an EQEmax of 22.0% at λEL of 606 nm. Crucially, the EQE100 of the 3DMAC-BP-based device remained as high as 17.5%. The same molecular design was also employed using PXZ as the donor.ref. ref510 Expectedly, increasing the number of PXZ units red-shifted the emission from 602 to 682 nm in toluene. The ΔE ST and ΦPL values for 1PXZ-BP are 0.25 eV and 73%, for 2PXZ-BP are 0.10 eV and 63%, and for 3PXZ-BP are 0.03 eV and ΦPL = 22%. Thus, as the number of PXZ groups increases the ΦPL decreases as does ΔE ST and also τd from 4.8 to 4.3 μs and 2.0 μs, respectively. The orange-red OLEDs with 1PXZ-BP, 2PXZ-BP, and 3PXZ-BP showed EQEmax of 26.3% (λEL of 590 nm), 19.2% (λEL of 606 nm) and 7.1% (λEL of 634 nm). However, compared to xDMAC–BP the efficiency roll-off of the xPXZ-BP series are all higher, which the authors speculated may be due to the inferior charge balance of the devices.
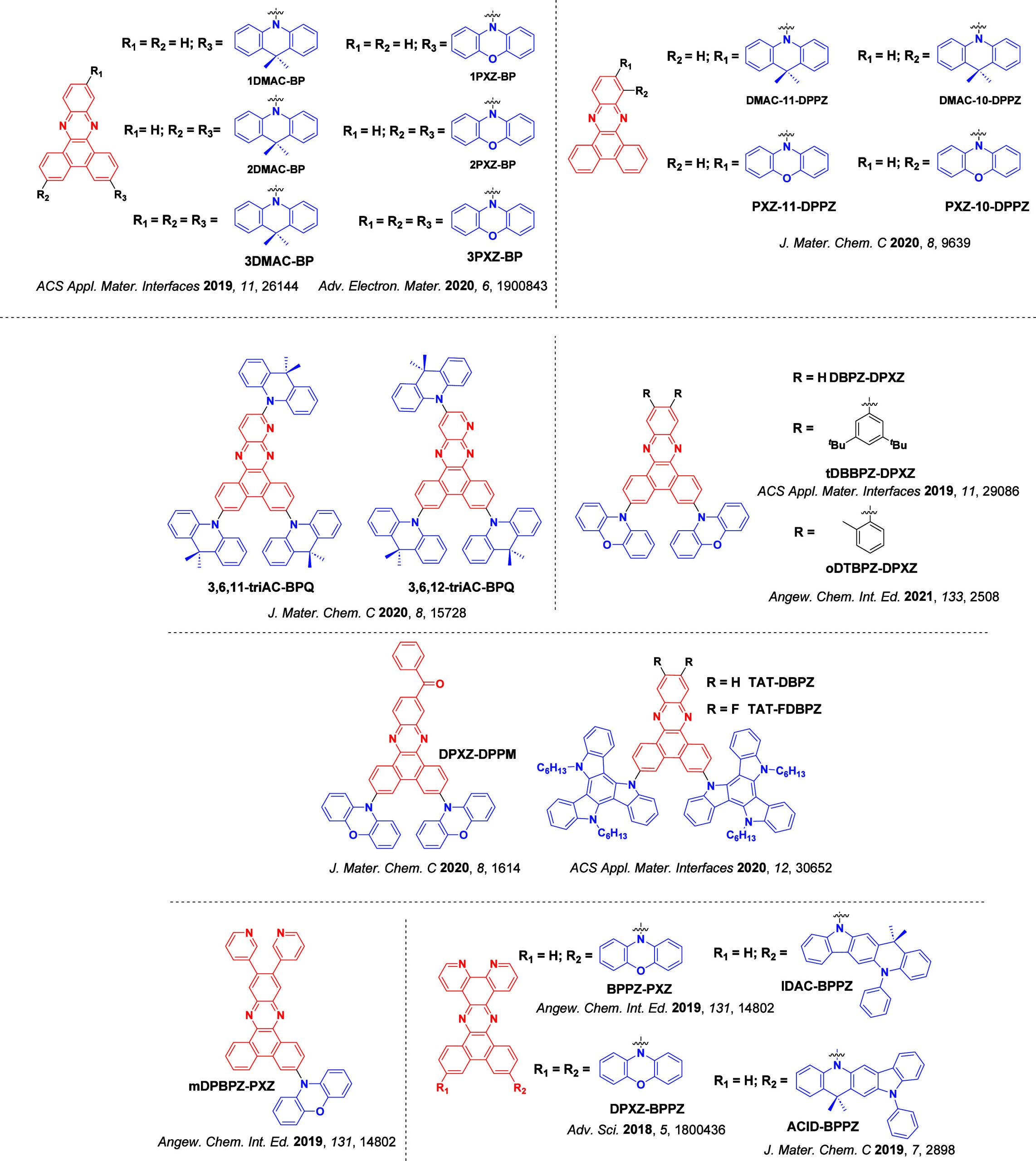
Xie et al. used a similar dibenzo[f,h]pyrido[2,3-b]quinoxaline (BPQ) acceptor coupled to three DMAC donors at either the 3-,6-,11-positions or 3-,6-,12-positions in 3,6,11-triAC-BPQ and 3,6,12-triAC-BPQ (Figure ).ref. ref511 In 15 wt% doped films in mCBP 3,6,11-triAC-BPQ and 3,6,12-triAC-BPQ emit at λPL of 516 and 611 nm in toluene, have ΦPL of 75 and 53%, and τd of 2.50 and 2.25 μs, all respectively. 3,6,11-triAC-BPQ was claimed to have HLCT character due to intramolecular hydrogen bonding between the isolated donor and the adjacent pyridine nitrogen, whilst the 3,6,12-triAC-BPQ displayed typical CT character. 3,6,11-triAC-BPQ and 3,6,12-triAC-BPQ have ΔE ST of 0.10 and 0.03 eV, and the corresponding devices showed EQEmax of 22.0% [λEL of 581 nm, CIE coordinates of (0.51, 0.48)] and 16.5% [λEL of 616 nm, CIE coordinates of (0.58, 0.39)], all respectively (Table S3).
Zhou et al. developed two pairs of emitters DMAC-11-DPPZ and DMAC-10-DPPZ, and PXZ-11-DPPZ and PXZ-10-DPPZ, differing only in the nature of the donor (DMAC or PXZ) connected through 10- or 11- positions on the acceptor BP moiety (Figure ).ref. ref512 The compounds substituted at the 11-position achieved much higher ΦPL (57.4 and 40.9%) than those substituted at the 10-position (28.6 and 5.3%), owing to suppressed non-radiative vibrational modes. The PL spectrum of DMAC-11-DPPZ in toluene has two emission peaks, at λPL of 567 and 490 nm, attributed to the coexistence of quasi-equatorial (QE) and quasi-axial (QA) conformers. DMAC-10-DPPZ emits at 620 nm, which is significantly red-shifted owing to the stronger CT state. The emission of PXZ-11-DPPZ shows a red emission peak at λPL of 630 nm, while by contrast is significantly red-shifted compared to PXZ-10-DPPZ, which unexpectedly exhibits a blue-shifted emission peak at λPL of 573 nm in toluene. The τd for DMAC-11-DPPZ, DMAC-10-DPPZ, PXZ-11-DPPZ, and PXZ-10-DPPZ are 1.53, 0.83, 0.72, and 0.51 μs, respectively. These values are in good agreement with the trend in the corresponding ΔE ST values of 0.112, 0.075, 0.062, and 0.057 eV, respectively. The DMAC-11-DPPZ based device showed orange emission at λEL of 588 nm [CIE coordinates of (0.53, 0.46)] with an EQEmax of 23.8%, while the device with DMAC-10-DPPZ showed a much lower EQEmax of 8.3%, albeit with a red-shifted λEL of 624 nm [CIE coordinates of (0.61, 0.38)]. Similarly, the PXZ-10-DPPZ device showed a red-shifted emission at λEL of 655 nm [CIE coordinates of (0.63, 0.37)] yet with a higher EQEmax of 8.7% compared to the device with PXZ-11-DPPZ at λEL of 627 nm [CIE coordinates of (0.65, 0.35)] and an EQEmax of only 0.8%.
Introduction of two 3,5-di-tert-butylphenyl groups in tDBBPZ-DPXZ improved the solubility compared to 2PXZ-BP (Figure ).ref. ref513 tDBBPZ-DPXZ emits at λPL of 617 nm, has a ΦPL of 83%, and a small ΔE ST of 0.03 eV in 10 wt% doped films in CBP. A solution-processed tDBBPZ-DPXZ OLED emitted at λEL of 620 nm and CIE coordinates of (0.62, 0.37), and showed an EQEmax of 10.1%. tDBBPZ-DPXZ has nearly the same photophysical properties as 2PXZ-BP (DBBPZ-DPXZ in that work). Vacuum-deposited devices with tDBBPZ-DPXZ and DBPZ-DPX both emitted at λEL 608 nm, corresponding to CIE coordinates of (0.58,0.42) and (0.57,0.43), and showed EQEmax of 17.0 and 17.8%, all respectively. In a subsequent report from the same group, a similar compound oDTBPZ-DPXZ containing o-tolyl groups instead of tert-butylphenyl groups shows comparable photophysics.ref. ref514 oDTBPZ-DPXZ emits at 622 nm, has a high ΦPL of 87%, and a small ΔE ST of 0.04 eV (Table S3). Solution-processed OLEDs with oDTBPZ-DPXZ achieved an EQEmax of 18.5% at λEL of 612 nm and CIE coordinates of (0.60, 0.40).
Liang et al. used a weakly electron-withdrawing benzoyl group attached to 2PXZ-BP to construct the emitter DPXZ-DPPM (Figure ).ref. ref495 DPXZ-DPPM doped in 5,5′-bis(carbazol-9-yl)-3,3′-bipyridine (DCzDPy) films emits at λPL of 630 nm, has a ΦPL of 61%, a ΔEST of 0.05 eV, and a τd of 3.53 μs. Compared to 2PXZ-BP (λEL of 606 nm), DPXZ-DPPM-based devices display a much redder emission at λEL of 630 nm corresponding to CIE coordinates of (0.61, 0.38), and showed an EQEmax of 11.5%. Fan et al. reported two similar TADF emitters, mDPBPZ-PXZ and BPPZ-PXZ, which have either substituted or annulated pyridyl groups on the acceptor.ref. ref515 In 14 wt% doped CBP films mDPBPZ-PXZ emits at λPL of 638 nm, has a small ΔE ST of 0.04 eV, and a high ΦPL of 95%, whereas the neat film has only a moderate ΦPL of 33% with a λPL of 607 nm (Table S3). This suggests the introduction of pyridine moieties somewhat relieves concentration-induced quenching. The OLEDs with mDPBPZ-PXZ in mCP showed an EQE of 21.7% at λEL of 624 nm and CIE coordinates of (0.62, 0.38). Non-doped devices showed a much lower EQE of 5.2% at λEL of 680 nm with CIE coordinates of (0.68, 0.32). The fused analogue BPPZ-PXZ emits at 607 nm and has a high ΦPL of 100%, ΔE ST of 0.03 eV, and a τd of 3.6 μs.ref. ref516 The OLED doped with BPPZ-PXZ showed an EQEmax of 25.2% at λEL at 604 nm, whereas the non-doped device showed a much lower EQEmax of only 2.5% at λEL at 656 nm. This contrast was attributed to more serious concentration quenching due to close molecular packing of this more planar emitter (compared to mDPBPZ-PZX with conformationally flexible pyridyl substituents). A disubstituted analogue DPXZ-BPPZ was also reported by Chen et al. and has similar optoelectronic properties.ref. ref517 The DPXZ-BPPZ OLED emitted at λEL of 612 nm and showed an EQEmax of 20.1%, and EQE100/EQE1000 that remained at ∼19.7/16.7% – an efficiency roll-off that was superior to the device with single-donor material BPPZ-PXZ. The superior performance of the devices was in part due to the excellent ΦPL of 97%, the reasonably fast k RISC of 2.24 × 105 s–1 and suppressed k nr of 0.5 × 104 s–1, the latter of which was attributed to the rigid nature of the molecule.
Chen et al. used the same acceptor in combination with fused donors in IDAC-BPPZ and ACID-BPPZ (Figure ).ref. ref518 Similar emission properties were observed for both compounds with λPL of 583 and 596 nm and ΦPL of 84 and 75%, respectively. IDAC-BPPZ and ACID-BPPZ have ΔE ST of 0.06 and 0.01 eV, and similar τd of 14 and 12 μs (Table S3). The OLEDs with IDAC-BPPZ showed EQEmax of 18.3% at λEL = 580 nm, as compared to only 14.7% for the device with ACID-BPPZ at λEL = 588 nm. A greater efficiency roll-off was observed for the device with IDAC-BPPZ, decreasing from maximum values by ∼39 and ∼68% at 1000 cd m–2 for the OLEDs with ACID-BPPZ and IDAC-BPPZ, respectively. This difference was ascribed to the faster τd alleviating triplet accumulation and quenching processes in ACID-BPPZ.
Liu et al. developed two red TADF emitters by incorporating triazatruxene (TAT) as the electron donor (Figure ).ref. ref519 Fluorine-substituted TAT-FDBPZ displayed a red-shifted emission (λPL = 601 nm) compared to that of TAT-DBPZ (λPL = 572 nm) as a result of the electron-withdrawing nature of the two fluorine atoms. The large steric hindrance between TAT and DBPZ was suggested to be responsible for a reduced ΔE ST value of 0.16 eV and suppressed ACQ, enabling AIE and high ΦPL in the 20 wt% doped films in CBP of these emitters (ΦPL of 76% for TAT-DBPZ and 62% for TAT-FDBPZ). TAT-DBPZ and TAT-FDBPZ indeed have small ΔE ST of 0.16 and 0.10 eV and short τd of 2.30 and 1.51 μs, respectively. Solution-processed OLEDs with TAT-DBPZ showed an EQEmax of 15.4% at λEL of 604 nm, while the TAT-FDBPZ based OLEDs showed a red-shifted at λEL of 611 nm and a smaller EQEmax of 9.2%. These values were accompanied by very low efficiency roll-off of only 1.0% at 100 cd m–2 and 19% at 1000 cd m–2.
Rather than installing fused pyridine groups onto phenanthrene, Xu et al. developed phenanthroline-based D-A red TADF emitters oTPA-DPPZ and pTPA-DPPZ (Figure fig65a and fig65b).ref. ref520 In a 30 wt% doped DBFDPO (4,6-bis(diphenylphosphoryl)-dibenzofuran) film, oTPA-DPPZ emits at λPL of 605 nm, has a ΦPL of 75%, a ΔE ST of 0.07 eV, and a τd of 12 μs (Table S3). OLEDs with oTPA-DPPZ showed an EQEmax of 18.5% at λEL of 600 nm. Through adjusting the position of the donor groups, the T-shaped pTPA-DPPZ emits to the red at λPL of 644 nm in neat film. The spatial arrangement of D and A groups in pTPA-DPPZ dramatically accelerated the rate of singlet emission without an increase in non-radiative decay, resulting in an increased ΦPL of 87% in the neat film. This change in optical properties was accompanied by remarkably improved carrier transport in the neat film. As a result, a high-efficiency bilayer non-doped OLED was demonstrated, displaying deep-red emission at λEL = 652 nm and CIE coordinates of (0.67, 0.33,) and showing an EQEmax of 12.3% with EQE1000 of 10.4%.
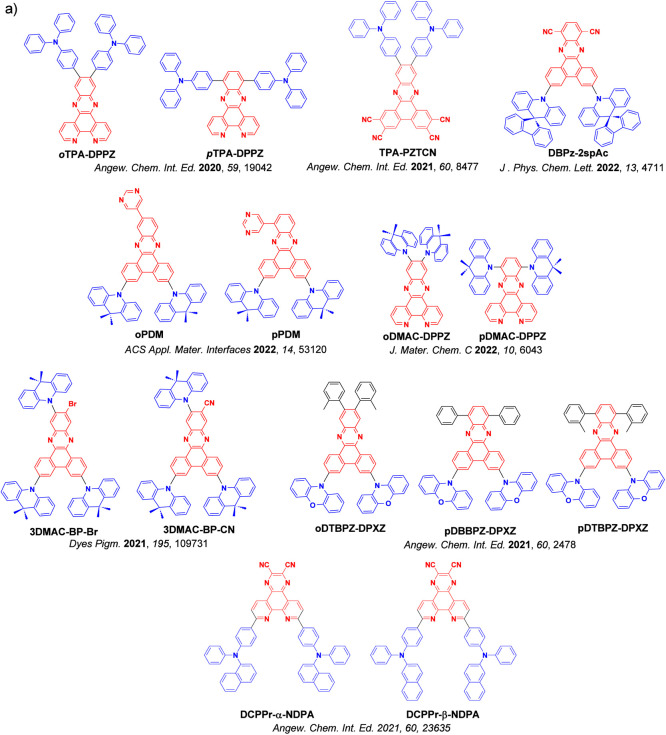
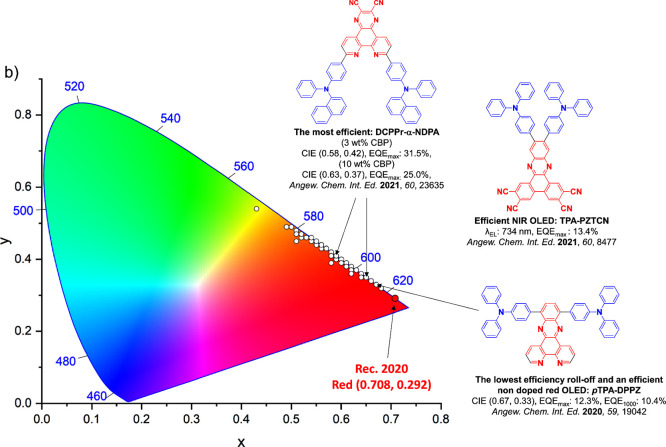
Zhang et al. reported the red TADF emitter DBPz-2spAc, (Figure ) based on an 8b,14a-dihydrodibenzo[a,c]phenazine-10,13-dicarbonitrile acceptor and containing two spiro-acridines as donors.ref. ref521 DBPz-2spAc has an ΦPL of 27% (λPL = 632 nm) in toluene and 65% (λPL = 632 nm) in 1 wt% doped films in CBP. OLEDs at 1 wt% doping ratio showed high EQEmax of 13.3%, with the λEL at 630 nm. However, the devices suffer from severe efficiency roll-off, were the EQE100 drops to about 1%, attributed to triplet–triplet annihilation (TTA).
Tan et al. reported two isomeric orange-red TADF emitters, oPDM and pPDM (Figure ), with the same basic donor-acceptor backbone but with pyrimidine (Pm) attached at different positions.ref. ref522 oPDM and pPDM emit at λPL of 582 and 573 nm, have moderate ΔE ST of 0.11 and 0.15 eV, and high ΦPL of ca. 100 and 88% in respective 8 wt% doped films in CBP (Table S3). OLEDs with oPDM or pPDM exhibited orange-red EL emission at λEL of 596 and 582 nm and CIE coordinates of (0.56, 0.44) and (0.52, 0.47), respectively. Despite similar PL properties, a significant difference in efficiency was seen in the two devices, with the OLEDs with oPDM or pPDM showing EQEmax of 28.2 and 11.8%, respectively. The difference was attributed to the differing molecular packing of the emitters in the aggregated state, resulting in very different charge transport performance.
Kothavale et al. reported red TADF emitters, oDMAC-DPPZ and pDMAC-DPPZ (Figure ), whose structures differ in the regiochemistry of the DMAC donor.ref. ref523 The ΦPL in toluene of the more red-shifted compound, pDMAC-DPPZ (λPL = 669 nm, ΦPL = 15%) is lower than that of oDMAC-DPPZ (λPL = 652 nm, ΦPL = 63%). In the bipolar host 2-phenyl-4,6-bis(12-phenylindolo[2,3-a]carbazole-11-yl)-1,3,5-triazine (PBICT, 1 wt%) the emission of oDMAC-DPPZ is blue-shifted to λPL = 614 nm with CIE coordinates (0.59, 0.40), while pDMAC-DPPZ emits at λPL of 638 nm with CIE coordinates (0.64, 0.35). Aligning with their ΦPL, the device with oDMAC-DPPZ showed a higher EQEmax of 13.4% at λEL of 614 nm, while the device with pDMAC-DPPZ displayed a lower EQEmax of 4.0%.
Huang et al. synthesized two orange-red TADF emitters with D3-A structures, and modified by either an inductively electron-withdrawing bromine atom (DMAC-BP-Br) or a cyano-group (3DMAC-BP-CN, Figure ).ref. ref524 3DMAC-BP-Br and 3DMAC-BP-CN emit at λPL of 612 and 617 nm in toluene, have ΦPL of 83 and 92%, ΔE ST of only 0.04 and 0.02 eV, and τd of 3.8 and 4.6 μs, all respectively in 15 wt% doped films in CBP. The OLEDs with 3DMAC-BP-Br and 3DMAC-BP-CN showed EQEmax of 18.9 and 22.4% at λEL of 596 and 586 nm, respectively (Table S3).
Balijapalli et al. reported a D-A deep-red/NIR emitting compound, TPA-PZTCN, featuring multiple cyano groups about the acceptor (Figure and fig65b).ref. ref525 TPA-PZTCN emits at λPL of 674 nm and has a ΦPL of 77% in toluene. Doped at 1 wt% in mCBP, the emission of TPA-PZTCN at 672 nm maintains a high ΦPL of 78%, while at 10 wt% loading the ΦPL decreases to 40% accompanied by a much red-shifted λPL at 729 nm. OLEDs with 1, 3, or 6 wt% TPA-PZTCN showed EQEmax of 19.3% (λEL = 651 nm), 17.7% (λEL = 671 nm), and 15.8% (λEL = 712 nm), respectively (Table S3).
Chen et al. reported red TADF emitters pDBBPZ-DPXZ, pDTBPZ-DPXZ, and oDTBPZ-DPXZ (Figure ) using phenoxazine as donors and differently functionalised acceptors.ref. ref514 pDBBPZ-DPXZ, pDTBPZ-DPXZ, and oDTBPZ-DPXZ have distinctive ΦPL of 49, 66, and 87% in respective 8 wt% doped CBP films. Despite the differing ΦPL, the photophysical properties of the three compounds are quite similar, (pDBBPZ-DPXZ with λPL = 622 nm, ΔE ST = 0.23 eV, and τd = 53.3 μs, pDTBPZ-DPXZ with λPL = 621 nm, ΔE ST = 0.10 eV, and τd = 5.6 μs, and oDTBPZ-DPXZ with λPL= 621, ΔE ST = 0.04, and τd = 3.3 μs), except for the differences in the energies of the T1 states. From pDBBPZ-DPXZ to pDTBPZ-DPXZ and oDTBPZ-DPXZ, the 3LEA energy levels gradually approach the CT states, from being deeply stabilized in pDBBPZ-DPXZ to near-isoenegetic with the 3CT state in oDTBPZ-DPXZ and leading to faster k RISC. The OLEDs with pDBBPZ-DPXZ, pDTBPZ-DPXZ, and oDTBPZ-DPXZ showed very similar red emission spectra, with λEL ∼ 604 nm and CIE coordinates of (0.59, 0.40), (0.58, 0.41), and (0.59, 0.41), respectively (Table S3). Following the ordering of ΦPL, the device with oDTBPZ-DPXZ showed the highest EQEmax of 20.1%, compared to the devices with pDTBPZ-DPXZ and pDBBPZ-DPXZ showing EQEmax of 16.0 and 8.0%, respectively.
A series of outstanding red emitters were reported by Cai et al., which were designed to contain varying electron-donating triarylamine moieties attached to a pyrazinylphenanthroline acceptor.ref. ref526 In this series, nitrogen atoms within the pyridine rings were conjectured to engage in hydrogen bonding with the hydrogen atoms found in phenyl rings from the donating moieties. This interaction produces a more planar confirmation and rigid molecule. Out of the series, the compounds DCPPr-α-NDPA and DCPPr-β-NDPA, Figure and fig65b, which contain N,N-diphenylnaphthalen-1-amine and N,N-diphenylnaphthalen-2-amine, respectively, as the donor groups showed the most interesting photophysics. DCPPr-α-NDPA and DCPPr-β-NDPA emit at similar λPL of 598 and 612, respectively, in toluene, whereas in the neat film the λPL were considerably red-shifted at 692 and 710 nm, respectively. DCPPr-α-NDPA has a superior ΦPL of 82%, compared to 74% for DCPPr-β-NDPA, which was attributed to the fact that the naphthalene is connected to the nitrogen atom via its α-position, which led to a suppressed molecular packing. On the other hand, a longer delayed lifetime of 42.7 μs was observed for DCPPr-α-NDPA, compared to 28.2 μs of DCPPr-β-NDPA, while the ΔE ST are similar at 0.07 and 0.08 eV, respectively in 3 wt% doped film in mCP. Importantly, the hydrogen bonding was asserted to be responsible to aid in the preferential horizontal orientation of the compounds in the vacuum-deposited films, which led to high-efficiency red OLEDs with CIE coordinates of (0.58, 0.42) for the device with DCPPr-α-NDPA and (0.59, 0.40) for the device with DCPPr-β-NDPA. Devices were fabricated using 3 wt% emitters in mCP, achieving an outstanding EQEmax of 31.5% (λEL = 606 nm) with DCPPr-α-NDPA, whereas the device with DCPPr-β-NDPA resulted in an EQEmax of 27.1% (λEL = 616 nm). No efficiency roll-off data were reported.
Phenanthro[4,5-abc]phenazine-11,12-dicarbonitrile or Phenanthro[4,5-fgh]quinoxaline Acceptors
The Phenanthro[4,5-abc]phenazine-11,12-dicarbonitrile (PPDCN) acceptor was combined with a TPA donor to form the D–A NIR TADF emitter TPA-PPDCN (Figure ).ref. ref527 By replacing the phenanthrene in the acceptor core of the previous examples with pyrene, the π-conjugation of the acceptor is increased. This substitution results in a significantly deeper LUMO energy and a red-shifted emission. The ΔE ST of TPA-PPDCN is 0.23 eV in toluene and the neat film emits at 725 nm, has a ΦPL of 21%, and a τd of 1.96 μs. The PL spectra of the doped films gradually red-shift from 650 to 687 nm with increasing doping concentration (5 to 20 wt% in CBP), indicating a shift from monomolecular emission to emission from aggregates. The highest ΦPL is 87% in the 10 wt% doped film in CBP, with λPL of 663 nm although the ΦPL is maintained at 77% when the doping concentration is as high as 20 wt%. The OLEDs with TPA-PPDCN (10 and 20% doped in CBP) showed deep red and NIR emission with respective λEL of 664 nm [CIE coordinates of (0.68, 0.32)] and 692 nm [CIE of (0.70, 0.30)], at EQEmax of 20.2 and 16.4%, all respectively (Table S3). However, all devices exhibited large efficiency roll-off, with EQE100 decreasing to 4.7 and 3.7%, also respectively.
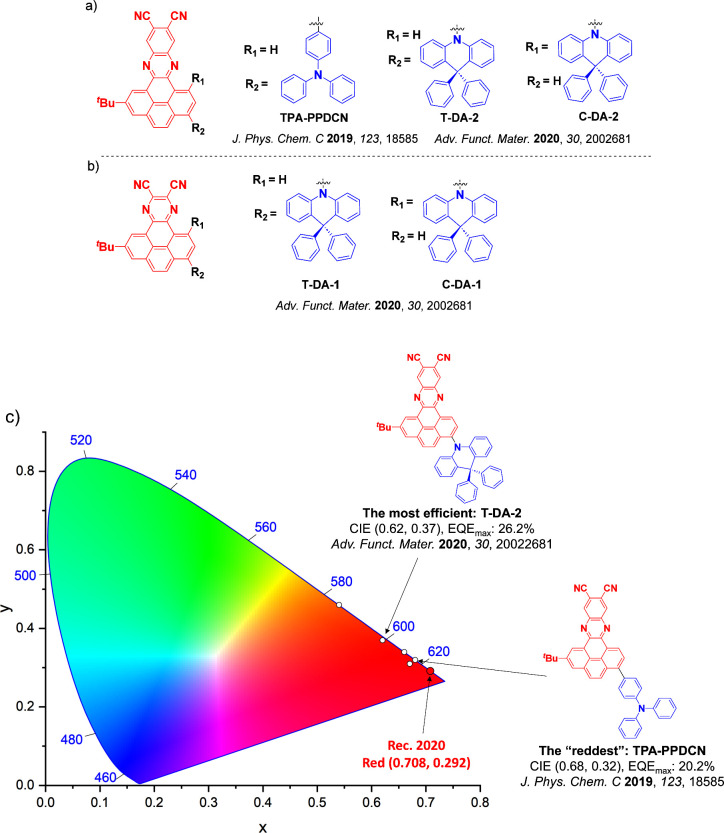
The same group reported two pairs of isomers employing either the same PPDCN as the acceptor or shortened analogue PDCN acceptor in combination with acridine donors attached at two different locations (T/C-DA-1/2, Figure ).ref. ref528 In 10 wt% doped films in mCBP, T-DA-1, T-DA-2, C-DA-1, and C-DA-2 emit at λPL of 601, 640, 640, and 689 nm, respectively. The ΦPL of the trans-isomers (T-DA-1 and T-DA-2, 78 and 89% respectively) are significantly higher than those of their corresponding cis-isomers (C-DA-1 and C-DA-2, 12 and 14%). The ΔE ST values are 0.16, 0.05, 0.02, and 0.02 eV for T-DA-1, T-DA-2, C-DA-1, and C-DA-2 respectively, in toluene at 77 K. The OLEDs with T-DA-1, T-DA-2, C-DA-1, and C-DA-2 doped at 10 wt% in mCBP showed orange-red to deep-red emission at λEL of 596, 640, 648, and 684 nm and CIE coordinates of (0.54, 0.46), (0.62, 0.37), (0.66, 0.34), and (0.67, 0.31), all respectively. Due to their low ΦPL, the devices with C-DA-1 (EQEmax of 3.5%) and C-DA-2 (EQEmax of 3.1%) showed much poorer efficiencies compared to the devices with T-DA-1 and T-DA-2, which instead showed EQEmax of 22.6 and 26.3% respectively (Table S3). Crucially, the EQE100 of the T-DA-2 based device still remained as high as 24%, corresponding to efficiency roll-off of just 8.8%.
1,8-Naphthalimide Acceptors
In addition to N-doped PAH acceptors discussed in the previous subsections, naphthalimide is another planar and strong acceptor with a deep LUMO. Incorporating a naphthalimide acceptor coupled to acridine donors, Zeng et al. reported efficient red emitters NAI-DMAC and NAI-DPAC (Figure ).ref. ref529 NAI-DMAC and NAI-DPAC emit at λPL of 582 and 570 nm, and have ΦPL of 59 and 71% respectively in 1.5 wt% doped films in mCPCN. Increasing this doping ratio to 6 wt% resulted in ACQ with ΦPL decreasing to 45% for NAI-DMAC. The concentration quenching effects were not observed for NAI-DPAC though, which retained a ΦPL of 72% even after increasing the doping concentration to 24 wt%. Both emitters showed preferential horizontal orientation of their TDMs, assisting the optical outcoupling to support high device EQEmax of 23.4 and 29.2% from 1.5 wt% NAI-DMAC and 6 wt% NAI-DPAC in mCPCN, at λEL of 597 and 584 nm, all respectively (Table S3). Although these compounds are not as deep red as some of the previously discussed examples, they represent some of the most efficient red TADF OLEDs to date. However, there was a large efficiency roll-off, with the EQE100 dropping to 13.6 and 13.0% for the devices with NAI-DMAC and NAI-DPAC, respectively, while the EQE dropped by 80 and 92% at 1000 cd m–2. The same group later reported two orange/red emitters, BFDMAc-NAI and BTDMAc-NAI (Figure ), by coupling fused heterocyclic DMAC donors to the NAI acceptor.ref. ref530 Both compounds showed red-shifted emission compared to those of NAI-DMAC and NAI-DPAC at 600 and 650 nm, respectively. BTDMAc-NAI in 1.5 wt% doped films in mCPCN has a lower ΦPL of 39%, while for BFDMAc-NAI the ΦPL is higher at 73%, similar to that of its parent compound NAI-DMAC. The lower ΦPL of BTDMAc-NAI was attributed to the introduction of the sulfur atom, which by virtue of the heavy atom effect increases both k RISC but also the competing phosphorescence rate constant. This compound also has a smaller ΔE ST of 0.07 eV compared to 0.16 eV for BFDMAc-NAI. OLEDs with BTDMAc-NAI emitted at λEL of 641 nm [CIE coordinates of (0.62, 0.38)], while the device with BFDMAc-NAI emitted at λEL of 590 nm [CIE coordinates of (0.54, 0.45)]. The redder BTDMAc-NAI-based device achieved an EQEmax of 9.2% (EQE100 dropping to 6.3%), while the orange BFDMAc-NAI-based device showed an EQEmax of 20.3%, but a large efficiency roll-off (EQE100 dropping to 10.6%).
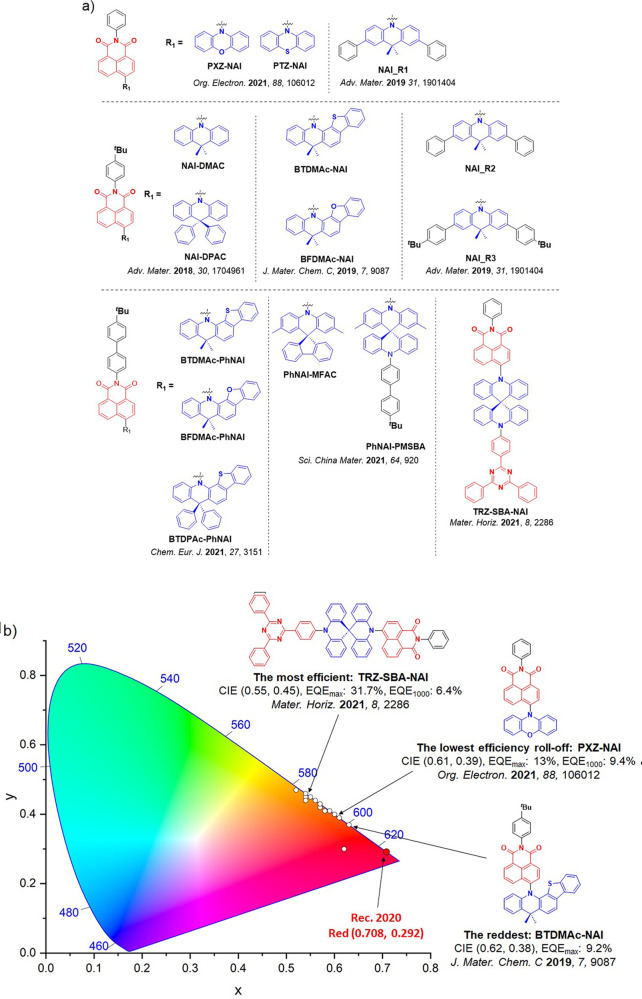
High-efficiency solution-processed OLEDs with NAI-based red emitters have been developed by Zeng et al..ref. ref531 NAI_R1, NAI_R2, and NAI_R3 contain phenyl disubstituted DMAC donors, and tert-butyl substitution on the 1,8-naphthalimide acceptor (Figure ). The phenyl extending units bestowed a stronger electron-donating ability and shallower HOMO level to these donors compared to DMAC, while also partially sterically protecting the emitter. The tert-butyl units were also attached at the para-position of the outer phenyl units of the donor and acceptor moieties, with the aim to both improve the solubility of the emitters and fine-tune their excited state energies. The emitters containing donors with tert-butyl substitution have significantly red-shifted emission (NAI_R3 at λPL of 639 nm compared to NAI_R1 and NAI_R2 both with λPL of 627 nm in toluene). NAI_R1, NAI_R2, and NAI_R3 have small ΔE ST of 90, 92, and 58 meV, and relatively high ΦPL of 63, 65, and 66%, respectively (Table S3). Solution-processed OLEDs were fabricated and displayed red λEL ranging from 610 to 622 nm, with the device with NAI_R3 exhibiting the reddest CIE coordinates of (0.60, 0.40) and also the highest EQEmax of 22.5%.
Wang et al. incorporated PXZ or PTZ donors onto an NAI acceptor, leading to λPL of 605 and 617 nm for PXZ-NAI and PTZ-NAI respectively (Figure ).ref. ref532 PXZ-NAI and PTZ-NAI have ΔE ST of 0.10 and 0.11 eV and short τd of 1.2 and 1.6 μs (Table S3). The OLEDs with PXZ-NAI and PTZ-NAI showed EQEmax of 13.0 and 11.4%, with λEL at 624 and 632 nm and CIE coordinates of (0.61, 0.39) and (0.63, 0.37), all respectively. Both devices exhibited relatively small efficiency roll-offs, with the EQE1000 values decreasing to 9.4 and 6.0%.
Zeng et al. reported a linear TADF molecule PhNAI-PMSBA (Figure ) bearing an NAI acceptor attached to a large spiro-acridan PMSBA donor, and employing a design strategy to control the orientation of the TDM of the emitter.ref. ref533 The properties and device performance were compared with a shortened reference emitter PhNAI-MFAC. PhNAI-PMSBA and PhNAI-MFAC emit at λPL of 606 and 603 nm, have ΦPL of 61 and 55%, ΔE ST of 0.06 and 0.05 eV, and short τd of 2.9 and 2.7 μs, in respective 1.5 wt% doped films (Table S3). The horizontal dipole ratio (Θ||) of PhNAI-PMSBA is 95%, enhanced compared to that of PhNAI-MFAC (Θ|| = 88%) and validating the emitter design strategy. The devices with PhNAI-MFAC and PhNAI-PMSBA emitted at λEL of 610 and 615 nm with corresponding CIE coordinates of (0.59, 0.41) and (0.60, 0.40). The reference device with PhNAI-MFAC showed an EQEmax of 22.5%, and despite its lower ΦPL the device with PhNAI-PMSBA achieved an EQEmax of 22.3%, supported by its higher outcoupling efficiency of 43.2%. Both devices exhibited severe efficiency roll-offs though, with EQE1000 values reducing to 7.6 and 5.7%, respectively. The same group also reported three orange–red TADF emitters, BFDMAc-PhNAI, BTDPAc-PhNAI, and BTDMAc-PhNAI (λPL of 600, 610, and 650 nm in toluene, Figure ), based on the same elongated acceptor coupled to three different fused heterocyclic donors.ref. ref534 All three emitters have ΔE ST < 0.16 eV and τd of around 45 μs, with ΦPL of 77, 63, and 42% in respective 1.5 wt% doped films in mCPCN. OLEDs with BTDPAc-PhNAI showed EQEmax of 18.7% with λEL at 601 nm, compared to 19.8% for the device with BFDMAc-PhNAI (λEL = 590 nm) and 10.1% for BTDMAc-PhNAI (λEL = 642 nm).
Zeng et al. reported an asymmetric linear A–D-A’ type TADF emitter, TRZ-SBA-NAI (Figure ), which contained a spiro-bisacridine donor core coupled to both an NAI and a triazine acceptor.ref. ref535 Due to the coexistence of two distinct charge-transfer excited states, dual emission was observed in toluene comprising a dominant orange-red emission and a sky-blue emission shoulder. In 3 wt% doped films in mCBPCN, TRZ-SBA-NAI has a single emission band at λPL of 577 nm, a high ΦPL of 87 %, ΔE ST of 0.16 eV, and a long τd of 398 μs. Similar in molecular design to PhNAI-PMSBA, TRZ–SBA–NAI has a Θ|| of 88% in the same films. The OLED with TRZ-SBA-NAI consequently demonstrated an outstanding EQEmax of 31.7% at λEL at 593 nm with CIE coordinates of (0.55, 0.45). The OLEDs suffered from severe efficiency roll-off though, with the EQE values reducing by 79.8% at a luminance of 1000 cd m–2.
In a more advanced molecular design, Hua et al. reported a series of emitters based on a trinaphtho[3,3,3]propellane (TNP) core that is derivatized with NAI-DMAC (Figure ).ref. ref536 The unique TNP hexagonal stacking architecture allows the D-A TADF units to be encapsulated in cavities between two adjacent TNPs, reducing quenching via aggregation and/or annihilation of long-lived triplet excitons on the active chromophore. In this series of emitters, tBu-S-mCP possesses the best photophysical properties, emitting at λPL of 604 nm with ΦPL of 70.9 % and a very small ΔE ST of 7 meV, which corresponds to a surprisingly long τd of 6.41 μs (Table S3). The solution-processed OLEDs with tBu-S-mCP showed an EQEmax of 24.7% at λEL of 594 nm, however, the devices exhibited a large efficiency roll-off, with EQE100 of 14.9%.
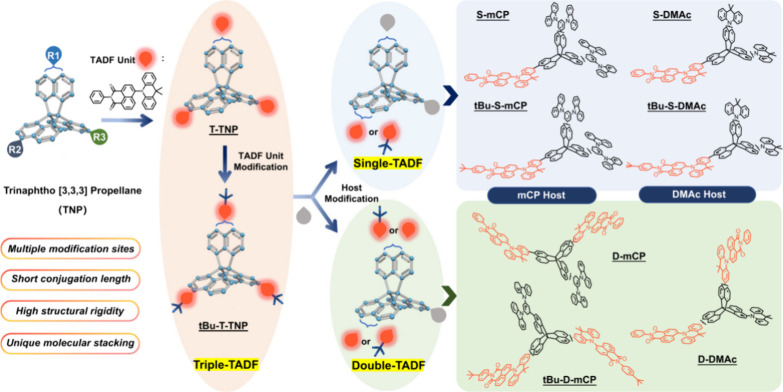
Other Miscellaneous Examples
The near-infinite scope of innovative and diverse design strategies associated with the development of red TADF emitters extends far beyond what this review can reasonably summarize. Apart from molecules using the previously discussed acceptor units, we also highlight here a collection of other notable design strategies. Kim et al. reported a highly efficient near-infrared TADF emitter, 2TPA-BF2 (Figure ) constructed from a boron difluoride curcuminoid acceptor and TPA donors.ref. ref485 By increasing the doping concentration from 2 to 60 wt% in CBP, the λPL shifted from 706 to 782 nm while the ΦPL decreased from 59 to 7.5%. The neat film also emits at 782 nm and has a ΦPL of 3.5% (Table S3). The highest ΦPL of 70% is for the 6 wt% doped film in CBP (λPL of 721 nm), which translated into a superior solution-processed device that showed outstanding NIR EQEmax of 10% at λEL of 721 nm. Quantum-chemical calculations revealed that the TADF mechanism was assisted by vibrational and spin–orbit coupling alongside a large oscillator strength, which was illustrated by the overlap of electron and hole wave functions together with a non-adiabatic coupling effect.
To further red-shift the electroluminescence, Ye et al. reported the dimeric bisborondifluoride curcuminoid dye 4TPA-2BF2 (Figure ) that emits from 760 to 801 nm, and has decreasing ΦPL (from 45.2 to 4.1%) as the doping concentration increases (from 2 to 40 wt%) in CBP.ref. ref537 From DFT calculations the ΔE ST is 0.3 eV and SOC between the S1 and T1 states is 0.13 cm–1. Solution-processed devices showed an EQEmax of 5.1% at λEL of 758 nm, supported by its high ΦPL of 45.2%. These advances in NIR OLEDs, though unsuitable for displays and lighting, can unlock new technological applications in sensing, LIDAR/optical wireless networking, and biological imaging through the tissue transmission window.ref. ref538 Utilizing the same difluoride curcuminoid acceptor with carbazole or DPA donors, Jin et al. reported the red TADF emitters DPhCzB and DTPAB.ref. ref539 Neat films of DPhCzB and DTPAB emit at λPL of 637 and 650 nm and have ΦPL of 54 and 56%, all respectively (Table S3). In 5 wt% doped films in mCP, the ΦPL increased to 87 and 97%, although the emission blue-shifted to λPL of 587 and 605 nm. Solution-processed OLEDs with DPhCzB and DTPAB showed EQEmax of 6.7 and 8.2% at λEL 587 and 605 nm, all respectively. Interestingly, both devices showed low efficiency roll-offs of 3.7 and 9.0% at 100 and 1000 cd m–2 which was attributed by the authors to the respective τd of the emitters (19.5 and 55.7 μs).
Kumar et al. reported the doubly boron-doped emitter DMAC2oDBA (Figure ), based on a 9,10-diboraanthracene (DBA) acceptor decorated with ortho-substituted acridine donors.ref. ref540 This compound emits at λPL of 602 nm and has a moderate ΦPL of 44% in 20 wt% doped film in CBP. The low ΔE ST of 54 meV was attributed to the very strongly twisted conformation, supported by the additional ortho-methyl substitution of the phenylene linkers. However, due to its ΦPL, the EQEmax was limited to 10.1% at λEL 615 nm. Utilizing the same DBA acceptor, Hsieh et al. reported orange-red emitters, dPhADBA, dmAcDBA, and SpAcDBA by attaching either DPA, DMAC, or spiro-acridine, respectively.ref. ref488 In contrast to ortho substitution, these para-substituted compounds showed increased ΦPL ranging from 53 to 85% and λPL between 570 to 614 nm in 12 wt% doped films in CBP (Table S3). These compounds have fast k RISC (1.3–2.4 ×105 s–1) resulting from the small ΔE ST values, ranging from 0.04 to 0.08 eV. The fast RISC and highly horizontal TDM orientation ratio of between 84–86% translated into devices with EQEmax ranging from 11.1 to 30.0%, tracking with the ΦPL of the emitters, at λEL from 567 to 613 nm.
Karthik et al. reported red TADF emitters PzTDBA and PzDBA (Figure ), constructed from rigid oxygen-bridged boron acceptors (DOBNA, see Section MR-TADF) and a central dihydrophenazine donor.ref. ref489 PzTDBA and PzDBA emit at λPL of 599 and 610 nm, have high ΦPL of 99.8 and 85.4%, small ΔE ST of 0.06 and 0.05 eV, short τd of 2.63 and 2.00 μs, and fast k RISC of 1.19 and 0.84 ×106 s–1, all respectively in 5 wt% doped films of TCTA/Bepp2 (1:1) mixed ambipolar host (Table S3). The devices with PzTDBA and PzDBA showed EQEmax of 30.3 and 21.8% and extremely low efficiency roll-off (reducing by 3.6 and 3.2% of maximum values at 1000 cd m–2) at λEL of 576 and 595 nm, respectively. Impressively, the devices with PzTDBA and PzDBA showed operating device lifetimes (LT50) of 159 and 193 h at 1000 cd m–2, also respectively.
Kumsampao et al. reported the NIR TADF D-A-D emitter TPACNBz (Figure ) based on strongly electron-deficient 5,6-dicyano[2,1,3]benzothiadiazole (CNBz) acceptor and TPA donors.ref. ref541 TPACNBz emits at λPL of 750 nm and has a ΦPL of 21% as a neat film (Table S3). The emission blue-shifts to 710 nm and the ΦPL increases to 52% in 30 wt% doped films in CBP, and the OLEDs showed an EQEmax of 6.6% at λEL of 712 nm. These results clearly demonstrate that this acceptor, commonly used in OPV dyes, is an excellent building block for creating low-band-gap emitters.
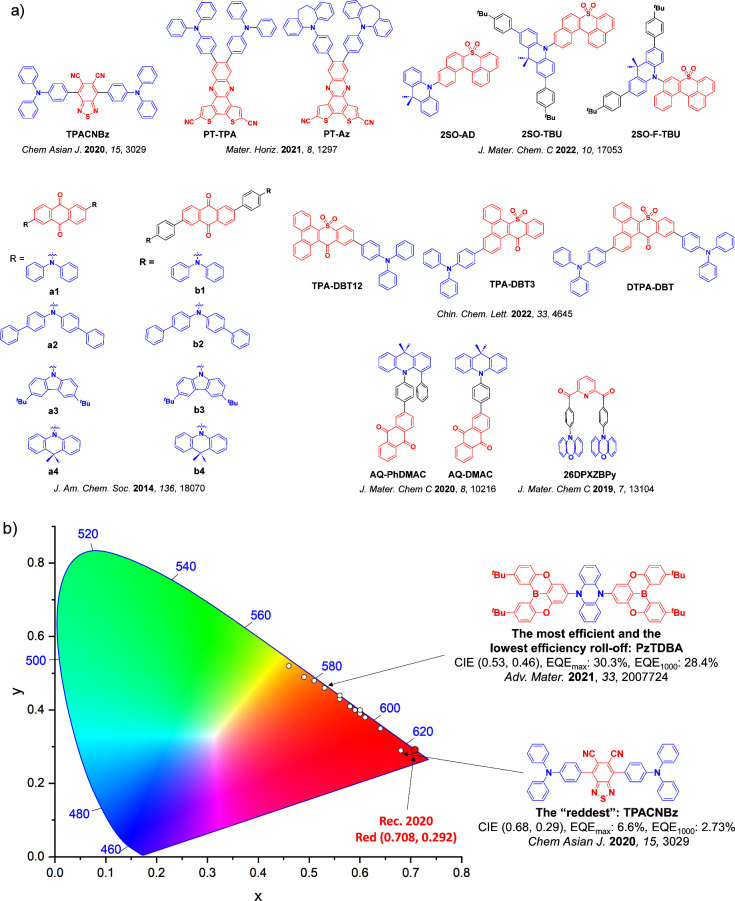
Wang et al. nicely demonstrated the impact of chromophore rigidity and/or flexibility on photophysics and the corresponding device performance.ref. ref542 Red emitters PT-TPA and PT-Az (Figure ) containing dithieno[3,2-a:2′,3′-c]phenazine acceptor and either a flexible TPA donor in PT-TPA or a relatively rigid Az donor in PT-Az were investigated. This structural variation in the donors of PT-TPA and PT-Az did not alter the energy levels of the S1 and T1 states to any appreciable extent, and in toluene both compounds have similar respective ΔE ST of 0.26 and 0.28 eV and ΦPL of 66.5 and 56.3% (Table S3). In PT-Az the rotation of terminal phenyl groups is constrained by an ethylene linker, leading to its inferior ΦPL. In contrast, PT-TPA with freely rotating phenyl groups has a low reorganization energy and a larger transition dipole moment for the S1–S0 transition, which resulted in a high k r of 2.31 × 107 s–1 (PT-Az k r = 2.33 × 106 s–1). In 15 wt% doped films in CBP PT-TPA has a near unity ΦPL of 99.7% (τd = 57.79 μs) while it is much lower at 52.7% (τd = 27.68 μs) for PT-Az, attributed to out-of-plane wagging vibration modes associated with the restricted Az units of the emitter contributing to increased non-radiative decay. OLEDs showed EQEmax of 29.7% (λEL = 632 nm) for PT-TPA and 14.1% (λEL = 612 nm,) for PT-Az.
Hu et al. reported the use of a rigid dibenzothioxanthone (DBT) acceptor that possesses a low-lying localized triplet excited state to facilitate effective RISC.ref. ref543 Isomeric D-A emitters TPA-DBT12, TPA-DBT3, and D-A-D DTPA-DBT (Figure ) emit at 597, 616, and 632 nm and have ΦPL of 44, 55, and 42%, in respective 5 wt% doped 35DCzPPY films (Table S3). Red OLEDs showed EQEmax of 14.5, 15.0, and 11.8% at λEL of 608, 612 and 628 nm, for the same emitters, respectively. Gao et al. employed a similar approach using a modified dibenzothioxanthene acceptor that had a low-lying localized triplet excited state.ref. ref544 2SO-AD, 2SO-TBU, and 2SO-F-TBU additionally contained bulky acridine donors to suppress ACQ (Figure ). The three compounds doped in 10 wt% 35DCzPPY films emit at 581, 615, and 591 nm and have ΦPL of 25, 58, and 53%, respectively. 2SO-AD, 2SO-TBU, and 2SO-F-TBU have ΔE ST values of 0.27, 0.14, and 0.20 eV and long τd of 553.0, 272.1, and 577.5 μs, all respectively. Red OLEDs with 2SO-AD, 2SO-TBU, 2SO-F-TBU showed EQEmax of 3.2, 16.3, and 14.5% with λEL of 599, 608, and 612 nm, also respectively.
Anthraquinone (AQ) has also been exploited as an acceptor unit in the design of red emitters owing to its deep LUMO (-2.80 eV). The first AQ-based TADF red emitter was reported by Zhang et al. where they synthesized four D-π-A-π-D type emitters (b1, b2, b3, and b4) with an AQ acceptor (Figure ).ref. ref545 They incorporated various donors such as DPA, BBPA, DTC, and DMAC, and employed phenyl (Ph) rings as π-bridges, respectively. The synthesized emitters were also compared to the corresponding D-A-D type emitters (a1, a2, a3, and a4). For all of these emitters the measured ΔE ST values are relatively small and showed a gradual decrease in magnitude with increasing donor strength from DPA to DMAC. For molecules a1–4, the ΔE ST values range from 0.08 to 0.29 eV, while for molecules b1–4, the values range from 0.07 to 0.24 eV, all in 1 wt% doped films in CBP. However, neither the λPL nor ΦPL strictly correlated with the donor strength. For molecules a1–4, the λPL (ΦPL) are 593 (0.5%), 603 (0.4%), 575 (0.5%), and 600 nm (0.1%) in the same respective films. By comparison, for molecules b1–4, the equivalent λPL (ΦPL) are 594 (0.8%), 601 (0.8%), 550 (0.7%), and 564 nm (0.5%). The b1– and b2-based OLEDs in 10 wt% CBP host emitted at λEL of 624 and 637 nm with corresponding CIE coordinates of (0.61, 0.39) and (0.63, 0.37), while b3 and b4 OLEDs emitted at λEL of 574 nm and 584 nm, all respectively. The devices with b1–4 showed EQEmax of 12.5, 9.0, 9.0, and 6.9%, but the devices with b1 and b2 exhibited severe efficiency roll-off, decreasing to 8.1 and 5.7% at a luminance of 100 cd m–2, and to 2.3 and 1.7% at a luminance of 1000 cd m–2, all respectively. This was attributed to the long τd of 416 and 185 μs observed for b1 and b2 in respective 1 wt% doped films in CBP. Material b4 having a much shorter τd of 6.5 μs translated in devices with much reduced efficiency roll-off of 6% at a luminance of 1000 cd m–2. Emitters b3 exists as a mixture of rotamers in the doped CBP films, with some having a short τd of 16 μs comparable to b4, while others having a long τd of 156 μs comparable to b2. Consequently, the efficiency roll-off of the OLEDs with b3-based fell between those of the devices with b2 and b4.
Hao et al. reported the emitter AQ-PhDMAC (Figure , containing a phenyl-substituted DMAC) and compared it to AQ-DMAC.ref. ref546 Owing to the steric effect of the α-phenyl ring, AQ-PhDMAC emits at λPL of 586 nm, has a ΔE ST of 0.22 eV, a τd of 63.6 μs, and a high ΦPL of 89%, trading off TADF performance for higher ΦPL compared with AQ-DMAC (λPL = 580 nm, ΔE ST = 0.02 eV, τd = 21.2 μs, and ΦPL = 63%) (Table S3). The orange-red OLED with AQ-PhDMAC showed an EQEmax of 18.1% which was higher than the device with AQ-DMAC (EQEmax of 13.9%). Both devices emitted similarly at λEL of 580 nm and with CIE coordinates of (0.49, 0.49); however, the AQ-PhDMAC-based device exhibited more serious efficiency roll-off arising from its longer τd.
Pandidurai et al. reported yellow-orange TADF emitter 26DPXZBPy (Figure ), containing dibenzoyl pyridine as the acceptor and PXZ as the donors.ref. ref547 26DPXZBPy emits at around 600 nm, has a ΦPL of 76%, a small ΔE ST of 0.04 eV, and a τd of 1 μs in 10 wt% doped films in mCBP (Table S3). Devices with 26DPXZBPy gave orange emission at λEL of 590 nm at CIE coordinates of (0.49, 0.49), and showed an EQEmax of 13.7%.
Outlook
TADF emitters based on N-doped PAH acceptors have drawn significant attention towards the development of efficient red OLEDs. There are now examples of devices with reported EQEmax exceeding 30%. As a representative example, OLEDs with DCPPr-α-NDPA achieved an EQEmax of 31.5% at λEL= 606 nm (Figure ).ref. ref526 To achieve deeper-red emission though, acceptors with a small degree of π-conjugation such as pyridine, quinoxaline, and acenaphtho[1,2-b]pyrazine require fortification with strong electron-withdrawing units like nitrile or fluorine to stabilize the CT singlet states. For example, pCNQ–TPA contains an acceptor decorated with two nitrile units, and the corresponding devices demonstrated deep red electroluminescence at λEL of 660 nm and an outstanding EQEmax of 30.3%.ref. ref525
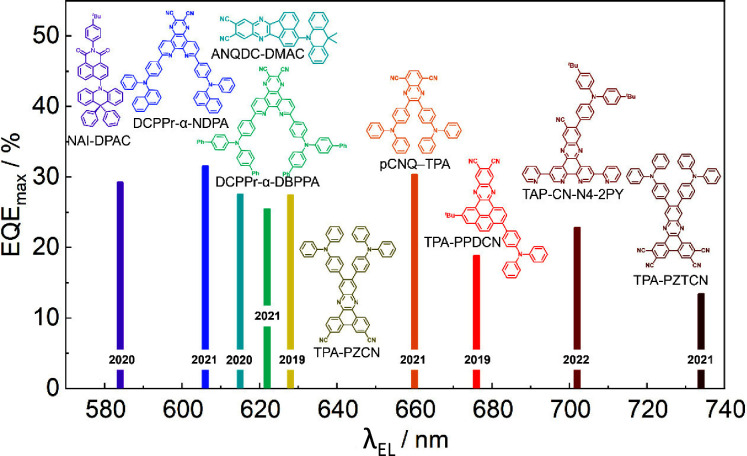
Extending past the visible spectrum, OLEDs with TPA-PZTCN, containing a π-expanded acceptor, exhibited intense NIR EL (EQEmax = 13.4%) at a λEL of 734 nm, which is particularly impressive in this wavelength region where the energy-gap law typically limits emission efficiency. This strategy of π-expansion can have drawbacks though, as acceptors with too large a π-conjugation like phenanthro[4,5-fgh]quinoxaline can result in D-A compounds with larger ΔE ST due to the presence of a too-stabilized acceptor LE state. Illustrating this balance, although the device with TPA-PPDCN showed an EQEmax of 18.8% at a λEL of 676 nm, there was also serious efficiency roll-off (EQE100 = 3.7%)ref. ref527 resulting from the moderate ΔE ST of 0.23 eV. Indeed, many of the examples in this section demonstrate the challenge of obtaining the desired deep-red emission while also preserving high EQE and low efficiency roll-off. There is certainly still pressing need for new emitter designs that address these deficiencies in device performance.
Aside from N-PAH acceptors, the recent use borondifluoride curcuminoids, diboraanthracene, dibenzothioxanthone, and 5,6-dicyano[2,1,3]benzothiadiazole acceptors have also shown promise in delivering deep-red and NIR emission. Additionally, while strong intermolecular interactions usually have a negative impact on device efficiency and stability, they are not always detrimental to performance (Section sec13 ), and can offer a route to red-shifted emission spectra. Since red TADF emitters with low T1 energies can take advantage of the full range of available OLED host materials (in contrast to high T1 blue emitters), we expect that development of red emitters in the coming years will follow in tandem with the development of similarly unrestricted green TADF emitters – although a few years behind in terms of raw performance metrics as more challenging energy-gap law and ACQ considerations are navigated by the research community. Red emission, lowest in energy in the visible wavelength range, can also take the fullest advantage of hyperfluorescence approaches (Section sec17 ), and thus benefit directly from performance advancements in materials of other colors. With multiple promising strategies clearly identifiable, the pace of progress and achievement in often-neglected red TADF OLEDs is hence likely to rise to match that of other colors in the near future.
White OLEDs Using TADF Materials
Introduction
White OLEDs (WOLEDs) show great potential for use in efficient and low power, flexible, large-area displays and lighting.ref548,ref549 Considering the now-established successes of high-performance red (R, Section sec5 ), green (G, Section sec4 ), and blue (B, Section sec3 ) TADF OLEDs, it is natural and promising to develop high-performance WOLEDs using TADF materials.ref. ref42 In this section we first summarize the distinct performance metrics and engineering challenges associated with WOLEDs (in comparison to single color devices), and then highlight the range of molecular and device design strategies that use TADF toward this goal.
Although WOLEDs understandably share many similarities with single color OLEDs, their performance is evaluated based on subtly different criteria, which include power efficiency, color quality, and color and operational stability. For WOLEDs, the power efficiency (PE, lm/W) is a more important parameter than EQE alone, which is closely related to the overall power consumption.ref. ref11 Whereas the EQE values of both single color and white TADF OLEDs have achieved the theoretical limits, it is not easy to optimize the PE values, especially for the intrinsically complicated WOLEDs. For both OLED categories, high PE value is often achieved by tuning the types and thicknesses of functional layers including transport layer and host, in order to make charge injection barriers between layers (and therefore the required driving voltages) as low as possible while also maintaining the EQE. To be competitive with commercial lighting luminaires such as those based on fluorescent tubes, a PE of greater than 90 lm/W, and ideally greater than 120 lm/W is expected. Furthermore, for lighting applications brightnesses of thousands of cd m–2 are typically required, with corresponding higher exciton densities and larger demand for triplet harvesting presenting a challenge for current TADF materials.
The second of these assessment criteria is the quality of the white light. As previously mentioned, there are two main kinds of white light: cool white and warm white. Cool white light has CIE coordinates of (0.33, 0.33), with a correlated color temperature (CCT, equating color to that of blackbody radiation at a set temperature) of around 5000 K. Warm white light has CIE coordinates of (0.448, 0.408) and a CCT corresponding to a lower temperature of 2856 K.ref. ref550 While ‘cool’ white corresponding to a hotter CCT appears at first contradictory, it is more readily understood when considered in the colloquial sense of ‘white-hot’ and less extreme ‘red-hot’ thermal emission. Warm white light therefore contains a smaller contribution from blue emission and is typically used in domestic lighting, whereas cool white light luminaires are more frequently found in commercial and industrial settings. Other types of white light with variable associated CCT exist both between and beyond these extremes, although these two have become de facto standards in research and industry.
An important associated parameter for the quality of the light is the color rendering index (CRI), ranging between 0 and 100. This index describes the degree to which the light source can resemble a ‘natural’ light source with a continuous blackbody emission spectrum, such as sunlight. Distinct from incandescence, WOLEDs can instead exhibit emission spectra that have some visible wavelengths overexaggerated, and others completely absent. Although such emission spectra may still be physiologically averaged to produce a perceived white CIE coordinate, illumination with such an OLED (with low CRI) will produce perceptible color changes in any illuminated objects. This is because some wavelengths that contribute to the normal reflectance and perceived color of the object are absent from the illumination source, and so balanced emission intensity at all visible wavelengths is required to achieve high CRI. For typical luminaires a CRI value of 80 is required, whereas for specialized applications such as art displays, in hospitals, and the textile industry, CRI values of over 90 are expected.
Finally, as with single-color OLEDs the stability of the WOLED is vital for commercialization. WOLEDs for luminaires must, however, show both color stability and device stability under continuous operation. As WOLEDs typically employ multiple emitter species, each with their own triplet harvesting performance and overall stability, the amount of emission from each and therefore also the overall color and CIE coordinates of the device can change significantly both at different brightnesses and over time. This can be assessed by CIE variation (both at different driving currents, and over time) and device lifetime, whereby smaller CIE variation and longer device lifetime are desired. For lighting applications, device stability is typically quantified in terms of LT50, which indicates the time at which the overall EL intensity is at 50% of its initial value (usually taken at 1000 cd m–2) under constant current driven conditions.
To fulfil all the above criteria, both high-performance emitters and rational designs for device structure are needed. Although all-phosphor based WOLEDs with maximum PE (PEmax) of over 100 lm/W have been reported, the poor stability of blue phosphorescent emitters renders them unsuitable for commercial applications.ref. ref551 Instead, a hybrid device structure currently enjoys widespread commercialization in which blue and the complementary colors (green, G, yellow/orange, Y/O and red, R) are generated from fluorescent and phosphorescent dyes, respectively.ref. ref552 In these vertically stacked multilayer devices, careful exciton management is crucial to excite the different layers in the correct ratios, harvest all the excitons, suppress unintentional exciton energy transfer, and ensure device operational stability. These simultaneous considerations result in a complicated device structure and doping scheme.ref553,ref554
The arrival of high-efficiency TADF materials has stimulated new strategies to manipulate excitons, optimize device structure, and ultimately improve WOLED device performance. There are several potential advantages and ways of using TADF materials in WOLEDs; they could serve as emitters, as hosts, as sensitizers, or combinations of these functions. Efficient exciton harvesting is certainly achievable, a prerequisite evidenced in single color TADF OLED devices. Indeed using TADF molecules, WOLEDs with EQEmax of 30% have already been achieved and surpassed, indicating that further advancement is limited now only by the light out-coupling efficiency.ref555−ref556ref557 Furthermore, by using an exciplex-type TADF host, WOLEDs with PEmax of over 80 lm/W have been reported.ref. ref558 Ultimately, the typical donor-acceptor molecular structure reported for TADF emitters and the associated capacity to fine tune the photophysical properties of the emitters provides a large freedom in materials design to generate white light systems. Dual-emission properties associated with this kind of D-A structure have also enabled a small number of examples of single molecule white TADF emitters, that have been explored in WOLEDs.ref. ref557 These emission properties – unrelated to TADF-activity but exceedingly rare for simple fluorescent molecules – provide an avenue to fabricate WOLEDs with considerably simpler device structures and therefore lower fabrication cost. Furthermore, contrary to the requirement for high color purity in displays, the use of CT emitters showing broad emission (FWHM: 70–120 nm) is desirable in WOLEDs to achieve a high CRI.
From all of these potential advances of using TADF materials in WOLEDs, there emerge two main design approaches to obtain white light emission: two-color and three-color systems. In two-color systems, the blue light originates in most cases from a TADF emitter, while the yellow or orange component comes from a separate phosphorescent, fluorescent, TADF exciplex or TADF emitter. This approach benefits from simpler device design, but without a dedicated green emitter often struggles to achieve high CRI. In three-color systems, TADF emitters have been used as one or more of the separate red, green, and blue components. For WOLEDs based on a three-color system, a CRI above 80 has been reported, which is still rare for most two-color systems.ref559,ref560
Using TADF components, the complexity of device structure and exciton management in WOLEDs can be somewhat mitigated as well. Examples of an emitting layer (EML) containing only TADF molecules as both the emitter and host, non-doped TADF EMLs, single EML TADF WOLEDs, and single molecule white TADF emitters have all been reported.ref561−ref562ref563ref564 The simplified device structure eases the device fabrication and reduces the number of associated optimization parameters, reducing costs for both research and development as well as for commercial production. Although device stability studies are quite limited, especially in terms of identifying the degradation mechanism, recent reports show encouraging evidence of stable TADF WOLEDs. The LT50 of hybrid TADF WOLEDs can now exceed 104 hours, while the LT80 of all-fluorescent TADF WOLEDs have reached over 8200 hours.ref565,ref566 Additional systematic studies are needed to thoroughly understand the degradation mechanisms in TADF WOLEDs. While such studies are both fundamentally and practically challenging to perform, the resulting insights will ultimately inform the design of materials leading to improved device performance.
Separating from their classification as two- or three-color devices, WOLEDs can be divided into three categories depending on the photophysical properties of the individual color components as shown in Figure . Hybrid TADF WOLEDs contain both phosphorescent and TADF emitters, all-fluorescent TADF WOLEDs contain either a combination of fluorescent and TADF emitters or TADF emitters only, and lastly single molecule TADF WOLEDs have also been demonstrated. In view of their importance and potential in industry, only vacuum-evaporated small molecule WOLEDs are considered in this section. Other related topics such as solution-processed WOLEDs, polymer-based WOLEDs, and out-coupling enhancement techniques are summarized in other sections and elsewhere.ref550,ref567,ref568 Unless indicated, the device characterization is performed in the forward-viewing mode and without the aid of a light out-coupling structures.
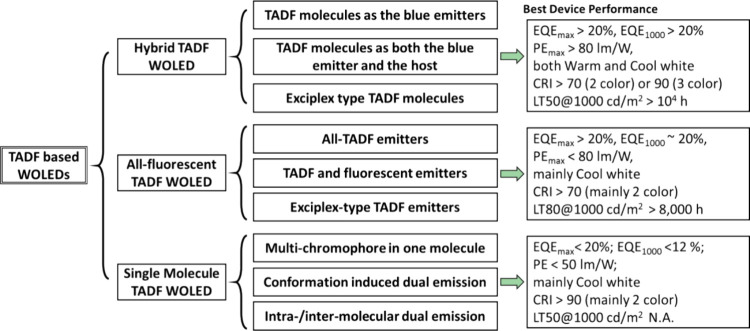
Hybrid TADF WOLEDs
The lack of available high-efficiency and stable blue emitters (and stable high triplet energy hosts) remains a bottleneck for high performance WOLEDs and displays.ref. ref21 Even today a hybrid strategy is adopted in industry, in which a stable blue fluorescent or TTA emitter and high-efficiency phosphorescent emitters of complementary colors are deposited inside the EML(s). To harvest all the excitons, the blue fluorescent emitter should have a higher triplet energy level than the phosphorescent emitters to avoid triplet energy trapping and subsequent triplet exciton quenching on the non-harvesting blue emitter. By careful control of the doping concentration, layer thickness, interlayer distance and EML architecture, the singlet excitons of the blue emitter can decay radiatively while its triplet excitons can diffuse to nearby phosphorescent emitters where they are harvested and radiatively decay efficiently. FRET transfer from the blue emitter to other color emitters can also occur, meaning that the balance of emission and overall color are extremely sensitive to dopant concentrations. Despite the desirable performance metrics of hybrid WOLEDs, the complicated device structure and delicate exciton management produce challenges for device fabrication and quality control. In addition, there are a limited number of blue fluorescent emitters that have sufficiently high triplet energy levels to be used within this device architecture, while the use of low-triplet TTA blue emitters only partially alleviated the issue of triplet quenching due to the fundamentally lower IQE limits of the TTA triplet harvesting channel.ref. ref569
High-efficiency blue TADF emitters can not only address the triplet harvesting issue – boosting the EQE of WOLEDs up to or even beyond 20% – but also enable new exciton manipulation strategies and device architectures serving as emitters, sensitizers, and/or hosts. Hybrid TADF WOLEDs with phosphorescent emitters and TADF components already show impressive device performance with reported EQE1000 greater than 20%, PEmax over 80 lm/W, and the CRI greater than 70 (two-color) or 90 (three-color). The device stability can also be promising despite widespread stability issues for blue TADF emitters and associated high-triplet hosts, with LT50 longer than 104 h demonstrated (though for commercial applications typically 20,000 h is required).ref565,ref570,ref571 Depending on the specific role of the TADF components in hybrid TADF WOLEDs, devices can be further subcategorised into those with a TADF blue emitter, those with TADF molecules as both the blue emitters and the host, and those with exciplex-type TADF emitters or hosts. The typical molecular structures are shown in Figure .
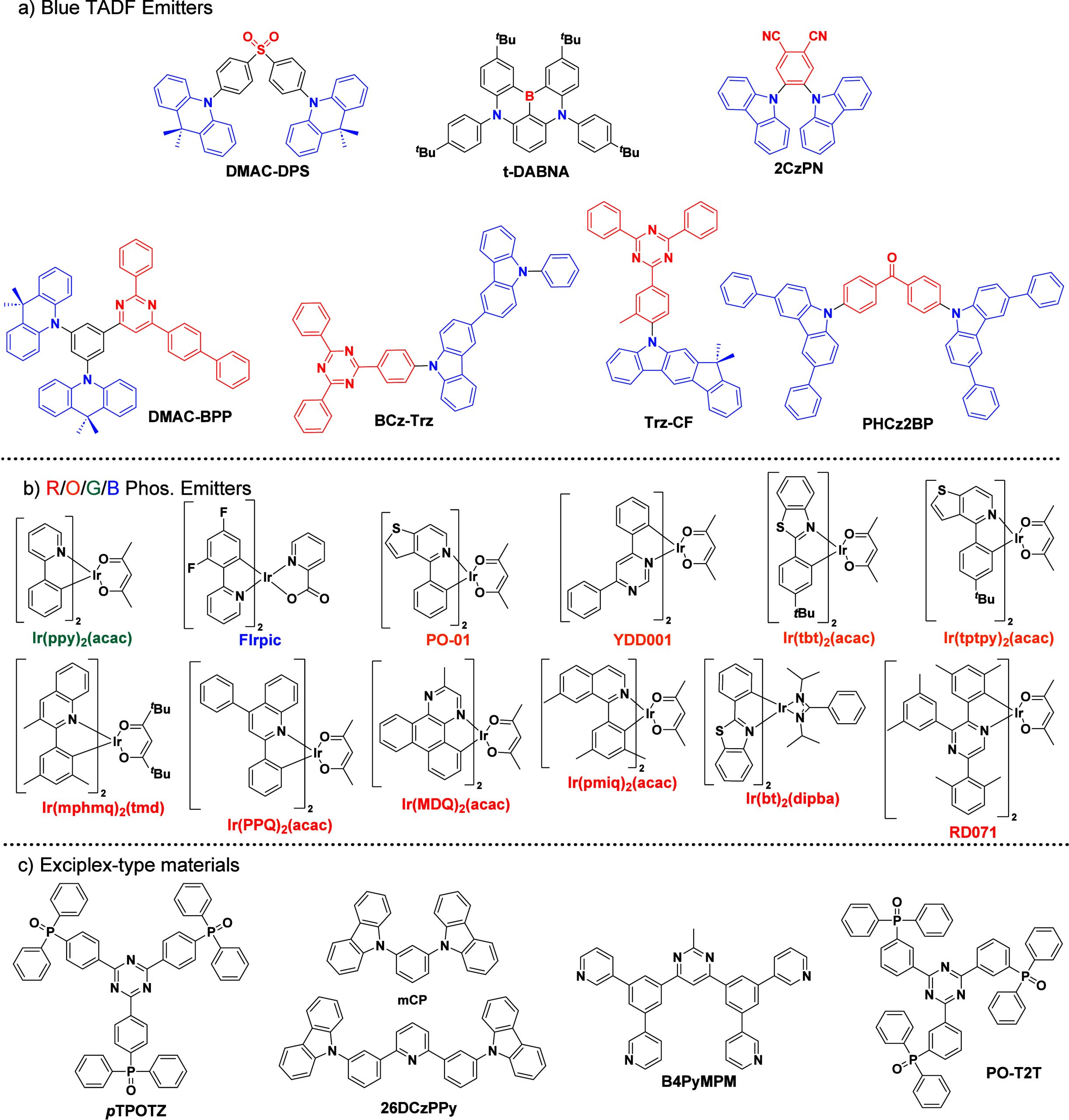
TADF Molecules as Blue Emitters
With high efficiency blue TADF emitters such as DMAC-DPS, 2CzPN, and t-DABNA (Figure ), singlet excitons formed by direct charge recombination can either radiatively decay to generate blue prompt fluorescence (PF), or transfer to lower energy phosphorescent emitters by FRET. Meanwhile, triplet excitons either undergo RISC to produce either blue delayed fluorescence (DF) or can diffuse to nearby phosphorescent emitters by a Dexter energy transfer process. In this manner, all the generated singlet and triplet excitons can be harvested, leading to IQEs of up to 100%. Typical phosphorescent emitters used in conjunction with blue TADF emitters in hybrid WOLEDs include red Ir(MDQ)2(acac), orange emitter PO-01, and green Ir(ppy)2(acac). To improve the device performance, much effort has been devoted to designing and optimizing the EML structure for efficient exciton energy transfer, confinement, and distribution; however, studying the energy transfer pathways directly remains challenging due to the number and complexity of processes involved.ref. ref104
Doped Single or Multiple EML
The most direct strategy for white light generation is to dope all the emitters (commonly blue and orange emitters) within a suitable host matrix into one single EML, i.e., S-EML. Through careful control of the doping concentration of each emitter, the extent of energy transfer and thus the ratio of blue and orange emission can be tuned, resulting in white light emission and high EQE. For an efficient exciton harvesting scheme, singlets should be confined to the TADF emitters, or transferred partly from the TADF emitters to the phosphorescent emitters via FRET. Triplet excitons formed on the TADF emitter are either up-converted to singlets by efficient RISC or diffuse to phosphorescent emitters by Dexter energy transfer. Therefore, as previously mentioned, the emission spectrum is exquisitely sensitive to the doping concentrations, which is usually kept lower than 0.5 wt% for the orange emitter to give balanced or warm white emission. As an illustrative example, using this strategy t-DABNA:PO-01 and DMAC-DPS:PO-01 co-doped S-EML WOLEDs were fabricated, showing efficient warm and cool white emission with CIE coordinates of (0.41, 0.47) and (0.33, 0.37), and high EQEmax (EQE1000) of 19.2% (15%), and 22.4% (18.3%), respectively.ref572,ref573,ref556 However, this all-in-one EML strategy with very low doping concentration of one emitter leaves little room for further device optimization.
Multiple emitting layer (M-EML) structures, including directly adjacent doped EML stacks or those separated by interlayers, provide more freedom and control to tune the emission spectrum, confine the excitons, and maintain efficiency and device lifetime. An example of such a WOLED used the TADF emitter DMAC-DPS as a sensitizer for the fluorescent blue emitter TBPe in a blue ‘hyperfluorescence’ EML, and a yellow emitter YDD001 in a yellow EML. This WOLED produced PEmax approaching 70 lm/W, EQEmax (EQE1000) of 20% (11.3%), and a device lifetime LT50 of over 1500 h at 1000 cd/m2.ref. ref574 Although the interlayer connecting the two EMLs was carefully tuned for better carrier balance, the best device still had a poor CRI of only 44. To improve the color quality, a three-color system was explored in another study. This time, WOLEDs comprising one TADF doped blue EML (B) of DMAC-DPS and one phosphorescent co-doped EML of Ir(PPQ)2(acac) (R) and Ir(ppy)2(acac) (G), demonstrated an EQEmax (EQE1000) of 23% (17.5%), and a CRI as high as 89.ref. ref559 Due to the well-confined excitons, all M-EML devices showed good color stability at high brightness.
Non-doped Multiple EML
An ultrathin non-doped M-EML structure can alleviate many limitations arising from host material selection, co-evaporation process, and dopant distribution. However, it requires the emitters to show negligible ACQ, and a careful control of the EML thickness. For example, using a DMAC-DPS (7 nm)/PO-01 (0.08 nm)/DMAC-DPS (7 nm) M-EML, warm white light devices with CIE coordinates of (0.44, 0.48) were generated with EQEmax (EQE1000) of 9.1% (7.1%)ref. ref575 Further increasing the number of EML, a seven-layer non-doped M-EML warm WOLED consisting of alternating DMAC-DPS (2.5 nm, B), Ir(MDQ)2(acac) (0.03 nm, R), and Ir(ppy)2(acac) (0.09 nm, G) layers, was fabricated with CIE coordinates of (0.42, 0.42), EQEmax (EQE1000) of 19.1% (17.3%), and a high CRI value of 83.ref. ref576 Similarly, a cool white light device with CIE coordinates of (0.26, 0.36) was generated using an ultrathin non-doped phosphorescent layer, Ir(tbt)2(acac) (0.1 nm, Y), sandwiched between two doped TADF layers, DPEPO:DMAC-DPS (9 nm, B). This device possessed an EQEmax of 15.7%, decreasing to 12.1% for EQE1000, and a stable EL spectrum at up to 104 cd/m2.ref. ref577
TADF Molecules Acting as Both the Blue Emitter and the Host
Blue TADF molecules resistant to ACQ effects (and so maintain high ΦPL in neat films) can serve as both the blue emitter and as a host for phosphorescent emitters in hybrid TADF WOLEDs. Not only does this simplify the EML structure, but it also facilitates direct exciton energy transfer between emitters, enabling improved device efficiency and stability. However, due to the rapid exciton energy transfer of both singlets and triplets from the blue TADF host to the phosphorescent emitters, the EL spectrum of the device once again depends sensitively on the doping concentration of the phosphorescent emitters, which is usually kept below 3 wt%.
Representative of this approach, with a low doping concentration of the orange PO-01 phosphorescent emitter in the blue TADF molecule Trz-CF (0.8 wt%), two-color S-EML WOLEDs showed CIE coordinates of (0.38, 0.45), low efficiency roll-off with EQEmax (EQE1000) of 20.3% (20.1%), and LT50 of over 1,000 h, which was attributed to the balanced bipolar carrier transport and efficient exciton harvesting of Trz-CF.ref. ref251 However, the dominant emission at around 560 nm from PO-01 results in an EL spectrum that deviates from a standard white light source, which can be improved by replacing PO-01 with another emitter or using a three-color system. With the red phosphorescent emitter, Ir2 (0.2 wt%) doped in a highly efficient blue TADF molecule, D-tCz-D-BP, S-EML WOLEDs showed slightly reduced EQEmax of 18.8%, but similar CIE coordinates of (0.41, 0.42), and CRI of around 80.ref. ref578
Iterating this same strategy, a M-EML WOLED was fabricated using red [Ir(pmiq)2(acac)] and yellow (PO-01) phosphorescent emitters doped separately into the blue TADF emitter DMAC-BPP. This device showed CIE coordinates of (0.50, 0.42), with EQEmax of 15.6% (EQE1000 of 14%), and a CRI of 86.ref. ref579 Similar results were reported by using a new blue bipolar TADF molecule PHCz2BP as the host for green [Ir(ppy)2(acac)] and red [Ir(bt)2(dipba)] phosphorescent emitters. The M-EML warm-white WOLEDs showed CIE coordinates of (0.41, 0.46), high EQE with low efficiency roll-off, i.e., EQEmax (EQE1000) of 25.6% (25.1%), and CRI of 85.ref. ref570 To simplify the EML structure, co-doping of green [Ir(ppy)2(acac)] and red [Ir(mphmq)2tmd] phosphorescent emitters together in the blue TADF molecule DMAC-DPS was proposed. S-EML WOLEDs generated efficient cool white light with EQEmax (EQE1000) of 20.2% (19.4%), CIE coordinates of (0.36, 0.39), and CRI of 85.ref. ref580
Exciplex Type TADF Molecules
Exciplex blends consisting of donor and acceptor molecules are ambipolar by nature, facilitating the transport of both holes and electrons, which is helpful for reducing the carrier injection barrier and balancing bipolar carrier transport in devices. These valuable transport properties – rarely possessed by individual TADF molecules or hosts – can improve device performance, especially in terms of power efficiency and device lifetime. This concept is covered thoroughly in Section sec8 . Exciplexes can be formed either through the mixing of donor and acceptor molecules (bulk exciplex), or by depositing layers of donor and acceptor molecules on top of each other (interfacial exciplex). By carefully matching the energy levels, balancing ambipolar carrier transport, and optimizing doping concentration (in the bulk exciplex), low turn-on voltage, high PE, and long device lifetime can be achieved in hybrid TADF WOLEDs. Because of the extreme decoupling of CT excitons that can form between exciplex D-A pairs, intrinsically low ΔE ST for these materials also often bestows them with TADF and triplet harvesting properties alongside any molecular TADF or phosphorescent dopants.
Wu et al. developed a co-doped mCP:B4PyMPM (Figure ) system, which by itself showed efficient bulk exciplex emission with a high triplet energy and TADF behavior.ref. ref571 S-EML WOLEDs with blue (FIrpic, 15 wt%) and orange (PO-01, 0.2 wt%) phosphorescent emitters co-doped into the mCP:B4PyMPM host were fabricated, showing PEmax as high as 105 lm/W, EQEmax (EQE1000) of 28.1% (21.5%), and CIE coordinates of (0.40, 0.48). However, the degradation of the warm white color into cool white was observed upon increasing the brightness, indicating an exciton-density dependant bottleneck in energy transfer to the orange emitter.
Besides serving as an efficient ambipolar host, some exciplex-type TADF hosts can directly provide blue emission, which further simplifies the EML structure. The bulk exciplex consisting of a co-doped mCP:pTPOTZ layer shows both blue PL and EL emission.ref. ref581 When doping PO-01 into a mCP:pTPOTZ layer, a warm white light was produced with CIE coordinates of (0.43, 0.49), the devices showing EQEmax (EQE1000) of 24.6% (22%), CRI of 71, and high PEmax of 90 lm/W. The EL spectrum was quite stable with increasing brightness.
The use of interfacial exciplexes has also been explored, for example using the PO-T2T and 26DCzPPy double layers.ref. ref582 The interfacial exciplex shows TADF behavior at λEL of 470 nm. By sandwiching non-doped ultrathin phosphorescent emitters (<0.5 nm) between 26DCzPPy and PO-T2T layers, high efficiency WOLEDs were fabricated. For a 2-color system, FIrpic (B) and Ir(tptpy)2acac (O) emitters separated by 3 nm thick 26DCzPPy were used to produce a white-emitting device, which has CIE coordinates of (0.46, 0.46), high PEmax of 83.2 lm/W, and EQEmax (EQE1000) of 19.6% (16.5%). In a 3-color system, FIrpic (B), Ir(ppy)2acac (G), and RD071 (R) were used, which enhanced the CRI from below 60 up to 86.
As the emission of exciplex-based devices alongside their dopants can support improved CRI, this approach was further investigated using a deep-blue emitter OCT as an excellent electron acceptor in combination with TAPC and m-MTDATA as electron donors.ref. ref583 Initially, single color green (λEL = 524 nm) devices using a TAPC:OCT exciplex and single color orange-red (λEL = 596 nm) devices using a m-MTDATA:OCT exciplex were fabricated. Due to the small ΔE ST of 0.03 eV efficient RISC was achieved, with the TAPC:OCT exciplex-based green devices exhibiting an adequate EQEmax of 10.6% suitable for use as a component in WOLEDs. An M-EML system with different exciplex pairs was employed, with TAPC:OCT, OCT, m-MTDATA:OCT, and m-MTDATA giving green, blue, red, and orange emissions, respectively. Although the resulting WOLEDs possessed a poor EQEmax of 1.7%, an impressive CRI of 97 was achieved in these devices.
Another encouraging result was the development of a 3-color tandem WOLED that included two sub-units. One sub-unit incorporated the blue TADF emitter BCz-Trz and the red [Ir(mphmq)2tmd] phosphorescent emitter co-doped into mCP as the host, and the other one employed the yellow (PO-01) and red [Ir(mphmq)2tmd] phosphorescent emitters co-doped into an exciplex host. Without optical extraction structure, this warm-white WOLED showed CIE coordinates of (0.47, 0.45), PEmax of 66.3 lm/W, and EQEmax (EQE1000) of 44.3% (42.3%). With an optical extraction structure, the optical outcoupling and device performance increased significantly, with PEmax of 162.9 lm/W, EQEmax (EQE1000) of 128.1% (126.2%), and CRI of 78. More impressively, a long device lifetime (LT50) of 12,600 h was achieved.ref. ref565
In summary, hybrid TADF WOLEDs successfully combine the advantages of both TADF and phosphorescent emitters, showing high performance in terms of efficiency, color quality, and stability. Nevertheless, the scarce and toxic heavy metal component remains an intrinsic shortcoming, which can be addressed by using metal-free all-fluorescent emitters.
All-Fluorescent TADF WOLEDs
The successes of high-efficiency primary color TADF molecules provides an avenue to fabricate high performance WOLEDs without the use of heavy metal complexes, i.e. all-fluorescent TADF WOLEDs. At present, most of the reported examples are simpler two-color systems, consisting of blue and yellow/orange emitters. Depending on the photophysical class of each emitter, all-fluorescent TADF WOLEDs can be subdivided into either all-TADF emitters, or TADF and fluorescent emitters, or exciplex-type TADF emitters. The typical molecular structures are shown in Figure , with some fluorescent structures also able to perform TTA in some cases (e.g., rubrene). Strategies used in the hybrid TADF WOLEDs to improve the device performance are also applicable here, such as S-EML, non-doped M-EML, and exciplex-type host. With the availability of an ever-increasing number of TADF emitters, we may soon see high-performance all-fluorescent TADF WOLEDs competitive with phosphorescent ones. The EQEmax of fluorescent TADF WOLEDs has indeed already reached and even surpassed the theoretical limit of 20%, with devices that show low efficiency roll-off and maintain EQE1000 at around 20%.ref. ref584 However, compared with the hybrid TADF WOLEDs, the efficiency roll-off of all-fluorescent TADF WOLEDs is typically more severe and their larger exciton energies (requiring higher driving voltages) means that reported PEmax remains low (below 70 lm W–1). In addition, due to the relatively strong blue emission in these two-color systems, the CIEx value is usually below 0.4, implying the generation of a cooler white light. Although long lifetime devices (LT80) of over 8,000 h have been reported, more studies are needed to assess and improve the stability of these fully organic all-fluorescent TADF WOLEDs.ref. ref566
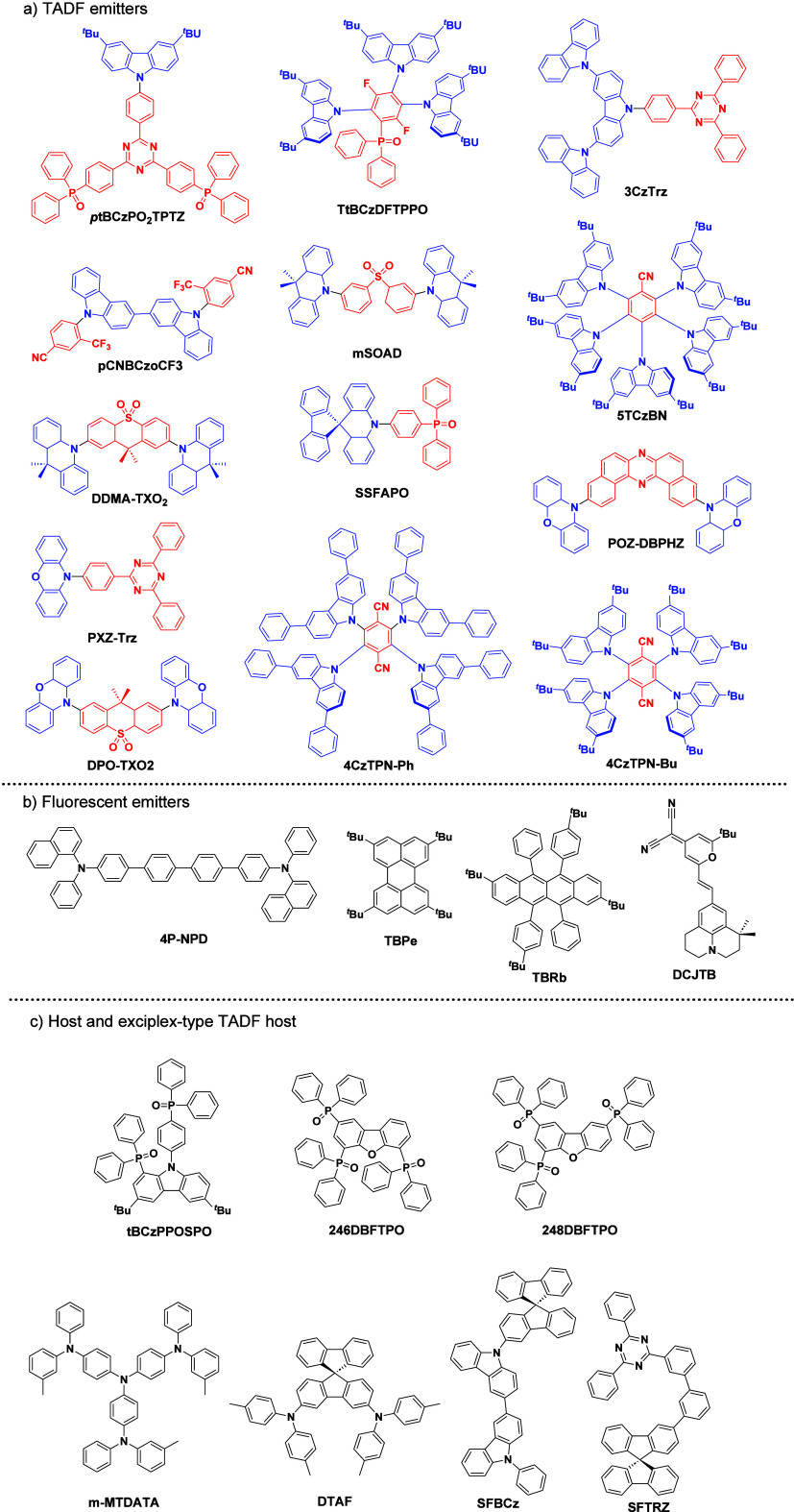
All-TADF Emitters
High-efficiency blue and yellow/orange TADF emitters play a key role in all-fluorescent TADF WOLEDs. Here, instead of enumerating all the new molecules and their photophysical properties, which have been discussed in other sections of this review, attention is devoted to the EML structure and the impact of the choice of host material. Although some of these WOLEDs show EQEmax of greater than 20%, the severe efficiency roll-off due to the long triplet exciton lifetime remains an issue common with single-color TADF OLEDs. In addition, most of these WOLEDs produce cool white light with low PEmax (< 70 lm W–1).
S-EML WOLEDs, using DMAC-DPS as both a blue emitter and as a host for the orange TADF molecule 4CzTPN-Ph, were fabricated. With a careful control of the doping concentration of 4CzTPN-Ph (0.8 wt%), the device showed cool white emission with CIE coordinates of (0.29, 0.39) and reasonable EQEmax (EQE1000) of 13.4% (9.4%).ref. ref585 With the same emitters but using a doped M-EML structure, i.e. B (DPEPO:DMAC-DPS)/Y (DMAC-DPS:4CzTPN-Ph)/B (DPEPO:DMAC-DPS), the emission spectrum was tuned to pure cool white with CIE coordinates of (0.33, 0.33), EQEmax of 12%, and CRI value of 82.ref. ref586 However, the orange emission became stronger with increasing brightness, and in both cases the EQE was low compared to optimised DMAC-DPS single-color devices (EQEmax ∼ 20%).
Replacing the phenyl groups in the 3- and 6-positions of carbazole in 4CzTPN-Ph with more sterically demanding tert-butyl groups gave 4CzTPN-Bu, with suppressed intermolecular interactions and improved device performance. In 4CzTPN-Bu:DMAC-DPS co-doped S-EML WOLEDs, the effects of phosphine oxide (PO)-based hosts were systematically studied. By carefully modifying the number, position, and symmetry of the PO-group, the triplet energy level and carrier transport properties were tuned, resulting in an improved ambipolar carrier transport, suppressed intermolecular interaction, and enhanced exciton confinement. The EQEmax (EQE1000) of the WOLEDs employing these PO-based hosts, i.e., 248DBFTPO, 246DBFTPO, and tBCzPPOSPO were 22.2% (19.8%), 21.9% (19.8%), and 21.1% (17.5%), respectively.ref587−ref588ref589 These devices also showed PEmax ranging from 63 to 77 lm W–1. Efficient exciton confinement and transfer resulted in controlled white light emission with CIE coordinates of (0.30, 0.40), (0.39, 0.48), and (0.36, 0.44), respectively.
The electron-withdrawing PO group was also explored as the acceptor in blue D-A TADF compounds, such as ptBCzPO2TPTZ, TtBCzDFTPPO, and xSFAPO, all of which were used giving excellent device performance.ref. ref584 S-EML WOLEDs were fabricated by doping 4CzTPN-Bu directly into ptBCzPO2TPTZ, and by varying the doping concentration of 4CzTPN-Bu (x wt%), cool white (x = 1.5%) and warm white (x = 2.0%) devices were fabricated with CIE coordinates of (0.34, 0.36) and (0.41, 0.42), EQEmax (EQE1000) of 23.6% (20.7%) and 20.3% (15.7%), and CRI of 87 and 73, respectively. Further, co-doping of 4CzTPN-Bu (1 wt%) with TtBCzDFTPPO (80 wt%) – or 4CzTPN-Bu (0.5 wt%) and SSFAPO (30 wt%) – into a DBFDPO host resulted in high-performance S-EML WOLEDs.ref590,ref591 These devices showed CIE coordinates of (0.28, 0.40) and (0.42, 0.50), EQEmax (EQE1000) of 22.3% (18%) and 25.1% (20.3%), and PEmax of 61.4 and 82.6 lm W–1, respectively.
To improve the PE of the WOLEDs, an orange-yellow TADF emitter DPPZ-DMAC was designed with DMAC as a donor unit and DPPZ as a strong acceptor unit.ref. ref592 The careful design of the molecule was claimed to suppress sensitizer-sensitizer interactions (SSI), improved the charge transport, and resulted in an efficient up-conversion of triplets transferred from the host. The monochrome OLEDs fabricated with 6 wt% of DPPZ-DMAC doped in CBP showed an EQEmax of 27.8%. However, the efficiency roll-off was large, especially in devices where the concentration of this dopant was higher. DPPZ-DMAC was combined with the blue TADF emitter 2tCz2CzBn (used as a co-host with mCBP) to produce WOLEDs. The devices showed an enhanced PEmax of > 80 lm W–1, an impressive EQEmax of ∼30%, and warm white emission with CIE coordinates of (0.40, 0.41). Nonetheless, the efficiency roll-off of these WOLEDs was severe, with a drop of the EQE1000 to 4.6%.
An attempt was made to reduce the efficiency roll-off of TADF WOLEDs by using compounds with fast k r and k RISC, thus reducing the triplet exciton population. The proposed pyridine-based emitters PyDCN-DMAC and PyDCN-PXZ have k r on the order of 107 s–1, and emit in the blue (λPL = 480 nm), and green (λPL = 532 nm), respectively.ref. ref446 The ΦPL for the 10 wt% doped film of PyDCN-DMAC in PPF is 82.8%, while the 15 wt% doped film of PyDCN-PXZ in CBP has a ΦPL of 89.6%. WOLEDs with a CIE of (0.39, 0.44) and CRI of 69 were fabricated using PyDCN-DMAC as a blue host and with an orange TADF molecule (PP-PXZ) as an emitter. Despite having a low CRI and a PE of 49 lm W–1, these WOLEDs showed an EQEmax of 18.5% and a Lmax of 9000 cd m–2. The efficiency roll-off was also reduced, where an EQE1000 of 12.6% was maintained by minimizing the Dexter energy transfer from PyDCN-DMAC to PP-PXZ due to an efficient k RISC in the host material.
Another approach for reducing the triplet loss via DET in S-EML WOLEDs involves the use of molecules with peripheral methyl substituents that weaken intermolecular interactions and increase intermolecular distances.ref. ref385 The sky-blue TADF emitter 5PCzCN was designed for this purpose, having a ΦPL of 96.5% and a high RISC efficiency of 99.3%. All-TADF WOLEDs were fabricated using 8 wt% of 5PCzCN with 0.7 wt% of the orange emitter 4CzTPN-Ph in a mCP host. The resulting devices showed an EQEmax (EQE1000) of 20.2% (16.9%), a lifetime LT50 of 10,010 h at a luminance of 100 cd m–2, but a low PE of 45.8 lm W–1. The CIE coordinates of the WOLEDs were found to be very stable with varying luminescence, with Δ(x, y) of only (0.01, 0.01) when increasing from 100 cd m–2 (0.31, 0.45) to 10,000 cd m–2 (0.30, 0.44). This was attributed to balanced exciton distributions throughout the emission layer.
In addition to two-color systems, three-color (R-G-B or Y-G-B) all-fluorescent TADF WOLEDs have been reported as well. With a doped M-EML structure based on 4CzTPNPh (O), 4CzPN (G), and 3CzTRZ (B) or POZ-DBPHZ (Y), DPO-TXO2 (G), and DDMA-TXO2 (B), M-EML WOLEDs were fabricated showing CIE coordinates of (0.30, 0.38) and (0.30, 0.40), EQEmax (EQE1000) of 17.1% (8.1%) and 16.1% (11%), respectively.ref593,ref594
To enhance the efficiency of carbazole-based TADF emitters, the number of carbazole groups on a molecule can be increased, in some cases leading to efficient RISC, enhanced excited-state mixing, and a delocalized HOMO across the carbazoles. This, however, can simultaneously lead to a randomised (more isotropic) orientation of molecules in the film and thus a lower light outcoupling efficiency. An alternative approach was proposed where a series of CzBN-based molecules with only two donors and a π-extended acceptor were designed to maintain a strongly horizontal orientation of the TDM.ref. ref595 Amongst the emitters in the study, 2PCzBN-FPh possessed the highest ΦPL of >90% and most strongly aligned horizontal TDM. As a result, blue OLEDs showed a EQEmax (EQE1000) of 35.7% (24.3%) at λEL of 469 nm. Due to these exceptional properties, 2PCzBN-FPh was used as a host in M-EML WOLEDs (two-color and three-color devices). The two-color devices used an orange MR-TADF emitter CNCz-BNCz and showed strong EQEmax of 29.3%, but poor CRI of 65 in this case hindered by the narrowband MR-TADF emission.ref. ref486 To improve the CRI, a three-color system with 2PCzBN-FPh as a blue TADF host, a green-yellow TADF emitter 4CzTPN-tBu along with a fluorescent red emitter RD were used. The CRI improved to 83 with CIE coordinates of (0.39, 0.41), although a lower EQEmax of 21.1% was obtained.
Red/yellow emitting TADF compounds containing more than one acceptor (A-D-A), such as DTXO-PhCz2, DTXO-PhCz4, DTXO-TPA2 and DTXO-TPA4, have been used as components in WOLEDs.ref. ref596 Among these emitters, the device with DTXO-TPA2 showed the best performance, with EQEmax (EQE1000) of 25.0% (10.06%), PEmax of 77.7 lm W–1 and a LT50 of 1392 hrs at 1000 cd m–2. The high efficiency of this device was attributed to the ΦPL of 70% of the emitter, good charge balance within the EML, and most importantly a strongly horizontally oriented TDM of DTXO-TPA2. The WOLEDs were made by combining DTXO-TPA2 with the blue TADF emitter 2SPAc-MPM, and the devices showed an EQEmax of 18.0% at CIE coordinates of (0.31, 0.31) with a CRI of 85.
TADF and Fluorescent Emitters
Despite the reduced IQEmax of around 25%, OLEDs using fluorescent emitters show high chemical/electrical stability and high brightness, owing in part to their chemical structures, low triplet energies, fast singlet radiative rates, and high ΦPL. These fluorescent emitters can be used in combinations with the TADF emitters to form M-EML WOLEDs with high stability and low efficiency roll-off. However, the low triplet energy states of the fluorescent materials can result in quenching of the triplet excitons of the TADF material. One of the strategies to solve the triplets and energy transfer losses is by the addition of an interlayer between the fluorescent and TADF emitters so that the excitons can be harvested adequately in their respective channels.ref. ref597 The interlayers of mCBP doped with different concentrations of Bepp2 were investigated where 30 wt% of Bepp2 presented the best results. A M-EML WOLED with two-color system but with double yellow EML was fabricated to better manage the exciton and charge distribution. For the first yellow EML, a fluorescent emitter 0.4 wt% TBRb with 6 wt% 4CzPN as a TADF assistant host doped in mCBP was used, while the second yellow EML contained 0.8 wt% TBRb:10 wt% 4CzPN in mCBP. For blue emission a fluorescent emitter DSA-Ph with 5 wt% in MADN host was used. The WOLEDs showed the highest EQEmax (EQE1000) of 15.1% (12.1) among all devices with CIE coordinates (0.35, 0.49) however, due to the absence of a red emitter, the CRI value of the WOLEDs was low (49). Hence, a three-color system was adopted where the first yellow EML was replaced with a red fluorescent emitter 0.4 wt% DBP:6 wt% CzPN in mCBP host. A moderate CRI of 68 with an EQEmax (EQE1000) of 14.7% (10.8%) was achieved.
Another approach involves a careful co-doping of TADF and fluorescent emitters into the EML, triplet excitons can be efficiently harvested on the TADF, resulting in enhanced device efficiency whilst maintaining good device stability. Long device lifetime WOLEDs have been achieved using this ‘hyperfluorescence’ strategy by balancing the completeness of FRET transfer from for example a blue TADF emitter to an orange or red fluorescent dye. However, as the lower energy dopant may not have any triplet harvesting properties, DET to this species as well as direct recombination must be avoided, enforcing low co-doping ratios.M-EML WOLEDs containing one co-doped EML with a fluorescent yellow emitter, TBRb, and a green TADF molecule, PXZ-TRZ in SF4-TPE as the host, alongside another doped EML of a fluorescent blue emitter, 4P-NPD in SF4-TPE as the host, showed CIE coordinates of (0.39, 0.39) and EQEmax (EQE1000) of 17.7% (15.5%). The CIE coordinates varied little between 300 to 13,000 cd m–2 [Δ(0.001, 0.012) for one of the systems], implying good color stability.ref. ref598 With the same fluorescent yellow emitter, TBRb and co-doped with the TADF blue emitter, 5TCzBN in an exciplex-type TADF host (SFBCz:SFTRZ), two-color S-EML WOLEDs showed CIE coordinates of (0.40, 0.51), EQEmax (EQE1000) of 21.7% (21.4%), PEmax of 78 lm W–1, and a long lifetime (LT80) of over 8200 h.ref. ref566 The long device lifetime was attributed to the advantages of both the exciplex-type host (bipolar carrier transport, TADF-type triplet harvesting) and the chosen emitters (inherent stability of fluorescent emitter, efficient triplet exciton harvesting of the TADF emitter).
In contrast to the low doping concentrations approach, the use of an ultrathin (< 1 nm) host-free blue fluorescent layer of TBPe and a TADF sensitizer assisted yellow fluorescent layer of TBRb with high concentration (3 wt%) was investigated.ref. ref599 The proposed system supported an efficient exciton harvesting by avoiding the dexter energy transfer to the blue emitter from the TADF host due to spatial separation while to the yellow emitter due to the large triplet gap. Two molecules DCzSPOTz and PhCzSPOTz were synthesised to be used as the hosts for the quasi-bilayer HF EML system. The resulting M-EML WOLEDs with PhCzSPOTz host showed an EQEmax (EQE1000) of 20.9% (17.7%), a high PEmax (PE1000) of 78.3 lmW–1 (38.0 lmW–1) with a CIE of (0.40, 0.52) and CCT of 4000K. Despite using an efficient approach, the devices failed to achieve an EQE higher than 20% which indicated that the triplet diffusion was still occurring in the system.ref. ref600 Hence, a HF system with very low yellow dopant concentrations was readopted for making efficient WOLEDs and was termed as a triplet-free exciton allocation system. Three TADF emitters ptBCzPO2TPTZ, 2CzPN, and DMAC-DPS were used for blue emission as well as sensitizers with a commonly used yellow fluorescent emitter TBRb. The WOLEDs with DBFDPO as a host and 40% ptBCzPO2TPTZ and 0.1% TBRb showed an impressive EQEmax (EQE1000) of 30.7% (27%), PEmax (PE1000) of over 100 lmW–1 (65 lmW–1) at CIE of (0.31, 0.37).
In a separate strategy that is already well-proven for white inorganic LEDs in industry and commercial applications, orange or green fluorescent emitters entirely external to the OLED can be used as partial down-conversion layers to produce white light from otherwise unaltered blue OLEDs. This approach was demonstrated for a blue TADF emitter, DMAC-TXO2 in DPEPO host, with layers of a polymer doped with green or orange perylene diimides spin coated directly atop the device.ref. ref601 The overall color could be controlled by the number of layer depositions, although with some complexity due to the radiative rather than FRET energy transfer between the OLED and external dyes. As the perylene dyes were external to the device, they completely avoid any formation of triplet excitons, with only the TADF emitter electrically excited. The balanced white WOLED itself maintained the good performance of the underlying blue OLED, with EQEmax of 17% and PEmax of 24.3 lm W–1, while also exhibiting perfect color stability at different driving voltages and CRI of 80.
Exciplex-Type TADF Emitters
Exciplex-type TADF emitters not only have low carrier injection barriers and balanced carrier transport but can also show efficient light emission properties. By carefully matching the energy levels of donor and acceptor molecules, exciplex-type TADF emitters can generate emission in the whole visible light range.ref. ref186 As previously mentioned, both bulk and interfacial exciplexes have been investigated to fabricate high performance WOLEDs. Though many other exciplex-type TADF emitters have been reported, the device performance using exciplex TADF emitters lags far behind other types of WOLEDs. In addition, due to the high exciton energy, high-efficiency blue exciplex-type TADF emitters are quite limited. Nonetheless, exciplex-type TADF emitters can be used along with fluorescent emitters or TADF emitters as documented above.
Doped layers of mCP:PO-T2T and DTAF:PO-T2T show exciplex-type TADF behavior with blue and orange emission, respectively. With a tandem device structure, WOLEDs were fabricated with CIE coordinates of (0.29, 0.35) and EQEmax (EQE1000) of 11.6% (10.5%).ref. ref554 It has also been demonstrated that some blue TADF emitters can form interfacial exciplexes with the adjacent organic layer, resulting in orange light emission and simplified device structure. Both mSOAD and pCNBCzoCF3 are efficient blue TADF emitters. When their non-doped layers are in contact with PO-T2T or m-MTDATA layers, respectively, orange interfacial exciplex-type emission is observed. mSOAD-based WOLEDs showed CIE coordinates of (0.49, 0.47) and EQEmax (EQE1000) of 11.6% (9.6%),ref. ref602 while pCNBCzoCF3-based WOLEDs showed CIE coordinates of (0.40, 0.44) and EQEmax (EQE1000) of 18.8% (17%).ref. ref603
Summarising the previous categories and examples, all-fluorescent TADF WOLEDs not only have a simpler EML structure, do not contain heavy-metal emitters, but are also showing improved device performance with examples of devices with EQEmax higher than 20%, PEmax approaching 80 lm/W, and LT80 of over 8200 h. However, due to the complicated exciton dynamics and long triplet excitons persisting in the EML, attention and progress is still required to further improve the efficiency roll-off, power efficiency, CRI, and color stability.
Single Molecule TADF WOLEDs
For even more simplicity in device design, it is desirable to achieve white light from single molecules.ref557,ref556 The most direct approach to achieve white emission is to integrate multiple chromophore units into one polymer chain. For individual small molecules this white emission property is typically rare, and at least dual-emission of blue and yellow/orange is needed. Nonetheless this can still be achieved by three main approaches: multiple chromophores within in one molecule; conformation induced dual-emission; and intra-/inter- molecular dual-emission. Examples of such molecules are shown in Figure .
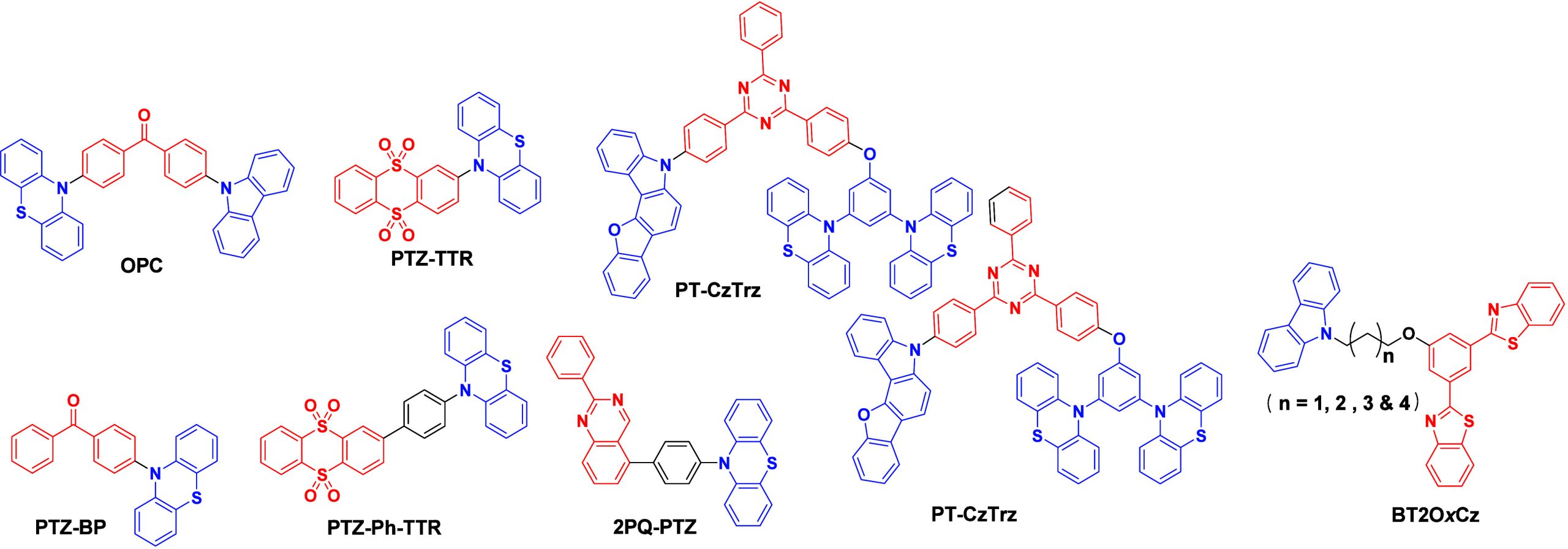
With an asymmetric D-A-D’ molecular design, a butterfly-shaped dual-emission white light emitter OPC was designed and synthesized.ref. ref604 When the Cz and PTZ donors are connected to a common benzophenone acceptor (BP), different CT states are formed, giving simultaneous blue and yellow emission from the bulk material. Though the blue component is fluorescent, delayed fluorescence was observed from the yellow emission. Under optical excitation, cool white emission with CIE coordinates of (0.35, 0.35) was observed from OPC, although no devices were fabricated in the report.
The strongly electron-donating PTZ can adopt two different conformations, quasi-axial or quasi-equatorial, which results in emission from different states that can be used to generate dual-emission. The PTZ-TTR molecule adopts both planar and orthogonal conformations, generating fluorescent blue and TADF-type yellow emissions, respectively.ref. ref564 By doping PTZ-TTR into CBP as the host, S-EML WOLEDs were fabricated with pure cool white emission with CIE coordinates of (0.33, 0.33) and a high CRI value of 92; however, EQEmax of the device was less than 3% and the EL spectrum was unstable. With a phenyl linker inserted between the PTZ and TTR moieties, PTZ-Ph-TTR preferentially adopts the orthogonal conformation, leading to greater TADF-type yellow emission that resulted in warm white light with CIE coordinates of (0.41, 0.48). The EQEmax of the device was significantly increased to 16.3% (EQE1000 = 11%), though the CRI value was lower at 64. Ultimately, this example reveals that the simplicity of single-molecule WOLEDs is also somewhat offset by a lack of control over their color.ref. ref564
PTZ-BP likewise shows dual-emission consisting of blue fluorescence and yellow TADF-type emission from LE and ICT states, respectively. Doping PTZ-BP into a DCzDPy host, S-EML WOLEDs showed CIE coordinates of (0.34, 0.46) and EQEmax (EQE1000) of 6.2% (2.8%).ref. ref605 With a similar design using a quinazoline (PQ) acceptor, the emitter 2PQ-PTZ shows white light emission (blue fluorescence and orange TADF emission emanating from quasi-axial and quasi-equatorial conformations, respectively).ref. ref606 Doping of 2PQ-PTZ into mCP, the S-EML WOLEDs produced cool white light with CIE coordinates of (0.32, 0.34), a CRI of 89, and an EQEmax of 10.1%.
Reiterating, despite their promise it is often difficult to tune the emission spectra of single molecule white light emitters. One solution is to simultaneously exploit both intramolecular and intermolecular CT emissions. An example of a molecule that does this is PT-CzTrz, which contains a twisted donor-acceptor moiety (pBFCz-Trz) responsible for blue emission and an electronically decoupled stronger donor moiety (mPTZ) that interacts intermolecularly with a second molecule of PT-CzTrz to produce a yellow-emitting exciplex (Figure )ref. ref607 By varying the doping concentration, the relative contributions of the blue and yellow components was tailored effectively. However, the device performance was still poor with CIE coordinates of (0.25, 0.31) and EQEmax of less than 2%.
In another approach, a series of emission-tunable molecules (BT2OxCz, x = 3, 4, 5, 6, where x refers to the number of aliphatic carbons) were designed with higher lying singlet and triplet states (Figure ).ref. ref556 The molecules contained a Cz donor and a BT2 acceptor connected through non-conjugated alkyl chains, and the emission color could be tuned by altering the length of the connecting alkyl chains. Here, the BT2O6Cz molecule is of the most interest as it provides a combination of TADF, room temperature phosphorescence and J/H-aggregates that emit in the blue, green, and red, respectively. The CIE coordinates for all the emitters are near pure white light emission (0.33, 0.33); however, the devices were not fabricated.
While still a developing area, dual-emissive single molecule white light emitters have shown great progress in recent years. This progress can be attributed in most cases to novel molecular design of different intra-/intermolecular CT states, conformation states, exciplex, and aggregate states. However, the overall device performance is still far behind other WOLED strategies, for the minority of examples where devices are demonstrated. Nonetheless, the appeal of massively simplified device design makes this an area of both practical and fundamental interest.
Outlook
In summary of this section, the device performance of WOLEDs using TADF materials as emitters, host, or sensitizers has significantly improved in efficiency, color quality and stability since their first reports in 2004.ref593,ref608,ref556,ref554 The hybrid TADF WOLEDs that show the best performance in terms of efficiency (up to ∼40% EQEmax), device lifetime, and CRI frequently rely on phosphorescent molecules doped in exciplex TADF hosts. All-fluorescent TADF WOLEDs show promise to have long device lifetime and are more environmentally friendly than hybrid TADF WOLEDs, though their power efficiencies and color quality still must improve to challenge phosphorescent devices. Single molecule WOLEDs are attractive as their device structures are significantly simpler; however, their efficiencies are the poorest of the WOLEDs that employ a TADF component in the EML, and are difficult to optimize from a given material structure. To further improve device performance, especially if organometallic phosphorescent co-dopants are to be avoided, judicious molecular design for high-performance TADF emitters as well as efficient exciton management are clearly still needed. Additionally, more insight and understanding of the degradation processes within WOLEDs to clarify the underlying mechanisms will help to improve the device lifetime towards industry requirements.
However, considering the technological underpinnings of WOLED use in displays and lighting, we predict that there will be a considerable decline in dedicated WOLED research in the coming years. This is because as the performance of monochromatic blue OLEDs continues to improve, WOLEDs will directly benefit in parallel. In the display industry this follows as a result of the ‘blue backplane’ concept,ref. ref21 using emissive color filters to achieve other colors from exclusively blue subpixel excitation. In lighting applications only blue and orange emission are required, which is once again most simply achieved through the use of external color downconversion filters,ref. ref601 which are already highly efficient. In both cases it therefore follows that the most impressive gains for WOLEDs can be achieved by exclusively focussing research on the underlying blue emitter, allowing simpler and longer-lived device architectures to be used inside the display or luminaire, and relying on photonic materials to generate other colors. Indeed, this is the currently dominant paradigm for now-widespread inorganic LED lighting, which has significant advantages over OLED in terms of efficiency, lifetime, and production cost. Apart from displays, which require small subpixels, and niche applications like aeronautical engineering, where weight is a critical concern, it seems unlikely that WOLEDs will be able to displace this now well-established technology.
Circularly Polarized Luminescence in TADF Emitters
Introduction
With the primary goal of increasing light output from the OLED, researchers have been focused not only on optimizing the intrinsic photophysics of the emitters but also devoting efforts to sidestep losses arising from external anti-glare polarising filters that are necessary in many display applications. Once such strategy is to employ materials that emit preferentially right- or left-circularly polarized emission. Indeed, circularly polarized luminescence (CPL) is the manifestation of preferential right- or left-circularly polarized emission emanating from materials that are either chiral or are influenced by their chiral environment. Chiral molecules emitting CPL have been widely investigated for their potential integration in optical data storageref. ref609 and optical spintronics applications.ref. ref610 This class of emitters has generated significant interest for their use in electroluminescent displays such as circularly polarized OLEDs (CP-OLEDs) with the promise of mitigating the significant efficiency losses associated with the presence of ‘anti-glare’ filters.ref. ref611 Many display technologies employ circular polarizing filters (a linear polarizer and a quarter-wave plate) to trap and attenuate reflections of surrounding unpolarized (randomly polarized) light sources (e.g. sunlight) that can otherwise cause glare.ref. ref612 This, however, also unavoidably blocks 50% of the unpolarized electroluminescence from exiting the display. CPL though can pass through such filters without loss, potentially doubling the external quantum efficiency and achievable brightness of these OLEDs while still preventing glare.ref47,ref613,ref614
The extent of CPL from a chiral emitter is quantified by the luminescence dissymmetry factor, glum or gPL, which is defined in equation eq16 :
where IL and IR are the intensities of left- and right-handed light, respectively. Thus, gPL values can range from −2 to +2 for perfectly right- or left-CP emission, respectively, and 0 for unpolarized or linearly polarized light. For CP-OLEDs the equivalent electroluminescence dissymmetry factor (gEL) is used, which is defined analogously to gPL.
The molecular origin of the emission dissymmetry is related to the relative orientation of the electric and magnetic transition dipole moments for the emissive transition, as defined in equation eq17 :
where μ and m are the respective electric and magnetic transition dipole moments between the excited and ground states (usually S1 and S0) and θ is the angle between the vectors of these TDMs. In closed shell systems like organic TADF emitters the electric transition dipole moment is typically large while the magnetic transition dipole moment is usually ∼100-fold smaller, and so CPL-active small organic chiral molecules often show low gPL values typically less than 10–2, limiting their practical applications. Much effort has been devoted to rationally design materials to tune the magnitude of μ and m to improve gPL at the molecular level.ref615−ref616ref617ref618
In the context of CP-OLEDs, not only should the device show high gEL but the intrinsic EQE must also remain competitively high. Consequently, chiral compounds that can also support triplet harvesting through TADF are an especially appealing class of emitters.ref619,ref620 We identify two key strategies used to construct CP-TADF molecules: (1) the design of molecules with an intrinsically chiral TADF skeleton (using point, axial, or planar chirality), or (2) the design of compounds that couple chiral groups to achiral TADF moieties (chiral perturbation). A number of recent reviews focusing on CP-TADF molecules have been published,ref47,ref613,ref614 and so here we highlight recent developments in CP-TADF emitter design. Key photophysical data of these chiral emitters are summarized in Table S4.
CP-TADF Emitters Containing Stereogenic Centers
The first example of a small molecule TADF CPL emitter, DPHN (Figure ), was developed by Imagawa, Hirata, et al. in 2015.ref. ref621 This compound contains a stereogenic carbon center linking the donor and the acceptor moieties. This molecule emits at λPL of 513 nm and has a moderate ΔE ST of 0.26 eV, a gPL of 1.1 × 10–3, and has a low ΦPL of only 4% and a τPL of 13.9 ns in toluene. DPHN also has a small ΦPL of 26% and a moderate ΔE ST of 0.19 eV in 9 wt% doped mCP films. Understandably from these low ΦPL, no CP-OLEDs were reported.
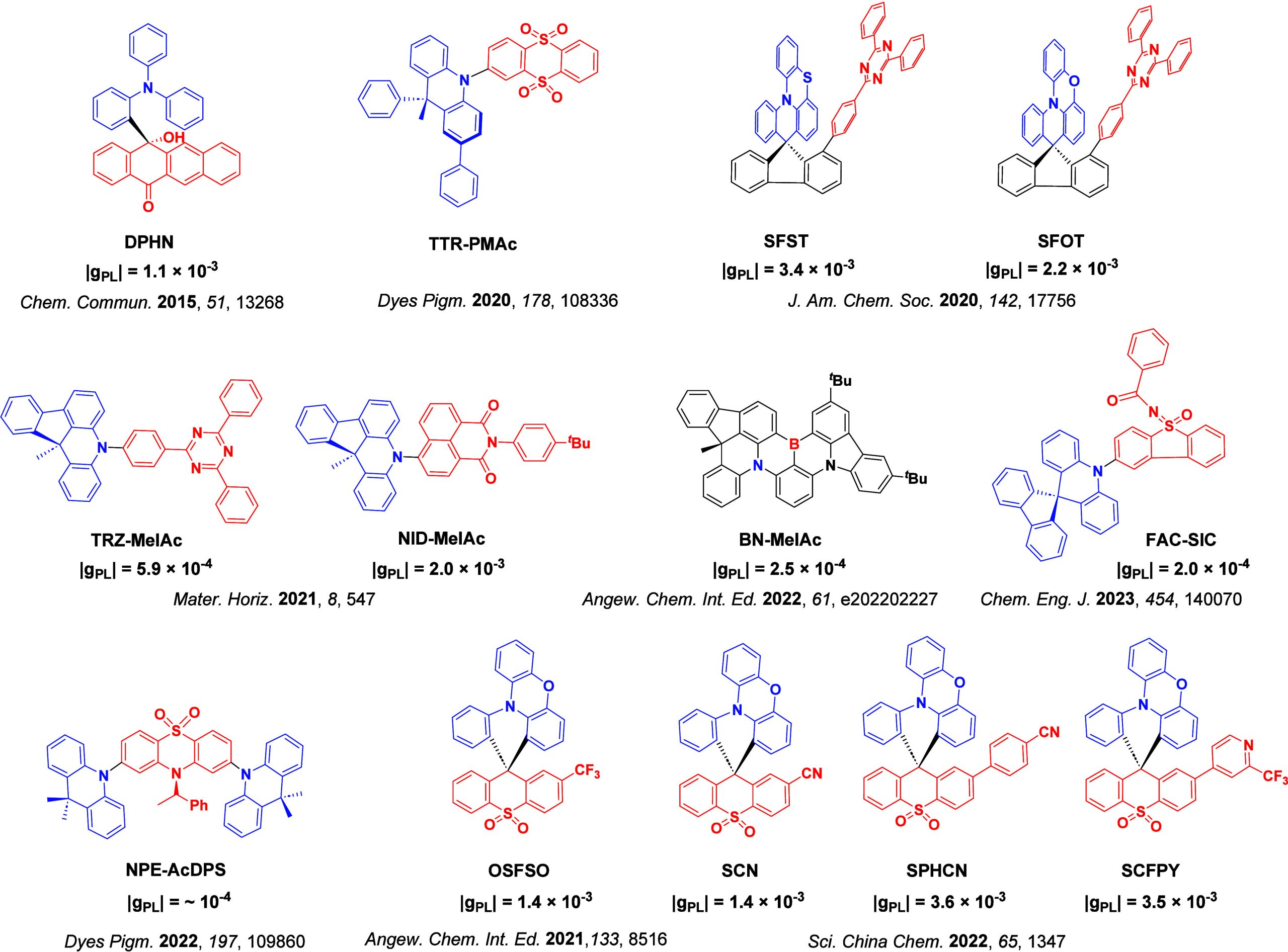
Using a similar strategy Hao et al. reported emitters (R)– and (S)-TTR-PMAc (Figure ) containing chiral donor units, (R)- and (S)-9-methyl-2,9-diphenyl-9,10-dihydroacridine (PMAc) linked to achiral acceptor thianthrene 5,5,10,10-tetraoxide.ref. ref622 This emitter exists in two distinct conformations, one that is near-planar and the other near-orthogonal, with associated calculated dihedral angles between the TTR and either (R)- or (S)-PMAc units of 173.46° and 85.57° respectively. Interestingly, it was demonstrated that in both enantiomer the CPL signals from the near-planar and near-orthogonal conformations showed dissymmetry factors of opposite sign. Both enantiomers display two broad and structureless emission bands at λPL of ∼430 and 577 nm. (R)-TTR-PMAc and (S)-TTR-PMAc have similar large ΔE ST of 0.36 and 0.39 eV, respectively for their near-planar conformers in 2-MeTHF. In contrast, only the orthogonal conformers are observed in neat films, which have much smaller associated ΔE ST of 0.02 and 0.05 eV, respectively.
Ni et al. later introduced a chiral rigid donor MeIAC, which was coupled to a triazine acceptor to give the sky-blue emitter TRZ-MeIAc (Figure ). This material emits at λPL of 473 nm with a ΦPL of 89%, ΔE ST of 0.19 eV, and a τd of 82.3 μs in 12 wt% doped films in mCPCN.ref. ref623 The same chiral MeIAc unit was also coupled to a naphthalimide acceptor in the orange emitter NID-MeIc. This compound emits at λPL of 565 nm, has a ΦPL of 86%, a ΔE ST of 0.22 eV, and thus a longer τd of 235.4 μs in 6 wt% doped films in mCPCN. The high ΦPL of these two emitters was attributed to the rigid molecular structure of the donor. CP-OLEDs with TRZ-MeIAc showed an EQEmax of 20.3%, while the device with NID-MeIc showed an EQEmax of 23.7%. The CP-OLED based on (R)-TRZ-MeIAc showed definite CPL although with a low gEL of 6.4 × 10–4, while the device based on (S)–NID-MeIc displayed a fourfold larger gEL of −2.4 × 10–3. It is fascinating, although entirely unclear, how the same chiral donor group can lead to significantly different CPL dissymmetry for the different emitters.
Subsequently, Yang et al. integrated the same MeIAc block into a B/N-doped aromatic skeleton to develop a pair of chiral green emitters (R)-BN-MeIAc and (S)-BN-MeIAc (Figure ), which featured an MR-TADF design strategy where the CPL properties originate from the chiral carbon centre.ref. ref624 The sp3-hybridized carbon atom in the structure not only serves as a configurationally stable stereocenter to induce CPL, but also locks the molecular geometry to guarantee high conformational stability. In addition, the fluorenyl unit within MeIAc extends the π-conjugation of the MR-TADF skeleton, which contributes to the simultaneous enhancement of the oscillator strength and the horizontal transition dipole orientation of the emitter in the devices. As a result of this rational design, BN-MeIAc displayed narrowband green emission with λPL of 497 nm, FWHM of 30 nm, gPL of +2.5 × 10–4 for (R)-BN-MeIAc and −2.5 × 10–4 for (S)-BN-MeIAc, and a small ΔE ST of 0.11 eV for both, all in toluene. These desirable photophysical properties also included a high ΦPL of 96%, a moderate τd of 28.1 μs, and a highly horizontal orientation of the TDM of 90% in 1 wt% doped films in DMIC-TRZ. The corresponding OLEDs showed EQEmax values up to 37.2%, although still with modest gEL of +2.7 × 10–4 for (R)-BN-MeIAc and −2.9 × 10–4 for (S)-BN-MeIAc, presumably limited by the intrinsic gPL of the emitters. This work expanded the application of the chiral acridan-derived building block used in chiral MR-TADF emitters, and although it also represents the highest device efficiency for all reported CP-OLEDs to date, it also highlights the need for greatly improved intrinsic molecular CPL properties to support higher gEL.
Yang et al. reported the first examples of through-space charge transfer (TSCT) CP-TADF emitters, SFST and SFOT (Figure ), containing either a PTZ or a PXZ donor attached alongside a triazine acceptor on a spiro-fluorene scaffold.ref. ref625 Both compounds showed a small ΔE ST of 0.05 eV and emit at λPL of 512 nm in toluene. The subtle difference in the structure of the donor brought about considerable changes in the secondary photophysical properties of the molecules though. A higher ΦPL of 89% and a much faster k RISC of 1.17 × 105 s–1 was observed for SFOT in 30 wt% doped films in mCBP, which led to devices with an EQEmax of 23.1% and EQE1000 of 21.3% (λEL at 508 nm). The larger sulfur atom in SFST instead distorted the molecular backbone of PTZ and altered the donor-acceptor distance with negative consequences on the TSCT interaction. This substitution resulted in a lower ΦPL of 53% and slower k RISC of 9.93 × 104 s–1 in 30 wt% doped films in mCBP, which translated into a device with a lower EQEmax of 12.5% (λEL at 508 nm). Both enantiomers of SFST presented higher |gPL| values than those of SFOT, up to 4.0 × 10–3 in toluene; in fact, they are almost double those of (S)-SFOT/(R)-SFOT (|gPL| up to 2.2 × 10–3). The increased CPL character was attributed to the large atomic radius of sulfur and consequently the more distorted and asymmetric structure of SFST. The CP-OLEDs based on (S)-SFST and (S)-SFOT showed gEL of 1.30 × 10–3 and 1.0 × 10–3, respectively.
Zhang et al. reported a similar example of a CP-TADF emitter containing a rigid spiro structure, (R)/(S)-OSFSO (Figure ).ref. ref626 The molecule possesses a similar PXZ-based donor motif as SFOT, while the acceptor thioxanthene moiety was linked directly to the donor across a spiro-center bridging atom. (Rac)-OSFSO has a small ΔE ST of 0.022 eV leading to a τd of 4.7 μs, emits at λPL of 470 nm, and has a ΦPL of 81% in 25 wt% doped films in DPEPO. The CP-OLEDs fabricated with both enantiomers showed not only an EQEmax of 20.0% (λEL of 472 nm), but also featured a remarkably low efficiency roll-off with an EQE1000 of 19%. The device gEL was 3.1 × 10–3, again small relative to application-relevant values but also somehow double the reported gPL (1.4 × 10–3 in toluene).
Hao et al. reported the first CP-TADF emitters containing heteroatomic stereocentres.ref. ref627 By combining sulfoximine-based acceptors and acridan-based donors within a highly twisted structure, a pair of chiral enantiomers [(R)-FAC-SIC and (S)-FAC-SIC, Figure ] were synthesized with the asymmetric sulfur atom serving as the stereocenter. The strongly twisted geometry facilitates a small ΔE ST and TADF, while intramolecular hydrogen bonding in the SIC acceptor helps to reduce non-radiative decay pathways by rigidifying the N-substituent. As a result, FAC-SIC emits at λPL of 507 nm and has a small ΔE ST of 0.075 eV in toluene, and a high ΦPL of 99% and a short τd of 5.8 μs in 10 wt% doped films in DBFPO, as well as gPL of +2.4 × 10–4 for (R)-FAC-SIC and −2.0 × 10–4 for (S)-FAC-SIC in toluene, respectively. The corresponding OLEDs with (R)-FAC-SIC showed an EQEmax of 28.5%, although the CPL signal was too weak to be detected.
Similar to having sulfur as the stereocenter, Huang et al. reported a pair of enantiomers, (S)-NPE-AcDPS and (R)-NPE-AcDPS (Figure ), that contained the commercially available chiral (S)-/(R)-1-phenylethylamine linked to the previously reported TADF emitter DMAC-DPS.ref. ref628 (S)-NPE-AcDPS emits at λPL of 451 nm, has a small ΔE ST of 0.05 eV, a τd of 3.4 μs, and a ΦPL of 86% in 12 wt% doped films in DBFPO, while the chirality conferred by the presence of the asymmetric nitrogen atom resulted in a gPL on the order of 10–4. The corresponding OLEDs with (S)-NPE-AcDPS showed an EQEmax of 18.5%, although again no obvious CPL signal was detected.
Finally, Zheng et al. developed three pairs of spiro-type TADF enantiomers with carbon stereocenters, similar to their previously reported OSFSO but with differently substituted acceptors: (R/S)-SCN, (R/S)-SPHCN, and (R/S)-SCFPY (Figure ).ref. ref629 SCN possesses a cyano group as the acceptor, SPHCN contains a benzonitrile as an elongated acceptor, and SCFPY uses 2-(trifluoromethyl)pyridine as a stronger acceptor. All three materials show green emission at λPL of 522 nm for (R/S)-SCN, 505 nm for (R/S)-SPHCN, and 526 nm for (R/S)-SCFPY. These compounds all have relatively small ΔE ST (in toluene) and high ΦPL (in 25 wt% doped films in 26DCzPPy): 0.01 eV and 89% for (R/S)-SCN, 0.16 eV and 67% for (R/S)-SPHCN, and 0.04 eV and 89% for (R/S)-SCFPY. The impact of the molecular structures on the CPL properties were then studied by comparing their chiroptical properties and device performances. (R/S)-SCN showed a |gPL| of 1.4 × 10–3 in toluene and the device showed an EQEmax of 23.0% with gEL of −1.4/1.8 × 10–3. For (R/S)-SPHCN with a longer acceptor, although the EQEmax decreased to 15.4% there is a larger |gPL| of 3.6 × 10–3 and |gEL| of −3.6 × 10–3. (R/S)-SCFPY, possessing an acceptor of similar size to (R/S)-SPHCN, has a similar |gPL| of 3.5×10–3 but the device showed a higher EQEmax of 23.3% (gEL of −3.7/3.6 × 10–3), which represents the highest efficiency spiro-type TADF material-based OLED to date. The authors therefore report that gPL and gEL can be enhanced by extending the length of the acceptor, which in this study caused a better alignment between μ and m (smaller θ), as confirmed by their calculations.
CP-TADF Emitters with Axial Chirality
The first examples of intrinsic axially chiral TADF emitters, (R/S)-1 and (R/S)-2 (Figure ), were developed by Wang et al. in 2019ref. ref630 and contained a stereogenic binaphthol (BINOL) unit. (R/S)-1 and (R/S)-2 show yellow or green emission at λPL of 568 and 530 nm and have ΦPL and ΔE ST of 18.5 and 15.7% and 0.059 and 0.076 eV, all respectively. (R)-1 has similar gPL of 1.6 × 10–3 in toluene, 8.2 × 10–4 in 15 wt% doped films in TCTA, and 9.2 × 10–4 as a neat film. Interestingly, (R/S)-2 did not show CPL, likely due to the rotatable benzophenone structure that limits the chirality transfer process from the binaphthyl to the peripheral D-A TADF chromophore. OLEDs with S-1 exhibited orange emission (λEL at 580 nm) with an EQEmax of 1.8% and gEL of +1.0 × 10–3.
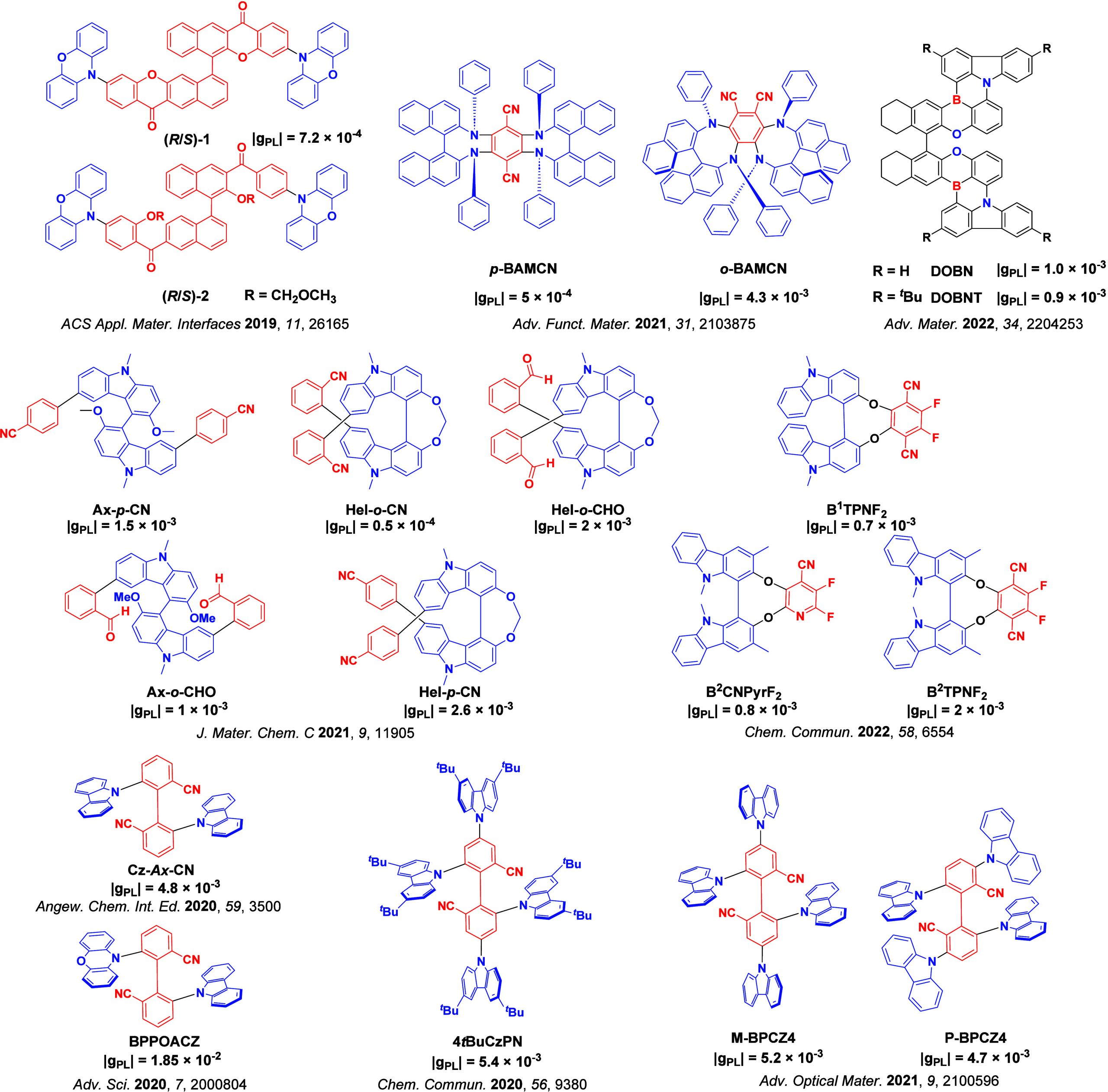
In 2021 Yan et al. designed two new chiral TADF materials, p-BAMCN and o-BAMCN (Figure ), containing modified chiral BINOL peripheral groups acting as axially chiral donors around either para or ortho substituted dicyanobenzene as the acceptor.ref. ref631 Both emitters showed narrowband green emission (FWHM of 61 nm for both), with λPL at 537 and 503 nm and ΦPL of 86 and 77% in either 8 wt% doped films in TCTA or in 26DCzPPy for p-BAMCN and o-BAMCN, all respectively. The ΔE ST were also similar for the pair of emitters at 0.18 eV for p-BAMCN and 0.15 eV for o-BAMCN. (S)-o-BAMCN showed higher but similar gPL in both toluene (5.3 × 10–3) and in the doped film (4.3 × 10–3) when compared to (S)-p-BAMCN (0.3 and 0.5 × 10–3), which was rationalized in terms of the different DFT-predicted angles between μ and m in the para– and ortho-derivatives. The OLED with (R)-p-BAMCN showed a high EQEmax of 27.6% although the CPEL of the device was too weak to be obtained. OLEDs with (R)-o-BAMCN showed an EQEmax of 20.5%. Semi-transparent devices were also fabricated to reduce the reflection of metallic cathodes and improve CPL performance, with (S)-o-BAMCN showing a gEL of 4.6 × 10–3 in line with its gPL.
To achieve narrower CPL emission, the same authors combined axial chirality with an MR-TADF design, leading to DOBN and DOBNT (Figure ).ref. ref632 DOBN and DOBNT emit at λPL of 453 and 459 nm, and both have FWHMs of 21 nm in toluene. Although both emitters exhibited gPL values lower than 0.2 × 10–4 in toluene, they showed high ΦPL of 91 and 96% and moderate gPL of 1.0 and 0.9 × 10–3 in 5 wt% doped films in 26DCzPPy. The CP-OLEDs with (R)-DOBN and (R)-DOBNT displayed narrowband blue emission at λEL of 459 and 464 nm with CIE coordinates of (0.14, 0.10) and (0.13, 0.12), and showed EQEmax of 23.9 and 25.6% with gEL of −0.9 and −1.0 × 10–3.
The axially chiral TADF emitter Cz-Ax-CN (Figure ), reported by Li et al., contains two coupled D-A 3-(9H-carbazol-9-yl)benzonitrile fragments.ref. ref633 Both enantiomers of Cz-Ax-CN exhibited dual TADF and AIE, emitting at 460 nm and have a small ΔE ST of 0.029 eV, a short τd of 12.6 ms, and a ΦPL of 68% in 15 wt% doped films in DPEPO. The gPL of (−)-(S)-Cz-Ax-CN in the film reached −4.8 × 10–3. The CP-OLED with (−)-(S)-Cz-Ax-CN showed blue electroluminescence at λEL of 468 nm, with an EQEmax of 12.5% and a gEL value of −1.2 × 10–2, which is larger than the gEL values of most other reported CP-TADF OLEDs.
In a separate report, the same group modified the nature and number of the donor moieties to further enhance CPL activity. 4tBuCzPN (Figure ) emits at λPL of 476 nm, has a ΦPL of 74%, and gPL values of 5.4 × 10–3 in toluene and a small ΔE ST of 0.05 eV, a short τd of 4 ms, and a gPL value of 5.2 × 10–3 in 25 wt% doped films in DPEPO.ref. ref382 The OLED fabricated with racemic 4tBuCzPN showed a significantly improved EQEmax of 20.8% compared to the device with Cz-Ax-CN (12.5%), and emitted at λEL of 500 nm. The authors did not however report gEL values for the devices since racemization of the enantiomers was discovered during the vacuum evaporation. Tu et al. employed the same design, substituting one carbazole for a phenoxazine to construct the emitter BPPOACZ (Figure ).ref. ref634 rac-BPPOACZ exhibited two emission bands peaking at 384 and 543 nm in toluene. Both enantiomers showed high ΦPL of 86%, |gPL| of 9.7 × 10–3, ΔE ST of 0.04 eV, and short τd of 1.1 ms in toluene, while only one emission peak at 527 nm appeared in 20 wt% doped films in 26DCzPPy at 527 nm, and the gPL in this host was higher at 1.85 × 10–2. The CP-OLED based on (S)-BPPOACZ displayed green electroluminescence (λEL = 537 nm) and showed an EQEmax of 17.8% and a low efficiency roll-off with EQE1000 of 15.2% and EQE10000 of 12.6%. However, the gEL was only 4.5 × 10–3 for reasons that remain unclear.
This same group also reported another two similar blue emitters, M-BPCZ4 and P-BPCZ4 (Figure ), which contain additional carbazole moieties but have different donor connectivity.ref. ref635 M-BPCZ4 and P-BPCZ4 both emit at 470 nm and show ΔE ST of 0.09 and 0.05 eV respectively in toluene. The emission is slightly red-shifted to λPL at 485 nm, and the ΦPL and τd are 64 and 76%, and 6.4 and 7.0 ms in 25 wt% doped films in DPEPO, respectively. (R)-M-BPCZ4 and (R)-P-BPCZ4 have high gPL of −5.0 and −4.7 × 10–3 in toluene. The blue CP-OLEDs with (R)-P-BPCZ4 showed a higher EQEmax of 18.3% (λEL = 480 nm) and a lower efficiency roll-off with EQE1000 of 17.2%, compared with (R)-M-BPCZ4 which showed an EQEmax of 16.7% and EQE1000 of 15.7%. This team also found that the position of the carbazole units affected the racemization temperatures and corresponding CPL properties of the enantiomer OLEDs greatly. The presence of a crowded set of carbazole donors in (R/S)-P-BPCZ4 results in a centralization of both the μ and m. Additionally, the steric congestion present in (R/S)-P-BPCZ4 prevents undesired racemization during vacuum deposition for device fabrication. Consequently, the device with (R/S)-P-BPCZ4 possesses a higher gEL value (-5.5 × 10–3) than that with (R/S)-M-BPCZ4 (-3.8 × 10–3).
Sumsalee et al. designed five chiral emitters Ax-p-CN, Ax-o-CHO, Hel-o-CN, Hel-p-CN, and Hel-o-CHO (Figure ) that each contain carbonyl-based acceptors with both axially or helically chiral bicarbazole electron donors.ref. ref636 Ax-o-CHO and Hel-o-CHO displayed TADF and emit at λPL of 460 and 439 nm in toluene and have τd of 1.04 and 0.80 μs in doped DPEPO films respectively; however, their ΦPL in toluene are very low at 3 and 2%, which also remained low at 10 and 5% along with ΔE ST of 0.19 and 0.37 eV in the DPEPO films, respectively. Interestingly the CPL properties changed with increasing solvent polarity, with (+)-Ax-o-CHO having a higher gPL of 1.0 × 10–3 in toluene than in chloroform (0.7 × 10–3) or DMF (0.5 × 10–3), while (+)-Hel-o-CHO showed a higher gPL of 2.0 × 10–3 in chloroform than in DMF (1.6 × 10–3) or toluene (∼0). This sensitivity of gPL to solvent polarity was rationalized as due to subtle reorganization of the intramolecular charge-transfer excited and ground states in the different solvents. These results also suggested that higher CPL intensity can be achieved with helical emitters, although these did also display lower TADF efficiency.
Subsequently, Poulard et al. reported TADF emitters B1TPNF2 , B2TPNF2 , and B2CNPyrF2 , containing axially chiral bicarbazole donors (Figure ).ref. ref637 All three showed green emission with λPL of 529, 530, and 492 nm, and ΦPL of 11, 29, and 23%, respectively. Their gPL values were determined to be 0.7, 2.0, and 0.8 × 10–3 in toluene. The higher gPL value for B2TPNF2 was attributed to a more favorable orientation between μ and m, likely a result of its helical structure.
CP-TADF Emitters with Planar Chirality
[2,2]Paracyclophane (PCP) and its derivatives have emerged as useful planar-chiral skeletons in the construction of CP-TADF emitters. Zhang et al. first introduced an electron-donating -NMe2 group and electron-withdrawing -Bmes2 group onto the two separate benzene rings of the PCP in g-BNMe2-Cp and m-BNMe2-Cp (Figure ).ref. ref638 These emit at λPL of 531 and 521 nm in toluene, respectively. The HOMOs and LUMOs were efficiently separated in these two compounds, resulting in ΔE ST of 0.17 and 0.12 eV in 2-MeTHF glass at 77 K. The powder ΦPL were moderate at 53 and 33% for g-BNMe2-Cp and m-BNMe2-Cp, respectively. The gPL value for g-BNMe2-Cp reached 4.24 × 10–3. The gPL for m-BNMe2-Cp was not mentioned and low energy barriers to racemization limited their application, with CP-OLEDs not explored.
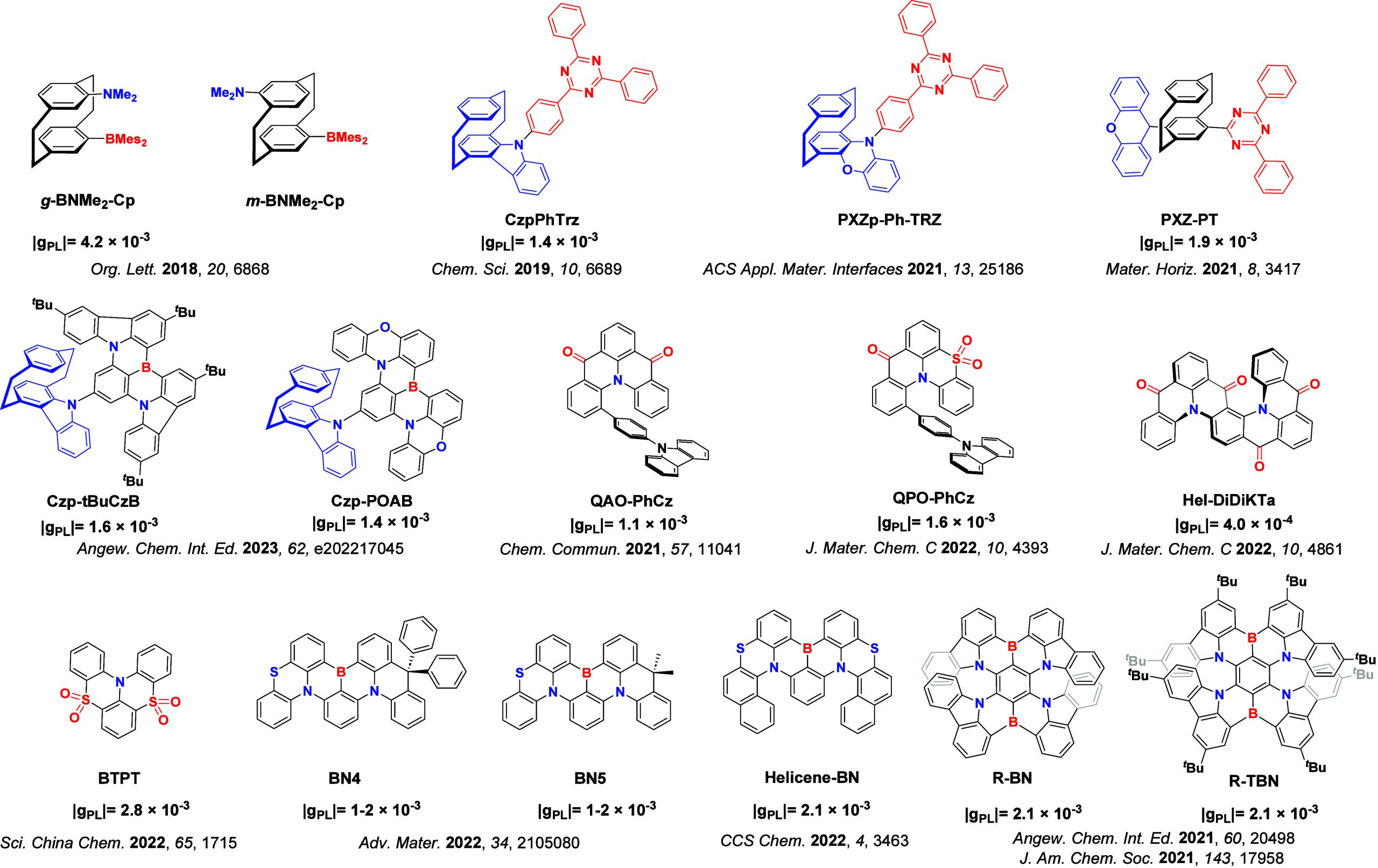
Sharma et al. soon after reported the first example of a carbazolophane (Czp) containing TADF emitter, CzpPhTrz (Figure ).ref. ref236 The increased steric bulk of the Czp unit induced an increased torsion angle between the donor and the phenylene bridge compared to the unsubstituted carbazole-containing analogue. This more twisted geometry coupled with a stronger electron-donor in the Czp compared to Cz resulted in a ΔE ST of 0.16 eV in 10 wt% doped film in DPEPO. (R)-CzpPhTrz emits at λPL of 470 nm and has a gPL value of 1.3 × 10–3 in toluene, while Rac-CzpPhTrz showed a ΦPL of 69% in 10 wt% doped DPEPO film. The OLEDs showed EQEmax of 17% at λEL of 480 nm, but CP-OLEDs were not pursued in this study. Liao et al. subsequently reported a structurally related CP-TADF molecule, PXZp-Ph-TRZ (Figure ), using a phenoxazine-based analogue to Czp.ref. ref639 The yellow emitter (λPL = 527 nm) has a much smaller ΔE ST of 0.03 eV compared to CzpPhTrz owing to the stronger donor, and has a ΦPL of 60% in 10 wt% doped films in CBP. The solution-processed CP-OLEDs displayed a gEL of 4.6 × 10–3 and showed an EQEmax of 7.8%.
Zhang et al. reported a pair of D-(chiral π)-A TADF emitters, (R/S)-PXZ-PT (Figure ), with a PCP skeleton attached to the central phenylene linker (but not to either the donor or acceptor).ref. ref640 This design strategy not only suppressed the racemization between the two enantiomers, making it possible to fabricate CP-OLEDs by vacuum-deposition, but also reduced non-radiative transitions that led to higher ΦPL. (R/S)-PXZ-PT emits at λPL of 565 nm and has a ΔE ST of 0.19 eV and a high ΦPL of 78% in 10 wt% doped films in CBP, while the gPL is ±1.9 × 10–3. The vacuum-deposited CP-OLEDs exhibited yellow emission [λEL of 557 nm, CIE coordinates of (0.44, 0.55)] and showed a higher EQEmax of 20.1% than those of the earlier reported devices with CzpPhTrz and PXZp-Ph-TRZ; the gEL was 1.5 × 10–3.
Liao et al. reported two pairs of Czp-substituted MR-TADF materials. Czp-tBuCzB and Czp-POAB (Figure ).ref. ref641 (R/S)-Czp-tBuCzB and (R/S)-Czp-POAB emit at 478 and 497 nm with narrow FWHMs of 23 and 36 nm, have ΔE ST of 0.09 and 0.13 eV and gPL of 0.54/-0.51 × 10–3 and 0.48/-0.46 ×10–3 in toluene. Both emitters have near unity ΦPL of 98 and 96%, and τd of 41.8 and 62.4 ms in doped films (5 wt% and 8 wt% doped films in 2,6DCzPPy), all respectively. The sky-blue CP-OLEDs with (R)-Czp-tBuCzB (λEL of 479 nm) showed a high EQEmax of 32.1%, EQE100 of 29.2%, EQE1000 of 30.9%, and the narrowest FWHM of 24 nm among reported CP-OLEDs alongside gEL of +1.54 × 10–3. Devices with (R)-Czp-POAB displayed near-pure green CP electroluminescence [CIE coordinates of (0.23, 0.65)] with EQEmax of 28.7%, EQE100 of 28.1%, EQE1000 of 20.4%, and gEL of +1.30 × 10–3. These studies demonstrate that the PCP unit can be used towards the construction of CPL-active D-A TADF and MR-TADF emitters, both showing modest gPL.
Helicenes are a class of fused polycyclic aromatic frameworks that possess a helical chirality. In helicenes larger than five rings the overlap between the opposite ends of the fused system renders the enantiomers kinetically stable towards racemization. Helicenes have attracted significant research interest due in part to their promising applications in CPL and CP-OLEDs. Yang et al. reported a blue CP-TADF emitter, QAO-PhCz, possessing a rigid hetero-helicene structure (Figure ).ref. ref642 The synergistic effects of the sterically hindered donor linkage and the rigid emissive core generated narrowband emission at λPL of 460 nm with FWHM of 29 nm. (P)-QAO-PhCz has a ΔE ST of 0.11 eV, τd of 40.36 ms, and a moderate ΦPL of 46.6% in 5 wt% doped films in mCBP. The corresponding CP-OLED showed a narrow FWHM of 36 nm (λEL of 467 nm) and an EQEmax of 14%. The enantiomers of QAO-PhCz displayed similar |gPL| and |gEL| of 1.1 and 1.5 × 10–3, respectively. Following this concept, the same group reported another pair of chiral hetero-helicene molecules (P/M)-QPO-PhCz (Figure ), this time with a carbonyl-/sulfone-bridged triarylamine structure.ref. ref643 Compared to QAO-PhCz, QPO-PhCz showed similar photophysical properties, emitting at 446 nm, having a ΔE ST of 0.23 eV, and a |gPL| of 1.2 × 10–3 in toluene. The compound has a long τd of 536 ms and a ΦPL of 51% in 18 wt% doped films in DPEPO. The CP-OLEDs with (M)-QPO-PhCz showed sky-blue emission (λEL of 488 nm) with EQEmax of 10.6%, and gEL of +1.6 × 10–3.
Extending the concept of helically chiral emitters further, Marques dos Santos et al. reported extended helical structure Hel-DiDiKTa (Figure ), which is an S-shaped double [4]helicene based on a pair of fused QAO (or equivalently DiKTa) cores.ref. ref644 The CPL-active MR-TADF molecule (P)-Hel-DiDiKTa emits in the sky-blue emission (λPL at 473 nm) and has a small ΔE ST of 0.15 eV, and τd of 5.4 ms in 1 wt% doped films in mCP. However, the gPL is only 4.0 × 10–4 and the ΦPL is low at 6.2% in 1 wt% doped PMMA film, which precluded devices from being investigated. Compared to previously reported DiKTa-based emitters, the molecular distortions present in this helical compound are thought to result in severe emission quenching.
Ning et al. reported a strikingly simple polycyclic aromatic heterocycle BTPT (Figure ) that contains sulfone groups at the two ortho-positions of a triphenylamine core and is helically chiral.ref. ref645 (P)-BTPT emits in the ultraviolet (λPL = 368 nm) with a narrow FWHM of 33 nm in toluene. In 1 wt% doped films in PMMA, (+)-(P)-BTPT has a ΔE ST of 0.14 eV, a τd of 109 ms, yet a ΦPL of only 9%. The enantiomeric crystals of BTPT not only displayed CPL with a gPL on the order of 10–3, but also showed room temperature phosphorescence.
Wu et al. developed another type of MR-TADF emitter with helical chirality, exemplified in BN4 and BN5 (Figure ).ref. ref646 These two compounds contain an asymmetrical peripheral lock to the well-known MR-TADF molecule DABNA-1, enhancing the helical nature of the B/N doped nanographene. Sulfur was chosen as the bridging atom of the rigid locked ring, and both compounds emit at λPL of 500 and 497 nm and have the same ΔE ST of 0.14 eV in toluene, alongside high ΦPL of 96 and 92% for BN4 and BN5 in 3 or 1 wt% doped films in mCPCN. BN4 and BN5 thus have similar k RISC of 3.7 and 3.3 × 104 s–1, all respectively. (R)/(S)-BN4 and (R)/(S)–BN5 in doped mCPCN films displayed gPL of +1.1/-1.0 and +1.3 /-1.0 × 10–3, respectively. The CP-OLEDs with BN4 and BN5 achieved narrowband emission at λEL of 510 and 506 nm (FWHM of 49 and 48 nm) and showed EQEmax of 20.6 and 26.5% with gEL of +3.7/-3.1 and +1.9/-1.6 × 10–3, respectively.
Yang et al. developed a pair of helicene-based enantiomers, (P)-helicene-BN and (M)-helicene-BN (Figure ), which merged helical chirality and the B/N/S doped polycyclic aromatic framework to concurrently exhibit CPL and MR-TADF behavior.ref. ref647 Helicene-BN emits at λPL of 525 nm, has a ΔE ST of 0.15 eV and a ΦPL of 100% in 1 wt% doped films in DMIC-TRZ. In toluene, the gPL values are +2.0 × 10–3 for (P)-helicene-BN and −2.1 × 10–3 for (M)-helicene-BN, while in the 1 wt% doped films in DMIC-TRZ, the glum values are +1.3 × 10–3 for (P)-helicene-BN and −2.0 × 10–3 for (M)-helicene-BN. CP-OLEDs with (P)-helicene-BN and (M)-helicene-BN showed EQEmax of 31.5% at CIE coordinates of (0.26, 0.66). The devices also exhibited gEL of +1.2 × 2.2 × 10–3, respectively.
Zhang et al. developed two similar helical deep-red MR-TADF emitters R-BN and R-TBN (Figure ) that emit at λPL of 662 and 692 nm and have ΦPL of 100%, ΔE ST of 0.18 and 0.16 eV, and τd of 16.6 and 46.4 ms in toluene, all respectively.ref. ref177 In 3 wt% doped films in CBP, they emits at 672 and 698 nm, and have τd of 0.31 and 0.71 ms, respectively. The OLEDs with R-BN and R-TBN showed EQEmax of 28.1 and 27.6%. Li, Wang et al. then explored the chiroptical properties of these two emitters, which have gPL of 2 × 10–3 in dichloromethane.ref. ref648 These examples illustrate state-of-art strategies to fabricate CP-TADF emitters with narrow emission based on a helical skeleton, but again illustrate the difficulties in discovering or designing molecules with gEL or gPL greater than 10–2.
CP-TADF Emitters Featuring Chiral Perturbation
The chiral perturbation strategy to construct CPL-active TADF compounds involves the introduction of a chiral peripheral group to an otherwise achiral TADF structure. The chiral unit does not directly participate in the emissive process. This strategy is now widely used because of the ease of the synthesis and enantiomer separation processes. The reported emitters using this strategy can maintain efficient TADF inherited from previously validated designs, while also exhibiting promising CPL behavior bestowed by the perturbing group.
Feuillastre et al. reported the first chiral perturbation TADF materials, (R)-1 and (S)-1 (Figure ), incorporating a BINOL unit to confer axial chirality to the molecule.ref. ref649 (R)-1 emits at 486 nm in cyclohexane with a gPL of 1.3 × 10–3, while the compound has a ΦPL of 53% and τd of 2.9 ms in toluene; however, the OLEDs based on (S)-1 displayed only a modest EQEmax of 9.1% with λEL at 535 nm. Song et al. used a similar design strategy to combine TADF, AIE, and CPL properties in the emitters BN-CF, BN-CCB, BN-DCB, and BN-AF (Figure ).ref. ref650 Compound (S)-BN-CF showed the highest gPL of 1.2 × 10–3 with λPL of 495 nm and ΦPL of 32% in toluene. It also showed the same ΦPL of 32% and τd of 24.33 ms in 10 wt% doped film in mCP. Surprisingly the gPL values of the neat films were amplified significantly, especially for (S)-BN-CF which achieved a very high gPL of 4.1 × 10–2. The CP-OLEDs using 10 wt% doped films in mCP as emitting layers showed an EQEmax of 9.3% and gEL of 2.6 × 10–2. The non-doped (S)-BN-CF OLED exhibited a further amplified gEL of 6 × 10–2. The higher gPL values of (S)-BN-CF than previous reported (R/S)-1 were attributed to the AIE properties. Huang et al. later also reported two BINOL-based chiral emitters, CPDCz and CPDCB (Figure ).ref. ref651 (S)-CPDCz and (S)-CPDCB emit at λPL of 511 and 533 nm, with ΔE ST of 0.08 and 0.04 eV in respective 10 wt% doped films in mCP. They also have ΦPL of 20 and 55%, τd of 18 and 10 ms, and gPL values of −3.3 and −4.0 × 10–4, respectively. The solution-processed CP-OLEDs with (S)-CPDCB showed an EQEmax of 10.6% and gEL of −3.9 × 10–3 compared to the device with (S)-CPDCz, which showed an EQEmax of 10.1% and gEL of −3.7 × 10–3.
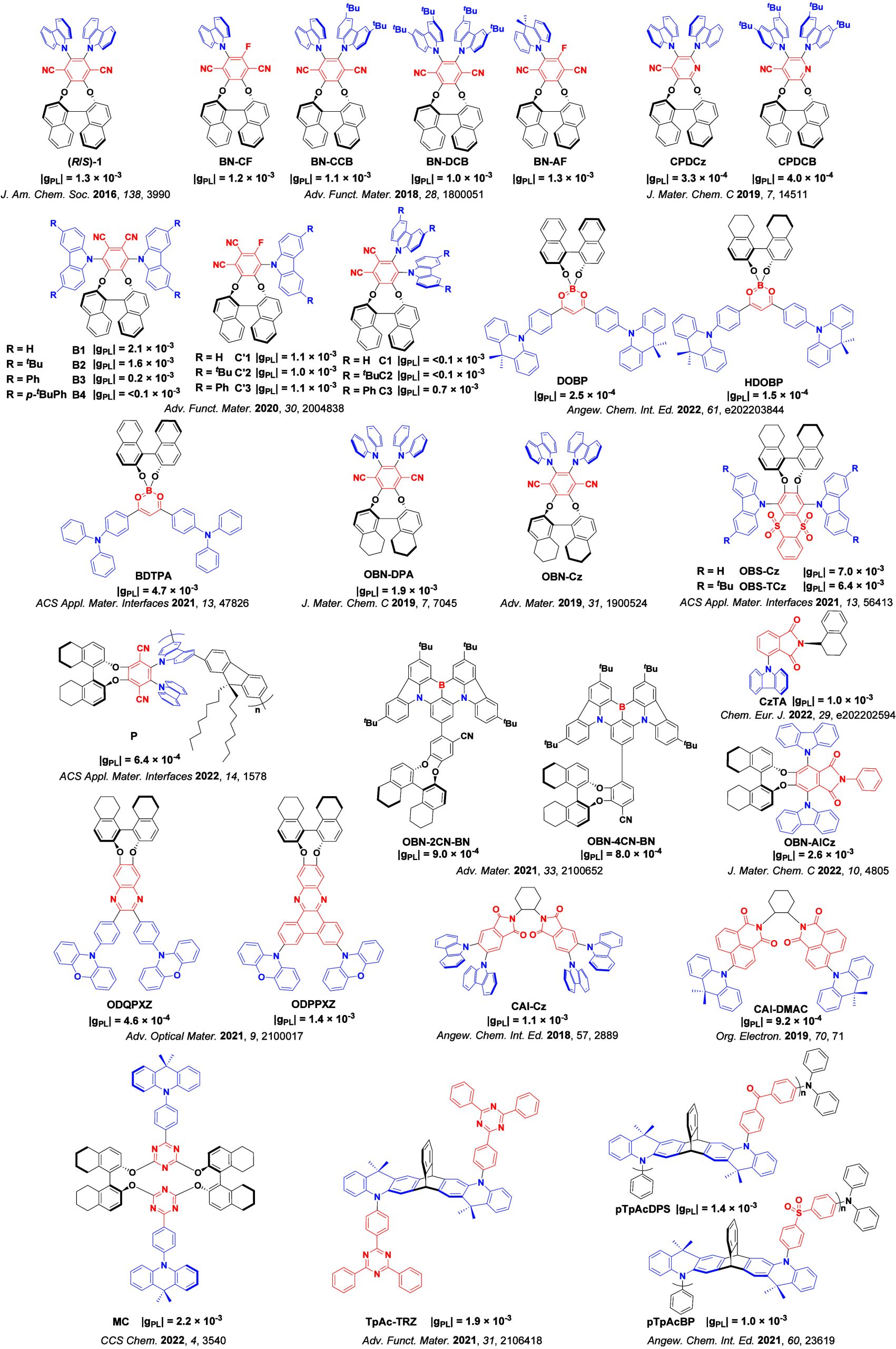
Pieters et al. reported three families of BINOL-based chiral TADF emitters (B, C, and C′, Figure ) with different numbers of donors at different positions and with different distances between the chromophore and the chiral perturbing unit.ref. ref652 For the B series, the molecule emits at λPL of 469–516 nm, having ΦPL of 7–30%, τd of 10–45–ms, and ΔE ST of 0.1–0.31 eV. For the C series, they emit at λPL of 493–519 nm, having ΦPL of 25–47%, τd of 18–40 ms, and ΔE ST of 0.1–0.22 eV. For the C′ series, they emit at λPL of 481–510 nm, having ΦPL of 29–42%, τd of 6–19 ms, and ΔE ST of 0.11–0.28 eV. The ΦPL are obtained in doped PMMA films and ΔE ST are estimated from spectra in 2-MeTHF, with other data are obtained in toluene. The B series has the smallest distance between two carbazole donors and the stereogenic unit, and showed improved CPL performance compared to the C and C′ families as predicted by the higher m and smaller θ from DFT calculations. Compound B1 exhibited the highest gPL of 2.1 × 10–3 (in toluene) of all the compounds in the study. As C’3 shows the best compromise between optical and chiroptical properties, it was used as the emitter in top emitting CP-OLED. The device showed green emission centered at λEL of 510 nm, a low EQE of only 0.8%, and gEL of 1.0 × 10–3.
Zhou et al. reported two pairs of enantiomers [(R/S)-DOBP and (R/S)-HDOBP, Figure ] that contain tetracoordinate boron atoms. These two compounds displayed concomitantly AIE, CPL, mechanochromism, and piezochromism.ref. ref653 (R/S)-DOBP and (R/S)-HDOBP emit at 536 and 534 nm and have large ΔE ST values of 0.28 and 0.23 eV in dilute toluene. In neat films the ΔE ST values decrease to 0.14 and 0.08 eV for (R)-DOBP and (R)-HDOBP, although the ΦPL are only 1 and 2%, all respectively. The gPL values are ±2.5 and ±1.5 × 10–4 for (R/S)-DOBP and (R/S)-HDOBP in 1,4-dioxane, respectively. The non-doped solution-processed OLEDs with (R)-DOBP showed NIR emission (λPL = 716 nm) and an EQEmax of 1.9%.
Xue et al. reported the emitter BDTPA that has a similar structure as the previous example (R/S)-DOBP, but with the DMAC donor replaced by a triphenylamine.ref. ref654 (R)-BDTPA emits at λPL of 560 nm and has a ΔE ST of 0.14 eV, a τd of 53.5 ms, and a significantly improved gPL of −1.7 × 10–3 in toluene. It is at present not clear why the gPL of (R)-BDTPA is so much higher than those of (R)-DOBP and (R)-HDOBP. (R)-BDTPA emits at 600 nm and has a ΦPL of 15.8% in 10 wt% doped mCP film. The solution-processed CP-OLED with (R)-BDTPA showed an EQEmax of 2.0% and a gEL value of −1.6 × 10–3 (λEL of 598 nm).
Wu et al. reported an analogue of (R)-1 that instead contained an octahydrobinaphthol unit, OBNCz (Figure ).ref. ref655 (R)-OBNCz has a gPL value of −1.55 × 10–3, emits with λPL of 504 nm, and has a ΦPL of 92% and a small ΔE ST of 0.037 eV in 10 wt% doped films in 26DCzPPy. CP-OLEDs with (R)-OBNCz showed an EQEmax of 32.6%, with very low efficiency roll-off (EQE1000 of 31.7% and EQE5000 of 30.6%) and gEL of 1.94 × 10–3 (λEL of 526 nm), making this example the best performing CP-OLED to date in terms of efficiency. Wu et al. also reported a pair of similar enantiomers, OBN-DPA, which replaced the carbazole in OBNCz with a diphenylamine moiety (Figure ).ref. ref656 (R)-OBN-DPA exhibited green emission peaking at 538 nm, a small ΔE ST of 0.09 eV, and a gPL of 1.88 × 10–3 in toluene. The 10 wt% doped film of (R)-OBN-DPA in 26DCzPPy has a ΦPL of 84.7% and a short τd of 13.5 ms along with a |gPL| value of 2.9 × 10–3. The doped and non-doped CP-OLEDs showed lower EQEmax of 12.3 and 6.6% compared to the device with OBNCz, though with somewhat higher gEL values of 2.9 and 2.3 × 10–3, respectively.
Liu et al. reported the compound (R)/(S)-OBS-TCz and the analogue (R)/(S)-OBS-Cz, both containing a 5,5,10,10-tetraoxide acceptor and the (R)/(S)-OBS group as the chiral perturbing unit (Figure ).ref. ref657 The enantiomers (R)/(S)-OBS-Cz and (R)/(S)-OBS-TCz emit at λPL of 504 and 520 nm in toluene, have small ΔE ST of 0.04 and 0.05 eV, short τd of 3.2 and 2.7 ms, ΦPL of 73 and 87%, and gPL of 8.7 and 6.4×10–4 in 15 wt% doped films in mCP, all respectively. CP-OLEDs with (R)/(S)-OBS-TCz showed higher EQEmax of 20.3% and EQE1000 of 20.1%, but smaller gEL values of +0.80/–1.00 × 10–3 than the devices with (R)/(S)-OBS-Cz (EQEmax of 15%, EQE1000 of 14.5%; gEL +5.00/–4.00 × 10–4).
Li et al. synthesized the first highly efficient green CP-MR-TADF molecules.ref. ref658 They introduced chiral (R)/(S)-octahydro-binaphthol ((R)/(S)-OBN) units onto the previously reported blue-green MR-TADF emitter (DtBuCzB) to induce CPL. The enantiomers (R)/(S)-OBN-2CN-BN and (R)/(S)-OBN-4CN-BN (Figure ) exhibit narrowband emission at 493 and 500 nm, with FWHM of 22 and 24 nm, and small ΔE ST of 0.12 and 0.13 eV in toluene, respectively. Both compounds have high ΦPL of 95 and 90%, and τd of 95.3 and 97.4 ms in 3 wt% doped films in PhCzBCz. Unfortunately, the gPL values are all rather low; +9.0 × 10–4 for (R)-OBN-2CN-BN, −9.1 × 10–4 for (S)-OBN-2CN-BN, +8.0 × 10–4 for (R)–OBN-4CN-BN, and −10.4 × 10–4 for (S)–OBN-4CN-BN. CP-OLEDs with (R)/(S)-OBN-2CN-BN and (R)/(S)-OBN-4CN-BN emitted at λEL of 496 and 508 nm and had small FWHMs of 30 and 33 nm, leading to CIE coordinates of (0.11, 0.52) and (0.14, 0.64), and EQEmax of 29.4 and 24.5% with gEL values of +1.43/-1.27 × 10–3 and +4.60/-4.76 × 10–4, all respectively. This report was the first example of a highly efficient narrowband-emitting CP-MR-TADF OLED. Despite these advances, the low gPL factors still indicate that there is significant space to design new molecules that show higher dissymmetry factors.
Teng et al. reported conjugated polymers (R)-P and (S)-P (Figure ) using the same strategy of chiral perturbation exemplified with the axial chiral binaphthyl units,ref. ref659 wherein the chirality is transferred from the stereogenic moiety to the D-A TADF monomers. As a result, the R and S polymers exhibited excellent TADF properties with small ΔE ST of 0.045 and 0.061 eV measured in 2-MeTHF glass, emit at λPL of 549 and 547 nm, and have similar ΦPL of 72 and 76% and short τd of 1.6 and 2.3 ms in 10 wt% doped films in mCP, all respectively. The k RISC are 6.28 and 6.31×10–5 s–1 based on their neat films. The polymers have gPL values of up to 1.9×10–3 obtained from the annealed doped films. The corresponding solution-processed CP-OLEDs with (R)-P and (S)-P emitted at λEL of 546 and 544 nm and showed EQEmax of 14.9 and 15.8% with gEL of −1.5 and +1.6×10–3, respectively. This work expanded the strategy of chiral perturbation with binaphthyl units to TADF polymers.
Xie et al. reported contrasting pairs of enantiomers, flexible (R/S)-ODQPXZ and rigid (R/S)-ODPPXZ, each containing (R/S)-octahydro-binaphthol as the stereogenic unit (Figure ).ref. ref660 (R)-ODQPXZ and (R)-ODPPXZ emit at λPL of 589 and 630 nm and have ΔE ST values of 0.16 and 0.07 eV in toluene, respectively. They also have high ΦPL of 92 and 89%, and short τd of 3.6 and 3.7 ms in 15 wt% doped films in CBP. (R/S)-ODPPXZ showed higher gPL values of 1.4/1.9×10–3 compared to (R/S)-ODQPXZ (gPL = −4.6/4.0×10–4). The yellow-emitting CP-OLED (λEL of 548 nm) with (R)-ODQPXZ showed higher EQEmax of 28.3%, EQE100 of 20.6%, and smaller gEL of 6.0×10–4, compared to (R)-ODPPXZ which showed EQEmax of 20.3% and EQE100 of 17.2%, with λEL of 600 nm and gEL of 2.4×10–3. The authors ascribed the more intense CPL in (R)-ODPPXZ to its more rigid structure wherein the phenyl groups are fused into a larger phenanthrene unit in the acceptor.
Zhao et al. applied a similar strategy in the design of CP-TADF macrocyclic enantiomers (+)-(R ,R)-MC and (−)-(S ,S)-MC, which combine two TADF skeletons with similar octahydro-binaphthol moieties (Figure ).ref. ref661 Macrocycle (+)-(R ,R)-MC emits at λPL of 505 nm, has a very small ΔE ST of 0.069 eV, ΦPL of 78%, and short τd of 1.76 ms as neat film. The gPL value was measured to be 2.2 × 10–3 in toluene. The solution-processed CP-OLED with (+)-(R ,R)-MC emitted at λEL of 522 nm and showed EQEmax of 17.1%, EQE1000 of 16.5%, and a gEL value of 1.5 × 10–3. This work documents the first example of a CP-TADF macrocycle. The same group also reported a pair of aromatic-imide-based TADF enantiomers, (R/S)-OBN-AICz, which contain (R/S)-octahydrobinaphthol attached to a D-A skeleton (Figure ).ref. ref662 (R)-OBN-AICz emits at λPL of 509 nm, has a ΦPL of 81%, and τd of 4.0 ms in 13 wt% doped film in mCBP. It also has ΔE ST of 0.08 eV as neat film. Clear mirror-image CPL with |gPL| values of up to 2.6 × 10–3 were reported in toluene. The CP-TADF OLEDs with (R)-OBN-AICz emitted at 514 nm, showed EQEmax of 19%, and had gEL of 4.7 × 10–4.
Instead of binaphthyl derivatives as the stereogenic unit, a separate strategy involved the use of chiral trans-1,2-diaminocyclohexane to link two imide-based D-A TADF emitters.ref. ref663 (+)-(S,S)-CAI-Cz (Figure ) has a |glum| value of 1.1 × 10–3 and emits at λPL of 528 nm with ΦPL of 98%, and has a small ΔE ST value of 0.06 eV, yet a rather long τd of 130 ms in 15 wt% doped films in mCBP. The CP-OLEDs showed an EQEmax of 19.8% at λEL of 520 nm and have gEL values of −1.7 and 2.3 × 10–3 for (+)-(S,S)-CAI-Cz and (−)-(R,R)-CAI-Cz, respectively. Using the same design but replacing the carbazole donor with DMAC, the same group reported another pair of enantiomers, CAI-DMAC (Figure ).ref. ref664 (−)-(R ,R)-CAI-DMAC emits at 583 nm in toluene and has a ΔE ST value of 0.07 eV, a τd of 37.4 ms, a low ΦPL of 39.9%, and gPL value of 9.2 × 10–4 in 6 wt% doped film in CBP. The OLEDs emitted at λEL of 592 nm and showed EQEmax of 12.4%, EQE100 of 9.7%, and EQE1000 of 4.1%; no gEL was reported for these devices. 1,2,3,4-Tetrahydro-1-naphthylamine is another stereogenic unit that has been used in CP-TADF emitter design, exemplified in (R/S)-CzTA (Figure ).ref. ref665 In the crystalline state (R)-CzTA and (S)-CzTA emit at λPL at 465 nm and have ΦPL of 48.7 and 45.3% and delayed lifetimes of 3.37 and 3.40 ms, respectively. The compounds both have ΔE ST of 0.13 eV, and the enantiomers have gPL of −1.03 × 10–3 for (S)-CzTA and +0.84 × 10–3 for (R)-CzTA in toluene. No CP-OLEDs were prepared.
Wang et al. developed a series CP-TADF emitters containing a chiral triptycene scaffold, exemplified by (S ,S)-/(R,R)-TpAc-TRZ (Figure ).ref. ref666 The enantiomers emit at λPL of 541 nm and have a small ΔE ST of 0.03 eV as neat films. The chiral triptycene scaffold mitigates intermolecular π–π stacking, which led to a ΦPL of 85% and short τd of 1.1 ms of the neat film. Obvious mirror-image CPL signals were also observed with gPL values of +1.9 and −1.8 × 10–3 for (S ,S)-(+)-TpAc-TRZ and (R ,R)-(−)-TpAc-TRZ as neat films, respectively. The solution-processed non-doped CP-OLEDs with (S ,S)-(+)-TpAc-TRZ showed EQEmax of 25.5%, EQE100 of 16.8%, and EQE1000 of 1.6% with gEL of +1.5 × 10–3. Using a similar triptycene scaffold the same group reported two pairs of chiral non-conjugated TADF polymers, (R ,R)-/(S,S)-pTpAcDPS and (R ,R)-/(S,S)-pTpAcBP (Figure ). The chiral triptycene donor subunit was introduced into the backbone of the polymers, and the well separated FMOs of the monomers produced a material that emits at λPL of 532 nm with a small ΔE ST of 0.01 eV, a high ΦPL of 92%, and a gPL value of −1.0 × 10–3 in 10 wt% doped films in mCP.ref. ref667 Solution-processed CP-OLED device with (R,R)-pTpAcBP showed an EQEmax of 22.1% and gEL of −1.0 × 10–3. This is the first report of CP-OLEDs based on a ‘main-chain’ chiral TADF polymer.
Other Strategies for Designing Chiral TADF Systems
TADF exciplexes are formed from a blend of hole and electron transporting materials, where the HOMO and LUMO are located on the two different molecules (See Section sec8 ). The completely separated FMOs produce a small μ while maintaining the same magnitude of m, which can be exploited for achieving higher gPL in chiral exciplexes. Favereau et al. reported exciplex emitters involving chiral bicarbazole donor 1 and achiral acceptor 5-fluoroisophthalonitrile A (Figure ).ref. ref668 The 1:A (1:2 ratio) blend emits at λPL of ≈520 nm with a ΔE ST of 0.16 eV and a ΦPL of 19%. Importantly, the gPL of 7 × 10–3 is ten times higher than the gPL of the chiral donor 1 alone (7 × 10–4).
Gu et al. designed a pair of chiral acceptors (R/S)-TRZ, and used the hole transporting material 4,4′-cyclohexylidenebis[N,N-bis(4-methylphenyl)]aniline (TAPC) as the donor to form another CPL-active TADF exciplex (Figure ).ref. ref669 The (R/S)-TRZ:TAPC blended film at the ratio of 1:1 emits at λPL of 520 nm, and has a very small ΔE ST of 0.012 eV with ΦPL of 39.5%. The |glum| of (R)-TRZ:TAPC increased from 2.07 to 2.73 × 10–3 when the ratio of the donor and chiral acceptor was changed from 2:1 to 1:2. The CP-OLEDs with (R)-TRZ:TAPC (1:1) film showed an EQEmax of 12.7%, and (S)-TRZ:TAPC (1:1) film showed an EQEmax of 9.3%. The (S)-TRZ:TAPC based device showed the higher gEL of −9.89 × 10–3 compared to (R)-TRZ:TAPC (gEL = 7.25 × 10–3).
Doping chiral emitters and introducing chiral groups into nematic liquid crystals have also been identified as good strategies to realize and amplify CPL properties. Yang et al. designed two green and yellow chiral TADF emitters, FAC-PDMLM and PXZ-PDMLM (Figure ), which have ΦPL of 18 and 13% and common ΔE ST of 0.02 eV in toluene.ref. ref670 Both emitters were virtually CPL-silent between 300 and 500 nm though, as the alkyl chains were unable to transfer chiroptical properties to the TADF moieties. However, when doping FAC-PDMLM and PXZ-PDMLM into the achiral liquid crystal 5CB, the co-assembly led to the formation of a chiral nematic liquid crystal phase with very high gPL values of 7.26 × 10–2 and 5.45 × 10–2, respectively.
Outlook
In this section we have systematically summarized the recent evolutions in the design of chiral TADF emitters, with CPL typically induced via intrinsically chiral emitter skeletons or via chiral perturbation. Significant progress has been made since the first report of chiral TADF emitters showing CPL, and OLEDs employing chiral emitters have seen improvements in gEL values from 10–4 to nearly 10–2. However, further improvements of these dissymmetry values are required for CP TADF OLEDs to be useful in chiroptical devices. The spread of gEL and EQEmax (for devices with EQEmax > 10%) of reported CP TADF OLEDs is plotted in Figure , with the current trend suggesting that it is extremely challenging to achieve both high EQEmax and significant gEL values simultaneously. Of the myriad structures presented herein, we highlight in particular (S)-Cz-Ax-CN, which showed a gPL of −4.8 × 10–3 in 15 wt% doped films in DPEPO, and with CP-OLEDs showing an EQEmax of 12.5% and gEL value of −1.2 × 10–2; the largest amongst CP TADF OLEDs reported to the end of 2022. Neat films of (S)-BN-CF, (S)-BN-CCB, (S)-BN-DCB and (S)-BN-AF also have high gPL of 4.1, 3.8, 3, and 2 × 10–2, respectively. CP TADF OLEDs with these emitters also exhibited very high gEL values, yet gave low EQEmax (6.0 × 10–2 and 3.5%; 5.4 × 10–2 and 2.3%; 6.7 × 10–2 and 2.9%; and 8.4 × 10–2 and 0.6%, respectively). Indeed, we note that most of the best performing CP-TADF emitters still show small gPL values of around 10–3, and there is currently no example of a CP-TADF OLED that shows both high EQEmax (> 20%) and gEL (> 0.1). The trend of decreasing gEL values with increasing EQEmax values may be fundamentally rationalized by equation eq17 , with a large electric transition dipole moment μ supporting a high ΦPL and thus EQEmax, but simultaneously limiting gEL.
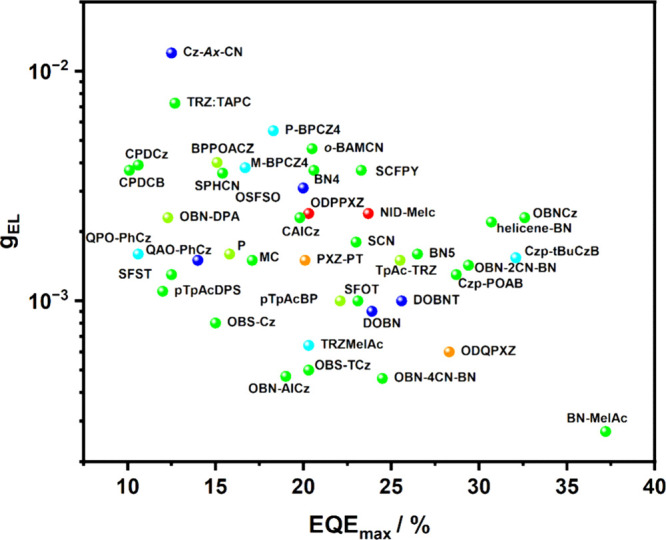
To overcome this apparent design limitation, we predict that strategies involving independent modulation of the electric transition dipole moment and magnetic transition dipole moment on separately optimized compounds (or systems) will become increasingly prominent in the coming years. As an example of the power of this approach, blend films comprising achiral polymers and chiral small molecule additives have resulted in the most robust chiroptical systems reported to date. Notably, gPL values exceeding 0.15 have been reported, mediated by CP-FRET between the conjugated polymer matrix and the chiral small molecule additives, although so far this has been demonstrated for fluorescent systems. Moreover, the integration of chiral macrocycles such as pillar[n]arenes and [n]cycloparaphenylenes with emissive subunits represents another unexplored potential strategy for forming highly luminescent chiroptical systems. This integration allows for the transfer of chiral information from the macrocycles to the emitter, without compromising the photophysical properties of the luminescent component. Integration of TADF emitters within macrocycles in this way could support simultaneous efficient triplet harvesting in OLEDs and high gPL, in a manner analogous to current hyperfluorescence approaches (Section sec17 ). Towards these outcomes, a more robust understanding of the design of chiral emitters (with or without TADF properties), chirality-preserving energy transfer, and optimized preparation processes for high-performance CP-TADF OLEDs will be essential to unlocking further performance and utility in these intriguing materials.
TADF Exciplex Emitters
Introduction
Sections sec3–sec5 showcased emitter designs based on twisted donor-acceptor compounds where electronic communication is mediated through bond across the π-network. Another strategy is to weakly couple donor and acceptor motifs through space by engineering π-stacking interactions. Similar to intramolecular CT states in covalently linked TADF molecules (see Section sec12 ), TADF can also arise from intermolecular CT states created by photo/electrical excitation of mixtures of distinct electron-donor and electron-acceptor molecules. The intermolecular CT state is formed by the transition of an electron from the LUMO of the excited-state donor to the LUMO of the acceptor, forming an exciton with the hole on the donor HOMO and electron on the acceptor LUMO. Since there is no interaction in the ground state, the emissive species is termed an excited-state complex (exciplex).ref. ref87 The mechanism behind exciplex TADF is then analogous to conventional TADF molecules, where a sufficiently small ΔE ST allows RISC to occur at ambient temperatures. This outcome is effectively an intermolecular analogue to the TSCT excited states that form when pseudo co-facially oriented donor and acceptor motifs, attached to a common scaffold, are electronically coupled (Figure ).ref. ref671 By modulating the energies of the HOMO of the donor and the LUMO of the acceptor and the distance between the two molecules, it is then possible to manipulate the exciplex emission wavelength in a straightforward, though perhaps less controllable, manner compared to TBCT or TSCT compounds. The T1 energy of the exciplex CT state is also typically lower than the LE triplet energies of the (donor) D or acceptor (A) molecules, assisting RISC by enforcing strong exciton confinement on the exciplex pair, although this confinement has been shown in some cases to be not complete and some diffusion can occur.ref. ref672
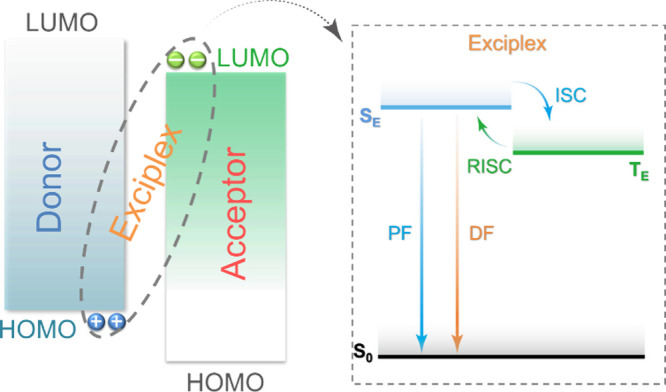
There are two key routes to prepare exciplex emitter films: bulk heterojunctions, and bilayer structures, also known as interfacial exciplexes. In the former, the donor and the acceptor materials are blended and/or co-deposited at a specific weight/volume ratio that is typically 1:1, allowing for the formation of interpenetrating networks of the materials with large contact surface area, facilitating the interaction between donor and acceptor compounds. For interfacial exciplexes, separate layers of the electron donor and acceptor materials are sequentially deposited. Interaction and exciplex formation can only occur at the interface between the two layers, although with the benefit of a considerably simpler fabrication.ref. ref671 In this review we denote bulk exciplexes by [Donor:Acceptor], with interfacial exciplexes represented by [Donor/Acceptor]. In both scenarios, as the electron-donating and accepting materials popular for exciplexes usually also possess excellent individual electron or hole transporting properties, the same materials are often used as charge transport layers, leading to simplified device architectures. Figure , at the end of this section, showcases the most efficient exciplex OLEDs in terms of EQEmax at specific color points that will be covered in this section.
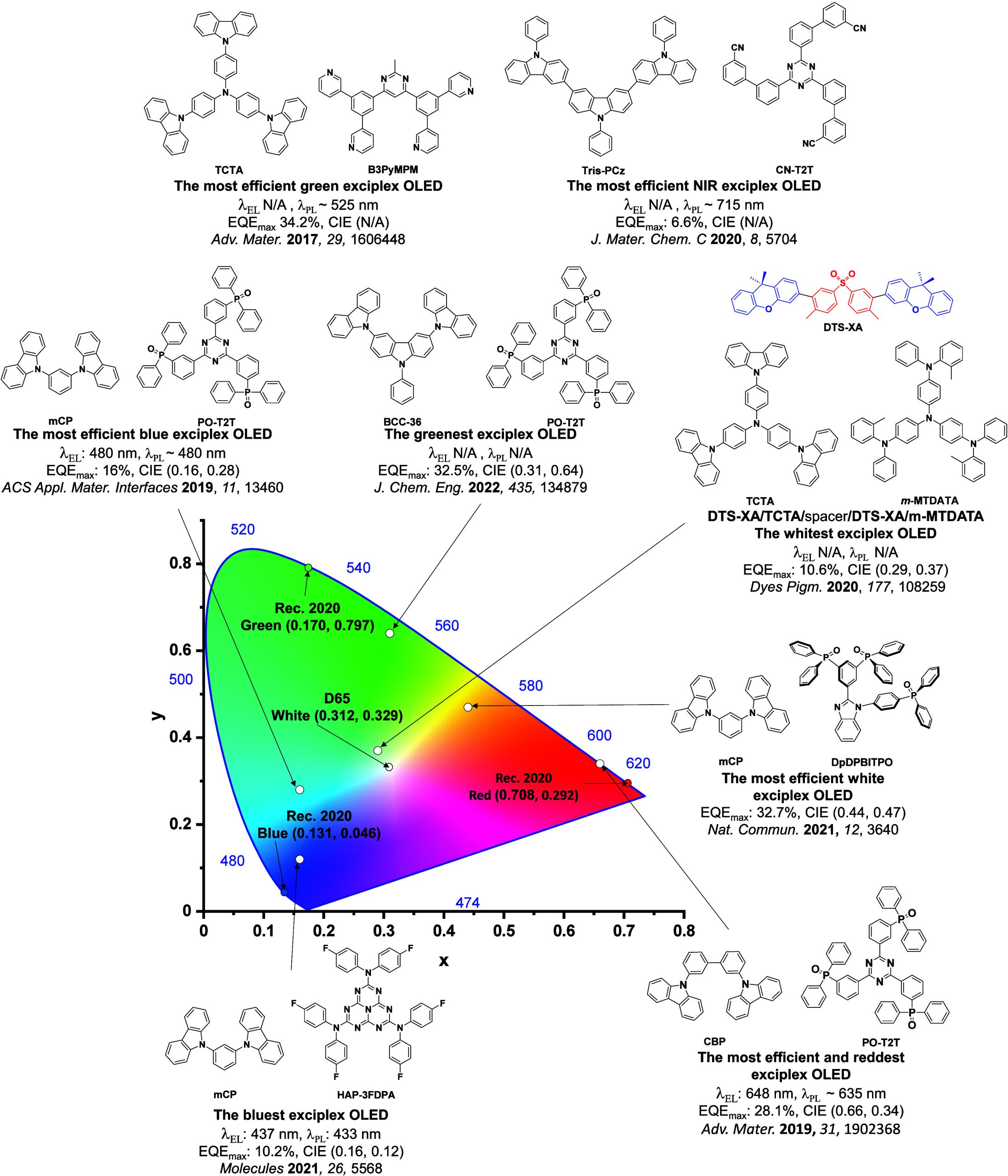
Materials Development
Not long after the pioneering studies of organic TADF OLEDs published by Adachi and co-workers in 2011 and 2012,ref76,ref673 the first exciplex TADF OLED was reported by the same group in a series of two reports.ref674,ref675 In these articles three bulk heterojunction devices were fabricated using the same donor m-MTDATA (Figure ), blended with one of three different acceptors: 3TPYMB, t-Bu-PBD, and PPT (Figure ) in 1:1 doping ratios. The device with m-MTDATA:t-Bu-PBD showed an EQEmax of 2.0%, while for the device with m-MTDATA:3TPYMB the EQEmax was higher at 5.4%, and for the device with m-MTDATA:PPT the EQEmax was the highest at 10%. While these values are low by current standards, the devices were considered promising at the time especially given the low film ΦPL of 20, 26, and 29%, respectively, indicating strong triplet harvesting ability of the exciplexes. The poor OLED performance was also partially explained by the energy levels of the exciplex allowing for exciton migration out of the emissive layer and into the m-MTDATA hole-transporting layer.
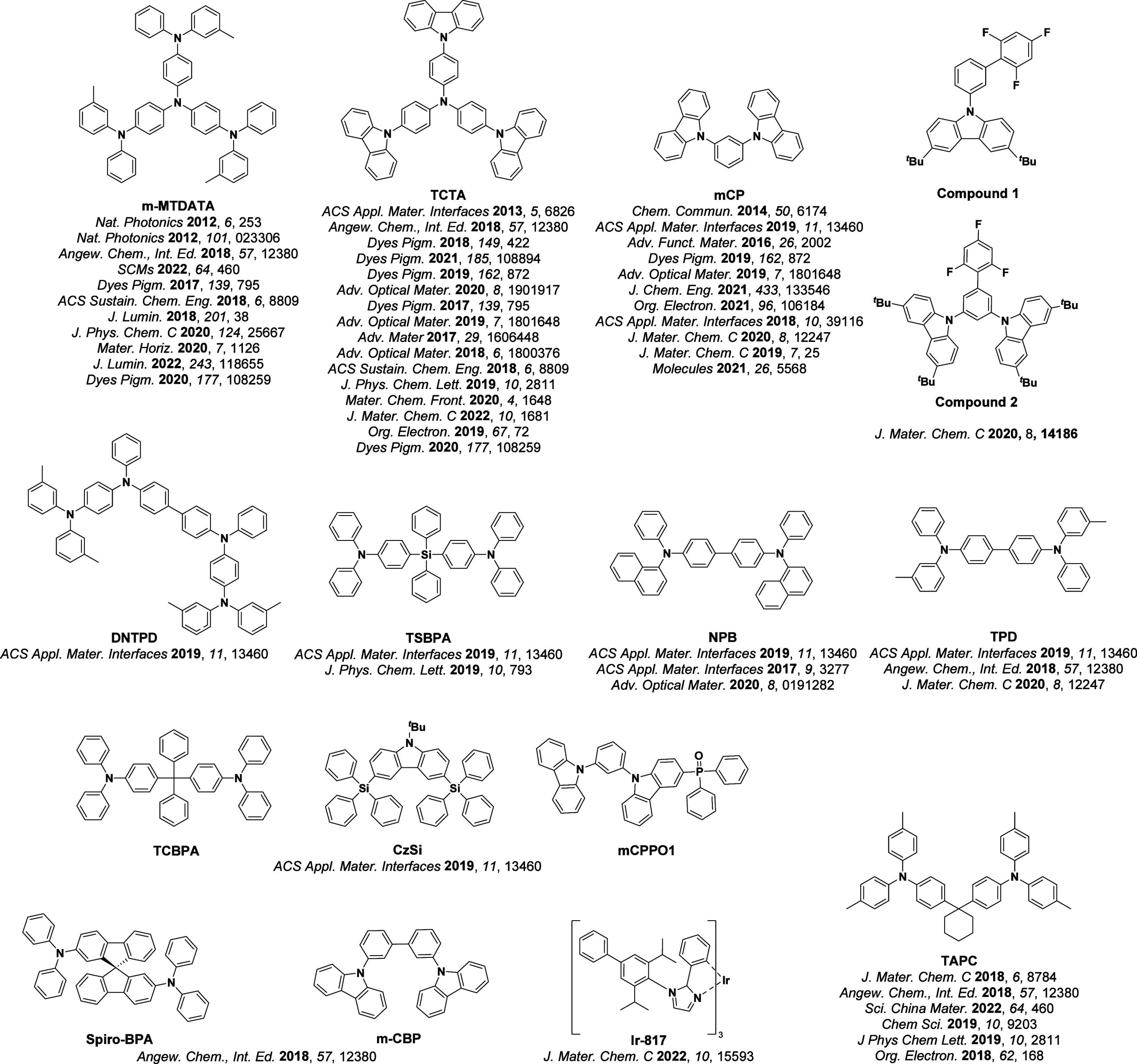
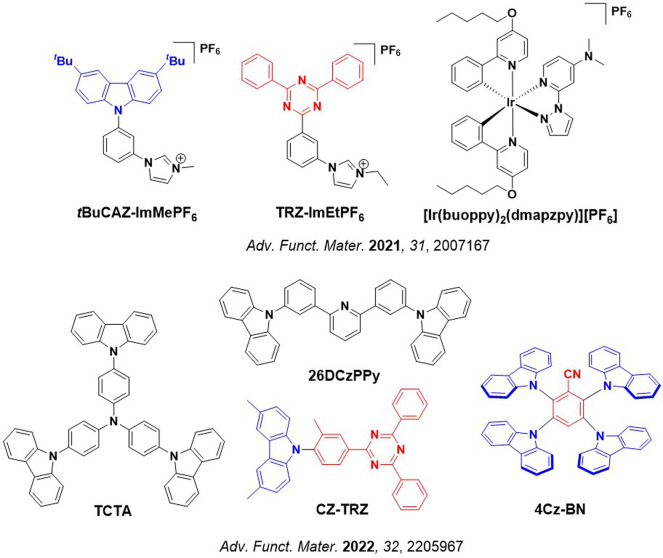
In 2013, Hung et al. published a study comparing the performance of TADF exciplex OLEDs with either interfacial or bulk heterojunction structures (1:1 ratio). The devices were produced with hole-transporting TCTA (Figure ) as the donor and electron-transporting 3P-T2T (Figure ) as the acceptor. The bilayer interface device showed yellow emission at λEL of 544 nm, an EQEmax of 7.7%, and very low efficiency roll-off with EQE1000 of 6.0%. The bulk heterojunction device displayed slightly higher EQEmax/1000 values of 7.8 and 7.7%, respectively. The greater surface area of the interpenetrating networks of donor and acceptor molecules in the bulk heterojunction device therefore resulted in a more efficient emitting layer when compared with the bilayer device.ref. ref676 Regardless, both types of devices showed comparable and promising performance, supported by the balanced hole- and electron-transporting properties of the donor and acceptor molecules of the exciplex.
The first bulk heterojunction exciplex TADF devices with different ratios of D and A molecules were reported by Li et al. in 2014. In this study the acceptor HAP-3MF (Figure ) was used in various doping percentages (8, 25, and 50 wt%, see Table S5) with the remainder made up by donor mCP (Figure ). The best results were obtained for the device with 92:8% mCP:HAP-3MF, which emitted at λEL of 538 nm and showed a EQEmax of 11.3%, supported by a ΦPL of 66.1% and τd of 1.7 and 5.7 μs (ΔE ST and CIE coordinates were not provided). The authors contended that by increasing the concentration of HAP-3MF beyond 8 wt%, deleterious concentration quenching would occur resulting in the lower EQE. These changes in composition likely also tune the charge transport within the emissive layer, and hence the size and position of the recombination zone.ref. ref677
Due to their typically lower EQEs compared to D-A TADF OLEDs, interest in exciplexes subsequently waned, reflected in the relatively small numbers of publications in the mid-to-late 2010s. However, in 2019 Chapran et al. published a study comparing a series of exciplex blends (1:1 wt% ratio) using the now-popular PO-T2T as the acceptor with different donors (Table S5). Aside from reporting an impressive EQEmax of 20% for the green-emitting TSBPA:PO-T2T devices (λEL = 528 nm, Figure ), the devices showed a highest reported maximum current efficiency of 60.9 cd A–1 and maximum power efficiency of 71 lm W–1.ref. ref186 The study also demonstrated that by changing the donor in the exciplex blend, the emission wavelength could be systematically modulated. The devices with NPB:PO-T2T and TPD:PO-T2T (Figure ) both showed orange-yellow emission (λEL = 585 nm) and EQEmax of 1.7 and 2.4% respectively. According to the authors, these low EQEmax values are related to the high rate of internal conversion, and therefore a low TADF contribution to the emission of the blends despite their near 0 ΔE ST.ref. ref186 The devices with TSPBA:PO-T2T and TCBPA:PO-T2T emitted at λEL = 528 and 542 nm, and had CIE color coordinates of (0.33, 0.57) and (0.38, 0.56), respectively. Interestingly, while these exciplex systems both showed excellent ΦPL of 100 and 93%, respectively, only the device using TSPBA:PO-T2T showed a relatively high EQEmax of 20.0% (EQEmax of 12.8% was reported for the TCBPA-PO-T2T devices). According to the authors, this difference in OLED performance is due to the formation of electron traps in the TCBPA:PO-T2T devices, which is detrimental to their performance. In the same study, the authors reported a sky-blue exciplex TADF OLED with mCP:PO-T2T, which showed CIE coordinates of (0.16, 0.28) and improved EQEmax of 16.0%, supported by the short τd ∼of 2 μs and ΔE ST < 0.01 eV reported for this blend. An exciplex TADF OLED showing a deeper blue emission [CIE coordinates of (0.16, 0.21)] was achieved using CzSi:PO-T2T (Figure ), though the EQEmax was only 6.1%. This exciplex blend has a ΔE ST of 0.10 eV leading to a τd of 6.3 μs. According to the authors, at the time of their report this device represented one of best known blue exciplex TADF OLEDs.ref. ref186 Blends with mCPPO1:PO-T2T and DNTPD:PO-T2T (Figure ) both emitted at λPL of 480 nm; however, only the former was TADF-active, which was surprising given the near-zero ΔE ST of both. Devices with mCPPO1:PO-T2T showed sky-blue emission with CIE coordinates of (0.16, 0.29) and an EQEmax of 6.5%.ref. ref186
Keruckiene et al. also employed PO-T2T to fabricate TADF exciplex systems with novel donors Compound 1 and Compound 2 (Figure ), bearing trifluorophenyl and carbazole moieties meta disposed to each other, through a phenylene spacer. These two exciplexes emit at λPL of 489 and 470 nm, and are TADF-active with τd of 865 and 879 ns (Table S5), all respectively. Even though the reported ΦPL for the blends are only 4 and 2% in air, the devices showed relatively high EQEs of 6.5 and 7.8%, values that the authors themselves noted do not easily correlate with each other. The device with an extra carbazole donor (Compound 2) presented the best overall efficiency of 7.8%, showing maximum CE of 24.8 cd A–1 and PE of 12.2 lm W–1.ref. ref678 As with much of the TADF exciplex research, it is difficult to disentangle whether the improved device performance arises from intrinsically superior exciplex photophysics, or from the tuning of charge transport properties inextricably linked to the use of the different donor materials.
Wu et al. reported exciplex blends between TAPC (Figure ) as the donor and two new acceptor molecules CbPyCN and CzPyCN (Figure ) that act both as the emitter and as host materials for PhOLEDs. The devices with TAPC:CzPyCN and TAPC:CbPyCN emitted at λEL of 530 and 520 nm and showed EQEmax of 7.4 and 9.1%, respectively. The higher device efficiency was ascribed to the more electron-deficient character of CbPyCN compared to CzPyCN, which led to better charge balance and thus less-active loss channels for excitons located in the middle of the emitting layer.ref. ref679
Mamada et al. outlined the importance of closely aligned LE and CT states in exciplex systems, using boron-based electron acceptors BFPPy-DPE (BFPD) and BPPy-DPE (BPD) (Figure ) in combination with m-MTDATA, TPD, TAPC, TCTA, Spiro-BPA, and m-CBP donors (Figure ). Not unexpectedly, the highest devices efficiencies (Table S5) were achieved when charge transfer (1CT and 3CT) and 3LE are closely aligned, allowing 3LE to be involved in the RISC process. The concentration ratio of the blend films also plays an important role, since it can modulate the energies of the CT states. The closest alignment of the 1CT, 3CT, and 3LE states was found in the BFPD:TAPC (1:1) exciplex system, which emits at λPL = 501 nm, has a ΦPL of 50.2%, and a ΔE ST of 0.04 eV. The device with this blend showed an EQEmax of 10.5% at λEL of 518 nm. However, severe efficiency roll-off was observed, with an achieved maximum luminance of only 1700 cd m–2.ref. ref680
Cao et al. reported cyano-substituted spiro[fluorine-9,9′-xanthene] (SFX) acceptors 2-carbonitrile-spiro[fluorene-9,9′-xanthene] and 2,7-dicarbonitrile-spiro[fluorene-9,9′-xanthene] (CNSFX and DCNSFX, Figure ) that were combined with TCTA to form bulk heterojunction exciplexes. Only the TCTA:DCNSFX blend showed TADF emission, with a ΔE ST of 0.05 eV and a τd of 4.67 μs. The blend emits at λPL of 520 nm and has a ΦPL of 31%. The optimized device emitted at λEL of 520 nm with CIE coordinates of (0.33, 0.52), and showed a relatively low EQEmax of 3.0%. The TCTA:CNSFX exciplex emits at λPL of 448 nm, has a ΦPL of 15%, a much larger ΔE ST of 0.32 eV, and a τPL of 65.9 ns.ref. ref681 Cao et al. later used similar SFX acceptors decorated with triazines (TRZSFX and DTRZSFX, Figure ) in combination with TCTA as the donor (Table S5). The exciplex TCTA:TRZSFX emits at λPL of 510 nm, has a ΔE ST of 0.03 eV, a very short τd of 0.18 μs, and high ΦPL of 81%, while the blend TCTA:DTRZSFX emits at λPL of 539 nm, has a ΔE ST of 0.06 eV, a comparably short τd of 0.28 μs, but a much lower ΦPL of 41%. Devices with TCTA:TRZSFX exhibited higher overall efficiency, with EQEmax, CEmax, and PEmax, of 22.5%, 79.6 cd A–1, and 78.1 lm W–1, respectively, at CIE coordinates of (0.35, 0.60). Not surprisingly considering its ΦPL, the devices with TCTA:DTRZSFX showed a much lower EQEmax of 9.7% at CIE coordinates of (0.44, 0.51).ref. ref682
Chapran et al. reported the use of a variety of electron-rich materials acting as donors, such as mCP and TCTA, together with phthalimide derivatives as acceptors to form a range of TADF exciplex systems. By varying both donor and acceptor components, a series of 20 exciplex OLEDs were studied (see Table S5). The highest efficiency device employed mCP:4-BpPht (Figure ), which emitted at CIE coordinates of (0.24, 0.41) (λPL of 497 nm) and showed an EQEmax of 2.9%. Despite this blend having a small ΔE ST of 0.06 eV and short of τd of 0.42 μs, its low ΦPL of 26% explains the low EQEmax of the device. Despite the low EQE, comparing to the ΦPL the authors nonetheless concluded that there must be active harvesting of triplet excitons in the device.ref. ref683
Zhang et al. reported the use of a sky-blue phosphorescent complex Ir-817 (Figure ) as donor motif in combination with the acceptor compounds B2PyMPM, B3PyMPM, and B4PyMPM (Figure ) to produce a series of TADF exciplex OLEDs showing deep-red-to-NIR emission. The decrease in non-radiative decay and higher emission efficiency observed in these blends was attributed to the strong spin-orbit coupling generated by the iridium atom, which contributes to faster k RISC as well as in promoting phosphorescent decay from T1 to S0. The blends with Ir-817:B2PyMPM, Ir-817:B3PyMPM, and Ir-817:B4PyMPM emit at λPL of 606, 632, and 642 nm, have ΦPL of 13.7, 7.2, and 4.6%, τd of 11.8, 9.6, and 9.1 μs, and ΔE ST of 0.01, 0.02, and 0.02 eV, all respectively. The OLEDs showed EQEmax of 3.1, 1.5, and 1.0%, emitting at λEL of 620, 640, and 672 nm [CIE coordinates of (0.58, 0.42), (0.62, 0.37), and (0.66, 0.33)]. By further increasing the strength of the acceptors in the use of TRZ-1SO2 , TRZ-2SO2 , and TRZ-3SO2 (Figure ), a stronger red-shift in the blends was observed with emission at λPL of 647, 666, and 698 nm, respectively and ΔE ST of 0.02 eV for all three blends. The NIR devices emitted at λEL of 658, 700, and 746 nm, and showed EQEmax of 0.26, 0.22, and 0.20%, respectively.ref. ref684
TADF Compounds Applied as Either Donors or Acceptors in Exciplex Systems
Beyond mixing separate fluorescent D and A molecules to form TADF exciplexes, D-A TADF materials can themselves be used, acting as either the donor or the acceptor in the blend. Lui et al. reported such an exciplex consisting of PO-T2T as the acceptor and MAC as a D-A TADF donor (Figure ). As an emitter in its own right, MAC, which is a composed of a DMAC donor and a 3-methyl-1H-isochromen-1-one acceptor, has a small ΔE ST of 0.02 eV. The study compared the EQEmax of a reference exciplex device using a blend of 1:1 mCP:PO-T2T, and a device with 1:1 MAC:PO-T2T. The former emits at λPL of 472 nm and has ΔE ST of 0.01 eV, while MAC:PO-T2T has a similar ΔE ST of 0.014 eV, but emitting at λPL of 514 nm. Even though the ΦPL of the blends are effectively the same under air (7.3 and 8.0%, respectively) there was a significant improvement in the device EQEmax, increasing from 8.6% for the device with mCP:PO-T2T [λEL of 476 nm and CIE coordinates (0.17, 0.26)] to 13.1% for the device with MAC:PO-T2T [λEL = 516 nm and CIE coordinates (0.31, 0.55)]. This improvement in EQEmax could be increased further to 17.8% by modifying the blend ratio to 7:3 wt% MAC:PO-T2T, which at the time of publication was one of the highest reported efficiencies for TADF exciplex OLEDs. This outstanding result was attributed to the parallel RISC processes in the TADF donor molecule and in the exciplex pair, resulting in higher triplet exciton harvesting efficiencies.ref. ref685
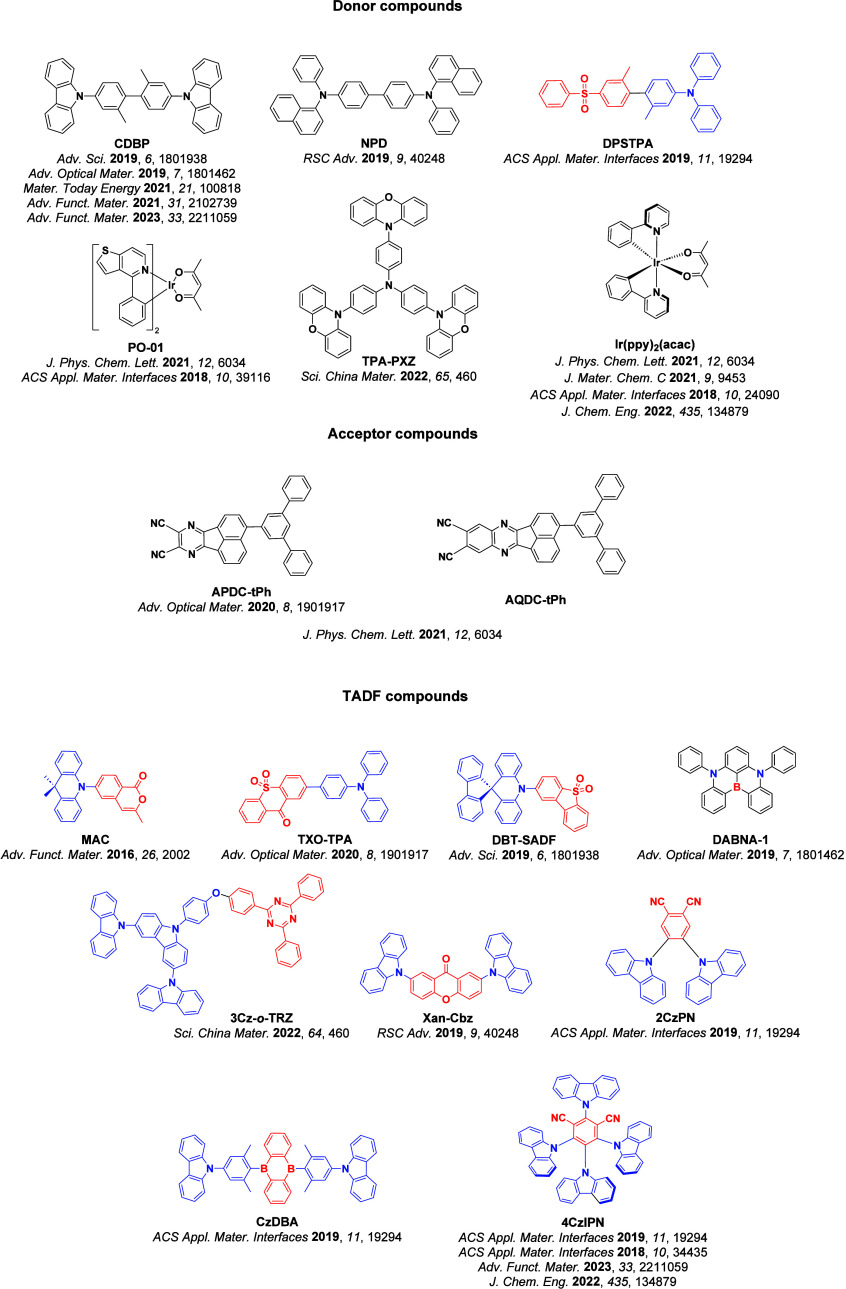
Zhang et al. proposed a similar strategy to improve exciton utilization in TADF exciplex emitters. A three-component exciplex featuring CDBP (Figure ) and the TADF compound DBT-SADF (Figure ) as the donors along with PO-T2T as the acceptor was investigated. Three separate RISC channels operating in DBT-SADF, DBT-SADF:PO-T2T, and CDBP:PO-T2T were posited to exist, evidenced by an increase of the ΦPL from 38% for DBT-SADF:PO-T2T to 61% (or the three-component exciplex. Additionally, a relatively fast combined k RISC of 14.2 × 105 s–1 was reported. Devices fabricated using the ternary mixture showed a low turn-on voltage of 2.4 V, EQEmax of 20.5%, CEmax of 60.0 cd A–1, and PEmax of 69.7 lm W–1.ref. ref686 A ternary TADF exciplex system was also reported by Jeon et al.ref. ref687 who employed a mixture of CDBP donor, PO-T2T acceptor, and different ratios of the MR-TADF emitter DABNA-1 as an additional donor (Figure ). The 1:1 blends of CDBP:PO-T2T and PO-T2T:DABNA-1 emit at λPL of 494 and 550 nm, have ΦPL of 53 and 46%, and τd of 17.2 and 14.7 μs respectively, and the ternary blends of CDBP:PO-T2T:DABNA-1 (Table S6) display similar photophysical properties. Ternary blends with ratios of 47.5:47.5:5, 45:45:10 and 40:40:20 all emit at λPL of 550 nm, have the same τd of 15.1 μs, and have ΦPL of 67, 69, and 50%, respectively (no ΔE ST values were reported). Devices with 47.5:47.5:5.0 CDBP:PO-T2T:DABNA-1 showed the highest EQEmax of the series at 17.5% [CIE coordinates of (0.31, 0.58)], while devices with 45:45:10 and 40:40:20 CDBP:PO-T2T:DABNA-1 achieved EQEmax of 16.9 and 13.4% [CIE coordinates of (0.34, 0.60) and (0.37, 0.60)], respectively. These efficiencies were partly attributed to energy transfer from the high energy exciplex (CDBP:PO-T2T, acting as a host), to the low-energy exciplex DABNA-1:PO-T2T acting as emissive dopant.
Siddiqui et al. (2019) demonstrated that the color emitted by an exciplex OLED could be modulated by simply changing the applied voltage. A carbazole-xanthone-based D-A-D TADF material (Xan-Cbz, Figure ), emitting at λPL of 470 nm and having a ΔE ST of 0.32 eV with a τd of 3.8 μs, was used as an acceptor alongside donor NPD (Figure ). The device fabricated using a bilayer structure of NPD/Xan-Cbz showed dual emission with λEL at 465 nm, associated with the TADF acceptor, as well as at 525 nm corresponding to the exciplex emission. By increasing the voltage, and hence changing the ratio of molecular/exciplex excitons across the interface, the ratio of two peaks changed along with the color of the device. Additional photophysical data for this interfacial exciplex was not provided by the authors, although similar voltage-dependant color changes in an interfacial exciplex using simpler materials were studied in detail previously.ref688,ref689
Wu et al. demonstrated the use of TADF compounds as electron acceptors in a study that showed how the intermolecular distancing between donor and acceptor molecules affects the overall efficiency of the exciplex OLEDs. Ambipolar DPSTPA (ΔE ST = 0.27 eV), was used as the donor alongside each of three TADF acceptors: 2CzPN, CzDBA, and 4CzIPN (Figure ). Devices with 3:1 wt% of DPSTPA:2CzPN or DPSTPA:CzDBA emitted at λEL of 544 and 592 nm, with EQEmax of 19.0 and 14.6%, CEmax of 59.9 and 29.6 cd A–1, and PEmax of 62.7 and 31.0 lm W–1, all respectively. However, devices with DPSTPA:4CzIPN (3:1 wt%) achieved a much lower EQEmax of 3.8%. This poor performance was attributed to the larger distances between DPSTPA and 4CzIPN molecules caused by the steric bulk of the carbazole (Cz) groups of 4CzIPN, which hinders the exciplex-forming interaction of the nitrile groups of 4CzIPN and the TPA donor of DPSTPA.ref. ref690
Hu et al. fabricated NIR exciplex OLEDs by combining APDC-tPh (Figure ) as the acceptor along with donors such as TCTA and TADF TXO-TPA (Figure ) in different weight percentages (Table S6). Due to the additional RISC pathway associated with the TADF acceptor, the 1:1 ratio device with TXO-TPA:APDC-tPh showed an EQEmax of 1.27% at λPL = 704 nm (a respectable efficiency at this NIR wavelength), contrasting with an EQEmax of 0.09% at λEL = 730 nm in the device with a 1:1 ratio of TCTA:APDC-tPh.ref. ref691 The same authors also explored different ratios of an exciplex blend consisting of the phosphorescent complexes Ir(ppy)2acac (Figure ) and PO-01 as the donors, while APDC-tPh and AQDC-tPh (Figure ) were employed as the acceptors (Table S6). The optimized device with 15:85 PO-01:AQDC-tPh emitted at λEL of 750 nm and showed an EQEmax of 0.23%, withthe triplet excitons being harvested by both the exciplex and the phosphor donor.ref. ref692
Yang et al. reported TADF material 3Cz-o-TRz containing a D-o-A structure (Figure ), which was employed as both a donor or acceptor in different exciplex blends (Table S6). A total of six blends were fabricated using the acceptors B3PyMPM, B4PyMPM, and PO-T2T, and with donor compounds TAPC, TPA-PXZ, and m-MTDATA (Figure ). The best performing devices had EML compositions of 3Cz-o-TRz:PO-T2T or TAPC:3Cz-o-TRz, both of which showed TADF at λPL of 510 nm and similar ΦPL and τd of 66 and 68%, and of 1.5 and 1.8 μs respectively. The respectively devices emitted at λEL of 516 and 520 nm and showed EQEmax of 11.8 and 12.1%. These results prompted fabrication of tandem OLEDs with the mixed heterojunction/interfacial structure TAPC:3Cz-o-TRz|3Cz-o-TRz|3Cz-o-TRz:PO-T2T, which emitted at λEL of 516 nm, had low turn-on voltage of 2.4 V, and showed an EQEmax of 14.1% with CEmax of 43.8 cd A–1.ref. ref693
Understanding and Improving Exciplex Efficiency
Hung et al. documented an interesting strategy to improve the ΦPL and thus the efficiency of exciplex TADF OLEDs.ref. ref694 The authors claimed that an enhancement in the performance can be achieved by introducing steric bulk onto the donors, thereby weakening the electronic coupling between donor and acceptor molecules. To demonstrate this, two reference blends (1:1 wt% ratio) using DTAF or CPF (Figure ) as donor molecules in combination with electron-acceptors 3N-T2T (Figure ) and PO-T2T, respectively, were fabricated. Their performance (Table S7) was compared to blends using bulkier congener donors DSDTAF and CPTBF (Figure ), containing either extra triphenylsilyl (SiPh3) groups or tert-butyl substituents. The DSDTAF:3N-T2T blend emits at λPL of 535 nm and has a ΦPL of 59%, with a τd of 2.54 μs and the corresponding device showing an EQEmax of 13.2%. The reference blend DTAF:3N-T2T emits at the same wavelength, has a modestly lower ΦPL of 51%, and has device EQEmax also slightly lower at 11.6%. Similarly, CPTBF:PO-T2T emits at λPL of 480 nm and has an ΦPL of 44% with τd of 5.86 μs, translating into a device EQEmax of 12.5%. The corresponding reference exciplex blend CPF:PO-T2T also emits at 480 nm, has similar ΦPL of 41% and τd of 2.8 μs, and yet the device showed a considerably reduced EQEmax of 9.5%. Although it is thought that the emission of the exciplex can be tuned as result changing the intermolecular distance of donor and acceptor, this work reported no shift in in the λPL.ref. ref695 These results, however, suggested that bulky tBu or SiPh3 substituents can improve the ΦPL of exciplex blends and their performance in OLEDs.
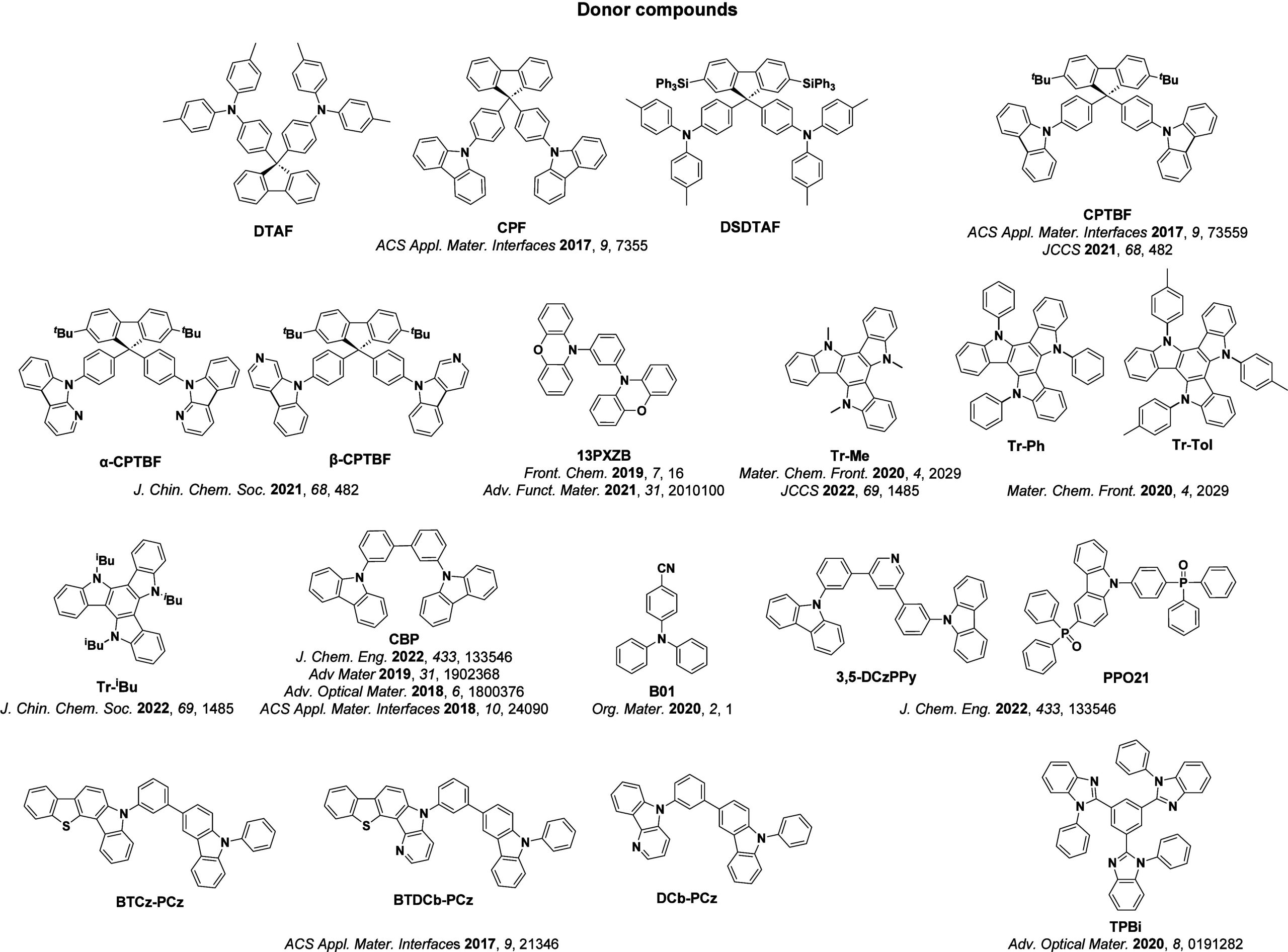
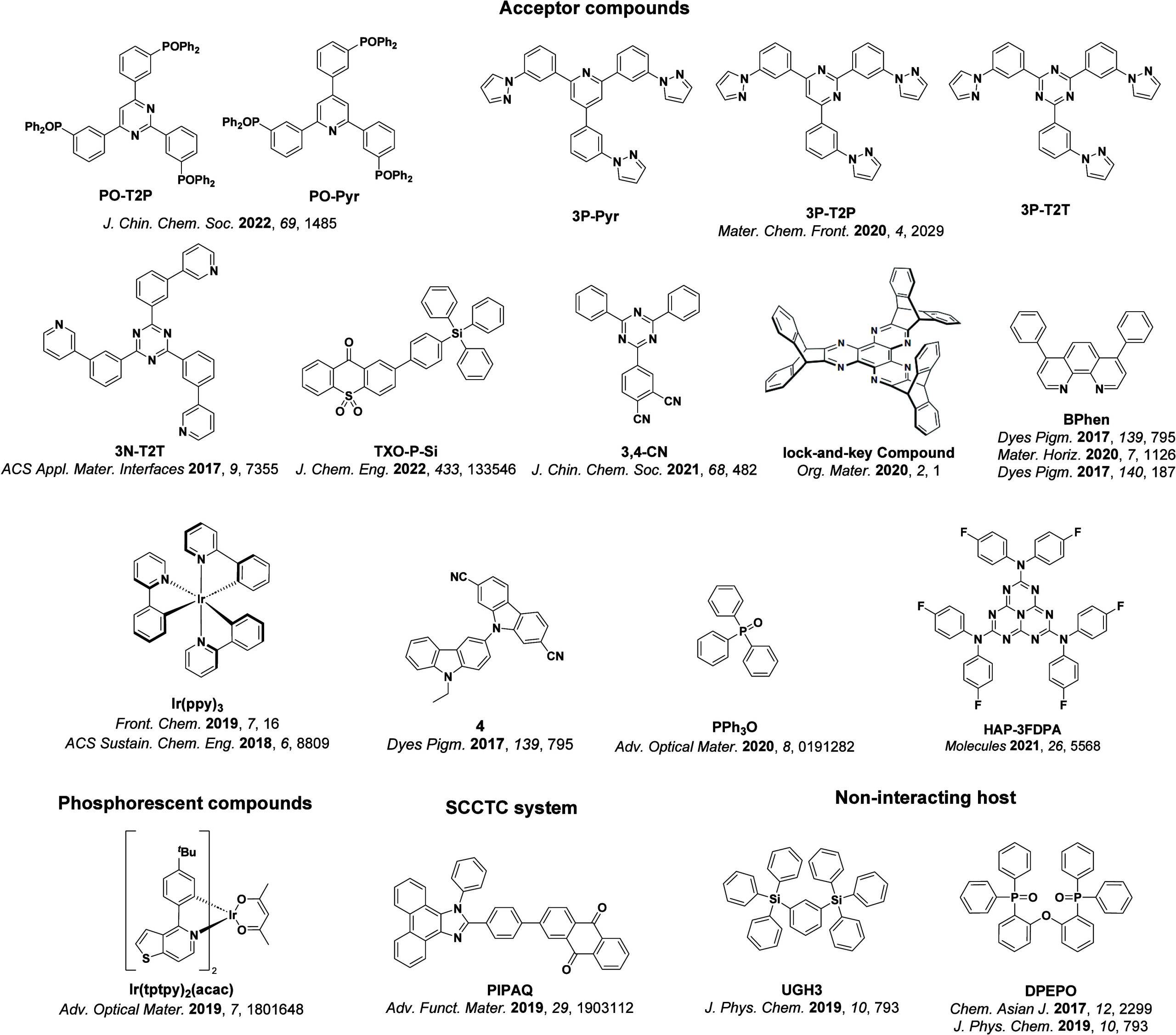
Skuodis et al. developed a new approach for the fabrication of TADF exciplex OLEDs in a device featuring both bilayer and bulk structures. A new carbazol-9-yl-substituted 9-ethylcarbazole derivative containing nitrile groups (material 4, Figure ) was employed as the acceptor along with standard donor materials. The exciplexes TCTA:4 and m-MTDATA:4 (1:1 ratio) emit at λPL of 490 and 584 nm, have ΦPL of 43.8 and 3.8%, and very short τd of 0.31 and 0.19 μs, respectively. The devices fabricated using the hybrid bulk/interfacial approach, TCTA:4/4/Bphen and m-MTDATA:4/4/Bphen, emitted at λEL of 490 and 600 nm, and showed comparable respective EQEmax of 4.2 and 3.2%.ref. ref696 By contrast, a device of 4/Bphen emitted at λEL of 475 nm and showed a lower EQEmax of 2.0%, demonstrating the contribution of the exciplex to the overall device performance.
Colella et al. demonstrated how to simultaneously induce a blue-shift in the emission of an exciplex TADF systems and also improve the ΦPL, leading to higher device EQE. This occurs due to weakened electronic coupling between donor and acceptor molecules in the exciplex blend as a third component is added, which reduces the Coulomb binding term of the exciton’s electron and hole and therefore increases the total energy of the emissive CT state. Previously reported blend TSPBA:PO-T2T (1:1 ratio) was diluted with different concentrations of a third non-interacting host, either UGH-3 or DPEPO (Figure ). By adding 90 vol% of UGH-3, the photoluminescence onset changed from 2.67 (for 1:1 exciplex films) to 2.85 eV. An increase in the ΦPL from 58% in undiluted exciplex films to 80% in a film with 50 vol% UGH-3 was also reported, although the source of this ΦPL enhancement remains a mystery and was later shown to not be universally translatable to improved OLED performance.ref. ref697 For concentrations higher than 50 vol% of the inert host material, the ΦPL began to decrease as the concentration of the exciplex-forming materials decreased. The highest device performance was achieved using 50 vol% of UGH-3, where the device showed an EQEmax of 19.2% in comparison with 14.8% for the undiluted exciplex.ref. ref698
Yuan et al. also explored the acceptor PO-T2T in order to show the importance of spatial distancing between donor and acceptor molecules in exciplex TADF systems.ref. ref695 According to the authors, the potential energy surfaces of the excited states have a strong dependence on this distance. By manipulating the separation between D and A compounds in a blend of TCTA:PO-T2T (1:1 weight ratio) using different weight concentrations mCP as a spacer within the bulk exciplex, an enhancement of up to 105% in the EQEmax was observed (Table S7). The best results were reported for the blend with a weight ratio of 1:1:3 TCTA:PO-T2T:mCP, where an EQEmax of 8.0% was achieved in comparison with only 3.9% for the device with TCTA:PO-T2T. The addition of the host also affected the ΔE ST, decreasing from 0.06 (without mCP) to 0.02 eV. As a result of the smaller ΔE ST there was an enhancement from 78 to 92% of the fraction of delayed fluorescence contributing to the total PL, and also an increase of the ΦPL from 13 to 37% for films with weight ratio of 1:1:0 and 1:1:3, respectively. It remains unclear though what role competing exciplex formation between PO-T2T:mCP plays in this performance enhancement. The authors then used the TCTA:PO-T2T:mCP blend as a host for orange phosphorescent emitter Ir(tptpy)2(acac) (Figure ), where the devices emitted at λEL of 555 nm and showed an EQEmax of 21.7%.ref. ref695
In a similar study, Pu et al. controlled the distance between donor and acceptor molecules of a TADF exciplex by incorporating an inert spacer layer of up to 70 nm between the layers of an interfacial exciplex system. TAPC and DCA were used as the respective donor and acceptor for the interfacial exciplex, while DMA and CBP (Figure ) were applied as the inert spacers (Table S7). The highest device efficiencies were achieved for the OLEDs with TAPC/DMA (70 nm)/DCA and TAPC/CBP (20 nm)/DCA, with EQEmax of 0.86 and 3.0% and emitting at λEL of 445 and 550 nm respectively. Exciplex formation at both the donor/spacer interface and at the spacer/acceptor interface resulted in long-distance spacer-mediated coupling between the donor and the acceptor.ref. ref699
Zhang et al. reported efficient red TADF exciplex devices using a phosphorescent complex as the acceptor moiety. Blends with PO-T2T acting unusually here as the donor and with fac–Ir(ppy)3 (Figure ) as the acceptor have small ΔE ST of 0.026 eV, τd of 2.8 μs, and ΦPL of 23.3%. The devices with PO-T2T:Ir(ppy)3 (92:8) emitted at λEL at 604 nm [CIE coordinates of (0.55, 0.44)] and showed an EQEmax of 5.0%, CEmax of 9.3 cd A–1, and PEmax of 11.6 lm W–1. By contrast, the use of 13PXZB (Figure ) as the donor, together with PO-T2T as the acceptor (60:40 13PXZB:PO-T2T) as the exciplex emitter resulted in a device that emitted at λEL at 592 nm [CIE coordinates of (0.52, 0.47)] and showed a lower EQEmax of 1.9%. The authors claimed that there is enhanced SOC within the PO-T2T:Ir(ppy)3 exciplex, associated with the iridium center, which is responsible for faster ISC and RISC processes, which leads to the higher ΦPL of the PO-T2T:Ir(ppy)3 blend (23.3%, compared to 8.6% 13PXZB:PO-T2T) and, hence, also an enhancement in the overall OLED efficiency.ref. ref700
Chen et al. published a study claiming to report the first example of a single-component charge transfer complex (SCCTC), showing deep-red-to-NIR TADF. A SCCTC is a molecule that has donor and acceptor moieties that only electronically couple to the respective acceptor and donor groups of neighboring molecules, essentially corresponding to a single-component bulk exciplex material. PIPAQ (Figure ) where the phenanthro[9,10-dimidazole (PI) and anthraquinone (AQ) are the respective donor and acceptor moieties is purported to be one such SCCTC compound. The isolated molecule has a moderate ΔE ST of 0.13 eV, yet forms co-facial head-to-tail dimers in neat films which result in exciplex emission at λPL of 650 nm, having a τd of 40.5 μs and a ΦPL of 12.3%.ref. ref701 Devices with PIPAQ showed an EQEmax of 2.1% at CIE coordinates of (0.64, 0.36).ref. ref702
Hu et al. showed that aggregation of the donor material can strongly affect device performance due to substantial residual emission from the aggregate.ref. ref703 In their study triazatruxene-based molecules Tr-Me, Tr-Ph, and Tr-Tol (Figure ) were used as donor materials alongside the acceptors 3P-T2T and its pyrimidine (3P-T2P) and pyridine (3P-Pyr) derivatives (Figure ). The blends using Tr-Me showed significant donor aggregation which prevented exciplex formation; however, the other donors blends were promising (Table S7), with the exciplex systems formed using 3P-T2P as the acceptor showing the best results. Blends of Tr-Ph:3P-T2P and Tr-Tol:3P-T2P showed similar photophysical properties, emitting at λPL of 526 and 525 nm, having τd of 1.77 and 2.39 μs, ΦPL of 41 and 40%, and ΔE ST of 0.18 and 0.10 eV, all respectively. The devices with Tr-Ph:3P-T2P and Tr-Tol:3P-T2P emitted in the green at CIE coordinates of (0.33, 0.54) and (0.35, 0.54) and showed EQEmax of 10.4 and 12.8%.ref. ref703 In a subsequent study, the same group analysed a series of exciplex blends with the goal to suppress donor aggregation. A total of six blends (Table S7) were fabricated using either Tr-Me or a triazatruxene-based analogue donor with larger alkyl substituents (Tr-iBu, Figure ) in combination with PO-T2T, PO-T2P, or PO-Pyr (Figure ) as acceptors. The blends using Tr-iBu showed suppressed donor aggregation and blue-shifted emission compared to those using Tr-Me, attributed to an increased intermolecular distance between donor and acceptor molecules leading to a destabilized charge transfer states. The highest performing OLEDs were obtained with Tr-iBu:PO-T2P (1:2 ratio), showing λEL at 560 nm [CIE coordinates of (0.43, 0.54)] and EQEmax of 8.3%. Devices with Tr-iBu:PO-Pyr (1:2 ratio), emitted at λEL of 516 nm [CIE coordinates of (0.27, 0.50)] and showed similar EQEmax of 7.5%. Tr-iBu:PO-Pyr was also used as an exciplex host for the emitter DPy2CN (Figure ). The highest efficiency device contained3 wt% DPy2CN, and showed an EQEmax of 6.3% at CIE coordinates of (0.63, 0.35).ref. ref704 Introducing silyl groups to similarly address molecular aggregation, Wei et al. reported a family of green-emissive TADF exciplexes consisting of TXO-P-Si (Figure ) as the acceptor and varying the donor compounds through mCP, CBP, 3,5-DCzPPy, and PPO21 (Figure ). The highest performance devices included mCP:TXO-P-Si (1:4) and 3,5-DCzPPy:TXO-P-Si (1:1), which showed EQEmax of 16.9 and 16.1% respectively.ref. ref705 This is due to their relatively higher respective ΦPL of 55.4 and 47.7% and small ΔE ST of 0.02 and 0.06 eV.
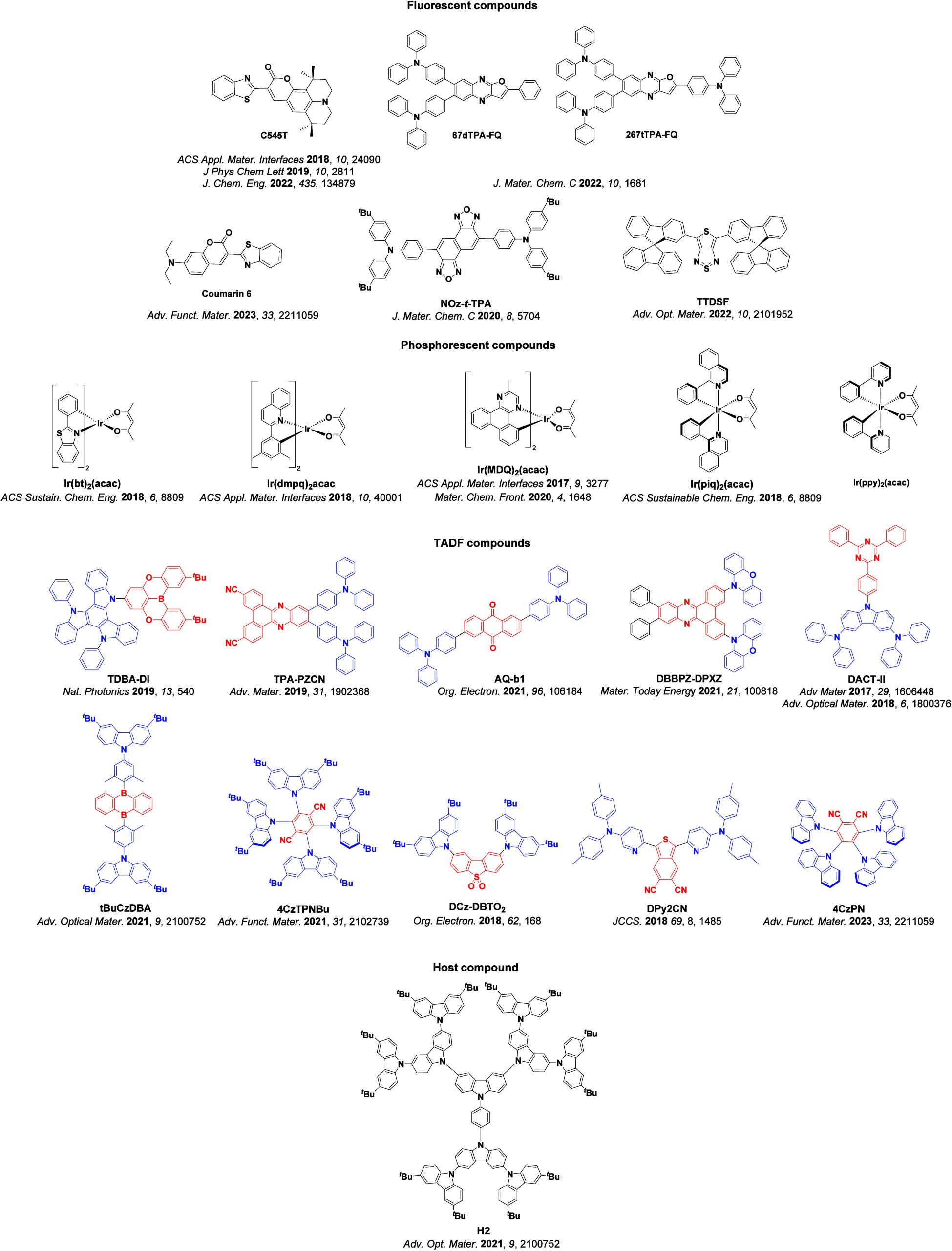
Chen et al. reported weak donor compounds α-CPTBF and β-CPTBF (Figure ), where the carbazole moiety of the model donor molecule CPTBF was replaced by either an α- or β-carboline. Used in conjunction with 3,4-CN (Figure ) as the acceptor, the 1:1 exciplex blends CPTBF:3.4-CN, α-CPTBF:3.4-CN, and β-CPTBF:3.4-CN each showed TADF emission with τd of 0.12, 0.10, and 0.10 μs, and ΦPL of 18.0, 20.0, and 21.0%, all respectively. The blends α-CPTBF:3.4-CN and β-CPTBF:3.4-CN both emit at λPL of 504 nm, which is blue-shifted in comparison with the reference blend CPTBF:3.4-CN (λPL= 522 nm). Such a blue-shift is not surprising, since the weaker carboline leads to a deeper HOMO of the donor molecules. The devices with α-CPTBF:3,4-CN emitted at CIE coordinates at (0.30, 0.56) and showed the highest EQEmax of the series at 7.6%, with CEmax of 25.2 cd A–1 and PEmax of 25.9 lm W–1. The superior performance was mainly attributed to the higher ΦPL and the faster RISC, reflected in the greater contribution of delayed fluorescence to the total emission of the device.ref. ref706
Zhang et al. demonstrated the value of introducing intermolecular hydrogen bonds between the donor and acceptor compounds, which were hypothesized to reduce inter- and intra-molecular vibrational relaxation and thus increase ΦPL.ref. ref707 Three exciplex systems composed of the donor 13PXZB (Figure ) and each of the acceptors B4PyMPM, B3PyMPM, and B2PyMPM were investigated. These were expected to have different numbers of hydrogen bonds between donor and acceptor groups: 13PXZB:B4PyMPM having the most intermolecular hydrogen bonds followed by 13PXZB:B3PyMPM, while 13PXZB:B2PyMPM does not have any hydrogen bonding between D and A. Correlated with this trend, the device with 13PXZB:B4PyMPM emitted at λEL of 560 nm [CIE coordinates of (0.41, 0.55)] and showed the highest EQEmax of 14.6% (CEmax of 43.1 cd A–1, PEmax of 48.3 lm W–1).
Voll et al. explored interlocking molecular donor-acceptor designs using a lock-and-key approach, where acceptor “key” and donor “lock” molecules were tailored to fit each other by supramolecular self-assembly. The acceptor contained a hexaazatriphenylene core flanked by three triptycene moieties, and was partnered with donors featuring triarylamines, triarylbenzenes, and triarylbenzotrithiophenes (lock and key compound, Figure ). Only one device was reported, fabricated using triarylamine donor B01 (Figure ) in a 1:1 weight ratio D:A blend. This device showed an EQEmax of 5.4% and emitted at λEL = 536 nm, which was significantly red-shifted compared to the film λPL of 461 nm (τd of 45.1 μs).ref. ref708
Towards developing elusive blue OLED emission, Wang et al. reported δ-carboline derivatives BTCz-PCz, BTDCb-PCz, and DCb-PCz as donors in both bulk and interfacial exciplexes with acceptor TmPyPB (Figure ).ref. ref709 The highest efficiency OLED used BTDCb-PCz:TmPyPB (99:1 wt% ratio), emitting at λEL of 468 [CIE coordinates (0.16, 0.21)] with ΔE ST of 0.05 eV, and with an EQEmax of 2.4%, CEmax of 4.64 cd A–1, and PEmax of 2.91 lm W–1. On the other hand, the interfacial exciplex device BTDCb-PCz/TmPyPB showed an EQEmax of only 1.1% at CIE coordinates (0.20, 0.31). The authors attributed the reduced efficiency of the interfacial exciplex device to the recombination zone being very close to the ETL. Guzauskas et al. documented that a device with interfacial exciplex mCP/PO-T2T emitted at λEL of 497 nm and showed an EQEmax of 8.2%. By thermally annealing the emitting layers after deposition, a red-shift to a λEL of 570 nm was observed while the EQEmax remained the same (Table S7).ref. ref710 Hippola et al. demonstrated that with an appropriate device structure, deep blue exciplex OLEDs could be fabricated with TPBi:PPh3O (Figure and Figure ). The OLED emitted at λEL of 435 nm and showed an EQEmax of 4.0%. According to the authors, the EL spectrum arose from the interfacial exciplex between NPB and the 5:1 TPBi:PPh3O blend.ref. ref711 Li et al. fabricated deep-blue devices with 92:8 wt% mCP:HAP-3FDPA (Figure ), which emitted at λPL of 433 nm, had ΦPL of 53.2%, and a ΔEST of 0.09 eV. The devices showed an EQEmax of 10.2% at CIE coordinates of (0.16, 0.12), making it the bluest exciplex OLED reported to date.ref. ref712
TADF Exciplex as Hosts
Using a sensitization approach that mirrors hyperfluorescence (see Section sec17 ), many studies now use TADF exciplexes as co-hosts and triplet harvesters for separate terminal emitters. This design allows harvesting of triplet excitons by the exciplex (and sometimes also TADF-active terminal emitters), while avoiding some of the undesirable photophysical properties of exciplexes such as broad CT emission spectra, slow radiative rates, and low ΦPL. This approach is enabled by Förster resonance energy transfer (FRET) from the exciplex to the emissive guest,ref713,ref714 and can be particularly effective in the design of NIR OLEDs. For instance, Huang et al. reported an OLED with EQEmax of 6.6% at λEL of 710 nm using NOz-t-TPA (Figure ) doped in the Tris-PCz:CN-T2T exciplex (Figure ),ref. ref713 while Chen et al. reported a device EQEmax of 5.3% at λEL of 774 with an EML of 7 wt% TTDSF (Figure ) doped in DPSF:CN-T2T (Figure ).ref. ref714 Zhang et al. similarly demonstrated the differences in device performance using a ‘passive’ conventional host (CBP) compared to a TADF-active exciplex host (CBP:PO-T2T) doped with the same red TADF emitter (TPA-PZCN, Figure ). With CBP:PO-T2T the device EQEmax was slightly improved (28.1%) along with a red-shifted electroluminescence indicating more complete energy transfer to the terminal emitter (λEL of 648 nm, Table S8) comparing favorably to the device with CBP as the host (λEL = 628 nm and EQEmax of 27.4%). No other photophysical data were provided.ref. ref507
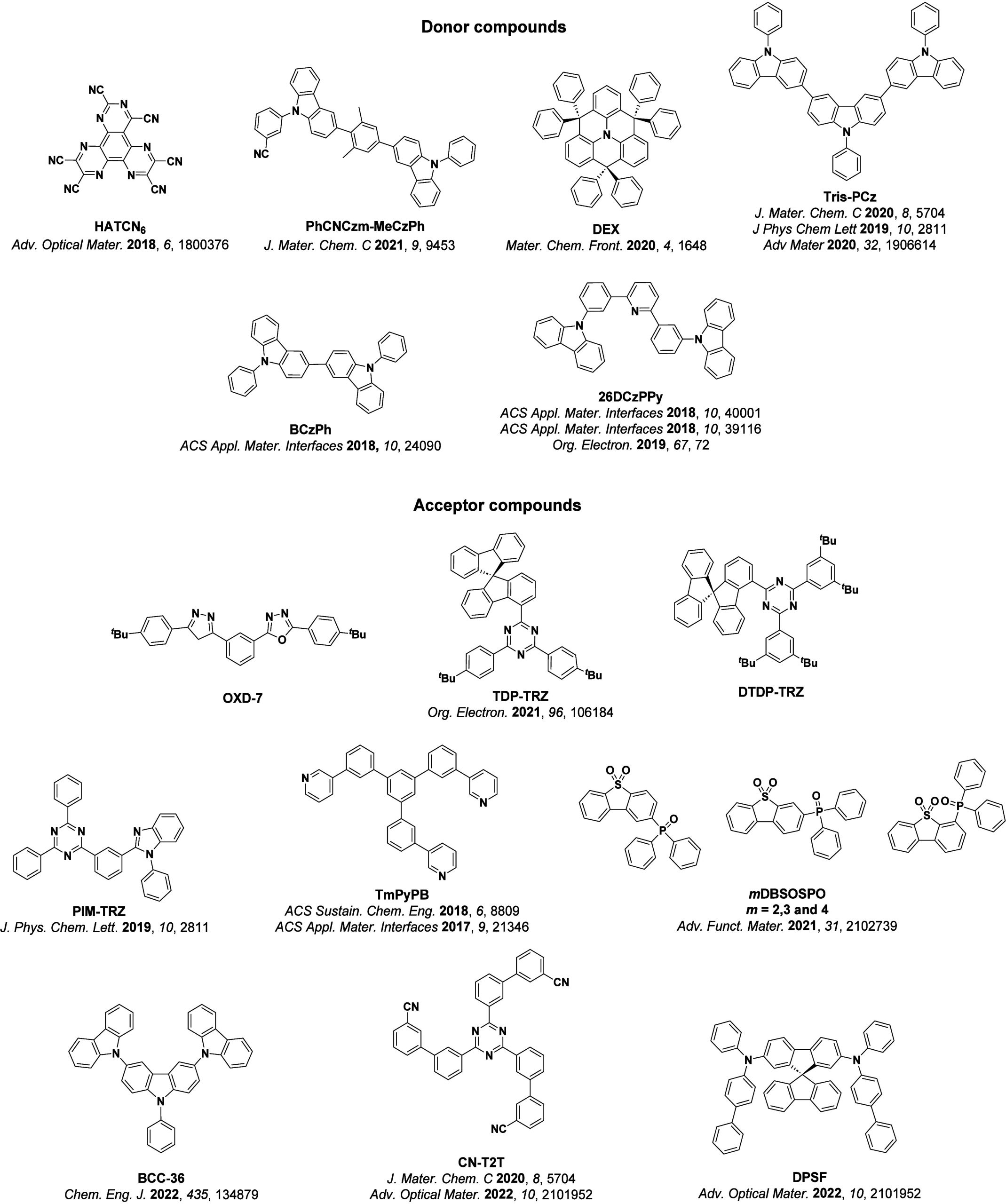
Examples of white, blue, and green TADF OLEDs using exciplex hosts have been demonstrated with high efficiencies.ref. ref673 For example, Chen et al. used different doping concentrations of the red TADF emitter DBBPZ-DPXZ (Figure ) in a blue emissive exciplex matrix (CDBP:PO-T2T) to achieve red and white emission. Using 0.2 wt% of red DBBPZ-DPXZ, a warm white OLED with CIE coordinates of (0.40, 0.38) and EQEmax of 20.7% was obtained. When the concentration of DBBPZ-DPXZ was increased to 6 wt% the energy transfer from host to dopant was more complete, resulting in a red OLED emitting at λEL of 628 nm and showing an EQEmax of 20.8%.ref. ref715 Similarly, Moon et al. reported green OLEDs using TCTA:B3PyMPM as the exciplex host and DACT-II (Figure ) as the TADF dopant. The blend emits at λPL ∼ 525 nm and has a near unity ΦPL of 96%. The devices showed an EQEmax of 34.2%, CEmax of 114 cd A–1 and a PEmax of 121.3 lm W–1.ref. ref716
The electron transport materials B4PyMPM and B3PyMPM (Figure ) are often employed as acceptors in TADF exciplex OLEDs. Sasabe et al. reported several interfacial devices using B3PyMPM, B4PyMPM, and B4PyPPM as the electron transport layer and CBP or TCTA as donors. DACT-II was employed as the emitter, doped at concentrations ranging from 4 to 20 wt% in the EML. The 20 wt% DACT-II:CBP/B4PyPPM device with structure ITO (100 nm) / HAT-CN6 (1 nm) / TAPC (65 nm) / TCTA (5 nm) / 20 wt% DACT-II-doped CBP (10 nm) / B4PyPPM (50 nm) / Liq (1 nm) / Al (80 nm)] emitted at 534 nm [CIE coordinates (0.38, 0.58)], and showed an EQEmax of 26.8% with PEmax of 122.2 lm W–1. However, the highest efficiency device was achieved with 9 wt% DACT-II-doped into the exciplex blend CBP:B4PyMPM as the EML. The device was fabricated with structure ITO (100 nm) / HAT-CN6 (1 nm) / TAPC (65 nm) / TCTA (5 nm) / 9 wt% DACT-II-doped CBP:B4PyMPM (10nm) / B4PyPPM (50 nm) / Liq (1 nm) / Al (80 nm), emitting at λEL of 534 nm [CIE coordinates (0.37, 0.58)] and showing an EQEmax of 29.2% with PEmax of 133.2 lm W–1.ref. ref717
A series of phenylcarbazole-based donors were used in conjunction with the acceptor B3PyMPM to form exciplex hosts for the phosphorescent green emitter Ir(ppy)2(acac) (Figure ). The devices with the highest efficiencies (Table S8) were obtained using PhCNCzm-MeCzPh (Figure ) as the exciplex donor, which adopts a twisted conformation with a relatively high triplet energy amongst the donors studied. The blend PhCNCzm-MeCzPh:B3PyMPM has ΔE ST of 0.32 eV and yet a very short τd of 0.15 μs. The devices showed an EQEmax of 31.5%, CEmax of 113.06 cd A–1, and PEmax of 99.41 lm W–1 at CIE coordinates of (0.32, 0.64).ref. ref718
Jia et al. fabricated a blue-emissive TADF exciplex using a 7:3 wt% ratio of m-MTDATA:TmPyPB (Figure ), which emits at λPL of 479 nm, has a modest ΦPL of 12.3%, and a small ΔE ST of 0.04 eV. According to the authors the suitably high triplet energy level of this exciplex made it a good candidate as a host for green, yellow, and red phosphorescent emitters (Table S8). With the yellow emitter Ir(bt)2(acac) (Figure ), the OLED showed EQEmax, CEmax, and PEmax, of 18.5%, 50.7 cd A–1, and 57.6 m W–1 respectively, at CIE coordinates of (0.51, 0.49). The green-emitting device with 2 wt% of fac-Ir(ppy)3 showed an EQEmax of 10.0% at CIE coordinates at (0.32, 0.61). When using the red phosphorescent complex Ir(piq)2(acac) (Figure ) the OLED showed an EQEmax of 10.0% at CIE (0.67, 0.33).ref. ref719
Shih et al. reported a device with the exciplex BCzPh:3P-T2T (2:1), (Figure ). which emits at λPL of 536 nm and has a ΦPL of 68%. The exciplex devices showed an EQEmax of 13.5%, and this exciplex was also used as a host for both fluorescent C545T (Figure ) and phosphorescent Ir(ppy)2(acac) green emitters. The exciplex film with 1 wt% of C545T emits at λPL of 516 nm and has a ΦPL of 97%, with the device achieving an EQEmax of 15.5%. The blend with 8 wt% of Ir(ppy)2(acac) emits at λPL of 523 nm and has ΦPL of 85%, with that device achieving an EQEmax of 29.7%.ref. ref720 This work demonstrates how the exciton harvesting capacity of the dopant can contribute significantly the efficiency, e.g. in the phosphorescent device. Furthermore, the very high EQEmax was also suggestive of some preferential horizontal orientation of the TDM of the dopant in the exciplex host. Liang et al. later reported an even higher efficiency device using C545T as a dopant in an exciplex blend host, consisting of TAPC as the donor and a bespoke acceptor containing benzimidazole and triazine units (PIM-TRZ, Figure ). The OLED with 0.6 wt% C545T in TAPC:PIM-TRZ showed an EQEmax of 20.2%, CEmax of 68.3 cd A–1, and PEmax of 86.4 lm W–1 at CIE coordinates of (0.29, 0.62). In comparison, the non-doped exciplex TAPC:PIM-TRZ device showed an EQEmax of 21.7%, CEmax of 71.2 cd A–1, and PEmax of 97.3 lm W–1 at CIE coordinates of (0.35, 0.58).ref. ref721
Colella et al. studied the energy-transfer from 26DCzPPy/PO-T2T interfacial exciplex (Figure ) to phosphorescent guest Ir(dmpq)2acac (Figure ), included in different ratios varying from 1 to 10 wt%. According to the study, both DET and FRET are operational between the exciplex and the dopant, with the former being the dominant energy transfer mechanism. The devices of 26DCzPPy:4 wt% Ir(dmpq)2aca/PO-T2T showed the highest efficiency with an EQEmax of 28.6%, and emitting at λEL of 630 nm.ref. ref722
Tian et al. reported an exciplex with DEX (Figure ), a bulky triphenyl amine donor of similar structure to HMAT (hexamethylazatriangulene, see Figure in Section sec21 ), with PO-T2T acceptor (Table S8). The device with DEX:PO-T2T emitted at λEL of 520 nm, and showed an EQEmax of 11.2%, CEmax of 36.0 cd A–1, and PEmax of 44.6 lm W–1. The PhOLED using DEX:PO-T2T as a host with 5 wt% Ir(MDQ)2(acac) (Figure ) as the dopant emitted at λEL of around 600 nm, and showed an EQEmax of 21.7%. A co-host system was formed once an extra layer of PO-T2T (15 nm) was introduced [device structure [ITO / HAT-CN6 (10 nm) / TAPC (30 nm) / DEX (10 nm) / DEX:PO-T2T:5 wt % Ir(MDQ)2(acac) (5 wt %, 20 nm) / PO-T2T (15 nm) / Bphen:0.1 wt% LiH / Al (120 nm)], leading to devices having the same λEL as above but showing improved EQEmax of 24.5%, CEmax of 36.0 cd A–1, and PEmax of 146.1 lm W–1.ref. ref723
Duan et al. reported an exciplex matrix fabricated using the hole-transporting molecule CDBP and varying the choice of phosphine-oxide-based acceptors (mDBSOSPO, m = 2, 3, and 4, Figure ). The exciplex blend doped with the yellow TADF emitter 4CzTPNBu (Figure ) emits at λPL of 570 nm, has a near unity ΦPL of 97%, a small ΔE ST of 0.02 eV, and a τd of 6.8 μs; the exciplex by itself emits at λPL of 471 nm, has a much lower ΦPL of only 26%, and a τd of 4.3 μs. A family of devices with different exciplex blends similarly doped with 3 wt% of 4CzTPNBu were fabricated (Table S7). Of these, the device with the highest efficiency consisted of CDBP:2DBSOSPO:4CzTPNBu, showing an EQEmax of 30.3%, a PEmax of 114.9 lm W–1, and emitting at CIE coordinates of (0.48, 0.49). By contrast, the device without the TADF dopant (CDBP:2DBSOSPO) showed an EQEmax of only 0.82% at CIE coordinates of (0.17, 0.23).ref. ref724
Zhou et al. investigated the changes in device performance of three OLEDs containing different green-emitting dopants with the same interfacial exciplex host CDBP/B4PyPPM. Devices with 5 wt% of the fluorescent material Coumarin 6 (Figure ), the phosphorescent complex Ir(ppy)2acac, or the TADF emitter 4CzIPN (Figure ) were fabricated. The highest efficiency device employed the TADF dopant and showed an EQEmax of 20% at λEL of 536 nm, supported by a near unity ΦPL of 98.9% of the dopant in this matrix as well as near 100% exciton utilization efficiency. Devices with the fluorescent or phosphorescent dopants showed EQEmax of just 4.0 and 7.9%, respectively. The low efficiency of the PhOLED is surprising considering that the ΦPL of the dopant is 93.0%. The enhanced performance of the device with 4CzIPN was attributed by the authors to the large electric dipole of this dopant molecule which assisted FRET, and to the short exciton lifetimes of 4CzPN which mitigated the build-up of triplet excitons and associated losses at a high current density.ref. ref725
Wang et al. reported fluorescent molecules 67dTPA-FQ and 267TTPA-FQ (Figure ), emitting at λPL of 532 and 526 nm with ΦPL of 91 and 100% (in toluene), and having T1 levels of 2.19 and 2.32 eV all respectively. These compounds were then used as dopants (1 wt%) in the bulk exciplex system TCTA:PO-T2T (8:2 wt% ratio); the blend itself emits at λPL of 538 nm and has a T1 level of 2.35 eV. Devices with just TCTA:PO-T2T emitted at λEL of 556 nm and showed EQEmax of 7.4%, while the devices with 67dTPA-FQ in TCTA:PO-T2T performed similarly, emitting at λEL of 552 nm and showing EQEmax of 8.4%. However, the device with 267TTPA-FQ in TCTA:PO-T2T emitted at λEL of 524 nm and showed a modest improvement in EQEmax to 9.6%. The improvement in the performance of the latter device was attributed partially to Förster energy transfer between the dopant and exciplex host, which was improved in the system (TCTA:PO-T2T):267TTPA-FQ.ref. ref726
Solution-Processed TADF Exciplexes
While the vast majority of reported exciplex OLEDs use thermal evaporation for the control over film composition and morphology that this method offers, solution-processing of exciplex emitters and hosts is also growing in prominence and necessary for molecules above a certain molecular weight. Chen et al. showed that small variations in the structure of isomeric acceptors significantly affected the energies of the CT excited states and device efficiencies using solution-processed interfacial exciplex host systems.ref. ref727 Oligocarbazole H2 (Figure ), was doped with TADF emitted tBuCzDBA, and used as donor in an interfacial exciplex with B3PyMPM or B4PyMPM as the acceptors (Table S7). The highest efficiency OLED consisted of H2:tBuCzDBA (10 wt%)/B3PyMPM, which emitted at λEL ∼ 550 nm [CIE coordinates of (0.42, 0.55)], with an EQEmax of 26.4% and a PEmax of 95.0 lm W–1. When the acceptor was switched to the isomeric B4PyMPM the emission wavelength did not change, but the performance of the devices decreased to an EQEmax of 20.0% and PEmax of 69.9 lm W–1. The lower efficiency of the latter device was attributed to the poorer hole-electron recombination ratio in H2:tBuCzDBA/B4PyMPM.ref. ref727
Xu et al. employed the red TADF emitter AQ-b1 (Figure ) as a dopant in a series of binary (1:1) and ternary (1:1:1) exciplex systems. mCP and OXD-7 (Figure ) were used as the respective exciplex donor and acceptor, while two molecules showing high electron mobility and containing spirofluorene and s-triazine moieties (TDP-TRZ and DTDP-TRZ, Figure ) were used as additional acceptors in the ternary blends. Solution-processed OLEDs with the binary (mCP:DTDP-TRZ) and ternary exciplex systems (mCP:OXD-7:DTDP-TRZ) doped with 10 wt% AQ-b1 showed EQEmax of 2.5 and 1.6% at CIE coordinates at (0.59, 0.39) and (0.60, 0.39), respectively. According to the study, multiple exciplex pairs in the ternary co-host contributed to improving the exciton harvesting efficiency and also provided balanced injection of charge carriers.ref. ref728
Colella et al. demonstrated that solution-processable TADF exciplex OLEDs can show similar efficiencies to vacuum-deposited devices, using a bulk exciplex consisting of TAPC as the donor and the D-A-D TADF molecule DCz-DBTO2 as the acceptor (Figure ) (70:30 wt% ratio). The authors used different solvents and spin-coating parameters to vary the thickness of the emissive exciplex layer (Table S7). The optimized device was fabricated using a 5:95 vol% solvent blend of chlorobenzene:chloroform, which produced an emissive layer thickness of 60 nm. The solution-processed device emitted at λEL of 550 nm, and showed EQEmax of 8.9%, a CEmax of 27.5 cd A–1, and a PEmax of 15 lm W–1.ref. ref729 The vacuum-deposited device was previously published by Jankus et al.ref. ref730, emitting at λpL of 540 nm and showing comparable EQE of 10.3%, CEmax of 32.3 cd/A, and PEmax of 26.7 lm W–1.
Kesavan et al. fabricated a solution-processed exciplex OLED that showed an EQEmax of 20% and CEmax of 41 cd A–1 at CIE coordinates of (0.29, 0.52). At the time of publication this was the highest-performing solution-processed exciplex OLED without the use of an additional emissive dopant. This exciplex consisted of carbazole-based donor BCC-36 (Figure ) with PO-T2T in a 5:1 ratio, which emits at λPL of 490 nm, has a ΦPL of 90%, a ΔE ST of 0.04 eV, and a τd of 1.1 μs. This exciplex was also used as a host for fluorescent (C545T), phosphorescent (Ir(ppy)2(acac)), and TADF (4CzIPN) compounds (Table S7). Devices using 1 wt% C545T showed EQEmax of 12.5% [CIE coordinates at (0.24, 0.57)], while devices with 7.5 wt% of 4CzIPN showed EQEmax of 26.5% [CIE coordinates at (0.26, 0.56)]. The devices doped with 12.5 wt% Ir(ppy)2(acac) showed the highest EQEmax of 32.5% [CIE coordinates at (0.31, 0.64)]. According to the authors, the strong spin-orbital coupling associated with the heavy metal in the phosphorescent material leads to an increased rate of ISC, increasing the energy transfer process from the host to the emitter, which contributes to highest device efficiency obtained for the device doped with the phosphorescent compound.ref. ref731
Fundamental Studies of TADF Exciplex Systems
As well as pursuing the highest performing devices, many studies have focused on exploring the fundamental mechanisms and decay pathways in TADF exciplex systems. For example, Huang et al. used transient photoluminescence and electroluminescence measurements to study the exciton dynamics in a 1:1 wt% blend of m-MTDATA:3TPYMB. According to the authors, exciplex excitons can stretch while remaining bound, and the recombination rate is determined by a local process involving the lateral motion of carriers that is related to the electron-hole separation.ref. ref732 A similar work published by Lin et al. measured steady-state and time-resolved IR spectroscopy and grazing incident X-ray diffraction (GIWAX) to gain in-depth insight into the structure and emission mechanisms associated with the TADF exciplex CN-Cz2:PO-T2T (Figure and Table S9). The devices using a 1:1 ratio showed the highest EQEmax of 16%, CEmax of 37.8 cd A–1, and PEmax of 47.5 lm W–1 at coordinates of CIE (0.20,0.40). The study reported the formation of polaron pairs in the exciplex blend, which could recombine to give charge-transfer emission or dissociate back to polarons. When dissociation occurs, positive and negative polarons would be created and their recombination for light generation would be prohibited, leading to losses.ref. ref733
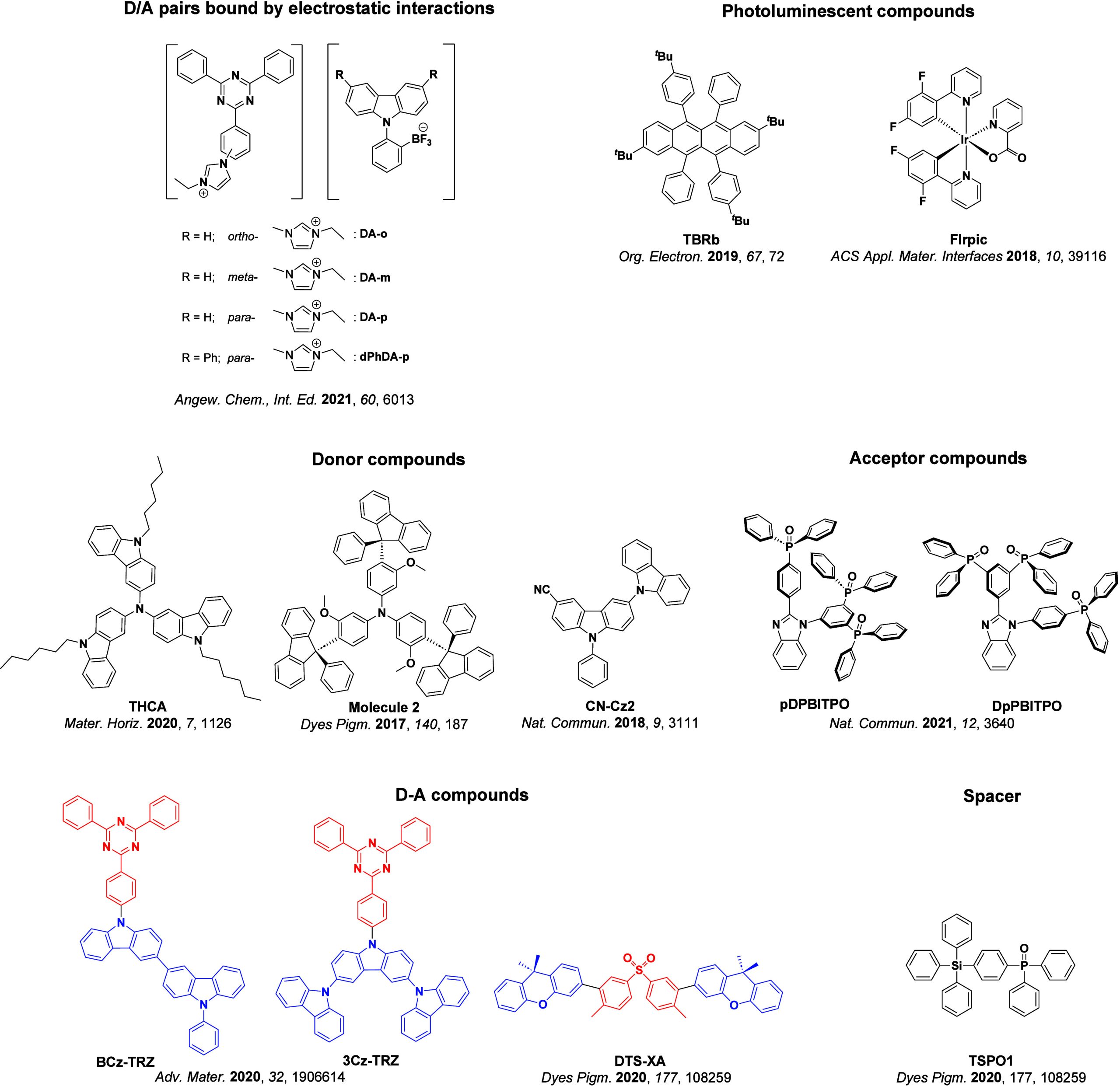
A kinetic model proposed by Grüne et al. is particularly suited for exciplexes and was applied to explain the photophysics of m-MTDATA:3TPYMB. The model accounted for the fact that triplet-triplet annihilation (TTA) is the main second-order effect, which contributes significantly to triplet depopulation. As the efficiency of the TTA is strongly influenced by temperature, this led to a constraint of the overall efficiency of the device at room temperature.ref. ref100
Moon et al. explored how the formation and photophysical properties of the TCTA:B4PyMPM CT state are influenced by the distance between the D and A molecules, their relative orientation, and the D:A ratio.ref. ref734 According to the study, the exciplex emission wavelength is determined by the configuration of the molecules in the system, which also strongly affects ΔE ST, kinetic rate constants, and emission dipole orientations. Short distances between donor and acceptor molecules results in lower exciplex energy due to the Coulomb interaction, which is proportional to r–1.
Bunzmann et al. used electron paramagnetic resonance (EPR) to study the involvement of different spin states in the RISC of TADF exciplex systems. Both electroluminescence and photoluminescence detected magnetic resonance (ELDMR, PLDMR) were used to probe the photophysics of three exciplex system: m-MTDATA:3TPYMB, m-MTDATA:Bphen, and THCA:BPhen (Figure , Table S9), which emit at λPL of 545, 560, and 570 nm, respectively. Of these three the exciplex m-MTDATA:3TPYMB has the highest ΦPL of 45%, and the devices with this exciplex emitted at λEL of 550 nm and showed EQEmax of 11.0%. However, the OLED performance was not the focus of this study, but rather it was whether the investigation of the activation energy for delayed fluorescence correlated with either the ΔE ST of the exciplex system or with the molecular triplet states of the donor or acceptor materials. The authors found that in all three systems exciplex states formed at the interface of donor and acceptor molecules in the blend led to TADF emission and that molecular (local) triplet exciton only formed under optical and not electrical excitation.ref. ref735
As with molecular TADF materials, most of the exciplex TADF studies published to date have not focused on device stability despite its importance for commercial applications. However, a few studies do exist that correlate device stability with the properties of the exciplex. For example, Nguyen et al. aimed to optimize the stability of exciplex OLEDs by varying the nature of the triazine-based acceptor with Tris-PCz as the donor. The acceptors were divided into three classes: molecules without a significant electron-donating group, D-A compounds that are not TADF, and D-A TADF compounds. The most stable devices featured exciplexes where the acceptor partner is itself TADF, and OLEDs with Tris-PCz:BCz-TRZ and Tris-PCz:3Cz-TRZ (Figure ) showed the longest device lifetimes (LT50 of 292 and 337 hours, respectively), ascribed to multichannel RISC processes. Both devices showed green electroluminescence at CIE coordinates at (0.26, 0.50) and (0.26, 0.53) respectively, however the device with Tris-PCz:BCz-TRZ showed the highest EQEmax of 11.9%, compared to 8.9% for the device with Tris-PCz:3Cz-TRZ. ref. ref736
A major challenge in exciplex design is the control of distance between donor and acceptor molecules. He et al. demonstrated a unique strategy that exploits electrostatic interactions (Coulombic attraction) rather than the circumstances of deposition to control these D-A distances. The exciplexes consisted of carbazole-based anionic donors ([CAZ-o-BF3 –] or dPhCAZ-o-BF3 –]) and 2,4,6-triphenyl-1,3,5-triazine-based cationic acceptors ([TRZ-o-ImEt+], [TRZ-m-Im-Et+] or [TRZ-p-ImEt+]), with the blends named DA-o, DA-m, DA-p, and dPhDA-p (Figure ). The films of these D/A pairs in doped at 1 wt% in PMMA films emit at λPL of 498, 481, 484, and 504 nm and have ΔEST of 0.02, 0.10, 0.18, and 0.20 eV, respectively, leading to τd in the range of 3.1–7.8 μs and associated k RISC in the range of 1.8–5.2 × 105 s–1. The authors documented that the distance and interaction between the ionic donor and the acceptor could be modified as a function of the position of the acceptor imidazolium moiety. This also tunes the overlap of the frontier orbitals and thus the radiative decay rate of the exciplex singlet, reflected in the differing τp/τd of 165 ns/3.1 ms, 114 ns/3.7 ms, 186 ns/6.2 ms, and 185 ns/7.8 ms for the blends with DA-o, DA-m, DA-p and dPhDA-p, respectively. The k RISC was found to decrease from DA-o (5.2 × 105 s–1) and DA-m (4.2 × 105 s–1), to DA-p (2.2 × 105 s–1) and to dPhDA-p (1.8 × 105 s–1), which follows the trend in their ΔE ST. The so-called isolated exciplexes exhibited a considerably higher ΦPL (24–52%) in PMMA film than neat exciplex blends (8–11%). As dPhDA-p has the highest ΦPL of 52% it was then evaluated as the emitter in a solution-processed OLED, which emitted at λEL of 510 nm [CIE coordinates of (0.25, 0.44)] and showed an EQEmax of 6.1%.ref. ref737
White TADF Exciplex OLEDs
Fabricating white organic light emitting diodes (WOLEDs) with high CRI, high efficiency, low operating voltage, and low efficiency roll-off is not a trivial task (Section sec6 ). Cekaviciute et al. demonstrated a new approach to fabricate a WOLED using multiple exciplexes. In this study a blue-emitting exciplex layer made of 3:7 Molecule 2:BPhen (Figure ) was sandwiched between two layers of the green exciplex m-MTDATA:BPhen. The maximum values of EQE, CE, and PE were as high as 2.55%, 6.34 cd A–1, and 4.09 lm W–1, respectively.ref. ref738 Another study by Tian et al. reported a multi-layer device using a bulk exciplex system composed of bipolar donor 26DCzPPy and acceptor B4PyMPM acting together as the host for FIrpic (Figure ) in sky-blue phosphorescent and white OLEDs. The blue OLED showed λEL of 472 nm [CIE coordinates of (0.17, 0.36)] and a PEmax of 48 lm W–1. The white device contained an extra layer of phosphorescent orange emitter PO-01 doped in 26DCzPPy, having the structure 26DCzPPy:PO-01/26DCzPPY:B4PyMPM: 15wt%FIrpic, and emitting at CIE coordinates of (0.45, 0.48) with EQE100 of 27.3% and corresponding CE of 79.0 cd A–1 and PE of 89.0 lm W–1.ref. ref739
Yao et al. documented a different design for fabricating high efficiency WOLEDs, where a blue emitting exciplex is used as a host for a yellow fluorescent compound. An additional hole transport layer is also inserted on top of the emissive layer, which is essential to improve the overall efficiency and efficiency roll-off at high luminance. The additional interfacial exciplex established in the EML regulates exciton distribution and enhances the energy transfer to fluorescent guest. The specific device contained blue emitter exciplex host 26DCzPPy:PO-T2T with fluorescent yellow dopant TBRb (Figure ), and a thin interlayer of TCTA. This device showed an EQEmax of 10.1% at CIE coordinates of (0.36,0.53), with CEmax of 32.6 cd A–1 and PEmax of 35.9, lm W–1 (Table S10).ref. ref740
Guo et al. demonstrated TADF exciplex WOLEDs by sandwiching a yellow exciplex layer between two blue exciplex layers. The emission spectra and device performance could then be tuned by changing the mass ratio of the intermediate yellow exciplex layer, and/or thickness of the two blue exciplex layers. The optimized device used mCP:PO-T2T (1:1, 4 nm)/PO-T2T:TPD (3:1, 3 nm)/Bphen:TPD (1:1, 4 nm), and showed EQEmax of 5.21%, CEmax of 12.78 cd A–1, and PEmax of 12.12 lm W–1 at CIE coordinates of (0.245, 0.320).ref. ref741
Tan et al. fabricated exciplex WOLEDs by layering separate blue and orange TADF interface exciplexes. A newly designed donor composed of a 4,4′-sulfonylbis(methylbenzene) central electron acceptor moiety and two peripheral 9,9-dimethyl-9H-xanthene groups (DTS-XA, Figure ) was combined with the donor compounds TCTA and m-MTDATA to form the interfacial exciplex systems DTS-XA/TCTA and DTS-XA/m-MTDATA. The devices based on DTS-XA/TCTA and DTS-XA/m-MTDATA emitted in the blue (λEL = 433 nm) and green-yellow (λEL = 524 nm) regions, showing EQEmax of 9.1 and 8.3%, respectively. WOLEDs were then fabricated by layering the two exciplexes using the following configuration: (DTS-XA/TCTA)/spacer/(DTS-XA/m-MTDATA), where the spacer consisted of a thin layer of diphenyl-4-triphenylsilylphenyl-phosphineoxide (TSPO1, Figure ) acting as a hole and electron-transporting modulator. The highest-efficiency WOLED showed an EQEmax of 10.6% at CIE coordinates of (0.29, 0.37).ref. ref742
Han et al. reported an exciplex WOLED composed of a single emissive layer featuring two phosphine oxide-based acceptors (pDPBITPO and DpPBITPO, Figure ). The large triplet energy gap (0.6 eV) between the mCP donor and these acceptors limited donor-acceptor triplet coupling, which in turn led to dual triplet levels accessible in the exciplex blend. The authors confirmed by transient emission spectroscopy that cascade triplet energy transfer takes place from the high-lying triplet level of the exciplex to the blue emitter, then to the low-lying triplet level of the acceptor, and finally to the yellow emitter. This arrangement and energy transfer between excited states led to 100% exciton harvesting, and, hence, the single-emissive layer design based on mCP:pDPBITPO and mCP:DpPBITPO produced TADF WOLEDs with a tantalizing EQEmax of 32.7%, PEmax of 108.2 lm W–1, and CIE coordinates of (0.31, 0.35).ref. ref743
Outlook
Exciplexes are intermolecular assemblies that frequently show TADF due to the intrinsic separation of HOMO and LUMO on separate molecules. The optoelectronic properties of these blends can also be straightforwardly manipulated through the choice of specific donor and acceptor materials. However, the very weak ‘through-space’ electronic coupling of chromophores in exciplexes tends to generate low ΦPL, which typically limits their intrinsic performance as emissive materials and hence also affected the relatively limited degree of attention this class of material has historically received from the research community.
Nonetheless, the performance of exciplex OLEDs has been more extensively studied in recent years and the overall stability and efficiency of these devices have progressively improved, with some now achieving performance metrics comparable to those of D-A TADF OLEDs (Figure ). For example, the most efficient exciplex OLEDs reported within the scope of this review include one with mCP:PO-T2T as the emitter that showed an EQEmax of 16% at CIE coordinates of (0.16, 0.28).ref. ref186 One of the most efficient green devices employed an exciplex host, showing an EQEmax of 34.2% using TCTA:B3PyMPM as the host and DACT-II as the TADF dopant.ref. ref716 Red OLEDs using exciplex hosts showed EQEmax as high as 28.1% at CIE coordinates of (0.66, 0.34) using CBP:PO-T2T host and TPA-PZCN as the TADF emitter.ref. ref507 An efficient WOLED with an EQEmax of 32.7% at CIE coordinates of (0.44, 0.47) was reported using the exciplex system mCP:DpPBITPO as the host, with DMAC-DPS as an assistant dopant and 4CzTPNBu as the terminal emitter in a single-emissive-layer device.ref. ref743 The high density of suitable recombination sites in exciplex emissive layers (analogous to high loading of TADF guests in conventional hosts) can also contribute to improved efficiency roll-off.ref. ref740
Despite this progress, we believe that applications of TADF exciplexes in OLEDs still have significant unrealised potential. As with D-A TADF molecules, color purity in exciplex OLEDs is frequently undermined by broad emission arising from the long-range charge-transfer character of the emissive excited state. Nonradiative decay processes intrinsic to intermolecular contact interfaces can also negatively impact device performance, particularly for red OLEDs. Most notably, the development of efficient deep-blue and blue exciplex OLEDs remains elusive, largely because of the challenge in designing (or discovering) donor and acceptor molecules with appropriate HOMO, LUMO, and T1 energy levels. Even with the use of donor/acceptor materials that can themselves harvest triplet excitons either by TADF or phosphorescence, many studies only employ conventional hole or electron transport materials as exciplex components, with this limited range of chemical space explored likely restricting recorded performance compared to more innovative D-A TADF, TSCT TADF, or MR-TADF emitter designs. A breakthrough specifically in blue emissive materials would be particularly valuable, allowing the use of TADF-active exciplexes as hosts for many other emissive species to generate narrowband blue or white emission (examples throughout Sections sec6 , 11, 17, 18), while providing the excellent charge transporting properties of the individual exciplex components.
Unique amongst other TADF materials, fabrication methods critically control the performance of exciplex OLEDs. The choice of bulk heterojunction or bilayer deposition influences the degree of interaction of the donor and acceptor molecules and thus the emission color and performance of the exciplex. Exploiting this feature, controlling the distance and/or orientation between donor and acceptor with a spacer layerref. ref689 or diluting materialref. ref698 influences the potential energy surfaces of the exited states, and can improve the EQEmax.ref695,ref699 Controlling the exciplex state using covalently bonded scaffolds now forms the basis of related TSCT emitters (Section sec12 ). We note that controlled self-assembly (Section sec19 ) of the donor and acceptor units to form the exciplex may become a powerful tool to achieve finer control of this in future, with currently only a few reports of self-assembled exciplexes.ref708,ref744−ref745ref746
These examples therefore highlight both the promise and current limitations of exciplexes as both hosts and emitters. While their often low ΦPL represents a major drawback as emitters in their own right, their balanced charge transport properties and ability to harvest both singlet and triplet excitons make them significantly more appealing than conventional ‘inactive’ OLED hosts. Indeed, we speculate that future uses of TADF exciplexes will increasingly focus on their use as hosts for other emissive materials, exploiting their ambipolar charge transporting properties while also largely circumventing their low ΦPL and broad emission.
Metal-Based TADF Emitters
Introduction
The majority of the sections of this review have focused on organic TADF molecules, reflecting their key advantage in their ability to harvest triplet excitons without the need for scarce and expensive heavy metals central to both the structure and function of organometallic phosphors. However, TADF emission is observed in a range of metal complexes as well, including those based on Earth-abundant metals. Indeed, the majority of the reported examples of TADF complexes are copper(I) complexes, although there are also numerous examples of silver(I), gold(I and III), palladium(II), and zinc(II) complexes. Examples based on each of these metals will be discussed in detail in this section. There are also examples of TADF emission emanating from complexes of abundant alkali metals, d0 transition metals, d10 transition metals, and main group compounds, which are also briefly discussed. An overview of the metals that have been incorporated into TADF compounds is shown in Figure . Like organic TADF emitters, organometallic TADF compounds have found wide applications in OLEDs, LEECs, and as photocatalysts. While this section focuses on metal-containing TADF emitters used in OLEDs, their use in LEECs and photocatalysis are covered in Sections sec16 and sec23.
The existence of TADF in metal complexes has been known since the pioneering work of McMillin, who identified that the time-resolved PL decays of [Cu(dmp)2]BF4 were temperature dependent, indicating interconversion between singlet and triplet excited states (Figure ).ref70,ref747 Following these initial reports, a number of copper(I) complexes with similar photophysical behavior were disclosed. There was little interest/application for these complexes though until 2004 when the first bright OLED was fabricated using [Cu(dnbp)(DPEphos)]BF4 , which showed a current efficiency of 10.5 cd A–1 and a maximum luminance of 1663 cd m–2.ref. ref74 This marked the starting point for a rapid expansion in research into emissive copper complexes and their use in OLEDs (Figure ).ref748−ref749ref750ref751ref752ref753ref754 A notable milestone in the steady improvement in OLED performance was the use of [Cu2I2(dppb)2], where the EQEmax reached 4.8%.ref. ref75 However, at that time no copper-based OLEDs surpassed the 5% EQE limit that would have permitted confident assertion that triplet excitons were being harvested for emission.
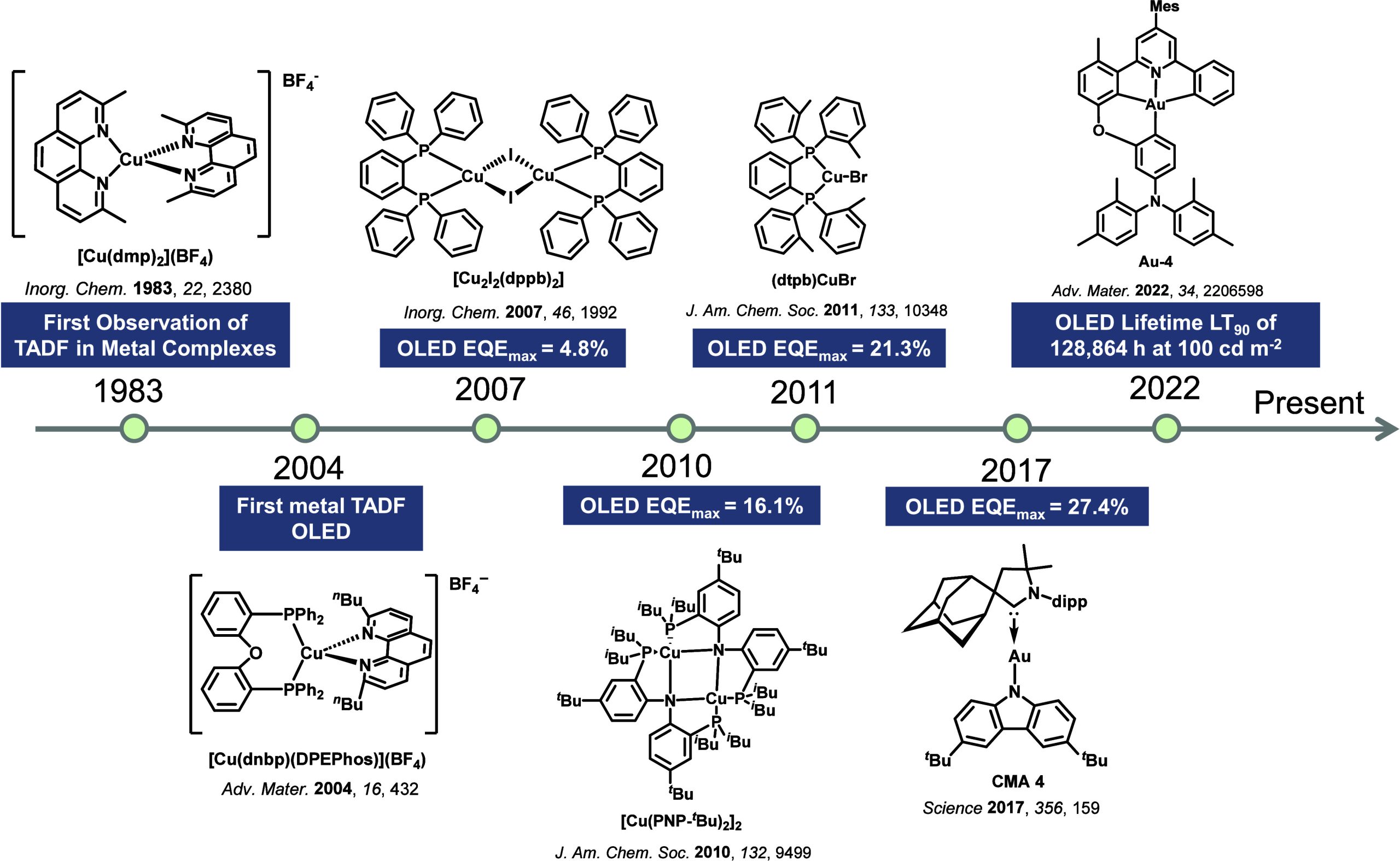
In 2010 Deaton et al. reported an OLED with [Cu(PNP-tBu)2]2 that showed an EQEmax of 16.1% (Figure ).ref. ref73 At this point TADF was well established as an operational photoluminescence emission mechanism for copper complexes, and this result also established the same for electroluminescence. Identification of TADF in other metal complexes rapidly expanded, especially in other coinage metal complexes.ref755,ref756 The development of copper(I) complexes as TADF emitters also continued, with the monometallic 3-coordinate complex (dtpb)CuBr used in an OLED that showed an EQEmax = 21.3% in 2011.ref. ref757 The report of highly emissive linear carbene metal amide (CMA) complexes of copper(I) and gold(I) in 2017 threw the field of metal-containing TADF materials into overdrive, as the solution-processed OLEDs with the gold(I) complex CMA-4 could reach an EQEmax = 27.4% and showed very low efficiency roll-off.ref. ref194 Since the first report of TADF emission from gold(III) complexes in 2015,ref. ref758 materials development has continued apace, exemplified currently by Au-4, where the OLED showed an EQEmax of 27.3% along with low efficiency roll-off and long lifetime.ref. ref759 These and other reports of similar performance put metal-containing TADF materials on equal footing with all-organic TADF emitters.
Analogous to all-organic TADF emitters, the emissive excited states in metal-containing TADF emitters have dominant charge-transfer character. However frequently these CT states involve transitions to/from metal-based orbitals. Categorized by the electronic and structural role of the metal center, there are several different classes of metal-TADF complexes, illustrated in Figure . The most common type of metal-containing TADF emitter benefits from a large contribution of metal d-orbitals to the excited state, either resulting in metal-to-ligand charge transfer (MLCT) or ligand-to-metal charge transfer (LMCT) states. The CT states of some metal complexes instead have no, or minimal, involvement of the metal d-orbitals, which are best described as ligand-to-ligand charge transfer (inter-ligand charge transfer, LLCT) excited states. In these complexes SOC from the metal center is subdued, and they behave comparably to organic Donor-Bridge-Acceptor TADF compounds with the metal center acting as the bridging element between different ligands acting as the donor and acceptor. A final class of metal-containing TADF emitter has the excited state localized on a single ligand in an intra-ligand charge transfer (ILCT) excited state, with this ligand itself effectively comprising a D-A TADF molecule. In these cases the metal acts as a Lewis acid, stabilizing the orbitals compared to those of the free ligand, and may also enhance the SOC between the excited states. Of particular interest are examples where the ligand is non-emissive, but coordination of a metal is capable of “turning on” both emission and TADF.ref. ref760
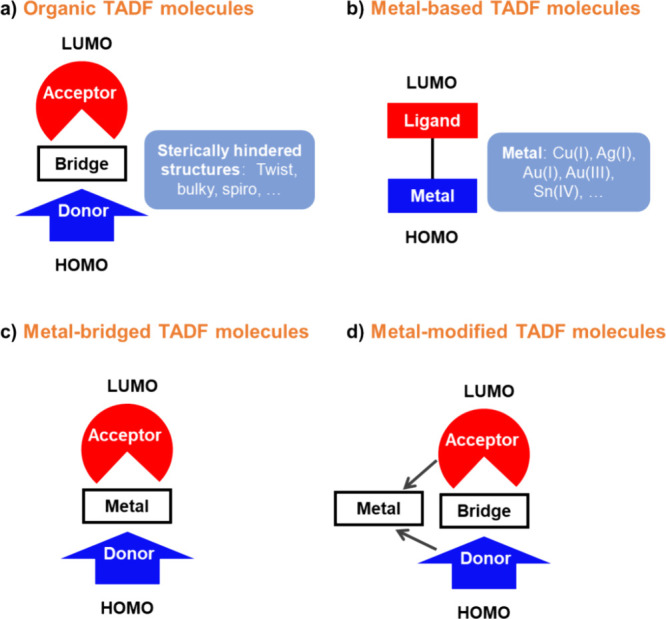
The presence of metals with very large atomic mass typically results in the SOC between low-lying singlet and triplet states becoming significantly larger than in purely organic TADF emitters. This impacts and simplifies the excited-state kinetics of these molecules in several ways. When SOC accelerates k ISC to > 1010 s–1, intersystem crossing can outcompete radiative emission from the S1 state. This results in the rate of TADF emission becoming independent of further small changes in k ISC and instead dependent primarily on the radiative decay rate (k S1) and the equilibrium constant for ISC/RISC cycling between S1 and T1 (K eq).ref. ref762 This situation mirrors the emission kinetics for organometallic phosphors, in which heavy-atom SOC also enables ultrafast initial ISC. SOC will also simultaneously increase radiative decay from the triplet state, and as a result phosphorescence can become a competitive radiative process alongside TADF in these materials, even at room temperature. Emission properties (time-resolved PL decays and others, see below) must be carefully analyzed to determine if emission is purely TADF, purely phosphorescence, or a combination of the two. A representation of the combined emission is seen in Figure .ref. ref763 The balance of these two processes has been studied in detail for a number of metal complexes, in particular copper(I) complexes.ref763−ref764ref765ref766ref767ref768ref769ref770ref771ref772
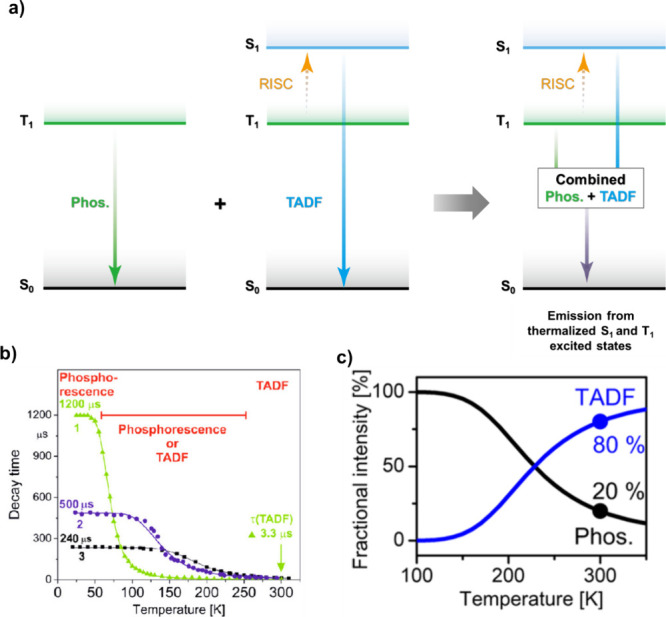
Determining the nature of the emission process occurring in a metal complex can be difficult. Both TADF and phosphorescence in metal complexes may have similar emission lifetimes and spectra, and even room temperature steady-state emission cannot always be unambiguously assigned to either singlet (TADF) or triplet (phosphorescence) excited states. In some cases, TADF can be inferred through comparison of the room temperature steady-state and delayed fluorescence spectra to the low temperature time-gated phosphorescence spectra, where these are distinguishable. In other cases, identifying TADF or phosphorescence emission (or mixed emission) in metal complexes relies on a detailed study of the temperature dependence of the emission lifetimes.ref763,ref764 The overall decay time of the system can be modelled according to equation eq18 , which models the decay kinetics according to a three-state model under the assumption of SOC-assisted rapid thermal equilibrium between the populations of T1 and S1.ref. ref774 At very low temperatures that deactivate TADF the measured lifetime of the system corresponds to the pure phosphorescence lifetime τ(T1), allowing the TADF lifetime τ(S1) and ΔE ST to be inferred by fitting the emission lifetime across different temperatures (for an example see Figure c).ref748,ref763,ref764,ref773,ref774
As well as the emission lifetime, the intensity contributions of TADF and phosphorescence emission have a strong temperature dependence, as illustrated in Figure c. Hence, when the spectra are readily resolvable, modelling this ratio at different temperatures can also be used to extract ΔE ST. There is a similar temperature dependence observed in organic TADF compounds as well; however, as the SOC in organic compounds is much smaller, the rate of phosphorescence is normally so slow that it is not observed at room temperature (or reasonably assumed to be negligible).ref. ref102 As SOC alone is also not able to instantly establish an equilibrium between singlets and triplets in all-organic TADF emitters following photoexcitation, more involved modelling procedures are typically required.ref99,ref102
Likely because of these challenges, many emissive complexes are reported without explicit assignment of the emission to either fluorescence, TADF, phosphorescence, or some combination thereof. Many reports also lack the photophysical data needed for a reader to reasonably infer this assignment. As a result, we propose that the number of TADF metal complexes is likely significantly under-reported, especially considering that many luminescent metal complexes emit from CT states, often with small anticipated ΔE ST. Despite these technical challenges the field of emissive metal complexes is sufficiently mature to have been covered in many reviews, with several reviews focused on TADF metal complexes.ref761,ref775−ref776ref777ref778ref779 Metal-containing TADF emitters are additionally discussed in a number of reviews that encompass either metal emittersref780−ref781ref782 or TADF emittersref35,ref86,ref783 more widely. Given the early discovery and extensive study of TADF emission from copper complexes, there are several reviews covering this topic specifically,ref748−ref749ref750ref751ref752ref753ref754 along with others that focus more widely on the photophysical properties of coinage metal complexes.ref784−ref785ref786
In this section, we review reported metal complexes where the authors have explicitly assigned the emission to TADF. The survey is divided into different sub-sections based on the metal in the complex. The copper, silver, and gold sub-sections cover selected complexes that demonstrate the history of coinage metal TADF emitters, highlighting key structural motifs or reports of particularly notable emission properties and OLED performance. Sub-sections concerning Carbene-metal-amide (CMA), palladium and platinum, zinc and other metals are comprehensive in scope and include all examples of metal complexes that have experimentally reported TADF emission. This section does not discuss the TADF properties of large metallic clustersref787−ref788ref789ref790 and coordination polymersref791,ref792 that have recently been shown to exhibit TADF (albeit likely with more exotic underlying emission mechanisms). The emission properties of the complexes discussed here are also summarized in Table S11, and the performance of OLEDs fabricated from metal-containing TADF emitters are collated in Table S12.
Copper
Since the first report of delayed fluorescence by McMillin and co-workers,ref. ref70 copper(I) complexes have emerged as the most abundant group of metal-containing TADF emitters. To give some indication of the scale of reported emissive copper complexes, a recent review provides absorption and emission data for more than 1200 photoactive monometallic copper(I) complexes,ref. ref753 although only a portion of these exhibit TADF. In 1999, the first report of a copper(I) emitter used in an OLED concerned the phosphorescent complex Cu4(C≡CPh)4(L)2 ;ref. ref793 however, by 2007 examples of copper(I) TADF OLEDs had been reported using [Cu(μ-I)(dppb)]2 (Figure ).ref. ref75
The majority of luminescent copper(I) emitters are 4-coordinate tetrahedral complexes like [Cu(dmp)2]BF4 reported by McMillin and co-workers (Figure ).ref70,ref748 The weakly emissive nature of many of these complexes is due to significant non-radiative decay, arising from Jahn-Teller distortion in the MLCT excited state as copper center becomes formally Cu(II).ref753,ref794−ref795ref796 Increasing the steric bulk of the ligands in these tetrahedral copper(I) complexes restricts this excited-state distortion (as well as addressing ligand dissociation and exciplex formation) improving the photophysical and emission properties of these complexes.ref753,ref774,ref797−ref798ref799 The tetrahedral complexes summarized here are further sub-divided into categories based on their structure: cationic bis-diimine complexes, cationic diimine/diphosphine complexes, switchable neutral/cationic complexes, and neutral complexes.
Following from the initial report of [Cu(dmp)2]BF4 , numerous other cationic bis-diimine copper(I) complexes have been reported that show weak emission in solution and moderately strong emission in the solid state (Figure ). The emission intensity in these [Cu(N∧N)2]+ complexes is increased when bulky ligands or solid-state interactions are used to restrict the Jahn-Teller distortion in the excited state. This was demonstrated in a series of complexes with substituents of increasing steric bulk at the 2- and 9-positions of a phenanthroline ligand, [Cu(bcp)2]BF4 , [Cu(dnp)2]BF4 , [Cu(tmbp)2]BF4 , [Cu(dpp)2]BF4 , and [Cu(tpp)2]BF4 .ref. ref800 In DCM solution the complexes are all red emitters (λPL = 710–750 nm) with ΦPL increasing from 0.03% for the weakly emissive [Cu(dmp)2]BF4 to 0.15% for the bulkiest complex [Cu(tpp)2]BF4 .
Further increases in the steric bulk of the diamine ligands result in [Cu(N∧N)2]+ complexes showing reduced geometric reorganization in the excited state and thus are yet more emissive. An example of how emissive these complexes can become is [Cu(dtbp)2]BF4 (Figure ), which has a ΦPL = 5.6% in DCM solution. This material, however, also undergoes ligand displacement more readily than other copper-phenanthroline complexes due to the steric demands of the tert-butyl groups, and thus the relatively weaker coordination of the dtbp ligands to the copper centre.ref. ref801 The photophysical properties of [Cu(dchtmp)2]PF6 with a bulky 2,9-dicyclohexyl-3,4,7,8-tetramethyl-1,10-phenanthroline ligand (ΦPL = 5.5% in DCM)ref. ref802 are similar to those of [Cu(dtbp)2]BF4 yet this material is more chemically inert and does not suffer from ligand dissociation to the same extent. To explore the limits of the steric bulk that can be installed in the 2- and 9-positions of the phenanthroline, the asymmetric ligand 2-isopropyl-9-tert-butyl-1,10-phenanthroline that is sterically in between those of the dtbp and dipp was investigated. The resulting complex [Cu(L1)2]PF6 ref. ref803 is surprisingly weakly emissive (τPL of 0.13 μs and ΦPL of 0.17%) but inert to ligand dissociation. The use of this asymmetric ligand was indeed found to lead to more distortion in the excited state, increasing non-radiative decay and resulting in a shorter lifetime and weaker emission compared to those of the reference emitter [Cu(dipp)2]BF4 (τPL = 0.34 μs; and ΦPL = 0.4%).ref. ref804
The impact of peripheral heavy atoms on the emission properties of [Cu(N∧N)2]+ complexes was explored by replacing the methyl groups in [Cu(dmp)2]BF4 with halide atoms to form [Cu(LCl)2]PF6 , [Cu(LBr)2]PF6 , and [Cu(LI)2]PF6 (Figure ).ref. ref805 The chloride atoms have very little impact on the emission properties of the complex; however, in DCM both the bromine and iodine complexes have higher ΦPL (0.08 and 0.09%) than non-halogenated [Cu(dmp)2]BF4 (ΦPL = 0.024%) and identical longer τPL of 0.11 μs, compared to 0.085 μs, due to increased phosphorescence radiative decay rates resulting from increased SOC. Additionally, while both [Cu(LCl)2]PF6 and [Cu(LBr)2]PF6 show TADF, [Cu(LI)2]PF6 emits only by phosphorescence as the high SOC of the iodine atoms significantly increased the rate of emission from T1.
Soon after the initial reports of emissive [Cu(N∧N)2]+ complexes, researchers started to explore the photophysical properties of heteroleptic complexes containing bulky phosphine (P) or diphosphine ligands (P∧P). Among several early reports of luminescent [Cu(N∧N)P2]+ complexes with photophysics incompatible with simple singlet emission, McMillin and co-workers identified TADF in [Cu(dmp)(PPh3)2]BF4 (Figure ).ref806,ref807 This complex is a weak green-yellow emitter (λPL = 560 nm, ΦPL = 0.14%, τPL = 330 ns) in methanol. After this initial report, there were numerous reports of phosphorescent and otherwise luminescent [Cu(N∧N)(P∧P)]+ complexes;ref74,ref798,ref808−ref809ref810 however, none of these reports claimed that the emission was TADF. In 2012, Yersin and co-workers reported that a related and previously studied complex, [Cu(dmp)(POP)]BF4 ,ref74,ref798 also emits by TADF (λPL = 538 nm, ΦPL = 80%, τPL = 18 μs) with ΔE ST of 110 meV.ref. ref811 Since this report many more emissive [Cu(N∧N)(P∧P)]+ complexes have been prepared,ref. ref753 with selected examples that have clearly identified TADF emission shown in Figure .
Yersin and co-workers later reported [Cu(dmp)(phanephos)]BF4 (Figure ), containing a bulky and rigid cyclophane-diphosphine ligand that showed green TADF emission (λPL = 530 nm, ΦPL = 80%, τPL = 14 μs) as a powder.ref. ref812 The temperature dependence of the emission lifetime was used to estimate the ΔE ST of 140 meV. The strong emission from this complex relative to other copper complexes (especially in DCM, ΦPL = 40%) was attributed to the rigid coordination environment provided by the phanephos ligand.
A series of substituted 2-pyridyl-pyrazoyl N∧N ligands was used to prepare complexes [Cu(pypz)(POP)]BF4 , [Cu(pympz)(POP)]BF4 , and [Cu(pytfmpz)(POP)]BF4 (Figure ).ref. ref813 The electron-rich pypz ligand was used to tune the emission of the complexes to the blue through LUMO destabilization. Of the three, [Cu(pympz)(POP)]BF4 has the most blue-shifted emission (λPL = 465 nm as a powder). The λPL values range from 465 to 492 nm, ΦPL range from 56 to 87%, and τPL range from 12.2 to 22.8 μs, despite all having the same calculated ΔE ST of 180 meV, which corresponds very well with the measured ΔE ST of 170 meV for [Cu(pytfmpz)(POP)]BF4 . Solution-processed OLEDs with [Cu(pypz)(POP)]BF4 in 26mCPy showed EQEmax of 3.2% at λEL of 516 nm, while [Cu(pympz)(POP)]BF4 in DPEPO showed EQEmax of 3.7% at λEL of 484 nm. The device with [Cu(pytfmpz)(POP)]BF4 in DPEPO showed a considerably higher EQEmax of 8.5% at λEL of 508 nm.
A family of five complexes with differently methyl- or trifluoromethyl-substituted pyridylpyrazoyl ligands, [Cu(L1)(POP)]BF4 to [Cu(L5)(POP)]BF4 (1 to 5 in that work, Figure ) show blue TADF emission in both powder (λPL from 464 to 481 nm, ΦPL from 82 to 99%, τPL from 4.1 to 16.9 μs) and doped films in PMMA.ref. ref814 The ΔE ST for the complexes ranged between 80 and 90 meV. A similar family of very bright blue-green emissive complexes with C3 (rather than N2) substituted pyridylpyrazolyl ligands, [Cu(tBupzmpy)(POP)]BF4 , [Cu(Phpzmpy)(POP)]BF4 , and [Cu(Adpzmpy)(POP)]BF4 were also prepared.ref. ref815 Both families of pyridylpyrazolyl-containing complexes have very similar photophysics, with powders emitting at λPL of between 498 to 523 nm, ΦPL ranging from 71 to 91%, and τPL of between 13.4 to 34.1 μs. This second set of complexes have ΔE ST values between 90 and 100 meV, again similar to the first set.
A pair of complexes containing a substituted bipyridine as the diimine ligand, [Cu(dmbpy)(POP)]BF4 and [Cu(tmbpy)(POP)]BF4 (Figure ), are also reported as TADF-active.ref. ref816 Of the two, the emission in [Cu(tmbpy)(POP)]BF4 is much stronger due to reduced excited-state distortion imposed by the additional methyl groups at the 6- and 6′-positions of the bipyridine ligand. [Cu(tmbpy)(POP)]BF4 emits at λPL of 555 nm, has a ΦPL of 74% and a τPL of 13 μs as a powder with an associated ΔE ST of 78 meV, the less hindered complex [Cu(dmbpy)(POP)]BF4 has a lower ΦPL of 9% as a powder.
A series of spiro-carbazole ligands was used to prepare [Cu(ECAF)(POP)]PF6 , [Cu(EHCAF)(POP)]PF6 , and [Cu(PCAF)(POP)]PF6 (Figure ).ref. ref817 These complexes are of particular interest as the bulky spirocarbazole ligands allow the cationic complexes to be sublimed to fabricate vacuum-deposited OLEDs. The complexes are green TADF emitters in the solid state (λPL = 525 to 528 nm, ΦPL = 31 to 33% in PMMA films), with ΔE ST of 90 meV for all three. The best performing OLEDs used 10 wt% [Cu(ECAF)(POP)]PF6 in mCP and showed EQEmax of 14.8% at λEL of 544 nm and CIE coordinates of (0.37, 0.55); however, the efficiency roll-off was severe (EQE4000 = 2%) and the turn-on voltage was high at 5.2 V, both attributed to poor electron confinement in the emissive layer featuring this uncommon ionic emitter.
An interesting strategy was employed for the design of [Cu(czpzpy)(PPh3)2]BF4 and [Cu(czpzpy)(POP)]BF4 , (Figure ),ref. ref818 with the carbazole-substituted pyridylpyrazoyl ligand also acting as a host material for OLEDs. The complexes are green TADF emitters as powders, with [Cu(czpzpy)(PPh3)2]BF4 emitting at λPL of 495 nm and having ΦPL of 45% and τPL of 134 μs, while [Cu(czpzpy)(POP)]BF4 emits at λPL of 518 nm, has ΦPL of 95% and a τPL of 23 μs. The TADF emission is supported by ΔE ST of 180 meV and 130 meV, respectively. Due to its higher ΦPL and shorter τPL, [Cu(czpzpy)(POP)]BF4 dispersed in additional ligand czpzpy as host was used as the emitter in a solution-processed OLED. The green OLED showed an EQEmax of 6.3% at CIE coordinates of (0.26, 0.49), while no efficiency roll-off out to 100 cd m–2 was observed. Interestingly, devices with the same performance could be obtained by spin-coating a solution of [Cu(NCMe)2(POP)]BF4 and czpzpy, showing that the copper complex could be formed in-situ during the solution-processing of the device. Related complexes [Cu(PNNA)(POP)]BF4 and [Cu(PNNA)(xant)]BF4 (Figure ) contain a diimine ligand decorated instead with a DMAC donor.ref. ref819 The diimine ligand acts as an electron acceptor in this case, which resulted in an ILCT state from the electron-donating DMAC group (more so than carbazole) and TADF emission emerging from the ligand itself. The two complexes only differ by fusing of the other POP/xanthene-based ligand, and so show similar photophysics as 20 wt% doped films in mCP (λPL = 482 to 492 nm, ΦPL = 70 to 74%, τPL ≈ 50 μs). The highest performance solution-processed OLED used [Cu(PNNA)(xant)]BF4 and showed an EQEmax = 7.4% at CIE coordinates of (0.21, 0.43).
A novel strategy to reduce the kinetic lability of ligands in [Cu(N∧N)(P∧P)]+ complexes involves the generation of a pseudorotaxane structure, wherein the ligated diphosphine is effectively encircled by a macrocycle containing the bound N∧N ligand.ref820,ref821 Of the complexes synthesized to explore this design, [Cu(m42)(POP)]BF4 (Figure ) was found to be the most kinetically inert. This complex has a ΦPL of 23% in DCM rising to 41% when doped at 1 wt% in PMMA, which is similar to reference compound [Cu(dmp)(POP)]BF4 (23 and 50%, respectively). The key difference between these complexes manifests in the OLED performance, where although the EQEmax are similar (at 10.5 and 9.5%, respectively) the device with [Cu(m42)(POP)]BF4 is found to be more stable and could attain a much higher maximum luminance of 12,800 cd m–2 vs. 7740 cd m–2 for the device with [Cu(dmp)(POP)]BF4 .
Using a more strongly π-accepting 2,2′-biquinoline N∧N ligand, dcbq, resulted in the red-emitting complexes [Cu(dcbq)(PPh3)2]PF6 , [Cu(dcbq)(POP)]PF6 , and [Cu(dcbq)(xant)]PF6 (Figure ).ref. ref822 These complexes emit at λPL ranging from 669 to 671 nm, have ΦPL ranging from 26 to 56%, and τPL ranging from 0.58 to 0.71 μs, while ΔE ST was not reported. With increasing size and rigidity of the phosphine ligand(s) and resulting suppression of Jahn-Teller distortion there was a progressive enhancement of the ΦPL.
The use of heavy chalcogens is another strategy to enhance SOC as illustrated by the use of thiadiazole and selenodiazole ligands in the complexes S1, S2, Se1 and Se2 (Figure ).ref. ref823 In 10 wt% doped films in PMMA, all four complexes are weakly yellow-orange emissive, with λPL between 577 and 605 nm, ΦPL of between 4 to 8%, and τPL ranging from 0.8 to 1.2 μs. Notably, k r in the Se-containing complexes is twice that of the sulfur-containing complexes. However, this was attributed not to enhanced SOC but rather to greater spatial separation of the frontier orbitals, resulting in a smaller ΔE ST. Supporting this interpretation the τPL of Se2 is only 0.8 μs, which was the shortest emission lifetime of any [Cu(N∧N)(P∧P)]+ complex at the time.
Bulky NHC ligands have been widely used in emissive complexesref194,ref781 but have seen limited use in tetrahedral copper(I) complexes. One of the few such reports describes complexes [Cu(Ph-BenIm-methPy)(POP)]PF6 and [Cu(Ph-Im-methPy)(POP)]PF6 (Figure ), that combine a pyridyl NHC ligand with a POP P∧P ligand.ref. ref772 The complexes are very bright sky-blue emitters as powders with λPL = 493 and 487 nm, ΦPL > 96% for both, and τPL = 63 and 56 μs, respectively. The ΔE ST are 128 and 108 meV, but temperature-dependent PL studies revealed that at room temperature only about 35% of the emission originates from the S1 state (TADF), while the remainder is concurrent phosphorescence from the T1 state.
Complexes [Cu(DMAC-PyPI)(POP)]BF4 , [Cu(DMAC-PyPI)(xant)]BF4 , [Cu(PXZ-PyPI)(POP)]BF4 , and [Cu(PXZ-PyPI)(xant)]BF4 (Figure ) contain N∧N ligands that possess both electron-donating (DMAC/PXZ) and electron-accepting (phenanthroimidazole) groups.ref. ref824 These four complexes emit at λPL between 534 and 564 nm, have ΦPL ranging from 42 to 71%, and τPL of between 4.3 and 24.1 μs. The photophysical properties of these complexes is entirely dependent on the nature of the D-A N∧N ligand and all complexes emit from an ILCT state with ΔE ST ranging from 50 and 110 meV. Interestingly, the the copper ion is not entirely decorative, with the ΔE ST of the free ligands larger at 450 and 310 meV for DAMC-PyPI and PXZ-PyPI, respectively. This study highlights how coordination to the copper can tune the energy levels of the orbitals localized on the N∧N ligand to enable TADF emission. Green emitting solution-processed OLEDs with [Cu(DMAC-PyPI)(xant)]BF4 and [Cu(PXZ-PyPI)(POP)]BF4 showed EQEmax ranging from 3.8 to 8.0% depending on emitter and doping concentration. The best performing device employed 16 wt% [Cu(PXZ-PyPI)(POP)]BF4 doped in PYD2 to achieve an EQEmax = 8.0%, which was maintained to EQE1000 > 5.0%.ref. ref824
Employing strongly π-accepting pyrazinyl sulfide N∧N ligands provided an effective strategy to achieve red emitting [Cu(N∧N)(P∧P)]+ complexes.ref. ref825 [Cu(pz-S-pz)(POP)]PF6 , [Cu(pq-S-pz)(POP)]PF6 , and [Cu(pz-S-CF3pm)(POP)]PF6 (Figure ) as powders emit at λPL ranging from 581 to 650 nm, have widely varying ΦPL of between 7.7 and 57.8%, and τPL of 6.47 to 10.5 μs. The ΔE ST values are between 60 and 130 meV. The most promising red emitter [Cu(pq-S-pz)(POP)]PF6 (λPL = 642 nm, and with highest ΦPL = 57.8% and shortest τPL = 6.47 μs) was used in LECs that showed very good performance for a red device (See Sections sec16 and sec5 for discussion of the challenges associated with this type of device and color).
As an alternative to the cationic copper(I) complexes described above, neutral complexes exhibiting TADF emission are of great interest. This is particularly because these non-ionic materials aree more readily evaporable and so are more compatible with vacuum deposition fabrication for OLEDs. Neutral tetrahedral copper(I) complexes can have a range of different ligand environments, from Cu(P∧P)(N∧N) bearing anionic diimine ligands, to the use of halido ligands in combination with one to three dative ligands. Representative examples are shown in Figure and are discussed below.
The first reported neutral copper(I) TADF complexes contained a POP ligand and a dipyrazolylborate ligand. Powders Cu(POP)(pz2BH2), Cu(POP)(pz4B), and Cu(POP)(pz2Bph2) (Figure ) are bright blue emitters (λPL = 436–464 nm, ΦPL up to 90% and τPL = 13–22 μs, ΔE ST = 99–161 meV).ref. ref774 A similar series of three complexes contain the similar dipyrazolyldiphenylborate ligand, Cu(Ph2Bpz2)(dppb), Cu(Ph2Bpz2)(dppb-F), and Cu(Ph2Bpz2)(dppb-CF3).ref. ref826 In doped mCP films (10 wt%), these complexes emit strongly in the green (λPL = 523 – 545 nm, ΦPL = 50 – 68% and τPL = 3.6 – 8.2 μs). The green-emitting (λEL = 528 – 552 nm) OLEDs showed EQEmax ranging from 11.9 to 17.7%. The photophysics of Cu(Ph2Bpz2)(dppb) and Cu(pz2Bph2)(POP) were subsequently studied in more detail, confirming assignment of the emission to TADF and measuring the ΔE ST to be 46 and 81 meV, respectively.ref. ref773
An anionic phosphinothiolato ligand was used to prepare Cu(PP)(PS) (Figure ).ref. ref756 As a powder the complex emits at λPL of 521 nm, has a ΦPL of 73%, and shows biexponential decay kinetics with τPL = 0.33 and 1.73 μs. Solution-processed OLEDs showed an EQEmax = 7.8% at CIE coordinates of (0.40, 0.53).
Beyond coordination environments with two bidentate ligands, 2-to-4-coordinate TADF copper complexes have been prepared using combinations of mono-, bi-, and tridentate ligands. TTPPCuCl, TTPPCuBr, and TTPPCuI (Figure ) are examples of this, containing a halido ligand in combination with a triphosphine ligand.ref. ref766 In neat films the complexes are bright green emitters (λPL = 521–530 nm, ΦPL = 76–83% and τPL = 11–19 μs) and have ΔE ST of 95–99 meV. While the Cl and Br complexes emit via TADF, the iodo complex showed mixed TADF/phosphorescence at room temperature (39% phosphorescence). The OLEDs with TTPPCuCl, TTPPCuBr, and TTPPCuI showed progressively increasing EQEmax from 9 to 12.2 and 16.3%, with the highest efficiency and lowest efficiency roll-off (6% decline at 1000 cd m–2) for the OLED with TTPPCuI attributed to the faster k r in that emitter.
Replacing the tridentate phosphine ligand above with a combination of a P∧P ligands and a triphenylphosphine results in the complexes CuCl(PPh3)(dpmb), CuBr(PPh3)(dpmb), and CuI(PPh3)(dpmb) (Figure ).ref. ref827 The complexes are sky-blue emitters as powders (λPL = 464–479 nm, ΦPL = 23–53% and τPL = 4.3–5.7 μs) and have calculated ΔE ST between 98 and 152 meV. Replacement of the bridging dimethylbenzyl group with a dimethylthiophene produced the series of complexes CuCl(PPh3)(dpmt), CuBr(PPh3)(dpmt), and CuI(PPh3)(dpmt) (Figure ).ref. ref828 The thiophene was chosen as an electron-rich heteroaryl ring in an attempt to raise the LUMO energy of the complexes and blue-shift the emission. This was only modestly successful, with powder emission blue-shifted by approximately 10 nm compared to the dpmb analogues (λPL between 459 and 484 nm), and the powders were less emissive (ΦPL ≤ 24%) while the calculated ΔE ST range from 64 to 198 meV. A later report examined the effects of removing the methyl groups on the bridging thiophene as in CuCl(PPh3)(dppt), CuBr(PPh3)(dppt), and CuI(PPh3)(dppt), or incorporate a trimethylsilyl group as in CuCl(PPh3)(dpts), CuBr(PPh3)(dpts) and CuI(PPh3)(dpts) (Figure ).ref. ref829 These complexes show bright sky-blue to yellow-green emission (λPL = 485–535 nm). Notably, the introduction of the trimethylsilyl group increased the solubility of the complexes in most organic solvents and reduced their k nr without affecting k r, resulting in both longer emission lifetimes (τPL increased from 4–10 μs to 20.8–48.9 μs) and higher ΦPL (increased from 3–18% to 29–52%). The non-doped solution-processed OLED with [CuBr(dpts)(PPh3)] showed an EQEmax of 7.7% at λEL of 564 nm.
Monodentate ligands need not be limited to halido groups. Two complexes containing tridentate phosphine ligands and a thiocyanato group, Cu(NCS)(P3) and Cu(NCS)(P4) (Figure ), showed green to yellow TADF emission.ref. ref830 As powders the two complexes emit at λPL of 520 and 543 nm, have ΦPL = 57 and 27% with τPL of 4.8 and 4.9 μs and ΔE ST of 62 and 80 meV, all respectively.
Examples of complexes bearing another tridentate N,P,P-ligand include CuCl(dmpzpp), CuBr(dmpzpp), CuI(dmpzpp), and CuSPh(dmpzpp) (Figure ).ref. ref831 CuCl(dmpzpp) is non-emissive, while the remaining complexes are bright green-yellow emitters as powders (λPL = 530–541 nm, ΦPL = 82–90% and τPL = 5–9 μs). OLEDs with CuI(dmpzpp) and CuSPh(dmpzpp) doped in a mixed TCTA:DPEPO host showed EQEmax between 10.8 and 16.4% across a range of doping concentrations (2–8%). The device with CuI(dmpzpp) showed the highest EQEmax of 16.4% and the lowest efficiency roll-off (EQE1000 = 10.2%), while the reduced performance of CuSPh(dmpzpp) was attributed to charge trapping in the emissive layer.
A complex with a thiophene-bridged diphosphine ligand and an anionic dipyrazolylborate ligand, Cu(pz2BH2)(3,4-dppTp) (Figure ) showed both mechanochromism and vapochromism.ref. ref832 The complex crystallizes in two polymorphs, which emit in the blue (1B) and yellow (1Y). Grinding 1B produced a new material 1G that is a green emitter. Exposing 1G to solvent vapors (dichloromethane or diethyl ether) returned the emission profile to that of 1B. The variable emission of the complex was attributed to intermolecular interactions that are modulated by grinding or exposing the material to solvent vapors.
Finally, Cu(thzbzi)(dppnc) (Figure ) contains an unusual anionic diphosphine-nido-carborane ligand.ref. ref833 As a powder this complex is a green-yellow emitter, with λPL of 547 nm, ΦPL of 16%, and τPL of 26 μs. Similarly when doped at 5 wt% in PMMA the emission has λPL of 542 nm, ΦPL of 10%, and τPL of 23 μs. The ΔE ST was found to be 114 meV (powder) and 127 meV (in the doped film). The similar photophysical properties in these two media imply little aggregation in the powder form.
There are a small number of reported TADF Cu(I) complexes that can switch between cationic and neutral forms following protonation of one of the ligands (Figure ). The first report of these switchable complexes included four neutral complexes containing (di)phosphine ligands and a pyridyltetrazolate ligand, Cu(P∧P)(PyrTet), where (P∧P) = (PPh3)2, POP, xantphos, or Me2Xantphos. When the tetrazole of the PyrTet ligand is protonated, the complexes become charged [Cu(P∧P)(PyrTetH)]BF4 .ref. ref834 All eight complexes are green to yellow emitters (λPL = 510–569 nm), while the neutral complexes show more efficient, longer-lived emission (ΦPL = 76–89% and τPL = 17.8–26.6 μs for the neutral complexes, compared to ΦPL = 4–46% and τPL = 5.2–15.3 μs for the cationic complexes). The less efficient emission from the cationic complexes was attributed to a change in the nature of the emissive state (1MLCT for neutral and mixed 1MLCT/LLCT for charged) as well as vibrational quenching effects of the N-H bond of the protonated tetrazole ring.
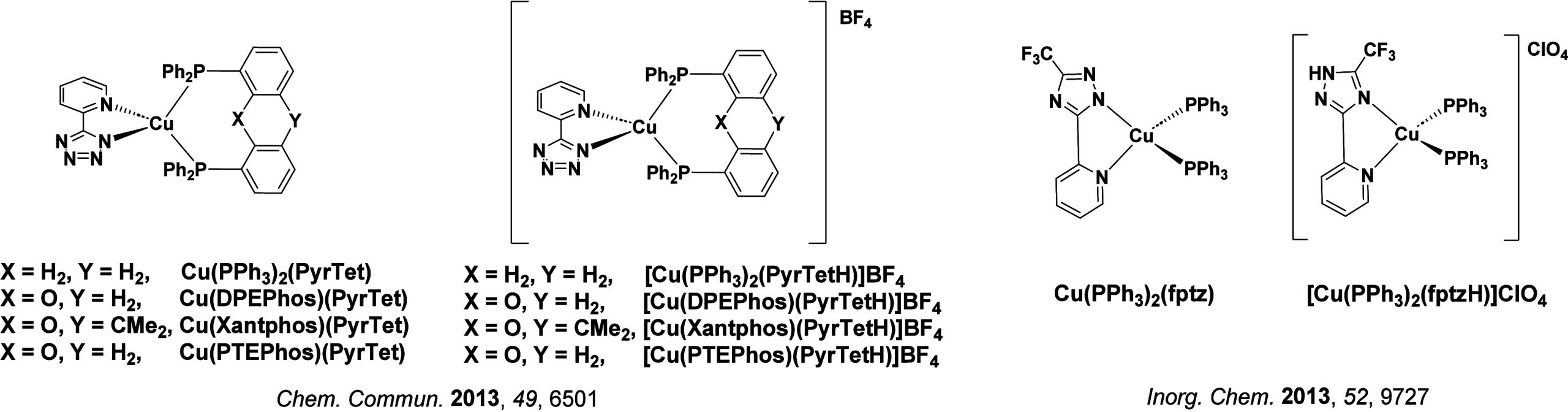
Another pair of interconvertible complexes, Cu(PPh3)2(fptz) and [Cu(PPh3)2(fptzH)]ClO4 (Figure ), also show interesting photophysical properties.ref. ref835 In this case conversion involves protonation and a ring inversion isomerism of the 1,2,4-triazole. Both complexes are emissive in solution and the solid state and moving from the cationic to the neutral complex results in a blue-shift in the solution-state emission but a red-shift in the solid-state emission. This change was attributed to the presence of the N-H bond raising the LUMO energy and blue-shifting the solution-state emission, while the more flexible structure of the neutral complex leads to greater excited-state relaxation and a lower energy excited state in the solid state. The contrasting electronic and geometric impact on the emission highlights the sensitivity of the photophysical properties of copper(I) complexes to the ligand environment.
TADF emission has also been reported for 3-coordinate trigonal planar copper(I) complexes, with selected examples shown in Figure . The use of bulky phosphine or carbene ligands is popular to restrict the pseudo Jahn-Teller Y-to-T excited-state distortion in 3-coordinate complexes, which contributes to non-radiative decay and ligand dissociation as in tetrahedral complexes.ref. ref836 The first report of TADF trigonal copper complexes featured (LMe)CuCl, (LMe)CuBr, and (LMe)CuI (LMe = dtpb = 1,2-bis(o-ditolylphosphino)benzene), although these were misattributed as phosphorescent likely due to the exceptional device performance (EQEmax = 21.3%, λEL = 517 nm for the bromo complex) that predated the key early reports of all-organic TADF OLEDs.ref757,ref837 The photophysics of these three complexes and additional related complexes (LEt)CuBr and (LiPr)CuBr were later analyzed in greater detail and confirmed to arise from TADF.ref. ref837 As powders all five complexes show bright sky-blue to green emission (λPL = 473–517 nm, ΦPL = 38–95%, τPL = 4.6–8.9 μs). (LEt)CuBr and (LiPr)CuBr were used in green OLEDs (EQEmax = 22.5 and 18.6%, and λEL = 529 and 515 nm respectively). The related complexes (LMe)Cu(SPh) and (LiPr)Cu(SPh) with thiolates replacing the halido ligands are also TADF-active and have near unity ΦPL as powders (ΦPL = 95%).ref. ref838 The emissive excited states in these materials were assigned to have LLCT character, in contrast to the MLCT states of (LMe)CuBr.ref. ref838
Cu(P∧P)X complexes employing bulky diphosphine ligands also have been shown to emit by TADF, for instance in the family of CuI(mpdp), CuBr(mpdp), and CuCl(mpdp) (mpdp = hexamethyl-bis(diphenylphosphino)-terphenyl, Figure ).ref. ref839 These complexes are only weakly emissive though (ΦPL = 1–5.4%), and OLEDs with CuI(mpdp) showed an unsurprisingly low EQEmax of 0.26%. The use of an unusual benzimidazole-linked diphosphine ligand instead resulted in two highly emissive complexes CuI(benzimPP) and CuBr(benzimPP) (named 1 and 2 in the initial publication, Figure ).ref. ref840 These complexes are orange-red emitters as powders, that show bright emission and long lifetimes (λPL = 630 and 615 nm, ΦPL = 65 and 72%, τPL = 143 and 228 μs, all respectively). Other trigonal planar copper(I) complexes using the bulky diimine ligand dtbp such as CuX(dtbp) (Figure ) have been shown to exhibit TADF, albeit with low ΦPL ≤ 15%.ref. ref841
Over time the sterically bulky class of NHC ligands have become the most popular for 3-coordinate copper complexes. The first example, (IPr)Cu(py2-BMe2) (Figure ), was initially reported as phosphorescent along with related complexes (BzI-3,5Me)Cu(py2-BMe2) and (PzI-3,5Me)Cu(py2-BMe2).ref. ref842 A subsequent study showed that (IPr)Cu(py2-BMe2) in fact emits by TADF, while (BzI-3,5Me)Cu(py2-BMe2) emits by phosphorescence.ref. ref764 It was determined that the emission mechanism is controlled by the different steric demands of the aryl groups on the NHC ligands – 2,6-diisopropylphenyl (dipp) vs 3,5-dimethylphenyl (xylyl). The bulkier dipp groups in (IPr)Cu(py2-BMe2) locked the ligands in a co-planar orientation, while the less bulky xylyl groups in (BzI-3,5Me)Cu(py2-BMe2) resulted in a perpendicular conformation. DFT calculations revealed that the ΔE ST is smaller in the co-planar ligand orientation (67 meV, enabling TADF) and higher when the ligands adopt an orthogonal conformation (459 meV), accounting for the different emission mechanisms observed in the complexes.
In addition to the neutral 3-coordinate copper(I) complexes described above, there are a number of cationic 3-coordinate copper(I) complexes that show TADF. Elie et al. documented the first examples of cationic trigonal copper(I) complexes in the structural form of [Cu(N∧N)(NHC)]PF6.ref. ref843 The 13 reported examples contained various combinations of 5 different NHC ligands and 6 different dipyridylamine ligands, and are blue to green emitters as powders (λPL = 455 to 521 nm). Varying the electronics of the NHC ligand had minimal impact on the emission color (λPL = 463 to 481 nm), while the emission color was sensitive to substitution of the pyridyl rings of the dipyridylamine ligands. Electron-withdrawing CF3 groups on the pyridyl rings stabilized the LUMO of the complexes, red-shifting the emission for [Cu(IPr)(L2)]PF6 and [Cu(IPr)(L3)]PF6 above 505 nm (Figure ), while electron-donating OMe groups stabilized the LUMO of [Cu(IPr)(L4)]PF6 , blue-shifting the emission to 420 nm. The ΦPL varied with the rigidity of the molecule, with ΦPL as high as 64% for [Cu(MeIPrMeO)(L1)]PF6 .
The same group later investigated a similar series of complexes by varying the bridge between the two pyridyl rings of the diimine ligand.ref. ref844 Of the compounds studied, powders of [Cu(IPr)(dpyp)]PF6 and [Cu(IPr)(Pphpy2)]PF6 (Figure ) have the highest ΦPL as these contained the most rigid bridges between the pyridyl rings (isopropyl and phenylphosphinyl, ΦPL = 73 and 86% respectively). These two compounds emit in the blue (λPL = 474 nm) and green (λPL = 503 nm), respectively.
2017 marked the first reports of linear copper(I) complexes that emit via TADF. Among these, linear carbene-metal-amide (CMA) complexes (including previously mentioned copper complex CMA2) showed the most desirable emission properties for OLED applications.ref. ref845 CMA complexes have consequently been the focus of an extensive research effort for the last 5 years, with notable early Cu containing materials summarized here. Aside from CMA complexes, a small number of other noteworthy linear copper(I) complexes emitting by TADF have also been reported (Figure ).
Romanov et al. made important early contributions to the research field by reporting copper complexes based on the adamantyl-substituted Cyclic Alkyl Amino Carbene (CAAC) ligand AdL.ref. ref845 While TADF is not explicitly mentioned in the paper, a number of the complexes, namely CuO1-CuO5, (AdL)CuSPh, CuN1, and CuN2 (Figure ) showed efficient emission and ΦPL up to 62% that is characterized by biexponential decay kinetics with associated prompt (τp = 2 to 9.7 ns) and delayed emission (τd = 0.29 to 0.66 μs) consistent with TADF. Further improvements in TADF performance have also been observed for CMA complexes employing other metal centers, particularly Au, and are summarized later in this section.
The sterically demanding trityl groups of the NHC ligand (ITr) were used in a series of linear copper(I) complexes with pyridyl and quinoline ligands. Of these complexes only two exhibited TADF, [Cu(ITr)(4-CN-py)]BF4 and [Cu(ITr)(4-CO-py)]BF4 (Figure ).ref. ref846 In 10 wt% doped films in PMMA the complexes emit with λPL of 525 and 545 nm, have modest ΦPL of 25 and 12%, and short τPL of 2.1 and 3.2 μs, all respectively.
Instead of co-doping a copper(I) complexes with a host material in the EML of the OLED, Thompson and co-workers pioneered an approach of co-depositing an OLED host that could coordinate directly with separate copper(I) precursors to form the emissive complex in-situ.ref. ref847 The initial report involved co-depositing mCPy and CuI to produce a green phosphorescent film (λPL = 528 nm, ΦPL = 64% and τPL = 10.7 μs). The OLED showed an EQEmax of only 4.4%; however, a number of the films were shown to be TADF-active (Figure ).
Wang et al. similarly prepared co-deposited films of CuI with two different carboline containing host materials.ref. ref848 One of the films, CuI:CzBPDCb (Figure ) showed green TADF (λPL = 520 nm, ΦPL = 22%, τd = 1.05 μs, ΔE ST = 120 meV). Many OLEDs were fabricated using different ratios of CuI:CzBPDCb, and the best device with 6 mol% CuI in the EML showed an EQEmax = 17.5%.ref. ref848
A spiro-bifluorene compound with a coordinating nitrogen in one of the aryl rings, aza-SBF, was similarly co-deposited with CuCl, CuBr, and CuI.ref. ref849 CuCl:aza-SBF, CuBr:aza-SBF, and CuI:aza-SBF (Figure ) all showed green TADF with doping ratios of between 5–11 mol% of CuX. There is a small blue-shift in the λPL as the halide increased in mass (λPL = 550, 537, 526 nm for the Cl, Br and I films, respectively), while all complexes showed similar delayed emission lifetimes (τd = 3.9 to 5.8 μs). The ΦPL varied significantly as a function of halide ligands and doping concentration, with the highest ΦPL being 78% for the 7 mol% CuBr:aza-SBF film. The highest performing device used 7 mol% CuBr:aza-SBF (EQEmax/100 = 13.6/13.6%). A subsequent study used aza-SBF analogues with the nitrogen atom in 4 different positions of the aryl ring to produce four different co-deposited films CuI:α-aza-SBF, CuI:β-aza-SBF, CuI:γ-aza-SBF, and CuI:δ-aza-SBF.ref. ref850 The co-deposited films showed yellow to red emission (λPL = 550 to 625 nm) with a wide range of range ΦPL (4.6 to 92.2%), the most efficient emitter being the yellow (λPL = 550 nm) emitting CuI:δ-aza-SBF. The emission at room temperature of all complexes was determined to be a mixture of TADF and phosphorescence (ranging from 20% phosphorescence contribution for CuI:α-aza-SBF to 58% for CuI:γ-aza-SBF). The most efficient OLED of the series used CuI(8 mol%):δ-aza-SBF (λEL = 540 nm) and achieved EQEmax of 16.8%.ref. ref850
Although the concept of TADF in simpler copper complexes has been observed for decades, the first TADF OLED in 2007 used a dinuclear copper complex with bridging iodo ligands, [Cu(μ-I)dppb]2 (Figure ).ref. ref75 As a powder [Cu(μ-I)dppb]2 showed two delayed fluorescence components with τPL of 1.5 and 4.0 μs, and has a small ΔE ST of 90 meV. Devices showed low EQEmax of 4.8% at λEL of 560 nm. The dinuclear structure nonetheless prevents the formation of a formally d9 copper center and large changes to the geometry in the excited, with the electron density being delocalized over both centersstate.ref73,ref851 Similar to the use of bulky ligands, this is a widely exploited tactic to produce highly efficient TADF emitting copper complexes. Selected dinuclear copper(I) complexes shown in Figure are discussed below, with additional polynuclear Cu complexes in Figure .
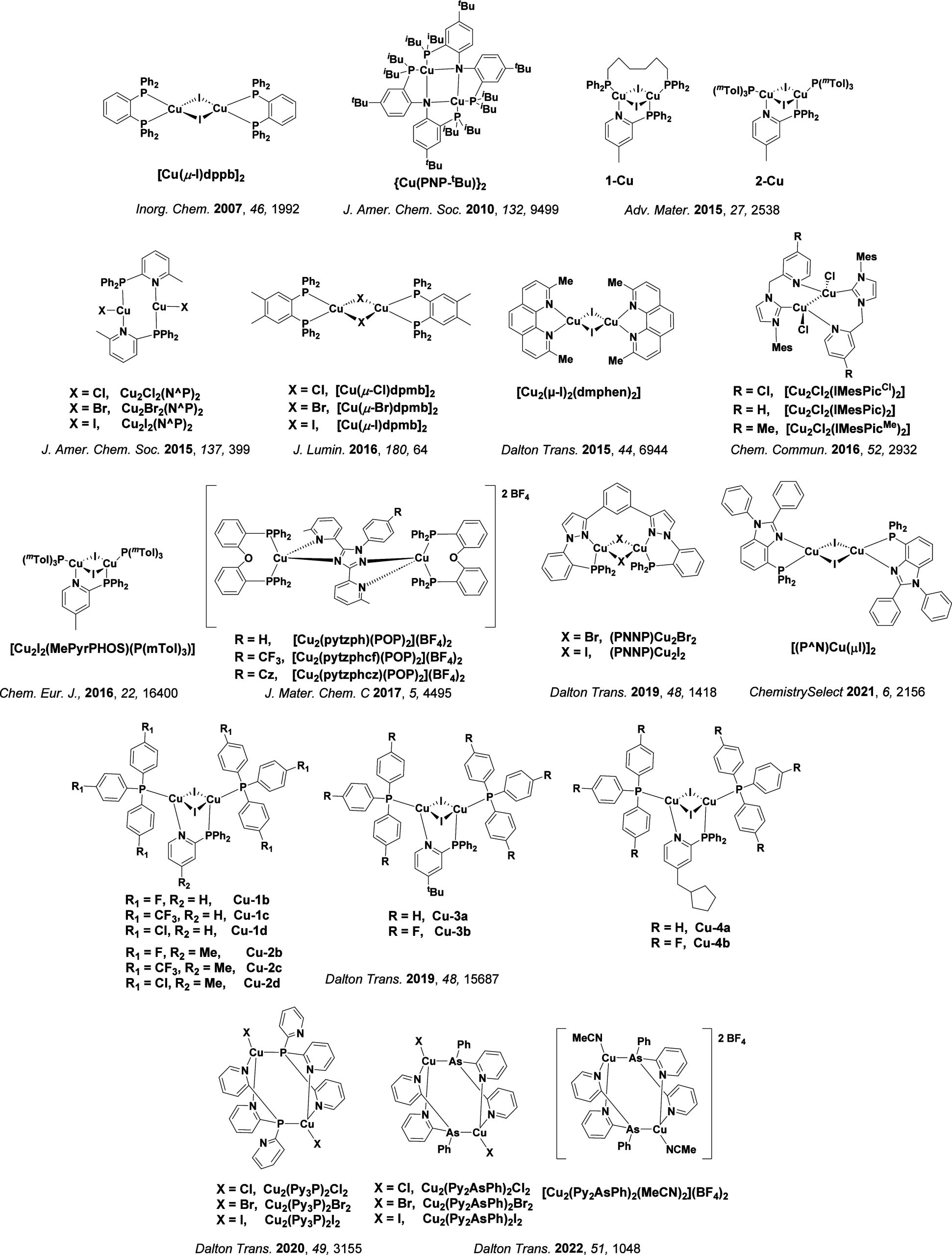
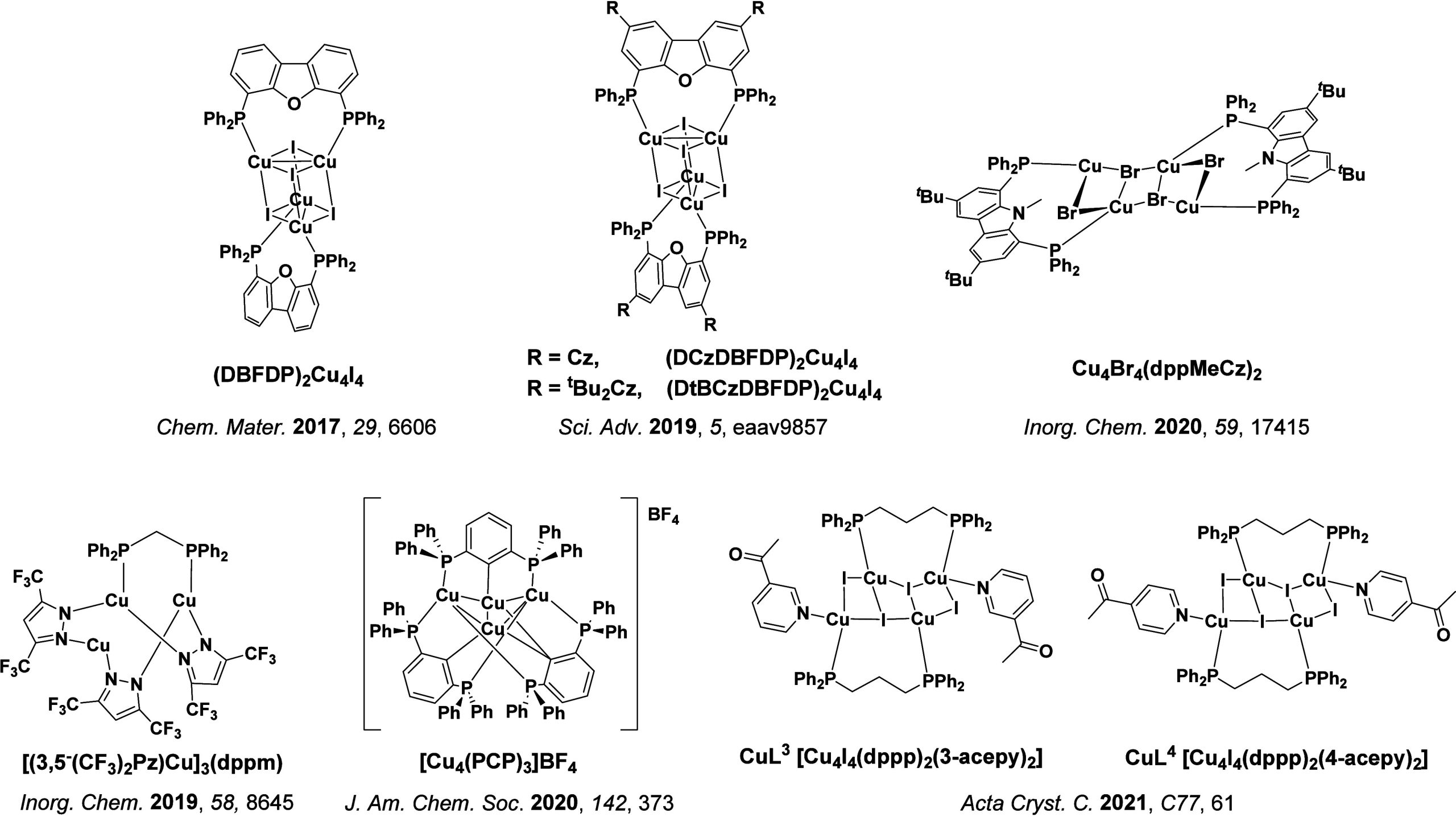
The first copper OLED to surpass the 5% EQE limit of fluorescent devices was another dinuclear emitter, [Cu(PNP-tBu)]2 (Figure ).ref. ref73 In 2-MeTHF the complex emits at λPL ≈ 510 nm, has a ΦPL of 57%, a τPL of 11.5 μs, and a ΔE ST of 100 meV. The OLED showed what was at the time a remarkable EQEmax of 16.1%, similar to the efficiencies of iridium(III)-based OLEDs. Volz et al. introduced additional dinuclear complexes 1-Cu and 2-Cu,ref. ref851 the former of which showed TADF in doped films (λPL = 540 nm, ΦPL = 92%, τd = 3.2 μs, and ΔE ST = 90 meV in 30 wt% doped PYD2 films). The solution-processed OLED with 1-Cu showed an EQEmax of 23% and a λEL of 555 nm. The dinuclear copper(I) complex Cu2Cl2(N∧P)2 and analogues Cu2Br2(N∧P)2 and Cu2I2(N∧P)2 all have high ΦPL and microsecond-long τPL (ΦPL = 52 to 92%, τPL = 7.3 to 12.4 μs).ref. ref763 Detailed photophysical studies of Cu2Cl2(N∧P)2 show that it emits from both S1 and T1 states with 80% TADF contribution and the remaining 20% being phosphorescence.
A family of dinuclear clusters containing the bidentate ligand 1,2-bis(diphenylphosphino)-4,5-dimethylbenzene (dpmb), [Cu(μ-Cl)dpmb]2, [Cu(μ-Br)dpmb]2 , and [Cu(μ-I)dpmb]2 (Figure ) are all green emitters as powders (λPL = 498–527 nm, ΦPL = 28–32%, τPL = 2.5–12.5 μs), with moderate ΔE ST ranging between 120 to 140 meV.ref. ref852 The green OLEDs using mCP as the host showed EQEmax ranging from 7.3 to 10.1%. The device with [Cu(μ-Cl)dpmb]2 in particular showed an EQEmax of 8.3%, and had moderate efficiency roll-off (EQE1000 = 4.9%) with CIE coordinates of (0.38, 0.51). The dmp-based dinuclear complex [Cu2(μ-I)2(dmp)2] showed very complicated photophysics owing to contributions from both TADF and phosphorescence (λPL = 667 nm, ΦPL = 18%, τPL = 6.4 μs).ref. ref841
NHC ligand-based dinuclear complexes [Cu2Cl2(IMesPicCl)2], [Cu2Cl2(IMesPicH)2], and [Cu2Cl2(IMesPicMe)2] all show TADF (Figure ).ref. ref853 These complexes emit at λPL = 520–550 nm, have moderate ΦPL of 49 to 68%, and similar τPL of 9.2 to 11.0 μs associated with similar ΔE ST of between 78 and 120 meV. Interestingly, the very closely related complex [Cu2Cl2(IMesPicOMe)2] is phosphorescent, highlighting that subtle changes in the ligand can completely change the emission mechanism of this class of complex.ref. ref853
Wallesch et al. reported the complex [Cu2I2(MePyrPHOS)(P(mTol)3].ref. ref854 This complex is a yellow-green emitter (λPL = 550 nm as a powder) with a ΦPL of 75% as a powder and 56% for the 50 wt% doped film in PMMA. Solution-processed OLEDs were fabricated by either spin coating or inkjet printing. The best performance was achieved with a spin-coated device and showed an EQEmax = 11.4%.ref. ref854
[Cu2(pytzph)(POP)2](BF4)2 , [Cu2(pytzphcf)(POP)2](BF4)2 , and [Cu2(pytzphcz)(POP)2](BF4)2 all contain a bridging dipyridyl-1,2,4-triazole ligand (Figure ).ref. ref855 The complexes emit at λPL = 503–519 nm, yet have distinct ΦPL of between 29 and 79%, τPL ranging from 5.5 to 16 μs, and ΔE ST of between 89 and 132 meV. The most efficient solution-processed OLEDs employed the complex [Cu2(pytzphcz)(POP)2](BF4)2 , which has the highest ΦPL and showed an EQEmax of 8.3% and minimal roll-off (EQE100 = 8.1%) at CIE coordinates of (0.29, 0.53).
The Cu2X2 copper halide core is supported by a single tetradentate ligand in complexes (PNNP)Cu2I2 and (PNNP)Cu2Br2 (Figure ).ref. ref856 The phenyl-bridged tetradentate ligand increases the rigidity of these complexes, reducing non-radiative decay by hindering excited-state distortions.ref766,ref851 (PNNP)Cu2I2 and (PNNP)Cu2Br2 have similar emission properties as powders, with (PNNP)Cu2I2 emitting at a λPL of 494 nm with a ΦPL of 42%, and τPL of 8.8 μs. (PNNP)Cu2Br2 emits at a slightly red-shifted at λPL of 517 nm and a slightly brighter ΦPL of 58%, and has a τPL of 13 μs. The complexes also have similar ΔE ST of 90 and 110 meV, respectively. A similar coordination environment was achieved with two unlinked ligands around the same copper halide core in [(P∧N)Cu(μI)]2 using an unusual diphenylphosphinobenzimidazole ligand.ref. ref857 The complex emits at λPL of 585 nm with a ΦPL of 37% and a τPL of 5.85 μs as a powder. Related complexes with methoxy groups at the 2-position of the phenyl ring of the benzimidazole were instead found to be phosphorescent. The solution-processed OLED with [(P∧N)Cu(μI)]2 showed an EQEmax of 3.0%.
Busch et al. synthesized a family of ten dinuclear copper(I) complexes containing bridging 2-phosphinopyridyl ligands.ref. ref858 The complexes Cu-1b through d, Cu-2b through d, Cu-3a and b, and Cu-4a and b (Figure ) are all bright green to yellow emitters as powders (λPL = 519 to 549 nm, ΦPL = 70 to 93%, τPL = 5.5 to 10.2 μs). These results are consistent with the properties of previously reported complexes using pyridylphosphine bridging ligands,ref851,ref854 and also show very high solubility in organic solvents due to the presence of the fluorinated and alkyl groups on the complexes, which makes them promising materials for solution-processed OLEDs.
A series of three further complexes with pyridyl phosphine ligands, [Cu2(Py3P)2X2] (X = Cl, Br, I) were prepared by mechanochemical synthesis.ref. ref771 These complexes are bright green emitters at room temperature, with the emission attributed to mixed phosphorescence and TADF. The trend in contributions of the two processes across the halide series is unusual, with more phosphorescence observed for the Cl complex (73%), while the least phosphorescence is observed for the I complex (39%). Normally, the increased SOC of the heavier halogens increases the radiative rate of the formally spin-forbidden phosphorescence.ref766,ref856 However, in these complexes ΔE ST also decreases with increasing halogen mass (from 186 to 155 to 124 meV) and the resulting increase in k RISC for the complexes with heavier halogen atoms outweighs any increase in phosphorescence rate due to enhanced SOC. In an effort increase the radiative rate and SOC of these dinuclear copper(I) complexes, a series of four similar complexes containing arsine ligands, [Cu2(Py2AsPh)2X2] (X = Cl, Br, I)ref. ref859 and [Cu2(Py2AsPh)2(MeCN)2](BF4)2 was prepared.ref. ref860 The three As-containing complexes are all bright green emitters in the solid state (λPL = 500 to 530 nm, ΦPL = 22 to 50%, τPL = 2.0 to 9.0 μs), with emission originating from TADF and phosphorescence. Due to the larger SOC, the emission lifetimes of the arsenic complexes are shorter than their isostructural phosphine analogues with similar photoluminescence quantum yields (τPL = 5 to 33 μs and ΦPL = 51–55%).ref. ref860
Beyond dinuclear copper(I) complexes, there are a number of larger copper clusters reported to display TADF emission. Selected examples of tri- and tetra-nuclear complexes are shown in Figure . At yet larger cluster sizes there are a number reported copper clusters that are luminescent; however, their electronic character moves away from molecular descriptions and are beyond the scope of this review.ref787,ref789 While there is an extensive body of work on tetranuclear copper clusters that display strong luminescence,ref861,ref862 it wasn’t until 2017 that the first TADF emitting cluster was reported.ref. ref767 Most of these clusters emit from a cluster-centered (3CC) excited state as phosphorescence. To enable TADF emitting tetranuclear cluster, ligands that can electronically couple to the core of the cluster to delocalize the electron density across the ligands are typically required.ref. ref863
The first tetranuclear copper(I) complex that showed TADF, (DBFDP)2Cu4I4 , contained two diphosphine ligands to stabilize a Cu4I4 cubic core (Figure ).ref. ref767 The complex is a weak blue-green emitter in DCM (λPL = 491 nm, ΦPL = 5%, τPL = 1.9 μs, ΔE ST = 160 meV), and analysis of temperature-dependent emission shows that the dominant contribution is TADF (80%) at room temperature. Solution-processed bilayer OLEDs showed dual emission from the 3CC and a higher energyref. ref1 (M-X)LCT state. The white OLED showed very low efficiency (EQEmax = 0.78%) as expected for the low ΦPL of the complex. Modification of the diphosphine ligand with carbazole groups to form donor-acceptor ligands DCzDBFDP and DtBCzDBFDP resulted in complexes (DCzDBFDP)2Cu4I4 and (DtBCzBFDP)2Cu4I4 .ref. ref863 Compared to the original complex without carbazole groups (DBFDP)2Cu4I4 , the emission is blue-shifted (λPL = 480 nm) and narrower (FWHM is reduced from 95 to 60 nm). Suppression of the 3CC excited state in the two carbazole-containing complexes leads to an increased ΦPL from 5 to 46–65% and reduced ΔE ST from 160 to 70–100 meV. Non-doped solution-processed OLEDs showed EQEmax of up to 7.9% at CIE coordinates of (0.22, 0.43).
Moving to emitters with yet higher Cu content, a TADF tri-nuclear copper(I) complex has been reported with a diphosphine ligand (dppm) completing the coordination spheres of a trimeric pyrazolate core in (dppm)[(3,5-(CF3)2Pz)Cu]3 (Figure ).ref. ref864 The complex is a green emitter as a powder (λPL = 514 nm, ΦPL = 41%, τPL = 32.7 μs) and has a ΔE ST = 131 meV. The emissive excited state is predominantly 1MLCT (metal to dppm ligand), highlighting the importance of the diphosphine ligand in generating TADF emission from the Cu3Pz3 core.
Interesting vapochromic emission was observed in a tetranuclear Cu4Br4-based complex with carbazole-bridged diphosphine ligands, Cu4Br4(dppMeCz)2 (Figure ).ref. ref865 Two polymorphs of the cluster, 1G and 1Y, are either green (λPL = 512 nm, ΦPL = 8%, τPL = 8.9 and 295 μs) or yellow (λPL = 550 nm, ΦPL = 13.8%, τPL = 6.7 and 473 μs) emitters, respectively. In both complexes the Cu4Br4 core has the same geometry, with the difference between the polymorphs due only to the orientation of the ligand relative to the cores. Crystals of the complex can be induced to change polymorphs by exposure of 1G to hexane vapor or 1Y to acetonitrile vapor. The emission of 1G was determined to be phosphorescence, while 1Y had a strong TADF component to the emission.
An organometallic cationic Cu4 cluster has been prepared with diphosphine ligands that also bond to the Cu atoms through a Caryl-Cu bond to form [Cu4(PCP)3]BArF 4 (Figure ).ref. ref866 The cluster is a bright green emitter in both solution and as a powder (λPL = 518 nm, ΦPL = 50%, τPL = 9.8 μs as a powder) with moderately narrow FWHM of 58 nm attributed to the rigid nature of the cluster. The ΔE ST is 72 meV and TADF was determined to be responsible for 92% of the emission at room temperature. The solution-processed OLED showed an EQEmax = 11.2% at CIE coordinates of (0.305, 0.637), with moderate roll-off (EQE1000 ≈ 8.5%).
The crystals of two isomeric clusters with diphosphine and pyridine ligands, [Cu4I4(dppp)2(3-acepy)2] (CuL3 ) and [Cu4I4(dppp)2(4-acepy)2] (CuL4 ) (Figure ) emit TADF with ΔE ST = 35 meV for both complexes).ref. ref867 Both emitters show yellow emission with moderate efficiency (λPL of 562 and 580 nm, ΦPL of 26 and 30%), and short τPL of 2.5 and 10.8 μs, respectively.
Silver
Silver(I) complexes, containing another d10 metal, have also gained interest recently due to their ability to engender TADF characteristics. SOC is expected to increase moving down group 11 from copper to silver, opening the potential for enhanced T1 → S0 transitions that can compete with RISC for this precious metal. This enhanced SOC may explain why only a few Ag(I) TADF emitters have been reported compared to analogous copper complexes. Excited states in Ag(I) complexes are also primarily ligand-centered rather than MLCT, resulting in large ΔE ST and phosphorescence facilitated by the increased SOC unless the ligands are carefully designed. Compound [Ag(dppb)(PS)] ref. ref756 is the first reported TADF silver complex, although emission was demonstrated to be a mixture of TADF and phosphorescence (Figure ). Similar to the mixed emission mechanisms in copper complexes discussed above, TADF is dominant at RT while phosphorescence prevails at lower temperatures. Complex [Ag(dppb)(PS)] has a ΦPL of 32% and shows biexponential decay kinetics with τPL of 0.6 μs and 2.2 μs, likely associated with delayed fluorescence and phosphorescence. Owing to its poor solubility, no devices were reported. Numerous three- and four-coordinate Ag(I) complexes have since been reported that exhibit TADF. Selected examples are shown in Figure , and generally emit in the blue-to-green with moderate to good ΦPL in the solid state (decreasing in solution). We note that very few of these complexes have been used as emitters in OLEDs.
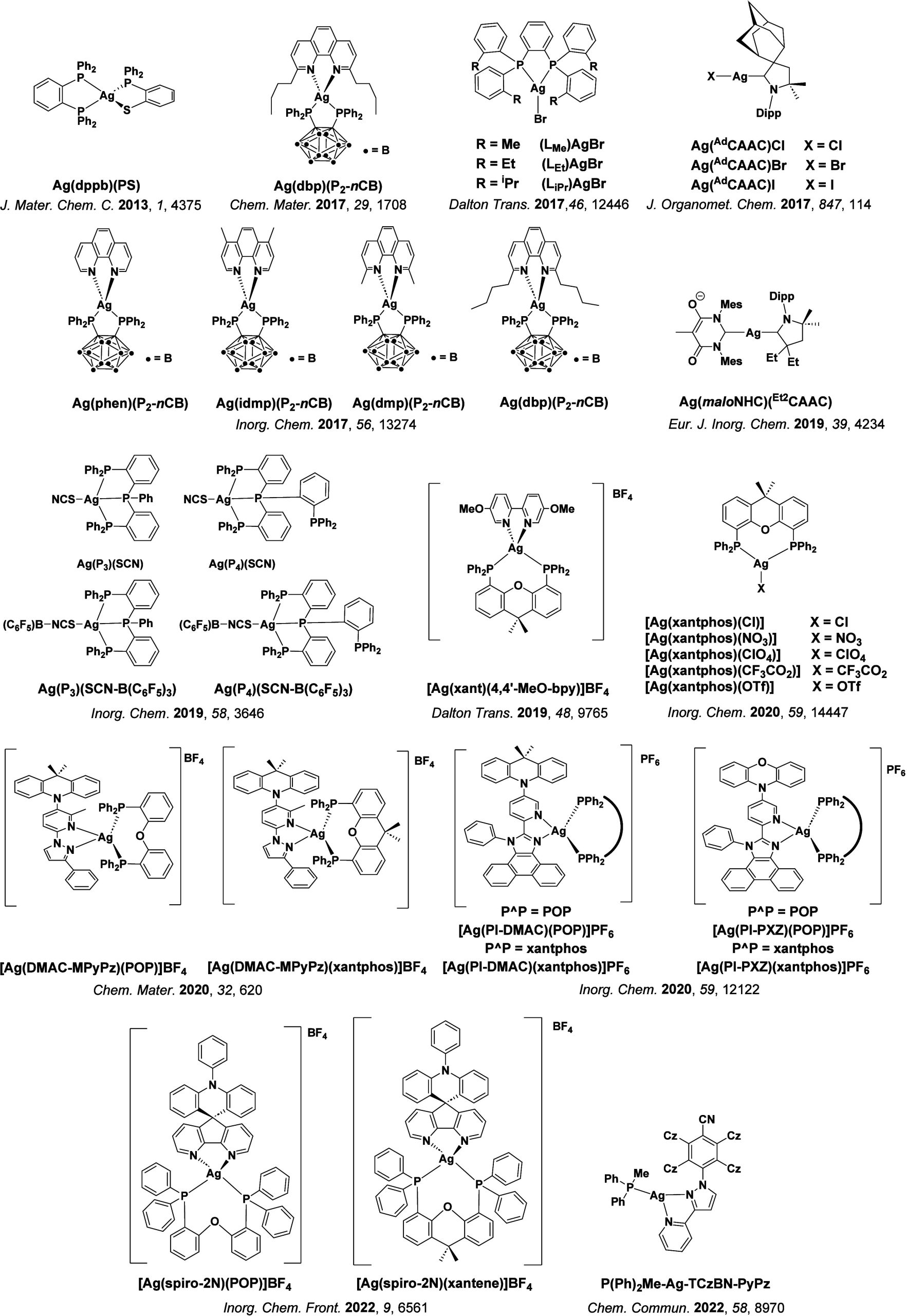
A family of complexes Ag(phen)(P2–nCB), Ag(idmp)(P2–nCB), Ag(dmp)(P2–nCB), and Ag(dbp)(P2–nCB) (Figure ) containing carborane ligands emit with λPL between 526 and 575 nm, and have powder ΦPL of between 36 and 100%.ref868,ref869 The complexes with the higher ΦPL use diimine ligands with increased steric bulk about the silver center, which reduces the capacity for the complex to distort its geometry in the excited state.
Complex [Ag(xant)(4,4′-MeO-bpy)]BF4 emits at λPL of 493 nm and has a ΦPL of 57.1% in DCM solution.ref. ref870 This is an increase in ΦPL over the analogous copper complex (vide supra), which was shown in theoretical studies to be due to the presence of additional low-lying non-emissive excited states in the copper complex.
A pair of silver complexes Ag(P3)(SCN) and Ag(P4)(SCN) (Figure ) containing tridentate phosphine ligands along with an isothiocyanate ligand have been prepared by Koshevoy and co-workers.ref. ref830 Ag(P3)(SCN) emits in the green (λPL = 538 nm, ΦPL = 32%) while Ag(P4)(SCN) showed interesting behavior with different crystalline forms (polymorphs and solvates) giving a range of emission colors (λPL from 525 to 630 nm) and ΦPL from 19 to 44%. Reaction of these complexes with B(C6F5)3 produced the isothiocyanatoborate complexes Ag(P3)(SCN-B(C6F5)3) and Ag(P4)(SCN-B(C6F5)3), which resulted in a blue-shift of the emission and a drop in the powder ΦPL. The emission of the powders is TADF in nature, with ΔE ST of 200 meV for Ag(P3)(SCN-B(C6F5)3) and 90 meV for Ag(P4)(SCN-B(C6F5)3).
Compounds (LMe)AgBr, (LEt)AgBr, and (LiPr)AgBr (Figure ), containing bidentate diphosphine ligands, emit in the sky-blue in both DCM solution and as powders.ref. ref871 The emission maxima of the solutions ranged from 492 to 499 nm with ΦPL of between 20 and 32%, while the powder emission was blue-shifted to 463 to 487 nm and considerably brighter with ΦPL increasing to 56 to 98%. In both solution and in the solid state the ΦPL increased with steric bulk of the P∧P ligand.
A number of interesting 4-coordinate silver(I) complexes exist where coordination of a silver(I)-diphosphine moiety to a separate donor-acceptor ligand turns on TADF from the ligand.ref760,ref872 Ag(DMAC-MPyPz)(POP) and Ag(DMAC-MPyPz)(xant) ref. ref760 (Figure ) emit at λPL of 502 and 500 nm in DCM, which are blue-shifted to 472 and 471 nm in 15 wt% doped films in PMMA, with ΦPL of up to 60% in solution and 99% in the same films, all respectively. The TADF nature of the emission was supported by the magnitude of the emission lifetime (τd = 6.3 and 6.5 μs, in PMMA) and a significant reduction in the ΔE ST (from 470 meV for the ligand to 170 meV and 150 meV in the complexes, respectively). Similar materials with D-A TADF ligands Ag(PI-DMAC)(POP), Ag(PI-DMAC)(xant)¸ Ag(PI-PXZ)(POP), and Ag(PI-PXZ)(xant) ref. ref872 all showed green to yellow emission in DCM (λPL = 502 to 533 nm) that red-shifted in 10 wt% doped films in DPEPO. Since the TADF in these compounds is ligand centered, there are few relevant geometric changes about the Ag(I) center and thus this is no longer a major contributor to non-radiative decay.ref. ref760 Solution-processed OLEDs with Ag(PI-PXZ)(POP) showed an EQEmax of 8.76% at CIE coordinates of (0.45, 0.62).
It has been demonstrated that k RISC can be dramatically increased when an already TADF-active donor-acceptor compound is coordinated to silver ions.ref. ref873 Both the free ligand TCzBN-PyPz and the silver complex P(Ph)2Me-Ag-TCzBN-PyPz (Figure ) show similar green emission (λPL = 536 and 522 nm, respectively), with the free ligand showing higher ΦPL (36 and 29%). The major difference in photophysics is in the τd, which decreased from 2074 μs in the ligand to 0.59 μs in the complex. This change is largely explained by the relative magnitudes of ΔE ST (160 meV for the ligand and 0.03 for the complex) as well as the much enhanced SOC from the Ag ion.ref. ref874 Similar in concept, [Ag(spiro-2N)(POP)]BF4 and [Ag(spiro-2N)(xanthene)]BF4 contain a spiro-type TADF emitter coordinated to Ag(I).ref. ref875 In 10 wt% doped films in PMMA both complexes exhibit strong blue-green emission, with λPL of 486 nm and ΦPL of 65% for [Ag(spiro-2N)(POP)]BF4 and 495 nm and 74% for [Ag(spiro-2N)(xanthene)]BF4 . DFT calculations revealed that the LUMO energy of the free ligand is considerably stabilized upon coordination with the Ag(I) center, with a concomitant reduction in calculated ΔE ST from 270 meV in the free ligand to 10 meV for [Ag(spiro-2N)(POP)]BF4 and 13 meV for [Ag(spiro-2N)(xanthene)]BF4 , in good agreement with the experimental ΔE ST of 90 and 50 meV, respectively. As with the previous study, the lifetime of the free ligand is significantly shortened upon coordination to the metal, accelerating from 539 ms (1 wt% doped phenyl benzoate film) in and also showing an afterglow of 5 s, to just 5.3 and 5.8 μs for [Ag(spiro-2N)(POP)]BF4 and [Ag(spiro-2N)(xanthene)]BF4 , respectively, in 10 wt% doped PMMA.
Several groups have investigated linear Ag(I) carbene complexes, analogues to corresponding high-performance Cu(I) CMAs. The first examples of these were a series of carbene-halide complexes Ag(AdCAAC)X (X = Cl, Br, I, Figure ).ref. ref876 These complexes emit at λPL of 432 to 443 nm and have poor ΦPL of 0.5 to 5% in the solid state, and were not emissive in solution. The TADF behavior of these complexes is supported by the observation of dual photoluminescence consisting of a prompt ns emission and long-lived microsecond emission. A related carbene ligand Et2CAAC was later used in combination with an unusual anionic carbene ligand maloNHC in the zwitterionic complex Ag(maloNHC)(Et2CAAC). This complexes was weakly blue emissive (ΦPL < 1%) as both a powder and a 5 wt% doped polystyrene film.ref. ref877 Beyond these complexes, a series of highly emissive linear silver CMA complexes are discussed together with other coinage metal CMA complexes further below.
While the previous examples of Ag(I) complexes are all mononuclear, there are also a range of reported dinuclear (Figure ) or larger (Figure ) TADF silver complexes. The interaction between the metal centers in these multinuclear silver(I) complexes can destabilize the antibonding d-orbitals of the silver(I) ions, thus increasing the energy of metal-centered (MC) states to such an extent that the emissive MLCT state is the lowest in energy.ref878,ref879 The first reported multinuclear silver(I) TADF emitters were a series of mixed phosphine/halide complexes with a bridging 1,2,4,5-tetrakis(diphenylphosphino)benzene ligand, [Ag(PPh3)(X)]2(tpbz) (Figure ).ref. ref880 These complexes were not emissive in the solution, but showed green emission (λPL = 517–531 nm) with ΦPL of up to 40% and τd of between 4.0–5.3 μs in 5 wt% doped films in PMMA. The emission as powders is blue-shifted (λPL = 471–495 nm) and the ΦPL are much higher (ΦPL = 74–98%). Interestingly there was little impact on the τPL when moving to iodide ions despite the expected increased SOC (X=Cl, τPL = 3.0 μs; X=I, τPL = 2.5 μs). As powders, all complexes have an ΔE ST < 200 meV. The same bridging ligand was used to prepare a dinuclear complex with the silver atoms supported by a diphosphinocarborane ligand [Ag2(tpbz)(P2–nCB)2].ref. ref881 This complex emits strongly at λPL of 555 nm (ΦPL = 70%) as a powder and has a small ΔE ST of 59 meV.
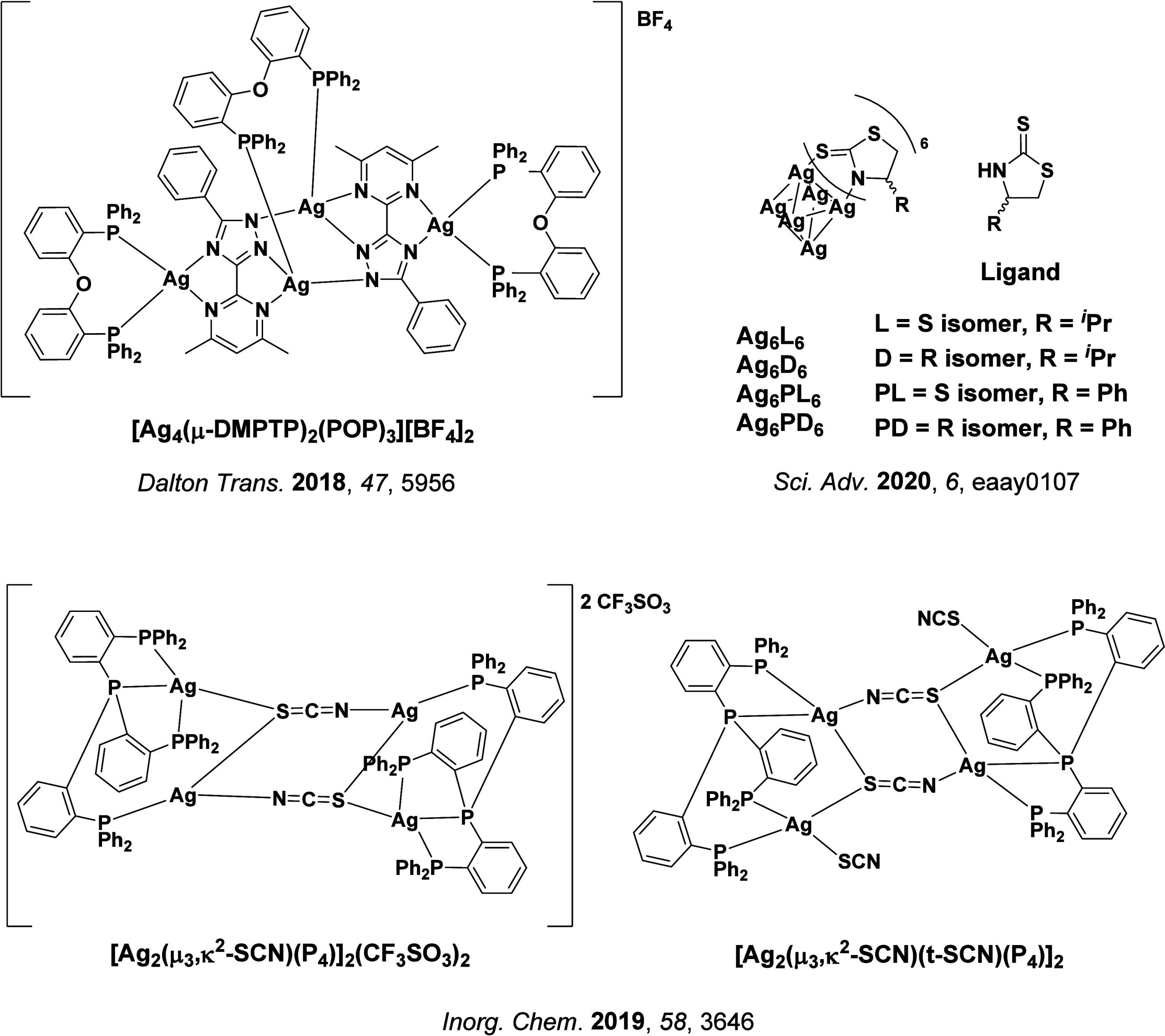
A halide bridged dinuclear CAAC complex [Ag(Et2CAAC)Cl]2 ref. ref876 (Figure ) showed weak blue emission (λPL = 454 nm, ΦPL = 5%), with a τd of 18.9 μs. Isothiocyanates can similarly act as bridging ligands, as in the case of [Ag(μ2-κ2-SCN)(dppb)]2 , which emits at λPL of 505 nm (ΦPL = 35%, τd = 12 μs) as a powder and has a small calculated ΔE ST of 90 meV.ref. ref830 A considerably different bonding motif of similar subunits is adopted in [Ag2(Py3P)3(SCN)2],ref. ref882 which contains two close lying silver(I) atoms bridged by three pyridylphosphine ligands, while the isothiocyanates are terminal. This complex exhibited a solvent-induced enhancement of the solid-state emission. The desiccated complex emits at λPL of 469 nm and has a low ΦPL of 16%, but upon exposure to CH2Cl2 or CHCl3 vapors, ΦPL increases to ≈ 70% along with a small (ca. 10 nm) red-shift in the emission. This process was reversible through heating the complex to 130 °C. Bridging isothiocyanate ligands are also present in tetranuclear silver(I) complexesref. ref830 [Ag2(μ3,κ2-SCN)(P4)]2(CF3SO3)2 and [Ag2(μ3,κ2-SCN)(t-SCN)(P4)]2 (Figure ) that show sky-blue emission (λPL = 468 to 475 nm) and have moderate ΦPL of up to 43% and τd of between 3.7 to 6.4 μs as powders.
A large series of complexes with phosphine ligands and bridging (pseudo-)halide ligands have been prepared that tune the emission color from sky blue to red,ref. ref883 with a subset of these complexes exhibiting TADF. The dppb-terminated complexes [Ag(dppb)(X)]2 (Figure ) show sky blue-to-green emission in 5 wt% doped films in PMMA (λPL = 476–515 nm) and have τPL ranging from 6 to 63 μs and ΦPL of up to 53%. The ΔE ST of the triflate-bridged complex was experimentally found to be 74 meV.
Moving beyond dinuclear Ag(I) complexes, the tetranuclear complex [Ag4(μ-DMPTP)2(POP)3][BF4]2 .ref. ref878 shows very bright green emission (λPL = 527 nm, ΦPL = 76%) and has a very short lifetime (τPL = 0.65 μs), associated with its small ΔE ST of 80 meV.
Lastly there are examples of reported hexameric clusters Ag6L6 ref. ref884 containing enantiopure thiazolidine-2-thione ligands that show TADF (Figure ). These clusters emit at λPL ranging from 556 to 575 nm in the solid state with ΦPL of between 56 and 95% and τPL of 16 to 18 μs at room temperature. The ΔE ST are 96 meV for Ag6L6 and 41 meV for Ag6PL6 . The chiral clusters also show moderate luminescence dissymmetry factors (g lum = ± 4.42 × 10–3) and are the first silver chiral TADF emitters.
Gold
Unlike copper and silver, there are examples of both emissive d10 gold(I) and d8 gold(III) complexes, many of which are TADF-active. The very first reports of TADF from gold-based materials were based on nano-clusters,ref884−ref885ref886 however this review will only focus on organometallic gold complexes. Further down group 11, the larger SOC of gold in comparison to the lighter coinage metals results in many emissive gold complexes showing phosphorescence rather than TADF.ref. ref887 Here we summarize gold complexes with emission that has been explicitly identified as TADF.
As with copper and silver complexes, there are a number of tri- and tetra-coordinated gold(I) complexes that show TADF (Figure ). Indeed, many of these examples have the same ligand environment as copper(I) TADF complexes, and similar to these the low-lying excited states in the gold(I) complexes can best be described as MLCT states with the gold is directly involved in the transition. The first reported TADF gold(I) complex was Au(dppb)(PS),ref. ref756 a gold analogue of a known copper(I) TADF emitter.ref. ref756 The complex emits in the orange as a powder (λPL = 610 nm, ΦPL = 12%, τPL = 1.66 μs), with the emission significantly red-shifted (90 nm) compared to the analogous copper(I) complex.
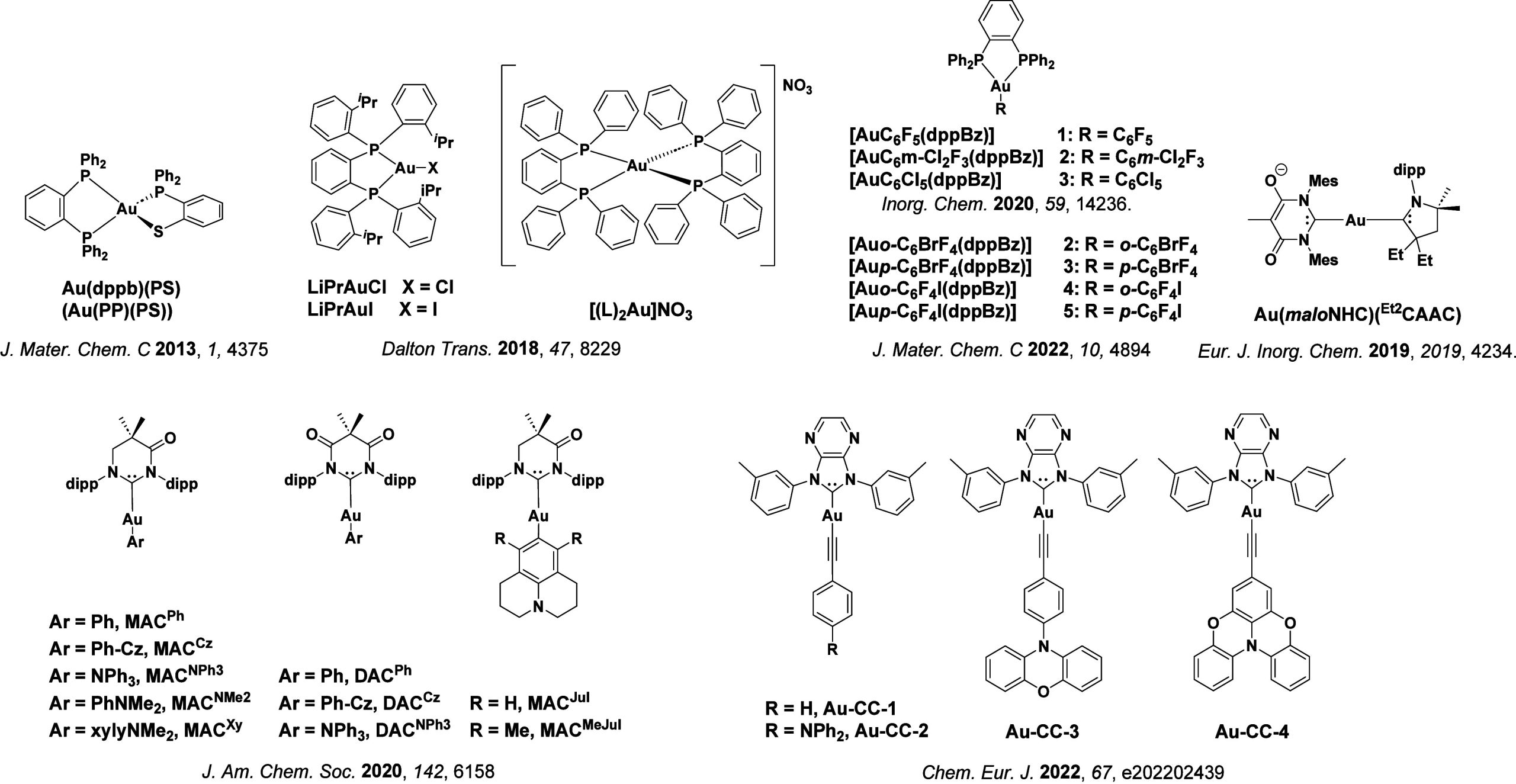
A pair of trigonal planar complexes (LiPr)AuX and a cationic tetrahedral complex [(dppb)2Au](NO3) (Figure ) all show very bright sky-blue to green emission as crystals (λPL = 485–558 nm, ΦPL > 82%).ref. ref888 The microsecond-long lifetimes (τPL = 3.8–13 μs) and small ΔE ST of 80 to 120 meV support the assignment to TADF. The complexes are much less emissive in 2-MeTHF; the neutral complexes are weak orange emitters (λPL = 596 to 607 nm, ΦPL = 2 to 4%) while [(dppb)2Au](NO3) is not emissive at all in solution. This red-shift and lower ΦPL are attributed to the more polar solvent stabilizing the CT excited state in the former and the faster k nr resulting from increased vibrational motion in fluid solution for the latter.
The trigonal planar series of complexes [(dppBz)Au(Ar)] (Figure ) contain perhalophenyl ligands, and are yellow-emissive (λPL = 545 to 560 nm, ΦPL = 11 to 29%, τPL = 10 to 21 μs) with ΔE ST ranging from 58 to 144 meV.ref. ref889 A subsequent study explored the impact of the addition of bromine and iodine atoms to the perfluorophenyl group at the ortho and para positions to determine if the presence of the additional heavy atom influenced the properties of the gold complexes. Ultimately there was little impact on the emission properties of the complexes, as the central gold atom already ensured very strong SOC that was not significantly increased through the use of ancillary heavy halogen atoms.ref. ref890
The linear gold(I) carbene complex Au(maloNHC)(Et2CAAC) (Figure ) contains an unusual zwitterionic mixed carbene structure and exhibits weak sky-blue emission as powder (λPL = 461 nm, ΦPL = 2.7%). In 5 wt% doped PS films the emission properties are similar (λPL = 464 nm, ΦPL = 3%), and biexponential decay kinetics typical of TADF were reported (τPL = 1.5 and 22 μs in the PS film).ref. ref877
The emission of a series of linear carbene-aryl gold(I) complexes (DACaryl and MACaryl , named 1a–c and 2a–e in the original work, Figure ) can be tuned from blue to orange (λPL = 460 to 620 nm in 1 wt% doped PS films, with ΦPL of up to 77%) as a function of the structure of the aryl group.ref. ref891 The complexes with either a phenyl ring or a phenyl carbazole for the aryl group (DACPh , DACCz , MACPh , and MACCz ) have MLCT excited states that emit via phosphorescence. In contrast, the remaining complexes have LLCT excited states that lead to emission via TADF. Most of the complexes showed only modest luminescent efficiencies (ΦPL < 40%), attributed to high non-radiative decay rates resulting from free rotation around the Au-Caryl bond. Addition of two methyl groups ortho to the Au-Caryl bond in either the dimethylaniline ligand in MACNMe2 or the julolidine ligand in MACJul blocks this rotation around the Au-Caryl bond and locks these two ligands in a co-planar configuration. The resulting reduction in the non-radiative decay rate for the complexes MACXy and MACMeJul leads to a significant increase in the ΦPL to between 35–75%. The ΔE ST range between 86 and 203 meV for all the TADF emissive complexes.
A series of four linear carbene-alkynyl complexes (1–4 in the original work and renamed Au-CC-1 to Au-CC-4 here, Figure ) are bright blue to green emitters.ref. ref892 The complexes have ΦPL ranging from 36 to 76%, and have τPL of up to 60 μs in 5% doped films in PMMA with ΔE ST ranging from 82 to 162 meV. The green solution-processed OLEDs with Au-CC-2 showed an EQEmax of 20.4% at CIE coordinates of (0.32, 0.54), however the efficiency roll-off was severe with an EQE1000 of 9.7%.
MR-TADF emitters are an emerging class of TADF materials that have garnered great interest (see Section sec11 ). One critical issue with MR-TADF emitters is their relatively slow k RISC compared to D-A analogues. In an effort to increase RISC several studies have added heavy atoms to the skeleton, including coordination to gold atoms.ref180,ref191,ref893 Cai et al. reported a series of MR-TADF emitters with a coordinated gold(I) NHC moiety.ref. ref894 (SIPr)AuBN, (BzIPr)AuBN, (PyIPr)AuBN, (PzIPr)AuBN, (Ipr)AuBN, and (BzIPr)AuBNO (Figure ) all have very high ΦPL of up to 99%, and show narrowband emission (FWHM of 30–37 nm) in both thin films (2 wt% in PMMA) and in THF. The OLEDs with these Au(I)-MR-TADF complexes show good performance, with (BzIPr)AuBN achieving an EQEmax of 30.3% at CIE coordinates of (0.16, 0.68), with very low efficiency roll-off (EQE1000 = 28.1%). Further, this device showed good stability with an LT60 of 1210 h at 1000 cd m–2.ref. ref894
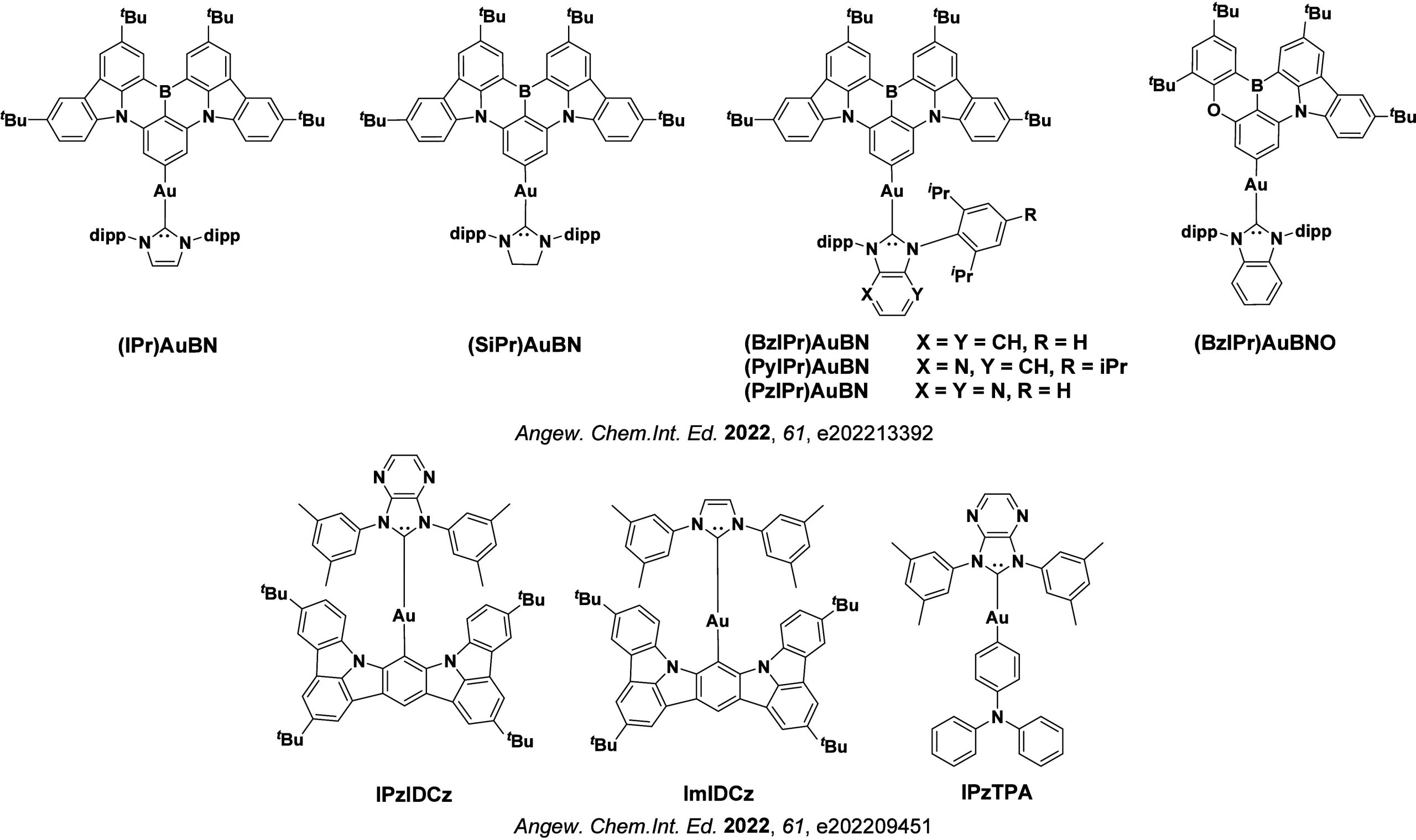
Feng et al. have also reported complexes with gold(I) carbene moieties coordinated to the narrowband fluorescent meta-diindolocarbazole coreref161,ref895. IPzIDCz emits by TADF while ImIDCz is phosphorescent, the mechanism controlled by the carbene ligand used (Figure ).ref. ref896 IPzIDCz shows intense green emission in 10 wt% doped films in DPEPO, with ΦPL = 66% and τPL = 2.9 μs. A green OLED with IPzIDCz (1 wt% doped in DMIC-TRZ) showed an EQEmax = 23.9% at CIE coordinates of (0.37, 0.57) and had only minimal efficiency roll-off (EQE = 20% at 47 000 cd m–2) yet, surprisingly, only a moderate LT95 of 27.4 h at 1000 cd m–2. These results suggest that coordination of gold(I) carbenes to MR-TADF compounds is a promising tactic for high performance OLEDs.
Unlike Au(I) complexes, all TADF Au(III) emitters are 4-coordinate complexes containing either a di-anionic tridentate pincer ligand with a monodentate halide, aryl, alkynyl, or amide ligand (Figure ), or a tri-anionic tetradentate ligand (Figure ). The first reported gold(III) TADF emitters were a series of complexes based on a diphenylpyrazine pincer ligand.ref. ref758 These complexes are all weakly emissive in the neat film and in DCM (ΦPL < 8%), with highly structured emission ranging from green to orange suggestive of emission from a LC state. The first OLEDs containing a gold(III) emitter were reported from complexes made with a diphenylpyridine pincer ligand.ref. ref897 Of the family of eight complexes prepared, the most promising contain triphenylamine groups, (C∧N∧C)Au(PhN(Ph)2) and (CF2 ∧NOEt∧CF2)Au(PhN(Ph)2). In 4 wt% doped films in PMMA these two compounds emit at λPL of 523 and 517 nm, have short τd of 1.35 and 0.72 μs, and high ΦPL of 66 and 84%, respectively. Solution-processed OLEDs with (C∧N∧C)Au(PhN(Ph)2) and (CF2 ∧NOEt∧CF2)Au(PhN(Ph)2) showed EQEmax of 14.8% and 23.8% (and efficiency roll-off of only 1 or 31% at 1000 cd m–2) at CIE coordinates of (0.32,0.55) and (0.27,0.51), all respectively. The efficiency roll-off for the latter was reduced to 8% when a larger band gap host (PYD2) was used but the EQEmax simultaneously dropped to 15.7%.
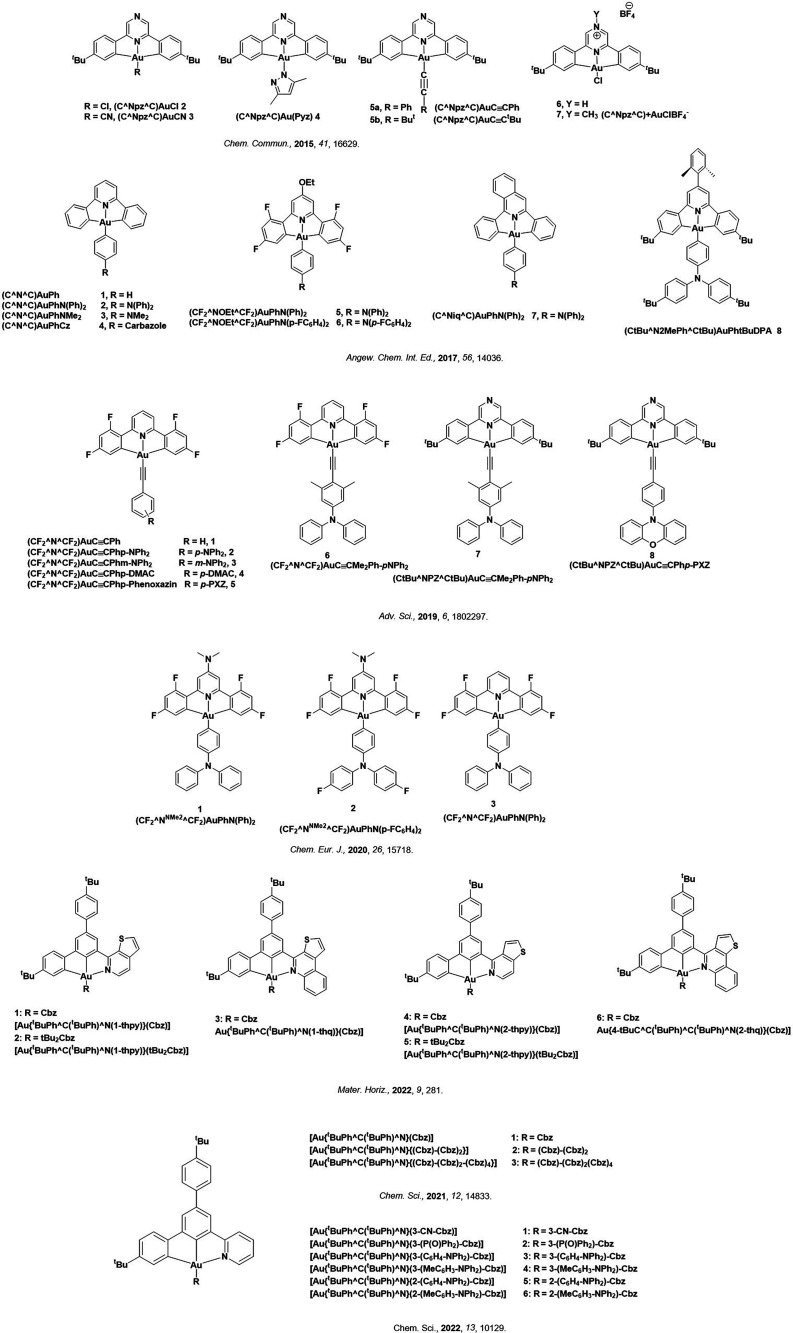
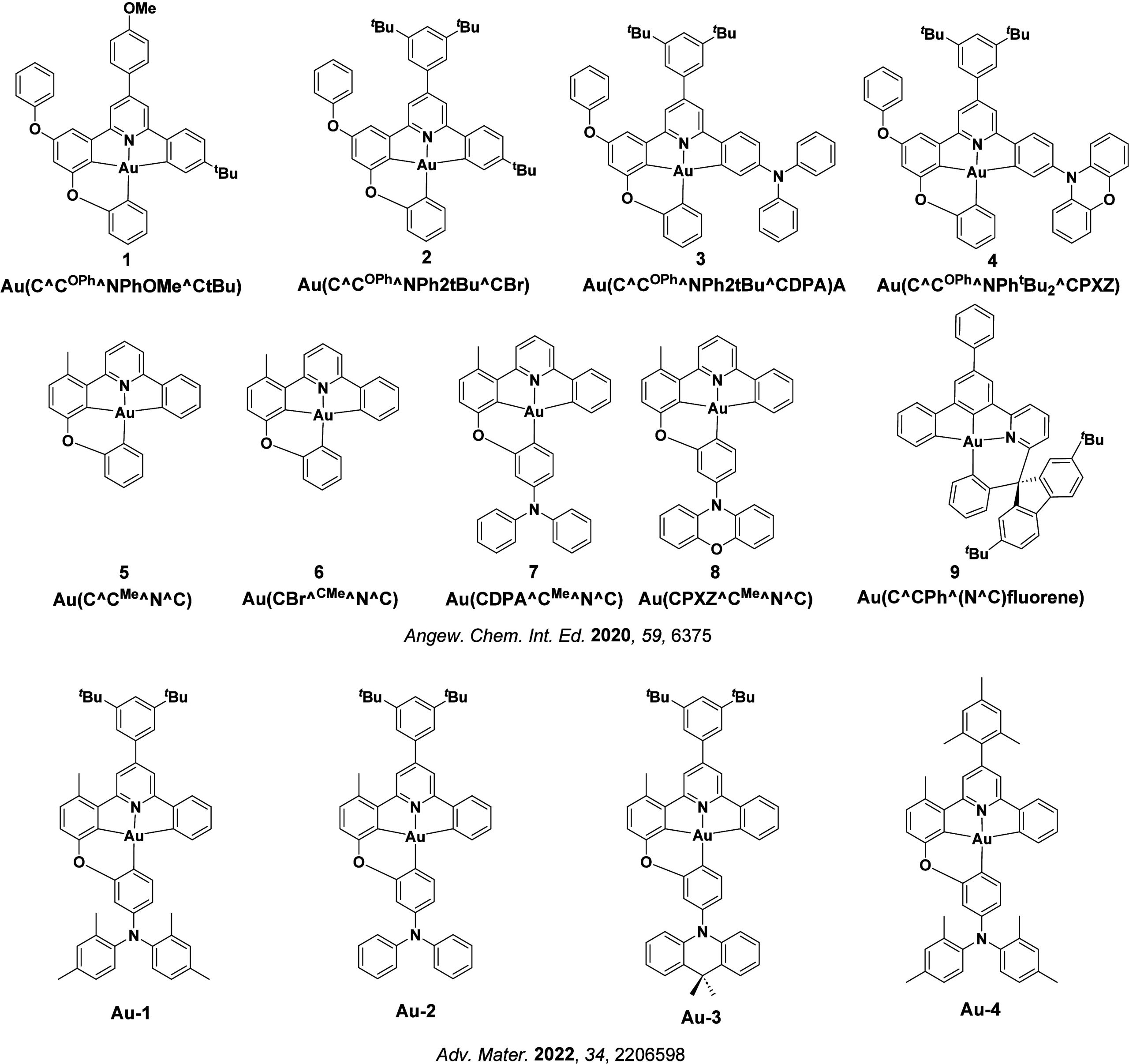
A similar set of complexes, (CF2 ∧NNme2 ∧CF2)Au(PhN(Ph)2) and (CF2 ∧NNme2 ∧CF2)Au(PhN(p-FC6H4)2) (Figure ), display a blue-shifted emission in 4 wt% doped PMMA films (λPL of 484 and 470 nm, ΦPL of 34 and 82%, and τPL = 0.97 and 0.95 μs, all respectively) due to the increased electron density on the central pyridine ring of the acceptor pincer ligand associated with the donating dimethylamine group. Analogue (CF2 ∧N∧CF2)Au(PhN(Ph)2) without this substituent exhibits a red-shifted emission (λPL = 550 nm, ΦPL = 81% and τPL = 0.69 μs).ref. ref898 The solution-processed OLEDs with the sky-blue emitters showed reasonably narrow electroluminescence (FWHM = 64–67 nm) with CIE coordinates of (0.16, 0.25) and (0.16, 0.23) and EQEmax of 6.8 (CF2 ∧NNme2 ∧CF2)Au(PhN(Ph)2) and 15.3% (CF2 ∧NNme2 ∧CF2)Au(PhN(p-FC6H4)2) although with considerable efficiency roll-off of 63 and 35% at 100 cd m–2, respectively. The solution-processed OLED with (CF2 ∧N∧CF2)Au(PhN(Ph)2) showed an EQEmax of 24.3% at CIE coordinates of (0.35, 0.56) and moderate efficiency roll-off of 24% 1000 cd m–2.
A series of green-to-yellow-emitting gold(III) complexes containing diphenylpyridine pincer ligands and substituted alkynyl ligands ((CF2 ∧N∧CF2)AuC≡CPh) and others, Figure ) have ΦPL up to 88% and τd from 0.33 to 1.46 μs in solution and doped PMMA films.ref. ref899 Calculations indicated that the lowest excited states are LLCT in nature where a diphenylamine (or a related ring-closed system such as phenoxazine or acridan) acts as the donor and the pincer ligand is the acceptor. The calculated ΔE ST is dependent on the conformation of the arylamine with respect to the plane of the pincer ligand bound to the gold center, and varies from 2 to 330 meV, suggesting that certain conformers are TADF-active. Of the OLEDs tested, the one with (CF2 ∧N∧CF2)AuC≡C-Me2Ph-pNPh2) showed the best performance with an EQEmax of 23.4% at CIE coordinates of (0.40, 0.55,) and an efficiency roll-off of 5% at 1000 cd m–2.ref. ref899 The OLEDs were also relatively stable with LT95 = 500 h at 100 cd m–2.
Beyond gold(III) aryl and acetylide complexes there are also gold(III) complexes bearing amide ligands. Yellow-to-red-emitting complexes [Au{tBuPh∧C(tBuPh)∧N(1-thpy)}(Cbz)] and [Au{tBuPh∧C(tBuPh)∧N(2-thpy)}(Cbz)] (λPL = 554 and 557 nm, respectively, Figure ) are examples that employ pincer ligands based on isomeric thienopyridine and thienoquinoline in combination with a carbazolate ligands.ref. ref900 In 5 wt% doped films in mCP these compounds have ΦPL of 83 and 81% and τd of 4 and 7 μs, respectively. The best performing OLEDs with [Au{tBuPh∧C(tBuPh)∧N(1-thpy)}(Cbz)] showed EQEmax of 14.5% at CIE coordinates of (0.60, 0.40), and also showed excellent device stability (LT70 = 63 258 h at 100 cd m–2).
Yam and co-workers reported a series of highly efficient TADF gold(III) complexes containing carbazolate donor dendrons.ref. ref901 A reference gold(III) TADF complex [Au{tBuPh∧C(tBuPh)∧N}(Cbz)] (G0 ), the first-generation dendrimer [Au{tBuPh∧C(tBuPh)∧N}{(Cbz)-(Cbz)2}] (G1 ), and the second-generation dendrimer [Au{tBuPh∧C(tBuPh)∧N}{(Cbz)-(Cbz)2-(Cbz)4}] (G2 , Figure ) emit at λPL of 547, 532, and 535 nm in 10 wt% doped mCP films. The dendrimer complexes G0 , G1 , and G2 have ΦPL of 82, 74, and 75% and τPL of 3.5, 1.2, and 1.4 μs, respectively. The solution-processed OLED based on [Au{tBuPh∧C(tBuPh)∧N}{(Cbz)-(Cbz)2-(Cbz)4}] showed an EQEmax of 15.8% at CIE coordinates of (0.38, 0.57).ref. ref901 Addition of triphenylamine groups to the carbazolate ligand of G0 results in a family of six further complexes showing bright green-yellow emission in 10 wt% doped films in mCP. Of these complexes, [Au{tBuPh∧C(tBuPh)∧N}(2-(MeC6H3-NPh2)-Cbz)] showed the highest ΦPL of 79% at a λPL of 535 nm, and has a τPL of 5.9 μs.ref. ref902
Finally, Zhou et al. explored gold(III) complexes containing a single tetradentate ligand, prepared via microwave synthesis.ref. ref903 Three of these complexes, Au(C∧COPh ∧NPh2tBu2 ∧CPXZ), Au(CDPA∧CMe ∧N∧C), and Au(CPXZ∧CMe ∧N∧C) (Figure ) show TADF (λPL = 520–568 nm, ΦPL = 71–89% and τd = 1.69–2.54 μs). Green OLEDs with Au(CDPA∧CMe ∧N∧C) and Au(CPXZ∧CMe ∧N∧C) showed EQEmax of 23 and 20% at CIE coordinates of (0.26, 0.54) and (0.34, 0.56), respectively. The OLED with Au(C∧COph ∧NPhtBu2 ∧CPXZ) showed an EQEmax of 25% with CIE coordinates of (0.43, 0.54), and showed low efficiency roll-off (EQE1000 = 22%) with very good stability (LT95 = 5 280 h at 100 cd m–2).
Zhou et al. later prepared another series of four green-emitting TADF tetradentate gold(III) complexes with trianionic (C∧C∧N∧C) ligands that showed excellent photophysical properties.ref. ref759 Complexes Au-1, Au-2, Au-3, and Au-4 emit between λPL of 525 to 547 nm with ΦPL of up to 88% in toluene. Doped at 4 wt% in DPEPO/TCTA films (1:1 host), these complexes emit between λPL of 513 to 530 nm with ΦPL up to 99%. The τd are between 0.47 and 0.69 μs in both toluene and doped films, although Au-3 alone is not TADF-active. The OLEDs with Au-1, Au-2, and Au-4 are bright and efficient with EQEmax > 24%, reaching 27.3% at CIE coordinates of (0.36, 0.60) Au-4.the devices showed relatively low efficiency roll-off of < 28% for all devices, and their stabilities are outstanding with the longest reported device lifetimes of any metal-based TADF OLEDs. For instance, the device with Au-1 had an LT90 of 128,864 h at 100 cd m–2. These results confirm that TADF gold(III) emitters are a very promising class of emitter materials for OLEDs.
Carbene Metal Amides (CMAs)
CMA complexes (Figure and Figure ) have rapidly come to the fore as arguably the most promising class of TADF coinage metal complexes. The pioneering work of Di et al. in 2017 demonstrated outstanding performance for solution-processed OLEDs with Au(I) CMA complexes,ref. ref194 and since this first study there has been a growing number of reported coinage metal CMA TADF emitters. The emissive excited states in these complexes are best characterized as LLCT, with the metal acting as a bridge between the amide donor and the carbene acceptor, but also contributing to SOC and RISC.
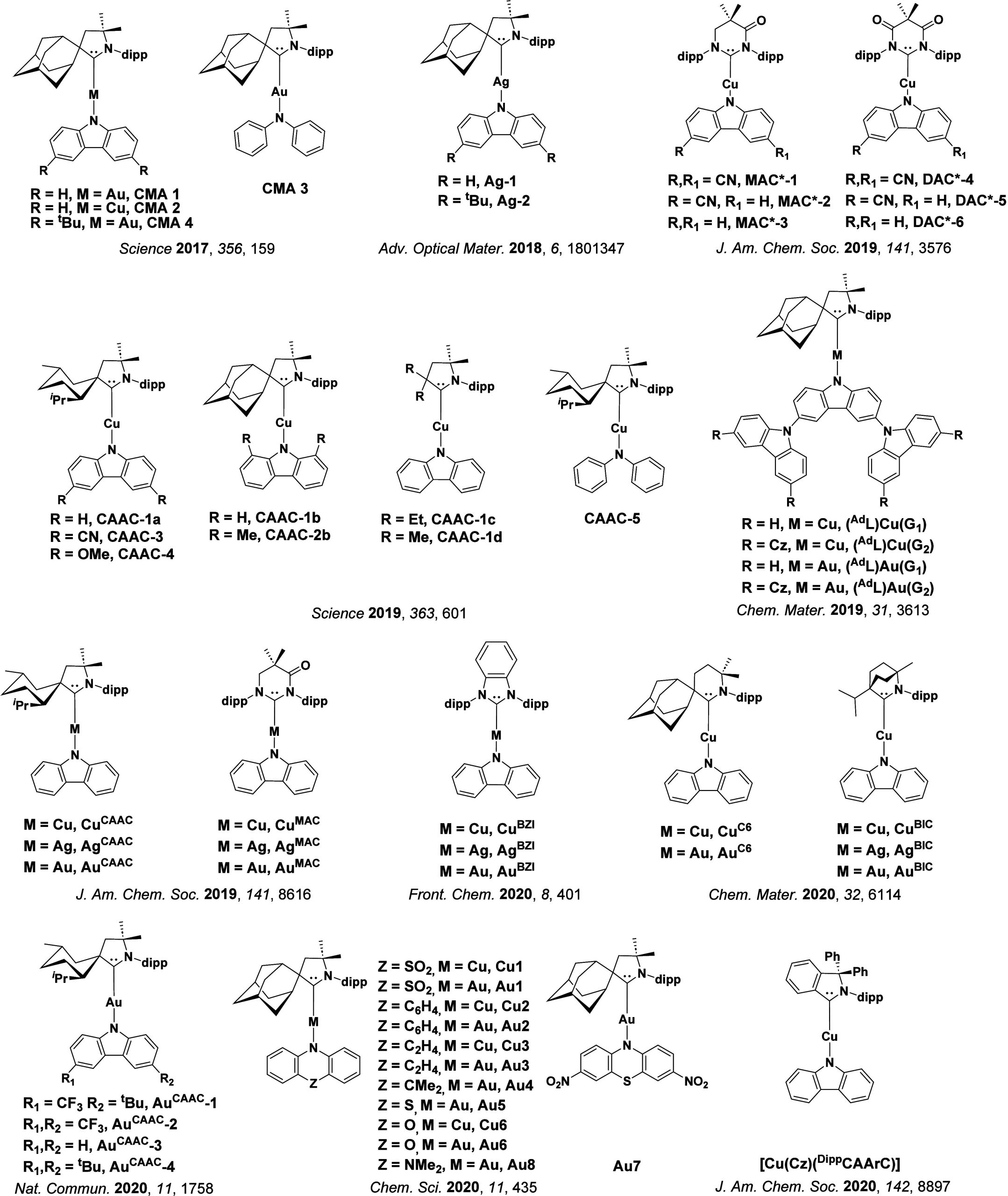
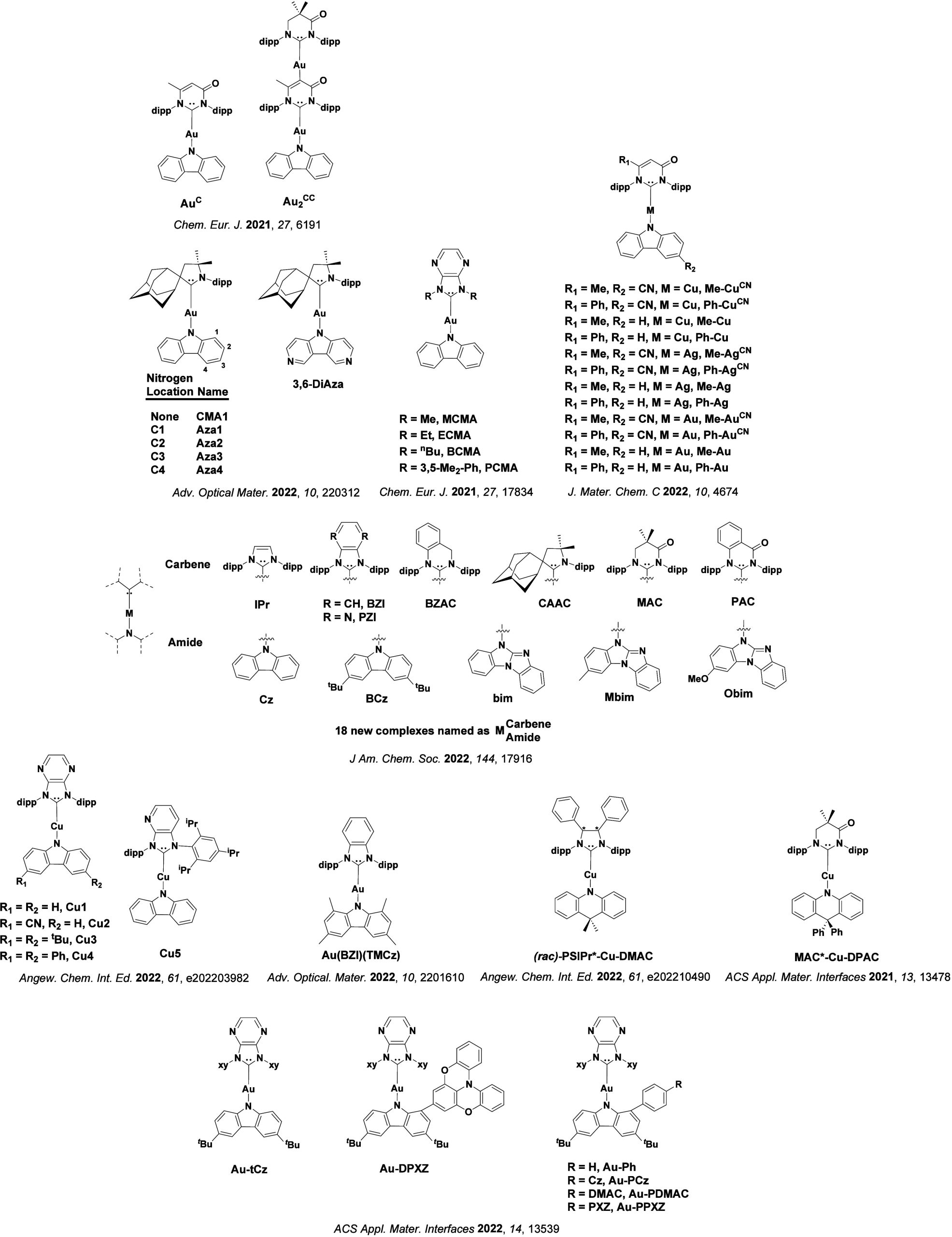
The first report of TADF CMA complexes contained an adamantyl-substituted cyclic alkyl amino carbene (CAAC) ligand as part of four separate gold and copper compounds.ref. ref194 These complexes showed green-to-yellow emission with near unity ΦPL in the solid state and very fast emission lifetimes (τPL = 0.35 μs). Solution-processed OLEDs with the gold emitter CMA 4 (Figure ) showed outstanding performance, with EQEmax of 27.5% at CIE coordinates of (0.26, 0.48). The devices with CMA 3 and CMA 1 showed similarly impressive EQEmax of 17.9% and 26.3% respectively, while copper-containing CMA 2 showed somewhat lower EQEmax of 9.7%. Impressively, the devices with all four complexes showed excellent efficiency roll-off of < 5% at 1000 cd m–2. It was proposed that rotation between the two ligands in the excited state modified singlet and triplet energies to give a negative ΔE ST, which the authors rationalized to explain both the short lifetimes and high ΦPL. However, the TD-DFT calculations upon which this conclusion was built were later found to be flawed, and it was later demonstrated that these complexes possess near-zero ΔE ST.ref195,ref196 Building on from this work, vacuum-deposited devices using CMA 1 showed slightly improved EQEmax of 26.9% at CIE coordinates of (0.24, 0.43), with efficiency roll-off at 1000 cd m–2 of only 7%. Non-doped device of the same showed impressive EQEmax of 23.1% at CIE coordinates of (0.24, 0.46) with an efficiency roll-off of 5% at 1000 cd m–2, making these some of the best performing non-doped TADF OLEDs to date.ref. ref904
Silver-containing complexes Ag-1 and Ag-2 (Figure ) show similar photophysical properties to their gold analogues CMA-1 and CMA-4,ref. ref905 emitting at λPL of 521 and 546 nm in toluene and having ΦPL of 74 and 55% with short τd of 0.46 and 0.305 μs, all respectively. These complexes were used in both solution-processed and vacuum-deposited devices. The solution-processed device with Ag-2 showed an EQEmax of 11.0% at CIE coordinates of (0.36, 0.56), with the EQE1000 decreasing to 8.2%. The vacuum-deposited device of the same showed an improved EQEmax of 13.7% at similar CIE coordinates of (0.31, 0.50), retaining a higher EQE1000 of 10.0%. Devices with Ag-1 showed much poorer performance with EQEmax < 4% for both solution-processed and vacuum-deposited devices, demonstrating the sensitivity of device performance to the choice of ligand in these CMA emitters.
Focusing on copper CMA complexes, Hamze et al. explored the impact of different carbenes and substitution of the carbazolate group in eight new complexes.ref. ref906 Decreasing the steric bulk of the carbene ligand (CAAC-1c and CAAC-1d, Figure ) results in increased non-radiative decay, manifesting in a lower ΦPL (decreasing from 56 to 11%, for CAAC-1c and CAAC-1d). Addition of electron-withdrawing CN groups to the carbazolide ligand in CAAC-3 results in localization of the excited state onto the carbazole ligand and fluorescence from a ligand-centered state (λPL = 428 nm in 2-MeTHF). Addition of electron-donating OMe groups to the carbazolide ligand results in a red-shift in the emission by destabilizing the HOMO of the donor carbazole group. Complexes with bulky ligands showed near unity ΦPL as 1 wt% doped films in polystyrene, with emission ranging from λPL = 426 to 558 nm. The OLED with CAAC-1a showed a leading EQEmax of 9.0% at λEL of 460 nm.ref. ref906
Six-membered heterocyclic carbenes MAC* and DAC* were used in a series of six new copper CMA complexes, MAC*-1–3 and DAC*-4–6 (Figure ).ref. ref907 The move from the monocarbonyl MAC ligand to the dicarbonyl DAC ligand resulted in stabilization of the LUMO by ≈ 1 eV, resulting in red-shifted emission for the DAC complexes. The addition of one or two CN groups to the carbazole ligand stabilized the HOMO by up to 0.5 eV, resulting in a blue-shift of the emission compared to complexes with just Cz. Between these two structural modifications, the emission of 1 wt% doped films in PS could be tuned across the full visible spectrum, from a λPL of 432 nm for MAC*-1 to 704 nm for DAC*-6. The OLED with MAC*-3 showed an EQEmax of 19.4% at a λEL of 543 nm.
Complexes (AdL)Cu(G1), (AdL)Au(G1), and (AdL)Au(G2) (Figure ) contain carbazole-based donor dendrons as ligands.ref. ref908 These complexes showed similar photophysical properties to the parent complexes, CMA-1 and CMA-2, with slightly reduced ΦPL arising from the greater conformational motion within the donor dendron units. Solution-processed OLEDs with (AdL)Au(G1) showed EQEmax of 10.6% at CIE coordinates of (0.39, 0.58) with negligible efficiency roll-off (EQE1000 = 10.0%), while the devices with the other two emitters showed poorer performance.
A number of reports have explored different carbene ligands in combination with carbazole in CMA complexes of copper, silver, and gold.ref909−ref910ref911ref912 Hamze et al. explored the difference in photophysical properties between 5-membered CAAC carbenes and 6-membered MAC ligands.ref. ref909 The use of the 6-membered MAC carbenes induced a 35 to 40 nm red-shift in the emission of the complexes (1 wt% doped PS films), to λPL of 506, 512, and 512 nm for CuMAC , AgMAC , and AuMAC compared to the CAAC complexes at λPL of 470, 472 and 472 nm for CuCAAC , AgCAAC , and AuCAAC , all respectively (Figure ). All six complexes have near unity ΦPL in the same 1 wt% doped PS films, with shorter τPL for the MAC complexes of 2.8, 0.50, and 1.14 μs for CuMAC , AgMAC , and AuMAC compared to the CAAC complexes with τPL of 1.40, 0.33, and 0.83 μs for CuCAAC , AgCAAC , and AuCAAC , all respectively. The OLEDs with AuMAC showed EQEmax of 18% and EQE1000 of 15% with λEL at 510 nm.ref. ref909 Changing the carbene from CAAC to BZI in a subsequent report resulted in a blue-shift of ca. 40 nm for each of CuBZI , AgBZI , and AuBZI , with λPL of 434, 438, and 432 nm, respectively.ref. ref910 The complexes retained their high ΦPL of > 85%, although the emission lifetime increased to τPL of 3.2 to 4.9 μs. Deep-blue OLEDs with AuBZI showed an EQEmax of 12% at CIE coordinates of (0.16, 0.06).ref. ref910
A similar and direct comparison has been reported between CMA complexes using CAAC ligands with a monocyclic 6-membered ring (C6 ligand) and those using bicyclic 6-membered rings (BIC) in the series CuC6 , AuC6 , CuBIC , AgBIC , and AuBIC (Figure ).ref. ref911 In 1 wt% doped Zeonex films the BIC complexes are sky blue emitters with λPL of 490 to 496 nm, while the C6 complexes are green emitters with λPL of 519 and 523 nm for CuC6 and AuC6 , respectively. The more rigid BIC ligand reduces non-radiative decay in the complexes resulting in more efficient emission (ΦPL = 82 to 100% for BIC complexes, compared to 3 to 22% for the C6 complexes) as well as longer τd.ref. ref911
In a bid to blue-shift the emission, replacement of the tBu groups on the carbazole of CMA-4 with an electron-withdrawing CF3 moiety leads to complexes AuCAAC-1 and AuCAAC-2 (Figure , named simply 1 and 2 in the original work).ref. ref913 The emission of the complexes in toluene is blue-shifted from 552 nm in CMA-4 to 495 and 456 nm respectively in AuCAAC-1 and AuCAAC-2. Despite the blue emission, no OLED was fabricated with AuCAAC-2 due to its long emission lifetime (τd > 10 μs) and moderate efficiency (ΦPL = 61%). The OLED with AuCAAC-1, however, showed an EQEmax of 20.9% at CIE coordinates of (0.17, 0.17), and moderate efficiency roll-off with EQE100 of 17.8%.
Changing the amide from carbazole to 6-membered heterocycles based on acridine resulted in a series of conformationally flexible complexes of both copper and gold, including Cu1–3, Cu5, and Au1–8 (Figure ).ref. ref914 The complexes emit across a wide range of wavelengths in toluene (λPL = 489 to 689 nm) and in 5 wt% doped PS films (λPL = 458 to 649 nm), with the color tuned by the electronics of the atom or bridging group at the 9-position of the acridine ligand. The bluest emitters are Cu1 and Au1 (λPL = 489 and 505 nm respectively, in toluene) with a strongly electron-withdrawing SO2 group resulting in a weakly donating amide ligand, and consequently a larger HOMO-LUMO gap. Complexes with hydrocarbon bridging groups, Cu2–3 and Au2–4, all emit similarly in toluene (λPL from 589 to 629 nm). Stronger donating amines with electron-donating O and S atoms, Cu6 and Au5–7, or electron-donating Nme2 groups, Au8, show the most red-shifted emission in toluene (λPL of 654 to 689 nm). The ΦPL decreases significantly as the emission color moves from blue to red (from 90% to < 0.1%), consistent with the energy gap law. Complex Au1 also showed mechanochromic behavior, moving from warm-white to sky-blue emission upon grinding. The dual emission of Au1 was exploited in solution-processed white OLEDs, which showed EQEmax of 4.6% at CIE coordinates of (0.18, 0.31).ref. ref914
The emission color of most CMA complexes has been limited to blue to green, although Gernert et al. showed that it is possible to tune the emission of complexes bearing aryl-fused CAAC ligands to the red.ref. ref915 Of their reported complexes only [Cu(Cz)(DippCAArC)] showed TADF, with deep-red emission (λPL = 621 nm, ΦPL = 32%, and τPL = 0.37 μs).
Over time the sophistication and performance of CMA design strategies have naturally increased, and notably so since 2021. In an attempt to increase the radiative decay rate Li et al. designed bimetallic CMA complex Au2 CC (Figure ) that has a faster kr in comparison to the monometallic complex AuMAC .ref. ref916 The emission was blue-shifted to 480 nm, with ΦPL = 80% and τd = 0.52 μs. Increasing the substituent steric bulk of the imidazopyrazine carbene ligands in the series MCMA, ECMA, BCMA, and PCMA (Figure ) had surprisingly little impact on the green emission color in 1 wt% doped films in PMMA, with λPL ranging between 510 to 520 nm for the four complexes. PCMA has the highest ΦPL of 89% and shortest τd of 0.35 μs of the series, suggesting that this is nonetheless a promising new type of carbene ligand for CMA complexes.ref. ref912
Instead of decorating the archetypal Cz with electron-withdrawing groups to blue-shift the emission of CMA complexes, an alternative strategy involves the use of more weakly electron-donating carboline derivatives.ref. ref917 For instance, compared to CMA-1 (λPL = 498 nm) the emission of 3,6-DiAza (Figure ) is blue-shifted to a λPL of 419 nm. Of the derivatives studied, the one with the most promising photophysical properties is Aza3, which emits at λPL of 450 nm with a ΦPL = 66% and a τPL of 2.1 μs in 3 wt% doped film in PS.ref. ref917
To understand in greater detail the excited state kinetics within CMA complexes, Li et al. performed a combined experimental and theoretical study on 12 CMA complexes that featured MAC ligands with either methyl or phenyl substituents and carbazole groups with or without a cyano group in the 3-position (Figure ).ref. ref918 All these complexes were bright emitters (ΦPL > 50%), with emission ranging from sky blue (λPL = 476 nm) to yellow (λPL = 558 nm) and having short emission lifetimes (τPL < 1.5 μs). The theoretical study identified a ‘sweet spot’ where an NTO overlap of around 0.25 to 0.3 produced the best balance of between ΔE ST and f to ensure both fast TADF and k r.
Muniz et al. recently described a series of 18 MAmide Carbene complexes containing both existing and new (BZAC and PAC) carbenes, along with established and new (bim, Mbim, and Obim) amides (Figure ).ref. ref919 In 1 wt% doped PS films the emission spans from λPL = 400 to 594 nm, and 15 of the complexes have ΦPL > 75%. Importantly, this study revealed strong correlation between the theoretical electron-hole distance in the CMA complexes (determined from NTO calculations) and the experimentally observed kr . Of the complexes investigated Aubim PZI , Aubim BZI , Aubim PAC , and Aubim BZAC have the shortest τPL of 0.24, 0.25, 0.27, and 0.28 μs respectively.
The use of bulky pyrazine- and pyridine-annulated NHC ligands has been further explored in a series of copper CMA complexes Cu1-Cu5 (Figure ).ref. ref920 The complexes emit across a wide range from sky-blue to orange (λPL = 470 to 660 nm in 2 wt% doped films in mCP). The use of the pyrazine-fused ligand PzIPr in Cu1-Cu4 resulted in red-shifted emission, especially when combined with an unsubstituted (Cu1) or substituted carbazolate ligand (Cu3 and Cu4), resulting in λPL ranging from 567 to 581 nm. Introduction of a weakly donating carbazolate ligand containing electron-withdrawing cyano groups in Cu2 led to a blue-shifted emission with λPL of 508 nm. Moving from a pyrazine-containing carbene to a weaklier accepting pyridine-based carbene in Cu5 led to a further blue-shift in the emission of 5 wt% doped films in mCP, to λPL of 470 nm. All the complexes showed short emission lifetimes (τd = 0.36 to 0.47 μs in 2 wt% doped films in mCP) and moderate to good efficiency (ΦPL = 52 to 88% in the same). OLEDs with Cu5 showed EQEmax of 23.6% at CIE coordinates of (0.14, 0.22), with low efficiency roll-off of 12% at 10,000 cd m–2 and very long device lifetimes (LT90 of up to 1300 h at 1000 cd m–2). These are the best-performing copper CMA OLEDs reported to date.ref. ref920
Given the excellent performance of CMA TADF emitters, they have also been explored as assistant dopants in HF-OLEDs (See Section sec17 ). In addition to using a known CMA complex (BZI)Au(Cz),ref. ref910 Heo et al. designed the related complex (BZI)Au(TMCz) containing a sterically modified carbazole (Figure ).ref. ref921 This modification resulted in a red-shift in the emission of the 5 wt% doped film in Zeonex to λPL = 466 nm, while retaining a high ΦPL of 95% and short τPL of 0.38 μs that makes this complex amenable as an assistant dopant for green HF-OLED.
In a later study focusing on the impact of restricting rotation around the Au-N bond, Yang et al. investigated a series of six CMA complexes with substituents in the 1-position of the 4,7-di-tBu-carbazole ligand to lock the CMA complexes into a twisted conformation.ref. ref922 The reference complex with no substituent on the carbazole ligand, Au-tCz (Figure ), has a dihedral angle between the carbene and carbazole of only 0.4°. In contrast, the other five complexes have much larger dihedral angles of between 66–74°. The high luminescence efficiency of CMA complexes is generally attributed to the high oscillator strength and fast radiative decay arising from a co-planar arrangement of the ligands around the metal centre.ref194,ref906 However in these twisted complexes the ΦPL remains high at between 73 to 94% in 5 wt% doped films in mCP. This outcome is attributed to a decrease in the ΔE ST and an increase in SOC of the T1 → S1 RISC process supported by the large dihedral angles, which counteracts the decrease in oscillator strength for the S1 → S0 transition moving away from a co-planar conformation.
To target CPL emission, the first chiral CMA complexes (R,R)-PSIPr*-Cu-DMAC and (S,S)-PSIPr*-Cu-DMAC (Figure ) employed a chiral analog of the SIPr NHC ligand.ref. ref923 Both enantiomers and the racemate showed TADF, with identical λPL = 531 nm, τPL = 0.14 μs, and ΦPL = 24% in toluene solution. In dilute solution the enantiopure complexes showed no CPL signal, however in both the powder and crystal the complexes showed strong CPL signals with |g lum| values of up to 2.7 × 10–2 for the crystals. An extensive DFT study highlighted that aggregation-induced CPL was induced by limiting the rotation of the ligands in the solid state.ref. ref923
Finally, Ying et al. replaced the common carbazole donor with the stronger electron-donor diphenylacridine to produce the red-emitting copper complex MAC*-CuDPAC (Figure ).ref. ref924 In toluene this complex emits at λPL of 638 nm and has a poor ΦPL of 12% with a short τPL of 0.11 μs. In 1.5 wt% doped films of mixed CBP:TPBi (1:1 co-host) it emits at λPL of 599 nm, has a much higher ΦPL of 65%, and a τPL of 0.95 μs. Notably the ΦPL of 65% in CBP:TPBi films is very good for a red TADF emitter, supported by the rigid ligands reducing non-radiative decay. The complex was also shown to have a strongly horizontally orientated TDM in the same films. As a result, despite the modest ΦPL the OLEDs with MAC*-Cu-DPAC showed an EQEmax of 21.1% at CIE coordinates of (0.58, 0.42), and the efficiency roll-off was also very low (EQE1000 = 20.1%).
Palladium and Platinum
Due to the very high SOC constant of platinum, most platinum(II) complexes are phosphorescent and have been widely used previously as emitters in OLEDs.ref106,ref925 Indeed, first report of triplet harvesting in an OLED used a platinum(II) porphyrin emitter.ref. ref14 There have recently also been reports of platinum and palladium emitters that exhibit metal-assisted delayed fluorescence (MADF), where both delayed fluorescence and phosphorescence have been detected.ref. ref926 In these examples the ΔE ST can be moderately large (> 150 meV) but the large SOC constants of Pd and Pt nonetheless enable rapid ISC/RISC resulting in some TADF emission. In addition, a small number of bimetallic platinum(II) complexes have recently been shown to exhibit TADF.ref927−ref928ref929
The first report of TADF in palladium(II) complexes presented PdN3N and PdN3O (Figure ), containing rigid and planar tetradentate ligands.ref. ref926 The small ΔE ST in these ‘phosphorescent’ complexes allows an additional high-energy component in their emission spectra to emerge, that is enhanced at higher temperatures. This emission band has been attributed to fluorescence from a thermally accessible S1 state – i.e. TADF. This balance of emission mechanisms is of interest as it may provide a route to achieve blue emission from complexes with lower energy triplet excited states, that may also translate into more stable devices. PdN3N and PdN3O both emit at λPL of 534 nm, have ΦPL of 76%, and τPL of 142 and 205 μs, respectively. The OLEDs with PdN3N and PdN3O showed EQEmax of 20.9 and 20.4%, however the efficiency roll-off was large at 32% at 100 cd m–2, attributed to the long emission lifetimes. Despite the efficiency roll-off, the OLED with PdN3N demonstrated excellent device stability with a very long lifetime (LT90 > 20 000 h at 100 cd m–2). There is a higher TADF contribution (30%) in PdN3N due to its smaller ΔE ST of 150 meV, compared to 180 meV for PdN3O. The mechanism of the mixed TADF and phosphorescence process in PdN3N was later investigated computationally,ref. ref930 and found to be supported by calculated T1 → S1 and T2 → T1 → S1 RISC rates comparable to the T1 → S0 phosphorescence rate at 300 K.
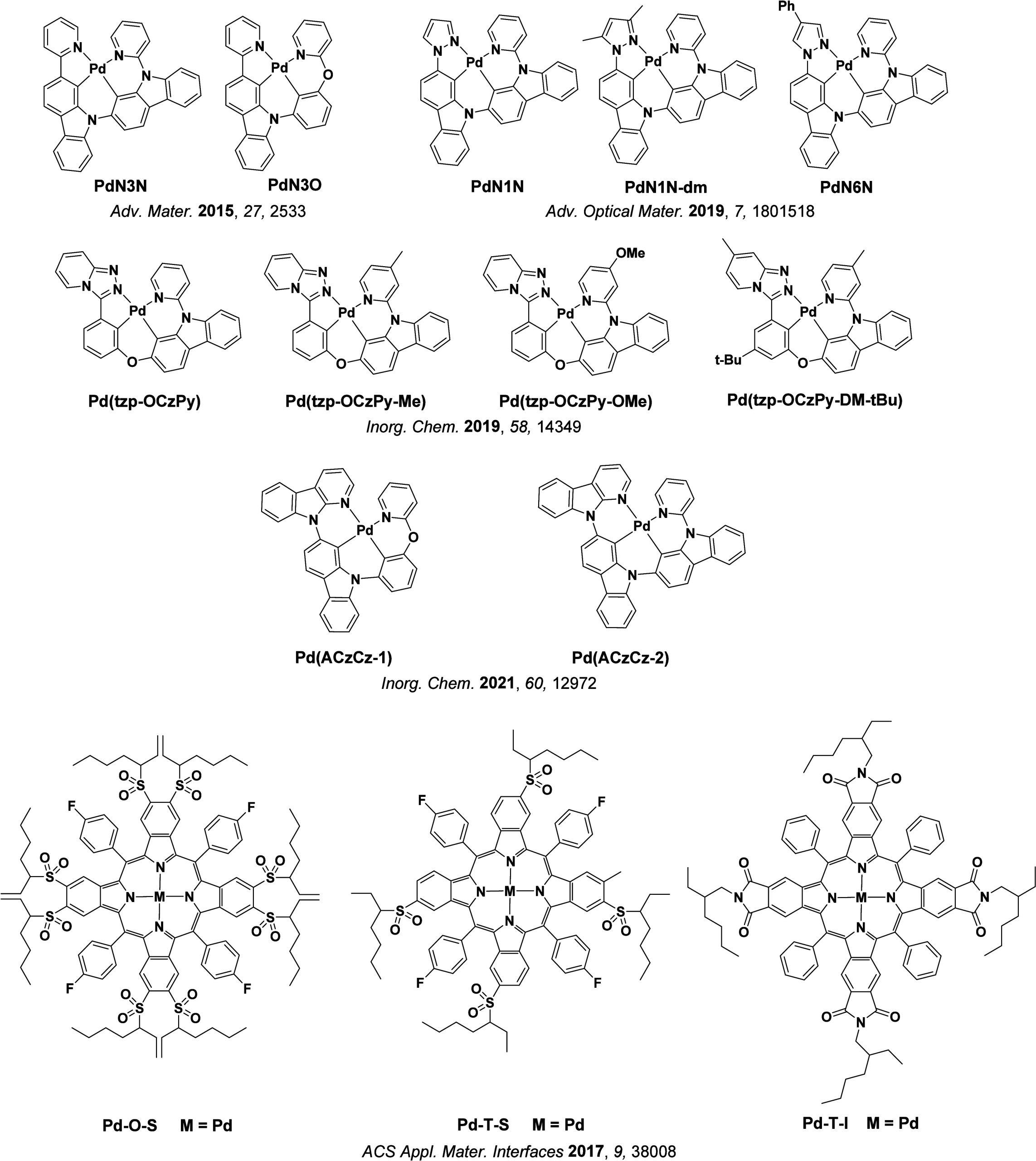
Subsequently, a series of three complexes were reported in which one of the pyridyl groups of PdN3N was replaced with a pyrazolyl group to destabilize the LUMO and blue-shift the emission of PdN1N, PdN1N-dm, and PdN6N (Figure ).ref. ref931 The complex PdN1N-dm has the highest ΦPL of 77% and the best thermal stability of the three complexes in the study, and so was used as the emitter in OLEDs. The devices showed an EQEmax of 25.1% at CIE coordinates of (0.14, 0.25), although there was very high efficiency roll-off (EQE100 = 11.1%). Another series of six complexes were also prepared in which the pyrazole within the ligand was replaced with a triazole group.ref. ref932 The complex emitting with the highest TADF component is Pd(tzp-OczPy-Ome), despite its ΔE ST of 228 meV. The use of the same ligands to form a platinum complex produced a phosphorescent emitter with no observed TADF contribution. Similarly fusing rings to form an azacarbazolylcarbazole-based tetradentate ligand in complexes Pd(AczCz-1) and Pd(AczCz-2) led to either sky blue emission for the former (λPL = 479 nm) or green (λPL = 506 nm) for the latter. These complexes have small ΔE ST of 57 and 112 meV, respectively in DCM, however are weak emitters with ΦPL = 10–11%.ref. ref933
A series of eight Pd and Pt porphyrin-based complexes (Figure and Figure ) have been developed for oxygen and temperature sensing.ref. ref934 Again due to the higher SOC constant, the Pt complexes possess a smaller TADF contribution (and thus a stronger phosphorescence contribution) than the Pd complexes.ref. ref931 The complexes all emit in the red to NIR region with well-resolved TADF (λPL = 620 to 652 nm) and phosphorescence (λPL = 742 to 800 nm) in the steady-state toluene PL spectra. The ΦPL of the complexes ranged from 3 to 30%, while the τd of the platinum complexes ranged from 12 to 47 μs while those of the palladium complexes ranged from 53 to 286 μs. The complexes with the largest TADF contribution to the emission are Pd-O-S and Pd-T-I, which showed an increase in the TADF:fluorescence ratio from 0.16 and 0.26 to 3.2 and 4.6 (respectively) as the temperature was increased from 5 to 80 °C, allowing optical readout.
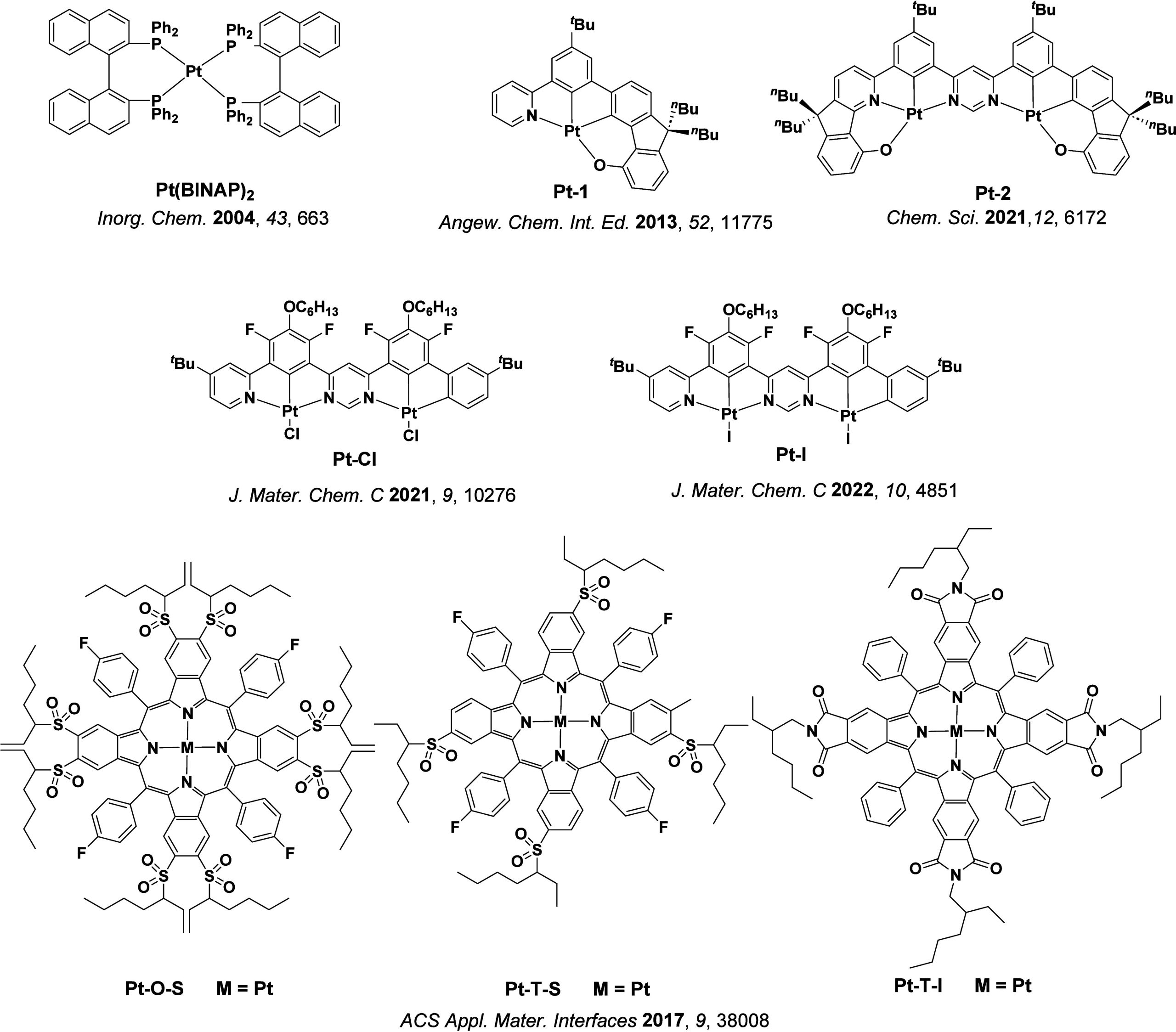
Delayed fluorescence was first identified in a platinum(0) complex in 2004.ref. ref935 The complex Pt(BINAP)2 (Figure ) emits at λPL of 763 nm in toluene and has a ΦPL of 12%. TADF was assigned from both the biexponential decay kinetics (τp = 3.2 ps, τd = 1.025 μs) and the temperature dependent intensity of the delayed emission. DFT calculations indicated that the unusual emission properties of this complex were due to rapid ISC/RISC between the 3MLCT and 1MLCT states owing to the small ΔE ST of 149 meV (calculated ΔE ST = 184 meV). To date, no other TADF platinum(0) emitters have been reported.
Pander et al. have nonetheless shown that moving from mono- to dinuclear platinum complexes leads to much smaller ΔE ST values, such that TADF outcompetes phosphorescence.ref. ref927 This was initially demonstrated by comparison of the photophysical properties of Pt-2 with its phosphorescent mononuclear analogue Pt-1 (Figure ),ref. ref936 where computations revealed that the ΔE ST decreased from 370 meV for the latter to 180 meV for the former. The smaller ΔE ST in Pt-2 is partially due to the use of the stronger electron-accepting pyrimidine (which coordinated both Pt atoms) compared to pyridine in the mononuclear complex, while simultaneously the calculated S1–T1 spin–orbit coupling matrix element (SOCME) drops from 88 cm–1 for Pt-1 to 10 cm–1 for Pt-2. This lower SOCME value in the latter implies that directly spin-forbidden processes (such as phosphorescence) become slower in this material, giving TADF (which becomes spin-allowed through vibronic coupling) a window of opportunity to dominate the emission mechanism. Dissolved in MCH Pt-2 emits at 602 nm, has a FWHM of 22 nm, and a small Stokes shift of only 7 nm, which is rare for third-row transition metal complexes and suggests both a small ΔE ST and small reorganization energy in the excited state. The solution-state ΦPL is 83% and the τPL is 2.1 μs. The OLEDs of Pt-2 showed an EQEmax of only 7.4% at CIE coordinates (0.62, 0.37), but the emission is broadened compared to solution (FWHM of 75 nm). The low efficiency and spectral broadening were ascribed to the formation of aggregates in the film.
Related dinuclear Pt complexes bearing ancillary halogen ligands (Pt-Cl ref. ref928 and Pt-I ref. ref929, Figure ) showed similar λPL of 635 and 633 nm with ΦPL of 51 and 57% respectively in chlorobenzene. Pt-Cl exhibits a longer τPL (5.0 μs) in both chlorobenzene solution and 0.1 wt% doped films in PS than Pt-I (1.7 μs in chlorobenzene solution, 2.3 μs in PS). These results were rationalized by the smaller ΔE ST of Pt-I (60 meV) compared to Pt-Cl (200 meV). Based on theoretical calculations, this variation in ΔE ST was proposed to originate from a much smaller HOMO-LUMO overlap in Pt-I, with an MO pattern and chemical structure resembling an MR-TADF emitter. Solution-processed OLEDs with both Pt-I and Pt-Cl showed EQEmax of 3.1 and 2.6%, respectively. One of the devices using Pt-Cl at a very high doping concentration (33 wt%) is notably the first example of an excimer-based Pt(II) solution-processed OLED with NIR emission beyond 800 nm.
Zinc
Given the number of d10 coinage metal emitters discussed so far it is surprising that d10 zinc(II) complexes have received relatively little attention as TADF emitters. This comes despite the first example of a TADF zinc complex being reported in 2015.ref. ref937 All the TADF zinc(II) complexes reported to date and discussed here show ligand-centered emission, with the zinc atom only minimally contributing to the excited states (charge transfer or otherwise). Adachi and co-workers reported the first zinc(II) TADF emitters Zn(p-PX-BOX)2 and Zn(m-PX-BOX)2 (Figure ),ref. ref937 using ligands that were themselves known D-A TADF emitters with phenoxazine donor and phenylbenzoxazole acceptor. The calculated ΔE ST are very small at 17 and 37 meV, respectively, while the HOMO and LUMO were expected to be located on the phenoxazine and phenylbenzoxazole with no involvement from the metal center. Zn(p-PX-BOX)2 and Zn(m-PX-BOX)2 emit at λPL of 523 nm (ΦPL = 78%) and λPL of 542 nm (ΦPL = 58%) and have ΔE ST of 60 and 180 meV, respectively, in 6 wt% doped films in mCBP. These ΔE ST values are notably smaller than the metal-free methoxy-substituted ligand, p-OMe-PX-BOX (ΔE ST = 310 meV), with the decrease attributed to an increase in the dihedral angle between the donor and acceptor upon coordination to the zinc. The green OLEDs with Zn(p-PX-BOX)2 showed EQEmax of 19.6% at λEL of 542 nm.ref. ref937
The same strategy of metal complexation enhancing the TADF in donor-acceptor ligands was also invoked in the dimeric zinc complexes [Zn(PhOPy-PXZ)2]2 and [Zn(PhOPy-DMAC)2]2 (Figure ).ref. ref938 These two complexes showed interesting luminescence polymorphism, with different emission observed for powders (λPL of 538 and 497 nm,), crystals (500 and 444 nm), and ground powders (532 and 473 nm, all respectively). Pristine powders of [Zn(PhOPy-PXZ)2]2 and [Zn(PhOPy-DMAC)2]2 emit at λPL of 538 and 497 nm, have ΦPL of 13 and 50%, and τPL of 2.09 and 2.45 μs, respectively. The calculated ΔE ST of 70 and 100 meV and the temperature dependence of the TRPL both indicate that these complexes are TADF-active.ref. ref938
The complex Zn(HL)Cl2 (Figure ) shows excitation-wavelength dependent emission, ESIPT, and TADF.ref. ref939 As a powder, low energy excitation at 480 nm results in yellow TADF emission (λPL = 565 nm, ΦPL = 7%, τPL = 6.0 μs, and ΔE ST = 11 meV), while excitation at 380 nm results in emission at λPL of 640 nm accompanied by a significant drop in ΦPL to 0.02%. Computational studies showed that either the keto or enol tautomer can be the most stable form of the molecule, depending on the excited state. Excitation at 480 nm to S1 of the enol form leads to ESIPT to the keto form, followed by both prompt fluorescence and TADF. In contrast, upon excitation at 380 nm to Sn>1, the enol form of the complex undergoes rapid ISC to the enol form’s Tn and then subsequent ESIPT the keto T1. As there is no initial photoexcitation of the keto form S1 excited state there is no prompt emission, and all the emission is either phosphorescence from keto T1 and/or TADF from keto S1.
Chen et al. reported the use of the chiral TADF Zn(II) salen complex Zn((R/S)-6-MeOsalen) (Figure ) as an emitter in CP-OLEDs.ref. ref940 In THF the complex emits at λPL of 491 nm while in the neat film it showed dual emission consisting of a shoulder at 490 nm and a more intense band at λPL of 576 nm. The low-energy band was assigned to excimer emission, and was much longer-lived than the nanosecond fluorescence of the band at 490 nm, with τPL of 8.42 μs for Zn((R)-6-MeOsalen) and 7.39 μs for Zn((S)-6-MeOsalen). CP-OLED devices showed an EQEmax of only ca. 0.04%, however the gEL values were high on the order of 10–2.
Two zinc(II) Schiff base complexes with microsecond-long τPL were used as emission lifetime based optical temperature sensors.ref. ref941 Both complexes are composed of a phthalonitrile acceptor unit and either an N,N’-dialkylaniline (Zn-Schiff-1) or a di-tert-butylcarbazole (Zn-Schiff-2) donor (Figure ). Use of a stronger donor moiety in Zn-Schiff-2 compared to Zn-Schiff-1 resulted in a small red-shift in the emission (from λPL = 542 to 547 nm), a smaller ΔE ST (from 310 to 280 meV) and a shorter τPL (from 2.1 ms to 435 μs). Delayed fluorescence was estimated to make up 30% of the total ΦPL for Zn-Schiff-2, while this was more difficult to quantify for Zn-Schiff-1 due to its long excited state lifetime but was nonetheless estimated to make up approximately 16% of the total ΦPL. More recently, similarly structured complexes of were studiedref. ref942 containing a phenyl spacer between the donor and the Schiff base ligand backbone. The incorporation of the phenyl spacer in ZnPH-Cz and ZnPH-Ph-Cz results in a larger ΔE ST compared with Zn-Schiff-1 and Zn-Schiff-2, while introduction of a stronger 2,3-pyrazinedicarbonitrile acceptor in ZnPZ-Cz and ZnPZ-Ph-Cz produces a smaller ΔE ST. Complexes ZnPZ-Cz and ZnPZ-Ph-Cz have shorter τPL of 114 and 236 μs compared with ZnPH-Cz and ZnPH-Ph-Cz (τPL = 945 and 1040 μs, all respectively), both of which exhibited τPL closer to those of Zn-Schiff-1 and Zn-Schiff-2. The presence of the strong acceptor also significantly reduced the ΦPL, particularly when combined with the phenyl spacer, falling from 37% for ZnPH-Cz to 1.9% for ZnPZ-Ph-Cz. This behavior can be rationalized by the larger ΔE ST in ZnPZ-Ph-Cz and increased non-radiative decay in this complex due to greater conformational flexibility in the ligand.
A zinc porphyrin complex, Zn-OS (Figure ), was used as a dual oxygen and temperature sensor.ref. ref943 This complex emits at λPL of 667 nm and has a τd > 1 ms. As the τd and the intensity of the prompt and delayed fluorescence (I DF/I PF) vary differently with temperature and oxygen quenching, an empirical model was built to measure both parameters from a single measurement.
Goswami et al. reported blue-emitting (λPL = 480 nm) iminophosphonamide zinc complex Zn-NPN-5 (Figure ) that has a ΔE ST of 120 meV.ref. ref944 Interestingly, a very similar complex Zn-NPN-6 was found to have no delayed emission. Analysis of single crystals of both complexes revealed that the Zn(II) center of Zn-NPN-6 adopts a square planar geometry while Zn-NPN-5 appears to coordinate in a distorted tetrahedron. The square planar geometry was proposed to be the origin of the poor triplet formation in Zn-NPN-6, hindering intraligand charge transfer. Interestingly, TADF was observed in a planar dinuclear copper complexes in the same study, for which the authors attributed the improved ISC/RISC to an enhanced SOC in this system.
Other Metals
Examples of TADF emitters containing metals other than those discussed above exist in smaller numbers. Among the first of these were a series of tin(IV) porphyrin complexes (Figure ).ref. ref32 A sufficiently small ΔE ST of 240 meV in SnF2-OEP resulted in dominant TADF emission, although some phosphorescence was also detected at room temperature. Temperature-dependent ΦPL was used to assign the emission as TADF (ΦPL increasing from 0.6% at 300 K to 2.4% at 400 K). Devices were fabricated and although no EQEmax was mentioned, upon electrical excitation a prompt and temperature-dependent delayed emission were observed. This report by Adachi and co-workers was one of the first examples of the TADF mechanism being applied (knowingly) to OLEDs.
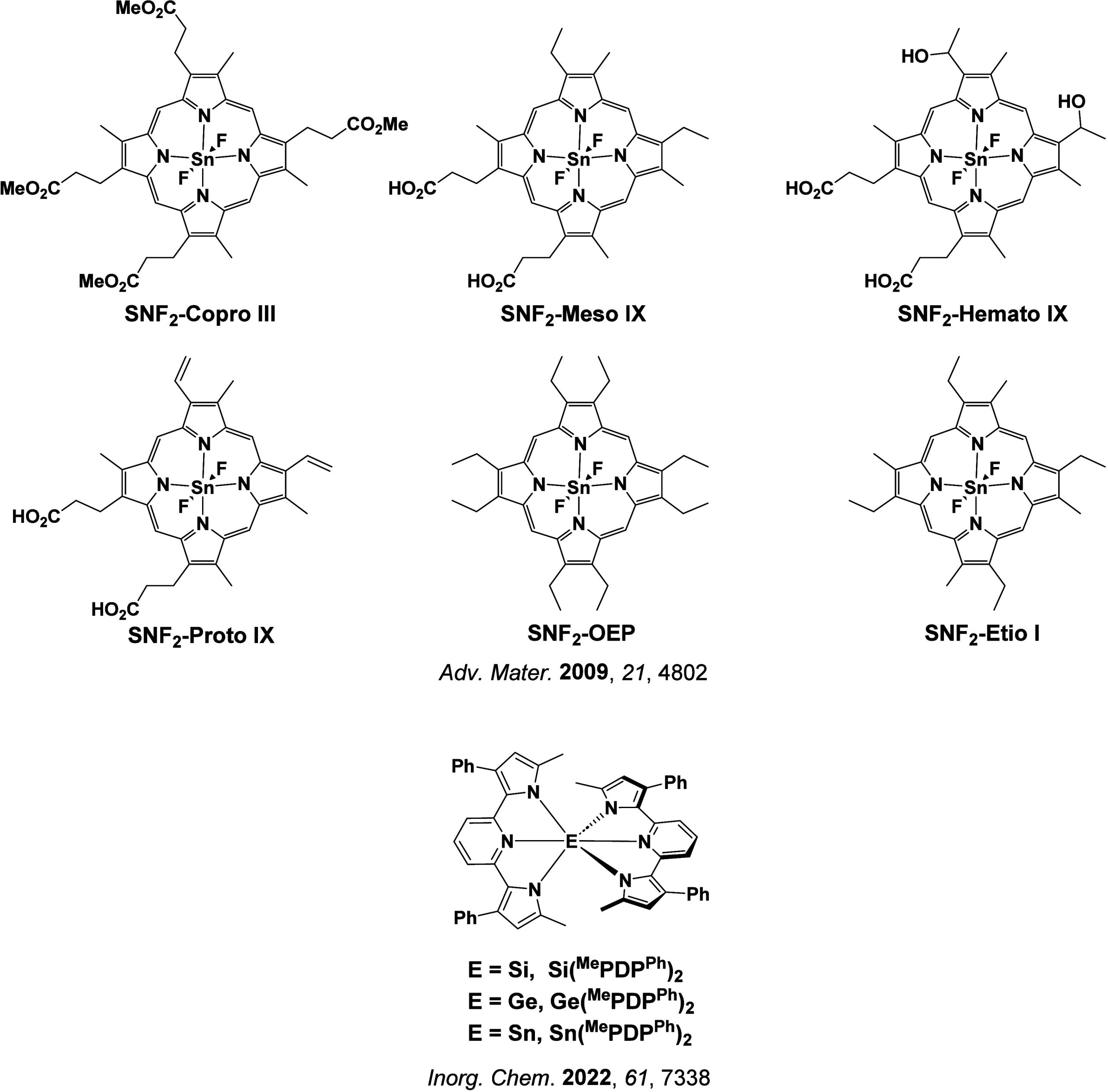
The only other report of TADF Sn complexes since this early discovery comes from Gowda et al., who prepared a series of three main group polypyrrole complexes, Si(MePDPPh)2 , Ge(MePDPPh)2 , and Sn(MePDPPh)2 (Figure ).ref. ref945 The complexes are green emitters in THF and all show TADF. Moving down the group the emission color blue-shifts from a λPL of 527 nm for Si(MePDPPh)2 to 512 nm for Sn(MePDPPh)2 , and the τPL increases from 0.9 ms for Si(MePDPPh)2 to 2.0 ms for Sn(MePDPPh)2 while the ΦPL ranges between 32 and 49% for the three complexes. The ΔE ST of the three complexes are 243, 260, and 313 meV for the lightest to the heaviest analogue. The k ISC for the three complexes was also measured by transient absorption spectroscopy, increasing from 3.23 × 108 to 4.0 × 109 s–1 when moving from Si to Sn. This recent study is the first comparing complexes of the different group 14 elements as TADF emitters, and one of few investigating the photophysical properties of metalloid and post transition metal complexes.
There are also a small number of early transition metal complexes that exhibit TADF (Figure ). A series of tungsten(0) isocyanide complexes of the form w(CNdippR)6 were shown to have yellow to red emission and were used as photocatalysts due to their large excited state reduction potentials.ref. ref946 In toluene these complexes have ΦPL of around 40% and τPL of ca. 1.5 μs. The emission was shown to originate from a MLCT excited state with temperature-dependent emission lifetimes. The tungsten(VI) Schiff base complex W(O)2(N-Ar3-Salen) also emits via TADF, from a mixed LLCT/MLCT excited state.ref. ref947 The calculated small ΔE ST of 93 meV provides support for the assignment of TADF. The presence of methyl groups on the xylyl linker were essential to promote a much more strongly twisted conformation, and to spatially separate the electron densities of the HOMO and LUMO, and an analogous complex using a phenyl linker between the Salen and diarylamine has a much larger ΔE ST of 347 meV. OLEDs with W(O)2(N-Ar3-Salen) showed EQEmax of 15.6% at CIE coordinates of (0.49, 0.49), and moderate efficiency roll-off (EQE1000 = 9.7%).
Millsman and co-workers reported a TADF-active zirconium(IV) complex. Zr(MesPDPPh )2 (Figure ) shows bright yellow emission in solution (λPL = 581 nm and ΦPL = 45%) and has a long emission lifetime of 350 μs.ref. ref948 Calculations revealed that the emissive excited state has mixed IL/LMCT character, and the negligible calculated TDM in the excited state helped to explain the lack of solvatochromism. Temperature-dependent emission studies supported the identification of TADF, alongside a ΔE ST of 200 meV. The complex was used as a photocatalyst in a number of different reactions,ref. ref948 and the same group subsequently reported six TADF complexes containing different substituents on the same pyridyl-dipyrrolide core.ref. ref949 One of these, Zr(MePDPPh)2 , had previously been reported without identification of its emission mechanism as TADF.ref. ref950 These complexes are yellow to orange emitters in benzene with λPL of 568 to 629 nm, are moderately emissive (ΦPL of 10 to 38%), and have long τPL of 190 to 576 μs.
Examples of TADF emitters incorporating both alkali metals and aluminum have also been reported (Figure ). Compounds Mg(p-PX-BOX)2 and Li(p-PX-BOX) emit at λPL of 510 and 516 nm, respectively and show a slightly blue-shifted emission relative to previously discussed Zn(p-PX-BOX)2 (λPL = 542 nm) in 6 wt% doped films in mCBP. All three complexes have high ΦPL, ranging from 70 to 78%, and very small ΔE ST of 60 to 80 meV.ref. ref937 Green OLEDs with Mg(p-PX-BOX)2 and Li(p-PX-BOX) showed EQEmax of 16.5 and 12.9% respectively, slightly lower than the EQEmax of 19.6% reported for Zn(p-PX-BOX). The same study documented the first aluminum TADF emitter [Al(p-PX-BOX)2(μ-OH)2] (λPL = 530 nm and ΦPL= 86.7% in 6 wt% doped films in mCBP) that showed temperature dependent intensity of the delayed emission. The ΔE ST of [Al(p-PX-BOX)2(μ-OH)2] is 60 meV in 2-MeTHF glass. The aluminum complex is thermally unstable though, and all films and devices were prepared by solution processing in contrast to the thermal evaporation methods used for the zinc, lithium, and magnesium complexes with the same ligand. The OLED showed an EQEmax of 6.8% at λEL of 505 nm.ref. ref937
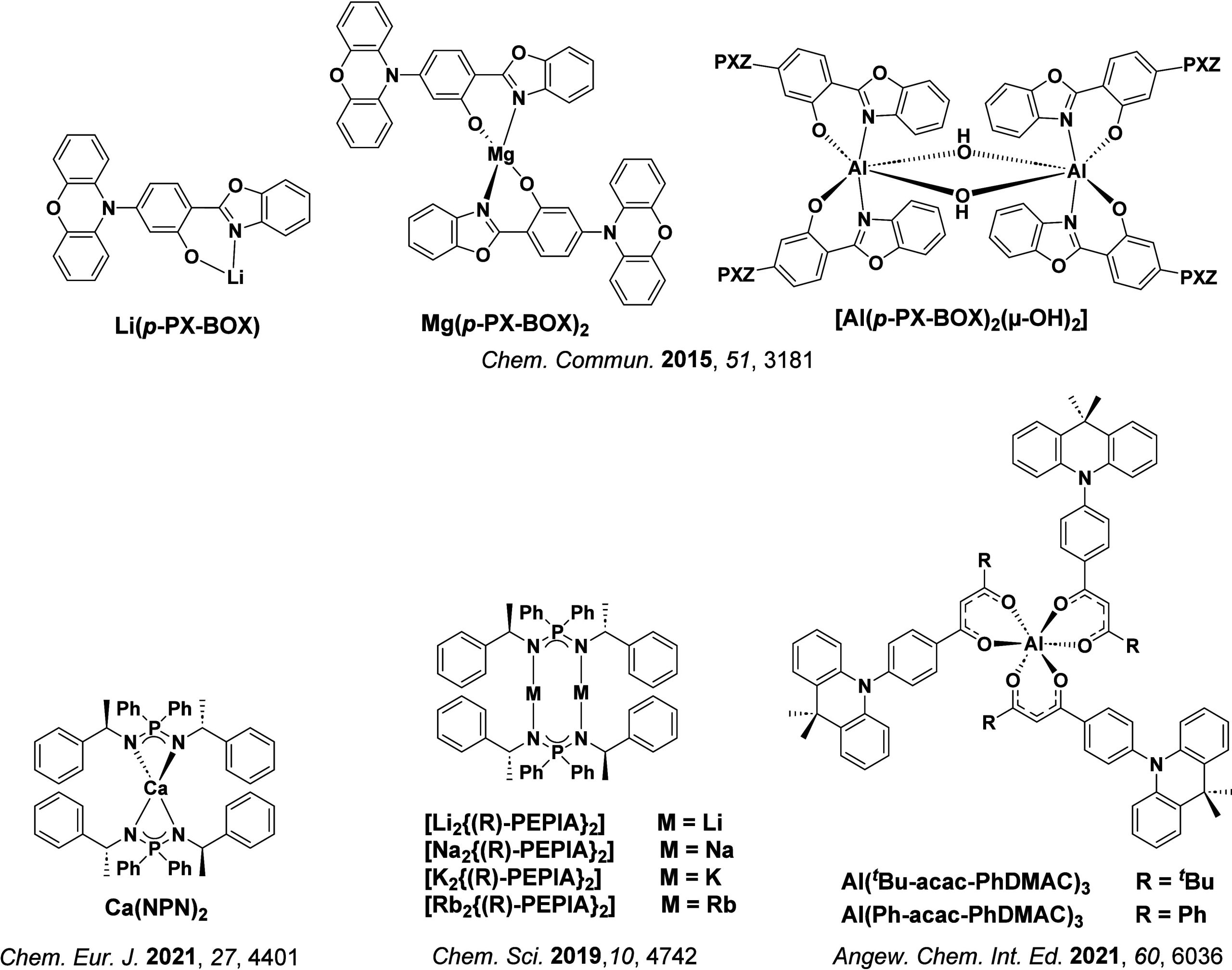
A series of emitters based on dimeric alkali metal complexes with enantiopure iminophosphonamide ligands of the form [M2((R)-PEPIA)2] have been reported (Figure ).ref. ref951 The complexes are blue emitters as neat films with low ΦPL ranging from 8–21%, and τPL between 4.1 to 14.8 μs with small ΔE ST values ranging from 73 to 90 meV. There were no clear trends in the reported optoelectronic properties of the complexes. The same iminophosphonamide ligand was later used in a monometallic calcium complex Ca((R)-PEPIA)2 [or Ca(NPN)2 ] that also showed TADF.ref. ref952 The complex is a blue-green emitter as a neat film, has a ΦPL of 22%, τPL of 24 μs and a ΔE ST of −148 meV – approximately double that of the related dinuclear alkali metal complexes.
The first family of mononuclear aluminum complexes to show TADF, of the form Al(R-acac-PhDMAC)3 , has recently been reported (Figure ).ref. ref953 The asymmetric acetylacetonate ligands showed weak TADF emission without complexation in 30 wt% doped films in CBP. Upon coordination to the aluminum, changes in the dihedral angle between the DMAC donor and the remainder of the molecule result in a decrease of the ΔE ST. The complexes are green emitters (λPL = 495–534 nm) in toluene, and 30 wt% doped CBP films have τPL < 4 μs with ΦPL ranging from 32 to 79%. Solution-processed OLEDs with Al(Ph-acac-PhDMAC)3 showed an EQEmax of 17.5% at CIE coordinates of (0.43, 0.55), and had small efficiency roll-off (EQE1000 = 14.7%).
Iridium(III) complexes are typically employed as phosphorescent emitters in OLEDs (PhOLEDs) due to their large SOC values.ref. ref954 However, introduction of TADF-emitting ligands can produce some interesting dual-emissive complexes (Figure ). Benjamin et al. reported complexes Ir-5 and Ir-6 that show dual TADF and phosphorescent emission in polar solvents, originating from 3CT and 3MLCT states respectively.ref. ref955 The blue-emitting OLEDs with Ir-5 and Ir-6 however showed very low EQEmax of 1.5 and 2.1%, respectively.
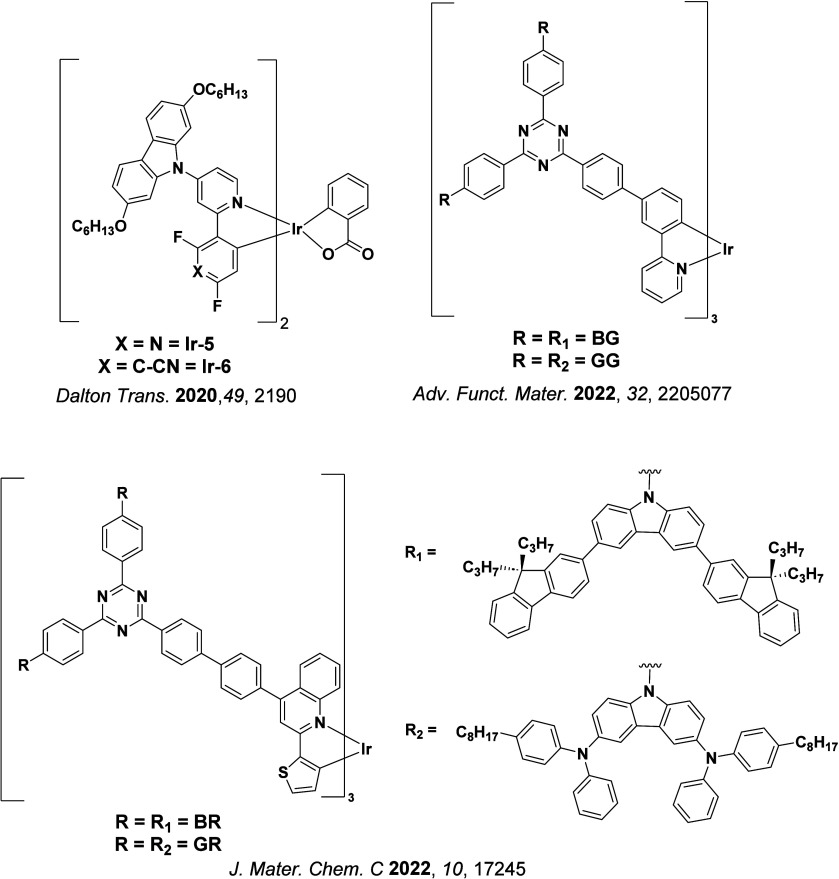
Thamarappalli et al. reported two dual-emitting complexes that incorporate TADF dendrimers onto a Ir(ppy)3 core: BG and GG (Figure ).ref. ref956 In solution the complexes showed dual TADF and phosphorescent emission, and as the solvent polarity increased from cyclohexane to toluene to DCM, the contribution of the TADF emission increased as the CT excited state associated with the TADF dendrimer moiety was stabilized. An energy transfer process between the TADF donor dendrons and the phosphorescent core was confirmed by PL measurements in solutions of varying solvent and temperature. This suggests that energy transfer from long-lived excitons in TADF dendrimers can be harvested and managed through faster radiative decay of a phosphorescent core. Both non-doped and doped OLEDs showed green emission, with the non-doped OLEDs of GG showing an EQEmax of 4.7% at CIE coordinates of (0.48, 0.51). The device with BG showed similar EQEmax of 4.0% at CIE coordinates of (0.42, 0.56). Devices using doped emissive layers (0.4 mol% in mCPCN) showed higher efficiencies, with the GG device showing an EQEmax of 9.8% at CIE coordinates of (0.41, 0.56), and BG showing an EQEmax of 15.1% at CIE coordinates of (0.32, 0.63).
Similar structures have also been reported by Jang et al. employing blue-emitting (BR) and green-emitting (GR) TADF dendrons attached to a fac-tris[2-(thiophen-2-yl)-4-(p-tolyl)quinolinato]iridium(III) (TQIr) phosphorescent core (Figure ).ref. ref957 In solution dual emission was observed from both the TADF donor dendrons and the iridium core, with the ratio between the emissions varying as a function of the polarity of the solvent. Both non-doped and doped OLEDs in TCTA showed red emission, with the non-doped BR OLEDs having an EQEmax of 2.6% at CIE coordinates of (0.68, 0.32), while the devices with GR showed an EQEmax of 0.9% at CIE coordinates of (0.67, 0.33). Devices using doped emissive layers (0.1 mol% in TCTA) showed higher efficiencies, with the BR OLED showing an EQEmax of 13.5% at CIE coordinates of (0.66, 0.34), and GR showing an EQEmax of 11.0% at CIE coordinates of (0.66, 0.35).
Outlook
Metal complexes whose reported emission originates in whole or in part from TADF are growing rapidly in number and show promise as emissive materials for OLEDs as well as in other applications such as photocatalysis (Section sec23 ) and LECs (Section sec16 ). In comparison to organic TADF materials, metal complexes can have much faster kISC and kRISC, which can lead to very short emission lifetimes even with relatively light (and abundant) metals. The complicated origins of the emission from metal complexes, often a mixture of phosphorescence and TADF, means that special care must be taken when interpreting and reporting their photophysics. Nonetheless, the photophysical properties of metal complexes can be readily tuned by varying the electronic and steric properties of the ligand(s), giving rise to a rainbow of interesting and useful luminescent materials.
Currently the most promising classes of complexes for use in OLEDs are the coinage metal CMA and gold(III) complexes. Both have excellent optoelectronic properties that translate to high-efficiency devices on-par with state-of-the-art iridium OLEDs. Since the first report of coinage metal CMA complexes in 2017,ref. ref194 a significant body of work on the design, synthesis, and characterization of these complexes has demonstrated their high optoelectronic performance as well as color tuning. This is exemplified in the complexes Aubim PZI , Aubim BZI ,Aubim PAC , and Aubim BZAC that have τPL of 240 to 280 ns, as well as by OLEDs with CMA4 that showed an EQEmax of 27.5% at CIE coordinates of (0.36, 0.54).ref. ref194 Separately, while gold(III) complexes have long been thought to be purely phosphorescent, efforts to elicit TADF activity have led to OLEDs using Au-4 showing an EQEmax of 26.8% at CIE coordinates of (0.36, 0.60), and long device lifetime of LT90 = 674 h (at an initial 1,000 cd m–2).
Separately, metal TADF complexes that show switchable emission based upon metal coordination are particularly relevant in the context of bioimaging and sensing (Sections sec20 and sec21). Underpinning such utility, several families of silver, zinc, and alkali metal complexes summarized in this section feature a geometry change of the ligand upon coordination of a metal centre, resulting in TADF-inactive ligands ‘turning on’ in response to external stimuli. Further refinement of this strategy should enable the design of new sensors, detectors, and bioimaging reagents that optically report on changes in solvent environment, or metal ion concentrations.
Combining TADF properties with the synthetic flexibility and tunable emission properties of organometallic complexes in TADF metal complexes may indeed be a viable path towards achieving the impressive performance and color targets currently available in PHOLEDs. By instead exploiting RISC and TADF, either in the ligands or in CT states, this performance may also be achievable in materials that do not rely on the heaviest and scarcest elements that promote SOC and phosphorescence, providing clear advantages in terms of device cost and sustainability.
Macromolecules TADF
TADF Polymers
Sections sec3–sec9 have largely focused on low molecular weight, small molecule emitters that can be vacuum-deposited during device fabrication. This section, by contrast, summarizes the advances in macromolecules, polymers and dendrimer that show TADF, which have been designed to be processed from solution, such as by spin-coating or inkjet printing, in the context of solution-processed OLEDs. TADF polymers have emerged as a promising class of emitter materials that can be used to achieve high-performance solution-processed OLEDs (SP-OLEDs). The use of polymers as emitters had previously been explored widely for both solution-processed fluorescent and phosphorescent emitters. The major advantage of SP-OLEDs is a considerable reduction in energy use and production costs compared to thermally vacuum-deposited small molecule-based OLED materials. Furthermore, polymers can easily be designed and synthesized to incorporate various optoelectronic functional units into or pendant from the polymer backbone, such as emitters, hosts, spacers, solubilizing groups, and charge-transporting moieties. Precise control of the polymerization then allows adjustable ratios of these units to be achieved in such a way that phase separation can be avoided within the polymer chains and where the polymer can be used neat within the EML. Polymeric materials also frequently display excellent film-forming properties, allowing them in some cases to outperform SP-OLEDs containing low molecular weight emitters (i.e., small molecules). Beside their use as emitters, TADF polymers can also act as host matrices for small molecule emitter dopants, enabling triplet-harvesting from the host in these SP-OLEDs.ref. ref958
There are a range of design strategies for TADF polymers, typically involving combinations of donor (D) and acceptor (A) components. How these subunits are engineered to interact varies according to a collection of identifiable strategies: 1) A known D-A type TADF emitter can be coupled directly to a non-conjugated or weakly conjugated polymer backbone, thus acting as a functional pendant group; 2) The polymer backbone itself can be composed of repeating donor units, which are directly coupled with acceptor components acting as pendant groups to form D-A emissive sites; 3) The polymer main chain can be composed of alternating donor and acceptor units; 4) Both donor and acceptor groups can be installed as separate pendant groups on a non-conjugated backbone, producing TADF by a through-space charge transfer interaction. Other functional groups can also be added either as pendant groups or as part of the chain to act as hosts or as non-conjugated spacers, ensuring good charge balance and triplet confinement. While these specific strategies are the most frequently reported and hence the ones highlighted in this section, this list, with properties summarized in Table S13, is by no means exhaustive. The considerable breadth and sophistication of modern polymer chemistry combined with the combinatorial nature of D-A TADF emitters allows for practically limitless innovation in this area.ref. ref959
Polymers with Pendant TADF Emitters
One of the simplest design strategies for TADF polymers is to attach known small-molecule TADF emitters as pendant groups to the main chain of the polymer. This main chain can be optoelectronically inert or active, and the final material can be prepared by either post-synthetic modification of the main chain polymer, or by polymerizing monomers that contain an embedded TADF motif. For example, by grafting a TADF emitter PXZ-DP-Cz onto a polycarbazole backbone, Xie et al.ref. ref960 reported a series of efficient bluish-green polymers PCzDP-x (Figure ). The polycarbazole backbone in this case not only improved the charge transport compared to aliphatic chains, additionally it also acts as a host due to its high T1 level so as not to quench triplet excitons of the TADF emitter group. Furthermore, incorporating N-ethylhexylcarbazole or N-hexylcarbazole substituted monomers into the main chain electronically isolates the pendant TADF emitter and prevented aggregation-caused quenching (ACQ) in the ‘self-hosting’ non-doped polymer. Polymer PCzDP-10 contained 10% mole fraction of TADF-containing monomers and showed the highest delayed contribution to the total emission (72%, τd = 2 μs). The ΦPL are as high as 67% in toluene and 74% in the neat film, indicating their promise as materials in SP-OLEDs. The non-doped SP-OLED with PCzDP–10 showed an EQEmax of 2.8%, although this increased to 5.9% when PCzDP–10 was used in conjunction with mCP in a 1:1 ratio. By incorporating an additional small molecule TADF emitter DMAC-DP-Cz as a sensitizer a considerably higher EQEmax of 16.1% at a luminance of around 100 cd·m–2 was achieved (Table S13), all while maintaining CIE coordinates clustered around (0.24, 0.40).
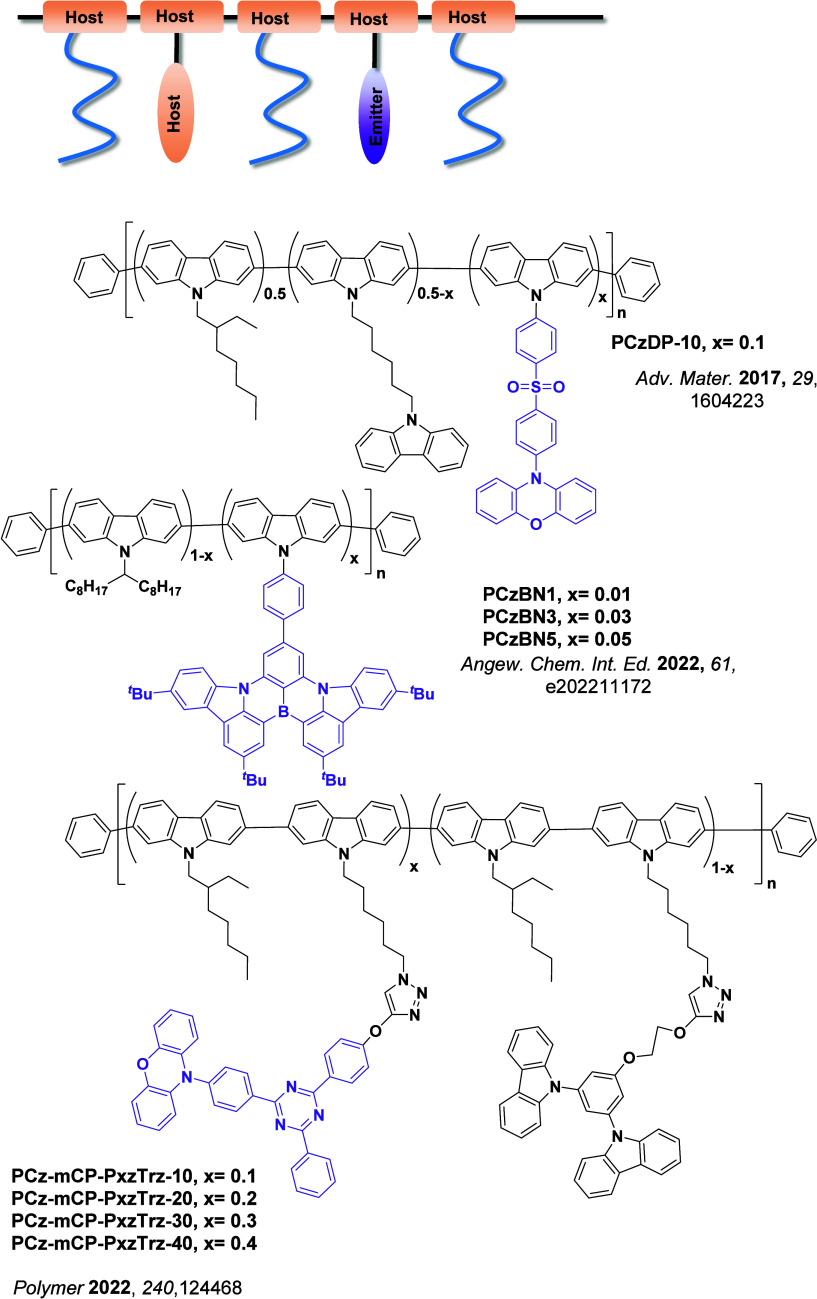
Using a similar approach, the same group reported the design and synthesis of a MR-TADF polymer, using a green MR-TADF emitter (BN) as a pendant group onto a polycarbazole backbone with polymers of molecular weights ranging from 4.4–10.0 kDa and polydispersity indices (PDI) of between 1.40–1.87. Among the doped SP-OLEDs (60 wt% polymer in mCP), those with polymers PCzBN1 and PCzBN3, with 1 or 3% of emitter-containing monomers, showed the best performance with EQEmax of 17.8 and 17.3%, respectively (Table S13). The SP-OLED with PCzBN5, containing 5% emitter monomer, showed a lower EQEmax of 13.3%. This decrease was attributed to increased ACQ due to the lower content of alkyl-carbazole monomers, which act to separated emissive monomers and improved solubility. The CIE coordinates of the SP-OLEDs with PCzBN1, PCzBN3, and PCzBN5 were (0.10, 0.43), (0.12, 0.54), and (0.11, 0.53), respectively. Compared to an EQEmax of 16.3% for SP-OLED based on just the use of BN as the emitter, the EQEmax for the polymer-based devices with PCzBN1 and PCzBN3 increased to 17.8 and 17.3%, respectively. This enhancement in device performance was attributed to the improved solubility and film morphology of the polymer compared to its monomer counterpart, while maintaining the same emission color and TADF performance. The intrinsic advantage of MR-TADF emitters was also conferred to the polymer OLEDs, with a resulting narrowband emission (FWHM of around 30 nm) for all fabricated devices.ref. ref961
Following the same design strategy, Zong et al.ref. ref962 grafted both TADF and host units as pendants onto a polycarbazole backbone in an effort to decrease ACQ. Together with small-molecule TADF emitter PXZ-TRZ, “self-hosted” TADF polymers containing a modified mCP monomer (Figure ) were synthesized with molecular weights of between 7.5–8.0 kDa and PDIs of between 1.5–1.6. Polymers PCz-mCP-PxzTrz-x with varying portions of emitter molecules (x, ranging from 10 to 40%) were produced, with PCz-mCP-PxzTrz-30 (30% emitter) showing the highest ΦPL of 39% in the neat film (in air) at a λPL of 540 nm. The non-doped SP-OLED showed an EQEmax of 15.3% and CIE coordinates of (0.35, 0.53). These polymer properties compare favorably to the intrinsic TADF performance of the PXZ-TRZ, reflected in the lower EQEmax of 12.5% in a doped vacuum-deposited device (6 wt% in CBP).ref. ref963
To raise the triplet energy of the polymer backbone, which is especially important for blue emission using conjugated polymers like polyfluorene, Yang et al.ref. ref964 inserted a 3,3′-dimethyldiphenyl ether group into the backbone to regulate the conjugation length. As a result, the triplet energy of the polymer increased from 2.16 to 2.58 eV for PFDMPE-R01 to PFDMPE-R10 (Figure ) as the ratio of the nonconjugated ether component increased. The synthesized polymers had molecular weights of between 83–132 kDa with PDIs of between 1.6–1.8. When a red TADF emitter ROC8 ref. ref964 was introduced as a pendant unit, effective energy transfer from the backbone to the grafted emitter was achieved, with all the polymers showing red emission similar to that of ROC8. The ΦPL of neat films of the polymers improved from 18 to 55% with increasing ether component. The device performance with PFDMPE-R05 (Figure , with 5% mole fraction of emitter-containing monomers) was the best in the study, with an EQEmax of 5.6% at λEL of 606 nm. Severe efficiency roll-off, decreasing by 82% at 500 cd·m–2, was reported and attributed to the long τd of 126 μs, thus allowing triplet quenching processes to dominate at higher luminance.
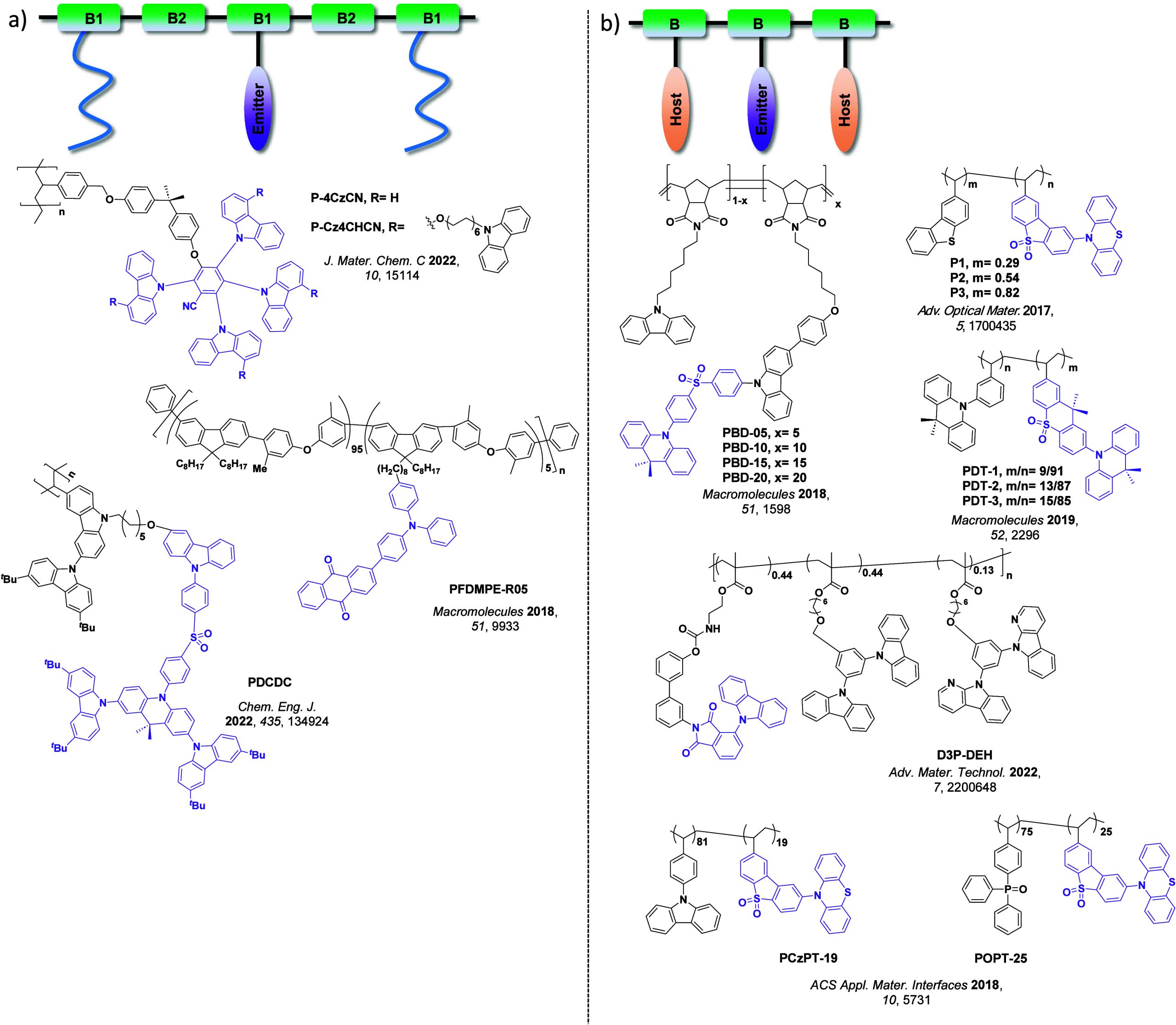
Ban et al.ref. ref965 similarly used a non-conjugated alkyl backbone alongside modified 4CzCN units to create a TADF polymer with a molecular weight of 7.5 kDa and a PDI of 1.5. The bulky 4CzCN units also showed AIE and bestowed the polymer P-4CzCN (Figure ) with neat film ΦPL of 37% at λPL of 472 nm and a τd of 1.5 μs. Non-doped SP-OLEDs showed an EQEmax of only 3.6% with CIE coordinates of (0.23, 0.39). However, when non-conjugated alkyl-carbazole were attached to the emitter unit in a semi-dendritic fashion (P-Cz4CzCN, Figure ), the increased encapsulation of the TADF-core in the self-hosting monomers led to much higher neat film ΦPL of 65% at λPL of 489 nm (Table S13). The EQEmax of the non-doped SP-OLEDs also increased significantly to 11.5%, with somewhat similar CIE coordinates of (0.24, 0.47). As well as presumably alleviating ACQ between emissive monomers, films of the encapsulated dendrimeric TADF polymer were also much more resistant to application of orthogonal solvent, which potentially simplifies the production of fully SP-OLEDs by allowing layers above the EML to also be processed from solution.
A similar approach was reported by Li et al.ref. ref966 using a non-conjugated backbone decorated with carbazole groups, themselves terminally functionalized with an asymmetric dendritic TADF emitter unit (DMAC-DPS-Cz). The resulting polymer PDCDC (Figure ), with a molecular weight of 12.7 kDa and a narrow PDI of only 1.16, showed a greenish-blue emission at 496 nm and a ΦPL of 68% in neat films (Table S13). The non-doped SP-OLEDs showed an EQEmax of 9.0% at CIE coordinates of (0.23, 0.39). Comparing this material to previous examples, it becomes clear that careful tuning of the overall carbazole content – either through monomer constitution by incorporating host moieties like mCP in the polymer by copolymerization, or simply by blending the polymers with a supporting host – is vital to ensure bright TADF polymer films.
Polyolefin is an alternative non-conjugated backbone that has also been explored in TADF polymers. Using a monomer containing the emitter DBTO2-PTZ, Li et al.ref. ref967 developed a series of TADF copolymers PCzPT-x and POPT-x containing either hole-transporting carbazole and electron-transporting phosphine oxide spacer monomers, respectively. These copolymers showed relatively small ΔE ST values between 0.05–0.13 eV and ΦPL up to 36%. POPT-25 and PCzPT-19 (with number representing mole fraction of emitter monomer, Figure ) were identified as the best performing of their respective series among the POPT-x and PCzPT-x polymers, with molecular weights of 27 and 16 kDa and PDIs of 1.5 and 1.8, respectively. The phosphine oxide pendant of the polymer POPT-25 was designed to work similarly to the common polar host, DPEPO. A moderate ΦPL of 52% at was obtained for POPT-25 in toluene, while the control polymer PCzPT-19, with only a donor pendant, only exhibited a ΦPL of 25% (Table S13). Even though both polymers share the same pendant emitter moiety, the k RISC of the two polymers deviated considerably, at 1.9 × 105 s–1 for PCzPT-19 and 8.1 × 105 s–1 for POPT-25, revealing that the polarity of host pendants has a significant impact on the TADF kinetics. A yellow SP-device with POPT-25 (10 wt% in mCP) showed an EQEmax of 5.2% with CIE coordinates of (0.36, 0.50). A lower EQEmax of 1.2% was obtained for the SP-OLED with PCzPT-19, attributed to the improved charge balance and higher ΦPL for POPT-25 (36% vs 21%) in 10 wt% doped mCP films. Non-doped devices of both polymers were also prepared; however, they displayed much lower efficiencies of under 1% EQEmax.
Polymers P1, P2, and P3 (Figure ) all based on the same emitter DBTO2-PTZ were also developed by Li et al.,ref. ref968 here using dibenzothiophene instead of carbazole as the host monomer. Polymers P1, P2, and P3 had molecular weights of 12, 30 and 23 kDa with PDIs of 1.3, 1.7 and 1.7, and ΦPL in neat films of 10.4%, 23.5% and 19.5%, respectively. This sequence of ΦPL indicated that the monomer ratio in P2 (approximately equal in TADF emitter and host monomers) gives the best environment for the TADF pendant group. The SP-OLEDs with P2 also had a performance very close to its carbazole-based analogue PCzPT-19, indicating only subtle differences in hosting environment when using the co-monomer based on carbazole and dibenzothiophene.
Li et al.ref. ref969 again adopted the same design strategy for blue TADF polymers PDT-x. These polymers incorporated a pendant 9,9-dimethyl-10-phenylacridine (BDMAc) with high triplet energy (E T = 3.38 eV) to act as host and spacer unit, in conjunction with the intrinsically high-performance blue TADF emitter DMA-TXO2.ref. ref970 The polymers PDT-1, PDT–2 and PDT-3 have molecular weights of 10, 11 and 21 kDa, with PDIs of 1.4, 1.7 and 1.7, respectively. In contrast to the high ΦPL of this emitter (ΦPL = 80% in 11 wt% in DPEPO), the ΦPL of the polymer neat films decreased to 42, 54, 46% for PDT-1, PDT-2, PDT-3, respectively (Table S13), most likely due to concentration quenching despite the excess of BDMAc spacer monomers. The best SP-OLED based on neat PDT-2 achieved an EQEmax of 5.3% at λEL of 436 nm and CIE coordinates of (0.15, 0.09), in comparison to an EQEmax of around 20% for the vacuum-deposited device based on DMA-TXO2.
Polynorbornene backbones have also been used for the development of TADF polymers. The high triplet energy (2.95 eV) of this subunit is suitable to prevent the quenching of triplets from TADF units to the polymer backbone, crucial for the design of high-triplet blue TADF polymers. A series of blue TADF polymers was reported by Zeng et al.,ref. ref971 with carbazole-containing monomers acting as hole injection units, and DMAC-DPS derivatives as emissive monomers linked to a norbornene backbone (Figure ). This design was chosen to avoid conjugation along the backbone and therefore suppress any red-shifting as monomer photophysics is effectively localized. The polymer molecular weight and branching can also be well-controlled because of the ring-opening metathesis polymerization conditions. The molar ratio of TADF monomers was varied (PBD-0, PBD-5, PBD-10, PBD-15, and PBD-20) to give polymers of molecular weight ranging from 5.8–8.0 kDa. The neat films of these polymers all showed blue emission at around 460 nm. The non-doped SP-OLEDs with PBD-10 showed an EQEmax of 7.3% at CIE coordinates of (0.20, 0.29). The SP-OLEDs with PBD-5, PBD-15 and PBD-20 showed EQEmax of 6.0, 7.1 and 6.7%, respectively, where the EQEmax tracked with the ΦPL of the polymers. The efficiency roll-off was severe though, with the EQE100 decreasing by between 45 to 53%, correlating with the relatively slow values of k RISC.
Cole et al.ref. ref972 engineered the polymer D3P-DEH, which contains a non-conjugated alkyl backbone grafted with three different side chains (Figure ). The emissive side chain contains a TADF emitter, while the second side chain contains the hole-transporting host material, mCP, and the third side chain contains a modified mCP unit (NmCP1), which is designed to act as an electron-transporting host material. The introduction of both electron and hole transporting units along the polymer backbone obviates the need for an external host material. The authors demonstrated this claim by comparing the performance of non-doped and doped devices. Even though the non-doped device exhibited a low EQEmax of 1.6% at CIE (0.27, 0.50), it still outperformed the doped device (30 wt% in 26DCzPPy) with an EQEmax of 0.6% (Table S13). This study also demonstrated the first report of a TADF “self-hosted” polymer SP-OLED deposited by inkjet-printing.
Donor Backbone with Acceptor Pendants
Zhu et al.ref. ref973 reported a TADF polymer, PAPTC, comprised of a donor-containing backbone where some of the donors are covalently linked to pendant acceptor groups. The conjugated backbone of PAPTC (Figure ) consists of acridan and carbazole groups linked via the 3- and 6-positions. A pendant triazine acceptor was linked to each of the acridan monomers to form the TADF subunits, while the carbazole groups provide both spacing to avoid ACQ of the TADF emitters as well as (presumably) hole transport properties. DFT calculations confirmed that the HOMO is delocalized over the entire polymer backbone, while the LUMO is localized on the pendant acceptor. PAPTC has a ΔE ST of 0.13 eV, and the ΦPL in toluene was increased from 22 to 40% after bubbling with N2. Non-doped SP-OLEDs showed an EQEmax of 12.6% with λEL at 521 nm (Table S13).
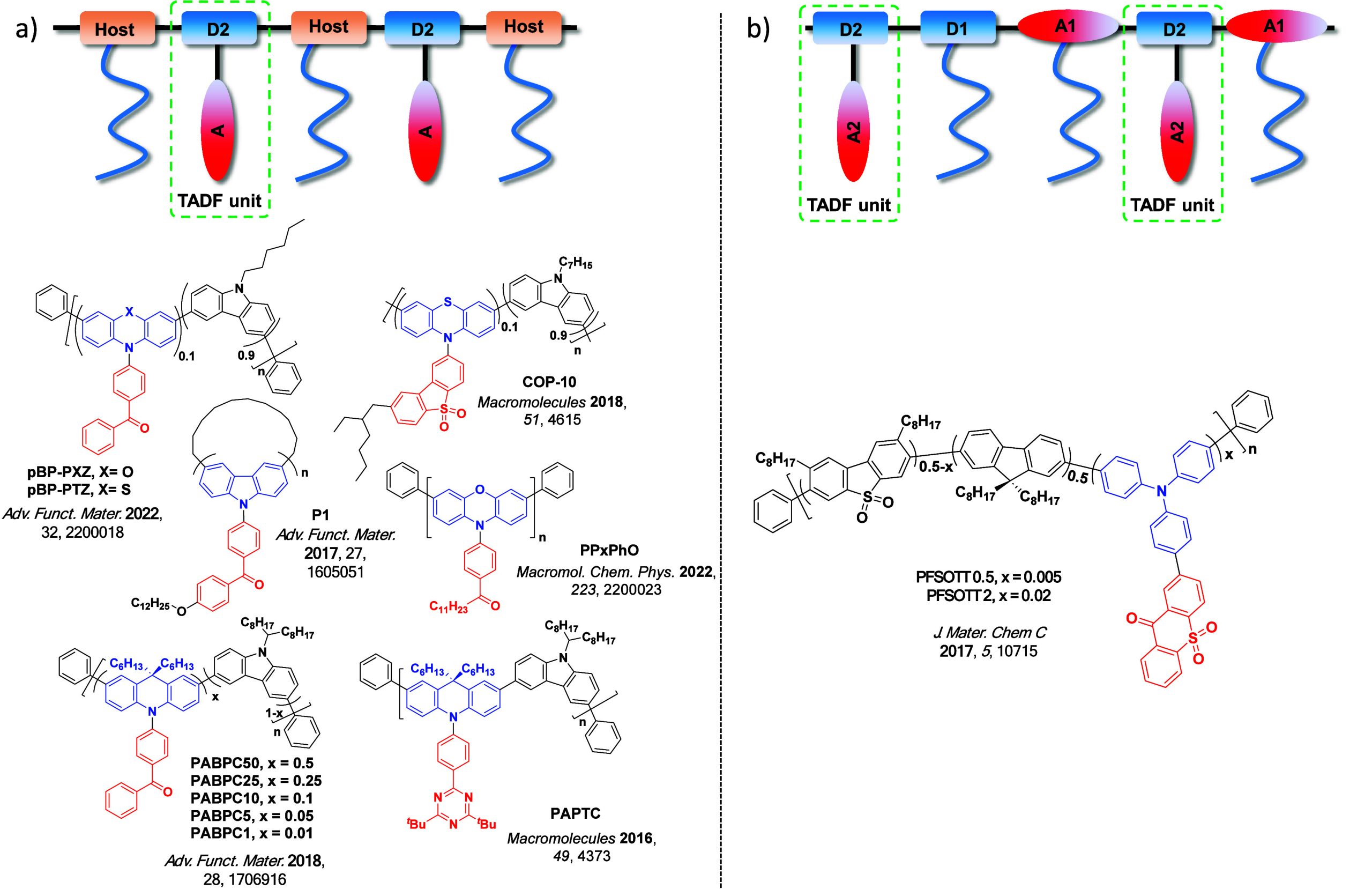
Yang et al.ref. ref974 employed a similar strategy to produce a series of TADF polymers with molecular weights of between 6.3–17.6 kDa, using the same acridan-carbazole backbone but instead incorporating a benzophenone acceptor coupled to the acridan. The best performing polymer, PABPC5 (Figure ), contained 5% of the TADF monomer and displayed a high ΦPL of 77% (Table S13). A non-doped SP-OLED using PABPC5 showed an EQEmax of 18.1% with CIE coordinates of (0.40, 0.56), and low efficiency roll-off with an EQE1000 of 17.8%.
A copolymer COP-10 (Figure ) using 10% DBTO2-PTZ as the TADF monomer and phenothiazine-carbazole backbone was reported by Liu et al.ref. ref975 The SP-OLEDs with COP-10 doped at 10 wt% in a mixed TCTA:TAPC (65 wt%:25 wt%) co-host showed an EQEmax of 15.7%. However, significant efficiency roll-off was observed, with a reduction in EQE of 76% at a luminance of 100 cd m–2.
A similar approach was used by Zhao et al.ref. ref976 who reported polymers pBP-PXZ and pBP-PTZ consisting of alkyl-substituted carbazoles copolymerized with 10% of either PXZ or PTZ donors. These donors were themselves coupled to benzophenone to give TADF emissive subunits within the polymers (Figure ), which both have a molecular weight of around 16 kDa and a PDI of 1.7. In neat films both polymers showed λPL at 550 nm yet diverging ΦPL of 82% (pBP-PXZ) and 48% (pBP-PTZ) (Table S13). The oxygen containing pBP-PXZ showed a slight faster τDF of 1.29 μs compared to 1.55 μs for pBP-PTZ. The non-doped SP-OLED with pBP-PXZ showed a higher EQEmax of 13.7% [CIE coordinates of (0.52, 0.48)] compared to its sulfur-containing counterpart with an EQEmax of just 7.9% at CIE coordinates of (0.50, 0.49). The non-doped device with pBP-PXZ showed a lower efficiency roll-off of 35% at 1000 cd·m–2 while the pBP-PTZ devices were not able to achieve a brightness of 1000 cd·m–2. In devices using 10 wt% pBN-PXZ doped in CBP, the EQEmax increased to 23.1% and the efficiency roll-off was reduced to 16%, with the EQE1000 still exceeding 19%. These reported devices are the best performing SP-OLED using a TADF polymer as emitter to date, and again highlight the importance of tuning the host-monomer content.
Wei et al.ref. ref977 demonstrated a new polymer P1 with macrocycle design to achieve TADF (Figure ), based on the linking together of non-TADF monomers and where the macrocycle gains TADF activity by virtue of the increased donor conjugation in the cyclized material. DFT calculations and photophysical characterization of P1 (2 wt% in polystyrene) showed that this material has a ΦPL of 71% (3% for the monomer) of which delayed fluorescence (ΦDF) contributed 51% with ΔE ST of 0.19 eV. Unfortunately, no OLEDs were prepared to test the performance of P1, although a subsequent study investigated the stepwise effects of this conjugation expansion in a series of non-cyclic oligomers.ref. ref977
Zhang et al.ref. ref978 reported a polymer consisting exclusively of TADF monomers, PxPhO (Figure ), with a poly(phenoxazine) backbone each bearing a ketone acceptor unit. The polymer maintained a small ΔE ST of 0.07 eV, similar in magnitude to its monomer unit (ΔE ST = 0.05 eV), although the emission of the polymer in toluene solution (λPL = 557 nm) is red-shifted compared to the monomer by approximately 70 nm (Table S13). A comparison of the fluorescence lifetimes (prompt and delayed) between monomer and polymer showed that the τp of the polymer is three times shorter than its the monomer (3.63 and 12.02 ns), while the claimed τd of the monomer was almost seven times longer than its polymer (0.054 and 0.008 μs). The ΦPL of the polymer (> 60%) and monomer (∼50%) are both similar, implying that these differences in lifetimes did not arise from faster non-radiative decay in the polymer, but instead due to faster RISC. Furthermore, in doped SP-OLEDs with an EML consisting of 80 wt% mCP (host) and 20 wt% emitter (monomeric PxPhO or polymeric PPxPhO) the device with the polymeric emitter showed a higher EQEmax of 11.8% (λEL = 550 nm) compared to the device with PxPhO (EQEmax = 8.8%; λEL = 520 nm). The authors attributed the enhanced performance of the polymer OLED to the better film-forming properties of PPxPhO compared to PxPhO.
Wang et al.ref. ref979 prepared a series of TADF polymers PFSOTT-x (Figure ) composed of a TADF monomer containing a triphenylamine donor and a thioxanthone-dioxide acceptor, alongside an alternating fluorene and dibenzothiphene-S,S-dioxide backbone. The resulting polymers have a molecular weight of 48.2–58.3 kDa with a broad PDI of over 2. With increasing proportion of the TADF monomer, the PL spectra of the polymers gradually red-shifted from blue to orange in the neat films. Despite a remarkably high ΦPL of 89% in the neat film for PFSOTT0.5 (0.5% of the TADF unit), the non-doped OLED achieved an EQEmax of only 2.6% with CIE coordinates of (0.49, 0.49) (Table S13), an indication of poor charge balance in the device. Indeed, the EL performance was significantly improved when PFSOTT2, (2% TADF unit) was dispersed with 40 wt% in an mCP matrix, giving an EQEmax of 19.4% at λEL at 592 nm, which was consistent with the near unity ΦPL of this polymer emitter in mCP. This result once again highlights the importance of designing TADF polymers that include functional groups to support both TADF emission and charge transport in a device context.
Chiral small molecule TADF emitters have been shown to emit circularly polarized luminescence (CPL) (See Section sec7 ). Hu et al.ref. ref980 developed the first chiral conjugated poly(carbazole-ran-acridine) polymer P10 which contained a stereogenic alanine pendant groups alongside achiral TADF co-monomers. The polymer has a molecular weight of 10 kDa and a PDI 1.7 (Figure ). By using a polymeric emitter instead of a small molecule emitter, better thermal stability and easier solution-processability was achieved. The g lum of P10 was −1.39 × 10–3. AIE was reported in the solid state, and neat films of P10 exhibited a green emission with ΦPL of 10.3% and τd of 1.3 μs (Table S13). The doped SP-OLED (5 wt% in mCP) showed green emission with CIE coordinates of (0.36, 0.52); however, the EQEmax was a rather low 0.87%, consistent with the low ΦPL.
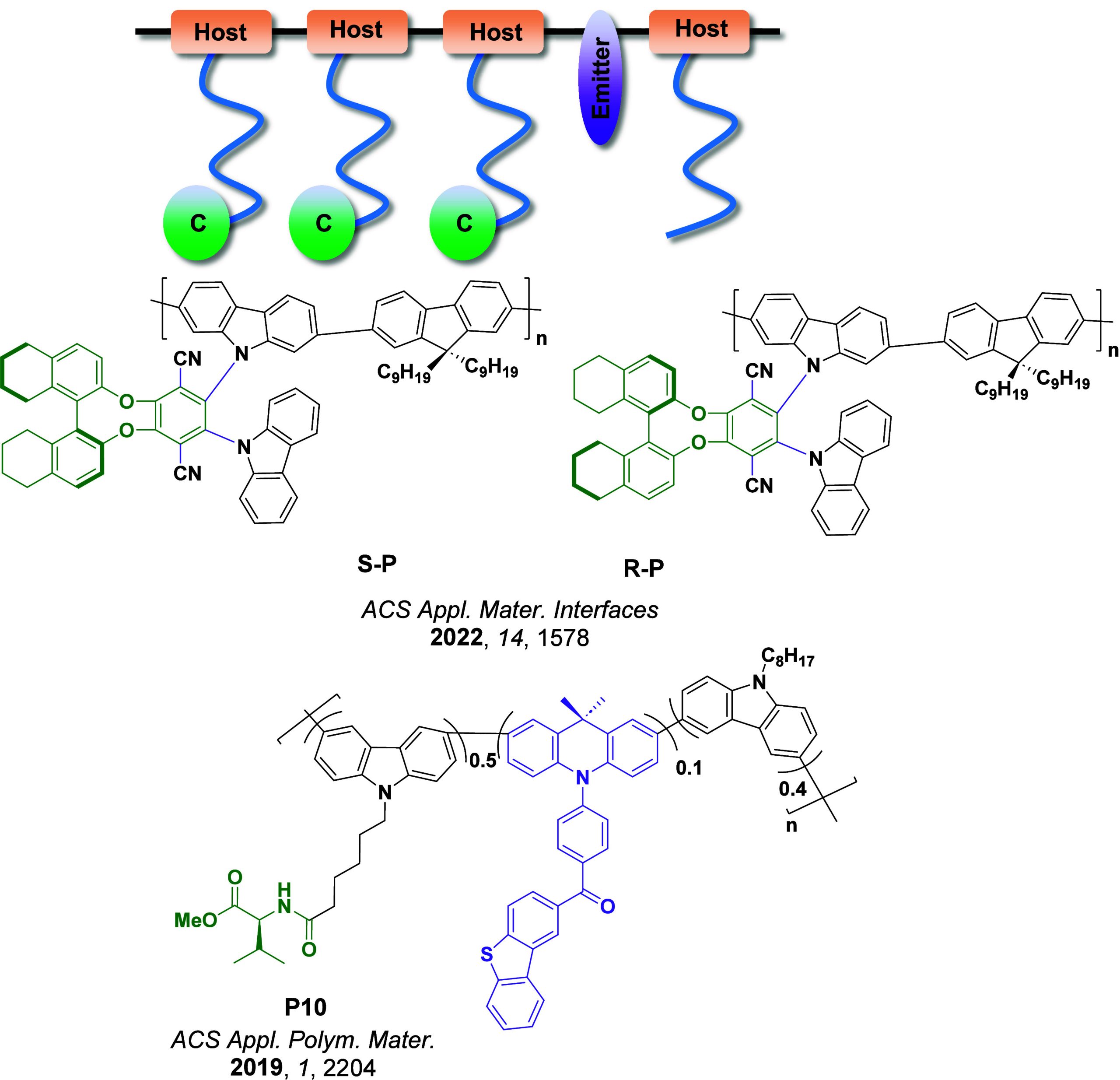
Inspired by this work, Teng et al.ref. ref981 reported CP-TADF polymers and SP-OLEDs. In contrast to the previous example, in this case the TADF unit was itself intrinsically chiral and polymerized by one of its carbazole donor into the main chain alongside a fluorene co-monomer (S-P and R-P, Figure ). Photophysical investigations of the neat film showed practically identical λPL of 560 and 562 nm for the S-P and R-P enantiomers, respectively. The emission of the polymers as 10 wt% doped films in mCP were blue-shifted by 13 nm compared to the neat film (ΦPL of 76% for S-P and 72% for R-P, with τd of 2.3 and 1.6 μs, respectively). The g lum values are 1.9 × 10–3 (S-P) and −1.9 × 10–3 (R-P). Doped SP-OLED devices (10 wt% in mCP) showed EQEmax of 15.8% for S-P and 14.9% for R-P, with a low efficiency roll-off at 1000 cd·m–2 of 22 and 15%, respectively. Both devices showed yellow-greenish emission with CIE coordinates of (0.41, 0.57), while the g EL values were +1.6 × 10–3 (S-P) and −1.5 × 10–3 (R-P).
Freeman et al.ref. ref982 proposed a new strategy to induce TADF in conjugated polymers by including an orthogonal acceptor group at the bridgehead position of alternating spiro-fluorene repeat units. In this way the pendant acceptor group is not directly attached to the donor units in the main chain, and instead the CT states and TADF are achieved as a result of through-space interactions. In ASFCN (Figure ), the electron and hole wave functions were consequently spatially separated due to the non-conjugated sp3 connection between acceptor pendants and donor backbone units. The low ΦPL of 16% at λPL of around 520 nm (Table S13) was attributed to a relatively high rate of non-radiative decay from a 3LE state and a low radiative decay rate of the singlet state, a result of the near zero overlap between frontier molecular orbitals.
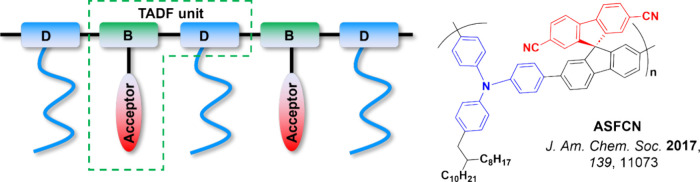
Main-Chain D-A Type TADF Polymers
Nikolaenko et al.ref. ref983 proposed a series of main-chain TADF polymers based on an “intermonomer TADF” strategy that induces TADF properties from the linked donor and acceptor repeating units (Figure ). The TADF polymer LEP (Figure ) was synthesized via Suzuki polymerization using a feed ratio of 5%:50%:45% of the three monomers, containing donor (amine), acceptor (triazine), and spacer (alkyl chain) units. The triazine unit in this case supported both the TADF emission by interacting with the amine group as well as contributed to charge transport to tune the recombination zone. As highlighted in previous examples, the large content of spacer monomers helped to maintain a uniform dispersion of TADF-emitting units in the polymer and mitigate ACQ. A moderate ΦPL of 43% and ΔE ST = 0.22 eV were obtained for LEP. The green non-doped SP-OLED device showed an EQEmax of 10% at CIE coordinates of (0.32, 0.56).
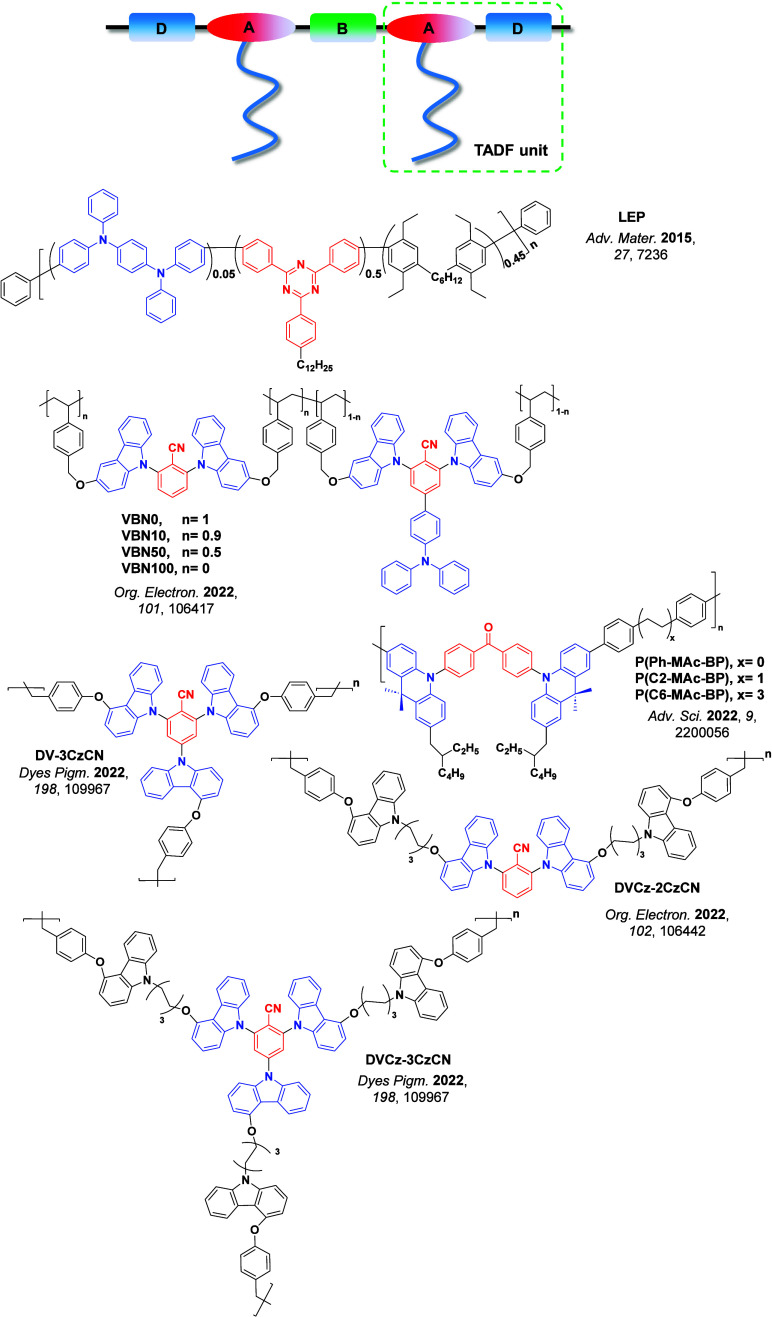
To observe the impact of including different spacers on the TADF properties of a main-chain-emissive polymer, Philipps et al.ref. ref984 synthesized a group of three polymers consisting of a benzophenone acceptor unit and two acridan donor units, connected in each monomer with non-conjugated spacers of different length (Figure ). Non-doped SP-OLEDs with longer non-conjugated spacer units displayed improved device performance, with EQEmax of 2.9% using P (Ph-Mac-BP) with a fully conjugated phenyl spacer, increasing to 6.7% for P(C2-Mac-BP) with an ethyl spacer and 7.1% for P(C6-Mac-BP) with a hexyl spacer. Furthermore, the SP-OLED with P(C6-Mac-BP) showed the best efficiency roll-off, with the EQE dropping by only 8% at 100 cd m–2 and 30% at 1000 cd m–2 (Table S13).
In contrast to linear polymers, Sun et al.ref985,ref986 prepared branched polymers comprised of a carbazole-benzonitrile emissive center and carbazole spacer units, that were thermally crosslinked by annealing after spin-coating. The resulting cross-linked polymer films showed blue emission at around 470 nm, with ΦPL values of 36, 54 and 68% for DV-3CzCN (without carbazole spacing units), DVCz-3CzCn, and DVCz-2CzCN (a linear analogue, Figure ), respectively. Non-doped thermally annealed devices exhibited sky-blue emission with CIE coordinates of (0.16, 0.31), (0.15, 0.30) and (0.16, 0.21). The SP-OLEDs with DVCz-2CzCN and DVCz-3CzCn showed an EQEmax of around 6%, while the device with DV-3CzCN only showed an EQEmax of 0.8% (Table S13). This difference in performance was explained as arising from the isolation of the TADF core units in the branched polymers, which minimized ACQ.
A second approach was also reported by Sun et al.,ref. ref987 using two small molecule TADF subunits, 2CzBn and 2CzTBn. These were linked with vinyl-benzyl groups to build a copolymer with different ratios of the emitter units (VBNx, Figure ). Two polymers containing different ratios of 2CzBn and 2CzTBn, VBN10 containing 90% 2CzBn and 10% 2CzTBn and VBN50 containing 50% 2CzBn and 50% 2CzTBn were reported, as well as control polymers containing each of the TADF subunits individually. According to the authors the 2CzBN units act primarily as hosts while the 2CzTBN units act as guest emitters in this polymer. Both copolymers showed similar photophysical properties in the neat film, with λPL at 488 nm and 497 nm and ΦPL of 74% and 70% for VBN10 and VBN50, respectively. The non-doped SP-OLEDs with VBN10 exhibited an enhanced performance with an EQEmax of 11.4% at CIE coordinates of (0.20, 0.38) compared to the device with VBN50, which showed an EQEmax of 9.1% at CIE coordinates of (0.21, 0.41). The similar emission spectra of the two polymers indeed supported the hypothesis that one TADF subunit acts as host for the other. Comparing to control polymers containing only one of the monomers (VBN0, VBN100) the copolymers also showed enhanced ΦPL and shorter τd in the neat film. Accordingly, comparison devices using the homopolymers with for VBN0 and VBN100 showed much lower performance with EQEmax of only 3.1 and 4.9%, respectively.
Through Space TADF Polymer
In recent years it has become clear that TADF can also arise effectively as a result of through-space interactions between donor and acceptor moieties (See Section sec12 ). Shao et al.ref. ref988 applied this strategy to produce blue TADF polymers based on a nonconjugated polyethylene backbone, with TSCT interactions between the pendant acridan-based donors and the triazine acceptors. The copolymers P-Ac95-TRZ05 and P-TBAc95-TRZ05 (Figure ) were synthesized accordingly, although only P-Ac95-TRZ05 exhibited TADF, associated with a small ΔE ST of 0.019 eV and a ΦPL of 60% in the neat film. As a control system, P-TBAc95-TRZ05 showed no TADF due to the too large inter-chromophore distances between the TBAc and TRZ pendants. The non-doped SP-OLEDs with P-Ac95-TRZ05 showed sky-blue electroluminescence at CIE (0.18, 0.27), an EQEmax of 12.1% and low efficiency roll-off with an EQE1000 of 11.5% (Table S13).
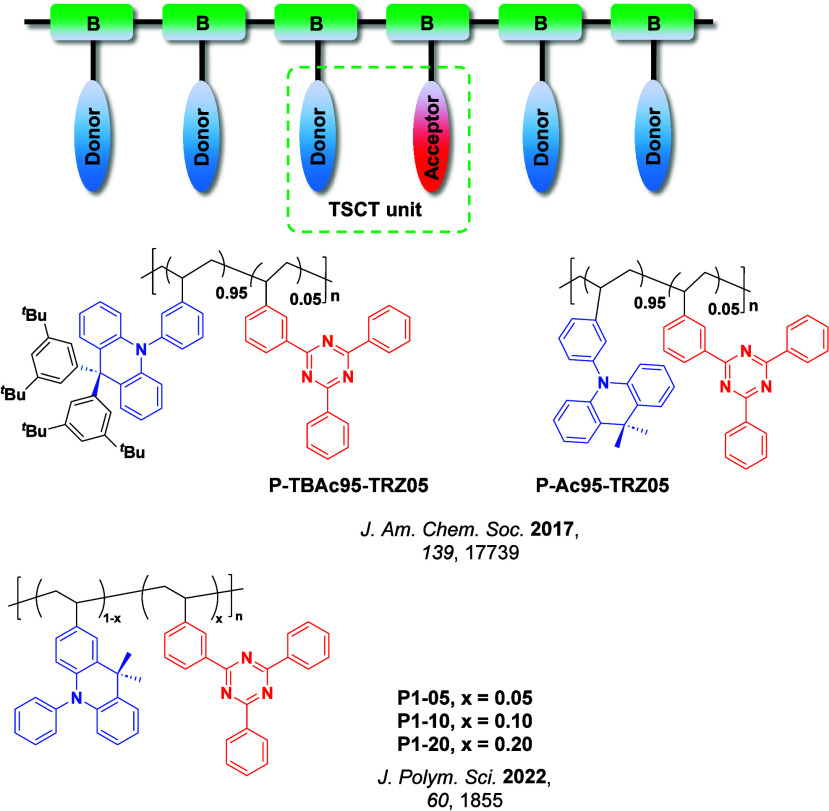
The same group reported a second class of TSCT-TADF polymersref. ref989 with the same donor and acceptor building blocks but different connectivity from the acridine donor to the main chain (P1-x series, Figure ). Compared to their previous work where the donor unit was attached to the backbone via a phenyl ring attached to the N-atom of the acridine ring (10-phenyl), here the donor unit was connected directly at the 2-position of the acridine ring. Compared to P-Ac95-TRZ05, the co-polymers P1-05, P1-10 and P1-20 (with 5, 10 and 20% TRZ, respectively) showed red-shifted emission at 485, 489 and 492 nm as neat films and lower ΦPL values of 54, 47 and 45%, respectively (Table S13). The ΔE ST values for the P1-x copolymers where also twice as large as for P-Ac95-TRZ05, at around 0.04 eV. In good agreement with these optical properties, non-doped device performance using the standout P1-05 polymer showed an EQEmax of 11.3% at CIE coordinates of (0.20, 0.37). This study demonstrates nicely that, just as in small molecule D-A TADF emitters, the relative geometries of the donor and acceptor groups play a crucial role in the design of TADF polymers. Understandably, this is all the more crucial for TSCT materials, where these geometries cannot be as directly controlled by covalent design and bond placement.
In summary, a wide range of TADF polymer design strategies has emerged in recent years, with selected relevant examples presented here to highlight their typical photophysical and electroluminescent properties. Since polymers can exhibit the advantage of reduced ACQ and superior solution processability, they remain a promising class of emitters for high-performance SP-OLEDs. However, to compete with SP-OLEDs with small molecule emitters–especially in terms of properties like color purity (FWHM) and EQEmax–still more materials development is still required.
TADF Dendrimers
Fluorescent, phosphorescent and now TADF polymer-based emitters for SP-OLEDs have all been widely reported. Despite their performance and suitability for SP-OLEDs, control purity and batch-to-batch variation in materials composition, polydispersity, branching and other structural defects during polymerization are intolerable concerns for commercial display production, and polymers are themselves nearly impossible to purify following synthesis to the level required by industry by standard methods.
One alternative method that can avoid these issues associated with polymers is to instead employ dendrimers. These are large and readily solution-processable macromolecules, but which also have well-defined molecular structures that can be purified in the same manner as low molecular weight small molecules. The large and globular nature of these dendrimer emitters also aids in protecting an emissive core from aggregation and thus in suppression of non-radiative decay pathways, opening the possibility of efficient non-doped devices. TADF dendrimers are indeed usually composed of a core emissive unit surrounded by optically inert dendron units that both shield the core from intermolecular interactions, and yet can also contribute to modulating charge transport within the film. The dendron units themselves can also act as donors, thus producing D-A TADF materials. Some donor dendrons can be coupled directly to a central acceptor core in a conjugated manner (i.e. conjugated dendrimers, Figure , while in other cases conjugation between the donor dendrons units and the emissive core is broken for example by using alkyl chains (non-conjugated dendrimers, Figure ). In this section we will review recent progress towards high efficiency SP-OLEDs using dendrimer materials and summarize their photophysics and device performances in Table S14.
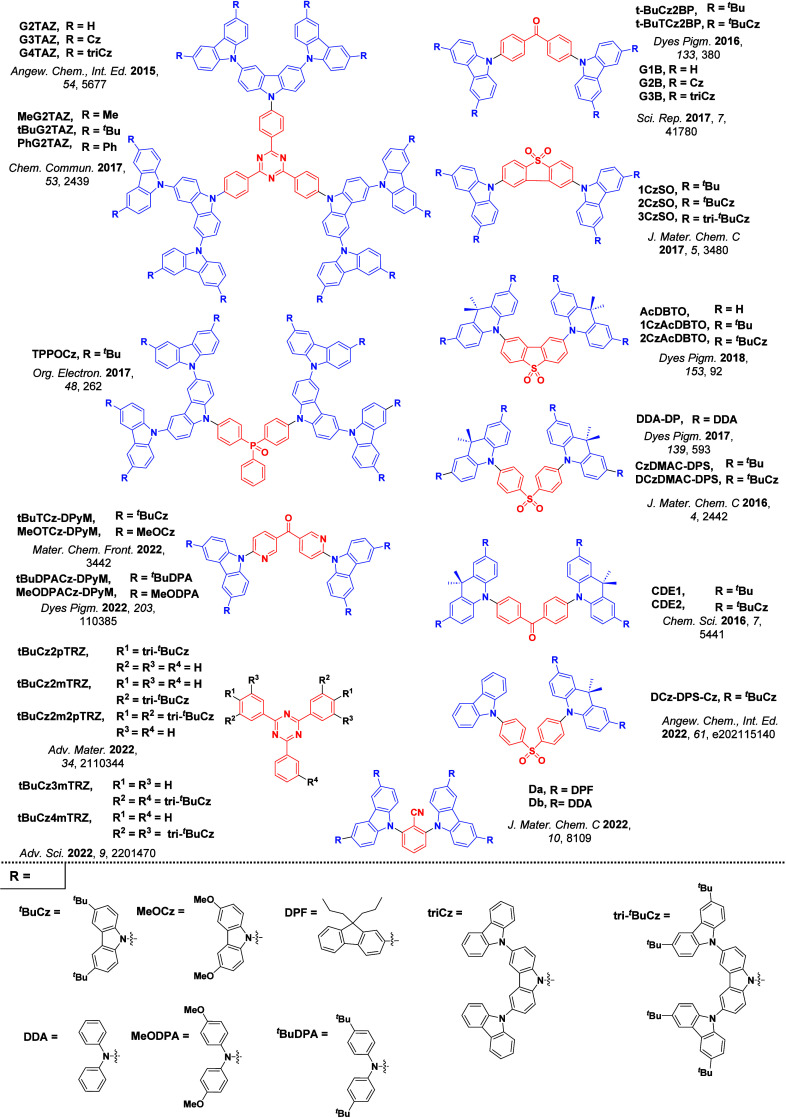
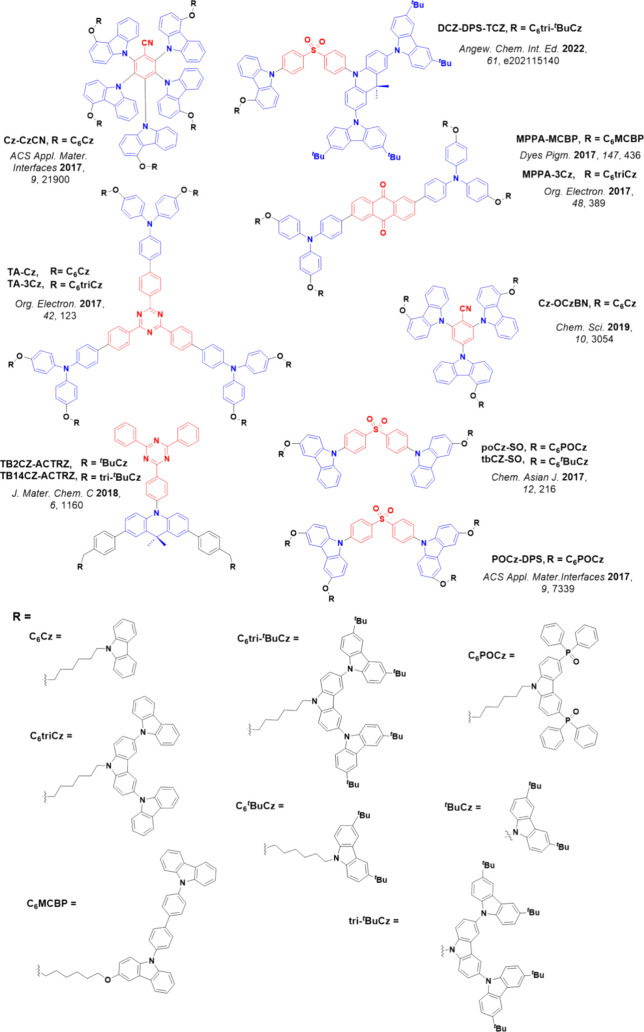
Conjugated Dendrimers
The first TADF dendrimers, reported by Albrecht et al. in 2015, contained carbazole donor dendrons coupled to a central triazine acceptor.ref. ref990 Dendrimers with one of three generations of donor dendrons, G2TAZ, G3TAZ and G4TAZ (Figure ), were reported, with the most promising G3TAZ producing non-doped SP-OLEDs with a modest EQEmax of 3.4%. The efficiency roll-off at 100 cd·m–2 was ∼19%, but this increased significantly at 1000 cd·m–2 where the efficiency roll-off was ∼56% (Table S14). G3TAZ has a ΦPL of 56% in the neat film, while when placed in dilute toluene solution, it increases to 100%, which suggested that the design of this dendrimer structure did not completely preventing ACQ in the neat film.
The use of an alternate donor dendron to G2TAZ with tert-butyl groups (tBu) decorating the peripheral carbazole units, tBuG2TAZ (Figure ),ref. ref991 gave a much-improved device performance with an EQEmax of 9.5% and an efficiency roll-off of just 1% at 100 cd·m–2. SP-OLEDs using congener dendrimers where the tBu substituents were replaced with Me, Ph, and H, (Figure ), produced EQEmax of 9.4%, 8.2% and 6.0%, respectively, demonstrating the value of these steric blocking groups at keeping the emitter dendrimers suitably isolated from each other.ref990,ref991 Further, each of these devices displayed excellent efficiency roll-off at 100 cd·m–2 where the EQE decreased by only 1%, 0% and 0%. There was also little variation in emission color between each of these devices, with λEL ranging from ∼500 nm to ∼505 nm (Table S14). Building upon the device with tBuG2TAZ (EQEmax 9.5%), a much higher EQEmax of 17.0% was achieved for non-doped devices based on emitter tBuG2B,ref. ref992 in which a diphenylketone acceptor was used instead of triazine as the acceptor and core of the dendrimer (Figure ). The ΦPL of 74% in the neat film was higher than that measured in toluene solution (47%). The relatively low efficiency roll-off of 19% at 1000 cd·m–2 in the device was attributed to both the small ΔE ST of 0.08 eV and the fast τd of 2.2 μs. The same emitter was also previously reported as t-BuTCz2BP by Huang et al.ref. ref993 exhibiting a lower ΦPL of 41% (in air), a shorter τd of 0.57 μs, and a lower EQEmax of only 4.3%, likely due to a less elaborate and optimized device stack. The other compounds in the study from Albrecht et al.ref. ref992 involved replacement of tBu for Me (MeG2B), OMe (MeOG2B) and Ph (PhG2B) (Figure ). The lower ΦPL (34, 17 and 41%) of these dendrimers led to lower EQEmax of 9.0, 6.4 and 8.8%, respectively, in the SP-OLEDs. The devices suffered from serious efficiency roll-off, with both MeG2B and PhG2B unable to achieve a brightness of 1000 cd·m–2, while the EQE decreased by 69% for the device with MeOG2B at this brightness. ACQ was surmised to be responsible for the low efficiency of MeOG2B. The non-doped SP-OLED with G2B (Figure ),ref. ref994 showed an EQEmax 5.7%. The use of the higher generation dendrimer G3B led to a further decrease in the EQEmax to 2.9%, again highlighting the importance of carefully balanced charge transport in the device EML, and the role of the dendron shell in supporting this.ref. ref994
Three dendrimers based on a triazine acceptor core and tBuTCz donor dendrons have been recently reported by our group that showed outstanding photophysics and device performance.ref. ref995 Two of the dendrimers are regioisomers, tBuCz2pTRZ, tBuCz2mTRZ (Figure ) while the third possessing a combination of both meta and para donor dendrons tBuCz2m2pTRZ. Dendrimers tBuCz2pTRZ, tBuCz2mTRZ exhibited similar photophysical properties in neat films, with λPL at 481 nm and 483 nm and ΦPL of 61% and 59% for each respectively. In contrast, tBuCz2m2pTRZ showed a red-shifted emission at 520 nm and a higher ΦPL of 86% (Table S14). The non-doped SP-OLEDs with tBuCz2pTRZ and tBuCz2mTRZ showed EQEmax of 18.5 and 19.9% and associated efficiency roll-offs at 100 cd·m–2 of 13% and 40%, at CIE coordinates of (0.23, 0.46) and (0.27, 0.53), all respectively. The device with tBuCz2m2pTRZ, however, showed an excellent EQEmax of 28.7% at CIE coordinates of (0.37, 0.57); however, the efficiency roll-off was suboptimal at 26%. Devices that incorporated 30 wt% OXD-7 within the EML showed a much-improved efficiency roll-off of 2% at 100 cd·m–2 while the EQEmax was maintained above 28%.
In a separate study,ref. ref996 we reported two dendrimers based on the same TRZ acceptor and different numbers of meta-connected tBuCz donor dendrons, tBuCz3mTRZ and tBuCz4mTRZ (Figure ). The non-doped SP-OLEDs with tBuCz3mTRZ and tBuCz4mTRZ showed improved performance compared to devices with tBuCz2mTRZ and tBuG2TAZ, with EQEmax of 23.7 and 23.8% at CIE coordinates of (0.35, 0.57) and (0.36, 0.58), respectively (Table S14). These examples showed that attaching the donor dendrons at the meta position and in large numbers is a good design strategy to increase device performance of TADF dendrimers, as this linking topology likely helps increase RISC.ref. ref119
A similar structure to tBuG2B was reported by Zhang et al.ref. ref997 who exchanged the diphenylketone acceptor with a dipyridinylketone acceptor (DpyM) and attached two 9́H-9,3́:6́,9́́-tercarbazole (triCz) donor dendrons containing either peripheral tBu or OMe groups (tBuTCz-DpyM, MeOTCz-DpyM, Figure ). In neat films the dendrimers emit at 495 nm for tBuTCz-DpyM and 514 nm for MeOTCz-DpyM, with ΦPL of 64 and 55%, respectively (Table S14). Both compounds exhibit delayed fluorescence with τd of 9.5 μs for tBuTCz-DpyM and 7.9 μs for MeOTCz-DpyM. The SP-OLED with tBuTCz-DpyM (8 wt% doped in mCBP) showed an EQEmax of 20.4% at CIE coordinates of (0.25, 0.48), while the device with MeOTCz-DpyM showed an EQEmax of only 9.2% at CIE coordinates of (0.37, 0.54).
A very closely related structure was reported by He et al.,ref. ref998 which contains diphenylamine-carbazole donor dendrons and either tBu or OMe groups decorating the periphery. The dendrimers tBuDPACz-DpyM and MeODPACz-DpyM (Figure ) both exhibit a red-shifted emission in neat film of 596 nm and 645 nm and a much-decreased ΦPL of only 11 and 3%, respectively, compared to their carbazole counterparts tBuTCz-DpyM and MeOTCz-DpyM. As a result of the low ΦPL, the EQEmax of the non-doped SP-OLEDs were low at 1.8% and 0.17%, respectively (Table S14).
Using the same design strategy, Li et al.ref. ref999 reported a dendrimer containing donor dendrons with DMAC as the innermost donor unit and carbazole peripheral groups. The neat film of dendrimer CDE1 (Figure ) emits at λPL of 520 nm and has a ΦPL of 77%. The non-doped devices showed an EQEmax of 13.8% and low efficiency roll-off at 1000 cd·m–2 of only 4%, which can be explained in part by the very short delayed lifetime of 0.52 μs. The CIE coordinates were (0.40, 0.54). The device emission mechanism was identified as a mixture of typical D-A TADF, along with exciplex emission occurring at the interface between the emitter and the TmPyPB electron transporting layer. A dendrimer with a higher generation donor dendron, CDE2 (Figure ) emits at λPL of 499 nm and has a comparable ΦPL of 75% (Table S14), but did not display the extra exciplex emission. Without this exciplex contribution the non-doped SP-OLEDs showed a much smaller EQEmax of 5.2%. The reduction in EQEmax was assigned to a mismatch of the work functions of the HTL and ETL in the device.
Replacing ketone with sulfone as the central acceptor unit but using the same donor dendron as in CDE1 gave the blue-emitting dendrimer CzDMAC-DPS (Figure ), which emits at λPL of 492 nm and has a ΦPL of 68% in the neat film. CzDMAC-DPS also has a small ΔE ST of 0.09 eV and a short τd of 1.5 μs. The SP-OLEDs showed an EQEmax of 12.2% at CIE coordinates of (0.22, 0.44) (Table S14), although this was accompanied by a much stronger efficiency roll-off (63% decrease at 1000 cd·m–2). The non-doped device with DCzDMAC-DPS, an analogue dendrimer but with a higher generation of donor dendron (Figure ), displayed a further blue-shift with CIE coordinates of (0.18, 0.27), but accompanied with a yet lower EQEmax of 2.2%.ref. ref1000
A similar approach was reported by Gong et al.,ref. ref397 who instead used peripheral diphenylamine units on the DMAC-based donor dendrons in DDA-DP (Figure ). This compound has a small ΔE ST of 0.04 eV as a neat film and emits at λPL at 549 nm, while in dilute toluene solution it has a ΦPL of only 12.4%. Unlike the SP-OLEDs using the two previous dendrimer examples, devices with DDA-DP maintained the emission color of the DMAC-DPS core, with CIE coordinates of (0.36, 0.56). The EQEmax of the device was 8.1% and the efficiency roll-off was very low at only 1% at 1000 cd·m–2 (Table S14).
In order to address concentration quenching, Li et al.ref. ref1001 designed a half-dendronized derivative of CzDMAC-DPS, DCz-DPS-Cz (Figure ), which consists of a DPS acceptor core with a carbazole donor attached to one side and the CzDMAC donor dendron on the other side. While DCz-DPS-Cz maintained the color of CzDMAC-DPS with a neat film λPL at 494 nm (and similar CIE coordinates), the EQEmax of the SP-OLED was almost doubled to 23.3% (Table S14), and the efficiency roll-off at 100 cd·m–2 was halved to 23.3%, likely due to optimized charge transfer properties using the half-dendrimer material.
Huang et al.ref. ref1002 designed dendrimers containing a related acceptor to DPS, dibenzothiophene-5,5-dioxide (DBTO). Attaching donor dendrons of varying size to this DBTO core afforded two green emitters, 1CzAcDBTO and 2CzAcDBTO (Figure ), which in the neat film emit at λPL of 559 nm and 540 nm, have ΦPL of 41 and 54%, and small ΔE ST of 0.02 and 0.04 eV, all respectively (Table S14). Non-doped SP-OLEDs showed EQEmax of 3.9 and 4.5%, at CIE coordinates of (0.43, 0.54) and (0.36, 0.54), with evidence of large current leakage but low efficiency roll-off at 1000 cd·m–2 of 3.3% and 4.4%, all respectively.
Investigating the effects of different generation for carbazole-based dendrons, Li et al.ref. ref1003 designed two green-emitting dendrimers 2CzSO and 3CzSO (Figure ). As observed with other examples, the use of higher generation donor dendrons leads to a smaller ΔE ST of 0.08 eV for 3CzSO compared to 0.16 eV for 2CzSO (Table S14), but the ΦPL decreased from 43% (2CzSO) to 21% (3CzSO) due to the decreased oscillator strength for the S0-S1 transition in the latter. Non-doped SP-OLEDs with 2CzSO and 3CzSO showed EQEmax of 10.7 and 7.3% at CIE coordinates of (0.27, 0.52) and (0.31, 0.53), respectively, demonstrating conclusively that the use of larger donor dendrons does not always equate to better performance in the SP-OLED.
Wang et al.ref. ref1004 reported a dendrimer (TPPOCz, Figure ) containing tri-tBuCz donor dendrons and a phosphine oxide acceptor that emits at λPL of 400 nm with ΦPL of 33% in the neat film (Table S14). The non-doped single-layer SP-OLED showed a poor EQEmax of only 0.27% at CIE coordinates of (0.18, 0.13). The low EQEmax was attributed to hindered charge injection into the emitting layer caused by a mismatch of transport layer work functions. To overcome this, the authors fabricated an SP-OLED with TmPyPB/TPBi acting as the ETL, which increased EQEmax to 2.0%, but also resulted in a large red-shift of the electroluminescence, with CIE coordinates of (0.26, 0.31).
Puttock et al.ref. ref1005 reported two TADF dendrimers based on a benzonitrile acceptor core surrounded by two ortho-carbazole donor dendrons functionalised with either fluorene (Da, Figure ) or diphenylamine groups (Db). Neat films of dendrimer Da emits at λPL of 463 nm and has a ΦPL of 27% in the neat film, whereas Db, containing the stronger donor dendrons, emits at λPL of 526 nm with a ΦPL of 21%. SP-OLEDs with both dendrimers showed low EQEmax, either in non-doped or doped devices (4 wt% in mCP). The 4 wt% doped mCP device of Db showed the highest EQEmax amongst the devices in this study of 5.8% at CIE coordinates of (0.25, 0.48). The same group has also investigated the use of hybrid dendrons that themselves contain D-A TADF subunits, which then feed excitons to a central organometallic phosphorescent centre.ref957,ref1006
Rather than basing the dendrimer design about a central acceptor moiety, Wang et al.ref. ref1007 developed a series of π-stacked dendrimers composed of cofacially aligned and alternating dendritic teracridan donor dendrons and triazine acceptors (Figure ). The closely spaced donors and acceptors around a central benzene ring led to efficient TSCT-TADF properties. By regulating the strength of the TSCT via substituent effects on the acceptor, the emission color of the dendrimers was tuned from blue to yellow/red. The PL spectra of BD-Cy, YD–TF and RD–2TF in toluene exhibit broad CT emission at λPL of 487, 552 and 590 nm (Figure ), respectively, a trend in line with the increased electron-withdrawing strength of the triazine acceptors containing increasing numbers of trifluoromethyl groups. The spatial separation between donor dendrons and acceptors reduces the overlap of the frontier molecular orbitals, thus leading to small ΔE ST of 0.05, 0.04 and 0.04 eV for BD–Cy, YD–TF and RD–2TF, respectively. The SP-OLEDs with BD–Cy, YD-TF and RD–2TF showed EQEmax of 18.2, 21.9, and 10.3%, respectively, in good agreement with their corresponding ΦPL values of 74, 86 and 49% as 10 wt% doped films in polystyrene.
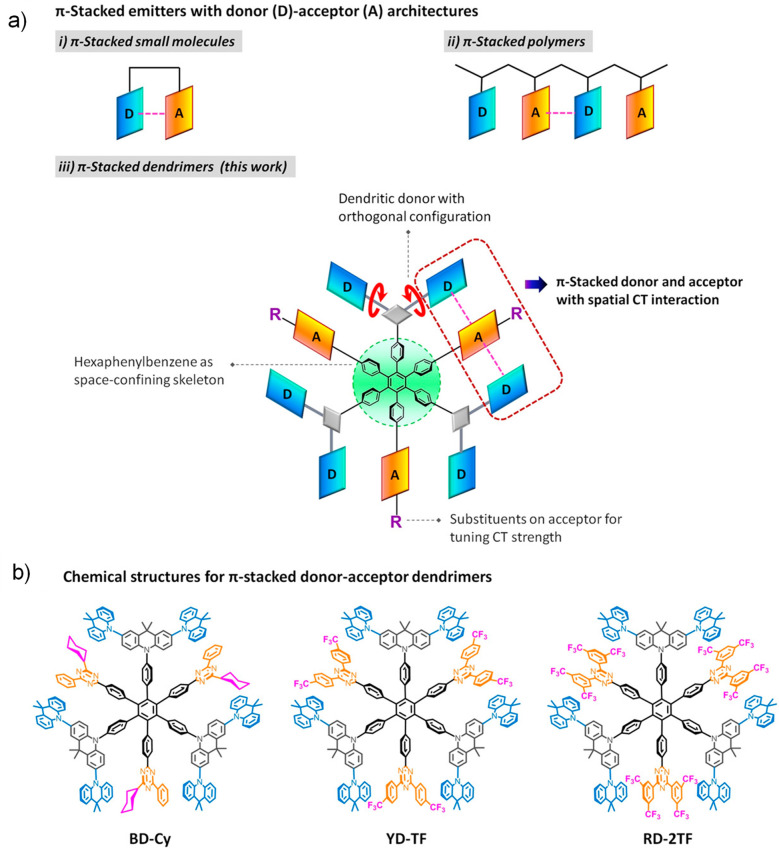
Non-conjugated Dendrimers
Cz-CzCN is a dendrimer consisting of a 5CzBN TADF core decorated with carbazole at the periphery of alkyl chain tethers (Figure ).ref. ref1008 The neat film emits at λPL of 509 nm and has a ΦPL of 52%, which is much higher than the 21% measured for 5CzBN. The τd is short at 2.3 μs while the ΔE ST is moderate at 0.17 eV. Non-doped SP-OLEDs with Cz-CzCN showed EQEmax of 17.1% at CIE coordinates of (0.26, 0.52), which showed a low efficiency roll-off of 11% at 1000 cd·m–2.
Of a similar concept, Cz-OCzBN (Figure ), using a 3CzBN core and the same dendronized carbazole, emits at λPL of 498 nm and has a ΦPL of 58% in the neat film (Table S14). The non-doped SP-OLED with the blue emitter showed an EQEmax of 6.6% at CIE coordinates of (0.18, 0.29) and had an efficiency roll-off of ∼26% at 1000 cd·m–2. This dendrimer was then also used in conjunction with a red phosphorescent emitter to produce solution processed white OLEDs. SP-WOLEDs with an EML consisting of Cz-OCzBN co-doped with 0.6 wt% of the iridium-based phosphorescent emitter PO-01 showed an EQEmax of 17% at warm white CIE coordinates of (0.34, 0.44) and an efficiency roll-off of 30% at 1000 cd·m–2, which makes them one of the highest performing hybrid SP-WOLEDs to date.ref. ref1009
Sun et al.ref. ref1010 reported a dendrimer with diphenylamine donors joined to a triazine core, with triCz at the periphery (TA-3Cz, Figure ). Conjugation between the core and outer dendron units was broken using hexyl chains. TA-3Cz emits at λPL of 541 nm, has a ΦPL of 71% and a short τd of 0.8 μs in the neat film (Table S14). The non-doped SP-OLEDs showed an EQEmax of 11.8%, and although efficiency roll-off at 1000 cd·m–2 was ∼58%, a high maximum luminance of 23,145 cd·m–2 was achieved. The strong efficiency roll-off was attributed to the fact that triCz is unipolar, resulting in poor charge balance within the non-doped EML. The device with a smaller derivative capped with only a single generation of carbazole, TA-Cz (Figure ), showed lower efficiencies with EQEmax 5.5%, a result of alleviated ACQ in TA-3Cz. An improved efficiency roll-off of ∼24% at 1000 cd·m–2 was also observed.
Godumala et al.ref. ref1011 reported a dendrimer, TB2CZ-ACTRZ (Figure ) in which a methylene bridge was used to break conjugation between the peripheral tBuCz groups and the DMAC donor, itself connected to a triazine acceptor. In addition, a derivative with one additional generation of carbazole substituents, TB14CZ-ACTRZ (Figure ) was also reported. TB2CZ-ACTRZ emits at λPL of 520 nm, with a ΦPL of 69% in neat film (Table S14), while TB14CZ-ACTRZ showed a blue-shifted emission in the neat film at λPL of 494 nm and a decreased ΦPL of 56%. Further, TB14CZ-ACTRZ has a longer τd of 25 μs compared to 2.9 μs for TB2CZ-ACTRZ. The devices with TB2CZ-ACTRZ showed an EQEmax of 9.9%, which was maintained at 100 cd·m–2, although at 1000 cd·m–2 the efficiency roll-off grew to ∼52%; a related device without any hole transporting or injection layers showed an EQEmax of 9.5%. The device incorporating TB14CZ-ACTRZ showed a lower EQEmax of 5.5%. Notably, for this material a device without hole transporting layers showed an improved EQEmax of 8.1%. The devices for both of these dendrimers and absent of HTLs showed large efficiency roll-off, with efficiency values at 100 cd m–2 dropping by ∼58% and ∼63%, respectively.
In order to address charge imbalance, Ban et al.ref. ref1012 developed the self-host dendrimer emitter POCz-DPS (Figure ), containing a dicarbazole diphenylsulfone emitting core surrounded by alkyl chains, and with phosphine oxide functionalized carbazole acting as the peripheral host unit. The neat film emits at λPL of 460 nm and has a ΦPL of 61% (Table S14). The non-doped SP-OLED showed an EQEmax of 7.3% at CIE coordinates of (0.18, 0.30) and the efficiency roll-off at 1000 cd·m–2 was only ∼19%, although the leakage current was very high.
The same group also compared the performance of two similar self-hosting emitters, one with bipolar carbazole and phosphine oxide containing dendrimers, poCz-SO, and the other with just tBu-carbazole dendrimers, tbCz-SO (Figure ).ref. ref1013 poCz-SO and tbCz-SO emit at λPL of 458 nm and 440 nm and have very short τd of only 0.2 μs and 0.1 μs, respectively, as neat films (Table S14). The differences in EQEmax of 6.2 and 2.6%, for the devices respective with poCz-SO and tbCz-SO were attributed to improved charge balance in the former material with bipolar dendrons. This tuning of the charge transport properties also improved efficiency roll-off at 100 cd·m–2, which were 11% and 46% for the devices with poCz-SO and tbCz-SO, respectively.
Li et al.ref. ref1001 subsequently reported an asymmetric dendrimer based on tbCz-SO, but replacing one of the tBuCz donor dendrons with a CzDMAC donor dendron to give DCz-DPS-TCz (Figure ). The neat film emission of DCz-DPS-TCz red-shifted to a λPL of 500 nm, while the ΦPL approached unity at 96%. The τd is 1.4 μs, which is associated with the small ΔE ST of 0.03 eV (Table S14). The non-doped SP-OLED with this dendrimer showed an EQEmax of 24% at CIE coordinates of (0.24, 0.45). The efficiency roll-off at 100 cd·m–2 was decreased significantly to only 2%, while at 1000 cd·m–2 it was still low at 11%. These results demonstrate that significantly enhanced device performance can be achieved by using dendrimers with asymmetrical donor dendrons that carefully balance TADF and charge transport properties.
Most of the materials highlighted so far are green emitters. An exception is the dendrimer reported by Sun et al.ref1014,ref1015 who developed a red self-host dendrimer consisting of CBP peripheral groups attached to a central TPA-anthraquinone D-A TADF core (MPPA-MCBP, Figure ). This dendrimer emits at λPL of 690 nm but has a low ΦPL of 10% (Table S14). Non-doped devices showed NIR emission with λEL at 698 nm and an EQEmax 0.62%. The efficiency of the SP-OLED was improved compared to devices using just the TADF core (EQEmax = 0.11%).
A similar emitter based on the same core but with triCz as the donor dendron, MPPA-3Cz (Figure ), emits further to the red at λPL of 708 nm, and has a ΦPL of 8% (Table S14). Devices incorporating MPPA-3Cz showed even worse EQEmax of 0.25% at λEL of 715 nm compared to MPPA-MCBP, owing to the reduced charge balance in this material, a result of replacing the CBP groups with carbazole.
Outlook
A wide range of TADF polymer design strategies has emerged in recent years. Selected relevant representative examples have been presented in this review to highlight their typical photophysical and electroluminescent properties. The majority of the OLEDs with the presented polymers emit within the sky-blue to green color region, with those with PDT-1, PDT-2, and PDT-3 ref. ref969 representing the only devices to achieving blue CIE coordinates of (0.15, 0.08), (0.15, 0.09) and (0.17, 0.14), respectively; however, their EQEmax values were low at around 5%. There are as of the end of 2022 no reported red or near-infrared emissive TADF polymers. Of the polymer TADF OLEDs present, arguably the best device performance has been achieved using the green-emitting PABPC5,ref. ref974 which showed an EQEmax of 18.1% and very low efficiency roll-off, with an EQE1000 of 17.8%. However, the emission spectrum is broad due to a combination of a CT emissive state and a distribution of polymers in the sample. Such broad emission from TADF polymers can be addressed by the incorporation of an MR-TADF emitter within the polymers, as exemplified in PCzBN1, PCzBN3, and PCzBN5,ref. ref961 which all showed small FWHM of 27, 34 and 30 nm, respectively. Devices with PCzBN1 and PCzBN5 showed EQEmax of 17.8 and 17.5%, respectively, demonstrating the potential of this approach.
Another big challenge in this field remains batch-to-batch variation endemic to polymer synthesis, evidenced by the rather large PDI. Finer control of the polymerization is needed in order to achieve polymers with a narrow size distribution. In addition, the monomer ratio (or in this case emitter to host ratio) is a crucial parameter to optimize to obtain suitably high-performance devices, as demonstrated in many of the reports summarized herein. It is also clear that polymers can exhibit reduced ACQ if the ratio of monomers is chosen correctly. Polymer materials do have the advantage of producing high-quality amorphous films and so as a whole they remain a promising class of emitters for high-performance SP-OLEDs. However, to compete with SP-OLEDs using small molecule emitters – especially in terms of properties like color purity (FWHM) and EQEmax – still more materials development efforts are required.
Three different classes of TADF dendrimers have been illustrated as an alternative family of macromolecular materials suitable for SP-OLEDs and key relevant examples have been highlighted. The outstanding issues for SP-OLED is a generally lower EQEmax and their typically inferior efficiency roll-off compared to vacuum-deposited devices, as seen for a lot of the devices employing polymers and dendrimers as emitters discussed in this section. For dendrimers it has been shown that certain designs can help to address these issues. As a first example, devices incorporating the conjugated dendrimer tBuCz2m2pTRZ ref. ref995 showed the highest EQEmax of 28.7% of all the reported dendrimers at green CIE coordinates of (0.37, 0.57), demonstrating that dendrimer TADF-SP-OLEDs can compete with small-molecule TADF SP-OLEDs in terms of their performance. Devices containing the TSCT-dendrimer YD-TF ref. ref1007 as the emitter showed an EQEmax of 21.9% and a low efficiency roll-off to 18.6% at a luminance of 1000 cd m–2 at CIE coordinates of (0.41, 0.54) when doped into a dendrimeric host. An impressive device performance using a non-conjugated dendrimer as the emitter, employed the asymmetrically substituted dendrimer DCz-DPS–TCz,ref. ref1001 which showed an EQEmax of 24.0% and an EQE500 of 21.3% at CIE coordinates of (0.24, 0.45). These three examples demonstrate that dendrimers represent a potent alternative class of emitters compared to small molecules and polymers in SP-OLEDs.
Dendrimers possess a balance of desirable properties that makes them attractive for SP-OLEDs. Due to their size, they are amenable for solution-processing fabrication techniques like polymers. Additionally, ACQ can be mitigated in all three classes of TADF dendrimers, exemplified by the performance of several examples of non-doped devices highlighted in this section. While likewise having good film-forming properties as do polymers, dendrimers also enjoy having a well-defined molecular structure, so there is no batch-to-batch variation and purification can be readily achieved. Dendrimers can also employ donor dendrons that have embedded charge transport units to help address charge balance in the SP-OLED, and the considerable flexibility in terms of dendron design will likely support both imaginative future material strategies as well as improved overall device performance in this area.
Multi-resonance TADF
Introduction
The most popular TADF emitter design strategy relies on highly twisted donor-acceptor architectures to reduce the exchange integral and hence ΔE ST. The most prominent recent examples have been documented in Sections sec3 –sec5. Though there are many examples of high-efficiency OLEDs using emitters with this design, the emission spectrum is frequently broad and unstructured, reflective of the CT nature of the excited state and inherent conformational flexibility. To account for varying bandshapes, the width of the emission is primarily quantified at half the emission intensity maximum (FWHM, Figure ), with D-A TADF materials typically having FWHM of 80–100 nm. Narrower emission spectra can more easily achieve high color saturation, which is required for commercial displays to meet industry-standard color space coverage. Standard red blue green (sRBG) coordinates have defined CIE coordinates of (0.64, 0.33), (0.15, 0.06) and (0.30, 0.60) respectively, which for emitters with large FWHM require subtractive filtering to achieve, sacrificing overall emission efficiency.ref. ref9 The more recent standard Rec. 2020 defines blue, green, and red CIE coordinates to be (0.13, 0.05), (0.17, 0.80) and (0.71, 0.29), respectively, which are even more challenging for D-A TADF emitters to acheive.ref. ref10
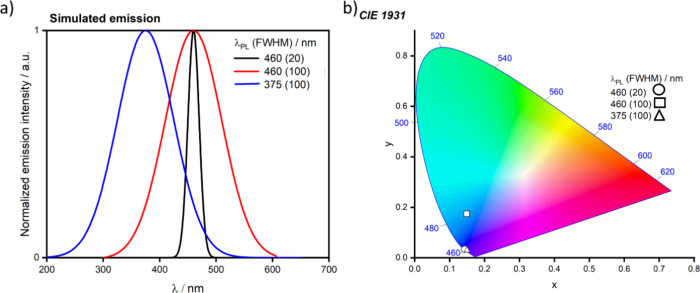
To demonstrate this challenge, Figure shows two simulated emission spectra with the same maximum at 460 nm, and with FWHM of 20 nm or 100 nm. These emission spectra correspond to CIE coordinates of (0.14, 0.03) and (0.15, 0.17), with the narrower spectrum having a far more saturated blue emission despite a considerably lower high-energy onset – itself a significant benefit for device host choice and lifetime. To achieve similar CIE coordinates of (0.14, 0.04) with an emission spectrum with a 100 nm FWHM, the λPL would need to be 375 nm (Figure ). An emitter design with these metrics would be very challenging and no host exits with a suitably high triplet energy to accommodate such an emitter.
In contrast to CT emission in D-A TADF molecules, narrowband emission can be achieved within an exciting sub-class of TADF materials based mainly on p- and n-doped nanographene fragments, termed multiresonant TADF (MR-TADF) emitters. Pioneered by Hatakeyama and co-workers,ref. ref118 these materials exploit complementary resonance effects, with the electron density distribution of the HOMO and LUMO localized on neighboring atoms in the heteroacene, setting up short-range charge transfer (SRCT) excitons and ensuring a sufficiently small ΔE ST to turn on TADF (Figure ).ref. ref45 Crucially, MR-TADF emitters possess a rigid structure with little change in the geometry from the ground to the excited state, resulting in small Stokes shifts and narrowband emission profiles with only minor contributions from vibronic bands.ref. ref162 The emissive SRCT excited states also have considerably subdued solvatochromism compared to the long-range CT states in D-A compounds.ref138,ref162
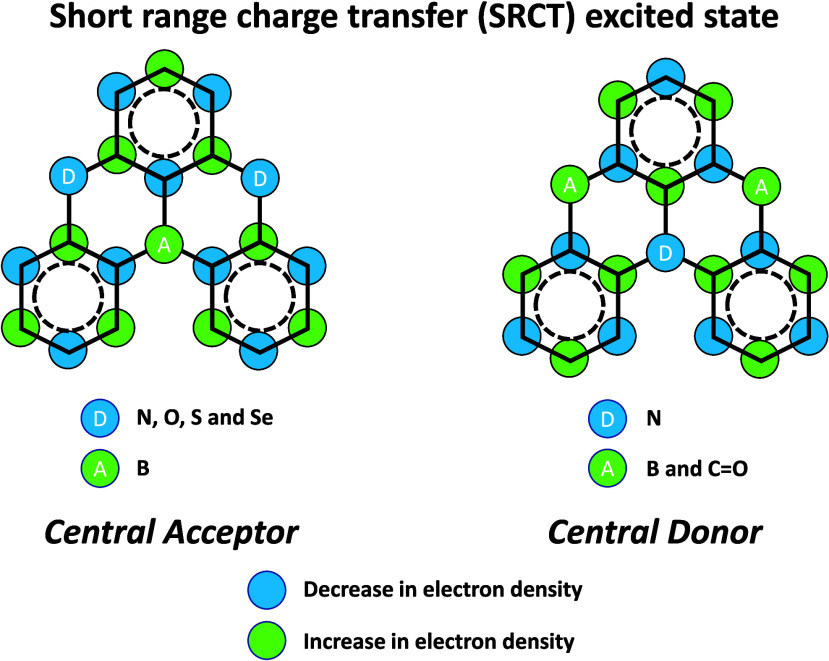
The SRCT excited states in MR-TADF materials can be clearly visualised using computed difference density plots. In these, the alternating pattern of increasing and decreasing electron density on neighboring atoms in the excited states (relative to the ground state) reveals the alternating charge-transfer interactions (Figure ). This is in stark contrast to LRCT excited states in D-A materials, in which electron density migrates large distances from the donor part of the molecule to the acceptor (See Section sec2 ). This different category of excited states, and particularly the confinement of electrons and holes in the nanographene fragment (with significant correlation and exchange interaction), necessitates the use of multireference methods rather than simpler DFT when calculating properties of these materials.
The structural diversity of MR-TADF emitters remains small at present. However, there are nonetheless now more than 250 reported examples, including some that approach the Rec. 2020 standard for each of blue, green, and red emission. In some cases, these materials also have exceptional efficiencies in devices, especially when supported by assistant dopants in hyperfluorescence OLEDs. Examples highlighted in this section have been grouped together based on common structural motifs, with their properties discussed and OLED performance cross-compared (Table S15).
Central Acceptor Structures
Early MR-TADF Emitters, DABNA and Its Derivatives
The most common design for MR-TADF emitters incorporates a central boron as the acceptor atom with oxygen or nitrogen atoms acting as the donors (Figure ). The first examples of MR-TADF emitters reported by Hatakeyama and co-workersref. ref232 possess this motif as exemplified by compound 2a (Figure , later called DOBNA or BOO).ref. ref179 Measurements in solution showed a ΔE ST of 0.15 eV (fluorescence measured in DCM and phosphorescence measured in EtOH); however, no time-resolved PL was collected to substantiate TADF activity.ref. ref232 The photophysics of this compound was revisited recently,ref. ref179 and the authors reported data for 1 wt% PMMA films, with a ΔE ST of 0.18 eV and λPL of 398 nm with a τd of 66 μs. No devices were fabricated in either of these reports, likely owing to the near-UV emission and lack of suitable host. Other derivatives of DOBNA are discussed in the “DOBNA derivatives” sub section.
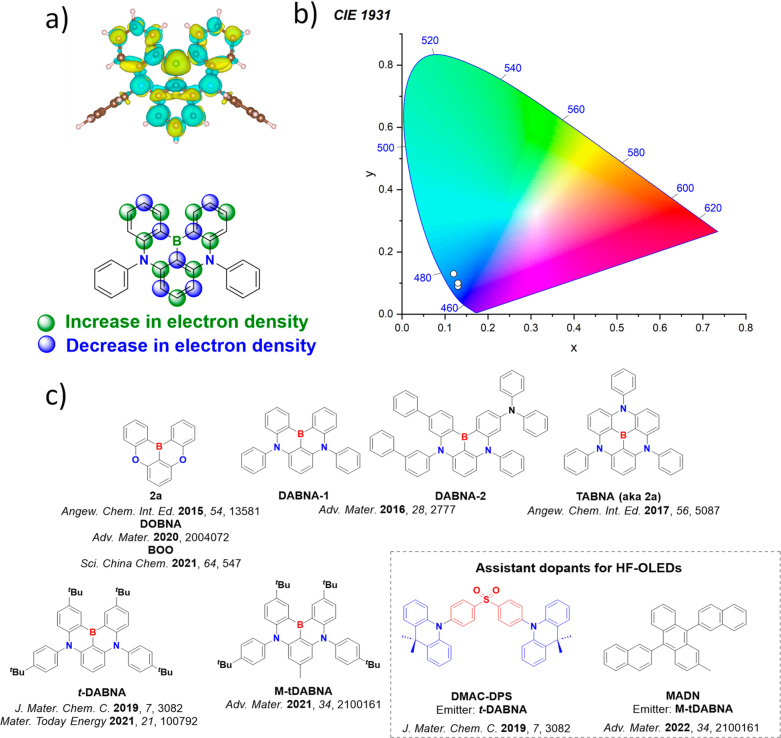
Hatakeyama and co-workers subsequently reported the emitters DABNA-1 and DABNA-2, where the oxygen donor atoms were replaced by nitrogen atoms within DPA groups fused to a central boron atom (Figure ), and with DABNA-2 featuring additional DPA and phenyl substituents.ref. ref118 Excellent ΦPL values in 1 wt% mCBP films of 88 and 90% were achieved for DABNA-1 and DABNA-2, respectively, with λPL red-shifted compared to DOBNA (λPL of 398 nm in 1 wt% PMMA)ref. ref179 at 460 nm for DABNA-1 and 469 nm for DABNA-2 in 1 wt% mCBP.ref. ref118 As will become evident for most MR-TADF compounds, moderately large ΔE ST of 0.18 and 0.14 eV and associated long τd of 94 and 65 μs were reported for DABNA-1 and DABNA-2, respectively. Vacuum-deposited OLEDs with DABNA-1 and DABNA-2 as the emitter showed EQEmax of 13.5 and 20.2%, respectively. The most attractive feature of these OLEDs is their narrow FWHM at 28 nm, that ensured pure blue emission with CIE coordinates of (0.13, 0.09) and (0.12, 0.13) for the devices with DABNA-1 and DABNA-2, respectively. This report was the first example of MR-TADF emitters employed in devices; however, despite the promising EQEmax values, the devices suffered from severe efficiency roll-off with EQE100 of 6.3 and 13.3%. A luminance of 1,000 cd m–2 was not achieved by either device. This efficiency roll-off stems from the large ΔE ST and associated long τd of these materials, a feature still commonly reported for MR-TADF materials to this day. A related D 3-symmetric derivative of DABNA-1, TABNA (named 2a in the original report, Figure )ref. ref1016 showed a moderate ΦPL of 54% in 1 wt% PMMA and a comparable ΔE ST of 0.21 eV to DABNA-1. A narrow FWHM of 28 nm at a λPL of 399 nm was observed, although no devices were reported.
t-DABNA, a tert-butyl decorated analogue of DABNA-1, was reported by Han et al. (Figure ).ref. ref1017 This compound was used as both an emitter in an OLED and as the terminal emitter in a hyperfluorescence OLED with DMAC-DPS (Figure ) as the assistant dopant. In 5 wt% DPEPO films the ΦPL of t-DABNA is 85%, the ΔE ST is 0.17 eV (similar to DABNA-1 with ΔE ST = 0.18 eV), and the τd is 83 μs. The OLED showed a promising EQEmax of 25.1%, but the efficiency roll-off was again severe (EQE100 of 6.0%) owing to the slow RISC and long delayed lifetime. The EQEmax of the HF device was 31.4% and the efficiency roll-off improved considerably, with EQE100 of 27%. The HF OLED strategy and mechanism are discussed in detail in Section sec17 and has proven popular for OLEDs employing MR-TADF terminal emitters, as this strategy can mitigate the slow k RISC in these compounds while preserving their valuable narrow FWHM emission. A related derivative of t-DABNA with a methyl substituent para to the boron, M-tDABNA,ref. ref1018 shows improved orientation of its TDM (Figure ). M-tDABNA has a λPL of 461 nm, a ΦPL of 84%, and a τd of 195 μs in 3 wt% mCBP films, while the ΔE ST in toluene is 0.11 eV. An OLED employing a TTA assistant dopant, MADN (Figure ), showed an EQEmax of 8.6% and CIE coordinates of (0.14, 0.08) using this MR-TADF terminal emitter.
Substituted DABNA Derivatives
t-DABNA has since been revisited by Kim et al., where it was investigated alongside a donor-decorated derivative (t-DAB-DPA, Figure ).ref. ref1019 Improved device performance was demonstrated compared to the previous report when t-DABNA was doped in a mixed host system (mCBP:mCBP-CN). The OLED showed an EQEmax of 28.4%, but the efficiency roll-off was large (EQE100 of 14.8%). Increasing the concentration of the emitter from 3 to 10 wt% resulted in ACQ and the EQEmax reached only 21.3%. The addition of peripheral tBuCz groups to the t-DABNA core as in TBN-TPA suppressed the ACQ (Figure and Figure );ref. ref1020 notably, the structure of the emitter was initially wrongly identified,ref. ref1021 and it was subsequently shown that the structure was in fact that of CzDABNA-NP-TB (Figure ). This emitter is discussed in detail alongside CzBN derivatives (vide infra).
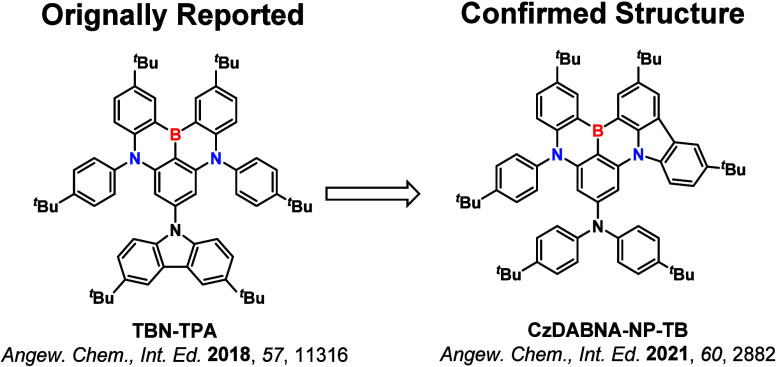
Two derivatives of DABNA-1 were reported that contained a third diarylamine donor as well as pendant methyl or tBu groups on the diarylamines: DABNA-NP-M and DABNA-NP-TB (Figure ).ref. ref1021 The addition of the third donor group in this case had minimal impact on the photophysical properties, with similar λPL of 460 and 453 nm and ΦPL of 88 and 83% in 1 wt% PMMA, respectively for DABNA-NP-M and DABNA-NP-TB, compared to DABNA-1 (λPL of 460 nm and ΦPL 88% in 1 wt% mCBP).ref. ref118 The ΔE ST value for both emitters in 1 wt% PMMA film was 0.17 eV and each showed similar τd of 89 and 90 μs,ref. ref1021 respectively, which are again similar to the values reported for DABNA-1 (ΔE ST = 0.18 eV and τd = 94 μs).ref. ref118 This demonstrates that these structural changes can in some cases have only a minimal impact, contrary to the conclusions drawn in the original report of the wrongly identified emitter TPN-TPA.ref. ref1020 Devices with DABNA-NP-TB showed an EQEmax of 19.5% at CIE (0.14, 0.11) and relatively low efficiency roll-off of 10% at 100 cd m–2,ref. ref1021 which was an improvement compared to the OLED with DABNA-1 (EQEmax = 13.5%, efficiency roll-off at 100 cd m–2 = 53%).ref. ref118
An investigation into the effect of tBu substitution was recently conducted by Wang et al.ref. ref1022 Three emitters including DABNA-NP-TB were presented, containing differing numbers of tBu substituents with PAB having none, 2tPAB containing four tBu groups on the DABNA-1 core, and 3tPAB (identical to DABNA-NP-TB) substituted on both the DABNA-1 core and the DPA donor (Figure ). The introduction of the additional tBu groups resulted in a very small red-shift of the emission across the series, with λPL of 453, 457, and 458 nm for PAB, 2tPAB, and 3tPAB, respectively, alongside similar τd and ΔE ST (τd of between 56 and 77 μs and ΔE ST ranging from 0.06 to 0.10 eV). Aside from intrinsic optical properties, addition of the tBu groups helped to suppress ACQ, evidenced by the steady increase in ΦPL from 61 and 67 to 75% for PAB, 2tPAB, and 3tPAB, respectively, in 3 wt% mCP films. Devices with PAB and 2tPAB showed EQEmax of 14.7 and 16.8% at CIE (0.15, 0.08), while the device with for 3tPAB showed a higher EQEmax of 19.3% at CIE (0.14, 0.08), which was correlated with the ΦPL.
3tPAB (renamed as t-DAB-DPA) was subsequently investigated alongside t-DABNA by Kim et al. (Figure and Figure ).ref. ref1019 The authors focused on reducing ACQ by decorating a peripheral donor group of t-DAB-DPA. Moving from 3 to 10 wt%, the OLEDs with t-DABNA saw the EQEmax decrease from 28.4 to 21.3%, while for devices with t-DAB-DPA the decrease in EQEmax was attenuated, decreasing from 27.6 to 25.9%. For the 3 wt% emitter doped devices, there was a corresponding modest blue-shift of the emission, reflected in the CIE coordinates of (0.13, 0.10) and (0.14, 0.08) for the devices with t-DABNA and t-DAB-DPA, respectively. As will be made clear by following examples, addressing the strong ACQ in MR-TADF emitters that arises from their large, planar and electron-rich structures has become a key concern for improving the overall performance across the research community.
A similar strategy to suppress ACQ was used by Park et al., where a bulky di-tert-butyl phenyl substituent was added para to the boron atom in t-DABNA-dtB (Figure ).ref. ref1023 A near unity ΦPL of 99% was reported for the 3 wt% doped film in mCBP, which also showed ΔE ST of 0.19 eV and τd of 110 μs. ACQ was found to be more severe for reference compound t-DABNA, where the ΦPL of the 3 wt% film in mCBP was 87% but dropped to 65% for the 7 wt% doped film, while the ΦPL of the 7 wt% doped film with t-DABNA-dtB remained as high as 96%. The corresponding OLEDs showed an EQEmax of 25.5%, but the EQE100 was only 5.4% and the LT95 < 1 hour at 100 cd m–2. When employed in a HF-OLED in conjunction with an anthracene-based TTA assistant dopant (HOST, Figure ), the EQEmax was much lower at 11.4% (limited by the TTA assistant dopant) yet the device stability improved markedly with LT95 of 13,124 hours at 100 cd m–2.
Lee et al. investigated the effect of heavy atom inclusion on MR-TADF emission using derivatives of PAB (renamed MR here, Figure ),ref. ref1024 incorporating chlorine (Cl-MR, Figure ) and bromine (Br-MR, Figure ). Calculations revealed the profound impact of the substituents on SOC between S1 and T2, increasing from 0.19 cm–1 in MR to 0.68 cm–1 in Cl-MR and 2.21 cm–1 in Br-MR. A small red-shift in the emission with halogen substitution was observed in 10 wt% doped DPEPO films, with λPL of 456, 474, and 474 nm for MR, Cl-MR, and Br-MR, respectively, while each showed identical ΔE ST of 0.13 eV. The influence of the heavy atoms was evident in the TADF kinetics, with increasing k RISC of 2.8, 9.8, and 59 × 104 s–1 for MR, Cl-MR, and Br-MR, respectively. Despite the higher k RISC and comparable ΦPL of 75–85%, the devices with MR and Cl-MR showed comparable EQEmax of 16 and 17%, while the device with Br-MR the EQEmax was only 4.2%. The decreased EQEmax of Br-MR was attributed to lower bond dissociation enthalpy values within the emitter, leading to its degradation under electrical stress.
Wang et al. reported a derivative of t-DABNA containing a DPAC donor attached to the t-DABNA core, tDPAC-BN (Figure ).ref. ref1025 The 1 wt% doped film of tDPAC-BN in PMMA emits at λPL of 454 nm, has a ΔE ST of 0.17 eV in toluene and a τd of 114 μs in 1 wt% doped film. The OLED showed a modest EQEmax of 12.4% at CIE (0.14, 0.08), dropping to 1.6% at 100 cd m–2. Hyperfluorescent devices with DMAC-DPS (Figure ) as the assistant dopant showed an improved EQEmax of 21.6% and reduced efficiency roll-off (EQE100 = 15.3%).
Cheon et al. developed a derivative of DABNA-1 containing bulky groups designed to suppress ACQ, pBP-DABNA-Me (Figure ).ref. ref1026 Biphenyls were added to preventing intermolecular π-stacking, while xylyl groups were added to further reduce ACQ and supress rotational vibrations. pBP-DABNA-Me shows both a narrower FWHM of 22 nm and higher ΦPL of 98% compared to DABNA-1 (30 nm and 79%) in the same 5 wt% DPEPO:mCBP host, but emits at λPL of 462 nm, identical to DABNA-1. Despite a similar ΔE ST of 0.18 eV to DABNA-1 (0.17 eV) the k RISC was also faster for pBP-DABNA-Me at 6.85 × 104 s–1, compared to 0.99 × 104 s–1 for DABNA-1. The enhanced k RISC was attributed to the introduction of closely lying 3LE states of the biphenyl groups, which according to computations facilitated RISC via spin-vibronic coupling. Devices showed EQEmax of 23.4% at CIE (0.13, 0.09). Non-doped OLEDs showed much poorer performance, with an EQEmax of 10.1%; however, there was little evidence of aggregation as the CIE coordinates were only slightly red-shifted to (0.14, 0.10). When utilized in HF-OLEDs with TDBA-SAF (Figure ) as the assistant dopant the EQEmax rose to 30.1%.
A similar derivative was reported by the same group,ref. ref1027 where the phenyl substituents of pBP-DABNA-Me were instead positioned meta to the nitrogen donor atoms to give mBP-DABNA-Me (Figure ). In 5 wt% mCP:DPEPO films the λPL is 467 nm and the ΦPL is 97%, similar to pBP-DABNA-Me. The k RISC of mBP-DABNA-Me was determined to be 1.95 × 104 s–1, slower than that of pBP-DABNA-Me and indicating that the contribution of LE biphenyl triplet states was less effective over meta linkages than para ones. The OLEDs showed an EQEmax of 24.3% at λEL 468 nm, while the EQE1000 dropped to 9.1%. Owing to the bulky nature of the emitter, the CIE coordinates remained impressively constant at (0.12, 0.14) at 0.5, 5, and 25 wt% emitter doping in the EML of the OLEDs.
Two carbazole- and biphenyl-decorated DABNA-1 derivatives, TBE01 and TBE02 (Figure ), were developed to improve the performance of HF-OLEDs compared to t-DABNA.ref. ref1028 These emitters were designed to have larger Förster radii compared to t-DABNA, while their bulkier size would help to suppress Dexter energy transfer. TBE01 and TBE02 showed identical λPL of 459 nm and FWHM of 21 nm, with ΦPL of 91 and 89%, respectively in toluene. Compared to t-DABNA (0.21 eV), these derivatives have smaller ΔE ST of 0.16 and 0.14 eV, respectively, in toluene solution, which translates into faster k RISC of 0.27, 0.51, and 1.03 × 104 s–1 for t-DABNA, TBE01, and TBE02, respectively (in exciplex host SiCzCz:SiTrzCz2 at 0.4 wt% emitter doping). HF-OLEDs with PtON-TBBI (Figure ) acting as the assistant dopant showed similar EQEmax (EQE1000) of 27.9% (25.4%) and 29.1% (25.8%) for the devices with TBE01 and TBE02, respectively, compared to 28.1% (23.7%) for the device with t-DABNA. The devices with the substituted emitters were considerably more stable though, with LT95 of 42.3 and 72.9 hours, compared to 19.8 hours for t-DABNA.
DOBNA Derivatives
There have now been numerous derivatives of DOBNA reported since the initial paper by Harai et al. (Figure ),ref. ref179 with sulfur also used as a donating atom. Using DOBNA as the MR-TADF core (renamed BOO, Figure ), Chen et al. reported a series of polymers containing this unit as well as two sulfur-containing analogues, BOS and BSS (Figure ).ref. ref1029 The monomer emitters BOO, BOS, and BSS emitted at λPL of 396, 434, and 457 nm respectively, with ΔE ST 0.18, 0.17, and 0.15 eV in toluene. In 1 wt% polystyrene each monomer showed ΦPL of 70, 63, and 58%, while their k RISC steadily increased from 1.1 to 6.1 and 11.8 × 104 s–1 for BOO, BOS, and BSS, respectively owing to the heavy atom effect. These were next incorporated into non-conjugated polymers PS-BOO, PS-BOS, and PS-BSS (Figure ). Neat films of PS-BOO, PS-BOS, and PS-BSS emitted at λPL of 398, 435, and 456 nm, respectively and showed τd of 133.8, 104.9, and 67.0 μs, but no devices were fabricated. Copolymerisation with an acridan monomer to act as a hole-transporting host afforded polymers PAc-BOO, PAc-BOS, and PAc-BSS (Figure ). The neat films of PAc-BOO and PAc-BOS showed significant spectral changes compared to PS-BOO and PS-BOS, with λPL of 457 and 434/475 nm, respectively, as a new LRCT state between electron-donating PAc and the MR-TADF core gave emission peaks at 457 and 475 nm; such behavior was not observed for PAc-BSS, which retained narrowband emission centred around 455 nm. Solution-processed devices of PAc-BSS likewise emitted at λEL of 458 nm and FWHM of 31 nm, with corresponding CIE coordinates of (0.16, 0.12) and an EQEmax of 13.1%. Several works applying MR-TADF emissive subunits within polymers are highlighted in Section sec10 .
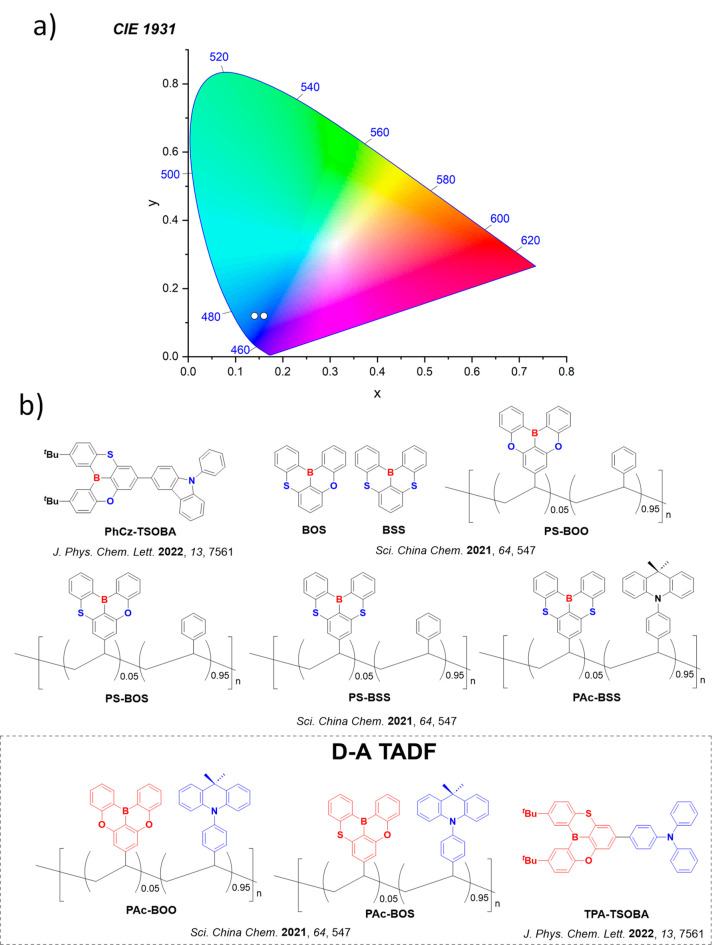
Gao et al. reported two similar emitters based on BOS containing either triphenylamine and phenylcarbazole substituted para to the boron, TPA-TSOBA and PhCz-TSOBA (Figure ).ref. ref317 Based on calculations and the large observed positive solvatochromism they assigned the emission of TPA-TSOBA to LRCT typical of D-A compounds, while PhCz-TSOBA was classified as MR-TADF. PhCz-TSOBA emits at λPL of 444 nm (FWHM of 32 nm) and has a ΔE ST of 0.23 eV in toluene. As a 10 wt% doped film in 2,6-DczPPy the ΦPL is 61% and the k RISC was measured to be 4.1 × 104 s–1. The OLEDs showed an EQEmax of 16.7% at CIE (0.14, 0.12), while the EQE100 was 6.7%. Despite TPA-TSOBA being assigned as a D-A emitter, it showed very similar properties in both films and devices to PhCz-TSOBA, suggesting this assignment may not apply when dispersed in solid OLED hosts.
DABNA Derivatives with Multiple Acceptor Atoms
Beyond materials based on the DABNA or DOBNA cores, a number of π-extended systems have been reported featuring an expanded network of donating or withdrawing atoms, Figure . In 2018, the group of Hatakeyama introduced this extended design strategy,ref. ref1030 wherein they altered the number of boron atoms across the emitters B2, B3, and B4 (Figure ). These three compounds showed moderate ΦPL values of 53, 33, and 57%, with ΔE ST of 0.19, 0.15, and 0.15 eV as 1 wt% doped films in PMMA. B2, B3, and B4 all showed blue emission with λPL of 455, 441, and 450 nm and FWHM of 32, 34, and 38 nm, respectively. Of the three, only B2 was used as an emitter in a device, which performed similarly to the device with DABNA-2 with EQEmax of 18.3% and EQE100 of 12.6% compared to 20.2 and 12.4%, respectively, for the OLED with DABNA-2.ref. ref118
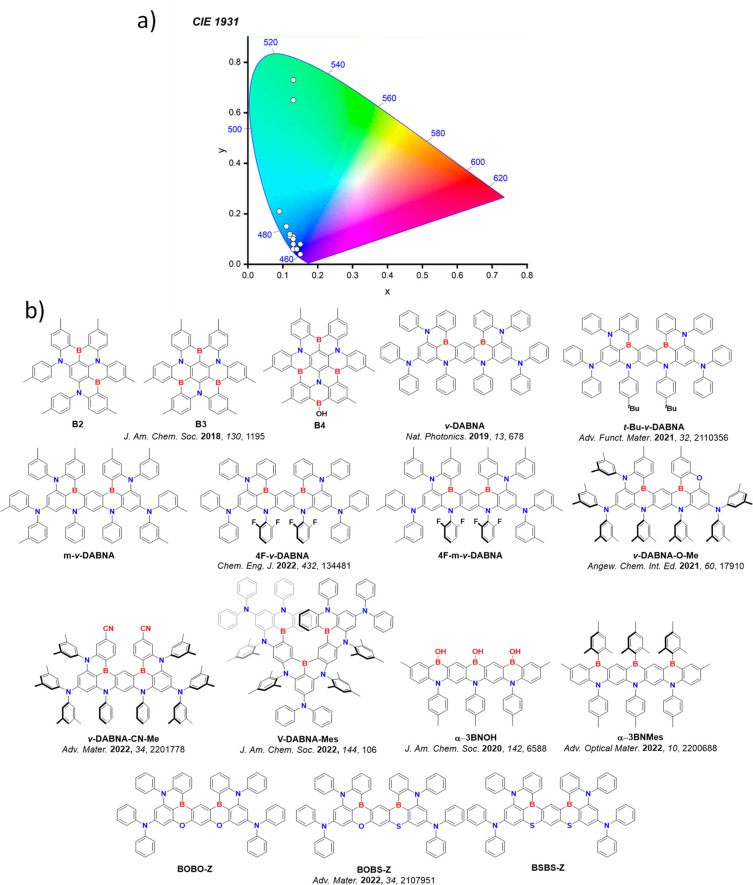
The blue-emissive linearly extended emitter, v-DABNA (Figure ),ref. ref1031 was subsequently reported by the same group. This compound emits sharply at λPL of 467 nm, has a ΦPL of 90% and a small ΔE ST of 0.02 eV at 1 wt% in the bespoke host DOBNA-OAr. DOBNA-OAr is an arylated derivative of DOBNA and is a rare example of an MR-TADF compound used as a host. Unlike most other MR-TADF emitters, v-DABNA showed a relatively efficient k RISC of 2.0 × 105 s–1 likely due to its small ΔE ST; most other MR-TADF emitters have ΔE ST of > 0.10 eV and RISC in the range of 1–10 × 104 s–1. There was no explanation initially provided for why this compound shows such a small ΔE ST; however, subsequent work has hypothesized that having an extended π-network is key to a small ΔE ST in this class of compounds.ref. ref138 The OLEDs showed an EQEmax of 34.4% at CIE (0.12, 0.11), representing one of the most efficient blue TADF emitters to date.ref. ref1031 Further, the device showed minimal efficiency roll-off, with an EQE1000 of 26.1% owing to the efficient k RISC that results in decreased triplet quenching that often plagues MR-TADF OLEDs. The narrowband emission of the v-DABNA combined with its excellent performance (supported by spontaneous horizontal emitter TDM alignment in films) has since sparked significant further research effort.
Following its introduction to the field, v-DABNA has been frequently used as a terminal emitter material in HF-OLEDs.ref253,ref1032 When a triplet harvesting assistant dopant HDT-1 (Figure ) was used alongside v-DABNA acting as the terminal emitter, the OLED showed an EQEmax of 41% at CIE (0.13, 0.16).ref. ref1032 The device stability was improved to LT95 of 18 hours at 1,000 cd m–2, compared to < 1 hour in the parent device at 100 cd m–2. The k RISC of HDT-1 at 8.6 × 105 s–1 is faster than that of v-DABNA (k RISC = 2.0 × 105 s–1), supporting efficient triplet harvesting separate to emission. When PPCzTrz and PCzTrz (Figure ) were used as assistant dopants, the OLEDs showed EQEmax of 33.0 and 33.5%, respectively.ref. ref253 In each device the efficiency roll-off was low, with EQE1000 of 25.2 and 23.8%, respectively. The device stability improved as well, with LT50 at 1000 cd m–2 of 151 and 113 hours for PPCzTrz and PCzTrz, respectively. In another report, HF devices with DMTDac-Me (Figure ) as the assistant dopant performed better than the one that only included v-DABNA in the EML; the EQEmax of the device with 1 wt% v-DABNA was 13.3%, while the 0.5 wt% device showed an EQEmax of 22.2%.ref. ref1033 This work highlighted that although the isolated performance of v-DABNA is exceptional, it still suffers considerably from ACQ and excimer formation at practical concentrations. Using the exciplex host 3Cz-TRZ:Tris-PCz (Figure ) alongside v-DABNA, Nguyen et al. reported stable devices with an LT50 of over 300 hours at an initial luminance of 1260 cd m–2.ref. ref736 The exciplex host contributed to the triplet harvesting, although broadening of the emission compared to v-DABNA alone indicates that energy transfer was not complete.
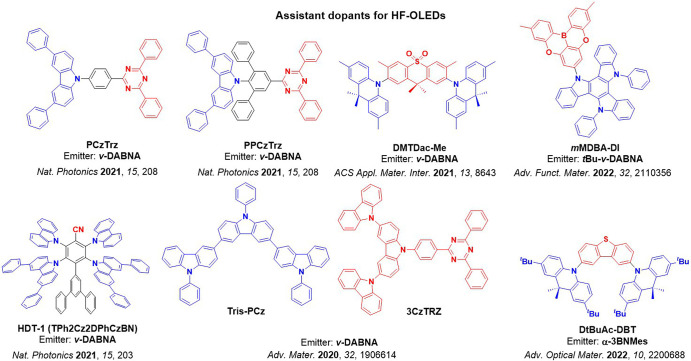
The tert-butyl decorated v-DABNA derivative t-Bu-v-DABNA (Figure ) shows comparable photophysical properties to v-DABNA in 5 wt% DBFPO films, with ΦPL of 92% and τd of 2.9 μs translating to a k RISC of 2.5 × 105 s–1.ref. ref306 In toluene the λPL is 467 nm and the ΔE ST is 0.04 eV. OLEDs using t-Bu-v-DABNA showed EQEmax of 36.3% at CIE coordinates of (0.11, 0.15). The same OLED stack using v-DABNA showed a slightly lower EQEmax of 35.2% at the same CIE coordinates. Unfortunately, the efficiency roll-off of the OLED with t-Bu-v-DABNA was large, with the EQE1000 of 16.5%. When used in conjunction with the assistant TADF dopant mMDBA-DI (Figure ) the EQEmax reached 39.1% while the EQE1000 remained high at 34.3%.
Efforts by the same group to blue-shift emission towards desired Rec. 2020 coordinates focussed first on the introduction of weakly donating methyl substituents para to the boron atoms to destabilize the LUMO in m-v-DABNA (Figure ).ref. ref1034 A second strategy saw the incorporation of fluorine atoms ortho to the nitrogen atoms, which stabilized the HOMO in 4F-v-DABNA (Figure ). The authors also designed a third emitter that combined both modifications in 4F-m-v-DABNA (Figure ). Compared to v-DABNA, all three compounds emit slightly bluer, with λPL in toluene of 464, 457, and 455 nm for m-v-DABNA, 4F-v-DABNA, and 4F-m-v-DABNA, respectively, compared to 468 nm for v-DABNA. m-v-DABNA, 4F-v-DABNA, and 4F-m-v-DABNA showed comparable ΦPL of 91, 90, and 89% and τd of 3.1, 3.1, and 3.2 μs, respectively, as 3 wt% doped films in DBFPO. The ΔE ST values of 0.05–0.07 eV in toluene are similar to that for v-DABNA (ΔE ST of 0.02 eV in 1 wt% DOBOA-OAr),ref. ref1031 leading to comparably fast k RISC ranging between 2.1–2.3 × 105 s–1 in 3 wt% doped films in DBFPOref. ref1034 (k RISC in v-DABNA was 2.0 × 105 s–1 in 1 wt% DOBNA-OAr).ref. ref1031 The OLEDs showed EQEmax of 36.2, 35.8, and 33.7% at CIE coordinates of (0.12, 0.12), (0.13, 0.08), and (0.13, 0.06) for m-v-DABNA, 4F-v-DABNA, and 4F-m-v-DABNA, respectively.ref. ref1034 Despite the impressive k RISC the efficiency roll-off was still large across all three of these emitters.
A strategy to access different color spaces by red-shifting the emission of v-DABNA involved decoration with electron-withdrawing cyano groups to generate v-DABNA-CN-Me (Figure ).ref. ref171 This high-performance green emitter has λPL of 498 nm, ΦPL of 86%, small ΔE ST of 0.01 eV, and τd of 10 μs in toluene. Like v-DABNA, k RISC is fast at 1.0 × 105 s–1. Green devices showed CIE coordinates of (0.13, 0.65) and an EQEmax of 31.6% with EQE1000 of 28.6%.
A V-shaped extended design was reported by Oda et al.ref. ref170 where three boron atoms and six nitrogen atoms were incorporated within the nanographene core to create the helical structure v-DABNA-Mes (Figure ), which is essentially three DABNA-1 units fused together. This compound has λPL of 484 nm, ΦPL of 80%, τd of 2.4 μs, and ΔE ST of 0.009 eV as a 1 wt% doped PMMA film, which is a red-shifted emission compared to v-DABNA (λPL of 467 nm and ΔE ST of 0.02 eV for in 1 wt% DOBNA-OAr).ref. ref1031 Owing to the smaller ΔE ST and ΔE T2‑T1 (calculated for v-DABNA-Mes to be 0.10 eV compared to 0.14 eV in v-DABNA), an improved k RISC of 4.4 × 105 s–1 was reported compared to 2.0 × 105 s–1 in v-DABNA. Solution-processed devices were reported, likely due to the high molecular weight of the emitter at 1774.7 g mol–1 preventing vacuum deposition and showed EQEmax of 22.9% and EQE100 of 20.3% at CIE coordinates of (0.09, 0.21).
An alternative strategy to blue-shift the emission of v-DABNA was presented by Tanaka et al. using v-DABNA-O-Me (Figure ),ref. ref173 in which reduction in HOMO delocalisation was realized when one of the nitrogen donor atoms was replaced with an oxygen atom. This compound has a slightly blue-shifted λPL of 464 nm compared to 467 nm in v-DABNA and a similar ΔE ST value of 0.03 eV in a 1 wt% doped film in PMMA (0.02 eV for v-DABNA in 1 wt% DOBNA-OAr). The devices showed a similar EQEmax of 29.5% while the efficiency roll-off was improved (EQE1000 of 26.9%); the CIE coordinates of the device with v-DABNA-O-Me were (0.13, 0.10), which are close to those of the device with v-DABNA (0.12, 0.11). Importantly, there is a vastly improved device lifetime (LT50 of 314 hours at 100 cd m–2) compared to the device with v-DABNA (LT50 of 31 hours). The differences in device lifetimes were attributed to a larger calculated SOC between S1 and T2 in v-DABNA-O-Me compared to v-DABNA, producing a more efficient TADF process.
A similar strategy to blue-shift the emission of MR-TADF compounds was reported by Park et al.ref. ref1035 who replaced nitrogen atoms with oxygen or sulfur. Three emitters containing either two oxygen atoms (BOBO-Z), one oxygen and one sulfur atom (BOBS-Z), and two sulfur atoms (BSBS-Z) showed progressively red-shifted emission with λPL of 445, 457, and 464 nm, respectively as 3 wt% doped mCBP films (Figure ). Each of these compounds shows a blue-shifted emission compared to v-DABNA, which has a λPL of 474 nm in the same medium. The ΦPL values of the same films varied widely at 64, 93, and 88%, while the ΔE ST and τd values in toluene were all similar at 0.15, 0.16, and 0.14 eV, and 7.7, 7.6, and 6.7 μs, all respectively. Due to the heavy atom effect k RISC was enhanced with more sulfur atoms, at 0.7, 8.6, and 16 × 105 s–1 for BOBO-Z, BOBS-Z, and BSBS-Z, respectively, in the 3 wt% doped films in mCBP. The devices with BOBO-Z, BOBS-Z, and BSBS-Z showed EQEmax of 13.6, 26.9, and 26.8%, respectively, at CIE coordinates of (0.15, 0.04), (0.14, 0.06), and (0.13, 0.08). The latter two devices outperformed the OLED with v-DABNA using the same stack [EQEmax of 24.6% at CIE coordinates of (0.12, 0.12)]. The efficiency roll-off was also modest, with EQE100 of 9.8, 24.0, and 24.0%, respectively.
Recently, our group introduced a linear boron and nitrogen-containing MR-TADF heptacene system, α-3BNOH (Figure ), that emits at a λPL of 398 nm in THF and has a FWHM of 31 nm.ref. ref164 Although this compound had a large measured ΔE ST of 0.31 eV, a small TADF contribution was nonetheless observed with a τd of 450 ns in THF. Interestingly the activation energy for T1 to S1 conversion was much lower at 0.07 eV, with RISC here believed to involve intermediate triplet states as corroborated by calculations. At room temperature the triplet harvesting pathways are a combination of TADF and TTA. Devices were reported subsequently using an EML consisting of 10 wt% of α-3BNOH doped in DPEPO.ref. ref1036 Compared to emission in THF, λEL was red-shifted and broadened (λEL at 410 nm and FWHM of 47 nm). The EQEmax was less than 1%, attributed to the formation of aggregates, which is consistent with the broadened and red-shifted EL spectrum. Replacement of the OH substituents with mesityl groups resulted in a red-shift of the emission in α-3BNMes (Figure ).ref. ref1037 In THF the λPL shifted from 398 nm for α-3BNOH to 442 nm for α-3BNMes. In 1 wt% doped PMMA films the ΔE ST of α-3BNMes was identical to that of α-3BNOH, at 0.28 eV for each. The photophysics is complex, reflected in the presence of two lifetimes in the delayed emission (τd of 9.1 μs and 7.1 ms), the shorter one associated with a mixture of aggregate and monomer emission and the longer one linked to pure monomer emission. RISC is thus inefficient, with k RISC of only 5.9 × 102 s–1, and the OLED performance was poor with an EQEmax of 1.7%. However, when used as the terminal emitter in conjunction with DtBuAc-DBT (Figure ) as the assistant dopant in a HF-OLED, the EQEmax improved to 15%, with CIE coordinates of (0.15, 0.10).
Central Boron MR-TADF Compounds with a Carbazole Scaffold
A separate design strategy has emerged in parallel with those described above, replacing the DPA groups embedded within DABNA-1 with other N-heterocycles. The first such derivative, DtBuCzB (Figure ),ref. ref1038 contained fused tert-butylcarbazole and displayed sky blue emission with λPL of 493 nm and ΦPL of 88% in 1 wt% doped mCBP films, with ΔE ST of 0.13 eV and τd of 69 μs. Compared to DABNA-1, the emission is red-shifted and the ΔE ST is smaller (λPL = 460 nm and ΔE ST = 0.18 eV for DABNA-1 in 1 wt% doped mCBP films)ref. ref118 owing to increased conjugation afforded by the fused structure.ref. ref1039 The OLEDs showed an EQEmax of 21.6% at CIE coordinates of (0.10, 0.42). The same material was also reported as BBCz-SB,ref. ref1040 wherein a slightly improved device performance was reported with EQEmax of 27.8%. Xu et al. presented solution processed HF-OLEDs, with an EQEmax of 16.3% at λEL of 490 nm reported for DtBuCzB, which was renamed BCzBN here when used alongside the assistant dopant CzAcSF (Figure ).ref. ref1039
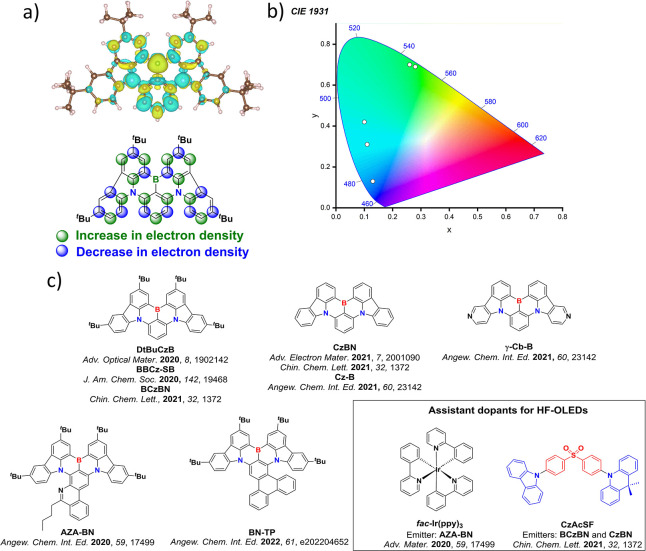
Developing from this fused-carbazole core, an analogue without tert-butyl substituents has been reported by three groups, named CzBN ref1039,ref1041 and Cz-B ref. ref1042 (Figure ). Cz-B was presented alongside a carboline analogue, γ-Cb-B (Figure ).ref. ref1042 The emission of γ-Cb-B is blue-shifted at λPL of 461 nm compared to λPL of 484 nm in Cz-B in 1 wt% doped films in mCBP and oCBP, respectively, a result of weaker electron-donating character for the carboline. The ΦPL and τd of Cz-B and γ-Cb-B are 97 and 89% and 32 and 44 μs, respectively, while both compounds show similar ΔE ST values of 0.14 and 0.12 eV in toluene. The devices with Cz-B and γ-Cb-B showed EQEmax of 22.1 and 19.0% at CIE coordinates of (0.11, 0.31) and (0.13, 0.13), respectively. Both OLEDs showed large efficiency roll-off though, with EQE1000 of 6.9 and 7.7% attributed to their long-delayed lifetimes. HF solution processed OLEDs were presented with CzBN emitter and CzAcSF assistant dopant by Xu et al., with a EQEmax of 14.7% presented at a 2 wt% doping of the emitter, with λEL of 480 nm.ref. ref1039
A fused derivative containing an azaphenanthrene-type structure, AZA-BN (Figure ), was reported by Zhang et al.ref. ref1043 The increased conjugation produced a red-shifted emission in toluene (λPL of 522 nm) compared to DtBuCzB (λPL = 481 nm).ref. ref1038 The ΔE ST in toluene is 0.18 eV while the ΦPL is essentially unity, reported as 99.7%. In the 4 wt% doped mCBP films the ΦPL is 94% and there is a long τd of 160 μs. The OLEDs showed EQEmax of 25.7% at CIE coordinates of (0.28, 0.69), while the EQE1000 dropped to 9%. This is unsurprising owing to the long-delayed lifetime and was addressed in HF-OLEDs using fac–Ir(ppy)3 (Figure ) as the phosphorescent assistant dopant, which then achieved EQEmax of 28.2% and a higher EQE1000 at 19.1%.
Triphenylene derivative BN-TP (Figure ) was reported by Xu et al.ref. ref1044 and prepared via a Scholl oxidative ring closing reaction. Compared to the parent DtBuCzB (λPL = 481 nm in toluene)ref. ref1038 there was a significant red-shift to 523 nm for BN-TP due to the increased π-conjugation in the backbone.ref. ref1044 In 3 wt% doped PhCzBCz films BN-TP has a ΦPL of 96% and a k RISC of 2.1 × 104 s–1. The device showed an impressive EQEmax of 35.1% at CIE coordinates of (0.26, 0.70), which was attributed to strong horizontally aligned TDM enhancing the light outcoupling. The EQE100 was maintained at 32.4%, while a promising LT50 of 28.8 hours was reported at 4,000 cd m–2.
Substituted with Peripheral Acceptor Units
Substituted analogs of DtBuCzB have proven to be a popular design strategy (Figure and Figure ) especially for color tuning of narrowband MR-TADF emission. Zhang et al.ref. ref1045 demonstrated the first examples of green MR-TADF emitters 2F–BN, 3F-BN, and 4F-BN with λPL of 501, 498, and 493 nm (Figure ), respectively. The electron-withdrawing fluorophenyl groups act to stabilize the LUMO compared to the parent, reducing the emission energy from blue to green. The ΔE ST values of 0.16, 0.08, and 0.11 eV remained similar to DtBuCzB while the τd and ΦPL ranged between 16.7–25.6 μs, and 83–91%, respectively. Green HF-OLED devices using 2F–BN, 3F-BN, and 4F-BN with 5TCzBN (Figure ) as the assistant dopant showed EQEmax of 22.0, 22.7, and 20.9% and efficiency roll-off of between 7–32% at 1000 cd m–2 at CIE coordinates (0.16, 0.60), (0.20, 0.58) and (0.12, 0.48), respectively. In a similar vein, direct substitution of a cyano group para to boron produced the emitter CN-BCz-BN (Figure ).ref. ref486 This compound has a modestly red-shifted emission in toluene (λPL = 496 nm) compared to DtBuCzB (λPL = 481 nm). No further studies were undertaken on this material.
Substitution with strong acceptors para to boron was also the subject of a study by Xu et al.ref. ref1046 The acceptors included triazine, phenyltriazine, phenylpyrimidine, and cyanopyrimidine, producing the emitters DtCzB-DPTRZ, DtCzB-TPTRZ, DtCzB-PPm, and DtCzB-CNPm, respectively (Figure ). The red-shifted emission maxima in toluene were 521, 501, 499, and 515 nm, respectively. The ΔE ST for these analogues measured in toluene ranged between 0.08–0.17 eV,ref. ref1046 and the ΦPL remained high at between 87–95% in 3 wt% doped PhCzBCz films. Devices with DtCzB-DPTRZ, DtCzB-TPTRZ, DtCzB-PPm, and DtCzB-CNPm showed EQEmax of 24.6, 29.8, 28.6, and 25.0%, at CIE coordinates of (0.33, 0.63), (0.18, 0.67), (0.16, 0.66), and (0.35, 0.63), respectively. Contrasting device performances were noted, with extremely high efficiency roll-off of 70% and 42% at 100 cd m–2 for the devices with DtCzB-DPTRZ and DtCzB-CNPm, compared to 11% and 15% for the devices with DtCzB-TPTRZ and DtCzB-PPM. This difference was attributed to faster k RISC for the latter two emitters (ca. 1 × 104 s–1 compared to ca. 1 × 103 s–1 for the former). The materials with more efficient k RISC had slightly smaller ΔE ST of 0.11 and 0.08 eV, compared to 0.17 and 0.12 eV.
Two chiral MR-TADF compounds (R/S)-OBN-2CN-BN and (R/S)-OBN-4CN-BN containing phenylcyano substituents showed narrowband CPL (Figure ).ref. ref658 The phenylcyano substitution red-shifted the emission compared to DtBuCzB, with λPL of 498 and 510 nm for (R/S)-OBN-2CN-BN and (R/S)-OBN-4CN-BN, respectively in 3 wt% doped PhCzBCz films. The ΔE ST of 0.12 and 0.15 eV are for the R-isomers in toluene while the τd are 95 and 97 μs in 3 wt% doped films in PhCzBCz; the S-isomers show similar photophysical behavior.ref. ref658 Devices of R and S isomers of OBN-2CN-BN and OBN-4CN-BN were fabricated, with similar properties between R and S isomers. The CIE coordinates of OBN-2CN-BN and OBN-4CN-BN were (0.11, 0.52) and (0.14, 0.64), respectively, for both the R and S isomers. The device EQEmax was 29.4 and 28.8% for OBN-2CN-BN R and S isomers, respectively, with a modest decrease for the device with OBN-4CN-BN at 24.5 and 24.3% for R and S isomers, respectively. Each showcased large efficiency roll-off with EQE100 of 19.8% (19.2%) and 8.0% (7.9%) for OBN-2CN-BN and OBN-4CN-BN, respectively, for their R and (S) isomers. HF OLEDs of (R)-OBN-2CN-BN and (R)-OBN-4CN-BN were fabricated using 5CzBN and 5tBuCzBN (Figure ) as assistant dopants, respectively, with similar EQEmax at 29.8 and 24.7% compared to the 29.4 and 24.5% previously reported. However, the efficiency roll-off dramatically improved, with EQE100 of 27.2 and 23.5%. Their chiral optical properties are discussed in more detail in Section sec7 .
Substituted with Peripheral Donor Units
Alongside acceptor substitution of MR-TADF emitters, there are now several reported examples of adding electron-donating substituents to DtBuCzB to modulate or enhance its properties. A tert-butylcarbazole unit coupled meta to the boron in m-Cz-BNCz (Figure )ref. ref210 produced a red-shifted emission with λPL of 519 nm in toluene and λPL of 528 nm in 10 wt% doped PhCzBCz films. The stabilization of the S1 state is the result of a destabilized HOMO arising from the electron-donating tert-butylcarbazole. The ΔE ST is 0.08 eV in toluene, producing a remarkably rapid delayed emission with τd of 0.86 μs in 10 wt% doped PhCzBCz films and efficient k RISC of 1.0 × 106 s–1.ref. ref210 However, the emission spectrum is broadened by the inclusion of the donating unit, reflected in a larger FWHM of 38 nm compared to 22 nm for DtBuCzB in toluene and likely arising from a hybrid SRCT/LRCT character. ref. ref1038 Efficient devices showed an EQEmax of 31.4% at CIE coordinates of (0.26, 0.68), and there was only a modest efficiency roll-off with EQE100 of 29%.ref. ref210
The corresponding para-substituted derivative TCz-BN ref. ref1045 (or p-Cz-BNCz, Figure )ref. ref210 has been investigated computationallyref. ref209 and used in HF-OLEDs;ref. ref1045 however, there is little documentation of its intrinsic photophysical properties. A blue-shifted emission in toluene (λPL of 477 nm) is apparent compared to m-Cz-BNCz (λPL of 519 nm).ref. ref210 This difference is attributed to the fact that meta substitution leads to a destabilized HOMO more so than para substitution.ref. ref209 HF-OLEDs with TCz-BN as the terminal emitter and 4T (Figure ) as the assistant dopant showed EQEmax of 18.9% at CIE coordinates of (0.13, 0.20).ref. ref1045
Based on these initial reports, two derivatives with additional carbazoles were designed (BBCz-Y and BBCz-G, Figure ).ref. ref1040 In the former the two meta positions are occupied, while in the latter both meta and the para positions are occupied. Compared to m-Cz-BNCz (λPL = 519 nm), the emission of BBCz-Y is red-shifted in toluene to 549 nm, while the λPL of BBCz-G surprisingly remains at 517 nm. For BBCz-Y, the second donating meta-substituted tBuCz further destabilizes the HOMO leading to further red-shift of the emission. For BBCz-G the same destabilizing interaction of the HOMO is achieved concomitantly with a destabilized LUMO resulting from the para-substituted tBuCz, producing minimal net effect compared to m-Cz-BNCz. Both compounds have identical ΔE ST of 0.14 eV, while the τd are similar at 13 and 11 μs in 2 wt% doped mCBP films. OLEDs with BBCz-Y and BBCz-G showed EQEmax of 29.3 and 31.8% at CIE coordinates of (0.37, 0.61) and (0.26, 0.68), respectively, and the EQE100 remained high at 25.8 and 29.5%.
Coupling of a Me-diphenylamine group para to boron in Cz2DABNA-NP-M/TB (Figure )ref. ref1021 resulted in a destabilized LUMO and a slight blue-shift of emission (λPL at 478 nm in 1 wt% doped PMMA films) compared to DtBuCzB,ref. ref1038 similar to that observed for TCz-BN (vide supra).ref. ref210 The similarity of the ΔE ST and τd (0.15 eV and 19 μs, respectively) to those of previously reported donor-substituted MR-TADF emitters suggest that the MR-TADF mechanism is largely unaffected by the nature or the regiochemistry of the donor in these materials; instead, the main impact is restricted to the emission color.ref. ref1021 The blue devices using Cz2DABNA-NP-M/TB showed EQEmax of 21.8% at CIE coordinates of (0.11, 0.23) while the EQE decreases by only 6% at 100 cd m–2.
A related tert-butyldiphenylamine derivative tDPA-DtCzB (Figure ) was reported by Yan et al.ref. ref1047 and direct comparison were made with the parent DtBuCzB, (named DtCzB in this study). In toluene the emission was blue-shifted from 481 to 470 nm for DtCzB and tDPA-DtCzB, respectively, along with a modest reduction in ΔE ST from 0.13 to 0.11 eV. In 1 wt% doped PhCzBCz films the two emitters show comparable ΦPL of 89 and 85%, while k RISC increased from 0.74 × 104 s–1 for DtCzB to 2.45 × 104 s–1 for tDPA-DtCzB. The OLEDs with DtCzB and tDPA-DtCzB showed EQEmax of 23.2 and 25.0%, respectively, but efficiency roll-off was severe with EQE100 of 13.0 and 16.4%. HF-OLEDs using either 5CzBN or Firpic (Figure ) as the assistant dopant were also fabricated, and the highest performing HF-OLED showed an EQEmax of 31.0% with the latter.
The emitter CzBNCz similarly contains a carbazole attached para to the CzBN core (Figure ).ref. ref1041 Similar to previously discussed donor substitutions, addition of Cz para to the boron produced a modest blue-shift with λPL shifting from 485 nm for CzBN to 470 nm for CzBNCz in 1 wt% doped films in mCBP. The ΦPL remains very high at 99% for CzBN and 95% for CzBNCz in the same films, while the ΔE ST increase from 0.15 to 0.18 eV (both in toluene). The larger ΔE ST resulted in a longer τd, increasing from 75 for CzBN to 92 μs for CzBNCz. HF-OLEDs with CzBNCz and CzBN using TPh2Cz2DPhCzBN (Figure ) as the assistant dopant showed EQEmax of 21.9 and 20.6%, respectively, at CIE coordinates of (0.16, 0.31) and (0.14, 0.31). The efficiency roll-off was modest with EQE100 of 21.0 and 19.4%, respectively, although the devices showed low stability with LT90 at 1000 cd m–2 of 39 hours and 29 hours for CzBNCz and CzBN, respectively.
Incorporation of mildly donating tBu-Phenyl groups onto DtBuCzB produced DtBuPhCzB (Figure ),ref. ref1038 which shows a red-shifted emission of λPL 508 nm in 1 wt% doped mCBP films (compared to 493 nm for DtBuCzB in the same). Despite the change in emission color, this substitution had a minimal effect on the TADF characteristics with ΔE ST of 0.10 eV and τd of 61 μs (compared to 0.13 eV and 69 μs reported for DtBuCzB). Green devices showed an EQEmax 23.4% at CIE coordinates of (0.15, 0.61). The EQEmax could be enhanced to 26.5% with the use of exciplex host TCTA:PIM-TRZ (at 3 wt% emitter doping), but the emission color red-shifted to CIE coordinates of (0.25, 0.65). The improvements in device performance were attributed to two factors: firstly, the exciplex host is itself TADF (see Section sec8 ), which allowed for triplet harvesting on the host as well as the emitter; secondly, the exciplex host displayed improved charge balance within the emissive layer compared to simpler mCBP devices.
Yang and co-workers reported a series of MR-TADF emitters where sequentially stronger donors were coupled para to the nitrogen atom.ref. ref1048 Compounds BN1, BN2, and BN3 contained either two carbazoles, two diphenylamines, or four diphenylamine donor groups (Figure ). Changing the nature and number of donors had a significant impact on color, with λPL red-shifting from 499 to 538 and 563 nm in 1 wt% doped mCBP films for BN1, BN2, and BN3, respectively. There were only minor changes to the other photophysical properties, with a modest broadening of the emission spectrum (FWHM increasing from 38 to 44 nm from BN1 to BN3) along with a slight decrease in the ΦPL from 93 to 86%, while the ΔE ST (0.09–0.13 eV) and the k RISC (1.9–1.4 × 105 s–1) were largely unaffected, all respectively. Devices with BN1, BN2, and BN3 showed EQEmax of 17.0, 20.7, and 21.4%, although the efficiency roll-off was severe with EQE1000 of 8.5 and 3.3% for the devices with BN1 and BN2 (luminance of 1,000 cd m–2 was not reached for BN3). When mCBP:PO-T2T exciplex host was employed, the EQEmax increased to 24.3, 24.5, and 24.7% for the devices with BN1, BN2, and BN3, at CIE coordinates of (0.15, 0.63), (0.38, 0.61), and (0.47, 0.52). Efficiency roll-off also improved, with EQE1000 of 18.4, 15.8, and 17.6%, respectively.
Two derivatives of BN3 containing bulkier substituents were recently reported, BN-1 and BN-8 (Figure ).ref. ref1049 In toluene these two emitters showed similar photophysical properties, with λPL of 566 and 568 nm for BN-1 and BN-8 respectively, and ΦPL of 95% for both. However, the ΔE ST values were distinct at 0.11 and 0.03 eV for BN-1 and BN-8, respectively. No further investigations were undertaken for these emitters.
A similar emitter design was reported by Yang et al. who attached carbazole (TCz-B, Figure ) and tetramethyldiphenylamine donors (DACz-B, Figure ) on to the core emitter Cz-B.ref. ref1042 The presence of the donor groups led to a red-shifting of the emission, with λPL of 484, 517, and 576 nm for Cz-B, TCz-B, and DACz-B, respectively in 1 wt% doped mCBP films. Similar ΔE ST of 0.14, 0.09, and 0.14 eV in toluene were obtained, while donor substitution produced a modest decrease in ΦPL from 97 to 89 and 87% in 1 wt% doped mCBP films. There was also an increase in τd with donor substitution, from 44 to 71 and 118 μs for Cz-B, TCz-B, and DACz-B, respectively. OLEDs with TCz-B and DACz-B showed EQEmax of 29.2 and 19.6% at CIE coordinates of (0.16, 0.71) and (0.47, 0.51). The long τd translated to a large efficiency roll-off with EQE1000 dropping to 9.4 and 4.8%.
Substituted with Peripheral Donor and Acceptor Units
The use of both donor and acceptor substitution can provided further control of emission color. This control was demonstrated by a range of derivatives of BN-1 and BN-8 (Figure ), previously reported by Cai et al.ref. ref1049 These two parent emitters were decorated with various acceptor groups para to the boron, including pyrimidine and triazine derivatives. The addition of the acceptor units red-shifts the emission, with λPL shifting from 566 to 586, 598, 612, 627, 618, and 629 nm for new emitters BN-2, BN-3, BN-4, BN-5, BN-6, and BN-7, respectively (Figure ). The largest red-shifts were observed for the emitters with the strongest triazine electron acceptors. The same trend was captured with materials based on BN-8, with λPL shifting from 568 to 585, 595, 608, and 624 nm for new emitters BN-9, BN-10, BN-11, and BN-12, respectively (Figure ). Addition of these acceptor units also broadened the emission (FWHM ranging between 35–47 nm). The ΔE ST values ranged from 0.03–0.12 eV, while the emitters possessed near nearly identical ΦPL of between 94 and 96%. No devices were fabricated.
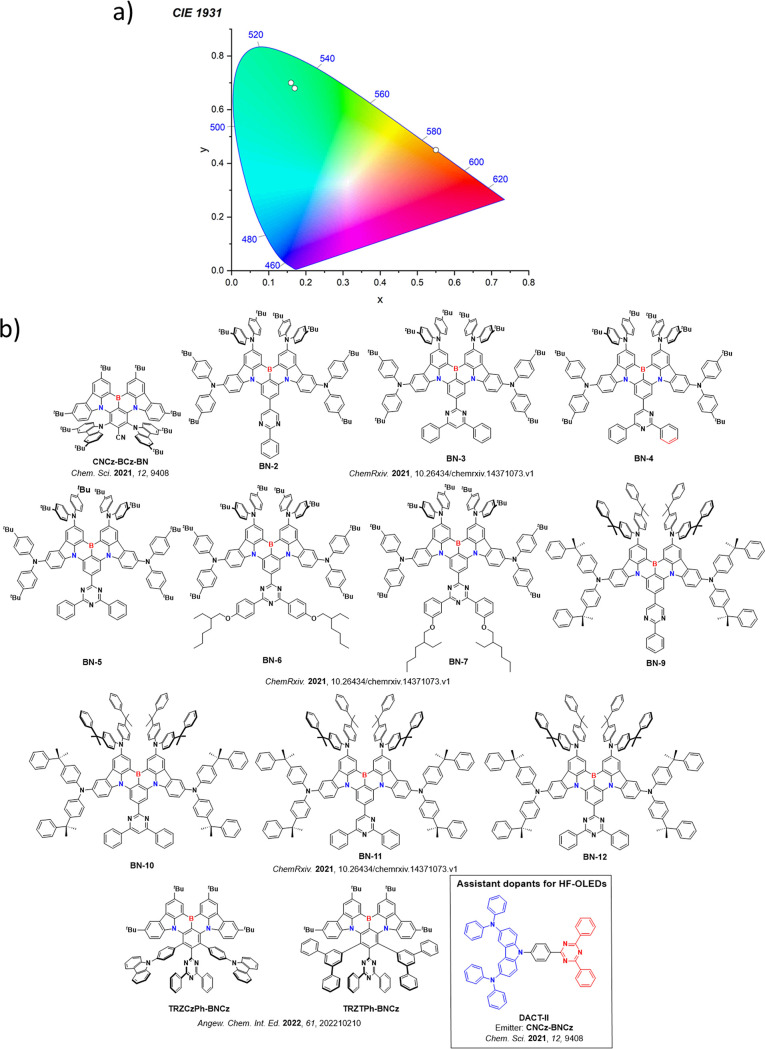
TRZCzPh-BNCz and TRZTPh-BNCz are further derivatives of DtBuCzB containing donor and acceptor substituents (Figure ).ref. ref1050 By replacing carbazole for triphenylene in TRZTPh-BNCz the authors aimed to introduce intermediate triplet states that could help mediate RISC while maintaining a similar steric environment. Narrowband green emission at λPL of 514 and 513 nm with corresponding FWHM of 34 and 29 nm were observed for TRZCzPh-BNCz and TRZTPh-BNCz, respectively. Solvatochromic studies highlighted that donor and acceptor substitution of these emitters had little impact on the SRCT nature of the excited state. TRZCzPh-BNCz and TRZTPh-BNCz have similar ΔE ST values of 0.13 and 0.11 eV in toluene, and high ΦPL of 98 and 99% in 3 wt% doped CBP films, respectively. Compared to DtCzB-DPTRZ (Figure ), without carbazole or triphenylene substitutents, calculations revealed T2 and T3 to be close in energy to S1, and in addition T2 was calculated to have a significant SOC > 1.4 cm–1 in both compounds. This contributed to efficient k RISC of 8.8 and 7.5 × 105 s–1 for TRZCzPh-BNCz and TRZTPh-BNCz, respectively, in 3 wt% doped CBP films. OLEDs with TRZCzPh-BNCz and TRZTPh-BNCz showed EQEmax of 32.5 and 31.4%, respectively, which are higher than the device with DTCzB-DPTRZ (EQEmax = 20.2%). The efficient k RISC also contributed to low efficiency roll-off, with the EQE100 remaining as high as 30.5 and 29.5%, while for the reference emitter the EQE100 dropped to 7.8%.
Based on BBCz-Y (Figure ), Liu et al.ref. ref486 reported the compound CNCz-BNCz that additionally contained a cyano group para to the boron centre (Figure ). The emission was red-shifted from 549 nm in BBCz-Y to 581 nm in CNCz-BNCz, which was more pronounced than the red-shift from 481 nm in BBCz-BN to 496 nm in CN-BCz-BN. The addition of the cyano had minimal impact on the FWHM (42 nm for both BBCz-Y and CNCz-BNCz). CNCz-BNCz has a ΔE ST of 0.18 eV, a τd of 60.4 μs, and the ΦPL is 96% in 3 wt% doped CBP films. Devices showed an EQEmax of 23.0% at CIE coordinates of (0.55, 0.45), although there was large efficiency roll-off with EQE100 dropping to 10.8%. HF-OLEDs using DACT-II (Figure ) as the assistant dopant showed improved EQEmax of 33.7% while the EQE100 remained high at 27.7%.
Substituted with Peripheral Electronically Inert Substituents
A number of examples exist where there is the introduction of electronically inert substituents that are designed to have minimal impact upon the emission color (Figure ). In most cases these groups are added to mitigate ACQ (without impacting the SRCT character of the S1 state) to prevent broadening of the emission spectrum at higher doping concentrations. Jiang et al. reported two such compounds BN-CP1 and BN-CP2, containing phenyl groups para-substituted on the MR-TADF core (Figure ),ref. ref1051 and featuring two carbazole moieties either ortho (BN-CP1) or meta (BN-CP2) to the phenyl substituents (Figure ). The two compounds showed similar photophysical properties in toluene with λPL of 490 nm for both and ΔE ST of 0.12 and 0.13 eV for BN-CP1 and BN-CP2, respectively. In 5 wt% doped DMIC-TRZ films the ΦPL are 98 and 95%, while τd are 65 and 58 μs for BN-CP1 and BN-CP2, respectively. Even at 30 wt% doping the ΦPL remained at 84 and 61%, highlighting how these substituents can mitigate ACQ while maintaining the narrowband emission (FWHM = 23 nm for each in toluene). Devices at doping concentrations ranging between 1–30 wt% emitter in the EML were prepared for each material, with 5 wt% doping showing the highest efficiencies. The OLEDs with BN-CP1 and BN-CP2 showed EQEmax of 40.0 and 36.4% at CIE coordinates of (0.09, 0.50) and (0.10, 0.53). The EQE100 remained high at 34.0 and 32.6%, respectively. HF-OLEDs with BN-CP1 were also fabricated using 5TCzBN (Figure ) as the assistant dopant. Despite a small drop in the EQEmax of 38.1%, the efficiency roll-off lessened with the EQE100 at 37.6%.
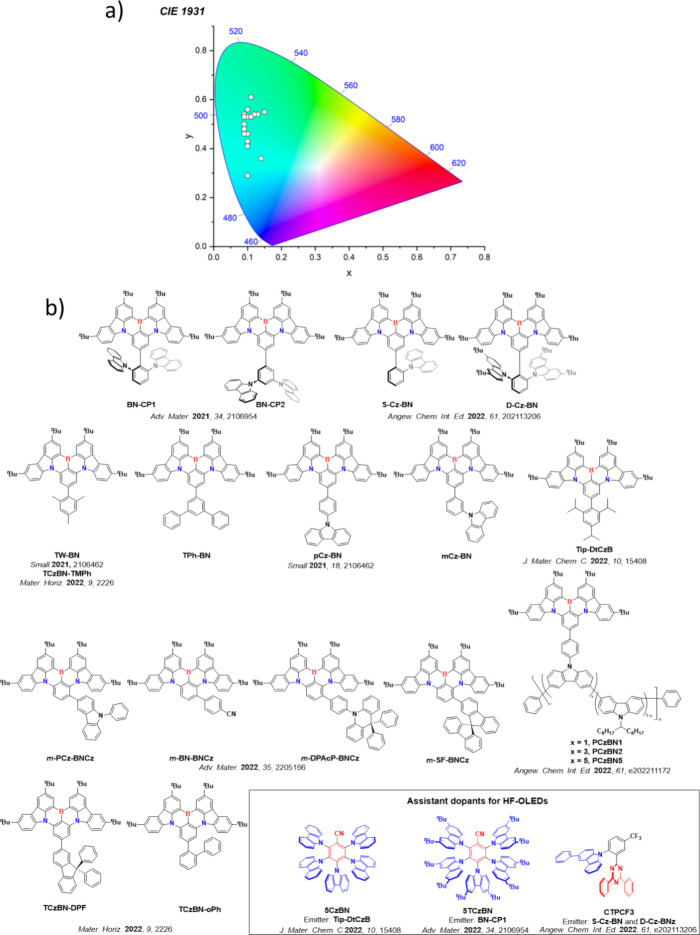
A similar design based on BN-CP1 was presented by Zhang et al.,ref. ref1052 where the DtCzBN core was substituted with an ortho-Czphenyl group in S-Cz-BN or an ortho,ortho-diDtCzphenyl group in D-Cz-BN (Figure ). In toluene both compounds emit at λPL of 490 nm and show similar τd and ΔE ST in 5 wt% doped CBP films at 42 and 44 μs and 0.16 and 0.14 eV for S-Cz-BN and D-Cz-BN, respectively. For S-Cz-BN the ΦPL decreased only modestly from 95 to 84% for 1–30 wt% doped films, while for D-Cz-BN there was an even smaller attenuation in ΦPL from 98 to 90% across the same doping range. In neat films the ΦPL were 47 and 54%, yet the emission remained narrow with FWHM of 40 and 26 nm for S-Cz-BN and D-Cz-BN, respectively. Devices were fabricated at a range of doping concentrations, with 5 wt% doping offering the best performances with EQEmax of 22.1 and 28.7% at CIE coordinates of (0.10, 0.42) and (0.10, 0.41) for the devices with S-Cz-BN and D-Cz-BN, respectively. At 1,000 cd m–2 the roll-off was significant, with EQE1000 of 12.4 and 11.4%, owing largely to their inefficient k RISC of 3.0 × 104 s–1 for both. Non-doped devices with S-Cz-BN and D-Cz-BN showed EQEmax of 12.8 and 14.8% at CIE coordinates of (0.16, 0.59) and (0.10, 0.42), with the latter showing the narrowest emission of any non-doped TADF OLED to date with a FWHM of 21 nm. Additionally, both emitters were used in HF-OLEDs with CTPCF3 (Figure ) as the assistant dopant. The HF-OLEDs with S-Cz-BN and D-Cz-BN showed EQEmax of 30.5 and 37.2%, respectively, while the EQE1000 remained high at 26.2 and 34.3%.
A similar series of emitters containing mesityl (TW-BN), triphenyl (TPh-BN), para-phenylcarbazole (pCz-BN), and meta-phenylcarbazole (mCz-BN) designed to mitigate ACQ and maintain narrowband emission have been reported (Figure ).ref. ref1053 In 3 wt% doped mCBP films all four compounds emit similarly, with λPL of 485–495 nm (FWHM 25–30 nm) and ΔE ST minimally varying at 0.12, 0.09, 0.15, and 0.14 eV. The spread of τd is larger at 112, 62, 89, and 95 μs for TW-BN, TPh-BN, pCz-BN, and mCz-BN, all respectively. The OLEDs with TW-BN, TPh-BN, pCz-BN, and mCz-BN showed EQEmax of 27.8, 28.9, 27.2, and 25.9% at CIE coordinates of (0.14, 0.36), (0.10, 0.46), (0.13, 0.54), and (0.15, 0.55), respectively. Efficiency roll-off of each was significant, with EQE1000 dropping to 10.7, 15.6, 12.2, and 14.0%, for the same devices. Device lifetimes were also assessed where the LT50 from an initial luminance of 500 cd m–2 were 10.4, 36.5, 27.3, and 18.6 hours, respectively.
A similar derivative was reported containing bulky triisopropyl (Tip) groups on a benzene substitutent para to the boron, Tip-DtCzB (Figure ).ref. ref1047 Compared to the parent emitter DtBuCzB, there is a modest blue-shift of the emission from 481 nm for DtBuCzB to 477 nm for Tip-DtCzB while both compounds have identical ΔE ST of 0.13 eV in toluene. Their ΦPL are also similar at 89 and 95%, as are the k RISC values of 8.3 and 9.6 × 103 s–1, respectively for DtBuCzB and Tip-DtCzB. The OLEDs showed improved efficiencies with the EQEmax increasing from 23.2% for the device with DtBuCzB to 28.9% for the device with Tip-DtCzB; both devices showed large efficiency roll-off, with EQE100 of 13.0 and 18.2%, respectively. Not surprisingly the efficiency roll-off in the HF-OLED with Tip-DtCzB and 5CzBN (Figure ) as assistant dopant improved, with EQEmax of 29.0% and EQE100 of 23.2%.
An alternative strategy using meta positioning of bulky groups has also been pursued, exemplified by m-PCz-BNCz, m-DPAcP-BNCz, m-BN-BNCz, and m-SF-BNCz (Figure ).ref. ref1054 Owing to their highly twisted geometry and use of phenyl spacer, the functional groups were weakly coupled to the MR-TADF core, resulting in a modest red-shift of the emission compared to the parent emitter DtBuCzB (481 nm in toluene),ref. ref1038 with λPL in toluene ranging from 488–494 nm.ref. ref1054 The four compounds possess similar ΔE ST of 0.14–0.16 eV and high ΦPL of 93–97%. Despite moderate k RISC of around 1 × 104 s–1 for each, the devices with m-PCz-BNCz, m-DPAcP-BNCz, m-BN-BNCz, and m-SF-BNCz showed very high EQEmax of 36.8, 42.0, 35.0, and 41.1%. The high EQEmax was supported by preferentially horizontally aligned TDM that enhanced the light-outcoupling in the devices. Efficiency roll-off was considerable though with the EQE1000 dropping to 19.0, 17.5, 10.9, and 17.9%, respectively.
Extending this approach, Wang et al. developed a family of conjugated polymers consisting of a polycarbazole backbone with pendant MR-TADF emitter DtBuCzB molar ratios of 1, 3, and 5% for polymers PCzBN1, PCzBN3 and PCzBN5, respectively (Figure ).ref. ref1055 Beyond mitigation of ACQ the polymers facilitate the fabrication of solution-processed OLEDs. The neat film emissions of the three polymers are very similar, with λPL of 491–501 nm and FWHM of 33–43 nm. Both PCzBN1 and PCzBN3 also showed emission from the polycarbazole backbone; however, complete energy transfer occurred from the polycarbazole to the DtBuCzB in PCzBN5. The ΦPL of the neat films ranged from 43–58%, while k RISC varied between 8.2–21.8 × 104 s–1. Non-doped solution-processed OLEDs with PCzBN1, PCzBN3, and PCzBN5 showed only EQEmax of 3.7, 5.3, and 10.3%. Positively, these devices showed narrowband EL (FWHM of 33, 41, and 43 nm, respectively), representing rare examples of non-doped OLEDs possessing saturated color. The authors demonstrated that the EQE could be improved when using a doped EML with the polymer dispersed in mCP as the host. Optimal doping concentrations of 60, 70, and 40% for the devices with PCzBN1, PCzBN3, and PCzBN5, respectively, were identified. At these concentrations the ΦPL increased to 65, 71, and 77%, while the EQEmax increased to 17.8, 17.5, and 13.3%, respectively, at CIE coordinates of (0.10, 0.43), (0.12, 0.54), and (0.11, 0.53). Additional examples of both D-A and MR-TADF emitters incorporated into polymers are presented in Section sec10.1 .
A series of compounds were investigated that contained different substituents para to the boron atom of DtBuCzB, designed to probe the origins of spectral broadening and annihilation pathways at higher emitter doping ratios.ref. ref1056 The authors incorporated diphenylfluorene (TCzBN-DPF), mesityl (identical structure to TW-BN, Figure , but renamed TCzBN-TMPh here) and biphenyl (TCzBN-oPh, Figure ). Together with reference emitter DtBuCzB these four compounds showed similar photophysics, with λPL 483, 491, 486, and 489 nm, and with ΔE ST of 0.14, 0.13, 0.12, and 0.13 eV for DtBuCzB, TCzBN-DPF, TCzBN-TMPh, and TCzBN-oPh, all respectively in toluene. The changes in photophysics of the emitters was investigated as a function of the doping concentration (from 1–20 wt%) in SF3TRZ. A small degree of emission broadening was observed for DtBuCzB and TCzBN-DPF, which was less pronounced for TCzBN-oPh and absent for TCzBN-TMPh. The observed broadening was attributed to exciplex formation and decreases in ΦPL mirrored the increasing emission contribution from the exciplex, where the ΦPL of DtBuCzB and TCzBN-TMPh decreased from 93 and 94%, respectively at 1 wt% loading, to 70 and 74% at 20 wt% loading. In TCzBN-DPF and TCzBN-oPh the decrease was less pronounced, falling from 97 and 96%, to 92 and 86% at the same concentrations. An optimal doping ratio of 5 wt% was identified for the devices with DtBuCzB and TCzBN-DPF, which showed EQEmax of 26.3 and 26.4%, respectively. Instead, a doping ratio of 1 wt% was needed for the devices with TCzBN-TMPh and TCzBN-oPh, where the EQEmax were 25.1 and 26.0%, respectively. Even at the optimal doping concentrations, the devices showed strong efficiency roll-off, with EQE1000 dropping to 9.0, 12.0, 6.5, and 10.4% for the devices with DtBuCzB, TCzBN-DPF, TCzBN-TMPh, and TCzBN-oPh, respectively.
Substitution of CzBN to Modulate Its Photophysics or Stability
Other examples have emerged where substitution of CzBN has been employed to alter other properties, including improving the energy transfer efficiency in HF-OLEDs, increasing the stability and enhancing the SOC. In examples of conspicuously non-inert substituents, Lee et al.ref. ref1057 demonstrated how the coupling of naphthalene (CzBNNa) and pyrene (CzBNPyr) para to CzBN improved the stability of HF-OLEDs (Figure ). The ΔE ST in toluene of CzBN and CzBNNa remained the same at 0.15 eV, but that of CzBNPyr was significantly larger at 0.61 eV due to the low T1 localised on the pyrene unit. CzBNNa emits at λPL of 487 nm in 1 wt% doped films in mCBP, which is modestly red-shift compared to that of CzBN (λPL = 483 nm), while the ΦPL of both compounds are near unity at 99 and 98% for CzBN and CzBNNa, respectively. Both compounds have similar k RISC of 1.2 and 3.1 × 104 s–1. CzBNPyr displayed no TADF behavior owing to its large ΔE ST but did show similar λPL and ΦPL of 485 nm and 90%, respectively, in 1 wt% doped films in mCBP to those of CzBN and CzBNNa. The devices with CzBN, CzBNNa, and CzBNPyr showed modest EQEmax of 6.3, 5.6, and 2.4%, respectively. The low EQEmax for the device with CzBNPyr was due to it not being TADF. In HF-OLEDs using HDT-1 (Figure ) as the assistant dopant, the EQEmax improved to between 19.4–22.0%. Interestingly and likely the intended outcome of the work, the device with CZBNPyr showed the best device stability, with LT95 of 29.1 hours compared to 4.7 and 6.8 hours for the other two devices. The improved device stability was attributed to the rapid clearing of long-lived triplets by the pyrene group, with detrimental impacts on EQE but at least alleviating device degradation mechanisms.
The influence of peripheral decoration on a MR-TADF core was further investigated by Xue et al.,ref. ref1058 for three derivatives of DtBuCzB that contained PhOH (BN-Ph-OH), PhOMe (BN-Ph-OCH3 ), or PhNMe2 (BN-PhN(CH3)2 ) substituents para to the boron (Figure ). The three compounds emit similarly in toluene with λPL of 485, 485, and 486 nm for BN-PhOH, BN-PhOCH3 , and BN-PhN(CH3)2 , respectively, all with FWHM of 24–26 nm, and ΔE ST between 0.14 and 0.15 eV. The k RISC ranged from 2.9 to 3.0 and 8.1 × 104 s–1 for BN-PhOH, BN-PhOCH3 , and BN-PhN(CH3)2 , respectively in 3 wt% doped mCBP films. The OLEDs with BN-PhOH, BN-PhOCH3 , and BN-PhN(CH3)2 showed EQEmax of 19.0, 25.6, and 24.1%, with similar λEL between 491–493 nm. The device degradation mechanism was investigated using a combination of UV-vis absorption, transient PL, and Raman spectroscopies. BN-PhOH was found to be the least stable following UV irradiation, while BN-PhN(CH3)2 was determined to be the most stable, with conformational and packing structure changes between the two ascribed as the key factor for differing degradation rates.
Cai et al. developed a strategy to improve the k RISC of DtBuCzB derivates by incorporating as a ligand of a Au(I) NHC complex, designed to enhance SOC.ref. ref894 Five analogues containing differing NHC ligands, (SIPr)AuBN, (IPr)AuBN, (BzIPr)AuBN, (PyIPr)AuBN, and (PzIPr)AuBN (Figure ) were reported. All five emitters show similar λPL of 513–515 nm and FWHM of 30–31 nm in 2 wt% doped PMMA films, which were modestly red-shifted compared to DtBuCzB (λPL of 505 nm in 1 wt% doped PMMA film and FWHM of 26 nm in THF). The five emitters have similar ΦPL, ΔE ST, and τd of 87–92%, 0.08–0.09 eV, and 5.5–5.9 μs, revealing that the specific NHC ligand used has a minimal impact on the photophysical properties of the emitters. Nonetheless, due to the enhanced SOC associated with the gold atom (computed SOC between S1 and T1 increased from 0.05 cm–1 in DtBuCzB to 1.62 cm–1 in (BzIPr)AuBN), only delayed emission was observed in the 2 wt% doped PMMA films, implying that k ISC ≫ k r. This is in contrast to the reference emitter DtBuCzB, which shows dual emission with τp and τd of 13.8 ns and 114 μs, respectively. The enhanced SOC contributed to fast k RISC of 3.2–5 × 106 s–1 for each of (IPr)AuBN, (BzIPr)AuBN, (PyIPr)AuBN, compared to 2.9 × 104 s–1 for DtBuCzB in MeCN. Devices using 0.5, 0.5, 2, 4, and 1 wt% of (SIPr)AuBN, (IPr)AuBN, (BzIPr)AuBN, (PzIPr)AuBN, and (PyIPr)AuBN (respectively) showed EQEmax of 24.8, 24.0, 30.3, 24.0, and 27.6%. Owing to their short triplet lifetimes, the EQE1000 remained greater than 20% (20.2–28.1%) in all cases. By contrast, the OLED with DtBuCzB showed an EQEmax of 13.6% (1 wt% doping), which decreased to 7.8% at 1,000 cd m–2. The LT95 at 1,000 cd m–2 of the devices with (SIPr)AuBN and (BzIPr)AuBN were 47.4 and 50.2 hours, respectively.
Fused Indolocarbazole Emitters
Recently, a range of MR-TADF emitters have been published where a central boron is used alongside a fused indolocarbazole unit (Figure ). Zhang et al. reported two emitters also based on the fusing of carbazole units to a DtBuCzB core: an unsubstituted compound BN-ICz-1 and carbazole-substituted BN-ICz-2 (Figure ).ref. ref1059 Both emit at λPL of 520 nm as 3 wt% doped mCBP films, and both showed narrow FWHM of 21 and 22 nm in toluene. In 3 wt% mCBP films ΦPL are 95 and 93% with τd of 239 and 160 μs for BN-ICz-1 and BN-ICz-2, respectively, and the ΔE ST are similar at 0.22 and 0.18 eV in toluene. Devices with BN-ICz-1 and BN-ICz-2 showed EQEmax of 24.1 and 22.2%, respectively, at CIE coordinates of (0.24, 0.73) and (0.23, 0.72); the EQE1000 decreased to 10.6 and 14.4%. Using 3CTF (Figure ) as the assistant dopant, HF-OLEDs with BN-ICz-1 and BN-ICz-2 showed EQEmax of 30.5 and 29.8%, and improved efficiency roll-off with EQE1000 of 17.2 and 26.1%.
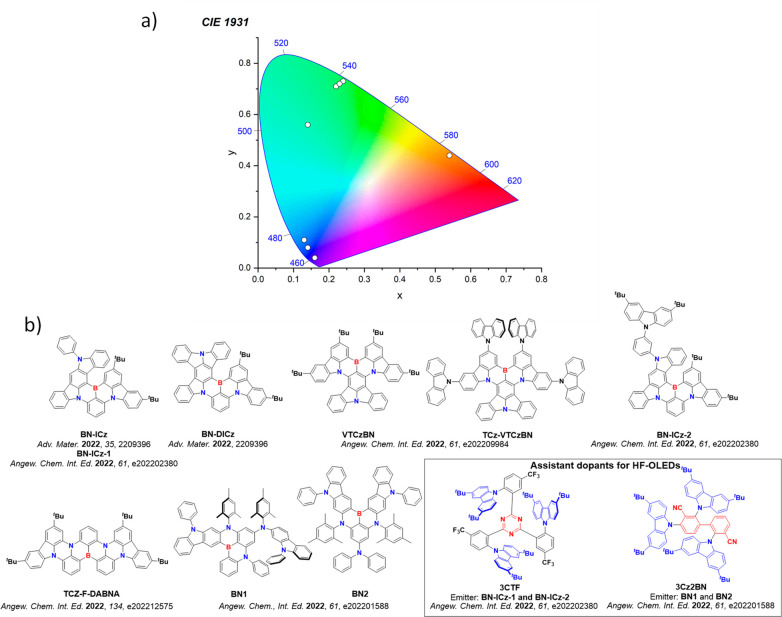
The same group reported a derivative of BN-ICz using instead an extended diindolocarbazole, BN-DICz (Figure ).ref. ref1060 This compound emits at λPL (FWHM) of 533 nm (20 nm), a modest red-shift compared to BN-ICz-1 (renamed BN-ICz here), which emits at 517 nm (21 nm), both in toluene. In toluene the ΔE ST was 0.26 eV, translating to delayed lifetimes of 496 μs and k RISC 7.8 × 104 s–1, while the ΦPL is 99%. HF-OLEDs with BN-DICz using 3CTF (Figure ) as the assistant dopant showed very high EQEmax of 31.5% at CIE coordinates of (0.30, 0.58).
Luo et al. reported an emitter containing indolocarbazole (ICz) embedded centrally within the core of the emitter, and where the nitrogen atom of the ICz is positioned para to the boron. VTCzBN and a second analogue relacing the tBu groups with additional carbazole donors, TCz-VTCzBN, were both investigated (Figure ).ref. ref169 Compared to DtBuCzB, both compounds show a red-shifted emission in toluene with λPL at 496 nm for VTCzBN and 521 nm for TCz-VTCzBN,ref. ref169 while the respective ΔE ST are very small at 0.06 and 0.01 eV for leading to k RISC of 1.0 and 0.9 × 106 s–1. Both compounds have high ΦPL of 98% in 4 wt% doped 2,6-DCzppy films. OLEDs with VTCzBN and TCz-VTCzBN showed EQEmax of 31.7 and 32.2% at CIE coordinates of (0.14, 0.56) and (0.22, 0.71), respectively, which decreased to 24.8% (19.8%) and 18.0% (16.0%) at 100 cd m–2 (1,000 cd m–2). The high EQEmax of the devices was attributed to both the high ΦPL and strongly horizontally orientated TDM of the emitters.
Cheng et al. designed a derivative of DABNA, TCZ-F-DABNA (Figure ),ref. ref1061 where two tert-butylcarbazoles were fused onto the DABNA core. This increased the conjugation within the emitter, red-shifting λPL to 558 nm in toluene, while its highly twisted structure ensured that ACQ was suppressed (vide infra). The ΔE ST in toluene was 0.12 eV, while in 8 wt% doped PhCzBCZ films the ΦPL was 99% and the τd was 20.2 μs. Promisingly, the ΦPL remained high at 92% in 40 wt% doped films, and the k RISC in the film was measured to be 7.8 × 104 s–1. Due to the preferential horizontally oriented TDM of the emitter, the OLEDs showed exceptionally high EQEmax of 39.2% at CIE coordinates of (0.54, 0.44); however, significant efficiency roll-off was apparent, with EQE decreasing to 24.4 and 7.84% at 100 cd m–2 and 1,000 cd m–2 arising from the still relatively small k RISC.
Lv et al. reported a series of emitters, BN1, BN2, and BN3 (Figure ), accessed by changing the stoichiometry of borylating reagent used.ref. ref1062 These three compounds emit at λPL of 454, 464, and 456 nm and show narrow FWHM of 18, 15, and 17 nm, all respectively in toluene. The influence of the different π-frameworks becomes apparent in the ΔE ST, which decrease from 0.20 to 0.16 and 0.15 eV for BN1, BN2, and BN3, respectively. In 1 wt% doped DBFPO films there is a progressive increase in ΦPL from 91 to 93 and 98% that is concurrent with an increased delay emission contribution (from 35, 45, and 76%) and faster k RISC of 1.3, 2.6, to 25.5 × 104 s–1 for BN1, BN2, and BN3, respectively. BN3 is discussed in more detail in the “Central donor” sub-section. OLEDs with BN1 and BN2 showed EQEmax of 30.0 and 32.9%, respectively; however, the efficiency roll-off was severe, with the EQE100 dropping to 8 and 14.7%. To address the efficiency roll-off, HF-OLEDs using 3Cz2BN (Figure ) as the assistant dopant showed much improved EQE100 of 18.3 and 25.5%.
CzBN Derivatives with Multiple Acceptor Atoms
A number of doubly borylated MR-TADF compounds containing carbazole-based skeletons have also been designed (Figure ). Similar in concept to the emitter B2 (Figure ),ref. ref1030 compounds CzB2-M/TB and CzB2-N/P contain 3 nitrogen and 2 boron atoms, and 1 carbazole, while Cz2B2-M/TB has an extra fused carbazole unit (Figure ).ref. ref1021 CzB2-M/TB, CzB2-N/P, and Cz2B2-M/TB emit at λPL of 491, 504, and 483 nm in 1 wt% doped PMMA films. The addition of phenyl substituents in CzB2-N/P results in a smaller ΔE ST of 0.06 eV compared to 0.12 and 0.11 eV for CzB2-M/TB and Cz2B2-M/TB. The OLED with CzB2-N/P showed an EQEmax of 26.7% at CIE coordinates of (0.15, 0.57). The efficiency roll-off was relatively low, decreasing by only 9%, at 100 cd m–2.
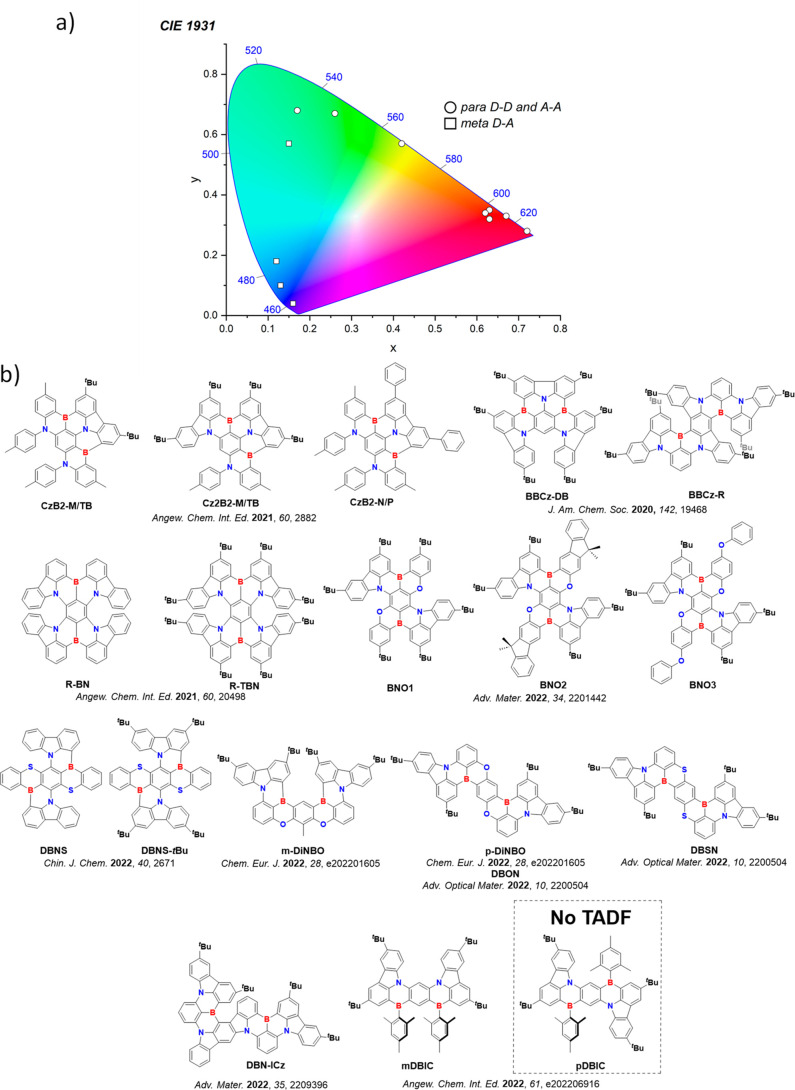
An emerging motif to achieve relatively rare red MR-TADF emitters is to install two boron atoms para to each other.ref177,ref1040 Illustrative of the impact of the regiochemistry of the boron substitution, BBCz-DB and BBCz-R (Figure )ref. ref1040 have strongly contrasting emission colors of 471 and 619 nm in 2 wt% doped mCBP films. The synergistic para disposition of both the electron-donating nitrogen atoms and electron-accepting boron atoms results in a destabilized HOMO and stabilized LUMO, thus decreasing the band gap, while the opposite effect is observed when the nitrogen and boron atoms are meta-disposed. Hence a blue-shift in the emission is observed for BBCz-DB (λPL = 471 nm) compared to BBCz-SB (λPL = 490 nm, Figure ) while for BBCz-R the emission is strongly red-shifted (λPL = 619 nm, all 2 wt% doped mCBP films). Compounds BBCz-DB and BBCz-R possess ΔE ST of 0.15 and 0.19 eV in toluene, and τd of 35 and 53 μs in the same respective films. Devices with BBCz-DB and BBCz-R showed EQEmax of 29.3 and 22.0% at CIE coordinates of (0.12, 0.18) and (0.67, 0.33), respectively, with the latter being the first reported red MR-TADF emitter. Very severe efficiency roll-off was observed for BBCz-R though, which could not attain 1000 cd m–2. Similarly, the EQE1000 dropped precipitously to 5.5% for the device with BBCz-DB.
The same approach to red-shift emission was also adopted by Zhang et al.ref. ref177 with the emitters R-BN and R-TBN (Figure ). These two compounds emit at λPL of 672 and 698 nm and have unity ΦPL in 3 wt% doped CBP films. The same films of R-BN and R-TBN have ΔE ST of 0.18 and 0.16 eV in toluene, and very long τd of 310 and 710 μs, respectively. The OLEDs showed EQEmax of 25.6 and 24.7% at CIE coordinates of (0.72, 0.28) for both devices. HF-OLEDs using the assistant dopant Ir(mphmq)2tmd (Figure ) emitted at identical CIE coordinates and with EQEmax of 28.4 and 28.1% for the devices with R-BN and R-TBN, respectively.
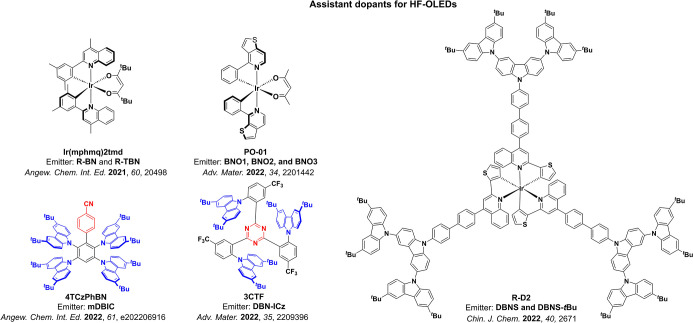
A similar design concept with para boron atoms, para nitrogen atoms, and para oxygen atoms alongside substituents tBu (BNO1), fluorene (BNO2), and phenoxy (BNO3) was also explored by Zou et al. (Figure ).ref. ref1063 Similarly to other reports, the presence of para-disposed donors/acceptors ensured red emission with λPL of 610, 618, and 624 nm for BNO1, BNO2, and BNO3, respectively in 1 wt% doped DMIC-TRZ films. These three compounds have large ΔE ST of between 0.25 and 0.27 eV, with τd over 100 ms for each. Devices with BNO1, BNO2, and BNO3 showed EQEmax of 14.9, 12.0, and 15.1%, respectively, and had significant efficiency roll-off (EQE1000 < 5.0% for each). Nonetheless, HF-OLEDs using PO-01 (Figure ) as the assistant dopant showed very high EQEmax of 35.6, 34.4, and 36.1%, and low efficiency roll-off (EQE1000 of 31.1, 29.8, and 32.1%) for the devices with BNO1, BNO2, and BNO3, respectively.
In a later report from Wang et al. DBNS and DBNS-tBu (Figure )ref. ref1064 containing para-disposed boron, nitrogen, and sulfur atoms were shown to emit similarly in the red at λPL of 631 and 641 nm in toluene, and to have similar ΔE ST and ΦPL in DCM of 0.20 and 0.19 eV, and 80 and 85%, respectively. Attributed to the presence of the heavier sulfur atoms that increase SOC, there is reasonably fast k RISC of 2.1 and 2.2 × 105 s–1 for these MR-TADF emitters. Solution-processed HF-OLEDs using red iridium dendrimeric sensitiser R-D2 (Figure ) showed surprisingly low EQEmax of 5.8 and 7.8% at CIE (0.64, 0.34) and (0.65, 0.34), respectively.
Two “dimeric” derivatives that contain either meta-disposed or para-disposed boron atoms (m-DiNBO and p-DiNBO, Figure ) were reported by Liu et al.ref. ref1065. Compared to the parent emitter NBO (Figure ) a slight red-shift of the emission was observed, from λPL of 448 to 456 nm for m-DiNBO, and more so for p-DiNBO at 500 nm, all in toluene. These extended structures also led to narrowed emission, with FWHM decreasing from 25 nm in NBO to 17 and 19 nm for m-DiNBO and p-DiNBO, respectively, attributed to suppression of vibronic coupling in the latter two. The ΔE ST decreased slightly from 0.10 eV for NBO to 0.06 eV for both m-DiNBO and p-mDiNBO, while comparable k RISC were reported for all emitters in the range of 1.2–3.1 × 104 s–1 in 3 wt% doped mCBP films. The OLEDs with NBO, m-DiNBO, and p-DiNBO showed EQEmax of 16.8, 24.2, and 21.6%, respectively, with the enhanced EQE in the dimers due to enhanced light outcoupling and improved charge balance within the EML; however, efficiency roll-off was still severe (an EQE1000 of 9.2% was reported only for the device with p-DiNBO).
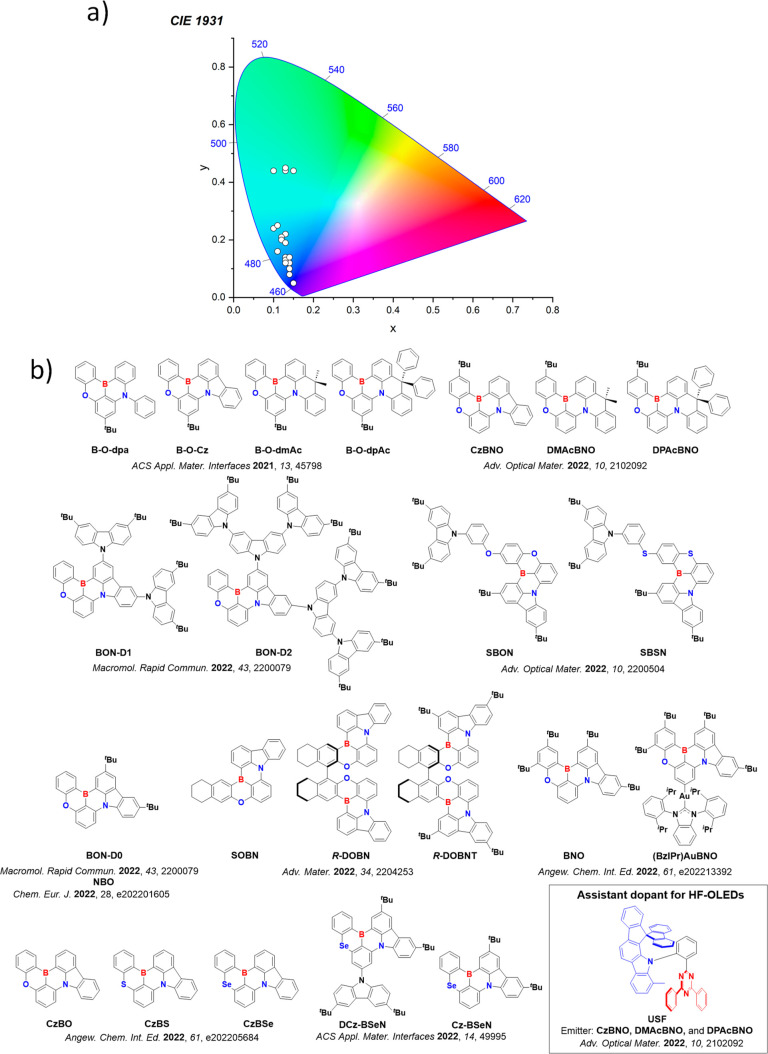
p-DiNBO was reported again by Luo et al. renamed DBON, they also presented the S-π-S (DBSN) derivative Figure ,ref. ref1066 with λPL in toluene of 505 and 553 nm, respectively. The mono-borylated analogues of these, SBON and SBSN, showed a blue-shifted emission with λPL of 463 and 489 nm, respectively. DBON and DBSN have identical ΔE ST of 0.13 eV in toluene and near unity ΦPL of 98% in 4 wt% doped mCBP films. k RISC was found to be faster in DBSN due to the larger SOC associated with the heavier chalcogen (k RISC of 0.8 and 1.9 × 105 s–1 for DBON and DBSN, respectively). OLEDs with DBON and DBSN showed EQEmax of 26.7 and 21.8% at CIE coordinates of (0.17, 0.58) and (0.42, 0.57), respectively. Despite the higher EQEmax for DBON device, it also showed larger efficiency roll-off with an EQE1000 of 12.0%, compared to 16.9% for the device with DBSN.
Zhang et al. reported an indolocarbazole di-borylated emitter which was two DtBuCzB fused together, DBN-ICz (Figure ).ref. ref1060 In toluene they report a λPL (FWHM) 542 nm (18 nm) and ΔE ST of 0.20, while in 3 wt% doped in mCBP they reported a τd of 48 μs and a ΦPL of 96%. HF-OLEDs with DBN-ICz using 3CTF (Figure ) as the assistant dopant showed very high EQEmax of 37.4% at CIE coordinates of (0.36, 0.59).
Wang et al. reported the deep blue emitter mDBIC (Figure ) that contains meta-disposed pairs of boron and nitrogen atoms.ref. ref175 The compound emits at λPL of 431 nm and has a ΦPL of 68% in 3 wt% doped mCP films; however, in toluene the ΔE ST is large at 0.31 eV, which translates to a slow k RISC of 5.0 × 103 s–1. The congener that instead has each of the boron and nitrogen atoms para to each other, pDBIC (Figure ), was also reported and has an even larger ΔE ST of 0.35 eV and no observable TADF. Illustrative of the challenges faced by MR-TADF emitters with slow RISC, devices with mDBIC showed a low EQEmax of 5.7% at nonetheless desirable CIE coordinates of (0.16, 0.04). HF-OLEDs with 4TCzPhBN (Figure ) as the assistant dopant showed much improved EQEmax of 13.5%.
Other Bridging Atoms and Groups
Instead of simply substituting or extending established MR-TADF core groups, several derivatives where carbazole or diphenylamine are replaced by other donor groups such as DMAC, PXZ, and PTZ have been explored (Figure ). Jiang et al.ref. ref1067 reported the DMAC and DPAC congeners of DtBuCzB, BN-DMAC and BN-DPAC (Figure ), which emit at λPL of 485 and 490 nm in toluene and have ΔE ST of 0.14 and 0.11 eV, respectively. In 1 wt% doped mCBP films they showed ΦPL of 63 and 86% and τd of 13.9 and 11.6 μs. OLEDs with BN-DMAC and BN-DPAC showed EQEmax of 21.1 and 28.2% at CIE coordinates of (0.14, 0.54) and (0.14, 0.56), respectively, while the efficiency roll-off was severe, with EQE1000 decreasing to 12.5 and 19.1%. When TADF exciplex host mCBP:PO-T2T was employed the EQEmax rose to 25.5 and 30.2% for the same devices, while the EQE1000 remained as high as 16.0 and 22.1%. In this exciplex host the LT80 were 82 and 8 hours at 500 cd m–2 for the devices with BN-DMAC and BN-DPAC, respectively.
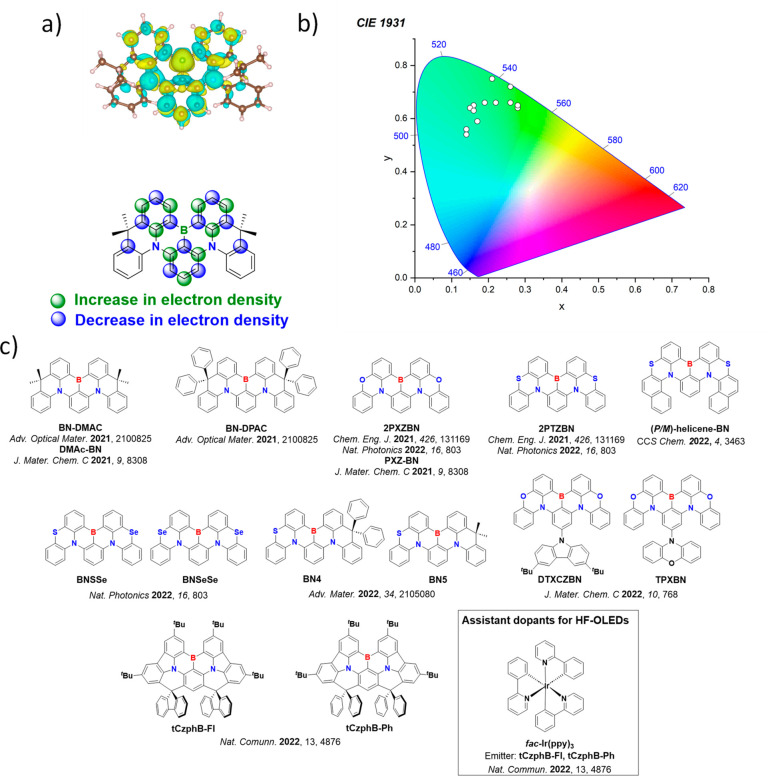
PXZ and PTZ analogues 2PXZBN and 2PTZBN (Figure ) were reported by Hua et al.ref. ref1068 Both emit in the green, with λPL of 515 and 519 nm and ΦPL of 84 and 80%, respectively, in 1 wt% mCBP:PO-TCTA doped films. In toluene both compounds have similar ΔE ST of 0.19 and 0.15 eV, but the incorporation of the heavier sulfur decreases the τd from 25.3 to 16.1 μs and improves the k RISC from 0.56 to 1.17 × 105 s–1. The improved k RISC translated into improved device performance with EQEmax of 17.1 and 25.5% for the devices with 2PXZBN and 2PTZBN, respectively, at CIE coordinates of (0.28, 0.65) and (0.28, 0.64). Efficiency roll-off is also less severe in the device with 2PTZBN (EQE1000 = 17.2% compared to 7.4% in the device with 2PXZBN).
Both BN-DMAC and 2PXZBN were included in a subsequent report,ref. ref1069 each renamed DMAc-BN and PXZ-BN, (Figure ) where some small differences in their photophysics were recorded, likely owing to the different media.ref1067,ref1068 Improvements in device performance were observed for the device with PXZ-BN with EQEmax increasing to 23.3% (17.2% previously) while the device with DMAc-BN was lower than before, at 20.3% (21.1% previously).ref1067−ref1068ref1069 A similar study from Hu et al.ref. ref180 extended the series containing 2PXZBN and 2PTZBN with analogues containing either mixed S/Se or double Se insertion, BNSSe and BNSeSe (Figure ). The Se atoms were added to help improve RISC via a further enhanced SOC due to the ‘heavier’ atom effect of Se compared to S. The four compounds emit similarly at λPL of 523, 525, 520 and 514 nm for 2PXZBN, 2PTZBN, BNSSe, and BNSeSe in respective 1 wt% DMIC-TRZ doped films. The four emitters also have similar ΔE ST between 0.12–0.15 eV. There was a progressive increase in ΦPL from 71 to 91, 99, and 100% for 2PXZBN, 2PTZBN, BNSSe, to BNSeSe, respectively, and a concomitant decrease in τd from 38.1 to 20.7, 12.7, and 9.9 μs. The higher ΦPL of BNSSe and BNSeSe was attributed to supressed ACQ due to their twisted geometry. The most striking difference between the emitters was their k RISC rates, being 0.04 and 0.19 × 106 s–1 for 2PXZBN and 2PTZBN and increasing to 0.6 and 2.0 × 106 s–1 for BNSSe and BNSeSe, respectively, owing to their enhanced SOC from Se and confirmed by calculations. OLEDs with BNSSe and BNSeSe showed EQEmax of 35.7 and 36.8% at CIE coordinates of (0.22, 0.66) and (0.19, 0.66). Strikingly, the EQE1000 remained very high at 32.0 and 34.0%. The devices with 2PXZBN and 2PTZBN showed lower EQEmax of 30.7 and 34.6%, which dropped to 24.0 and 29.5%at 1,000 cd m–2. Interestingly, despite their higher k RISC, the LT50 of the devices with 2PTZBN, BNSSe, and BNSeSe were much lower than that with 2PXZBN, at 5.6, 4.8, 4.1, and 158 hours respectively. A HF-OLED using BNSeSe as the assistant dopant and BN3 (Figure ) as the terminal emitter showed an outstanding EQEmax of 40.5%.
A CPL-active derivative of 2PTZBN containing naphthalene groups that induce chirality, (P/M)-helicene-BN (Figure ).ref. ref647 In dilute toluene (P/M)-helicene-BN emits at λPL of 520 nm, has a rather large FWHM for an MR-TADF emitters of 46 nm and a ΔE ST of 0.18 eV in the same medium. No reason was provided for the broader emission. The photophysical properties of the M isomer were investigated in 1 wt% doped films in DMIC-TRZ, with ΦPL of 98%, τd of 71.8 μs and a corresponding k RISC of 4.6 × 104 s–1. OLEDs of both P and M isomers were reported with EQEmax of 31.5% and 30.7%, respectively, at identical CIE coordinates of (0.26, 0.66). Each showed large efficiency roll-off, with EQE1000 of 18.7 and 17.9% for P and M isomers respectively. Its CPL properties are discussed in Section sec7 .
Another CPL active series was presented by Wu et al. where they presented two similar asymmetric compounds featuring S/N, BN4 and BN5.ref. ref646 These emitters incorporated a sulfur bridge on one side (similar to PTZBN, Figure ) alongside DPAC (BN4) and DMAC (BN5) units on the other side (Figure ). Both BN4 and BN5 have similar photophysical properties, with λPL of 522 and 512 nm, τd of 25 μs for both, ΦPL of 96 and 92%, and ΔE ST of 0.20 and 0.14 eV as 3 or 1 wt% doped mCPCN films, respectively. As with PTZBN, increased SOC from the heavy sulfur atom produced enhanced k RISC of 1.6 and 0.7 × 105 s–1 for BN4 and BN5, respectively in toluene. Devices with both emitters and both enantiomers were fabricated, with EQEmax of 20.6% (19.0%) and 22.0% (26.5%) reported for +(−)-BN4 and +(−)-BN5, respectively. The OLEDs were green with CIE coordinates of (0.19, 0.63) and (0.21, 0.64) for + and – BN4 and (0.17, 0.59) and (0.17, 0.60) for + and – BN5, respectively. Their chiroptical properties are discussed in Section sec7 .
Employing PXZ-BN as the core, Hu et al. reported of emitters where tBuCz and PXZ donors were positioned para to the boron centres (TPXZBN and DPXZCZBN, Figure ).ref. ref1070 These two compounds emit at λPL of 502 and 500 nm in toluene and have ΦPL of 99 and 94% in 5 wt% doped mCBP films. They also have similar τd of 27 and 15 μs, which are correlated with their similar ΔE ST of 0.16 and 0.13 eV in toluene and k RISC of 0.48 and 1.11 × 105 s–1. Devices with TPXZBN and DPXZCZBN showed EQEmax of 21.3 and 19.2%, respectively, at CIE coordinates of (0.16, 0.65) and (0.15, 0.64). The efficiency roll-off was modest where the EQE1000 was maintained at 17.4 and 17.2%.
An alterative bridging strategy was reported by Liu et al, where DtBuCzB was modified with spiro bridging units, locking all the rings.ref. ref1071 They reported a bis phenyl spiro bridging unit, tCzphB-Ph, and a fluorene bridge derivative, tCzphB-Fl (Figure ). The spiro linkage was added to prevent the in-plane phenyl distortion reported in DtBuCzB, generating planar compounds, with near pure green emission. In 2 wt% doped TPSS films, λPL of 527 nm and 535 nm was reported for tCzphB-Ph and tCzphB-Fl, respectively, with narrow FWH of 23 and 25 nm, possible due to supressed vibronic modes. Despite their small ΔE ST of 0.04 eV, long τd of 372 and 412 μs for for tCzphB-Ph and tCzphB-Fl reported, respectively were reported, in 2 wt% doped TPSS films. Slow τd was attributed to their small Huang-Rhys factors from a result of their rigid structure. OLED devices were reported, with high EQEmax of 29.3% and 26.2% at CIE (0.21, 0.75) and (0.26, 0.72) for tCzphB-Ph and tCzphB-Fl, respectively. Coordinates of (0.21, 0.75) for tCzphB-Ph are the closest to Rec. 2020 for green (0.17, 0.80) of any reported MR-TADF emitter. Long delayed lifetime resulted in large roll-off with EQE1000 of 9.2% for tCzphB-Ph and 8.2% for tCzphB-Fl. HF-OLEDs using Ir(ppy)3 phosphorescent sensitizer improved roll-off, with tCzphB-Ph having an EQE at 10,000 cd m–2 of 30.6%.
Asymmetric MR-TADF Emitters
Asymmetric MR-TADF Emitters with Nitrogen Donor Atoms
While the designs of most MR-TADF emitters are symmetrical in and around the central core unit, there are also now a range of unsymmetric analogues of DABNA-1 or DtBuCzB reported (Figure ). Qui et. al.ref. ref1072 reported a family of compounds, DPACzBN1, DPACzBN2, and DPACzBN3 (Figure ) based on a fused carbazole and different substituted diphenylamines around the same MR-TDAF core. DPACzBN1 emits at λPL of 479 nm in 3 wt% doped 26DczPPy films, which lies between DABNA-1 (λPL = 460 nm in 1 wt% doped mCBP films),ref. ref118 and DtBuCzBN (λPL = 493 nm in the same)ref. ref1038 Substitution of the DPA moiety resulted in a modest blue-shift of the emission, with λPL of 470 and 475 nm for DPACzBN2 and DPACzBN3, respectively. The ΔE ST of 0.11–0.13 eV in toluene are similar to that of DtBuCzB (0.13 eV in toluene)ref. ref1038 and smaller than that of DABNA-1 (0.18 eV in 1 wt% doped mCBP film).ref. ref118 The τd decreased from 116 μs in DPACzBN1 to 54 and 69 μs in DPACzBN2 and DPACzBN3, translating to accelerated k RISC in the latter two compounds, from 1.2 to 2.9 and 2.1 × 104 s–1. The ΦPL range between 92–98% in 3 wt% doped 26DczPPy films. The OLEDs with DPACzBN1, DPACzBN2, and DPACzBN3 showed EQEmax of 23.6, 24.0, and 27.7% at CIE coordinates of (0.14, 0.30), (0.13, 0.16), and (0.12, 0.18). Slow and inefficient k RISC was the primary cause of the large efficiency roll-off, with EQE1000 of 9.6, 14.3, and 6.3%, respectively.
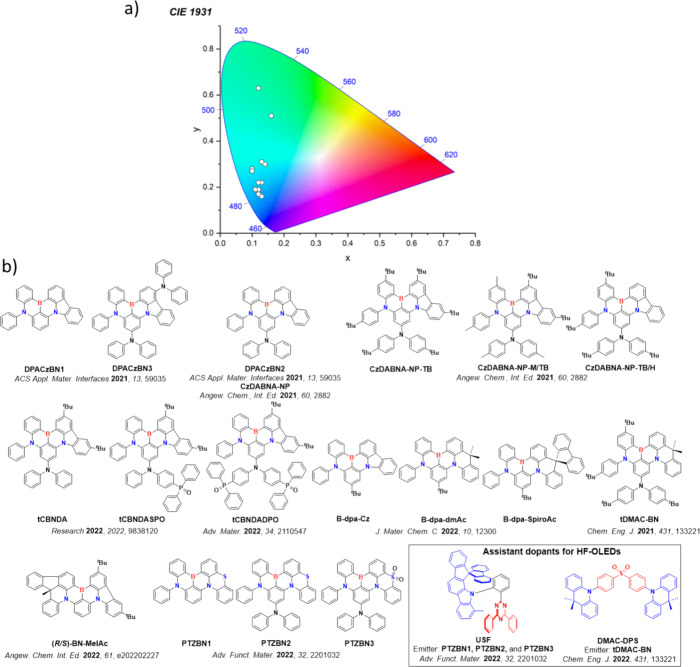
A series of similar derivatives of DPACzBN2; CzDABNA-NP-M/TB, CzDABNA-NP-TB/H, and CzDABNA-NP (Figure ) have also been reported.ref. ref1021 The substitution pattern around the periphery had negligible effect on the emission spectra, with these three compounds emit narrowly with λPL ranging from 461–468 nm. Similarly the ΦPL range from 80–86% and all three have the same ΔE ST of 0.18 eV. No devices were fabricated, however CzDABNA-NP-TB is the actual structure of the previously incorrectly identified TBN-TPA (Figure ).ref1020,ref1021 The correct structure was confirmed by later NMR spectroscopy studies, and although the structure was wrongly identified in the initial report, the data pointed to an emitter with excellent potential, with λPL at 470 nm in toluene, a high ΦPL of 98%, and a small ΔE ST of 0.14 eV in 8 wt% doped 26-DCzppy films where the τd is 51 μs.ref. ref1020 The OLEDs using this emitter showed an EQEmax of 32.1% at CIE coordinates (0.12, 0.19), while efficiency roll-off was moderate with a loss of 15% at 100 cd m–2.
Another derivative of CzDABNA-NP-TB contained fewer tBu substituents, tCBNDA and tCBNDASPO with a phosphine oxide group (Figure ) were investigated by Bian et al..ref. ref1073 These two compounds emit at λPL of 467 nm and with the same FWHM of 28 nm in DCM. tCBNDA and tCBNDASPO likewise have similar ΔE ST of 0.05 and 0.04 eV in 7 and 20 wt% doped DBFDPO films, respectively. The presence of the phosphine oxide led to an increase in ΦPL from 72 to 92% for tCBNDASPO. The presence of the phosphine oxide group also supressed ACQ, with ΦPL of tCBNDA in 20 wt% films dropping to 29% compared to 92% for tCBNDASPO. The sky-blue devices have identical CIE coordinates of (0.12, 0.17), but the EQEmax values differed at 20.2 and 28.0%, reflecting the shorter τd, higher ΦPL, and faster k r of tCBNDASPO. The OLEDs showed moderate roll-off with EQE100 of 12.4 and 20.6% for the devices with tCBNDA and tCBNDASPO, respectively.
The same group reported a similar emitter, tCBNDADPO, which contains two phosphine oxide units attached to the DPA unit of tCzBNDA (Figure ).ref. ref1074 Addition of the phosphine oxide unit was expected to increase the ambipolar character of the emitter, assisting exciton and charge trapping. A similar λPL of 466 nm for tCBNDADPO compared to 467 nm in tCBNDA and tCBNDASPO was reported in DCM, with the second phosphine oxide unit having a minimal impact on the emission. At an optimal doping of 30 wt% emitter in DBFDPO, tCBNDADPO emits at λPL of 472 nm, has a ΦPL 99%, a ΔE ST of 0.04 eV and a k RISC of 2.4 × 104 s–1. Devices showed a similar emission to previous emitters, with CIE coordinates of (0.14, 0.22) here compared to (0.12, 0.17) previously reported, but with an improved EQEmax of 30.8%. The EQE100 was 23.3% reflecting a similar efficiency roll-off to tCBNDA and tCBNDASPO (vide supra).
A related emitter design incorporated both DPA and PTZ groups where PTZBN1 is the parent in this series, PTZBN2 contains a DPA para to the boron, and PTZBN3 is an oxidized version of PTZBN2 (Figure ).ref. ref1075 The emission of PTZBN2 is blue-shifted at λPL of 483 nm compared to that of PTZBN1 (490 nm in toluene), ascribed to destabilisation of the LUMO in the former, while PTZBN3 emits at λPL of 468 nm. Interestingly, the emission spectrum of PTZBN3 has the smallest FWHM of 30 nm compared to 41 nm in the other two, attributed to reduced structural relaxation in the excited state for this compound. All three compounds have similar ΔE ST of 0.15–0.17 eV in toluene, along with high ΦPL of 95–98% and τd of 22.4–33.5 μs in 2 wt% doped 2,6-DCzppy films. Owing to the presence of the heavy sulfur atom, SOC was enhanced and reflected in the k RISC of 1.11, 4.51, and 1.08 × 105 s–1 for PTZBN1, PTZBN2, and PTZBN3, respectively. Devices showed EQEmax of 26.9, 30.5, and 19.9%, respectively, at CIE coordinates of (0.16, 0.51), (0.13, 0.31), and (0.13, 0.22). The HF-OLEDs using USF (Figure ) as the assistant dopant showed yet higher EQEmax of 32.7, 34.8, and 32.0%.
Park et al. reported three structurally related emitters B-dpa-Cz, B-dpa-dmAc, and B-dpa-SpiroAc (Figure ), in which fused ring extensions of carbazole, dimethylacridine, or spirofluroacridine were compared.ref. ref1076 These three compounds emit at λPL of 469, 476, and 476 nm in 3 wt% doped mCBP films, and have high ΦPL of 92–98%. They all have similar ΔE ST of 0.14–0.16 eV, while the k RISC of the DMAC analogues improved slightly at 2.7, 4.5, and 5.0 × 104 s–1 for B-dpa-Cz, B-dpa-dmAc, and B-dpa-SPiroAc, respectively. Devices showed EQEmax of 20.1, 24.2, and 25.1% at CIE coordinates of (0.11, 0.19), (0.10, 0.28), and (0.10, 0.27).
A similar molecular design from Wang et al. generated tDMAC-BN (Figure ),ref. ref1025 and borylation of the DPAc derivative produced tDPAc-BN (Figure ) discussed earlier in this section. tDMAC-BN emits at λPL of 468 nm and has a ΔE ST of 0.15 eV in toluene, while the ΦPL is 90% and the τd is 64 μs in 1 wt% doped PMMA films. Devices showed an EQEmax of 19.8% at CIE coordinates of (0.12, 0.22), but the efficiency roll-off was severe. HF-devices with DMAC-DPS as the assistant dopant showed an EQEmax to 22.3%, and efficiency roll-off improved (EQE1000 = 10.4%).
An unusual CPL-active emitter containing a carbazole moiety and a chiral acridine unit, (R/S)-BN-MeIAc (Figure ), was reported by Yang et al.ref. ref624 Unlike other reported CP-MR-TADF emitters, the chirality was achieved from a stereocenter, which also, according to calculations, suggested a large SOC between S1 and T2. In toluene, (R/S)-BN-MeIAc emits at λPL of 497 nm (FWHM of 30 nm), and has a small ΔE ST of 0.11 eV in 2-MeTHF. In 1 wt% doped films in DMIC-TRZ, it has a high ΦPL of 96% and a k RISC of 6.3 × 104 s–1. The devices with the R and S isomers showed EQEmax of 37.2 and 36.1%, respectively, with highly horizontally orientated TDMs the key to their impressive efficiencies. At 1,000 cd m–2 the EQE was maintained at 26.1 and 25.1% for the R and S isomers, respectively. Their chiroptical properties are discussed in detail in Section sec7 .
Asymmetric MR-TADF Emitters with Mixtures of Donating Atoms
As well as asymmetric substituents or fixed extensions, there are also now several examples of boron-based MR-TADF emitters that contain both nitrogen and oxygen donor atoms in the main core. Compound B-O-dpa and its congeners B-O-Cz, B-O-dmAC, and B-O-dpAc exemplify this design strategy (Figure ).ref. ref1077 For these materials the ΔE ST decrease from 0.18 eV in B-O-dpa to 0.15, 0.11, and 0.06 eV for B-O-Cz, B-O-dmAc, and B-O-dpAc in frozen THF. The ΦPL range from 86–94% in 10 wt% doped DPEPO films. The emission in PhMe red-shifts progressively from λPL = 433 nm for B-O-dpa to λPL of 441, 461, and 463 nm for B-O-Cz, B-O-dmAc, and B-O-dpAc, respectively. In the same DPEPO films all four compounds have long τd of 224, 51, 123, and 83 μs for B-O-dpa, B-O-Cz, B-O-dmAc, and B-dpAc, respectively, with the shorter τd of the latter three attributed to their smaller ΔE ST. Devices with B-O-dpa, B-O-Cz, B-O-dmAc, and B-O-dpAc showed EQEmax of 16.3, 13.4, 16.2, and 17.0% at CIE coordinates of (0.15, 0.05), (0.13, 0.22), (0.12, 0.21), and (0.12, 0.20), respectively. Efficiency roll-off was significant for all devices, with EQE100 of 2.2, 5.9, 8.4, and 9.6%, respectively. The devices were not stable either, as LT50 at 10 cd m–2 did not surpass 20 minutes, attributed to the instability of the host and the charge transport materials.
An analogous series of emitters was reported by Han et al., and contained a tBu substituent instead para to the oxygen atom (CzBNO, DMAcBNO, and DPAcBNO, Figure ).ref. ref1078 These three compounds emit at λPL of 450, 470, and 468 nm with ΦPL of 96, 99, and 98%, respectively in 3 wt% doped 26DczPPy films. The ΔE ST in toluene are 0.21, 0.23, and 0.19 eV, which are larger than those of the previous series, however the τd are on average shorter at 48, 129, and 100 μs, translating to k RISC of 3.5, 1.4, and 1.8 × 104 s–1 for CzBNO, DMAcBNO, and DPAcBNO, respectively. The blue devices the same showed EQEmax of 13.6, 20.4, and 23.0% at CIE coordinates of (0.14, 0.08), (0.13, 0.19), and (0.13, 0.14), respectively, and the EQE1000 decreased to only 5.0, 8.6 and 9.1%. In HF-OLEDs using USF (Figure ) as the assistant dopant the EQEmax increased to 25.9, 28.3, and 29.6%, while the EQE1000 improved to 23.0, 16.7, and 23.1%.
Based on a similar core to B-O-Cz, Liu et al.ref. ref1079 investigated the impact of donor dendronisation by comparing the performance of BON-D0 with BON-D1 and BON-D2 featuring 1st– and 2nd– generation carbazole-based donor dendrons (Figure ). The use of these donor dendrons produces a red-shift of the emission from 450 nm for BON-D0 to 476 and 472 nm for BON-D1 and BON-D2, respectively in toluene. An increase in ΦPL from 85% for BON-D0 to 94 and 98% for BON-D1 and BON-D2 was attributed to a reduction in ACQ in the larger species. Similar k RISC of 6.7, 7.7, and 9.8 × 104 s–1 were reported for BON-D0, BON-D1, and BON-D2 as 5 wt% doped mCP films. Solution-processed devices with BON-D0, BON-D1, and BON-D2 showed EQEmax of 9.7%, 13.4, and 14.9% at CIE coordinates of (0.14, 0.12), (0.13, 0.44), and (0.15, 0.45), respectively. No explanation was provided by the authors to explain the significant color change from that observed in the PL. Further examples of TADF emitters containing donor dendrons are summarised in Section sec10 .
BON-D0, also reported by Liu et al. and renamed NBO here, and was presented alongside two emitters with multiple acceptor atoms, m-DiNBO and p-DiNBO (Figure ).ref. ref1065 The photophysics of NBO were reported in 3 wt% doped in mCBP, with λPL of 458 nm, ΦPL of 99%, τd of 87 μs, k RISC of 1.2 × 104 s–1 and ΔE ST of 0.10 eV, each comparable to the previous report.ref. ref1079 Using their OLED stack, an improved EQEmax of 16.8% at CIE coordinates of (0.14, 0.14); however, efficiency roll-off was large, with EQE at 1,000 cd m–2 not reported. The differences in EQEmax may be attributed to the differing fabrication methods, with vacuum deposition used here, compared to solution processing in the previous report.
Luo et al. reported a decorated analogue of BON-D0 and a sulfur-based congener, SBON and SBSN, both of which contain a pendant DtBuCzPh group (Figure ).ref. ref1066 These were formed as by-products of incomplete borylation during the synthesis of DBON and DBSN, respectively (Figure ). SBON and SBSN emit at λPL of 463 and 489 nm, respectively in toluene, with FWHM of 24 and 27 nm. The diborylated analogues show significantly red-shifted emission, with λPL of 505 nm for DBON and 553 nm for DBSN. SBON and SBSN have ΔE ST of 0.16 and 0.10 eV in toluene, ΦPL of 74 and 76%, and associated k RISC of 0.5 and 1.5 × 105 s–1 respectively, in 4 wt% doped mCBP films. The higher k RISC for SBSN was attributed to a combination of increased SOC due to the presence of the sulfur atoms, and its smaller ΔE ST. Devices with SBON and SBSN showed EQEmax of 13.7 and 17.6% at CIE coordinates of (0.13, 0.13) and (0.10, 0.44), respectively, while the EQE1000 decreased to 6.7 and 12.0%.
A CPL-active family of compounds was reported by Yan et al.ref. ref632 based on a dimerized version of SOBN. Compounds R-DOBN and R-DOBNT (Figure ) differ only in that the latter contains tBu substituents. A modest red-shift in the emission compared to SOBN was observed for both, with λPL of 449, 453, and 459 nm for SOBN, R-DOBN, and R-DOBNT, respectively, while ΔE ST decreased from 0.19 to 0.14 and 0.12 eV in toluene. The CPL properties of R-DOBN and R-DOBNT are discussed in Section sec7 . In 5 wt% doped 2,6-DCzPPy films the ΦPL of SOBN is 82% and the k RISC is 1.4 × 104 s–1. OLEDs with SOBN showed an EQEmax of 14.6% at CIE coordinates of (0.14, 0.14). Higher EQEmax of 23.9% and 25.6% were obtained for R-DOBN, and R-DOBNT, respectively, attributed to the preferential horizontal orientation of their TDMs. The efficiency of the device deceased to an EQE100 of 10.2% for SOBN.
The influence of the size of the chalcogen atom was probed by Park et al.ref. ref893 across a family of oxygen (CzBO), sulfur (CzBS), and selenium containing (CzBSe) emitters (Figure ). Calculations revealed that SOC between S1 and T1 expectedly increased with heteroatom size CzBO < CzBS < CzBSe, while the increase in SOC was even larger between S1 and T2. In 1 wt% doped mCBP films the three compounds emit at λPL of 448, 472, and 479 nm, with a slight broadening of the emission spectrum across the series from 29 to 30 and 34 nm, all respectively. The three compounds have comparable ΔE ST of between 0.14 and 0.16 eV, but their TADF properties are remarkably different. CzBO and CzBS showed prompt fluorescent quantum yields of 83 and 15%, respectively, while it was only ∼0.1% for CzBSe as k ISC is very fast in the latter. With increasing SOC, k RISC increases from 0.9 to 22 × 104 s–1 and 1.8 × 108 s–1 for CzBO, CzBS, and CzBSe, respectively, with the latter, if accurate, being the fastest k RISC reported to date in MR-TADF systems and one that is comparable to the most efficient D-A TADF compounds. Devices with CzBO, CzBS, and CzBSe showed EQEmax of 13.4, 23.1, and 23.9%, respectively, at CIE coordinates of (0.15, 0.05), (0.11, 0.16), and (0.10, 0.24). The efficiency roll-off followed the same trend as k RISC, with EQE1000 of 3.5, 15.0, and 20.0% for the same devices.
Li et al. reported two derivatives of CzBSe, Cz-BSeN and DCz-BSeN (Figure ).ref. ref1080 As in the previous example, inclusion of Se was designed to increase SOC, a hypothesis that was corroborated by calculations. In toluene Cz-BSeN and DCz-BSeN emit at λPL of 479 and 472 nm, with FWHM of 30 and 28 nm, while ΔE ST are 0.15 and 0.14 eV, all respectively. Despite their ΔE ST values, fast k RISC of 7.5 and 8.8 × 106 s–1 were measured for 1 wt% doped PMMA films in of Cz-BSeN and DCz-BseN, respectively. OLEDs containing 1 wt% emitter in mCBP with Cz-BSeN and DCz-BSeN showed EQEmax of 17.7 and 19.1%, respectively, at CIE coordinates of (0.10, 0.39) and (0.11, 0.16). When the emitter doping was increased to 5 wt% the EQEmax increased to 20.3 and 22.3%, but this was accompanied by a red-shift of the emission with CIE coordinates of (0.13, 0.45) and (0.11, 0.25). Both Cz-BSeN and DCz-BSeN showed improved efficiency roll-off of 32.5 and 30.0% at 500 cd m–2 compared to a comparable device with 5 wt% DtBuCzB doped in mCBP (efficiency roll-off = 62.9%), attributed to the enhanced k RISC resulting from the Se heavy atom effect and associated increased SOC.
An alternative approach to increase SOC was introduced by Cai et al., where the authors prepared Au(I) complexes with the gold centre attached para to the boron of the BNO core.ref. ref894 The linear coordination sphere of the Au(I) was completed with a bulky NHC ligand, producing (BzIPr)AuBNO (Figure ). Compared to free ligand BNO, (BzIPr)AuBNO emits at slightly longer wavelength with λPL of 471 nm compared to 454 nm for BNO, while narrowband emission was conserved with FWHM of 28 and 30 nm in THF for BNO and (BzIPr)AuBNO, respectively. The ΔE ST are 0.17 and 0.11 eV for BNO and (BzIPr)AuBNO in 2 wt% doped PMMA films, and the enhanced SOC brought by the gold atom in the latter results in k RISC accelerating from 3.7 to 110 × 104 s–1. No devices were reported using these emitters.
Four-Coordinate Boron Emitters
A new family of central boron MR-TADF emitters was reported by Wang and co-workers where the central boron is four-coordinate instead of the usual trigonal planar 3-coordinate geometry (BN1, TCz-BN1, BN2, and TCz-BN2, Figure ).ref. ref163 BN1 and TCz-BN1 emit at λPL of 492 and 491 nm in 2 wt% doped mCBP films, while BN2 and TCz-BN2 emit at 559 and 560 nm in 5 wt% doped mCBP films. The FWHM of these four compounds are larger (FWHM = 82–108 nm) than most other MR-TADF systems while their MR-TADF character was inferred from the calculated difference density plots. The ΔE ST values range from 0.17–0.20 eV in 5 wt% doped PMMA films, while the ΦPL are moderate at 53–75%. The τd are shorter in BN1 and TCz-BN1 at 4.5 and 3.0 μs, respectively (2 wt% doped films mCBP) compared to 20.4 and 15.1 μs, for BN2 and TCz-BN2 in mCBP, all respectively. The k RISC of this series of four-coordinate boron compounds are faster than most MR-TADF systems at 2.10–4.67 × 105 s–1. OLEDs with BN1, TCz-BN1, BN2, and TCz-BN2 showed low EQEmax of 5.5, 5.7, 6.7, and 7.8%, respectively, at CIE coordinates of (0.27, 0.49), (0.27, 0.45), (0.40, 0.57), and (0.41, 0.56). The HF-devices using either TCTPCF3 (with BN1 and TCz-BN1, Figure ) and DACT-II (with BN2 and TCz-BN2, Figure ) as assistant dopants showed improved EQEmax of 9.9, 11.5, 19.9, and 25.5%, respectively, while the EQE1000 remained at 7.0, 10.2, 13.5, and 18.7%.
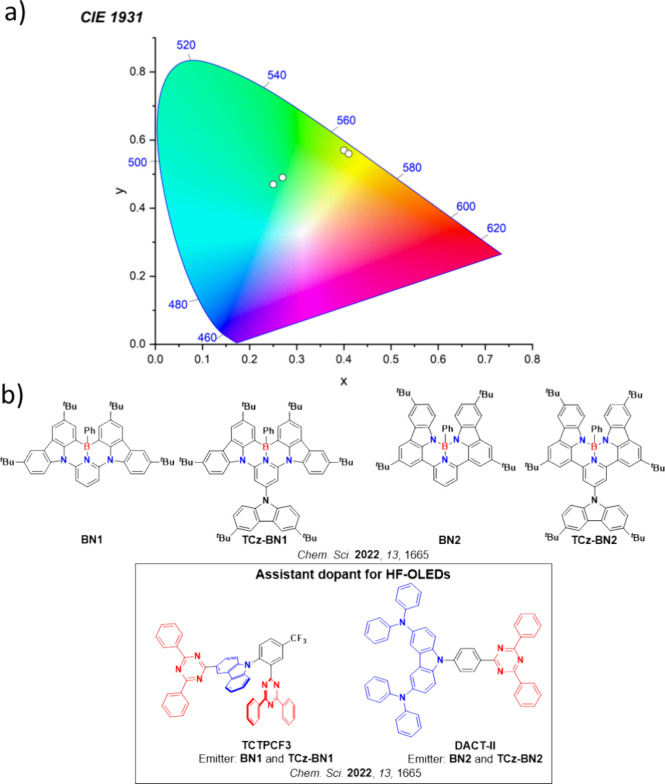
Central Donor Structures with Nitrogen Donor and Boron Acceptor Compounds
An ‘inversion’ of DABNA-1 that contains a central nitrogen donor and peripheral boron acceptors was reported by Hatakeyama and co-workers (Figure ).ref. ref1081 Compared to DABNA-1 with central boron (λPL = 460 nm in mCBP), the emitters ADBNA-Me-Mes and ADBNA-Me-Tip (Figure ) showed a red-shifted emission with λPL of 482 and 479 nm, in respective 1 wt% doped DOBNA-OAr films. ADBNA-Me-Mes and ADBNA-Me-Tip have similar ΔE ST of 0.18 eV, and τd of 165 and 147 μs, respectively, similar to DABNA-1 (0.18 eV and 94 μs). Sky-blue OLEDs showed EQEmax of 16.2 and 21.4% at CIE coordinates of (0.10, 0.27) and (0.11, 0.29), respectively. The superior performance of ADBNA-Me-Tip was ascribed to reduced concentration quenching due to the presence of the bulkier Tip groups.
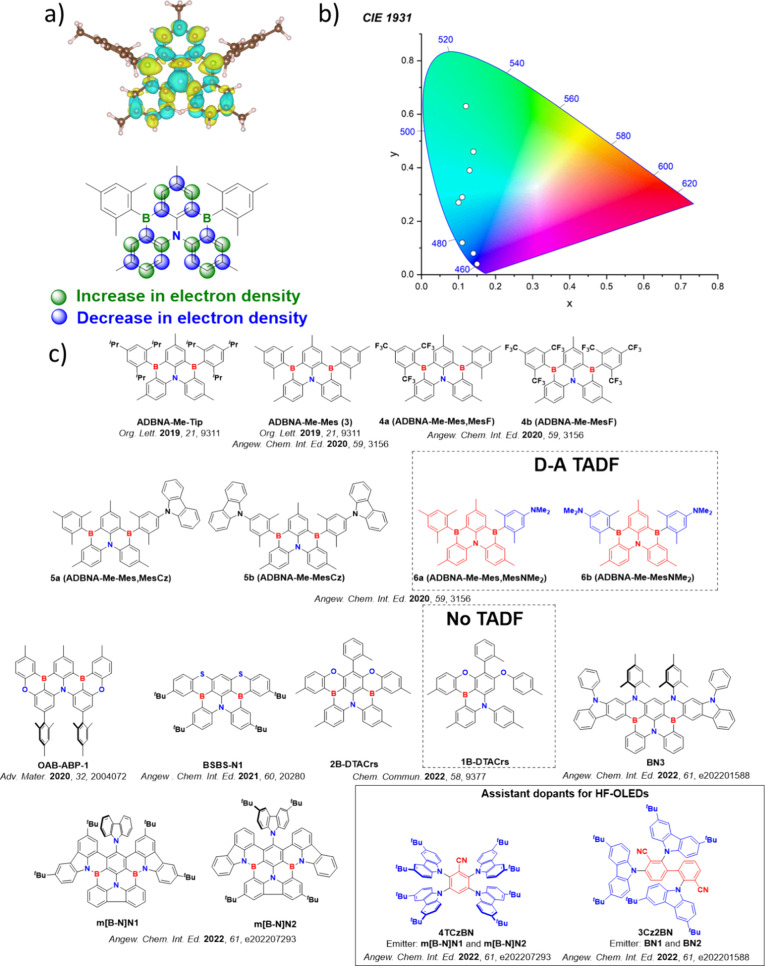
Further exploring this inverted design, symmetric (4b, 5b and 6b) and asymmetric (4a, 5a and 6a) derivatives of these compounds were reported, in which the methyl groups of the mesityl substituent were replaced either entirely with CF3 groups or by carbazole or NMe2 at positions para to the borons (Figure ).ref. ref150 Notably, the photophysical behavior of 6a and 6b was markedly different to that of the other derivatives, with emission spectra that are much broader and with much lower ΦPL values, reflecting a change in the nature of the excited states from SRCT to LRCT, a reflection of the D-A structure of these compounds. The change in nature of the S1 state was also reflected in the much more pronounced positive solvatochromism. Indeed, the use of moieties previously identified as having MR-TADF character as acceptors in D-A TADF systems has since been reported frequently.ref138,ref304,ref310,ref314,ref1082,ref1083 Compounds 4a, 4b, 5a and 5b showed red-shifted emission compared to DABNA-1 with λPL in 1 wt% doped PMMA films between 485–491 nm, and with similar ΦPL of 86–93% and ΔE ST of 0.17–0.19 eV. No devices were fabricated in this study.
Ikeda et al.ref. ref179 reported the green-emitting compound OAB-ABP-1 (λPL = 506 nm) that contains both oxygen and nitrogen donor atoms in conjunction with two boron acceptor atoms (Figure ). In 1 wt% doped DOBNA-OAr films this compound has a ΔE ST of 0.12 eV, a τd of 32 μs, and a ΦPL of 90%. Solution-processed OLEDs showed an EQEmax of 21.8% at CIE coordinates of (0.12, 0.63). The OLEDs showed excellent efficiency roll-off with the EQE1000 remaining as high as 17.4%, while the lifetime was measured to be 11 hours (LT50) at a luminance of 300 cd m–2. This report showed that color tuning by extension of the π-system was indeed possible with central-donor MR-TADF emitters, similar to the central-acceptor MR-TADF materials discussed previously. Nagata et al.ref. ref1084 reported a similar structure BSBS-N1 (Figure ) that contains two sulfur donor atoms in addition to the central nitrogen donor. In 2 wt% doped mCBP films this compound emits at λPL of 478 nm, has a ΔE ST of 0.14 eV, a ΦPL of 89%, and a short τd of 5.6 μs. As a result of the enhanced SOC associated with the heavy S atoms, the k RISC is much faster than most MR-TADF emitters at 1.9 × 106 s–1. The devices showed an EQEmax of 21.0% at CIE coordinates of (0.11, 0.22), yet despite the efficient k RISC, the reported EQE100 was only 16.3%.
Nagata et al.ref. ref1084 reported the similar structure BSBS-N1 (Figure ) that contains two sulfur donor atoms in addition to the central nitrogen donor. In 2 wt% doped mCBP films this compound emits at λPL of 478 nm, has a ΔE ST of 0.14 eV, a ΦPL of 89%, and a short τd of 5.6 μs. As a result of the enhanced SOC associated with the heavy S atoms, the k RISC is much faster than most MR-TADF emitters at 1.9 × 106 s–1. The devices showed an EQEmax of 21.0% at CIE coordinates of (0.11, 0.22), yet despite the efficient k RISC, the reported EQE100 was only 16.3%.
We likewise reported a similar structure, 2B-DTACrs, in which oxygen donors were used instead of sulfur (Figure ).ref. ref159 This compound emits in the deep blue, with λPL and FWHM of 448 nm and 24 nm and with ΦPL of 74% in 5 wt% doped mCBP films. Interestingly, while TADF was evident in 2B-DTACrs (τd of 13.1 μs and k RISC of 1.3 × 105 s–1), the corresponding mono-borylated emitter 1B-DTACrs (Figure ) did not show TADF. OLEDs with 2B-DTACrs showed EQEmax of 14.8% with CIE coordinates of (0.15, 0.04), while the TADF-inactive devices with 1B-DTACrs showed EQEmax of only 1.3%. Despite the promising EQEmax and k RISC for 2B-DTACrs, efficiency roll-off was severe and an EQE1000 could not be recorded.
Lv et al. reported an emitter, BN3, with indolocarbazole in the skeleton (Figure ), which was accessed by changing the stoichiometry of the borylating reagent (two other emitters, BN1 and BN2, are discussed in detail in the Fused Indolocarbazole Emitters section).ref. ref1062 BN3 emits at λPL of 456 nm and has a narrow FWHM 17 nm in toluene. Compared to BN1 and BN2, BN3 displays the smallest ΔE ST of 0.15 eV owing to its longer π-framework (0.20 and 0.16 eV for BN1 and BN2, respectively). In 1 wt% doped DBFPO films BN3 has a larger ΦPL of 98% and faster k RISC of 2.55 × 105 s–1 compared to BN1 and BN2 (91 and 93% for ΦPL and 1.3 and 2.6 × 104 s–1 for k RISC, all respectively). The RISC efficiency in BN3 particularly benefits not only from the smallest ΔE ST but also from significant SOC between S1 and a closely lying T2 state. OLEDs with BN3 showed EQEmax 36.8%, larger than the others, and representing the joint highest EQEmax of a blue MR-TADF emitter.ref. ref306 However, the efficiency roll-off was severe, with the EQE100 dropping 19.0%. To address the efficiency roll-off, HF-OLEDs using 3Cz2BN (Figure ) as the assistant dopant showed much improved EQE100 of 34.0% for the same emitter.
Most boron-based MR-TADF compounds contain three C-B bonds. By contrast, two emitters containing B-N bonds, m[B-N]N1 and m[B-N]N2, were reported by Meng et al. (Figure ).ref. ref1085 m[B-N]N1 and m[B-N]N2 differ only in the positions of the peripheral tBu groups, and both emit in the sky blue with λPL of 481 and 490 nm in toluene, respectively, where the red-shifted emission of m[B-N]N2 was attributed to the stronger donating ability of tBuCz donor compared to Cz. Both compounds have comparable ΔE ST of 0.15 and 0.13 eV and high ΦPL of 91 and 90% in 2 wt% doped mCPBC films, which translate into comparable k RISC of 1.59 and 1.44 × 104 s–1. Devices with m[B-N]N1 and m[B-N]N2 showed EQEmax of 18.1 and 17.3% at CIE coordinates of (0.13, 0.39) and (0.14, 0.46), respectively. Unfortunately, both devices showed severe efficiency roll-off, with EQE1000 of around 6%. In HF-OLEDs with 4TCzBN (Figure ) as the assistant dopant the EQEmax increased to 36.0 and 33.4% while the EQE1000 improved to 27.6 and 24.7%. Further, the LT50 at 1,000 cd m–2 for the HF-OLEDs were an impressive 602 and 535 hours.
Single Donor Acceptor Atoms
So far most of the emitters contain at least three functional dopant atoms within the PAH skeleton, but recently two papers have emerged where the MR-TADF skeleton contains only one boron and one nitrogen atom (Figure ). Bae et al.ref. ref1086 reported a family, BN1, BN2, BN3, and BN4 (Figure ). These four compounds emit at λPL of 401, 415, 420, and 417 nm, respectively, with modest FWHM of between 25–36 nm. Their ΔE ST in 1 wt% doped DPEPO films are large at 0.36, 0.28, 0.29, and 0.24 eV for BN1, BN2, BN3, and BN4, respectively. No delayed emission was observed for BN1, however long τd of 16.2, 13.3, and 4.2 ms were measured for BN2, BN3, and BN4, corresponding to slow k RISC of 0.4, 0.6, and 2.0 × 102 s–1. Devices with BN4 showed an EQEmax of 9.1% at CIE coordinates of (0.17, 0.04), however the efficiency roll-off was severe and an EQE100 was not reported.
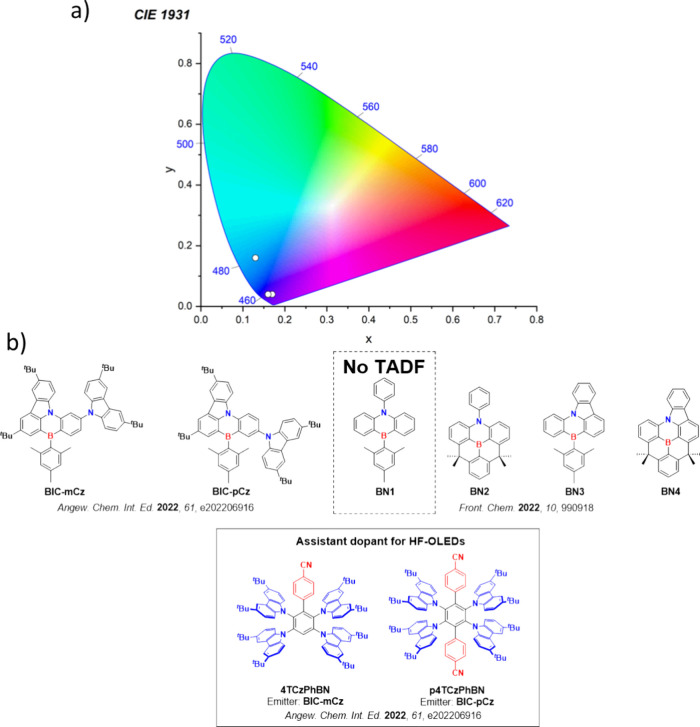
A similar series was reported by Wang et al.,ref. ref175 but where the compounds contained an extra carbazole donor decorated either meta or para to the nitrogen (BIC-mCz and BIC-pCz, Figure ). BIC-mCz and BIC-pCz emit at λPL of 432 and 471 nm in 2 wt% doped mCP films, respectively, while the ΔE ST are 0.29 and 0.15 eV in toluene. The origins of the unexpected contrast in ΔE ST were not discussed, and both compounds showed slow but surprisingly similar k RISC at 4.0 and 3.1 × 103 s–1. The devices showed EQEmax of 7.0 and 13.3% at CIE coordinates of (0.16, 0.04) and (0.13, 0.16), accompanied by strong efficiency roll-off. An EQE1000 was not observed for the device with BIC-mCz, and was only 1.2% for the device with BIC-pCz. In HF-OLEDs with BIC-mCz and BIC-pCz using 4TCzPhBN (with BIC-mCz) and p4TCzPhBN (with BIC-pCz) as assistant dopants (Figure ), the EQEmax increased to 19.4 and 39.8%, respectively, at CIE coordinates of (0.16, 0.05) and (0.14, 0.16).
Central Nitrogen Donor with Ketone Acceptor
QAO and Substituted QAO
Another important class of MR-TADF emitters contain carbonyl groups as electron-acceptors instead of boron atoms (Figure to Figure ). The electron-accepting planar carbonyl groups act in concert with a central donating nitrogen atom to ensure the complimentary HOMO-LUMO pattern that supports MR-TADF emission. The first carbonyl-containing MR-TADF compound was reported in 2019 in the form of QAO ref. ref1087 (also known as QAD ref. ref1088 and DiKTa,ref. ref162 Figure ). This compound emits at λPL of 466 nm (FWHM of 32 nm), has a ΔE ST of 0.18 eV in toluene, and τd of 93 μs in 5 wt% doped mCP films,ref. ref1087 with comparable ΔE ST of 0.19 eV and τd of 23 μs elsewhere reported in toluene.ref. ref162 Devices showed an EQEmax of 19.4% at CIE coordinates of (0.13, 0.18).ref. ref1087 However, the efficiency roll-off was severe with an EQE100 of 9.2%. The EQEmax was even lower using an alternate stack,ref. ref162 at 14.5%, however the maximum luminance was vastly improved from 1,100 cd m–2 in the original report,ref. ref1087 to 10,385 cd m–2 in the subsequent study.ref. ref162
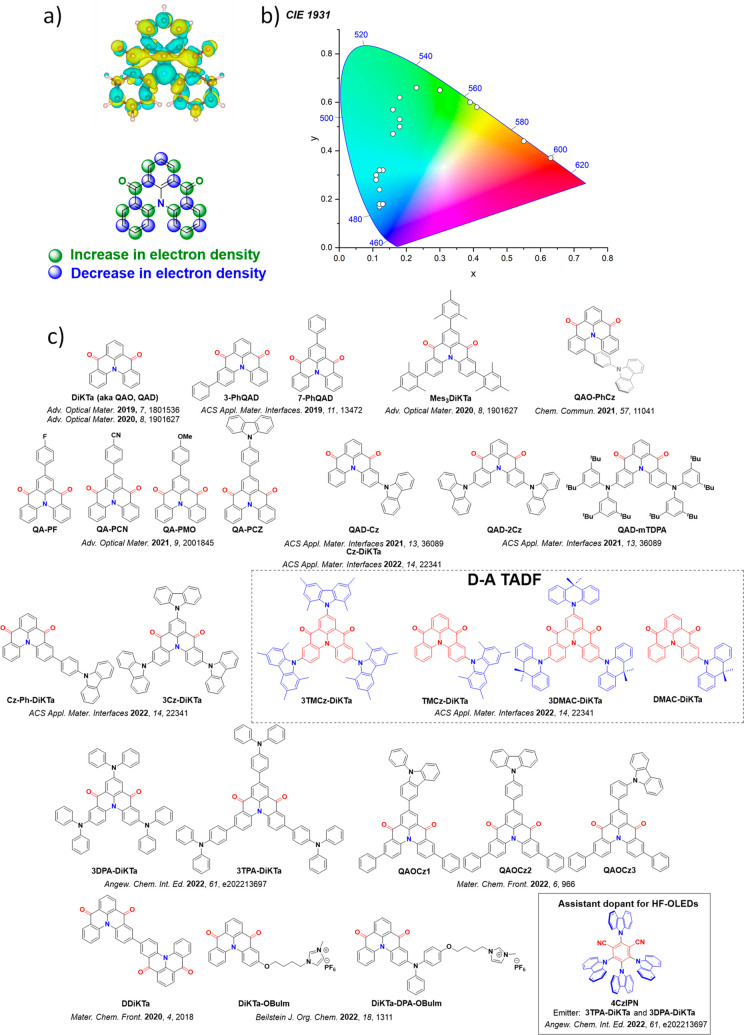
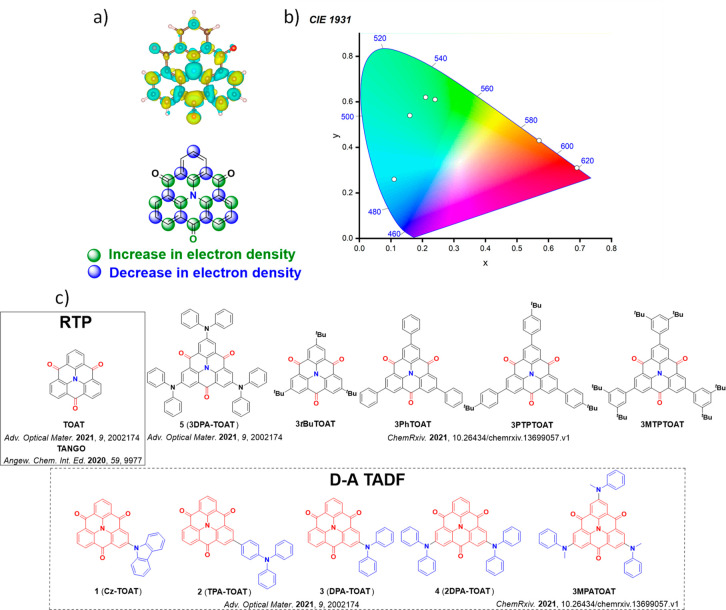
Two similar emitters were reported with phenyl groups located about the periphery, 3-PhQAD and 7-PhQAD (Figure ).ref. ref1088 The location of the phenyl substituent did not significantly affect the λPL, the ΔE ST or the ΦPL. QAD, 3-Ph-QAD, and 7-Ph-QAD emit narrowly with λPL ranging between 464–466 nm, similar ΔE ST of 0.18–0.19 eV in toluene, and ΦPL of 68–73% in 2 or 5 wt% doped mCP films. However, in the OLEDs the phenyl-substituted derivatives experienced a red-shifted and broader emission with λEL of 480 and 472 nm and FWHM of 44 and 34 nm for the devices with 3-Ph-QAD and 7-Ph-QAD, respectively, compared to 466 and 32 nm for the device with QAD. The presence of the phenyl substituent did not impact tangibly the EQEmax though, with values of 19.1 and 18.7% for devices with 3-Ph-QAD and 7-Ph-QAD respectively, compared to 19.4% reported for the device with the parent compound. Efficiency roll-off was again substantial at 46 and 71%, respectively at 100 cd m–2.
A strategy to mitigate ACQ in carbonyl-based MR-TADF OLEDs was introduced by our group.ref. ref162 The decoration of three mesityl groups about the core DiKTa structure in Mes3DiKTa were key to both suppression of aggregate emission and reducing ACQ (Figure ). However, the mesityl groups promote a modest red-shift in the emission, with λPL shifting from 453 nm in DiKTa to 468 nm in Mes3DiKTa (in toluene), while having a minimal effect on both ΔE ST at 0.20 and 0.19 eV and τd of 23 and 33 μs for the same, respectively. The effect of mesityl substitution was most apparent in the changes of ΦPL with concentration. The ΦPL of DiKTa decreased substantially with increasing concentration, while for Mes3DiKTa the ΦPL remained high up to about a 10 wt% doping. Furthermore, in neat films a distinct excimer emission was observed for DiKTa, which was not seen for Mes3DiKTa. Mirroring the changes in PL, the device with Mes3DiKTa showed a red-shifted emission with CIE coordinates of (0.12, 0.32) compared to that of the device with DiKTa (0.14, 0.18). The devices with Mes3DiKTa showed an improved EQEmax of 21.1% compared to those with DiKTa at 14.7%, while the efficiency roll-off was also improved to 31% at 100 cd m–2 compared to 43% for the DiKTa-based device.
A series of aryl-substituted derivatives were introduced onto the QAO core in QA-PF, QA-PCN, QA-PMO, and QA-PCz (Figure ) in an effort to tune the photophysical properties by external substitution.ref. ref203 The incorporation of electron-withdrawing substituents induced a modest blue-shift in emission, with λPL of 478 and 477 nm in QA-PF and QA-PCN compared to 485 and 480 nm in QA-PMO and QA-PCZ, respectively, and with respective ΦPL of 89, 68, 66, and 71% in 3 wt% doped mCP films. The ΔE ST and τd values ranged between 0.18 and 0.25 eV and 224 and 484 μs. The effect of substitution on the broadness of the emission was also probed computationally, where the authors suggested that the addition of these peripheral groups helped to suppress high-energy vibrations responsible for the broadened emission. OLEDs with QA-PF, QA-PCZ, QA-PMO, and QA-PCz showed similar EQEmax of 16.8, 16.9, 15.0, and 17.5% at CIE coordinates of of (0.12, 0.17), (0.12, 0.18), (0.11, 0.30), and (0.11, 0.28), all respectively. Efficiency roll-off was severe though, at between 44 and 77% at 100 cd m–2.
A chiral derivative, QAD-PhCz (Figure ) was reported where the chirality was induced as a result of its helical structure.ref. ref642 Although emission was attributed to a TSCT state, it would appear that it is in fact from the MR-TADF core, owing to its similar emission properties to those of QAO. In toluene QAD-PhCz emits at λPL of 460 nm while QAO emits at 453 nm, while the subdued observed solvatochromsim is again suggestive of an excited state of SRCT character.ref. ref162 Owing to these observations it is included alongside MR-TADF materials. In 5 wt% mCBP films, the ΦPL is 47% and τd is 40 μs, while ΔE ST is 0.11 eV in toluene, representing modest changes compared to those reported for QAO (72%, 93 μs and 0.19 eV in 5 wt% mCP films).ref. ref1087 The devices showed an EQEmax of 14.0% at CIE (0.13, 0.18); however, the efficiency roll-off was large, with an EQE100 of 8.6%. Its chiroptical properties are discussed in Section sec7 .
The effects of donor substitution were investigated by Huang et al.,ref. ref1089 where the QAD core was substituted with one carbazole (QAD-Cz), two carbazoles (QAD-2Cz), or tert-butyldiphenylamines (QAD-mTDPA), Figure . The emission of QAD-Cz, QAD-2Cz, and QAD-mTDPA is red-shifted compared to DiKTa (λPL = 463 nm in 3.5 wt% mCP)ref. ref162 at λPL of 500, 526, and 587 nm in 1 or12 wt% doped mCP films, or 1.5 wt% doped CBP films respectively. Similar to B/N MR-TADF compounds, addition of peripheral donor groups leads to broadened emission spectra, with FWHM of 50, 50, and 62 nm, respectively, attributed to a combination of increased structural flexibility and increased LRCT character of the excited state. QAD-Cz, QAD-2Cz, and QAD-mTDPA have very high ΦPL of 100, 100, and 97%, while the ΔE ST values are 0.17, 0.17, and 0.33 eV in the doped films. Devices with QAD-Cz, QAD-2Cz, and QAD-mTDPA showed EQEmax of 20.3, 27.3, and 26.3% at CIE coordinates of (0.16, 0.47), (0.30, 0.65), and (0.55, 0.44), respectively. The EQE100 diverged considerably at 5.4, 23.9, and 12.9%, respectively. Investigation of the efficiency roll-off identified a combination of TTA and SPA as the primary detrimental factors, allowed by the inefficient k RISC in QAD-Cz and QAD-mTDPA.
Recently we demonstrated how the strength of the peripheral donor group impacts the nature of the emissive excited state.ref. ref1090 With DMAC (DMAC-DiKTa and 3DMAC-DiKTa) and tetramethylcarbazole (TMCz-DiKTa and 3TMCz-DiKTa) as the donor substituents (Figure ) the excited state character become LRCT and the emission resembled D-A TADF systems, reflected in the broad FWHM (71–116 nm) in 2 wt% doped mCP films. However, when carbazole (Cz-DiKTa and 3Cz-DiKTa) and 4-N-carbazolylphenyl (Cz-Ph-DiKTa) were used, narrowband emission (FWHM 47–54 nm) associated with SRCT excited states was observed (Figure ). The emission also red-shifted from λPL of 502 nm in mono-substituted (Cz-DiKTa (same structure as QAD-Cz) to 539 nm in tri-substituted 3Cz-DiKTa in 2 wt% doped mCP films. The presence of the phenyl spacer in Cz-Ph-DiKTa instead contributed to a blue-shifted emission at λPL of 486 nm. Each of these carbazole-based derivatives has a similar ΔE ST, ranging between 0.10–0.16 eV, and τd ranging from 153 to 286 μs. These lifetimes are substantially longer than the D-A TADF DMAC and tetramethylcarbazole derivatives, that ranged from 3.0 to 22 μs. The devices with Cz-DiKTa, Cz-Ph-DiKTa, and 3Cz-DiKTa showed EQEmax of 24.9, 23.0, and 24.4%, respectively, while the efficiency roll-off was moderate with EQE100 of 20.4, 19.3, and 17.3%, for the same.
Building upon this work, we reported two new emitters, each with three diphenylamine (3DPA-DiKTa, Figure ) or triphenylamine (3TPA-DiKTa, Figure ) donors about a DiKTa core.ref. ref165 In 2 wt% doped mCP films 3TPA-DiKTa and 3DPA-DiKTa emit at λPL of 551 and 617 nm, respectively. The emission spectra are relatively broad, with FWHM of 58 and 56 nm reflecting excited states of mixed LRCT and SRCT character. 3TPA-DiKTa and 3DPA-DiKTa have ΔE ST of 0.13 and 0.20 eV and τd of 131 and 323 μs, which translate to k RISC of only 0.14 and 2.49 × 104 s–1, respectively. Devices with 3TPA-DiKTa and 3DPA-DiKTa nonetheless showed EQEmax of 30.8 and 16.7%, respectively, at CIE coordinates of (0.41, 0.58) and (0.63, 0.37)–efficiencies amongst the highest reported for carbonyl-containing MR-TADF emitters. The devices suffered from severe roll-off though, with EQE100 dropping to 18.1 and 3.4%, likely due to their inefficient k RISC. HF-OLEDs using 4CzIPN (Figure ) as the assistant dopant showed much improved efficiency roll-off, with the EQE100 of 27.4 and 8.7%. The exceptional efficiency of the device with 3TPA-DiKTa was partly attributed to the preferential horizontal alignment of its TDM, which was less pronounced for 3DPA-DiKTa.
The impact of changing donor strength on photophysical properties of DiKTa analogues was also investigated by Liu et al.ref. ref1091 In QAOCz1 carbazole was attached directly to the QAO core, while in QAOCz2 and QAOCz3 phenyl spacers were set between the QAO core and the carbazole donors, with QAOCz2 containing a para disposed carbazole and QAOCz3 having a meta linked carbazole (Figure ). QAOCz1, QAOCz2, and QAOCz3 emit at λPL of 502, 500, and 492 nm, respectively, in toluene, with the blue-shift trend in emission linked by the authors to a decreasing D-A interaction within the molecules. The ΔE ST was similarly reported to decrease from 0.26 to 0.18 and 0.16 eV, respectively. The ΦPL in 5 wt% doped CBP films were 86, 87, and 99%, with the increase attributed to enhanced rigidity across the series. OLEDs with QAOCz1, QAOCz2, and QAOCz3 showed EQEmax of 16.9, 19.4, and 21.1% respectively, with the increase in line with both the trends in ΦPL and ΔE ST. All of the devices showed significant efficiency roll-off though, with EQE1000 < 3% for each.
Two emitters based on DiKTa with charged side groups were presented by us for use in LECs.ref. ref166 One was directly coupled via an oxygen bridge to an alkyl-imidazolium ionic group (DiKTa-OBuIm, Figure ), while the other was coupled via a diphenylamine donor (DiKTa-DPA-OBuIm, Figure ). In 1 wt% mCP the λPL were 500 and 578 nm, with ΦPL of 71 and 61% for DiKTa-OBuIm and DiKTa-DPA-OBuIm respectively. Their FWHM was broad for MR-TADF emitters at 66 and 95 nm, with both oxygen and DPA acting as donating groups. TADF was observed for both emitters in 1 wt% mCP films, with very similar ΔE ST of 0.19 and 0.20 eV and slow k RISC of 2.9 and 3.0 × 103 s–1. Their LEC properties are discussed further in the LEC section.
A dimeric analogue of DiKTa, (DDiKTa, Figure ) was reported by us and showed blue-green emission at λPL 490 nm in 9 wt% doped DPEPO films.ref. ref55 This extended design produced a red-shifted emission and a suppression of the ACQ that is apparent in the parent DiKTa (λPL = 463 nm).ref. ref162 DDiKTa has ΔE ST of 0.16 eV and a relatively fast τd of 1.2 μs in the same films.ref. ref55 The activation energy for triplet to singlet up-conversion was even smaller at 0.04 eV, suggestive of the involvement of intermediate triplet states in the RISC process and confirmed by calculations.ref. ref83 Devices showed EQEmax of 19.0% at CIE coordinates of (0.18, 0.53), which at the time of publication was only the second example of a green-emitting MR-TADF OLED.ref. ref55 The efficiency roll-off was severe and the OLED could not attain a brightness of 1,000 cd m–2.
Exotic QAO Derivatives
Other ketone-based MR-TADF emitters with more elaborate structures have also been developed over time (Figure ). For example, rigid and planar analogues have been designed that embed one of a DMAC, PXZ, or PTZ groups within the QAO skeleton,ref. ref1092 producing DQAO, OQAO, and SQAO (Figure ). The introduction of O and S atoms resulted in a substantial red-shift and a modest broadening of the emission, with λPL (FWHM) in toluene of 465 (33), 520 (36), and 552 (54) nm, respectively. SQAO also showed a more pronounced positive solvatochromism, suggesting that the excited state contains greater LRCT character than the other two compounds. DQAO, OQAO, and SQAO have similar ΔE ST of 0.19, 0.16, and 0.16 eV, while τd varied more considerably at 111, 205, and 78 μs, with the latter likely shorter due to increased SOC from the heavier sulfur atom. The devices with DQAO, OQAO, and SQAO showed EQEmax of 15.2, 20.3, and 17.8%, respectively, at CIE coordinates of (0.12, 0.18), (0.32, 0.65), and (0.47, 0.52). In terms of efficiency roll-off, the EQE100 decreased somewhat to 8.5, 15.1, and 13.6%.
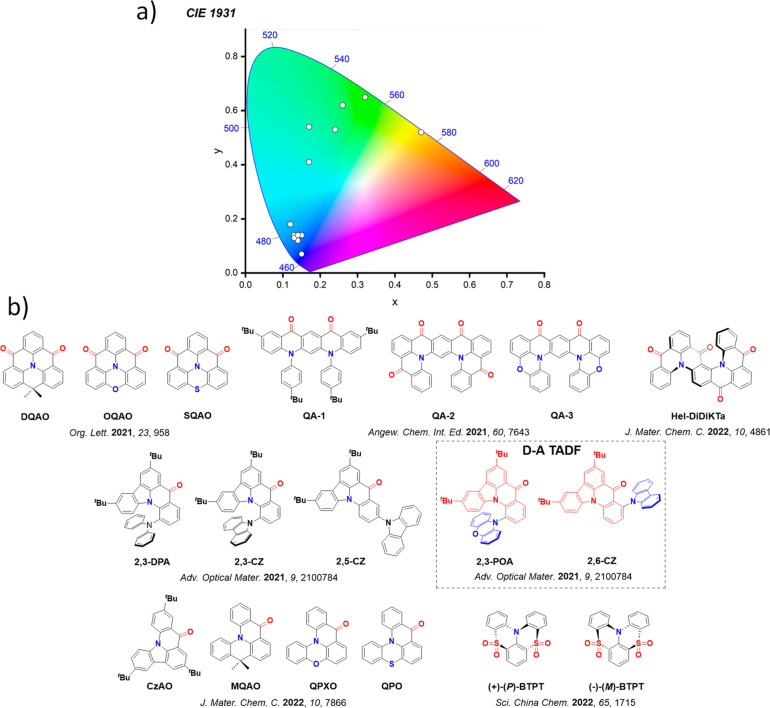
Yasuda et al. reported a family of linearly extended emitters, QA-1, QA-2, and QA-3 (Figure ).ref. ref1093 In 3 wt% doped PPCz films their ΔE ST are 0.29, 0.19, and 0.19 eV respectively, with associated τd of 655, 48, and 307 μs. The long τd for QA-1 can be rationalized by its much larger ΔE ST, while the differences in delayed lifetimes between QA-2 and QA-3 were attributed to the presence of intermediate triplet states in QA-2 that contribute to enhanced k RISC. QA-1 and QA-2 emit at λPL of 457 and 465 nm, while replacing two of the carbonyl groups with oxygen atoms led to a red-shifted emission at λPL of 523 nm for QA-3. Devices with QA-1, QA-2, and QA-3 showed EQEmax of 17.1, 19.0, and 16.6% respectively, at CIE coordinates of (0.14, 0.12), (0.13, 0.14), and (0.26, 0.62). The very long τd for QA-1 contributed to significant efficiency roll-off of 93% at 100 cd m–2, with TTA and STA quenching pathways proposed as the key culprits. The efficiency roll-off was considerable but less severe for both QA-2 and QA-3, at 42 and 40% at 100 cd m–2.
A helical isomer of QA-2, Hel-DiDiKTa, was reported by dos Santos et al.ref. ref160 (Figure ). In 1 wt% doped films in mCP, the compound emits at λPL of 480 nm, has a ΔE ST of 0.15 eV and a τd of 5.4 μs. However, as the ΦPL is very low at 4.1%, k RISC is very slow at 4.1 × 102 s–1 and no devices were reported. Its chiroptical properties are discussed in Section sec7 .
Replacing the ketone functionalities of QAO with sulfone moieties produced the near-UV emitter BTPT (Figure ).ref. ref645 In 1 wt% doped PMMA films this compound emits at λPL of 400 nm with modest FWHM of 56 nm, while its ΔE ST is 0.14 eV and it has a τd of 109 μs. In the crystal this compound shows both RTP and CPL from different enantiomers, although with TADF not apparent. No devices were fabricated.
A series of compounds containing only one carbonyl groups was reported by Luo et al.ref. ref464 Of the five compounds in the study, three were demonstrated to be MR-TADF (2,3-CZ, 2,5-CZ, and 2,3-DPA, Figure ), while 2,6-CZ and 2,3-POA behaved as D-A TADF compounds (Figure ). MR-TADF was inferred from the smaller FWHM of 36, 41, and 57 nm for 2,3-CZ, 2,5-CZ, and 2,3-DPA, respectively in toluene, compared to 80 and 92 nm for 2,6-CZ and 2,3-POA in the same medium. Further, HOMO-LUMO density distribution of 2,3-CZ, 2,5-CZ, and 2,3-DPA each showed patterns reminiscent of MR-TADF. The measured ΔE ST in toluene are 0.26, 0.29, and 0.19 eV for 2,3-CZ, 2,5-CZ, and 2,3-DPA, respectively, while τd are 436, 619, and 373 μs for the same in 3.5 wt% doped mCBP films. 2,3-CZ, 2,5-CZ, and 2,3-DPA emit at λPL of 449, 459, and 496 nm in toluene and have ΦPL of 40, 81, and 51% in 3.5 wt% doped mCBP films. Shorter delayed lifetimes of 6.2 and 28.1 μs for 2,6-CZ and 2,3-POA in the same films were attributed to their much smaller ΔE ST of 0.00 and 0.01 eV. Devices of 2,3-CZ and 2,5-CZ in mCBP showed EQEmax of 6.3 and 22.3% at CIE coordinates of (0.15, 0.14) and (0.13, 0.13). When the EML of the OLED instead consisted of 10 wt% 2,3-CZ or 2,3-DPA in 26DCzPPy, the EQEmax increased to 8.1 and 11.7% at CIE coordinates of (0.13, 0.15) and (0.17, 0.54).
Huang et al.ref. ref1094 investigated a related series of mono-ketone compounds, CzAO, MQAO, QPXO, and QPO (Figure ). A progressive red-shifting of the emission was observed across the series, which emit at λPL of 431, 447, 485, and 501 nm respectively, while their FWHM also increased from 36 to 61, 76, and 86 nm. Calculations confirmed that this trend in FWHM was mainly due to increasing LRCT content. Large ΔE ST of 0.27–0.40 eV led to relatively inefficient TADF, reflected in τd of 1.0–2.4 ms. Devices with CzAO, MQAO, QPXO, and QPO hence showed relatively low EQEmax of 8.6, 10.3, 7.1, and 15.3%, respectively.
Tri-ketone Emitters
The tri-ketone fused derivative of QAO, TOAT (Figure ), has generated similar levels of attention as a MR-TADF emitter with λPL of 417 nm and a large ΔE ST of 0.34 eV in toluene.ref. ref1095 Previous reports have highlighted the same core (TANGO) as an RTP emitter in the crystalline form.ref. ref1096 The addition of peripheral donating groups can alter the nature of the emissive S1 state from SRCT to LRCT associated with D-A TADF systems.ref. ref1095
For example, upon addition of one carbazole (originally 1, here renamed Cz-TOAT, Figure ) and triphenylamine (2, here TPA-TOAT, Figure ) the emission broadened, with the authors classifying these materials to be D-A TADF rather than MR-TADF. Increasing the number of DPA units instead induced a narrowing of the emission in 3, 4, and 5 (here DPA-TOAT, 2DPA-TOAT, and 3DPA-TOAT, Figure ), with FWHM decreasing from 84 to 75 and 45 nm, respectively. It was concluded that 3DPA-TOAT was indeed MR-TADF while the other species were of D-A character. The emission of 3DPA-TOAT in toluene is red-shifted at λPL of 590 nm compared to that of TOAT (λPL of 417 nm), accompanied by an increase in ΦPL from < 1% for TOAT to 7% for 3DPA-TOAT. The ΦPL similarly increased to 46% in 3 wt% doped mCBP films. In toluene the ΔE ST of 3DPA-TOAT is 0.34 eV, identical to that of TOAT, which resulted in a long τd of 2.1 ms and low k RISC of 9.3 × 102 s–1. Devices with 3DPA-TOAT showed very low EQEmax of 1.2% at CIE coordinates of (0.57, 0.43). Devices with the D-A TADF emitters, Cz-TOAT, TPA-TOAT, DPA-TOAT, and 2DPA-TOAT showed much improved EQEmax of up to 17% for the device with 2TPA-TOAT. The authors attributed the low efficiency of the device with 3DPA-TOAT to poor charge balance and undesired host-guest interactions.
A similar series of compounds using the same TOAT core but with different substituents (tBu, phenyl, p–tBu-Phenyl, m-DitBu-Phenyl, and NMePh in 3tBuTOAT, 3PhTOAT, 3PTPTOAT, 3MTPTOAT, and 3MPATOAT respectively, Figure ) was reported by Wang et al.ref. ref1097 In toluene 3tBuTOAT, 3PhTOAT, 3PTPTOAT, and 3MTPTOAT emit at λPL ranging from 439–468 nm and have large ΔE ST ranging from 0.30–0.40 eV; a large red-shift in the emission was observed for 3MPATOAT with λPL at 580 nm, as this system is a D-A TADF emitter due to the strongly electron-donating NPhMe groups. In doped films large changes in photophysical properties were observed, which the authors attribute to the formation of dimer species. These differences were particularly apparent in the phenyl series of 3PhTOAT, 3PTPTOAT, and 3MTPTOAT. In 15 wt% doped mCP films the emission of 3PhTOAT, 3PTPTOAT, and 3MTPTOAT was red-shifted compared to toluene solution, from 442, 449, and 446 nm to 516, 520, and 502 nm, respectively, with the ΦPL increasing from 16, 23, and 20% to 97, 93, and 92%. The large changes in ΦPL were rationalized in terms of the differences in HOMO-LUMO electron density distributions between the monomolecular and the dimer species. As an isolated species, the S1 state has n−π* character, with low oscillator strength, while for the dimeric species the excited state is π–π* with much larger oscillator strength. The ΔE ST decreased from 0.32, 0.32, and 0.30 eV in toluene to 0.12, 0.16, and 0.14 eV in the 15 wt% doped mCP films. The devices with 3PhTOAT, 3PTPTOAT, and 3MTPTOAT hence showed EQEmax of 29.2, 27.6, and 31.2%, respectively, at CIE coordinates of (0.16, 0.54), (0.24, 0.61), and (0.21, 0.62), compared to the much lower efficiencies of 13.0 and 11.3% for the devices with 3tBuTOAT and 3MPATOAT at CIE coordinates of (0.11, 0.26) and (0.69, 0.31). Strongly horizontal orientated TDMs of 3PTPTOAT and 3MTPTOAT in the film contributed to the high EQEmax values in the devices.
Acceptor-Free MR-TADF Emitters
Recently three publications have emerged that demonstrate that MR-TADF compounds need not necessarily contain acceptor groups (Figure ).ref161,ref167,ref172 These reports all centre on diindocarbazole units with para-disposed nitrogen atoms, with the differences in structure only extending to the peripheral substituents at present. Patil et al. reported BisICz, tBisICz, and tPBisICz, containing no substitution, tert-butyl groups, and di-tert-butylphenyl substituents, respectively (Figure ).ref. ref172 tBisICz and tPBisICz are MR-TADF with long τd of 12.5 and 1.7 ms, respectively, owing to their large ΔE ST of 0.29 and 0.27 eV in 1 wt% doped mCP:TSPO1 films. High ΦPL of 95 and 91% and narrow blue λPL of 442 and 450 nm suggested that these two compounds would be promising materials for blue OLEDs. RISC was inferred to proceed via a T2 state as SOC between S1 and T1 was calculated to be very small, however both tBisICz and tPBisICz show inefficient k RISC of 0.15 and 1.47 × 103 s–1, with the difference in magnitude attributed to a calculated smaller ΔE T2T1 in tPBisICz. Deep blue OLEDs with tBisICz and tPBisICz showed EQEmax of 15.1 and 23.1% at CIE coordinates of (0.16, 0.05) and (0.15, 0.05). However, owing to their inefficient k RISC, efficiency roll-off was catastrophic with EQE100 dropping to 3.0 and 4.8% respectively.
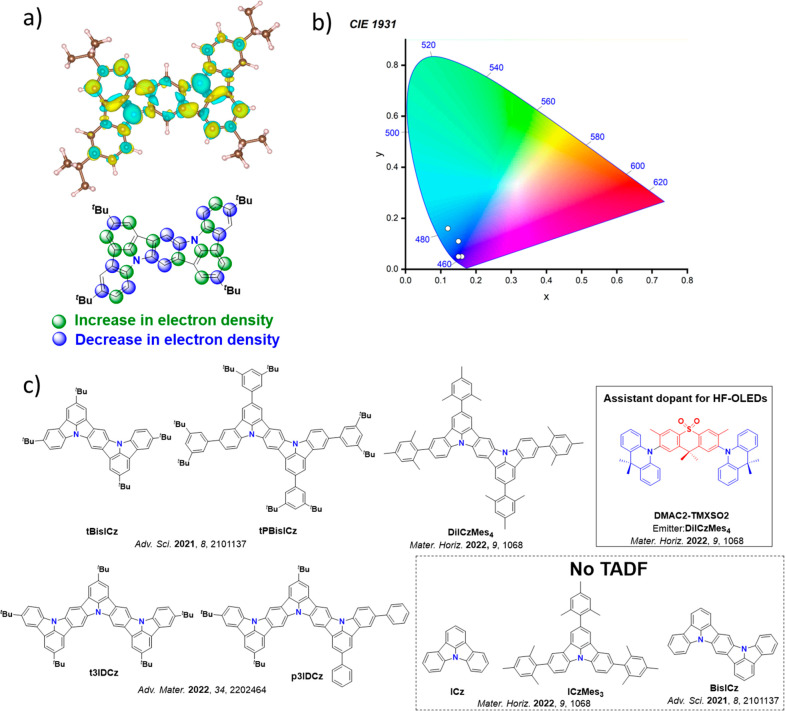
We reported a mesityl-substituted diindolocarbazole as part of a wider study that contrasted the photophysics of ICz, ICzMes3 , and DiICzMes4 (Figure ).ref. ref161 Red-shifted emission and progressively decreasing ΔE ST were observed across the series, with λPL of 374, 387, and 441 nm and ΔE ST of 0.47, 0.39, and 0.26 eV in ICz, ICzMes3 , and DiICzMes4 , respectively. As predicted computationally, there is an increase in ΦPL across the series from 58 to 66 and 70% in toluene. Owing to their large ΔE ST, ICz and ICzMes3 are not TADF-active; however, when doped in 3 wt% mCP films TADF is apparent in DiICzMes4 with ΔE ST of 0.26 eV, τd of 433 μs, and ΦPL of 82% and a k RISC of 1.9 × 102 s–1. OLEDs with DiICzMes4 showed an EQEmax of only 3.0% at CIE coordinates of (0.15, 0.11), but the device performance was measurably improved in HF-OLEDs using DMAC2-TMXSO2 (Figure ) as the assistant dopant where the EQEmax increased to 16.5% at the same CIE coordinates.
A further extension of the ICz core with three nitrogen atoms was reported by Lee et al.ref. ref167 Emitters t3IDCz and p3IDCz (Figure ) were reported, the first with all tBu substituents and the latter with two phenyl substituents. These two larger systems showed a red-shifted emission at λPL of 470 nm compared to tBisICz, (λPL of 442 nm) in doped films.ref. ref172 The ΔE ST are smaller though at 0.21 and 0.19 eV for t3IDCz and p3IDCz, respectively in THF. Coupled with the high ΦPL of 92 and 100%, k RISC in t3IDCz and p3IDCz reached 0.84 and 1.1 × 104 s–1 in 1 wt% doped mCP:mCBP-1CN films. The OLEDs with t3IDCz and p3IDCz showed EQEmax of 30.0 and 30.9% at CIE coordinates of (0.12, 0.16) for both. Despite the improved k RISC, efficiency roll-off was still high with EQE1000 of just 5.0 and 4.7%.
Outlook
Despite their very recent rise to prominence the efficiencies achievable by MR-TADF devices are already impressive, with EQEmax regularly exceeding 30%. We particularly highlight representative, green, and red MR-TADF OLEDs employing m-DPAcP-BNCz, and TCZ-F-DABNA, with EQEmax of 36.1, 42.0, and 39.2%, respectively, while for blue two emitters, t-Bu-v-DABNA and BN3 have identical EQEmax of 36.3% (Figure ). However, the efficiencies at display-relevant luminances are often undercut by significant efficiency roll-off for these devices, with few reports of MR-TADF OLEDs having EQE1000 above 20%. It is unclear at present whether the relatively slow k RISC in reported MR-TADF emitters (typically ∼100 times smaller than leading D-A emitters) is an intrinsic limitation, or merely reflective of the limited region of chemical space thus-far explored.
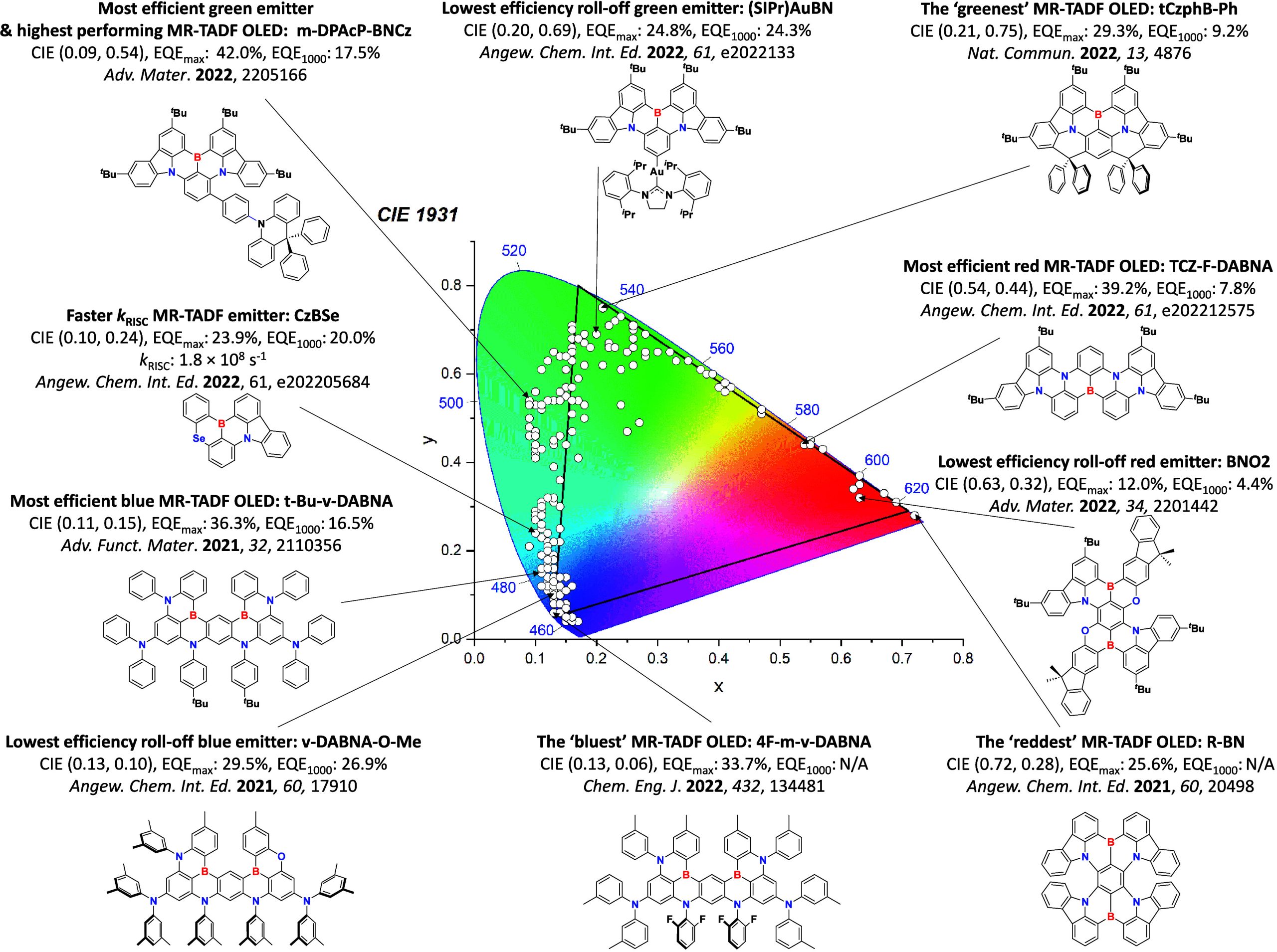
Promising current MR-TADF design strategies include extending the π-system as exemplified by v-DABNA and its derivatives (Section sec11.2.4 ), although with this singular approach reaching an apparent ceiling for k RISC of ∼105 s–1. Inclusion of heavy sulfur and selenium atoms can also accelerate k RISC, for example in CzBSe (Figure ) with a reported k RISC of 108 s–1, surpassing even the most efficient D-A TADF systems. Mechanistically, we note that the role of upper triplet states in mediating RISC for MR-TADF emitters has only recently begun to be appreciated and explored, with further fundamental and computational studies likely to inspire and refine new design insights. In the meantime, the relatively slow RISC of reported MR-TADF emitters is frequently overcome through the use of established D-A assistant dopants in hyperfluorescence devices (Section sec17 ).
In parallel with raw efficiency, while there are now examples of MR-TADF compounds that emit across the entire visible spectrum, challenges and mysteries remain with respect to color tuning. Indeed, one of the key benefits of narrowband MR-TADF emitters is their ability to reach highly saturated color coordinates that are practically inaccessible to D-A CT emitters, for example with red R-TBN and blue 4F-m-v-DABNA nearing the Rec. 2020 CIE coordinatesion of (0.13,0.05) and (0.71, 0.29), respectively (Figure ). tCzphB-Ph does provide the greenest MR-TADF emitter, nearing Rec. 2020 for green (0.17, 0.80), however, progress to this color point is still behind the red and blue counterparts with further development needed (Figure ). The emission color can be tuned by judicious decoration of (or substitution within) the MR-TADF core. On a fundamental level, mixing of some LRCT character into the SRCT emissive excited state can both tune the color (most commonly to the red) and increase RISC, albeit at the expense of a somewhat broadened emission.ref. ref150 Here we note that wavefunction-based computational methods, recently demonstrated to be necessary for the accurate modelling of MR-TADF excited-state energies (Section sec2 ), will become more popular in the coming years to inform molecular design regarding color tuning. This will be a particularly welcome development, as outside the visible spectrum there exist precious few near-UV and near-IR MR-TADF emitters, with potential for such materials to advance significant applications in, for instance, sensing, security, and (bio)imaging.
Lastly, we note that most of the OLEDs summarized in this section were fabricated by vacuum deposition. Owing to their planar structures and strong propensity to aggregate, MR-TADF OLEDs typically require evaporative doping fabrication at low emitter concentration (<5 wt%) to retain high device performance. Nonetheless, recent works using polymers or very bulky derivatives have sought to mitigate this issue and there are now examples of bright, narrowband MR-TADF emitter films at doping concentrations exceeding 40 wt%. Similar strategies can also enable MR-TADF emitters to be solution processable, and there are now a small but increasing number of reports of solution-processed MR-TADF OLEDs. Mirroring earlier developments for D-A TADF emitters, we now also see diversification in the properties and uses of MR-TADF materials, including with chiral centers and CPL emission (Section sec7 ), in LECs (Section sec16 ), organic lasing (Section sec22 ), and as photocatalysts (Section sec23 ). Ultimately MR-TADF materials – acting either as hosts, emitters, or otherwise – are an exciting class of compounds with potential yet to be fully realized.
Through-Space Charge Transfer (TSCT) Interactions in TADF
Introduction
Arguably the fundamental criteria for designing effective and efficient TADF OLED materials is to realize both fast k RISC, necessitating a small ΔE ST, and large ΦPL. The former requires a small exchange integral, while the latter relies on high oscillator strength for the excited states involved in emission. Reflecting the vast majority of reported TADF emitters, Sections sec2–sec7 highlight examples of highly twisted donor-acceptor molecular designs to achieve a small ΔE ST, forming emissive charge transfer states with “through bond” electronic conjugation through the π-network.
An alternative strategy to achieve weak electronic coupling between donor and acceptor motifs is to exploit “through-space” (TS) conjugation, where the π-systems of donor and acceptor moieties are aligned and interact without direct covalent bonding. Similar to TADF exciplex blends of separate donor and acceptor molecules (see Section sec8 ), molecular scaffolding can be used to controllably engineer through-space charge transfer (TSCT) states that translate into molecules with small ΔE ST. There are also a small number of compounds where donor and acceptor groups are electronically coupled through homoconjugated linkers, which also leads to a small ΔE ST. The number of reported TSCT TADF molecules has increased rapidly, especially so since 2020, offering examples with fast k RISC and outstanding OLED performance. In this section we summarise recent developments in the design and understanding of TSCT materials, categorized based on the structural units used as scaffolds to mediate the interaction of the donor and acceptor groups.
TSCT Featuring Non-conjugated Bridges
Many TSCT TADF emitters are constructed using a non-conjugated bridge to hold donor and acceptor subunits in a co-facial arrangement. Tsujimoto et al. were the first to explore this concept in a series of compounds containing a xanthene bridge that co-orients a triazine acceptor with donor units (phenothiazine for XPT, carbazole for XCT, or tert-butyl-carbazole for XtBuCT, Figure ).ref. ref1098 The distances between donor and acceptor were 3.3–3.5 Å, allowing TSCT states to exist. With increasing donor strength, a progressive red-shift in the emission was observed for XCT, XtBuCT, and XPT (λPL of 419 to 451 and 562 nm, respectively, in toluene). When doped at 10 wt% in DPEPO the emission of XtBuCT and XPT red-shift slightly to 453 and 566 nm, with ΦPL of 35 and 66%, and τd of 4.1 and 3.3 μs, respectively (no ΔE ST values were reported). The resulting OLEDs emitted at λEL of 488 and 584 nm for XtBuCT and XPT, and showed a modest EQEmax of 4 and 10%, respectively.
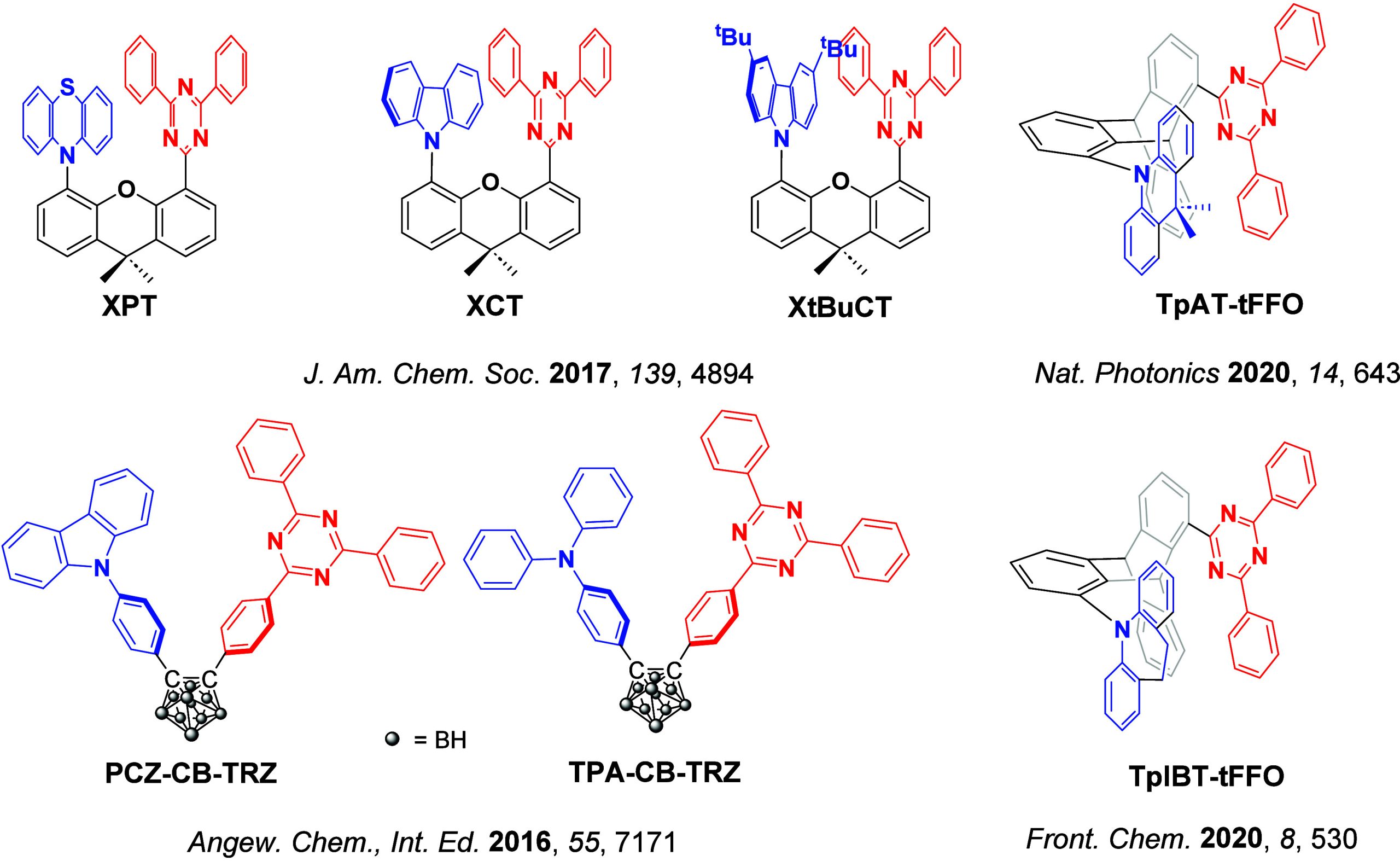
o-Carboranes are another group that can bridge donor and acceptor to achieve TSCT.ref. ref1099 Two emitters, PCZ-CB-TRZ and TPA-CB-TRZ (Figure ), displayed small DFT-calculated ΔE ST of 0.003 and 0.018 eV, respectively. The emitters also showed AIE (see Section sec13 for more discussion), exhibiting yellow emission with λPL of 557 and 571 nm and having ΦPL of 97 and 94% in neat film, all respectively. The non-doped OLEDs with PCZ-CB-TRZ and TPA-CB-TRZ emitted at λEL of 631 and 586 nm, and showed EQEmax of 11 and 9.2%, respectively. These EQEmax are still lower than what might be expected from the high ΦPL, suggesting further optimization in the device structure is needed.
Wada et al. reported three ‘tilted face-to-face with optimal distance’ (tFFO) TADF emitters, TpAT-tFFO, TpMAT-tFFO, and TpPXT-tFFO (Figure ).ref. ref98 Near-degenerate 1CT, 3CT, and 3LE states were realized by controlling the distance and orientation between the donors and acceptors using a triptycene scaffold, and large spin–orbit coupling values were realized with the donors and acceptors not perfectly co-facially oriented. With this strategy the three emitters all showed very fast k RISC; for example, TpAT-tFFO has a remarkably fast k RISC of 1.2×107 s–1 alongside a high ΦPL of 76% in 25 wt% doped films in mCBP. An OLED with TpAT-tFFO showed blue emission at λEL of 462 nm with an EQEmax of 19.2%. The same group further exploited this strategy using 10,11-dihydro-5H-dibenzo[b, f]azepine (IB) as the donor, giving the structure TpIBT-TFFO.ref. ref1100 This compound also has a very fast k RISC of 6.9 × 106 s–1, emits at λPL at 477 nm, and has a ΔE ST of 0.076 eV in toluene, while the ΦPL is 71.4% and the τd is 6.7 μs in 9 wt% doped films in CzSi (Table S16). A device with TpIBT-TFFO showed an EQEmax of 12.2%, emitting at λEL of 462 nm, with CIE coordinates of (0.16, 0.26).
TSCT Featuring Spiro-fluorene Bridges
Spiro-fluorenes can also be used to build TSCT skeletons, taking advantage of the perpendicular attachment point offered by the spiro centre. Attaching the donor or acceptor at the spiro position also leads to rigid structures with limited vibrational flexibility, thereby decreasing non-radiative decay and resulting in narrower emission bands. Tang et al. developed a series of pseudo co-facial TSCT emitters, DM-B, DM-Bm, and DM-G (Figure ), where the spacing and relative orientation of the donor and acceptor subunits were controlled by using a rigid spiro-fluorene as a linker.ref. ref1101 Using this approach, ground-state electronic coupling was strengthened, and non-radiative decay channels were suppressed. The resulting molecules DM-B, DM-Bm, and DM-G emit at 493, 495, and 504 nm, have short τd of 5.0, 4.5, and 3.3 μs, high ΦPL of 96, 92, and 88%, and have reported ΔE ST of 0.17, −0.08, and −0.11 eV in 20 wt% doped films in DPEPO. These apparent inverted singlet-triplet gaps likely reflect an energy gap between singlet and triplet states of different species. The OLEDs with DM-B, DM-Bm, and DM-G showed EQEmax of 27.4, 21.7, and 18.5% and low efficiency roll-off of only 10.9, 9.2 and 16.8% at 1000 cd m–2, at CIE coordinates of (0.20, 0.44), (0.22, 0.48), and (0.24, 0.50), respectively.
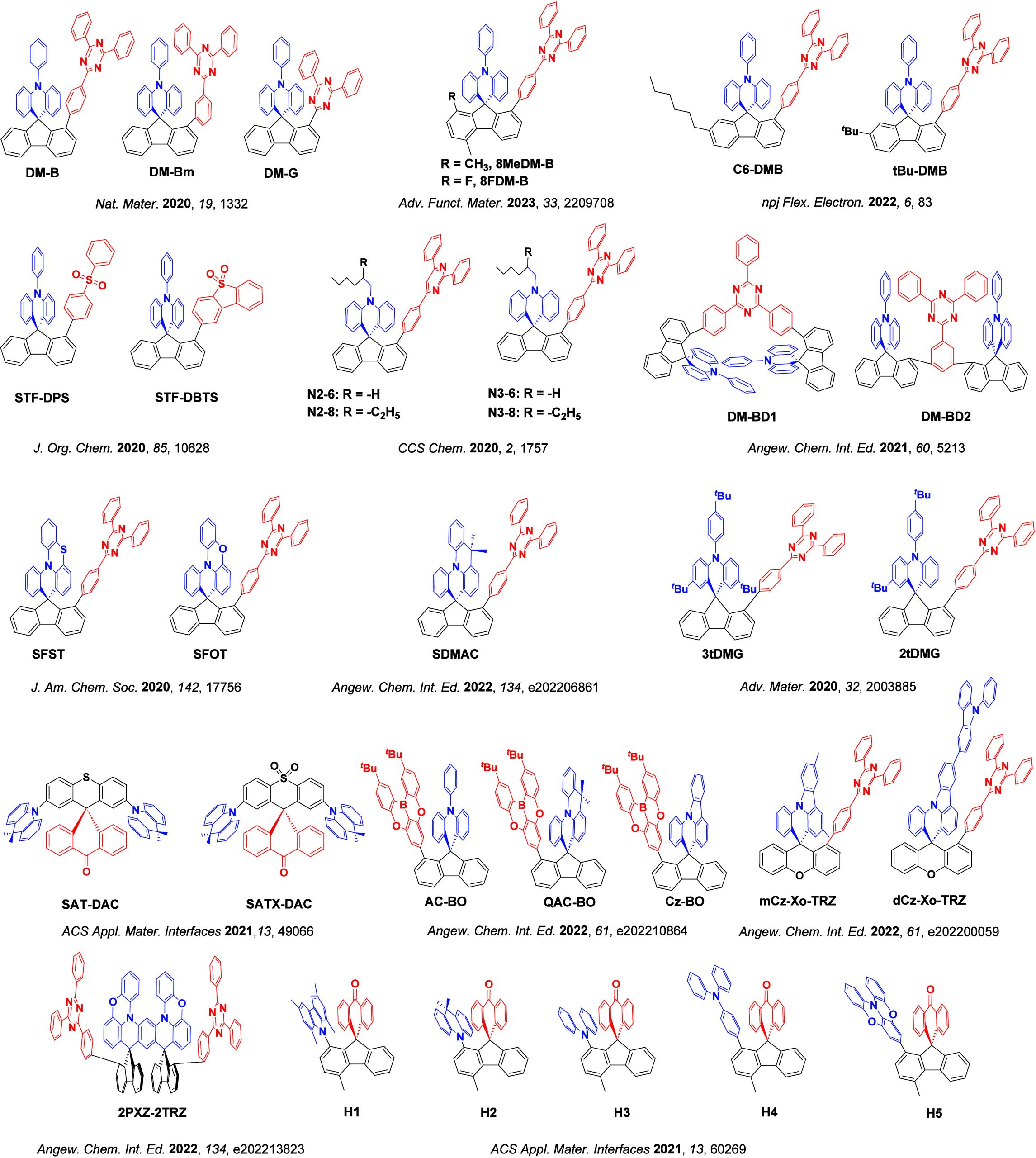
The same group subsequently reported two similar emitters, 8MeDM-B and 8FDM-B (Figure ), with methyl or fluorine groups substituted at the C8 site of the spiro-fluorene bridge.ref. ref1102 Interestingly, the presence of fluorine atoms gave stronger electrostatic repulsion than the methyl group, distorting the TPA unit away from the C8 position. Therefore, compared to 8MeDM-B, 8FDM-B has an additional interaction between the D and A groups and the acceptor adopts a more planar conformation. According to their DFT study, the HOMOs and LUMOs of both compounds are primarily located on the respective donors and acceptors, whereas there is nearly no electron density on the fluorene bridge. 8FDM-B emission is slightly red-shifted compared to 8MeDM-B (λPL of 483 and 480 nm in toluene), which was attributed to stronger charge transfer due to the shorter donor-acceptor distance. Both compounds have a ΔE ST of 0.15 eV in toluene glass at 77 K. In a 30 wt% doped PPF matrix, 8MeDM-B and 8FDM-B showed similar τd of 4.6 and 4.0 μs and very high ΦPL of 97 and 98%, respectively. The photophysical properties of 8MeDM-B and 8FDM-B supported exceptional device performance, with high EQEmax of 28.8 and 31.7%, and similar λEL of 492 and 496 nm, respectively. Both compounds exhibited similar efficiency roll-off of around 17.7% at 1000 cd m–2.
Zheng et al. introduced solubilizing tert-butyl and n-hexyl groups at the C7 position of the spiro-fluorene core to construct C6-DMB and tBu-DMB (Figure ).ref. ref1103 The solubilizing substituents had minimal influence on the photophysical properties of the parent emitter DM-B, with λPL of 447 and 446 nm and the same ΔE ST of 0.17 eV in toluene. Both emitters exhibited short τd of 5.99 and 5.58 μs, and high ΦPL of 89 and 98% in 30 wt% doped films in mCP, respectively. Solution-processed OLEDs with C6-DMB and tBu-DMB showed EQEmax of 21.0 and 21.7%, and the same CIE coordinates of (0.21, 0.38). Both devices, however, also showed severe efficiency roll-off of 60 and 63% at 1000 cd m–2.
Following this report, the same group developed dibenzothiophene sulfone as an acceptor to construct the TSCT emitter STF-DBTS (Figure ).ref. ref1104 A more conformationally flexible acceptor, diphenylsulfone, was also included in reference emitter STF-DPS. STF-DBTS emits at λPL of 460 nm, has a rather large ΔE ST of 0.27 eV in toluene, a ΦPL of 53%, and yet a τd of only 24.3 μs in 30 wt% doped films in CBP. This contrasts with STF-DPS which did not show TADF and has a low ΦPL of 16.0%, and shows a blue-shifted λPL at 441 nm in toluene. The rigid structure of STF-DBTS gave a shorter distance between the donor and acceptor (3.586 Å), which allowed for a more effective TSCT state to form than in STF-DPS, with a donor-acceptor separation of 3.752 Å. OLEDs with STF-DBTS displayed sky-blue emission at λEL of 488 nm and achieved an EQEmax of 10.3%.
Using the same spiro-fluorene scaffold, the same group developed four additional emitters containing alkyl chains of different lengths, N2-6, N3-6, N2-8, and N3-8.ref. ref1105 The different alkyl chains, n-hexyl (noted in emitter name with a −6) and 2-ethylhexyl (noted with a −8), were introduced to modulate the donor-acceptor distance and to improve the solubility of the emitters for solution-processed OLEDs. Different acceptors 2,4,6-triphenylpyrimidine (N2) and 2,4,6-triphenyl-1,3,5-triazine (N3) were also compared. The emitters N2-6 and N2-8 have slightly blue-shifted emission (λPL of 461 and 470 nm, respectively) compared with N3-6 and N3-8 (λPL of 485 and 495 nm) in toluene solution due to the weaker electron-withdrawing ability of N2 group. The ΔE ST of N2-6, N2-8, N3-6, and N3-8 are 0.27, 0.16, 0.18, and 0.14 eV respectively, revealing that shorter donor-acceptor distance can be beneficial for narrowing the ΔE ST. Also attributed to the short donor-acceptor distances, N2-8 and N3-8 have higher ΦPL (82 and 91%) than those of N2-6 and N3-6 (76 and 83%). All emitters showed very short τd: 1.01 μs for N2-6, 1.18 μs for N2-8, 1.29 μs for N3-6, and 1.50 μs for N3-8. Solution-processed OLEDs with N3-8 (λEL of 488 nm) and N2-8 (λEL of 479 nm) consequently showed superior device performances with EQEmax of 18.9 and 17.6%, relative to 14.2 and 14.7% for devices based on N2-6 (λEL of 480 nm) and N3-6 (λEL of 490 nm), respectively.
Wang et al. later refined their emitter design further, reporting sandwich type TSCT D-A-D systems DM-BD1 and DM-BD2 (Figure ).ref. ref1106 These compounds contain a multi-layer π-stacked arrangement that spatially confines the central acceptor between one or two peripheral donor groups. DM-BD1 possesses a bilayer structure with both donor groups on the same side of the acceptor, while DM-BD2 has a tri-layer structure. The congested geometry in each of the two emitters results in a short distance between the donor and acceptor units of 3.11 and 3.05 Å for DM-BD1 and DM-BD2, respectively, and the same λPL at around 495 nm in toluene. Similar ΦPL of 94.2 and 92.8% and τd of 3.1 and 2.8 μs were reported in 30 wt% doped films in DPEPO. OLEDs with DM-BD1 and DM-BD2 exhibited λEL at around 500 nm with CIE coordinates of (0.21, 0.47) and (0.20, 0.46), and achieved EQEmax of 28.0 and 26.6%, while the EQE1000 decreased to 18.9 and 15.8%, respectively.
Chiral emitters SFST and SFOT were reported by the same group using a similar spiro-skeleton containing an sp3-hybridized spiro carbon (Figure ).ref. ref1107 Sulfur and oxygen atoms were introduced into the donor to tune the photophysical and chiroptical properties (see Section sec7 ). Compared with SFOT, the larger sulfur atom in SFST resulted in enhanced SOC and led to a distortion of the molecular backbone that lengthened the donor-acceptor distance, resulting in a lower ΦPL and faster non-radiative decay. SFST and SFOT both emit at λPL of around 512 nm, have small ΔE ST of 0.052 and 0.053 eV in toluene, and ΦPL of 53.1 and 89.7% in 30 wt% doped films in mCBP, respectively. The OLEDs with SFST and SFOT both emitted at λEL of 508 nm and showed EQEmax of 12.5 and 23.1%, reflecting their differing ΦPL. The devices also showed low efficiency roll-off of 9.6 and 7.8% at 1000 cd m–2, respectively.
Yang et al. replaced the oxygen atom with a bridging Me2C group in the multi-stimulus response-active emitter SDMAC (Figure ).ref. ref1108 SDMAC exhibited aggregation-induced emission enhancement (AIEE), solvatochromism, piezochromism, and CPL under different external stimuli. SDMAC emits in the sky-blue at λPL of 468 nm and has a small ΔE ST of 0.034 eV in toluene. In 30 wt% doped films in PPF the ΦPL is 90% and the τd is 4.17 μs, leading to k RISC of 1.94×10–5 s–1. The device with SDMAC showed an EQEmax of 28.4%, with λEL of 492 nm and CIE coordinates of (0.18, 0.41). The same group reported two other derivatives using the same backbone, 2tDMG and 3tDMG (Figure ).ref. ref1109 t-Butyl groups were introduced at different positions and in different numbers to improve the emitter solubility. Both compounds emit similarly at λPL of 502 and 505 nm and have ΔE ST of 0.03 and 0.01 eV in toluene, all respectively. In 40 wt% doped films in DPEPO, 2tDMG and 3tDMG have τd of 3.43 and 2.28 μs. The OLEDs with 2tDMG and 3tDMG exhibited λEL at 504 and 518 nm and showed EQEmax of 30.8 and 26.3%, all respectively. Notably, the respective EQE1000 remained high at 28.5 and 23.2%.
Using an unconjugated spiro-anthrone backbone as the acceptor and DMAC as the donor, Wang et al. reported emitters SAT-DAC and SATX-DAC (Figure ).ref. ref470 With the ketone as the primary accepting unit both inter- (exciplex) and intramolecular (TSCT) excited states were inferred from close contacts revealed in the X-ray diffraction studies, with SAT-DCA and SATX-DAC emitting at λPL of 510 and 517 nm in toluene and having ΦPL of 76.8 and 68.1% in 10 wt% doped DPEPO films in DPEPO, respectively. In 1 wt% doped PMMA films in PMMA, both emitters have the same small ΔE ST of 0.02 eV. The OLEDs with these two emitters showed green emission with λEL of 520 and 524 nm and high EQEmax of 22.6 and 20.9%. The EQE1000 also remained high at 17.9 and 17.0%.
Zhao et al. reported three blue emitters by combining a spiro-fluorene skeleton with a boron/oxygen heterocycle acceptor (BO, aka DOBNA).ref. ref1110 To improve the rigidity of the donor unit in AC-BO, the conformation of the amine donor was locked using either with a single bond (Cz-BO) or Me2C (QAC-BO, Figure ). Thus, the minimum donor-acceptor distance could be tuned from 3.1 Å in AC-BO, to 3.0 Å in Cz-BO, and 2.6 Å in QAC-BO, which enabled progressively stronger π-orbital overlap between the donor and acceptor moieties leading to higher ΦPL. AC-BO, QAC-BO, and Cz-BO emit at λPL of 446, 428, and 411 nm, and show ΦPL of 76.9, 82.8, and 88.7%, respectively in 10 wt% doped films in PMMA (Table S16). However, the ΔE ST (in toluene) also increased from 0.13 eV in AC-BO to 0.20 and 0.29 eV for QAC-BO and Cz-BO. The largest ΔE ST of Cz-BO hindered RISC and this compound did not show TADF. AC-BO has a τd of 11.7 μs, while QAC-BO showed a surprisingly much shorter τd of 0.11 μs. Benefiting from both its high ΦPL and very short τd, QAC-BO showed a very high k RISC of 1.6 × 107 s–1 – almost two orders of magnitudes larger than for AC-BO (2.3×105 s–1). Even though the emitters exhibited impressive photophysical properties, the device with QAC-BO showed an EQEmax of only 15.8% and serious efficiency roll-off of 70% at 100 cd m–2, which likely implies that the reported τd is not accurate. The device showed emission at λEL of 448 nm and with CIE coordinates of (0.145, 0.076). As a purely fluorescent dopant, the device with Cz-BO showed an EQEmax of 5.5% at λEL of 412 nm with CIE coordinates of (0.163, 0.034). The device with AC-BO showed the highest EQEmax of 19.3% with λEL of 456 nm and CIE coordinates of (0.148, 0.122).
Based on a spiro-xanthene bridging unit, Huang et al. reported the TSCT emitters mCz-Xo-TRZ and dCz-Xo-TRZ (Figure ).ref. ref1111 In these compounds the triazine acceptor is almost perpendicular to the xanthene bridge and co-planar with the triphenylamine donor, resulting in short donor-acceptor distances in the range of 2.7 to 3.3 Å for mCz-Xo-TRZ and 2.8 to 3.3 Å for dCz-Xo-TRZ. dCz-Xo-TRZ emits at λPL of 461 nm in toluene, which is red-shifted compared to mCz-Xo-TRZ (λPL of 456 nm) due to the stronger electron-donating ability of the dCz group compared to the mCz donor. mCz-Xo-TRZ and dCz-Xo-TRZ have ΔE ST of 0.16 and 0.24 eV, with τd of 7.2 and 7.5 μs and high ΦPL of 90 and 92%, respectively in 30 wt% doped films in PPT. Devices with mCz-Xo-TRZ and dCz-Xo-TRZ showed EQEmax of 21.0 and 27.8% at λEL of 464 and 477 nm, with CIE coordinates of (0.15, 0.20) and (0.16, 0.29), all respectively. Crucially, low efficiency roll-off was observed with EQE1000 of 17.1 and 23.9% for the two devices.
Xie et al. reported the TSCT material 2PXZ-2TRZ, involving the linkage of a central biphenoxazine (2PXZ) donor and two triazine acceptors across two spiro-fluorene bridges in a so called “twin-locking” strategy (Figure ).ref. ref1112 This design efficiently suppresses intramolecular rotations and vibrations, and 2PXZ-2TRZ emits at λPL of 509 nm with a small ΔE ST of 0.01 eV as the neat film. Due to its small ΔE ST, 2PXZ-2TRZ has a τd of 5.3 μs and a high ΦPL of 94% in 30 wt% doped films in PPF. The doped and non-doped OLEDs showed EQEmax of 27.1 and 10.2%, at λEL of 508 and 518 nm with CIE coordinates of (0.26, 0.54) and (0.31, 0.58), all respectively. The EQE of the doped device decreased to 18.4% at 500 cd m–2, representing a rather sever efficiency roll-off.
Song et al. used spirofluorene-linked benzophenone as the acceptor unit and installed different donors at the C1 position of the fluorene to obtain emitters H1, H2, H3, H4, and H5 (Figure ).ref. ref1113 The single crystal X-ray structures of H1–H5 have donor-acceptor distances ranging from 3.3–3.8 Å, indicative of strong face-to-face π-π stacking interactions. The compounds emit with λPL of 493–550 nm, have ΦPL ranging from 55–92%, τd ranging from 3.3–6.8 μs, and ΔE ST all smaller than 0.07 eV. OLEDs with H1–5 showed sky-blue to yellow emission with λEL of 494, 527, 503, 507, and 550 nm and CIE coordinates of (0.20, 0.42), (0.31, 0.56), (0.22, 0.48), (0.24, 0.50), and (0.41, 0.55), and with EQEmax of 20.9, 16.1, 17.7, 20.0, and 13.2%, all respectively. The EQE1000 remained as high as 13.7, 13.5, 13.3, 15.6, and 11.7% for devices with H1–5, demonstrating the versatility of this kind of molecular design towards different donor groups.
TSCT Featuring Carbazole Bridges
Carbazole and its derivatives have somewhat similar molecular structures to fluorene and have thus similarly been used as scaffolds for TSCT emitters. Moreover, the C1, C8, and N9 positions of carbazole are chemically accessible to decorate, allowing for facile syntheses of a diverse range of targets. It should be noted though that the donating ability of carbazole can in some cases provide a competing through-bond CT state, and the single C-N linkage is significantly more vibrationally active than the spiro linkages in the previous fluorene examples.
Wu et al. linked donor and acceptor units via a carbazole bridge to construct the TSCT emitters PXZ-CTZ, DPXZ-CTZ, and DPXZ-BO (Figure ).ref. ref1114 To explore the changes in the photophysical properties as a function of donor and acceptor structure, the donor was varied from PXZ to DPXZ and the acceptor was varied from CTZ to BO moieties. Through this modification the D-A conformations were tuned from orthogonal (PXZ-CTZ) to co-facial (DPXZ-BO), leading to closer π-π stacking in DPXZ-BO and suppressing non-radiative decay. PXZ-CTZ, DPXZ-CTZ, and DPXZ-BO emit at λPL at 525, 524, and 511 nm in toluene. In 20 wt% doped films in DPEPO, these emitters have ΦPL of 55, 78, and 99%, and small ΔE ST of 0.07, −0.03, and 0.03 eV, with short τd of 3.41, 3.38 and 11.3 μs, all respectively. The apparent negative ΔE ST of DPXZ-CTZ is likely the result of spectroscopic measurements of different conformers in its fully relaxed singlet and triplet. OLEDs with PXZ-CTZ, DPXZ-CTZ, and DPXZ-BO showed similar green emission at λEL of ca. 528, 530, and 537 nm with CIE coordinates of (0.33, 0.56), (0.39, 0.57) and (0.26, 0.58), and EQEmax of 16.6, 19.7, and 24.0%, all respectively. For DPXZ-BO the EQE1000 remained above 20%, showing a small efficiency roll-off of 16%.
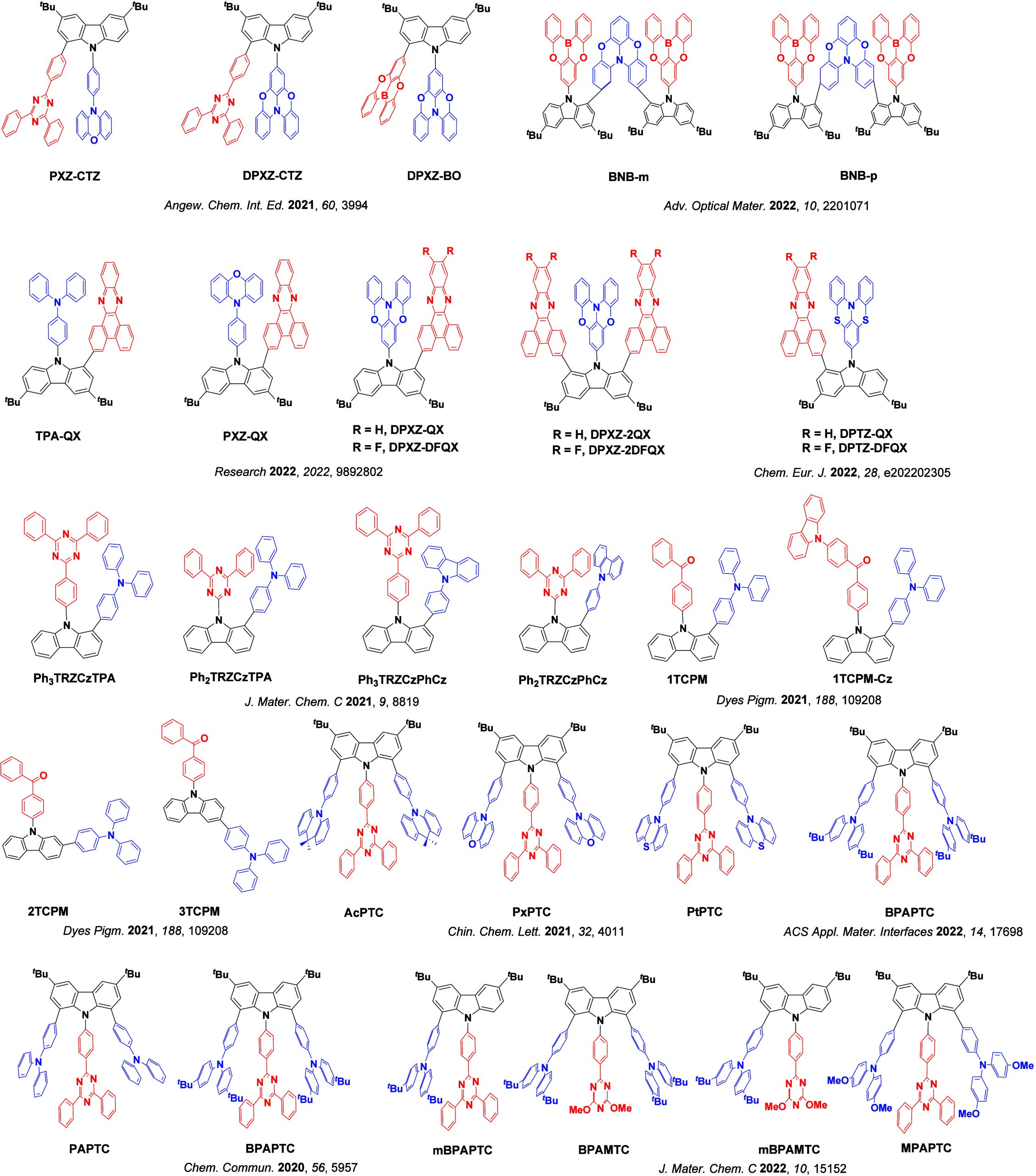
Using a similar strategy Wang et al. reported two sandwich-type derivatives, BNB-m and BNB-p (Figure ), containing a planar DPXZ donor connected through carbazole groups to two BO acceptors at either the meta– or para– position of DPXZ unit.ref. ref1115 BNB-m and BNB-p emit at λPL of 502 and 518 nm and have high ΦPL of 100 and 86%, respectively in 10 wt% doped films in mCP. The ΔE ST are 0.03 and 0.11 eV, with τd of 11.2 and 25.4 μs and k RISC of 16.0 and 9.88×104 s–1, also respectively. The OLED with BNB-m showed green emission at λEL of 508 nm and CIE coordinates of (0.23, 0.54), and an outstanding EQEmax of 34.9% and EQE1000 of 27.4%.
The same group also used carbazole as a bridge to investigate a combinatorial series of TSCT emitters featuring three new donor and two new acceptor groups (TPA-QX, PXZ-QX, DPXZ-QX, DPXZ-DFQX, DPXZ-2QX, and DPXZ-2DFQX, Figure ).ref. ref1116 By increasing the electron-donating ability of the donor unit, the emission could be red-shifted from 535 nm for TPA-QX to 582 nm for DPXZ-QX (in 5 wt% doped films in mCP), which was accompanied by an increase in the ΦPL from 44 to 74% and a decrease in the ΔE ST from 0.38 to 0.01 eV, therefore leading to a shorter τd of 6.8 μs for TPA-QX compared to 26.9 μs for DPXZ-QX. Modulation of the acceptor strength likewise increased the k RISC to 4.33×105 s–1 for DPXZ-DFQX compared to 1.86×105 s–1 for DPXZ-QX. With a goal to further accelerating k RISC, sandwich A-D-A structures DPXZ-2QX and DPXZ-2DFQX were synthesized. These two compounds emit at λPL of 594 and 599 nm, have ΦPL of 87 and 91%, and have τd of 8.7 and 4.9 μs correlated to their ΔE ST of 0.02 and −0.05 eV, all respectively, leading to fast k RISC of 8.21 and 4.64 × 105 s–1; again, the apparent negative ΔE ST is likely a reflection of not accurately measuring the phosphorescence energy, where at 77 K delayed fluorescence may still also exist. The OLEDs with DPXZ-QX, DPXZ-DFQX, DPXZ-2QX, and DPXZ-2DFQX emitted at λEL of 597, 602, 609, and 616 nm, and the device with DPXZ-2QX showed the best performance with an EQEmax of 23.2% (6 wt% doped in mCBP matrix). When the emitter concentration was increased to 12 wt% the EQEmax was maintained at a comparable value of 21.1%, and these devices retained a higher EQE1000 of 19.9%, compared to 14.4% for the 6 wt% device; however, the higher doping was accompanied by a red-shifted λEL of 616 nm and CIE coordinates of (0.60, 0.39).
The same group also investigated the impact of the addition of heavy atoms on TSCT-TADF properties by substituting the oxygen atom in the DPXZ donor with sulfur in DPTZ-QX and DPTZ-DFQX (Figure ).ref. ref1117 In 5 wt% mCP, DPTZ-DFQX and DPTZ-QX emit at λPL of 565 and 561 nm, which are blue-shifted relative to their PXZ analogues. Simultaneously, the T1 states became more LE in nature, inducing larger ΔE ST of 0.14 and 0.15 eV respectively. Consequently, longer τd of 255.0 and 114.3 μs and lower ΦPL of 49 and 61% were observed for DPTZ-QX and DPTZ-DFQX, compared with τd of 26.9 and 6.8 μs, and ΦPL of 74 and 71% for the previously reported DPXZ-QX and DPXZ-DFQX,ref. ref1116 all respectively. This work emphasizes the importance of fully considering the multifaceted influences of heavy atoms on TSCT excited states and RISC.
Using the same carbazole bridge, Miranda-Salinas et al. reported four TSCT emitters using triphenylamine and phenylcarbazole donors and TRZ as the acceptor.ref. ref1118 By increasing the electron-donating strength of the donor groups, the dominant CT state was tuned from through-bond between the carbazole bridge and the TRZ acceptor to through-space between the co-facially aligned decorated donor and acceptor groups. Compounds Ph3TRZCzTPA and Ph2TRZCzTPA (Figure ) showed onsets of their respectively emission spectra at 2.89 and 2.79 eV, and have small ΔE ST of 90 and −130 meV in 10 wt% doped films in DPEPO; again, the apparent negative ΔE ST likely reflects that the S1 and T1 energies were measured for different species as there is no photophysical reason for inverted singlet-triplet gaps in this class of material. The devices with Ph3TRZCzTPA and Ph2TRZCzTPA showed green emissions at λEL of 522 and 529 nm and EQEmax of 13.3 and 16.3%, respectively, while the QE1000 decreased to 9.7 and 10.9%.
Ma et al. reported four emitters containing carbazole bridges, but with substituents connected at different positions to permit fine-tuning of the CT interaction from through-bond to through-space.ref. ref1119 1TCPM, 2TCPM, and 1TCPM-Cz all show dual-emissions at 411/510 nm, 425/464 nm and 411/521 nm, while 3TCPM emits at λPL of 478 nm in toluene. As neat films the ΔE ST of 1TCPM (0.02 eV) and 1TCPM-Cz (0.04 eV) are much smaller than 3TCPM (0.26 eV) and 2TCPM (0.37 eV, Figure ), arising from the greater separation of electron density on the donor and acceptor groups. Restricted intramolecular motion in 1TCPM and 1TCPM-Cz suppresses the non-radiative decay pathways, resulting in higher ΦPL of 50 and 57% compared to 35 and 20% for 3TCPM and 2TCPM in neat film, respectively. All emitters have short τd of 2.1, 2.3, 1.7, and 1.6 μs for 3TCPM, 2TCPM, 1TCPM, and 1TCPM-Cz, respectively. The devices with 3TCPM, 2TCPM, 1TCPM, and 1TCPM-Cz showed EQEmax of 3.4, 1.8, 7.6, and 13.3% at λEL of 478, 471, 489, and 505 nm, respectively.
Li et al. reported a series of D-A-D sandwich TSCT emitters by employing carbazole as the bridge and decorating different donors on either side of a central TRZ acceptor, giving the compounds AcPTC, PxPTC, and PtPTC (Figure ).ref. ref1120 DFT calculations and single crystal X-ray diffraction analysis revealed that the three emitters all showed clear edge-to-face π-π interactions. By increasing the electron donating ability of the donors, the emission is red-shifted from 485 nm for AcPTC to 522 nm for PxPTC and 561 nm for PtPTC, all in 20 wt% doped films in SimCP2. AcPTC, PxPTC, and PtPTC have high ΦPL of 73, 61, and 51%, small ΔE ST of 0.05, 0.03, and 0.03 eV, along with τd of 10.5, 3.0, and 11.4 μs, all respectively. The devices with AcPTC, PxPTC, and PtPTC achieved EQEmax of 10.0, 11.0, and 5.6% at λEL of 483, 533, and 564 nm, respectively. Using the same sandwich D-A-D strategy the same group also investigated the use of triphenylamine or 4,4′-di-(tert-butyl)triphenylamine as the donors in PAPTC and BPAPTC (Figure ).ref. ref406 The introduction of the tert-butyl groups shortened the donor-acceptor distance to 3.081 from 3.139 Å, leading to improved TADF properties. In 20 wt% doped films in SimCP2 BPAPTC showed a red-shifted emission at 519 nm, a higher ΦPL of 90%, and similar ΔE ST of 0.06 eV and τd of 0.62 μs compared with PAPTC (λPL at 509 nm, ΦPL of 78%, ΔE ST of 0.07 eV, and τd of 0.61 μs). The solution-processed devices with PAPTC and BPAPTC showed EQEmax of 17.4 and 24.3% at identical λEL of 520 nm. Notably, the device with PAPTC still retained an EQE of 11.6% at 3000 cd m–2, and the device with BPAPTC retained 19.8% at 3000 cd m–2 and even 13.7% at 10000 cd m–2. The vastly superior performance of the device with BPAPTC results from the slight difference in the molecular structure instilled by the tert-butyl group.
To compare the sandwich D-A-D concept to equivalent D-A ‘open sandwich’ materials, the same group reported another set of emitters: mBPAPTC, BPAMTC, mBPAMTC, and MPAPTC (Figure ).ref. ref1121 The D-A-D sandwich compounds MPAPTC and BPAMTC showed slightly red-shifted emission profiles with λPL of 546 and 492 nm, compared to the open sandwich analogues with λPL of 510 and 491 nm for mBPAPTC and mBPAMTC, all in respective 20 wt% doped films in SimCP2. From an analysis of these λPL values it is evident that introduction of methoxy groups significantly red-shifts the emission. The ΔE ST values range from 0.002 to 0.13 eV, which are all sufficiently small to support TADF. The ΦPL are 90% for BPAPTC (a relevant structure from the previous examples) and 90% for mBPAPTC, 63% for BPAMTC, 69% for mBPAMTC, and 44% for MPAPTC. The ΦPL of BPAPTC/BPAMTC D-A-D sandwich compounds are therefore higher than those of the corresponding open sandwich emitters mBPAPTC/mBPAMTC, revealing useful practical design rules for this class of emitter. The origin of the higher ΦPL was attributed to their shorter π-π interaction distances and more rigid structures. The OLEDs with BPAPTC, mBPAPTC, BPAMTC, mBPAMTC, and MPAPTC emitted at λEL of 520, 520, 486, 484, and 564 nm, respectively. The devices with BPAPTC and BPAMTC showed higher EQEmax of 23.3 and 14.7%, compared to devices with mBPAPTC and mBPAMTC possessing EQEmax of 17.8 and 9.5% respectively, which were correlated with the higher ΦPL of the former. The device with MPAPTC showed a lower EQEmax of 9.1%, while the EQE of the devices with BPAPTC, mBPAPTC, BPAMTC, mBPAMTC, and MPAPTC decreased to 20.4, 13.2, 8.8, 5.3 and 6.2% at 1000 cd m–2.
TSCT featuring other bridges
In some of these TSCT materials the lowest-energy excited state can be described as a combination of both TSCT and through-bond CT (TBCT), as the bridging unit itself can be directly involved in the electronic transitions. In parallel, as TSCT becomes more deeply understood over time, examples of TBCT materials can sometimes be ‘rediscovered’ as having TSCT character.ref. ref474 As an illustrative example of this evolving understanding, Chen et al. reported B-oCz and B-oTC (Figure ).ref. ref1122 These two emitters have either a carbazole or a tert-butylcarbazole donor group that is ortho-disposed to an aryl boron acceptor. Such a structure may simply be assumed to be a TBCT emitter, although this structure also arranges the donor and acceptor in a co-facial array. Indeed, the crystal structures revealed a short intramolecular donor-acceptor distance of 2.76–3.55 Å.ref. ref119 B-oCz and B-oTC emit at λPL of 465 and 476 nm and have ΔE ST of 0.06 and 0.05 eV as neat films, respectively. The ΦPL of B-oCz is 61% as the neat film but significantly increases to 94% when the Cz donor is replaced with the sterically bulkier tert-butylcarbazole in B-oTC. Although this substitution does impact the donor strength, it is implausible for this alone to result in such large changes in ΦPL. The higher ΦPL for B-oTC was instead attributed to the increased steric bulk of the donor that inhibits both intermolecular and intramolecular π–π stacking, favoring the TBCT excited state. The solution-processed non-doped OLEDs with B-oCz and B-oTC showed blue emission with λEL of 463 and 474 nm and CIE coordinates of (0.15, 0.17) and (0.50, 0.26), and EQEmax of 8.0 and 19.1% respectively. However, these two devices exhibited serious efficiency roll-off with low EQE1000 of only 2.6 and 9.7%.
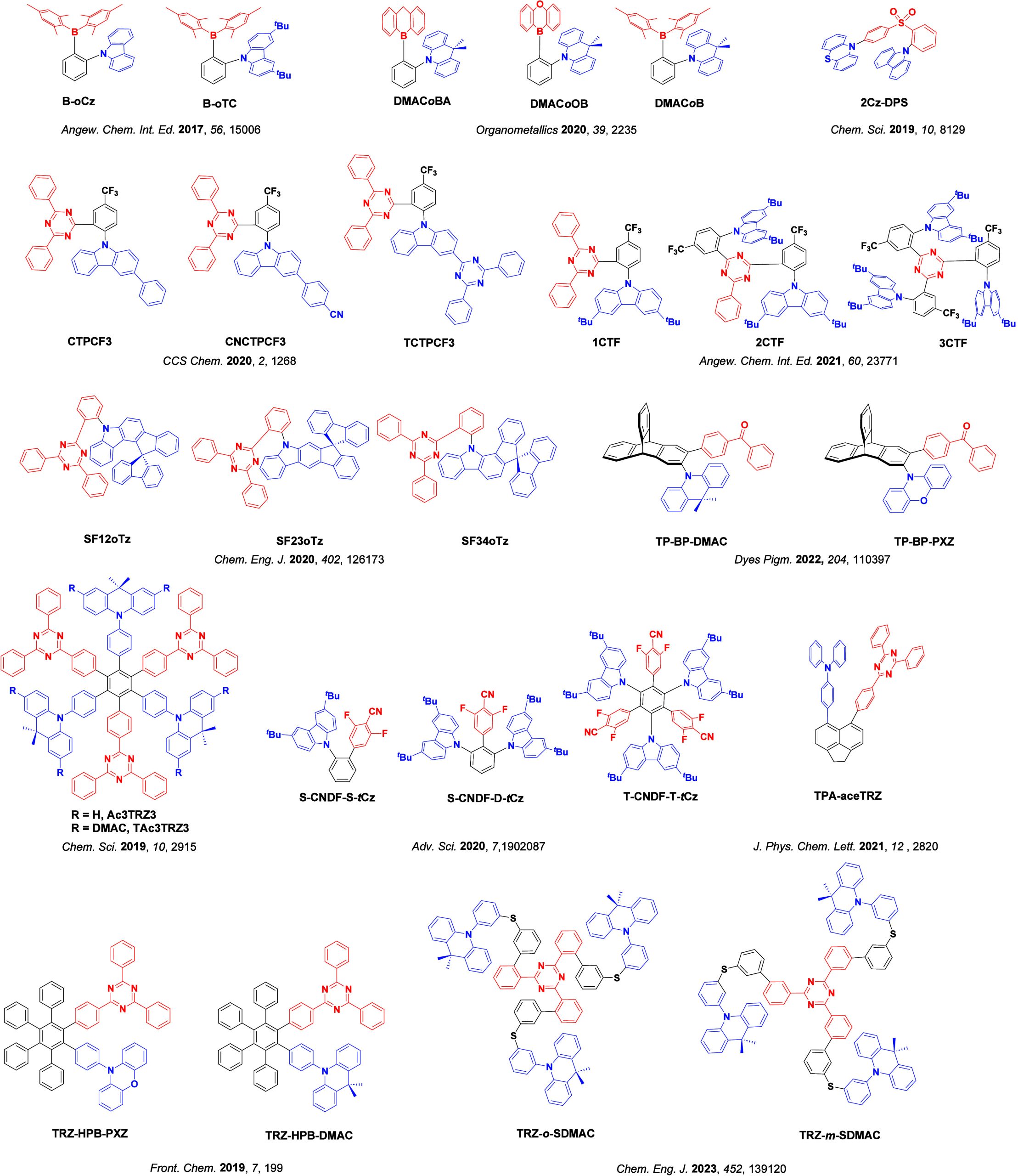
Kim et al. reported similar ortho-D-A compounds containing different boryl acceptors: DMACoBA, DMACoOB, and DMACoB (Figure ).ref. ref1123 Compared with DMACoB, DMACoBA and DMACoOB exhibited weaker N–B and C–H·π non-bonding interactions between the DMAC donor and the boryl moieties. Due to the planar structure of the cyclic boryl acceptors of DMACoBA and DMACoOB, these fragments are orientated perpendicular relative to the phenylene ring in these compounds, which leads to an excited state of almost purely TSCT character. The boryl plane in DMACoB is instead tilted relative to the phenyl ring, which according to the authors leads to mixed TSCT and TBCT character. Compared to the blue emission of DMACoBA and DMACoOB (λPL of 488 and 481 nm, Table S16), DMACoB hence shows red-shift emission at λPL of 518 nm, and all three emitters show high ΦPL of 100% in 10 wt% doped films in PMMA. DMACoBA, DMACoOB, and DMACoB have τd of 15.1, 10.8, and 8.5 μs in the same films, while their respective ΔE ST are 0.014, 0.004, and 0.001 eV in toluene at 77 K. No devices were fabricated with these emitters.
Yang et al. reported a derivative of PTZ-DPS that places a carbazole donor at the ortho position of sulfone acceptor, 2Cz-DPS (Figure ).ref. ref1124 Through this approach, both TSCT (with the carbazole) and TBCT (with the PTZ) excited states co-exist. The steric congestion resulting from the ortho-carbazole donor also leads to reduced molecular motion and suppressed non-radiative decay pathways. 2Cz-DPS emits in the green at λPL of 520 nm as the neat film, implying an excited state of mainly TBCT character (as the TSCT state would be expected to be higher in energy by comparison to the previously reported of dtCzDPS).ref. ref230 2Cz-DPS has a large ΔE ST of 0.32 eV and a high ΦPL of 91.9% as a neat film. Even with this large ΔE ST, the emission decays with τp and τd of 4.4 ns and 19.1 μs. The 2Cz-DPS-based non-doped OLED emitted at λEL of 518 nm and showed an EQEmax of 28.7%, with EQE300 decreased significantly to 8.4%.
Duan and co-workers reported three TSCT emitters with ortho-disposed Cz and TRZ units attached to a (trifluoromethyl)benzene linker.ref. ref1125 The dipole moments are reduced with the addition of electron-deficient substituents on the donor group. Through this strategy, emitters possessing combined TBCT and TSCT character were developed; however, owing to the highly twisted conformation between donor and acceptor groups the TSCT state often becomes dominant. CTPCF3, CNCTPCF3, and TCTPCF3 (Figure ) all have donor-acceptor distances sufficiently short to enable TSCT (2.85–3.81 Å), and emit at λPL of 494, 475, and 468 nm, respectively. The trends in the emission spectra can be attributed to the increasing strength of the electron-withdrawing groups on the donor, weakening the overall electron-donating ability of the carbazole. The ΔE ST are 0.04, 0.28, and 0.06 eV for CTPCF3, CNCTPCF3, and TCTPCF3, respectively, where the larger ΔE ST of CNCTPCF3 was attributed to the increased LE character of its triplet excited state. CTPCF3, CNCTPCF3, and TCTPCF3 have τd of 3.21, 6.04, and 2.52 μs (Table S16) that mirror the trend in ΔE ST, and have high ΦPL of 96, 67, and 65%, suggesting that the twisted structure can effectively suppress concentration-quenching effects. Benefitting from the efficient TADF of these three emitters, HF devices employing CTPCF3, CNCTPCF3, and TCTPCF3 as sensitizers and 2F-BN as the MR-TADF terminal emitter showed green emission at λEL of 495, 497, and 495 nm, and high EQEmax of 33.1, 25.6, and 23.2%, respectively.ref. ref1126 The CTPCF3 based device showed modest efficiency roll-off, with EQE1000 remaining at 27%.
The same group developed three other related emitters with different numbers of tert-butylcarbazole groups as donors and TRZ as the acceptor, 1CTF, 2CTF, and 3CTF (Figure ).ref. ref1127 A secondary trifluoromethyl (CF3) acceptor group was also incorporated to modulate the contributions from the TSCT and TBCT states. Benefiting from the steric clash of the Cz-donor perpendicularly linked to the acceptor plane, 2CTF and 3CTF both show face-to-face donor-acceptor interactions. Only edge-to-face donor-acceptor interactions were observed for 1CTF as a result of the less crowded steric environment. 1CTF, 2CTF, and 3CTF emit at λPL of 500, 507, and 514 nm in toluene and have near unity ΦPL of 99, 98, and 99%, small ΔE ST of 0.03, 0.03, and 0.04 eV, and short τd of 2.4, 1.8, and 1.2 μs, all respectively. Devices with 1CTF, 2CTF, and 3CTF showed good EQEmax of 17.5, 19.8, and 22.6% at λEL of 490, 503, and 508 nm with CIE coordinates of (0.23, 0.45), (0.26, 0.54), and (0.29, 0.57), all respectively. The efficiency roll-off of the devices was modest, with EQE1000 of 14.5, 17.6, and 21.0% for the devices with 1CTF, 2CTF, and 3CTF.
Lv et al. reported three emitters, SF12oTz, SF23oTz, and SF34oTz, consisting of spiro-fluorene-fused carbazole donors attached a the ortho-position of TRZ acceptors (Figure ).ref. ref1128 By changing the position of the fused fluorene, the molecular geometry and subsequent ratio of TBCT/TSCT character for each molecule could be modulated. DFT calculations predicted that SF34oTz has dominant TSCT character (96.8%), whereas SF23oTz and SF12oTz contain mixed TBCT and TSCT character, with the ratio of TBCT increasing from 21 to 32% in the latter. Due to the presence of a stronger donor, SF34oTz has the most red-shifted emission with λPL of 479 nm in toluene, whereas SF23oTz and SF12oTz exhibit dual-emission in toluene with respective λPL of 383/473 and 371/491 nm; the higher energy λPL at 383/371 nm arises from LE emission of the donors. The ΔE ST values in 20 wt% doped films in DPEPO are 0.29 eV for SF34oTz, 0.08 eV for SF23oTz, and 0.05 eV for SF12oTz, leading to τd of 8.2, 4.3, and 4.6 μs for the same. High ΦPL of 92 and 86% were observed for SF12oTz and SF23oTz, respectively, while SF34oTz has a lower ΦPL of 65%. The resulting solution-processed green OLEDs with SF12oTz, SF23oTz, and SF34oTz exhibited EQEmax of 22.4, 19.6, and 14.6% at λEL of 496, 484, and 482 nm, all respectively. The devices with SF12oTz and SF23oTz showed minimal efficiency roll-off, with EQE1000 of 20.0 and 15.9%, respectively. Due to the long delayed lifetimes, the device with SF34oTz showed more serious efficiency roll-off with an EQE1000 of 3.1%.
Huang et al. reported two other emitters, TP-BP-DMAC and TP-BP-PXZ (Figure ), in which a benzophenone acceptor and DMAC or PXZ donor units are attached at adjacent positions on a triptycene bridge.ref. ref1129 The ortho-linkage of the donor and acceptor leads to face-to-face alignment and strong intramolecular donor-acceptor interactions. The non-planar triptycene scaffold was chosen to limit concentration-related aggregation and quenching, and to improve film quality. TP-BP-DMAC and TP-BP-PXZ emit at λPL of 508 and 531 nm in 20 wt% doped films in DPEPO, respectively. The RISC activation energies, determined using an Arrhenius analysis of the variable-temperature time-resolved PL decays, are 6.7 and 10.8 meV, while the ΦPL of TP-BP-DMAC and TP-BP-PXZ are 80 and 40%, all respectively. OLEDs with TP-BP-DMAC and TP-BP-PXZ showed EQEmax of 20.5 and 13.8%, which remained as high as 9.6 and 9.3% at 1000 cd m–2, while the EL spectra are consistent with the λPL at λEL of 488 and 531 nm with CIE coordinates of (0.21, 0.38) and (0.35, 0.53), all respectively.
The strategy of attaching donor and acceptor units ortho to each other has also been expanded with the use of hexaphenylbenzene scaffolds (HPB). HPB has a non-planar propeller shaped structure, where peripheral groups sit orthogonal to the central benzene due to steric constraints. This conformation forces the peripheral donor and acceptor groups to adopt co-facial arrangements, which in turn enables TSCT states. Two examples of this design strategy are the emitters Ac3Trz3 and TAc3Trz3 (Figure ), which emit at λPL of 505 and 535 nm, respectively in 10 wt% doped films in AC-6 as the host.ref. ref1130 Both materials have small ΔE ST of 0.08 and 0.04 eV and moderate ΦPL of 54 and 63%, respectively. The resulting solution-processed green OLEDs with Ac3Trz3 and TAc3Trz3 showed EQEmax of 11.0 and 14.2% at λEL of 520 and 538 nm, while the EQE100 remained at 10.4 and 13.5%, all respectively.
Zheng et al. employed a similar design strategy, reporting a series of emitters that build step-wise to the fully substituted HPB: S-CNDF-S-tCz, S-CNDF-D-tCz, and T-CNDF-T-tCz (Figure ).ref. ref327 This multi-chromophore approach was claimed by the authors to increase k RISC by exploiting the presence of degenerate triplet states that form on the different donors and acceptors. The emitter T-CNDF-T-tCz contains three donor and three acceptor units and emits in the sky-blue at λPL of 472 nm and has the smallest ΔE ST of 0.03 eV of the series of compounds studied (Table S16), a high ΦPL of 76%, and k RISC of 5.07 ± 0.65 × 105 s–1 as the neat film. Non-doped OLEDs with T-CNDF-T-tCz showed an EQEmax of 21% at λEL of 466 nm.
Using the same HPB scaffold to bridge triazine to different donors (acridine and phenoxazine), Tang and co-workers reported the two emitters TRZ-HPB-PXZ and TRZ-HPB-DMAC (Figure ).ref. ref1131 These two compounds emit at λPL of 576 and 484 nm, reflective of the relative strength of the donor group, have ΦPL of 61.5 and 51.8%, and small ΔE ST of 0.02 and 0.09 eV, all respectively as neat films. The non-doped devices with TRZ-HPB-PXZ and TRZ-HPB-DMAC showed EQEmax of 12.7 and 6.5%, which decreased to 12.3 and 6.0% at 1000 cd m–2. The device with TRZ-HPB-PXZ and TRZ-HPB-DMAC showed λEL of 544 and 521 nm and CIE coordinates of (0.39, 0.57) and (0.28, 0.58), all respectively. These results indicate that the HBP-based TSCT emitters can effectively suppress exciton annihilation processes by inhibiting aggregation.
Li et al. reported a series of propeller-shaped isomers with a triazine acceptor and three donor units linked via diphenylsulfides.ref. ref1132 Highlighting two of these compounds, TRZ-o-SDMAC and TRZ-m-SDMAC (Figure ) emit at λPL of 496 and 499 nm and both have small ΔE ST of 0.01 eV as neat films. TRZ-m-SDMAC has a ΦPL of 52% while that of TRZ-o-SDMAC is much lower at ΦPL of 13%, likely due increased non-radiative decay processes arising from the donors being connected meta to the triazine. Devices with TRZ-m-SDMAC exhibited blue-green emission at λPL of 510 nm with CIE coordinates of (0.24, 0.49) and an EQEmax of 20.3% but with a very large efficiency roll-off of 78.5% at 1000 cd m–2. The TRZ-o-SDMAC device showed inferior EQEmax of only 1.1% at λEL of 518 nm with CIE coordinates of (0.30, 0.47).
Zysman-Colman, Monkman, and co-workers have also used acenaphthene as a scaffold, employing TPA as a donor and TRZ as an acceptor in the emitter TPA-ace-TRZ (Figure ).ref. ref1133 The structure of TPA-ace-TRZ places the donor and acceptor highly coplanar and at quite short distances compared to other examples in this section. The spectroscopic study evidenced conclusively the presence of both TSCT and TBCT states, while the TSCT interaction is frequently only inferred from a combination of DFT calculations and structural information derived from X-ray structure analysis in other works. TPA-ace-TRZ emits at λPL of 518 nm and has a ΦPL of only 17% in toluene.ref. ref1133 In 1 wt% zeonex film TPA-ace-TRZ emits at λPL of 505 nm but has a large ΔE ST of 0.48 eV and low ΦPL of only 12%. No delayed emission lifetime was observed due to the large ΔE ST.
TADF and CT States Featuring Homoconjugation
Somewhat distinct from both TBCT and TSCT states, in homoconjugated systems the donor and acceptor moieties are connected via a bridge where the electronic coupling is mediated by co-aligned sigma bonds, while the distances between these fragments are too large to mediate direct TSCT interactions via their π-network. Triptycene is a specific bridge that permits this type of homoconjugation to occur and this strategy was first explored by Swager and co-workers in the compounds TPA-QNX(CN)2 and TPA-PRZ(CN)2 (Figure ).ref. ref1134 In these materials the triphenylamine donor and the dicyanoquinoxaline or dicyanopyrazine acceptor units are fixed at 120° relative to one another across the three arms of the bridge. The homoconjugated CT excited states resulted in predicted ΔE ST of 0.11 and 0.08 eV, respectively. TPA-QNX(CN)2 emits at λPL of 487 nm, has a moderate ΦPL of 44%, and a τd of 2.4 μs in cyclohexane. The OLEDs showed a significantly red-shifted emission at λEL = 573 nm and CIE coordinates of (0.45, 0.54), but nonetheless showed an EQEmax of 9.4% (10 wt% doped in mCP). The large red-shift was ascribed by the authors to the sensitivity of the CT state to the polarizability of the surrounding medium.
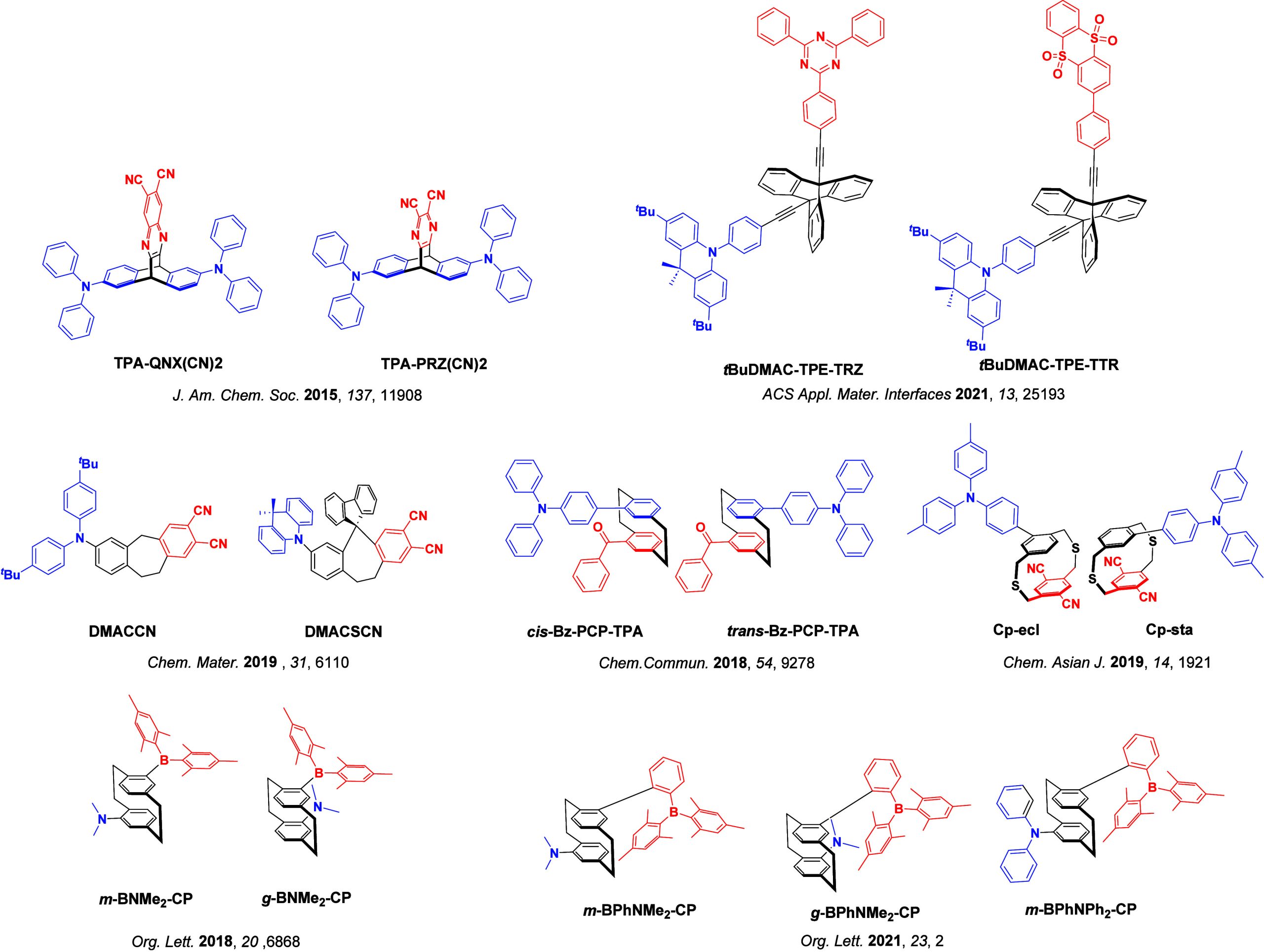
Zhang and co-workers reported the emitters tBuDMAC-TPE-TRZ and tBuDMAC-TPE-TTR (Figure ), where the donor and acceptor units were also separated with a triptycene bridge.ref. ref1135 Uniquely though, the donors and acceptors are positioned more remote from the triptycene, with the donor separated by an ethynyl bridge to the bridgehead carbon of the triptycene, and the acceptor attached to one of the arms. tBuDMAC-TPE-TRZ emits at 500 nm and has a ΦPL of 43.7% (Table S16). Using transient PL measurements at different concentrations in PMMA the authors demonstrated that intra and intermolecular CT channels both play roles in the emission process. The non-doped devices with tBuDMAC-TPE-TRZ and tBuDMAC-TPE-TTR showed green and red emission at λEL of 532 and 600 nm and showed EQEmax of 10.0 and 1.3%, respectively.
Yersin et al. bridged a TPA donor and a dicyanobenzene acceptor through a non-conjugated alkyl spacer in the compound DMACCN (originally named 1, renamed here for clarity, Figure ), and also reported a derivative that contains a spiro-fluorene between donor and acceptor groups to mediate a TSCT interaction in DMACSCN (originally named 2).ref. ref1136 DMACCN and DMACSCN have very small calculated ΔE ST of 6 and 2 meV, respectively. DMACCN emits at λPL of 476 nm and has a τd of 9 μs in toluene solution while DMACSCN emits at λPL of 468 nm, has a ΦPL of 65%, and a very short τd of 420 ns. The authors claimed that the introduction of a plurality of high lying states for coupling resulted in the apparent disappearance of any long-lived TADF due to the very fast RISC between the pseudo-degenerate 1CT and 3CT states.
Spuling et al. explored intramolecular TSCT using a [2.2]paracyclophane (PCP) bridging unit (Figure ).ref. ref1137 The reduced Van der Waals distance of 3.09 Å between the the two benzenes of the PCP is sufficiently small to mediate electronic communication between the donor and acceptor groups positioned on the benzene rings. The structure of the cis-linked (pseudo geminal) cis-Bz-PCP-TPA, or the trans-linked (pseudo anti) trans-Bz-PCP-TPA has a significant impact on the optical properties. Cis-Bz-PCP-TPA and trans-Bz-PCP-TPA exhibited blue emission in solution (two peaks of 404/492 and 404/455 nm in toluene, respectively), while the 15 wt% doped films in mCP emit λPL of 480 and 465 nm and have ΔE ST of 0.13 and 0.17 eV (Table S16), leading to small τd of 1.8 and 3.6 μs, all respectively. Unfortunately, the ΦPL of these emitters remained quite low in the solid state (12% for cis-Bz-PCP-TPA and 15% for trans-Bz-PCP-TPA in 15 wt% mCP film), and thus OLEDs were not fabricated. Adachi and co-workers reported emitters using the related dithia[3.3]paracyclophane bridging moiety, Cp-ecl and Cp-sta (Figure ).ref. ref1138 Cp-ecl and Cp-sta each emit at λPL of ∼520 nm and have ΦPL of 61 and 2%, with ΔE ST of 0.03 and 0.05 eV, all respectively.
Zhang et al. reported a series of structurally similar chiral green TADF molecules containing PCP bridging units, g-BNMe2-Cp and m-BNMe2-Cp (Figure ). These emit at λPL of 531 (with ΦPL = 72% in cyclohexane, ΔE ST = 0.17 eV, and τd = 0.38 ms in toluene) and at 521 nm (with ΦPL = 39% in cyclohexane, ΔE ST = 0.12 eV, and τd = 0.22 ms in toluene), respectively.ref. ref638 Recently the same group introduced a phenylene spacer between the PCP and the acceptor moiety to obtain sky-blue emitters showing an enhanced ΦPL in cyclohexane of 83% for g-BPhNMe2-Cp (λPL = 488 m), 93% for m-BPhNMe2-Cp (λPL = 461 nm), and 82% for g-BPhNPh2-Cp (λPL = 455 nm).ref. ref1139 To our knowledge there are not yet any reports of efficient OLEDs using PCP bridged TADF materials, widely stymied by low ΦPL.
Outlook
This section offers a comprehensive overview of TSCT TADF materials, providing an in-depth analysis of optoelectronic properties and their performance as emitters in OLEDs. The field of TSCT TADF design has witnessed significant advancements since its initial report by Tsujimoto et al. in 2017,ref. ref1098 marked by the development of emitters with near-unity ΦPL, and with examples covering the entire visible spectrum.
Triazine, which is frequently used in other classes of TADF compounds, stands out as the most commonly used acceptor in the TSCT donor-acceptor motif. This preference is due to its planar geometry, readily forming co-facial or tilted co-facial interactions with the donor moiety. To date, the acceptor triazine has showcased its versatility in creating high-efficiency emitters on diverse backbones from non-conjugated bridges like xanthene and triptycene to conjugated counterparts like carbazole and spirofluorene. In these reported examples, fine-tuning the donor and acceptor structures has been instrumental in exploring and optimizing the CT strength between them. Differing from this design, Kaji and coworkers have delved into the impact of the distance and orientation between the donor and acceptor units on emitter performance, employing DMAC and triazine as the donor and the acceptor, respectively, attached to a triptycene bridge. This work provides a clue as to how the alignment of the 3LE state relative to the 3CT and 1CT states affects the RISC rate. However, it is worth noting that, similar to the conventional donor-acceptor TADF design, due to their long-range CT nature, TSCT emitters seem unavoidably to show broad emission, posing challenges in terms of the color purity of the device. Therefore, a promising avenue for future exploration lies in improving color purity, by supressing molecular vibration and possibly by incorporation of emissive excited state of SRCT character, like the strategies employed in MR-TADF emitter design.
Spirofluorene and carbazole by far have been used as the most popular backbones to anchor the electron donor and acceptor units, with the aim of achieving efficient TSCT. These advances have pushed the EQEmax of the devices beyond the theoretical value of 25–30%. For example, Wang et al.ref. ref1115 demonstrated that the devices featuring sandwich-like emitters, BNB-m and BNB-p, achieving an impressive EQEmax of approximately 35%. Despite these remarkable achievements, there remains a need for research that explores the impact of backbone rigidity and stability on device performance, particularly in terms of device roll-off, color purity, and operational lifetime, particularly as most TSCT TADF emitter reports focus on decorating donors and acceptors with the objective to improve device efficiency.
In many of the examples presented here, the emissive excited states possess mixed TBCT/TSCT character; further, it is difficult to spectroscopically disentangle the contributions, if any, from these two excited states. In forming these states, the magnitude of the electronic coupling between donor and acceptor moieties in TSCT TADF compounds is mediated not only by the distance between the two but also their relative orientation, both of which are modulated by the choice of bridging scaffold. As one of the most recently popularized classes of TADF emitters, it is particularly exciting to imagine the novel and innovative molecular designs that will arise in this area in the coming years.
Compounds Displaying Both Aggregation-Induced Emission (AIE) and TADF
Introduction
One of the main challenges in luminophore design is their propensity to form aggregates, both in high-concentration solutions and during film deposition. This frequently leads to aggregation-caused quenching (ACQ), which results frequently in a significant decrease in the ΦPL and a red-shifted emission. ACQ is observed to some extent in most aromatic emitters in the solid state, unless dispersed at low doping concentration into a host medium to disrupt intermolecular interactions between emitter molecules. This is a primary reason why the vast majority of examples reported in Sections sec3 –sec7 and sec9–sec12 involve TADF molecules doped into a host matrix within the emissive layer of the device. The host molecules effectively keep the emitter molecules separated, preventing the short-range π-system overlap that drives ACQ; however, use of a host increases the complexity of OLED fabrication as well as the cost.
In 2001 a new mechanism to circumvent ACQ was introduced by Tang and co-workers.ref. ref1140 Molecules with flexible functional groups, which were poorly emissive in solution due to non-radiative decay associated with molecular motion (rotations and vibrations), were shown to become very emissive in the solid state where these rotations are restricted. Aggregation of these emitters hinders these motions, limiting non-radiative decay, and hence enhances the emission of the aggregate – the complete opposite of ACQ. This phenomenon is known as aggregation-induced emission (AIE). In recent years this effect has been incorporated into TADF emitter design, offering the potential to deliver efficient non-doped OLEDs and sidestep the technical challenges and limitations associated with hosts.
Sulfone-Based AIE-TADF Emitters
Sulfone-based TADF emitters represent a large class of those that also show AIE. The structures of emitters containing a sulfone acceptor moiety are shown in Figure and relevant photophysical and device data are tabulated in Table S17. The first AIE-TADF emitters containing a sulfone acceptor moiety, TXO-TPA and TXO-PhCz, were reported by Wang and co-workers.ref. ref1141 The compounds were poorly emissive in toluene, with ΦPL of 24 and 25% at λPL of 586 and 522 nm, respectively. AIE was demonstrated through changes in the emission color and intensity in acetonitrile/water mixtures, a now commonplace technique that allows the properties of the isolated and aggregated molecules to be determined as the mixed solvent is gradually changed from ‘good’ to ‘poor’ in terms of its capacity to solubilize the emitter. The neat films of each emitter showed enhanced ΦPL of 36 and 93% at 625 and 570 nm for TXO-TPA and TXO-PhCz, respectively. Green-emitting OLEDs [CIE coordinates of (0.45, 0.53) and (0.31, 0.56), respectively] were fabricated incorporating both emitters and showed EQEmax of 18.5 and 21.5%, although these devices had EML consisting of 5 wt% emitters doped in mCP. This is a rather common theme for most of the reported AIE-TADF emitters; frequently, only doped devices are investigated, even when AIE is present, while non-doped OLEDs are neglected. We speculate that this arises from a desire to publish the highest possible EQEmax values for new emitters, with non-doped devices frequently struggling to surpass the performance doped devices.
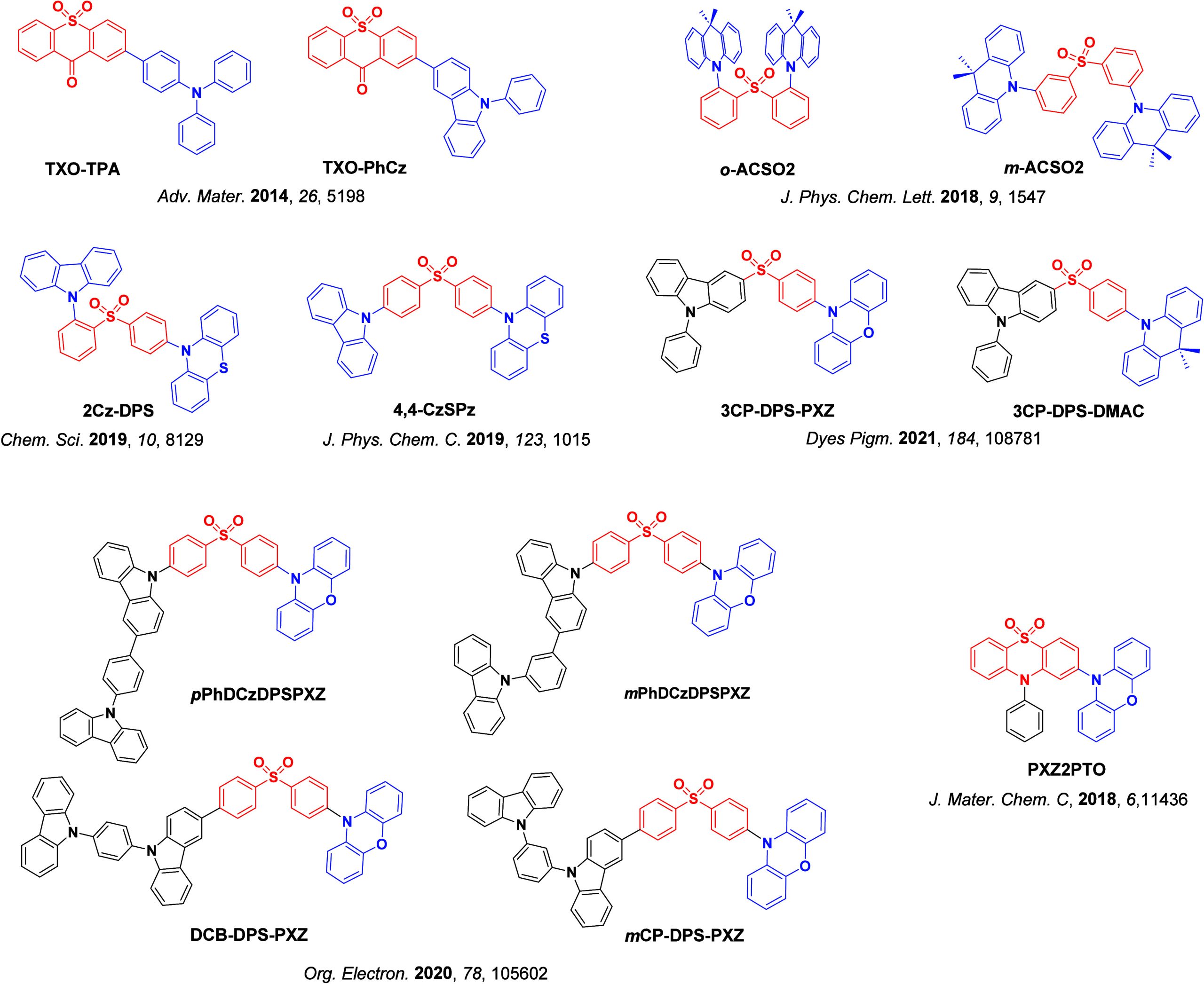
A blue non-doped solution-processed AIE-TADF device (λEL = 486 nm) using m-ACSO2 (Figure ) showed an EQEmax of 17.2% and a mild efficiency roll-off of 5% at 100 cd m–2. The high EQEmax results from a ΦPL of 76% in neat films, along with a relatively short τd of 3.2 μs and a small ΔE ST of 0.07 eV.ref. ref1142 Devices with the analogue o-ACSO2 showed much poorer performance in non-doped devices with an EQEmax of only 5.9%, which cannot be fully explained by the lower ΦPL of 66%. This was observed despite the emitter having a short τd of 1.8 μs and a small ΔE ST of 0.04 eV in the neat film, which highlights the challenges in rational molecular design of AIE-TADF emitters, which must simultaneously perform emission, triplet harvesting, and charge transport in non-doped devices.
Chi and co-workers reported 4,4-CzSPz, an emitter containing two different donor groups, which has a near unity ΦPL of 97.3% in the neat film (Figure ).ref. ref1143 A non-doped OLED based on 4,4-CzSPz reached an EQEmax of 20.7% at λEL of 526 nm, attributed to the dual AIE and TADF character of the emitter. A doped device (10 wt% in CBP) did perform better with an EQEmax of 26.2% at λEL of 518 nm. Another example of a highly efficient non-doped OLED was reported with the similar emitter 2Cz-DPS, which places the Cz donor at the ortho position to the sulfone. The non-doped device of 2Cz-DPS showed a record-high EQEmax of 28.7% at λEL of 518 nm. The contributing factors for the excellent performance are the high ΦPL of 91.9% at λPL of 520 nm, and the relatively short τd of 19.1 μs in the neat film. The inclusion of a carbazole donor also likely contributed to the improved intrinsic charge transport properties in the non-doped emissive layer. However, the non-doped devices suffered from a severe efficiency roll-off, with the EQE decreasing by 71% at 300 cd m–2.ref. ref1124
Guo et al.ref. ref1144 reported two AIE-TADF emitters, 3CP-DPS-PXZ and 3CP-DPS-DMAC (Figure ), composed of a diphenylsulfone core within an asymmetrical D-A-D′ configuration. These two emitters showed ΦPL of 52% at λPL of 518 nm and 65% at λPL of 472 nm in the neat film, respectively. The non-doped device with 3CP-DPS-PXZ showed an EQEmax of 17.9% at λEL of 508 nm, which remained as high as 14.5% at 1000 cd m–2. The EQEmax of the 3CP-DPS-DMAC-based blue non-doped OLED was comparatively lower at 9.1% (λEL = 484 nm). These examples highlight the popular yet poorly understood design strategy of using non-identical donors to achieve high performance in non-doped devices.
Leng et al.ref. ref1145 consciously integrated host-like substituents (DCB, mCP, pPhDCz and mPhDCz) with AIE-TADF chromophores to generate the ‘self-hosting’ TADF emitters DCB-DPS-PXZ, mCP-DPS-PXZ, mPhDCzDPSPXZ and pPhDCzDPSPXZ (Figure ) that have ΦPL of 40, 47, 56 and 55%, with λPL of 547, 547, 548, and 548 nm, respectively, in the neat film. The host moieties were found not to be involved in CT transitions, and instead effectively dispersed the luminophoric centres, which led to the realization of high-performance non-doped OLEDs with EQEmax of 13.9, 14.7, 18.1 and 17.1% and λEL of 520, 520, 523 and 521 nm for the devices with DCB-DPS-PXZ, mCP-DPS-PXZ, mPhDCzDPSPXZ and pPhDCzDPSPXZ, respectively. The efficiency roll-off was found to be lower for the device with mPhDCzDPSPXZ (7.7%) than for the device with pPhDCzDPSPXZ (9.9%) at 1000 cd m–2; however, more severe efficiency roll-off was observed for the devices with DCB-DPS-PXZ (20.8%) and mCP-DPS-PXZ (17.7%) at 1000 cd m–2. While it is not clear whether these materials were intrinsically AIE-active, the strategy of using peripheral substitutions that preserve emission in the solid state overlaps strongly with the AIE approach.
The potential of 10-phenyl-10H-phenothiazine 5,5-dioxide (2PTO) as an acceptor for AIE-TADF emitters was demonstrated by Wang and co-workers.ref. ref1146 An emitter comprised of 2PTO and phenoxazine donors, PXZ2PTO (Figure ), has a ΦPL of 61.5% at λPL of 512 nm in the neat film. The non-doped device showed an EQEmax of 16.4% at λEL of 504 nm. Interestingly, the doped device (80 wt% doped in DPEPO) showed nearly the same EQEmax of 16.3% at 500 nm, demonstrating the utility of the AIE approach. Both devices exhibited low-efficiency roll-off of 4.9% for the doped and 7.9% for the non-doped device at 100 cd m–2.
Carbonyl-Based AIE-TADF Emitters
Although the reasons are at present unclear, many of the reported high-performance AIE-TADF materials feature carbonyl-based acceptor groups such as benzophenone and xanthone. The structures of the emitters are shown in Figure and Figure , and relevant photophysical and device data are tabulated in Table S17. Tang and co-workers reported the asymmetric D-A-D′ emitter DBT-BZ-DMAC, which decorates a benzoyl core with and a dibenzothiophene and has an acridine donor.ref. ref1147 This compound has a ΦPL of 8.3% in THF, which increases to ΦPL of 66% in 6 wt% doped CBP film and to 80% as the neat film, a clear indication of AIE. The EQEmax of the device containing 6 wt% DBT-BZ-DMAC in CBP was 17.9%, compared to 14.2% for the non-doped device. The non-doped device shows a lower efficiency roll-off, with an EQE1000 of 10.9% and 14.2% for the 6 wt% and non-doped devices, respectively.
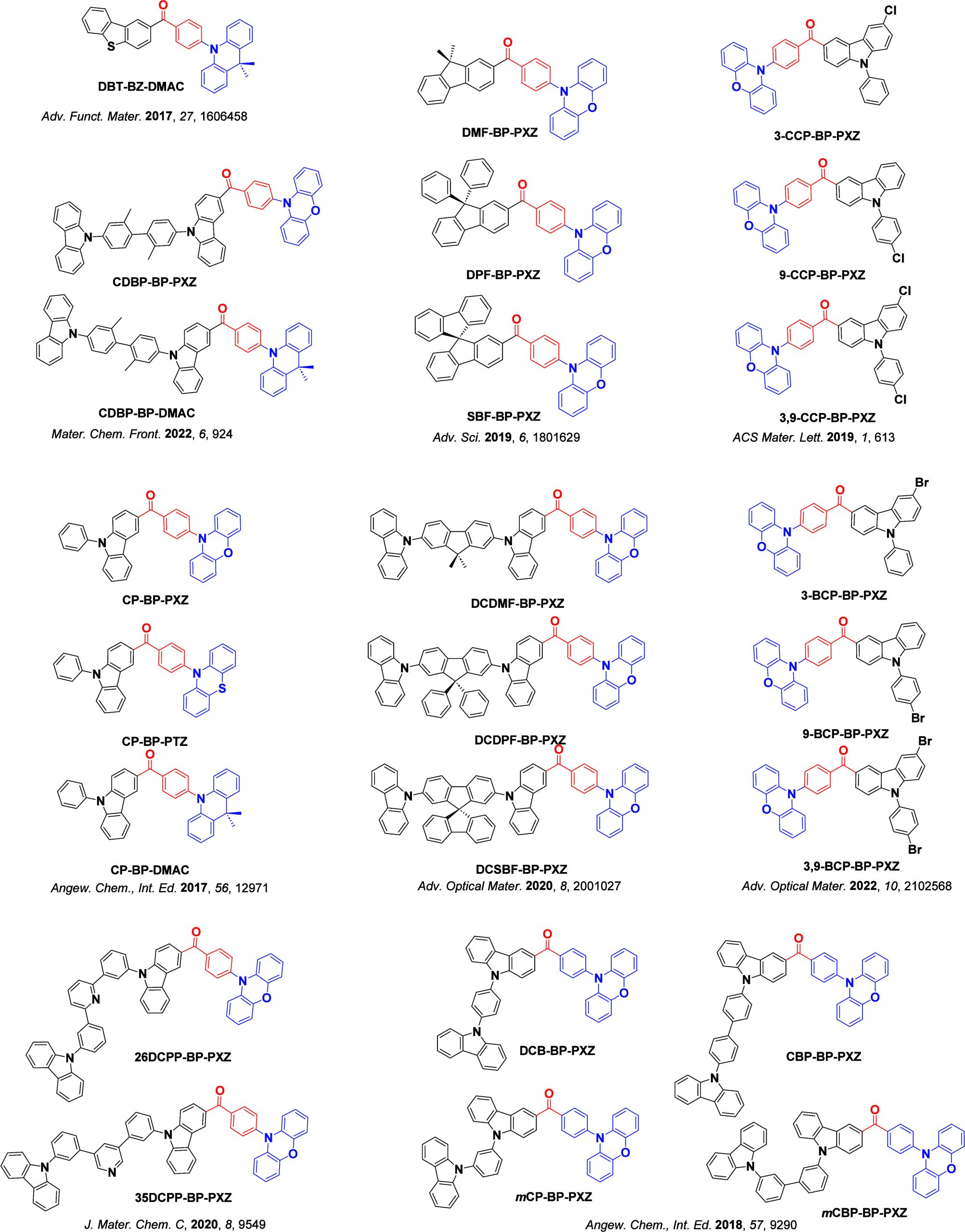
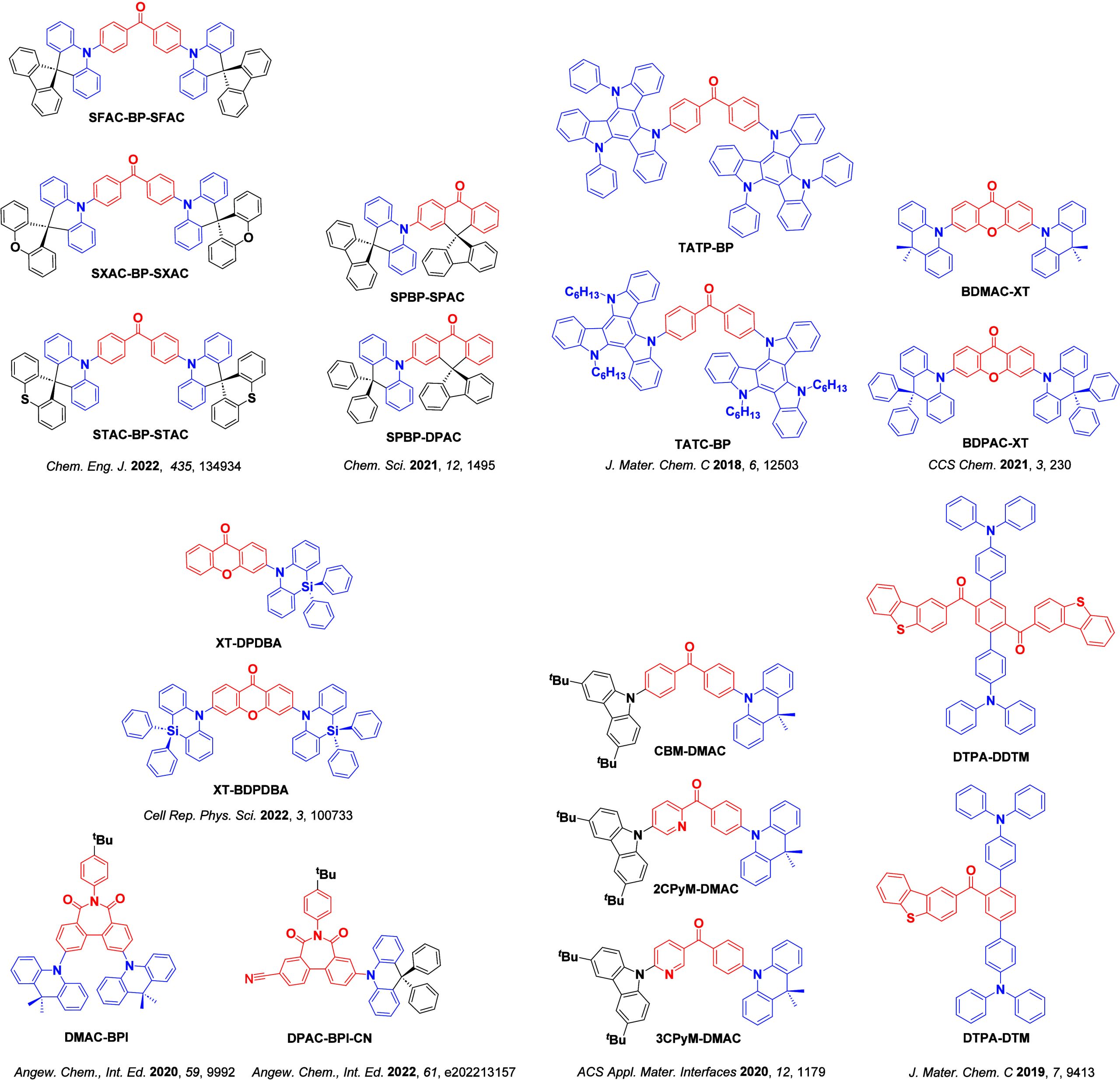
Chen et al.ref. ref1148 designed two emitters, CDBP-BP-PXZ and CDBP-BP-DMAC (Figure ) using the same asymmetric D-A-D′ configuration strategy and appending a CDBP unit that has good OLED hosting properties. In the neat film the ΦPL is 77.4% (λPL = 523 nm) for CDBP-BP-PXZ and 59.2% (λPL = 488 nm) for CDBP-BP-DMAC. The non-doped devices with CDBP-BP-PXZ and CDBP-BP-DMAC showed EQEmax of 15.5% at λEL of 536 nm and 9.5% at λEL of 496 nm, respectively, with corresponding efficiency roll-off of 0.6 and 2.1% at 1000 cd m–2. Huang et al.ref. ref1149 reported the emitters CP-BP-PXZ, CP-BP-PTZ, and CP-BP-DMAC (Figure ), which showed AIE activity in THF/water mixtures. Further, the magnitude of the delayed fluorescence contribution increased upon aggregate formation, thus revealing aggregation-induced delayed fluorescence (AIDF) acting not just on ΦPL but also on RISC in these compounds. These compounds have ΦPL ranging from 45.3 to 67.4% and λPL ranging from 490 to 538 nm in the neat films. The non-doped OLEDs showed EQEmax and λEL of 18.4% and 548 nm for the device with CP-BP-PXZ, 15.5% and 554 nm for the device with CP-BP-PTZ, and 15% and 502 nm for the device with CP-BP-DMAC. The devices exhibited a relatively small efficiency roll-off of 1.2, 16.7 and 0.2% at 1000 cd m–2, respectively, attributed to greatly suppressed emission quenching in the neat films.
Tang and co-workers reported three compounds DMF-BP-PXZ, DPF-BP-PXZ, and SBF-BP-PXZ (Figure ) all containing a PXZ donor and carbonyl acceptors with progressively bulkier fluorene substituents.ref. ref1150 The three compounds all emit similarly at λPL of 548–551 nm, have ΦPL ranging from 45 to 49% in the neat film and short τd of 1.1–1.4 μs. Solution-state TAS showed no signal while the neat films showed a broad excited-state absorption in the range of 800–1000 nm, indicating the formation of a triplet state upon aggregation. From theoretical calculations and experimental observations, the authors claimed that AIDF originates from the S2 excited state rather than the S1 excited state, implying an anti-Kasha behavior of the compounds. The non-doped devices showed EQEmax ranging from 12.3 to 14.3%, with small efficiency roll off of 0.8–6% at 1000 cd m–2. Using a similar molecular design Liu et al.ref. ref1140 reported three AIDF emitters DCDMF-BP-PXZ, DCDPF-BP-PXZ, and DCSBF-BP-PXZ (Figure ) that have ΦPL of 88.5, 89.0, and 39.6% and λPL of 540, 530, and 527 nm, respectively, in neat film. The lower ΦPL of DCSBF-BP-PXZ was attributed to the relatively poor π–conjugation as well as strong intermolecular π–π interactions. Non-doped OLEDs with DCDMF-BP-PXZ and DCDPF-BP-PXZ showed EQEmax of 19.0% at λEL of 540 nm and 18.5% at λEL of 544 nm, respectively. The device with DCSBF-BP-PXZ showed a much lower EQEmax value of 3.3% (λEL at 548 nm) due to both the low ΦPL and unbalanced carrier transport within the EML.
Fu et al.ref. ref1151 developed AIDF materials 35DCPP-BP-PXZ and 26DCPP-BP-PXZ (Figure ) by integrating an AIDF moiety, 4-(phenoxazin-10-yl)benzoyl, with the bipolar carrier transport materials, 3,5-bis((9H-carbazol-9-yl)-3,1-phenylene)pyridine (35DCPP) and 2,6-bis(3-(9H-carbazol-9-yl) phenyl)pyridine (26DCPP). In neat films these two compounds have ΦPL of 66.5 and 67.9% at λPL of 530 and 533 nm, respectively. In contrast, and demonstrating their AIE-activity, the respective ΦPL are very low in THF at 2.2 and 2.7%. Non-doped OLEDs with 35DCPP-BP-PXZ and 26DCPP-BP-PXZ showed EQEmax of 17.3 and 16.1% at λPL of 538 and 542 nm, respectively. Remarkably, the former device showed a very low efficiency roll-off of 0.6, 7.5 and 16.2%, at 1000, 5000 and 10000 cd m–2, respectively, which can be partially attributed to the balanced charge transfer ability embedded within the emitter design.
Zhao et al.ref. ref1152 reported an emitter that combines AIDF with enhanced SOC through the incorporation of heavy halogen atoms that leads to faster k RISC. The three AIDF emitters 3-CCP-BP-PXZ, 9-CCP-BP-PXZ and 3,9-CCP-BP-PXZ (Figure ) contain the popular PXZ-BP core coupled to suitably halogen-decorated Cz donors. These compounds have ΦPL of 73.0, 70.4 and 72.6% at λPL of 541, 543 and 536 nm, respectively, as neat films. Exceptionally short τds ranging from 0.42 to 0.76 μs result from the fast k RISC of between 1.73 × 106 – 3.10 × 106 s–1, while the control compound without the halogen substituents possesses a longer τd of 2.10 μs and slower k RISC of 0.63 × 106 s–1. Non-doped OLEDs with 3-CCP-BP-PXZ, 9-CCP-BP-PXZ and 3,9-CCP-BP-PXZ showed EQEmax of 21.7, 20.4 and 20.6% at λEL of 540, 537 and 541 nm, respectively; the corresponding efficiency roll-offs in the devices were 4.4–8.7% at 1000 cd m–2. Replacement of the chloro substituents for bromine produced analogs 3-BCP-BP-PXZ, 9-BCP-BP-PXZ and 3,9-BCP-BP-PXZ.ref. ref1153 These three compounds have slightly attenuated ΦPL of 61.0, 53.4 and 50.7% and modestly blue-shifted λPL at 521, 531 and 540 nm, respectively in the neat films. Reflecting the lower ΦPL, the non-doped devices with 3-BCP-BP-PXZ, 9-BCP-BP-PXZ and 3,9-BCP-BP-PXZ showed EQEmax of 19.5, 14.3 and 16.4% with λEL of 544, 540 and 544 nm, respectively. The efficiency roll-off of these devices was also low at between 3.5–6.1% at 1000 cd m–2.
Although AIE is an important property to consider when designing non-doped emitters, ensuring balanced transport and efficient charge recombination is paramount to obtaining efficient devices. Similar to the strategy of Leng et al.ref. ref1145, the addition of host-like components to a TADF emitter helped to prevent ACQ and support non-doped device performance for DCB-BP-PXZ, CBP-BP-PXZ, mCP-BP-PXZ and mCBP-BP-PXZ (Figure ).ref. ref1154 These compounds are poorly emissive in THF solution, with ΦPL of 3.9, 3, 3.1 and 2.8% respectively; however, their neat films showed much higher ΦPL of 69, 71.6, 66 and 71.2%, respectively. Notably, the incorporation of the host-like groups negligibly impacted the λPL, with the compounds displaying nearly identical emission maxima of between 529–532 nm in the neat film. The ΔE ST values of the four compounds are around 0.02 eV in the neat film, which was correlated with the short τd of between 2.3–2.6 μs. Increased delayed emission was also found for aggregates in water-rich THF/water mixtures, demonstrating AIDF. These optical and aggregation properties in turn produced excellent green devices (λEL = 542–548 nm), with EQEmax of 22.6% for the device with DCB-BP-PXZ and 21.4% for the device with CBP-BP-PXZ. The OLEDs showed low efficiency roll-off of between 9.9 and 11.4% at 5000 cd m–2. These excellent results were attributed to the combination of AIDF and ambipolar charge transport in the emitter materials.
The use of symmetric D-A-D emitters also works well to obtain highly efficient non-doped OLEDs. Zhao et al.ref. ref1155 reported three AIDF emitters, SFAC-BP-SFAC, SXAC-BP-SXAC, and STAC-BP-STAC (Figure ) constructed from spiro-acridine-based donors and a benzophenone acceptor. These three compounds have ΦPL of 52–58% at λPL of 500–511 nm in the neat films. A relatively short τd of 3.6–4.0 μs linked to the miniscule ΔE ST ranging from 36–52 meV were observed for these emitters in the neat films. Non-doped OLEDs based on these emitters showed EQEmax ranging from 17.1 to 18.6% at λEL of between 504–508 nm. The devices with 30 wt% emitter doped in PPF showed higher EQEmax ranging from 34.3 to 35.3% due to the preferentially horizontally oriented TDM of the emitter.
Triazatruxene-based TATC-BP and TATP-BP (Figure ) exhibited combined TADF, AIE and MCL.ref. ref1156 Although the ΦPL of TATC-BP and TATP-BP in THF solution are quite low at 0.8 and 1.9%, respectively, these increased considerably to 22.0 and 24.2% in the neat film, which was attributed to their AIE activity (λPL of 524 and 520 nm, respectively). Solution-processed non-doped OLEDs with TATC-BP and TATP-BP showed EQEmax of 5.9 and 6.0%, respectively; however, the λEL were red-shifted to 549 and 541 nm, respectively. The doped OLEDs using H2 (a dendritic oligocarbazole host) showed an enhanced EQEmax of 15.9 and 15.4%, respectively for TATC-BP and TATP-BP. In what is a widely observed trend, the efficiency roll-off for the non-doped OLEDs of TATP-BP (3.3%) is much lower than that of many doped devices, likely due to the large number of TADF molecules being able to harvest triplets more rapidly. The efficiency roll-off of the device with TATC-BP was 18.6% at 1000 cd m–2.
Fusing the benzophenone with an oxygen bridge to give xanthenone (XT), produces a more rigid acceptor that should translate to higher ΦPL. Chen et al.ref. ref1157 reported the AIDF emitters BDMAC-XT and BDPAC-XT (Figure ) that have high ΦPL of 96% at λPL of 518 nm and 94% at λPL of 495 nm in the neat film, respectively. Non-doped OLEDs with BDMAC-XT and BDPAC-XT showed EQEmax of 21% at λEL of 526 nm and 21% at λEL of 496 nm, respectively. The devices also showed negligible efficiency roll-off where the EQE1000 remained remarkably high at 21 and 18%. He et al.ref. ref1158 reported two similar blue AIDF emitters, XT-DPDBA and XT-BDPDBA, composed of a XT acceptor and weak electron-donor 10-dihydrodibenzo[b,e][1,4]azasiline groups. Compounds XT-DPDBA and XT-BDPDBA have ΦPL of 77 and 86% at λPL of 472 and 480 nm, respectively, in the neat film. The non-doped OLEDs showed EQEmax values of 8.9 and 13.1% at λEL of 472 and 488 nm, respectively, with corresponding efficiency roll-off at 1000 cd m–2 of 10% and 16%. In a similar effort to increase the rigidity of the emitter, Wu et al.ref. ref1159 designed SPBP-DPAC and SPBP-SPAC (Figure ) containing a carbonyl acceptor with a fused spirofluorene bridging group. Compounds SPBP-DPAC and SPBP-SPAC have ΦPL of 93 and 98% at λPL of 495 and 504 nm, in the neat films and non-doped OLEDs showed EQEmax of 22.8 and 21.3% at λEL of 504 and 516 nm, all respectively. Once again and typical of efficient non-doped OLEDs, the devices showed extremely small respective efficiency roll-off of 1.8 and 2.3% at 1000 cd m–2.
Fulong et al.ref. ref1160 rationally designed a series of AIE-TADF emitters by employing phenyl(pyridyl)methanone as the acceptor moiety that contained intramolecular H-bonding, and compared this to a control phenyl-linked compound where H-bonding cannot occur. Compounds 3CPyM-DMAC (ΦPL = 66.8%; λPL= 514 nm; ΔE ST = 0.04 eV) and 2CPyM-DMAC (ΦPL = 53.3%; λPL = 536 nm; ΔE ST = 0.03 eV) showed higher ΦPL and smaller ΔE ST in the neat film compared to the parent emitter CBM-DMAC (ΦPL = 46.7%; λP L= 501 nm; ΔE ST = 0.1 eV). Solution-processed non-doped OLEDs with 3CPyM-DMAC (EQEmax = 11.4%; λEL = 532 nm) and 2CPyM-DMAC (EQEmax = 9.1%; λEL = 544 nm) showed better performance than the device with CBM-DMAC (EQEmax = 6.7%; λEL = 499 nm), demonstrating the effective role that the intramolecular H-bonds may play in enhancing the ΦPL of the emitter – although some doubt remains on this interpretation.ref. ref122
Huang et al.ref. ref1161 reported an AIDF emitter based on a new heptagonal diimide acceptor (BPI). DMAC-BPI (Figure ) has a ΦPL of 95.8% at λPL of 510 nm in the neat film, which decreased to 16.2% in THF, reflecting its AIE activity. In the neat film, DMAC-BPI has a τd of 3.1 μs linked to a small ΔE ST of 0.02 eV (determined from toluene solution). The non-doped OLED showed an EQEmax of 24.7% at λEL of 511 nm and had an exceptionally low efficiency roll-off of 1% at 1000 cd m–2. Using the same acceptor, these authors also rationally designed DPAC-BPI-CN, based on a “medium-ring”-lock strategy, which has a ΦPL of 90.1% at λPL of 525 nm, a τd of 3 μs and a ΔE ST of 0.35 eV in the neat film. The non-doped device showed an EQEmax of 26.2% at λEL of 531 nm.ref. ref476
Finally, Qi et al.ref. ref1162 reported AIE-TADF emitters with dual charge-transfer states (TBCT and TSCT), DTPA-DTM and DTPA-DDTM (Figure ). These compounds have moderately large ΔE ST of 0.18 and 0.17 eV in toluene yet retain relatively high ΦPL of 38.6 and 60.5% in the neat film, all respectively. The higher ΦPL of DTPA-DDTM is due to effective suppression of intramolecular vibrational relaxation, resulting from the enhanced intramolecular D–A interaction with the additional donor. The ΦPL of DTPA-DTM and DTPA-DDTM in THF are only 8.4 and 5.1%, respectively. Non-doped device of DTPA-DTM exhibited green emission with λEL at 494 nm and a low EQEmax of 4.4%, while the device with DTPA-DDTM exhibited an EQEmax of 8.2% and yellow emission with λEL at 555 nm, in line with their respective ΦPL. Doped devices with DTPA-DTM and DTPA-DDTM (30 wt% doped in mCP) showed moderately improved performance, with EQEmax of 7.1 and 13.6%, respectively.
AIE-TADF Emitters Based on Other Acceptors
The structures of AIE-TADF emitters with other assorted acceptors are shown in Figure , and the relevant photophysical and device data are shown in Table S17. Wang et al.ref. ref1163 reported two AIDF emitters, CzTAZPO and sCzTAZPO, composed of carbazole donor dendrons and a triazine acceptor that is decorated with a secondary phosphine oxide acceptor to improve the electron transport properties of the emitters. The two compounds have ΦPL of 71 and 57% and λPL at 512 and 502 nm, respectively, in the neat film. The non-doped solution-processed OLEDs with CzTAZPO and sCzTAZPO showed EQEmax of 12.8 and 9.6% at λEL of 537 and 531 nm, with remarkably low efficiency roll-off at 1.8 and 0.97% at 1000 cd m–2, all respectively. This level of performance was attributed to their small ΔE ST of 0.08 and 0.10 eV and short τd of 1.1 and 0.81 μs, respectively.
Park et al.ref. ref1164 reported two large, three-armed structures, IAcTr-in and IAcTr-out (Figure ), composed of triazine and indenoacridine moieties that showed dual AIE and TADF. IAcTr-in has a higher ΦPL (64.5%) at λPL of 525 nm than IAcTr-out (ΦPL = 47.7% and λPL = 524 nm). IAcTr-in and IAcTr-out both have short τd of 1.6 and 1.3 μs and associated small ΔE ST of 0.069 and 0.052 eV as neat films. The non-doped solution-processed OLED with IAcTr-in showed an EQEmax of 10.9%, increasing to 18.4% in the doped device (35 wt% emitter in mCP). An even more pronounced change in EQEmax was observed for the devices with IAcTr-out, with the doped device showing an efficiency of 17.5%, while the non-doped device showed an EQEmax of only 3.8%. The poor efficiency of the non-doped devices was attributed in part to the lower ΦPL (64.5 vs. 47.7%), and mainly to the poorer charge balance in IacTr-out associated with its different ratio of donor/acceptor subunits, making charge recombination less favourable.
Zhang et al.ref. ref1165 designed AIE-TADF emitters containing a novel acridine–carbazole fused donor, combined with either a pyrimidine or triazine as the acceptor to give 34AcCz-PM and 34AcCz-Trz (Figure ). The compounds have short τd of 0.64 and 0.75 μs at λPL of 538 and 556 nm in the neat film, respectively. 34AcCz-PM has a higher ΦPL of 67% and faster k RISC of 8.97 × 105 s–1 than 34AcCz-Trz (ΦPL = 42%; k RISC = 1.79 × 105 s–1). Consequently, the non-doped device with 34AcCz-PM showed superior performance with EQEmax of 14.1% at λEL of 548 nm, while the device with 34AcCz-Trz showed an EQEmax of 7.3% at λEL 576 nm.
Yasuda et al.ref. ref1166 reported the three carborane-based AIDF emitters PCZ-CB-TRZ, TPA-CB-TRZ, and 2PCZ-CB (Figure ). In neat film these have ΦPL of 97, 55 and 94% at λPL of 557, 624 and 571 nm, respectively. Despite the strongly varying ΦPL values, the non-doped devices with PCZ-CB-TRZ, TPA-CB-TRZ, and 2PCZ-CB all showed similar EQEmax of 11.0, 10.1 and 9.2%, respectively. The emitters SFDBQPXZ and DFDBQPXZ also showed combined AIE and TADF behavior, having neat film ΦPL of 43.4 and 33.2% at λPL of 546 and 551 nm, respectively. The corresponding non-doped devices showed EQEmax of 10.1 (λEL = 584 nm) and 9.8% (λEL = 584 nm). However, the doped OLEDs (10 wt% SFDBQPXZ and DFDBQPXZ doped in mCP) showed much improved performance due to the much higher ΦPL of 99.6 and 88.3%, giving EQEmax of 23.5 and 16.8%, all respectively.ref. ref1167
Three quinoline-based TADF emitters, DMAC-QL, PXZ-QL and PTZ-QL (Figure ) have moderate ΦPL of 32.6, 64.7 and 52.3%, and emit at λPL of 489, 531 and 537 nm in the neat films, all respectively.ref. ref1168 Of these, RISC was most efficient in PXZ-QL, which has the shortest τd (1.86 μs) compared to DMAC-QL (2.15 μs) and PTZ-QL (15.76 μs). The non-doped OLEDs with DMAC-QL, PXZ-QL and PTZ-QL showed EQEmax of 7.7, 17.3 and 14.8%, respectively, at λEL of 522, 536 and 546 nm. With the fastest RISC the efficiency roll-off was most attenuated in the PXZ-QL device, with a decrease of only 12% at 1000 cd m–2. Zhang and co-workersref. ref1169 reported similar quinoline-based AIDF emitters, Fene, Fens and Yad that have ΦPL ranging from 36.1 to 79.6% at λPL ranging from 544 to 591 nm, and small ΔE ST ranging from 0.03 to 0.04 eV as neat films. The non-doped OLEDs showed EQEmax ranging from 13.1 to 17.4% at λEL of between 534–570 nm. These results illustrate the potential of quinoline-based AIDF emitters for non-doped OLEDs.
Finally, Kim et al.ref. ref1170 reported two blue AIDF emitters TB-tCz and TB-tPCz bearing organoboron-based cores as acceptors and 3,6-substituted carbazoles as donors. Compounds TB-tCz and TB-tPCz have ΦPL of 41.4 and 51.9% at λPL of 433 and 445 nm, respectively, in the neat films. Owing to the closely aligned 1CT and 3LE states, both emitters exhibit relatively fast kRISC (∼106 s–1). Solution-processed non-doped OLEDs with TB-tCz and TB-tPCz showed EQEmax of 8.21 and 15.8% along with narrowband emission, with λEL at 416 (FWHM = 44 nm) and 428 nm (FWHM = 42 nm), respectively. The higher performance of the device with TB-tPCz is due in part to its faster RISC and more efficient upconversion of triplet into singlet excitons.
Outlook
This section has highlighted the recent advances in AIE-TADF and AIDF emitter design, and particularly their application towards non-doped OLEDs. While the majority of AIDF emitters contain sulfonyl- or carbonyl-based acceptors, diverse strategies including asymmetric D-A-D′ configurations, incorporation of intramolecular hydrogen bonding, and integration of host moieties within the emitters have all been explored in efforts to enhance the photophysical performance of the emitter and hence the device performance. Among sulfonyl-containing derivatives, the non-doped device with 2Cz-DPS showed the highest EQEmax of 28.7% at λEL of 518 nm amongst this family of emitters. Among carbonyl-containing derivatives, the non-doped device with DPAC-BPI-CN showed the highest EQEmax of 26.2% at λEL of 531 nm. Though many examples of carbonyl-containing AIE-TADF emitters also employ a phenoxazine donor, it remains at present difficult to identify general design rules for the construction of AIE-TADF emitters.
Promising AIE-TADF or AIDF emitters must show high ΦPL along with small ΔEST and short τd as neat films. However, promising photophysical properties do not always translate to high performance non-doped OLEDs – charge transport is also critical, and difficult to assess from optical measurements alone. AIDF emitters nonetheless provide a promising route to non-doped OLEDs, and frequently show significant resistance to efficiency roll-off at high luminance. We also note that most of the AIDF emitters discussed in this section emit in the blue and green spectral region, while there is an apparent paucity of recognized examples of red/deep red AIDF emitters. This need not be a serious limitation though, as the alternate use of AIDF emitters as hosts and sensitizers for other terminal emitters can readily access longer wavelengths (Section sec17 and sec18). Ultimately, this progress in the area of AIDF emitters demonstrates the ability of the TADF research community to weaponize apparently inescapable molecular properties (ACQ) and exploit new and unexpected understanding (e.g, the existence of AIE) towards enhanced material properties and performance.
Excited-State Intramolecular Proton Transfer (ESIPT) Based TADF
Introduction
Excited-state intramolecular proton transfer (ESIPT) is a photochemical process that produces a tautomer with a different electronic structure from the initial ground state.ref1171,ref1172 ESIPT emission in this context involves the rapid photo-induced tautomerization of a molecule in its electronic excited state and subsequent emission from this second tautomer, or in some cases from both tautomers; the latter case is often described as a dual ESIPT-based emission. The most frequently reported systems are those that show a tautomerization between enol and ketone-type molecules (A and B, respectively, in Figure ), with the enol species frequently being the most stable in the ground state and the ketone tautomer the most stable in the excited state. This tautomerization occurs faster than the radiative decay from the vertical excited state, particularly when no other geometric reorganization is required prior to the proton transfer. Hence the radiative decay occurs from the B* species and not from the A* species (Figure ), each with distinct energy levels and orderings.
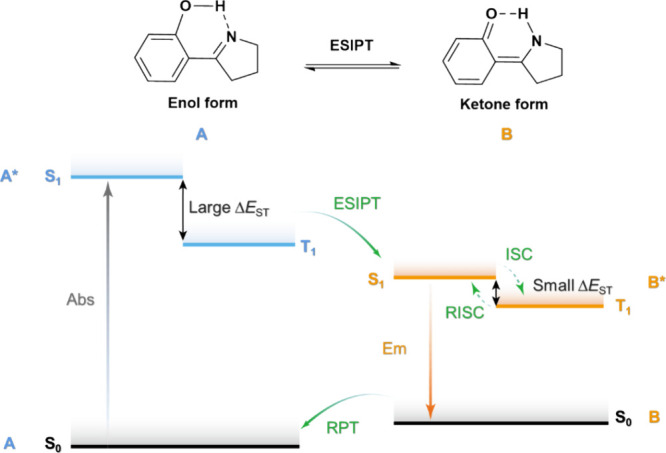
Indeed, a key consequence of ESIPT is that the electron density distribution of the frontier molecular orbitals can change significantly between the two tautomeric forms, leading to changes in both the singlet and triplet energies and thus also ΔE ST, which can then induce TADF and vary its efficiency.ref1172,ref1173 Hence, ESIPT-induced TADF represents a distinct, alternative pathway to achieving the well-separated HOMO-LUMO distributions that are established by either highly twisted D-A conformations (See Sections sec3–sec5), by engineering π-stacking interactions between donor and acceptor motifs in either an intermolecular (Section sec8 ) or intramolecular (Section sec12 ) design, or in systems possessing alternating networks of donating and accepting atoms (Section sec11 ). Due to the large electronic effects associated with proton transfer, ESIPT luminescence is characterized by very large Stokes shift (as absorption and emission occur from distinct tautomers) and an emission that can often be tuned via the local environment. Due to these photophysical properties, ESIPT molecules are attractive for fluorescence sensing,ref1174,ref1175 bioimaging,ref1174,ref1176 NIR emitters,ref1177−ref1178ref1179 latent fingerprint detection,ref. ref1178 UV absorbersref. ref1180 as well as for lighting materials.ref1181−ref1182ref1183ref1184 A small number of reports exist that use ESIPT-based fluorophores as emitters in OLEDs; however, the performance of these devices is generally poor in part due to their inefficient harvesting of triplet excitons.ref1181,ref1183,ref1185−ref1186ref1187ref1188
ESIPT Materials Development
In 2007 the first example of a molecule exhibiting both ESIPT and TADF (HPI-Ac) was reported and compared with the non-ESIPT derivative (MeOPI-Ac), in which the phenolic proton was replaced with a non-labile methyl substituent to prevent the ESIPT (Figure and Table S18).ref. ref1189 Surprisingly, in MeOPI-Ac no TADF behavior was observed, which was rationalized by the absence of the phenolic proton and, thus, the inhibition of ESIPT. By contrast, in HPI-Ac a delayed lifetime of 25 μs was observed in CHCl3, along with a λPL of 465 nm and a modest ΦPL of 22% (reduced to 18% in air). Unfortunately, no OLEDs were fabricated based on either of these two emitters, although understandably so as this report predated the key work establishing the utility of TADF in OLEDs by several years.ref. ref31
Mamada et al. reported the first use of a TADF ESIPT emitter not based on a donor-acceptor system, triquolonobenzene (TQB, Figure ), in an OLED.ref. ref1190 TQB displayed a λPL at 516 nm with ΦPL = 55% in 10 wt% CzSi doped film, while the OLED achieved an EQEmax of 14% at λEL of 518 nm. The ground state species (A) has a large ΔE ST (> 0.5 eV), a consequence of the large spatial overlap of the electron densities of the HOMO and LUMO for this tautomer. ESIPT leads to the formation of B*, a tautomer that has spatially separated HOMO and LUMO orbitals and hence a small ΔE ST (< 0.2 eV), which enables RISC to occur (Figure and Table S18). In subsequent studies, Cao et al. demonstrated through computations that the proton in TQB is transferred within 20 femtoseconds upon photoexcitation, suggesting the direct action of proton transfer itself plays little role in triplet harvesting. However, proton transfer dynamics from TQB-TA to TQB-TB provides access to multiple triplet states, with a decisive influence on the efficiency of the triplet harvesting (3 TQB-TA → 1 TQB-TB).ref. ref1173 Supporting Cao’s theoretical study, Long et al. performed transient absorption and time-resolved photoluminescence studies on TQB and demonstrated that the RISC in TQB occurs from T2 to S1, alongside induced absorptions and quenching bands associated with tautomers from secondary and additional proton transfers.ref. ref1191
Recently Wu et al. reported TADF emitters PXZPDO and DMACPDO, and claimed that these compounds also showed an ESIPT (Figure and Table S18).ref. ref1192 The symmetry of the two compounds and the presence of the enol tautomer in the ground state, however, preclude ESIPT as an operational mechanism. The vacuum-deposited OLEDs employing these two emitters achieved an EQEmax of 18.8% at 560 nm and 23.3% at 536 nm, respectively. In the same report, the control non-ESIPT TADF emitters PXZDMePDO and DMACDMePDO were also synthesized and used for comparison. The TADF efficiency was not affected by the methylation; however, the EQEmax was lower but still high overall (12.2% for PXZDMePDO at 544 nm and 14.6% for DMACDMePDO at 518 nm). The improved device performance for PXZPDO and DMACPDO was attributed to the presence of the intramolecular hydrogen bond that was proposed to produce a more rigid structure. As a result, superior ΦPL and k RISC could be achieved; for example, PXZPDO has a k RISC of 1.3 × 106 s–1 compared to 2.2 × 105 s–1 for the non-ESIPT emitter (PXZDMePDO), in 1 wt% CBP films. Similarly, in 6 wt% CBP films a k RISC of 8.8 × 105 s–1 for DMACPDO and 4.5 × 105 s–1 for non-ESIPT emitter (DMACDMePDO), was observed. Each of these OLEDs showed a similar efficiency roll-off at 100 cd m–2; indeed, only a slight improvement was observed in the efficiency roll-offs of 6, 7, 11 and 18% for the OLEDs using PXZPDO, PXZDMePDO, DMACPDO and PXZMePDO, respectively (Table S18).
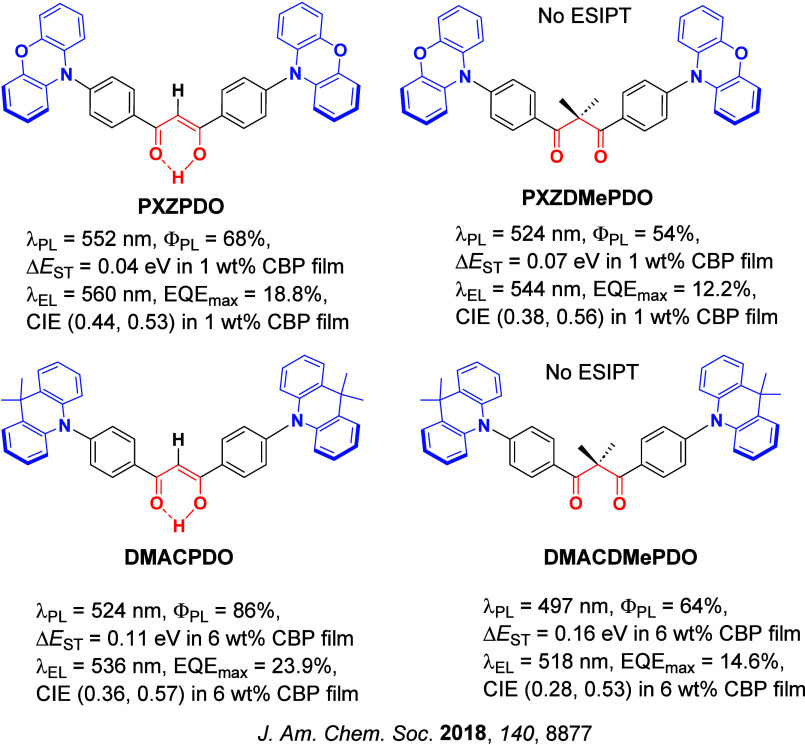
Inspired by this work, Gupta et al. reported a new ESIPT-based TADF emitter (TPXZBM) that contains phenoxazine donor groups in combination with a β-triketone – a stronger acceptor moiety than the one found in PXZPDO.ref. ref1193 The molecular design produced a more acidic methine proton, which pushed the equilibrium position in the ground state towards the presence of both tautomers, unlike that observed for PXZPDO (Figure and Table S18) where only the enol tautomer was observed by 1H NMR spectroscopy. ESIPT was observed for TPXZBM, which showed a red-shifted emission at 650 nm in comparison to PXZPDO (604 nm) in toluene. Cross-comparison of the optoelectronic properties and OLED device performance using this compound revealed significant differences to those of PXZPDO and of the β-tetraketone non-ESIPT reference emitter, BPXZBM. The latter compound exists as only one tautomer due to the absence of an enolizable proton but retains TADF activity, presumably arising through its D-A-D structure. The emitter TPXZBM showed both ESIPT and TADF, with the enol tautomer dominant in the excited state, resulting in a ΔE ST of 0.020 eV, ΦPL of 30% and τ d of 1.44 μs in 1 wt% CBP host. The solution-processed OLEDs of TPXZBM showed an EQEmax = 12.7% at 582 nm with a low efficiency roll-off (the EQE at 10,000 cd m–2 reached 4.7%), while for PXZPDO, a much better device performance was observed (EQEmax = 20.1%, comparable to thermally evaporated devices of PXZPDO previously) with low efficiency roll-off (the EQE at 10,000 cd m–2 reached 12.7%). The non-ESIPT control emitter BPXZBM showed poor ΦPL= 17% and a τ d = 1.01 μs in 1 wt% in 1 wt% CBP, and thus the device performance suffered, with an EQEmax of 7% at 598 nm. This was also the first report of a solution-processed ESIPT-based TADF OLED.
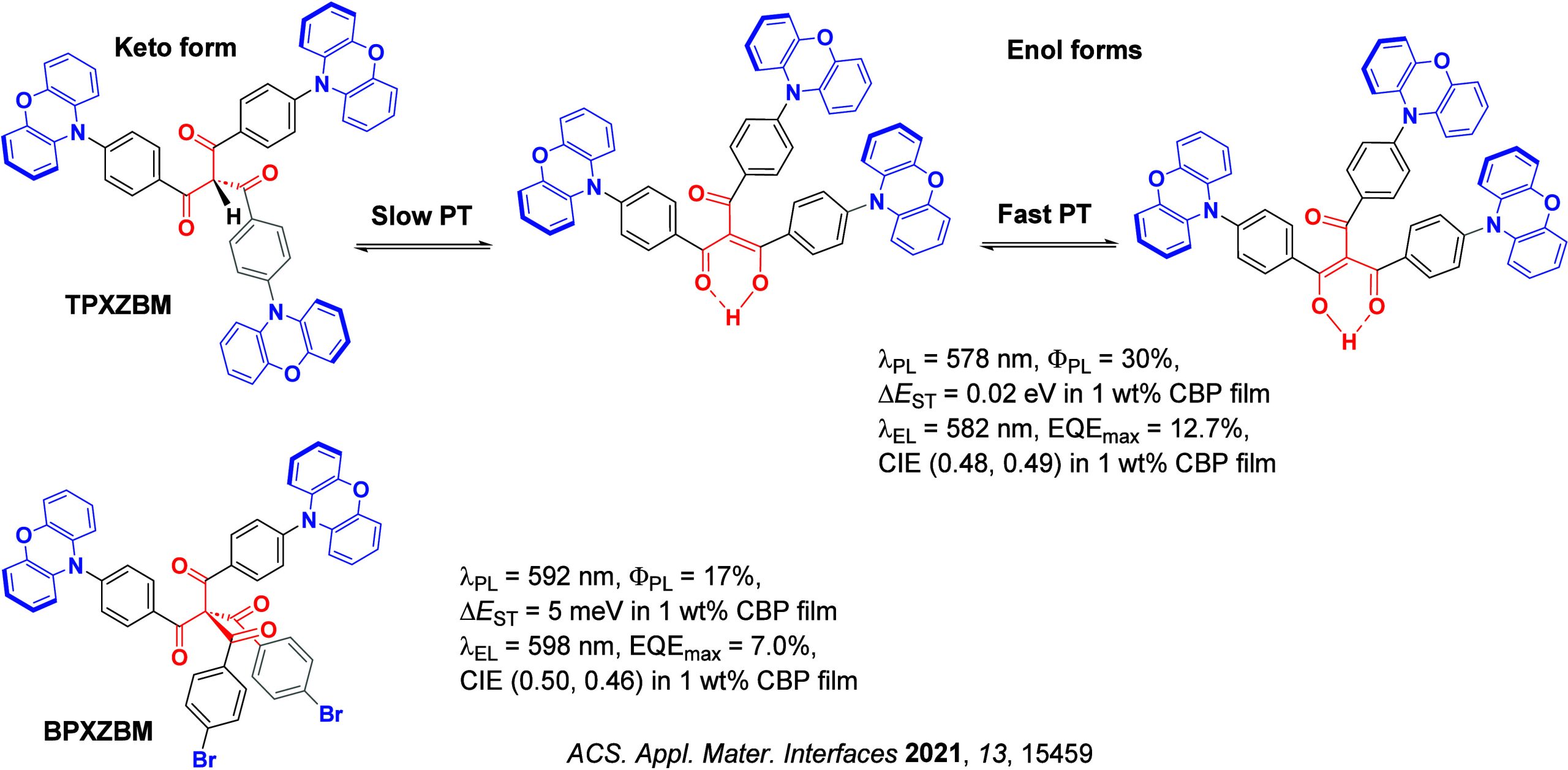
The high sensitivity of ESIPT emitters to their surrounding environment inspired Kim and co-workers to devise ESIPT-based compounds that could switch between room temperature phosphorescence (RTP) and TADF, depending on the substitution about the core structure.ref. ref1194 The introduction of both an aromatic carbonyl and an adjacent bromo substituent to (2′-hydroxyphenyl)benzimidazole (HBI), as in BrA-HBI (Figure ), increased the SOC and resulted in RTP from the enol form. In contrast, the keto form of BrA-HBI exhibited a mixture of prompt fluorescence and TADF with λPL of 450 nm, ΦPL that grew from 10 to 31% upon degassing, and with an associated τ d of 1.90 ms in 1 wt% PMMA doped film at room temperature. At 77 K, the same BrA-HBI film showed a new emission band at around 505 nm, which was assigned to phosphorescence (τph ≈ 13 ms) from the enol form of BrA-HBI, and the ΔE ST was measured to be 0.31 eV. The phosphorescence from the enol-form was further confirmed by doping 1 wt% BrA-HBI in polyacrylic acid (PAA), which can inhibit intramolecular proton transfer through competitive intermolecular hydrogen bonding. The non-ESIPT control molecule “methylated BrA-HBI” showed similar photophysical behavior to the enol form of BrA-HBI. In contrast, the non-functionalized parent molecule HBI showed high ΦPL of 70%, but no delayed emission. However, the authors assigned the emission of aldehyde-substituted A-HBI to be TADF (albeit with a reduced ΦPL of 53%), while Br-HBI mostly showed phosphorescence with ΦPL of 23% independent of temperature (Figure ). The authors then used Br-HBI in a photochromic photo-patterning system, and as hydrogen chloride vapor detection system with optical readout.
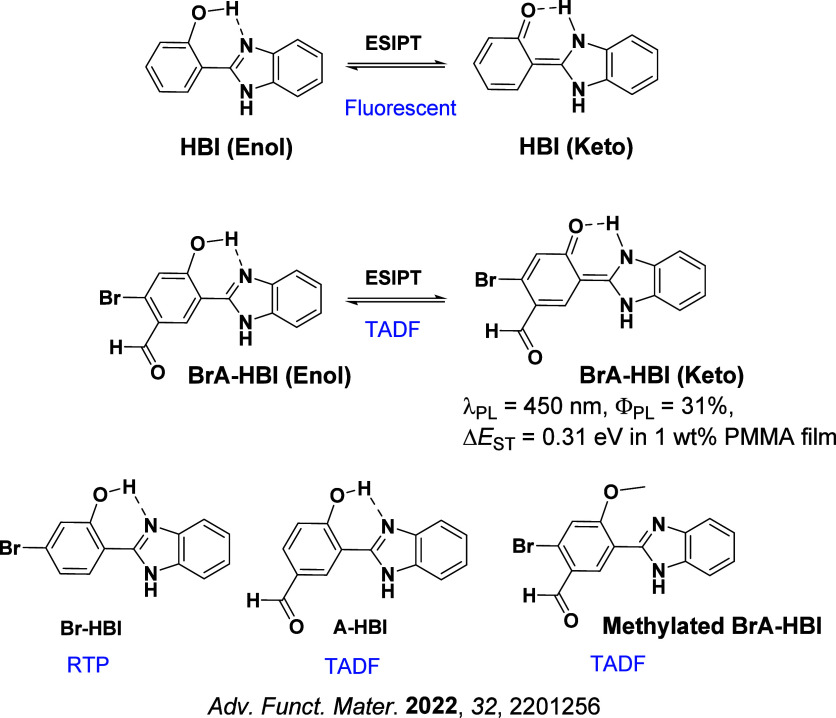
Berezin et al. demonstrated TADF behavior in the pyrimidine-based ESIPT ligand 2-[6-(3,5-dimethyl-1H-pyrazol-1-yl)pyrimidin-4-yl]phenol (HL) and its Zn complex [Zn(HL)]Cl2 (Figure ).ref. ref939 The HL ligand features a short O-H···N intramolecular H-bond (O···N ca 2.6 Å) that enables the ESIPT, and a separate N,N-chelating pocket for binding metal ions. Complex [Zn(HL)]Cl2 showed excitation wavelength-dependent emission, ESIPT, and TADF, while HL alone also showed both ESIPT and TADF. DFT calculations revealed that the presence of the Zn2+ ions facilitate S2 → T2 → T1 and S2 → T1 ISC. The neat powder of HL emits with λPL of 555 nm; however, [Zn(HL)Cl2] showed emission at 640 nm which shifted to 565 nm on changing the excitation wavelength from 420 to 480 nm. Compound HL showed a short τp of 2 ns and a τd of 890 μs at 300 K, the latter of which increased to 1500 μs at 220 K. The DFT calculated a small ΔE ST of 0.10 eV, which explains the TADF behavior of [Zn(HL)Cl2]. Based on a theoretical study, the authors suggested that the ESIPT process in both compounds is barrierless and results in an abnormal anti-Kasha fluorescence (S2 → S0) and anti-Kasha phosphorescence (T2 → S0) associated with relatively low S2 → S1 and T2 → S1 internal conversion rates.
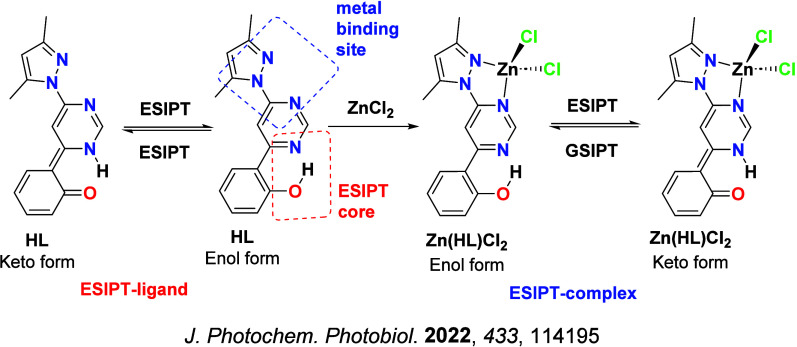
Outlook
The examples summarized in this section reveal that it is possible to design molecules in which ESIPT supports TADF. The ESIPT process paves the way for HOMO and LUMO separation and a small singlet-triplet gap, in a way that is fundamentally distinct from the strongly twisted conformation adopted in most D-A TADF emitters. For example, TQB is a compound far outside the donor-acceptor design paradigm (Figure ), which was employed in green OLEDs that showed an EQEmax of 14.2%. Looking to the future, it is an open question whether the relatively small molecular reorganisation energies associated with ESIPT might eventually enable faster RISC rates than the large-amplitude dihedral motions associated with vibronic coupling in D-A TADF emitters.
ESIPT-active chromophores can also be flexibly deployed in hybrid designs, for example combined with electron-donating fragments in PXZPDO, DMACPDO, and TPXZBM (Figure ). These display ESIPT in their acceptor moiety and each have small ΔE ST, which translated to devices with higher efficiency than non-ESIPT counterparts. The large Stokes shifts inherent in ESIPT-based emitters may also be harnessed towards the design of deep-red emitters. Especially considering their unique RISC pathway, we find it surprising that ESIPT materials have not received more research attention. Although impossible to predict, it may well be that a few high-performance materials – potentially discovered just outside the small regions of chemical space currently explored – could ignite global efforts and rapid development in this area.
Mechanochromism/Mechanoluminescence and TADF
Introduction
Mechanochromism and mechanochromic luminescence (MCL) involve changes of the emission spectrum and color of a material when external force is applied. This is distinct from triboluminescence, in which mechanical force directly causes the emission of light. The applied force in this context produces a change in the bulk material, usually in the packing arrangement such as a transition from the crystalline to the amorphous state or a crystal-to-crystal phase transition, which impacts the electronic structure of the molecules and hence their emission color.ref. ref1195 The force that triggers these changes in packing can be applied physically, such as by shearing and grinding, indirectly through heating, or involve various crystallization techniques including changing the solvent system or exposure to solvent vapor. Some of these structural changes can be reversible, resulting in switching behavior that is valuable in sensing and other applications.
Because of their sensitivity to D-A molecular geometries, as exemplified in emitters throughout Sections sec3 –sec5, mechanochromism has been observed in a number of D-A TADF materials, which are summarized in this section. In some examples the excited-state decay mechanism may change entirely depending on the packing arrangement, for example switching from TADF to fluorescence; however, most reports neglect to probe the operational emission mechanism of each of the different morphologies. Most of the reported examples also exhibit both AIE and MCL, with both properties arising from changes in molecular geometry.ref. ref1196 To date, there are only a few reports of TADF materials that have been observed to be mechanoresponsive (Figure –Figure ). A subset of these have also been used as emitters in OLEDs. Table S19 summarizes materials and their photophysical properties for which no OLEDs were fabricated, while Table S20 collates TADF compounds that show MCL and which were also used in or towards OLED applications.
Materials Development
The first reported example of a TADF emitter undergoing MCL was the D-A-D′ compound OPC (Figure ). It displayed dual-channel white emission with bands at 456 nm and 554 nm, the latter of which exhibited TADF.ref. ref604 The dual emission was found to be due to the coexistence of two different excited-state conformers associated with quasi–axial or quasi–equatorial conformations that are now commonly observed for the PTZ donor group. The emission at 456 nm is from the quasi-axial conformer that has a calculated ΔE ST of 0.56 eV, while the emission at 554 nm is from the quasi-equatorial conformer with a calculated ΔE ST of 0.01 eV. Upon grinding the quasi-axial conformer was converted to the quasi-equatorial conformer, culminating in exclusive emission from this lower energy species in the ground powder. In single crystals the quasi-equatorial conformer is the dominant species, with two emission bands once again observed.
Using a similar D-A design, Xie et al. reported dual emission for a series of PTZ-ketone emitters,ref. ref1197 where additional π-conjugated groups such as naphthalene (OPNa), pyrene (OPPy) and anthracene (OPAn) were also coupled to the acceptor (Figure ). In the crystalline state each compound displayed dual emission, with a high-energy high-intensity fluorescent band at between 429–454 nm. A second low-energy band located at around 570–587 nm was assigned to arise from TADF. Upon grinding the crystals, the high-energy band decreased in intensity and the low-energy band dominated the emission spectrum. This spectral change was assigned to increased intermolecular hydrogen bonding between the donor and acceptor components across neighboring molecules, turning on an intermolecular CT transition in the ground powder state. This CT state and low-energy emission band was indeed found to be TADF-active for OPNa, OPPy and OPAn. Additionally, these bulky groups proved to be essential to achieve MCL, as with just phenyl substitution only the low-energy intermolecular CT band was present, even in the crystal.
The D-A compound PTZ-AQ (Figure ) displays five different crystal morphologies, each with different photophysical properties.ref. ref1198 The five morphologies were described by their yellow, orange or red color and labelled as Y-Solid, Y-crystal, O-Crystal, R-crystal and R-solid by the authors, with λPL of 545, 554, 568, 606 and 649 nm, respectively. Each of the samples was obtained using different crystal growth techniques, while heating of the R-solid yielded the Y-solid. The reverse transition (Y-solid to R-solid) was possible through exposure to CH2Cl2 vapours. Color changes in the crystals were understood to arise from alteration in the π–π interactions in these systems. Interestingly, each solid displayed distinct ΔE ST and ΦPL values, with ΔE ST varying between 0.01 and 0.42 eV and ΦPL varying between 3 and 85% (Table S19). TADF was observed in the Y-Solid, Y-crystal, O-Crystal and R-crystal, while R-solid showed no TADF owing to its larger ΔE ST of 0.42 eV (0.01–0.25 eV for the others).
Two phosphine oxide-containing emitters, CPzPO and SPzPO (Figure ), showed dual emission in the crystalline state with λPL of 459 and 564 nm for CPzPO and 433 and 546 nm for SPzPO.ref. ref1199 The two emission bands displayed different emission mechanisms, with the lower energy bands showing TADF and τd of 62 and 29 μs, for CPzPO and SPzPO respectively. The higher energy bands were simply fluorescent in both materials. Upon grinding to an amorphous state, the intensity of the higher energy fluorescence band decreased. The contrasting intensities and TADF behaviour of the high and low energy bands were a result of changes in the packing arrangements, with intermolecular hydrogen bonding becoming more prominent in the ground species. Similar to the previous examples with OPC and related emitters, this enhancement of intramolecular interactions was proposed to be responsible for the enhancement of the low-energy TADF-active emission channel.
A similar effect was reported by Xu et al. for the emitter SCP (Figure ),ref. ref1200 where again dual emission was observed in the pristine form. Emission bands at 415 nm and 545 nm were observed, where emission from the high-energy band is purely fluorescent while emission from the low-energy band is TADF-active. Upon grinding, the intensity of the two peaks changed, with the longer wavelength TADF band dominating the spectrum, resulting in a significant color change. The ratio of these emission bands could be tuned to achieve white light emission. The high-energy band at 415 nm was assigned to the Cz-Ph → sulfone transition, while the emission at 545 nm was attributed to the PTZ → sulfone transition. Two contrasting calculated ΔE ST values of 0.99 eV (Cz-Ph CT state) and 0.44 eV (PTZ CT state), explain the differences in TADF properties, with TADF only observed in the latter despite the relatively large ΔE ST. The Cz-Ph conformation was proposed to planarize upon grinding, affecting the photophysical properties associated with this fragment and increasing the probability of energy transfer to the PTZ-centred excited CT state associated with the low-energy band, which then dominates emission.
Once again exploiting the two accessible conformers of PTZ,ref. ref1201 two MCL compounds using nitrogen-rich acceptors were developed by Okazaki et al., (1 and 2, Figure ), which emitted in the green and deep red, respectively. Using different solvent systems, distinct yellow or orange crystals of 1 were grown (1_Y and 1_O) with λPL of 568 nm and 640 nm, respectively. Upon grinding of either 1_Y or 1_O, a different red-emitting form 1_R was generated with λPL of 673 nm. Thermal annealing of 1_R produced 1_O2 (λPL of 646 nm), while exposure of 1_R to CH2Cl2 generated 1_YO (λPL of 646 nm). Grinding of either of 1_O2 or 1_YO reformed 1_R. The substantial color changes for 1 with different processing conditions were explained as a result of changing one or both of the PTZ conformation in each crystalline form, with 1_Y and 1_YO composed of axial-axial donors, 1_O2 and 1_O having axial-equatorial PTZ conformations, and 1_R being equatorial-equatorial (Figure ). Compound 2 has a similar structure but with –tBu substitution on the PTZ donors and showed a total of four colored forms. 2_YG (λPL of 547 nm) was obtained from recrystallization from hexane:CHCl3, 2_R (λPL 663 nm) was obtained from grinding of 2_YG, heating of 2_R to 240 °C formed 2_R2, while exposure of 2_R to CH2Cl2 vapor resulted in the formation of 2_Y. All generated samples reverted to 2_YG upon recrystallization from hexane:CHCl3. Both emitters 1 and 2 displayed TADF when doped at 10 wt% in CBP films, and efficient devices with EQEmax of 16.8% and 11.2% for the OLEDs with 1 and 2, respectively were demonstrated.
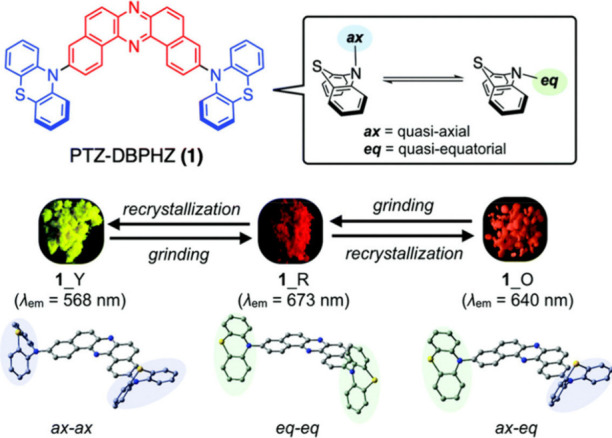
A TSCT emitter, XPT (Figure ),ref. ref1098 was also reported to undergo MCL. A red-shift of the emission from λPL of 536 nm to 569 nm was observed upon grinding of the single crystal to form a powder. Sublimation of XPT produced a similar spectral shift with λPL of 566 nm, similar to the powder form. In both ground and sublimed samples the original λPL of 536 nm could be reconstituted upon exposure to CH2Cl2 vapor. Compound XPT was used as the emitter in an OLED (EML: 10 wt% XPT in DPEPO) that showed an EQEmax of 10%. Although TADF was claimed in this report, it is not clear whether the TADF was also observed in the powder samples displaying MCL.
The same strategy of using axial and equatorial conformation changes to induce MCL was employed using a phosphine derivative of PTZ in the compound DPPZS-DBPHZ (Figure ).ref. ref1203 This compound showed strong color tuning from 496 nm to 704 nm between different conformers. Reversible color tuning was also demonstrated by recrystallization of the 1-BG conformer from four other accessible conformers, themselves accessed by various combinations of grinding, heating, or solvent vapor fuming. The conformers 1-BG, 1-G1, 1-G2, 1-Y and 1-DR emitted at λPL of 497, 518, 520, 534 and 740 nm (and with ΦPL of 6%, 9%, 9%, 16% and 3%) respectively. Different combinations of equatorial and axial donors were responsible for the different emission colors, with 1-BG equatorial-equatorial, 1-Y axial-equatorial, and 1-G1 and 1-G2 ascribed to be axial-axial with potential axial-equatorial conformers also present. The decay mechanism of each conformer was not investigated, although the compound in a 10 wt% Zeonex matrix showed both TADF and RTP.
Pashazadeh et al., documented MCL in samples of OIDBQx (Figure ),ref. ref1204 where the λPL of the powder red-shifted from 494 to 522 nm upon grinding. The original emission was restored upon exposure to CH2Cl2 vapor. Only fluorescence was observed for this compound in the powder forms, although both TADF and RTP were present in 1 wt% Zeonex films with an average delayed lifetime of 128 ms.
A series of materials presenting reversible MCL properties was reported by Yang et al., composed of a combination of planar acceptors (DPP and DPQ) and donors (DMAC and PXZ).ref. ref1205 DPP-DMAC, DPQ-DMAC, DPP-PXZ and DPQ-PXZ (Figure ) were each ground, fumed with CH2Cl2 or heated to achieve distinct color changes in each example. Switching between the colors was shown to be completely reversible for each emitter. Following grinding, a loss of crystallinity was observed and the materials became amorphous, while subsequent fuming and heating produced new crystalline packing motifs compared to the original sample. The most significant color change occurred following grinding, where a red-shift of the emission was observed from 554 to 608 nm, 548 to 571 nm, 589 to 616 nm and 628 to 682 nm for DPP-DMAC, DPQ-DMAC, DPQ-PXZ and DPP-PXZ, respectively. In each solid-state environment a delayed emission component was observed in the μs regime, with lifetimes ranging from 1.1 to 8.3 μs and assigned to TADF. Using DPQ-DMAC the authors demonstrated ‘ink-free rewritable paper’, where with the application of mechanical pressure it was possible to write text using the color change of the material. Upon exposure to CH2Cl2 vapor it was also possible to restore the color of the ‘written’ material, thus deleting the text. The potential of DPQ-DMAC as an emitter for OLEDs was also assessed, with the material doped at 10 wt% in DPEPO showing an EQEmax of 11.3% at 556 nm, and exhibiting low efficiency roll-off (EQE100 of 10.5%).
Another D-A system (material 2, Figure ) composed of diphenylamine as the donor and a boron atom linked to two anthracene units as the acceptor was reported by Pandey et al..ref. ref1206 This compound displayed dual emission as pristine powder with λPL of 455 and 530 nm. Upon grinding, only a single peak centred at around 540 nm remained. Exposing this new form to CH2Cl2, CHCl3 or hexane vapor though restored the original dual emission. No explanation was offered for this behavior, however different powder XRD patterns were observed for each form. The compound also displayed dual emission in PhMe solution, with both emission peaks at 430 and 530 nm being TADF-active, with τd of 5.9 and 5.8 μs.
MCL was observed for the emitter TPA-DQP (Figure ), which showed two distinct polymorphs. Crystal-Y and Crystal-R have λPL of 576 and 694 nm, respectively, and Crystal-Y was identified as the thermodynamic product.ref. ref1207 For Crystal-Y, CH−π interactions between the acceptor and the donor as well as π–π stacking between the acceptor groups were identified, while for Crystal-R, the packing structure was composed entirely of π–π stacking between the acceptor units. Upon grinding Crystal-Y the emission red-shifted from 576 to 698 nm, and the emission at 576 nm could be restored upon heating the sample. For Crystal-R, the color shifts were much less pronounced, with grinding red-shifting λPL from 694 to 706 nm, which was restored to 694 nm upon CH2Cl2 fuming. MCL observed in both polymorphs was rationalized as due to transferring from crystalline to an amorphous packing arrangement. Both crystals displayed prompt and delayed emission assigned as TADF, with τd of 1.1 and 2.4 μs for the Y and R-crystals, respectively. TADF was also observed in 10 wt% doped films in Bepp2, with a corresponding small ΔE ST of 0.11 eV. OLEDs fabricated with TPA-DQP showed an EQEmax of 18.3% at CIE coordinates of (0.67, 0.32).
For crystalline Cz-AQ,ref. ref1208 two emission peaks with λPL of 604 and 541 nm were documented corresponding to two distinct crystal packing regimes (R-crystal and Y-crystal, Figure ). The red-shifted emission was associated with a morphology featuring strongly π–π overlapped H-aggregates, while the higher energy band was linked to weaker J-aggregates. Both R-crystal and Y-crystal were interconvertable, with heating of R-crystal producing Y-crystal, while grinding or haloalkane fuming of Y-crystal recovering R-crystal. TADF was observed for both Y-crystal and R-crystal forms with τd of 1.8 and 1.9 μs while the ΦPL were 59 and 28%, respectively. The different packing regimes were subsequently exploited in solution-processed OLEDs. When dichloroethane was used to spin-coat the films, a device λEL of 680 nm was observed, while when a dichloroethane:ethanol (1:1) solution was employed, λEL was 600 nm. The EQEmax of the non-doped devices were low at 0.75 and 1.15%, respectively, and while doped devices showed higher EQEmax they did not have the color tuning potential of the non-doped devices. The change in both the λEL and the λPL was attributed to different aggregation states in the neat thin films, analogous to the Y-crystal and R-crystal forms.
A similar derivative using DPA as the donor and thioxanthone as the acceptor, TXDM (Figure ), was reported by Mane et al.. Exploiting both the MCL and oxygen sensitivity of this material, this work reported a logic gate based on the PL of this compound.ref. ref1209 Supporting this application, starkly contrasting photophysical properties were obtained for different morphologies, with the crystalline form of TXDM showing significantly quenched emission and no TADF (λPL of 470 nm, ΦPL of 1.8%). In the amorphous state the emission was much brighter (λPL of 486 nm, ΦPL of 27%) and exhibited TADF. These changes in the photophysics were ascribed to suppression of π–π stacking interactions in the amorphous state. ΔE ST was 0.30 eV for the amorphous form and increased to 0.42 eV for the crystal, with these differing ΔE ST responsible for the contrasting TADF activity. MCL was achieved upon grinding, heating, and fuming, with each of these external forces accessing a different output in the logic gate system.
Zhou et al. reported two pairs of enantiomeric emitters,ref. ref653 each containing a tetracoordinate boron acceptor, and chiral binaphthol or octahydro-binaphthol and DMAC donor groups. R/S-DOBP and R/S-HDOBP (Figure ), showed multifunctional properties, including CPL, mechanochromism, and piezochromism (Figure ). A significant spectral change was observed for R-DOBP upon grinding, with the emission red-shifted from 580 nm for the crystalline sample to 647 nm for the ground form. Application of pressure from 0 to 5.9 GPa the crystalline R-DOBP in a diamond anvil cell also led to a red-shifted emission with increased intensity. A further red-shift of the emission accompanying a gradual decrease of its intensity was observed when the pressure exceeded 5.9 Gpa. Interestingly, the emission spectrum associated with atmospheric pressure could be gradually recovered when the pressure was released. By contrast, R/S-HDOBP did not show any mechanochromism. The authors also fabricated solution-processed non-doped near infrared OLEDs that showed EQEmax of 1.9 and 0.7% with λEL of 716 and 700 nm using R-DOBP and R-HDOBP, respectively.
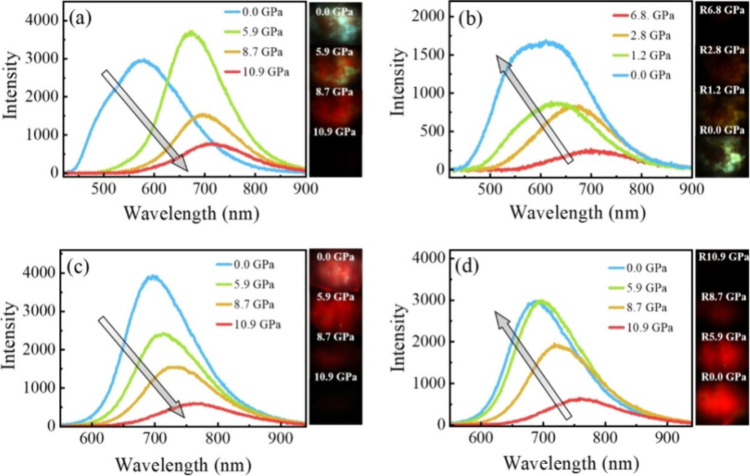
Two additional MCL-active TADF emitters, XT-T and XT-OT, were constructed from a xanthone acceptor and triphenylamine (T) or 4,4′-dimethoxytriphenylamine (OT) donors (Figure )ref. ref1210. The crystals of XT-OT showed a large red-shift from blue-green (λPL = 466 nm, ΦPL = 42.5%) to yellow (λPL = 567 nm, ΦPL = 53.4%) upon grinding. However, crystals of XT-T exhibited a much more attenuated spectral shift from blue (λPL = 478 nm, ΦPL = 38.1%) to green emission (λPL = 510 nm, ΦPL = 40.7%) upon grinding. The ground powders were fumed with DCM solvent vapors which restored the original emission. PXRD analysis indicated that the change in emission color was due to a crystalline-to-amorphous transition caused by the grinding. This study highlights how slight differences in chemical composition can have a large impact on the conformation of the compounds, on the intermolecular interactions and packing arrangements in the crystal and ground forms, and thus on the extent of MCL response. OLEDs were also explored using 10 wt% emitter doping in CBP host. Devices with XT-OT showed superior performance, with EQEmax 9.4% and λEL of 532 nm compared to XT-T (EQEmax 3.3% and λEL of 488 nm). This performance dichotomy was likely caused by the larger ΔE ST and longer τd of XT-T.
The compound Py-BZTCN is another example of a TADF material where the emission mechanism changes upon grinding (Figure ).ref. ref1211 The pristine crystalline powder showed orange-yellow fluorescence at λPL of 581 nm (ΦPL of 52.8%; τp 2.37 ns). The emission of the ground powder was red-shifted to λPL of ∼676 nm with an associated smaller ΦPL of 5.3%, and also showed TADF (τp = 7.97 ns; τd = 2.30 μs) with a small ΔEST of 0.087 eV. Similar to other systems, grinding disturbs the ordered π–π stacking of the pristine crystalline powder, converting it into an amorphous form as evidenced by the changes in the PXRD pattern. The original emission is again recovered by solvent fuming or heat treatment.
Beyond MCL, the broad category of mechanoluminance (ML) also encompasses triboluminance and fractoluminance.ref. ref1212 These emission categories progress in the same way as photoluminescence, with the only difference being the method of exciton generation.ref. ref1213 At present, compound 1 is the only example of ML exhibited by a TADF material (Figure ).ref. ref1214 Emission was observed upon scratching the powder sample, which shared the same spectrum as typical photoluminescence. While delayed emission was not explicitly measured for this material upon mechanical stimulus, it was shown to be TADF-active in the powder form upon photoexcitation. In the powder, the compound emits at λPL of 518 nm, with τd of 1.2 ms and has a ΔE ST of 0.20 eV.
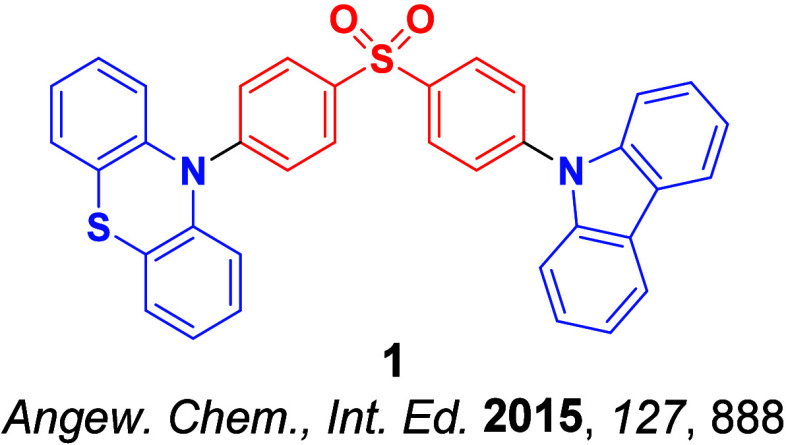
Outlook
As explored in this section, MCL and mechanoluminescence have been reported for a moderate number of TADF emitter systems. Although materials containing PTZ donors feature heavily, MCL can arise across a diverse range of emitter structures, and it remains difficult to predict a priori which compounds will show MCL or what underlying conformational changes alter the photophysics. This phenomenon also highlights how solid-state packing of TADF molecules can significantly and unpredictably impact the optical properties, which are so closely tied to molecular geometry adopted by D-A TADF emitters.
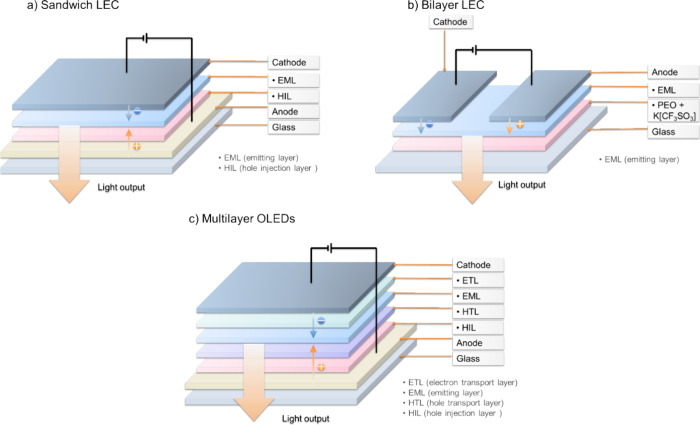
We note that although some niche applications have been demonstrated, the observation of MCL remains largely an academic curiosity. It is not clear how this property might ever be utilised in thin, fragile, and encapsulated OLEDs. Nonetheless, materials exhibiting MCL and TADF properties can have independent applications of each property, and we propose that harnessing both simultaneously may unlock future utility. For example, since the lifetime of a TADF-MCL emitter can be used to distinguish between different forms of the material, time-resolved measurements could present a quantitative detection method with potential in anti-tampering and advanced anti-counterfeiting applications beyond simple ratiometric colorimetry. Pressure sensitive materials are also desirable considering the demand for stress/strain sensors, and TADF materials could present appealing candidates for in situ optical readout in both emission color and kinetics.
Despite the current lack of compelling applications for this class of compounds, they should be recognized as occupying the crossover region between optoelectronic and mechanically responsive material. Additionally, we speculate that a vast number of reported TADF emitters may have as-yet undiscovered MCL activity, which would escape the notice of the wider OLED community. Indeed, even venerable 4CzIPN shows such properties, which were not discovered until relatively recentlyref. ref125 – likely because grinding and solvent vapor exposure are simply not widespread characterization techniques. It is therefore reasonable to expect that MCL may support innovative applications in the future beyond our current imaginations, with a wide library of candidate materials already available to be deployed.
TADF Light-Emitting Electrochemical Cells (LECs)
Introduction
In contrast to the OLEDs that are the electroluminescent device of focus in the previous sections, light-emitting electrochemical cells (LECs) have emerged as an alternative class of electroluminescent devices. Although a few examples of analogous single-layer OLEDs exist (e.g., single-layer PhOLEDs),ref. ref1215 LECs offer an overall simplified device structure and are generally fabricated using solution-processing techniques. The most common architecture is a sandwich LEC, with an active layer and a hole injection layer (HIL) sandwiched between an air-stable cathode and a transparent anode (Figure a). The active layer is typically a blend of luminescent materials, ion transporting materials, and inorganic salts. There are also examples of LECs using polymers,ref. ref1216 ionic transition-metal complexes,ref1217,ref1218 and organic small molecules (SMs) as emitter materials.ref. ref1219 Similar to solution-processed OLEDs, the HIL in LECs is typically a blend of poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT:PSS). This water-soluble polymer is coated onto a glass substrate with transparent indium tin oxide (ITO) anode, and provides a smooth electrode surface with increased work function (WF) to promote charge injection, while also being impervious to subsequent depositions from organic solvents.ref. ref1220 A metallic cathode such as aluminium is then deposited on top of the active layer to complete the device, typically by thermal evaporation.
A key feature of LECs that distinguishes them from OLEDs is the use of ions in the active layer to achieve charge transport, rather than relying on direct transport of electrons and holes through static layers. When an external bias is applied to the LEC, the separation of the ions in the active layer reduces the injection barrier, which enables the use of air-stable cathodes and established an in situ electrochemical doping of the organic semiconductor, forming a p-n junction across the active material.ref. ref1216 The addition of salts into the emitter layer (EML) enables balanced electron and hole flow, translating into a high recombination rate of these particles into excitons. However, this operational mechanism also leads to increased exciton-polaron annihilation, affecting device performance much more acutely than in OLEDs. Thus, it remains an open research question whether a LEC can show high efficiency at high luminance.ref. ref1221
In 2010 Sandström et al. reported a second LEC device structure based on a planar bilayer architecture (Figure b) that is similar to a bottom-gate top-contact transistor.ref. ref1222 Thanks to this architecture the luminescent materials is largely separated from the electrolyte, which typically consists of a mixture of K[CF3SO3] and poly(ethylene oxide) (PEO). This planar structure with charge transport along rather than through the layers also permits observation of the temporal evolution of the luminance of the device, and insights into the device degradation mechanism.ref1223,ref1224
For LECs there are two principal models that explain the microscopic working mechanism (Figure ).ref. ref1225 The first is known as the electrochemical doping model (ECD, Figure a).ref. ref1226 Upon application of a voltage, electrolyte anions start migrating towards the positively charged electrode while holes are injected into emitter molecules, producing radical cations. The opposite processes occur at the negatively charged electrode, forming a very thin electric double layer (EDL) of approximately 1 nm on each side of the device. The presence of these EDLs causes a substantial drop of the electric potential at the electrodes and facilitates further charge injection into the active layer. At the cathode the injection of electrons is compensated by diffusion of cations, which results in the formation of an n-type doped region. At the opposite electrode the extraction of electrons at the anode attracts anions and forms a p-type doped region. Such p- and n-doped type regions grow from the electrodes towards the centre of the cell, where radiative recombination takes place and steady-state emission is eventually established. The reliance on diffusion and growth of doped regions in the device aligns with their relatively long experimental turn-on times (ton), typically reaching maximum brightness over a few seconds or minutes.ref. ref1227
The second LEC model is known as the electrodynamical model (ED) (Figure b).ref. ref1228 As in the ECD model, charge injection is also made possible by the formation of the EDL at the electrodes. When the applied voltage is high enough, electrons and holes can additionally travel through the LUMO and HOMO levels of the semiconductors, respectively, and recombine to form excitons in the central field-free region and emit light.
Although the mechanisms are microscopically different, experimentally both models have been shown to be feasible.ref1229,ref1230 Indeed, van Reenen et al. revealed that a changeover in operating regimes occurs depending on the ability of the device to form non injection-limited ohmic contacts.ref. ref1218 When ohmic contacts are formed the LEC follows the electrochemical doping model, but when the injection of charge carriers is limited the device instead follows the electrodynamical model.
As with an OLED, the EQE of a LEC is defined in terms of equation eq19 :
where b is the fraction of holes and electrons that recombine to excitons (analogous to γ for OLEDs, Section sec1 equation eq1 , φ is the fraction of electrically produced excitons that can decay radiatively (β·Φ PL for OLEDs), and describes the outcoupling efficiency with n the glass substrate refractive index.ref. ref1231 Typically, there is unitary recombination of holes and electrons in LECs (b = 1),ref. ref1232 and therefore the EQE will depend primarily on the emitter’s inherent ability to harvest excitons and convert these to light – i.e., its ability to harvest singlets and triplets, and its emission efficiency. Clearly the use of emitters that are capable of harvesting both singlet and triplet excitons is highly desirable, and therefore TADF materials have attracted increasing attention within the LEC community. This section reviews progress in the development of both all-organic TADF emitters and copper(I) TADF complexes for LECs. Data of the emitters and devices summarised in this section are also collected in Table S21.
Ionic TADF LECs
In parallel with the development of OLEDs, LECs have historically employed cationic phosphorescent emitters to manage triplet excitons, mostly based on Ru(II) and Ir(III) complexes.ref1233,ref1234 There have also been a few examples involving the use of cationic organic fluorescent compounds as emitters in LECs.ref. ref1235 Our group reported the first example of an LEC using a cationic organic TADF emitter in 2015 (Figure ).ref. ref1236 The emitter skeleton was derivative of 2CzPN with pendant imidazolium groups linked to the carbazole donors, 2CzPN-LEC (originally named 2 in that work). This compound has a ΦPL of 90% as a 10 wt% doped film in PMMA and emits with λPL of 536 nm. In neat film the ΦPL drops to 21% but with unchanged λPL. LECs using a neat film of 2CzPN-LEC as the active layer showed an EQEmax of 0.4% at λEL = 538 nm. The LEC showed a very low luminance of 12 cd m–2, as well as a decreasing driving voltage and luminance with time. An LEC incorporating the ionic liquid [Bmim][PF6] as additional electrolyte performed even more poorly, with an EQEmax of 0.12%. This result was surprising considering literature precedents of improved LEC performance when ionic transition metal complex emitters (iTMC) are co-doped with ionic liquids in the EML.ref. ref1237 The same ionic TADF emitter was also used as a host in combination with a yellow fluorescent cyanine dye.ref. ref1238 The reported EQEmax for this host-guest device was 1.9%, implying very efficient exciton utilization and high FRET efficiency from the TADF host to the cyanine dye, mirroring the hyperfluroescence approach developed for OLEDs. We later developed a blue-emitting LEC using the same cationic carbazole donor in combination with a weaker sulfone acceptor, imCzDPS (Figure ).ref. ref1239 The LEC emits at λEL = 470 nm but showed a very low maximum luminance of 2.5 cd m–2 under an average current density of 200 A m–2, and an EQEmax of 1.14%. The low luminance was attributed to the electrochemical instability of the emitter.
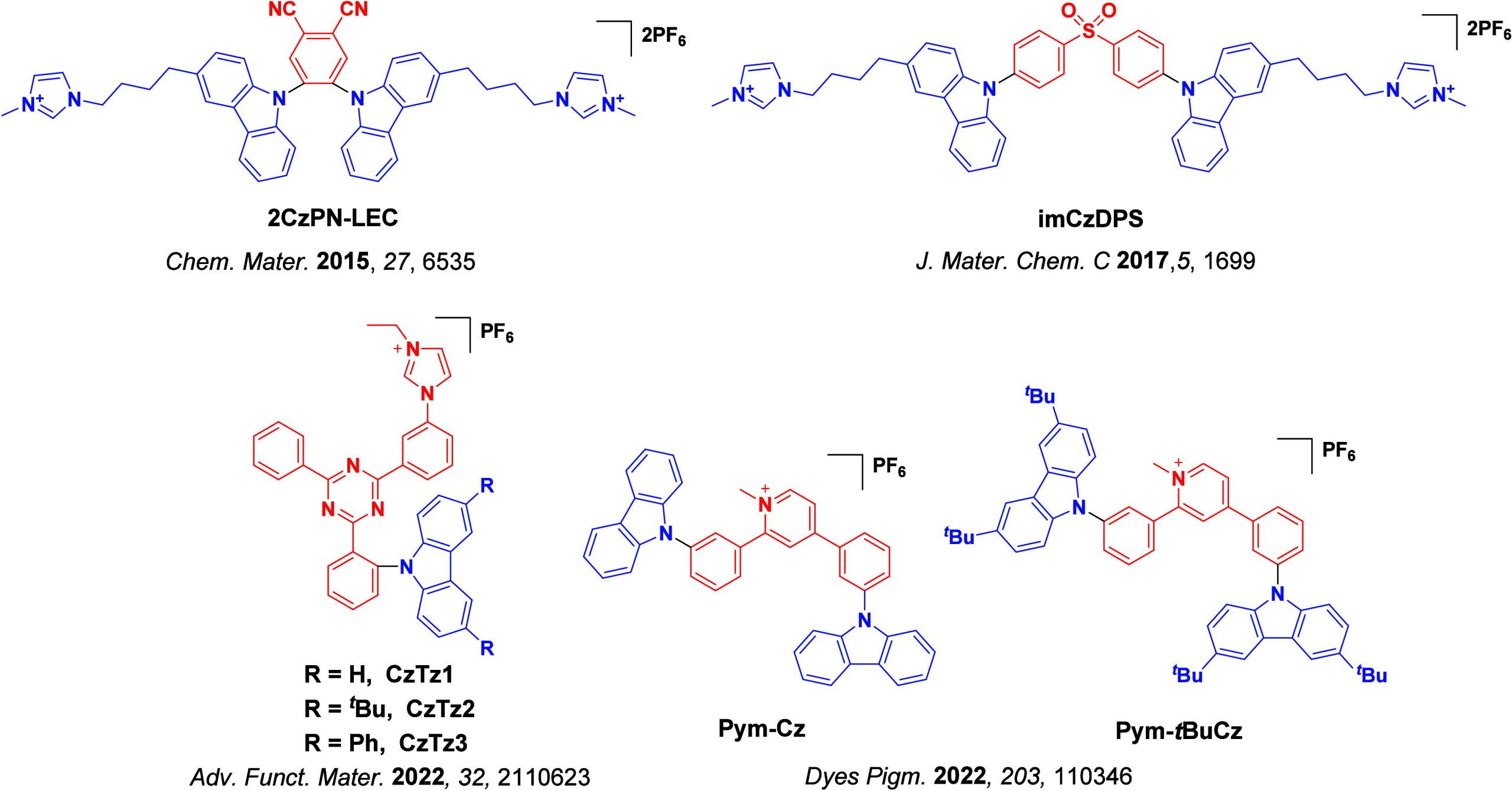
In 2022 a series of ionic D-A TADF compounds, CzTz1, CzTz2 and CzTz3 (originally named 1, 2 and 3 in that work) were reported by Yu et al. as green emitters in LECs (Figure ).ref. ref1240 The architecture of the devices was ITO/PEDOT:PSS/neat emitter/Al, and the most efficient LEC with CzTz2 as the emitter showed an EQEmax of 6.8%, and Lummax of 572 cd m–2 at CIE coordinates of (0.34,0.57). The device LT50 was 11.4 h under the at 4V and could be increased to 218 h when the device was driven at a lower constant current of 10 A m–2. The ton of the CzTz2-based LEC under a constant driving voltage of 4.0 V was 740 min, which was rationalized due to the slow motion of the [PF6]− anions, while under a constant current of 50 A m–2, the ton for the same device was only 5.5 min.
Another two ionic emitters, Pym-CZ and Pym-tBuCZ (Figure ) were used in orange-red emitting LECs by Shen et al.ref. ref1241 The design strategy of these materials was distinct, as rather than appending an ionic group to a TADF-active core, instead an ionic methylpyridinium unit also formed the central acceptor of the D-A TADF structure. Orange-red emission from aggregates of Pym-CZ was present at the high doping concentrations used in the EML. The most efficient device with Pym-CZ showed an EQEmax of 1.19%, a CEmax of 2.48 cd A–1 under 3.0 V, and a Lummax of 8.69 cd m–2 under 4.0 V. The ton of this device under 4 V was about 9 min, longer than the device with Pym-tBuCZ (about 5 min).
Neutral TADF LECs
A green-emitting device was reported by Lundberg et al. in 2017, utilising 4CzIPN (Figure ).ref. ref1242 In this example the emissive layer contained a mixture of host CBP, electrolyte (K[CF3SO3] in PEO), and polystyrene which helped to produce a homogeneous film. The optimal ratio of materials in the emissive layer was found to be 10:3:2.6:0.78:1.81 for CBP:4CzIPN:PEO:K[CF3SO3]:polystyrene. The inclusion of a layer of PEDOT:PPS between the ITO and the emitting layer proved essential to prevent short-circuiting of the devices. The LEC showed an EQEmax of 0.17% under a constant current of 770 A m–2 and an impressive Lummax of 760 cd m–2 during a voltage ramp, which constitutes a much-improved brightness compared to TADF LECs using charged emitters. The ton of this device was also less than 15 s, while the low EQE was attributed to the high electrolyte loading of 18.5 wt%.
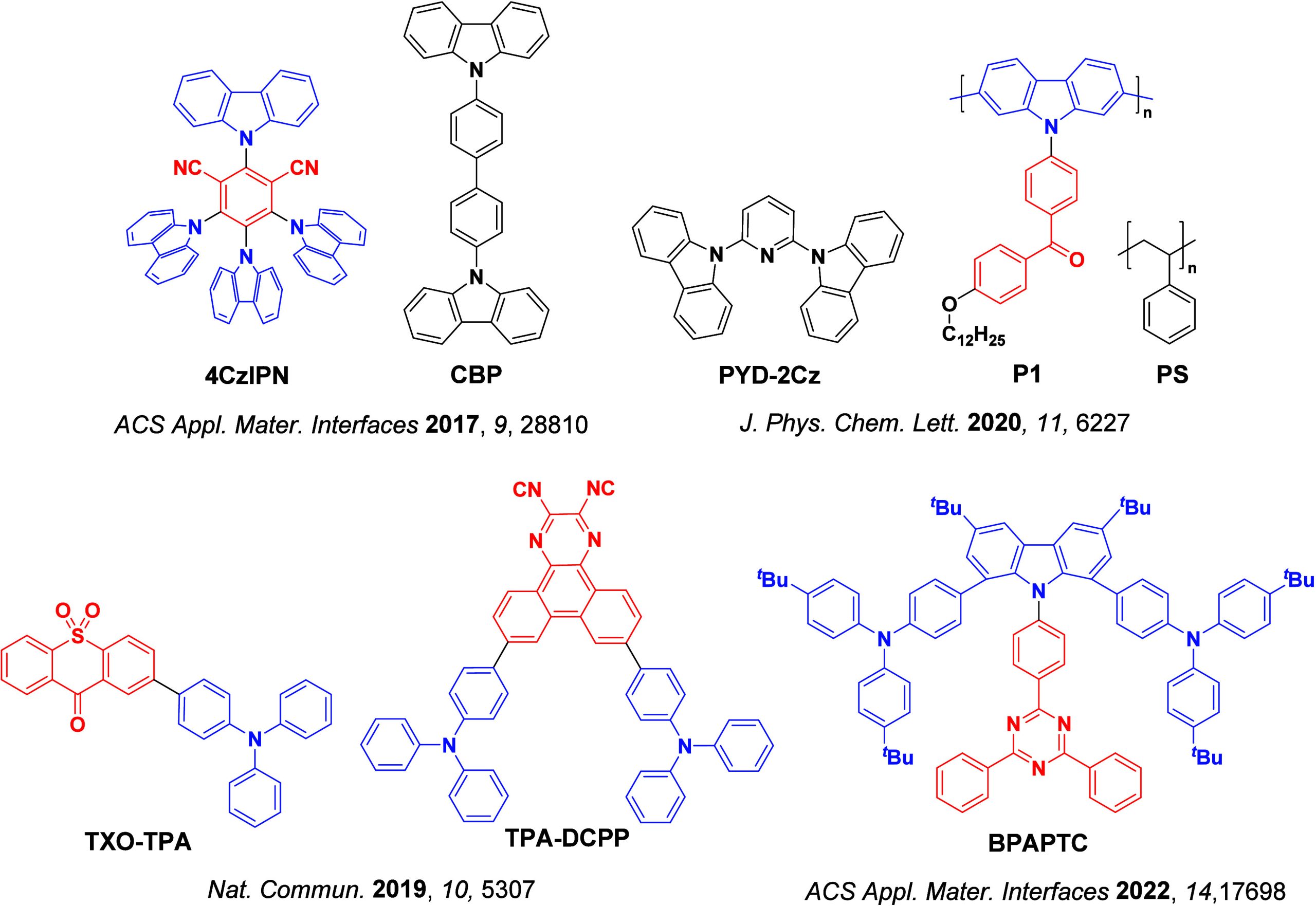
The same group later reported the first example of a LEC employing a TADF polymer as the emitter.ref. ref1243 The EML of the LEC incorporated the ambipolar host, PYD-2Cz, and the TADF polymer emitter, P1, along with ionic liquid tetrahexylammonium tetrafluoroborate (THABF4) as electrolyte in a ratio of 66:17:8:9 (Figure ). The LEC showed a luminance of 96 cd m–2 at 4 V, a CE of 1.4 cd A–1, and >600 cd m–2 at 6 V.
Exciplexes are excited states that can form when mixtures of donor and acceptor molecules interact to produce intermolecular charge-transfer (CT) excited states.ref. ref671 Exciplexes frequently show TADF (see Section sec8 ) and have been widely employed as both hosts and emitters in OLEDs. In 2019, Lundberg et al. demonstrated highly efficient LECs using each of the TADF emitters 4CzIPN, TXO-TPA and TPA-DCPP (Figure ), all using the same polymer exciplex host material composed of a blend of p-type PVK and n-type OXD-7 and driven by 100 A m–2.ref. ref1244 The balanced hole and electron transport from the host blend significantly improved the efficiency of the devices by reducing exciton–polaron quenching.ref. ref1221 The most efficient LEC in this report was obtained with TXO-TPA as the emitter, and showed an EQEmax of 7.0% and a CE of 16.0 cd A–1 at 120 cd m–2 with CIE coordinates of (0.46, 0.50). In comparison, the OLED with TXO-TPA showed an EQEmax of 18.5%, a CEmax of 43.3 cd A–1, and a Lummax up to 16300 cd cm–2,ref. ref1141 illustrating that LECs still require significant development to match the efficiency of corresponding OLED. The device with TPA-DCPP showed the highest performance for a red TADF LEC to date, with λEL = 618 nm, CIE coordinates of (0.54, 0.44), and an EQEmax of ca. 4% and a Lummax of 380 cd m–2. The turn-on time to a luminance of 100 cd m–2 (ton100) ranged between 20–25 s for all three devices under a current density of 100 A m–2.
Similarly, using PVK:OXD-7 as the co-host materials, Ye et al. reported a green TADF LEC with BPAPTC (Figure ) as the emitter.ref. ref1245 The active layer in the most efficient device used a blend of PVK:OXD-7:BPAPTC:THABF4 in a 23.4:15.6:9:2 ratio. This LEC showed an EQEmax of 7.67%, a Lummax of 3696 cd m–2 and a CEmax of 23.64 cd A–1 at a λEL of 533 nm, representing the highest EQEmax and Lummax reported for TADF LECs to date. The exceptional performance in this device was attributed to intramolecular π–π stacking and hydrogen bond interactions in BPAPTC, which contribute to reduced ACQ. As such, a greater emitter doping concentration (18 wt%) could be exploited, translating to improve exciton-harvesting efficiency and higher EQE and luminance.
Bai et al. reported the first example of an LEC using an ionic exciplex system as a host material (Figure ).ref. ref1246 The exciplex host was formed between cationic donor ([tBuCAZ-ImMe][PF6]) and acceptor ([TRZ-ImEt][PF6]), which was used in combination with [Ir(buoppy)2(dmapzpy)][PF6] as an emissive dopant in the EML of the LEC. The device with only the exciplex showed green emission and an EQEmax of 2.6%, a CEmax of 6.4 cd A–1 under 5.0 V, and a Lummax of 231 cd m–2 under a constant current of 50 A m–2. The best device with the iridium complex co-dopant in the EML showed an EQEmax of 11.5% at 4.0 V, a low Lummax of 45 cd m–2, but a high current efficiency of 25.8 cd A–1, at λEL of 473 nm.
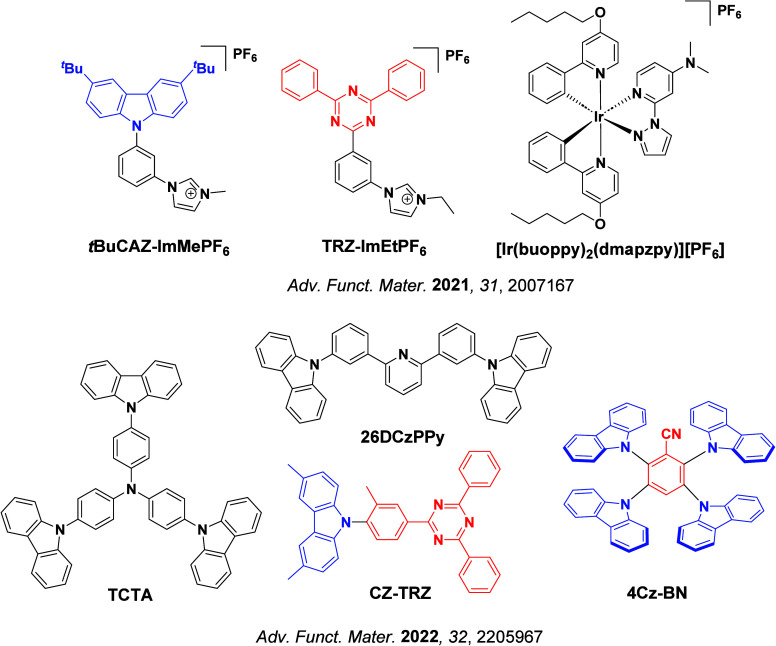
Adopting an EML more reminiscent of an OLED, several blue host-guest TADF LECs were reported by Tang et al. These devices used TCTA and 26DczPPy as host materials and CZ-TRZ or 4CZ-BN as TADF emitters (Figure ),ref. ref1247 with the best device consisting of 31.2:31.2:31.2:6.4 for TCTA:26DczPPy:CZ-TRZ:THABF4, and TCTA and 26DczPPy acting as co-hosts. This LEC showed an EQEmax of 5.0% at a constant current density of 7.7 mA cm–2, a Lummax of 740 cd m–2, and a CE of 9.6 cd A–1. Emitting at λEL of 475 nm, this device shows the highest performance for a blue TADF LEC to date due to the use of the exciplex-forming co-host materials that can harvest both singlet and triplet excitons and then transfer these to the guest TADF emitter by FRET.ref. ref1248
MR-TADF LECs
Due to their narrowband emission MR-TADF emitters have quickly become a hot topic for OLED applications (see Section sec11 ). With LEC development once again mirroring OLEDs, we were the first to use ionic MR-TADF compounds (DiKTa-ObuIm and DiKTa-DPA-ObuIm, Figure ) as emitters in LECs.ref. ref1249 The device with DiKTa-ObuIm showed a Lummax of 15 cd m–2 at 5 V and emits at λEL = 534 nm, while the Lummax of the device with DiKTa-DPA-ObuIm was only 2 cd m–2 at 8 V and emitted at λEL = 656 nm. Much like the initially inferior performance of MR-TADF emitters in OLEDs, we expect that use of these in LECs will rapidly progress as design and application strategies are discovered. The red-emitting device based on DiKTa-DPA-ObuIm should be highlighted, as examples of red LECs using organic emitters are rare.
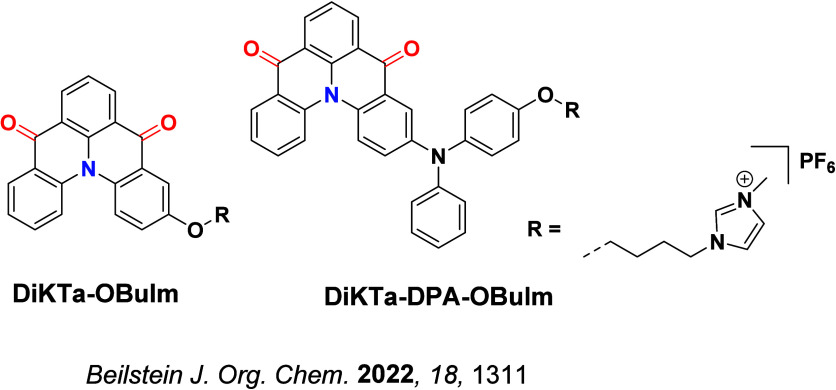
Cu(I) Complex TADF LECs
In addition to fully organic TADF molecules, copper-based ionic transition metal complexes that display TADF (Cu-iTMCs) are also attractive as triplet-harvesting LEC emitters.ref. ref1250 Similar to some TADF Carbene-Metal-Amides used in OLEDs (see Section sec9 ), instead of relying only on the large spin–orbit coupling conferred by a heavy metal centre in phosphorescent materials, many copper(I) complexes emit via TADF or a combination of TADF and phosphorescence as a result of the small singlet–triplet energy gap (ΔE ST) and the MLCT nature of the lowest-lying excited states.ref. ref752 Unlike other 3d-metal complexes, the d10 electronic configuration in Cu(I) means that there are no non-radiative d→d* electronic transitions, rendering this family of complexes unusually luminescent. The smaller SOC coefficient of Cu compared to 4d- and 5d- elements results in relatively slower phosphorescence and ISC rates, which allows TADF to become a competitive processes in the Cu(I) complexes.ref. ref1251 All these characteristics make copper(I) complexes an attractive alternative to iridium complexes for their use as emitters in electroluminescent devices.
One consequence of the low-lying MLCT states is that upon excitation the Cu(I) formally is oxidized to Cu(II), which then undergoes Jahn-Teller distortion to a flattened geometry that is both more susceptible to nucleophilic attach and which leads to greater non-radiative decay. Such nucleophilic reaction leads to a pentacoordinate excited complexes, which also relaxes via non-emissive deactivation paths that lead to a reduction in the ΦPL.ref. ref1252 The solution to both concerns is to use bulky ligands that limit the degree of geometric distortion in the excited state, preserving the tetrahedral geometry of the ground state and sterically shielding the metal centre from additional coordination. The most widely investigated family of copper complexes consequently contain both a diimine (N∧N) and a diphosphine (P∧P) ligand, [Cu(N∧N)(P∧P)]+, within which the P∧P ligands are typically very bulky bis(2-(diphenylphosphino)phenyl)ether (POP aka DPEPhos) or 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (xantphos) derivatives (Figure a). The N∧N ligand is usually a derivative of 2,2′-bipyridine (bpy) or 1,10-phenanthroline (phen) (Figure b).
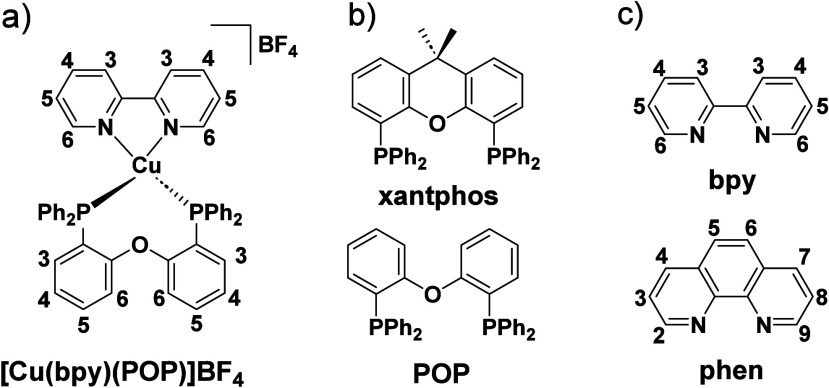
In [Cu(N∧N)(P∧P)]+ complexes, substitution at the 6- and 6′-positions of bpy (the 2- and 9-positions of phen, Figure b) is often desirable to further limit geometric changes in the excited state.ref1253,ref1254 Such restriction leads to a blue-shifted emission, higher ΦPL and longer excited state lifetime than complexes that do not contain substituents at these positions,ref. ref1255 as substitution at these positions help to prevent relaxation to lower energy geometries with more active non-radiative decay.ref. ref816
Keller et al. explored the impact of alkyl substitution of the N∧N ligand of [Cu(N∧N)(P∧P)]][PF6] complexes on the performance of the LECs.ref. ref1256 They found that [Cu(6,6′-Me2bpy)(POP)] (Figure , λPL = 564 nm, dissolved in CH2Cl2) had a more blue-shifted emission than [Cu(4,5,6-Me3bpy)(POP)] (λPL = 598 nm). Meanwhile [Cu(6,6′-Me2bpy)(xantphos)] (λPL = 606 nm) had a more red-shifted emission than [Cu(4,5,6-Me3bpy)(xantphos)] (λPL = 582 nm). For thin films consisting of 4:1 [Cu(N∧N)(P∧P)]+:[Emim][PF6], [Cu(6,6′-Me2bpy)(POP)] showed the highest ΦPL of 38%, [Cu(xantphos)(6,6′-Me2bpy)], [Cu(4,5,6-Me3bpy)(xantphos)] and [Cu(4,5,6-Me3bpy)(POP)] had ΦPL of 22, 19 and 16%, respectively. A possible explanation for these differences in ΦPL is that for complexes with POP, which has a more flexible structure than xantphos, two methyl groups (or other substituents) next to the nitrogen atoms of the bpy are necessary to efficiently stabilize the tetrahedral complex geometry. However, for xantphos, a single alkyl substituent on the N∧N chelating ligand is sufficient to stabilize the geometry. Further, the total number of alkyl groups on the N∧N chelating ligand had a stronger impact on the HOMO–LUMO gap than their position. The best performing devices in this report used [Cu(4,5,6-Me3bpy)(xantphos)][PF6] and [Cu(2-Etphen)(POP)][PF6]. The device with [Cu(4,5,6-Me3bpy)(xantphos)][PF6] showed an EQEmax of 1.7% and Lummax of 462 cd m–2 under an average current density of 100 A m–2 at λEL = 570 nm, and had a ton of 13 min. Under the same average current density, the LEC based on [Cu(2-Etphen)(xantphos)][PF6] showed an EQEmax of 1.8% and a Lummax of 451 cd m–2 at λEL = 582 nm, with a ton of 25 min. However, this latter device was more stable and had a longer LT50 of 34.0 h. This improved stability was attributed to the similar structure of 2-Etphen and 6-Etbpy ligands, the later of which has been proven to lead to a long lifetime.ref. ref1257 Although the electron-donating ability of the alkyl substituent at the α-position to the nitrogen atom of the N∧N ligand typically leads to a blue-shifted emission and higher ΦPL for the complex compared to analogues without this substituent, substitution with a bulky tBu group led to a lower ΦPL and shorter excited state lifetime due to the steric crowding about the metal centre. This excessive crowding results in elongated Cu–N bonds that affects both the non-radiative decay rates and the LUMO level that is localized on the N∧N ligand and demonstrates that careful management of the steric environment is required for this category of materials.
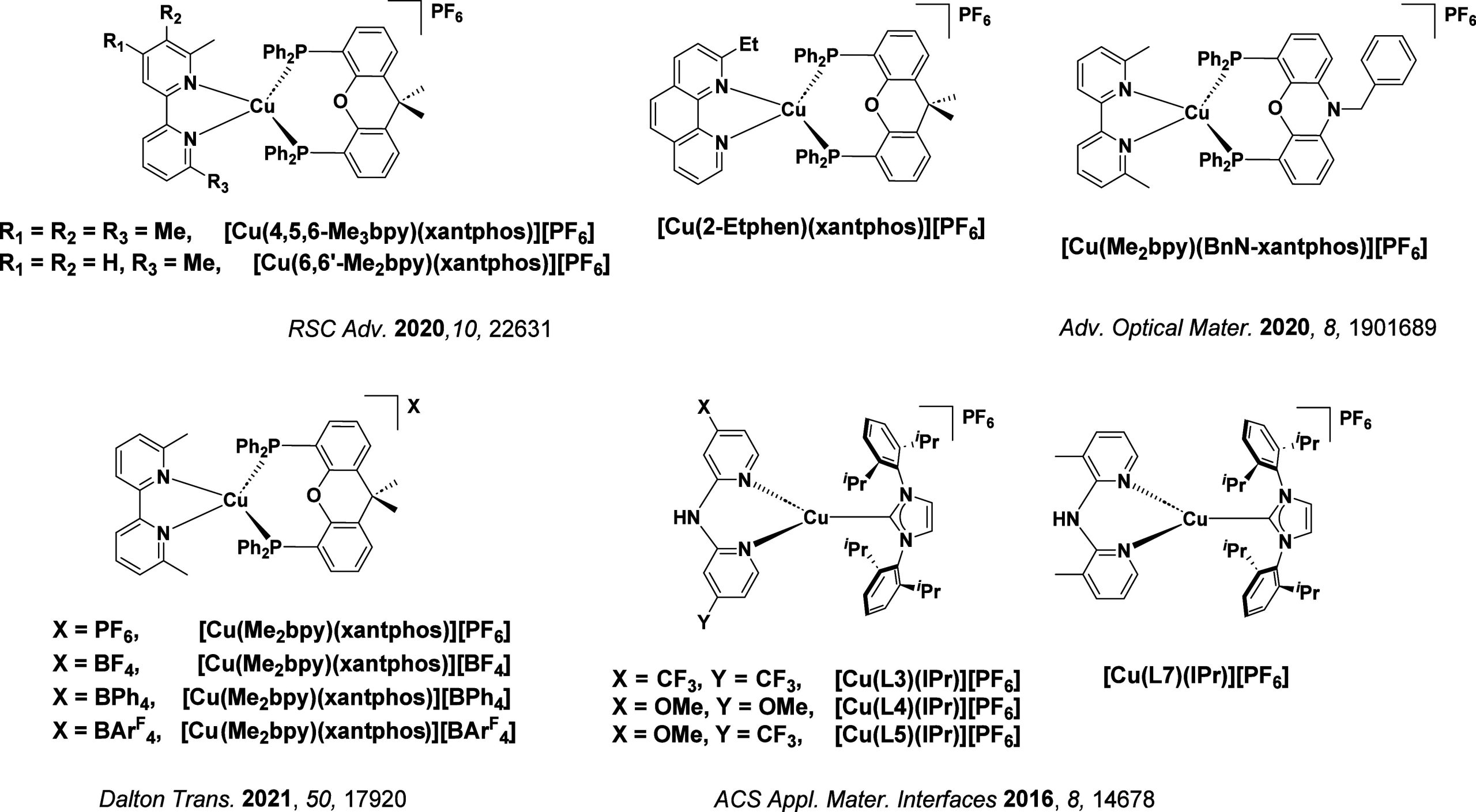
Arnosti et al. investigated the influence of hole injection layers on the efficiency of Cu(I) LEC devices.ref. ref1220 Different compositions of PEDOT:PSS (CLEVIOS P VP CH 8000, PEDOT:PSS = 1:20 w/w and CLEVIOS P VP AI 4083, PEDOT:PSS = 1:6 w/w) were employed as hole injection layers in devices with [Cu(Me2bpy)(BnN-xantphos)][PF6] as the emitter (Figure ). The device using the CLEVIOS P VP CH 8000 film – which has a higher PSS content, lower conductivity and a higher work function (WF) – showed the best performance with an EQEmax of 1.2% and Lummax of 355 cd m–2 under a current density of 100 A m–2 at λEL = 567 nm. The improved performance of this device was attributed to the larger injection layer WF that can facilitate hole injection from the anode to the adjacent layers. The lower conductivity of the CLEVIOS P VP CH 8000 was also hypothesized to decrease the rate of non-radiative recombination at the PEDOT:PSS interface, which may result in lower exciton quenching.
The influence of counterions on the performance of Cu(I) LECs was investigated by Meyer and co-workers.ref1220,ref1258 A family of [Cu(N∧N)(P∧P)]+ complexes where the counterion differed between [BF4]−, [PF6]−, [BPh4]− and [BarF 4]− were studied, and the complexes with the larger [BPh4]− and [BarF 4]− counterions were found to be more loosely packed. Π-Stacking interactions between copper complexes has been shown to enable higher ΦPL and can assist with restricting molecular geometry distortions, and these interactions can be disrupted with bulkier counteranions.ref. ref1259 The LEC devices with [Cu(Me2bpy)(xantphos)]+ (Figure ) as the emitter and smaller counterions [BF4]− and [PF6]− showed rapid turn-on times (ton of 15 s and 58 s to reach a luminance of 100 cd m–2), while the devices with larger counterions [BPh4]− and [BarF 4]− failed to turn on at all, presumably because of the poor charge injection caused by the lower ionic mobility of the large counterions.
A series of 3-coordinate Cu(I) complexes have also been developed, employing N-heterocyclic carbenes (NHCs) as the monodentate ligand along with a bidentate N∧N ligand. Unlike most transient carbenes, the lone pair located in the plane of the heterocyclic ring of NHCs makes these compounds nucleophilic, excellent σ-donors, and able to easily bind transition metals.ref. ref1260 NHCs have therefore become an attractive class of ligands in copper(I) complexes due to strong bonds to the metal and the ability to easily modify its structure, allowing for wide emission color tunability.ref. ref842 Previous research has also demonstrated that the combination of NHC and dipyridylamine (dpa) in copper(I) complexes can lead to high-efficiency emitters.ref. ref1261
To further investigate the impact of ligand modification on the photophysical properties, Elie et al. reported several emitters with both NHC and dpa-type ligands for blue LECs.ref. ref1262 The study revealed that the dpa ligands have a more significant impact on emission in Cu(I) complexes than the NHC. The λem in emitters with the same NHC ligand and different dpa ligands indeed varied widely from 420 to 550 nm. With the same dpa ligand, different NHC ligands instead had little effect on λem, ranging only from 465 to 481 nm. However, the different NHCs did lead to significant changes in ΦPL, varying from 17 to 64%. This implies that substituting the NHC ligand could potentially increase the radiative rate constant and/or reduce the non-radiative rate constant, thereby increasing ΦPL without affecting λem in the blue region. By comparing [Cu(L3)(Ipr)][PF6], [Cu(L4)(Ipr)][PF6] and [Cu(L5)(Ipr)][PF6] (Figure ), which have different substituents at the same 4,4′-position of dpa, it was additionally demonstrated that this asymmetrical substitution leads to a significant enhancement in ΦPL. Specifically, the ΦPL was found to be < 5% in [Cu(L3)(Ipr)][PF6] and [Cu(L4)(Ipr)][PF6], but increased to 20% in [Cu(L5)(Ipr)][PF6]. This impact of asymmetrical substitution at the 4,4′-position of dpa, which the authors termed a push-pull effect, was attributed to the more distinct intraligand charge transfer character in [Cu(L5)(Ipr)][PF6]. The best device in this report used [Cu(L7)(Ipr)][PF6] and showed green emission at 497 nm, a Lummax of 80.3 cd m–2 at 33.2 mA cm–2 and a high current efficiency of 0.29 cd A–1 at 16.65 mA cm–2. A long LT50 of 16.5 min was also achieved at a low constant current of 9.97 mA cm–2. The observed red-shift of the EL spectrum over time and an inability for the device to relight after power cycling reflects strong degradation of the emitters in the device.
Outlook
The development of organic TADF emitters for LECs has evolved rapidly since the first report in 2015ref. ref1236 while there are now a large number of three- and four-coordinate Cu(I) complexes that have been used in LECs. LECs offer the promise of a low-cost alternative to OLEDs due to their simpler device structure.ref. ref1263 Of the organic TADF LECs reported to date, the highest EQEmax devices employed BPAPTC as the emitter, showing an EQE of 7.7% and λEL of 533 nm.ref. ref1245 However, the performance of LECs still lags significantly behind that of OLEDs, even when using the same emissive material. For instance, one of the most efficient LECs was reported with the emitter TXO-TPA, showing an EQEmax of 7.0% at CIE coordinates of (0.46, 0.50 (Figure ).ref. ref1244 The OLED with the same emitter showed an EQEmax of 18.5.ref. ref1244 Issues surrounding ACQ and exciton polaron annihilation will need to be addressed for LECs performance to begin to rival that of OLEDs. Similar to that observed with OLEDs, the performance of blue and red LECs is much poorer than for green devices. The highest efficiency blue LEC employed CZ-TRZ as the emitter, showing an EQEmax of 5.0% and emitting at 475 nm.ref. ref1247 The champion red LEC [CIE coordinates of (0.54, 0.44)] used TPA-DCPP as the emitter and showed an EQEmax of 4%.ref. ref1244 Indeed, porting over successful OLED device strategies to LECs, such as the use of exciplex hosts and HF, are certainly worth deeper exploration in a bid to improve the performance of these devices. The relatively small number of reports to date make it hard to predict the potential value of TADF emitters in LECs. However, the fact that there are already examples that rival some of the highest efficiency iridium-based LECs should provide impetus to continue to develop improved organic emitter materials and device architectures for this alternative electroluminescence technology.
Alongside organic TADF LECs, recent works using cationic Cu(I) complexes have focused on correlating structure to device performance in the case of four-coordinate complexes and exploring the potential of three-coordinate complexes as a superior class of organometallic emitters. Most of the copper(I) complexes incorporating bidentate N∧N, N∧P or P∧P ligands are red, orange, or yellow emitters, meanwhile examples of blue and green copper complexes used in LEC are much more scarce and frequently contain strongly σ-donating NHC ligands. There are, up to the end of 2022, no reports of deep blue or near-infrared copper LECs using Cu(I) complexes. Nonetheless, the performance of Cu(I)-based LECs presently rival that of the well-studied iridium(III)-based LECsref. ref1233 and thus still drives interest in this area.
TADF Assistant Dopant and Hyperfluorescence
Introduction
There is an inescapable compromise in the design of D-A TADF emitters for OLED applications. While decoupling of the HOMO and LUMO in orthogonal conformations helps to minimise ΔEST and promote RISC, it can also inhibit emission by decoupling S1 from S0, attenuating the oscillator strength of the emissive transitions. This fundamental trade-off means that D-A emitters typically excel at either RISC or ΦPL, or attempt to balance both. Inadequate performance in either aspect has detrimental impact on device performance, either in terms of the EQEmax (relying on high ΦPL) or the efficiency roll-off at higher current densities (relying on fast k RISC).
One solution that has gained prominence is to decouple exciton harvesting from emission by employing separate materials, each individually optimized to handle these processes within the emission layer. In this context the TADF material acts as an assistant dopant or sensitiser in the OLED, typically supporting singlet emission from another fluorescent emitter in the EML (TAF or TSF OLEDs);ref. ref1264 this same strategy has been coined by Adachi as hyperfluorescence (HF).ref1265−ref1266ref1267 Upon electrical excitation, RISC occurs on the TADF assistant dopant, harvesting triplet excitons, followed by Förster resonant energy transfer (FRET) from the singlet state of the TADF assistant dopant to the terminal emitter (itself either purely fluorescent or TADF), with resultant radiative decay from the latter (Figure ). Particularly effective in this regard is the use of MR-TADF compounds as the terminal emitters (Section sec11 ), which can provide a solution to producing devices having narrowband emission and a horizontally aligned transition dipole moment in the HF-OLED without undermining performance through otherwise slow K RISC. The key advantage of this mechanism is that the TADF assistant dopant is no longer required to simultaneously possess two fundamentally incompatible photophysical properties (i.e., fast k RISC and high ΦPL).ref. ref1268
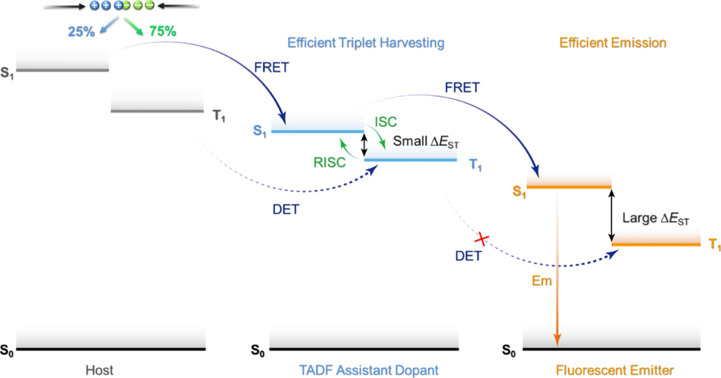
Paramount to achieving efficient HF-OLEDs is the requirement for rapid FRET between the assistant TADF dopant and the terminal emitter, which is most favorable with strong overlap between the emission spectrum of the assistant dopant and the absorption spectrum of the terminal emitter. This concept is not new, having been exploited in PhOLEDs using a phosphorescent assistant dopant coupled with a fluorescent terminal emitter;ref. ref1269 however, since the first promising example of this strategy using a TADF assistant dopant,ref. ref1270 there has been a surge in the number of reports of HF-OLEDs. It must be noted, however, that competing processes such as Dexter energy transfer (DET) or direct hole-electron recombination to form triplet excitons on the terminal emitter can also take place. These processes open new quenching channels not applicable to regular TADF OLEDs, leading to sometimes poorer efficiencies in HF-OLEDs that are particularly challenging to study due to the complexity of the multi-component emissive layer.ref. ref1271 Nevertheless, these competing processes can be somewhat mitigated by lowering the doping concentration of the terminal emitter.ref. ref1269 A summary of the device performance of the examples discussed in this section is summarized in Table S22.
Materials Development
The first examples of HF-OLEDs were reported in 2014, where a series of emitters was used by Nakanotan et al. covering blue, green, yellow, and red emission.ref. ref1270 The four systems involved combinations of fluorescent terminal emitters (TBPe, TTPA, TBRb, and DBP) paired appropriately with TADF assistant dopants (ACRSA, ACRXTN, PXZ-TRZ, and Tri-PXZ-TRZ) (Figure ) to ensure the appropriate spectral overlap and thus efficient FRET. To mitigate DET, the fluorescent terminal emitters were doped at 1 wt% concentration, whereas the TADF assistant dopants were used at higher concentrations optimized separately in normal TADF-OLEDs. Blue-emitting HF devices consisted of TBPe with 15 wt% TADF assistant dopant ACRSA in the DPEPO host and showed an EQEmax of 13.4% at CIE coordinates of (0.17, 0.30). This was much higher than the performance of an OLED containing only the terminal emitter, although less than what had been previously reported for the device with 20 wt% ACRSA by itself in DPEPO, which showed an EQEmax of 16.5%.ref. ref472 The green-emitting HF devices contained TTPA and 50 wt% ACRXTN as the TADF assistant dopant in mCP and showed an EQEmax of 15.8% at CIE coordinates of (0.29, 0.49). The yellow-emitting devices were obtained using TBRb with 25 wt% PXZ-TRZ assistant dopant in mCBP and showed an EQEmax of 18.0% at CIE coordinates of (0.45, 0.53). Finally, DBP with 15 wt% of assistant dopant Tri-PXZ-TRZ in CBP produced red-emitting devices with an EQEmax of 17.5% at CIE coordinates of (0.61, 0.39). Efficiency roll-off was low-to-moderate at 32, 26, 4, and 38% for the blue-, green-, yellow-, and red-emitting devices, respectively, at 1000 cd m–2. Further, the device stability improved, exemplified by the blue-emitting OLED LT50 of 194 hours at an initial luminance of 3225 cd m–2, suggesting rapid utilization of excitons.
Despite these promising results, the color of these HF-devices became less saturated where, for instance, the HF device containing TBPe had CIE coordinates of (0.17, 0.30), redder than the assistant dopant [CIE coordinates of (0.15, 0.21)]. With the later advent of narrowband MR-TADF materials and their use as terminal emitters (examples below and in Section sec11 ), it has since become possible for HF-OLEDs to possess a more saturated emission color compared to the TADF assistant dopant, and even to ‘upconvert’ the perceived emission color as the emission spectrum narrows (with lower energy onset).ref1272−ref1273ref1274 This approach may even help address current challenges in designing appropriate host materials for blue TADF OLEDsref. ref1275 (see Section sec18 ).
A different TADF assistant dopant, CzAcSF (Figure ), was used by Lee et al.ref. ref1276 With an EML composed of 50 wt% CzAcSF and 0.1 wt% TBPe (Figure ) in a DPEPO host, an EQEmax of 18.1% was achieved, while the color point improved due to efficient FRET, reflected in the CIE coordinates of (0.15, 0.22) having become much closer to those of the fluorescent device. The improved efficiency was attributed to not only the more efficient FRET for this HF pair, but also reduction of charge trapping on the terminal emitter resulting from the low doping concentration (0.1 wt% compared to 1.0 wt%) and higher doping concentration of the TADF assistant dopant (50 wt% compared to 15 wt% in the previous example).
Ahn et al.ref. ref1277 reported an HF-OLED using 0.4 wt% BPPyA (Figure ) as the terminal emitter in conjunction with 40 wt% DMAC-DMT (Figure ) as the assistant dopant, all in DBFPO host. These devices showed an EQEmax of 19.0% at CIE coordinates of (0.14, 0.15) along with a low-efficiency roll-off of 8% at 500 cd m–2 and improved device lifetime (LT50 = 2.8 h at an initial luminance of 400 cd m–2), compared to the device with DMAC-DMT alone (LT50 = 0.7 h) that showed an EQEmax of 22.5%. This study illustrated that the HF strategy could reduce the probability of singlet-triplet annihilation (STA) and triplet-triplet annihilation (TTA) processes, with rapid FRET from the assistant dopant to the terminal emitter in the HF device effectively reducing the triplet exciton population and thus the chance of multi-excitonic quenching. This was evidenced by the shorter τd of 2.49 ms in the BPPyA:DMAC-DMT:DBFPO emissive system compared to DMAC-DMT:DBFPO system, although this analysis has since been demonstrated to be unexpectedly complex in a similar HF system.ref. ref1271
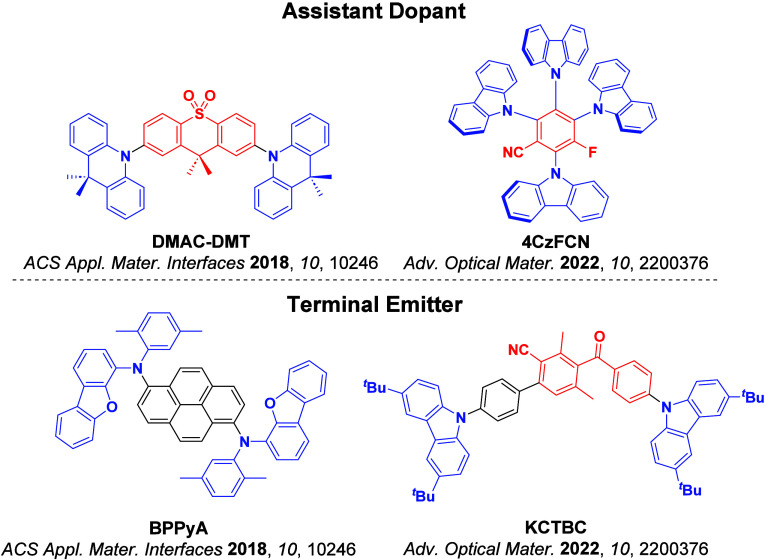
HF-OLEDs can also be fabricated using solution-processing methods, as demonstrated by Alam et al.ref. ref1278 The blue-emitting device contained 3 wt% KCTBC (Figure ) as the terminal emitter and 12.5 wt% 4CzFCN (Figure ) as the assistant dopant in CBP host, and showed an improved EQEmax of 13.9% compared to that of the device with 3 wt% KCTBC alone, which exhibited an EQEmax of 9%. As well as reducing production costs, in the context of HF-OLEDs solution-processing also significantly simplifies the challenging 3-way co-deposition processes for the EML compared to vacuum deposition, which becomes particularly challenging for ultralow terminal emitter doping ratios <1%.
Delicately modulating the concentrations of both the assistant dopant and the terminal emitter is paramount for controlling energy transfer and achieving optimal results in HF-OLEDs. However, from a molecular design standpoint, the introduction of bulky functional moieties such as tert-butyl groups can also help to control intermolecular spacing, and so reduce the likelihood of undesired DET processes. Following this principle, Yun et al. designed a molecule, FTrzTCz (Figure ), using 3,6-di-tert-butylcarbazole as the donor and triazine as the acceptor.ref. ref1279 The HF-OLED using 20 wt% FTrzTCz as the assistant dopant and 1 wt% 6tBPA as terminal emitter showed an EQEmax of 17.9% with CIE coordinates of (0.24, 0.58). The same group further investigated the influence of steric hindrance by introducing three 3,6-tert-butylcarbazole donors about the triazine unit to produce tert-butyl-functionalized donor-acceptor compound TbCzTrz.ref. ref1280 As a comparison, TmCzTrz was also synthesized, which contained methyl substituents on the carbazole donor as opposed to tert-butyl groups. The TADF device based on TbCzTrz showed a much lower intrinsic EQEmax than that based on TmCzTRz (13.7% vs. 28.1%); however, using 20 wt% TbCzTrz or TmCzTrz as assistant dopants with 0.5 wt% 6tBPA (Figure ) as the terminal emitter gave EQEmax values of 14.6 and 18.5%, respectively. This demonstrated the utility of the tBu substitution outside of the direct TADF performance of the TbCzTrz emitter. HF-OLEDs using C545T as the terminal emitter similarly showed EQEmax of 16.1 and 15.9%, again higher for the device employing the more sterically shielded TADF assistant dopant, which presumably contributed to suppressing DET between the assistant dopant and the fluorescent terminal emitter.
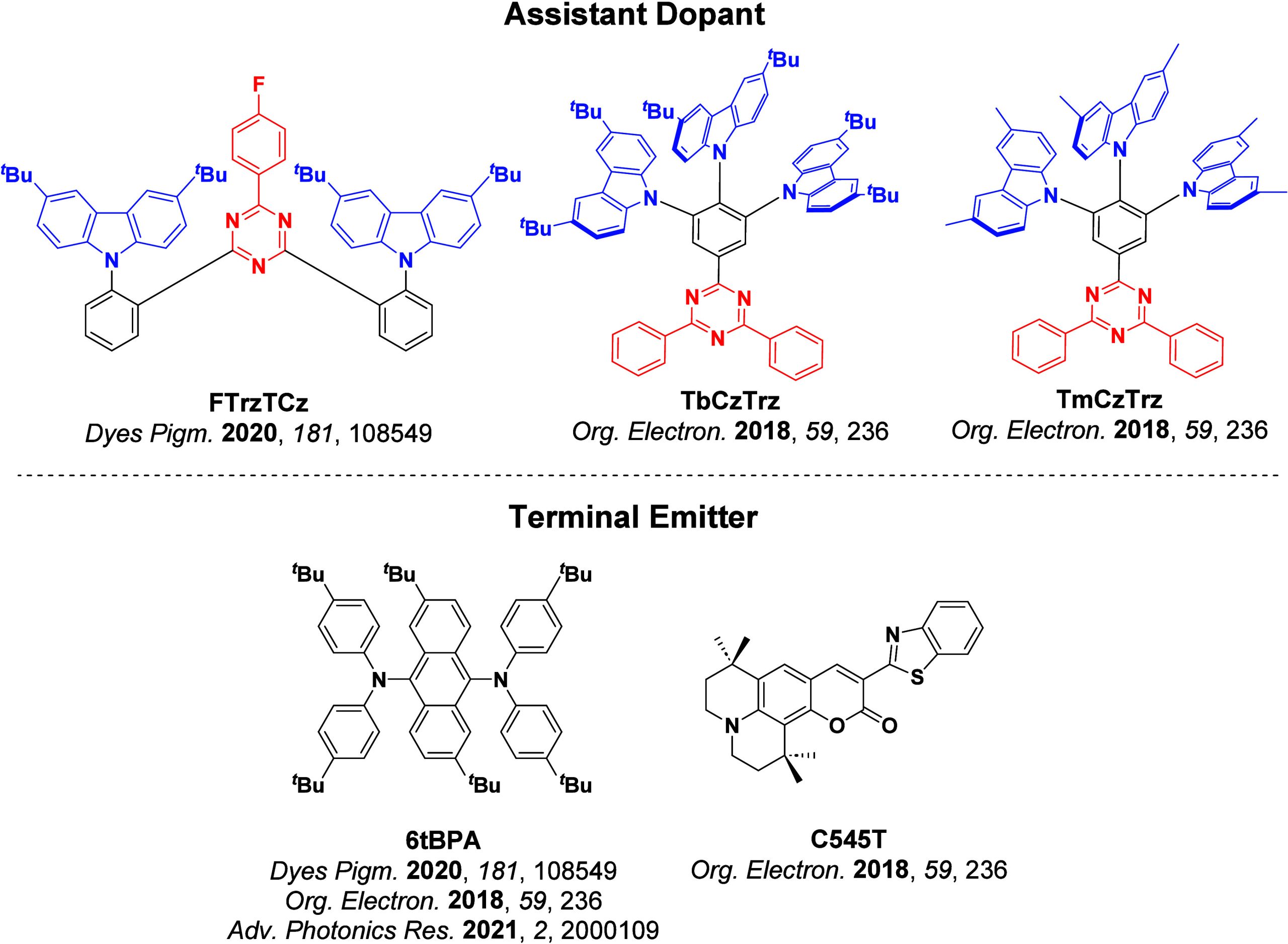
Blocking undesired DET in HF-OLEDs was also studied by Xie et al.ref. ref1281 using TADF compounds PXZ-DBPZ and FPXZ-DBPZ (Figure ) as assistant dopants. Photophysical investigations and Kinetic Monte Carlo simulations revealed that the inert phenyl-fluorene substituents on FPXZ-DBPZ could effectively suppress DET process compared to PXZ-DBPZ. The device with 9 wt% FPXZ-DBPZ as the assistant dopant and 0.6 wt% DBP as terminal emitter in CBP showed an EQEmax of 18.1%, which was higher than that with PXZ-DBPZ (EQEmax = 15.2%) as the assistant dopant.
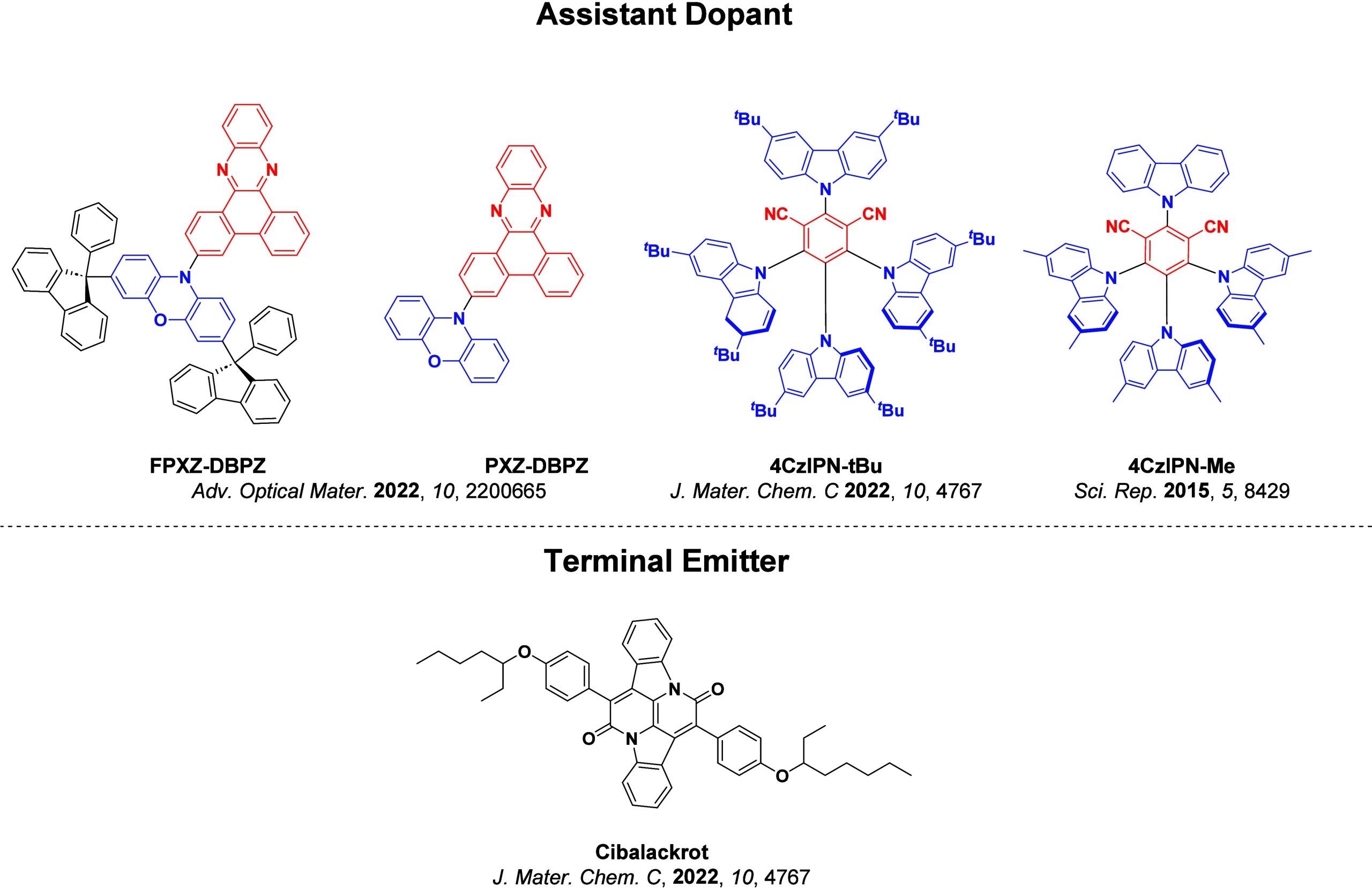
Similarly to reduce the DET, a tert-butyl-functionalized 4CzIPN-tBu (Figure ), was employed as TADF assistant dopant in HF-OLEDs by Wallwork et al.ref. ref1282 The best-performing solution-processed device with 0.5 mol% cibalackrot (Figure ) as the terminal emitter and 29.5 mol% 4CzIPN-tBu as the assistant dopant in mCP showed an EQEmax of 15.3%, and EQE100 of 14.9%.
Taking the structure of the fluorescent emitter into account, the TADF compound 4CzIPN-Me (Figure ) was used in a similar study by Furukawa et al.,ref. ref1283 alongside the emitter TBRb (Figure ), which contains four tert-butyl groups. 4CzIPN-Me showed an efficient k RISC of 7.7 × 105 s–1 and the yellow-emitting HF device with 0.65 wt% TBRb and 6.3 wt% 4CzIPN-Me in mCBP showed an EQEmax of 19.1% at CIE coordinates of (0.43, 0.54), with EQE1000 of 16.7%. Importantly, the LT50 at initial 1000 cd m–2 was 1470 h for the device with only 4CzIPN-Me, which increased to 3775 hours when the HF-OLED architecture was used. The authors attributed the improved device stability to the rapid and efficient FRET, which effectively reduced the triplet population on 4CzIPN-Me and alleviates device degradation.
TBRb was also employed as the terminal emitter in conjunction with the TADF sensitizer 34AcCzTrz (Figure ) and a TADF host 3CzPhpPM by Lv et al..ref. ref287 The yellow-emitting HF device with 4 wt% 34AcCzTrz and 1 wt% TBRb in the emissive layer displayed an EQEmax of 19.1% with a near zero efficiency roll-off at 1000 cd m–2. This HF device performance was also remarkably improved compared to the non-HF counterpart with 3 wt% 34AcCzTRz alone as the emitter, which showed an EQEmax of 14.5% and had an efficiency roll-off 9.1% at 1000 cd m–2.
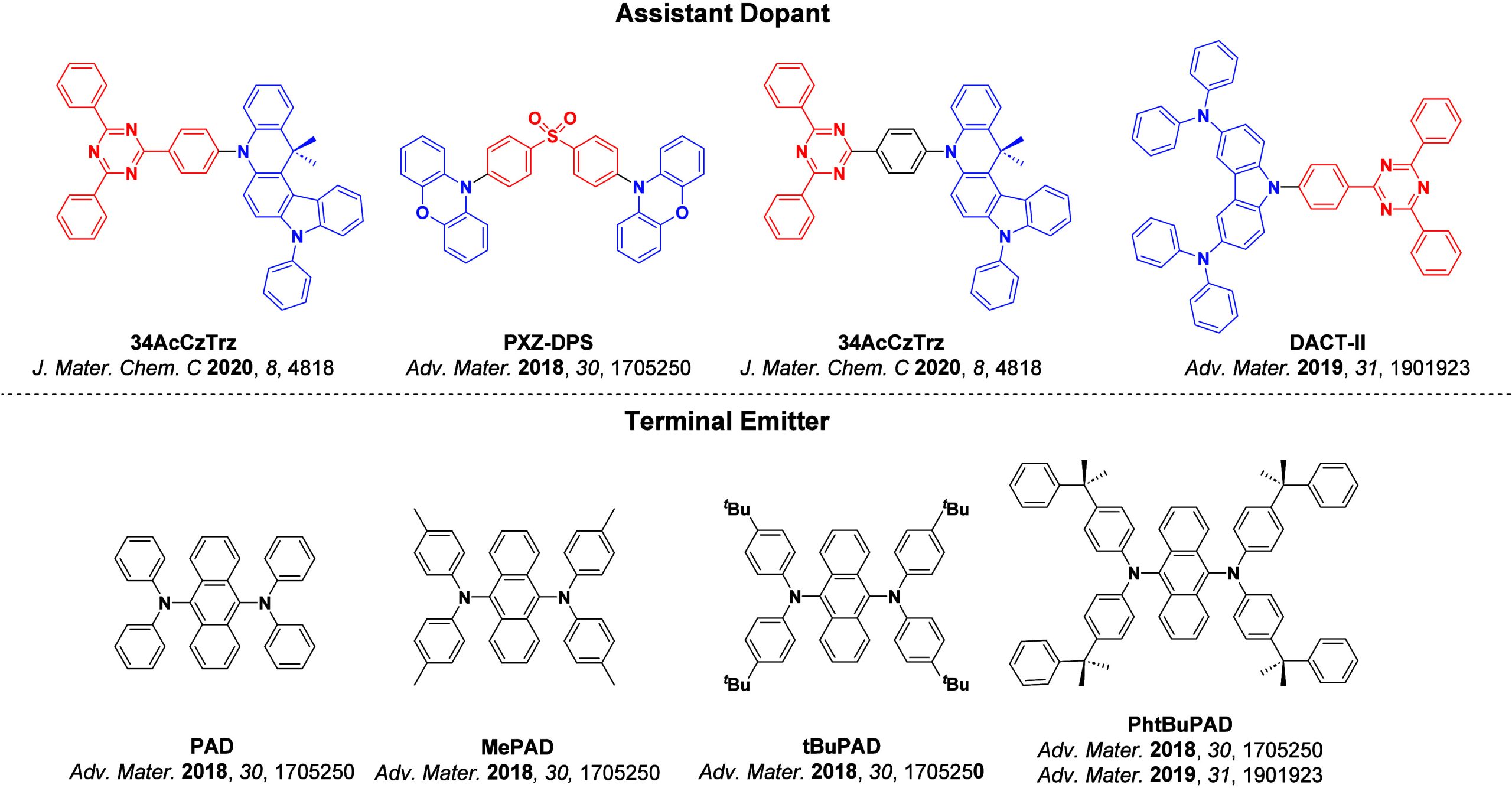
Zhang et al. adopted a similar strategy by introducing sequentially bulkier groups on the terminal emitter to suppress DET pathways.ref. ref1264 Four fluorescent dyes, PAD, MePAD, tBuPAD, and PhtBuPAD (Figure ), were investigated as terminal emitters in combination with TADF assistant dopant PXZ-DPS (Figure ). The green HF-OLEDs with 1 wt% PAD, MePAD, tBuPAD, and PhtBuPAD in combination with 30 wt% PXZ-DPS as the assistant dopant showed increasing EQEmax of 18.6, 20.2, 22.7 and 24.0%, respectively, with λEL ranging from 525–540 nm. When the doping concentration of the terminal emitter was between 5–8%, excellent efficiency roll-off out to 5,000 cd m–2 was observed for all devices. The same group also reported a device with 3 wt% PhtBuPAD as the terminal emitter and 40 wt% DACT-II (Figure ) as the assistant dopant that showed EQEmax and PEmax of 23.2% and 76.9 lm W–1, respectively, where the EQE5000 remained as high as 20.0%, and with CIE coordinates of (0.36, 0.60).ref. ref1284
Kim et al. reported four orange-colored TADF emitters, tBIQAC, tBIQAP, DtBIQAC, and DtBIQAP (Figure ). Compared to DMAC-decorated tBIQAC and tBIQAP, DtBIQAC, and DtBIQAP possess a bulkier diphenylacridan donor. Their relative device performance as assistant dopants in conjunction with DBP (Figure ) as the terminal emitter in PBICT host was investigated.ref. ref1285 The DtBIQAP– and DtBIQAC-based devices showed the highest EQEmax of 18.2% and 17.5% with CIE coordinates of (0.62, 0.38) and (0.64, 0.36), respectively, both higher than tBIQAC– and tBIQAP-based devices with EQEmax of 16.8 and 14.7% at CIE coordinates of (0.63, 0.37), respectively. These results again demonstrate that the use of bulky groups on the assistant dopant can effectively increase the EQE of the TADF-assisted fluorescent OLEDs.
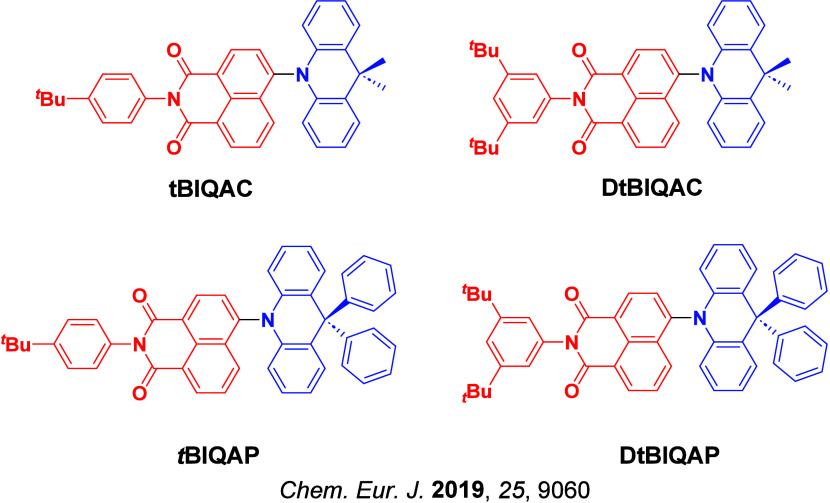
Besides introducing steric blocking groups, the physical separation of the assistant dopant and the terminal emitter has been used to suppress DET channels. Han et al. engineered a multi-layered emissive layer in the OLED, where the assistant dopant and the terminal emitter in DPEPO were alternately deposited.ref. ref1286 The device with 50 wt% DMAC-DPS (Figure ) as the assistant dopant and 1 wt% TBPe (Figure ) as the terminal emitter showed an EQEmax of 18.8% at CIE coordinates of (0.14, 0.25), higher than that of the conventional HF-device (EQEmax = 13.1%), where DMAC-DPS and TBPe were co-deposited simultaneously. Later, Chen et al. used DMAC-DPS as the assistant dopant at the optimized doping concentration of 20 wt% in combination with 1 wt% TBPe to produce a blue HF-OLED. The device only exhibited an EQEmax of 14.1% at CIE coordinates of (0.14, 0.17).ref. ref574 The results indicate that the strategy employing alternate deposition of assistant dopant and terminal emitter is a possible solution to suppressing DET.
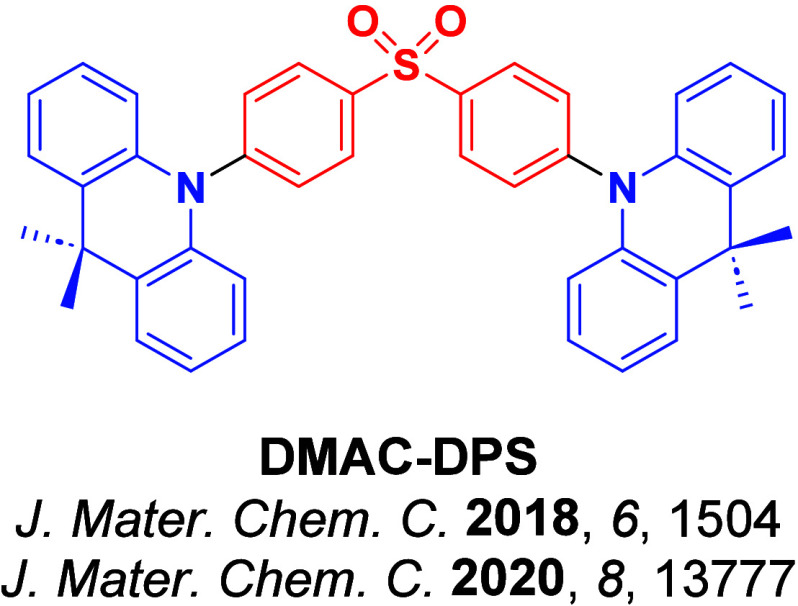
Somewhat different from the conventional sensitization strategy, Ma et al. reported an OLED using Pr-1 (Figure ) as the assistant dopant and fluorescent luminophore DCJTB (Figure ) as the terminal emitter, both doped in the TADF exciplex mCBP:PO-T2T.ref. ref1287 A competing exciplex was also formed between Pr-1 and PO-T2T in the mCBP:PO-T2T exciplex host. Therefore, three RISC channels in this system could act simultaneously to mitigate TTA and DET, each then feeding into FRET to the terminal emitter. The OLED with 10 wt% Pr-1 and 1 wt% DCJTB in the mCBP:PO-T2T exciplex host as the emissive layer showed CEmax, PEmax and EQEmax of 22.6 cd A–1, 29.5 lm W–1, and 13%, respectively, with an LT50 at 1000 cd m–2 reaching 415 hours.
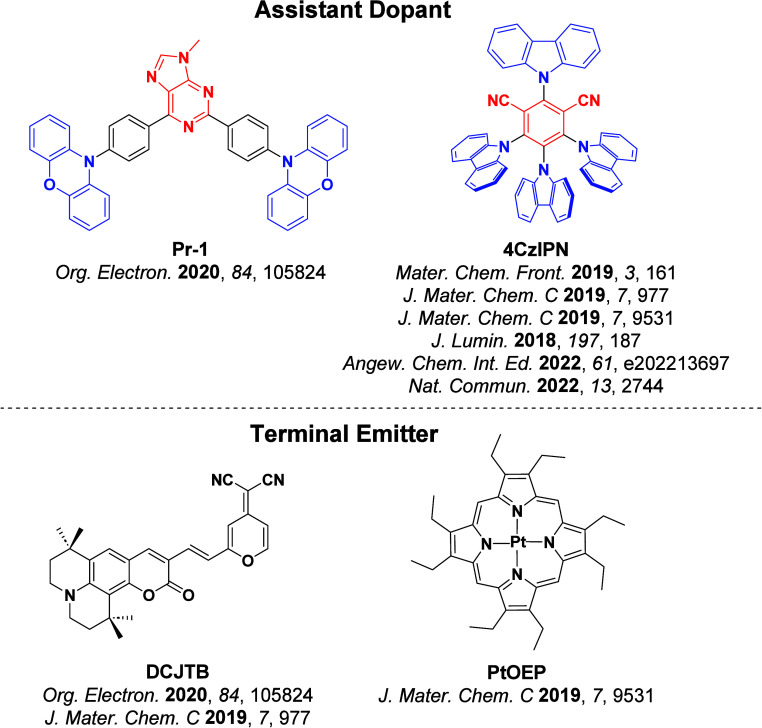
Li et al.ref. ref1266 similarly explored the use of multiple TADF materials, with 4CzIPN (Figure ) and a TADF exciplex TCTA:B4PyMPM host used together with fluorescent terminal emitter, DCJTB. Three FRET processes were proposed by the authors, which occur between the exciplex and DCJTB (FRET1), the exciplex and 4CzIPN (FRET2) and 4CzIPN and DCJTB (FRET3), all of which contributed to the harnessing of triplet excitons. The red-emitting device with 2 wt% 4CzIPN as the assistant dopant and 0.5 wt% DCJTB as the terminal emitter showed EQEmax of up to 12.9% at CIE coordinates of (0.58, 0.41). A negligible efficiency roll-off of 3.9% was recorded at 100 cd m–2. By contrast, a device without the co-assistant dopant 4CzIPN reached an EQEmax of only 7.3%, and a non-HF device with only 4CzIPN as the TADF dopant exhibited an EQEmax of only 6.6%.
Employing the same exciplex co-host methodology, Liao et al. reported a novel deep red-emitting (∼650 nm) HF-OLED where the EML was composed of exciplex co-host (CBP:B4PyMPM, 1:1), TADF emitter 4CzIPN (Figure ) as the assistant dopant, and a phosphorescent complex PtOEP (Figure ) as the terminal emitter.ref. ref1288 In this design, the excitons first form on the exciplex co-host, followed by the energy transfer to 4CzIPN and to PtOEP. The triplet harvesting ability of the phosphorescent terminal emitter also means that this kind of device does not suffer quenching through DET channels. The optimized device used 4 wt% 4CzIPN and 4 wt% PtOEP in CBP:B4PyMPM as the EML, and showed an EQEmax of 21.5%. The EQEmax of the device with just 4 wt% PtOEP in CBP:B4PyMPM and the device with 6 wt% PtOEP in CBP were instead ∼17 and 9.1%, respectively. Furthermore, the LT50 at 550 cd m–2 of an HF-OLEDs using a staircase-doping strategy was improved to 90 hours, double that of the device using a uniformly doped emitting layer.
Jang et al. investigated the impact of the dihedral angle of the TADF sensitizer on FRET efficiency, and hence the performance of the HF-OLED devices.ref. ref1289 Two TADF emitters, BPAc and BPAcCz (Figure ), both containing a benzophenone acceptor and either DMAc/DMAc or DMAc/carbazole donor groups were used as assistant dopants. The calculated molecular geometries showed a dihedral angle of 89° between DMAc and benzophenone for BPAc, while a more planar conformation was observed between carbazole and benzophenone (dihedral angle 49°) in BPAcCz. The devices with 20 wt% BPAc or BPAcCz and 0.5 wt% 6tBPA (Figure ) as the terminal emitter in DPEPO showed EQEmax of 16.6 and 15.0%, respectively. The authors asserted that the planar geometry of BPAcCz should be responsible for enhanced DET in the HF-OLEDs, this geometry allowing increased short-distance interactions in the emissive layer. To validate this hypothesis, 1 wt% of the blue emitter AnTP (Figure ) was also dispersed in DPEPO alongside 20 wt% of BPAc or BPAcCz. The AnTP-doped system was chosen as it only allowed for DET to occur but, with inhibited FRET due to the large singlet energy of AnTP. Time-resolved decay measurements verified that the more perpendicularly structured BPAc could indeed suppress DET compared to more planarized BPAcCz.
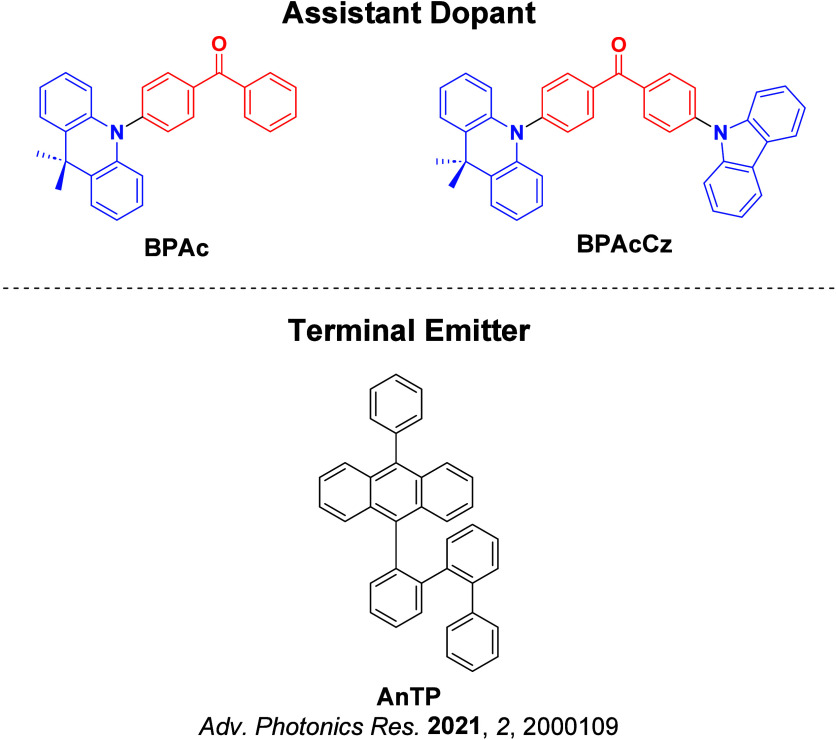
To date, it remains highly challenging to achieve both large singlet radiative rate (kr s) and small electron exchange energy (J) to produce high-performance red emitters for OLEDs (see Section sec5 ). To overcome this issue, the TADF sensitization strategy was employed by Chen et al.ref. ref1290 Solution-processed red-emitting HF-OLEDs using conventional red fluorescent emitter DBP (2 wt%) (Figure ) and the green-emitting TADF assistant dopant DC-TC (15 wt%) (Figure ) in CBP host showed an EQEmax of 8.0% at CIE coordinates of (0.61, 0.38). An alternative TADF emitter, DC-ACR (Figure ), was also used as an alternative assistant dopant, which demonstrated a much lower efficiency (EQEmax = 4.25%) due to trapping of excitons on the DBP emitter directly, which limited the FRET process.
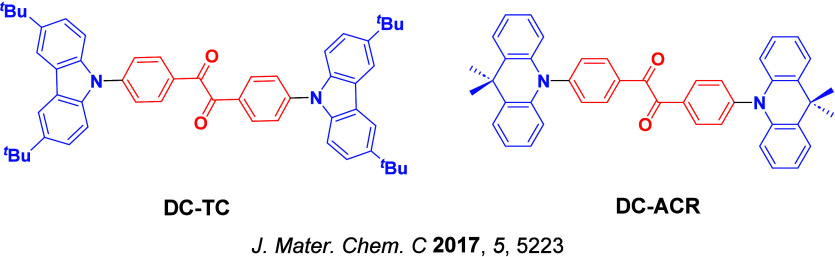
Wang et al. fabricated a red-emitting device (λEL = 612 nm) using 0.5 wt% OTPA-BT-CN (Figure ) as the terminal emitter alongside 25 wt% OSTFB (Figure ) as the assistant dopant in mCP, resulting in an EQEmax of 12.4%.ref. ref1265 The use of 4CzIPN (Figure ) as an alternative assistant dopant, resulted in an HF-OLED with much lower EQEmax of 6.3%, despite having similar spectral overlap with the terminal emitter. The higher k r in OSTFB was determined to be the main reason for these differences in device performance, which generally leads to a high-efficiency energy transfer.
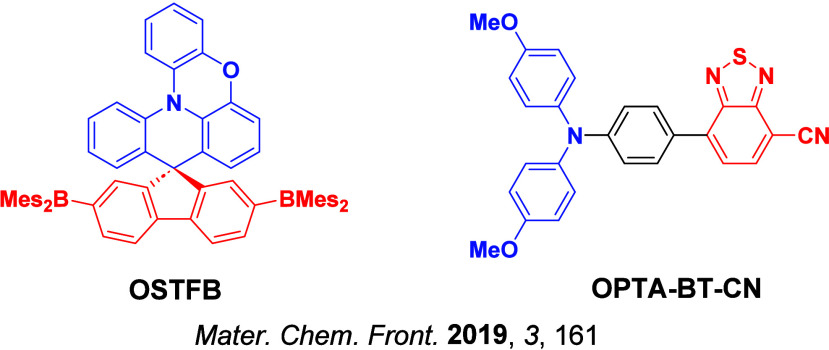
TADF assistant dopants have also been used with phosphorescent complexes as terminal emitters, where energy transfer still occurs via FRET between the S1 state of the TADF compound to the S1 state of the phosphorescent emitter. Singlet FRET is followed by ISC to the T1 state and phosphorescence emission, while DET to the phosphorescent emitter (or direct recombination) can also lead to emission. Exotic triplet-to-singlet energy transfer may also be active in such systems.ref1291,ref1292 Liao et al. reported a solution-processed WOLED using 10 wt% of TADF dendrimer BPS (Figure ) as the assistant dopant and 0.5 wt% Ir(bt)2acac (Figure ) as the emitter in co-host DCzPPy:OXD-7 (100:40 ratio).ref. ref1293 The device achieved an EQEmax of 6.6%, CEmax of 17.34 cd A–1 and the CIE coordinates varied by only (0.02, 0.02) across the luminance range of 100 to 10000 cd m–2, indicating good color stability and energy transfer within the device. Another example of this strategy involved combination of the red phosphorescent terminal emitter, Hex-Ir(phq)2(acac) (Figure ), and 4CzIPN (Figure ) together in CBP host to produce red phosphorescent OLEDs.ref. ref1294 The device with 1.5 wt% of terminal emitter and 7.5 wt% of TADF assistant dopant showed an EQEmax of 9.8% and an impressive maximum brightness of 52,204 cd m–2, compared to 7.9% and 12,200 cd m–2 in devices without 4CzIPN. The enhanced brightness resulted from improved exciton utilisation resulting from efficient triplet harvesting. However, FRET was incomplete with some emission still observed directly from 4CzIPN (a trait also observed by Wang et al. in a previously discussed example).ref. ref1265
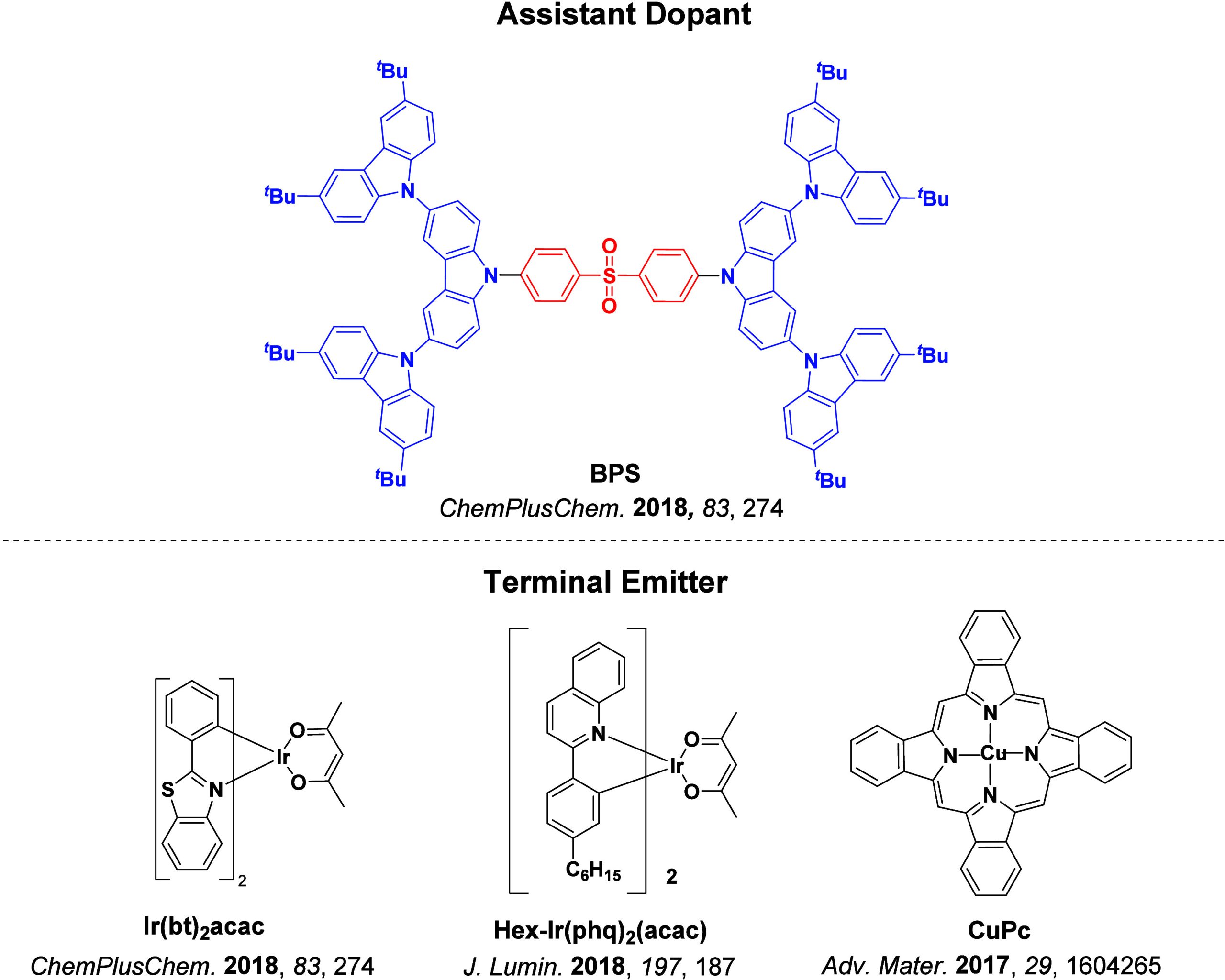
Aside from iridium complexes, copper complexes have also been used as terminal phosphorescent emitters in OLEDs of this type, though some copper complexes can also emit via TADF or dual TADF/phosphorescence (see Section sec9 ). Nagata et al. used PXZ-TRZ (Figure ) as the assistant dopant and CuPc (Figure ) as NIR phosphorescent terminal emitter,ref. ref1295 both dispersed into mCBP host to produce an OLED that showed an EQEmax of 0.037%. Despite further advances in NIR OLEDs in the years since (see Section sec5 ), this low EQE performance was still impressive at the time. Due to the large distance between the assistant dopant and the CuPc terminal emitter at the relative doping concentrations used, and the large separation between the triplet levels (T1_PXZ‑TRZ – T1_CuPc = 1.1 eV), the main energy transfer route between the two was assigned to FRET.
NIR OLEDs are of particular interest for applications as light sources for optical communication, medical and biological imaging systems, and for military use, including night vision goggles. Shahalizad et al. designed the red TADF emitter, TPAM-BF2 (Figure ), which emits variously at λPL of 746 nm, 752 nm, and 764 nm with associated ΦPL of 41.9, 25.0, and 13.7% when doped in CBP at concentrations of 6, 10 and 20 wt%, all respectively.ref. ref1296 When 20 wt% TPAM-BF2 in CBP was used as the assistant dopant in conjunction with 0.5 wt% of the NIR fluorescent emitter, BPPC-Ph (Figure ), the solution-processed OLED showed an EQEmax of 3.5% and with notably narrowband emission (FWHM < 40 nm) at 840 nm. BPPC-Ph was also used in another report,ref. ref1297 now named as BPPC, as the terminal emitter at 0.8 wt% doping in combination with 20 wt% TPA-DCPP (Figure ) as the TADF assistant dopant in B3PYMPM host, which gave an impressive EQEmax of 5.4% at λPL of 790 nm.ref. ref1297 Notably, most organic NIR OLEDs have a considerable fraction of their spectral power density (>50%) in the visible range, while the TPA-DCPP-based NIR device displayed narrowband NIR emission with 90% of total emission beyond 750 nm, and an NIR cut-on wavelength (corresponding to 10% of the peak PL intensity) of 790 nm.
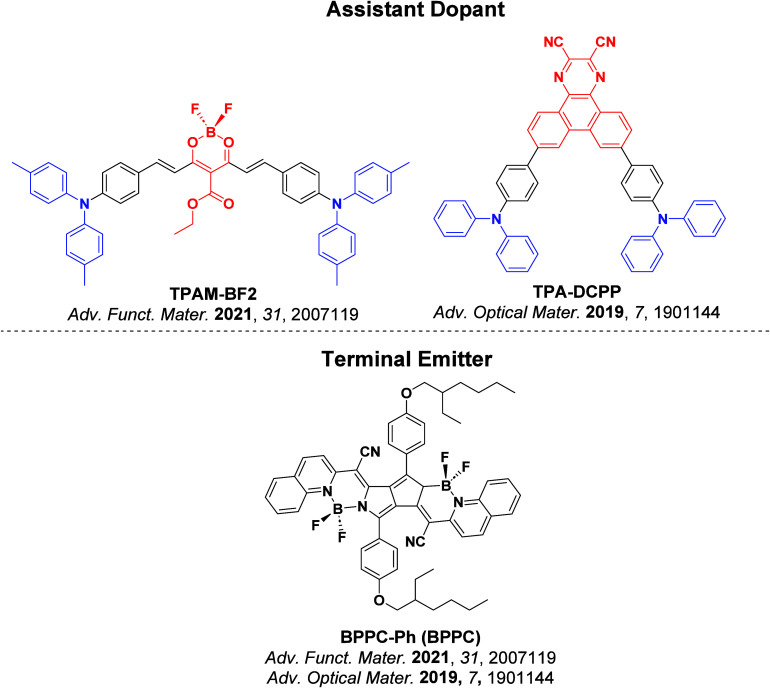
Bartkowski et al.ref. ref1298 employed a D-A TADF compound (tBuCz-σ-NI, 2 in that work) as the assistant dopant and structurally analogous but rigidified fused-aromatic emitter (tBuCz-π-NI, 5 in that work) (Figure ) as the terminal emitter to realize a narrowband emitting HF-OLED. The green-emitting device with 10 wt% tBuCz-σ-NI and 0.6 wt% as tBuCz-π-NI in mCP showed an EQEmax of 27% and FWHM of 40 nm. A similar popular strategy of obtaining narrowband emission is to use MR-TADF compounds as terminal emitters. Examples of HF-OLEDs using MR-TADF compounds as terminal emitters are summarised in Section sec11 .
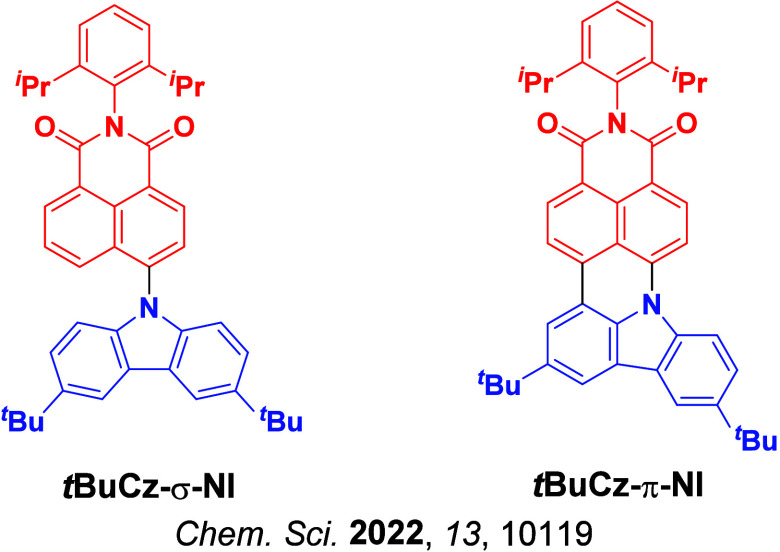
Although most HF-OLEDs contain purely organic TADF assistant dopants, organometallic TADF complexes have also been used as co-dopants. Zhan et al. employed a copper-based CMA complex (MAC*)Cu(Cz) (Figure ) at 20 wt% doping with 1 wt% TBRb (Figure ) as the terminal emitter in mCBP host.ref. ref1299 The yellow-emitting HF-OLED (λEL = 566 nm) showed an EQEmax of 14.6%, with a very low-efficiency roll-off of 12% at 1000 cd m–2. The LT50 of the device was 767 hours at an initial luminance of 100 cd m–2. Further, a device with MR-TADF emitter BN3 (Figure ) instead of TBRb exhibited an improved EQEmax of 26.5%, which only decreased to 10.5% at a luminance of 10,000 cd m–2.
A second example of the same strategy saw the use of a gold (III) TADF complex Au-1 (Figure ) as the assistant dopant.ref. ref1300 The blue-emitting HF device with 10 wt% of Au-1 as the sensitizer and 0.5 wt% v-DABNA (Figure ) as the terminal emitter in PYD2 host showed an EQEmax of 16.6%, which remained as high as 14.4% at 1000 cd m–2.
Finally, TADF assistant dopants have recently been used in conjunction with doublet organic radical emitters in an HF-OLED. The red-emitting HF-OLED contained 3 wt% of the radical emitter TTM-3PCz (Figure ) and 25 wt% 4CzIPN (Figure ) in CBP,ref. ref1301 and showed an EQEmax of 16.4% with a broad emission band ranging from 680–800 nm. The LT50 at 0.4 mA cm–2 of the HF-OLED was only 42 min, likely due to the instability of the radical species. This was nonetheless a higher efficiency than the device with only TTM-3PCz, which showed an EQEmax of 10.7%, although theoretically doublet OLEDs entirely avoid the problem of triplet harvesting, and so may not need to rely on HF-OLED strategies once sufficiently developed.
Outlook
From an assessment of the performance of HF-OLEDs compared to normal TADF OLEDs, it is clear that this mixed-materials approach can achieve significant improvements in efficiency, efficiency roll-off, and color purity. As an emblematic example, EQEmax of over 38% at blue CIE coordinates of (0.12,0.15) have been reported using leading D-A TADF sensitizers with the MR-TADF terminal emitter ν-DABNA (Figure and Figure ).ref. ref1302 Similarly high efficiency green and red devices have been fabricated. For instance, an EQEmax of 27.0% at CIE coordinates of (0.38,0.59) was achieved for a green OLED with the D-A TADF sensitizer tBuCz-σ-NI and the fluorescent terminal emitter tBuCz-π-NI,ref. ref1303 while the highest efficiency red OLED showed an EQEmax of 21.5% at CIE coordinates of (0.72, 0.30) with the D-A TADF sensitizer 4CzIPN and the phosphorescent terminal emitter PtOEP.ref. ref1288 This trend in efficiency results from the fact that fast k s r and high ΦPL of the terminal emitter can be decoupled from the exciton harvesting efficiency provided by the TADF assistant dopant, allowing the HF systems to benefit from advances in both separate fields.ref. ref1288 Despite the additional challenges associated with multiple material depositions and complex energy transfer pathways, it is likely that HF-TADF OLEDs will continue to claim record device efficiencies, particularly at higher brightness, for the foreseeable future. This includes at extreme color coordinates, a recent development supported with the use of MR-TADF materials as narrowband terminal emitters and explains the prominence of the HF-OLED strategy seen across MR-TADF research activity (see Section sec11 ).
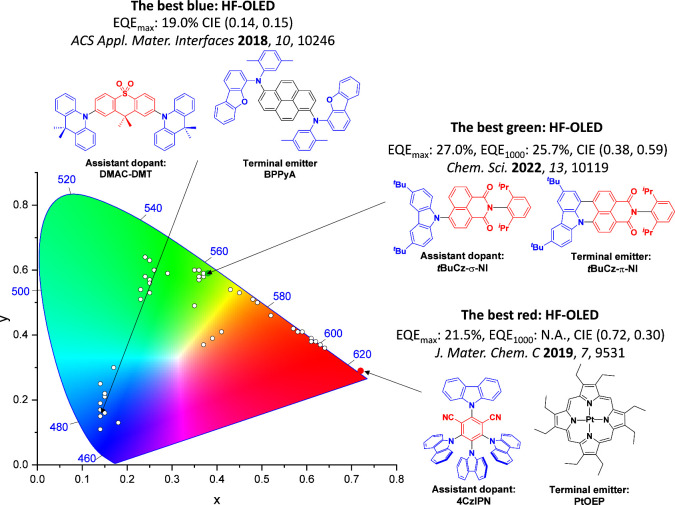
As well as enabling progressive improvements in efficiency and color purity, it has recently been shown that HF sensitiser/emitter pairs featuring surprisingly low FRET overlap can nonetheless efficiently drive blue OLED emission using green-emitting sensitisers. This development allows the use of lower energy (and intrinsically more stable) emitters and hosts for blue OLEDs and may hence unlock significant gains in device stability that have persistently eluded research efforts at this wavelength range.ref1037,ref1304 At the same time, the underlying processes that control HF-OLED device performance remain poorly understood and is an area ripe for new experimental methods to be developed to yield new insights into device and materials design. In this context, we anticipate that HF-OLED development will continue to grip the attention of applied, fundamental, and computational research in the short and medium term.
TADF Materials as Hosts
Introduction
Due to their ambipolar character resulting from these materials comprising both electron-donating and electron-accepting moieties, many of the D-A TADF materials employed as emitters in previous sections are potentially also useful as host materials in OLEDs. The bipolar nature of TADF materials as hosts allows them to promote balanced charge transport into the EML.ref. ref1305 The TADF host can also assist in exciton harvesting via RISC, followed by FRET to the guest emitter.ref1306,ref1307 This FRET process remains feasible even at low doping concentrations of the terminal emitter, which is particularly beneficial for improving the efficiency of OLEDs employing fluorescent emitters in the hyperfluorescence category of devices (see Section sec17 ). In phosphorescent devices, the use of TADF host systems has also been demonstrated to lead to devices with improved efficiency and stability, even at <1 wt% doping of the emitter.ref. ref1308 Of course, the capacity to support guest emitters of a particular energy necessitates that the TADF host itself has a sufficiently high triplet energy, so that excitons are confined on the terminal emitter. There are two classes of TADF compounds that have been explored as hosts: D-A TADF compounds and exciplexes (intermolecular donor-acceptor mixtures, see Section sec8 ). Examples of OLEDs using D-A TADF hosts are examined here and are split into three groups based on the nature of the emissive material: phosphorescent, fluorescent, and TADF. The device performance for the OLEDs discussed in this section are collated in Table S23.
TADF Hosts with Phosphorescent Emitters
The first reported OLEDs with TADF materials used as hosts featured phosphorescent emitters (Figure ). Zhang et al. demonstrated early on that the device lifetime is less sensitive to the doping concentration of fac–Ir(ppy)3 (≤3 wt%) when a TADF host is used, compared to a conventional host such as CBP.ref. ref1248 The authors compared to the TADF host PBICT (λPL = 488 nm, ET = 2.66 eV in neat film, and ΔE ST = 0.10 eV in DCM), consisting of an indolocarbazole donor and triazine acceptor, which was doped with the phosphorescent green emitter (λPL = 507 nm in CHCl3). In thin films with very low emitter doping concentrations (0.5 – 3.0 wt%), the energy transfer process is mainly governed by long-range FRET from the TADF host to the phosphorescent guest. This supports the improved efficiency of the PBICT:Ir(ppy)3 devices (EQEmax = 23.9% at 3 wt% of the emitter) compared to the CBP:Ir(ppy)3 devices (EQEmax = 14.5% at 3 wt% of the emitter).ref. ref1248 The efficiency could be further improved by employing DIC-TRZ as the TADF host, in part due to an even smaller ΔE ST (0.06 eV in DCM). The authors compared the EQE among devices featuring differing dopant concentrations (EQEmax,x%) with respect to the highest EQEmax recorded (EQEmax,all). In the case of devices using DIC-TRZ, the EQEmax,all was achieved at a dopant concentration of 2 wt%. On the other hand, for devices using PBICT, the highest EQEmax,all was attained at a dopant concentration of 3 wt%. Moreover, at very low doping levels (0.5 wt%), the devices using DIC-TRZ attained ∼92% of the EQEmax,all. In contrast, for the devices using PBICT only 80% of the EQEmax,all was attained at such a low loading (0.5 wt%). The authors thus concluded that a faster RISC rate and a higher RISC efficiency were enabled by the smaller ΔE ST of DIC-TRZ, supporting greater device efficiency for the eventual phosphorescence emission.
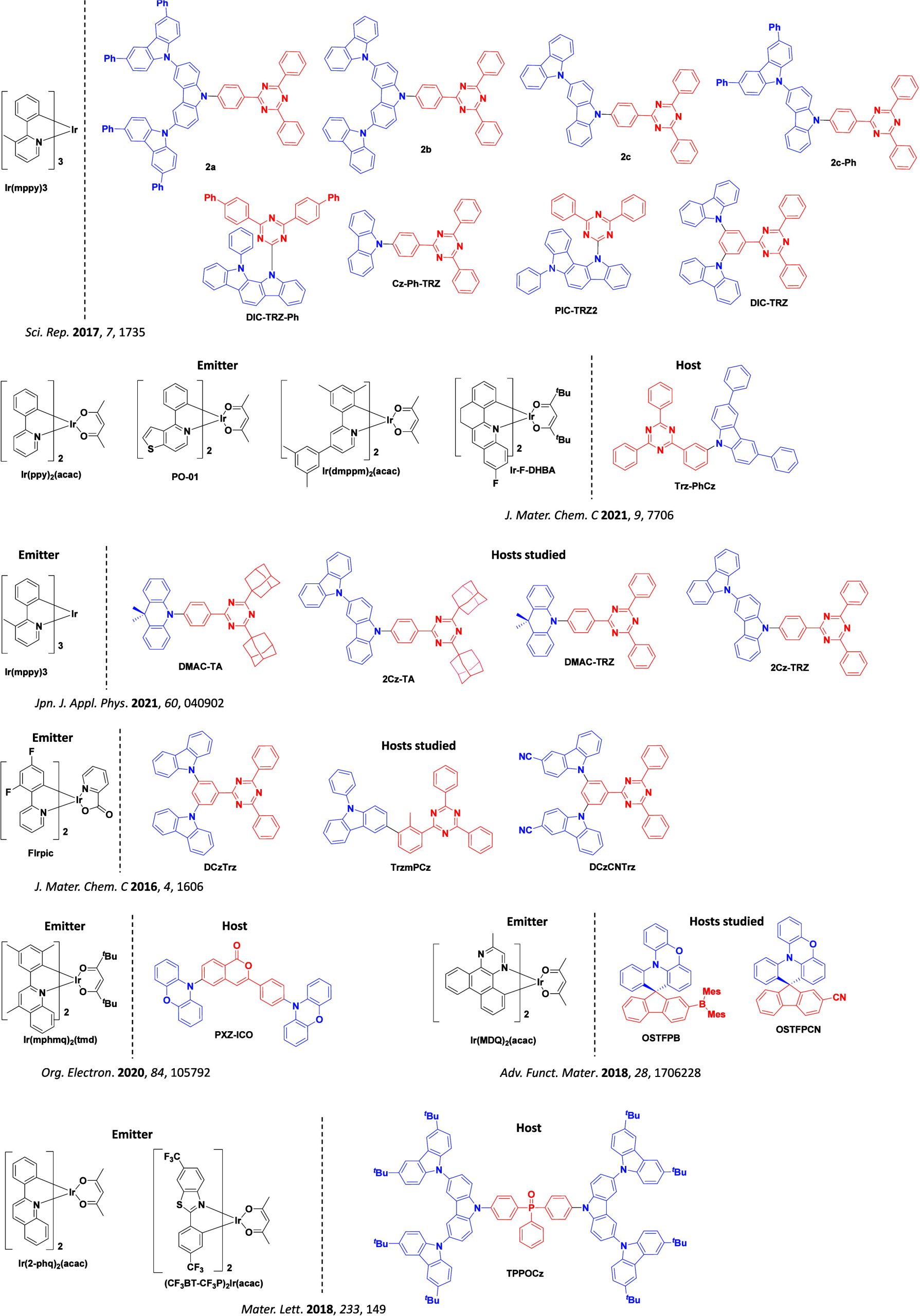
In a subsequent study the same group investigated the device performance using a phosphorescent orange emitter (PO-01, Figure ) doped in a series of indolocarbazole-triazine TADF hosts.ref. ref1309 POBICT, BICT, PBICT, and BBICT have ΔE ST/E T of 0.34/2.70, 0.28/2.70, 0.10/2.66 and 0.06/2.47 eV, respectively, in DCM. The devices were compared using the same doping concentration of 10 wt% PO-01 in the hosts, and it was found that PBICT with a combination of low ΔE ST and high ET translated into the best device performance. A low efficiency roll-off was also observed for this OLED, with an EQEmax = 24.5%, EQE1000 = 24.2%, and EQE10,000 = 23.8%. Despite having the smallest ΔE ST, the device employing BBICT showed poorer performance (EQEmax = 13.7%, EQE1000 = 13.6% and EQE10,000 = 13.1%), attributed to the energy mismatch with PO-01. The LUMO level of BBICT, ELUMO = −2.80 eV, is lower than that of PO-01, ELUMO = −2.70 eV, which leads to inefficient charge recombination and confinement on the phosphorescent emitter. Encouraged by their preliminary success with PBICT as a host material, the same group sought to further optimize the host through the addition of nitrile groups on the phenyl rings of the triazine to generate BCPICT.ref. ref1310 The OLED using BCPICT (λPL = 575 nm, E T = 2.76 eV, and ΔE ST = 0.08 eV in toluene) as the host showed an EQEmax = 10.5% and an EQE1000 = 9.9% in combination with the phosphorescent red emitter Ir(mphmq)2(tmd) at 2 wt% doping concentration.
Duan and co-workers have investigated the blue TADF emitter DMAC-DPS (Figure ) as a host material.ref. ref562 In their study white OLEDs were fabricated by combining the blue-emitting TADF host with orange-emitting phosphor PO-01, and controlling the degree of energy transfer from host to guest and overall EL color by modulating the doping level. The best white OLED, doped with 0.8 wt% PO-01, showed EQEmax/EQE1000 of 20.8/19.6% and PE1000 of 38.7 lm W–1 at CIE coordinates of (0.398, 0.456), and even at 5000 cd m–2 the EQE remained above 15%.ref. ref562 DMAC-DPS was also used as a host to demonstrate efficient green, red, and white OLEDs using green Ir(ppy)2(acac) and red Ir(mphmq)2(tmd) phosphorescent dopants.ref. ref580 The red and green devices showed similarly high EQEmax of 22.4 and 19.5%, with the efficiencies remaining as high as 19.6 and 18.7%, respectively, at 5000 cd m–2. The EML of the white devices contained DMAC-DPS with 0.2 wt% of both the green Ir(ppy)2(acac) and red Ir(mphmq)2(tmd) dopants, and displayed CIE coordinates of (0.360, 0.390), (0.352, 0.387) and (0.364, 0.390) at voltages of 5, 7, and 9 V, respectively. The EQEmax reported for this white device was 20.2%, and the efficiency roll-off was very low with an EQE1000 of 19.4%.
A meta-linked isomeric variant of DMAC-DPS, mSOAD (Figure ), was used by Wang et al. as a host for the red phosphorescent emitter Ir(pq)2acac.ref. ref1311 mSOAD possesses a high triplet energy of 2.91 eV, a small ΔE ST of 0.01 eV, and a short td of 2.11 μs in the crystalline state.ref. ref1312 The best red device was achieved using 4 wt% of the emitter within the EML, and showed an EQEmax of 20.3% and an EQE1000 of 10.8%.ref. ref1311 By reducing the concentration of the red emitter to between 0.4–1.5 wt%, incomplete energy transfer occurs and white emission is produced from the combined emissions of the blue host and red guest. The EQEmax of the WOLEDs ranged from 12.2–17.4%, and the EQE1000 varied from 4.8–13.0%, depending strongly on the emitter doping concentration. The emission CIE coordinates for devices with dopant concentration of 1.5, 0.8, and 0.4 wt% were (0.549, 0.399), (0.448, 0.400) and (0.032, 0.415), respectively. Further evaluation of sulfone-based TADF compounds as host materials was conducted by Xia et al.,ref. ref1313 using both mSOAD or the carbazole analogue tBu-mSOCz (λPL = 440 nm, E T = 2.88 eV and ΔEST = 0.42 eV in toluene). These hosts were combined with sky-blue (FIrpic, λEL = 471 nm), green (fac-Ir(ppy)3, λEL = 515 nm), or red (Ir(pq)2acac, λEL = 606 nm) phosphorescent dopants to produce PhOLEDs.ref. ref1313 The best blue device employed tBu-mSOCz with 6 wt% of FIrpic, and showed L max, EQEmax, and EQE1000 of 6176 cd m–2, 14.7%, and 13.3%, respectively. The best green devices employed mSOAD with 4 wt% of fac-Ir(ppy)3, and achieved L max, EQEmax, and EQE1000 of 35,530 cd m–2, 19.0%, and 18.4%. mSOAD was also the host of choice for the red PhOLEDs with 4 wt% of Ir(pq)2acac, with corresponding L max, EQEmax, and EQE1000 of 19,420 cd m–2, 20.3%, and 10.6%. Related A-D-D-A carbazole-sulfone TADF material BCz-2SO (λPL = 410 nm, E T = 2.91 eV, and ΔE ST = 0.35 eV in toluene) has also been explored as a host in PhOLEDs with the sky-blue emitter FIrpic.ref. ref1314 The solution-processed device with 1 wt% doping of the emitter showed an EQEmax of 7.8% and a L max of 16,537 cd m–2.
Lin et al. reported deep-blue TADF materials BT-01 (λPL = 396 nm, E T = 3.00 eV, and ΔE ST = 0.45 eV in neat film) and BT-02 (λPL = 375 nm, E T = 3.03 eV, and ΔE ST = 0.52 eV in neat film, Figure ) and demonstrated their potential as hosts for phosphorescent and TADF OLEDs.ref. ref1315 Both compounds are composed of a sulfone acceptor and carbazole donor that are electronically decoupled through a m-bitolyl bridge. The cyano group attached to the carbazole in BT-02 explains its blue-shifted emission compared to BT-01, and likely also contributes to charge transport as an OLED host material. Both molecules showed delayed emission despite their large ΔE ST values (0.45 and 0.52 eV for BT-01 and BT-02, respectively, in neat film). This result implies the involvement of higher-lying triplet states enabling RISC, which was further supported by lower measured TADF activation energies of 0.067 and 0.109 eV, and surprisingly short td of 1.3 and 1.8 μs for BT-01 and BT-02, respectively. Devices with FIrpic as the emitter using BT-01 or BT-02 as the host showed EQEmax/EQE1000 of 31.8/31.2% and 30.7/29.9%, respectively. The combination of a rather high dopant concentration of 10 wt%, bipolar charge transport by the host, and orbital alignment between host and guest led not only to these high efficiencies but also to the very low efficiency roll-off. Indeed, the efficiency roll-off was much higher when the emitter was switched to 2CzPN, with the efficiency sharply decreasing from EQEmax of 25.5 and 22.3% to EQE1000 of 10.0 and 6.2% for BT-01 and BT-02 as hosts, respectively.
The first use of pyrimidine-based TADF compounds as hosts was reported by Wang et al., containing either acridine (DMAC-BPP: λPL = 502 nm, E T = 2.50 eV, and ΔE ST = 0.03 eV in toluene) or δ-carboline (DCb-BPP: λPL = 452 nm, E T = 2.54 eV, and ΔE ST = 0.20 eV in toluene, Figure ) as the donor units.ref. ref1316 These hosts were used in conjunction with 5 wt% of phosphorescent emitter PO-01. The device using DCb-BPP as the host exhibited an EQEmax of 21.5% and an EQE of 17.7% at 10,000 cd m–2. In addition, the OLED showed a long operational lifetime with an LT50 of 424 h at initial brightness of 1000 cd m–2. The device using DMAC-BPP as the host showed similar performance (EQEmax of 19.8% and EQE10,000 of 17.9%) to that of DCb-BPP; however, the lifetime of the DMAC-BPP device was only about 5% of the DCb-BPP device.
The influence of the host on the operational stability of OLEDs was assessed by Fukagawa et al. by investigating a series of triazine-containing TADF hosts with the same green phosphorescent emitter fac-Ir(mppy)3.ref. ref233 Across hosts 2a, 2b, 2c, 2c-Ph, Cz-Ph-TRZ, PIC-TRZ2, DIC-TRZ, and DIC-TRZ-Ph (Figure ), the authors found that the k RISC of the host strongly indicates the device lifetime. This conclusion applies most strongly when the emitter is doped at very low concentration within the EML, as the FRET rate between host and guest, k FRET, also affects the device lifetimes. The highest performing device used 2c as the host and had a k FRET of 10.0 × 108 s–1; the authors did not however provide the value for the host k RISC. This device showed an EQEmax of 21.5% as well as an excellent lifetime (LT50) of 20,000 h from an initial 1000 cd m–2. To better understand the role that the TADF host plays in the success of the guest emitter, the authors also investigated TADF-inactive Cz-Ph-TRZ as a reference host. The LT50 of the PhOLED using Cz-Ph-TRZ, in which triplet up-conversion on the host is suppressed, is about 500 hours: 40-fold shorter than the device using 2c as the host. Additionally, triazine-containing hosts were shown to form exciplexes with the platinum-based emitter PtN7N, which adversely impacts the device performance and color. To overcome this problem, sterically hindered triazines within the host material can be employed to suppress exciplex formation. Accordingly, devices with PIC-TRZ2 as the host did not suffer the same degree of exciplex formation as was observed with DIC-TRZ.
The related triazine-containing TADF host material Trz-PhCz, was reported by Sun et al., containing a 3,6-diphenyl-9H-carbazole donor linked at meta position on the TRZ phenylene linker (Figure ).ref. ref1317 Trz-PhCz (λPL = 470 nm, E T = 2.85 eV, and ΔE ST = 0.19 eV in 2-MeTHF) exhibits a short τd of 2.5 μs in neat film, which should help to support triplet harvesting and reduce efficiency roll-off in devices. This host was employed with Ir(ppy)2(acac), PO-01, Ir(dmppm)2(acac), and Ir-F-DHBA to fabricate green, yellow, orange, and red PhOLEDs, respectively. All the resultant devices showed extremely low efficiency roll-off, with EQEs of over 20% at 10,000 cd m–2. Notably, the orange device showed a record-high efficiency and low roll-off with EQEmax = 31.4%, and EQE10,000 = 25.5%.
Ito et al. reported a comparative study on the operational lifetime of PhOLEDs using one of four triazine-based TADF host materials with the phosphorescent green dopant Ir(mppy)3 at 3 wt% loading: DMAC-TA, 2Cz-TA, DMAC-TRZ, and 2Cz-TRZ (Figure ).ref. ref1318 The LT50 of the devices were found to be 45, 180, 2500, and 13000 h for the different respective hosts, from an initial brightness of 1000 cd m–2. Such significant variations in the LT50 were correlated with the bond dissociation energies (BDE) of the C-N and C-C bonds in the host.
Jeon et al. studied a range of triazine-based hosts in combination with the sky-blue iridium complex FIrpic (E T = 2.65 eV) to gauge the importance of the singlet and triplet energies of the host in relation to the triplet energy of the emitter.ref. ref1319 TADF compounds DCzTrz (E T = 2.64 eV) and TrzmPCz (E T = 2.79 eV, Figure ), and the fluorescent compound DCzCNTrz (E T = 2.68 eV) were investigated along with the commercially available host mCP (E T = 2.90 eV). The EQEmax/EQE1000 values of the devices were found to be 15.6/15.3, 15.7/15.0, 16.3/14.1, and 2.8%/1.3% for the devices using mCP, DCzTrz, TrzmPCz, and DCzCNTrz, respectively, showing similarity in performance across all hosts except for DCzDNTrz. According to the authors, in the charge trapping process triplet excitons of FIrpic may decay directly to the ground state or transfer energy to triplet excitons of DCzTrz because of the high triplet energy of FIrpic (E T = 2.65 eV) compared to that of DCzTrz (E T = 2.64 eV). In addition, the smaller bandgap host DCzTrz performed the best in terms of lower driving voltage and higher current density of the device, with this outcome proposed to arise from its shallow HOMO and deep LUMO, which facilitates suitable hole and electron injection. In the DCzTrz host, emission arises exclusively from the phosphorescent dopant (based on the spectra) but with a fitted delayed emission component that is suggested to be associated with TADF. This indicates that TADF hosts can upconvert triplets to singlets and transfer energy to the near-isoenergetic guest by FRET, supporting the device performance.
Qian et al. reported the TADF host PXZ-ICO (Figure ), consisting of a phenoxazine donor and an isocoumarin acceptor.ref. ref1320 This host has a small ΔE ST of 0.14 eV (λPL = 560 nm) in 2-MeTHF glass and a τd of 343 μs in 10 wt% doped films in DPEPO. The OLED with 3.5 wt% doping of red phosphorescent emitter Ir(mphmq)2(tmd) in PXZ-ICO host exhibited CIE coordinates of (0.62, 0.37) and showed an EQEmax of 18.6%. These device metrics surpass those of a similar OLED using the conventional host CBP (EQEmax = 15.3%).ref. ref1320
Dendritic TADF molecules have also been explored as hosts, for example TPPOCz (Figure )ref. ref1004 with sky-blue FIrpic, orange Ir (CF3BT-CF3P)2(acac), and red Ir(2-phq)2(acac).ref. ref1321 TPPOCz contains a second-generation carbazole donor dendron and a central phosphine oxide acceptor, and has a high E T of 2.98 eV with ΔE ST of 0.22 eV and λPL of 400 nm, all in neat film. Of the devices reported using 4 wt% doping of FIrpic as the emitter, the highest EQEmax was 20.4% and the maximum luminance was 13,235 cd m–2. The devices employing 3 wt% of either Ir(CF3BT-CF3P)2(acac) or Ir(2-phq)2(acac) showed EQEmax of 14.9 and 12.4%, respectively. This work is one of the rare examples of a solution-processed TADF dendrimer host for OLEDs, with dendrimer TADF materials explored further in Section sec10 .ref. ref1321
Two spiro-based TADF hosts, OSTFPB (λPL = 495 nm, E T = 2.59 eV, and ΔE ST = 0.21 eV in toluene) and OSTFPCN (λPL = 460 nm, E T = 2.68 eV, and ΔE ST = 0.20 eV in toluene, Figure ) were used by Wang et al. in red PhOLEDs with Ir(MDQ)2(acac).ref. ref4 Devices using 2 wt% of the dopant in OSTFPB or OSTFPCN as hosts showed very high EQEmax of 29.1 and 31.2%, and low efficiency roll-off at 100 cd m–2 of 0.3 and 2.6%, respectively.ref. ref4
TADF Hosts with Fluorescent Emitters
In addition to PhOLEDs employing TADF materials as hosts, several groups have worked to produce efficient devices using the same strategy for fluorescent emitters. The hosts and emitters used in these devices are shown in Figure . For example, Zhang et al. used DIC-TRZ (E T = 2.82 eV and ΔE ST = 0.06 eV)ref. ref1322 and PIC-TRZ (E T = 2.70 eV and ΔE ST = 0.11 eV; 6 wt% in mCP)ref. ref76 as TADF host materials for 1 wt% DDAF in yellow OLEDs (Figure ).ref. ref1268 The device based on DIC-TRZ:DDAF achieved an EQEmax of 12.2% and an EQE1000 of 5.5%, while the combination of PIC-TRZ:DDAF resulted in EQEmax of just 4.7% and EQE1000 of 3.9%. This lower efficiency can be attributed to the larger ΔE ST of PIC-TRZ in comparison to DIC-TRZ, leading to inefficient triplet harvesting from the host.
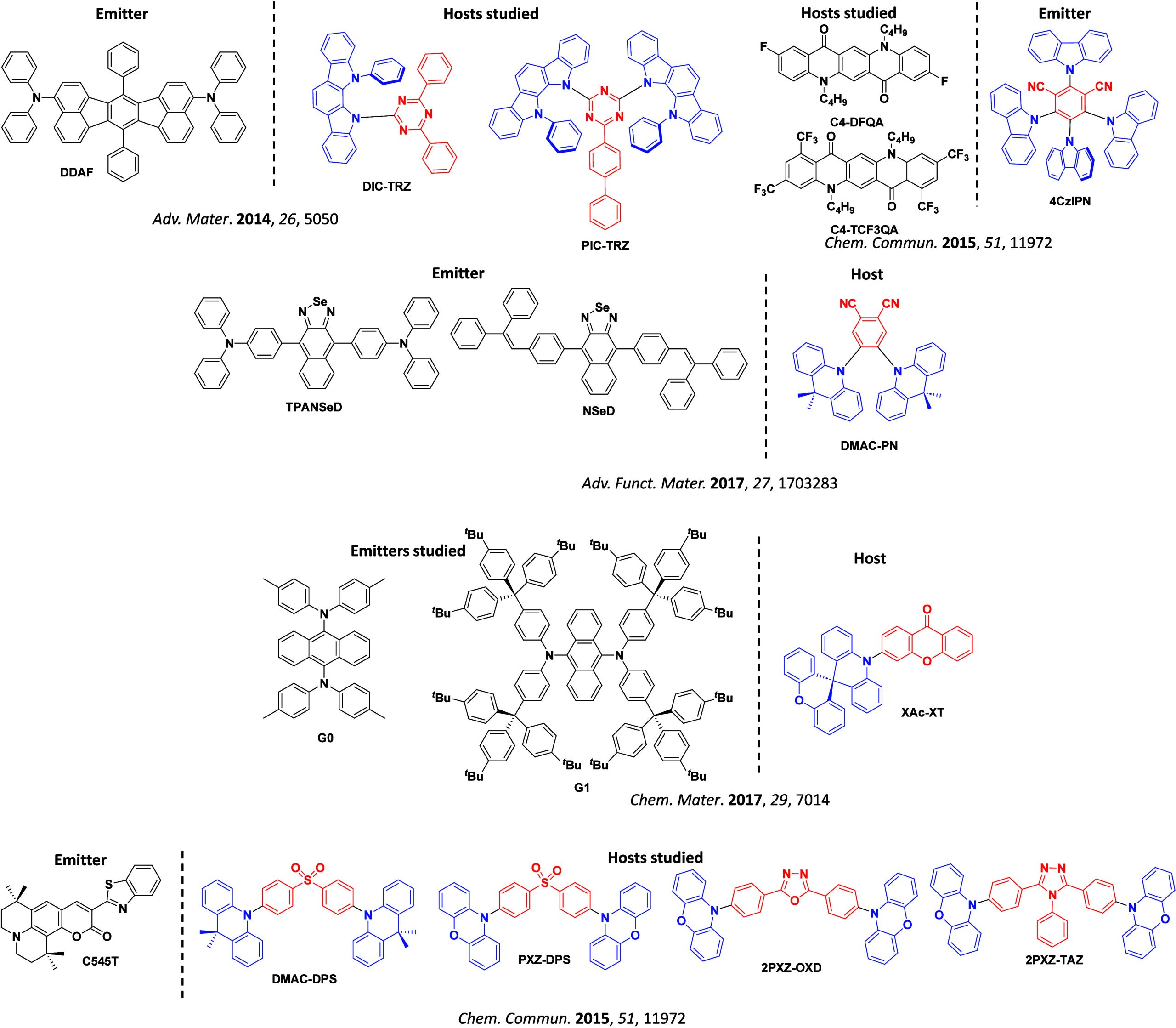
The orange TADF compound DMAC-PN, with λPL = 557 nm in 5 wt% doped CBP films and ΔE ST = 0.27 eV in toluene,ref. ref1323 was used as a host for two near infra-red (NIR) dyes containing naphthoselenadiazole moieties (TPANSeD and NSeD, Figure ).ref. ref1324 The devices with 4 wt% TPANSeD showed an EQEmax of 2.7% at λEL of 730 nm and an L max of 10,569 cd m–2. Upon replacing the side group of 4-(diphenylamino)phenyl in TPANSeD with the bulkier 4-(2,2-diphenylvinyl)phenyl) in NSeD, non-radiative DET pathways were suppressed resulting in higher EQEmax of 3.8% at λEL 664 nm, and an L max 16,956 cd m–2.ref. ref1324
Wang et al. used 4CzIPN as a host in combination with structurally similar quinacridone derivatives C4-DFQA and C4-TCF3QA (Figure ) as yellow-green fluorescent dopants.ref. ref1325 At 0.5 wt% loading the devices showed EQEmax of 13.5 and 14.6% respectively, and even at these very low doping concentrations only emission from the dopant was observed, indicating very efficient energy transfer between the host and guest. Excellent efficiency roll-off at 1000 and 5000 cd m–2 was noted for both emitters due to efficient RISC within the host and subsequent FRET from the host to the guest. The 4CzIPN:C4-DFQA and 4CzIPN:C4-TCF3QA devices showed EQE1000/EQE5000 of 12.6/11.0 and 13.7/12.3% respectively.ref. ref1325
A dual TADF sensitizing strategy was used to transfer energy within OLEDs to fluorescent green emitter C545T through FRET.ref. ref1326 Initial devices were fabricated using a series of TADF hosts (DMAC-DPS, PXZ-DPS, 2PXZ-OXD, and 2PXZ-TAZ, Figure ); however, only DMAC-DPS and PXZ-DPS were selected for further investigation due to their superior performance in the preliminary studies. The FRET and DET energy transfer rates were measured with and without the auxiliary PXZ-DPS sensitizer to understand its effect in the energy transfer process, with the final compared devices composed of DMAC-DPS:PXZ-DPS (30 wt%):C545T (1.5 wt%), and DMAC-DPS:C545T (1.5 wt%). Changes in the prompt and delayed emission components in thin films were measured to understand the FRET and DET rates. After introducing the second TADF host, the FRET rates increased from 9.26 × 107 to 1.43 × 108 s–1, while the authors quote DET rates to be on the order of 106 s–1. The devices consequently showed an EQEmax of 11.1% in the dual host system, and only 9.0% in the absence of the second TADF host.
Aizawa et al. designed dendritic fluorescent emitter G1 (Figure ), with the aim of preventing Dexter energy transfer to the emitter from TADF hosts XAc-XT.ref. ref1327 The solution-processed OLEDs with 1 mol% of reference emitter G0 showed an EQEmax of only 3.2% (at 442 cd m–2), while with G1 the device performance improved measurably to EQEmax = 5.2% (at 417 cd m–2).
Exciplex Hosts with Fluorescent Emitters
Exciplex-forming co-host systems with TADF properties have been used in conjunction with phosphorescent emitters to generate efficient and highly stable EL.ref1328−ref1329ref1330 Inspired by this, Liu et al. employed exciplex TADF host TAPC:DPTPCz (Figure ) in combination with fluorescent dopant C545T.ref. ref1331 Devices were fabricated using a range of doping concentrations (0.2–1.0 wt%), and surprisingly the best efficiency was obtained with only 0.2 wt% of emitter. The devices achieved EQEmax and EQE100 of 14.5 and 12.0%, respectively. However, at such low doping concentrations the color purity of the device was low due incomplete energy transfer to the emitter, with residual exciplex emission observed in the EL. At the higher doping concentration of 1 wt% of C545T, only its emission was observed although the EQEmax and EQE100 were significantly lower at 7.5 and 5.3%, respectively.
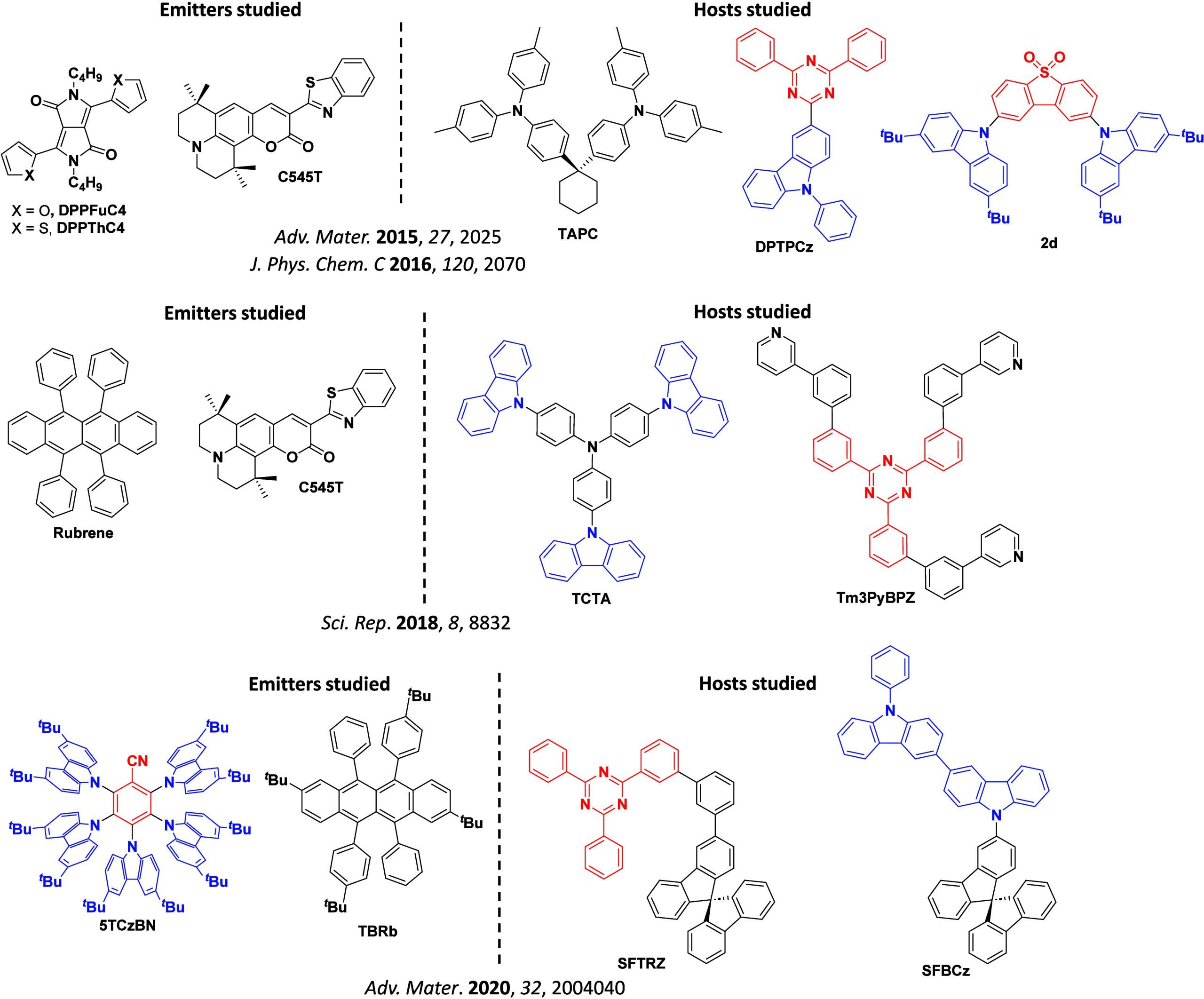
A multichannel exciplex-TADF host composed of TAPC:2d (7:3), was used alongside a series of diketodipyrolopyrole emitters (Figure ).ref. ref1332 In this case both the exciplex itself and the exciplex component 2d are TADF active, giving multiple channels for RISC and triplet harvesting to occur. The most efficient green devices used DPPFuC4 as the fluorescent dopant with an EQEmax/EQE100 of 12.1/11.2%. The efficiency increased when the dopant was a thiophene analogue (DPPThC4), perhaps due to more efficient triplet harvesting of the exciplex influenced by an external heavy atom effect from the sulfur of the thiophene, with an EQE100 of 11.1%, and a high L max of 9983 cd m–2.
Zhang et al. also reported fluorescent devices using a TADF exciplex host.ref. ref1333 In this case TCTA:Tm3PyBPZ (λPL = 514 nm in 1:1 film, Figure ) was used to transfer energy to the green and red emitters C545t and rubrene. The green devices with 1 wt% of C545t showed L max, EQEmax, and EQE1000 of 20,640 cd m–2, 10.4%, and 7.9%, respectively. The red rubrene devices showed comparable values of 22,170 cd m–2, 10.0%, and 8.4%. All the devices exhibited higher efficiencies than previous reports which used the non-doped TADF exciplex as the emitter (1:1 ratio), which had L max, EQEmax, and EQE1000 of 12,800 cd m–2, 13.1%, and 8.8%, respectively.ref. ref1334
A pair of π-D and π-A exciplex forming materials were designed for WOLED purposes, envisioned to operate by partial energy transfer from the co-host to both a blue TADF sensitizer (5TCzBN) and a yellow fluorescent emitter (TBRb, Figure ).ref. ref566 This reported SFTRZ:SFBCz exciplex system also incorporates a bulky bipolar π-group as a spacer, which achieves two objectives: i) an increased separation distance between the D and A subunits of the π–D and π–A molecules, resulting in a blue-shifted emission; and ii) the retention of the superior charge transporting ability characteristic of exciplex systems. When SFTRZ:SFBCz was used in combination with 20 wt% 5TCzBN (λEL = 485 nm), 0.2 wt% TBRb (λEL = 552 nm), and 0.05 wt% red fluorescent dopant, RD (RD = 1,3,7,9-tetrakis(4-(tert-butyl)phenyl)-5,5-difluoro-10-(2-methoxyphenyl)-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinine) (λEL = 614 nm), the best performing device achieved EQEmax, EQE1000, and lifetime (LT80 at initial 5000 cd m–2) of 16.7, 16.5, and 203 h, respectively, at warm white CIE coordinates of (0.439, 0.452).
TADF Hosts with TADF Emitters
The aforementioned examples demonstrate the value of TADF hosts used in combination with phosphorescent and fluorescent emitters. Similar device performance improvements can also be achieved in OLEDs that employ TADF hosts for other TADF emitters (Figure ). In a study by Duan and co-workers, the TADF host DMIC-TRZ was used in combination with TADF emitters 5TCzBN (blue, λEL = 486 nm), DMAC-BP (green, λEL = 513 nm) and 4TCzTPN (orange, λEL ∼548 nm) to produce efficient devices across the visible spectrum.ref. ref1335 The devices showed EQEmax of 19.2, 21.0, and 23.2% respectively, and the efficiency roll-offs at 2,000 cd m–2 were excellent, ranging from 3–7%. Reference devices produced with conventional hosts CBP and 26DCzPPy showed reduced performance and increased efficiency roll-off compared to those with the triplet-harvesting TADF hosts.
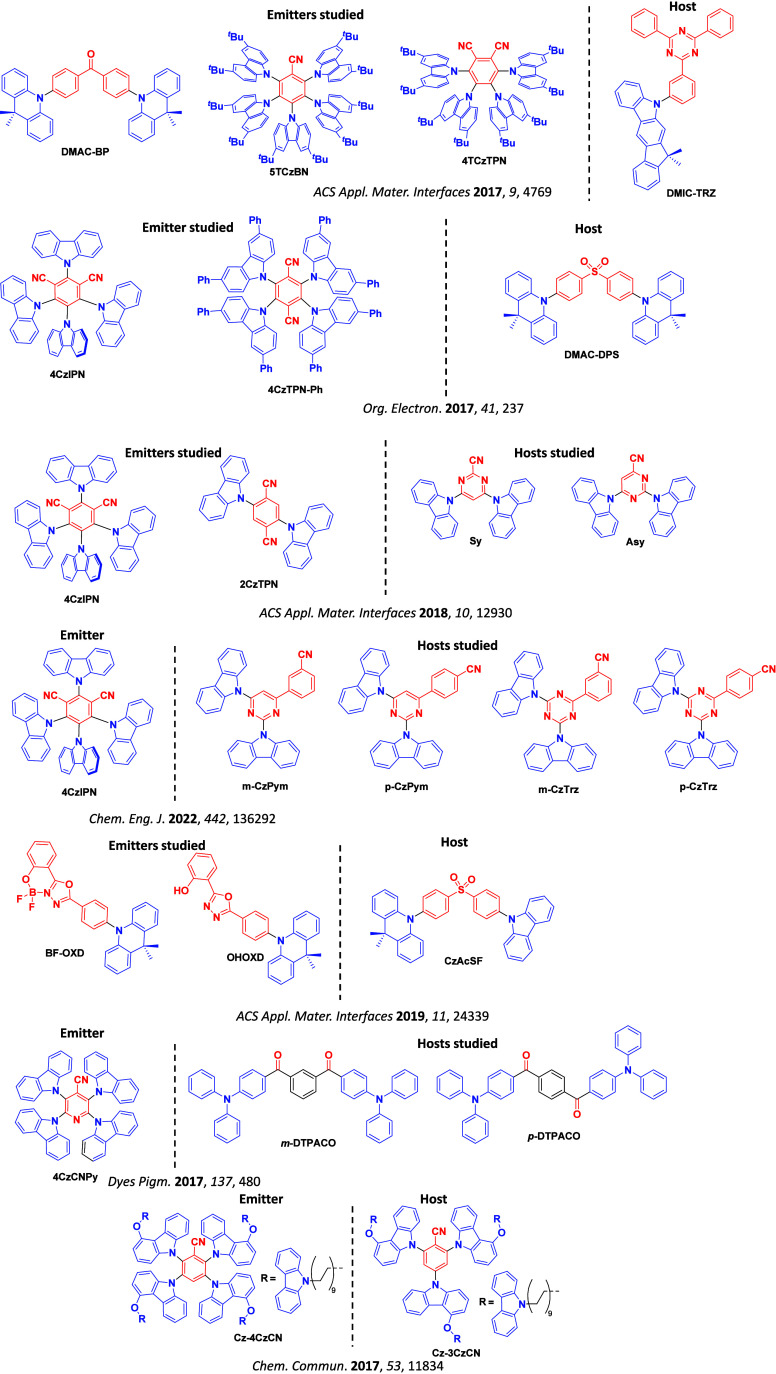
Using DMAC-DPS as a host, Liu et al. fabricated a series of green and orange devices with 4CzIPN or 4CzTPN-Ph as the TADF emitters (Figure ).ref. ref585 The best device with 4CzIPN was achieved using 6 wt% doping, showing an EQEmax of 10.9% and an EQE1000 of 9.1%. By contrast, the best device with 4CzTPN-Ph required a higher doping concentration of 9 wt% and showed an EQEmax of 11.0% and an EQE1000 of 10.1%. By exploiting partial energy transfer from the host to the guest, white-emitting devices were also fabricated, the best of which using 0.2 wt% 4CzTPN-Ph in DMAC-DPS showed an EQEmax of 14.7% and EQE1000 of 12.0% with CIE coordinates of (0.25, 0.37).
Symmetric and asymmetric hosts Sy and Asy (Figure ) were reported by Li et al. incorporating carbazole and cyanopyrimidine.ref. ref1336 These were used along with 2CzTPN (blue)ref. ref1337 and 4CzIPN (green) emitters.ref. ref31 Both the blue and green devices using the Sy host (λPL = 471 nm, E T = 3.06 eV, and ΔE ST = 69 meV in toluene) performed better than using Asy (λPL = 518 nm, E T = 2.92 eV, ΔE ST = 114 meV in toluene). In the device with 10 wt% 2CzTPN in Sy the L max, EQEmax, and EQE1000 were 122,100 cd m–2, 20.4, and 16.9%, while the corresponding values for the 4CzIPN:Sy device were 221,500 cd m–2, 24.0, and 22.1%. Factors such as its smaller ΔE ST and more efficient energy transfer to the emitters may explain the improved performance of the Sy devices. Notably, the TDMs of both emitters were preferentially horizontally aligned (88% in both hosts), contributing to the high efficiencies.
Chen et al. reported TADF host materials m-CzPym, p-CzPym, m-CzTrz, and p-CzTrz (Figure ). Comprised of carbazole as the donor and benzonitrile-substituted heteroarenes (triazine or pyrimidine) as the acceptor, these had measured ΔE ST in toluene of 0.44, 0.46, 0.31, and 0.40 eV respectively.ref. ref1338 High-performance green TADF OLEDs were fabricated using these hosts in combination with 4CzIPN as the emitter.ref. ref31 Amongst all the devices, m-CzPym was found to be the best host with the device showing an EQEmax of 31.5%, PEmax of 116.5 lm W–1, a turn-on voltage of 2.5 V, and low efficiency roll-off (EQE1000 = 29.0%). The high EQEmax was linked to the outstanding light outcoupling efficiency of over 31–35%, as verified through angle-dependent PL intensity measurement.
Zhou et al. reported two new TADF emitters that contain a DMAC donor and oxadiazole acceptor either with or without a chelated BF2 group. OHOXD (Figure ) has λPL = 473 nm, ΔE ST = 0.16 eV, and τd = 1.9 μs in toluene with ΦPL = 30% in 10 wt% doped CzAcSF films, while boron-chelated BFOXD has identical λPL = 473 nm, smaller ΔE ST = 0.09 eV, yet slower τd= 4.3 μs, in toluene, with much larger ΦPL = 66% in the same CzAcSF host.ref. ref1339 Solution-processed devices with OHOXD showed L max, EQEmax, and EQE1000 of 1520 cd m–2, 12.1, and 4.3%, which rose considerably for BFOXD at 4518 cdm–2, 20.1, and 12.7%.ref. ref1339
Hu et al. reported two isomeric phthaloyl/triphenylamine TADF materials as hosts for solution-processed devices.ref. ref1340 m-DTPACO and p-DTPACO (Figure ) consist of triphenylamine as end-capping electron-donating groups and isophthaloyl or terephthaloyl as the central electron-withdrawing moieties. m-DTPACO (λPL = 477 nm as the neat film) and p-DTPACO (λPL = 522 nm as the neat film) have ΔE ST of 0.21 and 0.05 eV, and td of 8.29 and 9.60 μs with ΦPL of 75 and 39%, respectively. Non-doped solution-processed devices with m-DTPACO and p-DTPACO as emitters exhibited L max of 10,005 and 7354 cd m–2, and EQEmax of 2.4 and 3.7% respectively. Their potential as host materials was then investigated by doping green TADF emitter 4CzCNPy ref. ref1341 at 10 wt%.ref. ref1340 The emission spectrum of m-DTPACO showed better overlap with the absorption spectrum of 4CzCNPy, allowing more efficient energy transfer from the host to the guest. This is reflected in the solution-processed device performance, with high L max of 22,322 cd m–2 and EQEmax/EQE1000 of 13.0/10.3% for the device using m-DTPACO. By contrast, the device performance was lower using p-DTPACO with L max and EQEmax/EQE1000 values of 15,510 cd m–2 and 9.0/5.6%, respectively.
Lastly, Ban et al. employed encapsulated TADF materials as both host (Cz-3CzCN, λPL = 445 nm as the neat film) and guest (Cz-4CzCN, λPL = 475 nm as the neat film, Figure ) in solution-processed devices.ref. ref1342 Alkyl chains connected to a peripheral carbazole donor were used to insulate the emissive 3CzCN and 4CzCN cores. Cz-3CzCN and Cz-4CzCN have promising ΔE ST values of 0.24 and 0.22 eV and ΦPL of 25 and 78%, respectively, in toluene. Solution-processed green devices with 4 wt% Cz-4CzCN in Cz-3CzCN showed an EQEmax of 23.5%; however, the efficiency roll-off was significant, with EQE100/EQE1000 of 15.5 and 7.8%, respectively. Reference devices were also made with conventional non-encapsulated TADF host (3CzBN) and guest (4CzBN) for comparison. These non-encapsulated host-guest devices showed greatly reduced EQEmax, EQE100, and EQE1000 values of only 5.9, 3.1 and 1.9%, respectively, demonstrating the effectiveness of the encapsulation strategy for improving device performance.
Outlook
This section details examples of TADF-active molecules or exciplex blends acting as promising host materials for both vacuum-deposited and solution-processed OLEDs. The intrinsically electron-donating and electron-accepting chemical groups associated with the charge-transfer excited states of these TADF materials provide balanced charge transport as well as RISC pathways for triplet harvesting for separate fluorescent, phosphorescent, or TADF guest emitters. This application of TADF materials can therefore support improvements in device lifetime and efficiency, with strong conceptual overlap to hyperfluorescence (Section sec17 ), AIE emitters (Section sec13 ), and both exciplex and through-space charge transfer emitters (Sections sec8 and sec12). Emblematic of the examples of this concept summarized here, we re-highlight the use of m-CzPym as a host for the emitter 4CzIPN.ref. ref1338 While 4CzIPN doped into standard hosts such as CBP can achieve EQEmax of ∼20%, with the added support of the TADF active m-CzPym host the the device with the same emitter can have an EQEmax that exceeds 30% and also shows remarkably low efficiency roll-off.
Given the promise of this approach, it seems evident that significant future performance gains across a wide range of OLED technologies will likely be enabled by a better understanding and the application of TADF materials not just as emitters, but also as hosts. While this is already demonstrated for various green and red terminal emitters, we note that this concept is yet to be fully realised for blue emission, which would nominally require very high triplet energy hosts (>3.0 eV). Rather than an intrinsic limitation though, we anticipate that the as-yet undiscovered host materials required to replace DPEPO and related phosphine oxide compounds that are the most commonly used hosts in supporting deep-blue and UV TADF OLEDs will likely arise from this area of research, with D-A TADF emitters (and likely poor emitters) finding successful repurposing as hosts that directly contribute to triplet management.
Supramolecular Assemblies of TADF Materials
Introduction
Supramolecular chemistry is now widely recognised as a powerful and fascinating strategy to bestow molecules with new structural features and properties outside the scope of covalent bonding.ref1343−ref1344ref1345ref1346ref1347 This provides an additional dimension of materials design compared to the combinatorial strategies associated with D-A TADF emitters (Sections sec3–sec5), and the still evolving understanding of MR-TADF compounds (Section sec11 ). Naturally, researchers have applied supramolecular strategies to TADF emitters with the aim to significantly alter their photophysical properties, in some cases leading to emergent properties unseen in their discrete counterparts. We recently reviewed this area in detail.ref. ref56 It is worth noting that despite the wide range of supramolecular structures shown to exhibit TADF, the intersection of these research fields is still relatively young and so there are at present still few examples from each class of supramolecular system.
Here, we firstly discuss a TADF core comprised of carbazole and benzophenone as an illustrative example that has been incorporated in three different supramolecular systems, each showing vastly different properties and functionalities. We then divide and summarise other notable examples of TADF supramolecular assemblies into two categories: architectures that involve co-ordination to metal centres, and non-co-ordinating systems operating via aggregation and/or encapsulation.
CzBP – One Core in Three Systems
Three distinct supramolecular structures have been formed using the same parent TADF emitter, CzBP: gels, metallocages, and rotaxanes. Each structure possesses different photophysical properties, highlighting the potential of supramolecular chemistry to modulate to properties of other TADF emitters when integrated into distinct assemblies.
The first examples of TADF gels were formed by appending 4-pyridyl groups to the carbazole moiety to give 4PyCzBP and mixing this compound with diacids (Figure ).ref. ref1348 4PyCzBP itself shows blue emission in both degassed DCM (λPL = 477 nm, ΦPL = 52%) and in 10 wt% PMMA-doped films (λPL = 449 nm, ΦPL = 21%). Mixing 4PyCzBP with one equivalent of succinic acid gave a yellow/green gel with enhanced emission at λPL = 500 nm, with the pyridine moieties hydrogen bonding with the diacid hydrogen atoms, though the gel was only weakly bound with a critical gel concentration (CGC) of 5 mg mL–1. A stronger gel formed when using (L)-tartaric acid due to the greater number of hydrogen bonds that could be formed, with a red-shifted emission at λPL = 510 nm and a CGC of 3 mg mL–1. Compared to isolated 4PyCzBP, there was an 11-fold enhancement in the emission when using 0.5 equivalents of the (L)-tartaric acid and a 60-fold enhancement when using 1 equivalent of the same. However, using a greater excess of diacid resulted in a decrease of the emission intensity due to disruption of the intramolecular hydrogen bonding within the gel structure. The TADF nature of the 1:1 4PyCzBP:(L)-tartaric acid was confirmed by transient photoluminescence measurements, which showed biexponential decay kinetics with τPL = 20 ns and 2.3 μs. The ΦPL of the xerogel is six times higher than the neat film (ΦPL = 36% vs. 6%).
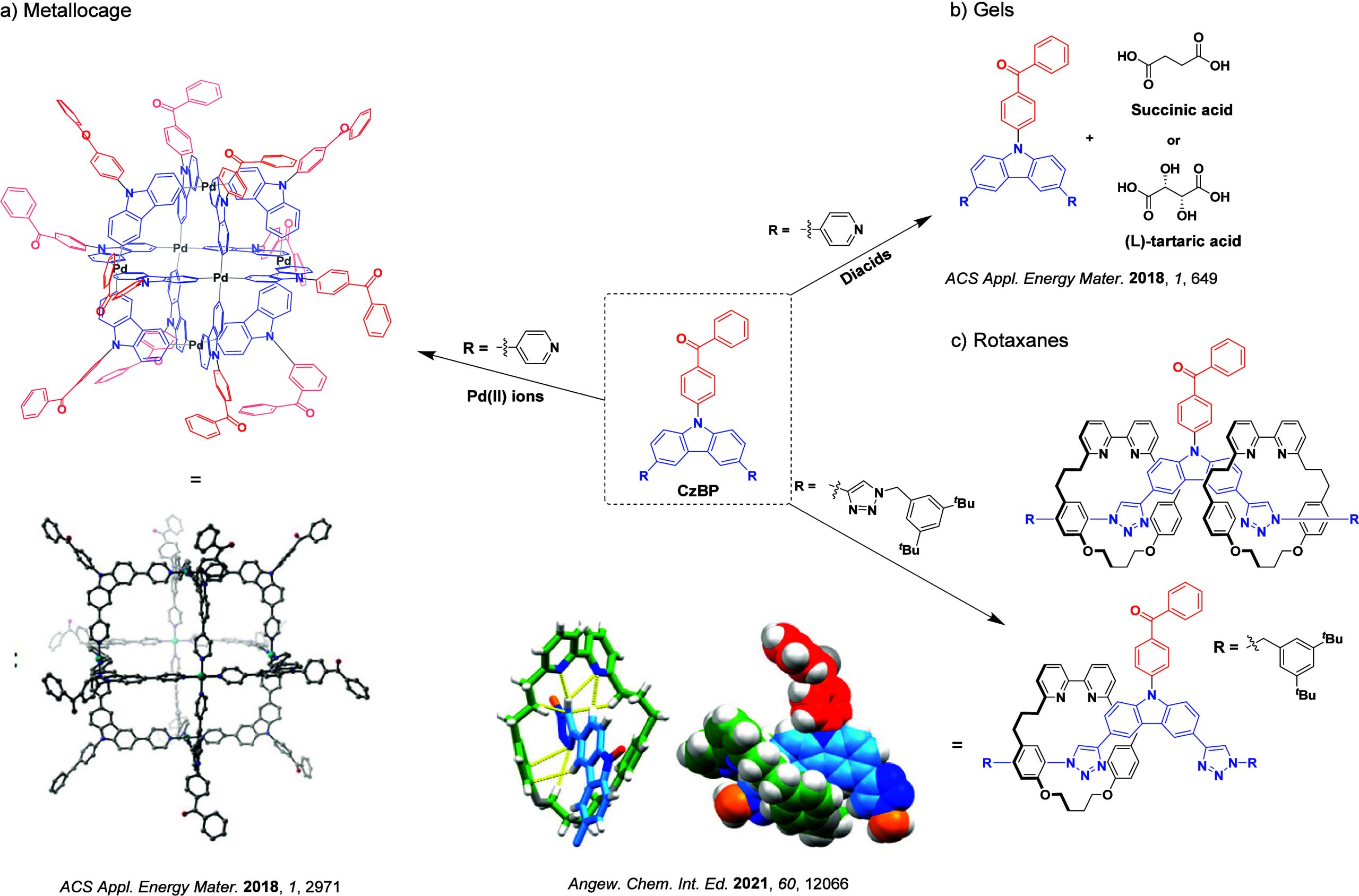
The same 4PyCzBP was separately employed as a ligand in conjunction with Pd2+ to give the M6L12 metallocage, 4PyCzBP-Pd (Figure ).ref. ref1349 This metallocage geometry formed from the combination of the square planar palladium centres along with the angle between co-ordinating pyridines of 4PyCzBP being 93.5°, which has previously been shown by Fujita and co-workers to facilitate M6L12 metallocages.ref. ref1350 The resulting cuboctahedron was calculated to have an internal volume of 6400 Å3, showed a significant reduction in ΦPL and an accompanying red-shift (PyCzBP-Pd; λPL = 555 nm, ΦPL = 4% in DCM) compared to the free emitter in degassed DCM (4PyCzBP; λPL = 477 nm, ΦPL = 52%). No delayed emission was observed for 4PyCzBP-Pd, with only prompt biexponential lifetimes of τPL = 3 ns and 30 ns. This significant change in optical properties was rationalised by DFT calculations, showing that while the HOMO was still distributed over the carbazole moiety, the LUMO became localized at the palladium(II) centres rather than the benzophenone acceptor. The ligand-to-metal charge transfer into antibonding d-orbitals was therefore identified as the likely source of emission quenching. We note that the dynamic nature of supramolecular structure association often allows them to support reversible stimulus-responsive behaviour. The demonstrated ability of these assemblies to then modulate emissive and TADF properties therefore provides an attractive pathway to optical readout of such stimulus responses.
The cavity of the PyCzBP-Pd metallocage was also used to host two emissive xanthene dyes: fluorescein and Rose Bengal. Electrospray ionization mass spectrometry (ESI-MS) revealed that up to three molecules of neutral fluorescein can be held within 4PyCzBP-Pd. This host-guest complex also engaged in photoinduced electron transfer (PET) from host to guest, giving [F].+[4PyCzBP]− and completely quenching the emission of both species. ESI-MS analysis of the Rose Bengal complex showed that two molecules of the open dianion quinoid form of Rose Bengal are held within 4PyCzBP-Pd. This host-guest complex similarly engaged in photoinduced energy transfer (PEnT) in DMSO solution, where the green emission of the host was quenched along with emergence of orange Rose Bengal emission. Förster energy transfer was proposed as the proposed PEnT mechanism, quantified by a quenching rate constant of k q = 4.07 x 1011 M–1 s–1.
Incorporation of triazole groups to extend the carbazole of the CzBP core was employed to produce mechanically interlocked macrocycle rotaxanes TzCzBP⊂R1 and TzCzBP⊂R2 (Figure ).ref. ref1351
Through-space interactions between the triazole protons and the macrocyclic bipyridine nitrogen lone pairs led to an increase in the ΦPL under N2, a decrease in ΔE ST, and increased photostability under UV irradiation in toluene (TrCzBP; ΦPL = 11%, TzCzBP⊂R1 ; ΦPL = 31%, TzCzBP⊂R2 ; ΦPL = 30%). DFT calculations revealed that the LUMO of the rotaxanes remains on the benzophenone moiety, whereas the carbazole-based HOMO is destabilised due to the aforementioned hydrogen bonding. This example demonstrates the ability of supramolecular approaches to finely modulate the photophysical properties of the emitter via rotaxane formation.
Of the remaining examples of supramolecular TADF materials reviewed here, we may broadly categorize these into those which are co-ordinated or covalently bound to give a supramolecular assembly, and assemblies formed non-covalently through aggregation or through-space interactions.
Coordinatively Bound Supramolecular TADF Assemblies
As seen for 4PyCzBP-Pd, TADF emitters with moieties capable of co-ordination may form supramolecular assemblies templated by metal ion vertices. Metallocages are not the only assembled structures that can form between co-ordinating emitters and metal centres though; indeed, other TADF emissive supramolecular systems exist, including one Zr(IV) and two Zn(II) metal organic frameworks (MOFs), a platinum(II) metallocycle, and a cobalt-containing dendritic photocatalyst.
MOFs
The first TADF MOF was reported by Adachi and co-workers using Zr(IV) centres and an organic diacid linker. This linker, A, was chosen due to its small calculated ΔE ST of 0.2 eV (measured 0.24 eV) (Figure ).ref. ref1350 A red-shift of the emission and a decrease in the delayed lifetime of MOF Zr-A-MOF in the solid state (λPL = 501 nm, τPF = 17 ns, τDF = 180 μs, ΦPL = 30% (N2), 18% (air)) was observed compared to the emission of the free linker A in 2 wt% doped PMMA films [λPL = 481 nm, τPF = 18 ns, τDF = 199 ms, ΦPL = 39% (N2), 32% (air); Zr-A-MOF in the solid state]. The decrease of the MOF ΦPL in air was ascribed to more active quenching of the triplet excited state by oxygen. The decrease in lifetime and red-shift of emission in Zr-A-MOF compared to A was attributed to co-ordination to the electron-poor Zr(IV) centres, acting similarly to an auxiliary acceptor in D-A TADF materials.
Haldar et al. formed a TADF MOF between a diphenylaminoteraphenylethylene linker, B, and Zn(II) ions, Zn-B-MOF (Figure ).ref. ref1352 In dilute ethanol solution, linker B is poorly emissive due to non-radiative decay via free rotation of the phenyl groups (τPL = 1.5 ns). This process is supressed in powder samples of B, (τPL ≈ 200 μs). Crystalline and orientated thin-films of Zn-B-MOF showed the same delayed lifetime of ≈ 200 μs, with a ΦPL of 14%. The TADF nature of the emission was confirmed using variable temperature photoluminescence studies. This thin-film MOF system was furthermore incorporated into an OLED, although this displayed a high turn-on voltage of 5.8 eV and a low maximum luminance of 270 cd m–2 at 14 V. Using time-dependant DFT, the authors inferred that the electroluminescence actually originated from a hot-exciton mechanism, made possible by the small energy gap between the T2 and S1 states compared to the relatively large energy gap for coupling between T1 and T2.
Small changes in the structure of a system may have substantial effect on the mode of emission, as demonstrated by Gutiérrez et al. (Figure ).ref. ref1353 MOFs formed from Pb(II) and terephthalic acid C were synthesised to study what effect crystallizing the MOF from either water, (MOF-C-H2O), or DMF (MOF-C-DMF) would have on their optoelectronic properties. Green photoluminescence centred at ∼525 nm, was observed for MOF-C-H2O, with an associated ΦPL of 59% for the powder. For MOF-C-DMF, the emission blue-shifts to 480 nm, with a ΦPL of 32% in the solid state. The authors hypothesized that differences in the crystal packing were the source of the difference in the photophysical properties, with XRD analysis of MOF-C-H2O revealing a more densely packed structure. At 77 and 298 K MOF-C-H2O showed similar multiexponential emission decay kinetics, with lifetimes of τ PL of 39 ns, 145 μs, and 1.53 ms. The lifetime for MOF-C-DMF was instead temperature-dependant, with τ PL of 43 ns, 0.29 μs, and 15 μs at 298 K; however, at 77 K only the nanosecond lifetime component was observed. The authors proposed that the more densely packed MOF-C-H2O features many inter-linker interactions, leading to relatively temperature-insensitive RTP dominating the emission. In the less densely packed MOF-C-DMF, larger inter-linker distances begin to favour TADF, as evidenced by the temperature-dependant nature of emission.
Bie et al. synthesised an organic diacid linker, 4,4′′-(10H,10′′H-9,9′′-spirobi[acridine]-10,10′′-diyl)dibenzoic acid (D) and coordinated this with zirconium clusters to give MOF-D, which displayed oxygen-insensitive TADF (Figure ).ref. ref1354 The design of D is an A-D-σ-D-A structure. The rigid backbone prevents rotation and increases the rigidity of the system, with the two electronically isolated A-D units separated by a quaternary sp3-carbon in the centre and additionally giving a twist to the compound.ref. ref1355 The linker D has an emission centred at around 453 nm, and is a promising TADF material in its own right with a ΔE ST value of 0.02 eV and a τd of 1.18 μs in degassed THF, with this long-lived emission disappearing upon exposure to air. A slight increase in the ΔEST was noted, from 0.02 eV to 0.14 eV, along with a red-shift to 490 nm in the emission of MOF-D compared to the free linker. The rigid design was chosen to counteract the decrease in emission lifetime observed by Adachi and co-workers for their emitter A upon incorporation into a MOF,ref. ref1356 which the authors of that work believed to be driven by the flexibility of the linker within the MOF. Indeed, for MOF-D the τd is 0.72 μs in the powder, only modestly different compared to 0.23 μs for D in the solid state. Indeed, the lengthening of the emission lifetime upon complexation is likely due to the increased rigidity of the linker, resulting in a suppression of the non-radiative pathways.
Liu et al. reported the encapsulation of an electron-rich triphenylene donor within a Cd(II) MOF (NKU-11) containing electron-poor triazine panels, giving rise to exciplex-like TADF between the panels and the guest (Figure ).ref. ref1357 The host⊂guest MOF system, herein called MOF-E, was formed via the solvothermal synthesis of Cd(II) ions, triazine, triphenylene, and terephthalic acid. The triphenylene sits within triangular prism cages in the MOF, formed of two triazine ligands and three terephthalic acid linkers. MOF-E shows a broad, featureless emission centred around 492 nm, pointing to emission originating from a charge-transfer state. Temperature-dependant photoluminescence studies showed a 20-fold increase in the emission intensity upon heating from 77 K to 297 K, supporting a TADF mechanism. MOF-E displayed a triexponential excited-state lifetime with τPL of 17.5 ns, 1.29 μs, and 4.21 μs indicating the presence of both prompt and delayed emission, and with a ΔE ST of 0.11 eV.
A silver cluster-containing MOF was formed upon co-ordination of 2-mercaptonic acid, F, with Ag(I) ions to give hexameric silver clusters, which may organise into a MOF upon complexing with Ca2+ ions, MOF-F.ref. ref1358 The discrete silver nanoclusters possessed poor ΦPL of ∼2%, but upon complexation to the calcium ions a 10-fold increase in the ΦPL to ∼20% was seen. MOF-F proved to be pH-sensitive; protonation occurs on the carbonate co-ordinating moieties on the silver clusters at low pH thus breaking MOF-F apart, which can then reform under basic conditions. MOF-F showed green emission centred at around 590 nm in the thin film with τPL of 557 ns, 8.61 μs. The intensity of the delayed emission was found to be temperature-dependent, confirming the TADF behaviour of this system.
Metallocycles
A TADF platinum (II) metallocycle with coordinating organic ligand, BTZPy, was reported by Lv et al., showing promise as a photodynamic therapy and chemotherapy drug (Figure ).ref. ref1359 Compound BTZPy by itself showed efficient fluorescence with ΦPL = 78%, τPL = 8.65 ns and λPL = 569 nm in degassed ethanol solution. Co-ordination of BTZPy to Pt(II) centres, cPt, afforded the triangular metallocycle BTZPy-Pt, which also showed a high ΦPL but with a blue-shifted emission (ΦPL = 60%, λPL = 550 nm, and an average τPL = 8.65 ns). Nanosecond transient absorption spectroscopy revealed the lifetimes of G and PtG to be 1.87 μs and 1.76 μs, respectively (λexc = 532 nm), with temperature-dependant emission studies of G and PtG uncovering a higher delayed emission intensity with increasing temperature, again suggesting the metallocycle is TADF-active. Both G and PtG displayed excellent singlet oxygen generating ability in ethanol solution [measured relative to a meso-tetrakis(p-sulfonato-phenyl) standard], with quantum yields of singlet oxygen generation of 95% and 86%, respectively.
Cobalt-Containing Dendrimeric Antenna Complex
Combining a typical D-A TADF core 4CzPN with terminal pyridine groups (in this context H), and binding to cobaloxime centres gave the photocatalytic assembly H-Co. This material was used to promote the catalytic acceptorless dehydrogenation (CAD) of secondary amines (Figure ).ref. ref1360 DFT calculations of H revealed the HOMO is centred on the carbazole moieties and the LUMO is localised on the phthalonitrile core as expected from the D-A structure, with a ΔEST of 0.13 eV. Compound H emits at λPL = 591 nm with CT emission profile, and has a τd of 17.4 μs and a ΦPL of 7% in degassed CH2Cl2 at 298 K. Upon complexation to the cobaloxime groups, a decrease in both the lifetime and photoluminescence quantum yield is observed (τPL = 13.8 μs, ΦPL = 2.9%), arising from PET from H to the cobalt centres that supports its catalytic activity. The ability of H-Co to generate hydrogen using blue LEDs (450 nm ± 10 nm, 3W) was demonstrated with a 0.04% catalyst loading of H-Co in dry degassed THF and gave a turnover number (TON) of 305 after 12 h. An uncomplexed mixture of cobaloxime and H under the same conditions gave a TON of only 53, with the increase in performance upon complexation attributed to more efficient absorption and electron transfer between adjacent subcomponents of the well-defined bound structure.
Non-coordinatively Bound Supramolecular TADF Systems
As well as covalent or coordinate bonding interactions, supramolecular assemblies may form through non-bonding interactions such as aggregation or encapsulation. Both approaches have the potential to significantly modulate the photophysical properties of the photoactive TADF emitters compared to isolated molecules.
Aggregation-Based TADF Assemblies
There are a small number of reported examples of organic and carbon dots TADF emitters. These are small micelle-like spherical particles that form in poor solvents such as water and are then decorated with water-solubilising side chains. Common organic emitters used for dot preparation include derivatives of 4CzIPN and structurally similar emitters,ref1361−ref1362ref1363ref1364 as well as materials using phenoxazine and phenothiazine,ref1365,ref1366 anthraquinone,ref. ref1367 and benzophenoneref. ref1368 (Figure ). Polyethylene glycol (PEG) chains are commonly used as water-solubilising groups and may be covalently linked to the organic emitter or mixed with the particles in solution to self-assemble into a particle coating that prevents further particle aggregation and sedimentation. Such assemblies are not only water soluble, but their excited states are sufficiently long-lived to outcompete biological autofluorescence. As the emitters are shielded within micelle or hydrophilic coating, the presence of oxygen in these biological systems does not necessarily contribute to the quenching of the excited state. In cases where oxygen does diffuse into the micelle, emission intensity can even be used as an optical probe for cellular oxygen concentration.ref. ref1369 One such micellar system was reported by Zhu et al. who employed peptide chains as the water-solubilising groups, allowing the material to pass through cellular and nuclear membranes and further demonstrating their versatility and suitability as biological probes.ref. ref1370
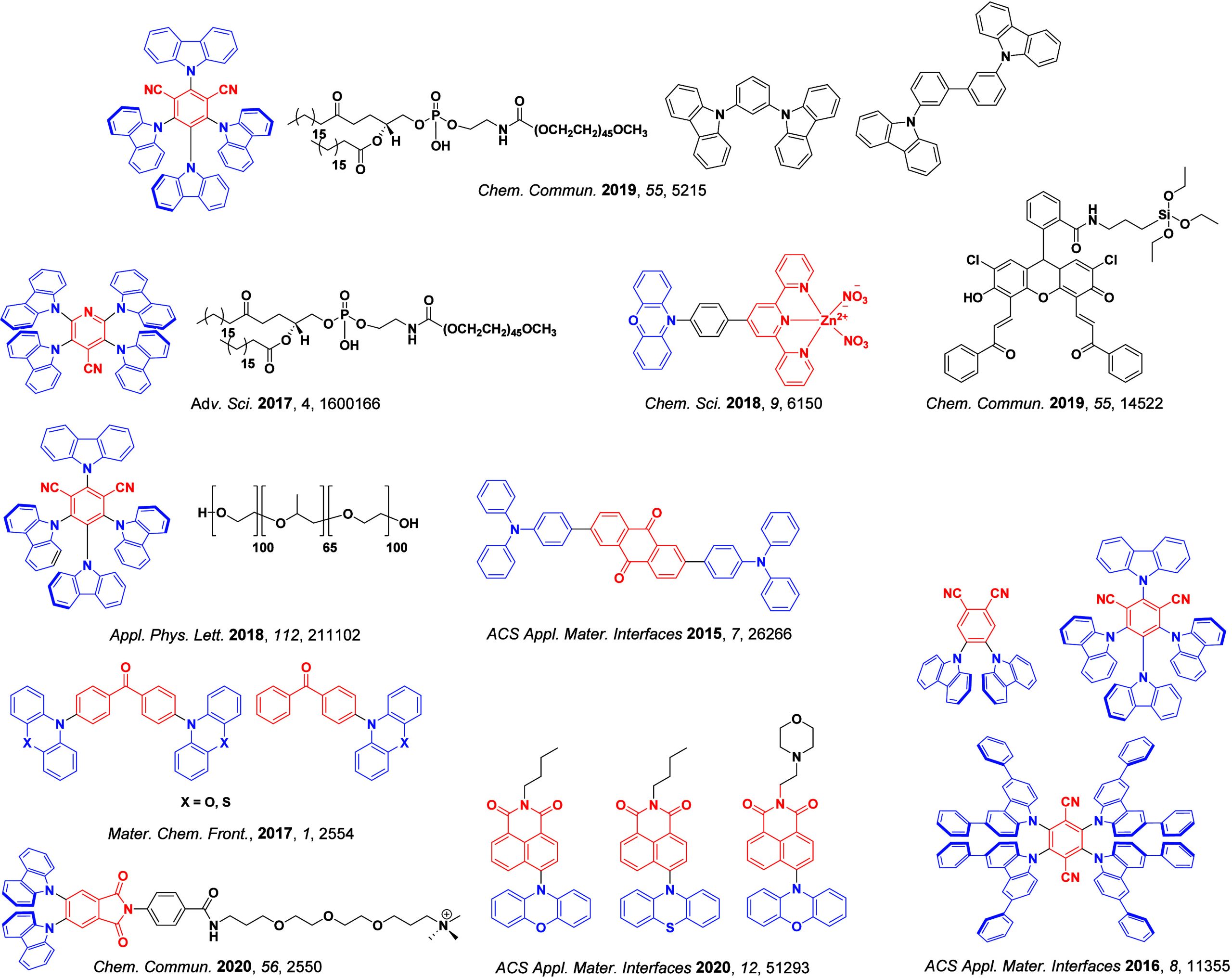
Another example of a supramolecular TADF system formed via aggregation was reported by Qi et al., where the emitter, CDPA, formed nanorod needles in thin neat films that were several hundred nanometres thick and several hundred micrometres long (Figure ).ref. ref1371 The emission of these nanorod needles in the thin film (λPL of 645 nm) is slightly red-shifted relative to the powder (λPL of 640 nm), and the needles showed an enhanced ΦPL of 26% compared to 13% for the unassembled thin film. Transient photoluminescence decay measurements of the thin film show prompt (2.3 ns) and delayed (10.0 μs) emission, the latter of which showed a temperature dependence.ref. ref1372
TADF from Zeolite-Encapsulated Emitters
Other non-covalently bound supramolecular TADF emitter systems can be formed by the encapsulation of carbon dots into a zeolite host. Multiple reports from Yu, Li, and co-workers have demonstrated this self-assembly approach, forming a zeolite host and carbon dot in a one-pot reaction, leading to trapped dots within the zeolitic framework. This encapsulation leads to millisecond excited state lifetimes and high photoluminescence quantum yields of the materials, assisted by shielding from external atmospheric oxygen and restricting internal conformational and vibrational degrees of freedom of the carbon dots, thus suppressing non-radiative decay.
The authors first reported three dots-in-zeolite systems, CD1, CD2, and CD3, formed under solvothermal conditions.ref. ref1373 System CD1 used triethylamine, aluminum tri-iso-propoxide, phosphoric acid, triethylene glycol, and hydrofluoric acid to form dots trapped within a zeolite matrix. System CD2 was formed under similar conditions, but with 4,7,10-trioxa-1-13-tridecanediamine in place of triethylamine, and no hydrofluoric acid. System CD3 had a similar preparation to CD2, but with the addition of dimagnesium phosphate. Upon excitation at 370 nm all three systems displayed deep-blue emission [(C1: λPL = 430 nm, ΦPL = 15%, CIE (0.17, 0.13); C2: λPL = 440 nm, ΦPL = 52%, CIE (0.17, 0.14); C3: λPL = 425 nm, ΦPL = 23%, CIE (0.17, 0.13)]. Additionally, all three systems showed delayed emission, with τd = 350 ms, 197 ms, and 216 ms the characteristic temperature dependence associated with TADF for CD1, CD2, and CD3, respectively. The measured ΔE ST values for these systems are 0.22 eV, 0.23 eV, and 0.22 eV, respectively.
The authors then reported another dot-in-zeolite system showing TADF using 4,7,10-trioxa-1-13-tridecaneamine as a template, CD4.ref. ref1374 Excitation at 370 nm led to emission at λPL = 440 nm with ΦPL = 29%, and excitation wavelength-dependent emission intensity (excitation at 350 nm led to the brightest emission). Temperature-dependent transient photoluminescence decay measurements showed an increase in the delayed emission intensity with increasing temperature. Combined with the delayed emission (τd = 153 ms) and the measured ΔE ST of 0.18 eV, these data confirmed TADF as the emission mechanism. By contrast, the unconfined carbon dot in the mother liquor shows no delayed emission at room temperature, and a slightly larger ΔE ST of 0.21 eV. The authors suggested that encapsulation of the carbon dot within the zeolite led to the suppression of nonradiative decay along with a stabilisation of the triplet state, switching on TADF.
Yu, Li, and co-workers have also reported four dots-in-zeolites systems, CD5–8, whereby two dots are encapsulated within the same zeolite framework in varying ratios.ref. ref1375 The ratio of two carbon dot templates, m-phenylenediamine and 4,7,10,-trioxa-1-13-tridecanediamine, was varied from 0:1, 0.007:1, 0.0014:1, and 0.042:1 in the starting mixtures for self-assembly to give four systems; CD5, CD6, CD7, and CD8. As the ratio of m-phenylenediamine to 4,7,10-trioxa-1-13-tridecanediamine increased, the emission red-shifted (CD5–8; λPL = 425 nm, 484 nm, 498 nm, 515 nm, respectively) while the delayed lifetime increased and the prompt lifetime decreased (CD5: τPL = 24.43 ns, 271 ms, CD6: τPL = 37.54 ns, 578 ms, CD7: τPL = 11.76 ns, 801 ms, CD8: τPL = 7.70 ns, 860 ms). The photoluminescence quantum yields also increased with increasing m-phenylenediamine inclusion (CD5–8: ΦPL = 20.9%, 25.1%, 37.1%, 42.0%, respectively). Temperature-dependant time-resolved emission studies of the dots-in-zeolites systems confirmed TADF, with ΔEST for CD5 and CD8 measured between 0.20 and 0.14 eV. The authors propose that FRET from the 4,7,10-trioxa-1-13-tridecanediamine dots to the m-phenylenediamine dots within the confined matrix occurs, with the m-phenylenediamine dots emitting via TADF.
In a similar manner, Koninti et al. demonstrated TADF behavior from benzophenone once encapsulated within mesoporous silica nanostructures (MSN).ref. ref1376 Benzophenone is known to phosphoresce at 77 K, but once encapsulated the non-radiative pathways are supressed and ΔE ST is reduced from 0.11 eV (in MeCN) to between 0.048 eV and 0.060 eV, depending on the MSN used. The PL spectrum of free benzophenone in aerated MeCN shows weak fluorescence at around 450 nm, owing to rather efficient ISC followed by quenching of the T1 state by oxygen, as well as non-radiative deactivation by free rotation of the phenyl rings. Upon adding MSN to the MeCN solution, the emission intensity increases due to the effects of encapsulation of benzophenone: reducing the ΔE ST, increasing the RISC rate to give emission from the S1, and suppressing non-radiative decay via molecular rotations/vibrations due to increased environmental rigidity. The emission intensity is further increased in degassed solution, further supporting the expectation of triplet involvement in the emission. It should be noted that the presence of MSN being linked to the increase of the emission intensity suggests some shielding from oxygen upon encapsulation within the MSN framework. A τd of between 22 and 44 μs was observed, depending on the MSN used, with ΦPL of 1.7% in all systems compared with a ΦPL of 0.2% for benzophenone in MeCN; the τd in each of the MSN-based systems was 5 μs.
Co-crystallized Host⊂Guest Donor-Acceptor TADF
A TADF host⊂guest system of α-cyclodextrin (α-CD) and diphenylacetylene (DPA) was reported by Huang et al., which exhibited TADF through ‘long-range charge transportation’ giving ‘long persistent luminescence’ (Figure ).ref. ref1377 Three related systems were synthesised, using unsubstituted DPA (DPA-1), 4-(4-fluorophenylethynyl)phenol (DPA-2), and 4-4′-difluorodiphenylacetylene (DPA-3). For each DPA derivative a 1:1 aqueous solution with α-CD was prepared and crystals grown via slow evaporation, while an alternate method of forming the host⊂guest complexes was also explored involving grinding the host and guest with a small amount of water. Using either method, host⊂guest complexes α-CD-DPA-n (n = 1, 2, or 3) were formed, each showing dual TADF and RTP with an afterglow of more than 2 seconds. The complexes showed two oxygen-sensitive emission maxima at 360 nm and 460 nm, with the 360 nm peak (associated with the TADF) having very long τd of 134 ms, 282 ms, and 256 ms, for α -CD-DPA-1, 2, and 3, respectively. The emission at 460 nm results from phosphorescence and exhibits similarly long lifetimes of 354 ms, 292 ms, and 245 ms for α -CD-DPA-1, 2, and 3, respectively, with associated ΦPL values of 47.4%, 33.5%, and 29%. The authors ascribed the TADF to originate from the α-CD host, while the phosphorescence is from the DPA guest, giving the observed dual emission.
Recently the co-crystallisation of a calix[3]-acridian ring (C[3]A) with dicyanobenzene (DCB) was shown to produce a host⊂guest complex exhibiting TADF (Figure ).ref. ref1378 Dissolving equimolar C[3]A and DCB in n-hexane/CHCl3 gave green crystals upon evaporation, with bright green/blue photoluminescence under UV light. X-ray crystallography showed multiple C-H···π interactions between host and guest, producing a tightly bound C[3]A⊂DCB complex. The crystals of C[3]A⊂DCB crystals showed an absorption maximum at around 400 nm and an emission at 500 nm, which is significantly red-shifted compared to C[3]A (λPL = 385 nm). Transient photoluminescence measurements revealed biexponential decay kinetics, with a τp of 152 ns and a τd of 5.2 μs, where the delayed emission demonstrated the expected temperature dependence associated with TADF. The ΦPL = 70% was notably large, which the authors attributed to the rigidity of the crystal structure inhibiting non-radiative decay processes. The ΔE ST was measured to be 0.014 eV, which translated into a k RISC of 9.42 x 104 s–1.
Outlook
Incorporating a TADF unit within a supramolecular assembly can have an impact on the photophysical properties that are as diverse as the different supramolecular structures themselves. Given the small but growing number of examples to date, it is difficult to project what functional properties each of these classes of assemblies may ultimately unlock.
The CzBP core, which featured in gels, rotaxanes, and a metallocage, provides insight into the wide range of supramolecular assemblies discrete TADF emitters may be incorporated into. The gel formed from 4PyCzBP allowed for the production of xerogel films with higher photoluminescence quantum yields than their neat film counterparts, while incorporation into a rotaxane gave fine control over the emission wavelength from the CzBP core and though the metallocage constructed from 4PyCzBP and Pd2+ ions was poorly emissive, nonetheless it could act as a photoactive host where either photoinduced energy or electron transfer could occur depending on the nature of the encapsulated substrate. These emergent properties are as diverse as the supramolecular structures which give rise to them and demonstrate the vast potential this field may hold for the many TADF emitters that already exist. What is required at present is an increased effort to explore this field. Considerable work will be needed to correlate the properties of discrete emitters and their surpramolecular counterparts to identify trends and emergent properties. What is certain is that this is an area ripe for further exploration and innovation.
TADF Sensors
Introduction
As a consequence of the underpinning photophysics, TADF emission is acutely sensitive towards both temperature and oxygen, which has been exploited in sensing applications.ref. ref1379 Indeed, establishing and calibrating correlations between emission properties (spectrum, intensity, or lifetime) and temperature is the foundation of optical molecular thermometry.ref. ref1380 For example, the temperature-dependent nature of RISC and thus the ratio of delayed fluorescence to phosphorescence of a TADF emitter can be exploited for optical readout of temperature. Similarly the quenching of triplet excitons of TADF materials by oxygen and the associated drop in emission intensity can be harnessed for use as an oxygen sensor.ref1381,ref1382 Oxygen sensing is highly relevant in medicine, where the oxygen level in exhaled air, patient blood, or even within cells is a key physiological parameter that sometimes requires continuous monitoring. Similarly, the measurement of the oxygen concentration is important in industries that use metabolizing organisms, such as yeast for brewing and baking,ref. ref1383 and in biotechnology, where microorganisms are used to produce antibiotics and anticancer drugs.ref. ref1384
Materials Development
Firster et al. demonstrated the first TADF-based temperature sensor in 1995, using acridine yellow embedded within a saccharide host matrix for optical thermometry (Figure ).ref. ref1380 One of the major advantages of employing TADF materials as thermosensors is that the delayed emission lifetimes follow an Arrhenius behavior, in contrast to the more complex models required in other fluorescence-based sensors.ref1380,ref1385 As well as from delayed emission lifetimes, temperature can also be inferred from the relative emission intensities of delayed fluorescence and phosphorescence. For acridine yellow the average temperature sensitivities of the delayed fluorescence lifetime and of the delayed fluorescence-to-phosphorescence intensity ratio were 2.5 and 4.5% °C–1, respectively (confirmed across −50 to 50 °C). According to the authors, these sensitivities were ∼10 times higher than typical optical thermometer materials available at that time. Beyond this temperature range the error increases as material stability deteriorates at higher temperatures.
C70 doped in PtBMA polymer acts as a temperature sensor with an expanded temperature range, evaluated from the ratio of intensities between delayed fluorescence and phosphorescence.ref. ref1386 The working device possessed sensitivity of 0.5% K–1 across a temperature range of −80 °C to 140 °C. The device could also undergo numerous heating/cooling cycles, showing less than 2% change in readout over several weeks. In this system degassing is also essential given that the triplet excitons undergoing RISC are oxygen sensitive. By taking advantage of this oxygen sensitivity, oxygen sensors were also developed using 13 C70 , showing detection limits down to parts per billion (ppb).ref1382,ref1387 This was the first example of an oxygen sensor with an optical readout. Reversibility and reusability of the sensor film was emphasized along with good stability over many months.
DeRosa et al. reported biological oxygen sensing using TADF- and RT-active derivatives of difluoroboron-β-diketonate-poly(lactic acid) (BF2bdk-PLA) and difluoroborondibenzoylmethane-poly(lactic acid) [BF2dbm (X)PLA (X = H, Br, I)] (Figure ).ref. ref1388 The non-halogenated BF2dnmPLA is a highly efficient TADF emitter under N2 atmosphere at room temperature, while the halogenated polymers BF2dnm(Br)PLA and BF2dnm(I)PLA are phosphorescent due the higher SOC resulting from the presence of these heavy atoms. The polymer BF2dnm(I)PLA enabled ratiometric oxygen-sensing and imaging due to its distinguishable dual-emission in fluorescence (green) and phosphorescence channels (orange), depicted in Figure . Further, BF2dnm(I)PLA nanoparticles were used to detect differences in intracellular oxygen concentrations demonstrated for in vitro ratiometric imaging of T41 mouse mammary cells.ref. ref1388
Steinegger et al. reported a series of carbazole-substituted dicyanobenzenes (b, c, d1, and d2) and diphenylamine-substituted anthraquinones (a1–a7 and e) for use as oxygen and temperature sensors (Figure ).ref. ref1389 In toluene the dicyanobenzene-based emitters emit strongly between 506 and 546 nm and have ΦPL of between 61 and 79%, while the series of anthraquinone-based emitters emit weakly between 609 and 678 nm (ΦPL of between 0.1 and 15%). The τd of the dicyanobenzene-based emitters varies from 5 to 15 μs while the τd of the anthraquinone-based emitters varies over a much wider range (τPL of between 11 and 583 μs). For the preparation of oxygen-sensitive materials, 1 wt% dyes were immobilized in oxygen-permeable polystyrene (PS). The emission bands of these emitters in PS shift hypsochromically to between 577 and 614 nm for the anthraquinone-based emitters and to between 493 and 531 nm for the dicyanobenzene-based emitters, coupled with increases in their respective τ d (42 μs to 5.5 ms for anthraquinones and 9 μs to 40 μs for dicyanobenzenes) and in their ΦPL (26 to 48% for anthraquinones and 59 to 96% for dicyanobenzenes). Oxygen sensitivity was calibrated using Stern-Volmer (SV) quenching analysis. The oxygen sensitivity of these materials varied from moderate to very high and was proportional to the τd of the compound. For temperature sensing these emitters were incorporated into gas-impermeable poly(vinylidene chloride-co-acrylonitrile) [P(VDC-co-AN)] and temperature was calibrated against corresponding change in τ d. These temperature sensors demonstrated sensitivities from −1.4 to −3.7% K–1, determined from the change of τ d per unit change in temperature. Further, the authors also prepared a fiber-optic mini sensor by using d2 as the temperature reporting emitter incorporated in P(VDC-co-AN), enabling rapid and high-resolution temperature monitoring. Additionally, the authors made temperature-sensitive nanoparticles based on c and d2 for use in temperature imaging at the cellular level.
Zach et al. designed a family of Pt(II) and Pd(II) tetraphenyltetrabenzoporphyrin (TPTBP)-based TADF complexes for oxygen and temperature sensing (Figure ).ref. ref934 The Pt(II) and Pd(II) benzoporphyrin complexes were decorated with four (tetra-) or eight (octa-) alkylsulfonyl groups (Pt-T-S, Pd-T-S Pt-O-S and Pd-O-S), although this was eventually shown to have minimal effects on the sensing properties. Related imide-modified Pt(II) and Pd(II) benzoporphyrin complexes (Pt-T-I and Pd-T-I) were also studied, and the performance of each of these complexes was compared to the parent Pt(II) and Pd(II) benzophorphyrin complexes (Pt-TPTBP and Pd-TPTBP).ref. ref1390 At room temperature, all Pt(II) and Pd(II) complexes emit between 620 and 652 nm and simultaneously show phosphorescence ranging between 742–786 nm and thus have ΔE ST ranging from 0.29–0.36 eV in degassed toluene at 25 °C. The ΦPL of the Pt(II) complexes (8.2 to 34%) were higher than those of the corresponding Pd(II) complexes (3.2 to 10.5%), although the phosphorescence lifetimes (τ Ph) of the Pd complexes (τPh of between 53 and 286 μs) were much longer than of those of the Pt complexes (ranging from 12 to 47 μs), all in toluene at 25 °C. The Pt(II) complexes also show much less efficient delayed fluorescence than the Pd(II) analogues, and faster deactivation of the T1 state via phosphorescence. For optical temperature and oxygen sensing, these complexes were immobilized in PS at between 1–2.5 wt% doping ratios. When increasing temperature from 23 °C to 133 °C the intensity of the red TADF dramatically increased while the phosphorescence intensity decreased (Figure ). Simultaneously, the delayed lifetimes of all complexes were significantly affected by temperature, and the observed temperature sensitivity was found to be in the range of 0.102% K–1 to 0.537% K–1. The phosphorescence lifetime was significantly affected by the presence of oxygen, and due to the greater intensity and longer lifetime of the phosphorescence band in the Pd(II) complexes, their oxygen sensitivity was found to be higher than that of Pt(II) complexes. While PS optical sensors based on Pt(II) dyes are suitable for measurement from 1 to 1000 hPa O2, those based on the Pd(II) complexes permit much lower oxygen partial pressure readouts. The authors further demonstrated the applicability of these dyes for simultaneous oxygen and temperature measurements.ref. ref934
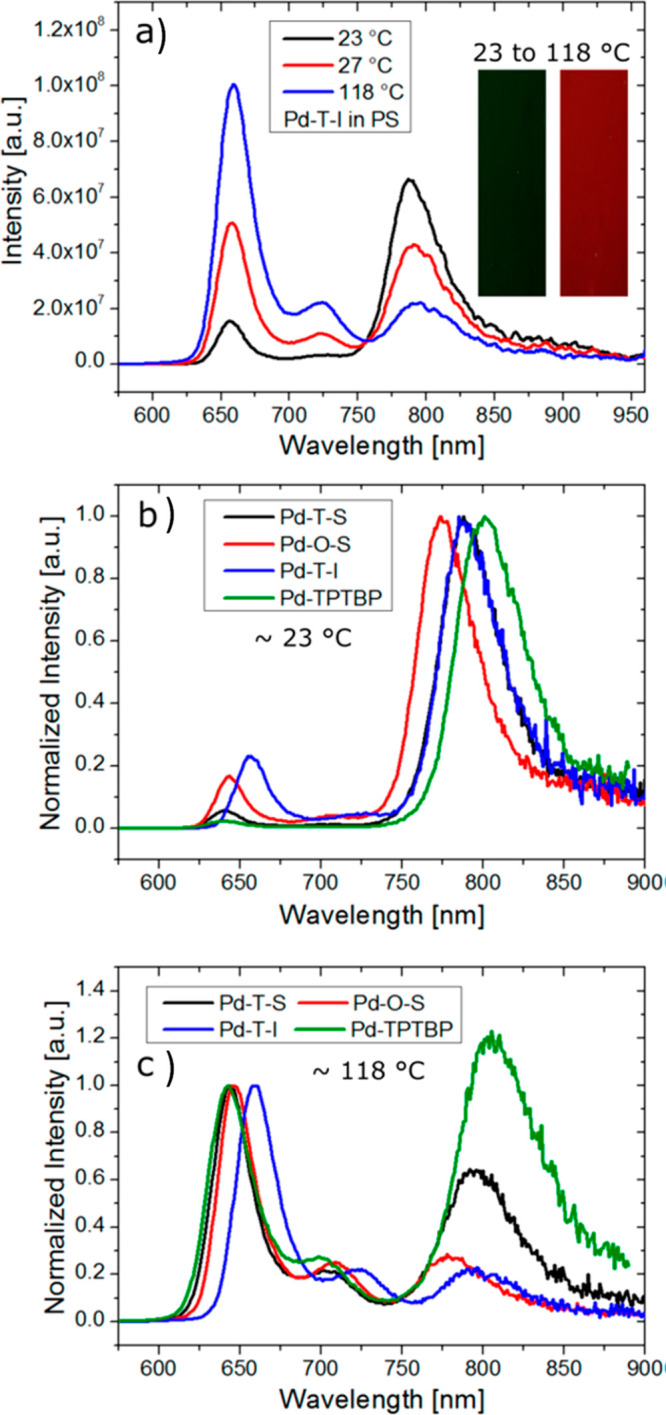
Zieger et al. prepared a related Zn(II) benzoporphyrin-based TADF complex, Zn-OS for temperature and oxygen sensing (Figure ).ref. ref943 Complex Zn-OS emits at 667 nm in toluene and has a τ d ≥ 1 ms. An optical oxygen sensor containing Zn-OS (1 wt%) immobilized in poly(styrene-co-acrylonitrile (PSAN) emits at 675 nm and has a ΦPL of 3.3% and a τ d = 7.87 ms under degassed conditions. The prompt fluorescence was affected by neither molecular oxygen nor temperature and used as internal reference. However, the τ d and delayed fluorescence intensity (I DF) decreased significantly due to dynamic quenching by oxygen, with this quenching calibrated to achieve an optical readout of the oxygen concentration. The limit of detection at 26 °C was estimated to be 0.002 hPa O2. With increasing temperature, the τ d of Zn-OS decreases whereas I DF was enhanced (Figure ). Zn-OS could therefore be used for simultaneous sensing of oxygen and temperature using a single material with a single-wavelength readout.
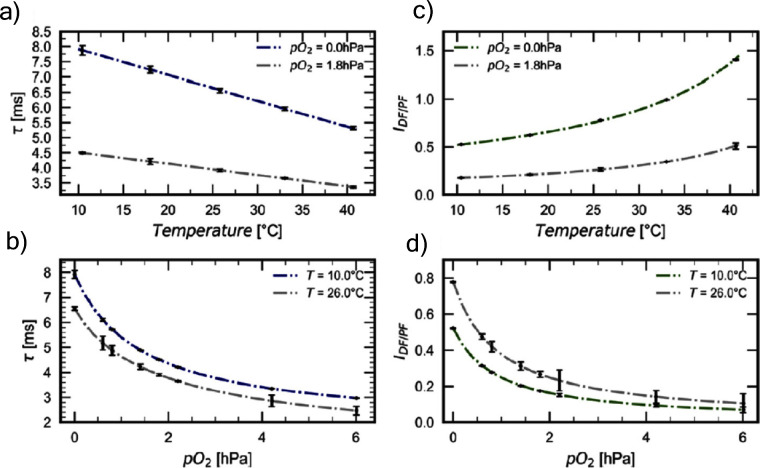
TADF Schiff base complexes of Zn(II) (Zn-1 and Zn-2, Figure ) have similarly been developed as temperature sensors.ref. ref941 Immobilized in PS films, Zn-1 emits at 542 nm and has a ΦPL of 30% and a τd of 7.41 ms, whereas carbazole-containing Zn-2 emits at 547 nm and has a ΦPL of 65% and a τd of 1.45 ms. The temperature sensitivities at 25 °C were 3.7 and 3.5% K–1 based on the changes in delayed lifetime, respectively, with temperature resolution of at least 0.03 °C. To eliminate competitive oxygen quenching, the PS-immobilized Zn-1 was covered with an additional layer of off-stoichiometry thiol–ene polymer (OSTE) as an oxygen-consuming layer and then a layer on P(VDC-co-AN). Changes in the τd of the Zn-1/PS/OSTE/P(VDC-co-AN) device were tracked as a function of temperature, with sensitivities of 4.1%K–1 over a temperature range of 5–45 °C. The probe was stable to oxygen quenching for more than 60 days during storage under ambient air.ref. ref941
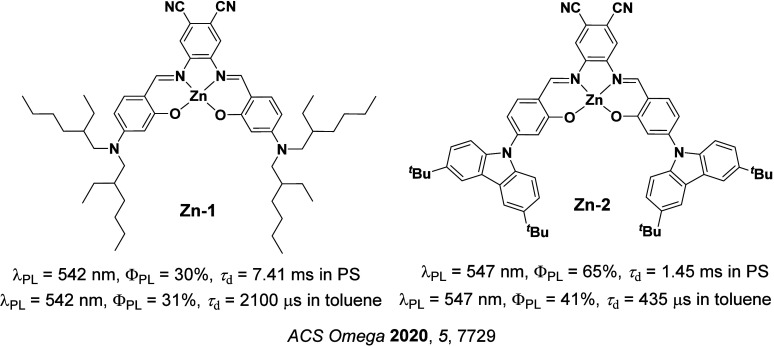
Christopherson et al. demonstrated that TADF polymers containing acrylate-functionalized oxadiazole-based donor–acceptor monomers such as ACR-ODA, PXZ-ODA, PTZ-ODA, PAZ-ODA, and TTAC-ODA can be used as oxygen sensors (Figure ).ref1391,ref1392 These monomers were copolymerized with a carbazole host co-monomer (CzBA) using Cu(0) reversible deactivation radical polymerization (RDRP). These polymers have high molecular weight (M n > 20 kDa), with polydispersities ranging from 1.11 to 1.45. As neat films the TADF polymers emit from 449–457 nm for ACR-ODA, 496–507 nm for PXZ-ODA, 510–517 nm for PTZ-ODA, 566–584 nm for PAZ-ODA, and 490–502 nm for TTAC-ODA. These emission wavelengths are each dependent on the attached donor moiety and on the doping concentration of the TADF-active ODA monomer, which ranged from 5 to 15 wt%. Of these polymers, TTAC-ODA has the highest ΦPL (42% for TTAC-ODA0.15 ) which can be explained by the rigidity of the diphenylamine-carbazole donor dendron. The calculated ΔE ST for these polymers is <0.011 eV, except for TTAC-ODA where the calculated ΔE ST is much larger at 0.21 eV. The overall emission intensity of the polymers typically decreased as the film was coOLED and was aerated, indicating that TADF and not phosphorescence was the operational emission mechanism at room temperature.ref. ref1391 However, PAZ-ODA0.15 showed no delayed emission, which the authors attributed to the low triplet energy of the strongly donating PAZ moiety. PTZ-ODA0.15 was shown to act as a single-component ratiometric oxygen sensor, able to be calibrated from the changing ratio of prompt and delayed fluorescence as a function of O2 concentration. This ratiometric emission behavior of PTZ-ODA0.15 arose from the presence of the pseudoaxial and pseudoequatorial conformers of the phenothiazine donor (Figure ). The PTZ-ODA0.15 film was demonstrated to be able to sense oxygen concentrations from 0 to 50% and was also incorporated into water-soluble polymer dots (Pdots) to sense O2 in biological systems.
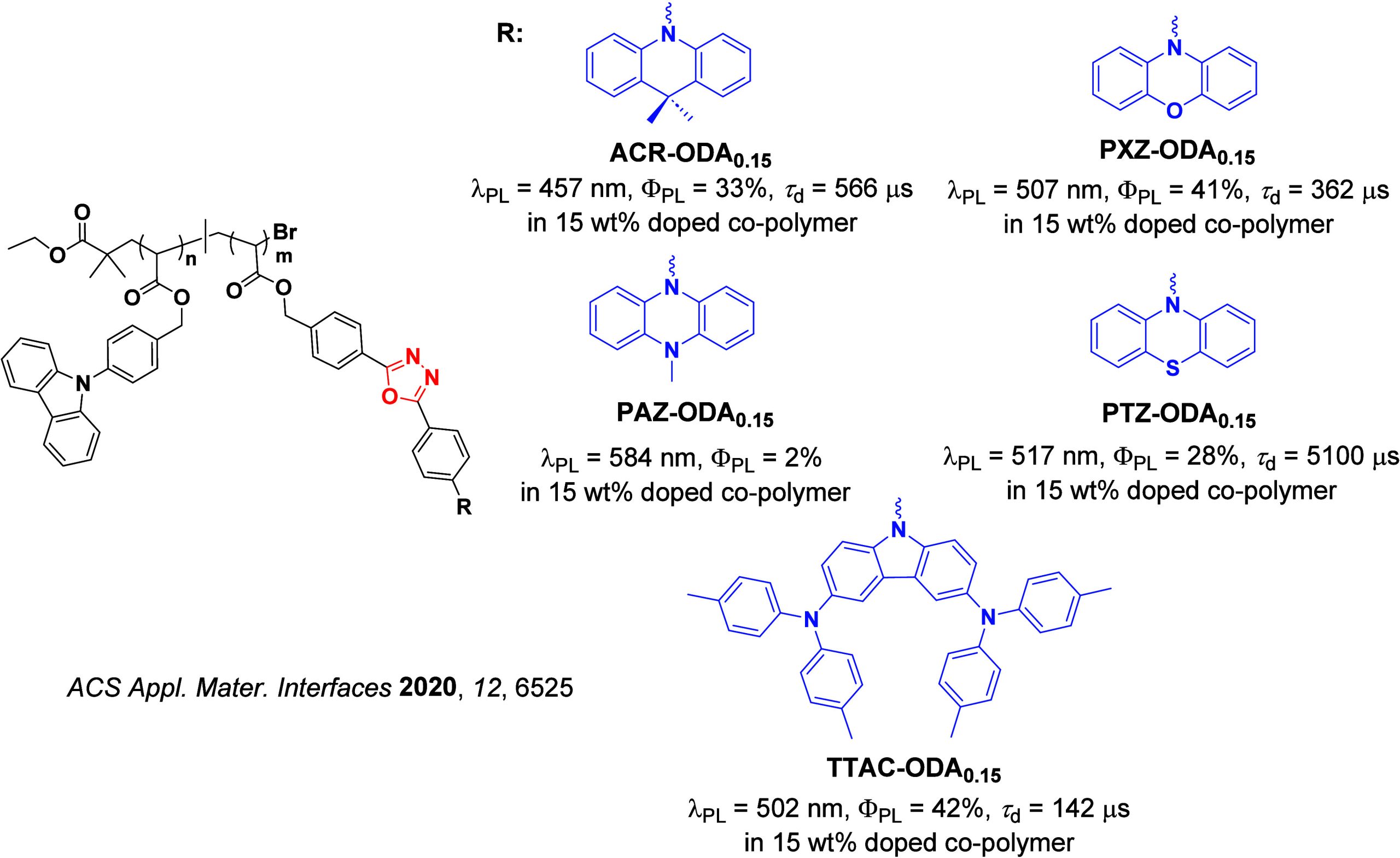
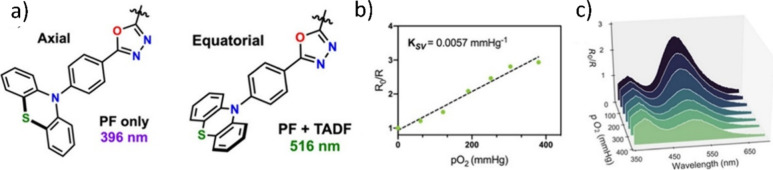
Christopherson et al. developed additional temperature sensing materials by co-polymerizing naphthalimide (NAI)-based red-emissive TADF acrylic monomers (NAI-DMAC, NAI-PTZ and NAI-POZ) with an acrylate-functionalized 1,3-bis(N-carbazolyl)benzene (mCPA) co-monomer as a host (Figure ).ref. ref1392 Both star-shaped and linear polymers (P1-P17) were synthesized, showing high molecular weight (12,000 < M n < 22,000) and narrow polydispersities between 1.07 and 1.25. The star-shaped polymers (P1-P8) were obtained from polymerization of the mCPA host monomer and TADF dopant monomers (1–12 mass%) with a four-arm initiator (4-BriBu), while the linear polymers (P9-P11) were obtained from polymerization of the mCPA host monomer and TADF dopant monomers (12 mass%) with ethyl α-bromoisobutyrate (EBiB) as the initiator.ref. ref1393 All three TADF monomers show broad CT emission in toluene at 630, 582, and 594 nm for NAI–DMAC, NAI–PTZ, and NAI–POZ, respectively. Each monomer emission spectrum also exhibits peaks at 343 and 362 nm, attributed to fluorescence from the LE state of the NAI core. The star-shaped polymer with 12 mass% NAI-DMAC (P5) emits at 638 nm and has a ΦPL of 58%, 12 mass% NAI-PTZ (P7) emits at 700 nm and has a ΦPL of 11%, and 12 mass% NAI-POZ (P8) emits at 675 nm and has a ΦPL of 4.5% in toluene. Surprisingly, only P5 showed evidence of delayed fluorescence, with τd of 6.1 μs in toluene. However, all three polymers P5, P7 and P8 as neat films show delayed fluorescence with emission at around λPL of 610, 650, and 660 nm, and with τd of 6.14, 76.66, and 51.97 μs, respectively, which track with their respective ΔE ST of 0.12, 0.22 and 0.21 eV. Like their monomers, these polymers exhibited dual-emission consisting of a high-energy fluorescence from the NAI acceptor (λPL = 340 nm in toluene) and a lower-energy long-lived TADF from a CT excited state (λPL = 633–711 nm in toluene).
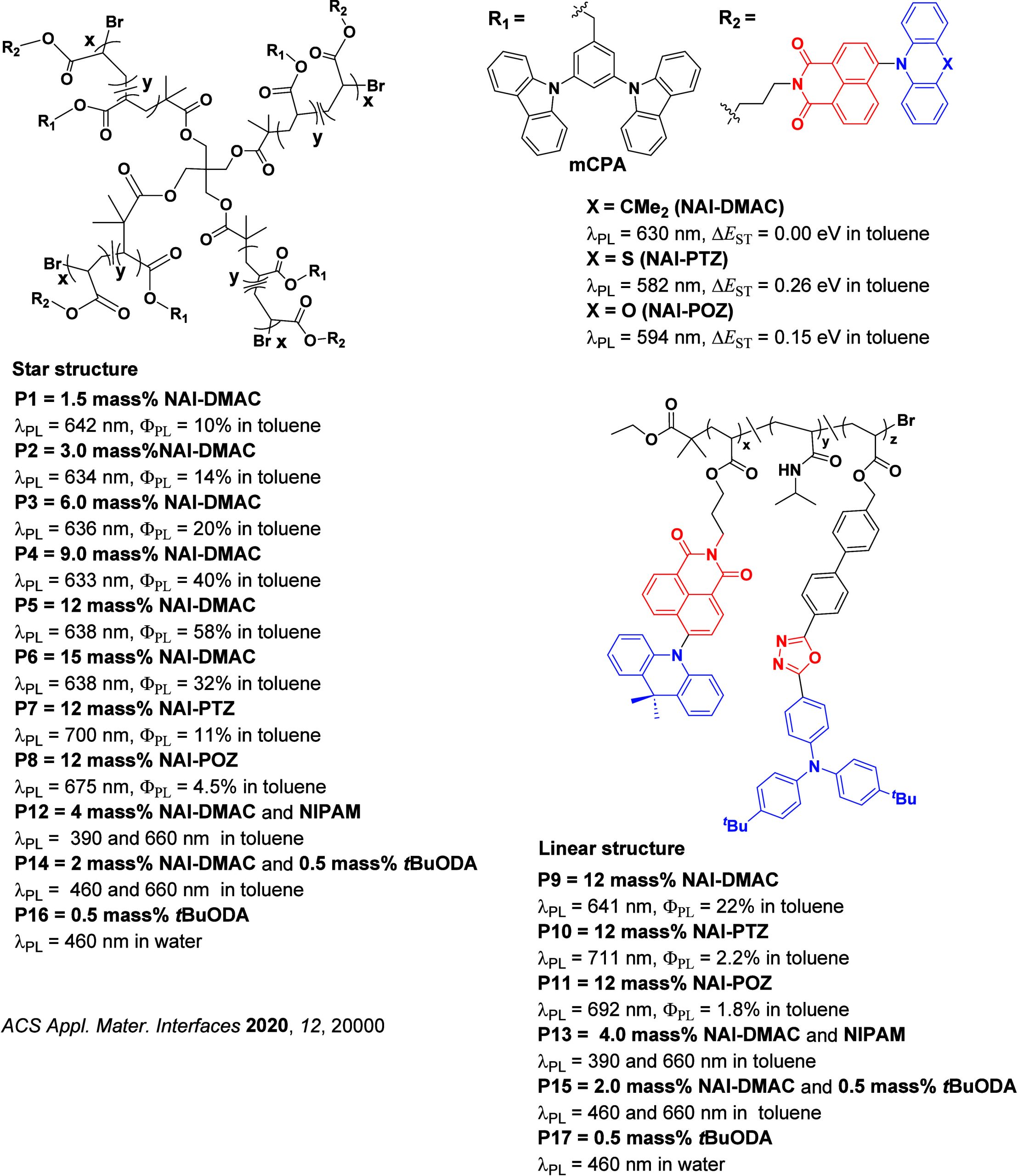
The dual-emission behavior of the NAI-DMAC monomer was exploited to develop ratiometric temperature-responsive polymers P12 and P13 (Figure ). Star-shaped polymer P12 and linear polymer P13 were developed by copolymerizing 4.0% NAI-DMAC and N-isopropylacrylamide (NIPAM) with 4-BriBu and EBiB initiators, respectively. The I 390/ I 660 ratio of emission peaks at 390 nm (NAI emission) and 660 nm (CT state) increases linearly with temperature from 20 to 70 °C for P12 and P13 (Figure ). To make an all-visible sensor the authors employed a second blue triphenylamine-oxadiazole co-dopant tBuODA that would emit as a result of FRET from the UV-emitting NAI-based LE state, and prepared star-shaped polymer P14 by co-polymerizing 2.0% NAI-DMAC and 0.5 mass% tBuODA with 4-BriBu initiator. A linear analog P15 contained 0.5 mass% tBuODA and 2.0% NAI-DMAC and used EBiB initiator. These two polymers show dual-emission with characteristic TADF from NAI-DMAC at 660 nm and fluorescence from tBuODA at 460 nm at 20 °C. Upon increasing the temperature to 70 °C a large increase in the blue emission intensity was registered in both polymers, which was attributed to increased FRET from the NAI-DMAC to tBuODA (Figure ). The ratiometric optical response to temperature of P14 is 32 ± 4% K–1 and 30 ± 6% K–1 for P15; fluorescent star-shaped P16 and linear polymer P17 with only tBuODA monomers did not show such a temperature-dependent behavior.
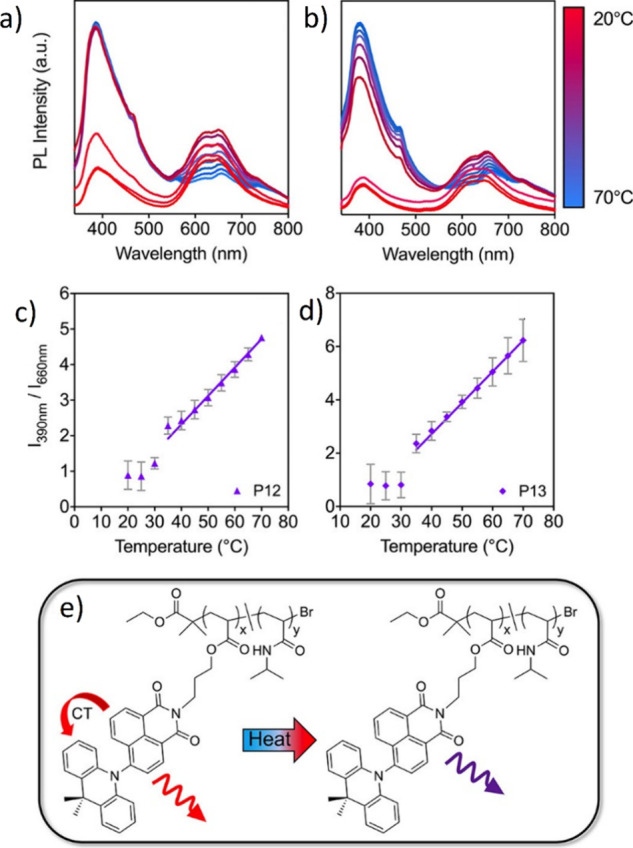
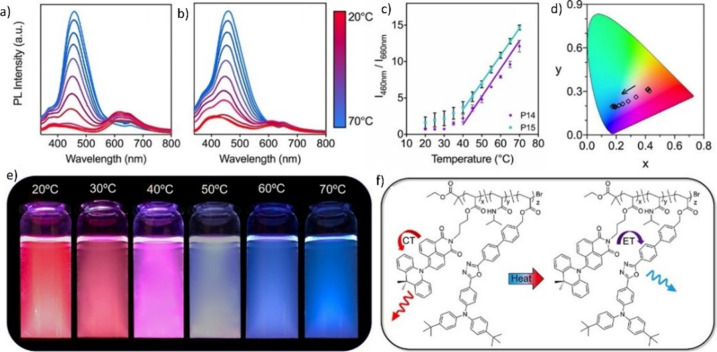
Li et al. developed three emitters containing diphenylsulfone (DPS) as an acceptor attached to different donors such as, 5-nitroindole (1), 5-aminoindole (2) and 5-acetaminoindole (AMID, 3), with the goal of developing sensors for solvent polarity (Figure ).ref. ref1394 Emitters 1 and 2 are purely fluorescent while emitter 3 is TADF-active. Compound 3 shows dual-emissions at 332 nm (strong LE fluorescence) and 435 nm (weak CT emission with TADF) in DCM under air. Upon degassing the ΦPL of 3 increased from 12 to 33% and the τd increased from 55 μs to 167 μs, linked to a small ΔE ST of 0.17 eV that is with TADF; the τPL of 332 nm band is 22 ns and is insensitive to oxygen.ref. ref1394 Using the invariant LE fluorescence as an internal reference, the ratio of the intensities of the LE and CT bands as well as the ratios of the prompt and delayed lifetimes were used to calibrate against solvent polarity (Figure ). Increasing solvent polarity from hexane to DMF caused the ratio of emission wavelengths for the CT and LE states to increase from 1.17 to 1.45, and the lifetime decreased from 55 μs in toluene to 1.6 μs in DMF, showing that 3 can act as a sensitive optical probe of polarity. Further, the authors employed a 3-D ratiometric luminescent sensing strategy to detect the microenvironment polarity in a biological membrane.
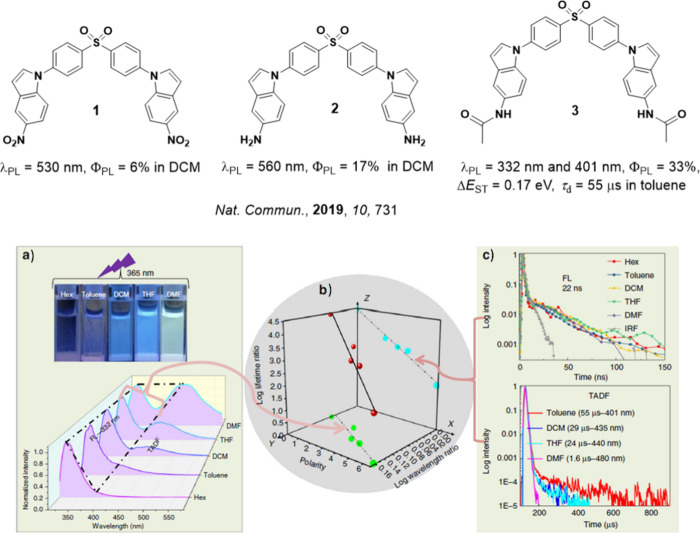
Aside from temperature and oxygen sensing, TADF compounds have also been used for anion and cation sensing. Yin et al. developed a fluorescein-based fluorescence turn-on chemosensor DCF-MPYM-lev for sulfite ion (SO3 2–) detection (Figure ).ref. ref1395 Compound DCM-MPYM-lev is very weakly emissive; however, on the addition of [SO3]2– into the 3.0 μM DCM-MPYM-lev solution in CH3CN/PBS buffer (1/1), the fluorescence intensity significantly increases and dual-emissions at 535 nm (weak emission) and 640 nm (strong emission) was observed, providing a detection limit of 2.98 μM of [SO3]2–. The mode of action of this sensor is the sulfite-mediated deprotection of the levulinyl group, thereby releasing the luminescent DCF-MPYM (Figure ). Previously, DCF-MPYM was reported to be TADF, with a τ d = 22.11 μs in deaerated ethanol and a ΔE ST of 28 meV. This compound was also used as a bioimaging reagent of breast cancer MCF-2 cells.ref. ref1396 Additionally, DCF-MPYM was used as a chemosensor to detect cysteine and hypochlorite.ref1397,ref1398 Compound DCF-MPYM-lev was used to monitor the exogenous [SO3]2– concentration in living cells.
Qiu et al. reported carbazole-triazine TADF emitter PhTRZ-OCHO (Figure ) as a fluorescence turn-off/fluorescence quenching sensor for the detection of Na+, Mg2+ and Fe3+ ions.ref. ref1399 PhTRZ-OCHO emits at 470 nm in THF with a ΦPL of 58% and τ d of 0.32 μs, while a control TADF compound PhTRZ-OCH3 emits at 490 nm. PhTRZ-OCHO has a ΔE ST 0.25 eV while PhTRZ-OCH3 has a ΔE ST of 0.17 eV. Though the emission intensity of PhTRZ-OCHO at 470 nm decreased upon the addition of many of the metal ions tested (Ba+, Ca+, Cd2+, Co2+, Cr2+, Cu2+, Fe3+, Hg2+, K+, Mg2+, Mn2+, Na+, Ni2+, Pb+), the strongest emission quenching occurred upon addition of Na+, Mg2+ and Fe3+, with detection limits of 7.03 × 10–7, 6.7 × 10–7 and 5.9 × 10–7 mol/L, respectively. The authors hypothesized that the excellent fluorescence quenching behavior was due to the presence of the metal-binding aldehyde group in PhTRZ-OCHO, which stabilized the CT band and becomes non-emissive upon complexation. As such an interaction is not possible in the control emitter PhTRZ-OCH3 , it does not show any selective sensing of these cations.
Recently, Ma et al. reported an unusual application of TADF emitters DMAC-TRZ, 4CzIPN, and 4CzTPN-Bu (Figure c) as scintillators for the detection and imaging of X-ray radiation.ref. ref1400 X-ray photons initially interact with atoms in organic molecules through both the photoelectric effect and Compton scattering, which causes ejection of high-energy electrons. These high-energy electrons further interact with emitter molecules and generate a cascade of secondary lower-energy electrons that ionize or excite other molecules to generate electron–hole pairs (Figure ). Directly analogous to exciton formation following electrical excitation in OLEDs, in the scintillation process X-ray irradiation and subsequent recombination of ionized molecules (holes) with uncorrelated ejected electrons favors triplet states over singlet states in a 1:3 ratio. While fluorescent scintillators waste these triplet excitons, the use of phosphorescent emitters in scintillators is undesirable as it leads to significant deadtime between detector events (due to long exciton lifetimes). In TADF emitters, these triplet excitons can be harvested through rapid RISC and increase the amount of light available to the detector electronics (Figure b). As scintillators, DMAC-TRZ, 4CzIPN, and 4CzTPN-Bu embedded in 10 wt% sucrose octaacetate (SO) exhibit internal X-ray-to-light conversion efficiencies of 73,500 ± 400, 33,200 ± 60 and 44,900 ± 210 photons MeV–1, supported both by efficient conversion of triplet excitons to produce light, and reduced self-absorption. The limit of detection (LOD) of the TADF scintillator for DMAC-TRZ is 103.2 ± 2.9 nGyair s–1, 250 ± 12 nGyair s–1 for 4CzIPN, and 208 ± 4 nGyair s–1 for 4CzTPN-Bu, which are much lower than a competing TTA compound (anthracene, 506 ± 21 nGyair s–1 in SO). To demonstrate the practical application of TADF scintillators for X-ray imaging, the TADF emitters were embedded in a SO matrix at 0.5 to 10 wt% doping to produce solid-state scintillator screens (Figure ). The radioluminescence (RL) intensity of these TADF emitters was 612–743% higher than that of anthracene in SO. The 0.5% DMAC-TRZ:SO scintillator screen was the used to produce X-ray images of industrial and biological samples at a high resolution of 16.6 line pairs (lp) mm–1 (Figure ).
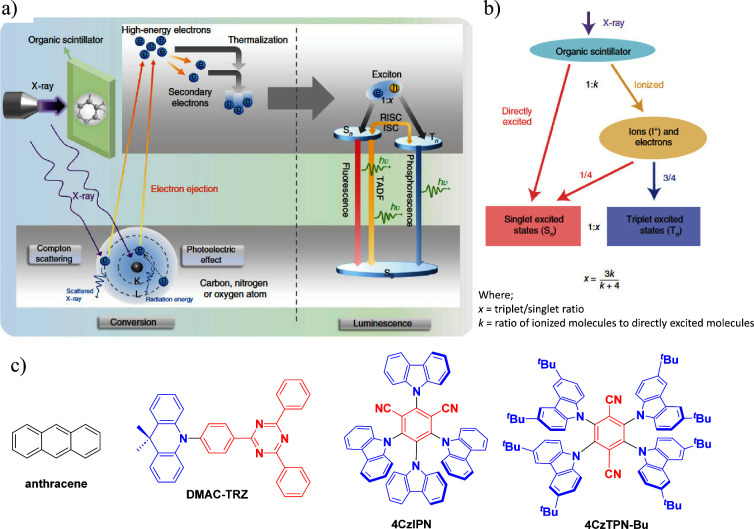
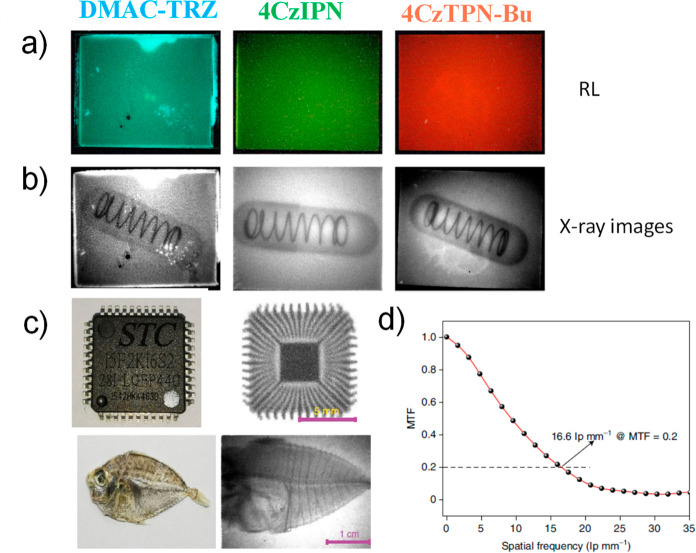
Highly efficient and reliable scintillators with low detection limits could be achieved by using organic scintillation materials with high X-ray absorption capability, high exciton utilization efficiency, and high photoluminescence quantum yield. Recognizing that larger atoms usually have larger X-ray absorption cross-sections, Wang et al. introduced heavy halogen atoms (Cl, Br and I) generating the 4CzIPN derivatives TADF-H, TADF-Cl, TADF-Br, and TADF-I, which were used to fabricate organic scintillator screens for X-ray imaging (Figure ).ref. ref1401 The four emitters each emit at approximately 505 nm in 1 wt% doped PMMA films. Additionally, the τd of 4.53 μs for TADF-H, 2.99 μs for TADF-Cl, 2.22 μs for TADF-Br, and 1.42 μs for TADF-I systematically decrease due to the heavy-atom effect enhancing SOC and accelerating RISC. To illustrate the application of these TADF emitters in the detection and imaging of X-rays, scintillation screens consisting of 60 wt% emitter doped in PMMA were fabricated with different thicknesses (0.1 to 0.5 mm). Due to presence of the heavy atoms, the X-ray absorptivity of the films with TADF-I and TADF-Br is higher than the others, and the relative light yields also increase (∼18000 photons MeV–1 for TADF-I and TADF-Br, 7076 photons MeV–1 for TADF-Cl, and 1892 photons MeV–1 for TADF-H, Figure b-c). The role of the increased X-ray cross section is highlighted by relatively uniform ΦPL of the scintillation screens, ranging from 44–65% and highest for TADF-H. Due to the high relative light yield in TADF-I and TADF-Br, the LOD is significantly improved in these (both ∼45 nGy s–1) in comparison to TADF-H (438.5 nGy s–1 and TADF-Cl (100.6 nGy s–1), and is comparable to a reference scintillator material LYSO:Ce (34.8 nGy s–1). The RL intensities of these TADF scintillators was also found to be linearly correlated with the X-ray dosages, allowing for X-ray imaging applications (Figure d). TADF-Br exhibited high X-ray imaging resolution of 12.0 lp mm–1 in comparison to TADF-H (5.1 lp mm–1), TADF-Cl (6.8 lp mm–1), and TADF- I (9.4 lp mm–1, Figure e-f).
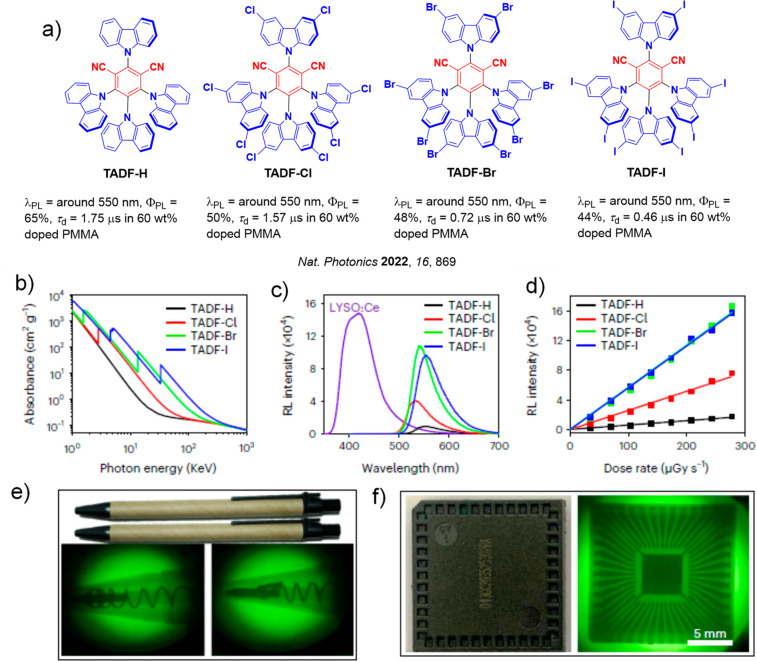
To develop reabsorption-free X-ray imaging scintillators (required for high-quality images at low detection limits) along with achieving air and light stability, Wang et al. reported the design of a nanocomposite film (Zr-fcu-BADC-MOF-TADF), Figure , consisting of a combination of a luminescent MOF (Zr-fcu-BADC-MOF) and TADF chromophores (4CzTPN-Bu, Figure c).ref. ref1402 The authors demonstrated that there was nearly 100% energy transfer from the fluorescent MOF to the TADF co-dopant, which, coupled with direct harnessing of singlet and triplet excitons mediated by the co-dopant, translated into a remarkable enhancement of the radioluminescence upon X-ray irradiation (Figure ). The detection limit of the optimized D-A0.4 nanocomposite film (D = MOF, A = 4CzTPN-Bu) at 256 nGy s–1 is significantly improved compared to the undoped Zr-fcu-BADC-MOF film (15,000 nGy s–1) and a reference 4CzTPN-Bu film at (1,600 nGy s–1). This detection limit is approximately 22 times lower than the standard dosage for X-ray diagnostics (5.5 mGy s–1) (Figure ), while the D-A0.4 nanocomposite film simultaneously exhibits excellent photostability (Figure e). The D-A0.4 nanocomposite film-based scintillator could thus be used for imaging of a steel famework (Figure g).
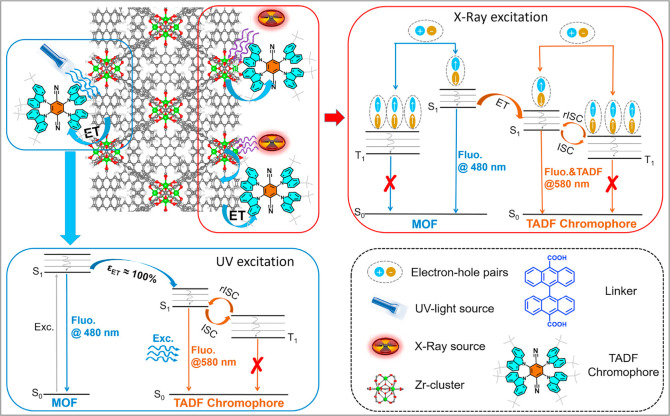
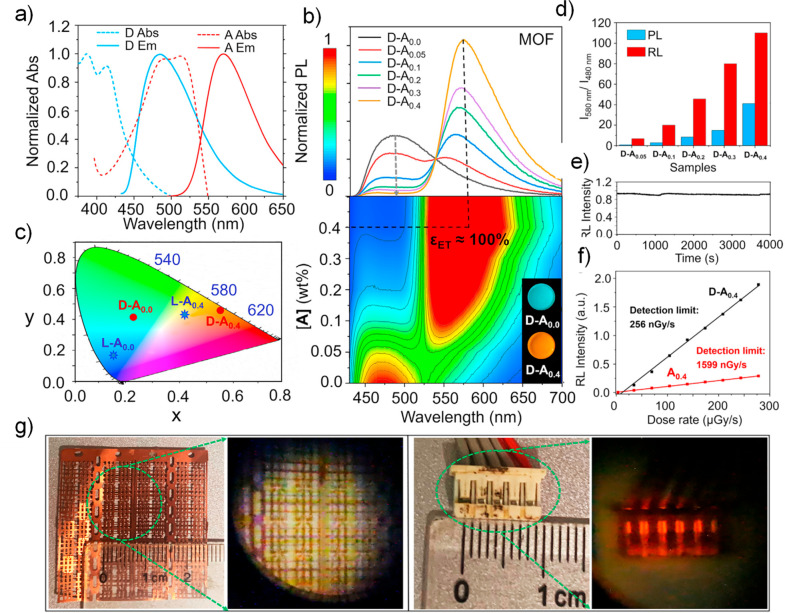
Abraham et al. reported polyvinyltoluene (PVT) based cross-linked plastic scintillators for the detection of γ-rays, containing 1 wt% 4CzIPN or tBuCzDBA as a TADF dye alongside various amounts (0, 5, 10, 40 wt%) of (caproted)di-(methacrylate)bismuth (CMB) cross-linkable compounds, and either with (4.5 wt%) or without cross-linker divinyl benzene (DVB, Figure ).ref. ref1403 The TADF dye acts to harvest triplet excitons via RISC to achieve 100% luminescence quantum yield, and thus increase the light yield. The plastic scintillator containing 4CzIPN without CMB emits close to 500 nm and has a τd of 3.3 μs, while the emission of 4CzIPN is slightly red-shifted around 515 nm (τd = 3.0 μs) in the plastic scintillator containing 40 wt% CMB. Similar optical behavior of tBuCzDBA was observed in the plastic scintillator (λPL = 550 nm and τd = 2.9 μs, without CMB), while the τd decreased slightly to 2.6 μs in scintillator containing 40 wt% CMB. The scintillator without CMB but containing tBuCzDBA exhibited higher relative light yield (0.25) than 4CzIPN (0.11). The authors suggested that the higher light yield in tBuCzDBA could be due to either a higher fraction of horizontal emitting dipoles or reduced internal scattering within the bulk for the tBuCzDBA sample. Another reason for the higher light yield could be more efficient energy transfer from the PVT matrix to the tBuCzDBA dye. Even though the CMB loading adversely affected the light yield, the cross-linking approach nonetheless improved the mechanical robustness with a uniaxial yield strength up to ≈ 66 MPa for the scintillators loaded with 40% CMB.
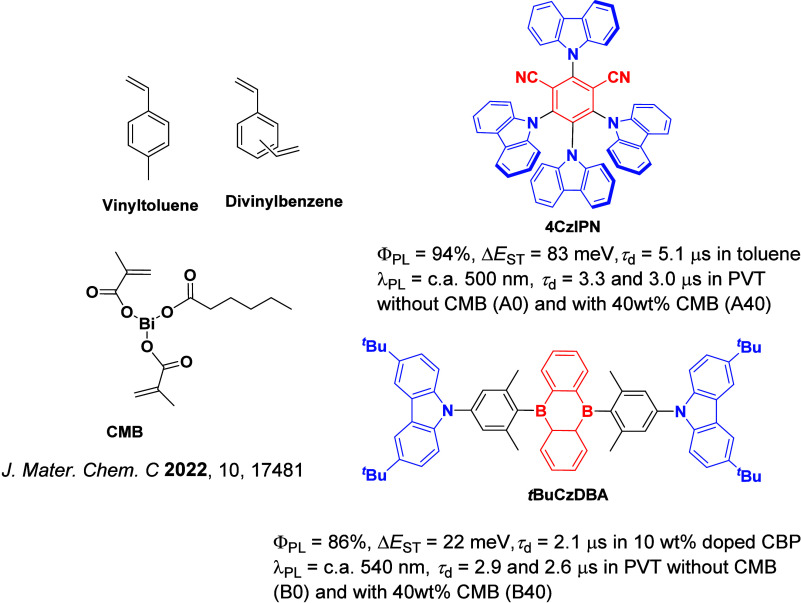
Outlook
The sensitivity of the spectral response of TADF materials to temperature and O2 make them particularly attractive for sensing applications in comparison to typical fluorescent probes. Simultaneously, many of the reported TADF sensors are dual emissive, with one of the bands insensitive to oxygen and/or temperature and so act as a convenient internal reference for the other band. Aside from the examples highlighted here, we note that the long-lived nature of TADF itself can be exploited to increase signal-to-noise in sensing applications, and for use in time-resolved fluorescence imaging (see Section sec21 ). There are now also multiple reports of TADF materials employed as optical sensors for a number of chemical analytes, as well as for X-ray detection.
Despite the relative infancy of TADF sensors, the examples to date nonetheless demonstrate the potential of this class of materials. Considering that any optical sensing ability arises from changes to photophysical in response to external environment, we predict that there will be additional demonstrations of sensing utility developed in the coming years. These may include solvent polarity and trace concentrations of aqueous hydrocarbons (through CT emission red-shift), viscosity (through impacts on vibronically-coupled RISC), pH (especially in ESIPT TADF materials, see Section sec14 ), and as electrochromic redox sensors. The sensing utility of TADF materials is primarily limited by our understanding of their intricate photophysical mechanisms, with both expected to continue growing and developing over time.
TADF Bioimaging Agents
Introduction
On the scale of individual cells, most living tissue is both optically transparent and has minimal intrinsic contrast (in refractive index or otherwise) between different cellular components. Bioimaging dyes and stains are therefore a frequently necessary tool for observing cell structures, offering the potential to visualize internal organelles and biological processes optically, and often without damaging the cell.ref1404,ref1405
While conventional fluorescent emitters are established as contrast agents in bioimaging, issues can arise as a result of the autofluorescence of cells. Autofluorescence is the emission from photoactive materials endogenous to the cell itself, which can mask the desired signal from the contrast bioimaging agent.ref. ref1406 One strategy to overcome this issue is to employ phosphorescent emitters, as by virtue of their long-lived emission they can allow autofluorescence and phosphorescence to be distinguished in the time-domain. However, while phosphorescent metal complexes have the additional complication of potential toxicity,ref46,ref1407−ref1408ref1409 all-organic TADF materials can potentially also address autofluorescence with their suitably long-lived emission. Similar to their desirability in replacing organometallic complexes in electroluminescent devices, the use of all-organic TADF materials as bioimaging reagents also carries benefits in terms of sustainability, bioavailability, and cost (which dictate accessibility in biomedical contexts). The large Stokes shifts for D-A TADF materials can also potentially allow autofluorescence to be addressed and eliminated in the spectral domain. However, for both phosphorescent and TADF materials, quenching of triplet states and any delayed emission by physiological dissolved oxygen must also be carefully considered.
To date, there is a small but rapidly growing body of work in which organic TADF compounds have been developed for bioimaging applications, including for time-resolved luminescence imaging (TRLI) for living cells. Many TADF compounds can also emit in the red to NIR region, which is especially transparent to living tissue, even in bulk. These wavelengths are therefore advantageous for in vivo bio-imaging because of reduced photo-damage to the biological samples, greater deep tissue penetration allowing optical signal to emerge, and minimal interference from background (typically blue) autofluorescence from biomolecules in the living systems. In this section we will discuss recent examples of TADF emitters that have been used as bioimaging agents.
TADF Emitters Capped with Bovine/Human Serum Albumin (BSA/HSA)
One strategy to circumvent the quenching of TADF emission by oxygen is to use either human serum albumin (HSA) or bovine serum albumin (BSA). Both contain tryptophan, which is a chromophoric amino acid that can react with singlet oxygen, preventing the quenching of the triplet excited states and thus the delayed fluorescence of emitters.ref. ref1396 BSA has been used in living-cell imaging experiments to enhance the signal originating from the bioimaging agent and also to help cellular uptake by masking the hydrophobic TADF molecule and rendering the TADF-BSA assembly more hydrophilic,ref1410,ref1411 increasing their solubility and stability in aqueous media.ref1396,ref1411 In addition, BSA can also protect the emitters from degradation by cellular enzymes and improve their biological compatibility, making them less toxic to cells. In 2014,ref1396,ref1412 Xiong et al. were the first to propose a TADF emitter, DCF-MPYM (Figure a), that was used in conjunction with BSA. This adduct was employed as the contrast agent in TRLI of MCF-7 cells, and showed long-lived luminescence (τ PL = 22.11 μs in deaerated ethanol) at λPL of 649 nm with a small ΔE ST (0.03 eV) in 5:4 MeOH:EtOH (v/v).ref1396,ref1410 The BSA protein provides a hydrophobic cavity and a reductive environment that shields the emitter from oxygen, thus permitting the long-lived delayed emission of DCF-MPYM to persist in the cells. TRLI of MCF-7 cancer cells using this contrast agent much stronger red luminescence signals and significantly suppressed background signal in time-resolved imaging mode (Figure b), compared to equivalent images obtained in steady-state mode (Figure c).
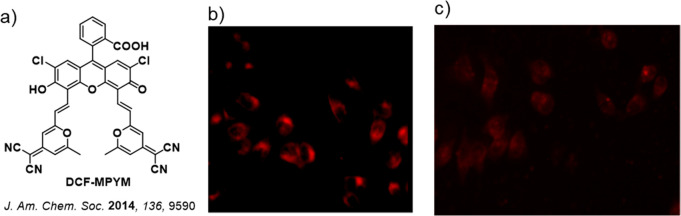
The same group later developed two derivatives of DCF-MPYM through the introduction of aromatic carbonyl groups, with the goal of enhancing the ISC process to increase the population of triplet excitons and the DF contribution to total emission by augmenting SOC (Figure ).ref. ref1413 Indeed, derivatives DCF-MPYM-Ph and DCF-MPYM-Th possess much longer τd of 31.29 μs and 52.05 μs, respectively, than that of DCF-MPYM (τ d = 22.11 μs). With the assistance of HSA, these two emitters were also used in the TRLI of MCF-7 cells.ref. ref1413
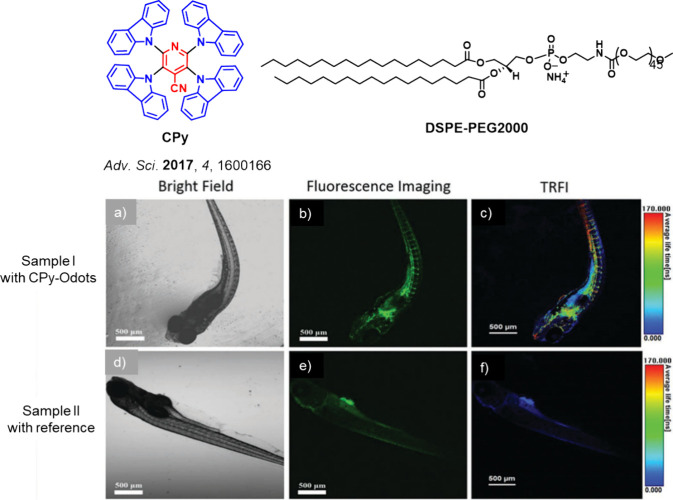
Another family of TADF emitters used as contrast agents through encapsulation with BSA include BP-PXZ, BP-2PXZ, BP-PTZ, and BP-2PTZ (Figure ). These compounds reflect typical D-A TADF emitter designs developed for OLED applications, containing benzophenone (BP) as the acceptor (A) and PXZ or PTZ as donor. As documented in Section sec13 , this motif also confers AIE properties to the molecule, especially active in aqueous environments.ref. ref12 BP-2PXZ, BP-PXZ, BP-2PTZ, and BP-PTZ in the neat films possess τ d of 0.73, 0.96, 0.66 and 1.36 μs at λPL of 558, 546, 551 and 544 nm, respectively. After their encapsulation within BSA, the obtained water-soluble nanoparticles demonstrated strong green or yellow luminescence, low cytotoxicity, and good performance in fluorescence lifetime imaging which provided a clear map of intracellular viscosity.ref1365,ref1368
Organic Dots (Odots)
Organic dots (Odots) have emerged as a class of fluorescent nanoprobes for biological imaging as they are very bright, possess good photostability, do not blink, and are nontoxic.ref1414−ref1415ref1416 Currently, Odots have mainly been used in cell imaging, biosensing, drug and gene delivery, photothermal and photodynamic therapy, and two-photon-excited fluorescence imaging.ref1417−ref1418ref1419 However, these applications largely rely on the fluorescence intensity signals instead of their fluorescence lifetime.ref1419−ref1420ref1421 Odots based on TADF emitters would combine the merits of fluorescent Odots but also feature much longer-lived fluorescence suitable for time-domain microscopy.ref1361,ref1363,ref1370,ref1422,ref1423 Li et al. fabricated CPy-based Odots (CPy-Odots) by encapsulating the high-performance TADF emitter CPyref. ref1341 into DSPE-PEG2000, an amphiphilic and biocompatible polymer that was chosen as the encapsulation matrix due to its ability to encapsulate small, neutral, organic compounds (Figure ).ref. ref1424 The CPy-Odots are water-soluble and bright (ΦPL of 38% in Milli-Q water), with a τ d = 9.3 μs under ambient atmosphere. CPy-Odots were consequently employed in time-resolved and confocal fluorescence imaging of living Hela cells and in living zebrafish. As shown in Figure , by comparing the images captured with fluorescence lifetime imaging microscopy (FLIM), the vivid green-to-red signals of the CPy-Odots were easily distinguished from the autofluorescence (bioluminescence) as the latter possesses a τPL shorter than 3 ns (λex = 405 nm). This study demonstrated that CPy-Odots can be used as bright microangiography agents for FLIM in living zebrafish.ref. ref1422
In addition to CPy, another well-known TADF emitter 4CzIPN was reported to show two-photon absorption as an Odot in HeLa cells.ref. ref1425 Odots of 4CzIPN were formed upon encapsulation into PEG-b-PPG-b-PEG (Figure ). This Odot material possessed a τd ≈ 1.47 μs and has good biocompatibility and biodegradability, low toxicity, and shows specificity for uptake into malignant cells that were imaged by confocal fluorescence imaging in living cells.ref. ref1425 Ran and co-workers similarly prepared nanoprobe micelles by coating Al-Cz (Figure ) in glucose-PEG2000-DSPE, which were then used for malignant cell imaging diagnosis.ref. ref1426 The Glucose-PEG2000-DSPE TADF micelles emitted at λPL ∼ 500 nm and were nontoxic, biocompatible, and even biodegradable. They could be efficiently transported into the cancer cells by an over-expressed glucose transporter on the tumor cell membrane, and then once taken into the HepG2 tumor cells localized in the lysosome.
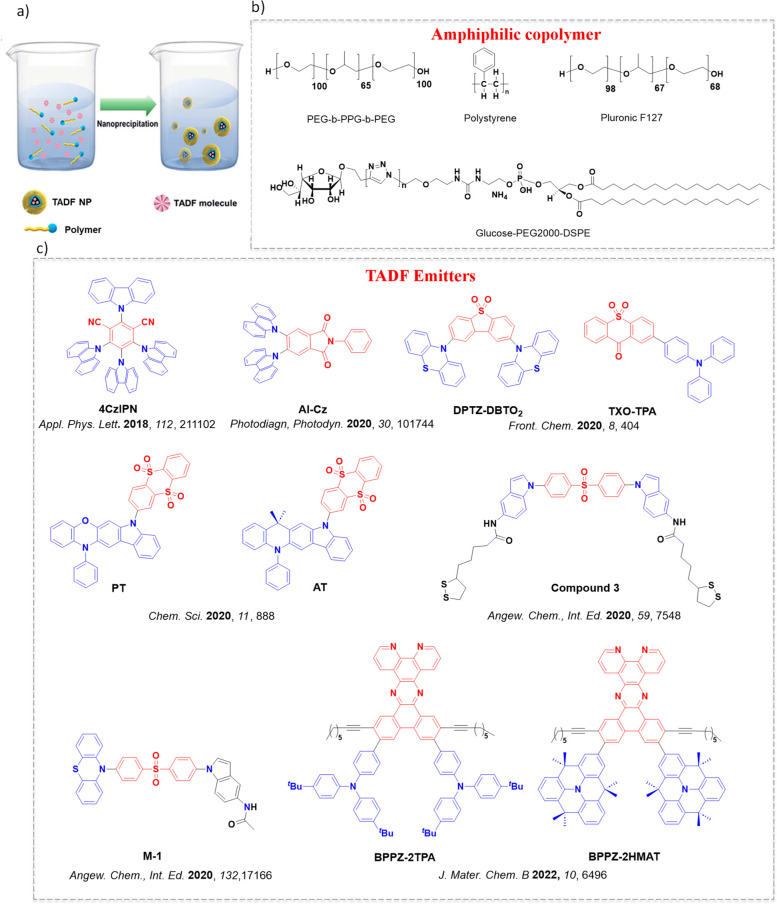
Xiao et al. prepared two TADF molecules, PT and AT, containing different electron-donating moieties to demonstrate a rational design of photosensitizers and fluorescence imaging agents, respectively. The proposed TADF emitters exhibit a tailored balance between two-photon singlet oxygen generation and fluorescence emission (Figure ).ref1426,ref1427 PT possesses both a smaller calculated ΔE ST of 0.06 eV and f of 0.03 compared to a larger calculated ΔE ST of 0.1 eV and an f of 0.07 for AT. In a mixture of 1:99 THF:water, the ΦPL of PT and AT were 2.2% and 9.1%, respectively, while in the corresponding neat thin films, the ΦPL of PT and AT increased to 7.9% and 17%, respectively. In this study, DSPE-PEG2000 was employed to encapsulate AT and PT to produce nanoparticles (PT NPs and AT NPs) which improved both the stability and biocompatibility of PT and AT in aqueous environment. The cell studies further indicated that, in line with their contrasting ΔE ST and ΦPL values, the PT NPs show much stronger singlet oxygen generation capability and photodynamic therapy (PDT) performance compared to the AT NPs, while the AT NPs produced a much brighter fluorescence image.ref. ref1426
Besides DSPE-PEG2000, polystyrene has also been used to encapsulate TADF emitters. In one study DPTZ-DBTO2 and TXO-TPA (Figure ) were encapsulated in order to conserve their photophysical properties as nanoparticles in biological media.ref. ref29 While for DPTZ-DBTO2 the effect of encapsulation on its photophysical properties was not significant (e.g., λPL = 563 nm and λPL–NP = 556 nm), for TXO-TPA the emission was markedly blue-shifted when the dye was incorporated in the nanomaterial (e.g., λPL = 629 nm and λPL‑NP = 555 nm). Both NPs possessed microsecond-long τ d = 2.89 μs for DPTZ-DBTO2 NP and τ d = 9.56 μs for TXO-TPA, respectively. The authors found that upon using amino-modified NPs the reagents could be more efficiently internalised with more uniform dispersion inside the cells.
Zhu and co-workers designed an asymmetric donor-acceptor-donor compound that showed dual TADF emission resulting from CT states from each of the phenothiazine and N-(1H-indole-5-yl) acetamide donors with the diphenylsulfone acceptor (Figure ).ref. ref1428 The two emission bands of M-1 were at λPL = 420 nm and λPL = 580 nm, each showing distinct τd of 5.2 μs and 12.9 μs, respectively with a total ΦPL of 20.1% in THF. Compound M-1 was encapsuled within the amphiphilic block copolymer Pluronic F-127, and dispersion of M-1 in the cell culture medium led to an enhanced average τ PL of 33 μs and 36 μs, respectively, in the dual-channel luminescence imaging studies [the DAPI (4′,6-diamidino-2-phenylindole) and FITC (fluorescein isothiocyanate) channels, dual-channel luminescence imaging here referring to capturing separate images from different spectral bands, usually blue (DAPI) and green (FITC)]. By calibrating the two time-resolved signals, serialized and integrated intracellular local imaging information could also be observed.ref. ref1428 The same group also designed a new TADF emitter based on an indole-derived D-A-D skeleton linked with long α-lipoic alkyl chains (Figure ). Compound 3 exhibited blue emission at λPL = 487 nm with DF in both pure DMF (τ PL = 1.4 μs, ΦPL of 35.3%) and DMF:H2O 1:99 mixtures (τ PL = 3.6 μs, ΦPL of 30.8%). Both the aggregates of Compound 3 and NPs formed by encapsulation into Pluronic F-127 were investigated as imaging reagents by TRLI, which demonstrated that the dual-emission was conserved in the cells.ref. ref1429
Moving away from emitters in non-doped aggregated states, Tsuchiya et al. recently reported an alternative strategy where the Odot is composed of an emitter (4CzIPN), a host (mCP) and a surfactant (DSPE-PEG2k).ref1361,ref1430 This design mitigates possible aggregation-caused quenching (ACQ) by effectively diluting the emitter within the micelle in an analogous manner to the emissive layers in OLEDs. These Odots showed near unity ΦPL of 94% and an associated τ d of 3.1 μs under air-free conditions in water. The conditions and ratios involved in the preparation of the ODots affected the properties, where oxygen-free processing gave ODots with higher ΦPL and greater photostability. Further, upon using a host to surfactant ratio of 10:1, the best photostability was achieved, with photo-degradation causing emission to drop to 50% of the initial intensity after 360 mins, which was superior to a reference blue quantum dot sample. Once the Odot is formed, the photophysics was observed to be insensitive to external oxygen. HEK293 cell imaging was demonstrated and the Odots remained stable for at least 7 days after uptake into the cells (Figure ).ref. ref1361
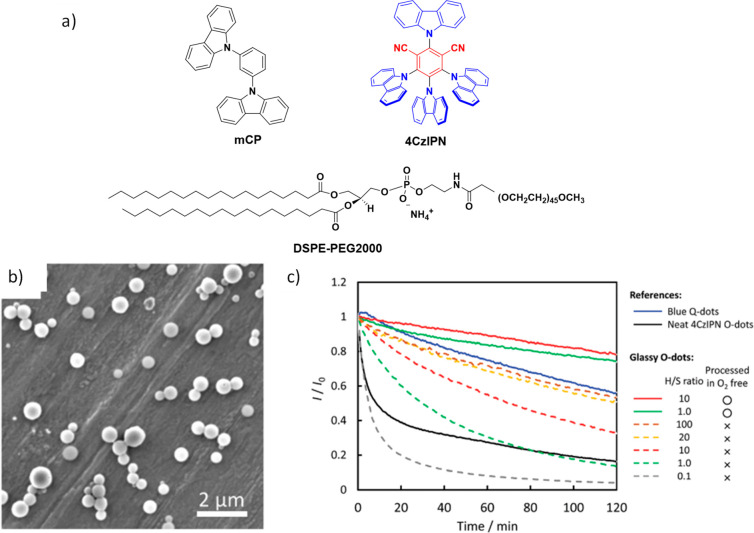
Using a similar methodology as Tsuchiya and co-workers, Hudson and co-workers developed two TADF emitters, BPPZ-2TPA and BPPZ-2HMAT (Figure ). Based on a rigid and strongly electron-withdrawing dibenzo[a,c]dipyrido[3,2-h:20-30-j]phenazine-12-yl (BPPZ) motif, they demonstrated two approaches for the encapsulation of these emitters to yield water-dispersible nanoparticles suitable for TRLI.ref. ref1431 Although Odots prepared with the undoped emitters did not show long-lived emission, their prompt fluorescence lifetimes were long, ranging from 8.5 to 11.9 ns in aqueous solution. Glassy organic nanoparticles (g-Odots) were also prepared with 5 wt% doped emitters in mCP surrounded by the amphiphilic polymer DSPE-PEG2000. The g-Odots by contrast showed long-lived emission in aerated aqueous solutions, with τPL of 123 ms for TPA g-Odots, and 85 ms for HMAT g-Odots. Both approaches yielded nanoparticles suitable for imaging of human cervical (HeLa) and liver (HepG2) cancer cell lines.
Hudson and co-workers also explored other g-Odots based on heptazine-type TADF emitters (Figure ).ref. ref1430 In this study three s-heptazine TADF materials, HAP-3Cz, HAP-3MeOTPA, and HAP-3MeOCz, showed green to deep-red emission (λPL = 525–664 nm) and variable ΦPL (ΦPL = 73% for 2 wt% HAP-3Cz, 33% for 2 wt% HAP-3MeOTPA, and 7% for 2 wt% HAP-3MeOCz in poly-mCP). For HAP-3MeOCz and HAP-3Cz, the g-Odots synthesized in air showed both shorter emission lifetimes and substantially lower ΦPL values (30–41%) relative to those synthesized under nitrogen (ΦPL = 99–100%). By contrast, unity ΦPL was observed for the HAP-3MeOTPA g-Odots for samples synthesized both under air and under nitrogen. Similar delayed fluorescence lifetimes were observed for the HAP-3MeOTPA (50 μs under air, 52 μs under nitrogen) and HAP-3Cz g-Odots (1.1 μs under air, 1.2 μs under nitrogen), but no delayed fluorescence was observed for HAP-3MeOCz g-Odots. These g-Odots were then used as biological imaging probes of immortalized human kidney cancer (HEK293) cells, and both for single- and multi-photon excited microscopy coupled with time-gated luminescence measurements (Figure ). This work therefore not only described new routes to efficient heptazine-based TADF materials, but also demonstrated their potential as nanoparticle-based bioimaging probes.
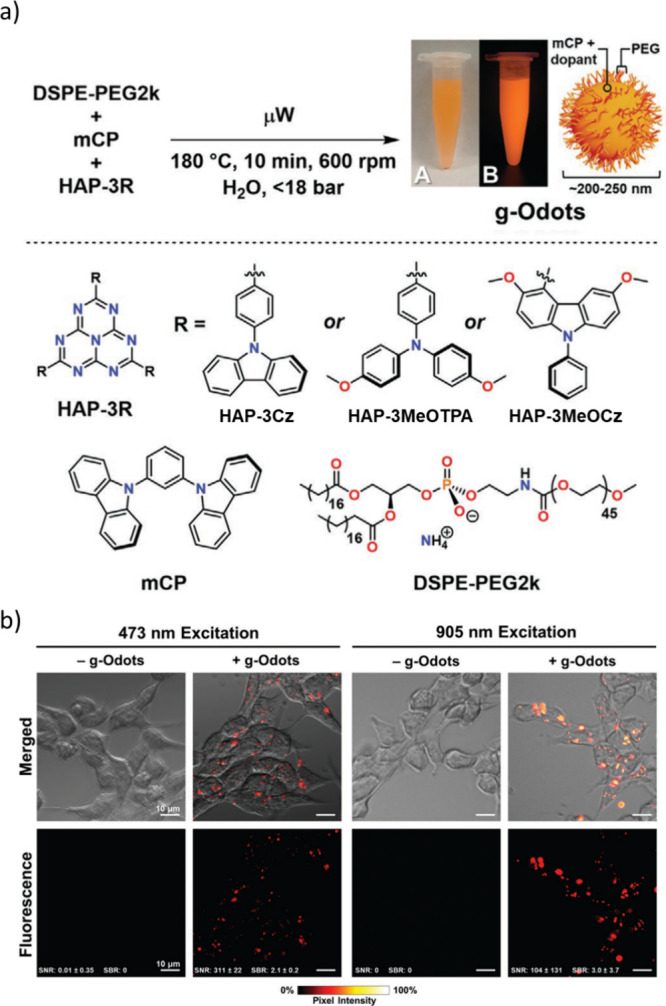
Hudson and co-workers also reported two boron difluoride curcuminoid (BFC)-based polymers, CzBN-co-DtaB and CzBN-co-HmatB (Figure a), exhibiting TADF in the deep red/NIR region with λPL of 694 nm and 717 nm in toluene, respectively. CzBN-co-DtaB and CzBN-co-HmatB showed τd of 4.7 and 5.2 μs and ΦPL of 18% and 12%, respectively, in the solid state. Both polymers were incorporated into water-soluble Odots using the amphiphilic polymer poly(styrene-co-maleic anhydride) [PSMA; PS11-co-MA6)] that had an average diameter of 65 nm and 58 nm for the Odots with CzBN-co-DtaB and CzBN-co-HmatB, respectively. There was only a small red-shift in the emission noted for the Odots compared to the neat films (λPL=730 and 752 nm and 731 and 764 nm in the neat film and in the Odots for CzBN-co-DtaB and CzBN-co-HmatB, respectively), while the delayed lifetimes were considerably shortened compared to those in the solid state, with τd of 0.86 μs and 0.95 μs, respectively. These Odots were used in specific extracellular immunolabeling experiments with human SK-BR3 cells and showed nonspecific binding.ref. ref1432 Using a similar experimental design strategy, Hsu et al. prepared serval types of NIR-II emissive Odots using polymer TADF emitters, with a DMAC-TRZ derivative as a TADF monomer and 3-alkoxy-substituted thiophene as conjugated linker, encapsulated within amphiphilic lipids (Figure b). These Odots exhibited λPL of 1064–1100 nm and ΦPL of 0.40–1.58% in aqueous solution, a significant departure from the typical properties of the DMAC-TRZ monomer. Although no delayed fluorescence was detected for these Odots, they were nonetheless used successfully in in vivo whole-body vascular imaging and 3D bond mapping.ref. ref1433
Besides using amphiphilic molecules or polymers to encapsulate luminophores, amphiphilic peptides have also been used as a delivery vector in the construction of NPs containing TADF emitters. Zhu et al. reported the use of the amphiphilic cell-penetrating peptide (CPP), [F6G6(rR)3R2] (Figure ), to transport hydrophobic fluorophores across cellular barriers. Three known TADF molecules, 4CzIPN, NAI-DPAC, and BTZ-DMAC, were incorporated in well-dispersed nanoparticles (NPs) employing CPP in aqueous solution. The CPP-functionalized NPs of 4CzIPN, NAI-DPAC, and BTZ-DMAC showed much lower ΦPL of 12%, 2.5% and 0.8% in aqueous solution at λPL of 555, 607, and 657 nm, respectively, compared to that observed for the emitters in dilute toluene or doped thin films [(4CzIPN: λPL = 507 nm, ΦPL = 94%),ref. ref1432 (NAI-DPAC: λPL = 570 nm, ΦPL = 94% in 6 wt% doped into mCPCN film),ref. ref1433 [(BTZ-DMAC: λPL = 638 nm, ΦPL = 56% in 3 wt% doped CBP film)ref. ref31]. These three NPs still maintained long-lived luminescence with τ d of 1.8, 6.1 and 31.0 μs for the NPs based on 4CzIPN, NAI-DPAC, and BTZ-DMAC, respectively. The low cytotoxicity and high cytomembrane permeability of the NPs enabled them to be exploitated for TRLI of living cells.ref. ref1370 These findings expanded the applications of cell-penetrating peptides for delivery of molecules and NPs using only noncovalent interactions.
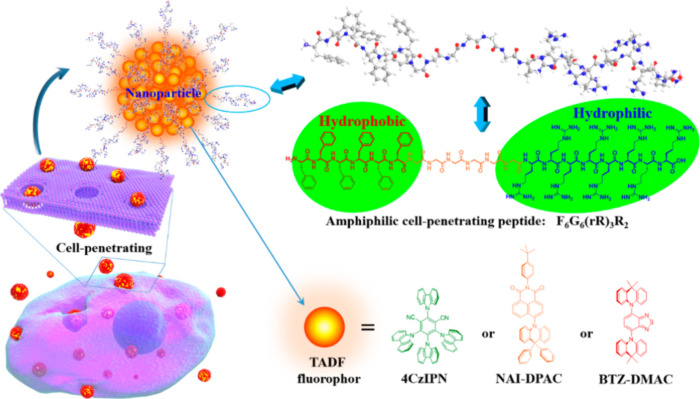
Silica Nanoparticles
Silica-based nanoparticles (SiNPs) have been extensively used as delivery vectors of conventional fluorescent dyes in optical imaging and sensing applications.ref1434−ref1435ref1436 Avó et al. described a method for producing TADF emitter-doped SiNPs that conserve their delayed luminescence in aqueous media. SiNPs (Figure a) were prepared using a modified Stöber process from tetraethoxysilane and Compound 3 in water.ref. ref1437 The SiNPs emitted at ca. 585 nm and with a τ d of 1.20 ms and a ΦPL of 6% in H2O. To address the low ΦPL of the SiNPs, a modified silica source bearing small PEG chains was prepared. The ΦPL of PEG-SiNPs was higher at 20% and these SiNPs possess a longer τ d of 1.25 ms, but with an accompanied red-shift in the λPL to 610 nm. The TADF PEG-SiNPs were effectively internalized by human cells, even at low incubation concentration, localizing primarily in the cytosol and enabling fluorescence microscopy imaging at low dye concentrations.ref. ref1438 Mo et al. encapsulated fluorine and nitrogen co-doped carbon dots (FCDs, NCDs) within amorphous silica using a sol–gel method to obtained TADF materials in aqueous solution (F, NCDs@SiO2 ).ref. ref1439 The presence of a hydrogen bond network between the CDs and the amorphous silica contributed to reducing non-radiative transitions and producing a long-lived afterglow. The F, NCDs@SiO2 had a ΔE ST of 0.32 eV, a high ΦPL of 58.8%, and a τ d = 0.48 s. This versatile material was used separately in optical information encryption, temperature monitoring, and TRLI studies.
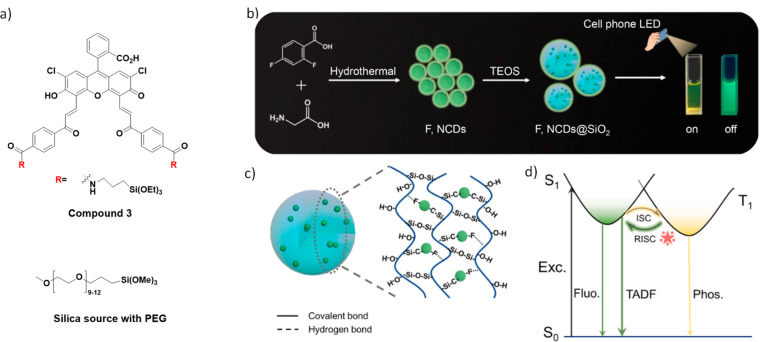
Self-Assembled Nanoparticles
Small-molecule fluorescent organic nanoprobes (FONs) have emerged as promising competitors to inorganic semiconductor quantum dots and fluorescent polymer dots in terms of their wide structural variability, low toxicity, and good biodegradability.ref1417,ref1440−ref1441ref1442 Self-assembled water-dispersible TADF nanostructures based on three known TADF emitters (2CzPN, 4CzIPN, and 4CzIPN-Ph) were reported by Lee et al. that relied on the limited water solubility of these compounds to form the nanostructures (Figure ).ref. ref39 The FONs were made by self-assembly of each of these three TADF emitters into water dispersible NPs/nanorods (NRs), with sizes ranging from 80 to 200 nm. Under nitrogen environment the reference λPL (and ΦPL and τd) of 2CzPN, 4CzIPN, and 4CzIPN-Ph in toluene solutions are λPL = 473 nm (ΦPL = 21.5%; τd = 166 μs), 507 nm (ΦPL = 33.5%; τd = 5.1 μs), and 577 nm (ΦPL = 6.6% τd = 1.1 μs), respectively. All three FONs showed a slight red-shift in their λPL compared to those in toluene (λPL = 503 nm for 2CzPN NRs, λPL = 518 nm for 4CzIPN NPs and λPL = 588 nm for 4CzTPN-Ph NPs) coupled with a slight decrease of their respective ΦPL (ΦPL = 19.4% for 2CzPN NRs, ΦPL = 11.9% for 4CzIPN NPs and ΦPL = 3.6% for 4CzTPN-Ph NPs). In order to evaluate the imaging capabilities of the three FONs, one- and two-photon fluorescence images were obtained in an A549 cell using fluorescence microscopy and laser scanning confocal fluorescence, respectively. Figure b shows the strong cytoplasmic blue, green, and red fluorescence signals from the 2CzPN, 4CzIPN, and 4CzTPN-Ph nanoprobes, respectively. Two-photon fluorescence imaging for FONs showed greater cytoplasmic details and no fluorescence signal from the nucleus, indicating that the FONs do not penetrate into the cell nucleus. These results suggest that self-assembled nanostructures of carbazole-containing TADF emitters are also promising high-performance fluorescence probes for bioimaging.ref. ref1443
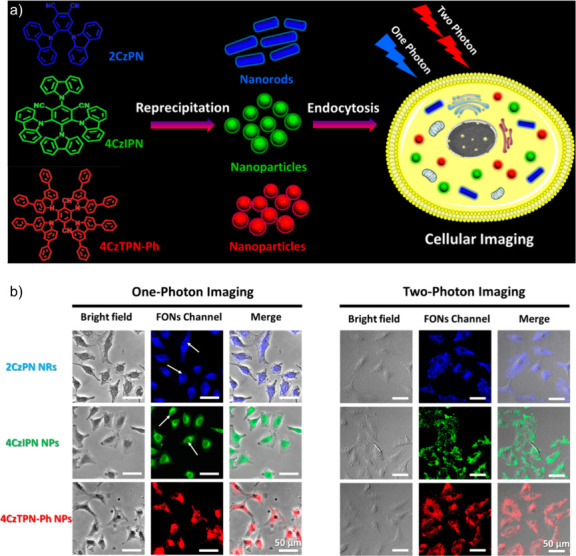
Another reported self-assembled TADF amphiphilic monomer (AI-Cz-AM) is based on the coupling of the lipophilic aromatic imide-based TADF emitter (AI-Cz) with a hydrophilic chain containing a positively charged ammonium terminus.ref. ref1444 Its amphiphilic nature allowed this TADF monomer to spontaneously form a water-soluble and biocompatible nanoprobe, AI-Cz-NP (Figure ). The λPL of AI-Cz-AM and AI-Cz-NP were nearly identical at 517 and 514 nm, and the two materials had moderately small and similar ΔE ST values of 0.10 and 0.12 eV although with very low ΦPL of 1.36% and 0.94%, respectively. Interestingly, the τd increased from 6.08 μs for AI-Cz-AM in degassed THF to 10.68 μs for AI-Cz-NP in oxygenic aqueous solution, indicating significant resistance to ambient oxygen quenching. The latter material was subsequently used in FLIM studies of HepG2cells where long-lived fluorescence signals lasting about 15 ms were detected. This study illustrated how a self-assembly strategy could be used to effectively eliminate emission quenching by oxygen in living cells, without the need for either a polymerization step or biooligomer encapsulation.
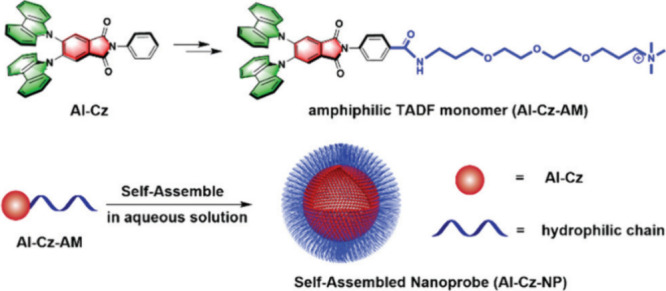
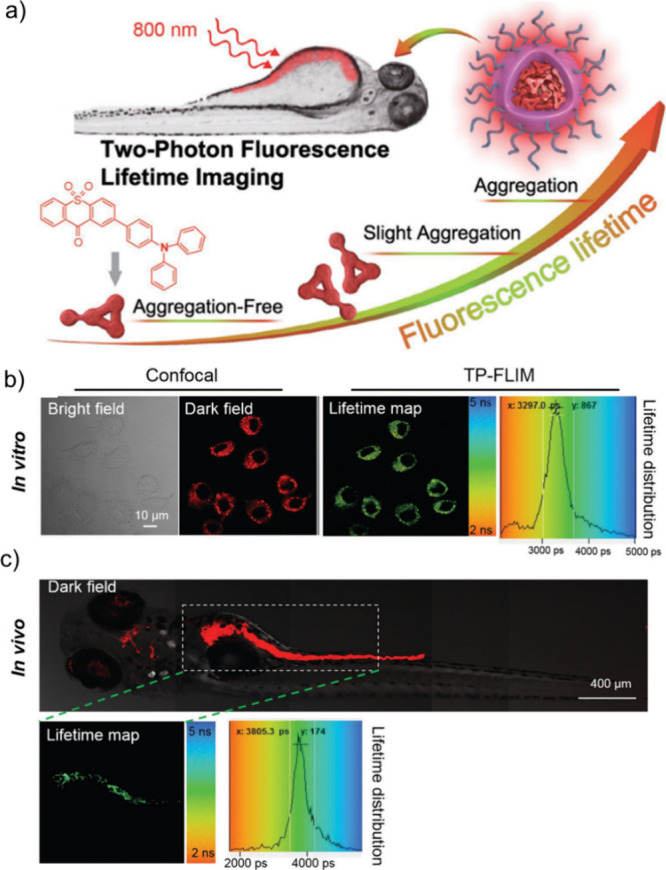
Aggregation-Induced Delayed Fluorescence
One strategy to bypass the oxygen sensitivity of delayed fluorescence in TADF emitters is to use aggregated states. The quenching of emission by oxygen can be suppressed due to its limited ability to make physical contact with the emitter in the aggregated state, as demonstrated in some of the previous examples (Figure and Figure ).ref. ref1446 As noted in Section sec13 , ACQ often takes place in TADF emitters in their aggregated states though, which would be detrimental for bioimaging applications. Therefore, emitters that enjoy AIE instead of suffering from ACQ (Figure a) are viewed as particularly advantageous. For example, the known AIDF emitter TXO was encapsulated in the amphiphilic polymer PEG-b-PPG-b-PEG.ref. ref1447 As shown in Figure b, TXO NPs can readily enter the cytoplasm and exhibit strong red emission by two-photon confocal fluorescence imaging. The two-photon FLIM of TXO NPs revealed localization in the cytoplasm, where the lifetime of the TXO NPs was distributed over a range from ∼2.8 to 3.8 ns. Furthermore, TXO NPs were used for in vivo two-photon FLIM of living zebrafish.
Qi et al. reported the use of three AIDF emitters, PXZ-NI, PTZ-NI and lysosome-targeting Lyso-PXZ-NI (Figure ), each based on a 1,8-naphthalimide (NI) acceptor.ref. ref1448 The τd of the 10 wt% PMMA films of PXZ-NI, PTZ-NI, and Lyso-PXZ-NI were 1.0, 1.7, and 1.3 μs, respectively. In aqueous solutions that produced the aggregated form, all three TADF materials demonstrated markedly enhanced delayed fluorescence when concentrations were increased. Subsequently, confocal fluorescence and lifetime imaging studies were performed using laser scanning microscopy and time-resolved fluorescence microscopy. The confocal fluorescence and lifetime images of HeLa cells after incubation with PXZ-NI, PTZ-NI, and Lyso-PXZ-NI for 2 h were captured and exhibited not only strong red fluorescence signals but also long fluorescence lifetime signals in the HeLa cells.ref. ref1449 In another report, Xu and co-workers presented two phosphine oxide-decorated TADF molecules, CzPOTCF and tBCzPOTCF (Figure ).ref. ref1439 CzPOTCF and tBCzPOTCF in neat films exhibited red emission with λPL = 634 and 647 nm, small ΔE ST values of 0.05 and 0.07 eV, moderate ΦPL of 24.5% and 32.7%, and τ d of 8.95 and 8.69 μs, respectively. Steady-state and time-resolved luminescence imaging of HeLa cells was demonstrated using these emitters in their aggregated form. The delayed lifetimes were slightly shortened compared to the neat films at τ d = 6.69 and 7.41 μs for CzPOTCF and tBCzPOTCF, respectively, yet nonetheless leading to high signal-to-noise ratios in the microscopy applications.ref. ref1450
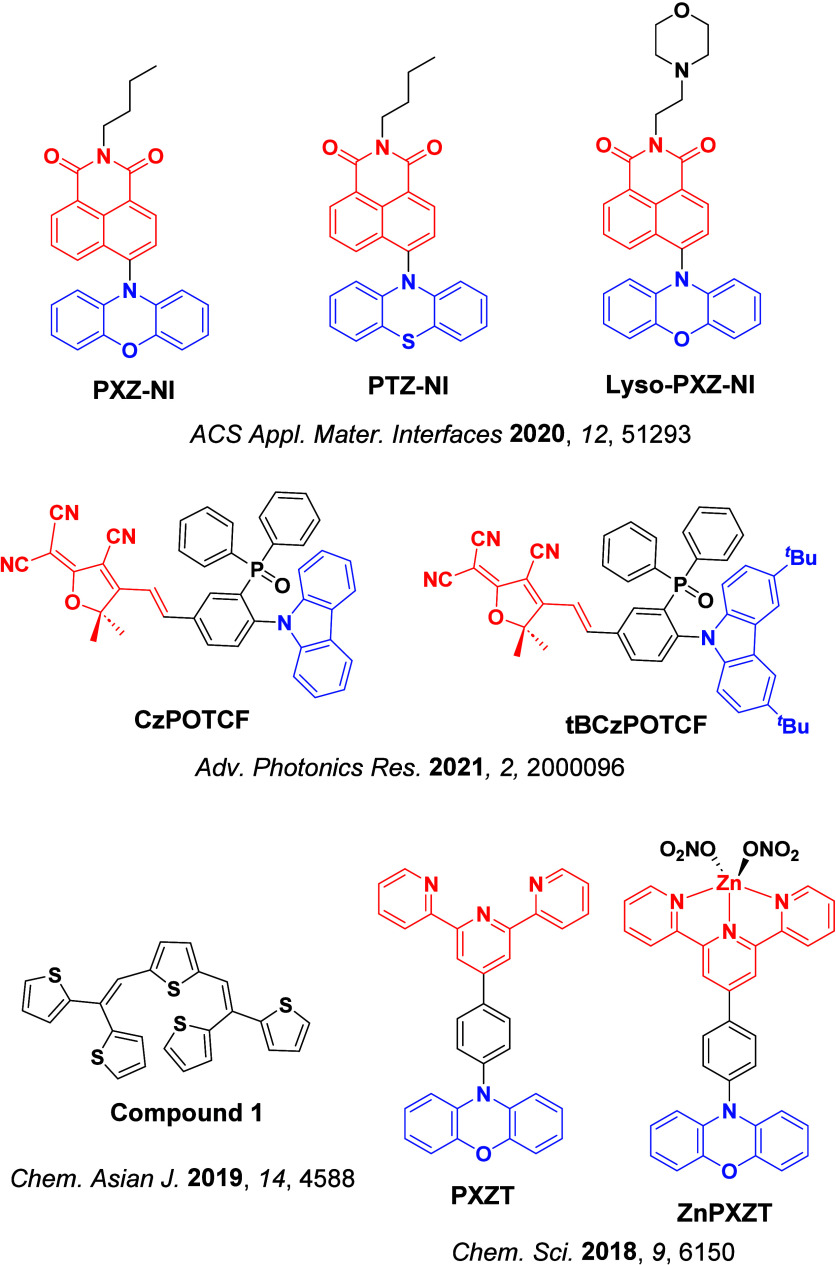
Sarkar et al. reported the first example of a TADF material that is not a D-A structure but rather an oligothiophene derivative, Compound 1 (Figure ).ref. ref1451 In DMSO solution the compound acts as a conventional fluorophore, however in a DMSO/H2O mixture the emitter aggregates exhibiting AIDF at λPL = 600 nm (τ PL = 4.2 μs and 8.0 μs, ΦPL = 11%). Time-dependent luminescence imaging and cytotoxicity studies of Compound 1 were carried out in HeLa cells, showing low cytotoxicity to the cells and excellent signal-to-noise ratios.ref. ref1451
Instead of preparing aggregates before entry into cells as described in the previous examples, Ni et al. proposed a strategy where aggregates would only form within the cells.ref1365,ref1408 With an increase of the water content in THF/water mixtures, the lifetimes and the fraction of delayed emission contribution both increased for PXZT (Figure ), demonstrating AIDF. In 20 wt% PMMA films, PXZT likewise showed a long-lived emission with a τd of 1.4 μs. However, as with most organic TADF emitters this compound was insoluble in water, which limits its applications to biological microscopy. Incorporation of [Zn(NO3)2]4– resulted in the formation of a water-soluble complex, although the emission was also quenched. It was proposed that once in the cell the complex becomes kinetically labile and the zinc ions dissociate when the complex is close to a channel or protein that acts upon zinc. Dissociation of the metal complex thus leads to precipitation of the ligand, and the formation of TADF aggregates which can then be visualized. When the compound was added to HeLa cells and allowed to incubate for 5 hours TADF could indeed be observed, suggesting zinc complex dissociation. The same method was then used for detection of chelating ligand EDTA, as EDTA complexes strongly with zinc leading to dissociation of the zinc from the TADF complex and turning on that ligand’s own TADF.ref. ref1365
Other TADF Bioimaging Reagents
Another strategy to render small molecule TADF emitters biocompatible for imaging studies is to develop hydrophilic TADF luminophores, which can be achieved through the introduction of a hydrophilic group.ref11,ref46 Ni et al., for instance, designed a hydrophilic TADF luminophore (NID-TPP) by introducing a triphenylphosphonium (TPP+) group onto 6-(9,9-dimethylacridin-10(9H)-yl)-2-phenyl-1H-benzo[de]isoquinoline-1,3(2H)-dione (NID), Figure a.ref. ref1452 The pristine NID exhibits TADF with an emission at λPL of 610 nm, a small ΔE ST of 0.03 eV, and a τ d of 5.58 μs in toluene. NID-TPP possesses the same ΔE ST value of 0.03 eV, but with a shorter τ d of 902 ns in the solid state. The ΦPL of the NID-TPP is 0.015% in aqueous solution; however, a 40-fold enhancement was observed (ΦPL = 0.6%) upon addition of sodium tetraphenylborate. Due to the strong electrostatic interactions between the TPP+ group and BPh4 –, NID-TPP aggregates and there is a resulting AIE associated with an emission peak at 618 nm and τ d of 1.2 μs. In both the plasma and mitochondria the membrane potential is negatively charged, allowing positively charged species such as NID-TPP to gradually accumulate in the cytoplasm as well as into the mitochondrial matrix through passive transport. Thus, NID-TPP was utilized for TRLI and two-photon luminescence imaging of HeLa cells and their substructures (Figure b). As shown in Figure , at short incubation times no fluorescence signal could be detected in the extracellular medium, while with longer incubation time enhanced fluorescence signals could be observed in HeLa cells.ref. ref46
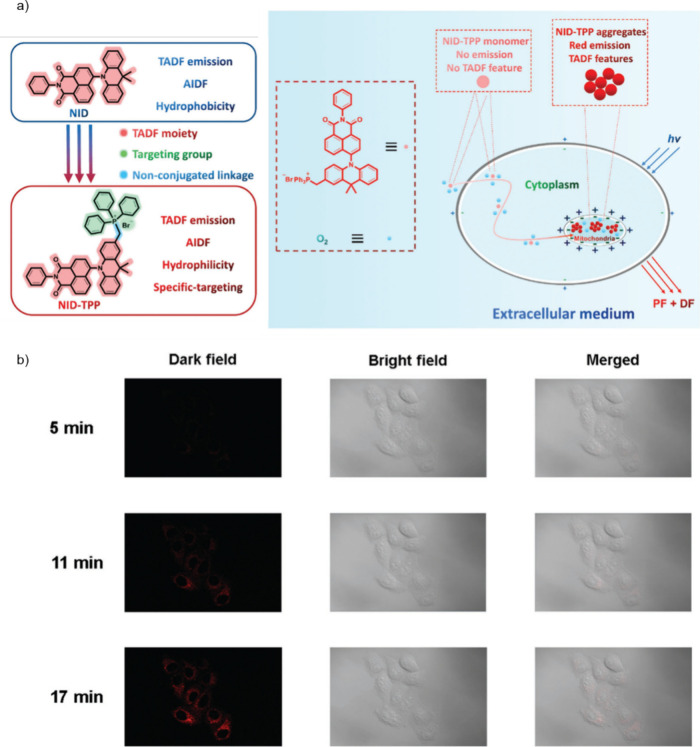
Hudson and co-workers developed multifunctional compartmentalized nanoparticles based on block copolymers, with a hydrophilic cell-penetrating corona surrounding the TADF-active co-monomer block (Figure ).ref. ref1453 This was the first system to employ a single polymer as both the emitter and the cell-penetrating moiety. The polymer nanoparticles (Pdot), BGN10–b-P20Pdot, exhibited a ΦPL of up to 19% in water and significant delayed fluorescence (τd > 26 μs) under both air and inert atmospheres. These all-organic polymer nanoparticles were shown to efficiently enter HeLa, CHO, and HepG2 cells within 30 min, with cell viabilities remaining high for Pdot concentrations of up to 25 mg mL–1. When used for fixed cellular imaging, Pdot-incubated cells showed high signal-to-background ratios compared to control samples with no Pdot exposure. Using time-resolved spectroscopy, the delayed emission of the Pdots was effectively separated from that of both a biological serum as well as from a secondary fluorescent dye.
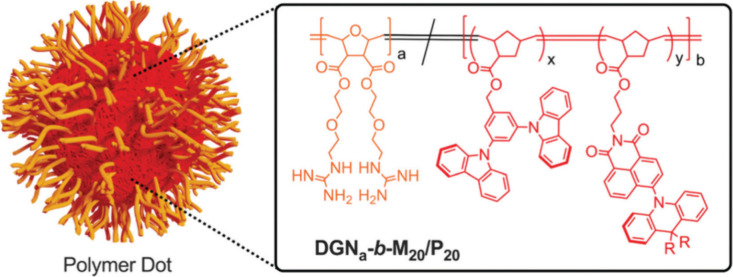
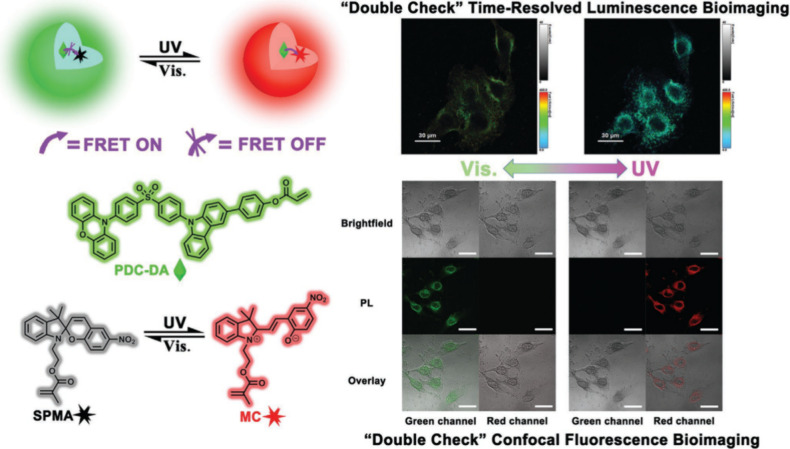
By covalently incorporating a TADF monomer (PDC-DA) and a photochromic spiropyran derivative (SPMA), Yang and co-workers reported a two-component photoswitchable TADF polymeric nanoparticle (PDFPNs) (Figure )ref. ref1454. The polymerizable luminophore, PDC-DA, was used as the energy donor while the photoresponsive SPMA was employed as the energy acceptor. The green emission of PDC-DA can be converted into red fluorescence when the SP unit is converted into its red-emissive ring-open merocyanine (MC) state using 365 nm UV light, enabling FRET from PDC-DA to MC. Subsequently, 525 nm visible light can be used to efficiently recyclize the SPMA into a FRET-inactive form, recovering the green emission of PDC-DA. The PDFPNs possesses a τd of 3.3 μs under degassed conditions and a shorter lifetime of 2.73 μs in aerated aqueous solution. After being irradiated by 365 nm light, the τd was remarkably reduced to 1.61 μs, which the authors ascribed to an efficient FRET process that was switched on between the donor and acceptor. These nanoparticles showed negligible oxygen-sensitivity, high FRET efficiency, rapid and reversible photo responsiveness, and long-term fluorescence stability. They were thus used to realize reversible dual-color confocal and time-resolved luminescence imaging.
Outlook
Organic TADF compounds with long-lived excited states have emerged as highly promising bioimaging agents. Their distinctive advantage, compared to fluorescent emitters, stems from their ability to eliminate interference from short-lived autofluorescence background signals in TRLI. By developing materials with a suitable delay time between short-lived biological autofluorescence and the long-lived TADF emission of the dye, accurate detection and imaging of various biologically relevant species is enabled. The examples included above illustrate the promise and several direct applications of TADF materials as versatile cellular and tissue stains.
Aside from long-lived emission arising from relatively slow RISC (in contrast to the requirements of TADF materials for OLEDs), good biocompatibility and tolerance of both atmospheric and intracellular oxygen are required in these applications. To achieve these properties, the design strategies reviewed in this section include: 1) TADF emitters capped with BSA/HSA; 2) TADF-based Odots formed by encapsulating TADF emitters in an amphiphilic polymer; 3) Silica-based nanoparticles as hosts for the encapsulation of the TADF emitter; 4) Self-assembled nanoparticles; 5) Aggregation-induced delayed fluorescence; and 6) Other TADF bioimaging agents such as water-soluble TADF polymers. While successful examples of each of these strategies exist, because these target properties are so different from those sought by ‘mainstream’ TADF OLED research, design rules to produce optimal imaging agents are still rapidly developing. Consequently, TADF materials offer significant opportunities for future innovation, although various unique challenges must be addressed before deployment in preclinical/clinical context. Of these we highlight in particular the following: 1) improving the inherent water solubility and poor bioavailability of these organic emitters; 2) designing high brightness deep red or NIR TADF emitters for deep-tissue theranostics, thereby mitigating optical tissue attenuation in vivo; 3) Design of a wider library of TADF bioimaging agents that show targeted uptake for imaging of specific organelles. In contrast to collaboration between physicists and chemists underpinning much of the work in other sections of this review, in this arena it will be growing collaboration between chemists, biologists, and medical researchers that spurs deeper understanding, progress, and utility of these materials.
Organic Solid-State Laser Using TADF Components
Introduction
Including vertical excitation, the four-level energy structure of TADF materials makes it practical to achieve population inversion of excited states, which is the very first step for lasing. In addition, TADF emitters and organic semiconductor materials more widely possess distinct advantages over their inorganic counterparts, such as wide range of tunability for their emission spectrum, light weight, mechanical flexibility, and potential for low-cost fabrication of large-area arrays. Their strong optical transitions lead to high gain, and they can have high ΦPL in the solid state. These properties (purely fluorescent and not yet involving TADF activity) have driven the recent interest in organic solid-state lasers (OSSLs), which are promising devices with applications in scanners, printers, sensors, and as cutting-edge light sources with high spectral, spatial, and temporal resolution.ref39,ref1455,ref1456
Although the OSSL has been demonstrated under pulsed or even quasi-continuous-wave optical excitation, producing an electrically operated OSSL remains a challenging but tantalizing goal.ref. ref1457 Development of commercial applications for OSSL requires overcoming this bottleneck. Compared to optical excitation, where the majority of generated excitons are singlets that possess a fast radiative decay rates, under electrical excitation spin-statistics of uncorrelated charge recombination governs the exciton ratio between the emissive singlets and dark triplets. As with OLED applications discussed in most of the previous sections, this leads to a 1:3 singlet:triplet exciton ratio under electrical excitation, with triplets typically unable to contribute to lasing. As a result, to achieve amplified spontaneous emission (ASE) from the gain medium, extremely high current densities (> kA/cm2) are needed to produce the singlet exciton densities required to reach the lasing threshold under optical excitation.ref1458,ref1459 These high current densities and long-lived triplet excitons induce detrimental effects on the ASE process and material. These effects include 1) exciton loss due to the bi-excitonic interactions, such as exciton-exciton annihilation, and exciton-polaron quenching; 2) gain loss due to the excited state absorption of singlet, triplet, polaron, and other species; 3) material degradation through Joule heat generation, and photo- and electrochemical bond cleavage under high current and excitation density. For all the above, managing the population of triplet exciton plays a crucial role in maintaining an achievably low lasing threshold.
There are two approaches for the management of triplet excitons. The first is triplet quenching, in which triplet excitons are actively quenched by a doped triplet scavengers in the gain medium, such as oxygen, anthracene, and cyclooctatetraene (COT).ref1460−ref1461ref1462 These scavengers have a higher singlet exciton energy but lower triplet exciton energy than the dye molecules, so that they do not interfere with lasing of the singlet excitons and only quench the triplets. Direct removal of triplet excitons helps alleviate some of the gain loss and material degradation pathways that rely on these, but does not avoid their initial formation, meaning that high current densities are still required. The second approach to managing triplets is harvesting, in which triplet excitons are either made emissive with the help of heavy metal atoms such as those found in phosphorescent metal complexes, or are converted to singlets by RISC in TADF materials, allowing the triplet excitons (and up to 100% of total excitons) to be used in the same way this is achieved in OLEDs.
Although reports of ASE directly from the triplet states of organic phosphorescent molecules are rare,ref. ref1463 ASE has been observed in TADF molecules. In this section we document the recent progress on OSSLs employing TADF molecules, categorized key examples as: 1) TADF molecules used as the gain medium; 2) TADF molecules used as the triplet harvester; and 3) lasing from TADF molecules (see material structures in Figure ).
TADF Molecules as the Gain Medium
A promising gain medium must have gain exceeding loss. Typically this involves materials with sufficiently high ΦPL, absorption separated from emission, and low waveguide loss, leading to low threshold for ASE. Applying these conditions, a number of TADF materials have been reported.
The effects of the molecular structure on the ASE were investigated in a comparative study between MR-TADF (DABNA-2) and D-A-TADF (3CzTrz) molecules.ref. ref1464 Although both compounds have similar ΦPL in 6 wt% doped mCBP films (∼80%), similar peak λPL (∼470 nm), and similar singlet exciton lifetime (∼5 ns), ASE was only observed in the DABNA-2 (Figure a) doped thin films under pulsed optical excitation. As the excitation energy increased above the threshold energy (E th = 1.6 μJ cm–2), an increase in PL intensity accompanied by a spectral narrowing was observed, confirming an ASE process. The ASE wavelength (λASE) was 494 nm, corresponding to the 0-1 electronic transition. The absence of ASE in 3CzTrz was attributed to its much broader PL spectrum (FWHM = 80 nm) compared to that of DABNA-2 (FWHM = 30 nm), which decreased the stimulated emission cross-section, thereby increasing the possibility of both self-absorption and triplet absorption.
Recently, the molecular properties of 17 different BN-cored molecules were theoretically computed to give molecular design rules for TADF-based gain materials.ref. ref1465 Four key screening parameters including the oscillator strength, net optical emission cross-section, singlet lifetime, and k RISC were considered, resulting in four promising candidates for lasing, DABNA-2, m-Cz-BNCz, ADBNA-Me-Mes, and ADBNA-Me-Tip. Depending on the amplification modes, specific molecular design strategies were proposed to minimize the self-absorption; either to introduce additional vibrational modes for the 0-1 transition, or to optimize the substitution position to induce a large Stokes shift for the 0-0 transition.
To engineer high ΦPL, conventional D-A-TADF emitters can be rigidified using a proposed intramolecular-lock strategy as in PXZN-B and DMACN-B (Figure a), in which a diphenylmethylene group was inserted to adjust the torsion angle and restrict the intramolecular relaxation.ref. ref1466 The locked TADF emitters not only showed high ΦPL of 93% and 90%, but also narrow PL emission with FWHM of 48 nm and 29 nm in doped films for PXZN-B and DMACN-B, respectively. Under pulsed optical excitation, the doped thin films of PXZN-B and DMACN-B in mCP showed ASE behavior with λASE at 470 nm and 448 nm, and low ASE thresholds (E th) of 4.0 and 12.0 μJ cm–2, respectively.
Since 2018, high efficiency solution-processable red TADF emitters based on borondifluoride curcuminoid (BC) derivatives have drawn increasing attention, which also exhibit ASE behavior.ref. ref1467 These molecules possess a linear donor-acceptor-donor (D-A-D) structure and usually show photophysical properties that are highly dependent on the doping concentration, implying strong intermolecular interaction. In TPABC (Figure ) doped thin films in CBP, ASE behavior was observed across a wide range of doping concentrations from 2 wt% to 40 wt%, with E th all below 100 μJ cm–2. The lowest E th of 7.5 μJ cm–2 (λASE = 750 nm) was obtained in the film, which had the lowest doping concentration (2 wt%) and also the highest ΦPL of 70%. The origin of ASE was subsequently identified by the same group as resulting from a low energy dimeric structure.ref. ref485 In addition, the gain loss due to the triplet absorption was found to be negligible in those molecules. To push the λASE beyond 800 nm, a thiophene ring was inserted between the D and A moieties to give TPATBC (Figure a), which reduced the HOMO–LUMO energy gap.ref. ref1468 When doped into a low triplet energy host, F8BT, TPATBC showed a ΦPL of about 45%, a short singlet lifetime of 1.3 ns, and λPL at 724 nm. Under pulsed optical excitation, ASE was observed with E th of 13.3 μJ cm–2 and λASE of 807 nm. The large red-shift of the λASE with respect to the PL was attributed to the strong singlet-singlet absorption of TPATBC, which inhibits the electronic transition to the lowest vibrational level of the ground state. With a second-order DFB (distributed feedback) resonator, lasing was observed, with a further reduced E th of 6.2 μJ cm–2.
TADF Molecules as the Triplet Harvester
The potential of TADF molecules for ASE has also been explored in co-doped thin films, in which triplet excitons are harvested by TADF molecules and then transferred to a fluorescent laser dye via FRET. Excitons accumulate on the laser dye, allowing ASE to occur, in so-called TADF-assisted ASE with strong parallels to HF-OLED approaches. In this manner, ASE has been observed with the same λASE but reduced E th compared with the system without the inclusion of the TADF molecules. This approach is highly appealing for applications in electrically-pumped lasing, where the TADF material may be able to convert triplet excitons to singlets before FRET, thus theoretically reducing the threshold current density by as much as a factor of 4. As an additional consideration, for efficient FRET to occur there must be a sufficient spectral overlap between the emission spectrum of TADF molecules and absorption spectrum of laser dyes.
The high efficiency TADF molecule ACRXTN possesses considerable FRET overlap with the green laser dye C545T (Figure b), implying that an efficient FRET process can occur. Indeed, when co-doped into an mCBP host (ACRXTN: 6 wt%, C545T: 1 wt%), E th was decreased from 1.2 μJ cm–2 (C545T only) to 0.8 μJ cm–2 (TADF assisted), with λASE of 535 nm in both cases.ref. ref1469 A similar effect was observed when using a sky-blue fluorescent molecule BUBD-1 (Figure b) as the laser dye.ref. ref1470 A slightly different strategy was adopted wherein instead of co-doping, the TADF molecule DMAC-DPS (Figure b) was itself used as the host. The doped thin films of BUBD-1 (2 wt%) showed similar high ΦPL of 82% in both CBP and DMAC-DPS hosts. However, E th was reduced from 1.51 μJ cm–2 in CBP to 1.19 μJ cm–2 in DMAC-DPS, with λASE of 500 nm for both. It was proposed that the TADF host could not only harvest triplet excitons, but also promote FRET through better overlap with the dopant, together resulting in a lower E th. Recently, the co-doping strategy was explored with the use of a red laser dye, dithiophenyl-diketopyrrolopyrrole (DT-DPP).ref. ref1471 The green TADF emitter Cz-DBA (10 wt%) was used as the assistant dopant with DT-DPP (1 wt%) together in CBP as the bulk host (Figure b). With the same λASE of 620 nm, ΦPL and E th were both improved from 65% and 7.3 μJ cm–2 in the simple doped film to 77% and 4.0 μJ cm–2 in the TADF-assisted thin film.
With the co-doped TADF molecule acting as a triplet harvester, a lower E th was achieved in fluorescent dye molecules without varying λASE. These results show that TADF molecules can not only minimize the detrimental effect of triplet excitons on ASE, but also promote exciton energy transfer to dye molecules, resulting in higher ΦPL and lower E th. A brief synopsis of ASE parameters for TADF molecules is shown in Table S24. In all of these examples though it should be noted that the TADF assistant dopant is only harvesting the relatively small number of triplet excitons that are generated by photoexcitation (some of which form directly on or because of the TADF emitter itself). In the ultimate application of this strategy in electrically-pumped OSSLs, the density of triplet excitons will be orders of magnitude larger, requiring TADF materials with outstanding RISC rates in order to convert these triplets sufficiently quickly to avoid quenching and material damage. Although this concept is already thoroughly demonstrated in high k RISC OLEDs with small efficiency roll-off, the higher exciton densities required for lasing operation means that such a device remains yet to be demonstrated.
Lasing from TADF Molecules
With well-designed optical resonators that act as cavities to tune wavelengths and more easily reach threshold, lasing has been observed from several TADF molecules. So far, the optical resonators used have been a microring array,ref. ref1472 a microsphere array,ref. ref1473 and a Fabry–Pérot type microcrystal/microwire structureref. ref1474 (Figure ). All these resonators show strong optical confinement with high Q-factors near or above 1000. When the excitation energy reaches above threshold energy (E th laser), lasing with characteristically narrow FWHM (< 1 nm) can be observed.
By using a confined solution-growth method, a whispering-gallery mode (WGM) microring resonator array was fabricated with a high Q-factor of 1300 at 683 nm.ref. ref1472 The gain material was a red BC derivative, CAZ-A (Figure c), which was doped in CBP host. With the strong optical confinement of the microring resonator, lasing was observed with E th laser of 3.69 μJ cm–2, λlasing of 683 nm as a single narrow peak (FWHM = 0.52 nm). By careful tuning of the microring size from 11.5 to 29.0 μm, the lasing mode spacing (Δλ) was successfully modulated from 8.12 nm to 2.85 nm. It was found that E th laser increased with decreasing temperature, which was attributed to the slower k RISC at lower temperature, resulting in more accumulated triplets. It should be noted that complex thermal effects on the lasing material, cavity, or otherwise can often confound the accurate identification of lasing behavior,ref. ref1475 and so whether a change in E th arises as a consequence of changes in RISC requires further study.
Using an emulsion-solvent-evaporation method, another WGM resonator was fabricated based on polymeric microspheres with circular boundaries and smooth surfaces. The gain medium was a green TADF molecule, 4CzTPN (Figure c) doped in PS (polystyrene).ref. ref1473 Under pulsed optical excitation, lasing at 563 nm with FWHM of 0.21 nm was observed when the excitation energy increased above 88 μJ cm–2. A similar resonator size dependence of Δλ was observed in microsphere as well, confirming the lasing is indeed WGM-type. From the transient absorption spectrum and temperature dependence of E th laser, the authors concluded that triplet absorption was negligible and fast RISC had a positive influence on the lasing threshold.
Recently, faceted microcrystals of boron difluoride-based TADF molecules were fabricated by a facile reprecipitation method.ref. ref1474 These microcrystals not only covered emission wavelengths from red (MOON), yellow (ON), to green (MOCN) (Figure c), but also possessed a high-quality intrinsic Fabry-Pérot resonator with Q factor larger than 1000. Under pulsed optical excitation, lasing and lasing oscillation were observed with E th laser (λlasing) of 3.04 μJ cm–2 (650 nm), 4.96 μJ cm–2 (561 nm), and 3.49 μJ cm–2 (525 nm) for MOON, ON, and MOCN, respectively. Beside the temperature dependence of E th laser, triplet up-conversion was supported by transient PL measurements, in which a plateau structure was attributed to the extra generated singlet exciton via RISC.
Besides microcrystals, single-crystalline microwires of the TADF material DCzBF2 (Figure c) were also fabricated using a solution self-assembly method.ref. ref1476 The molecular geometry changed from a highly twisted D-A structure in solution to a nearly coplanar conformation in the microwires, which showed AIE originating from locally excited states. The 1D microwires, exhibiting herringbone-like molecular packing, smooth surface, and high ΦPL of 48%, had a uniform diameter of about 1 μm and a length of 10–100 μm. These microwires thus served as natural Fabry-Pérot resonators with estimated Q factor of 930. Under pulsed optical excitation, a series of sharp cavity mode peaks at around 465 nm were observed with E th laser of 3.74 μJ cm–2 and FWHM below 0.5 nm. The temperature dependence of E th laser and the microwire length-modulated cavity modes confirmed the TADF behavior and the internal microcavity effect, respectively. For a summary of TADF materials with lasing properties, see Table S25.
Outlook
Although ASE has been observed in both TADF molecules and in TADF-assisted laser dye systems, key advancements that RISC is expected to support in this area are yet to be achieved – most notably the harvesting of triplet excitons for electrically driven lasing. Even exploiting well-designed resonators such as microcrystal/microwire Fabry-Pérot cavities, DFBs, microrings, and microspheres, lasing thresholds from TADF molecules are typically an order higher than those of conventional fluorescent laser dyes. This is likely a consequence of their slower radiative rates and their active ISC channels (generating triplet excitons) outweighing any beneficial ability to harvest triplets by RISC. The relatively low absorption cross section in the lowest charge-transfer bands of D-A TADF emitters also hinders population inversion, as does the broad emission bands of their CT emission for ASE. In contrast though, the strong absorption as well as efficient narrowband emission and high radiative rates of MR-TADF materials we speculate will find increasing use for optically-driven lasing films in the coming years, sharing these advantageous properties with purely fluorescent emitters.
Returning to electrically driven lasing, the large accumulation of triplets when operating at the necessary high currents is detrimental to stability, and likely incompatible with the delicate materials currently deployed in TADF OLEDs. While this appears discouraging for the use of TADF materials in laser systems, further enhancements in RISC (as sought by the OLED research community) could eventually overcome this issue. Some promising results have already been seen with the aid of LE states,ref. ref1476 proving there is a sizeable contribution from RISC to ASE and lasing, but more significant advances in increasing RISC rates will be required in order for these materials to contribute meaningfully towards the prized development of electrically-pumped OSSLs. Considering the significant applications such a technology could unlock, we anticipate only an intensification of research activity in this area over the remainder of the decade.
TADF Materials as Photocatalysts
Introduction
The use of organic donor-acceptor (D-A) TADF compounds as photocatalysts (PCs) has gained considerable attention since the first report in 2016.ref. ref1477 Visible light photocatalysis has been known since the late 19th century, but has seen a resurgence of interest over the last 15 years, especially as a tool for developing ‘green’ chemistry.ref1478−ref1479ref1480ref1481 From water splittingref. ref1482 to degradation of pollutants,ref. ref1483 the applications of photocatalysis are vast and potentially deeply impactful. Historically employing transition metal-based complexes, the use of TADF PCs, however, has thus far only been investigated with respect to small molecule photocatalysis or photopolymerization.
Photocatalysis proceeds by recruiting the excited state of the PC, generated by electronic excitation upon absorption of light, to engage in either energy or electron transfer with an organic substrate (Figure ). In the photoinduced energy transfer (PEnT) mechanism, the substrate receives energy from the excited PC through a Förster or Dexter energy transfer process, regenerating the ground state of the PC. In this way the PC is able to undergo many catalytic cycles, with overall turnover limited by its own intrinsic photostability. When the PC is instead involved in a photoinduced electron transfer (PET) this is termed photoredox catalysis and can occur through either an oxidative or reductive quenching mechanism. A second single electron transfer (SET) step is subsequently required to close the photocatalytic cycle. It is common, though not essential, for sacrificial electron donors or acceptors to be employed in reactions to allow catalytic turnover of the photoredox catalytic cycle.
Traditionally, iridium(III) and ruthenium(II) complexes (Figure ) have been used as PCs and dominate much of the literature in homogeneous photocatalysis.ref1484,ref1485 However, the search for cheaper and less toxic PCs has led to investigations into both Earth-abundant metal complexes,ref1486,ref1487 and purely organic compounds as suitable alternatives.ref36,ref1488 To be a useful PC, the compound should exhibit appreciable light absorption (preferably in the visible region) to allow for selective photoexcitation of the PC in the presence of the organic reagents (that typically only absorb in the UV). Excited state lifetimes on the order of at least a few nanoseconds are necessary to allow for diffusion of the excited PC to encounter and undergo PEnT or PET with the substrate in solution. For photoredox catalysis, a wide redox window is also important to facilitate SET with a large range of organic molecules, while for PEnT an appropriate spectral overlap between the emission of the PC and the absorption of the substrate is necessary for efficient energy transfer. From these required properties it is unsurprising that TADF compounds, both organic (see for instance Sections sec3 –sec5) and organometallic (Section sec9 ) have been used to great effect as PCs. It is, however, unclear at this point what intrinsic value TADF provides with respect to photocatalysis aside from generating PCs with excited-state lifetimes with sufficiently long-lived excited states to enable the photochemistry. Indeed, different photophysical properties may prove to be ideal for photocatalysis (fast ISC and slow RISC), as opposed to the OLEDs where high photoluminescence quantum yield and fast RISC are desired traits of the emitter..
Eosin Y (Figure ) was the first TADF compound to be used as a PC, where it was employed to photocatalyze the reduction of phenacylonium salts by 1,4-dihydropyridines,ref. ref1489 and to this day remains a staple in the library of commonly used visible-light PCs. Since 2016, organic D-A TADF compounds based on the carbazolyl dicyanobenzene (CDCB) family and related derivatives have been shown to act as potent PCs,ref. ref1477 with 4CzIPN proving to be a viable replacement for cationic heteroleptic iridium(III) complexes across a diverse range of organic reactions (Figure and Table S26).ref. ref1490 Other organic D-A TADF compounds beyond the CDCB family are yet to receive comparable interest in small molecule photocatalysis, with only two examples known to date: an imidazoacridine-based structure (Figure ) used in [2+2] PEnT cycloadditions,ref. ref1491 and a pyrimidyl sulfone compound (Figure ), which showed a broad range of mechanistically distinct photocatalytic reactions.ref. ref1492 A small selection of other D-A TADF compounds have also found applicability in photopolymerizations reactions.ref. ref1493 One of the particular benefits of using D-A TADF compounds as PCs is that facile tuning of the redox properties is possible through judicious and combinatorial choice of each of the donor or acceptor moieties. This kind of tunability has thus far been difficult to achieve with many other organic PCs, where the HOMO and LUMO are not as obviously inferred from the molecular structure.ref. ref1494 Organometallic TADF compounds based on, for instance, copper(I), zirconium(IV) and gold(I) have additionally been explored as Earth-abundant metal PCs.ref948,ref1495−ref1496ref1497
Recently, we reported that MR-TADF compounds can also be employed as PCs. Both DiKTa and Mes3DiKTa (Figure ) have been shown to be effective PCs across a wide variety of PEnT and PET reactions, rivalling the efficiency and applicability of 4CzIPN.ref. ref1498 One particular advantage of using MR-TADF compounds over D-A TADF emitters is that the former displays only modest positive solvatochromism, translating to less solvent stabilization of the excited state, and hence preserving more of the excited state energy for driving reactions. This may contribute to the greater reactivity of MR-TADF PCs, particularly in polar aprotic solvents such as MeCN and DMF which are typically employed in photocatalysis reactions. In a follow up study, DiKTa was found to be the most efficient PC in a dual NHC/photoredox reaction for the synthesis of unsymmetric 1,4-diketones.ref. ref1499
Throughout the myriad of reports that use TADF compounds as PCs, it should be noted that the vast majority make no attempt to correlate PC activity with other key TADF photophysical properties. Consequently, no real mechanistic investigation has been undertaken to understand what, if anything, TADF activity contributes to their success as PCs. Indeed, it is reasonable to expect that small ΔE ST and fast RISC – prized for OLED applications – would be counterproductive in photocatalysis as these properties would more rapidly depleted excited states through emission channels that compete with PEnT and PET. Regardless, we here present a select few examples of TADF compounds used as PCs, primarily to highlight their versatility. The focus of these examples is based upon the use of CDCB TADF compounds and particularly 4CzIPN, since these compounds have become notably popular in the photocatalysis literature over the last seven years. A comprehensive overview of the wide range of photocatalytic transformations mediated by organic TADF compounds and a summary of the different organic D-A TADF compounds employed as PCs has been recently reviewed by Bryden and Zysman-Colman.ref. ref36
TADF Compounds as PCs
From a survey of the photocatalyst literature it becomes apparent that 4CzIPN has entered the pantheon of common photocatalysts assessed for photochemical transformations since the first report of its use in 2016.ref. ref1477 The popularity of 4CzIPN as a PC is therefore clearly evident, although its excellent properties for OLED applications means that its photophysical properties can be further refined for photocatalysis.ref. ref1490 The related material 4DPAIPN is rapidly becoming a popular alternative, given the stronger reducing capacity of 4DPAIPN relative to 4CzIPN (Table S26).ref. ref1500
The scope of these reported TADF photocatalyzed reactions encompasses a large range of organic transformations, from polymerizationsref1501,ref1502 to cyclizations,ref1503,ref1504 although the most frequently encountered class of reactions involves C(sp2)-C(sp3) cross-coupling reactions. A wide range of radical precursors such as carboxylic acids,ref1505,ref1506 trifluoroborate saltsref. ref1507 and 4-alkyl-1,4-dihydropyridine derivatives (DHPs),ref. ref1508 can be used to reductively quench the excited PC, releasing a C(sp3)-centred alkyl radical. This requires the PC to be a relatively strong photooxidant (e.g., Eox of carboxylates typically ranges from 1.2–1.5 V vs SCE).ref. ref1509 Through a radical addition or radical coupling mechanism, a C-C bond is formed, usually facilitated through SET from the reduced PC. Given the strong photooxidizing ability of 4CzIPN in comparison to other common PCs (E*red = 1.35 V vs SCE, see Table S26), this compound typically is able to act as a photocatalyst for this class of reactions and more widely. For example, in the hydrosilylation of alkenes (Figure a), 4CzIPN yielded 82% of product, while the next best PC in the study, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, affording only 45% of product.ref. ref1510 This difference in yield is likely due to the suitable redox potentials of 4CzIPN to oxidize the silacarboxylate radical precursor, as this step is thermodynamically difficult for this iridium PC (e.g., Eox(Ph2MeSiCO2 •/Ph2MeSiCO2 –) = 1.32 V vs SCE and E*red = 1.35 V and 1.21 V vs SCE for 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively).
The reductive dehalogenation of aryl chlorides is a challenging transformation that has also been shown to proceed efficiently using 4CzIPN and related D-A cyanoarenes as PCs, this time through a proposed consecutive photoinduced electron transfer (conPET) mechanism.ref. ref1511 The PC is initially reductively quenched by a sacrificial electron donor, before the resultant radical anion of the PC is photoexcited again to yield an incredibly strong photoreductant capable of reducing substrates such as 4-chloroanisole (Ered = −2.90 V vs SCE).ref. ref1512 The aryl radical formed can then be coupled with a variety of partners, including boronate esters, phosphines and phosphites. While no alternative PCs outside of the CDCB family were screened in this reaction, a total of 13 4CzIPN-based PCs were trialled, all of which could borylate 4-chlorotoluene (Figure b) to varying degrees (6–96%).
Although less well documented, TADF compounds can act as PCs in energy transfer processes, such as the E/Z isomerisation of alkenes,ref. ref1513 which proceeds through a Dexter energy transfer mechanism. The Z/E ratio can be optimized by tuning the triplet energy, ET, of the PC, while moderate ISC provides a suitable quantity of triplet excitons from photoexcitation. In the E/Z isomerisation of stilbene for example (Figure c), 4CzTPN showed the highest selectivity, even greater than that of the literature PC [Ru(bpy)3](PF6)2 (Z/E = 8.56/1 and 6.69/1 and ET = 2.34 eV and 2.03 eV, respectively). Other CDCB PCs, such as 2,4,6-3CzBN (ET = 2.87 eV), with unsuitably high ET, tended to show poorer selectivity with this substrate. More recently, 4CzIPN has been used to photocatalyze the intramolecular [2+2] cycloaddition reaction of enynes to generate 1,3-diene-quinolinone products (Figure d), affording the same yield as [Ir(ppy)2(dtbbpy)]PF6, but at a fraction of the cost.ref. ref1514 In the endeavour to develop more sustainable photocatalytic protocols, the authors also demonstrated the recyclability of 4CzIPN, with no loss in the yield of product even after reuse of the PC three times.
Similar to the use of TADF materials as PC for energy transfer reactions, these materials are also gaining recognition as useful triplet sensitisers for other photophysical processes. In TTA upconversion solutions, triplet excitons are generated from low energy photons typically using metalloporphyrin sensitisers. Excitons are then transferred to a separate emitter species, pairs of which diffuse and undergo TTA to generate anti-Stokes shifted emission.ref. ref1515 Recently it has been shown that TADF materials can be used in a similar way,ref. ref1516 with their significantly higher triplet energies (compared to metalloporphyrins) particularly useful for generating UV TTA emission.ref1517,ref1518 MR-TADF emitters also show promise for this kind of application,ref. ref1519 expanding significantly the tunability and range of potential designs for triplet photosensitisers. An example of this in photocatalysis involves 4CzIPN, which undergoes a DET to the benzene-based annihilator 1,4-bis((tri-iso-propylsilyl)ethynyl)benzene (bTIPS-Bz).ref. ref1520 Subsequently, 1bTIPS-Bz* is formed through TTA, which can then undergo FRET sensitization of UVB-absorbing carbonyls, such as pinacolene, to generate isobutylene (Figure e). Although proof of the necessity of both 4CzIPN and 1bTIPS-Bz to the reaction was shown, no yields were provided, nor any comparison of performance with other PCs.
TADF Compounds as PCs in Dual Catalysis
There is now a wide body of literature discussing the overlap of transition metal catalysis with photocatalysis, aptly termed metallaphotocatalysis or metallophotoredox catalysis.ref. ref1521 Specifically, 4CzIPN has been shown to be compatible with this form of synergistic dual catalysis, working in tandem with nickel(II),ref1522−ref1523ref1524ref1525 palladium(II),ref1526−ref1527ref1528 cobalt(II),ref1529−ref1530ref1531ref1532 titanium(IV),ref1533−ref1534ref1535 iron(II and III),ref1536,ref1537 chromium(II)ref1538,ref1539 and copper(II)ref1540−ref1541ref1542 catalysts. Of these metal-based co-catalysts, examples with Nickel catalysts have been the most widely documented, typically involving C(sp2)-C(sp3) cross-coupling reactions. These dual metallaphotoredox catalysis reactions have been reported to work via a reductive quenching mechanism for the CDCB PCs. 4CzIPN typically performs well for this class of reactionsref1522,ref1543,ref1544 as it is capable of reducing the in-situ Ni(I) species (Ered ≈ −1.1 V vs SCE).ref. ref1545
CDCB PCs can additionally perform well in oxidative quenching cycles, as is in operation when the co-catalyst used is the titanium(IV) complex TiCp2Cl2. For instance, in the Barbier allylation of aldehydes (Figure f),ref. ref1533 4CzIPN can replicate the success of iridium(III) PCs, such as [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, since photoreduction of TiCp2Cl is facile (Ered = −0.22 V vs SCE). Regeneration of the PC occurs through SET with a sacrificial reductant such as a Hantzsch ester (Hantzsch ester Eox = 1.10 V vs SCE).ref. ref1546
Aside from dual catalysis with transition metals, CDCB compounds have been documented to work alongside organic catalysts, including hydrogen atom transfer (HAT) catalysts,ref1547−ref1548ref1549ref1550 N-heterocyclic carbenes (NHCs)ref1499,ref1551,ref1552 and bromine catalysts like cinnamyl bromide.ref. ref1553 The photocatalytic formation of carbanion equivalents has received attention over the last few yearsref. ref1554 as an accessible way to form C-C bonds without the need for stoichiometric reductants like low-valent metals. An example of this involves photocarboxylation of benzylic C-H bonds (Figure g), which Meng et al. found to be possible using CDCB compounds as the PC in the presence of tri-iso-propylsilanethiol as the HAT catalyst,ref. ref1555 whereas both cationic and neutral iridium(III) PCs were incapable of completing the transformation. The success of the CDCB compounds here is thought to be due to their ability to form a strongly reducing in-situ PC, 2,3,4,6-tetra(9H-carbazol-9-yl)-5-(1-phenylethyl)benzonitrile, by photosubstitiution of one of the cyano groups. The resultant photosubstituted photoactive product, possessing significantly different photophysical and electrochemical properties, is hypothesised to be the active photocatalyst in reactions such as this.ref. ref1556
Outlook
From these examples it is clear that although the use of organic TADF compounds as PCs is still in its relative infancy, especially compared to organometallic PCs, the results obtained thus far are promising. These early studies, using TADF materials unoptimized for photocatalytic activity, indicate that organic TADF compounds can act routinely as replacements for heavy metal PCs. Unquestionably, the CDCB family of D-A TADF PCs, most commonly exemplified by 4CzIPN, has been most influential in this research area; however, there have been an increasing number of other examples, both D-A and MR-TADF systems, that suggests a generalisability of using TADF compounds, originally designed for use in OLEDs, as PCs. The wealth of TADF compounds available to synthetic chemists should provide broad scope for tuning the reactivities of a TADF PC for a specific transformation, providing similar versatility to the status quo organometallic PCs.
Additionally, the enhanced light absorption typically displayed by TADF materials in the visible light region compared to organometallic ruthenium(II) and iridium(III) polypyridyl complexes should be beneficial to improve reaction kinetics. Due to the vast number of published structures, it is likely that high-throughput experimentation would be needed to efficiently explore which TADF materials would be the most useful as PCs. Understanding how to appropriately tune the excited-state properties of TADF PCs to best support the desired photocatalytic pathways is imperative and will require detailed mechanistic understanding of both the TADF material and the reactions/substrates that they act upon. It will also be important to understand exactly how the TADF mechanism is implicated and influences photocatalysis reactions; indeed, investigating whether the triplet or singlet excited states, or both, are involved will require thorough mechanistic and spectroscopic studies. PC stability under photocatalytic reaction conditions is also an important consideration: popular organometallic and TADF PCs have been observed to photodegrade during photocatalytic reactions (a phenomenon that is easily observed via UV-Vis absorption spectroscopy).ref1556−ref1557ref1558 It would be highly desirable for new PCs to be designed showing improved photocatalytic stability. Alternatively, suspension on solid supports could enable the design of more recyclable heterogeneous PCs. With an enhanced understanding of the factors governing both the operational photocatalysis mechanism and the stability of the PC, it is expected that it would be possible to design TADF compounds for their explicit use as PCs, rather than simply repurposing known emitters that were developed for OLED applications.
Conclusions and Outlook
Over the last decade TADF has dominated the optoelectronic materials and applications literature, with over 3,500 articles and 1,000 patents published up to the end of 2022.ref. ref1559 The majority of these reports have focussed on the design and exploitation of TADF materials for use in OLEDs, building upon the seminal 2012 work of Adachi and co-workers.ref. ref31 Indeed, steady and sometimes breakthrough improvements in device performance have been made, with examples of blue, green, red, and white TADF OLED efficiencies (Sections sec3–sec6) now rivalling PhOLED counterparts, especially at low brightness. Attention has also increasingly shifted to addressing outstanding barriers to commercialization, such as poor device lifetime and efficiency roll-off at practical brightnesses. Of particular note, specifically during the scope of this review, a solution to the undesirably broad emission spectra of D-A TADF emitters has emerged with the emergence of MR-TADF materials (Section sec11 ), and especially their tandem application in hyperfluorescence device architectures (Sections sec17 and sec18).
Beyond the visible spectrum there is still considerable work required to improve the performance of near-IR TADF OLEDs, for which there are currently few studies or standout materials. This improved performance is required to unlock the distinct applications of near-IR emission, particularly for night vision, biological sensing and imaging. On the other wavelength extreme, despite a tremendous research effort focussed on developing high-performance deep-blue TADF OLEDs, their efficiencies, color purity, and roll-off behavior remain sub-optimal (or at best, individually optimized). The recent rise to prominence of the ‘blue-backplane’ concept makes progress at this particular color point all the more valuable as it can support next-generation performance in both display and lighting applications. Simultaneously, performance gains in UV-emitting OLEDs could support new applications in biological sterilisation, security, and lithography applications. Despite current challenges, we predict that sustained experimental effort, increasingly guided by theoretical studies, will result in improved emitter designs as the years progress, and a more nuanced understanding of the structural features that control the efficiency of the TADF process.
Alongside the mainstream research efforts linked to OLEDs, this review also documents the increasingly widespread use of TADF materials in broader research spheres. Improved performance of related LEC devices (Sections sec16 ) employing TADF emitters continues in parallel with OLEDs, albeit with a more limited materials set leading to a more relaxed pace of development. TADF materials have also made promising early inroads as active materials in bioimaging (Section sec21 ) and sensing (Section sec20 ), and we predict a significant expansion of multi-disciplinary research activity in this specific area in the near future. The utility of TADF compounds in photocatalysis has also now become widely recognized (Section sec23 ); however, the chemical space explored in terms of photocatalyst design remains stubbornly limited around a small set of established phthalonitrile-based OLED emitters. Assessing the wider panoply of reported TADF materials as photocatalysts will likely result in powerful additions to the synthetic chemist’s toolbox. Indeed, as the wide scope of this review attests, investigations into TADF materials design and various applications are only broadening and accelerating as this field reaches adolescence. We are certainly excited to witness further developments on what is surely a bright horizon.
Supplementary Materials
References
- OLED Adoption by Smartphones to Slow Down in 2022.
- Smartwatch Market Size, Share & Trends Analysis Report by Price Band, by Display Technology (LCD, OLED), by Region, and Segment Forecasts, 2022 – 2030.
- DSCC: The OLED Market Will Grow 2% in 2022, Led by Strong Demand for IT Panels.
- Automotive OLED Market Size to Reach Usd 9 Billion 2030.
- A. Köhler, H. Bässler. Triplet States in Organic Semiconductors. Mater. Sci. Eng., R, 2009. [DOI]
- S. R. Forrest, D. D. C. Bradley, M. E. Thompson. Measuring the Efficiency of Organic Light-Emitting Devices. Adv. Mater., 2003. [DOI]
- W. Brütting, J. Frischeisen, T. D. Schmidt, B. J. Scholz, C. Mayr. Device Efficiency of Organic Light-Emitting Diodes: Progress by Improved Light Outcoupling. Phys. Status Solidi A, 2013. [DOI]
- B. Geffroy, P. le Roy, C. Prat. Organic Light-Emitting Diode (OLED) Technology: Materials, Devices and Display Technologies. Polym. Int., 2006. [DOI]
- M. Fröbel, F. Fries, T. Schwab, S. Lenk, K. Leo, M. C. Gather, S. Reineke. Three-Terminal RGB Full-Color OLED Pixels for Ultrahigh Density Displays. Sci. Rep., 2018. [DOI | PubMed]
- M. Pedzisz. Beyond Bt.709. SMPTE Motion Imaging J., 2014. [DOI]
- S. Reineke, M. Thomschke, B. Lüssem, K. Leo. White Organic Light-Emitting Diodes: Status and Perspective. Rev. Modern Phys., 2013. [DOI]
- C. Murawski, K. Leo, M. C. Gather. Efficiency Roll-Off in Organic Light-Emitting Diodes. Adv. Mater., 2013. [DOI | PubMed]
- C. W. Tang, S. A. VanSlyke. Organic Electroluminescent Diodes. Applied Physics Letters, 1987. [DOI]
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson, S. R. Forrest. Highly Efficient Phosphorescent Emission from Organic Electroluminescent Devices. Nature, 1998. [DOI]
- C. Adachi, M. A. Baldo, M. E. Thompson, S. R. Forrest. Nearly 100% Internal Phosphorescence Efficiency in an Organic Light-Emitting Device. J. Appl. Phys., 2001. [DOI]
- Universal Display and Samsung Display Sign a Long-Term Material Supply Agreement.
- D. Y. Kondakov. Triplet-Triplet Annihilation in Highly Efficient Fluorescent Organic Light-Emitting Diodes: Current State and Future Outlook. Philos. Trans. Royal Soc., 2015. [DOI]
- P. Rajamalli, N. Senthilkumar, P. Gandeepan, P.-Y. Huang, M.-J. Huang, C.-Z. Ren-Wu, C.-Y. Yang, M.-J. Chiu, L.-K. Chu, H.-W. Lin, C.-H. Cheng. A New Molecular Design Based on Thermally Activated Delayed Fluorescence for Highly Efficient Organic Light Emitting Diodes. J. Am. Chem. Soc., 2016. [DOI | PubMed]
- D. Lee, J. Park, W. Lee, H. S. Kim, S. Yoo. Realization of Flexible Ultraviolet Organic Light-Emitting Diodes: Key Design Issues. Adv. Photon. Res., 2021. [DOI]
- D. G. Congrave, B. H. Drummond, P. J. Conaghan, H. Francis, S. T. E. Jones, C. P. Grey, N. C. Greenham, D. Credgington, H. Bronstein. A Simple Molecular Design Strategy for Delayed Fluorescence toward 1000 Nm. J. Am. Chem. Soc., 2019. [DOI | PubMed]
- A. Monkman. Why Do We Still Need a Stable Long Lifetime Deep Blue OLED Emitter?. ACS Appl. Mater. Interfaces, 2022. [DOI | PubMed]
- C. Wang, X. Li, Y. Pan, S. Zhang, L. Yao, Q. Bai, W. Li, P. Lu, B. Yang, S. Su, Y. Ma. Highly Efficient Nondoped Green Organic Light-Emitting Diodes with Combination of High Photoluminescence and High Exciton Utilization. ACS Appl. Mater. Interfaces, 2016. [DOI | PubMed]
- W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang, Y. Ma. A Hybridized Local and Charge-Transfer Excited State for Highly Efficient Fluorescent OLEDs: Molecular Design, Spectral Character, and Full Exciton Utilization. Adv. Optical Mater., 2014. [DOI]
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang, Y. Ma. Employing ∼100% Excitons in OLEDs by Utilizing a Fluorescent Molecule with Hybridized Local and Charge-Transfer Excited State. Adv. Funct. Mater., 2014. [DOI]
- J. Sanz-Rodrigo, G. Ricci, Y. Olivier, J. C. Sancho-Garcia. Negative Singlet-Triplet Excitation Energy Gap in Triangle-Shaped Molecular Emitters for Efficient Triplet Harvesting. J. Phys. Chem. A, 2021. [DOI | PubMed]
- P. de Silva. Inverted Singlet-Triplet Gaps and Their Relevance to Thermally Activated Delayed Fluorescence. J. Phys. Chem. Lett, 2019. [DOI | PubMed]
- J. Ehrmaier, E. J. Rabe, S. R. Pristash, K. L. Corp, C. W. Schlenker, A. L. Sobolewski, W. Domcke. Singlet-Triplet Inversion in Heptazine and in Polymeric Carbon Nitrides. J. Phys. Chem. A, 2019. [DOI | PubMed]
- N. Aizawa, Y. J. Pu, Y. Harabuchi, A. Nihonyanagi, R. Ibuka, H. Inuzuka, B. Dhara, Y. Koyama, K. I. Nakayama, S. Maeda. Delayed Fluorescence from Inverted Singlet and Triplet Excited States. Nature, 2022. [DOI | PubMed]
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend, F. Li. Efficient Radical-Based Light-Emitting Diodes with Doublet Emission. Nature, 2018. [DOI | PubMed]
- A. Abdurahman, T. J. H. Hele, Q. Gu, J. Zhang, Q. Peng, M. Zhang, R. H. Friend, F. Li, E. W. Evans. Understanding the Luminescent Nature of Organic Radicals for Efficient Doublet Emitters and Pure-Red Light-Emitting Diodes. Nat. Mater., 2020. [DOI | PubMed]
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura, C. Adachi. Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence. Nature, 2012. [DOI | PubMed]
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato, C. Adachi. Thermally Activated Delayed Fluorescence from Sn4+-Porphyrin Complexes and Their Application to Organic Light Emitting Diodes – a Novel Mechanism for Electroluminescence. Adv. Mater., 2009. [DOI | PubMed]
- T. Chatterjee, K. T. Wong. Perspective on Host Materials for Thermally Activated Delayed Fluorescence Organic Light Emitting Diodes. Adv. Optical Mater., 2019. [DOI]
- A. M. Polgar, Z. M. Hudson. Thermally Activated Delayed Fluorescence Materials as Organic Photosensitizers. Chem. Commun., 2021. [DOI]
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch, S. Brase. A Brief History of OLEDs – Emitter Development and Industry Milestones. Adv. Mater., 2021. [DOI]
- M. A. Bryden, E. Zysman-Colman. Organic Thermally Activated Delayed Fluorescence (TADF) Compounds Used in Photocatalysis. Chem. Soc. Rev., 2021. [DOI | PubMed]
- F. Fang, L. Zhu, M. Li, Y. Song, M. Sun, D. Zhao, J. Zhang. Thermally Activated Delayed Fluorescence Material: An Emerging Class of Metal-Free Luminophores for Biomedical Applications. Adv. Sci., 2021. [DOI]
- Thermally Activated Delayed Fluorescence Emitters for Light-Emitting Diodes and Sensing Applications. In. Fluorescence in Industry; Springer Series on Fluorescence;, 2019
- Y. Jiang, Y. Y. Liu, X. Liu, H. Lin, K. Gao, W. Y. Lai, W. Huang. Organic Solid-State Lasers: A Materials View and Future Development. Chem. Soc. Rev., 2020. [DOI]
- M. Y. Wong, E. Zysman-Colman. Purely Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes. Adv. Mater., 2017. [DOI]
- P. L. dos Santos, M. K. Etherington, A. P. Monkman. Chemical and Conformational Control of the Energy Gaps Involved in the Thermally Activated Delayed Fluorescence Mechanism. J. Mater. Chem. C, 2018. [DOI]
- Y. Liu, C. Li, Z. Ren, S. Yan, M. R. Bryce. All-Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes. Nat. Rev. Mater., 2018. [DOI]
- M. Godumala, S. Choi, M. J. Cho, D. H. Choi. Recent Breakthroughs in Thermally Activated Delayed Fluorescence Organic Light Emitting Diodes Containing Non-Doped Emitting Layers. J. Mater. Chem. C, 2019. [DOI]
- X. Liang, Z.-L. Tu, Y.-X. Zheng. Thermally Activated Delayed Fluorescence Materials: Towards Realization of High Efficiency through Strategic Small Molecular Design. Chem. Eur. J., 2019. [DOI | PubMed]
- S. Madayanad Suresh, D. Hall, D. Beljonne, Y. Olivier, E. Zysman-Colman. Multiresonant Thermally Activated Delayed Fluorescence Emitters Based on Heteroatom-Doped Nanographenes: Recent Advances and Prospects for Organic Light-Emitting Diodes. Adv. Funct. Mater., 2020. [DOI]
- F. Ni, N. Li, L. Zhan, C. Yang. Organic Thermally Activated Delayed Fluorescence Materials for Time-Resolved Luminescence Imaging and Sensing. Adv. Optical Mater., 2020. [DOI]
- L. Frédéric, A. Desmarchelier, L. Favereau, G. Pieters. Designs and Applications of Circularly Polarized Thermally Activated Delayed Fluorescence Molecules. Adv. Funct. Mater., 2021. [DOI]
- S. Achelle, M. Hodée, J. Massue, A. Fihey, C. Katan. Diazine-Based Thermally Activated Delayed Fluorescence Chromophores. Dyes Pigm., 2022. [DOI]
- J. Han, Y. Chen, N. Li, Z. Huang, C. Yang. Versatile Boron-Based Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes. Aggregate, 2022. [DOI]
- Y. Jin. The Recent Progress and State-of-Art Applications for Thermally Activated Delayed Fluorescence (TADF). HSET, 2022. [DOI]
- D. Sun, C. Si, T. Wang, E. Zysman-Colman. 1,3,5-Triazine-Functionalized Thermally Activated Delayed Fluorescence Emitters for Organic Light-Emitting Diodes. Adv. Photon. Res., 2022. [DOI]
- H. J. Kim, T. Yasuda. Narrowband Emissive Thermally Activated Delayed Fluorescence Materials. Adv. Optical Mater., 2022. [DOI]
- Y. J. Yu, F. M. Liu, X. Y. Meng, L. Y. Ding, L. S. Liao, Z. Q. Jiang. Carbonyl-Containing Thermally Activated Delayed Fluorescence Emitters for Narrow-Band Electroluminescence. Chem. Eur. J., 2023. [DOI | PubMed]
- Y. Xiaodong, Z. Xiaokang, D. Hailiang, S. Jing, W. Hua. Research Progress of Circularly Polarized Thermally Activated Delayed Fluorescence Materials and Devices. Chin. J. Org. Chem., 2022. [DOI]
- D. Sun, S. M. Suresh, D. Hall, M. Zhang, C. Si, D. B. Cordes, A. M. Z. Slawin, Y. Olivier, X. Zhang, E. Zysman-Colman. The Design of an Extended Multiple Resonance TADF Emitter Based on a Polycyclic Amine/Carbonyl System. Mater. Chem. Front., 2020. [DOI]
- T. A. Comerford, E. Zysman-Colman. Supramolecular Assemblies Showing Thermally Activated Delayed Fluorescence. Small Sci., 2021. [DOI | PubMed]
- F. Perrin. La Fluorescence Des Solutions – Induction Moléculaire. – Polarisation Et Durée D’émission. – Photochimie. Annales de Physique, 1929. [DOI]
- G. N. Lewis, D. Lipkin, T. T. Magel. Reversible Photochemical Processes in Rigid Media. A Study of the Phosphorescent State. J. Am. Chem. Soc., 1941. [DOI]
- S. Boudin. Phosphorescence Des Solutions Glycériques D’éosine Influence Des Iodures. J. Chim. Phys. Phys.-Chim. Biol., 1930. [DOI]
- C. A. Parker, C. G. Hatchard. Triplet-Singlet Emission in Fluid Solutions. Phosphorescence of Eosin. Trans. Faraday Soc., 1961. [DOI]
- C. A. Parker. Sensitized P-Type Delayed Fluorescence. Proc. R. Soc. London, A, 1963. [DOI]
- C. A. Parker, C. G. Hatchard. Delayed Fluorescence of Pyrene in Ethanol. Trans. Faraday Soc., 1963. [DOI]
- J. Saltiel, H. C. Curtis, L. Metts, J. W. Miley, J. Winterle, M. Wrighton. Delayed Fluroescence and Phosphorescence of Aromatic Ketones in Solution. J. Am. Chem. Soc., 1970. [DOI]
- R. E. Brown, L. A. Singer, J. H. Parks. Prompt and Delayed Fluorescence from Benzophenone. Chem. Phys. Lett., 1972. [DOI]
- M. W. Wolf, K. D. Legg, R. E. Brown, L. A. Singer, J. H. Parks. Photophysical Studies on the Benzophenones. Prompt and Delayed Fluorescences and Self-Quenching. J. Am. Chem. Soc., 1975. [DOI]
- A. Maciejewski, M. Szymanski, R. P. Steer. Thermally Activated Delayed S1 Fluorescence of Aromatic Thiones. J. Phys. Chem., 1986. [DOI]
- M. N. Berberan-Santos, J. M. M. Garcia. Unusually Strong Delayed Fluorescence of C70. J. Am. Chem. Soc., 1996. [DOI]
- F. A. Salazar, A. Fedorov, M. N. Berberan-Santos. A Study of Thermally Activated Delayed Fluorescence in C60. Chem. Phys. Lett., 1997. [DOI]
- W. L. Parker, G. A. Crosby. Assignment of the Charge-Transfer Excited States of Bis(N-Heterocyclic) Complexes of Copper(I). J. Phys. Chem., 1989. [DOI]
- J. R. Kirchhoff, R. E. Gamache, M. W. Blaskie, A. A. Del Paggio, R. K. Lengel, D. R. McMillin. Temperature Dependence of Luminescence from Cu(Nn)2 + Systems in Fluid Solution. Evidence for the Participation of Two Excited States. Inorg. Chem., 1983. [DOI]
- R. M. Everly, D. R. McMillin. Reinvestigation of the Absorbing and Emitting Charge-Transfer Excited States of [Cu(Nn)2]+ Systems. J. Phys. Chem., 1991. [DOI]
- Yersin, H. ; Monkowius, U. 2008.
- J. C. Deaton, S. C. Switalski, D. Y. Kondakov, R. H. Young, T. D. Pawlik, D. J. Giesen, S. B. Harkins, A. J. M. Miller, S. F. Mickenberg, J. C. Peters. E-Type Delayed Fluorescence of a Phosphine-Supported Cu2(Μ-NAr2)2 Diamond Core: Harvesting Singlet and Triplet Excitons in OLEDs. J. Am. Chem. Soc., 2010. [DOI | PubMed]
- Q. Zhang, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing, F. Wang. Highly Efficient Green Phosphorescent Organic Light-Emitting Diodes Based on Cu(I) Complexes. Adv. Mater., 2004. [DOI]
- A. Tsuboyama, K. Kuge, M. Furugori, S. Okada, M. Hoshino, K. Ueno. Photophysical Properties of Highly Luminescent Copper(I) Halide Complexes Chelated with 1,2-Bis(Diphenylphosphino)Benzene. Inorg. Chem., 2007. [DOI | PubMed]
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki, C. Adachi. Efficient up-Conversion of Triplet Excitons into a Singlet State and Its Application for Organic Light Emitting Diodes. Appl. Phys. Lett., 2011. [DOI]
- F. B. Dias, T. J. Penfold, A. P. Monkman. Photophysics of Thermally Activated Delayed Fluorescence Molecules. Methods Appl. Fluoresc., 2017. [DOI | PubMed]
- M. Kasha. Characterization of Electronic Transitions in Complex Molecules. Discuss. Faraday Soc., 1950. [DOI]
- A. Niwa, T. Kobayashi, T. Nagase, K. Goushi, C. Adachi, H. Naito. Temperature Dependence of Photoluminescence Properties in a Thermally Activated Delayed Fluorescence Emitter. Appl. Phys. Lett., 2014. [DOI]
- S. K. Lower, M. A. El-Sayed. The Triplet State and Molecular Electronic Processes in Organic Molecules. Chem. Rev., 1966. [DOI]
- T. J. Penfold, E. Gindensperger, C. Daniel, C. M. Marian. Spin-Vibronic Mechanism for Intersystem Crossing. Chem. Rev., 2018. [DOI | PubMed]
- M. A. El-Sayed. The Triplet State: Its Radiative and Nonradiative Properties. Acc. Chem. Res., 1968. [DOI]
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold, A. P. Monkman. Revealing the Spin-Vibronic Coupling Mechanism of Thermally Activated Delayed Fluorescence. Nat. Commun., 2016. [DOI | PubMed]
- Z. R. Grabowski, K. Rotkiewicz, W. Rettig. Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures. Chem. Rev., 2003. [DOI | PubMed]
- Y. Olivier, B. Yurash, L. Muccioli, G. D’Avino, O. Mikhnenko, J. C. Sancho-García, C. Adachi, T. Q. Nguyen, D. Beljonne. Nature of the Singlet and Triplet Excitations Mediating Thermally Activated Delayed Fluorescence. Phys. Rev. Mater., 2017. [DOI]
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang, W. Huang. Thermally Activated Delayed Fluorescence Materials Towards the Breakthrough of Organoelectronics. Adv. Mater., 2014. [DOI | PubMed]
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi, M. P. Aldred. Recent Advances in Organic Thermally Activated Delayed Fluorescence Materials. Chem. Soc. Rev., 2017. [DOI | PubMed]
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro, C. Adachi. Evidence and Mechanism of Efficient Thermally Activated Delayed Fluorescence Promoted by Delocalized Excited States. Sci. Adv., 2017. [DOI | PubMed]
- E. Zysman-Colman. Molecular Designs Offer Fast Exciton Conversion. Nat. Photonics, 2020. [DOI]
- J. Gibson, A. P. Monkman, T. J. Penfold. The Importance of Vibronic Coupling for Efficient Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Molecules. ChemPhysChem, 2016. [DOI | PubMed]
- H. Noda, H. Nakanotani, C. Adachi. Excited State Engineering for Efficient Reverse Intersystem Crossing. Sci. Adv., 2018. [DOI | PubMed]
- P. L. dos Santos, J. S. Ward, D. G. Congrave, A. S. Batsanov, J. Eng, J. E. Stacey, T. J. Penfold, A. P. Monkman, M. R. Bryce. Triazatruxene: A Rigid Central Donor Unit for a D-A3 Thermally Activated Delayed Fluorescence Material Exhibiting Sub-Microsecond Reverse Intersystem Crossing and Unity Quantum Yield Via Multiple Singlet-Triplet State Pairs. Adv. Sci., 2018. [DOI]
- L.-S. Cui, A. J. Gillett, S.-F. Zhang, H. Ye, Y. Liu, X.-K. Chen, Z.-S. Lin, E. W. Evans, W. K. Myers, T. K. Ronson. Fast Spin-Flip Enables Efficient and Stable Organic Electroluminescence from Charge-Transfer States. Nat. Photonics, 2020. [DOI]
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias, M. R. Bryce. The Interplay of Thermally Activated Delayed Fluorescence (TADF) and Room Temperature Organic Phosphorescence in Sterically-Constrained Donor-Acceptor Charge-Transfer Molecules. Chem. Commun., 2016. [DOI]
- M. Hempe, N. A. Kukhta, A. Danos, M. A. Fox, A. S. Batsanov, A. P. Monkman, M. R. Bryce. Vibrational Damping Reveals Vibronic Coupling in Thermally Activated Delayed Fluorescence Materials. Chem. Mater., 2021. [DOI | PubMed]
- K. Masui, H. Nakanotani, C. Adachi. Analysis of Exciton Annihilation in High-Efficiency Sky-Blue Organic Light-Emitting Diodes with Thermally Activated Delayed Fluorescence. Org. Electron., 2013. [DOI]
- T. Serevicius, R. Skaisgiris, G. Kreiza, J. Dodonova, K. Kazlauskas, E. Orentas, S. Tumkevicius, S. Jursenas. TADF Parameters in the Solid State: An Easy Way to Draw Wrong Conclusions. J. Phys. Chem. A, 2021. [DOI | PubMed]
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka, H. Kaji. Organic Light Emitters Exhibiting Very Fast Reverse Intersystem Crossing. Nat. Photonics, 2020. [DOI]
- N. Haase, A. Danos, C. Pflumm, A. Morherr, P. Stachelek, A. Mekic, W. Brütting, A. P. Monkman. Kinetic Modeling of Transient Photoluminescence from Thermally Activated Delayed Fluorescence. J. Phys. Chem. C, 2018. [DOI]
- J. Grüne, N. Bunzmann, M. Meinecke, V. Dyakonov, A. Sperlich. Kinetic Modeling of Transient Electroluminescence Reveals TTA as an Efficiency-Limiting Process in Exciplex-Based TADF OLEDs. J. Phys. Chem. C, 2020. [DOI]
- S. Sem, S. Jenatsch, K. Stavrou, A. Danos, A. P. Monkman, B. Ruhstaller. Determining Non-Radiative Decay Rates in TADF Compounds Using Coupled Transient and Steady State Optical Data. J. Mater. Chem. C, 2022. [DOI]
- Y. Tsuchiya, S. Diesing, F. Bencheikh, Y. Wada, P. L. Dos Santos, H. Kaji, E. Zysman-Colman, I. D. W. Samuel, C. Adachi. Exact Solution of Kinetic Analysis for Thermally Activated Delayed Fluorescence Materials. J. Phys. Chem. A, 2021. [DOI | PubMed]
- B. Yurash, H. Nakanotani, Y. Olivier, D. Beljonne, C. Adachi, T. Q. Nguyen. Photoluminescence Quenching Probes Spin Conversion and Exciton Dynamics in Thermally Activated Delayed Fluorescence Materials. Adv. Mater., 2019. [DOI]
- N. Haase, A. Danos, C. Pflumm, P. Stachelek, W. Brutting, A. P. Monkman. Are the Rates of Dexter Transfer in TADF Hyperfluorescence Systems Optically Accessible?. Mater. Horiz., 2021. [DOI | PubMed]
- X. Yang, X. Xu, G. Zhou. Recent Advances of the Emitters for High Performance Deep-Blue Organic Light-Emitting Diodes. J. Mater. Chem. C, 2015. [DOI]
- X. Yang, G. Zhou, W.-Y. Wong. Functionalization of Phosphorescent Emitters and Their Host Materials by Main-Group Elements for Phosphorescent Organic Light-Emitting Devices. Chem. Soc. Rev., 2015. [DOI | PubMed]
- C. Sekine, Y. Tsubata, T. Yamada, M. Kitano, S. Doi. Recent Progress of High Performance Polymer OLED and OPV Materials for Organic Printed Electronics. Sci. Technol. Adv. Mater., 2014. [DOI | PubMed]
- H. B. Wu, J. H. Zou, F. Liu, L. Wang, A. Mikhailovsky, G. C. Bazan, W. Yang, Y. Cao. Efficient Single Active Layer Electrophosphorescent White Polymer Light-Emitting Diodes. Adv. Mater., 2008. [DOI]
- S. Gong, C. Yang, J. Qin. Efficient Phosphorescent Polymer Light-Emitting Diodes by Suppressing Triplet Energy Back Transfer. Chem. Soc. Rev., 2012. [DOI | PubMed]
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns, A. B. Holmes. Light-Emitting Diodes Based on Conjugated Polymers. Nature, 1990. [DOI]
- P. L. Burn, S. C. Lo, I. D. Samuel. The Development of Light-Emitting Dendrimers for Displays. Adv. Mater., 2007. [DOI]
- F. Tenopala-Carmona, O. S. Lee, E. Crovini, A. M. Neferu, C. Murawski, Y. Olivier, E. Zysman-Colman, M. C. Gather. Identification of the Key Parameters for Horizontal Transition Dipole Orientation in Fluorescent and TADF Organic Light-Emitting Diodes. Adv. Mater., 2021. [DOI | PubMed]
- T. D. Schmidt, T. Lampe, M. R. Daniel Sylvinson, P. I. Djurovich, M. E. Thompson, W. Brütting. Emitter Orientation as a Key Parameter in Organic Light-Emitting Diodes. Phys. Rev. Applied, 2017. [DOI]
- J. J. Burke, G. I. Stegeman, T. Tamir. Surface-Polariton-Like Waves Guided by Thin, Lossy Metal Films. Phys. Rev. B, 1986. [DOI]
- Non-Isotropic Emitter Orientation and Its Implications for Efficiency Analysis of Organic Light-Emitting Diodes.. Organic Photonics V;, 2012
- D. Yokoyama, A. Sakaguchi, M. Suzuki, C. Adachi. Horizontal Orientation of Linear-Shaped Organic Molecules Having Bulky Substituents in Neat and Doped Vacuum-Deposited Amorphous Films. Org. Electron., 2009. [DOI]
- T.-L. Wu, S.-H. Lo, Y.-C. Chang, M.-J. Huang, C.-H. Cheng. Steric Switching for Thermally Activated Delayed Fluorescence by Controlling the Dihedral Angles between Donor and Acceptor in Organoboron Emitters. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono, T. Ikuta. Ultrapure Blue Thermally Activated Delayed Fluorescence Molecules: Efficient HOMO-LUMO Separation by the Multiple Resonance Effect. Adv. Mater., 2016. [DOI | PubMed]
- N. A. Kukhta, H. F. Higginbotham, T. Matulaitis, A. Danos, A. N. Bismillah, N. Haase, M. K. Etherington, D. S. Yufit, P. R. McGonigal, J. V. Gražulevičius, A. P. Monkman. Revealing Resonance Effects and Intramolecular Dipole Interactions in the Positional Isomers of Benzonitrile-Core Thermally Activated Delayed Fluorescence Materials. J. Mater. Chem. C, 2019. [DOI]
- A. Danos, D. Gudeika, N. A. Kukhta, R. Lygaitis, M. Colella, H. F. Higginbotham, A. N. Bismillah, P. R. McGonigal, J. V. Grazulevicius, A. P. Monkman. Not the Sum of Their Parts: Understanding Multi-Donor Interactions in Symmetric and Asymmetric TADF Emitters. J. Mater. Chem. C, 2022. [DOI]
- G. Haykir, M. Aydemir, A. Danos, S. Gumus, G. Hizal, A. P. Monkman, F. Turksoy. Effects of Asymmetric Acceptor and Donor Positioning in Deep Blue Pyridyl-Sulfonyl Based TADF Emitters. Dyes Pigm., 2021. [DOI]
- M. Hempe, N. A. Kukhta, A. Danos, A. S. Batsanov, A. P. Monkman, M. R. Bryce. Intramolecular Hydrogen Bonding in Thermally Activated Delayed Fluorescence Emitters: Is There Evidence Beyond Reasonable Doubt?. J. Phys. Chem. Lett, 2022. [DOI | PubMed]
- D. Hall, P. Rajamalli, E. Duda, S. M. Suresh, F. Rodella, S. Bagnich, C. L. Carpenter-Warren, D. B. Cordes, A. M. Z. Slawin, P. Strohriegl. Substitution Effects on a New Pyridylbenzimidazole Acceptor for Thermally Activated Delayed Fluorescence and Their Use in Organic Light-Emitting Diodes. Adv. Optical Mater., 2021. [DOI]
- L. Salah, M. K. Etherington, A. Shuaib, A. Danos, A. A. Nazeer, B. Ghazal, A. Prlj, A. T. Turley, A. Mallick, P. R. McGonigal. Suppressing Dimer Formation by Increasing Conformational Freedom in Multi-Carbazole Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C, 2021. [DOI]
- M. K. Etherington, N. A. Kukhta, H. F. Higginbotham, A. Danos, A. N. Bismillah, D. R. Graves, P. R. McGonigal, N. Haase, A. Morherr, A. S. Batsanov. Persistent Dimer Emission in Thermally Activated Delayed Fluorescence Materials. J. Phys. Chem. C, 2019. [DOI]
- M. K. Etherington. Thermally Activated Delayed Fluorescence: Beyond the Single Molecule. Front. Chem., 2020. [DOI | PubMed]
- A. Dreuw, M. Head-Gordon. Single-Reference Ab Initio Methods for the Calculation of Excited States of Large Molecules. Chem. Rev., 2005. [DOI | PubMed]
- D. Giavazzi, F. Di Maiolo, A. Painelli. The Fate of Molecular Excited States: Modeling Donor-Acceptor Dyes. Phys. Chem. Chem. Phys., 2022. [DOI | PubMed]
- A. Carreras, D. Casanova. Theory of Exciton Dynamics in Thermally Activated Delayed Fluorescence. ChemPhotoChem, 2022. [DOI]
- T. J. Penfold, J. Eng. Tailoring Donor-Acceptor Emitters to Minimise Localisation Induced Quenching of Thermally Activated Delayed Fluorescence. ChemPhotoChem, 2023. [DOI]
- D. K. A. Phan Huu, S. Saseendran, R. Dhali, L. G. Franca, K. Stavrou, A. Monkman, A. Painelli. Thermally Activated Delayed Fluorescence: Polarity, Rigidity, and Disorder in Condensed Phases. J. Am. Chem. Soc., 2022. [DOI | PubMed]
- D. K. A. Phan Huu, S. Saseendran, A. Painelli. Effective Models for TADF: The Role of the Medium Polarizability. J. Mater. Chem. C, 2022. [DOI]
- T. J. Penfold. On Predicting the Excited-State Properties of Thermally Activated Delayed Fluorescence Emitters. J. Phys. Chem. C, 2015. [DOI]
- Y. Olivier, J. C. Sancho-Garcia, L. Muccioli, G. D’Avino, D. Beljonne. Computational Design of Thermally Activated Delayed Fluorescence Materials: The Challenges Ahead. J. Phys. Chem. Lett, 2018. [DOI | PubMed]
- A. J. Gillett, A. Pershin, R. Pandya, S. Feldmann, A. J. Sneyd, A. M. Alvertis, E. W. Evans, T. H. Thomas, L. S. Cui, B. H. Drummond. Dielectric Control of Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Emitters. Nat. Mater., 2022. [DOI | PubMed]
- R. Gómez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D. G. Ha, T. Wu. Design of Efficient Molecular Organic Light-Emitting Diodes by a High-Throughput Virtual Screening and Experimental Approach. Nat. Mater., 2016. [DOI | PubMed]
- A. Pershin, D. Hall, V. Lemaur, J. C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne, Y. Olivier. Highly Emissive Excitons with Reduced Exchange Energy in Thermally Activated Delayed Fluorescent Molecules. Nat. Commun., 2019. [DOI | PubMed]
- D. Hall, J. C. Sancho-García, A. Pershin, G. Ricci, D. Beljonne, E. Zysman-Colman, Y. Olivier. Modeling of Multiresonant Thermally Activated Delayed Fluorescence Emitters – Properly Accounting for Electron Correlation Is Key!. J. Chem. Theory Comput., 2022. [DOI | PubMed]
- H. Sun, C. Zhong, J.-L. Brédas. Reliable Prediction with Tuned Range-Separated Functionals of the Singlet-Triplet Gap in Organic Emitters for Thermally Activated Delayed Fluorescence. J. Chem. Theory Comput., 2015. [DOI | PubMed]
- D. Valverde, S. Mai, A. V. Sanches de Araujo, S. Canuto, L. Gonzalez, A. C. Borin. On the Population of Triplet States of 2-Seleno-Thymine. Phys. Chem. Chem. Phys., 2021. [DOI | PubMed]
- S. Hirata, M. Head-Gordon. Time-Dependent Density Functional Theory within the Tamm-Dancoff Approximation. Chem. Phys. Lett., 1999. [DOI]
- M. Moral, L. Muccioli, W. J. Son, Y. Olivier, J. C. Sancho-Garcia. Theoretical Rationalization of the Singlet-Triplet Gap in OLEDs Materials: Impact of Charge-Transfer Character. J Chem Theory Comput, 2015. [DOI | PubMed]
- J.-L. Brédas. Organic Electronics: Does a Plot of the Homo-Lumo Wave Functions Provide Useful Information?. Chem. Mater., 2017. [DOI]
- T. Le Bahers, C. Adamo, I. Ciofini. A Qualitative Index of Spatial Extent in Charge-Transfer Excitations. J. Chem. Theory Comput., 2011. [DOI | PubMed]
- M. J. Peach, P. Benfield, T. Helgaker, D. J. Tozer. Excitation Energies in Density Functional Theory: An Evaluation and a Diagnostic Test. J. Chem. Phys., 2008. [DOI | PubMed]
- M. J. G. Peach, D. J. Tozer. Illustration of a TDDFT Spatial Overlap Diagnostic by Basis Function Exponent Scaling. J. Mol. Struct. Theochem, 2009. [DOI]
- C. A. Guido, P. Cortona, B. Mennucci, C. Adamo. On the Metric of Charge Transfer Molecular Excitations: A Simple Chemical Descriptor. J. Chem. Theory Comput., 2013. [DOI | PubMed]
- D. Hall, J. C. Sancho-García, A. Pershin, D. Beljonne, E. Zysman-Colman, Y. Olivier. Benchmarking DFT Functionals for Excited-State Calculations of Donor-Acceptor TADF Emitters: Insights on the Key Parameters Determining Reverse Inter-System Crossing. J. Phys. Chem. A, 2023. [DOI | PubMed]
- D. Hall, J. C. Sancho-Garcia, A. Pershin, D. Beljonne, E. Zysman-Colman, Y. Olivier. Benchmarking DFT Functionals for Excited-State Calculations of Donor Acceptor TADF Emitters: Insights on the Key Parameters Determining Reverse Inter System Crossing. J. Phys. Chem. A, 2023. [DOI | PubMed]
- J. A. Knoller, G. Meng, X. Wang, D. Hall, A. Pershin, D. Beljonne, Y. Olivier, S. Laschat, E. Zysman-Colman, S. Wang. Intramolecular Borylation Via Sequential B-Mes Bond Cleavage for the Divergent Synthesis of B,N,B-Doped Benzo[4]Helicenes. Angew. Chem., Int. Ed., 2020. [DOI]
- M. Moral, L. Muccioli, W.-J. Son, Y. Olivier, J.-C. Sancho-Garcia. Theoretical Rationalization of the Singlet-Triplet Gap in OLEDs Materials: Impact of Charge-Transfer Character. J. Chem. Theory Comput., 2015. [DOI | PubMed]
- K. Lee, D. Kim. Local-Excitation Versus Charge-Transfer Characters in the Triplet State: Theoretical Insight into the Singlet-Triplet Energy Differences of Carbazolyl-Phthalonitrile-Based Thermally Activated Delayed Fluorescence Materials. J. Phys. Chem. C, 2016. [DOI]
- Y. Olivier, M. Moral, L. Muccioli, J.-C. Sancho-Garcia. Dynamic Nature of Excited States of Donor-Acceptor TADF Materials for OLEDs: How Theory Can Reveal Structure-Property Relationships. J. Mater. Chem. C, 2017. [DOI]
- P. K. Samanta, D. Kim, V. Coropceanu, J.-L. Brédas. Up-Conversion Intersystem Crossing Rates in Organic Emitters for Thermally Activated Delayed Fluorescence (TADF): Impact of the Nature of Singlet Vs. Triplet Excited States. J. Am. Chem. Soc., 2017. [DOI | PubMed]
- L. Kunze, A. Hansen, S. Grimme, J. M. Mewes. Pcm-Roks for the Description of Charge-Transfer States in Solution: Singlet-Triplet Gaps with Chemical Accuracy from Open-Shell Kohn-Sham Reaction-Field Calculations. J. Phys. Chem. Lett, 2021. [DOI | PubMed]
- T. Cardeynaels, S. Paredis, J. Deckers, S. Brebels, D. Vanderzande, W. Maes, B. Champagne. Finding the Optimal Exchange-Correlation Functional to Describe the Excited State Properties of Push-Pull Organic Dyes Designed for Thermally Activated Delayed Fluorescence. Phys. Chem. Chem. Phys., 2020. [DOI | PubMed]
- J. Eng, B. A. Laidlaw, T. J. Penfold. On the Geometry Dependence of Tuned-Range Separated Hybrid Functionals. J. Comput. Chem., 2019. [DOI | PubMed]
- R. Scholz, P. Kleine, R. Lygaitis, L. Popp, S. Lenk, M. K. Etherington, A. P. Monkman, S. Reineke. Investigation of Thermally Activated Delayed Fluorescence from a Donor-Acceptor Compound with Time-Resolved Fluorescence and Density Functional Theory Applying an Optimally Tuned Range-Separated Hybrid Functional. J. Phys. Chem. A, 2020. [DOI | PubMed]
- C. Y. Chan, S. Madayanad Suresh, Y. T. Lee, Y. Tsuchiya, T. Matulaitis, D. Hall, A. M. Z. Slawin, S. Warriner, D. Beljonne, Y. Olivier. Two Boron Atoms Versus One: High-Performance Deep-Blue Multi-Resonance Thermally Activated Delayed Fluorescence Emitters. Chem. Commun., 2022. [DOI]
- J. M. dos Santos, D. Sun, J. M. Moreno-Naranjo, D. Hall, F. Zinna, S. T. J. Ryan, W. Shi, T. Matulaitis, D. B. Cordes, A. M. Z. Slawin. An S-Shaped Double Helicene Showing Both Multi-Resonance Thermally Activated Delayed Fluorescence and Circularly Polarized Luminescence. J. Mater. Chem. C, 2022. [DOI]
- D. Hall, K. Stavrou, E. Duda, A. Danos, S. Bagnich, S. Warriner, A. M. Z. Slawin, D. Beljonne, A. Kohler, A. Monkman. Diindolocarbazole – Achieving Multiresonant Thermally Activated Delayed Fluorescence without the Need for Acceptor Units. Mater. Horiz., 2022. [DOI | PubMed]
- D. Hall, S. M. Suresh, P. L. dos Santos, E. Duda, S. Bagnich, A. Pershin, P. Rajamalli, D. B. Cordes, A. M. Z. Slawin, D. Beljonne. Improving Processability and Efficiency of Resonant TADF Emitters: A Design Strategy. Adv. Optical Mater., 2020. [DOI]
- G. Meng, L. Liu, z. He, D. Hall, X. Wang, T. Peng, X. Yin, P. Chen, D. Beljonne, Y. Olivier. Multi-Resonant Thermally Activated Delayed Fluorescence Emitters Based on Tetracoordinate Boron-Containing Pahs: Colour Tuning Based on the Nature of Chelate. Chem. Sci., 2022. [DOI | PubMed]
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bassler, D. Beljonne, M. Buck, Y. Olivier. A Deep Blue B,N-Doped Heptacene Emitter That Shows Both Thermally Activated Delayed Fluorescence and Delayed Fluorescence by Triplet-Triplet Annihilation. J. Am. Chem. Soc., 2020. [DOI | PubMed]
- S. Wu, A. Kumar Gupta, K. Yoshida, J. Gong, D. Hall, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel, E. Zysman-Colman. Highly Efficient Green and Red Narrowband Emissive Organic Light-Emitting Diodes Employing Multi-Resonant Thermally Activated Delayed Fluorescence Emitters. Angew. Chem., Int. Ed., 2022. [DOI]
- M. Karaman, A. Kumar Gupta, S. Madayanad Suresh, T. Matulaitis, L. Mardegan, D. Tordera, H. J. Bolink, S. Wu, S. Warriner, I. D. Samuel, E. Zysman-Colman. Ionic Multiresonant Thermally Activated Delayed Fluorescence Emitters for Light Emitting Electrochemical Cells. Beilstein J. Org. Chem., 2022. [DOI | PubMed]
- H. L. Lee, S. O. Jeon, I. Kim, S. C. Kim, J. Lim, J. Kim, S. Park, J. Chwae, W. J. Son, H. Choi, J. Y. Lee. Multiple-Resonance Extension and Spin-Vibronic-Coupling-Based Narrowband Blue Organic Fluorescence Emitters with over 30% Quantum Efficiency. Adv. Mater., 2022. [DOI]
- X. F. Luo, H. X. Ni, H. L. Ma, Z. Z. Qu, J. Wang, Y. X. Zheng, J. L. Zuo. Fused Π-Extended Multiple-Resonance Induced Thermally Activated Delayed Fluorescence Materials for High-Efficiency and Narrowband OLEDs with Low Efficiency Roll-Off. Adv. Optical Mater., 2022. [DOI]
- X. F. Luo, S. Q. Song, H. X. Ni, H. Ma, D. Yang, D. Ma, Y. X. Zheng, J. L. Zuo. Multiple-Resonance-Induced Thermally Activated Delayed Fluorescence Materials Based on Indolo[3,2,1-Jk]Carbazole with an Efficient Narrowband Pure-Green Electroluminescence. Angew. Chem., Int. Ed., 2022. [DOI]
- S. Oda, B. Kawakami, Y. Yamasaki, R. Matsumoto, M. Yoshioka, D. Fukushima, S. Nakatsuka, T. Hatakeyama. One-Shot Synthesis of Expanded Heterohelicene Exhibiting Narrowband Thermally Activated Delayed Fluorescence. J. Am. Chem. Soc., 2022. [DOI | PubMed]
- S. Oda, T. Sugitani, H. Tanaka, K. Tabata, R. Kawasumi, T. Hatakeyama. Development of Pure Green Thermally Activated Delayed Fluorescence Material by Cyano Substitution. Adv. Mater., 2022. [DOI]
- V. V. Patil, H. L. Lee, I. Kim, K. H. Lee, W. J. Chung, J. Kim, S. Park, H. Choi, W. J. Son, S. O. Jeon, J. Y. Lee. Purely Spin-Vibronic Coupling Assisted Triplet to Singlet up-Conversion for Real Deep Blue Organic Light-Emitting Diodes with over 20% Efficiency and Y Color Coordinate of 0.05. Adv. Sci., 2021. [DOI]
- H. Tanaka, S. Oda, G. Ricci, H. Gotoh, K. Tabata, R. Kawasumi, D. Beljonne, Y. Olivier, T. Hatakeyama. Hypsochromic Shift of Multiple-Resonance-Induced Thermally Activated Delayed Fluorescence by Oxygen Atom Incorporation. Angew. Chem., Int. Ed., 2021. [DOI]
- S. Uemura, S. Oda, M. Hayakawa, R. Kawasumi, N. Ikeda, Y. T. Lee, C. Y. Chan, Y. Tsuchiya, C. Adachi, T. Hatakeyama. Sequential Multiple Borylation toward an Ultrapure Green Thermally Activated Delayed Fluorescence Material. J. Am. Chem. Soc., 2023. [DOI | PubMed]
- X. Wang, Y. Zhang, H. Dai, G. Li, M. Liu, G. Meng, X. Zeng, T. Huang, L. Wang, Q. Peng. Mesityl-Functionalized Multi-Resonance Organoboron Delayed Fluorescent Frameworks with Wide-Range Color Tunability for Narrowband OLEDs. Angew. Chem., Int. Ed., 2022. [DOI]
- F. Liu, Z. Cheng, Y. Jiang, L. Gao, H. Liu, H. Liu, Z. Feng, P. Lu, W. Yang. Highly Efficient Asymmetric Multiple Resonance Thermally Activated Delayed Fluorescence Emitter with EQE of 32.8% and Extremely Low Efficiency Roll-Off. Angew. Chem., Int. Ed., 2022. [DOI]
- Y. Zhang, D. Zhang, T. Huang, A. J. Gillett, Y. Liu, D. Hu, L. Cui, Z. Bin, G. Li, J. Wei, L. Duan. Multi-Resonance Deep-Red Emitters with Shallow Potential-Energy Surfaces to Surpass Energy-Gap Law*. Angew. Chem., Int. Ed., 2021. [DOI]
- S. M. Pratik, V. Coropceanu, J.-L. Brédas. Purely Organic Emitters for Multiresonant Thermally Activated Delay Fluorescence: Design of Highly Efficient Sulfur and Selenium Derivatives. ACS Mater. Lett., 2022. [DOI]
- N. Ikeda, S. Oda, R. Matsumoto, M. Yoshioka, D. Fukushima, K. Yoshiura, N. Yasuda, T. Hatakeyama. Solution-Processable Pure Green Thermally Activated Delayed Fluorescence Emitter Based on the Multiple Resonance Effect. Adv. Mater., 2020. [DOI | PubMed]
- Y. X. Hu, J. Miao, T. Hua, Z. Huang, Y. Qi, Y. Zou, Y. Qiu, H. Xia, H. Liu, X. Cao, C. Yang. Efficient Selenium-Integrated TADF OLEDs with Reduced Roll-Off. Nat. Photonics, 2022. [DOI]
- W. Sotoyama. Simulation of Low-Lying Singlet and Triplet Excited States of Multiple-Resonance-Type Thermally Activated Delayed Fluorescence Emitters by Delta Self-Consistent Field (Deltascf) Method. J. Phys. Chem. A, 2021. [DOI | PubMed]
- K. L. Woon, P. A. Nikishau, G. Sini. Fast and Accurate Determination of the Singlet-Triplet Gap in Donor-Acceptor and Multiresonance TADF Molecules by Using Hole-Hole Tamm-Dancoff Approximated Density Functional Theory. Adv. Theory Simul., 2022. [DOI]
- N. Aizawa, A. Matsumoto, T. Yasuda. Thermal Equilibration between Singlet and Triplet Excited States in Organic Fluorophore for Submicrosecond Delayed Fluorescence. Sci. Adv., 2021. [DOI | PubMed]
- J. M. Kaminski, A. Rodriguez-Serrano, F. Dinkelbach, H. Miranda-Salinas, A. P. Monkman, C. M. Marian. Vibronic Effects Accelerate the Intersystem Crossing Processes of the through-Space Charge Transfer States in the Triptycene Bridged Acridine-Triazine Donor-Acceptor Molecule Tpat-Tffo. Chem. Sci., 2022. [DOI | PubMed]
- N. Sharma, M. Y. Wong, D. Hall, E. Spuling, F. Tenopala-Carmona, A. Privitera, G. Copley, D. B. Cordes, A. M. Z. Slawin, C. Murawski. Exciton Efficiency Beyond the Spin Statistical Limit in Organic Light Emitting Diodes Based on Anthracene Derivatives. J. Mater. Chem. C, 2020. [DOI]
- M. Chapran, P. Pander, M. Vasylieva, G. Wiosna-Salyga, J. Ulanski, F. B. Dias, P. Data. Realizing 20% External Quantum Efficiency in Electroluminescence with Efficient Thermally Activated Delayed Fluorescence from an Exciplex. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- A. J. Gillett, C. Tonnele, G. Londi, G. Ricci, M. Catherin, D. M. L. Unson, D. Casanova, F. Castet, Y. Olivier, W. M. Chen. Spontaneous Exciton Dissociation Enables Spin State Interconversion in Delayed Fluorescence Organic Semiconductors. Nat. Commun., 2021. [DOI | PubMed]
- I. Kim, K. H. Cho, S. O. Jeon, W. J. Son, D. Kim, Y. M. Rhee, I. Jang, H. Choi, D. S. Kim. Three States Involving Vibronic Resonance Is a Key to Enhancing Reverse Intersystem Crossing Dynamics of an Organoboron-Based Ultrapure Blue Emitter. JACS Au, 2021. [DOI | PubMed]
- K. Shizu, H. Kaji. Comprehensive Understanding of Multiple Resonance Thermally Activated Delayed Fluorescence through Quantum Chemistry Calculations. Commun. Chem., 2022. [DOI | PubMed]
- L. Lin, J. Fan, L. Cai, C.-K. Wang. Excited State Dynamics of New-Type Thermally Activated Delayed Fluorescence Emitters: Theoretical View of Light-Emitting Mechanism. Mol. Phys., 2018. [DOI]
- S. M. Pratik, V. Coropceanu, J.-L. Brédas. Enhancement of Thermally Activated Delayed Fluorescence (TADF) in Multi-Resonant Emitters Via Control of Chalcogen Atom Embedding. Chem. Mater., 2022. [DOI]
- C. M. Marian. Mechanism of the Triplet-to-Singlet Upconversion in the Assistant Dopant Acrxtn. J. Phys. Chem. C, 2016. [DOI]
- H. Noda, X.-K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J.-L. Brédas, C. Adachi. Critical Role of Intermediate Electronic States for Spin-Flip Processes in Charge-Transfer-Type Organic Molecules with Multiple Donors and Acceptors. Nat. Mater., 2019. [DOI | PubMed]
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti. High-Performance Light-Emitting Diodes Based on Carbene-Metal-Amides. Science, 2017. [DOI | PubMed]
- J. Foller, C. M. Marian. Rotationally Assisted Spin-State Inversion in Carbene-Metal-Amides Is an Artifact. J. Phys. Chem. Lett, 2017. [DOI | PubMed]
- E. J. Taffet, Y. Olivier, F. Lam, D. Beljonne, G. D. Scholes. Carbene-Metal-Amide Bond Deformation, Rather Than Ligand Rotation, Drives Delayed Fluorescence. J. Phys. Chem. Lett, 2018. [DOI | PubMed]
- R. Dhali, D. K. A. Phan Huu, F. Terenziani, C. Sissa, A. Painelli. Thermally Activated Delayed Fluorescence: A Critical Assessment of Environmental Effects on the Singlet-Triplet Energy Gap. J. Chem. Phys., 2021. [DOI | PubMed]
- E. W. Evans, Y. Olivier, Y. Puttisong, W. K. Myers, T. J. H. Hele, S. M. Menke, T. H. Thomas, D. Credgington, D. Beljonne, R. H. Friend, N. C. Greenham. Vibrationally Assisted Intersystem Crossing in Benchmark Thermally Activated Delayed Fluorescence Molecules. J. Phys. Chem. Lett, 2018. [DOI | PubMed]
- G. D’Avino, L. Muccioli, C. Zannoni, D. Beljonne, Z. G. Soos. Electronic Polarization in Organic Crystals: A Comparative Study of Induced Dipoles and Intramolecular Charge Redistribution Schemes. J. Chem. Theory Comput., 2014. [DOI | PubMed]
- S. Verlaak, P. Heremans. Molecular Microelectrostatic View on Electronic States near Pentacene Grain Boundaries. Phys. Rev. B, 2007. [DOI]
- T. Northey, J. E. Stacey, T. J. Penfold. The Role of Solid State Solvation on the Charge Transfer State of a Thermally Activated Delayed Fluorescence Emitter. J. Mater. Chem. C, 2017. [DOI]
- J. Fan, L. Lin, C.-K. Wang. Excited State Properties of Non-Doped Thermally Activated Delayed Fluorescence Emitters with Aggregation-Induced Emission: A Qm/Mm Study. J. Mater. Chem. C, 2017. [DOI]
- X. Qiu, G. Tian, C. Lin, Y. Pan, X. Ye, B. Wang, D. Ma, D. Hu, Y. Luo, Y. Ma. Narrowband Emission from Organic Fluorescent Emitters with Dominant Low-Frequency Vibronic Coupling. Adv. Optical Mater., 2021. [DOI]
- J. Cerezo, F. Santoro. Fcclasses3: Vibrationally-Resolved Spectra Simulated at the Edge of the Harmonic Approximation. J. Comput. Chem., 2023. [DOI | PubMed]
- F. Santoro, A. Lami, R. Improta, J. Bloino, V. Barone. Effective Method for the Computation of Optical Spectra of Large Molecules at Finite Temperature Including the Duschinsky and Herzberg-Teller Effect: The Qx Band of Porphyrin as a Case Study. J. Chem. Phys., 2008. [DOI | PubMed]
- G. Ricci, J.-C. Sancho-García, Y. Olivier. Establishing Design Strategies for Emissive Materials with an Inverted Singlet-Triplet Energy Gap (Invest): A Computational Perspective on How Symmetry Rules the Interplay between Triplet Harvesting and Light Emission. J. Mater. Chem. C, 2022. [DOI]
- E. Di Donato, D. Vanzo, M. Semeraro, A. Credi, F. Negri. Tuning Fluorescence Lifetimes through Changes in Herzberg-Teller Activities: The Case of Triphenylene and Its Hexamethoxy-Substituted Derivative. J. Phys. Chem. A, 2009. [DOI | PubMed]
- J.-L. Brédas, D. Beljonne, V. Coropceanu, J. Cornil. Charge-Transfer and Energy-Transfer Processes in Π-Conjugated Oligomers and Polymers: A Molecular Picture. Chem. Rev., 2004. [DOI | PubMed]
- Z. Pei, Q. Ou, Y. Mao, J. Yang, A. Lande, F. Plasser, W. Liang, Z. Shuai, Y. Shao. Elucidating the Electronic Structure of a Delayed Fluorescence Emitter Via Orbital Interactions, Excitation Energy Components, Charge-Transfer Numbers, and Vibrational Reorganization Energies. J. Phys. Chem. Lett, 2021. [DOI | PubMed]
- Y. Xu, C. Li, Z. Li, Q. Wang, X. Cai, J. Wei, Y. Wang. Constructing Charge-Transfer Excited States Based on Frontier Molecular Orbital Engineering: Narrowband Green Electroluminescence with High Color Purity and Efficiency. Angew. Chem., Int. Ed., 2020. [DOI]
- S. Madayanad Suresh, L. Zhang, D. Hall, C. Si, G. Ricci, T. Matulaitis, A. M. Z. Slawin, S. Warriner, Y. Olivier, I. D. W. Samuel, E. Zysman-Colman. A Deep-Blue-Emitting Heteroatom-Doped MR-TADF Nonacene for High-Performance Organic Light-Emitting Diodes. Angew. Chem., Int. Ed., 2023. [DOI]
- J. Gibson, T. Penfold. Nonadiabatic Coupling Reduces the Activation Energy in Thermally Activated Delayed Fluorescence. Phys. Chem. Chem. Phys., 2017. [DOI | PubMed]
- T. Northey, T. J. Penfold. The Intersystem Crossing Mechanism of an Ultrapure Blue Organoboron Emitter. Org. Electron., 2018. [DOI]
- Y. Shu, B. G. Levine. Simulated Evolution of Fluorophores for Light Emitting Diodes. J. Chem. Phys., 2015. [DOI | PubMed]
- M. H. Lee. Identification of Host-Guest Systems in Green TADF-Based OLEDs with Energy Level Matching Based on a Machine-Learning Study. Phys. Chem. Chem. Phys., 2020. [DOI | PubMed]
- H. Shi, W. Jing, W. Liu, Y. Li, Z. Li, B. Qiao, S. Zhao, Z. Xu, D. Song. Key Factors Governing the External Quantum Efficiency of Thermally Activated Delayed Fluorescence Organic Light-Emitting Devices: Evidence from Machine Learning. ACS Omega, 2022. [DOI | PubMed]
- K. H. Lin, G. A. H. Wetzelaer, P. W. M. Blom, D. Andrienko. Virtual Screening of TADF Emitters for Single-Layer OLEDs. Front. Chem., 2021. [DOI | PubMed]
- N. C. Forero-Martinez, K. H. Lin, K. Kremer, D. Andrienko. Virtual Screening for Organic Solar Cells and Light Emitting Diodes. Adv. Sci., 2022. [DOI]
- K. Zhao, Ö. H. Omar, T. Nematiaram, D. Padula, A. Troisi. Novel Thermally Activated Delayed Fluorescence Materials by High-Throughput Virtual Screening: Going Beyond Donor-Acceptor Design. J. Mater. Chem. C, 2021. [DOI]
- Y. Zhu, S. Vela, H. Meng, C. Corminboeuf, M. Fumanal. Donor-Acceptor-Donor “Hot Exciton” Triads for High Reverse Intersystem Crossing in OLEDs. Adv. Optical Mater., 2022. [DOI]
- Z. Tan, Y. Li, Z. Zhang, X. Wu, T. Penfold, W. Shi, S. Yang. Efficient Adversarial Generation of Thermally Activated Delayed Fluorescence Molecules. ACS Omega, 2022. [DOI | PubMed]
- Itu-R Bt.2020-2, ed.; International Telecommunications Union: Geneva, 2015, https://www.itu.int/rec/R-REC-BT.2020-2-201510-I/en (accessed 26-07-2023).
- J.-H. Lee, C.-H. Chen, P.-H. Lee, H.-Y. Lin, M.-k. Leung, T.-L. Chiu, C.-F. Lin. Blue Organic Light-Emitting Diodes: Current Status, Challenge, and Future Outlook. J. Mater. Chem. C, 2019. [DOI]
- X. Cai, S. J. Su. Marching toward Highly Efficient, Pure-Blue, and Stable Thermally Activated Delayed Fluorescent Organic Light-Emitting Diodes. Adv. Funct. Mater., 2018. [DOI]
- T. T. Bui, F. Goubard, M. Ibrahim-Ouali, D. Gigmes, F. Dumur. Recent Advances on Organic Blue Thermally Activated Delayed Fluorescence (TADF) Emitters for Organic Light-Emitting Diodes (OLEDs). Beilstein J. Org. Chem., 2018. [DOI | PubMed]
- H. Lee, D. Karthik, R. Lampande, J. H. Ryu, J. H. Kwon. Recent Advancement in Boron-Based Efficient and Pure Blue Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes. Front. Chem., 2020. [DOI | PubMed]
- C. Yin, D. Zhang, L. Duan. A Perspective on Blue TADF Materials Based on Carbazole-Benzonitrile Derivatives for Efficient and Stable OLEDs. Appl. Phys. Lett., 2020. [DOI]
- S. S. Kothavale, J. Y. Lee. Three- and Four-Coordinate, Boron-Based, Thermally Activated Delayed Fluorescent Emitters. Adv. Optical Mater., 2020. [DOI]
- J. W. Sun, K. H. Kim, C. K. Moon, J. H. Lee, J. J. Kim. Highly Efficient Sky-Blue Fluorescent Organic Light Emitting Diode Based on Mixed Cohost System for Thermally Activated Delayed Fluorescence Emitter (2czpn). ACS Appl. Mater. Interfaces, 2016. [DOI | PubMed]
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki, C. Adachi. Design of Efficient Thermally Activated Delayed Fluorescence Materials for Pure Blue Organic Light Emitting Diodes. J. Am. Chem. Soc., 2012. [DOI | PubMed]
- W.-L. Tsai, M.-H. Huang, W.-K. Lee, Y.-J. Hsu, K.-C. Pan, Y.-H. Huang, H.-C. Ting, M. Sarma, Y.-Y. Ho, H.-C. Hu. A Versatile Thermally Activated Delayed Fluorescence Emitter for Both Highly Efficient Doped and Non-Doped Organic Light Emitting Devices. Chem. Commun., 2015. [DOI]
- H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta, T. Hatakeyama. One-Step Borylation of 1,3-Diaryloxybenzenes Towards Efficient Materials for Organic Light-Emitting Diodes. Angew. Chem., Int. Ed., 2015. [DOI]
- H. Fukagawa, T. Shimizu, Y. Iwasaki, T. Yamamoto. Operational Lifetimes of Organic Light-Emitting Diodes Dominated by Förster Resonance Energy Transfer. Sci. Rep., 2017. [DOI | PubMed]
- S. Y. Byeon, J. Kim, D. R. Lee, S. H. Han, S. R. Forrest, J. Y. Lee. Nearly 100% Horizontal Dipole Orientation and Upconversion Efficiency in Blue Thermally Activated Delayed Fluorescent Emitters. Adv. Optical Mater., 2018. [DOI]
- H. Nakanotani, K. Masui, J. Nishide, T. Shibata, C. Adachi. Promising Operational Stability of High-Efficiency Organic Light-Emitting Diodes Based on Thermally Activated Delayed Fluorescence. Sci. Rep., 2013. [DOI | PubMed]
- N. Sharma, E. Spuling, C. M. Mattern, W. Li, O. Fuhr, Y. Tsuchiya, C. Adachi, S. Bräse, I. D. W. Samuel, E. Zysman-Colman. Turn on of Sky-Blue Thermally Activated Delayed Fluorescence and Circularly Polarized Luminescence (CPL) Via Increased Torsion by a Bulky Carbazolophane Donor. Chem. Sci., 2019. [DOI | PubMed]
- B. Liu, J. Li, D. Liu, Y. Mei, Y. Lan, K. Song, Y. Li, J. Wang. Electron-Withdrawing Bulky Group Substituted Carbazoles for Blue TADF Emitters: Simultaneous Improvement of Blue Color Purity and RISC Rate Constants. Dyes Pigm., 2022. [DOI]
- Q. Feng, K. Tan, X. Zheng, S. Xie, K. Xue, Y. Bo, H. Zhang, D. Lin, J. Rao, X. Xie. Simultaneous and Significant Improvements in Efficiency and Stability of Deep-Blue Organic Light Emitting Diodes through Friedel-Crafts Arylmethylation of a Fluorophore. ChemPhotoChem, 2020. [DOI]
- S. Xiang, X. Lv, S. Sun, Q. Zhang, Z. Huang, R. Guo, H. Gu, S. Liu, L. Wang. To Improve the Efficiency of Thermally Activated Delayed Fluorescence OLEDs by Controlling the Horizontal Orientation through Optimizing Stereoscopic and Linear Structures of Indolocarbazole Isomers. J. Mater. Chem. C, 2018. [DOI]
- X. Lv, R. Huang, S. Sun, Q. Zhang, S. Xiang, S. Ye, P. Leng, F. B. Dias, L. Wang. Blue TADF Emitters Based on Indenocarbazole Derivatives with High Photoluminescence and Electroluminescence Efficiencies. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- D. J. Shin, J. Lim, J. Y. Lee. Combinatorial Donor Engineering for Highly Efficient Blue Thermally Activated Delayed Fluorescence Emitters with Low Efficiency Roll-Off. J. Mater. Chem. C, 2021. [DOI]
- W.-W. Tao, K. Wang, J.-X. Chen, Y.-Z. Shi, W. Liu, C.-J. Zheng, Y.-Q. Li, J. Yu, X.-M. Ou, X.-H. Zhang. Dibenzofuran/Dibenzothiophene as the Secondary Electron-Donors for Highly Efficient Blue Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C, 2019. [DOI]
- D.-H. Wang, J.-F. Yao, H.-Z. Li, G.-Z. Li, F.-M. Xie, Y.-Q. Li, Y.-Y. Hu, J.-X. Tang, X. Zhao. New Strategy for Developing Deep Blue TADF Materials with Narrow Emission and Ciey < 0.06 Employing Host Materials as Donors. New J. Chem., 2023. [DOI]
- H. L. Lee, K. H. Lee, J. Y. Lee, W. P. Hong. Management of Thermally Activated Delayed Fluorescence Using a Secondary Electron Accepting Unit in Thermally Activated Delayed Fluorescent Emitters. J. Mater. Chem. C, 2019. [DOI]
- J. H. Yun, K. H. Lee, J. Y. Lee. Propeller Type Dibenzofurocarbazole as a New Rigid Donor Moiety for Highly Efficient and Long Living Thermally Activated Delayed Fluorescence Emitters. Chem. Eng. J., 2020. [DOI]
- M. Jung, K. H. Lee, J. Y. Lee. Molecular Engineering of Isomeric Benzofurocarbazole Donors for Photophysical Management of Thermally Activated Delayed Fluorescence Emitters. Chem. Eur. J., 2020. [DOI | PubMed]
- M. M. Raikwar, S. C. Kim, J. Y. Lee. Highly Efficient Thermally Activated Delayed Fluorescence Emitter Based on the 5h-Benzo[D]Benzo[4,5]Imidazo[1,2-a]Imidazole Donor. Mater. Chem. Front., 2022. [DOI]
- L. S. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani, C. Adachi. Controlling Singlet-Triplet Energy Splitting for Deep-Blue Thermally Activated Delayed Fluorescence Emitters. Angew. Chem., Int. Ed., 2017. [DOI]
- W. Huang, M. Einzinger, T. Zhu, H. S. Chae, S. Jeon, S.-G. Ihn, M. Sim, S. Kim, M. Su, G. Teverovskiy. Molecular Design of Deep Blue Thermally Activated Delayed Fluorescence Materials Employing a Homoconjugative Triptycene Scaffold and Dihedral Angle Tuning. Chem. Mater., 2018. [DOI]
- W. Huang, M. Einzinger, A. Maurano, T. Zhu, J. Tiepelt, C. Yu, H. S. Chae, T. Van Voorhis, M. A. Baldo, S. L. Buchwald. Large Increase in External Quantum Efficiency by Dihedral Angle Tuning in a Sky-Blue Thermally Activated Delayed Fluorescence Emitter. Adv. Optical Mater., 2019. [DOI]
- Q. Wang, Y.-X. Zhang, Y. Yuan, Y. Hu, Q.-S. Tian, Z.-Q. Jiang, L.-S. Liao. Alleviating Efficiency Roll-Off of Hybrid Single-Emitting Layer WOLED Utilizing Bipolar TADF Material as Host and Emitter. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- H. Min, I. S. Park, T. Yasuda. Dipolar and Quadrupolar Luminophores Based on 1,8-Dimethylcarbazole-Triazine Conjugates for High-Efficiency Blue Thermally Activated Delayed Fluorescence OLEDs. ChemPhotoChem, 2020. [DOI]
- S. O. Jeon, K. H. Lee, J. S. Kim, S.-G. Ihn, Y. S. Chung, J. W. Kim, H. Lee, S. Kim, H. Choi, J. Y. Lee. High-Efficiency, Long-Lifetime Deep-Blue Organic Light-Emitting Diodes. Nat. Photonics, 2021. [DOI]
- W. Li, X. Cai, B. Li, L. Gan, Y. He, K. Liu, D. Chen, Y.-C. Wu, S.-J. Su. Adamantane-Substituted Acridine Donor for Blue Dual Fluorescence and Efficient Organic Light-Emitting Diodes. Angew. Chem., Int. Ed., 2019. [DOI]
- S. J. Woo, Y. Kim, S. K. Kwon, Y. H. Kim, J. J. Kim. Phenazasiline/Spiroacridine Donor Combined with Methyl-Substituted Linkers for Efficient Deep Blue Thermally Activated Delayed Fluorescence Emitters. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- J. W. Sun, J. Y. Baek, K.-H. Kim, C.-K. Moon, J.-H. Lee, S.-K. Kwon, Y.-H. Kim, J.-J. Kim. Thermally Activated Delayed Fluorescence from Azasiline Based Intramolecular Charge-Transfer Emitter (Dtpdda) and a Highly Efficient Blue Light Emitting Diode. Chem. Mater., 2015. [DOI]
- J. W. Sun, J. Y. Baek, K. H. Kim, J. S. Huh, S. K. Kwon, Y. H. Kim, J. J. Kim. Azasiline-Based Thermally Activated Delayed Fluorescence Emitters for Blue Organic Light Emitting Diodes. J. Mater. Chem. C, 2017. [DOI]
- S.-J. Woo, Y. Kim, Y.-H. Kim, S.-K. Kwon, J.-J. Kim. A Spiro-Silafluorene-Phenazasiline Donor-Based Efficient Blue Thermally Activated Delayed Fluorescence Emitter and Its Host-Dependent Device Characteristics. J. Mater. Chem. C, 2019. [DOI]
- W. Li, B. Li, X. Cai, L. Gan, Z. Xu, W. Li, K. Liu, D. Chen, S. J. Su. Tri-Spiral Donor for High Efficiency and Versatile Blue Thermally Activated Delayed Fluorescence Materials. Angew. Chem., Int. Ed., 2019. [DOI]
- W. Li, W. Li, L. Gan, M. Li, N. Zheng, C. Ning, D. Chen, Y. C. Wu, S. J. Su. J-Aggregation Enhances the Electroluminescence Performance of a Sky-Blue Thermally Activated Delayed-Fluorescence Emitter in Nondoped Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- Y. Lee, S.-J. Woo, J.-J. Kim, J.-I. Hong. Blue Thermally Activated Delayed Fluorescence Emitter Using Modulated Triazines as Electron Acceptors. Dyes Pigm., 2020. [DOI]
- H. Kang, S.-G. Ihn, I. Kim, Y. S. Chung, S. O. Jeon, M. Sim, J. Kim, H. Lee, Y. Son, W.-J. Son. Designing Stable Deep-Blue Thermally Activated Delayed Fluorescence Emitters through Controlling the Intrinsic Stability of Triplet Excitons. Adv. Optical Mater., 2022. [DOI]
- Y. Wada, S. Kubo, H. Kaji. Adamantyl Substitution Strategy for Realizing Solution-Processable Thermally Stable Deep-Blue Thermally Activated Delayed Fluorescence Materials. Adv. Mater., 2018. [DOI]
- X. Fan, C. Li, Z. Wang, Y. Wei, C. Duan, C. Han, H. Xu. Enhancing Reverse Intersystem Crossing Via Secondary Acceptors: Toward Sky-Blue Fluorescent Diodes with 10-Fold Improved External Quantum Efficiency. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- C. S. Oh, D. D. S. Pereira, S. H. Han, H.-J. Park, H. F. Higginbotham, A. P. Monkman, J. Y. Lee. Dihedral Angle Control of Blue Thermally-Activated Delayed Fluorescent Emitters through Donor Substitution Position for Efficient Reverse Intersystem Crossing. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- C. S. Oh, H. L. Lee, S. H. Han, J. Y. Lee. Rational Molecular Design Overcoming the Long Delayed Fluorescence Lifetime and Serious Efficiency Roll-Off in Blue Thermally Activated Delayed Fluorescent Devices. Chem. Eur. J., 2019. [DOI | PubMed]
- J.-R. Cha, C. W. Lee, M.-S. Gong. Effect of Increasing Electron Donor Units for High-Efficiency Blue Thermally Activated Delayed Fluorescence. Dyes Pigm., 2017. [DOI]
- G. H. Kim, R. Lampande, J. B. Im, J. M. Lee, J. Y. Lee, J. H. Kwon. Controlling the Exciton Lifetime of Blue Thermally Activated Delayed Fluorescence Emitters Using a Heteroatom-Containing Pyridoindole Donor Moiety. Mater. Horiz., 2017. [DOI]
- K. M. Youn, D. H. Ahn, G. H. Kim, D. Karthik, J. Y. Lee, J. H. Kwon. Blue Thermally Activated Delayed Fluorescence Emitters with a Δ-Pyridoindole Donor Moiety. New J. Chem., 2018. [DOI]
- D. R. Lee, S. H. Han, C. W. Lee, J. Y. Lee. Bis(Diphenyltriazine) as a New Acceptor of Efficient Thermally Activated Delayed Fluorescent Emitters. Dyes Pigm., 2018. [DOI]
- H. L. Lee, K. H. Lee, J. Y. Lee. Transformation from Nonthermally Activated Delayed Fluorescence Molecules to Thermally Activated Delayed Fluorescence Molecules. Adv. Optical Mater., 2020. [DOI]
- C. Y. Lin, C. H. Lu, K. H. Kuo, M. Wang, Y. Tang, Y. Dou, B. Hu, C. C. Wu, K. T. Wong. Highly Efficient Blue Thermally Activated Delayed Fluorescence Emitters with a Triphenylamine-Based Macrocyclic Donor. Adv. Optical Mater., 2023. [DOI]
- S. K. Pathak, Y. Xiang, M. Huang, T. Huang, X. Cao, H. Liu, G. Xie, C. Yang. Fused Tetracyclic Tris[1,2,4]Triazolo[1,3,5]Triazine as a Novel Rigid Electron Acceptor for Efficient Thermally Activated Delayed Fluorescence Emitters. RSC Adv., 2020. [DOI | PubMed]
- S. Wang, X. Wang, K. H. Lee, S. Liu, J. Y. Lee, W. Zhu, Y. Wang. Blue Thermally Activated Delayed Fluorescence Based on Tristriazolotriazine Core: Synthesis, Property and the Application for Solution-Processed OLEDs. Dyes Pigm., 2020. [DOI]
- F. Hundemer, E. Crovini, Y. Wada, H. Kaji, S. Bräse, E. Zysman-Colman. Tris(Triazolo)Triazine-Based Emitters for Solution-Processed Blue Thermally Activated Delayed Fluorescence Organic Light-Emitting Diodes. Mater. Adv., 2020. [DOI]
- R. Cristiano, H. Gallardo, A. J. Bortoluzzi, I. H. Bechtold, C. E. Campos, R. L. Longo. Tristriazolotriazines: A Core for Luminescent Discotic Liquid Crystals. Chem. Commun., 2008. [DOI]
- Z. Fang, S. Wang, J. Liao, X. Chen, Y. Zhu, W. Zhu, Y. Wang. Asymmetric Sky-Blue Thermally-Activated Delayed Fluorescence Emitters Bearing Tris(Triazolo)Triazine Moiety for Solution-Processable Organic Light-Emitting Diodes. J. Mater. Chem. C, 2022. [DOI]
- S. Krotkus, T. Matulaitis, S. Diesing, G. Copley, E. Archer, C. Keum, D. B. Cordes, A. M. Z. Slawin, M. C. Gather, E. Zysman-Colman, I. D. W. Samuel. Fast Delayed Emission in New Pyridazine-Based Compounds. Front. Chem., 2020. [DOI | PubMed]
- R. Komatsu, T. Ohsawa, H. Sasabe, K. Nakao, Y. Hayasaka, J. Kido. Manipulating the Electronic Excited State Energies of Pyrimidine-Based Thermally Activated Delayed Fluorescence Emitters to Realize Efficient Deep-Blue Emission. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- T. Serevičius, J. Dodonova, R. Skaisgiris, D. Banevičius, K. Kazlauskas, S. Juršénas, S. Tumkevičius. Optimization of the Carbazole-Pyrimidine Linking Pattern for Achieving Efficient TADF. J. Mater. Chem. C, 2020. [DOI]
- B. Li, Z. Li, T. Hu, Y. Zhang, Y. Wang, Y. Yi, F. Guo, L. Zhao. Highly Efficient Blue Organic Light-Emitting Diodes from Pyrimidine-Based Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C, 2018. [DOI]
- I. S. Park, H. Komiyama, T. Yasuda. Pyrimidine-Based Twisted Donor-Acceptor Delayed Fluorescence Molecules: A New Universal Platform for Highly Efficient Blue Electroluminescence. Chem. Sci., 2017. [DOI | PubMed]
- S. Sohn, M. W. Ha, J. Park, Y. H. Kim, H. Ahn, S. Jung, S. K. Kwon, Y. H. Kim. High-Efficiency Diphenylpyrimidine Derivatives Blue Thermally Activated Delayed Fluorescence Organic Light-Emitting Diodes. Front. Chem., 2020. [DOI | PubMed]
- K. Nakao, H. Sasabe, R. Komatsu, Y. Hayasaka, T. Ohsawa, J. Kido. Significant Enhancement of Blue OLED Performances through Molecular Engineering of Pyrimidine-Based Emitter. Adv. Optical Mater., 2017. [DOI]
- Q. Zhang, S. Sun, X. Lv, W. Liu, H. Zeng, R. Guo, S. Ye, P. Leng, S. Xiang, L. Wang. Manipulating the Positions of Ch···N in Acceptors of Pyrimidine-Pyridine Hybrids for Highly Efficient Sky-Blue Thermally Activated Delayed Fluorescent OLEDs. Mater. Chem. Front., 2018. [DOI]
- P. Ganesan, D.-G. Chen, J.-L. Liao, W.-C. Li, Y.-N. Lai, D. Luo, C.-H. Chang, C.-L. Ko, W.-Y. Hung, S.-W. Liu. Isomeric Spiro-[Acridine-9,9′-Fluorene]-2,6-Dipyridylpyrimidine Based TADF Emitters: Insights into Photophysical Behaviors and OLED Performances. J. Mater. Chem. C, 2018. [DOI]
- X. Lv, S. Sun, Q. Zhang, S. Ye, W. Liu, Y. Wang, R. Guo, L. Wang. A Strategy to Construct Multifunctional TADF Materials for Deep Blue and High Efficiency Yellow Fluorescent Devices. J. Mater. Chem. C, 2020. [DOI]
- Q. Zhang, S. Xiang, Z. Huang, S. Sun, S. Ye, X. Lv, W. Liu, R. Guo, L. Wang. Molecular Engineering of Pyrimidine-Containing Thermally Activated Delayed Fluorescence Emitters for Highly Efficient Deep-Blue (Cie Y < 0.06) Organic Light-Emitting Diodes. Dyes Pigm., 2018. [DOI]
- M. Cai, M. Auffray, D. Zhang, Y. Zhang, R. Nagata, Z. Lin, X. Tang, C.-Y. Chan, Y.-T. Lee, T. Huang. Enhancing Spin-Orbital Coupling in Deep-Blue/Blue TADF Emitters by Minimizing the Distance from the Heteroatoms in Donors to Acceptors. Chem. Eng. J., 2021. [DOI]
- D. Banevičius, G. Kreiza, R. Klioštoraitis, S. Juršénas, T. Javorskis, V. Vaitkevičius, E. Orentas, K. Kazlauskas. Enhanced Blue TADF in a D-a-D Type Naphthyridine Derivative with an Asymmetric Carbazole-Donor Motif. J. Mater. Chem. C, 2022. [DOI]
- M. Mahmoudi, D. Gudeika, S. Kutsiy, J. Simokaitiene, R. Butkute, L. Skhirtladze, K. L. Woon, D. Volyniuk, J. V. Grazulevicius. Ornamenting of Blue Thermally Activated Delayed Fluorescence Emitters by Anchor Groups for the Minimization of Solid-State Solvation and Conformation Disorder Corollaries in Non-Doped and Doped Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2022. [DOI | PubMed]
- J. Shi, Z. Ran, F. Peng, M. Chen, L. Li, L. Ji, W. Huang. High-Performance Three-Coordinated Organoboron Emitters for Organic Light-Emitting Diodes. J. Mater. Chem. C, 2022. [DOI]
- S. Madayanad Suresh, D. Hall, D. Beljonne, Y. Olivier, E. Zysman-Colman. Multiresonant Thermally Activated Delayed Fluorescence Emitters Based on Heteroatom-Doped Nanographenes: Recent Advances and Prospects for Organic Light-Emitting Diodes. Adv. Funct. Mater., 2020. [DOI]
- Y. H. Lee, S. Park, J. Oh, J. W. Shin, J. Jung, S. Yoo, M. H. Lee. Rigidity-Induced Delayed Fluorescence by Ortho Donor-Appended Triarylboron Compounds: Record-High Efficiency in Pure Blue Fluorescent Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- I. S. Park, K. Matsuo, N. Aizawa, T. Yasuda. High-Performance Dibenzoheteraborin-Based Thermally Activated Delayed Fluorescence Emitters: Molecular Architectonics for Concurrently Achieving Narrowband Emission and Efficient Triplet-Singlet Spin Conversion. Adv. Funct. mater., 2018. [DOI]
- K. Matsuo, T. Yasuda. Blue Thermally Activated Delayed Fluorescence Emitters Incorporating Acridan Analogues with Heavy Group 14 Elements for High-Efficiency Doped and Non-Doped OLEDs. Chem. Sci., 2019. [DOI | PubMed]
- I. S. Park, K. Matsuo, N. Aizawa, T. Yasuda. High-Performance Dibenzoheteraborin-Based Thermally Activated Delayed Fluorescence Emitters: Molecular Architectonics for Concurrently Achieving Narrowband Emission and Efficient Triplet-Singlet Spin Conversion. Adv. Funct. Mater., 2018. [DOI]
- D. H. Ahn, H. Lee, S. W. Kim, D. Karthik, J. Lee, H. Jeong, J. Y. Lee, J. H. Kwon. Highly Twisted Donor-Acceptor Boron Emitter and High Triplet Host Material for Highly Efficient Blue Thermally Activated Delayed Fluorescent Device. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- H. Min, I. S. Park, T. Yasuda. Blue Thermally Activated Delayed Fluorescence with Sub-Microsecond Short Exciton Lifetimes: Acceleration of Triplet-Singlet Spin Interconversion Via Quadrupolar Charge-Transfer States. Adv. Optical mater., 2022. [DOI]
- T. Agou, K. Matsuo, R. Kawano, I. S. Park, T. Hosoya, H. Fukumoto, T. Kubota, Y. Mizuhata, N. Tokitoh, T. Yasuda. Pentacyclic Ladder-Heteraborin Emitters Exhibiting High-Efficiency Blue Thermally Activated Delayed Fluorescence with an Ultrashort Emission Lifetime. ACS Mater. Lett., 2020. [DOI]
- K. Matsuo, T. Yasuda. Boronate- and Borinate-Based Pi-Systems for Blue Thermally Activated Delayed Fluorescence Materials. Chem. Commun., 2019. [DOI]
- G. Li, W. Lou, D. Wang, C. Deng, Q. Zhang. Difluoroboron-Enabled Thermally Activated Delayed Fluorescence. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta, T. Hatakeyama. One-Step Borylation of 1, 3-Diaryloxybenzenes Towards Efficient Materials for Organic Light-Emitting Diodes. Angew. Chem., Int. Ed., 2015. [DOI]
- D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee, J. H. Kwon. Highly Efficient Blue Thermally Activated Delayed Fluorescence Emitters Based on Symmetrical and Rigid Oxygen-Bridged Boron Acceptors. Nat. Photonics, 2019. [DOI]
- D. H. Ahn, J. H. Maeng, H. Lee, H. Yoo, R. Lampande, J. Y. Lee, J. H. Kwon. Rigid Oxygen-Bridged Boron-Based Blue Thermally Activated Delayed Fluorescence Emitter for Organic Light-Emitting Diode: Approach Towards Satisfying High Efficiency and Long Lifetime Together. Adv. Optical Mater., 2020. [DOI]
- K. R. Naveen, H. Lee, R. Braveenth, D. Karthik, K. J. Yang, S. J. Hwang, J. H. Kwon. Achieving High Efficiency and Pure Blue Color in Hyperfluorescence Organic Light Emitting Diodes Using Organo-Boron Based Emitters. Adv. Funct. Mater., 2022. [DOI]
- Y. Liu, B. Du, X. Han, X. Wu, H. Tong, L. Wang. Intramolecular-Locked Triazatruxene-Based Thermally Activated Delayed Fluorescence Emitter for Efficient Solution-Processed Deep-Blue Organic Light Emitting Diodes. Chem. Eng. J., 2022. [DOI]
- H. J. Kim, M. Godumala, S. K. Kim, J. Yoon, C. Y. Kim, H. Park, J. H. Kwon, M. J. Cho, D. H. Choi. Color-Tunable Boron-Based Emitters Exhibiting Aggregation-Induced Emission and Thermally Activated Delayed Fluorescence for Efficient Solution-Processable Nondoped Deep-Blue to Sky-Blue OLEDs. Adv. Optical Mater., 2020. [DOI]
- D. Karthik, D. H. Ahn, J. H. Ryu, H. Lee, J. H. Maeng, J. Y. Lee, J. H. Kwon. Highly Efficient Blue Thermally Activated Delayed Fluorescence Organic Light Emitting Diodes Based on Tercarbazole Donor and Boron Acceptor Dyads. J. Mater. Chem. C, 2020. [DOI]
- J. U. Kim, I. S. Park, C. Y. Chan, M. Tanaka, Y. Tsuchiya, H. Nakanotani, C. Adachi. Nanosecond-Time-Scale Delayed Fluorescence Molecule for Deep-Blue OLEDs with Small Efficiency Rolloff. Nat. Commun., 2020. [DOI | PubMed]
- H. J. Tan, G. X. Yang, Y. L. Deng, C. Cao, J. H. Tan, Z. L. Zhu, W. C. Chen, Y. Xiong, J. X. Jian, C. S. Lee, Q. X. Tong. Deep-Blue OLEDs with Rec.2020 Blue Gamut Compliance and EQE over 22% Achieved by Conformation Engineering. Adv. Mater., 2022. [DOI]
- I. S. Park, H. Min, J. U. Kim, T. Yasuda. Deep-Blue OLEDs Based on Organoboron-Phenazasiline-Hybrid Delayed Fluorescence Emitters Concurrently Achieving 30% External Quantum Efficiency and Small Efficiency Roll-Off. Adv. Optical Mater., 2021. [DOI]
- T. Hua, Y.-C. Liu, C.-W. Huang, N. Li, C. Zhou, Z. Huang, X. Cao, C.-C. Wu, C. Yang. High-Efficiency and Low Roll-Off Deep-Blue OLEDs Enabled by Thermally Activated Delayed Fluorescence Emitter with Preferred Horizontal Dipole Orientation. Chem. Eng. J., 2022. [DOI]
- G. Meng, X. Chen, X. Wang, N. Wang, T. Peng, S. Wang. Isomeric Bright Sky-Blue TADF Emitters Based on Bisacridine Decorated Dbna: Impact of Donor Locations on Luminescent and Electroluminescent Properties. Adv. Optical Mater., 2019. [DOI]
- J. Han, Z. Huang, J. Miao, Y. Qiu, Z. Xie, C. Yang. Narrowband Blue Emission with Insensitivity to the Doping Concentration from an Oxygen-Bridged Triarylboron-Based TADF Emitter: Nondoped OLEDs with a High External Quantum Efficiency up to 21.4. Chem. Sci., 2022. [DOI | PubMed]
- X.-Y. Meng, Z.-Q. Feng, Y.-J. Yu, L.-S. Liao, Z.-Q. Jiang. Highly Efficient Blue Thermally Activated Delayed Fluorescence Emitters Based on Multi-Donor Modified Oxygen-Bridged Boron Acceptor. Molecules, 2022. [DOI | PubMed]
- H. Gao, S. Shen, Y. Qin, G. Liu, T. Gao, X. Dong, Z. Pang, X. Xie, P. Wang, Y. Wang. Ultrapure Blue Thermally Activated Delayed Fluorescence (TADF) Emitters Based on Rigid Sulfur/Oxygen-Bridged Triarylboron Acceptor: Mr TADF and D-a TADF. J. Phys. Chem. Lett, 2022. [DOI | PubMed]
- C. Y. Chan, L. S. Cui, J. U. Kim, H. Nakanotani, C. Adachi. Rational Molecular Design for Deep-Blue Thermally Activated Delayed Fluorescence Emitters. Adv. Funct. Mater., 2018. [DOI]
- Z. Cheng, Z. Li, Y. Xu, J. Liang, C. Lin, J. Wei, Y. Wang. Achieving Efficient Blue Delayed Electrofluorescence by Shielding Acceptors with Carbazole Units. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- H.-J. Park, H. L. Lee, H. J. Lee, K. H. Lee, J. Y. Lee, W. P. Hong. Peripheral Decoration of Dibenzofuran with Donors and Acceptors as a New Design Platform for Thermally Activated Delayed Fluorescence Emitters. Chem. Mater., 2019. [DOI]
- C.-Y. Chan, M. Tanaka, H. Nakanotani, C. Adachi. Efficient and Stable Sky-Blue Delayed Fluorescence Organic Light-Emitting Diodes with Ciey Below 0.4. Nat. Commun., 2018. [DOI | PubMed]
- S. J. Zou, F. M. Xie, M. Xie, Y. Q. Li, T. Cheng, X. H. Zhang, C. S. Lee, J. X. Tang. High-Performance Nondoped Blue Delayed Fluorescence Organic Light-Emitting Diodes Featuring Low Driving Voltage and High Brightness. Adv. Sci., 2020. [DOI]
- F.-M. Xie, Z.-D. An, M. Xie, Y.-Q. Li, G.-H. Zhang, S.-J. Zou, L. Chen, J.-D. Chen, T. Cheng, J.-X. Tang. Tert-Butyl Substituted Hetero-Donor TADF Compounds for Efficient Solution-Processed Non-Doped Blue OLEDs. J. Mater. Chem. C, 2020. [DOI]
- D. Zhang, X. Song, A. J. Gillett, B. H. Drummond, S. T. E. Jones, G. Li, H. He, M. Cai, D. Credgington, L. Duan. Efficient and Stable Deep-Blue Fluorescent Organic Light-Emitting Diodes Employing a Sensitizer with Fast Triplet Upconversion. Adv. Mater., 2020. [DOI]
- M. Mamada, H. Katagiri, C. Y. Chan, Y. T. Lee, K. Goushi, H. Nakanotani, T. Hatakeyama, C. Adachi. Highly Efficient Deep-Blue Organic Light-Emitting Diodes Based on Rational Molecular Design and Device Engineering. Adv. Funct. Mater., 2022. [DOI]
- W. Zhang, Y.-X. Zhang, X.-Q. Zhang, X.-Y. Liu, J. Fan, L.-S. Liao. Blue Thermally Activated Delayed Fluorescence Materials Based on Bi/Tri-Carbazole Derivatives. Org. Electron., 2018. [DOI]
- X. Zheng, R. Huang, C. Zhong, G. Xie, W. Ning, M. Huang, F. Ni, F. B. Dias, C. Yang. Achieving 21% External Quantum Efficiency for Nondoped Solution-Processed Sky-Blue Thermally Activated Delayed Fluorescence OLEDs by Means of Multi-(Donor/Acceptor) Emitter with through-Space/-Bond Charge Transfer. Adv. Sci., 2020. [DOI]
- C. Adachi, T. Tsutsui, S. Saito. Blue Light-Emitting Organic Electroluminescent Devices. Appl. Phys. Lett., 1990. [DOI]
- A. R. Brown, D. D. C. Bradley, J. H. Burroughes, R. H. Friend, N. C. Greenham, P. L. Burn, A. B. Holmes, A. Kraft. Poly(P-Phenylenevinylene) Light-Emitting Diodes: Enhanced Electroluminescent Efficiency through Charge Carrier Confinement. Appl. Phys. Lett., 1992. [DOI]
- G. Hughes, M. R. Bryce. Electron-Transporting Materials for Organic Electroluminescent and Electrophosphorescent Devices. J. Mater. Chem., 2005. [DOI]
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook, J. Y. Lee. Molecular Design Strategy of Organic Thermally Activated Delayed Fluorescence Emitters. Chem. Mater., 2017. [DOI]
- M. Y. Wong, S. Krotkus, G. Copley, W. Li, C. Murawski, D. Hall, G. J. Hedley, M. Jaricot, D. B. Cordes, A. M. Z. Slawin. Deep-Blue Oxadiazole-Containing Thermally Activated Delayed Fluorescence Emitters for Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- Z. Li, W. Li, C. Keum, E. Archer, B. Zhao, A. M. Z. Slawin, W. Huang, M. C. Gather, I. D. W. Samuel, E. Zysman-Colman. 1,3,4-Oxadiazole-Based Deep Blue Thermally Activated Delayed Fluorescence Emitters for Organic Light Emitting Diodes. J. Phys. Chem. C, 2019. [DOI]
- M. W. Cooper, X. Zhang, Y. Zhang, S. O. Jeon, H. Lee, S. Kim, C. Fuentes-Hernandez, S. Barlow, B. Kippelen, S. R. Marder. Effect of the Number and Substitution Pattern of Carbazole Donors on the Singlet and Triplet State Energies in a Series of Carbazole-Oxadiazole Derivatives Exhibiting Thermally Activated Delayed Fluorescence. Chem. Mater., 2018. [DOI]
- M. W. Cooper, X. Zhang, Y. Zhang, A. Ashokan, C. Fuentes-Hernandez, S. Salman, B. Kippelen, S. Barlow, S. R. Marder. Delayed Luminescence in 2-Methyl-5-(Penta(9-Carbazolyl)Phenyl)-1,3,4-Oxadiazole Derivatives. J. Phys. Chem. A, 2022. [DOI | PubMed]
- Y. Tan, B. Rui, J. Li, Z. Zhao, Z. Liu, Z. Bian, C. Huang. Blue Thermally Activated Delayed Fluorescence Emitters Based on a Constructing Strategy with Diversed Donors and Oxadiazole Acceptor and Their Efficient Electroluminescent Devices. Opt. Mater., 2019. [DOI]
- Y. Z. Song, K. X. Wei, J. S. Lv. An Experimental and Theoretical Method for Determination of Standard Electrode Potential for the Redox Couple Diphenyl Sulfone/Diphenyl Sulfide. Russ. J. Phys. Chem. A, 2013. [DOI]
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka, C. Adachi. Efficient Blue Organic Light-Emitting Diodes Employing Thermally Activated Delayed Fluorescence. Nat. Photonics, 2014. [DOI]
- L. Zhan, Y. Xiang, Z. Chen, K. Wu, S. Gong, G. Xie, C. Yang. Fine-Tuning the Photophysical Properties of Thermally Activated Delayed Fluorescent Emitters Using Torsion Angles: High Performance Sky-Blue OLEDs. J. Mater. Chem. C, 2019. [DOI]
- C. H. Ryoo, J. Han, J. h. Yang, K. Yang, I. Cho, S. Jung, S. Kim, H. Jeong, C. Lee, J. E. Kwon. Systematic Substituent Control in Blue Thermally Activated Delayed Fluorescence (TADF) Emitters: Unraveling the Role of Direct Intersystem Crossing between the Same Charge-Transfer States. Adv. Optical Mater., 2022. [DOI]
- Y. Zhu, S. Zeng, W. Gong, X. Chen, C. Xiao, H. Ma, W. Zhu, J. Yeob Lee, Y. Wang. Molecular Design of Blue Thermally Activated Delayed Fluorescent Emitters for High Efficiency Solution Processable OLED Via an Intramolecular Locking Strategy. Chem. Eng. J., 2022. [DOI]
- J. Wang, J. Zhang, C. Jiang, C. Yao, X. Xi. Effective Design Strategy for Aggregation-Induced Emission and Thermally Activated Delayed Fluorescence Emitters Achieving 18% External Quantum Efficiency Pure-Blue OLEDs with Extremely Low Roll-Off. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- P. Sharif, E. Alemdar, S. Ozturk, O. Caylan, T. Haciefendioglu, G. Buke, M. Aydemir, A. Danos, A. P. Monkman, E. Yildirim. Rational Molecular Design Enables Efficient Blue TADF-OLEDs with Flexible Graphene Substrate. Adv. Funct. Mater., 2022. [DOI]
- M. Liu, R. Komatsu, X. Cai, H. Sasabe, T. Kamata, K. Nakao, K. Liu, S.-J. Su, J. Kido. Introduction of Twisted Backbone: A New Strategy to Achieve Efficient Blue Fluorescence Emitter with Delayed Emission. Adv. Optical Mater., 2017. [DOI]
- N. Jürgensen, A. Kretzschmar, S. Höfle, J. Freudenberg, U. H. F. Bunz, G. Hernandez-Sosa. Sulfone-Based Deep Blue Thermally Activated Delayed Fluorescence Emitters: Solution-Processed Organic Light-Emitting Diodes with High Efficiency and Brightness. Chem. Mater., 2017. [DOI]
- D. Chen, K. Liu, X.-L. Li, B. Li, M. Liu, X. Cai, Y. Ma, Y. Cao, S.-J. Su. Engineering Excited-State Properties of Intramolecular and Intermolecular Charge Transfer Purely Organic Emitters Towards High-Performance Fluorescent OLEDs. J. Mater. Chem. C, 2017. [DOI]
- J. Rao, C. Zhao, Y. Wang, K. Bai, S. Wang, J. Ding, L. Wang. Achieving Deep-Blue Thermally Activated Delayed Fluorescence in Nondoped Organic Light-Emitting Diodes through a Spiro-Blocking Strategy. ACS Omega, 2019. [DOI | PubMed]
- X. Zeng, K.-C. Pan, W.-K. Lee, S. Gong, F. Ni, X. Xiao, W. Zeng, Y. Xiang, L. Zhan, Y. Zhang. High-Efficiency Pure Blue Thermally Activated Delayed Fluorescence Emitters with a Preferentially Horizontal Emitting Dipole Orientation Via a Spiro-Linked Double D-a Molecular Architecture. J. Mater. Chem. C, 2019. [DOI]
- P. L. dos Santos, J. S. Ward, M. R. Bryce, A. P. Monkman. Using Guest-Host Interactions to Optimize the Efficiency of TADF OLEDs. J. Phys. Chem. Lett, 2016. [DOI | PubMed]
- Y. P. Jeon, B. K. Kong, E. J. Lee, K.-H. Yoo, T. W. Kim. Ultrahighly-Efficient and Pure Deep-Blue Thermally Activated Delayed Fluorescence Organic Light-Emitting Devices Based on Dimethylacridine/Thioxanthene-S,S-Dioxide. Nano Energy, 2019. [DOI]
- P. Stachelek, J. S. Ward, P. L. dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce, A. P. Monkman. Molecular Design Strategies for Color Tuning of Blue TADF Emitters. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- Y. Luo, S. Li, Y. Zhao, C. Li, Z. Pang, Y. Huang, M. Yang, L. Zhou, X. Zheng, X. Pu, Z. Lu. An Ultraviolet Thermally Activated Delayed Fluorescence OLED with Total External Quantum Efficiency over 9%. Adv. Mater., 2020. [DOI]
- S. Sun, R. Guo, Q. Zhang, X. Lv, P. Leng, Y. Wang, Z. Huang, L. Wang. Efficient Deep-Blue Thermally Activated Delayed Fluorescence Emitters Based on Diphenylsulfone-Derivative Acceptor. Dyes Pigm., 2020. [DOI]
- J. Wang, Y. Yang, C. Jiang, M. He, C. Yao, J. Zhang. Ultrapure Deep-Blue Aggregation-Induced Emission and Thermally Activated Delayed Fluorescence Emitters for Efficient OLEDs with Ciey < 0.1 and Low Efficiency Roll-Offs. J. Mater. Chem. C, 2022. [DOI]
- Y. Hu, J. Yao, Z. Xu, Z. Wang, L. Li, S. J. Su, D. Ma, F. Huang. Three-Dimensional Organic Cage with Narrowband Delayed Fluorescence. Sci. China Chem., 2020. [DOI]
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang, C. Adachi. Luminous Butterflies: Efficient Exciton Harvesting by Benzophenone Derivatives for Full-Color Delayed Fluorescence OLEDs. Angew. Chem., Int. Ed., 2014. [DOI]
- J. Lee, N. Aizawa, T. Yasuda. Isobenzofuranone- and Chromone-Based Blue Delayed Fluorescence Emitters with Low Efficiency Roll-Off in Organic Light-Emitting Diodes. Chem. Mater., 2017. [DOI]
- K. Shizu, T. Miwa, Y. Wada, I. Ogata, H. Kaji. Thermally Activated Delayed Fluorescence Emitter with a Symmetric Acceptor-Donor-Acceptor Structure. J. Photopolym. Sci. Technol., 2017. [DOI]
- T. Miwa, S. Kubo, K. Shizu, T. Komino, C. Adachi, H. Kaji. Blue Organic Light-Emitting Diodes Realizing External Quantum Efficiency over 25% Using Thermally Activated Delayed Fluorescence Emitters. Sci. Rep., 2017. [DOI | PubMed]
- H. Min, I. S. Park, T. Yasuda. Blue Thermally Activated Delayed Fluorescence with Sub-Microsecond Short Exciton Lifetimes: Acceleration of Triplet-Singlet Spin Interconversion Via Quadrupolar Charge-Transfer States. Adv. Optical Mater., 2022. [DOI]
- D. Zhang, Y. Wada, Q. Wang, H. Dai, T. Fan, G. Meng, J. Wei, Y. Zhang, K. Suzuki, G. Li. Highly Efficient and Stable Blue Organic Light-Emitting Diodes Based on Thermally Activated Delayed Fluorophor with Donor-Void-Acceptor Motif. Adv. Sci., 2022. [DOI]
- P. Rajamalli, N. Senthilkumar, P. Y. Huang, C. C. Ren-Wu, H. W. Lin, C. H. Cheng. New Molecular Design Concurrently Providing Superior Pure Blue, Thermally Activated Delayed Fluorescence and Optical out-Coupling Efficiencies. J. Am. Chem. Soc., 2017. [DOI | PubMed]
- B. Sk, E. Ravindran, U. Deori, N. Yadav, G. P. Nanda, P. Rajamalli. A Deep Blue Thermally Activated Delayed Fluorescence Emitter: Balance between Charge Transfer and Color Purity. J. Mater. Chem. C, 2022. [DOI]
- P. Rajamalli, V. Thangaraji, N. Senthilkumar, C.-C. Ren-Wu, H.-W. Lin, C.-H. Cheng. Thermally Activated Delayed Fluorescence Emitters with Am, M-Di-Tert-Butyl-Carbazolyl Benzoylpyridine Core Achieving Extremely High Blue Electroluminescence Efficiencies. J. Mater. Chem. C, 2017. [DOI]
- G. Kreiza, D. Banevičius, J. Jovaišaité, K. Maleckaité, D. Gudeika, D. Volyniuk, J. V. Gražulevičius, S. Juršénas, K. Kazlauskas. Suppression of Benzophenone-Induced Triplet Quenching for Enhanced TADF Performance. J. Mater. Chem. C, 2019. [DOI]
- Y. Fu, H. Liu, D. Yang, D. Ma, Z. Zhao, B. Z. Tang. Boosting External Quantum Efficiency to 38.6% of Sky-Blue Delayed Fluorescence Molecules by Optimizing Horizontal Dipole Orientation. Sci. Adv., 2021. [DOI | PubMed]
- C. Han, Z. Zhang, H. Xu, J. Li, G. Xie, R. Chen, Y. Zhao, W. Huang. Controllably Tuning Excited-State Energy in Ternary Hosts for Ultralow-Voltage-Driven Blue Electrophosphorescence. Angew. Chem., Int. Ed., 2012. [DOI]
- C. Han, L. Zhu, J. Li, F. Zhao, Z. Zhang, H. Xu, Z. Deng, D. Ma, P. Yan. Highly Efficient Multifluorenyl Host Materials with Unsymmetrical Molecular Configurations and Localized Triplet States for Green and Red Phosphorescent Devices. Adv. Mater., 2014. [DOI | PubMed]
- Q. Liang, C. Han, C. Duan, H. Xu. Blue Thermally Activated Delayed Fluorescence-Emitting Phosphine Oxide Hosts for Ultrasimple and Highly Efficient White Organic Light-Emitting Diodes. Adv. Optical Mater., 2018. [DOI]
- F. Gao, R. Du, C. Han, J. Zhang, Y. Wei, G. Lu, H. Xu. High-Efficiency Blue Thermally Activated Delayed Fluorescence from Donor-Acceptor-Donor Systems Via the through-Space Conjugation Effect. Chem. Sci., 2019. [DOI | PubMed]
- L. Mei, J. Hu, X. Cao, F. Wang, C. Zheng, Y. Tao, X. Zhang, W. Huang. The Inductive-Effect of Electron Withdrawing Trifluoromethyl for Thermally Activated Delayed Fluorescence: Tunable Emission from Tetra- to Penta-Carbazole in Solution Processed Blue OLEDs. Chem. Commun., 2015. [DOI]
- W. Yuan, M. Zhang, X. Zhang, X. Cao, N. Sun, S. Wan, Y. Tao. The Electron Inductive Effect of Cf3 on Penta-Carbazole Containing Blue Emitters: Trade-Off between Color Purity and Luminescent Efficiency in TADF OLEDs. Dyes Pigm., 2018. [DOI]
- C. L. Yi, C. L. Ko, T. C. Yeh, C. Y. Chen, Y. S. Chen, D. G. Chen, P. T. Chou, W. Y. Hung, K. T. Wong. Harnessing a New Co-Host System and Low Concentration of New TADF Emitters Equipped with Trifluoromethyl- and Cyano-Substituted Benzene as Core for High-Efficiency Blue OLEDs. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- X. Liang, H.-B. Han, Z.-P. Yan, L. Liu, Y.-X. Zheng, H. Meng, W. Huang. Versatile Functionalization of Trifluoromethyl Based Deep Blue Thermally Activated Delayed Fluorescence Materials for Organic Light Emitting Diodes. New J. Chem., 2018. [DOI]
- J.-A. Seo, Y. Im, S.-H. Han, C. W. Lee, J. Y. Lee. Unconventional Molecular Design Approach of High Efficiency Deep Blue Thermally Activated Delayed Fluorescent Emitters Using Indolocarbazole as an Acceptor. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- V. V. Patil, K. H. Lee, J. Y. Lee. Universal Blue Emitters for High Efficiency Thermally Activated Delayed Fluorescence and Fluorescent Organic Light-Emitting Diodes. Dyes Pigm., 2020. [DOI]
- Y. Zhu, C. Qu, J. Ye, Y. Xu, Z. Zhang, Y. Wang. Donor-Acceptor Type of Fused-Ring Thermally Activated Delayed Fluorescence Compounds Constructed through an Oxygen-Containing Six-Membered Ring. ACS Appl. Mater. Interfaces, 2022. [DOI | PubMed]
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura, C. Adachi. Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence. Nature, 2012. [DOI | PubMed]
- G. Haykir, M. Aydemir, A. Tekin, E. Tekin, A. Danos, F. Yuksel, G. Hizal, A. P. Monkman, F. Turksoy. Effects of Donor Position and Multiple Charge Transfer Pathways in Asymmetric Pyridyl-Sulfonyl TADF Emitters. Mater. Today Commun., 2022. [DOI]
- D.-Q. Wang, M. Zhang, K. Wang, C.-J. Zheng, Y.-Z. Shi, J.-X. Chen, H. Lin, S.-L. Tao, X.-H. Zhang. Fine-Tuning the Emissions of Highly Efficient Thermally Activated Delayed Fluorescence Emitters with Different Linking Positions of Electron-Deficient Substituent Groups. Dyes Pigm., 2017. [DOI]
- Z.-P. Chen, D.-Q. Wang, M. Zhang, K. Wang, Y.-Z. Shi, J.-X. Chen, W.-W. Tao, C.-J. Zheng, S.-L. Tao, X.-H. Zhang. Optimization on Molecular Restriction for Highly Efficient Thermally Activated Delayed Fluorescence Emitters. Adv. Optical Mater., 2018. [DOI]
- Y. F. Wang, M. Li, W. L. Zhao, Y. F. Shen, H. Y. Lu, C. F. Chen. An Axially Chiral Thermally Activated Delayed Fluorescent Emitter with a Dual Emitting Core for a Highly Efficient Organic Light-Emitting Diode. Chem. Commun., 2020. [DOI]
- U. Balijapalli, M. Tanaka, M. Auffray, C. Y. Chan, Y. T. Lee, Y. Tsuchiya, H. Nakanotani, C. Adachi. Utilization of Multi-Heterodonors in Thermally Activated Delayed Fluorescence Molecules and Their High Performance Bluish-Green Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- S.-J. Woo, Y.-H. Ha, Y.-H. Kim, J.-J. Kim. Effect of Ortho-Biphenyl Substitution on the Excited State Dynamics of a Multi-Carbazole TADF Molecule. J. Mater. Chem. C, 2020. [DOI]
- M. Zhang, C.-J. Zheng, K. Wang, Y.-Z. Shi, H.-Y. Yang, H. Lin, S.-L. Tao, X.-H. Zhang. Efficient and Stable Single-Emitting-Layer White Organic Light-Emitting Diodes by Employing All Thermally Activated Delayed Fluorescence Emitters. Org. Electron., 2022. [DOI]
- G. Zhao, D. Liu, P. Wang, X. Huang, H. Chen, Y. Zhang, D. Zhang, W. Jiang, Y. Sun, L. Duan. Exceeding 30% External Quantum Efficiency in Non-Doped OLEDs Utilizing Solution Processable TADF Emitters with High Horizontal Dipole Orientation Via Anchoring Strategy. Angew. Chem., Int. Ed., 2022. [DOI]
- Y. J. Lien, T. C. Lin, C. C. Yang, Y. C. Chiang, C. H. Chang, S. H. Liu, Y. T. Chen, G. H. Lee, P. T. Chou, C. W. Lu, Y. Chi. First N-Borylated Emitters Displaying Highly Efficient Thermally Activated Delayed Fluorescence and High-Performance OLEDs. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- C.-C. Tsai, W.-C. Huang, H.-Y. Chih, Y.-C. Hsh, C.-W. Liao, C.-H. Lin, Y.-X. Kang, C.-H. Chang, Y. J. Chang, C.-W. Lu. Efficient Donor-Acceptor-Donor Borylated Compounds with Extremely Small Δest for Thermally Activated Delayed Fluorescence OLEDs. Org. Electron., 2018. [DOI]
- D.-G. Chen, T.-C. Lin, C.-L. Chen, Y.-T. Chen, Y.-A. Chen, G.-H. Lee, P.-T. Chou, C.-W. Liao, P.-C. Chiu, C.-H. Chang. Optically Triggered Planarization of Boryl-Substituted Phenoxazine: Another Horizon of TADF Molecules and High-Performance OLEDs. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- C. Qu, G. Xia, Y. Xu, Y. Zhu, J. Liang, H. Zhang, J. Wang, Z. Zhang, Y. Wang. Boron-Containing D-a-a Type TADF Materials with Tiny Singlet-Triplet Energy Splittings and High Photoluminescence Quantum Yields for Highly Efficient OLEDs with Low Efficiency Roll-Offs. J. Mater. Chem. C, 2020. [DOI]
- A. Kumar, W. Lee, T. Lee, J. Jung, S. Yoo, M. H. Lee. Triarylboron-Based TADF Emitters with Perfluoro Substituents: High-Efficiency OLEDs with a Power Efficiency over 100 Lm W-1. J. Mater. Chem. C, 2020. [DOI]
- T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu, C.-H. Cheng. Diboron Compound-Based Organic Light-Emitting Diodes with High Efficiency and Reduced Efficiency Roll-Off. Nat. Photonics, 2018. [DOI]
- M. Ouyang, L. Xing, Q. Chen, H. Huang, M. Zhu, K. Hu, Y. Liu, W.-C. Chen, Y. Huo, C. Yang. Highly Efficient Thermally Activated Delayed Fluorescence Emitters Enabled by Double Charge Transfer Pathways Via Ortho-Linked Triarylboron/Carbazole Hybrids. J. Mater. Chem. C, 2021. [DOI]
- Y. H. Lee, Y.-S. Shin, T. Lee, J. Jung, J.-H. Lee, M. H. Lee. Managing Local Triplet Excited States of Boron-Based TADF Emitters for Fast Spin-Flip Process: Toward Highly Efficient TADF-OLEDs with Low Efficiency Roll-Off. Chem. Eng. J., 2021. [DOI]
- T. L. Wu, J. Lei, C. M. Hsieh, Y. K. Chen, P. Y. Huang, P. T. Lai, T. Y. Chou, W. C. Lin, W. Chen, C. H. Yu. Substituent Engineering of the Diboron Molecular Architecture for a Nondoped and Ultrathin Emitting Layer. Chem. Sci., 2022. [DOI | PubMed]
- Y.-J. Shiu, Y.-T. Chen, W.-K. Lee, C.-C. Wu, T.-C. Lin, S.-H. Liu, P.-T. Chou, C.-W. Lu, I.-C. Cheng, Y.-J. Lien, Y. Chi. Efficient Thermally Activated Delayed Fluorescence of Functional Phenylpyridinato Boron Complexes and High Performance Organic Light-Emitting Diodes. J. Mater. Chem. C, 2017. [DOI]
- S. Gong, J. Luo, Z. Wang, Y. Li, T. Chen, G. Xie, C. Yang. Tuning Emissive Characteristics and Singlet-Triplet Energy Splitting of Fluorescent Emitters by Encapsulation Group Modification: Yellow TADF Emitter for Solution-Processed OLEDs with High Luminance and Ultraslow Efficiency Roll-Off. Dyes Pigm., 2017. [DOI]
- K. Wang, W. Liu, C.-J. Zheng, Y.-Z. Shi, K. Liang, M. Zhang, X.-M. Ou, X.-H. Zhang. A Comparative Study of Carbazole-Based Thermally Activated Delayed Fluorescence Emitters with Different Steric Hindrance. J. Mater. Chem. C, 2017. [DOI]
- K. Wang, C. J. Zheng, W. Liu, K. Liang, Y. Z. Shi, S. L. Tao, C. S. Lee, X. M. Ou, X. H. Zhang. Avoiding Energy Loss on TADF Emitters: Controlling the Dual Conformations of D-a Structure Molecules Based on the Pseudoplanar Segments. Adv. Mater., 2017. [DOI]
- M. Zhang, G.-L. Dai, C.-J. Zheng, K. Wang, Y.-Z. Shi, X.-C. Fan, H. Lin, S.-L. Tao, X.-H. Zhang. Novel D-D′-a Structure Thermally Activated Delayed Fluorescence Emitters Realizing over 20% External Quantum Efficiencies in Both Evaporation- and Solution-Processed Organic Light-Emitting Diodes. Org. Electron., 2021. [DOI]
- X. Wei, Z. Li, T. Hu, R. Duan, J. Liu, R. Wang, Y. Liu, X. Hu, Y. Yi, P. Wang, Y. Wang. Substitution Conformation Balances the Oscillator Strength and Singlet-Triplet Energy Gap for Highly Efficient D-a-D Thermally Activated Delayed Fluorescence Emitters. Adv. Optical Mater., 2019. [DOI]
- M. Ma, J. Li, D. Liu, D. Li, R. Dong, Y. Mei. Low Efficiency Roll-Off Thermally Activated Delayed Fluorescence Emitters for Non-Doped OLEDs: Substitution Effect of Thioether and Sulfone Groups. Dyes Pigm., 2021. [DOI]
- S. Gao, X. Chen, X. Ge, Z. Chen, J. Zhao, Z. Chi. Asymmetric Thermally Activated Delayed Fluorescence Materials Rendering High-Performance OLEDs through Both Thermal Evaporation and Solution-Processing. Chem. Res. Chin. Univ., 2022. [DOI]
- L. Gan, Z. Xu, Z. Wang, B. Li, W. Li, X. Cai, K. Liu, Q. Liang, S.-J. Su. Utilizing a Spiro TADF Moiety as a Functional Electron Donor in TADF Molecular Design toward Efficient “Multichannel” Reverse Intersystem Crossing. Adv. Funct. Mater., 2019. [DOI]
- C. Li, C. Duan, C. Han, H. Xu. Secondary Acceptor Optimization for Full-Exciton Radiation: Toward Sky-Blue Thermally Activated Delayed Fluorescence Diodes with External Quantum Efficiency of ≈30%. Adv. Mater., 2018. [DOI]
- K. Li, Y. Zhu, B. Yao, Y. Chen, H. Deng, Q. Zhang, H. Zhan, Z. Xie, Y. Cheng. Rotation-Restricted Thermally Activated Delayed Fluorescence Compounds for Efficient Solution-Processed OLEDs with EQEs of up to 24.3% and Small Roll-Off. Chem. Commun., 2020. [DOI]
- C. H. Ryoo, I. Cho, J. Han, J.-h. Yang, J. E. Kwon, S. Kim, H. Jeong, C. Lee, S. Y. Park. Structure-Property Correlation in Luminescent Indolo[3,2-B]Indole (Idid) Derivatives: Unravelling the Mechanism of High Efficiency Thermally Activated Delayed fluorescence (TADF). ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- J. H. Maeng, D. H. Ahn, H. Lee, Y. H. Jung, D. Karthik, J. Y. Lee, J. H. Kwon. Rigid Indolocarbazole Donor Moiety for Highly Efficient Thermally Activated Delayed Fluorescent Device. Dyes Pigm., 2020. [DOI]
- S. J. Yoon, H. J. Lee, K. H. Lee, J. Y. Lee. A Study on the Effect of a Pyridine Secondary Acceptor on the Emission Properties of Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C, 2020. [DOI]
- K. J. Kim, G. H. Kim, R. Lampande, D. H. Ahn, J. B. Im, J. S. Moon, J. K. Lee, J. Y. Lee, J. Y. Lee, J. H. Kwon. A New Rigid Diindolocarbazole Donor Moiety for High Quantum Efficiency Thermally Activated Delayed Fluorescence Emitter. J. Mater. Chem. C, 2018. [DOI]
- J. G. Yu, S. H. Han, H. L. Lee, W. P. Hong, J. Y. Lee. A Novel Molecular Design Employing a Backbone Freezing Linker for Improved Efficiency, Sharpened Emission and Long Lifetime in Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C, 2019. [DOI]
- Z. Liu, G. Li, H. Liu, C. Zhou, K. Li, Z. Wang, C. Yang. Side by Side Alignment of Donors Enabling High-Efficiency TADF OLEDs with Insensitivity to Doping Concentration. Adv. Optical Mater., 2021. [DOI]
- H. L. Lee, W. J. Chung, J. Y. Lee. Efficient up-Conversion Process by Isolation of Two Chromophores in Thermally Activated Delayed Fluorescent Emitters. Chem. Eng. J., 2021. [DOI]
- H. L. Lee, S. H. Han, W. P. Hong, O.-K. Song, J. Y. Π Lee. Linker Mediated Coupling of Two Emitting Units for Improved Efficiency in Thermally Activated Delayed Fluorescent Emitters. Dyes Pigm., 2019. [DOI]
- H. L. Lee, K. H. Lee, J. Y. Lee, H. J. Lee. Molecular Design Opening Two Emission Pathways for High Efficiency and Long Lifetime of Thermally Activated Delayed Fluorescent Organic Light-Emitting Diodes. J. Mater. Chem. C, 2021. [DOI]
- S. L. Zhang, Y. Z. Shi, K. Wang, X. C. Fan, J. Yu, X. M. Ou, X. H. Zhang. Pyridine-Substituted Triazine as an Acceptor for Thermally Activated Delayed Fluorescence Emitters Showing High Efficiency and Low Roll-Off in Organic Light-Emitting Diodes. Mater. Today Energy, 2021. [DOI]
- J. H. Yun, K. H. Lee, J. Y. Lee. Benzoylphenyltriazine as a New Acceptor of Donor-Acceptor Type Thermally-Activated Delayed-Fluorescent Emitters. J. Ind. Eng. Chem., 2021. [DOI]
- Q. Zhan, C. Cao, T. Huang, C. Zhou, Z. Xie, Y. Zou, C. S. Lee, C. Yang. 3d Triptycene-Fused Acridine Electron Donor Enables High-Efficiency Nondoped Thermally Activated Delayed Fluorescent OLEDs. Adv. Optical Mater., 2021. [DOI]
- C. Shi, D. Liu, J. Li, Z. He, K. Song, B. Liu, Q. Wu, M. Xu. Tert-Butyltriazine-Diphenylaminocarbazole Based TADF Materials: Π-Bridge Modification for Enhanced KRISC and Efficiency Stability. Dyes Pigm., 2022. [DOI]
- Q. Feng, Y. Qian, H. Wang, W. Hou, X. Peng, S. Xie, S. Wang, L. Xie. Donor Arylmethylation toward Horizontally Oriented TADF Emitters for Efficient Electroluminescence with 37% External Quantum Efficiency. Adv. Optical Mater., 2022. [DOI]
- X.-C. Fan, K. Wang, Y.-Z. Shi, D.-M. Sun, J.-X. Chen, F. Huang, H. Wang, J. Yu, C.-S. Lee, X.-H. Zhang. Thermally Activated Delayed Fluorescence Materials for Nondoped Organic Light-Emitting Diodes with Nearly 100% Exciton Harvest. SmartMat, 2023. [DOI]
- Y. Xiang, Y. Zhao, N. Xu, S. Gong, F. Ni, K. Wu, J. Luo, G. Xie, Z. Lu, C. Yang. Halogen-Induced Internal Heavy-Atom Effect Shortening Emissive Lifetime and Improving Fluorescence Efficiency of Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C, 2017. [DOI]
- Y. Xiang, P. Li, S. Gong, Y.-H. Huang, C.-Y. Wang, C. Zhong, W. Zeng, Z. Chen, W.-K. Lee, X. Yin. Acceptor Plane Expansion Enhances Horizontal Orientation of Thermally Activated Delayed Fluorescence Emitters. Sci. Adv., 2020. [DOI | PubMed]
- Y. Kato, H. Sasabe, Y. Hayasaka, Y. Watanabe, H. Arai, J. Kido. A Sky Blue Thermally Activated Delayed Fluorescence Emitter to Achieve Efficient White Light Emission through in Situ Metal Complex Formation. J. Mater. Chem. C, 2019. [DOI]
- T. Serevičius, R. Skaisgiris, J. Dodonova, L. Jagintavicius, D. Banevicius, K. Kazlauskas, S. Tumkevicius, S. Jursenas. Achieving Submicrosecond Thermally Activated Delayed Fluorescence Lifetime and Highly Efficient Electroluminescence by Fine-Tuning of the Phenoxazine-Pyrimidine Structure. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- Q. Zhang, Y. Wang, S. J. Yoon, W. J. Chung, S. Ye, R. Guo, P. Leng, S. Sun, J. Y. Lee, L. Wang. Fusing Acridine and Benzofuran/Benzothiophene as a Novel Hybrid Donor for High-Performance and Low Efficiency Roll-Off TADF OLEDs. J. Mater. Chem. C, 2020. [DOI]
- H.-J. Park, S. H. Han, J. Y. Lee, H. Han, E.-G. Kim. Managing Orientation of Nitrogens in Bipyrimidine-Based Thermally Activated Delayed Fluorescent Emitters to Suppress Nonradiative Mechanisms. Chem. Mater., 2018. [DOI]
- C. L. Yi, C. Y. Lin, Y. Tang, C. Y. Wang, C. W. Huang, X. Gong, S. Gong, C. C. Wu, K. T. Wong. A Rational Molecular Design Strategy of TADF Emitter for Achieving Device Efficiency Exceeding 36%. Adv. Optical Mater., 2022. [DOI]
- Y. Lee, J.-I. Hong. High-Efficiency Thermally Activated Delayed Fluorescence Emitters Via a High Horizontal Dipole Ratio and Controlled Dual Emission. J. Mater. Chem. C, 2020. [DOI]
- Y. Lee, S.-J. Woo, J.-J. Kim, J.-I. Hong. Linear-Shaped Thermally Activated Delayed Fluorescence Emitter Using 1,5-Naphthyridine as an Electron Acceptor for Efficient Light Extraction. Org. Electron., 2020. [DOI]
- H. Sasabe, N. Onuma, Y. Nagai, T. Ito, J. Kido. High Power Efficiency Blue-to-Green Organic Light-Emitting Diodes Using Isonicotinonitrile-Based Fluorescent Emitters. Chem. Asian J., 2017. [DOI | PubMed]
- H. Sasabe, Y. Hayasaka, R. Komatsu, K. Nakao, J. Kido. Highly Luminescent Π-Conjugated Terpyridine Derivatives Exhibiting Thermally Activated Delayed Fluorescence. Chem. Eur. J., 2017. [DOI | PubMed]
- L. Wang, Z. Huang, S. Xiang, Q. Zhang, X. Lv, S. Ye, S. Zhuang, R. Guo. Highly Efficient Green Organic Light Emitting Diodes with Phenanthroimidazole-Based Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C, 2018. [DOI]
- S. Kothavale, W. J. Chung, J. Y. Lee. Rational Molecular Design of Highly Efficient Yellow-Red Thermally Activated Delayed Fluorescent Emitters: A Combined Effect of Auxiliary Fluorine and Rigidified Acceptor Unit. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- X. Zeng, T. Zhou, J. Liu, K. Wu, S. Li, X. Xiao, Y. Zhang, S. Gong, G. Xie, C. Yang. Incorporating Thermally Activated Delayed Fluorescence into Mechanochromic Luminescent Emitters: High-Performance Solution-Processed Yellow Organic Light Emitting Diodes. Adv. Optical Mater., 2018. [DOI]
- H. Kim, Y. Lee, H. Lee, J. I. Hong, D. Lee. Click-to-Twist Strategy to Build Blue-to-Green Emitters: Bulky Triazoles for Electronically Tunable and Thermally Activated Delayed Fluorescence. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- Y. Chen, D. Zhang, Y. Zhang, X. Zeng, T. Huang, Z. Liu, G. Li, L. Duan. Approaching Nearly 40% External Quantum Efficiency in Organic Light Emitting Diodes Utilizing a Green Thermally Activated Delayed Fluorescence Emitter with an Extended Linear Donor-Acceptor-Donor Structure. Adv. Mater., 2021. [DOI]
- X. Zhang, M. W. Cooper, Y. Zhang, C. Fuentes-Hernandez, S. Barlow, S. R. Marder, B. Kippelen. Host-Free Yellow-Green Organic Light-Emitting Diodes with External Quantum Efficiency over 20% Based on a Compound Exhibiting Thermally Activated Delayed Fluorescence. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- D. Hu, M. Zhu, C. Shi, W. Yuan, N. Sun, B. Huang, Y. Tao. Manipulating Peripheral Non-Conjugated Substituents in Carbazole/Oxadiazole Hybrid TADF Emitters Towards High-Efficiency OLEDs. J. Mater. Chem. C, 2021. [DOI]
- J. X. Zhou, X. Y. Zeng, F. M. Xie, Y. H. He, Y. Q. Tang, Y. Q. Li, J. X. Tang. High-Efficiency Orange Thermally Activated Delayed Fluorescence by Secondary Acceptor Modification. Mater. Today Energy, 2021. [DOI]
- Y. Liu, J. Yang, Z. Mao, D. Ma, Y. Wang, J. Zhao, S. J. Su, Z. Chi. Donor or Acceptor: Molecular Engineering Based on Dibenzo[a,C]Phenazine Backbone for Highly Efficient Thermally-Activated Delayed Fluorescence Organic Light-Emitting Diodes. Adv. Optical Mater., 2023. [DOI]
- B. Li, Z. Wang, S.-J. Su, F. Guo, Y. Cao, Y. Zhang. Quinazoline-Based Thermally Activated Delayed Fluorecence for High-Performance OLEDs with External Quantum Efficiencies Exceeding 20%. Adv. Optical Mater., 2019. [DOI]
- S.-C. Ji, T. Zhao, Z. Wei, L. Meng, X.-D. Tao, M. Yang, X.-L. Chen, C.-Z. Lu. Manipulating Excited States Via Lock/Unlock Strategy for Realizing Efficient Thermally Activated Delayed Fluorescence Emitters. Chem. Eng. J., 2022. [DOI]
- Z. Chen, Z. Wu, F. Ni, C. Zhong, W. Zeng, D. Wei, K. An, D. Ma, C. Yang. Emitters with a Pyridine-3,5-Dicarbonitrile Core and Short Delayed Fluorescence Lifetimes of About 1.5 Μs: Orange-Red TADF-Based OLEDs with Very Slow Efficiency Roll-Offs at High Luminance. J. Mater. Chem. C, 2018. [DOI]
- Y. Z. Shi, K. Wang, X. C. Fan, J. X. Chen, X. M. Ou, J. Yu, J. S. Jie, C. S. Lee, X. H. Zhang. High-Performance Nondoped Organic Light-Emitting Diode Based on a Thermally Activated Delayed Fluorescence Emitter with 1d Intermolecular Hydrogen Bonding Interactions. Adv. Optical Mater., 2021. [DOI]
- R. Dong, D. Liu, J. Li, M. Ma, Y. Mei, D. Li, J. Jiang. Acceptor Modulation for Blue and Yellow TADF Materials and Fabrication of All-TADF White OLED. Mater. Chem. Front., 2021. [DOI]
- H. Liu, J. Li, W.-C. Chen, Z. Chen, Z. Liu, Q. Zhan, X. Cao, C.-S. Lee, C. Yang. Modulating the Acceptor Structure of Dicyanopyridine Based TADF Emitters: Nearly 30% External Quantum Efficiency and Suppression on Efficiency Roll-Off in OLED. Chem. Eng. J., 2020. [DOI]
- J. Li, W.-C. Chen, H. Liu, Z. Chen, D. Chai, C.-S. Lee, C. Yang. Double-Twist Pyridine-Carbonitrile Derivatives Yielding Excellent Thermally Activated Delayed Fluorescence Emitters for High-Performance OLEDs. J. Mater. Chem. C, 2020. [DOI]
- Y. K. Chen, J. Jayakumar, C. M. Hsieh, T. L. Wu, C. C. Liao, J. Pandidurai, C. L. Ko, W. Y. Hung, C. H. Cheng. Triarylamine-Pyridine-Carbonitriles for Organic Light-Emitting Devices with EQE Nearly 40. Adv. Mater., 2021. [DOI]
- Z. Xie, C. Cao, Y. Zou, X. Cao, C. Zhou, J. He, C. S. Lee, C. Yang. Molecular Engineering Enables TADF Emitters Well Suitable for Non-Doped OLEDs with External Quantum Efficiency of Nearly 30%. Adv. Funct. Mater., 2022. [DOI]
- H. L. Lee, C. S. Oh, K. H. Lee, J. Y. Lee, W. P. Hong. Lifetime-Extending 3-(4-Phenylbenzo[4,5]Thieno[3,2-D]Pyrimidin-2-Yl)Benzonitrile Acceptor for Thermally Activated Delayed Fluorescence Emitters. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- H. L. Lee, W. J. Chung, J. Y. Lee. Selective Efficiency Boosting in Thermally Activated Delayed Fluorescence Emitters by a Secondary Donor. Chem. Eng. J., 2021. [DOI]
- L. G. Franca, Y. Long, C. Li, A. Danos, A. Monkman. The Critical Role of Npi* States in the Photophysics and Thermally Activated Delayed Fluorescence of Spiro Acridine-Anthracenone. J. Phys. Chem. Lett, 2021. [DOI | PubMed]
- C.-F. Chen, M. Li, Y. Liu, R. Duan, X. Wei, Y. Yi, Y. Wang. Aromatic Imide Based Thermally Activated Delayed Fluorescence Materials for Highly Efficient Organic Light Emitting Diodes. Angew. Chem., Int. Ed., 2017. [DOI]
- Y. Xiang, Z.-L. Zhu, D. Xie, S. Gong, K. Wu, G. Xie, C.-S. Lee, C. Yang. Revealing the New Potential of an Indandione Unit for Constructing Efficient Yellow Thermally Activated Delayed Fluorescence Emitters with Short Emissive Lifetimes. J. Mater. Chem. C, 2018. [DOI]
- Y. Liu, Z. Yin, X. Wang, E. Baranoff, D. Zhou, K. Zhang, Z. Ren, S. Wang, W. Zhu, Y. Wang. A Novel Donor Moiety 9,9,9′9′-Tetramethyl-9,9′10,10′-Tetrahydro-2,10′-Biacridine Via One-Pot C-H Arylation for TADF Emitters and Their Application in Highly Efficient Solution-Processable OLEDs. J. Mater. Chem. C, 2020. [DOI]
- J. Zeng, J. Guo, H. Liu, Z. Zhao, B. Z. Tang. A Multifunctional Bipolar Luminogen with Delayed Fluorescence for High-Performance Monochromatic and Color-Stable Warm-White OLEDs. Adv. Funct. Mater., 2020. [DOI]
- P. Wang, J. Yu, S. Chen, H. Yu, X. Yan, Y. Guan, J. Chen, L. Li. 3-Benzoyl-4h-Chromen-4-One: A Novel Twisted Acceptor for Highly Efficient Thermally Activated Delayed Fluorescence Emitters. Dyes Pigm., 2020. [DOI]
- X. Chen, Z. Yang, Z. Xie, J. Zhao, Z. Yang, Y. Zhang, M. P. Aldred, Z. Chi. An Efficient Yellow Thermally Activated Delayed Fluorescence Emitter with Universal Applications in Both Doped and Non-Doped Organic Light-Emitting Diodes. Mater. Chem. Front., 2018. [DOI]
- M.-D. Bai, M. Zhang, K. Wang, Y.-Z. Shi, J.-X. Chen, H. Lin, S.-L. Tao, C.-J. Zheng, X.-H. Zhang. Novel Star-Shaped Yellow Thermally Activated Delayed Fluorescence Emitter Realizing over 10% External Quantum Efficiency at High Luminance of 30000 Cd M–2 in OLED. Org. Electron., 2018. [DOI]
- R. Dong, J. Li, D. Liu, D. Li, Y. Mei, M. Ma, J. Jiang. Self-Host Thermally Activated Delayed Fluorescence Material with Aggregation-Induced Emission Character: Multi-Functional Applications in OLEDs. Adv. Optical Mater., 2021. [DOI]
- Y.-Y. Jing, X.-D. Tao, M.-X. Yang, X.-L. Chen, C.-Z. Lu. Triptycene-Imbedded Thermally Activated Delayed Fluorescence Emitters with Excellent Film Morphologies for Applications in Efficient Nondoped and Doped Organic Light-Emitting Devices. Chem. Eng. J., 2021. [DOI]
- J.-X. Chen, W. Liu, C.-J. Zheng, K. Wang, K. Liang, Y.-Z. Shi, X.-M. Ou, X.-H. Zhang. Coumarin-Based Thermally Activated Delayed Fluorescence Emitters with High External Quantum Efficiency and Low Efficiency Roll-Off in the Devices. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- X. F. Luo, F. L. Li, J. W. Zou, Q. Zou, J. Su, M. X. Mao, Y. X. Zheng. A Series of Fused Carbazole/Carbonyl Based Blue to Yellow-Green Thermally Activated Delayed Fluorescence Materials for Efficient Organic Light-Emitting Diodes. Adv. Optical Mater., 2021. [DOI]
- J. Xu, X. Wu, J. Guo, Z. Zhao, B. Z. Tang. Sky-Blue Delayed Fluorescence Molecules Based on Pyridine-Substituted Acridone for Efficient Organic Light-Emitting Diodes. J. Mater. Chem. C, 2021. [DOI]
- Y. Mei, D. Liu, J. Li, H. Li, W. Wei. Acridin-9(10h)-One Based Thermally Activated Delayed Fluorescence Material: Simultaneous Optimization of RISC and Radiation Processes to Boost Luminescence Efficiency. J. Mater. Chem. C, 2021. [DOI]
- Y. Mei, D. Liu, J. Li, J. Wang. Thermally Activated Delayed Fluorescence Materials Based on Acridin-9(10h)-One Acceptor for Organic Light-Emitting Diodes. Dyes Pigm., 2022. [DOI]
- Y. Mei, D. Liu, J. Li, R. Dong, M. Ma, W. Wei, Y. Lan. Acridin-9(10h)-One-Based Blue Thermally Activated Delayed Fluorescence Materials: Improvement of Color Purity and Efficiency Stability. Mater. Today Chem., 2022. [DOI]
- K. Shizu, Y. Ren, H. Kaji. Promoting Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Via the Heavy-Atom Effect. J. Phys. Chem. A, 2023. [DOI | PubMed]
- R. Wang, Z. Li, T. Hu, L. Tian, X. Hu, S. Liu, C. Cao, Z. L. Zhu, J. H. Tan, Y. Yi. Two-Channel Space Charge Transfer-Induced Thermally Activated Delayed Fluorescent Materials for Efficient OLEDs with Low Efficiency Roll-Off. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- D. Yang, J.-S. Huh, J.-I. Hong. Spiro-Type TADF Emitters Based on Acridine Donors and Anthracenone Acceptor. Dyes Pigm., 2022. [DOI]
- K. Nasu, T. Nakagawa, H. Nomura, C.-J. Lin, C.-H. Cheng, M.-R. Tseng, T. Yasuda, C. Adachi. A Highly Luminescent Spiro-Anthracenone-Based Organic Light-Emitting Diode Exhibiting Thermally Activated Delayed Fluorescence. Chem. Commun., 2013. [DOI]
- I. Lyskov, C. M. Marian. Climbing up the Ladder: Intermediate Triplet States Promote the Reverse Intersystem Crossing in the Efficient TADF Emitter Acrsa. J. Phys. Chem. C, 2017. [DOI]
- L. G. Franca, A. Danos, A. Monkman. Donor, Acceptor, and Molecular Charge Transfer Emission All in One Molecule. J. Phys. Chem. Lett, 2023. [DOI | PubMed]
- L. G. Franca, A. Danos, A. Monkman. Spiro Donor-Acceptor TADF Emitters: Naked TADF Free from Inhomogeneity Caused by Donor Acceptor Bridge Bond Disorder. Fast RISC and Invariant Photophysics in Solid State Hosts. J. Mater. Chem. C, 2022. [DOI]
- Z. Huang, B. Lei, D. Yang, D. Ma, Z. Bin, J. You. Modified Intramolecular-Lock Strategy Enables Efficient Thermally Activated Delayed Fluorescence Emitters for Non-Doped OLEDs. Angew. Chem., Int. Ed., 2022. [DOI]
- J. S. Ward, A. Danos, P. Stachelek, M. A. Fox, A. S. Batsanov, A. P. Monkman, M. R. Bryce. Exploiting Trifluoromethyl Substituents for Tuning Orbital Character of Singlet and Triplet States to Increase the Rate of Thermally Activated Delayed Fluorescence. Mater. Chem. Front., 2020. [DOI]
- D. Chen, E. Zysman-Colman. Exploring the Possibility of Using Fluorine-Involved Non-Conjugated Electron-Withdrawing Groups for Thermally Activated Delayed Fluorescence Emitters by TD-DFT Calculation. Beilstein J. Org. Chem., 2021. [DOI | PubMed]
- W. Yuan, H. Yang, C. Duan, X. Cao, J. Zhang, H. Xu, N. Sun, Y. Tao, W. Huang. Molecular Configuration Fixation with C-H···F Hydrogen Bonding for Thermally Activated Delayed Fluorescence Acceleration. CHEM, 2020. [DOI]
- J.-X. Chen, H. Wang, X. Zhang, Y.-F. Xiao, K. Wang, L. Zhou, Y.-Z. Shi, J. Yu, C.-S. Lee, X.-H. Zhang. Using Fullerene Fragments as Acceptors to Construct Thermally Activated Delayed Fluorescence Emitters for High-Efficiency Organic Light-Emitting Diodes. Chem. Eng. J., 2022. [DOI]
- R. Englman, J. Jortner. The Energy Gap Law for Radiationless Transitions in Large Molecules. Mol. Phys., 1970. [DOI]
- M. Fakis, D. Anestopoulos, V. Giannetas, P. Persephonis. Influence of Aggregates and Solvent Aromaticity on the Emission of Conjugated Polymers. J. Phys. Chem. B, 2006. [DOI | PubMed]
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam, B. Z. Tang. Aggregation-Induced Emission: Together We Shine, United We Soar!. Chem. Rev., 2015. [DOI | PubMed]
- M. Li, C. F. Chen. TADF-Sensitized Fluorescent Enantiomers: A New Strategy for High-Efficiency Circularly Polarized Electroluminescence. Chem. Eur. J., 2022. [DOI | PubMed]
- D.-H. Kim, A. D’Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya. High-Efficiency Electroluminescence and Amplified Spontaneous Emission from a Thermally Activated Delayed Fluorescent near-Infrared Emitter. Nat. Photonics, 2018. [DOI]
- Y. Liu, X. Xiao, Y. Ran, Z. Bin, J. You. Molecular Design of Thermally Activated Delayed Fluorescent Emitters for Narrowband Orange-Red OLEDs Boosted by a Cyano-Functionalization Strategy. Chem. Sci., 2021. [DOI | PubMed]
- A. Klimash, P. Pander, W. T. Klooster, S. J. Coles, P. Data, F. B. Dias, P. J. Skabara. Intermolecular Interactions in Molecular Crystals and Their Effect on Thermally Activated Delayed Fluorescence of Helicene-Based Emitters. J. Mater. Chem. C, 2018. [DOI]
- C. M. Hsieh, T. L. Wu, J. Jayakumar, Y. C. Wang, C. L. Ko, W. Y. Hung, T. C. Lin, H. H. Wu, K. H. Lin, C. H. Lin. Diboron-Based Delayed Fluorescent Emitters with Orange-to-Red Emission and Superior Organic Light-Emitting Diode Efficiency. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- D. Karthik, Y. H. Jung, H. Lee, S. Hwang, B. M. Seo, J. Y. Kim, C. W. Han, J. H. Kwon. Acceptor-Donor-Acceptor-Type Orange-Red Thermally Activated Delayed Fluorescence Materials Realizing External Quantum Efficiency over 30% with Low Efficiency Roll-Off. Adv. Mater., 2021. [DOI]
- H. Liu, J. Li, W.-C. Chen, X. Lv, C. Zhou, C.-S. Lee, C. Yang. Efficient Yellow Thermally Activated Delayed Fluorescent Emitters Based on 3,5-Dicyanopyridine Acceptors. J. Phys. Chem. C, 2020. [DOI]
- C. Li, R. Duan, B. Liang, G. Han, S. Wang, K. Ye, Y. Liu, Y. Yi, Y. Wang. Deep-Red to near-Infrared Thermally Activated Delayed Fluorescence in Organic Solid Films and Electroluminescent Devices. Angew. Chem., Int. Ed., 2017. [DOI]
- S. Kothavale, K. H. Lee, J. Y. Lee. Isomeric Quinoxalinedicarbonitrile as Color-Managing Acceptors of Thermally Activated Delayed Fluorescent Emitters. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- S. Kothavale, W. J. Chung, J. Y. Lee. High Efficiency and Long Lifetime Orange-Red Thermally Activated Delayed Fluorescent Organic Light Emitting Diodes by Donor and Acceptor Engineering. J. Mater. Chem. C, 2021. [DOI]
- T. Huang, D. Liu, D. Li, W. Jiang, J. Jiang. Novel Yellow Thermally Activated Delayed Fluorescence Emitters for Highly Efficient Full-TADF WOLEDs with Low Driving Voltages and Remarkable Color Stability. New J. Chem., 2019. [DOI]
- J. Liang, C. Li, Y. Cui, Z. Li, J. Wang, Y. Wang. Rational Design of Efficient Orange-Red to Red Thermally Activated Delayed Fluorescence Emitters for OLEDs with External Quantum Efficiency of up to 26.0% and Reduced Efficiency Roll-Off. J. Mater. Chem. C, 2020. [DOI]
- Y. Yuan, Y. Hu, Y.-X. Zhang, J.-D. Lin, Y.-K. Wang, Z.-Q. Jiang, L.-S. Liao, S.-T. Lee. Light-Emitting Diodes: Over 10% EQE Near-Infrared Electroluminescence Based on a Thermally Activated Delayed Fluorescence Emitter. Adv. Funct. Mater., 2017. [DOI]
- S. Wang, X. Yan, Z. Cheng, H. Zhang, Y. Liu, Y. Wang. Highly Efficient near-Infrared Delayed Fluorescence Organic Light Emitting Diodes Using a Phenanthrene-Based Charge-Transfer Compound. Angew. Chem., Int. Ed., 2015. [DOI]
- J. Xue, Q. Liang, R. Wang, J. Hou, W. Li, Q. Peng, Z. Shuai, J. Qiao. Highly Efficient Thermally Activated Delayed Fluorescence Via J-Aggregates with Strong Intermolecular Charge Transfer. Adv. Mater., 2019. [DOI]
- C. Leng, S. You, Y. Si, H. M. Qin, J. Liu, W. Q. Huang, K. Li. Unraveling the Mechanism of near-Infrared Thermally Activated Delayed Fluorescence of Tpa-Based Molecules: Effect of Hydrogen Bond Steric Hindrance. J. Phys. Chem. A, 2021. [DOI | PubMed]
- X. Gong, P. Li, Y. H. Huang, C. Y. Wang, C. H. Lu, W. K. Lee, C. Zhong, Z. Chen, W. Ning, C. C. Wu. A Red Thermally Activated Delayed Fluorescence Emitter Simultaneously Having High Photoluminescence Quantum Efficiency and Preferentially Horizontal Emitting Dipole Orientation. Adv. Funct. Mater., 2020. [DOI]
- J.-F. Cheng, Z.-H. Pan, K. Zhang, Y. Zhao, C.-K. Wang, L. Ding, M.-K. Fung, J. Fan. Interrupted Intramolecular Donor-Acceptor Interaction Compensated by Strong through-Space Electronic Coupling for Highly Efficient near-Infrared TADF with Emission over 800 nm. Chem. Eng. J., 2022. [DOI]
- X. Gong, C.-H. Lu, W.-K. Lee, P. Li, Y.-H. Huang, Z. Chen, L. Zhan, C.-C. Wu, S. Gong, C. Yang. High-Efficiency Red Thermally Activated Delayed Fluorescence Emitters Based on Benzothiophene-Fused Spiro-Acridine Donor. Chem. Eng. J., 2021. [DOI]
- S. Wang, Z. Cheng, X. Song, X. Yan, K. Ye, Y. Liu, G. Yang, Y. Wang. Highly Efficient Long-Wavelength Thermally Activated Delayed Fluorescence OLEDs Based on Dicyanopyrazino Phenanthrene Derivatives. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- B. Wang, X. Qiao, Z. Yang, Y. Wang, S. Liu, D. Ma, Q. Wang. Realizing Efficient Red Thermally Activated Delayed Fluorescence Organic Light-Emitting Diodes Using Phenoxazine/Phenothiazine-Phenanthrene Hybrids. Org. Electron., 2018. [DOI]
- B. Wang, H. Yang, Y. Zhang, G. Xie, H. Ran, T. Wang, Q. Fu, Y. Ren, N. Sun, G. Zhao. Highly Efficient Electroluminescence from Evaporation- and Solution-Processable Orange-Red Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C, 2019. [DOI]
- R. Furue, K. Matsuo, Y. Ashikari, H. Ooka, N. Amanokura, T. Yasuda. Highly Efficient Red-Orange Delayed Fluorescence Emitters Based on Strong Π-Accepting Dibenzophenazine and Dibenzoquinoxaline Cores: Toward a Rational Pure-Red OLED Design. Adv. Optical Mater., 2018. [DOI]
- Y.-L. Zhang, Q. Ran, Q. Wang, Y. Liu, C. Hänisch, S. Reineke, J. Fan, L.-S. Liao. High-Efficiency Red Organic Light-Emitting Diodes with External Quantum Efficiency Close to 30% Based on a Novel Thermally Activated Delayed Fluorescence Emitter. Adv. Mater., 2019. [DOI]
- S. Kothavale, W. J. Chung, J. Y. Lee. Color Tuning of Dibenzo[a,C]Phenazine-2,7-Dicarbonitrile-Derived Thermally Activated Delayed Fluorescence Emitters from Yellow to Deep-Red. J. Mater. Chem. C, 2020. [DOI]
- F. M. Xie, H. Z. Li, G. L. Dai, Y. Q. Li, T. Cheng, M. Xie, J. X. Tang, X. Zhao. Rational Molecular Design of Dibenzo[a,C]Phenazine-Based Thermally Activated Delayed Fluorescence Emitters for Orange-Red OLEDs with EQE up to 22.0. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- F.-M. Xie, P. Wu, S.-J. Zou, Y.-Q. Li, T. Cheng, M. Xie, J.-X. Tang, X. Zhao. Efficient Orange-Red Delayed Fluorescence Organic Light-Emitting Diodes with External Quantum Efficiency over 26%. Adv. Electron. Mater., 2020. [DOI]
- F.-M. Xie, X.-Y. Zeng, J.-X. Zhou, Z.-D. An, W. Wang, Y.-Q. Li, X.-H. Zhang, J.-X. Tang. Intramolecular H-Bond Design for Efficient Orange-Red Thermally Activated Delayed Fluorescence Based on a Rigid Dibenzo[F,H]Pyrido[2,3-B]Quinoxaline Acceptor. J. Mater. Chem. C, 2020. [DOI]
- C. Zhou, W.-C. Chen, H. Liu, X. Cao, N. Li, Y. Zhang, C.-S. Lee, C. Yang. Isomerization Enhanced Quantum Yield of Dibenzo[a,C]Phenazine-Based Thermally Activated Delayed Fluorescence Emitters for Highly Efficient Orange OLEDs. J. Mater. Chem. C, 2020. [DOI]
- J. X. Chen, W. W. Tao, Y. F. Xiao, K. Wang, M. Zhang, X. C. Fan, W. C. Chen, J. Yu, S. Li, F. X. Geng. Efficient Orange-Red Thermally Activated Delayed Fluorescence Emitters Feasible for Both Thermal Evaporation and Solution Process. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- J. X. Chen, Y. F. Xiao, K. Wang, D. Sun, X. C. Fan, X. Zhang, M. Zhang, Y. Z. Shi, J. Yu, F. X. Geng. Managing Locally Excited and Charge-Transfer Triplet States to Facilitate up-Conversion in Red TADF Emitters That Are Available for Both Vacuum- and Solution-Processes. Angew. Chem., Int. Ed., 2021. [DOI]
- J. Fan, Y. Zhang, Y. Ma, Y. Song, L. Lin, Y. Xu, C.-K. Wang. The Role of Intermolecular Interactions in Regulating the Thermally Activated Delayed Fluorescence and Charge Transfer Properties: A Theoretical Perspective. J. Mater. Chem. C, 2020. [DOI]
- J. X. Chen, W. W. Tao, W. C. Chen, Y. F. Xiao, K. Wang, C. Cao, J. Yu, S. Li, F. X. Geng, C. Adachi. Red/near-Infrared Thermally Activated Delayed Fluorescence OLEDs with near 100% Internal Quantum Efficiency. Angew. Chem., Int. Ed., 2019. [DOI]
- J.-X. Chen, K. Wang, C.-J. Zheng, M. Zhang, Y.-Z. Shi, S.-L. Tao, H. Lin, W. Liu, W.-W. Tao, X.-M. Ou, X.-H. Zhang. Red Organic Light-Emitting Diode with External Quantum Efficiency Beyond 20% Based on a Novel Thermally Activated Delayed Fluorescence Emitter. Adv. Sci., 2018. [DOI]
- J.-X. Chen, W.-W. Tao, Y.-F. Xiao, S. Tian, W.-C. Chen, K. Wang, J. Yu, F.-X. Geng, X.-H. Zhang, C.-S. Lee. Isomeric Thermally Activated Delayed Fluorescence Emitters Based on Indolo[2,3-B]Acridine Electron-Donor: A Compromising Optimization for Efficient Orange-Red Organic Light-Emitting Diodes. J. Mater. Chem. C, 2019. [DOI]
- Y. Liu, Y. Chen, H. Li, S. Wang, X. Wu, H. Tong, L. Wang. High-Performance Solution-Processed Red Thermally Activated Delayed Fluorescence OLEDs Employing Aggregation-Induced Emission-Active Triazatruxene-Based Emitters. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- H. Xu, B. Zhao, H. Wang, C. Han, P. Ma, Z. Li, P. Chang. Highly Efficient Deep-Red Non-Doped Diodes Based on T-Shape Thermally Activated Delayed Fluorescence Emitter. Angew. Chem., Int. Ed., 2020. [DOI]
- K. Zhang, X. Zhang, J. Fan, Y. Song, J. Fan, C. K. Wang, L. Lin. Novel Deep Red Thermally Activated Delayed Fluorescence Molecule with Aggregation-Induced Emission Enhancement: Theoretical Design and Experimental Validation. J. Phys. Chem. Lett, 2022. [DOI | PubMed]
- J. H. Tan, J. M. Jin, W. C. Chen, C. Cao, R. Wang, Z. L. Zhu, Y. Huo, C. S. Lee. The Role of Balancing Carrier Transport in Realizing an Efficient Orange-Red Thermally Activated Delayed-Fluorescence Organic Light-Emitting Diode. ACS Appl. Mater. Interfaces, 2022. [DOI | PubMed]
- S. Kothavale, W. J. Chung, J. Y. Lee. Isomer Engineering of Dipyrido[3,2-A:3′,4′-C]Phenazine-Acceptor-Based Red Thermally Activated Delayed Fluorescent Emitters. J. Mater. Chem. C, 2022. [DOI]
- F.-X. Huang, H.-Z. Li, F.-M. Xie, X.-Y. Zeng, Y.-Q. Li, Y.-Y. Hu, J.-X. Tang, X. Zhao. Efficient Orange-Red Thermally Activated Delayed Fluorescence Material Containing a Cyano Group. Dyes Pigm., 2021. [DOI]
- U. Balijapalli, R. Nagata, N. Yamada, H. Nakanotani, M. Tanaka, A. D’Aleo, V. Placide, M. Mamada, Y. Tsuchiya, C. Adachi. Highly Efficient near-Infrared Electrofluorescence from a Thermally Activated Delayed Fluorescence Molecule. Angew. Chem., Int. Ed., 2021. [DOI]
- Z. Cai, X. Wu, H. Liu, J. Guo, D. Yang, D. Ma, Z. Zhao, B. Z. Tang. Realizing Record-High Electroluminescence Efficiency of 31.5% for Red Thermally Activated Delayed Fluorescence Molecules. Angew. Chem., Int. Ed., 2021. [DOI]
- T. Yang, B. Liang, Z. Cheng, C. Li, G. Lu, Y. Wang. Construction of Efficient Deep-Red/near-Infrared Emitter Based on a Large Π-Conjugated Acceptor and Delayed Fluorescence OLEDs with External Quantum Efficiency of over 20%. J. Phys. Chem. C, 2019. [DOI]
- T. Yang, Z. Cheng, Z. Li, J. Liang, Y. Xu, C. Li, Y. Wang. Improving the Efficiency of Red Thermally Activated Delayed Fluorescence Organic Light-Emitting Diode by Rational Isomer Engineering. Adv. Funct. Mater., 2020. [DOI]
- W. Zeng, H. Y. Lai, W. K. Lee, M. Jiao, Y. J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma. Organic Light-Emitting Diodes: Achieving Nearly 30% External Quantum Efficiency for Orange-Red Organic Light Emitting Diodes by Employing Thermally Activated Delayed Fluorescence Emitters Composed of 1,8-Naphthalimide-Acridine Hybrids. Adv. Mater., 2018. [DOI]
- T. Chen, C.-H. Lu, C.-W. Huang, X. Zeng, J. Gao, Z. Chen, Y. Xiang, W. Zeng, Z. Huang, S. Gong. Tuning the Emissive Characteristics of TADF Emitters by Fusing Heterocycles with Acridine as Donors: Highly Efficient Orange to Red Organic Light-Emitting Diodes with EQE over 20%. J. Mater. Chem. C, 2019. [DOI]
- W. Zeng, T. Zhou, W. Ning, C. Zhong, J. He, S. Gong, G. Xie, C. Yang. Realizing 22.5% External Quantum Efficiency for Solution-Processed Thermally Activated Delayed-Fluorescence OLEDs with Red Emission at 622 Nm Via a Synergistic Strategy of Molecular Engineering and Host Selection. Adv. Mater., 2019. [DOI]
- B. Wang, Y. Zheng, T. Wang, D. Ma, Q. Wang. 1,8-Naphthalimide-Based Hybrids for Efficient Red Thermally Activated Delayed Fluorescence Organic Light-Emitting Diodes. Org. Electron., 2021. [DOI]
- X. Zeng, Y.-H. Huang, S. Gong, X. Yin, W.-K. Lee, X. Xiao, Y. Zhang, W. Zeng, C.-H. Lu, C.-C. Lee. Rational Design of Perfectly Oriented Thermally Activated Delayed Fluorescence Emitter for Efficient Red Electroluminescence. Sci. China Mater., 2021. [DOI]
- T. Chen, C. H. Lu, Z. Chen, X. Gong, C. C. Wu, C. Yang. Modulating the Electron-Donating Ability of Acridine Donor Units for Orange-Red Thermally Activated Delayed Fluorescence Emitters. Chem. Eur. J., 2021. [DOI | PubMed]
- X. Zeng, Y. H. Huang, S. Gong, P. Li, W. K. Lee, X. Xiao, Y. Zhang, C. Zhong, C. C. Wu, C. Yang. An Unsymmetrical Thermally Activated Delayed Fluorescence Emitter Enables Orange-Red Electroluminescence with 31.7% External Quantum Efficiency. Mater. Horiz., 2021. [DOI | PubMed]
- L. Hua, Y. Liu, B. Liu, Z. Zhao, L. Zhang, S. Yan, Z. Ren. Constructing High-Efficiency Orange-Red Thermally Activated Delayed Fluorescence Emitters by Three-Dimension Molecular Engineering. Nat. Commun., 2022. [DOI | PubMed]
- H. Ye, D. H. Kim, X. Chen, A. S. D. Sandanayaka, J. U. Kim, E. Zaborova, G. Canard, Y. Tsuchiya, E. Y. Choi, J. W. Wu. Near Infrared Electroluminescence and Low Threshold Amplified Spontaneous Emission above 800 Nm from a Thermally-Activated Delayed Fluorescent Emitter. Chem. Mater., 2018. [DOI]
- A. Zampetti, A. Minotto, F. Cacialli. Near-Infrared (Nir) Organic Light-Emitting Diodes (OLEDs): Challenges and Opportunities. Adv. Funct. Mater., 2019. [DOI]
- J. Jin, W. Wang, P. Xue, Q. Yang, H. Jiang, Y. Tao, C. Zheng, G. Xie, W. Huang, R. Chen. Intermolecular Locking Design of Red Thermally Activated Delayed Fluorescence Molecules for High-Performance Solution-Processed Organic Light-Emitting Diodes. J. Mater. Chem. C, 2021. [DOI]
- A. Kumar, H. Y. Shin, T. Lee, J. Jung, B. J. Jung, M. H. Lee. Doubly Boron-Doped TADF Emitters Decorated with Ortho-Donor Groups for Highly Efficient Green to Red OLEDs. Chem. Eur. J., 2020. [DOI | PubMed]
- J. Kumsampao, C. Chaiwai, P. Chasing, T. Chawanpunyawat, S. Namuangruk, T. Sudyoadsuk, V. Promarak. A Simple and Strong Electron-Deficient 5,6-Dicyano[2,1,3]Benzothiadiazole-Cored Donor-Acceptor-Donor Compound for Efficient near Infrared Thermally Activated Delayed Fluorescence. Chem. Asian J., 2020. [DOI | PubMed]
- Y.-Y. Wang, K.-N. Tong, K. Zhang, C.-H. Lu, X. Chen, J.-X. Liang, C.-K. Wang, C.-C. Wu, M.-K. Fung, J. Fan. Positive Impact of Chromophore Flexibility on the Efficiency of Red Thermally Activated Delayed Fluorescence Materials. Mater. Horiz., 2021. [DOI | PubMed]
- X. Hu, Y. Qin, Z. Li, H. Gao, T. Gao, G. Liu, X. Dong, N. Tian, X. Gu, C.-S. Lee. Nearly 100% Exciton Utilization in Highly Efficient Red OLEDs Based on Dibenzothioxanthone Acceptor. Chin. Chem. Lett., 2022. [DOI]
- T. Gao, S. Shen, Y. Qin, H. Gao, X. Dong, Z. Pang, P. Wang, Y. Wang, X. Hu. Modulating up-Conversion and Non-Radiative Deactivation to Achieve Efficient Red Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C, 2022. [DOI]
- Q. Zhang, H. Kuwabara, W. J. Potscavage, S. Huang, Y. Hatae, T. Shibata, C. Adachi. Anthraquinone-Based Intramolecular Charge-Transfer Compounds: Computational Molecular Design, Thermally Activated Delayed Fluorescence, and Highly Efficient Red Electroluminescence. J. Am. Chem. Soc., 2014. [DOI | PubMed]
- F.-Y. Hao, Y.-Z. Shi, K. Wang, X.-C. Fan, L. Wu, J. Ye, C.-J. Zheng, Y.-Q. Li, X.-M. Ou, X.-H. Zhang. Forcing Dimethylacridine Crooking to Improve the Efficiency of Orange-Red Thermally Activated Delayed Fluorescent Emitters. J. Mater. Chem. C, 2020. [DOI]
- J. Pandidurai, J. Jayakumar, N. Senthilkumar, C.-H. Cheng. Effects of Intramolecular Hydrogen Bonding on the Conformation and Luminescence Properties of Dibenzoylpyridine-Based Thermally Activated Delayed Fluorescence Materials. J. Mater. Chem. C, 2019. [DOI]
- Z. Wu, D. Ma. Recent Advances in White Organic Light-Emitting Diodes. Mater. Sci. Eng., R, 2016. [DOI]
- Y. Yin, M. U. Ali, W. Xie, H. Yang, H. Meng. Evolution of White Organic Light-Emitting Devices: From Academic Research to Lighting and Display Applications. Mater. Chem. Front., 2019. [DOI]
- H. Liu, F. Liu, P. Lu. Multiple Strategies Towards High-Efficiency White Organic Light-Emitting Diodes by the Vacuum Deposition Method. J. Mater. Chem. C, 2020. [DOI]
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lussem, K. Leo. White Organic Light-Emitting Diodes with Fluorescent Tube Efficiency. Nature, 2009. [DOI | PubMed]
- J. Chen, F. Zhao, D. Ma. Hybrid White OLEDs with Fluorophors and Phosphors. Mater. Today, 2014. [DOI]
- B. Liu, X.-L. Li, H. Tao, J. Zou, M. Xu, L. Wang, J. Peng, Y. Cao. Manipulation of Exciton Distribution for High-Performance Fluorescent/Phosphorescent Hybrid White Organic Light-Emitting Diodes. J. Mater. Chem. C, 2017. [DOI]
- W.-Y. Hung, G.-C. Fang, S.-W. Lin, S.-H. Cheng, K.-T. Wong, T.-Y. Kuo, P.-T. Chou. The First Tandem, All-Exciplex-Based WOLED. Sci. Rep., 2014. [DOI | PubMed]
- B. Zhang, L. Liu, Z. Xie. Recent Advances in Solution-Processed White Organic Light-Emitting Materials and Devices. Isr. J. Chem., 2014. [DOI]
- D. Barman, M. Annadhasan, R. Chandrasekar, P. K. Iyer. Hot-Exciton Harvesting Via through-Space Single-Molecule Based White-Light Emission and Optical Waveguides. Chem. Sci., 2022. [DOI | PubMed]
- Z. Chen, C. L. Ho, L. Wang, W. Y. Wong. Single-Molecular White-Light Emitters and Their Potential WOLED Applications. Adv. Mater., 2020. [DOI]
- X.-K. Liu, Z. Chen, J. Qing, W.-J. Zhang, B. Wu, H. L. Tam, F. Zhu, X.-H. Zhang, C.-S. Lee. Remanagement of Singlet and Triplet Excitons in Single-Emissive-Layer Hybrid White Organic Light-Emitting Devices Using Thermally Activated Delayed Fluorescent Blue Exciplex. Adv. Mater., 2015. [DOI | PubMed]
- Z. Wu, J. Luo, N. Sun, L. Zhu, H. Sun, L. Yu, D. Yang, X. Qiao, J. Chen, C. Yang. High-Performance Hybrid White Organic Light-Emitting Diodes with Superior Efficiency/Color Rendering Index/Color Stability and Low Efficiency Roll-Off Based on a Blue Thermally Activated Delayed Fluorescent Emitter. Adv. Funct. Mater., 2016. [DOI]
- F. Li, G. Jiang, M. Li, J. Fan, Y. Song, C. K. Wang, L. Lin. Thermally Activated Delayed Fluorescence Emitters with Dual Conformations for White Organic Light-Emitting Diodes: Mechanism and Molecular Design. Phys. Chem. Chem. Phys., 2020. [DOI | PubMed]
- Y. J. Cho, K. S. Yook, J. Y. Lee. Cool and Warm Hybrid White Organic Light-Emitting Diode with Blue Delayed Fluorescent Emitter Both as Blue Emitter and Triplet Host. Sci. Rep., 2015. [DOI | PubMed]
- D. Zhang, M. Cai, Y. Zhang, D. Zhang, L. Duan. Highly Efficient Simplified Single-Emitting-Layer Hybrid WOLEDs with Low Roll-Off and Good Color Stability through Enhanced Forster Energy Transfer. ACS Appl. Mater. Interfaces, 2015. [DOI | PubMed]
- Q. Zhang, D. Tsang, H. Kuwabara, Y. Hatae, B. Li, T. Takahashi, S. Y. Lee, T. Yasuda, C. Adachi. Nearly 100% Internal Quantum Efficiency in Undoped Electroluminescent Devices Employing Pure Organic Emitters. Adv. Mater., 2015. [DOI | PubMed]
- K. Wang, Y. Shi, C.-J. Zheng, W. Liu, K. Liang, X. Li, M. Zhang, H. Lin, S. Tao, C.-S. Lee. Control of Dual Conformations: Developing TADF Emitters for Highly Efficient Single-Emitter White Organic-Light Emitting Diodes. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- C. Huang, Y. Zhang, J. Zhou, S. Sun, W. Luo, W. He, J. Wang, X. Shi, M. K. Fung. Hybrid Tandem White OLED with Long Lifetime and 150 Lm W-1 in Luminous Efficacy Based on TADF Blue Emitter Stabilized with Phosphorescent Red Emitter. Adv. Optical Mater., 2020. [DOI]
- C. Zhang, Y. Lu, Z. Liu, Y. Zhang, X. Wang, D. Zhang, L. Duan. A Pi-D and Pi-a Exciplex-Forming Host for High-Efficiency and Long-Lifetime Single-Emissive-Layer Fluorescent White Organic Light-Emitting Diodes. Adv. Mater., 2020. [DOI]
- L. Ying, C. L. Ho, H. Wu, Y. Cao, W. Y. Wong. White Polymer Light-Emitting Devices for Solid-State Lighting: Materials, Devices, and Recent Progress. Adv. Mater., 2014. [DOI | PubMed]
- M. C. Gather, S. Reineke. Recent Advances in Light Outcoupling from White Organic Light-Emitting Diodes. J. Photon. Energy, 2015. [DOI]
- G. Schwartz, K. Fehse, M. Pfeiffer, K. Walzer, K. Leo. Highly Efficient White Organic Light Emitting Diodes Comprising an Interlayer to Separate Fluorescent and Phosphorescent Regions. Appl. Phys. Lett., 2006. [DOI]
- J. Liang, C. Li, X. Zhuang, K. Ye, Y. Liu, Y. Wang. Novel Blue Bipolar Thermally Activated Delayed Fluorescence Material as Host Emitter for High-Efficiency Hybrid Warm-White OLEDs with Stable High Color-Rendering Index. Adv. Funct. Mater., 2018. [DOI]
- S.-F. Wu, S.-H. Li, Y.-K. Wang, C.-C. Huang, Q. Sun, J.-J. Liang, L.-S. Liao, M.-K. Fung. Organic Light-Emitting Diodes: White Organic Led with a Luminous Efficacy Exceeding 100 Lm W-1 without Light out-Coupling Enhancement Techniques. Adv. Funct. Mater., 2017. [DOI]
- S. H. Han, Y. H. Park, J. Y. Lee. Design of High-Efficiency and Long-Lifetime White Organic Light-Emitting Diodes by Selective Management of Singlet and Triplet Excitons Using a Triplet Exciton Manager. Adv. Optical Mater., 2018. [DOI]
- W. Song, J. Y. Lee. High-Power-Efficiency Hybrid White Organic Light-Emitting Diodes with a Single Emitting Layer Doped with Blue Delayed Fluorescent and Yellow Phosphorescent Emitters. J. Phys. D: Appl. Phys., 2015. [DOI]
- Y. Chen, Q. Sun, Y. Dai, D. Yang, X. Qiao, D. Ma. High Efficiency Blue and Color-Stable Hybrid Warm White Organic Light-Emitting Diodes Based on a Thermally Activated Delayed Fluorescent Material as an Assistant Host. J. Mater. Chem. C, 2020. [DOI]
- C. Xue, G. Zhang, W. Jiang, J. Lang, X. Jiang. High Performance TADF-Phosphorescence Hybrid Warm-White Organic Light-Emitting Diodes with a Simple Fully Doping-Free Device Structure. J. Appl. Phys., 2020. [DOI]
- C. Zhao, T. Zhang, J. Chen, D. Yan, D. Ma. High-Performance Hybrid White Organic Light-Emitting Diodes with Simple Emitting Structures and Low Efficiency Roll-Off Based on Blue Thermally Activated Delayed Fluorescence Emitters with Bipolar Transport Characteristics. J. Mater. Chem. C, 2018. [DOI]
- Y. Qi, Z. Wang, S. Hou, J. Yu. Color Stable and Highly Efficient Hybrid White Organic Light-Emitting Devices Using Heavily Doped Thermally Activated Delayed Fluorescence and Ultrathin Non-Doped Phosphorescence Layers. Org. Electron., 2017. [DOI]
- Z. He, J. Li, D. Liu, H. Wan, Y. Mei, C. Shi. Multichannel Charge Transfer Enhanced Radiative Decay and RISC in TADF Materials Containing Multiple Donors and Acceptors. J. Mater. Chem. C, 2022. [DOI]
- H. Wang, C. Zang, G. Shan, Z. Yu, S. Liu, L. Zhang, W. Xie, H. Zhao. Bluish-Green Thermally Activated Delayed Fluorescence Material for Blue-Hazard Free Hybrid White Organic Light-Emitting Device with High Color Quality and Low Efficiency Roll-Off. Adv. Optical Mater., 2019. [DOI]
- P. Wei, D. Zhang, M. Cai, X. Song, Z. Wang, L. Duan. Simplified Single-Emitting-Layer Hybrid White Organic Light-Emitting Diodes with High Efficiency, Low Efficiency Roll-Off, High Color Rendering Index and Superior Color Stability. Org. Electron., 2017. [DOI]
- C. Duan, C. Han, R. Du, Y. Wei, H. Xu. High-Efficiency Blue Dual-Emissive Exciplex Boosts Full-Radiative White Electroluminescence. Adv. Optical Mater., 2018. [DOI]
- S. Ying, S. Zhang, J. Yao, Y. Dai, Q. Sun, D. Yang, X. Qiao, J. Chen, D. Ma. High-Performance White Organic Light-Emitting Diodes with Doping-Free Device Architecture Based on the Exciton Adjusting Interfacial Exciplex. J. Mater. Chem. C, 2020. [DOI]
- G. Wang, M. Yin, Y. Miao, Y. Guo, H. Zhou, Q. Lu, J. Huang, H. Wang. Combining Intrinsic (Blue) and Exciplex (Green and Orange-Red) Emissions of the Same Material (Oct) in White Organic Light-Emitting Diodes to Realize High Color Quality with a Cri of 97. J. Mater. Chem. C, 2022. [DOI]
- D. Ding, Z. Wang, C. Li, J. Zhang, C. Duan, Y. Wei, H. Xu. Highly Efficient and Color-Stable Thermally Activated Delayed Fluorescence White Light-Emitting Diodes Featured with Single-Doped Single Emissive Layers. Adv. Mater., 2020. [DOI]
- Z. Liu, Y. Lei, C. Fan, X. Peng, X. Ji, G. E. Jabbour, X. Yang. Simple-Structure Organic Light Emitting Diodes: Exploring the Use of Thermally Activated Delayed Fluorescence Host and Guest Materials. Org. Electron., 2017. [DOI]
- W. Luo, T.-T. Wang, X. Chen, K.-N. Tong, W. He, S.-Q. Sun, Y.-J. Zhang, L.-S. Liao, M.-K. Fung. High-Performance Organic Light-Emitting Diodes with Natural White Emission Based on Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C, 2020. [DOI]
- C. Han, W. Yang, H. Xu. Asymmetrically Phosphorylated Carbazole Host for Highly Efficient Blue and White Thermally Activated Delayed Fluorescence Diodes. Chem. Eng. J., 2020. [DOI]
- J. Zhang, C. Han, F. Du, C. Duan, Y. Wei, H. Xu. High-Power-Efficiency White Thermally Activated Delayed Fluorescence Diodes Based on Selectively Optimized Intermolecular Interactions. Adv. Funct. Mater., 2020. [DOI]
- J. Zhang, Y. Wei, H. Xu. High-Power-Efficiency Thermally Activated Delayed Fluorescence White Organic Light-Emitting Diodes Based on Asymmetrical Host Engineering. Nano Energy, 2021. [DOI]
- J. Sun, J. Zhang, Q. Liang, Y. Wei, C. Duan, C. Han, H. Xu. Charge-Transfer Exciton Manipulation Based on Hydrogen Bond for Efficient White Thermally Activated Delayed Fluorescence. Adv. Funct. Mater., 2020. [DOI]
- Y. Li, Z. Li, J. Zhang, C. Han, C. Duan, H. Xu. Manipulating Complementarity of Binary White Thermally Activated Delayed Fluorescence Systems for 100% Exciton Harvesting in OLEDs. Adv. Funct. Mater., 2021. [DOI]
- X. Y. Zeng, J. X. Zhou, S. J. Zou, Y. Q. Tang, H. Z. Li, Y. H. He, Y. Q. Li, W. J. Wang, J. X. Tang. Management of Multi-Energy-Transfer Channels and Exciton Harvesting for Power-Efficient White Thermally Activated Delayed Fluorescence Diodes. Adv. Optical Mater., 2022. [DOI]
- J.-i. Nishide, H. Nakanotani, Y. Hiraga, C. Adachi. High-Efficiency White Organic Light-Emitting Diodes Using Thermally Activated Delayed Fluorescence. Appl. Phys. Lett., 2014. [DOI]
- D. d. S. Pereira, P. L. dos Santos, J. S. Ward, P. Data, M. Okazaki, Y. Takeda, S. Minakata, M. R. Bryce, A. P. Monkman. An Optical and Electrical Study of Full Thermally Activated Delayed Fluorescent White Organic Light-Emitting Diodes. Sci. Rep., 2017. [DOI | PubMed]
- X. Hong, D. Zhang, C. Yin, Q. Wang, Y. Zhang, T. Huang, J. Wei, X. Zeng, G. Meng, X. Wang. TADF Molecules with Π-Extended Acceptors for Simplified High-Efficiency Blue and White Organic Light-Emitting Diodes. CHEM, 2022. [DOI]
- X. Hu, N. Aizawa, M. Kim, M. Huang, Z. Li, G. Liu, H. Gao, T. Gao, X. Dong, Y. Zhang. Dual-Acceptor Thermally Activated Delayed Fluorescence Emitters: Achieving High Efficiency and Long Lifetime in Orange-Red OLEDs. Chem. Eng. J., 2022. [DOI]
- Z. Wang, X. L. Li, Z. Ma, X. Cai, C. Cai, S. J. Su. Exciton-Adjustable Interlayers for High Efficiency, Low Efficiency Roll-Off, and Lifetime Improved Warm White Organic Light-Emitting Diodes (WOLEDs) Based on a Delayed Fluorescence Assistant Host. Adv. Funct. Mater., 2018. [DOI]
- X. Tang, Y. Li, Y. K. Qu, C. C. Peng, A. Khan, Z. Q. Jiang, L. S. Liao. All-Fluorescence White Organic Light-Emitting Diodes Exceeding 20% EQEs by Rational Manipulation of Singlet and Triplet Excitons. Adv. Funct. Mater., 2020. [DOI]
- C. Duan, Y. Xin, Z. Wang, J. Zhang, C. Han, H. Xu. High-Efficiency Hyperfluorescent White Light-Emitting Diodes Based on High-Concentration-Doped TADF Sensitizer Matrices Via Spatial and Energy Gap Effects. Chem. Sci., 2021. [DOI | PubMed]
- D. Ding, Z. Wang, C. Duan, C. Han, J. Zhang, S. Chen, Y. Wei, H. Xu. White Fluorescent Organic Light-Emitting Diodes with 100% Power Conversion. Research, 2022. [DOI | PubMed]
- E. Aksoy, A. Danos, C. Varlikli, A. P. Monkman. Navigating Cie Space for Efficient TADF Downconversion WOLEDs. Dyes Pigm., 2020. [DOI]
- B. Zhao, Z. Liu, Z. Wang, H. Zhang, J. Li, H. Wang, B. Xu, Y. Wu, W. Li. Structural Design for Highly Efficient Pure Fluorescent Warm WOLEDs by Employing TADF Molecule as Blue Emitter and Exciplex Donor. Org. Electron., 2019. [DOI]
- G. Grybauskaite-Kaminskiene, K. Ivaniuk, G. Bagdziunas, P. Turyk, P. Stakhira, G. Baryshnikov, D. Volyniuk, V. Cherpak, B. Minaev, Z. Hotra. Contribution of TADF and Exciplex Emission for Efficient “Warm-White” OLEDs. J. Mater. Chem. C, 2018. [DOI]
- Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu, Z. Chi. White-Light Emission Strategy of a Single Organic Compound with Aggregation-Induced Emission and Delayed Fluorescence Properties. Angew. Chem., Int. Ed., 2015. [DOI]
- C. Li, J. Liang, B. Liang, Z. Li, Z. Cheng, G. Yang, Y. Wang. An Organic Emitter Displaying Dual Emissions and Efficient Delayed Fluorescence White OLEDs. Adv. Optical Mater., 2019. [DOI]
- B. Li, Z. Li, F. Guo, J. Song, X. Jiang, Y. Wang, S. Gao, J. Wang, X. Pang, L. Zhao, Y. Zhang. Realizing Efficient Single Organic Molecular White Light-Emitting Diodes from Conformational Isomerization of Quinazoline-Based Emitters. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- H. L. Lee, H. J. Jang, J. Y. Lee. Single Molecule White Emission by Intra- and Inter-Molecular Charge Transfer. J. Mater. Chem. C, 2020. [DOI]
- D. Zhang, L. Duan, Y. Li, D. Zhang, Y. Qiu. Highly Efficient and Color-Stable Hybrid Warm White Organic Light-Emitting Diodes Using a Blue Material with Thermally Activated Delayed Fluorescence. J. Mater. Chem. C, 2014. [DOI]
- C. Wang, H. Fei, Y. Qiu, Y. Yang, Z. Wei, Y. Tian, Y. Chen, Y. Zhao. Photoinduced Birefringence and Reversible Optical Storage in Liquid-Crystalline Azobenzene Side-Chain Polymers. Appl. Phys. Lett., 1999. [DOI]
- R. Farshchi, M. Ramsteiner, J. Herfort, A. Tahraoui, H. T. Grahn. Optical Communication of Spin Information between Light Emitting Diodes. Appl. Phys. Lett., 2011. [DOI]
- J. R. Brandt, F. Salerno, M. J. Fuchter. The Added Value of Small-Molecule Chirality in Technological Applications. Nat. Rev. Chem., 2017. [DOI]
- B. C. Kim, Y. J. Lim, J. H. Song, J. H. Lee, K.-U. Jeong, J. H. Lee, G.-D. Lee, S. H. Lee. Wideband Antireflective Circular Polarizer Exhibiting a Perfect Dark State in Organic Light-Emitting-Diode Display. Opt. Express, 2014. [DOI | PubMed]
- Y. Xu, Q. Wang, X. Song, Y. Wang, C. Li. New Fields, New Opportunities and New Challenges: Circularly Polarized Multiple Resonance Thermally Activated Delayed Fluorescence Materials. Chem. Eur. J., 2023. [DOI | PubMed]
- M. Li, C.-F. Chen. Advances in Circularly Polarized Electroluminescence Based on Chiral TADF-Active Materials. Org. Chem. Front., 2022. [DOI]
- H. Kubo, D. Shimizu, T. Hirose, K. Matsuda. Circularly Polarized Luminescence Designed from Molecular Orbitals: A Figure-Eight-Shaped [5]Helicene Dimer with D2 Symmetry. Org. Lett., 2020. [DOI | PubMed]
- Y. Nagata, T. Mori. Irreverent Nature of Dissymmetry Factor and Quantum Yield in Circularly Polarized Luminescence of Small Organic Molecules. Front. Chem., 2020. [DOI | PubMed]
- H. Tanaka, Y. Inoue, T. Mori. Circularly Polarized Luminescence and Circular Dichroisms in Small Organic Molecules: Correlation between Excitation and Emission Dissymmetry Factors. ChemPhotoChem, 2018. [DOI]
- H. Tanaka, M. Ikenosako, Y. Kato, M. Fujiki, Y. Inoue, T. Mori. Symmetry-Based Rational Design for Boosting Chiroptical Responses. Commun. Chem., 2018. [DOI]
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz, S. De La Moya. Circularly Polarized Luminescence from Simple Organic Molecules. Chem. Eur. J., 2015. [DOI | PubMed]
- J. E. Field, G. Muller, J. P. Riehl, D. Venkataraman. Circularly Polarized Luminescence from Bridged Triarylamine Helicenes. J. Am. Chem. Soc., 2003. [DOI | PubMed]
- T. Imagawa, S. Hirata, K. Totani, T. Watanabe, M. Vacha. Thermally Activated Delayed Fluorescence with Circularly Polarized Luminescence Characteristics. Chem. Commun., 2015. [DOI]
- F.-Y. Hao, Y.-Z. Shi, K. Wang, S.-Y. Xiong, X.-C. Fan, L. Wu, C.-J. Zheng, Y.-Q. Li, X.-M. Ou, X.-H. Zhang. Chiral Thermally Activated Delayed Fluorescence Emitters with Dual Conformations Based on a Pair of Enantiomeric Donors Containing Asymmetric Carbons. Dyes Pigm., 2020. [DOI]
- F. Ni, C.-W. Huang, Y. Tang, Z. Chen, Y. Wu, S. Xia, X. Cao, J.-H. Hsu, W.-K. Lee, K. Zheng. Integrating Molecular Rigidity and Chirality into Thermally Activated Delayed Fluorescence Emitters for Highly Efficient Sky-Blue and Orange Circularly Polarized Electroluminescence. Mater. Horiz., 2021. [DOI | PubMed]
- Y. Yang, N. Li, J. Miao, X. Cao, A. Ying, K. Pan, X. Lv, F. Ni, Z. Huang, S. Gong, C. Yang. Chiral Multi-Resonance TADF Emitters Exhibiting Narrowband Circularly Polarized Electroluminescence with an EQE of 37.2. Angew. Chem., Int. Ed., 2022. [DOI]
- S. Y. Yang, Y. K. Wang, C. C. Peng, Z. G. Wu, S. Yuan, Y. J. Yu, H. Li, T. T. Wang, H. C. Li, Y. X. Zheng. Circularly Polarized Thermally Activated Delayed Fluorescence Emitters in through-Space Charge Transfer on Asymmetric Spiro Skeletons. J. Am. Chem. Soc., 2020. [DOI | PubMed]
- Y. P. Zhang, X. Liang, X. F. Luo, S. Q. Song, S. Li, Y. Wang, Z. P. Mao, W. Y. Xu, Y. X. Zheng, J. L. Zuo, Y. Pan. Chiral Spiro-Axis Induced Blue Thermally Activated Delayed Fluorescence Material for Efficient Circularly Polarized OLEDs with Low Efficiency Roll-Off. Angew. Chem., Int. Ed., 2021. [DOI]
- Z. Hao, N. Li, J. Miao, Z. Huang, X. Lv, X. Cao. Chiral Sulfoximine-Based TADF Emitter for Circularly Polarized Luminescence and Highly Efficient OLEDs. Chem. Eng. J., 2023. [DOI]
- Z. Huang, C.-W. Huang, Y.-K. Tang, Z. Xiao, N. Li, T. Hua, X. Cao, C. Zhou, C.-C. Wu, C. Yang. Chiral Thermally Activated Delayed Fluorescence Emitters for Circularly Polarized Luminescence and Efficient Deep Blue OLEDs. Dyes Pigm., 2022. [DOI]
- Y.-P. Zhang, S.-Q. Song, M.-X. Mao, C.-H. Li, Y.-X. Zheng, J.-L. Zuo. Efficient Circularly Polarized Photoluminescence and Electroluminescence of Chiral Spiro-Skeleton Based Thermally Activated Delayed Fluorescence Molecules. Sci. China Chem., 2022. [DOI]
- Y. Wang, Y. Zhang, W. Hu, Y. Quan, Y. Li, Y. Cheng. Circularly Polarized Electroluminescence of Thermally Activated Delayed Fluorescence-Active Chiral Binaphthyl-Based Luminogens. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- Z. P. Yan, T. T. Liu, R. Wu, X. Liang, Z. Q. Li, L. Zhou, Y. X. Zheng, J. L. Zuo. Chiral Thermally Activated Delayed Fluorescence Materials Based on R/S-N2, N2′-Diphenyl-[1,1′-Binaphthalene]-2,2′-Diamine Donor with Narrow Emission Spectra for Highly Efficient Circularly Polarized Electroluminescence. Adv. Funct. Mater., 2021. [DOI]
- Z. P. Yan, L. Yuan, Y. Zhang, M. X. Mao, X. J. Liao, H. X. Ni, Z. H. Wang, Z. An, Y. X. Zheng, J. L. Zuo. A Chiral Dual-Core Organoboron Structure Realizes Dual-Channel Enhanced Ultrapure Blue Emission and Highly Efficient Circularly Polarized Electroluminescence. Adv. Mater., 2022. [DOI]
- M. Li, Y. F. Wang, D. D. Zhang, L. Duan, C. F. Chen. Axially Chiral TADF-Active Enantiomers Designed for Efficient Blue Circularly Polarized Electroluminescence. Angew. Chem., Int. Ed., 2020. [DOI]
- Z. L. Tu, Z. P. Yan, X. Liang, L. Chen, Z. G. Wu, Y. Wang, Y. X. Zheng, J. L. Zuo, Y. Pan. Axially Chiral Biphenyl Compound-Based Thermally Activated Delayed Fluorescent Materials for High-Performance Circularly Polarized Organic Light-Emitting Diodes. Adv. Sci., 2020. [DOI]
- Z. L. Tu, J. J. Lu, X. F. Luo, J. J. Hu, S. Li, Y. Wang, Y. X. Zheng, J. L. Zuo, Y. Pan. Blue Axially Chiral Biphenyl Based Thermally Activated Delayed Fluorescence Materials for Efficient Circularly Polarized OLEDs. Adv. Optical Mater., 2021. [DOI]
- P. Sumsalee, L. Abella, T. Roisnel, S. Lebrequier, G. Pieters, J. Autschbach, J. Crassous, L. Favereau. Axial and Helical Thermally Activated Delayed Fluorescence Bicarbazole Emitters: Opposite Modulation of Circularly Polarized Luminescence through Intramolecular Charge-Transfer Dynamics. J. Mater. Chem. C, 2021. [DOI]
- L. Poulard, S. Kasemthaveechok, M. Coehlo, R. A. Kumar, L. Frederic, P. Sumsalee, T. d’Anfray, S. Wu, J. Wang, T. Matulaitis. Circularly Polarized-Thermally Activated Delayed Fluorescent Materials Based on Chiral Bicarbazole Donors. Chem. Commun., 2022. [DOI]
- M.-Y. Zhang, Z.-Y. Li, B. Lu, Y. Wang, Y.-D. Ma, C.-H. Zhao. Solid-State Emissive Triarylborane-Based [2.2]Paracyclophanes Displaying Circularly Polarized Luminescence and Thermally Activated Delayed Fluorescence. Org. Lett., 2018. [DOI | PubMed]
- C. Liao, Y. Zhang, S. H. Ye, W. H. Zheng. Planar Chiral [2.2]Paracyclophane-Based Thermally Activated Delayed Fluorescent Materials for Circularly Polarized Electroluminescence. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- D. W. Zhang, J. M. Teng, Y. F. Wang, X. N. Han, M. Li, C. F. Chen. D-Pi*-a Type Planar Chiral TADF Materials for Efficient Circularly Polarized Electroluminescence. Mater. Horiz., 2021. [DOI | PubMed]
- X. J. Liao, D. Pu, L. Yuan, J. Tong, S. Xing, Z. L. Tu, J. L. Zuo, W. H. Zheng, Y. X. Zheng. Planar Chiral Multiple Resonance Thermally Activated Delayed Fluorescence Materials for Efficient Circularly Polarized Electroluminescence. Angew. Chem., Int. Ed., 2023. [DOI]
- S. Y. Yang, S. N. Zou, F. C. Kong, X. J. Liao, Y. K. Qu, Z. Q. Feng, Y. X. Zheng, Z. Q. Jiang, L. S. Liao. A Narrowband Blue Circularly Polarized Thermally Activated Delayed Fluorescence Emitter with a Hetero-Helicene Structure. Chem. Commun., 2021. [DOI]
- S.-Y. Yang, Q.-S. Tian, X.-J. Liao, Z.-G. Wu, W.-S. Shen, Y.-J. Yu, Z.-Q. Feng, Y.-X. Zheng, Z.-Q. Jiang, L.-S. Liao. Efficient Circularly Polarized Thermally Activated Delayed Fluorescence Hetero-[4]Helicene with Carbonyl-/Sulfone-Bridged Triarylamine Structures. J. Mater. Chem. C, 2022. [DOI]
- J. M. dos Santos, D. Sun, J. M. Moreno-Naranjo, D. Hall, F. Zinna, S. T. J. Ryan, W. Shi, T. Matulaitis, D. B. Cordes, A. M. Z. Slawin. An S-Shaped Double Helicene Showing Both Multi-Resonance Thermally Activated Delayed Fluorescence and Circularly Polarized Luminescence. J. Mater. Chem. C, 2022. [DOI]
- W. Ning, H. Wang, S. Gong, C. Zhong, C. Yang. Simple Sulfone-Bridged Heterohelicene Structure Realizes Ultraviolet Narrowband Thermally Activated Delayed Fluorescence, Circularly Polarized Luminescence, and Room Temperature Phosphorescence. Sci. China Chem., 2022. [DOI]
- X. Wu, J. W. Huang, B. K. Su, S. Wang, L. Yuan, W. Q. Zheng, H. Zhang, Y. X. Zheng, W. Zhu, P. T. Chou. Fabrication of Circularly Polarized MR-TADF Emitters with Asymmetrical Peripheral-Lock Enhancing Helical B/N-Doped Nanographenes. Adv. Mater., 2022. [DOI]
- W. Yang, N. Li, J. Miao, L. Zhan, S. Gong, Z. Huang, C. Yang. Simple Double Hetero[5]Helicenes Realize Highly Efficient and Narrowband Circularly Polarized Organic Light-Emitting Diodes. CCS Chem., 2022. [DOI]
- J. K. Li, X. Y. Chen, Y. L. Guo, X. C. Wang, A. C. Sue, X. Y. Cao, X. Y. Wang. B,N-Embedded Double Hetero[7]Helicenes with Strong Chiroptical Responses in the Visible Light Region. J. Am. Chem. Soc., 2021. [DOI | PubMed]
- S. Feuillastre, M. Pauton, L. Gao, A. Desmarchelier, A. J. Riives, D. Prim, D. Tondelier, B. Geffroy, G. Muller, G. Clavier, G. Pieters. Design and Synthesis of New Circularly Polarized Thermally Activated Delayed Fluorescence Emitters. J. Am. Chem. Soc., 2016. [DOI | PubMed]
- F. Song, Z. Xu, Q. Zhang, Z. Zhao, H. Zhang, W. Zhao, Z. Qiu, C. Qi, H. Zhang, H. H. Y. Sung. Highly Efficient Circularly Polarized Electroluminescence from Aggregation-Induced Emission Luminogens with Amplified Chirality and Delayed Fluorescence. Adv. Funct. Mater., 2018. [DOI]
- S. Sun, J. Wang, L. Chen, R. Chen, J. Jin, C. Chen, S. Chen, G. Xie, C. Zheng, W. Huang. Thermally Activated Delayed Fluorescence Enantiomers for Solution-Processed Circularly Polarized Electroluminescence. J. Mater. Chem. C, 2019. [DOI]
- L. Frédéric, A. Desmarchelier, R. Plais, L. Lavnevich, G. Muller, C. Villafuerte, G. Clavier, E. Quesnel, B. Racine, S. Meunier-Della-Gatta. Maximizing Chiral Perturbation on Thermally Activated Delayed Fluorescence Emitters and Elaboration of the First Top-Emission Circularly Polarized OLED. Adv. Funct. Mater., 2020. [DOI | PubMed]
- L. Zhou, F. Ni, N. Li, K. Wang, G. Xie, C. Yang. Tetracoordinate Boron-Based Multifunctional Chiral Thermally Activated Delayed Fluorescence Emitters. Angew. Chem., Int. Ed., 2022. [DOI]
- P. Xue, X. Wang, W. Wang, J. Zhang, Z. Wang, J. Jin, C. Zheng, P. Li, G. Xie, R. Chen. Solution-Processable Chiral Boron Complexes for Circularly Polarized Red Thermally Activated Delayed Fluorescent Devices. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- Z.-G. Wu, H.-B. Han, Z.-P. Yan, X.-F. Luo, Y. Wang, Y.-X. Zheng, J.-L. Zuo, Y. Pan. Chiral Octahydro-Binaphthol Compound-Based Thermally Activated Delayed Fluorescence Materials for Circularly Polarized Electroluminescence with Superior EQE of 32.6% and Extremely Low Efficiency Roll-Off. Adv. Mater., 2019. [DOI]
- Z.-G. Wu, Z.-P. Yan, X.-F. Luo, L. Yuan, W.-Q. Liang, Y. Wang, Y.-X. Zheng, J.-L. Zuo, Y. Pan. Non-Doped and Doped Circularly Polarized Organic Light-Emitting Diodes with High Performances Based on Chiral Octahydro-Binaphthyl Delayed Fluorescent Luminophores. J. Mater. Chem. C, 2019. [DOI]
- T. T. Liu, Z. P. Yan, J. J. Hu, L. Yuan, X. F. Luo, Z. L. Tu, Y. X. Zheng. Chiral Thermally Activated Delayed Fluorescence Emitters-Based Efficient Circularly Polarized Organic Light-Emitting Diodes Featuring Low Efficiency Roll-Off. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- Y. Xu, Q. Wang, X. Cai, C. Li, Y. Wang. Highly Efficient Electroluminescence from Narrowband Green Circularly Polarized Multiple Resonance Thermally Activated Delayed Fluorescence Enantiomers. Adv. Mater., 2021. [DOI]
- J. M. Teng, D. W. Zhang, Y. F. Wang, C. F. Chen. Chiral Conjugated Thermally Activated Delayed Fluorescent Polymers for Highly Efficient Circularly Polarized Polymer Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2022. [DOI | PubMed]
- F. M. Xie, J. X. Zhou, X. Y. Zeng, Z. D. An, Y. Q. Li, D. X. Han, P. F. Duan, Z. G. Wu, Y. X. Zheng, J. X. Tang. Efficient Circularly Polarized Electroluminescence from Chiral Thermally Activated Delayed Fluorescence Emitters Featuring Symmetrical and Rigid Coplanar Acceptors. Adv. Optical Mater., 2021. [DOI]
- W.-L. Zhao, Y.-F. Wang, S.-P. Wan, H.-Y. Lu, M. Li, C.-F. Chen. Chiral Thermally Activated Delayed Fluorescence-Active Macrocycles Displaying Efficient Circularly Polarized Electroluminescence. CCS Chem., 2022. [DOI]
- Y.-F. Wang, X. Liu, Y. Zhu, M. Li, C.-F. Chen. Aromatic-Imide-Based TADF Enantiomers for Efficient Circularly Polarized Electroluminescence. J. Mater. Chem. C, 2022. [DOI]
- M. Li, S.-H. Li, D. Zhang, M. Cai, L. Duan, M.-K. Fung, C.-F. Chen. Stable Enantiomers Displaying Thermally Activated Delayed Fluorescence: Efficient OLEDs with Circularly Polarized Electroluminescence. Angew. Chem., Int. Ed., 2018. [DOI]
- Y.-F. Wang, H.-Y. Lu, C. Chen, M. Li, C.-F. Chen. 1,8-Naphthalimide-Based Circularly Polarized TADF Enantiomers as the Emitters for Efficient Orange-Red OLEDs. Org. Electron., 2019. [DOI]
- Y. Zheng, L. Zhang, Z. Huang, S. Li, L. Zuo, Y. Liang, C. Liu, S. Luo, G. Shi, Z. Zhao. Bright Organic Mechanoluminescence and Remarkable Mechanofluorochromism from Circularly Polarized TADF Enantiomers with Aggregation-Induced Emission Properties. Chem. Eur. J., 2023. [DOI | PubMed]
- Y. F. Wang, M. Li, J. M. Teng, H. Y. Zhou, C. F. Chen. High-Performance Solution-Processed Nondoped Circularly Polarized OLEDs with Chiral Triptycene Scaffold-Based TADF Emitters Realizing over 20% External Quantum Efficiency. Adv. Funct. Mater., 2021. [DOI]
- Y. F. Wang, M. Li, J. M. Teng, H. Y. Zhou, W. L. Zhao, C. F. Chen. Chiral TADF-Active Polymers for High-Efficiency Circularly Polarized Organic Light-Emitting Diodes. Angew. Chem., Int. Ed., 2021. [DOI]
- P. Sumsalee, L. Abella, S. Kasemthaveechok, N. Vanthuyne, M. Cordier, G. Pieters, J. Autschbach, J. Crassous, L. Favereau. Luminescent Chiral Exciplexes with Sky-Blue and Green Circularly Polarized-Thermally Activated Delayed Fluorescence. Chem. Eur. J., 2021. [DOI | PubMed]
- Q. Gu, Z. Chen, W. Xie, W. Qiu, X. Peng, Y. Jiao, M. Li, Z. Liu, G. Sun, Y. Lu. Chiral Exciplex Acceptor Enables Circularly Polarized Electroluminescence with High Dissymmetry Factor Close to 10-2. Adv. Optical Mater., 2022. [DOI]
- Y. Zhu, Z. Chen, A. Ying, S. Gong, T. Wang, C. Yang. Nematic Liquid Crystals Induce and Amplify the Circularly Polarized Luminescence of Chiral TADF Emitters. J. Mater. Chem. C, 2022. [DOI]
- M. Sarma, K.-T. Wong. Exciplex: An Intermolecular Charge-Transfer Approach for TADF. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- H.-B. Kim, J.-J. Kim. Diffusion Mechanism of Exciplexes in Organic Optoelectronics. Phys. Rev. Applied, 2020. [DOI]
- M. Sarma, L.-M. Chen, Y.-S. Chen, K.-T. Wong. Exciplexes in OLEDs: Principles and Promises. Mater. Sci. Eng. R Rep., 2022. [DOI]
- K. Goushi, K. Yoshida, K. Sato, C. Adachi. Organic Light-Emitting Diodes Employing Efficient Reverse Intersystem Crossing for Triplet-to-Singlet State Conversion. Nat. Photonics, 2012. [DOI]
- K. Goushi, C. Adachi. Efficient Organic Light-Emitting Diodes through up-Conversion from Triplet to Singlet Excited States of Exciplexes. Appl. Phys. Lett., 2012. [DOI]
- W. Y. Hung, G. C. Fang, Y. C. Chang, T. Y. Kuo, P. T. Chou, S. W. Lin, K. T. Wong. Highly Efficient Bilayer Interface Exciplex for Yellow Organic Light-Emitting Diode. ACS Appl Mater Interfaces, 2013. [DOI | PubMed]
- J. Li, H. Nomura, H. Miyazaki, C. Adachi. Highly Efficient Exciplex Organic Light-Emitting Diodes Incorporating a Heptazine Derivative as an Electron Acceptor. Chem. Commun., 2014. [DOI]
- R. Keruckiene, M. Guzauskas, L. Lapienyte, J. Simokaitiene, D. Volyniuk, J. Cameron, P. J. Skabara, G. Sini, J. V. Grazulevicius. An Experimental and Theoretical Study of Exciplex-Forming Compounds Containing Trifluorobiphenyl and 3,6-Di-Tert-Butylcarbazole Units and Their Performance in OLEDs. J. Mater. Chem. C, 2020. [DOI]
- Q. Wu, M. Wang, X. Cao, D. Zhang, N. Sun, S. Wan, Y. Tao. Carbazole/Α-Carboline Hybrid Bipolar Compounds as Electron Acceptors in Exciplex or Non-Exciplex Mixed Cohosts and Exciplex-TADF Emitters for High-Efficiency OLEDs. J. Mater. Chem. C, 2018. [DOI]
- M. Mamada, G. Tian, H. Nakanotani, J. Su, C. Adachi. The Importance of Excited-State Energy Alignment for Efficient Exciplex Systems Based on a Study of Phenylpyridinato Boron Derivatives. Angew. Chem., Int. Ed., 2018. [DOI]
- H.-T. Cao, Y. Zhao, C. Sun, D. Fang, L.-H. Xie, M.-N. Yan, Y. Wei, H.-M. Zhang, W. Huang. Novel Electron Acceptor Based on Spiro[Fluorine-9,9′-Xanthene] for Exciplex Thermally Activated Delayed Fluorescence. Dyes Pigm., 2018. [DOI]
- H.-T. Cao, J. Wan, B. Li, H. Zhang, L.-H. Xie, C. Sun, Q.-Y. Feng, W.-J. Yu, W. Huang. Highly Efficient Exciplex-Emission from Spiro[Fluorene-9,9′-Xanthene] Derivatives. Dyes Pigm., 2021. [DOI]
- M. Chapran, R. Lytvyn, C. Begel, G. Wiosna-Salyga, J. Ulanski, M. Vasylieva, D. Volyniuk, P. Data, J. V. Grazulevicius. High-Triplet-Level Phthalimide Based Acceptors for Exciplexes with Multicolor Emission. Dyes Pigm., 2019. [DOI]
- M. Zhang, C.-J. Zheng, H.-Y. Zhang, H.-Y. Yang, K. Wang, Y.-Z. Shi, H. Lin, S.-L. Tao, X.-H. Zhang. Thermally Activated Delayed Fluorescence Exciplexes with Phosphor Components Realizing Deep-Red to near-Infrared Electroluminescence. J. Mater. Chem. C, 2022. [DOI]
- W. Liu, J. X. Chen, C. J. Zheng, K. Wang, D. Y. Chen, F. Li, Y. P. Dong, C. S. Lee, X. M. Ou, X. H. Zhang. Novel Strategy to Develop Exciplex Emitters for High-Performance OLEDs by Employing Thermally Activated Delayed Fluorescence Materials. Adv. Funct. Mater., 2016. [DOI]
- M. Zhang, W. Liu, C. J. Zheng, K. Wang, Y. Z. Shi, X. Li, H. Lin, S. L. Tao, X. H. Zhang. Tricomponent Exciplex Emitter Realizing over 20% External Quantum Efficiency in Organic Light-Emitting Diode with Multiple Reverse Intersystem Crossing Channels. Adv. Sci., 2019. [DOI]
- S. K. Jeon, H. J. Jang, J. Y. Lee. Ternary Exciplexes for High Efficiency Organic Light-Emitting Diodes by Self-Energy Transfer. Adv. Optical Mater., 2019. [DOI]
- Q. T. Siddiqui, A. A. Awasthi, P. Bhui, P. Parab, M. Muneer, S. Bose, N. Agarwal. TADF and Exciplex Emission in a Xanthone-Carbazole Derivative and Tuning of Its Electroluminescence with Applied Voltage. RSC Adv, 2019. [DOI | PubMed]
- H. A. Al Attar, A. P. Monkman. Electric Field Induce Blue Shift and Intensity Enhancement in 2d Exciplex Organic Light Emitting Diodes; Controlling Electron-Hole Separation. Adv. Mater., 2016. [DOI | PubMed]
- T. L. Wu, S. Y. Liao, P. Y. Huang, Z. S. Hong, M. P. Huang, C. C. Lin, M. J. Cheng, C. H. Cheng. Exciplex Organic Light-Emitting Diodes with Nearly 20% External Quantum Efficiency: Effect of Intermolecular Steric Hindrance between the Donor and Acceptor Pair. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- Y. Hu, Y. J. Yu, Y. Yuan, Z. Q. Jiang, L. S. Liao. Exciplex-Based Organic Light-Emitting Diodes with near-Infrared Emission. Adv. Optical Mater., 2020. [DOI]
- X. Q. Wang, Y. Hu, Y. J. Yu, Q. S. Tian, W. S. Shen, W. Y. Yang, Z. Q. Jiang, L. S. Liao. Over 800 Nm Emission Via Harvesting of Triplet Excitons in Exciplex Organic Light-Emitting Diodes. J. Phys. Chem. Lett, 2021. [DOI | PubMed]
- H.-Y. Yang, C.-J. Zheng, M. Zhang, J.-W. Zhao, Y.-Z. Shi, C.-P. Pu, H. Lin, S.-L. Tao, X.-H. Zhang. Novel Donor-Spacer-Acceptor Compound as the Multifunctional Component of Exciplexes for Efficient Organic Light-Emitting Diodes. Sci. China Mater., 2022. [DOI]
- W.-Y. Hung, T.-C. Wang, P.-Y. Chiang, B.-J. Peng, K.-T. Wong. Remote Steric Effect as a Facile Strategy for Improving the Efficiency of Exciplex-Based OLEDs. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- P. Yuan, X. Guo, X. Qiao, D. Yan, D. Ma. Improvement of the Electroluminescence Performance of Exciplex-Based OLEDs by Effective Utilization of Long-Range Coupled Electron-Hole Pairs. Adv. Optical Mater., 2019. [DOI]
- E. Skuodis, A. Tomkeviciene, R. Reghu, L. Peciulyte, K. Ivaniuk, D. Volyniuk, O. Bezvikonnyi, G. Bagdziunas, D. Gudeika, J. V. Grazulevicius. OLEDs Based on the Emission of Interface and Bulk Exciplexes Formed by Cyano-Substituted Carbazole Derivative. Dyes Pigm., 2017. [DOI]
- M. Colella, A. Danos, A. P. Monkman. Identifying the Factors That Lead to Plqy Enhancement in Diluted TADF Exciplexes Based on Carbazole Donors. J. Phys. Chem. C, 2019. [DOI]
- M. Colella, A. Danos, A. P. Monkman. Less Is More: Dilution Enhances Optical and Electrical Performance of a TADF Exciplex. J. Phys. Chem. Lett, 2019. [DOI | PubMed]
- Y. J. Pu, Y. Koyama, D. Otsuki, M. Kim, H. Chubachi, Y. Seino, K. Enomoto, N. Aizawa. Exciplex Emissions Derived from Exceptionally Long-Distance Donor and Acceptor Molecules. Chem. Sci., 2019. [DOI | PubMed]
- M. Zhang, K. Wang, C. J. Zheng, D. Q. Wang, Y. Z. Shi, H. Lin, S. L. Tao, X. Li, X. H. Zhang. Development of Red Exciplex for Efficient OLEDs by Employing a Phosphor as a Component. Front. Chem., 2019. [DOI | PubMed]
- J. Guo, Y. Zhen, H. Dong, W. Hu. Recent Progress on Organic Exciplex Materials with Different Donor-Acceptor Contacting Modes for Luminescent Applications. J. Mater. Chem. C, 2021. [DOI]
- W.-C. Chen, B. Huang, S.-F. Ni, Y. Xiong, A. L. Rogach, Y. Wan, D. Shen, Y. Yuan, J.-X. Chen, M.-F. Lo. Deep-Red/near-Infrared Electroluminescence from Single-Component Charge-Transfer Complex Via Thermally Activated Delayed Fluorescence Channel. Adv. Funct. mater., 2019. [DOI]
- Y.-C. Hu, Z.-L. Lin, T.-C. Huang, J.-W. Lee, W.-C. Wei, T.-Y. Ko, C.-Y. Lo, D.-G. Chen, P.-T. Chou, W.-Y. Hung, K.-T. Wong. New Exciplex Systems Composed of Triazatruxene Donors and N-Heteroarene-Cored Acceptors. Mater. Chem. Front., 2020. [DOI]
- Y. C. Hu, L. M. Chen, Z. L. Lin, J. W. Lee, W. C. Wei, T. Y. Ko, C. Y. Lo, W. Y. Hung, K. T. Wong. Suppressing Intermolecular Interactions for Enhancing the Performance of Exciplex-Based OLEDs. J. Chin. Chem. Soc., 2022. [DOI]
- X. Wei, T. Hu, Z. Li, Y. Liu, X. Hu, H. Gao, G. Liu, P. Wang, Y. Yi, Y. Wang. Rational Strategy of Exciplex-Type Thermally Activated Delayed Fluorescent (TADF) Emitters: Stacking of Donor and Acceptor Units of the Intramolecular TADF Molecule. Chem. Eng. J., 2022. [DOI]
- C.-H. Chen, J.-T. Cheng, W.-C. Ding, Z.-L. Lin, Y.-S. Chen, T.-L. Chiu, Y.-C. Lo, J.-H. Lee, K.-T. Wong. New Carboline-Based Donors for Green Exciplex-Forming Systems. J. Chin. Chem. Soc. (Taipei, Taiwan), 2021. [DOI]
- M. Zhang, C. J. Zheng, K. Wang, Y. Z. Shi, D. Q. Wang, X. Li, H. Lin, S. L. Tao, X. H. Zhang. Hydrogen-Bond-Assisted Exciplex Emitters Realizing Improved Efficiencies and Stabilities in Organic Light Emitting Diodes. Adv. Funct. Mater., 2021. [DOI]
- C.-C. A. Voll, G. Markopoulos, T. C. Wu, M. Welborn, J. U. Engelhart, S. Rochat, G. G. D. Han, G. T. Sazama, T.-A. Lin, T. Van Voorhis. Lock-and-Key Exciplexes for Thermally Activated Delayed Fluorescence. Org. Mater., 2020. [DOI]
- Z. Wang, H. Wang, J. Zhu, P. Wu, B. Shen, D. Dou, B. Wei. Manipulation of Thermally Activated Delayed Fluorescence of Blue Exciplex Emission: Fully Utilizing Exciton Energy for Highly Efficient Organic Light Emitting Diodes with Low Roll-Off. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- M. Guzauskas, D. Volyniuk, A. Tomkeviciene, A. Pidluzhna, A. Lazauskas, J. V. Grazulevicius. Dual Nature of Exciplexes: Exciplex-Forming Properties of Carbazole and Fluorene Hybrid Trimers. J. Mater. Chem. C, 2019. [DOI]
- C. Hippola, D. Danilovic, U. Bhattacharjee, C. Perez-Bolivar, K. A. N. Sachinthani, T. L. Nelson, P. Anzenbacher, J. W. Petrich, R. Shinar, J. Shinar. Bright Deep Blue TADF OLEDs: The Role of Triphenylphosphine Oxide in Npb/Tpbi:Pph3o Exciplex Emission. Adv. Optical Mater., 2020. [DOI]
- J. Li, H. Gong, J. Zhang, H. Liu, L. Tao, Y. Wang, Q. Guo. Efficient Exciplex-Based Deep-Blue Organic Light-Emitting Diodes Employing a Bis(4-Fluorophenyl)Amine-Substituted Heptazine Acceptor. Molecules, 2021. [DOI | PubMed]
- C.-Y. Huang, S.-Y. Ho, C.-H. Lai, C.-L. Ko, Y.-C. Wei, J.-A. Lin, D.-G. Chen, T.-Y. Ko, K.-T. Wong, Z. Zhang. Insights into Energy Transfer Pathways between the Exciplex Host and Fluorescent Guest: Attaining Highly Efficient 710 Nm Electroluminescence. J. Mater. Chem. C, 2020. [DOI]
- Y. S. Chen, D. Luo, W. C. Wei, B. L. Chen, T. H. Yeh, S. W. Liu, K. T. Wong. New Exciplex-Forming Co-Host System and Thienothiadazole-Based Fluorescent Emitter for High-Efficiency and Promising Stability near-Infrared OLED. Adv. Optical Mater., 2022. [DOI]
- J.-X. Chen, H. Wang, K. Wang, X. Zhang, L. Zhou, Y.-Z. Shi, J. Yu, X.-H. Zhang. High-Performance Red and White Organic Light-Emitting Diodes Based on a Novel Red Thermally Activated Delayed Fluorescence Emitter in an Exciplex Matrix. Mater. Today Energy, 2021. [DOI]
- C. K. Moon, K. Suzuki, K. Shizu, C. Adachi, H. Kaji, J. J. Kim. Combined Inter-and Intramolecular Charge-Transfer Processes for Highly Efficient Fluorescent Organic Light-Emitting Diodes with Reduced Triplet Exciton Quenching. Adv. Mater., 2017. [DOI]
- H. Sasabe, R. Sato, K. Suzuki, Y. Watanabe, C. Adachi, H. Kaji, J. Kido. Ultrahigh Power Efficiency Thermally Activated Delayed Fluorescent OLEDs by the Strategic Use of Electron-Transport Materials. Adv. Optical Mater., 2018. [DOI]
- C.-C. Lee, N. R. A. Amin, J.-J. Xu, B.-C. Wang, D. Luo, K. Sutanto, S. Biring, S.-W. Liu, C.-H. Chen. Structural Effect of Phenylcarbazole-Based Molecules on the Exciplex-Forming Co-Host System to Achieve Highly Efficient Phosphorescent OLEDs with Low Efficiency Roll-Off. J. Mater. Chem. C, 2021. [DOI]
- L. Jia, L. Jin, K. Yuan, L. Chen, J. Yuan, S. Xu, W. Lv, R. Chen. High-Performance Exciplex-Type Host for Multicolor Phosphorescent Organic Light-Emitting Diodes with Low Turn-on Voltages. ACS Sustain. Chem. Eng., 2018. [DOI]
- C. J. Shih, C. C. Lee, T. H. Yeh, S. Biring, K. K. Kesavan, N. R. A. Amin, M. H. Chen, W. C. Tang, S. W. Liu, K. T. Wong. Versatile Exciplex-Forming Co-Host for Improving Efficiency and Lifetime of Fluorescent and Phosphorescent Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- B. Liang, J. Wang, Z. Cheng, J. Wei, Y. Wang. Exciplex-Based Electroluminescence: Over 21% External Quantum Efficiency and Approaching 100 Lm/W Power Efficiency. J Phys Chem Lett, 2019. [DOI | PubMed]
- M. Colella, P. Pander, D. d. S. Pereira, A. P. Monkman. Interfacial TADF Exciplex as a Tool to Localize Excitons, Improve Efficiency, and Increase OLED Lifetime. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- Q.-S. Tian, X.-D. Zhu, L.-S. Liao. Highly Efficient Exciplex-Based OLEDs Incorporating a Novel Electron Donor. Mater. Chem. Front., 2020. [DOI]
- C. Duan, C. Han, J. Zhang, X. Zhang, C. Fan, H. Xu. Manipulating Charge-Transfer Excitons by Exciplex Matrix: Toward Thermally Activated Delayed Fluorescence Diodes with Power Efficiency Beyond 110 lm w-1. Adv. Funct. Mater, 2021. [DOI]
- Z. Zhou, R. Chen, P. Jin, J. Hao, W. Wu, B. Yin, C. Zhang, J. Yao. Interplay between Singlet and Triplet Excited States in Interface Exciplex OLEDs with Fluorescence, Phosphorescence, and TADF Emitters. Adv. Funct. Mater., 2023. [DOI]
- X. Wang, Y. Zhang, Z. Yu, Y. Wu, D. Wang, C. Wu, H. Ma, S. Ning, H. Dong, Z. Wu. Overcoming Energy Loss of Thermally Activated Delayed Fluorescence Sensitized-OLEDs by Developing a Fluorescent Dopant with a Small Singlet-Triplet Energy Splitting. J. Mater. Chem. C, 2022. [DOI]
- L. Chen, J. Lv, S. Wang, S. Shao, L. Wang. Dendritic Interfacial Exciplex Hosts for Solution-Processed TADF-OLEDs with Power Efficiency Approaching 100 lm w-1. Adv. Optical mater., 2021. [DOI]
- T. Xu, G. Xie, T. Huang, H. Liu, X. Cao, Y. Tang, C. Yang. Solution-Processed Multiple Exciplexes Via Spirofluorene and S-Triazine Moieties for Red Thermally Activated Delayed Fluorescence Emissive Layer OLEDs. Org. Electron., 2021. [DOI]
- M. Colella, P. Pander, A. P. Monkman. Solution Processable Small Molecule Based TADF Exciplex OLEDs. Org. Electron., 2018. [DOI]
- V. Jankus, P. Data, D. Graves, C. McGuinness, J. Santos, M. R. Bryce, F. B. Dias, A. P. Monkman. Highly Efficient TADF OLEDs: How the Emitter-Host Interaction Controls Both the Excited State Species and Electrical Properties of the Devices to Achieve near 100% Triplet Harvesting and High Efficiency. Adv. Funct. Mater., 2014. [DOI]
- K. Kishore Kesavan, J. Jayakumar, M. Lee, C. Hexin, S. Sudheendran Swayamprabha, D. Kumar Dubey, F.-C. Tung, C.-W. Wang, J.-H. Jou. Achieving a 32% EQE Solution-Processed Simple Structure OLED Via Exciplex System. Chem. Eng. J., 2022. [DOI]
- Q. Huang, S. Zhao, P. Wang, Z. Qin, Z. Xu, D. Song, B. Qiao, X. Xu. Investigating the Evolution of Exciplex States in Thermally Activated Delayed Fluorescence Organic Light-Emitting Diodes by Transient Measurement. J. Lumin., 2018. [DOI]
- T.-C. Lin, M. Sarma, Y.-T. Chen, S.-H. Liu, K.-T. Lin, P.-Y. Chiang, W.-T. Chuang, Y.-C. Liu, H.-F. Hsu, W.-Y. Hung. Probe Exciplex Structure of Highly Efficient Thermally Activated Delayed Fluorescence Organic Light Emitting Diodes. Nat. Commun., 2018. [DOI | PubMed]
- C.-K. Moon, J.-S. Huh, J.-M. Kim, J.-J. Kim. Electronic Structure and Emission Process of Excited Charge Transfer States in Solids. Chem. Mater., 2018. [DOI]
- N. Bunzmann, S. Weissenseel, L. Kudriashova, J. Gruene, B. Krugmann, J. V. Grazulevicius, A. Sperlich, V. Dyakonov. Optically and Electrically Excited Intermediate Electronic States in Donor:Acceptor Based OLEDs. Mater. Horiz., 2020. [DOI]
- T. B. Nguyen, H. Nakanotani, T. Hatakeyama, C. Adachi. The Role of Reverse Intersystem Crossing Using a TADF-Type Acceptor Molecule on the Device Stability of Exciplex-Based Organic Light-Emitting Diodes. Adv. Mater., 2020. [DOI]
- L. He, R. Bai, R. Yu, X. Meng, M. Tian, X. Wang. Donor/Acceptor Pairs Created by Electrostatic Interaction: Design, Synthesis, and Investigation on the Exciplex Formed within the Pair. Angew. Chem., Int. Ed., 2021. [DOI]
- M. Cekaviciute, J. Simokaitiene, D. Volyniuk, G. Sini, J. V. Grazulevicius. Arylfluorenyl-Substituted Metoxytriphenylamines as Deep Blue Exciplex Forming Bipolar Semiconductors for White and Blue Organic Light Emitting Diodes. Dyes Pigm., 2017. [DOI]
- Q. S. Tian, L. Zhang, Y. Hu, S. Yuan, Q. Wang, L. S. Liao. High-Performance White Organic Light-Emitting Diodes with Simplified Structure Incorporating Novel Exciplex-Forming Host. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- J. Yao, S. Ying, X. Qiao, D. Yang, J. Chen, T. Ahamad, S. M. Alshehri, D. Ma. High Efficiency and Low Roll-Off All Fluorescence White Organic Light-Emitting Diodes by the Formation of Interface Exciplex. Org. Electron., 2019. [DOI]
- Y. Guo, Y. Zhao, Y. Miao, L. Wang, T. Li, H. Wang, B. Xu, J. Yu. All-Exciplex-Based White Organic Light-Emitting Diodes by Employing an Interface-Free Sandwich Light-Emitting Unit Achieving High Electroluminescence Performance. J. Mater. Chem. C, 2020. [DOI]
- X. Tan, D. Volyniuk, T. Matulaitis, J. Keruckas, K. Ivaniuk, I. Helzhynskyy, P. Stakhira, J. V. Grazulevicius. High Triplet Energy Materials for Efficient Exciplex-Based and Full-TADF-Based White OLEDs. Dyes Pigm., 2020. [DOI]
- C. Han, R. Du, H. Xu, S. Han, P. Ma, J. Bian, C. Duan, Y. Wei, M. Sun, X. Liu, W. Huang. Ladder-Like Energy-Relaying Exciplex Enables 100% Internal Quantum Efficiency of White TADF-Based Diodes in a Single Emissive Layer. Nat. Commun., 2021. [DOI | PubMed]
- S. Heo, K. Y. Kim, H. Choi, S. G. Kang, W. Choi, S. S. Lee, S. H. Jung, J. H. Jung. Exciplex Emissive Supramolecular Polymer Formed by Tuning Molecular Conformation. Nanoscale, 2020. [DOI | PubMed]
- G. Yang, C. Lin, X. Feng, T. Wang, J. Jiang. Multi-Component Supramolecular Gels Induce Protonation of a Porphyrin Exciplex to Achieve Improved Collective Optical Properties for Effective Photocatalytic Hydrogen Generation. Chem. Commun., 2020. [DOI]
- A. Garci, Y. Beldjoudi, M. S. Kodaimati, J. E. Hornick, M. T. Nguyen, M. M. Cetin, C. L. Stern, I. Roy, E. A. Weiss, J. F. Stoddart. Mechanical-Bond-Induced Exciplex Fluorescence in an Anthracene-Based Homo[2]Catenane. J. Am. Chem. Soc., 2020. [DOI | PubMed]
- D. R. McMillin, M. T. Buckner, B. T. Ahn. A Light-Induced Redox Reaction of Bis(2,9-Dimethyl-1,10-Phenanthroline)Copper(I). Inorg. Chem., 1977. [DOI]
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier, H. Yersin. Cu(I) Complexes – Thermally Activated Delayed Fluorescence. Photophysical Approach and Material Design. Coord. Chem. Rev., 2016. [DOI]
- Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl, B. Dietzek. Heteroleptic Diimine-Diphosphine Cu(I) Complexes as an Alternative Towards Noble-Metal Based Photosensitizers: Design Strategies, Photophysical Properties and Perspective Applications. Coord. Chem. Rev., 2018. [DOI]
- M. Wallesch, D. Volz, D. M. Zink, U. Schepers, M. Nieger, T. Baumann, S. Brase. Bright Coppertunities: Multinuclear Cu(I) Complexes with N-P Ligands and Their Applications. Chem. Eur. J., 2014. [DOI | PubMed]
- C. Sandoval-Pauker, M. Santander-Nelli, P. Dreyse. Thermally Activated Delayed Fluorescence in Luminescent Cationic Copper(I) Complexes. RSC Adv., 2022. [DOI | PubMed]
- C. E. Housecroft, E. C. Constable. TADF: Enabling Luminescent Copper(I) Coordination Compounds for Light-Emitting Electrochemical Cells. J. Mater. Chem. C, 2022. [DOI]
- J. Beaudelot, S. Oger, S. Perusko, T. A. Phan, T. Teunens, C. Moucheron, G. Evano. Photoactive Copper Complexes: Properties and Applications. Chem. Rev., 2022. [DOI | PubMed]
- V. K.-M. Au. Organic Light-Emitting Diodes Based on Luminescent Self-Assembled Materials of Copper(I). Energy Fuels, 2021. [DOI]
- A. Barbieri, G. Accorsi, N. Armaroli. Luminescent Complexes Beyond the Platinum Group: The D10 Avenue. Chem. Commun., 2008. [DOI]
- M. Osawa, I. Kawata, R. Ishii, S. Igawa, M. Hashimoto, M. Hoshino. Application of Neutral D10 Coinage Metal Complexes with an Anionic Bidentate Ligand in Delayed Fluorescence-Type Organic Light-Emitting Diodes. J. Mater. Chem. C, 2013. [DOI]
- M. Hashimoto, S. Igawa, M. Yashima, I. Kawata, M. Hoshino, M. Osawa. Highly Efficient Green Organic Light-Emitting Diodes Containing Luminescent Three-Coordinate Copper(I) Complexes. J. Am. Chem. Soc., 2011. [DOI | PubMed]
- J. Fernandez-Cestau, B. Bertrand, M. Blaya, G. A. Jones, T. J. Penfold, M. Bochmann. Synthesis and Luminescence Modulation of Pyrazine-Based Gold(III) Pincer Complexes. Chem. Commun., 2015. [DOI]
- D. Zhou, G. S. M. Tong, G. Cheng, Y. K. Tang, W. Liu, D. Ma, L. Du, J. R. Chen, C. M. Che. Stable Tetradentate Gold(III)-TADF Emitters with Close to Unity Quantum Yield and Radiative Decay Rate Constant of up to 2 × 106 S–1: High-Efficiency Green OLEDs with Operational Lifetime (Lt90) Longer Than 1800 H at 1000 Cd M–2. Adv. Mater., 2022. [DOI]
- J.-H. Jia, D. Liang, R. Yu, X.-L. Chen, L. Meng, J.-F. Chang, J.-Z. Liao, M. Yang, X.-N. Li, C.-Z. Lu. Coordination-Induced Thermally Activated Delayed Fluorescence: From Non-TADF Donor-Acceptor-Type Ligand to TADF-Active Ag-Based Complexes. Chem. Mater., 2020. [DOI]
- G. U. Mahoro, J. Fernandez-Cestau, J. L. Renaud, P. B. Coto, R. D. Costa, S. Gaillard. Recent Advances in Solid-State Lighting Devices Using Transition Metal Complexes Exhibiting Thermally Activated Delayed Fluorescent Emission Mechanism. Adv. Optical Mater., 2020. [DOI]
- D. S. M. Ravinson, M. E. Thompson. Thermally Assisted Delayed Fluorescence (TADF): Fluorescence Delayed Is Fluorescence Denied. Mater. Horiz., 2020. [DOI]
- T. Hofbeck, U. Monkowius, H. Yersin. Highly Efficient Luminescence of Cu(I) Compounds: Thermally Activated Delayed Fluorescence Combined with Short-Lived Phosphorescence. J. Am. Chem. Soc., 2015. [DOI | PubMed]
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson, H. Yersin. Phosphorescence Versus Thermally Activated Delayed Fluorescence. Controlling Singlet-Triplet Splitting in Brightly Emitting and Sublimable Cu(I) Compounds. J. Am. Chem. Soc., 2014. [DOI | PubMed]
- X. L. Chen, R. Yu, X. Y. Wu, D. Liang, J. H. Jia, C. Z. Lu. A Strongly Greenish-Blue-Emitting Cu4cl4 Cluster with an Efficient Spin-Orbit Coupling (Soc): Fast Phosphorescence Versus Thermally Activated Delayed Fluorescence. Chem. Commun., 2016. [DOI]
- J. Zhang, C. Duan, C. Han, H. Yang, Y. Wei, H. Xu. Balanced Dual Emissions from Tridentate Phosphine-Coordinate Copper (I) Complexes toward Highly Efficient Yellow OLEDs. Adv. Mater., 2016. [DOI | PubMed]
- M. Xie, C. Han, J. Zhang, G. Xie, H. Xu. White Electroluminescent Phosphine-Chelated Copper Iodide Nanoclusters. Chem. Mater., 2017. [DOI]
- A. Schinabeck, M. J. Leitl, H. Yersin. Dinuclear Cu(I) Complex with Combined Bright TADF and Phosphorescence. Zero-Field Splitting and Spin-Lattice Relaxation Effects of the Triplet State. J. Phys. Chem. Lett, 2018. [DOI | PubMed]
- A. Schinabeck, N. Rau, M. Klein, J. Sundermeyer, H. Yersin. Deep Blue Emitting Cu(I) Tripod Complexes. Design of High Quantum Yield Materials Showing TADF-Assisted Phosphorescence. Dalton Trans., 2018. [DOI | PubMed]
- X. Li, J. Zhang, Z. Zhao, X. Yu, P. Li, Y. Yao, Z. Liu, Q. Jin, Z. Bian, Z. Lu, C. Huang. Bluish-Green Cu(I) Dimers Chelated with Thiophene Ring-Introduced Diphosphine Ligands for Both Singlet and Triplet Harvesting in OLEDs. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- A. Y. Baranov, A. S. Berezin, D. G. Samsonenko, A. S. Mazur, P. M. Tolstoy, V. F. Plyusnin, I. E. Kolesnikov, A. V. Artem’ev. New Cu(I) Halide Complexes Showing TADF Combined with Room Temperature Phosphorescence: The Balance Tuned by Halogens. Dalton Trans., 2020. [DOI | PubMed]
- J. Wang, H. Chen, S. Xu, Q. Su, F. Zhao, H. He. Highly Effective Luminescence Stemmed from Thermally Activated Delayed Fluorescence (TADF) and Phosphorescence for the New Four-Coordinate Copper(I) Complexes Containing N-Heterocyclic Carbene (Nhc) Ligands. J. Photochem. Photobiol., A, 2020. [DOI]
- R. Czerwieniec, H. Yersin. Diversity of Copper(I) Complexes Showing Thermally Activated Delayed Fluorescence: Basic Photophysical Analysis. Inorg. Chem., 2015. [DOI | PubMed]
- R. Czerwieniec, J. Yu, H. Yersin. Blue-Light Emission of Cu(I) Complexes and Singlet Harvesting. Inorg. Chem., 2011. [DOI | PubMed]
- G. Li, Z.-Q. Zhu, Q. Chen, J. Li. Metal Complex Based Delayed Fluorescence Materials. Org. Electron., 2019. [DOI]
- K. Li, Y. Chen, J. Wang, C. Yang. Diverse Emission Properties of Transition Metal Complexes Beyond Exclusive Single Phosphorescence and Their Wide Applications. Coord. Chem. Rev., 2021. [DOI]
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz, S. Bräse. Sustainable Metal Complexes for Organic Light-Emitting Diodes (OLEDs). Coord. Chem. Rev., 2018. [DOI]
- M. C. Tang, M. Y. Chan, V. W. Yam. Molecular Design of Luminescent Gold(III) Emitters as Thermally Evaporable and Solution-Processable Organic Light-Emitting Device (OLED) Materials. Chem. Rev., 2021. [DOI | PubMed]
- Z. Han, X. Y. Dong, S. Q. Zang. Crystalline Metal-Organic Materials with Thermally Activated Delayed Fluorescence. Adv. Optical Mater., 2021. [DOI]
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck, T. Fischer. The Triplet State of Organo-Transition Metal Compounds. Triplet Harvesting and Singlet Harvesting for Efficient OLEDs. Coord. Chem. Rev., 2011. [DOI]
- H. Amouri. Luminescent Complexes of Platinum, Iridium, and Coinage Metals Containing N-Heterocyclic Carbene Ligands: Design, Structural Diversity, and Photophysical Properties. Chem. Rev., 2023. [DOI | PubMed]
- X. Li, Y. Xie, Z. Li. Diversity of Luminescent Metal Complexes in OLEDs: Beyond Traditional Precious Metals. Chem. Asian J., 2021. [DOI | PubMed]
- T. Huang, W. Jiang, L. Duan. Recent Progress in Solution Processable TADF Materials for Organic Light-Emitting Diodes. J. Mater. Chem. C, 2018. [DOI]
- J. Hossain, R. Akhtar, S. Khan. Luminescent Coinage Metal Complexes of Carbenes. Polyhedron, 2021. [DOI]
- H. Yersin, R. Czerwieniec, M. Z. Shafikov, A. F. Suleymanova. TADF Material Design: Photophysical Background and Case Studies Focusing on Cu(I) and Ag(I) Complexes. ChemPhysChem, 2017. [DOI | PubMed]
- H. Takeda, A. Kobayashi, K. Tsuge. Recent Developments of Photoactive Cu(I) and Ag(I) Complexes with Diphosphine and Related Ligands. Coord. Chem. Rev., 2022. [DOI]
- L. L. M. Zhang, W. Y. Wong. Atomically Precise Copper Nanoclusters as Ultrasmall Molecular Aggregates: Appealing Compositions, Structures, Properties, and Applications. Aggregate, 2023. [DOI]
- X. S. Han, X. Luan, H. F. Su, J. J. Li, S. F. Yuan, Z. Lei, Y. Pei, Q. M. Wang. Structure Determination of Alkynyl-Protected Gold Nanocluster Au22(Tbuc≡C)18 and Its Thermochromic Luminescence. Angew. Chem., Int. Ed., 2020. [DOI]
- L. L. Zhang, G. Zhou, G. Zhou, H. K. Lee, N. Zhao, O. V. Prezhdo, T. C. W. Mak. Core-Dependent Properties of Copper Nanoclusters: Valence-Pure Nanoclusters as Nir TADF Emitters and Mixed-Valence Ones as Semiconductors. Chem. Sci., 2019. [DOI | PubMed]
- Z. R. Yuan, Z. Wang, B. L. Han, C. K. Zhang, S. S. Zhang, Z. Y. Zhu, J. H. Yu, T. D. Li, Y. Z. Li, C. H. Tung, D. Sun. Ag22 Nanoclusters with Thermally Activated Delayed Fluorescence Protected by Ag/Cyanurate/Phosphine Metallamacrocyclic Monolayers through in-Situ Ligand Transesterification. Angew. Chem., Int. Ed., 2022. [DOI]
- K. A. Vinogradova, N. A. Shekhovtsov, A. S. Berezin, T. S. Sukhikh, M. I. Rogovoy, A. V. Artem’ev, M. B. Bushuev. Coordination-Induced Emission Enhancement in Copper(I) Iodide Coordination Polymers Supported by 2-(Alkylsulfanyl)Pyrimidines. Dalton Trans., 2021. [DOI | PubMed]
- X. Hei, K. Zhu, G. Carignan, S. J. Teat, M. Li, G. Zhang, M. Bonite, J. Li. Solution-Processable Copper(I) Iodide-Based Inorganic-Organic Hybrid Semiconductors Composed of Both Coordinate and Ionic Bonds. J. Solid State Chem., 2022. [DOI]
- Y. Ma, C.-M. Che, H.-Y. Chao, X. Zhou, W.-H. Chan, J. Shen. High Luminescence Gold(I) and Copper(I) Complexes with a Triplet Excited State for Use in Light-Emitting Diodes. Adv. Mater., 1999. [DOI]
- M. Iwamura, H. Watanabe, K. Ishii, S. Takeuchi, T. Tahara. Coherent Nuclear Dynamics in Ultrafast Photoinduced Structural Change of Bis(Diimine)Copper(I) Complex. J. Am. Chem. Soc., 2011. [DOI | PubMed]
- M. W. Mara, K. A. Fransted, L. X. Chen. Interplays of Excited State Structures and Dynamics in Copper(I) Diimine Complexes: Implications and Perspectives. Coord. Chem. Rev., 2015. [DOI]
- G. Capano, M. Chergui, U. Rothlisberger, I. Tavernelli, T. J. Penfold. A Quantum Dynamics Study of the Ultrafast Relaxation in a Prototypical Cu(I)-Phenanthroline. J. Phys. Chem. A, 2014. [DOI | PubMed]
- A. Lavie-Cambot, M. Cantuel, Y. Leydet, G. Jonusauskas, D. M. Bassani, N. D. McClenaghan. Improving the Photophysical Properties of Copper(I) Bis(Phenanthroline) Complexes. Coord. Chem. Rev., 2008. [DOI]
- D. G. Cuttell, S.-M. Kuang, P. E. Fanwick, D. R. McMillin, R. A. Walton. Simple Cu(I) Complexes with Unprecedented Excited-State Lifetimes. J. Am. Chem. Soc., 2002. [DOI | PubMed]
- D. Felder, J.-F. Nierengarten, F. Barigelletti, B. Ventura, N. Armaroli. Highly Luminescent Cu(I)-Phenanthroline Complexes in Rigid Matrix and Temperature Dependence of the Photophysical Properties. J. Am. Chem. Soc., 2001. [DOI | PubMed]
- A. K. Ichinaga, J. R. Kirchhoff, D. R. McMillin, C. O. Dietrich-Buchecker, P. A. Marnot, J. P. Sauvage. Charge-Transfer Absorption and Emission of Cu(Nn)2 + Systems. Inorg. Chem., 1987. [DOI]
- O. Green, B. A. Gandhi, J. N. Burstyn. Photophysical Characteristics and Reactivity of Bis(2,9-Di-Tert-Butyl-1,10-Phenanthroline)Copper(I). Inorg. Chem., 2009. [DOI | PubMed]
- M. C. Rosko, K. A. Wells, C. E. Hauke, F. N. Castellano. Next Generation Cuprous Phenanthroline Mlct Photosensitizer Featuring Cyclohexyl Substituents. Inorg. Chem., 2021. [DOI | PubMed]
- L. Gimeno, B. T. Phelan, E. A. Sprague-Klein, T. Roisnel, E. Blart, C. Gourlaouen, L. X. Chen, Y. Pellegrin. Bulky and Stable Copper(I)-Phenanthroline Complex: Impact of Steric Strain and Symmetry on the Excited-State Properties. Inorg. Chem., 2022. [DOI | PubMed]
- C. S. Smith, K. R. Mann. Void Space Containing Crystalline Cu(I) Phenanthroline Complexes as Molecular Oxygen Sensors. Chem. Mater., 2009. [DOI]
- S. Brown-Xu, M. Fumanal, C. Gourlaouen, L. Gimeno, A. Quatela, C. Thobie-Gautier, E. Blart, A. Planchat, F. Riobe, C. Monnereau. Intriguing Effects of Halogen Substitution on the Photophysical Properties of 2,9-(Bis)Halo-Substituted Phenanthrolinecopper(I) Complexes. Inorg. Chem., 2019. [DOI | PubMed]
- C. E. A. Palmer, D. R. McMillin. Singlets, Triplets, and Exciplexes: Complex, Temperature-Dependent Emissions from Cu(Dmp)(Pph3)2 + and Cu(Phen)(Pph3)2 + in Solution. Inorg. Chem., 1987. [DOI]
- P. A. Breddels, P. A. M. Berdowski, G. Blasse, D. R. McMillin. Luminescence of Some CuI Complexes. J. Chem. Soc, Faraday Trans. 2, 1982. [DOI]
- N. Armaroli, G. Accorsi, M. Holler, O. Moudam, J. F. Nierengarten, Z. Zhou, R. T. Wegh, R. Welter. Highly Luminescent CuI Complexes for Light-Emitting Electrochemical Cells. Adv. Mater., 2006. [DOI]
- C. S. Smith, C. W. Branham, B. J. Marquardt, K. R. Mann. Oxygen Gas Sensing by Luminescence Quenching in Crystals of Cu(Xantphos)(Phen)+ Complexes. J. Am. Chem. Soc., 2010. [DOI | PubMed]
- A. Wada, Q. Zhang, T. Yasuda, I. Takasu, S. Enomoto, C. Adachi. Efficient Luminescence from a Copper(I) Complex Doped in Organic Light-Emitting Diodes by Suppressing C-H Vibrational Quenching. Chem. Commun., 2012. [DOI]
- Singlet Harvesting with Brightly Emitting Cu(I) and Metal-Free Organic Compounds;. Organic Photonics V;, 2012
- R. Czerwieniec, K. Kowalski, H. Yersin. Highly Efficient Thermally Activated Fluorescence of a New Rigid Cu(I) Complex [Cu(Dmp)(Phanephos)]+. Dalton Trans., 2013. [DOI | PubMed]
- X.-L. Chen, R. Yu, Q.-K. Zhang, L.-J. Zhou, X.-Y. Wu, Q. Zhang, C.-Z. Lu. Rational Design of Strongly Blue-Emitting Cuprous Complexes with Thermally Activated Delayed Fluorescence and Application in Solution-Processed OLEDs. Chem. Mater., 2013. [DOI]
- Q. Zhang, J. Chen, X. Y. Wu, X. L. Chen, R. Yu, C. Z. Lu. Outstanding Blue Delayed Fluorescence and Significant Processing Stability of Cuprous Complexes with Functional Pyridine-Pyrazolate Diimine Ligands. Dalton Trans., 2015. [DOI | PubMed]
- C. H. Huang, M. Yang, X. L. Chen, C. Z. Lu. Bright Bluish-Green Emitting Cu(I) Complexes Exhibiting Efficient Thermally Activated Delayed Fluorescence. Dalton Trans., 2021. [DOI | PubMed]
- C. L. Linfoot, M. J. Leitl, P. Richardson, A. F. Rausch, O. Chepelin, F. J. White, H. Yersin, N. Robertson. Thermally Activated Delayed Fluorescence (TADF) and Enhancing Photoluminescence Quantum Yields of [CuI(Diimine)(Diphosphine)]+ ComplexesPhotophysical, Structural, and Computational Studies. Inorg. Chem., 2014. [DOI | PubMed]
- F. Zhang, Y. Guan, X. Chen, S. Wang, D. Liang, Y. Feng, S. Chen, S. Li, Z. Li, F. Zhang. Syntheses, Photoluminescence, and Electroluminescence of a Series of Sublimable Bipolar Cationic Cuprous Complexes with Thermally Activated Delayed Fluorescence. Inorg. Chem., 2017. [DOI | PubMed]
- X.-L. Chen, C.-S. Lin, X.-Y. Wu, R. Yu, T. Teng, Q.-K. Zhang, Q. Zhang, W.-B. Yang, C.-Z. Lu. Highly Efficient Cuprous Complexes with Thermally Activated Delayed Fluorescence and Simplified Solution Process OLEDs Using the Ligand as Host. J. Mater. Chem. C, 2015. [DOI]
- D. Liang, X. L. Chen, J. Z. Liao, J. Y. Hu, J. H. Jia, C. Z. Lu. Highly Efficient Cuprous Complexes with Thermally Activated Delayed Fluorescence for Solution-Processed Organic Light-Emitting Devices. Inorg. Chem., 2016. [DOI | PubMed]
- M. Holler, B. Delavaux-Nicot, J. F. Nierengarten. Topological and Steric Constraints to Stabilize Heteroleptic Copper(I) Complexes Combining Phenanthroline Ligands and Phosphines. Chem. Eur. J., 2019. [DOI | PubMed]
- M. Mohankumar, M. Holler, E. Meichsner, J. F. Nierengarten, F. Niess, J. P. Sauvage, B. Delavaux-Nicot, E. Leoni, F. Monti, J. M. Malicka. Heteroleptic Copper(I) Pseudorotaxanes Incorporating Macrocyclic Phenanthroline Ligands of Different Sizes. J. Am. Chem. Soc., 2018. [DOI | PubMed]
- E. Fresta, M. D. Weber, J. Fernandez-Cestau, R. D. Costa. White Light-Emitting Electrochemical Cells Based on Deep-Red Cu(I) Complexes. Adv. Optical Mater., 2019. [DOI]
- G. Farias, C. A. M. Salla, R. S. Heying, A. J. Bortoluzzi, S. F. Curcio, T. Cazati, P. L. dos Santos, A. P. Monkman, B. d. Souza, I. H. Bechtold. Reducing Lifetime in Cu(I) Complexes with Thermally Activated Delayed Fluorescence and Phosphorescence Promoted by Chalcogenolate-Diimine Ligands. J. Mater. Chem. C, 2020. [DOI]
- T. Teng, J. Xiong, G. Cheng, C. Zhou, X. Lv, K. Li. Solution-Processed OLEDs Based on Thermally Activated Delayed Fluorescence Copper(I) Complexes with Intraligand Charge-Transfer Excited State. Molecules, 2021. [DOI | PubMed]
- E. Fresta, G. U. Mahoro, L. M. Cavinato, J. F. Lohier, J. L. Renaud, S. Gaillard, R. D. Costa. Novel Red-Emitting Copper(I) Complexes with Pyrazine and Pyrimidinyl Ancillary Ligands for White Light-Emitting Electrochemical Cells. Adv. Optical Mater., 2022. [DOI]
- S. Igawa, M. Hashimoto, I. Kawata, M. Yashima, M. Hoshino, M. Osawa. Highly Efficient Green Organic Light-Emitting Diodes Containing Luminescent Tetrahedral Copper(I) Complexes. J. Mater. Chem. C, 2013. [DOI]
- W.-J. Zhang, Z.-X. Zhou, L. Liu, X.-X. Zhong, A. M. Asiri, K. A. Alamry, F.-B. Li, N.-Y. Zhu, W.-Y. Wong, H.-M. Qin. Highly-Efficient Blue Neutral Mononuclear Copper(I) Halide Complexes Containing Bi- and Mono-Dentate Phosphine Ligands. J. Lumin., 2018. [DOI]
- Q. Wei, R. Zhang, L. Liu, X. X. Zhong, L. Wang, G. H. Li, F. B. Li, K. A. Alamry, Y. Zhao. From Deep Blue to Green Emitting and Ultralong Fluorescent Copper(I) Halide Complexes Containing Dimethylthiophene Diphosphine and Pph3 Ligands. Dalton Trans., 2019. [DOI | PubMed]
- B.-K. Guo, F. Yang, Y.-Q. Wang, Q. Wei, L. Liu, X.-X. Zhong, L. Wang, J.-K. Gong, F.-B. Li, W.-Y. Wong. Efficient TADF-OLEDs with Ultra-Soluble Copper(I) Halide Complexes Containing Non-Symmetrically Substituted Bidentate Phosphine and Pph3 Ligands. J. Lumin., 2020. [DOI]
- G. Chakkaradhari, T. Eskelinen, C. Degbe, A. Belyaev, A. S. Melnikov, E. V. Grachova, S. P. Tunik, P. Hirva, I. O. Koshevoy. Oligophosphine-Thiocyanate Copper(I) and Silver(I) Complexes and Their Borane Derivatives Showing Delayed Fluorescence. Inorg. Chem., 2019. [DOI | PubMed]
- M. Klein, N. Rau, M. Wende, J. Sundermeyer, G. Cheng, C.-M. Che, A. Schinabeck, H. Yersin. Cu(I) and Ag(I) Complexes with a New Type of Rigid Tridentate N,P,P-Ligand for Thermally Activated Delayed Fluorescence and OLEDs with High External Quantum Efficiency. Chem. Mater., 2020. [DOI]
- X. Yu, X. Li, Z. Cai, L. Sun, C. Wang, H. Rao, C. Wei, Z. Bian, Q. Jin, Z. Liu. Mechanochromic Properties in a Mononuclear Cu(I) Complex without Cuprophilic Interactions. Chem. Commun., 2021. [DOI]
- A. Alconchel, O. Crespo, P. Garcia-Orduna, M. C. Gimeno. Closo- or Nido-Carborane Diphosphane as Responsible for Strong Thermochromism or Time Activated Delayed Fluorescence (TADF) in [Cu(N∧N)(P∧P)]0/+. Inorg. Chem., 2021. [DOI | PubMed]
- L. Bergmann, J. Friedrichs, M. Mydlak, T. Baumann, M. Nieger, S. Brase. Outstanding Luminescence from Neutral Copper(I) Complexes with Pyridyl-Tetrazolate and Phosphine Ligands. Chem. Commun., 2013. [DOI]
- J. L. Chen, X. F. Cao, J. Y. Wang, L. H. He, Z. Y. Liu, H. R. Wen, Z. N. Chen. Synthesis, Characterization, and Photophysical Properties of Heteroleptic Copper(I) Complexes with Functionalized 3-(2’-Pyridyl)-1,2,4-Triazole Chelating Ligands. Inorg. Chem., 2013. [DOI | PubMed]
- K. A. Barakat, T. R. Cundari, M. A. Omary. Jahn-Teller Distortion in the Phosphorescent Excited State of Three-Coordinate Au(I) Phosphine Complexes. J. Am. Chem. Soc., 2003. [DOI | PubMed]
- M. Osawa, M. Hoshino, M. Hashimoto, I. Kawata, S. Igawa, M. Yashima. Application of Three-Coordinate Copper(I) Complexes with Halide Ligands in Organic Light-Emitting Diodes That Exhibit Delayed Fluorescence. Dalton Trans., 2015. [DOI | PubMed]
- M. Osawa. Highly Efficient Blue-Green Delayed Fluorescence from Copper(I) Thiolate Complexes: Luminescence Color Alteration by Orientation Change of the Aryl Ring. Chem. Commun., 2014. [DOI]
- L.-P. Liu, R. Zhang, L. Liu, X.-X. Zhong, F.-B. Li, L. Wang, W.-Y. Wong, G.-H. Li, H.-J. Cong, N. S. Alharbi, Y. Zhao. A New Strategy to Synthesize Three-Coordinate Mononuclear Copper(I) Halide Complexes Containing a Bulky Terphenyl Bidentate Phosphine Ligand and Their Luminescent Properties. New J. Chem., 2019. [DOI]
- X. Yin, C. Liu, S. Liu, M. Cao, J. M. Rawson, Y. Xu, B. Zhang. Structural Characterization and Luminescence Properties of Trigonal Cu(I) Iodine/Bromine Complexes Comprising Cation-Π Interactions. New J. Chem., 2022. [DOI]
- J. Nitsch, C. Kleeberg, R. Frohlich, A. Steffen. Luminescent Copper(I) Halide and Pseudohalide Phenanthroline Complexes Revisited: Simple Structures, Complicated Excited State Behavior. Dalton Trans., 2015. [DOI | PubMed]
- V. A. Krylova, P. I. Djurovich, B. L. Conley, R. Haiges, M. T. Whited, T. J. Williams, M. E. Thompson. Control of Emission Colour with N-Heterocyclic Carbene (Nhc) Ligands in Phosphorescent Three-Coordinate Cu(I) Complexes. Chem. Commun., 2014. [DOI]
- M. Elie, F. Sguerra, F. Di Meo, M. D. Weber, R. Marion, A. Grimault, J. F. Lohier, A. Stallivieri, A. Brosseau, R. B. Pansu. Designing Nhc-Copper(I) Dipyridylamine Complexes for Blue Light-Emitting Electrochemical Cells. ACS Appl. Mater. Interfaces, 2016. [DOI | PubMed]
- M. Elie, M. D. Weber, F. Di Meo, F. Sguerra, J. F. Lohier, R. B. Pansu, J. L. Renaud, M. Hamel, M. Linares, R. D. Costa, S. Gaillard. Role of the Bridging Group in Bis-Pyridyl Ligands: Enhancing Both the Photo- and Electroluminescent Features of Cationic (Ipr)Cu(I) Complexes. Chem. Eur. J., 2017. [DOI | PubMed]
- A. S. Romanov, C. R. Becker, C. E. James, D. Di, D. Credgington, M. Linnolahti, M. Bochmann. Copper and Gold Cyclic (Alkyl)(Amino)Carbene Complexes with Sub-Microsecond Photoemissions: Structure and Substituent Effects on Redox and Luminescent Properties. Chem. Eur. J., 2017. [DOI | PubMed]
- T. Holzel, A. Belyaev, M. Terzi, L. Stenzel, M. Gernert, C. M. Marian, A. Steffen, C. Ganter. Linear Carbene Pyridine Copper Complexes with Sterically Demanding N,N’-Bis(Trityl)Imidazolylidene: Syntheses, Molecular Structures, and Photophysical Properties. Inorg. Chem., 2021. [DOI | PubMed]
- Z. Liu, M. F. Qayyum, C. Wu, M. T. Whited, P. I. Djurovich, K. O. Hodgson, B. Hedman, E. I. Solomon, M. E. Thompson. A Codeposition Route to CuI-Pyridine Coordination Complexes for Organic Light-Emitting Diodes. J. Am. Chem. Soc., 2011. [DOI | PubMed]
- Z. Wang, J. Zhu, Z. Liu, P. Wu, H. Wang, Z. Zhang, B. Wei. Thermally Activated Delayed Fluorescence of Co-Deposited Copper(I) Complexes: Cost-Effective Emitters for Highly Efficient Organic Light-Emitting Diodes. J. Mater. Chem. C, 2017. [DOI]
- L. Yang, X. Xu, P. Zhang, M. Chen, G. Chen, Y. Zheng, B. Wei, J. Zhang. Photophysical Properties and Stability of Binuclear Emissive Copper(I) Complexes Co-Deposited with Cux (X = Cl, Br, I) and Aza-9,9′-Spirobifluorenes. Dyes Pigm., 2019. [DOI]
- J. Guo, Z. Zhang, P. Wu, J. Zhu, D. Dou, Z. Liao, R. Xia, K. Wang, Z. Wang. Co-Deposited Copper(I) Complexes Integrating Phosphorescence and TADF Properties for Highly Efficient OLEDs. J. Lumin., 2021. [DOI]
- D. Volz, Y. Chen, M. Wallesch, R. Liu, C. Fléchon, D. M. Zink, J. Friedrichs, H. Flügge, R. Steininger, J. Göttlicher. Bridging the Efficiency Gap: Fully Bridged Dinuclear Cu(I)-Complexes for Singlet Harvesting in High-Efficiency OLEDs. Adv. Mater., 2015. [DOI | PubMed]
- X. Hong, B. Wang, L. Liu, X.-X. Zhong, F.-B. Li, L. Wang, W.-Y. Wong, H.-M. Qin, Y. H. Lo. Highly Efficient Blue-Green Neutral Dinuclear Copper(I) Halide Complexes Containing Bidentate Phosphine Ligands. J. Lumin., 2016. [DOI]
- J. Nitsch, F. Lacemon, A. Lorbach, A. Eichhorn, F. Cisnetti, A. Steffen. Cuprophilic Interactions in Highly Luminescent Dicopper(I)-Nhc-Picolyl Complexes – Fast Phosphorescence or TADF?. Chem. Commun., 2016. [DOI]
- M. Wallesch, A. Verma, C. Flechon, H. Flugge, D. M. Zink, S. M. Seifermann, J. M. Navarro, T. Vitova, J. Gottlicher, R. Steininger. Towards Printed Organic Light-Emitting Devices: A Solution-Stable, Highly Soluble Cu(I)-Nhetphos. Chem. Eur. J., 2016. [DOI | PubMed]
- L. Lin, D.-H. Chen, R. Yu, X.-L. Chen, W.-J. Zhu, D. Liang, J.-F. Chang, Q. Zhang, C.-Z. Lu. Photo- and Electro-Luminescence of Three TADF Binuclear Cu(I) Complexes with Functional Tetraimine Ligands. J. Mater. Chem. C, 2017. [DOI]
- J. H. Jia, X. L. Chen, J. Z. Liao, D. Liang, M. X. Yang, R. Yu, C. Z. Lu. Highly Luminescent Copper(I) Halide Complexes Chelated with a Tetradentate Ligand (PNNP): Synthesis, Structure, Photophysical Properties and Theoretical Studies. Dalton Trans., 2019. [DOI | PubMed]
- M. Cao, Y. Zhao, M. Gu, C. Liu, Q. Zhu, Y. Chen, B. Wei, C. Du, B. Zhang. Syntheses, Crystal Structures and Photophysical Properties of Dinuclear Copper(I) Complexes Bearing Diphenylphosphino-Substituted Benzimidazole Ligands. ChemistrySelect, 2021. [DOI]
- J. M. Busch, D. M. Zink, P. Di Martino-Fumo, F. R. Rehak, P. Boden, S. Steiger, O. Fuhr, M. Nieger, W. Klopper, M. Gerhards, S. Brase. Highly Soluble Fluorine Containing Cu(I) Alkylpyrphos TADF Complexes. Dalton Trans., 2019. [DOI | PubMed]
- A. V. Artem’ev, Y. V. Demyanov, M. I. Rakhmanova, I. Y. Bagryanskaya. Pyridylarsine-Based Cu(I) Complexes Showing TADF Mixed with Fast Phosphorescence: A Speeding-up Emission Rate Using Arsine Ligands. Dalton Trans., 2022. [DOI | PubMed]
- A. Kobayashi, T. Hasegawa, M. Yoshida, M. Kato. Environmentally Friendly Mechanochemical Syntheses and Conversions of Highly Luminescent Cu(I) Dinuclear Complexes. Inorg. Chem., 2016. [DOI | PubMed]
- E. Cariati, X. Bu, P. C. Ford. Solvent- and Vapor-Induced Isomerization between the Luminescent Solids [CuI(4-Pic)]4 and [CuI(4-Pic)]∞ (Pic = Methylpyridine). The Structural Basis for the Observed Luminescence Vapochromism. Chem. Mater., 2000. [DOI]
- S. Perruchas, X. F. Le Goff, S. Maron, I. Maurin, F. Guillen, A. Garcia, T. Gacoin, J.-P. Boilot. Mechanochromic and Thermochromic Luminescence of a Copper Iodide Cluster. J. Am. Chem. Soc., 2010. [DOI | PubMed]
- M. Xie, C. Han, Q. Liang, J. Zhang, G. Xie, H. Xu. Highly Efficient Sky Blue Electroluminescence from Ligand-Activated Copper Iodide Clusters: Overcoming the Limitations of Cluster Light-Emitting Diodes. Sci. Adv., 2019. [DOI | PubMed]
- A. A. Titov, O. A. Filippov, A. F. Smol’yakov, I. A. Godovikov, J. R. Shakirova, S. P. Tunik, I. S. Podkorytov, E. S. Shubina. Luminescent Complexes of the Trinuclear Silver(I) and Copper(I) Pyrazolates Supported with Bis(Diphenylphosphino)Methane. Inorg. Chem., 2019. [DOI | PubMed]
- Y. Wu, J. Y. Wang, L. Y. Zhang, L. J. Xu, Z. N. Chen. Vapor-Triggered Green-to-Yellow Luminescence Conversion Due to the Variation of Ligand Orientations in Tetranuclear Copper(I) Complex. Inorg. Chem., 2020. [DOI | PubMed]
- M. Olaru, E. Rychagova, S. Ketkov, Y. Shynkarenko, S. Yakunin, M. V. Kovalenko, A. Yablonskiy, B. Andreev, F. Kleemiss, J. Beckmann, M. Vogt. A Small Cationic Organo-Copper Cluster as Thermally Robust Highly Photo- and Electroluminescent Material. J. Am. Chem. Soc., 2020. [DOI | PubMed]
- B. J. Cao, R. Li, X. H. Huang. Synthesis, Structure and Photophysical Properties of Two Tetranuclear Copper(I) Iodide Complexes Based on Acetylpyridine and Diphosphine Mixed Ligands. Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2021. [DOI]
- M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec, H. Yersin. Design Strategy for Ag (I)-Based Thermally Activated Delayed Fluorescence Reaching an Efficiency Breakthrough. Chem. Mater., 2017. [DOI]
- M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec, H. Yersin. Thermally Activated Delayed Fluorescence from Ag(I) Complexes: A Route to 100% Quantum Yield at Unprecedentedly Short Decay Time. Inorg. Chem., 2017. [DOI | PubMed]
- J. M. Carbonell-Vilar, E. Fresta, D. Armentano, R. D. Costa, M. Viciano-Chumillas, J. Cano. Photoluminescent Cu(I) Vs. Ag(I) Complexes: Slowing Down Emission in Cu(I) Complexes by Pentacoordinate Low-Lying Excited States. Dalton Trans., 2019. [DOI | PubMed]
- M. Osawa, M. Hashimoto, I. Kawata, M. Hoshino. Photoluminescence Properties of TADF-Emitting Three-Coordinate Silver(I) Halide Complexes with Diphosphine Ligands: A Comparison Study with Copper(I) Complexes. Dalton Trans., 2017. [DOI | PubMed]
- T. Teng, K. Li, G. Cheng, Y. Wang, J. Wang, J. Li, C. Zhou, H. Liu, T. Zou, J. Xiong. Lighting Silver(I) Complexes for Solution-Processed Organic Light-Emitting Diodes and Biological Applications Via Thermally Activated Delayed Fluorescence. Inorg. Chem., 2020. [DOI | PubMed]
- X. B. Cai, D. Liang, M. Yang, X. Y. Wu, C. Z. Lu, R. Yu. Efficiently Increasing the Radiative Rate of TADF Material with Metal Coordination. Chem. Commun., 2022. [DOI]
- M. Mońka, I. E. Serdiuk, K. Kozakiewicz, E. Hoffman, J. Szumilas, A. Kubicki, S. Y. Park, P. Bojarski. Understanding the Internal Heavy-Atom Effect on Thermally Activated Delayed Fluorescence: Application of Arrhenius and Marcus Theories for Spin-Orbit Coupling Analysis. J. Mater. Chem. C, 2022. [DOI]
- D. Liang, J.-H. Jia, X.-B. Cai, Y.-Q. Zhao, Z.-Q. Wang, C.-Z. Lu. Tuning Excited State Energy Levels by Achieving Coordination-Induced Thermally Activated Delayed Fluorescence. Inorg. Chem. Front., 2022. [DOI]
- A. S. Romanov, M. Bochmann. Synthesis, Structures and Photoluminescence Properties of Silver Complexes of Cyclic (Alkyl)(Amino)Carbenes. J. Organomet. Chem., 2017. [DOI]
- F. Chotard, A. S. Romanov, D. L. Hughes, M. Linnolahti, M. Bochmann. Zwitterionic Mixed-Carbene Coinage Metal Complexes: Synthesis, Structures, and Photophysical Studies. Eur. J. Inorg. Chem., 2019. [DOI]
- X. M. Gan, R. Yu, X. L. Chen, M. Yang, L. Lin, X. Y. Wu, C. Z. Lu. A Unique Tetranuclear Ag(I) Complex Emitting Efficient Thermally Activated Delayed Fluorescence with a Remarkably Short Decay Time. Dalton Trans., 2018. [DOI | PubMed]
- V. W. Yam, V. K. Au, S. Y. Leung. Light-Emitting Self-Assembled Materials Based on D8 and D10 Transition Metal Complexes. Chem. Rev., 2015. [DOI | PubMed]
- J. Chen, T. Teng, L. Kang, X. L. Chen, X. Y. Wu, R. Yu, C. Z. Lu. Highly Efficient Thermally Activated Delayed Fluorescence in Dinuclear Ag(I) Complexes with a Bis-Bidentate Tetraphosphane Bridging Ligand. Inorg. Chem., 2016. [DOI | PubMed]
- M. Z. Shafikov, A. F. Suleymanova, A. Schinabeck, H. Yersin. Dinuclear Ag(I) Complex Designed for Highly Efficient Thermally Activated Delayed Fluorescence. J. Phys. Chem. Lett, 2018. [DOI | PubMed]
- A. V. Artem’ev, M. Z. Shafikov, A. Schinabeck, O. V. Antonova, A. S. Berezin, I. Y. Bagryanskaya, P. E. Plusnin, H. Yersin. Sky-Blue Thermally Activated Delayed Fluorescence (TADF) Based on Ag(I) Complexes: Strong Solvation-Induced Emission Enhancement. Inorg. Chem. Front., 2019. [DOI]
- M. Calvo, O. Crespo, M. C. Gimeno, A. Laguna, M. T. Oliván, V. Polo, D. Rodríguez, J.-M. Sáez-Rocher. Tunable from Blue to Red Emissive Composites and Solids of Silver Diphosphane Systems with Higher Quantum Yields Than the Diphosphane Ligands. Inorg. Chem., 2020. [DOI | PubMed]
- Z. Han, X.-Y. Dong, P. Luo, S. Li, Z.-Y. Wang, S.-Q. Zang, T. C. W. Mak. Ultrastable Atomically Precise Chiral Silver Clusters with More Than 95% Quantum Efficiency. Sci. Adv., 2020. [DOI | PubMed]
- J. Xie, Y. Zheng, J. Y. Ying. Protein-Directed Synthesis of Highly Fluorescent Gold Nanoclusters. J. Am. Chem. Soc., 2009. [DOI | PubMed]
- X. Kang, M. Zhu. Tailoring the Photoluminescence of Atomically Precise Nanoclusters. Chem. Soc. Rev., 2019. [DOI | PubMed]
- K. T. Chan, G. S. M. Tong, W. P. To, C. Yang, L. Du, D. L. Phillips, C. M. Che. The Interplay between Fluorescence and Phosphorescence with Luminescent Gold(I) and Gold(III) Complexes Bearing Heterocyclic Arylacetylide Ligands. Chem. Sci., 2017. [DOI | PubMed]
- M. Osawa, M. A. Aino, T. Nagakura, M. Hoshino, Y. Tanaka, M. Akita. Near-Unity Thermally Activated Delayed Fluorescence Efficiency in Three- and Four-Coordinate Au(I) Complexes with Diphosphine Ligands. Dalton Trans., 2018. [DOI | PubMed]
- J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, M. Rodríguez-Castillo, I. Soldevilla, D. Sundholm, R. R. Valiev. Perhalophenyl Three-Coordinate Gold(I) Complexes as TADF Emitters: A Photophysical Study from Experimental and Computational Viewpoints. Inorg. Chem., 2020. [DOI | PubMed]
- I. Soldevilla, A. García-Camacho, R. T. Nasibullin, M. E. Olmos, M. Monge, D. Sundholm, R. R. Valiev, J. M. López-de-Luzuriaga, M. Rodríguez-Castillo. Influence of Perhalophenyl Groups in the TADF Mechanism of Diphosphino Gold(I) Complexes. J. Mater. Chem. C, 2022. [DOI]
- T. Y. Li, D. S. Muthiah Ravinson, R. Haiges, P. I. Djurovich, M. E. Thompson. Enhancement of the Luminescent Efficiency in Carbene-Au((I))-Aryl Complexes by the Restriction of Renner-Teller Distortion and Bond Rotation. J. Am. Chem. Soc., 2020. [DOI | PubMed]
- F. H. Yu, X. F. Song, G. H. Liu, X. Chang, K. Li, Y. Wang, G. Cui, Y. Chen. Highly Efficient Au(I) Alkynyl Emitters: Thermally Activated Delayed Fluorescence and Solution-Processed OLEDs. Chem. Eur. J., 2022. [DOI | PubMed]
- I. S. Park, H. Min, T. Yasuda. Ultrafast Triplet-Singlet Exciton Interconversion in Narrowband Blue Organoboron Emitters Doped with Heavy Chalcogens. Angew. Chem., Int. Ed., 2022. [DOI]
- S. Cai, G. S. M. Tong, L. Du, G. K. So, F. F. Hung, T. L. Lam, G. Cheng, H. Xiao, X. Chang, Z. X. Xu, C. M. Che. Gold(I) Multi-Resonance Thermally Activated Delayed Fluorescent Emitters for Highly Efficient Ultrapure-Green Organic Light-Emitting Diodes. Angew. Chem., Int. Ed., 2022. [DOI]
- H. L. Lee, W. J. Chung, J. Y. Lee. Narrowband and Pure Violet Organic Emitter with a Full Width at Half Maximum of 14 Nm and Y Color Coordinate of Below 0.02. Small, 2020. [DOI]
- X. Feng, J. G. Yang, J. Miao, C. Zhong, X. Yin, N. Li, C. Wu, Q. Zhang, Y. Chen, K. Li, C. Yang. Au···H-C Interactions Support a Robust Thermally Activated Delayed Fluorescence (TADF) Gold(I) Complex for OLEDs with Little Efficiency Roll-Off and Good Stability. Angew. Chem., Int. Ed., 2022. [DOI]
- W.-P. To, D. Zhou, G. S. M. Tong, G. Cheng, C. Yang, C.-M. Che. Highly Luminescent Pincer Gold(III) Aryl Emitters. Thermally Activated Delayed Fluorescence and Solution-Processed OLEDs with EQE and Luminance of up to 23.8% and 57340 Cd M–2. Angew. Chem., Int. Ed., 2017. [DOI]
- D. Zhou, G. Cheng, G. S. M. Tong, C. M. Che. High Efficiency Sky-Blue Gold(III)-TADF Emitters. Chem. Eur. J., 2020. [DOI | PubMed]
- D. Zhou, W.-P. To, Y. Kwak, Y. Cho, G. Cheng, G. S. M. Tong, C.-M. Che. Thermally Stable Donor-Acceptor Type (Alkynyl)Gold(III) TADF Emitters Achieved EQEs and Luminance of up to 23.4% and 70 300 Cd M–2 in Vacuum-Deposited OLEDs. Adv. Sci., 2019. [DOI]
- L. K. Li, C. C. Au-Yeung, M. C. Tang, S. L. Lai, W. L. Cheung, M. Ng, M. Y. Chan, V. W. Yam. Design and Synthesis of Yellow- to Red-Emitting Gold(III) Complexes Containing Isomeric Thienopyridine and Thienoquinoline Moieties and Their Applications in Operationally Stable Organic Light-Emitting Devices. Mater. Horiz., 2022. [DOI | PubMed]
- L. K. Li, W. K. Kwok, M. C. Tang, W. L. Cheung, S. L. Lai, M. Ng, M. Y. Chan, V. W. Yam. Highly Efficient Carbazolylgold(III) Dendrimers Based on Thermally Activated Delayed Fluorescence and Their Application in Solution-Processed Organic Light-Emitting Devices. Chem. Sci., 2021. [DOI | PubMed]
- C. Y. Wong, M. C. Tang, L. K. Li, M. Y. Leung, W. K. Tang, S. L. Lai, W. L. Cheung, M. Ng, M. Y. Chan, V. W. Yam. Carbazolylgold(III) Complexes with Thermally Activated Delayed Fluorescence Switched on by Ligand Manipulation as High Efficiency Organic Light-Emitting Devices with Small Efficiency Roll-Offs. Chem. Sci., 2022. [DOI | PubMed]
- D. Zhou, W. P. To, G. S. M. Tong, G. Cheng, L. Du, D. L. Phillips, C. M. Che. Tetradentate Gold(III) Complexes as Thermally Activated Delayed Fluorescence (TADF) Emitters: Microwave-Assisted Synthesis and High-Performance OLEDs with Long Operational Lifetime. Angew. Chem., Int. Ed., 2020. [DOI]
- P. J. Conaghan, S. M. Menke, A. S. Romanov, S. T. E. Jones, A. J. Pearson, E. W. Evans, M. Bochmann, N. C. Greenham, D. Credgington. Efficient Vacuum-Processed Light-Emitting Diodes Based on Carbene-Metal-Amides. Adv. Mater., 2018. [DOI]
- A. S. Romanov, S. T. E. Jones, L. Yang, P. J. Conaghan, D. Di, M. Linnolahti, D. Credgington, M. Bochmann. Mononuclear Silver Complexes for Efficient Solution and Vacuum-Processed OLEDs. Adv. Optical Mater., 2018. [DOI]
- R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand, M. E. Thompson. Eliminating Nonradiative Decay in Cu(I) Emitters: >99% Quantum Efficiency and Microsecond Lifetime. Science, 2019. [DOI | PubMed]
- S. Shi, M. C. Jung, C. Coburn, A. Tadle, M. R. Daniel Sylvinson, P. I. Djurovich, S. R. Forrest, M. E. Thompson. Highly Efficient Photo- and Electroluminescence from Two-Coordinate Cu(I) Complexes Featuring Nonconventional N-Heterocyclic Carbenes. J. Am. Chem. Soc., 2019. [DOI | PubMed]
- A. S. Romanov, L. Yang, S. T. E. Jones, D. Di, O. J. Morley, B. H. Drummond, A. P. M. Reponen, M. Linnolahti, D. Credgington, M. Bochmann. Dendritic Carbene Metal Carbazole Complexes as Photoemitters for Fully Solution-Processed OLEDs. Chem. Mater., 2019. [DOI]
- R. Hamze, S. Shi, S. C. Kapper, D. S. Muthiah Ravinson, L. Estergreen, M. C. Jung, A. C. Tadle, R. Haiges, P. I. Djurovich, J. L. Peltier. “Quick-Silver” from a Systematic Study of Highly Luminescent, Two-Coordinate, D10 Coinage Metal Complexes. J. Am. Chem. Soc., 2019. [DOI | PubMed]
- R. Hamze, M. Idris, D. S. Muthiah Ravinson, M. C. Jung, R. Haiges, P. I. Djurovich, M. E. Thompson. Highly Efficient Deep Blue Luminescence of 2-Coordinate Coinage Metal Complexes Bearing Bulky NHC Benzimidazolyl Carbene. Front. Chem., 2020. [DOI | PubMed]
- F. Chotard, V. Sivchik, M. Linnolahti, M. Bochmann, A. S. Romanov. Mono- Versus Bicyclic Carbene Metal Amide Photoemitters: Which Design Leads to the Best Performance?. Chem. Mater., 2020. [DOI]
- J. G. Yang, X. F. Song, J. Wang, K. Li, X. Chang, L. Y. Tan, C. X. Liu, F. H. Yu, G. Cui, G. Cheng. Highly Efficient Thermally Activated Delayed Fluorescence from Pyrazine-Fused Carbene Au(I) Emitters. Chem. Eur. J., 2021. [DOI | PubMed]
- P. J. Conaghan, C. S. B. Matthews, F. Chotard, S. T. E. Jones, N. C. Greenham, M. Bochmann, D. Credgington, A. S. Romanov. Highly Efficient Blue Organic Light-Emitting Diodes Based on Carbene-Metal-Amides. Nat. Commun., 2020. [DOI | PubMed]
- A. S. Romanov, S. T. E. Jones, Q. Gu, P. J. Conaghan, B. H. Drummond, J. Feng, F. Chotard, L. Buizza, M. Foley, M. Linnolahti. Carbene Metal Amide Photoemitters: Tailoring Conformationally Flexible Amides for Full Color Range Emissions Including White-Emitting OLED. Chem. Sci., 2020. [DOI | PubMed]
- M. Gernert, L. Balles-Wolf, F. Kerner, U. Muller, A. Schmiedel, M. Holzapfel, C. M. Marian, J. Pflaum, C. Lambert, A. Steffen. Cyclic (Amino)(Aryl)Carbenes Enter the Field of Chromophore Ligands: Expanded Π System Leads to Unusually Deep Red Emitting Cu(I) Compounds. J. Am. Chem. Soc., 2020. [DOI | PubMed]
- T. Y. Li, D. G. Shlian, P. I. Djurovich, M. E. Thompson. A Luminescent Two-Coordinate Au(I) Bimetallic Complex with a Tandem-Carbene Structure: A Molecular Design for the Enhancement of TADF Radiative Decay Rate. Chem. Eur. J., 2021. [DOI | PubMed]
- A. P. M. Reponen, F. Chotard, A. Lempelto, V. Shekhovtsev, D. Credgington, M. Bochmann, M. Linnolahti, N. C. Greenham, A. S. Romanov. Donor N-Substitution as Design Principle for Fast and Blue Luminescence in Carbene-Metal-Amides. Adv. Optical Mater., 2022. [DOI]
- T.-y. Li, J. Schaab, P. I. Djurovich, M. E. Thompson. Toward Rational Design of TADF Two-Coordinate Coinage Metal Complexes: Understanding the Relationship between Natural Transition Orbital Overlap and Photophysical Properties. J. Mater. Chem. C, 2022. [DOI]
- C. N. Muniz, J. Schaab, A. Razgoniaev, P. I. Djurovich, M. E. Thompson. Π-Extended Ligands in Two-Coordinate Coinage Metal Complexes. J. Am. Chem. Soc., 2022. [DOI | PubMed]
- R. Tang, S. Xu, T. L. Lam, G. Cheng, L. Du, Q. Wan, J. Yang, F. F. Hung, K. H. Low, D. L. Phillips, C. M. Che. Highly Robust Cu(I) -TADF Emitters for Vacuum-Deposited OLEDs with Luminance up to 222 200 Cd M–2 and Device Lifetimes (Lt90) up to 1300 Hours at an Initial Luminance of 1000 Cd M–2. Angew. Chem., Int. Ed., 2022. [DOI]
- S. Heo, Y. Jung, J. Kim, I. Kim, H. J. Bae, W. J. Son, H. Choi, Y. You. High-Performance Blue Electroluminescence Devices Based on Linear Gold(I) Complexes as Ultrafast Triplet Exciton Harvesters. Adv. Optical Mater., 2022. [DOI]
- J. G. Yang, X. F. Song, G. Cheng, S. Wu, X. Feng, G. Cui, W. P. To, X. Chang, Y. Chen, C. M. Che. Conformational Engineering of Two-Coordinate Gold(I) Complexes: Regulation of Excited-State Dynamics for Efficient Delayed Fluorescence. ACS Appl. Mater. Interfaces, 2022. [DOI | PubMed]
- A. Ying, Y. Ai, C. Yang, S. Gong. Aggregation-Dependent Circularly Polarized Luminescence and Thermally Activated Delayed Fluorescence from Chiral Carbene-Cu(I) -Amide Enantiomers. Angew. Chem., Int. Ed., 2022. [DOI]
- A. Ying, Y. H. Huang, C. H. Lu, Z. Chen, W. K. Lee, X. Zeng, T. Chen, X. Cao, C. C. Wu, S. Gong, C. Yang. High-Efficiency Red Electroluminescence Based on a Carbene-Cu(I)-Acridine Complex. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- C. Cebrian, M. Mauro. Recent Advances in Phosphorescent Platinum Complexes for Organic Light-Emitting Diodes. Beilstein J. Org. Chem., 2018. [DOI | PubMed]
- Z.-Q. Zhu, T. Fleetham, E. Turner, J. Li. Harvesting All Electrogenerated Excitons through Metal Assisted Delayed Fluorescent Materials. Adv. Mater., 2015. [DOI | PubMed]
- P. Pander, R. Daniels, A. V. Zaytsev, A. Horn, A. Sil, T. J. Penfold, J. A. G. Williams, V. N. Kozhevnikov, F. B. Dias. Exceptionally Fast Radiative Decay of a Dinuclear Platinum Complex through Thermally Activated Delayed Fluorescence. Chem. Sci., 2021. [DOI | PubMed]
- P. Pander, A. V. Zaytsev, A. Sil, J. A. G. Williams, P.-H. Lanoe, V. N. Kozhevnikov, F. B. Dias. The Role of Dinuclearity in Promoting Thermally Activated Delayed Fluorescence (TADF) in Cyclometallated, N∧C∧N-Coordinated Platinum(II) Complexes. J. Mater. Chem. C, 2021. [DOI]
- P. Pander, A. V. Zaytsev, A. Sil, J. A. G. Williams, V. N. Kozhevnikov, F. B. Dias. Enhancement of Thermally Activated Delayed Fluorescence Properties by Substitution of Ancillary Halogen in a Multiple Resonance-Like DiPlatinum(II) Complex. J. Mater. Chem. C, 2022. [DOI]
- Z. W. Li, L. Y. Peng, X. F. Song, W. K. Chen, Y. J. Gao, W. H. Fang, G. Cui. Room-Temperature Phosphorescence and Thermally Activated Delayed Fluorescence in the Pd Complex: Mechanism and Dual Upconversion Channels. J. Phys. Chem. Lett, 2021. [DOI | PubMed]
- Z. Q. Zhu, C. D. Park, K. Klimes, J. Li. Highly Efficient Blue OLEDs Based on Metal-Assisted Delayed Fluorescence Pd(Ii) Complexes. Adv. Optical Mater., 2019. [DOI]
- G. Li, Q. Chen, J. Zheng, Q. Wang, F. Zhan, W. Lou, Y. F. Yang, Y. She. Metal-Assisted Delayed Fluorescent Pd(Ii) Complexes and Phosphorescent Pt(Ii) Complex Based on [1,2,4]Triazolo[4,3-a]Pyridine-Containing Ligands: Synthesis, Characterization, Electrochemistry, Photophysical Studies, and Application. Inorg. Chem., 2019. [DOI | PubMed]
- Y. She, K. Xu, X. Fang, Y. F. Yang, W. Lou, Y. Hu, Q. Zhang, G. Li. Tetradentate Platinum(II) and Palladium(II) Complexes Containing Fused 6/6/6 or 6/6/5 Metallocycles with Azacarbazolylcarbazole-Based Ligands. Inorg. Chem., 2021. [DOI | PubMed]
- P. W. Zach, S. A. Freunberger, I. Klimant, S. M. Borisov. Electron-Deficient near-Infrared Pt(Ii) and Pd(Ii) Benzoporphyrins with Dual Phosphorescence and Unusually Efficient Thermally Activated Delayed Fluorescence: First Demonstration of Simultaneous Oxygen and Temperature Sensing with a Single Emitter. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- Z. Abedin-Siddique, T. Ohno, K. Nozaki, T. Tsubomura. Intense Fluorescence of Metal-to-Ligand Charge Transfer in [Pt(0)(Binap)2] [Binap = 2,2‘-Bis(Diphenylphosphino)-1,1‘-Binaphthyl]. Inorg. Chem., 2004. [DOI | PubMed]
- P. K. Chow, C. Ma, W. P. To, G. S. Tong, S. L. Lai, S. C. Kui, W. M. Kwok, C. M. Che. Strongly Phosphorescent Palladium(II) Complexes of Tetradentate Ligands with Mixed Oxygen, Carbon, and Nitrogen Donor Atoms: Photophysics, Photochemistry, and Applications. Angew. Chem., Int. Ed., 2013. [DOI]
- Y. Sakai, Y. Sagara, H. Nomura, N. Nakamura, Y. Suzuki, H. Miyazaki, C. Adachi. Zinc Complexes Exhibiting Highly Efficient Thermally Activated Delayed Fluorescence and Their Application to Organic Light-Emitting Diodes. Chem. Commun., 2015. [DOI]
- J. Xiong, K. Li, T. Teng, X. Chang, Y. Wei, C. Wu, C. Yang. Dinuclear Zn(II) Complexes Exhibiting Thermally Activated Delayed Fluorescence and Luminescence Polymorphism. Chem. Eur. J., 2020. [DOI | PubMed]
- A. Berezin, K. Vinogradova, V. Krivopalov, E. Nikolaenkova, V. Plyusnin, A. Kupryakov, N. Pervukhina, D. Naumov, M. B. Bushuev. Excitation-Wavelength-Dependent Emission and Delayed Fluorescence in a Proton Transfer System. Chem. Eur. J., 2018. [DOI | PubMed]
- Y. Chen, X. Li, N. Li, Y. Quan, Y. Cheng, Y. Tang. Strong Circularly Polarized Electroluminescence Based on Chiral Salen-Zn(II) Complex Monomer Chromophores. Mater. Chem. Front., 2019. [DOI]
- A. Steinegger, S. M. Borisov. Zn(II) Schiff Bases: Bright TADF Emitters for Self-Referenced Decay Time-Based Optical Temperature Sensing. ACS Omega, 2020. [DOI | PubMed]
- A. Russegger, L. Eiber, A. Steinegger, S. M. Borisov. Zinc Donor-Acceptor Schiff Base Complexes as Thermally Activated Delayed Fluorescence Emitters. Chemosensors, 2022. [DOI]
- S. E. Zieger, A. Steinegger, I. Klimant, S. M. Borisov. TADF-Emitting Zn(II)-Benzoporphyrin: An Indicator for Simultaneous Sensing of Oxygen and Temperature. ACS Sens., 2020. [DOI | PubMed]
- B. Goswami, T. J. Feuerstein, R. Yadav, S. Lebedkin, P. J. Boden, S. T. Steiger, G. Niedner-Schatteburg, M. Gerhards, M. M. Kappes, P. W. Roesky. Thermally Activated Delayed Fluorescence and Phosphorescence Quenching in Iminophosphonamide Copper and Zinc Complexes. Chem. Eur. J., 2021. [DOI]
- A. S. Gowda, T. S. Lee, M. C. Rosko, J. L. Petersen, F. N. Castellano, C. Milsmann. Long-Lived Photoluminescence of Molecular Group 14 Compounds through Thermally Activated Delayed Fluorescence. Inorg. Chem., 2022. [DOI | PubMed]
- W. Sattler, L. M. Henling, J. R. Winkler, H. B. Gray. Bespoke Photoreductants: Tungsten Arylisocyanides. J. Am. Chem. Soc., 2015. [DOI | PubMed]
- K. T. Chan, T. L. Lam, D. Yu, L. Du, D. L. Phillips, C. L. Kwong, G. S. M. Tong, G. Cheng, C. M. Che. Strongly Luminescent Tungsten Emitters with Emission Quantum Yields of up to 84 %: TADF and High-Efficiency Molecular Tungsten OLEDs. Angew. Chem., Int. Ed., 2019. [DOI]
- Y. Zhang, T. S. Lee, J. M. Favale, D. C. Leary, J. L. Petersen, G. D. Scholes, F. N. Castellano, C. Milsmann. Delayed Fluorescence from a Zirconium(IV) Photosensitizer with Ligand-to-Metal Charge-Transfer Excited States. Nature Chem., 2020. [DOI | PubMed]
- Y. Zhang, D. C. Leary, A. M. Belldina, J. L. Petersen, C. Milsmann. Effects of Ligand Substitution on the Optical and Electrochemical Properties of (Pyridinedipyrrolide)Zirconium Photosensitizers. Inorg. Chem., 2020. [DOI | PubMed]
- Y. Zhang, J. L. Petersen, C. Milsmann. A Luminescent Zirconium(IV) Complex as a Molecular Photosensitizer for Visible Light Photoredox Catalysis. J. Am. Chem. Soc., 2016. [DOI | PubMed]
- T. J. Feuerstein, B. Goswami, P. Rauthe, R. Köppe, S. Lebedkin, M. M. Kappes, P. W. Roesky. Alkali Metal Complexes of an Enantiopure Iminophosphonamide Ligand with Bright Delayed Fluorescence. Chem. Sci., 2019. [DOI | PubMed]
- B. Goswami, T. J. Feuerstein, R. Yadav, R. Koppe, S. Lebedkin, M. M. Kappes, P. W. Roesky. Enantiopure Calcium Iminophosphonamide Complexes: Synthesis, Photoluminescence, and Catalysis. Chem. Eur. J., 2021. [DOI | PubMed]
- K. Nakao, H. Sasabe, Y. Shibuya, A. Matsunaga, H. Katagiri, J. Kido. Novel Series of Mononuclear Aluminum Complexes for High-Performance Solution-Processed Organic Light-Emitting Devices. Angew. Chem., Int. Ed., 2021. [DOI]
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson, S. R. Forrest. Very High-Efficiency Green Organic Light-Emitting Devices Based on Electrophosphorescence. Appl. Phys. Lett., 1999. [DOI]
- H. Benjamin, Y. Zheng, V. N. Kozhevnikov, J. S. Siddle, L. J. O’Driscoll, M. A. Fox, A. S. Batsanov, G. C. Griffiths, F. B. Dias, A. P. Monkman, M. R. Bryce. Unusual Dual-Emissive Heteroleptic Iridium Complexes Incorporating TADF Cyclometalating Ligands. Dalton Trans., 2020. [DOI | PubMed]
- A. Thamarappalli, C. S. K. Ranasinghe, J. Jang, M. Gao, P. L. Burn, E. V. Puttock, P. E. Shaw. Properties of Dual Emissive Dendrimers Based on Thermally Activated Delayed Fluorescence Dendrons and a Phosphorescent Ir(Ppy)3 Core. Adv. Funct. Mater., 2022. [DOI]
- J. Jang, C. S. K. Ranasinghe, A. Thamarappalli, M. Gao, M. Koodalingam, P. L. Burn, E. V. Puttock, P. E. Shaw. Understanding the Emission from Dendrimers Composed of Thermally Activated Delayed Fluorescence-Based Dendrons and a Phosphorescent Fac-Tris[2-(Thiophen-2-Yl)-4-(Phenyl)Quinoline]Iridium(Iii) Core. J. Mater. Chem. C, 2022. [DOI]
- T. Zhou, K. Zhang, Q. Cao, H. Xu, X. Ban, P. Zhu, Q. Li, L. Shi, F. Ge, W. Jiang. Benzonitrile-Based AIE Polymer Host with a Simple Synthesis Process for High-Efficiency Solution-Processable Green and Blue TADF Organic Light Emitting Diodes. J. Mater. Chem. C, 2022. [DOI]
- P. Pander, S. Gogoc, M. Colella, P. Data, F. B. Dias. Thermally Activated Delayed Fluorescence in Polymer-Small-Molecule Exciplex Blends for Solution-Processed Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- G. Xie, J. Luo, M. Huang, T. Chen, K. Wu, S. Gong, C. Yang. Inheriting the Characteristics of TADF Small Molecule by Side-Chain Engineering Strategy to Enable Bluish-Green Polymers with High PLQYs up to 74% and External Quantum Efficiency over 16% in Light-Emitting Diodes. Adv. Mater., 2017. [DOI]
- T. Wang, Y. Zou, Z. Huang, N. Li, J. Miao, C. Yang. Narrowband Emissive TADF Conjugated Polymers Towards Highly Efficient Solution-Processible OLEDs. Angew. Chem., Int. Ed., 2022. [DOI]
- W. Zong, W. Qiu, P. Yuan, F. Wang, Y. Liu, S. Xu, S. J. Su, S. Cao. Thermally Activated Delayed Fluorescence Polymers for High-Efficiency Solution-Processed Non-Doped OLEDs: Convenient Synthesis by Binding TADF Units and Host Units to the Pre-Synthesized Polycarbazole-Based Backbone Via Click Reaction. Polymer, 2022. [DOI]
- H. Tanaka, K. Shizu, H. Miyazaki, C. Adachi. Efficient Green Thermally Activated Delayed Fluorescence (TADF) from a Phenoxazine-Triphenyltriazine (Pxz-Trz) Derivative. Chem. Commun., 2012. [DOI]
- Y. Yang, L. Zhao, S. Wang, J. Ding, L. Wang. Red-Emitting Thermally Activated Delayed Fluorescence Polymers with Poly(Fluorene-Co-3,3′-Dimethyl Diphenyl Ether) as the Backbone. Macromolecules, 2018. [DOI]
- X. Ban, T. Zhou, Q. Cao, K. Zhang, Z. Tong, H. Xu, A. Zhu, W. Jiang. Combining Molecular Encapsulation and an AIE Strategy to Construct an Efficient Blue TADF Polymer for Solution-Processed Multilayer White OLEDs. J. Mater. Chem. C, 2022. [DOI]
- C. Li, A. K. Harrison, Y. Liu, Z. Zhao, F. B. Dias, C. Zeng, S. Yan, M. R. Bryce, Z. Ren. TADF Dendronized Polymer with Vibrationally Enhanced Direct Spin-Flip between Charge-Transfer States for Efficient Non-Doped Solution-Processed OLEDs. Chem. Eng. J., 2022. [DOI]
- C. Li, Y. Wang, D. Sun, H. Li, X. Sun, D. Ma, Z. Ren, S. Yan. Thermally Activated Delayed Fluorescence Pendant Copolymers with Electron- and Hole-Transporting Spacers. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- C. Li, R. S. Nobuyasu, Y. Wang, F. B. Dias, Z. Ren, M. R. Bryce, S. Yan. Solution-Processable Thermally Activated Delayed Fluorescence White OLEDs Based on Dual-Emission Polymers with Tunable Emission Colors and Aggregation-Enhanced Emission Properties. Adv. Optical Mater., 2017. [DOI]
- C. Li, Z. Ren, X. Sun, H. Li, S. Yan. Deep-Blue Thermally Activated Delayed Fluorescence Polymers for Nondoped Solution-Processed Organic Light-Emitting Diodes. Macromolecules, 2019. [DOI]
- P. L. Dos Santos, J. S. Ward, M. R. Bryce, A. P. Monkman. Using Guest-Host Interactions to Optimize the Efficiency of TADF OLEDs. J. Phys. Chem. Lett, 2016. [DOI | PubMed]
- X. Zeng, J. Luo, T. Zhou, T. Chen, X. Zhou, K. Wu, Y. Zou, G. Xie, S. Gong, C. Yang. Using Ring-Opening Metathesis Polymerization of Norbornene to Construct Thermally Activated Delayed Fluorescence Polymers: High-Efficiency Blue Polymer Light-Emitting Diodes. Macromolecules, 2018. [DOI]
- C. M. Cole, S. V. Kunz, P. E. Shaw, C. Sampath, K. Ranasinghe, T. Baumann, J. P. Blinco, P. Sonar, C. Barner-Kowollik, S. D. Yambem. Inkjet-Printed Self-Hosted TADF Polymer Light-Emitting Diodes. Adv. Mater. Tech., 2022. [DOI]
- Y. Zhu, Y. Zhang, B. Yao, Y. Wang, Z. Zhang, H. Zhan, B. Zhang, Z. Xie, Y. Wang, Y. Cheng. Synthesis and Electroluminescence of a Conjugated Polymer with Thermally Activated Delayed Fluorescence. Macromolecules, 2016. [DOI]
- Y. Yang, S. Wang, Y. Zhu, Y. Wang, H. Zhan, Y. Cheng. Thermally Activated Delayed Fluorescence Conjugated Polymers with Backbone-Donor/Pendant-Acceptor Architecture for Nondoped OLEDs with High External Quantum Efficiency and Low Roll-Off. Adv. Funct. Mater., 2018. [DOI]
- Y. Liu, Y. Wang, C. Li, Z. Ren, D. Ma, S. Yan. Efficient Thermally Activated Delayed Fluorescence Conjugated Polymeric Emitters with Tunable Nature of Excited States Regulated Via Carbazole Derivatives for Solution-Processed OLEDs. Macromolecules, 2018. [DOI]
- Z. Zhao, Y. Liu, L. Hua, S. Yan, Z. Ren, Z. Zhao, L. Hua, S. Yan, Z. Ren, Y. Liu. Activating Energy Transfer Tunnels by Tuning Local Electronegativity of Conjugated Polymeric Backbone for High-Efficiency OLEDs with Low Efficiency Roll-Off. Adv. Funct. Mater., 2022. [DOI]
- Q. Wei, P. Kleine, Y. Karpov, X. Qiu, H. Komber, K. Sahre, A. Kiriy, R. Lygaitis, S. Lenk, S. Reineke, B. Voit. Conjugation-Induced Thermally Activated Delayed Fluorescence (TADF): From Conventional Non-TADF Units to TADF-Active Polymers. Adv. Funct. Mater., 2017. [DOI]
- J. Zhang, Q. Wei, L. Lyu, L. Cao, M. Zhao, N. Fei, T. Wang, Z. Ge. Thermally Activated Delayed Fluorescent (TADF) Mono-Polymeric OLED with Higher EQE over Its TADF Repeating Unit. Macromol. Chem. Phys., 2022. [DOI]
- Y. Wang, Y. Zhu, G. Xie, H. Zhan, C. Yang, Y. Cheng. Bright White Electroluminescence from a Single Polymer Containing a Thermally Activated Delayed Fluorescence Unit and a Solution-Processed Orange OLED Approaching 20% External Quantum Efficiency. J. Mater. Chem. C, 2017. [DOI]
- Y. Hu, F. Song, Z. Xu, Y. Tu, H. Zhang, Q. Cheng, J. W. Y. Lam, D. Ma, B. Z. Tang. Circularly Polarized Luminescence from Chiral Conjugated Poly(carbazole-ran-acridine)s with Aggregation-Induced Emission and Delayed Fluorescence. ACS Appl. Polym. Mater., 2019. [DOI]
- J. M. Teng, D. W. Zhang, Y. F. Wang, C. F. Chen. Chiral Conjugated Thermally Activated Delayed Fluorescent Polymers for Highly Efficient Circularly Polarized Polymer Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2022. [DOI | PubMed]
- D. M. E. Freeman, A. J. Musser, J. M. Frost, H. L. Stern, A. K. Forster, K. J. Fallon, A. G. Rapidis, F. Cacialli, I. McCulloch, T. M. Clarke. Synthesis and Exciton Dynamics of Donor-Orthogonal Acceptor Conjugated Polymers: Reducing the Singlet-Triplet Energy Gap. J. Am. Chem. Soc., 2017. [DOI | PubMed]
- A. E. Nikolaenko, M. Cass, F. Bourcet, D. Mohamad, M. Roberts. Thermally Activated Delayed Fluorescence in Polymers: A New Route toward Highly Efficient Solution Processable OLEDs. Adv. Mater., 2015. [DOI | PubMed]
- K. Philipps, Y. Ie, B. van der Zee, R. Q. Png, P. K. H. Ho, L. L. Chua, E. del Pino Rosendo, C. Ramanan, G. J. A. H. Wetzelaer, P. W. M. Blom, J. J. Michels. Role of Linker Functionality in Polymers Exhibiting Main-Chain Thermally Activated Delayed Fluorescence. Adv. Sci., 2022. [DOI]
- K. Sun, J. Wu, L. Zhu, H. Liu, Y. Zhou, W. Tian, Z. Cai, W. Jiang, Y. Sun. Highly Efficient Blue All-Solution-Processed Organic Light-Emitting Diodes Based on the Strategy of Constructing a Thermally Cross-Linkable TADF Dendrimer. Dyes Pigm., 2022. [DOI]
- K. Sun, W. Tian, H. Gao, C. Bi, J. Yao, Z. Wang, Z. Cai, W. Jiang. Creation of a Thermally Cross-Linkable Encapsulated TADF Molecule for Highly Efficient Solution-Processed Hybrid White OLEDs. Org. Electron., 2022. [DOI]
- K. Sun, W. Tian, C. Ge, F. Gu, Y. Zhou, W. Wang, Z. Cai, W. Jiang, Y. Sun. Creation of Efficient Solution-Processed OLEDs Via a Strategy of the Host-Guest System Constructing with Two Small Cross-Linkable TADF Molecules. Org. Electron., 2022. [DOI]
- S. Shao, J. Hu, X. Wang, L. Wang, X. Jing, F. Wang. Blue Thermally Activated Delayed Fluorescence Polymers with Nonconjugated Backbone and through-Space Charge Transfer Effect. J. Am. Chem. Soc., 2017. [DOI | PubMed]
- J. Hu, Y. Chang, F. Chen, Q. Yang, S. Shao, L. Wang. Design, Synthesis, and Properties of Polystyrene-Based through-Space Charge Transfer Polymers: Effect of Triplet Energy Level of Electron Donor Moiety on Delayed Fluorescence and Electroluminescence Performance. J. Polym. Sci., 2022. [DOI]
- K. Albrecht, K. Matsuoka, K. Fujita, K. Yamamoto. Carbazole Dendrimers as Solution-Processable Thermally Activated Delayed-Fluorescence Materials. Angew. Chem., Int. Ed., 2015. [DOI]
- K. Albrecht, K. Matsuoka, D. Yokoyama, Y. Sakai, A. Nakayama, K. Fujita, K. Yamamoto. Thermally Activated Delayed Fluorescence OLEDs with Fully Solution Processed Organic Layers Exhibiting Nearly 10% External Quantum Efficiency. Chem. Commun., 2017. [DOI]
- K. Matsuoka, K. Albrecht, A. Nakayama, K. Yamamoto, K. Fujita. Highly Efficient Thermally Activated Delayed Fluorescence Organic Light-Emitting Diodes with Fully Solution-Processed Organic Multilayered Architecture: Impact of Terminal Substitution on Carbazole-Benzophenone Dendrimer and Interfacial Engineering. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- B. Huang, X. Ban, K. Sun, Z. Ma, Y. Mei, W. Jiang, B. Lin, Y. Sun. Thermally Activated Delayed Fluorescence Materials Based on Benzophenone Derivative as Emitter for Efficient Solution-Processed Non-Doped Green OLED. Dyes Pigm., 2016. [DOI]
- K. Matsuoka, K. Albrecht, K. Yamamoto, K. Fujita. Mulifunctional Dendritic Emitter: Aggregation-Induced Emission Enhanced, Thermally Activated Delayed Fluorescent Material for Solution-Processed Multilayered Organic Light-Emitting Diodes. Sci. Rep., 2017. [DOI | PubMed]
- D. Sun, E. Duda, X. Fan, R. Saxena, M. Zhang, S. Bagnich, X. Zhang, A. Köhler, E. Zysman-Colman. Thermally Activated Delayed Fluorescent Dendrimers That Underpin High-Efficiency Host-Free Solution-Processed Organic Light-Emitting Diodes. Adv. Mater., 2022. [DOI]
- D. Sun, R. Saxena, X. Fan, S. Athanasopoulos, E. Duda, M. Zhang, S. Bagnich, X. Zhang, E. Zysman-Colman, A. Köhler. Regiochemistry of Donor Dendrons Controls the Performance of Thermally Activated Delayed Fluorescence Dendrimer Emitters for High Efficiency Solution-Processed Organic Light-Emitting Diodes. Adv. Sci., 2022. [DOI]
- C. Zhang, H. Yan, Y. He, Y. Chai, D. Zhou. Thermally Activated Delayed Fluorescence Dendrimers Achieving 20% External Quantum Efficiency for Solution-Processed OLEDs. Mater. Chem. Front., 2022. [DOI]
- Y. He, D. Zhou, C. Zhang, H. Yan, Y. Chai. Orange-Red and Saturated Red Thermally Activated Delayed Fluorescent Dendrimers for Non-Doped Solution-Processed OLEDs. Dyes Pigm., 2022. [DOI]
- Y. Li, G. Xie, S. Gong, K. Wu, C. Yang. Dendronized Delayed Fluorescence Emitters for Non-Doped, Solution-Processed Organic Light-Emitting Diodes with High Efficiency and Low Efficiency Roll-Off Simultaneously: Two Parallel Emissive Channels. Chem. Sci., 2016. [DOI | PubMed]
- J. Luo, S. Gong, Y. Gu, T. Chen, Y. Li, C. Zhong, G. Xie, C. Yang. Multi-Carbazole Encapsulation as a Simple Strategy for the Construction of Solution-Processed, Non-Doped Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C, 2016. [DOI]
- C. Li, A. K. Harrison, Y. Liu, Z. Zhao, C. Zeng, F. B. Dias, Z. Ren, S. Yan, M. R. Bryce. Asymmetrical-Dendronized TADF Emitters for Efficient Non-Doped Solution-Processed OLEDs by Eliminating Degenerate Excited States and Creating Solely Thermal Equilibrium Routes. Angew. Chem., Int. Ed., 2022. [DOI]
- M. Huang, Y. Li, K. Wu, J. Luo, G. Xie, L. Li, C. Yang. Carbazole-Dendronized Thermally Activated Delayed Fluorescent Molecules with Small Singlet-Triplet Gaps for Solution-Processed Organic Light-Emitting Diodes. Dyes Pigm., 2018. [DOI]
- Y. Li, T. Chen, M. Huang, Y. Gu, S. Gong, G. Xie, C. Yang. Tuning the Twist Angle of Thermally Activated Delayed Fluorescence Molecules Via a Dendronization Strategy: High-Efficiency Solution-Processed Non-Doped OLEDs. J. Mater. Chem. C, 2017. [DOI]
- J. Wang, J. Peng, W. Yao, C. Jiang, C. Liu, C. Zhang, M. He, R. Liu, X. Xia, C. Yao. Carbazole-Dendrite-Encapsulated Electron Acceptor Core for Constructing Thermally Activated Delayed Fluorescence Emitters Used in Nondoped Solution-Processed Organic Light-Emitting Diodes. Org. Electron., 2017. [DOI]
- E. V. Puttock, C. S. K. Ranasinghe, M. Babazadeh, J. C. M. Kistemaker, J. Jang, M. Gao, D. M. Huang, C. Adachi, P. L. Burn, P. E. Shaw. Thermally Activated Delayed Fluorescence Poly(Dendrimer)S – Detrapping Excitons for Reverse Intersystem Crossing. J. Mater. Chem. C, 2022. [DOI]
- A. Thamarappalli, C. S. K. Ranasinghe, J. Jang, M. Gao, P. L. Burn, E. V. Puttock, P. E. Shaw. Properties of Dual Emissive Dendrimers Based on Thermallyactivated Delayed Fluorescence Dendrons and a Phosphorescent Ir(Ppy)3 Core. Adv. Funct. Mater., 2022. [DOI]
- X. Wang, J. Hu, J. Lv, Q. Yang, H. Tian, S. Shao, L. Wang, X. Jing, F. Wang. Π-Stacked Donor-Acceptor Dendrimers for Highly Efficient White Electroluminescence. Angew. Chem., Int. Ed., 2021. [DOI]
- X. Ban, A. Zhu, T. Zhang, Z. Tong, W. Jiang, Y. Sun. Highly Efficient All-Solution-Processed Fluorescent Organic Light-Emitting Diodes Based on a Novel Self-Host Thermally Activated Delayed Fluorescence Emitter. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- X. Ban, F. Chen, Y. Liu, J. Pan, A. Zhu, W. Jiang, Y. Sun. Design of Efficient Thermally Activated Delayed Fluorescence Blue Host for High Performance Solution-Processed Hybrid White Organic Light Emitting Diodes. Chem. Sci., 2019. [DOI | PubMed]
- K. Sun, Y. Sun, T. Huang, J. Luo, W. Jiang, Y. Sun. Design Strategy of Yellow Thermally Activated Delayed Fluorescent Dendrimers and Their Highly Efficient Non-Doped Solution-Processed OLEDs with Low Driving Voltage. Org. Electron., 2017. [DOI]
- M. Godumala, S. Choi, H. J. Kim, C. Lee, S. Park, J. S. Moon, K. Si Woo, J. H. Kwon, M. J. Cho, D. H. Choi. Novel Dendritic Large Molecules as Solution-Processable Thermally Activated Delayed Fluorescent Emitters for Simple Structured Non-Doped Organic Light Emitting Diodes. J. Mater. Chem. C, 2018. [DOI]
- X. Ban, W. Jiang, K. Sun, B. Lin, Y. Sun. Self-Host Blue Dendrimer Comprised of Thermally Activated Delayed Fluorescence Core and Bipolar Dendrons for Efficient Solution-Processible Nondoped Electroluminescence. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- X. Ban, B. Lin, W. Jiang, Y. Sun. Constructing a Novel Dendron for a Self-Host Blue Emitter with Thermally Activated Delayed Fluorescence: Solution-Processed Nondoped Organic Light-Emitting Diodes with Bipolar Charge Transfer and Stable Color Purity. Chem. Asian J., 2017. [DOI | PubMed]
- K. Sun, Y. Sun, D. Liu, Y. Feng, X. Zhang, Y. Sun, W. Jiang. Cbp Derivatives Dendronized Self-Host TADF Dendrimer: Achieving Efficient Non-Doped near-Infrared Organic Light-Emitting Diodes. Dyes Pigm., 2017. [DOI]
- K. Sun, D. Chu, Y. Cui, W. Tian, Y. Sun, W. Jiang. Near-Infrared Thermally Activated Delayed Fluorescent Dendrimers for the Efficient Non-Doped Solution-Processed Organic Light-Emitting Diodes. Org. Electron., 2017. [DOI]
- S. Nakatsuka, H. Gotoh, K. Kinoshita, N. Yasuda, T. Hatakeyama. Divergent Synthesis of Heteroatom-Centered 4,8,12-Triazatriangulenes. Angew. Chem., Int. Ed., 2017. [DOI]
- S. H. Han, J. H. Jeong, J. W. Yoo, J. Y. Lee. Ideal Blue Thermally Activated Delayed Fluorescence Emission Assisted by a Thermally Activated Delayed Fluorescence Assistant Dopant through a Fast Reverse Intersystem Crossing Mediated Cascade Energy Transfer Process. J. Mater. Chem. C, 2019. [DOI]
- H. Lim, S. J. Woo, Y. H. Ha, Y. H. Kim, J. J. Kim. Breaking the Efficiency Limit of Deep-Blue Fluorescent OLEDs Based on Anthracene Derivatives. Adv. Mater., 2022. [DOI]
- J. H. Kim, W. J. Chung, J. Kim, J. Y. Lee. Concentration Quenching-Resistant Multiresonance Thermally Activated Delayed Fluorescence Emitters. Mater. Today Energy, 2021. [DOI]
- X. Liang, Z. P. Yan, H. B. Han, Z. G. Wu, Y. X. Zheng, H. Meng, J. L. Zuo, W. Huang. Peripheral Amplification of Multi-Resonance Induced Thermally Activated Delayed Fluorescence for Highly Efficient OLEDs. Angew. Chem., Int. Ed., 2018. [DOI]
- S. Oda, W. Kumano, T. Hama, R. Kawasumi, K. Yoshiura, T. Hatakeyama. Carbazole-Based DABNA Analogues as Highly Efficient Thermally Activated Delayed Fluorescence Materials for Narrowband Organic Light-Emitting Diodes. Angew. Chem., Int. Ed., 2021. [DOI]
- Y. Wang, Y. Duan, R. Guo, S. Ye, K. Di, W. Zhang, S. Zhuang, L. Wang. A Periphery Cladding Strategy to Improve the Performance of Narrowband Emitters, Achieving Deep-Blue OLEDs with Ciey < 0.08 and External Quantum Efficiency Approaching 20%. Org. Electron., 2021. [DOI]
- J. Park, K. J. Kim, J. Lim, T. Kim, J. Y. Lee. High Efficiency of over 25% and Long Device Lifetime of over 500 H at 1000 Nit in Blue Fluorescent Organic Light-Emitting Diodes. Adv. Mater., 2022. [DOI]
- Y. Lee, J.-I. Hong. Multiple Resonance Thermally Activated Delayed Fluorescence Enhanced by Halogen Atoms. J. Mater. Chem. C, 2022. [DOI]
- Y. Wang, K. Di, Y. Duan, R. Guo, L. Lian, W. Zhang, L. Wang. The Selective Regulation of Borylation Site Based on One-Shot Electrophilic C-H Borylation Reaction, Achieving Highly Efficient Narrowband Organic Light-Emitting Diodes. Chem. Eng. J., 2022. [DOI]
- H. J. Cheon, S. J. Woo, S. H. Baek, J. H. Lee, Y. H. Kim. Dense Local Triplet States and Steric Shielding of a Multi-Resonance TADF Emitter Enable High-Performance Deep-Blue OLEDs. Adv. Mater., 2022. [DOI]
- H. J. Cheon, Y. S. Shin, N. H. Park, J. H. Lee, Y. H. Kim. Boron-Based Multi-Resonance TADF Emitter with Suppressed Intermolecular Interaction and Isomer Formation for Efficient Pure Blue OLEDs. Small, 2022. [DOI]
- E. Kim, J. Park, M. Jun, H. Shin, J. Baek, T. Kim, S. Kim, J. Lee, H. Ahn, J. Sun. Highly Efficient and Stable Deep-Blue Organic Light-Emitting Diode Using Phosphor-Sensitized Thermally Activated Delayed Fluorescence. Sci. Adv., 2022. [DOI | PubMed]
- F. Chen, L. Zhao, X. Wang, Q. Yang, W. Li, H. Tian, S. Shao, L. Wang, X. Jing, F. Wang. Novel Boron- and Sulfur-Doped Polycyclic Aromatic Hydrocarbon as Multiple Resonance Emitter for Ultrapure Blue Thermally Activated Delayed Fluorescence Polymers. Sci. China Chem., 2021. [DOI]
- K. Matsui, S. Oda, K. Yoshiura, K. Nakajima, N. Yasuda, T. Hatakeyama. One-Shot Multiple Borylation toward Bn-Doped Nanographenes. J. Am. Chem. Soc., 2018. [DOI | PubMed]
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai, T. Hatakeyama. Narrowband Deep-Blue Organic Light-Emitting Diode Featuring an Organoboron-Based Emitter. Nat. Photonics, 2019. [DOI]
- C.-Y. Chan, M. Tanaka, Y.-T. Lee, Y.-W. Wong, H. Nakanotani, T. Hatakeyama, C. Adachi. Stable Pure-Blue Hyperfluorescence Organic Light-Emitting Diodes with High-Efficiency and Narrow Emission. Nat. Photonics, 2021. [DOI]
- K. Stavrou, A. Danos, T. Hama, T. Hatakeyama, A. Monkman. Hot Vibrational States in a High-Performance Multiple Resonance Emitter and the Effect of Excimer Quenching on Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- K. Rayappa Naveen, H. Lee, R. Braveenth, K. Joon Yang, S. Jae Hwang, J. Hyuk Kwon. Deep Blue Diboron Embedded Multi-Resonance Thermally Activated Delayed Fluorescence Emitters for Narrowband Organic Light Emitting Diodes. Chem. Eng. J., 2022. [DOI]
- I. S. Park, M. Yang, H. Shibata, N. Amanokura, T. Yasuda. Achieving Ultimate Narrowband and Ultrapure Blue Organic Light-Emitting Diodes Based on Polycyclo-Heteraborin Multi-Resonance Delayed Fluorescence Emitters. Adv. Mater., 2022. [DOI]
- S. M. Suresh, E. Duda, F.-J. Kahle, D. Hall, S. Bagnich, H. Bässler, D. Beljonne, Y. Olivier, A. Köhler, E. Zysman-Colman. Design of Multi-Resonance Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes. SID Dig. Tech. Pap., 2021. [DOI]
- K. Stavrou, S. Madayanad Suresh, D. Hall, A. Danos, N. A. Kukhta, A. M. Z. Slawin, S. Warriner, D. Beljonne, Y. Olivier, A. Monkman, E. Zysman-Colman. Emission and Absorption Tuning in TADF B,N-Doped Heptacenes: Toward Ideal-Blue Hyperfluorescent OLEDs. Adv. Optical Mater., 2022. [DOI]
- Y. Xu, Z. Cheng, Z. Li, B. Liang, J. Wang, J. Wei, Z. Zhang, Y. Wang. Molecular-Structure and Device-Configuration Optimizations toward Highly Efficient Green Electroluminescence with Narrowband Emission and High Color Purity. Adv. Optical Mater., 2020. [DOI]
- S. Xu, Q. Yang, Y. Zhang, H. Li, Q. Xue, G. Xie, M. Gu, J. Jin, L. Huang, R. Chen. Solution-Processed Multi-Resonance Organic Light-Emitting Diodes with High Efficiency and Narrowband Emission. Chin. Chem. Lett., 2021. [DOI]
- M. Yang, I. S. Park, T. Yasuda. Full-Color, Narrowband, and High-Efficiency Electroluminescence from Boron and Carbazole Embedded Polycyclic Heteroaromatics. J. Am. Chem. Soc., 2020. [DOI | PubMed]
- Y. T. Lee, C. Y. Chan, M. Tanaka, M. Mamada, U. Balijapalli, Y. Tsuchiya, H. Nakanotani, T. Hatakeyama, C. Adachi. Investigating Homo Energy Levels of Terminal Emitters for Realizing High-Brightness and Stable TADF-Assisted Fluorescence Organic Light-Emitting Diodes. Adv. Electron. Mater., 2021. [DOI]
- M. Yang, S. Shikita, H. Min, I. S. Park, H. Shibata, N. Amanokura, T. Yasuda. Wide-Range Color Tuning of Narrowband Emission in Multi-Resonance Organoboron Delayed Fluorescence Materials through Rational Imine/Amine Functionalization. Angew. Chem., Int. Ed., 2021. [DOI]
- Y. Zhang, D. Zhang, J. Wei, X. Hong, Y. Lu, D. Hu, G. Li, Z. Liu, Y. Chen, L. Duan. Achieving Pure Green Electroluminescence with Ciey of 0.69 and EQE of 28.2% from an Aza-Fused Multi-Resonance Emitter. Angew. Chem., Int. Ed., 2020. [DOI]
- Y. Xu, Q. Wang, J. Wei, X. Peng, J. Xue, Z. Wang, S. J. Su, Y. Wang. Constructing Organic Electroluminescent Material with Very High Color Purity and Efficiency Based on Polycyclization of the Multiple Resonance Parent Core. Angew. Chem., Int. Ed., 2022. [DOI]
- Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu, L. Duan. Multi-Resonance Induced Thermally Activated Delayed Fluorophores for Narrowband Green OLEDs. Angew. Chem., Int. Ed., 2019. [DOI]
- Y. Xu, C. Li, Z. Li, J. Wang, J. Xue, Q. Wang, X. Cai, Y. Wang. Highly Efficient Electroluminescent Materials with High Color Purity Based on Strong Acceptor Attachment onto B-N-Containing Multiple Resonance Frameworks. CCS Chem., 2022. [DOI]
- X. Yan, Z. Li, Q. Wang, Y. Qu, Y. Xu, Y. Wang. Achieving Highly Efficient Narrowband Sky-Blue Electroluminescence with Alleviated Efficiency Roll-Off by Molecular-Structure Regulation and Device-Configuration Optimization. J. Mater. Chem. C, 2022. [DOI]
- Y. Qi, W. Ning, Y. Zou, X. Cao, S. Gong, C. Yang. Peripheral Decoration of Multi-Resonance Molecules as a Versatile Approach for Simultaneous Long-Wavelength and Narrowband Emission. Adv. Funct. Mater., 2021. [DOI]
- X. Cai, Y. Xu, Q. Wang, C. Li, Y. Wang. Constructing Narrowband Thermally Activated Delayed Fluorescence Materials with Emission Maxima Beyond 560 Nm Based on Frontier Molecular Orbital Engineering. Angew. Chem., Int. Ed., 2023
- Y. Liu, X. Xiao, Z. Huang, D. Yang, D. Ma, J. Liu, B. Lei, Z. Bin, J. You. Space-Confined Donor-Acceptor Strategy Enables Fast Spin-Flip of Multiple Resonance Emitters for Suppressing Efficiency Roll-Off. Angew. Chem., Int. Ed., 2022. [DOI]
- P. Jiang, J. Miao, X. Cao, H. Xia, K. Pan, T. Hua, X. Lv, Z. Huang, Y. Zou, C. Yang. Quenching-Resistant Multiresonance TADF Emitter Realizes 40% External Quantum Efficiency in Narrowband Electroluminescence at High Doping Level. Adv. Mater., 2022. [DOI]
- Y. Zhang, J. Wei, D. Zhang, C. Yin, G. Li, Z. Liu, X. Jia, J. Qiao, L. Duan. Sterically Wrapped Multiple Resonance Fluorophors for Suppression of Concentration Quenching and Spectrum Broadening. Angew. Chem., Int. Ed., 2022. [DOI]
- F. Liu, Z. Cheng, L. Wan, Z. Feng, H. Liu, H. Jin, L. Gao, P. Lu, W. Yang. Highly Efficient Multi-Resonance Thermally Activated Delayed Fluorescence Material with a Narrow Full Width at Half-Maximum of 0.14 Ev. Small, 2022. [DOI]
- Q. Wang, Y. Xu, T. Yang, J. Xue, Y. Wang. Precise Functionalization of a Multiple-Resonance Framework: Constructing Narrowband Organic Electroluminescent Materials with External Quantum Efficiency over 40. Adv. Mater., 2023. [DOI]
- T. Wang, Y. Zou, Z. Huang, N. Li, J. Miao, C. Yang. Narrowband Emissive TADF Conjugated Polymers Towards Highly Efficient Solution-Processible OLEDs. Angew. Chem., Int. Ed., 2022. [DOI]
- F. Huang, X. C. Fan, Y. C. Cheng, H. Wu, Y. Z. Shi, J. Yu, K. Wang, C. S. Lee, X. H. Zhang. Distinguishing the Respective Determining Factors for Spectral Broadening and Concentration Quenching in Multiple Resonance Type TADF Emitter Systems. Mater. Horiz., 2022. [DOI | PubMed]
- Y. T. Lee, C. Y. Chan, M. Tanaka, M. Mamada, K. Goushi, X. Tang, Y. Tsuchiya, H. Nakanotani, C. Adachi. Tailor-Made Multi-Resonance Terminal Emitters toward Narrowband, High-Efficiency, and Stable Hyperfluorescence Organic Light-Emitting Diodes. Adv. Optical Mater., 2022. [DOI]
- W. Xue, H. Yan, Y. He, L. Wu, X. Zhang, Y. Wu, J. Xu, J. He, C. Yan, H. Meng. Identifying the Molecular Origins of Green Bn-TADF Material Degradation and Device Stability Via in Situ Raman Spectroscopy. Chem. Eur. J., 2022. [DOI | PubMed]
- Y. Zhang, G. Li, L. Wang, T. Huang, J. Wei, G. Meng, X. Wang, X. Zeng, D. Zhang, L. Duan. Fusion of Multi-Resonance Fragment with Conventional Polycyclic Aromatic Hydrocarbon for Nearly Bt.2020 Green Emission. Angew. Chem., Int. Ed., 2022. [DOI]
- Y. Zhang, J. Wei, L. Wang, T. Huang, G. Meng, X. Wang, X. Zeng, M. Du, T. Fan, C. Yin. Multiple Fusion Strategy for High-Performance Yellow OLEDs with Full Width at Half Maximums Down to 23 Nm and External Quantum Efficiencies up to 37.4. Adv. Mater., 2023. [DOI | PubMed]
- Y. C. Cheng, X. C. Fan, F. Huang, X. Xiong, J. Yu, K. Wang, C. S. Lee, X. H. Zhang. A Highly Twisted Carbazole-Fused DABNA Derivative as an Orange-Red TADF Emitter for OLEDs with Nearly 40% EQE. Angew. Chem., Int. Ed., 2022. [DOI]
- X. Lv, J. Miao, M. Liu, Q. Peng, C. Zhong, Y. Hu, X. Cao, H. Wu, Y. Yang, C. Zhou. Extending the Pi-Skeleton of Multi-Resonance TADF Materials Towards High-Efficiency Narrowband Deep-Blue Emission. Angew. Chem., Int. Ed., 2022. [DOI]
- Y. Zou, J. Hu, M. Yu, J. Miao, Z. Xie, Y. Qiu, X. Cao, C. Yang. High-Performance Narrowband Pure-Red OLEDs with External Quantum Efficiencies up to 36.1% and Ultralow Efficiency Roll-Off. Adv. Mater., 2022. [DOI]
- Y. Wang, K. Zhang, F. Chen, X. Wang, Q. Yang, S. Wang, S. Shao, L. Wang. Boron-, Sulfur- and Nitrogen-Doped Tridecacyclic Aromatic Emitters with Multiple Resonance Effect for Narrowband Red Emission. Chin. J. Chem., 2022. [DOI]
- G. Liu, H. Sasabe, K. Kumada, H. Arai, J. Kido. Nonbonding/Bonding Molecular Orbital Regulation of Nitrogen-Boron-Oxygen-Embedded Blue/Green Multiresonant TADF Emitters with High Efficiency and Color Purity. Chem. Eur. J., 2022. [DOI | PubMed]
- X.-F. Luo, H.-X. Ni, A.-Q. Lv, X.-K. Yao, H.-L. Ma, Y.-X. Zheng. High-Efficiency and Narrowband OLEDs from Blue to Yellow with Ternary Boron/Nitrogen-Based Polycyclic Heteroaromatic Emitters. Adv. Optical Mater., 2022. [DOI]
- P. Jiang, L. Zhan, X. Cao, X. Lv, S. Gong, Z. Chen, C. Zhou, Z. Huang, F. Ni, Y. Zou, C. Yang. Simple Acridan-Based Multi-Resonance Structures Enable Highly Efficient Narrowband Green TADF Electroluminescence. Adv. Optical Mater., 2021. [DOI]
- T. Hua, L. Zhan, N. Li, Z. Huang, X. Cao, Z. Xiao, S. Gong, C. Zhou, C. Zhong, C. Yang. Heavy-Atom Effect Promotes Multi-Resonance Thermally Activated Delayed Fluorescence. Chem. Eng. J., 2021. [DOI]
- G. Liu, H. Sasabe, K. Kumada, A. Matsunaga, H. Katagiri, J. Kido. Facile Synthesis of Multi-Resonance Ultra-Pure-Green TADF Emitters Based on Bridged Diarylamine Derivatives for Efficient OLEDs with Narrow Emission. J. Mater. Chem. C, 2021. [DOI]
- J.-J. Hu, X.-F. Luo, M.-X. Mao, H.-X. Ni, X. Liang, Y.-P. Zhang, Y.-X. Zheng. Green Multi-Resonance Induced Thermally Activated Delayed Fluorescence Emitters Containing Phenoxazine Units with Highly Efficient Electroluminescence. J. Mater. Chem. C, 2022. [DOI]
- J. Liu, Y. Zhu, T. Tsuboi, C. Deng, W. Lou, D. Wang, T. Liu, Q. Zhang. Toward a Bt.2020 Green Emitter through a Combined Multiple Resonance Effect and Multi-Lock Strategy. Nat Commun, 2022. [DOI | PubMed]
- Y. Qiu, H. Xia, J. Miao, Z. Huang, N. Li, X. Cao, J. Han, C. Zhou, C. Zhong, C. Yang. Narrowing the Electroluminescence Spectra of Multiresonance Emitters for High-Performance Blue OLEDs by a Peripheral Decoration Strategy. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- J. Bian, S. Chen, L. Qiu, N. Zhang, J. Zhang, C. Duan, C. Han, H. Xu. Synergetic Insulation and Induction Effects Selectively Optimize Multiresonance Thermally Activated Delayed Fluorescence. Research, 2022. [DOI | PubMed]
- J. Bian, S. Chen, L. Qiu, R. Tian, Y. Man, Y. Wang, S. Chen, J. Zhang, C. Duan, C. Han, H. Xu. Ambipolar Self-Host Functionalization Accelerates Blue Multi-Resonance Thermally Activated Delayed Fluorescence with Internal Quantum Efficiency of 100. Adv. Mater., 2022. [DOI]
- T. Hua, J. Miao, H. Xia, Z. Huang, X. Cao, N. Li, C. Yang. Sulfone-Incorporated Multi-Resonance TADF Emitter for High-Performance Narrowband Blue OLEDs with EQE of 32%. Adv. Funct. Mater., 2022. [DOI]
- J. Park, J. Moon, J. Lim, J. Woo, S. S. Yoon, J. Y. Lee. Fine-Tuned Asymmetric Blue Multiple Resonance Thermally Activated Delayed Fluorescence Emitters with High Efficiency and Narrow Emission Band. J. Mater. Chem. C, 2022. [DOI]
- J. Park, J. Lim, J. H. Lee, B. Jang, J. H. Han, S. S. Yoon, J. Y. Lee. Asymmetric Blue Multiresonance TADF Emitters with a Narrow Emission Band. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- J. Han, Z. Huang, X. Lv, J. Miao, Y. Qiu, X. Cao, C. Yang. Simple Molecular Design Strategy for Multiresonance Induced TADF Emitter: Highly Efficient Deep Blue to Blue Electroluminescence with High Color Purity. Adv. Optical Mater., 2022. [DOI]
- J. Liu, L. Chen, X. Wang, Q. Yang, L. Zhao, C. Tong, S. Wang, S. Shao, L. Wang. Multiple Resonance Dendrimers Containing Boron, Oxygen, Nitrogen-Doped Polycyclic Aromatic Emitters for Narrowband Blue-Emitting Solution-Processed OLEDs. Macromol. Rapid Commun., 2022. [DOI]
- Q. Li, Y. Wu, Q. Yang, S. Wang, S. Shao, L. Wang. Selenium-Doped Polycyclic Aromatic Hydrocarbon Multiresonance Emitters with Fast Reverse Intersystem Crossing for Narrowband Blue Emission. ACS Appl. Mater. Interfaces, 2022. [DOI]
- S. Oda, B. Kawakami, R. Kawasumi, R. Okita, T. Hatakeyama. Multiple Resonance Effect-Induced Sky-Blue Thermally Activated Delayed Fluorescence with a Narrow Emission Band. Org. Lett., 2019. [DOI | PubMed]
- D. Song, Y. Yu, L. Yue, D. Zhong, Y. Zhang, X. Yang, Y. Sun, G. Zhou, Z. Wu. Asymmetric Thermally Activated Delayed Fluorescence (TADF) Emitters with 5,9-Dioxa-13b-Boranaphtho[3,2,1-De]Anthracene (Oba) as the Acceptor and Highly Efficient Blue-Emitting OLEDs. J. Mater. Chem. C, 2019. [DOI]
- H. Lim, H. J. Cheon, S.-J. Woo, S.-K. Kwon, Y.-H. Kim, J.-J. Kim. Highly Efficient Deep-Blue OLEDs Using a TADF Emitter with a Narrow Emission Spectrum and High Horizontal Emitting Dipole Ratio. Adv. Mater., 2020. [DOI]
- M. Nagata, H. Min, E. Watanabe, H. Fukumoto, Y. Mizuhata, N. Tokitoh, T. Agou, T. Yasuda. Fused-Nonacyclic Multi-Resonance Delayed Fluorescence Emitter Based on Ladder-Thiaborin Exhibiting Narrowband Sky-Blue Emission with Accelerated Reverse Intersystem Crossing. Angew. Chem., Int. Ed., 2021. [DOI]
- G. Meng, H. Dai, T. Huang, J. Wei, J. Zhou, X. Li, X. Wang, X. Hong, C. Yin, X. Zeng. Amine-Directed Formation of B-N Bonds for Bn-Fused Polycyclic Aromatic Multiple Resonance Emitters with Narrowband Emission. Angew. Chem., Int. Ed., 2022. [DOI]
- J. Bae, M. Sakai, Y. Tsuchiya, N. Ando, X. K. Chen, T. B. Nguyen, C. Y. Chan, Y. T. Lee, M. Auffray, H. Nakanotani. Multiple Resonance Type Thermally Activated Delayed Fluorescence by Dibenzo [1,4] Azaborine Derivatives. Front. Chem., 2022. [DOI | PubMed]
- Y. Yuan, X. Tang, X. Y. Du, Y. Hu, Y. J. Yu, Z. Q. Jiang, L. S. Liao, S. T. Lee. The Design of Fused Amine/Carbonyl System for Efficient Thermally Activated Delayed Fluorescence: Novel Multiple Resonance Core and Electron Acceptor. Adv. Optical Mater., 2019. [DOI]
- X. Li, Y. Z. Shi, K. Wang, M. Zhang, C. J. Zheng, D. M. Sun, G. L. Dai, X. C. Fan, D. Q. Wang, W. Liu. Thermally Activated Delayed Fluorescence Carbonyl Derivatives for Organic Light-Emitting Diodes with Extremely Narrow Full Width at Half-Maximum. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- F. Huang, K. Wang, Y. Z. Shi, X. C. Fan, X. Zhang, J. Yu, C. S. Lee, X. H. Zhang. Approaching Efficient and Narrow RGB Electroluminescence from D-a-Type TADF Emitters Containing an Identical Multiple Resonance Backbone as the Acceptor. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- S. Wu, W. Li, K. Yoshida, D. Hall, S. Madayanad Suresh, T. Sayner, J. Gong, D. Beljonne, Y. Olivier, I. D. W. Samuel, E. Zysman-Colman. Excited-State Modulation in Donor-Substituted Multiresonant Thermally Activated Delayed Fluorescence Emitters. ACS Appl. Mater. Interfaces, 2022. [DOI | PubMed]
- J.-F. Liu, S.-N. Zou, X. Chen, S.-Y. Yang, Y.-J. Yu, M.-K. Fung, Z.-Q. Jiang, L.-S. Liao. Isomeric Thermally Activated Delayed Fluorescence Emitters Based on a Quinolino[3,2,1-De]Acridine-5,9-Dione Multiple Resonance Core and Carbazole Substituent. Mater. Chem. Front., 2022. [DOI]
- S. N. Zou, C. C. Peng, S. Y. Yang, Y. K. Qu, Y. J. Yu, X. Chen, Z. Q. Jiang, L. S. Liao. Fully Bridged Triphenylamine Derivatives as Color-Tunable Thermally Activated Delayed Fluorescence Emitters. Org. Lett., 2021. [DOI | PubMed]
- H. Min, I. S. Park, T. Yasuda. Cis-Quinacridone-Based Delayed Fluorescence Emitters: Seemingly Old but Renewed Functional Luminogens. Angew. Chem., Int. Ed., 2021. [DOI]
- J.-W. Huang, Y.-C. Hsu, X. Wu, S. Wang, X.-Q. Gan, W.-Q. Zheng, H. Zhang, Y.-Z. Gong, W.-Y. Hung, P.-T. Chou, W. Zhu. Influence of Charge Transfer Strength on Emission Bandwidth for Multiple-Resonance Emitters Via Systematically Tuning the Acceptor-Donor Assembly. J. Mater. Chem. C, 2022. [DOI]
- Y. Tsuchiya, Y. Ishikawa, S. H. Lee, X. K. Chen, J. L. Brédas, H. Nakanotani, C. Adachi. Thermally Activated Delayed Fluorescence Properties of Trioxoazatriangulene Derivatives Modified with Electron Donating Groups. Adv. Optical Mater., 2021. [DOI]
- E. Hamzehpoor, D. F. Perepichka. Crystal Engineering of Room Temperature Phosphorescence in Organic Solids. Angew. Chem., Int. Ed., 2020. [DOI]
- K. Wang, X.-C. Fan, Y. Tsuchiya, Y.-Z. Shi, M. Tanaka, Z. Lin, Y.-T. Lee, X. Zhang, W. Liu, G.-L. Dai. Efficient and High Colour Purity RGB OLEDs Employing Densely Packed Dimers. ChemRxiv, 2023. [DOI]
- H. Tsujimoto, D.-G. Ha, G. Markopoulos, H. S. Chae, M. A. Baldo, T. M. Swager. Thermally Activated Delayed Fluorescence and Aggregation Induced Emission with through-Space Charge Transfer. J. Am. Chem. Soc., 2017. [DOI | PubMed]
- R. Furue, T. Nishimoto, I. S. Park, J. Lee, T. Yasuda. Aggregation-Induced Delayed Fluorescence Based on Donor/Acceptor-Tethered Janus Carborane Triads: Unique Photophysical Properties of Nondoped OLEDs. Angew. Chem., Int. Ed., 2016. [DOI]
- Y. Kusakabe, Y. Wada, H. Nakagawa, K. Shizu, H. Kaji. Conformation Control of Iminodibenzyl-Based Thermally Activated Delayed Fluorescence Material by Tilted Face-to-Face Alignment with Optimal Distance (TFFO) Design. Front. Chem., 2020. [DOI | PubMed]
- X. Tang, L.-S. Cui, H.-C. Li, A. J. Gillett, F. Auras, Y.-K. Qu, C. Zhong, S. T. E. Jones, Z.-Q. Jiang, R. H. Friend, L.-S. Liao. Highly Efficient Luminescence from Space-Confined Charge-Transfer Emitters. Nat. Mater., 2020. [DOI | PubMed]
- Z. Q. Feng, S. Y. Yang, F. C. Kong, Y. K. Qu, X. Y. Meng, Y. J. Yu, D. Y. Zhou, Z. Q. Jiang, L. S. Liao. Indirect Control of Donor/Acceptor Interactions for Highly Efficient Space-Confined Thermally Activated Delayed Fluorescence Emitters. Adv. Funct. Mater., 2023. [DOI]
- Q. Zheng, X.-Q. Wang, Y.-K. Qu, G. Xie, L.-S. Liao, Z.-Q. Jiang. Solution-Processable through-Space Charge-Transfer Emitters Via Solubilizing Groups Modification. Npj Flex. Electron., 2022. [DOI]
- S. Y. Yang, Q. S. Tian, Y. J. Yu, S. N. Zou, H. C. Li, A. Khan, Q. H. Wu, Z. Q. Jiang, L. S. Liao. Sky-Blue Thermally Activated Delayed Fluorescence with Intramolecular Spatial Charge Transfer Based on a Dibenzothiophene Sulfone Emitter. J. Org. Chem., 2020. [DOI | PubMed]
- T.-T. Wang, G. Xie, H.-C. Li, S.-Y. Yang, H. Li, Y.-L. Xiao, C. Zhong, K. Sarvendra, A. Khan, Z.-Q. Jiang, L.-S. Liao. Π-Stacked Thermally Activated Delayed Fluorescence Emitters with Alkyl Chain Modulation. CCS Chem., 2021. [DOI]
- X.-Q. Wang, S.-Y. Yang, Q.-S. Tian, C. Zhong, Y.-K. Qu, Y.-J. Yu, Z. Jiang, L.-S. Liao. Multi-Layer Π-Stacked Molecules as Efficient Thermally Activated Delayed Fluorescence Emitters. Angew. Chem., Int. Ed., 2021. [DOI]
- S. Y. Yang, Y. K. Wang, C. C. Peng, Z. G. Wu, S. Yuan, Y. J. Yu, H. Li, T. T. Wang, H. C. Li, Y. X. Zheng. Circularly Polarized Thermally Activated Delayed Fluorescence Emitters in through-Space Charge Transfer on Asymmetric Spiro Skeletons. J. Am. Chem. Soc., 2020. [DOI | PubMed]
- S. Y. Yang, Z. Q. Feng, Z. Fu, K. Zhang, S. Chen, Y. J. Yu, B. Zou, K. Wang, L. S. Liao, Z. Q. Jiang. Highly Efficient Sky-Blue Pi-Stacked Thermally Activated Delayed Fluorescence Emitter with Multi-Stimulus Response Properties. Angew. Chem., Int. Ed., 2022. [DOI]
- C.-C. Peng, S.-Y. Yang, H.-C. Li, G.-H. Xie, L.-S. Cui, S.-N. Zou, C. Poriel, Z.-Q. Jiang, L.-S. Liao. Highly Efficient Thermally Activated Delayed Fluorescence Via an Unconjugated Donor-Acceptor System Realizing EQE of over 30%. Adv. Mater., 2020. [DOI]
- Z. Zhao, C. Zeng, X. Peng, Y. Liu, H. Zhao, L. Hua, S. J. Su, S. Yan, Z. Ren. Tuning Intramolecular Stacking of Rigid Heteroaromatic Compounds for High-Efficiency Deep-Blue through-Space Charge-Transfer Emission. Angew. Chem., Int. Ed., 2022. [DOI]
- T. Huang, Q. Wang, G. Meng, L. Duan, D. Zhang. Accelerating Radiative Decay in Blue through-Space Charge Transfer Emitters by Minimizing the Face-to-Face Donor-Acceptor Distances. Angew. Chem., Int. Ed., 2022. [DOI]
- F. M. Xie, H. Z. Li, K. Zhang, Y. Shen, X. Zhao, Y. Q. Li, J. X. Tang. A Dislocated Twin-Locking Acceptor-Donor-Acceptor Configuration for Efficient Delayed Fluorescence with Multiple through-Space Charge Transfer. Angew. Chem., Int. Ed., 2022. [DOI]
- Y. Song, M. Tian, R. Yu, L. He. Through-Space Charge-Transfer Emitters Developed by Fixing the Acceptor for High-Efficiency Thermally Activated Delayed Fluorescence. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- C. Wu, W. Liu, K. Li, G. Cheng, J. Xiong, T. Teng, C. M. Che, C. Yang. Face-to-Face Orientation of Quasiplanar Donor and Acceptor Enables Highly Efficient Intramolecular Exciplex Fluorescence. Angew. Chem., Int. Ed., 2021. [DOI]
- J. Wang, J. Miao, C. Jiang, S. Luo, C. Yang, K. Li. Engineering Intramolecular Π-Stacking Interactions of through-Space Charge-Transfer TADF Emitters for Highly Efficient OLEDs with Improved Color Purity. Adv. Optical Mater., 2022. [DOI]
- C. Jiang, J. Miao, D. Zhang, Z. Wen, C. Yang, K. Li. Acceptor-Donor-Acceptor Pi-Stacking Boosts Intramolecular through-Space Charge Transfer Towards Efficient Red TADF and High-Performance OLEDs. Research, 2022. [DOI | PubMed]
- D. Zhang, C. Jiang, Z. Wen, X. Feng, K. Li. Influence of Sulfur Atoms on TADF Properties from through-Space Charge Transfer Excited States. Chem. Eur. J., 2022. [DOI | PubMed]
- H. Miranda-Salinas, Y.-T. Hung, Y.-S. Chen, D. Luo, H.-C. Kao, C.-H. Chang, K.-T. Wong, A. Monkman. Controlling through-Space and through-Bond Intramolecular Charge Transfer in Bridged D-D′-a TADF Emitters. J. Mater. Chem. C, 2021. [DOI]
- F. Ma, H. Ji, D. Zhang, K. Xue, P. Zhang, Z. Qi, H. Zhu. Adjusting the Photophysical Properties of AIE-Active TADF Emitters from through-Bond to through-Space Charge Transfer for High-Performance Solution-Processed OLEDs. Dyes Pigm., 2021. [DOI]
- K. Li, T. Wang, B. Yao, Y. Chen, H. Deng, H. Zhan, Z. Xie, Y. Cheng. Carbazole Ring: A Delicate Rack for Constructing Thermally Activated Delayed Fluorescent Compounds with through-Space Charge Transfer. Chin. Chem. Lett., 2021. [DOI]
- K. Li, Y. Chen, B. Yao, K. Dou, T. Wang, H. Deng, H. Zhan, Z. Xie, Y. Cheng. Insight into through-Space Conjugation in Rotation-Restricted Thermally Activated Delayed Fluorescence Compounds. J. Mater. Chem. C, 2022. [DOI]
- X. L. Chen, J. H. Jia, R. Yu, J. Z. Liao, M. X. Yang, C. Z. Lu. Combining Charge-Transfer Pathways to Achieve Unique Thermally Activated Delayed Fluorescence Emitters for High-Performance Solution-Processed, Non-Doped Blue OLEDs. Angew. Chem., Int. Ed., 2017. [DOI]
- J. Kim, T. Lee, J. Y. Ryu, Y. H. Lee, J. Lee, J. Jung, M. H. Lee. Highly Emissive Ortho-Donor-Acceptor Triarylboranes: Impact of Boryl Acceptors on Luminescence Properties. Organometallics, 2020. [DOI]
- Z. Yang, Z. Mao, C. Xu, X. Chen, J. Zhao, Z. Yang, Y. Zhang, W. Wu, S. Jiao, Y. Liu. A Sterically Hindered Asymmetric D-a-D′ Thermally Activated Delayed Fluorescence Emitter for Highly Efficient Non-Doped Organic Light-Emitting Diodes. Chem. Sci., 2019. [DOI | PubMed]
- C. Yin, D. Zhang, Y. Zhang, Y. Lu, R. Wang, G. Li, L. Duan. High-Efficiency Narrow-Band Electro-Fluorescent Devices with Thermally Activated Delayed Fluorescence Sensitizers Combined through-Bond and through-Space Charge Transfers. CCS Chem., 2020. [DOI]
- Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu, L. Duan. Multi-Resonance Induced Thermally Activated Delayed Fluorophores for Narrowband Green OLEDs. Angew. Chem., Int. Ed., 2019. [DOI]
- T. Huang, Q. Wang, S. Xiao, D. Zhang, Y. Zhang, C. Yin, D. Yang, D. Ma, Z. Wang, L. Duan. Simultaneously Enhanced Reverse Intersystem Crossing and Radiative Decay in Thermally Activated Delayed Fluorophors with Multiple through-Space Charge Transfers. Angew. Chem., Int. Ed., 2021. [DOI]
- X. Lv, Y. Wang, N. Li, X. Cao, G. Xie, H. Huang, C. Zhong, L. Wang, C. Yang. Regulating the Photophysical Properties of Highly Twisted TADF Emitters by Concurrent through-Space/-Bond Charge Transfer. Chem. Eng. J., 2020. [DOI]
- Y. Huang, D.-H. Zhang, X.-D. Tao, Z. Wei, S. Jiang, L. Meng, M.-X. Yang, X.-L. Chen, C.-Z. Lu. Triptycene-Derived Thermally Activated Delayed Fluorescence Emitters with Combined through-Bond and through-Space Charge Transfers. Dyes Pigm., 2022. [DOI]
- X. Wang, S. Wang, J. Lv, S. Shao, L. Wang, X. Jing, F. Wang. Through-Space Charge Transfer Hexaarylbenzene Dendrimers with Thermally Activated Delayed Fluorescence and Aggregation-Induced Emission for Efficient Solution-Processed OLEDs. Chem. Sci., 2019. [DOI | PubMed]
- P. Zhang, J. Zeng, J. Guo, S. Zhen, B. Xiao, Z. Wang, Z. Zhao, B. Z. Tang. New Aggregation-Induced Delayed Fluorescence Luminogens with through-Space Charge Transfer for Efficient Non-Doped OLEDs. Front. Chem., 2019. [DOI | PubMed]
- J. Li, L. Zhou, J. He, Q. Xue, L. Xu, G. Xie. Propeller-Shape Isomers with Turn-on through-Space Charge Transfer for Solution-Processed Non-Doped Organic Light-Emitting Diodes. Chem. Eng. J., 2023. [DOI]
- S. Kumar, L. G. Franca, K. Stavrou, E. Crovini, D. B. Cordes, A. M. Z. Slawin, A. P. Monkman, E. Zysman-Colman. Investigation of Intramolecular through-Space Charge-Transfer States in Donor-Acceptor Charge-Transfer Systems. J. Phys. Chem. Lett, 2021. [DOI | PubMed]
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. Van Voorhis, M. A. Baldo, T. M. Swager. Thermally Activated Delayed Fluorescence Materials Based on Homoconjugation Effect of Donor-Acceptor Triptycenes. J. Am. Chem. Soc., 2015. [DOI | PubMed]
- G. Dai, M. Zhang, K. Wang, X. Fan, Y. Shi, D. Sun, W. Liu, J. Chen, J. Yu, X. Ou. Nonconjugated Triptycene-Spaced Donor-Acceptor-Type Emitters Showing Thermally Activated Delayed Fluorescence Via Both Intra- and Intermolecular Charge-Transfer Transitions. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- H. Yersin, L. Mataranga-Popa, R. Czerwieniec, Y. Dovbii. Design of a New Mechanism Beyond Thermally Activated Delayed Fluorescence toward Fourth Generation Organic Light Emitting Diodes. Chem. Mater., 2019. [DOI]
- E. Spuling, N. Sharma, I. Samuel, E. Zysman-Colman, S. Braese. (Deep) Blue through-Space Conjugated TADF Emitters Based on [2.2]Paracyclophanes. Chem. Commun., 2018. [DOI]
- M. Auffray, D. H. Kim, J. U. Kim, F. Bencheikh, D. Kreher, Q. Zhang, A. D’Aleo, J. C. Ribierre, F. Mathevet, C. Adachi. Dithia[3.3]Paracyclophane Core: A Versatile Platform for Triplet State Fine-Tuning and through-Space TADF Emission. Chem. Asian J., 2019. [DOI | PubMed]
- M. Y. Zhang, X. Liang, D. N. Ni, D. H. Liu, Q. Peng, C. H. Zhao. 2-(Dimesitylboryl)Phenyl-Substituted [2.2]Paracyclophanes Featuring Intense and Sign-Invertible Circularly Polarized Luminescence. Org. Lett., 2021. [DOI | PubMed]
- H. Liu, H. Liu, J. Fan, J. Guo, J. Zeng, F. Qiu, Z. Zhao, B. Z. Tang. An Effective Design Strategy for Robust Aggregation-Induced Delayed Fluorescence Luminogens to Improve Efficiency Stability of Nondoped and Doped OLEDs. Adv. Optical Mater., 2020. [DOI]
- H. Wang, L. Xie, Q. Peng, L. Meng, Y. Wang, Y. Yi, P. Wang. Novel Thermally Activated Delayed Fluorescence Materials-Thioxanthone Derivatives and Their Applications for Highly Efficient OLEDs. Adv. Mater., 2014. [DOI | PubMed]
- K. Wu, Z. Wang, L. Zhan, C. Zhong, S. Gong, G. Xie, C. Yang. Realizing Highly Efficient Solution-Processed Homojunction-Like Sky-Blue OLEDs by Using Thermally Activated Delayed Fluorescent Emitters Featuring an Aggregation-Induced Emission Property. J. Phys. Chem. Lett, 2018. [DOI | PubMed]
- J. Zhao, X. Chen, Z. Yang, T. Liu, Z. Yang, Y. Zhang, J. Xu, Z. Chi. Highly-Efficient Doped and Nondoped Organic Light-Emitting Diodes with External Quantum Efficiencies over 20% from a Multifunctional Green Thermally Activated Delayed Fluorescence Emitter. J. Phys. Chem. C., 2019. [DOI]
- R. Guo, P. Leng, Q. Zhang, Y. Wang, X. Lv, S. Sun, S. Ye, Y. Duan, L. Wang. Donor Engineering for Diphenylsulfone Derivatives with Both Thermally Activated Delayed Fluorescence and Aggregation-Induced Emission Properties. Dyes Pigm., 2021. [DOI]
- P. Leng, S. Sun, R. Guo, Q. Zhang, W. Liu, X. Lv, S. Ye, L. Wang. Modifying the AIE-TADF Chromophore with Host-Substituents to Achieve High Efficiency and Low Roll-Off Non-Doped OLEDs. Org. Electron., 2020. [DOI]
- S. Xiang, Z. Huang, S. Sun, X. Lv, L. Fan, S. Ye, H. Chen, R. Guo, L. Wang. Highly Efficient Non-Doped OLEDs Using Aggregation-Induced Delayed Fluorescence Materials Based on 10-Phenyl-10h-Phenothiazine 5,5-Dioxide Derivatives. J. Mater. Chem. C, 2018. [DOI]
- J. Guo, X.-L. Li, H. Nie, W. Luo, S. Gan, S. Hu, R. Hu, A. Qin, Z. Zhao, S.-J. Su, B. Z. Tang. Achieving High-Performance Nondoped OLEDs with Extremely Small Efficiency Roll-Off by Combining Aggregation-Induced Emission and Thermally Activated Delayed Fluorescence. Adv. Funct. Mater., 2017. [DOI]
- H. Chen, H. Liu, Y. Xiong, J. He, Z. Zhao, B. Z. Tang. New Aggregation-Induced Delayed Fluorescent Materials for Efficient OLEDs with High Stabilities of Emission Color and Efficiency. Mater. Chem. Front., 2022. [DOI]
- J. Huang, H. Nie, J. Zeng, Z. Zhuang, S. Gan, Y. Cai, J. Guo, S.-J. Su, Z. Zhao, B. Z. Tang. Highly Efficient Nondoped OLEDs with Negligible Efficiency Roll-Off Fabricated from Aggregation-Induced Delayed Fluorescence Luminogens. Angew. Chem., Int. Ed., 2017. [DOI]
- J. Guo, J. Fan, L. Lin, J. Zeng, H. Liu, C.-K. Wang, Z. Zhao, B. Z. Tang. Mechanical Insights into Aggregation-Induced Delayed Fluorescence Materials with Anti-Kasha Behavior. Adv. Sci., 2019. [DOI]
- Y. Fu, H. Liu, X. Zhu, J. Zeng, Z. Zhao, B. Z. Tang. Efficient Aggregation-Induced Delayed Fluorescent Materials Based on Bipolar Carrier Transport Materials for the Fabrication of High-Performance Nondoped OLEDs with Very Small Efficiency Roll-Off. J. Mater. Chem. C, 2020. [DOI]
- J. Xu, X. Zhu, J. Guo, J. Fan, J. Zeng, S. Chen, Z. Zhao, B. Z. Tang. Aggregation-Induced Delayed Fluorescence Luminogens with Accelerated Reverse Intersystem Crossing for High-Performance OLEDs. ACS Mater. Lett., 2019. [DOI]
- J. Xu, X. Wu, J. Li, Z. Zhao, B. Z. Tang. Regulating Photophysical Property of Aggregation-Induced Delayed Fluorescence Luminogens Via Heavy Atom Effect to Achieve Efficient Organic Light-Emitting Diodes. Adv. Optical Mater., 2022. [DOI]
- H. Liu, J. Zeng, J. Guo, H. Nie, Z. Zhao, B. Z. Tang. High-Performance Non-Doped OLEDs with Nearly 100% Exciton Use and Negligible Efficiency Roll-Off. Angew. Chem., Int. Ed., 2018. [DOI]
- R. Huang, H. Chen, H. Liu, Z. Zhuang, J. Wang, M. Yu, D. Yang, D. Ma, Z. Zhao, B. Zhong Tang. Creating Efficient Delayed Fluorescence Luminogens with Acridine-Based Spiro Donors to Improve Horizontal Dipole Orientation for High-Performance OLEDs. Chem. Eng. J., 2022. [DOI]
- Y. Chen, S. Wang, X. Wu, Y. Xu, H. Li, Y. Liu, H. Tong, L. Wang. Triazatruxene-Based Small Molecules with Thermally Activated Delayed Fluorescence, Aggregation-Induced Emission and Mechanochromic Luminescence Properties for Solution-Processable Nondoped OLEDs. J. Mater. Chem. C, 2018. [DOI]
- J. Chen, J. Zeng, X. Zhu, J. Guo, Z. Zhao, B. Z. Tang. Versatile Aggregation-Enhanced Delayed Fluorescence Luminogens Functioning as Emitters and Hosts for High-Performance Organic Light-Emitting Diodes. CCS Chem., 2021. [DOI]
- J. He, H. Chen, J. Li, J. Wang, J. Xu, Z. Zhao, B. Z. Tang. Aggregation-Induced Delayed Fluorescence Molecules with Mechanochromic Behaviors for Efficient Blue Organic Light-Emitting Diodes. Cell Rep. Phys. Sci., 2022. [DOI]
- L. Wu, K. Wang, C. Wang, X.-C. Fan, Y.-Z. Shi, X. Zhang, S.-L. Zhang, J. Ye, C.-J. Zheng, Y.-Q. Li. Using Fluorene to Lock Electronically Active Moieties in Thermally Activated Delayed Fluorescence Emitters for High-Performance Non-Doped Organic Light-Emitting Diodes with Suppressed Roll-Off. Chem. Sci., 2021. [DOI]
- F. Ma, G. Zhao, Y. Zheng, F. He, K. Hasrat, Z. Qi. Molecular Engineering of Thermally Activated Delayed Fluorescence Emitters with Aggregation-Induced Emission Via Introducing Intramolecular Hydrogen-Bonding Interactions for Efficient Solution-Processed Nondoped OLEDs. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- Z. Huang, Z. Bin, R. Su, F. Yang, J. Lan, J. You. Molecular Design of Non-Doped OLEDs Based on a Twisted Heptagonal Acceptor: A Delicate Balance between Rigidity and Rotatability. Angew. Chem., Int. Ed., 2020. [DOI]
- F. Ma, Y. Cheng, Y. Zheng, H. Ji, K. Hasrat, Z. Qi. Rational Design of Thermally Activated Delayed Fluorescence Emitters with Aggregation-Induced Emission Employing Combined Charge Transfer Pathways for Fabricating Efficient Non-Doped OLEDs. J. Mater. Chem. C, 2019. [DOI]
- J. Wang, C. Liu, C. Jiang, C. Yao, M. Gu, W. Wang. Solution-Processed Aggregation-Induced Delayed Fluorescence (Aidf) Emitters Based on Strong Π-Accepting Triazine Cores for Highly Efficient Nondoped OLEDs with Low Efficiency Roll-Off. Org. Electron., 2019. [DOI]
- S. Y. Park, S. Choi, G. E. Park, H. J. Kim, C. Lee, J. S. Moon, S. W. Kim, S. Park, J. H. Kwon, M. J. Cho, D. H. Choi. Unconventional Three-Armed Luminogens Exhibiting Both Aggregation-Induced Emission and Thermally Activated Delayed Fluorescence Resulting in High-Performing Solution-Processed Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- Q. Zhang, S. Sun, W. Liu, P. Leng, X. Lv, Y. Wang, H. Chen, S. Ye, S. Zhuang, L. Wang. Integrating TADF Luminogens with AIE Characteristics Using a Novel Acridine-Carbazole Hybrid as Donor for High-Performance and Low Efficiency Roll-Off OLEDs. J. Mater. Chem. C, 2019. [DOI]
- Aggregation-Induced Delayed Fluorescence Based on Donor/Acceptor-Tethered Janus Carborane Triads: Unique Photophysical Properties of Nondoped OLEDs.. Angew. Chem. Int. Ed., 2016. [DOI]
- L. Yu, Z. Wu, G. Xie, C. Zhong, Z. Zhu, D. Ma, C. Yang. An Efficient Exciton Harvest Route for High-Performance OLEDs Based on Aggregation-Induced Delayed Fluorescence. Chem. Commun., 2018. [DOI]
- Y.-F. Shen, M. Li, W.-L. Zhao, Y.-F. Wang, H.-Y. Lu, C.-F. Chen. Quinoline-Based TADF Emitters Exhibiting Aggregation-Induced Emission for Efficient Non-Doped Organic Light-Emitting Diodes. Mater. Chem. Front., 2021. [DOI]
- L. Zhang, Y.-F. Wang, M. Li, Q.-Y. Gao, C.-F. Chen. Quinoline-Based Aggregation-Induced Delayed Fluorescence Materials for Highly Efficient Non-Doped Organic Light-Emitting Diodes. Chin. Chem. Lett., 2021. [DOI]
- H. J. Kim, H. Kang, J. E. Jeong, S. H. Park, C. W. Koh, C. W. Kim, H. Y. Woo, M. J. Cho, S. Park, D. H. Choi. Ultra-Deep-Blue Aggregation-Induced Delayed Fluorescence Emitters: Achieving Nearly 16% EQE in Solution-Processed Nondoped and Doped OLEDs with Ciey < 0.1. Adv. Funct. Mater., 2021. [DOI]
- V. S. Padalkar, S. Seki. Excited-State Intramolecular Proton-Transfer (Esipt)-Inspired Solid State Emitters. Chem. Soc. Rev., 2016. [DOI | PubMed]
- J. Zhao, S. Ji, Y. Chen, H. Guo, P. Yang. Excited State Intramolecular Proton Transfer (Esipt): From Principal Photophysics to the Development of New Chromophores and Applications in Fluorescent Molecular Probes and Luminescent Materials. Phys. Chem. Chem. Phys., 2012. [DOI | PubMed]
- Y. Cao, J. Eng, T. J. Penfold. Excited State Intramolecular Proton Transfer Dynamics for Triplet Harvesting in Organic Molecules. J. Phys. Chem. A, 2019. [DOI | PubMed]
- A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian, J. Yoon. Excited-State Intramolecular Proton-Transfer (Esipt) Based Fluorescence Sensors and Imaging Agents. Chem. Soc. Rev., 2018. [DOI | PubMed]
- S. Sinha, B. Chowdhury, P. Ghosh. A Highly Sensitive Esipt-Based Ratiometric Fluorescence Sensor for Selective Detection of Al3+. Inorg. Chem., 2016. [DOI | PubMed]
- Q. Zheng, F. Ding, X. Hu, J. Feng, J. Shen, X. He. Esipt-Based Fluorescent Probe for Bioimaging and Identification of Group Iiia Ions in Live Cells and Zebrafish. Bioorg. Chem., 2021. [DOI | PubMed]
- A. K. Gupta, A. Kumar, R. Singh, M. Devi, A. Dhir, C. P. Pradeep. Facile Synthesis of an Organic Solid State near-Infrared-Emitter with Large Stokes Shift Via Excited-State Intramolecular Proton Transfer. ACS Omega, 2018. [DOI | PubMed]
- R. Singh, A. K. Gupta, C. P. Pradeep. Synthesis of a New Series of Organic Solid-State near-Infrared Emitters: The Role of Crystal Packing and Weak Intermolecular Interactions and Application in Latent Fingerprint Detection. Cryst. Growth Des., 2021. [DOI]
- N. Suzuki, K. Suda, D. Yokogawa, H. Kitoh-Nishioka, S. Irle, A. Ando, L. M. Abegão, K. Kamada, A. Fukazawa, S. Yamaguchi. Near Infrared Two-Photon-Excited and-Emissive Dyes Based on a Strapped Excited-State Intramolecular Proton-Transfer (Esipt) Scaffold. Chem. Sci., 2018. [DOI | PubMed]
- S. Kim, T. G. Hwang, J. W. Namgoong, H. M. Kim, J. P. Kim. Effect of Linker Moiety on Linear Dimeric Benzotriazole Derivatives as Highly Stable Uv Absorber for Transparent Polyimide Film. Dyes Pigm., 2020. [DOI]
- L. G. T. A. Duarte, J. C. Germino, J. F. Berbigier, C. A. Barboza, M. M. Faleiros, D. de Alencar Simoni, M. T. Galante, M. S. de Holanda, F. S. Rodembusch, T. D. Z. Atvars. White-Light Generation from All-Solution-Processed OLEDs Using a Benzothiazole-Salophen Derivative Reactive to the Esipt Process. Phys. Chem. Chem. Phys., 2019. [DOI | PubMed]
- J. E. Kwon, S. Y. Park. Advanced Organic Optoelectronic Materials: Harnessing Excited-State Intramolecular Proton Transfer (Esipt) Process. Adv. Mater., 2011. [DOI | PubMed]
- B. Li, G. Tang, L. Zhou, D. Wu, J. Lan, L. Zhou, Z. Lu, J. You. Unexpected Sole Enol-Form Emission of 2-(2′-Hydroxyphenyl) Oxazoles for Highly Efficient Deep-Blue-Emitting Organic Electroluminescent Devices. Adv. Funct. Mater., 2017. [DOI]
- S. Park, J. Seo, S. H. Kim, S. Y. Park. Tetraphenylimidazole-Based Excited-State Intramolecular Proton-Transfer Molecules for Highly Efficient Blue Electroluminescence. Adv. Funct. Mater., 2008. [DOI]
- B. Li, G. Tang, L. Zhou, D. Wu, J. Lan, L. Zhou, Z. Lu, J. You. Unexpected Sole Enol-Form Emission of 2-(2′-Hydroxyphenyl)Oxazoles for Highly Efficient Deep-Blue-Emitting Organic Electroluminescent Devices. Adv. Funct. Mater, 2017. [DOI]
- K.-C. Tang, M.-J. Chang, T.-Y. Lin, H.-A. Pan, T.-C. Fang, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu, P.-T. Chou. Fine Tuning the Energetics of Excited-State Intramolecular Proton Transfer (Esipt): White Light Generation in a Single Esipt System. J. Am. Chem. Soc., 2011. [DOI | PubMed]
- Z. Xie, Q. Huang, T. Yu, L. Wang, Z. Mao, W. Li, Z. Yang, Y. Zhang, S. Liu, J. Xu. Hydrogen-Bonding-Assisted Intermolecular Charge Transfer: A New Strategy to Design Single-Component White-Light-Emitting Materials. Adv. Funct. Mater, 2017. [DOI]
- Z. Zhang, Y.-A. Chen, W.-Y. Hung, W.-F. Tang, Y.-H. Hsu, C.-L. Chen, F.-Y. Meng, P.-T. Chou. Control of the Reversibility of Excited-State Intramolecular Proton Transfer (Esipt) Reaction: Host-Polarity Tuning White Organic Light Emitting Diode on a New Thiazolo[5,4-D]Thiazole Esipt System. Chem. Mater., 2016. [DOI]
- S. Park, O. H. Kwon, Y. S. Lee, D. J. Jang, S. Y. Park. Imidazole-Based Excited-State Intramolecular Proton-Transfer (Esipt) Materials: Observation of Thermally Activated Delayed Fluorescence (Tdf). J. Phys. Chem. A, 2007. [DOI | PubMed]
- M. Mamada, K. Inada, T. Komino, W. J. Potscavage, H. Nakanotani, C. Adachi. Highly Efficient Thermally Activated Delayed Fluorescence from an Excited-State Intramolecular Proton Transfer System. ACS Cent. Sci., 2017. [DOI | PubMed]
- Y. Long, M. Mamada, C. Li, P. L. dos Santos, M. Colella, A. Danos, C. Adachi, A. Monkman. Excited State Dynamics of Thermally Activated Delayed Fluorescence from an Excited State Intramolecular Proton Transfer System. J. Phys. Chem. Lett, 2020. [DOI | PubMed]
- K. Wu, T. Zhang, Z. Wang, L. Wang, L. Zhan, S. Gong, C. Zhong, Z.-H. Lu, S. Zhang, C. Yang. De Novo Design of Excited-State Intramolecular Proton Transfer Emitters Via a Thermally Activated Delayed Fluorescence Channel. J. Am. Chem. Soc., 2018. [DOI | PubMed]
- A. K. Gupta, W. Li, A. Ruseckas, C. Lian, C. L. Carpenter-Warren, D. B. Cordes, A. M. Z. Slawin, D. Jacquemin, I. D. W. Samuel, E. Zysman-Colman. Thermally Activated Delayed Fluorescence Emitters with Intramolecular Proton Transfer for High Luminance Solution-Processed Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces, 2021. [DOI | PubMed]
- W. Shao, J. Hao, H. Jiang, P. M. Zimmerman, J. Kim. Metal-Free Organic Triplet Emitters with on-Off Switchable Excited State Intramolecular Proton Transfer. Adv. Funct. Mater., 2022. [DOI]
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu, J. Xu. Recent Advances in Organic Mechanofluorochromic Materials. Chem. Soc. Rev., 2012. [DOI | PubMed]
- D. Barman, R. Gogoi, K. Narang, P. K. Iyer. Recent Developments on Multi-Functional Metal-Free Mechanochromic Luminescence and Thermally Activated Delayed Fluorescence Organic Materials. Front. Chem., 2020. [DOI | PubMed]
- Z. Xie, T. Su, E. Ubba, H. Deng, Z. Mao, T. Yu, T. Zheng, Y. Zhang, S. Liu, Z. Chi. Achieving Tunable Dual-Emissive and High-Contrast Mechanochromic Materials by Manipulating Steric Hindrance Effects. J. Mater. Chem. C, 2019. [DOI]
- B. Huang, W. C. Chen, Z. Li, J. Zhang, W. Zhao, Y. Feng, B. Z. Tang, C. S. Lee. Manipulation of Molecular Aggregation States to Realize Polymorphism, AIE, MCL, and TADF in a Single Molecule. Angew. Chem., Int. Ed., 2018. [DOI]
- Z. Xie, Q. Huang, T. Yu, L. Wang, Z. Mao, W. Li, Z. Yang, Y. Zhang, S. Liu, J. Xu. Hydrogen-Bonding-Assisted Intermolecular Charge Transfer: A New Strategy to Design Single-Component White-Light-Emitting Materials. Adv. Funct. Mater., 2017. [DOI]
- B. Xu, Y. Mu, Z. Mao, Z. Xie, H. Wu, Y. Zhang, C. Jin, Z. Chi, S. Liu, J. Xu. Achieving Remarkable Mechanochromism and White-Light Emission with Thermally Activated Delayed Fluorescence through the Molecular Heredity Principle. Chem. Sci., 2016. [DOI | PubMed]
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman, S. Minakata. Thermally Activated Delayed Fluorescent Phenothiazine-Dibenzo [a, J] Phenazine-Phenothiazine Triads Exhibiting Tricolor-Changing Mechanochromic Luminescence. Chem. Sci., 2017. [DOI | PubMed]
- P. Data, M. Okazaki, S. Minakata, Y. Takeda. Thermally Activated Delayed Fluorescence Vs. Room Temperature Phosphorescence by Conformation Control of Organic Single Molecules. J. Mater. Chem. C, 2019. [DOI]
- Y. Takeda, T. Kaihara, M. Okazaki, H. Higginbotham, P. Data, N. Tohnai, S. Minakata. Conformationally-Flexible and Moderately Electron-Donating Units-Installed D-a-D Triad Enabling Multicolor-Changing Mechanochromic Luminescence, TADF and Room-Temperature Phosphorescence. Chem. Commun., 2018. [DOI]
- R. Pashazadeh, P. Pander, A. Bucinskas, P. J. Skabara, F. B. Dias, J. V. Grazulevicius. An Iminodibenzyl-Quinoxaline-Iminodibenzyl Scaffold as a Mechanochromic and Dual Emitter: Donor and Bridge Effects on Optical Properties. Chem. Commun., 2018. [DOI]
- W. Yang, Y. Yang, L. Zhan, K. Zheng, Z. Chen, X. Zeng, S. Gong, C. Yang. Polymorphism-Dependent Thermally Activated Delayed Fluorescence Materials with Diverse Three Dimensional Supramolecular Frameworks. Chem. Eng. J., 2020. [DOI]
- U. P. Pandey, R. P. Nandi, P. Thilagar. Design, Synthesis, and Temperature-Driven Molecular Conformation-Dependent Delayed Fluorescence Characteristics of Dianthrylboron-Based Donor-Acceptor Systems. Front. Chem., 2020. [DOI | PubMed]
- H. Yu, X. Song, N. Xie, J. Wang, C. Li, Y. Wang. Reversible Crystal-to-Crystal Phase Transitions with High-Contrast Luminescent Alterations for a Thermally Activated Delayed Fluorescence Emitter. Adv. Funct. Mater., 2021. [DOI]
- B. Huang, Z. Li, H. Yang, D. Hu, W. Wu, Y. Feng, Y. Sun, B. Lin, W. Jiang. Bicolour Electroluminescence of 2-(Carbazol-9-Yl)Anthraquinone Based on a Solution Process. J. Mater. Chem. C, 2017. [DOI]
- S. K. B. Mane, Y. Mu, Z. Yang, E. Ubba, N. Shaishta, Z. Chi. A TADF Compound with High-Contrast Mechano-Responsive Fluorescence on/Off Switching for Both Sequential and Combinational Logic Gates. J. Mater. Chem. C, 2019. [DOI]
- Y. Fu, Z. Ye, J. Xiao, L. Liao, L. Chen, Y. Mu, S. Ji, Z. Zhao, H.-L. Zhang, Y. Huo. Large Effects of Tiny Structural Changes on the AIE-TADF Type Xanthone Derivatives in Mechano-Responsive Luminescence and Electroluminescence. Dyes Pigm., 2022. [DOI]
- X. Zhang, T. Lu, C. Zhou, H. Liu, Y. Wen, Y. Shen, B. Li, S.-T. Zhang, B. Yang. Thermally Activated Delayed Fluorescence of Aggregates Induced by Strong Π-Π Interactions and Reversible Dual-Responsive Luminescence Switching. CCS Chem., 2022. [DOI]
- J. I. Zink. Triboluminesence. Acc. Chem. Res., 1978. [DOI]
- B. P. Chandra, J. I. Zink. Triboluminescence and the Dynamics of Crystal Fracture. Phys. Rev. B, 1980. [DOI]
- S. Xu, T. Liu, Y. Mu, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, Y. Zhang, A. Lien, J. Xu. An Organic Molecule with Asymmetric Structure Exhibiting Aggregation-Induced Emission, Delayed Fluorescence, and Mechanoluminescence. Angew. Chem., Int. Ed., 2015. [DOI]
- C. Poriel, J. Rault-Berthelot. Designing Host Materials for the Emissive Layer of Single-Layer Phosphorescent Organic Light-Emitting Diodes: Toward Simplified Organic Devices. Adv. Funct. Mater., 2021. [DOI]
- Q. Pei, G. Yu, C. Zhang, Y. Yang, A. J. Heeger. Polymer Light-Emitting Electrochemical Cells. Science, 1995. [DOI | PubMed]
- K. M. Maness, R. H. Terrill, T. J. Meyer, R. W. Murray, R. M. Wightman. Solid-State Diode-Like Chemiluminescence Based on Serial, Immobilized Concentration Gradients in Mixed-Valent Poly[Ru(Vbpy)3](Pf6)2 Films. J. Am. Chem. Soc., 1996. [DOI]
- S. van Reenen, P. Matyba, A. Dzwilewski, R. A. J. Janssen, L. Edman, M. Kemerink. A Unifying Model for the Operation of Light-Emitting Electrochemical Cells. J. Am. Chem. Soc., 2010. [DOI | PubMed]
- Z. B. Hill, D. B. Rodovsky, J. M. Leger, G. P. Bartholomew. Synthesis and Utilization of Perylene-Based N-Type Small Molecules in Light-Emitting Electrochemical Cells. Chem. Commun., 2008. [DOI]
- N. Arnosti, F. Brunner, I. Susic, S. Keller, J. M. Junquera-Hernández, A. Prescimone, H. J. Bolink, M. Sessolo, E. Ortí, C. E. Housecroft, E. C. Constable. Remote Modification of Bidentate Phosphane Ligands Controlling the Photonic Properties in Their Complexes: Enhanced Performance of [Cu(Rn-Xantphos)(N∧N)][Pf6] in Light-Emitting Electrochemical Cells. Adv. Optical Mater., 2020. [DOI]
- S. Tang, A. Sandström, P. Lundberg, T. Lanz, C. Larsen, S. van Reenen, M. Kemerink, L. Edman. Design Rules for Light-Emitting Electrochemical Cells Delivering Bright Luminance at 27.5 Percent External Quantum Efficiency. Nat. Commun., 2017. [DOI | PubMed]
- A. Sandström, P. Matyba, O. Inganäs, L. Edman. Separating Ion and Electron Transport: The Bilayer Light-Emitting Electrochemical Cell. J. Am. Chem. Soc., 2010. [DOI | PubMed]
- S. Tang, W.-Y. Tan, X.-H. Zhu, L. Edman. Small-Molecule Light-Emitting Electrochemical Cells: Evidence for in Situ Electrochemical Doping and Functional Operation. Chem. Commun., 2013. [DOI]
- K. Birdee, S. Hu, J. Gao. Strong Doping and Electroluminescence Realized by Fast Ion Transport through a Planar Polymer/Polymer Interface in Bilayer Light-Emitting Electrochemical Cells. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- I. Verboven, W. Deferme. Printing of Flexible Light Emitting Devices: A Review on Different Technologies and Devices, Printing Technologies and State-of-the-Art Applications and Future Prospects. Progress in Materials Science, 2021. [DOI]
- D. J. Dick, A. J. Heeger, Y. Yang, Q. Pei. Imaging the Structure of the P-N Junction in Polymer Light-Emitting Electrochemical Cells. Adv. Mater., 1996. [DOI]
- E. Zysman-Colman, J. D. Slinker, J. B. Parker, G. G. Malliaras, S. Bernhard. Improved Turn-on Times of Light-Emitting Electrochemical Cells. Chem. Mater., 2008. [DOI]
- J. C. deMello, N. Tessler, S. C. Graham, R. H. Friend. Ionic Space-Charge Effects in Polymer Light-Emitting Diodes. Phys. Rev. B, 1998. [DOI]
- J. D. Slinker, J. A. DeFranco, M. J. Jaquith, W. R. Silveira, Y.-W. Zhong, J. M. Moran-Mirabal, H. G. Craighead, H. D. Abruña, J. A. Marohn, G. G. Malliaras. Direct Measurement of the Electric-Field Distribution in a Light-Emitting Electrochemical Cell. Nat. Mater., 2007. [DOI | PubMed]
- L. S. C. Pingree, D. B. Rodovsky, D. C. Coffey, G. P. Bartholomew, D. S. Ginger. Scanning Kelvin Probe Imaging of the Potential Profiles in Fixed and Dynamic Planar Lecs. J. Am. Chem. Soc., 2007. [DOI | PubMed]
- H. J. Bolink, E. Coronado, R. D. Costa, N. Lardiés, E. Ortí. Near-Quantitative Internal Quantum Efficiency in a Light-Emitting Electrochemical Cell. Inorg. Chem., 2008. [DOI | PubMed]
- G. G. Malliaras, J. C. Scott. The Roles of Injection and Mobility in Organic Light Emitting Diodes. J. Appl. Phys., 1998. [DOI]
- Henwood, A. F. ; Zysman-Colman, E. In Iridium(Iii) in Optoelectronic and Photonics Applications; John Wiley & Sons Ltd., 2017; Vol. 1.
- Henwood, A. F. ; Zysman-Colman, E. In Photoluminescent Materials and Electroluminescent Devices; Armaroli, N. , Bolink, H. J. , Eds.; Springer International Publishing: Cham, 2017.10.1007/978-3-319-59304-3_2
- Sharma, N. ; Wong, M. Y. ; Samuel, I. D. W. ; Zysman-Colman, E. In Highly Efficient OLEDs; Yersin, H. , Ed.; Wiley-VCH Verlag GmbH & Co: KGaA, 2018; Vol. 1.
- M. Y. Wong, G. J. Hedley, G. Xie, L. S. Kölln, I. D. W. Samuel, A. Pertegás, H. J. Bolink, E. Zysman-Colman. Light-Emitting Electrochemical Cells and Solution-Processed Organic Light-Emitting Diodes Using Small Molecule Organic Thermally Activated Delayed Fluorescence Emitters. Chem. Mater., 2015. [DOI]
- R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, S. Schaffner, M. Neuburger, C. E. Housecroft, E. C. Constable. Archetype Cationic Iridium Complexes and Their Use in Solid-State Light-Emitting Electrochemical Cells. Adv. Funct. Mater., 2009. [DOI]
- A. Pertegás, M. Y. Wong, M. Sessolo, E. Zysman-Colman, H. J. Bolink. Efficient Light-Emitting Electrochemical Cells Using Small Molecular Weight, Ionic, Host-Guest Systems. ECS J. Solid State Sci. Tech., 2016. [DOI]
- M. Y. Wong, M.-G. La-Placa, A. Pertegas, H. J. Bolink, E. Zysman-Colman. Deep-Blue Thermally Activated Delayed Fluorescence (TADF) Emitters for Light-Emitting Electrochemical Cells (LEECs). J. Mater. Chem. C, 2017. [DOI]
- R. Yu, Y. Song, K. Zhang, X. Pang, M. Tian, L. He. Intrinsically Ionic, Thermally Activated Delayed Fluorescent Materials for Efficient, Bright, and Stable Light-Emitting Electrochemical Cells. Adv. Funct. Mater., 2022. [DOI]
- H.-L. Shen, P.-W. Hsiao, R.-H. Yi, Y.-H. Su, Y. Chen, C.-W. Lu, H.-C. Su. Purely Organic Pyridium-Based Materials with Thermally Activated Delayed Fluorescence for Orange-Red Light-Emitting Electrochemical Cells. Dyes Pigm., 2022. [DOI]
- P. Lundberg, E. M. Lindh, S. Tang, L. Edman. Toward Efficient and Metal-Free Emissive Devices: A Solution-Processed Host-Guest Light-Emitting Electrochemical Cell Featuring Thermally Activated Delayed Fluorescence. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- P. Lundberg, Q. Wei, Z. Ge, B. Voit, S. Reineke, L. Edman. Polymer Featuring Thermally Activated Delayed Fluorescence as Emitter in Light-Emitting Electrochemical Cells. J. Phys. Chem. Lett, 2020. [DOI | PubMed]
- P. Lundberg, Y. Tsuchiya, E. M. Lindh, S. Tang, C. Adachi, L. Edman. Thermally Activated Delayed Fluorescence with 7% External Quantum Efficiency from a Light-Emitting Electrochemical Cell. Nat. Commun., 2019. [DOI | PubMed]
- J. Ye, Y. He, K. Li, L. Liu, C. Xi, Z. Liu, Y. Ma, B. Zhang, Y. Bao, W. Wang. Achieving Record Efficiency and Luminance for TADF Light-Emitting Electrochemical Cells by Dopant Engineering. ACS Appl. Mater. Interfaces, 2022. [DOI | PubMed]
- R. Bai, X. Meng, X. Wang, L. He. Color-Stable, Efficient, and Bright Blue Light-Emitting Electrochemical Cell Using Ionic Exciplex Host. Adv. Funct. Mater, 2021. [DOI]
- S. Tang, P. Lundberg, Y. Tsuchiya, J. Ràfols-Ribé, Y. Liu, J. Wang, C. Adachi, L. Edman. Efficient and Bright Blue Thermally Activated Delayed Fluorescence from Light-Emitting Electrochemical Cells. Adv. Funct. Mater., 2022. [DOI]
- D. Zhang, L. Duan, D. Zhang, Y. Qiu. Towards Ideal Electrophosphorescent Devices with Low Dopant Concentrations: The Key Role of Triplet up-Conversion. J. Mater. Chem. C, 2014. [DOI]
- M. Karaman, A. Kumar Gupta, S. Madayanad Suresh, T. Matulaitis, L. Mardegan, D. Tordera, H. J. Bolink, S. Wu, S. Warriner, I. D. Samuel, E. Zysman-Colman. Ionic Multiresonant Thermally Activated Delayed Fluorescence Emitters for Light Emitting Electrochemical Cells. Beilstein J Org Chem, 2022. [DOI | PubMed]
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi, N. Armaroli. Luminescent Ionic Transition-Metal Complexes for Light-Emitting Electrochemical Cells. Angew. Chem., Int. Ed., 2012. [DOI]
- G. U. Mahoro, J. Fernandez-Cestau, J.-L. Renaud, P. B. Coto, R. D. Costa, S. Gaillard. Recent Advances in Solid-State Lighting Devices Using Transition Metal Complexes Exhibiting Thermally Activated Delayed Fluorescent Emission Mechanism. Adv. Optical mater., 2020. [DOI]
- N. Armaroli. Photoactive Mono- and Polynuclear Cu(I)-Phenanthrolines. A Viable Alternative to Ru(Ii)-Polypyridines?. Chem. Soc. Rev., 2001. [DOI]
- M. K. Eggleston, D. R. McMillin, K. S. Koenig, A. J. Pallenberg. Steric Effects in the Ground and Excited States of Cu(Nn)2+ Systems. Inorg. Chem., 1997. [DOI]
- S. Keller, E. C. Constable, C. E. Housecroft, M. Neuburger, A. Prescimone, G. Longo, A. Pertegás, M. Sessolo, H. J. Bolink. [Cu(Bpy)(P∧P)]+ Containing Light-Emitting Electrochemical Cells: Improving Performance through Simple Substitution. Dalton Trans., 2014. [DOI | PubMed]
- S. Keller, A. Prescimone, H. Bolink, M. Sessolo, G. Longo, L. Martínez-Sarti, J. M. Junquera-Hernández, E. C. Constable, E. Ortí, C. E. Housecroft. Luminescent Copper(I) Complexes with Bisphosphane and Halogen-Substituted 2,2′-Bipyridine Ligands. Dalton Trans., 2018. [DOI | PubMed]
- S. Keller, A. Prescimone, M.-G. La Placa, J. M. Junquera-Hernández, H. J. Bolink, E. C. Constable, M. Sessolo, E. Ortí, C. E. Housecroft. The Shiny Side of Copper: Bringing Copper(I) Light-Emitting Electrochemical Cells Closer to Application. RSC Adv., 2020. [DOI | PubMed]
- M. Alkan-Zambada, S. Keller, L. Martínez-Sarti, A. Prescimone, J. M. Junquera-Hernández, E. C. Constable, H. J. Bolink, M. Sessolo, E. Ortí, C. E. Housecroft. [Cu(P∧P)(N∧N)][Pf6] Compounds with Bis(Phosphane) and 6-Alkoxy, 6-Alkylthio, 6-Phenyloxy and 6-Phenylthio-Substituted 2,2′-Bipyridine Ligands for Light-Emitting Electrochemical Cells. J. Mater. Chem. C, 2018. [DOI]
- M. Meyer, L. Mardegan, D. Tordera, A. Prescimone, M. Sessolo, H. J. Bolink, E. C. Constable, C. E. Housecroft. A Counterion Study of a Series of [Cu(P∧P)(N∧N)][a] Compounds with Bis(Phosphane) and 6-Methyl and 6,6′-Dimethyl-Substituted 2,2′-Bipyridine Ligands for Light-Emitting Electrochemical Cells. Dalton Trans., 2021. [DOI | PubMed]
- E. Leoni, J. Mohanraj, M. Holler, M. Mohankumar, I. Nierengarten, F. Monti, A. Sournia-Saquet, B. Delavaux-Nicot, J.-F. i. Nierengarten, N. Armaroli. Heteroleptic Copper(I) Complexes Prepared from Phenanthroline and Bis-Phosphine Ligands: Rationalization of the Photophysical and Electrochemical Properties. Inorg. Chem., 2018. [DOI | PubMed]
- M. N. Hopkinson, C. Richter, M. Schedler, F. Glorius. An Overview of N-Heterocyclic Carbenes. Nature, 2014. [DOI | PubMed]
- R. Marion, F. Sguerra, F. Di Meo, E. Sauvageot, J.-F. Lohier, R. Daniellou, J.-L. Renaud, M. Linares, M. Hamel, S. Gaillard. NHC Copper(I) Complexes Bearing Dipyridylamine Ligands: Synthesis, Structural, and Photoluminescent Studies. Inorg. Chem., 2014. [DOI | PubMed]
- M. Elie, F. Sguerra, F. Di Meo, M. D. Weber, R. Marion, A. Grimault, J.-F. Lohier, A. Stallivieri, A. Brosseau, R. B. Pansu. Designing NHC-Copper(I) Dipyridylamine Complexes for Blue Light-Emitting Electrochemical Cells. ACS Appl. Mater. Interfaces, 2016. [DOI | PubMed]
- K. Schlingman, Y. Chen, R. S. Carmichael, T. B. Carmichael. 25 Years of Light-Emitting Electrochemical Cells: A Flexible and Stretchable Perspective. Adv. Mater., 2021. [DOI]
- D. Zhang, X. Song, M. Cai, L. Duan. Blocking Energy-Loss Pathways for Ideal Fluorescent Organic Light-Emitting Diodes with Thermally Activated Delayed Fluorescent Sensitizers. Adv. Mater., 2018. [DOI]
- Y.-K. Wang, C.-C. Huang, S. Kumar, S.-F. Wu, Y. Yuan, A. K. Aziz Khan, Z.-Q. Jiang, M.-K. Fung, L.-S. Liao. The Roles of Thermally Activated Delayed Fluorescence Sensitizers for Efficient Red Fluorescent Organic Light-Emitting Diodes with D-a-a Type Emitters. Mater. Chem. Front., 2019. [DOI]
- D. Li, Y. Hu, L.-S. Liao. Triplet Exciton Harvesting by Multi-Process Energy Transfer in Fluorescent Organic Light-Emitting Diodes. J. Mater. Chem. C, 2019. [DOI]
- 10.1: Invited Paper: Hyperfluorescence; a Game Changing Technology of OLED Display;. SID Symposium Digest of Technical Papers;, 2019
- D. Zhang, L. Duan, C. Li, Y. Li, H. Li, D. Zhang, Y. Qiu. High-Efficiency Fluorescent Organic Light-Emitting Devices Using Sensitizing Hosts with a Small Singlet-Triplet Exchange Energy. Adv. Mater., 2014. [DOI | PubMed]
- M. Baldo, M. E. Thompson, S. Forrest. High-Efficiency Fluorescent Organic Light-Emitting Devices Using a Phosphorescent Sensitizer. Nature, 2000. [DOI | PubMed]
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda, C. Adachi. High-Efficiency Organic Light-Emitting Diodes with Fluorescent Emitters. Nat. Commun., 2014. [DOI | PubMed]
- N. Haase, A. Danos, C. Pflumm, P. Stachelek, W. Brütting, A. P. Monkman. Are the Rates of Dexter Transfer in TADF Hyperfluorescence Systems Optically Accessible?. Mater. Horiz., 2021. [DOI | PubMed]
- K. Stavrou, L. G. Franca, T. Böhmer, L. Duben, C. Marian, A. Monkman. Unexpected Quasi-Axial Conformer in Thermally Activated Delayed Fluorescence Dmac-Trz. Demonstrating How to Turn Green OLEDs into Blue. Adv. Funct. Mater., 2023. [DOI]
- K. Stavrou, S. Madayanad Suresh, D. Hall, A. Danos, N. A. Kukhta, A. M. Z. Slawin, S. Warriner, D. Beljonne, Y. Olivier, A. Monkman, E. Zysman-Colman. Emission and Absorption Tuning in TADF B,N-Doped Heptacenes: Toward Ideal-Blue Hyperfluorescent OLEDs. Adv. Optical Mater., 2022. [DOI]
- D. Hall, K. Stavrou, E. Duda, A. Danos, S. Bagnich, S. Warriner, A. M. Slawin, D. Beljonne, A. Köhler, A. Monkman. Diindolocarbazole-Achieving Multiresonant Thermally Activated Delayed Fluorescence without the Need for Acceptor Units. Mater. Horiz., 2022. [DOI | PubMed]
- I. A. Wright, A. Danos, S. Montanaro, A. S. Batsanov, A. P. Monkman, M. R. Bryce. Conformational Dependence of Triplet Energies in Rotationally Hindered N-and S-Heterocyclic Dimers: New Design and Measurement Rules for High Triplet Energy OLED Host Materials. Chem. Eur. J., 2021. [DOI | PubMed]
- I. H. Lee, W. Song, J. Y. Lee, S.-H. Hwang. High Efficiency Blue Fluorescent Organic Light-Emitting Diodes Using a Conventional Blue Fluorescent Emitter. J. Mater. Chem. C, 2015. [DOI]
- D. H. Ahn, J. H. Jeong, J. Song, J. Y. Lee, J. H. Kwon. Highly Efficient Deep Blue Fluorescent Organic Light-Emitting Diodes Boosted by Thermally Activated Delayed Fluorescence Sensitization. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- M. I. Alam, M. R. Nagar, S. R. Nayak, A. Choudhury, J.-H. Jou, S. Vaidyanathan. Acceptor Interlocked Molecular Design for Solution-Processed Stable Deep-Blue TADF and Hyper Fluorescence Organic Led Enabling High-Efficiency. Adv. Optical Mater., 2022. [DOI]
- J. H. Yun, K. H. Lee, J. Y. Lee. Fluorine Substituted Triazine Acceptor Based Thermally Activated Delayed Fluorescent Emitter as an Assistant Dopant of Fluorescent Emitter. Dyes Pigm., 2020. [DOI]
- J. S. Jang, S. H. Han, H. W. Choi, K. S. Yook, J. Y. Lee. Molecular Design of Sensitizer to Suppress Efficiency Loss Mechanism in Hyper-Fluorescent Organic Light-Emitting Diodes. Org. Electron., 2018. [DOI]
- W. Xie, X. Peng, M. Li, W. Qiu, W. Li, Q. Gu, Y. Jiao, Z. Chen, Y. Gan, K. k. Liu, S.-J. Su. Blocking the Energy Loss of Dexter Energy Transfer in Hyperfluorescence OLEDs Via One-Step Phenyl-Fluorene Substitution of TADF Assistant Host. Adv. Optical Mater., 2022. [DOI]
- N. R. Wallwork, M. Mamada, A. Shukla, S. K. McGregor, C. Adachi, E. B. Namdas, S.-C. Lo. High-Performance Solution-Processed Red Hyperfluorescent OLEDs Based on Cibalackrot. J. Mater. Chem. C, 2022. [DOI]
- T. Furukawa, H. Nakanotani, M. Inoue, C. Adachi. Dual Enhancement of Electroluminescence Efficiency and Operational Stability by Rapid Upconversion of Triplet Excitons in OLEDs. Sci. Rep., 2015. [DOI | PubMed]
- X. Song, D. Zhang, Y. Lu, C. Yin, L. Duan. Understanding and Manipulating the Interplay of Wide-Energy-Gap Host and TADF Sensitizer in High-Performance Fluorescence OLEDs. Adv. Mater., 2019. [DOI]
- J. H. Kim, K. H. Lee, J. Y. Lee. Design of Thermally Activated Delayed Fluorescent Assistant Dopants to Suppress the Nonradiative Component in Red Fluorescent Organic Light-Emitting Diodes. Chem. Eur. J., 2019. [DOI | PubMed]
- S. H. Han, J. Y. Lee. Spatial Separation of Sensitizer and Fluorescent Emitter for High Quantum Efficiency in Hyperfluorescent Organic Light-Emitting Diodes. J. Mater. Chem. C, 2018. [DOI]
- J. Yao, Z. Wang, X. Qiao, D. Yang, Y. Dai, Q. Sun, J. Chen, C. Yang, D. Ma. High Efficiency and Long Lifetime Fluorescent Organic Light-Emitting Diodes Based on Cascaded Energy Transfer Processes to Efficiently Utilize Triplet Excitons Via Sensitizer. Org. Electron., 2020. [DOI]
- D. Li, L.-S. Liao. Highly Efficient Deep-Red Organic Light-Emitting Diodes Using Exciplex-Forming Co-Hosts and Thermally Activated Delayed Fluorescence Sensitizers with Extended Lifetime. J. Mater. Chem. C, 2019. [DOI]
- J. S. Jang, S. H. Han, J. Lee. Design Rule of Assistant Dopant for High External Quantum Efficiency in Hyperfluorescence Organic Light-Emitting Diodes. Adv. Photon. Res., 2021. [DOI]
- D. Chen, X. Cai, X.-L. Li, Z. He, C. Cai, D. Chen, S.-J. Su. Efficient Solution-Processed Red All-Fluorescent Organic Light-Emitting Diodes Employing Thermally Activated Delayed Fluorescence Materials as Assistant Hosts: Molecular Design Strategy and Exciton Dynamic Analysis. J. Mater. Chem. C, 2017. [DOI]
- A. Cravcenco, M. Hertzog, C. Ye, M. N. Iqbal, U. Mueller, L. Eriksson, K. Börjesson. Multiplicity Conversion Based on Intramolecular Triplet-to-Singlet Energy Transfer. Sci. Adv., 2019. [DOI | PubMed]
- J. Jõgela, A. Uri, L.-O. Pålsson, E. Enkvist. Almost Complete Radiationless Energy Transfer from Excited Triplet State of a Dim Phosphor to a Covalently Linked Adjacent Fluorescent Dye in Purely Organic Tandem Luminophores Doped into Pva Matrix. J. Mater. Chem. C, 2019. [DOI]
- X. Liao, X. Yang, J. Cheng, Y. Li, X. Meng, J. Li, Q. Pei, L. Li. Solution-Processed Warm White Organic Light-Emitting Diodes Based on a Blue Thermally Activated Delayed Fluorescence Dendrimer. Chempluschem, 2018. [DOI | PubMed]
- J. Hu, S. Hu, C. Lu, Y. Huang, K. Xu, X. Wang. Assistant Dopant System in Red Phosphorescent OLEDs and Its Mechanism Reveal. J. Lumin., 2018. [DOI]
- R. Nagata, H. Nakanotani, C. Adachi. Near-Infrared Electrophosphorescence up to 1.1 μm Using a Thermally Activated Delayed Fluorescence Molecule as Triplet Sensitizer. Adv. Mater., 2017. [DOI]
- A. Shahalizad, A. Malinge, L. Hu, G. Laflamme, L. Haeberlé, D. M. Myers, J. Mao, W. G. Skene, S. Kéna-Cohen. Efficient Solution-Processed Hyperfluorescent OLEDs with Spectrally Narrow Emission at 840 Nm. Adv. Funct. Mater., 2021. [DOI]
- J. Brodeur, L. Hu, A. Malinge, E. Eizner, W. G. Skene, S. Kéna-Cohen. Highly Efficient and Spectrally Narrow near-Infrared Fluorescent OLEDs Using a TADF-Sensitized Cyanine Dye. Adv. Optical Mater., 2019. [DOI]
- K. Bartkowski, P. Zimmermann Crocomo, M. A. Kochman, D. Kumar, A. Kubas, P. Data, M. Lindner. Tandem Rigidification and Π-Extension as a Key Tool for the Development of a Narrow Linewidth Yellow Hyperfluorescent OLED System. Chem. Sci., 2022. [DOI | PubMed]
- L. Zhan, A. Ying, Y. Qi, K. Wu, Y. Tang, Y. Tan, Y. Zou, G. Xie, S. Gong, C. Yang. Copper(I) Complex as Sensitizer Enables High-Performance Organic Light-Emitting Diodes with Very Low Efficiency Roll-Off. Adv. Funct. Mater., 2021. [DOI]
- D. Zhou, S. Wu, G. Cheng, C.-M. Che. A Gold(III)-TADF Emitter as a Sensitizer for High-Color-Purity and Efficient Deep-Blue Solution-Processed OLEDs. J. Mater. Chem. C, 2022. [DOI]
- F. Li, A. J. Gillett, Q. Gu, J. Ding, Z. Chen, T. J. H. Hele, W. K. Myers, R. H. Friend, E. W. Evans. Singlet and Triplet to Doublet Energy Transfer: Improving Organic Light-Emitting Diodes with Radicals. Nat. Commun., 2022. [DOI | PubMed]
- R. Braveenth, H. Lee, J. D. Park, K. J. Yang, S. J. Hwang, K. R. Naveen, R. Lampande, J. H. Kwon. Achieving Narrow Fwhm and High EQE over 38% in Blue OLEDs Using Rigid Heteroatom-Based Deep Blue TADF Sensitized Host. Adv. Funct. Mater., 2021. [DOI]
- K. Bartkowski, P. Zimmermann Crocomo, M. A. Kochman, D. Kumar, A. Kubas, P. Data, M. Lindner. Tandem Rigidification and Pi-Extension as a Key Tool for the Development of a Narrow Linewidth Yellow Hyperfluorescent OLED System. Chem. Sci., 2022. [DOI | PubMed]
- Unexpected Quasi-Axial Conformer in Thermally Activated Delayed Fluorescence Dmac-Trz, Pushing Green OLEDs to Blue.. Adv. Funct. Mater., 2023. [DOI]
- J. Li, S.-C. Dong, A. Opitz, L.-S. Liao, N. Koch. Design Principles of Carbazole/Dibenzothiophene Derivatives as Host Material in Modern Efficient Organic Light-Emitting Diodes. J. Mater. Chem. C, 2017. [DOI]
- C. W. Tang, S. A. VanSlyke, C. H. Chen. Electroluminescence of Doped Organic Thin Films. J. Appl. Phys., 1989. [DOI]
- I. Tanaka, Y. Tabata, S. Tokito. Förster and Dexter Energy-Transfer Processes in Fluorescent Balq Thin Films Doped with Phosphorescent Ir(Ppy)3 Molecules. J. Appl. Phys., 2006. [DOI]
- Y. K. Wang, S. H. Li, S. F. Wu, C. C. Huang, S. Kumar, Z. Q. Jiang, M. K. Fung, L. S. Liao. Tilted Spiro-Type Thermally Activated Delayed Fluorescence Host for ≈100% Exciton Harvesting in Red Phosphorescent Electronics with Ultralow Doping Ratio. Adv. Funct. Mater., 2018. [DOI]
- D. Zhang, L. Duan, Y. Li, H. Li, Z. Bin, D. Zhang, J. Qiao, G. Dong, L. Wang, Y. Qiu. Towards High Efficiency and Low Roll-Off Orange Electrophosphorescent Devices by Fine Tuning Singlet and Triplet Energies of Bipolar Hosts Based on Indolocarbazole/1, 3, 5-Triazine Hybrids. Adv. Funct. Mater., 2014. [DOI]
- Y. Li, D. Zhang, Y. Zhang, M. Cai, L. Duan. Red Phosphorescent Organic Light-Emitting Diodes Based on a Novel Host Material with Thermally Activated Delayed Fluorescent Properties. Sci. China Chem., 2016. [DOI]
- Z. Wang, H. Zhang, Z. Wang, B. Zhao, L. Chen, J. Li, H. Wang, Y. Hao, W. Li. Efficient Management of Excitons in Red and White Organic Light-Emitting Diodes by Employing Blue Thermally Activated Delayed Fluorescent Emitter Based Acridine/Sulfone Derivative as the Host. Org. Electron., 2018. [DOI]
- J. Li, R. Zhang, Z. Wang, B. Zhao, J. Xie, F. Zhang, H. Wang, K. Guo. Zig-Zag Acridine/Sulfone Derivative with Aggregation-Induced Emission and Enhanced Thermally Activated Delayed Fluorescence in Amorphous Phase for Highly Efficient Nondoped Blue Organic Light-Emitting Diodes. Adv. Optical Mater., 2018. [DOI]
- Y. Xia, Z. Liu, J. Li, C. Fan, G. Li, B. Zhao, Y. Wu, H. Wang, K. Guo. TADF Material with Non-Conjugated Rigid Donor for High-Performance Full-Color Phosphorescent OLEDs: Effects of Triplet Harvest and Charge Transport on Efficiency. Org. Electron., 2020. [DOI]
- Y. Liu, J. Pan, F. Chen, K. Gao, A. Zhu, R. Wang, X. Yue, X. Ban. An Effective Thermally Activated Delayed Fluorescence Host Material for Highly Efficient Blue Phosphorescent Organic Light-Emitting Diodes with Low Doping Concentration. J. Photochem. Photobiol., A, 2020. [DOI]
- C.-C. Lin, M.-J. Huang, M.-J. Chiu, M.-P. Huang, C.-C. Chang, C.-Y. Liao, K.-M. Chiang, Y.-J. Shiau, T.-Y. Chou, L.-K. Chu. Molecular Design of Highly Efficient Thermally Activated Delayed Fluorescence Hosts for Blue Phosphorescent and Fluorescent Organic Light-Emitting Diodes. Chem. Mater., 2017. [DOI]
- H. Wang, H. Zhao, C. Zang, S. Liu, L. Zhang, W. Xie. Stable and Efficient Phosphorescent Organic Light-Emitting Device Utilizing a Δ-Carboline-Containing Host Displaying Thermally Activated Delayed Fluorescence. J. Mater. Chem. C, 2020. [DOI]
- B. Sun, K.-N. Tong, X. Chen, J.-L. He, H. Liu, M.-K. Fung, J. Fan. A Universal Thermally Activated Delayed Fluorescent Host with Short Triplet Lifetime for Highly Efficient Phosphorescent OLEDs with Extremely Low Efficiency Roll-Off. J. Mater. Chem. C, 2021. [DOI]
- H. Ito, T. Shimizu, Y. Wada, H. Kaji, H. Fukagawa. Comprehensive Study on Operational Lifetime of Organic Light-Emitting Diodes: Effects of Molecular Structure and Energy Transfer. Jpn. J. Appl. Phys., 2021. [DOI]
- S. K. Jeon, C. S. Oh, M. Kim, K. S. Yook, J. Y. Lee. Triplet Exciton Recycling of a Phosphorescent Emitter by an up-Conversion Process Using a Delayed Fluorescence Type Low Triplet Energy Host Material. J. Mater. Chem. C, 2016. [DOI]
- S. Qian, H. Zhang, J. Lan, Z. Bin. Facile Access to Isocoumarin-Based D-a-D Triad: A Thermally Activated Delayed-Fluorescence Host for Efficient Red Phosphorescent OLEDs. Org. Electron., 2020. [DOI]
- J. Wang, C. Jiang, C. Liu, H. Liu, C. Yao. Highly Efficient Blue, Orange and Red PhOLEDs with Low Roll-Off of Efficiency Using a Carbazole Dendritic Thermally Activated Delayed Fluorescence (TADF) Material as Host. Mater. Lett., 2018. [DOI]
- D. Zhang, L. Duan, D. Zhang, J. Qiao, G. Dong, L. Wang, Y. Qiu. Extremely Low Driving Voltage Electrophosphorescent Green Organic Light-Emitting Diodes Based on a Host Material with Small Singlet-Triplet Exchange Energy without P- or N-Doping Layer. Org. Electron., 2013. [DOI]
- Y. Zhang, D. Zhang, M. Cai, Y. Li, D. Zhang, Y. Qiu, L. Duan. Towards Highly Efficient Red Thermally Activated Delayed Fluorescence Materials by the Control of Intra-Molecular Pi-Pi Stacking Interactions. Nanotechnology, 2016. [DOI | PubMed]
- J. Xue, Q. Liang, Y. Zhang, R. Zhang, L. Duan, J. Qiao. High-Efficiency Near-Infrared Fluorescent Organic Light-Emitting Diodes with Small Efficiency Roll-Off: A Combined Design from Emitters to Devices. Adv. Funct. Mater., 2017. [DOI]
- S. Wang, Y. Zhang, W. Chen, J. Wei, Y. Liu, Y. Wang. Achieving High Power Efficiency and Low Roll-Off OLEDs Based on Energy Transfer from Thermally Activated Delayed Excitons to Fluorescent Dopants. Chem. Commun., 2015. [DOI]
- M. Li, J. Wang, Y. Dai, Y. Zhang, L. Chen, L. Jin, Y. Tao, R. Chen, W. Huang. Evoking Synergetic Effect of Dual Thermally Activated Delayed Fluorescent Hosts for High-Efficiency Sensitized Fluorescent Organic Light-Emitting Diodes. J. Phys. Chem. C, 2020. [DOI]
- N. Aizawa, S. Shikita, T. Yasuda. Spin-Dependent Exciton Funneling to a Dendritic Fluorophore Mediated by a Thermally Activated Delayed Fluorescence Material as an Exciton-Harvesting Host. Chem. Mater., 2017. [DOI]
- J.-H. Lee, H. Shin, J.-M. Kim, K.-H. Kim, J.-J. Kim. Exciplex-Forming Co-Host-Based Red Phosphorescent Organic Light-Emitting Diodes with Long Operational Stability and High Efficiency. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- S. Lee, K. H. Kim, D. Limbach, Y. S. Park, J. J. Kim. Low Roll-Off and High Efficiency Orange Organic Light Emitting Diodes with Controlled Co-Doping of Green and Red Phosphorescent Dopants in an Exciplex Forming Co-Host. Adv. Funct. Mater., 2013. [DOI]
- H. Shin, S. Lee, K. H. Kim, C. K. Moon, S. J. Yoo, J. H. Lee, J. J. Kim. Blue Phosphorescent Organic Light-Emitting Diodes Using an Exciplex Forming Co-Host with the External Quantum Efficiency of Theoretical Limit. Adv. Mater., 2014. [DOI | PubMed]
- X.-K. Liu, Z. Chen, C.-J. Zheng, M. Chen, W. Liu, X.-H. Zhang, C.-S. Lee. Nearly 100% Triplet Harvesting in Conventional Fluorescent Dopant-Based Organic Light-Emitting Devices through Energy Transfer from Exciplex. Adv. Mater., 2015. [DOI | PubMed]
- P. Data, A. Kurowska, S. Pluczyk, P. Zassowski, P. Pander, R. Jedrysiak, M. Czwartosz, L. Otulakowski, J. Suwinski, M. Lapkowski, A. P. Monkman. Exciplex Enhancement as a Tool to Increase OLED Device Efficiency. J. Phys. Chem. C, 2016. [DOI]
- L. Zhang, K. W. Cheah. Thermally Activated Delayed Fluorescence Host for High Performance Organic Light-Emitting Diodes. Sci. Rep., 2018. [DOI | PubMed]
- L. Zhang, C. Cai, K. F. Li, H. L. Tam, K. L. Chan, K. W. Cheah. Efficient Organic Light-Emitting Diode through Triplet Exciton Reharvesting by Employing Blended Electron Donor and Acceptor as the Emissive Layer. ACS Appl. Mater. Interfaces, 2015. [DOI | PubMed]
- D. Zhang, C. Zhao, Y. Zhang, X. Song, P. Wei, M. Cai, L. Duan. Highly Efficient Full-Color Thermally Activated Delayed Fluorescent Organic Light-Emitting Diodes: Extremely Low Efficiency Roll-Off Utilizing a Host with Small Singlet-Triplet Splitting. ACS Appl. Mater. Interfaces, 2017. [DOI | PubMed]
- S. W. Li, C. H. Yu, C. L. Ko, T. Chatterjee, W. Y. Hung, K. T. Wong. Cyanopyrimidine-Carbazole Hybrid Host Materials for High-Efficiency and Low-Efficiency Roll-Off TADF OLEDs. ACS Appl. Mater. Interfaces, 2018. [DOI | PubMed]
- T. Nishimoto, T. Yasuda, S. Y. Lee, R. Kondo, C. Adachi. A Six-Carbazole-Decorated Cyclophosphazene as a Host with High Triplet Energy to Realize Efficient Delayed-Fluorescence OLEDs. Mater. Horiz., 2014. [DOI]
- C.-H. Chen, S.-C. Lin, B.-Y. Lin, C.-Y. Li, Y.-C. Kong, Y.-S. Chen, S.-C. Fang, C.-H. Chiu, J.-H. Lee, K.-T. Wong. New Bipolar Host Materials for High Power Efficiency Green Thermally Activated Delayed Fluorescence OLEDs. Chem. Eng. J., 2022. [DOI]
- D. Zhou, D. Liu, X. Gong, H. Ma, G. Qian, S. Gong, G. Xie, W. Zhu, Y. Wang. Solution-Processed Highly Efficient Bluish-Green Thermally Activated Delayed Fluorescence Emitter Bearing an Asymmetric Oxadiazole-Difluoroboron Double Acceptor. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- J. Hu, X. Zhang, D. Zhang, X. Cao, T. Jiang, X. Zhang, Y. Tao. Linkage Modes on Phthaloyl/Triphenylamine Hybrid Compounds: Multi-Functional AIE Luminogens, Non-Doped Emitters and Organic Hosts for Highly Efficient Solution-Processed Delayed Fluorescence OLEDs. Dyes Pigm., 2017. [DOI]
- C. Tang, T. Yang, X. Cao, Y. Tao, F. Wang, C. Zhong, Y. Qian, X. Zhang, W. Huang. Tuning a Weak Emissive Blue Host to Highly Efficient Green Dopant by a Cn in Tetracarbazolepyridines for Solution-Processed Thermally Activated Delayed Fluorescence Devices. Adv. Optical Mater., 2015. [DOI]
- X. Ban, A. Zhu, T. Zhang, Z. Tong, W. Jiang, Y. Sun. Design of Encapsulated Host and Guest for Highly Efficient Blue and Green Thermally Activated Delayed Fluorescence OLEDs Based on Solution-Process. Chem. Commun., 2017. [DOI]
- J.-M. Lehn. Supramolecular ChemistryScope and Perspectives Molecules, Supermolecules, and Molecular Devices(Nobel Lecture). Angew. Chem., Int. Ed., 1988. [DOI]
- J. F. Stoddart. Mechanically Interlocked Molecules (Mims)-Molecular Shuttles, Switches, and Machines (Nobel Lecture). Angew. Chem., Int. Ed., 2017. [DOI]
- H. Chen, J. Fraser Stoddart. From Molecular to Supramolecular Electronics. Nat. Rev. Mater., 2021. [DOI]
- I. V. Kolesnichenko, E. V. Anslyn. Practical Applications of Supramolecular Chemistry. Chem. Soc. Rev., 2017. [DOI | PubMed]
- D. B. Amabilino, P. R. Ashton, A. S. Reder, N. Spencer, J. F. Stoddart. Olympiadane. Angew. Chem., Int. Ed., 1994. [DOI]
- P. Rajamalli, D. R. Martir, E. Zysman-Colman. Molecular Design Strategy for a Two-Component Gel Based on a Thermally Activated Delayed Fluorescence Emitter. ACS Appl. Energy Mater., 2018. [DOI]
- D. R. Martir, A. Pizzolante, D. Escudero, D. Jacquemin, S. L. Warriner, E. Zysman-Colman. Photoinduced Energy and Electron Transfer between a Photoactive Cage Based on a Thermally Activate Delayed Fluorescence Ligand and Encapsulated Fluorescent Dyes. ACS Appl. Energy Mater., 2018. [DOI]
- K. Suzuki, M. Tominaga, M. Kawano, M. Fujita. Self-Assembly of an M6l12 Coordination Cube. Chem. Commun., 2009. [DOI]
- P. Rajamalli, F. Rizzi, W. Li, M. A. Jinks, A. K. Gupta, B. A. Laidlaw, I. D. W. Samuel, T. J. Penfold, S. M. Goldup, E. Zysman-Colman. Using the Mechanical Bond to Tune the Performance of a Thermally Activated Delayed Fluorescence Emitter*. Angew. Chem., Int. Ed., 2021. [DOI]
- R. Haldar, M. Jakoby, M. Kozlowska, M. Rahman Khan, H. Chen, Y. Pramudya, B. S. Richards, L. Heinke, W. Wenzel, F. Odobel. Tuning Optical Properties by Controlled Aggregation: Electroluminescence Assisted by Thermally-Activated Delayed Fluorescence from Thin Films of Crystalline Chromophores. Chem. Eur. J., 2020. [DOI | PubMed]
- M. Gutiérrez, C. Martín, J. Hofkens, J.-C. Tan. Long-Lived Highly Emissive Mofs as Potential Candidates for Multiphotonic Applications. J. Mater. Chem. C, 2021. [DOI]
- B. Bie, L. Guo, M. Zhang, Y. Ma, C. Yang. Metal-Organic Framework Based Thermally Activated Delayed Fluorescence Emitter with Oxygen-Insensitivity for Cell Imaging. Adv. Optical Mater., 2022. [DOI]
- M. Hempe, A. K. Harrison, J. S. Ward, A. S. Batsanov, M. A. Fox, F. B. Dias, M. R. Bryce. Cyclophane Molecules Exhibiting Thermally Activated Delayed Fluorescence: Linking Donor Units to Influence Molecular Conformation. J. Org. Chem., 2021. [DOI | PubMed]
- H. Mieno, R. Kabe, M. D. Allendorf, C. Adachi. Thermally Activated Delayed Fluorescence of a Zr-Based Metal-Organic Framework. Chem. Commun., 2018. [DOI]
- X. T. Liu, W. Hua, H. X. Nie, M. Chen, Z. Chang, X. H. Bu. Manipulating Spatial Alignment of Donor and Acceptor in Host-Guest Mof for TADF. Nat. Sci. Rev., 2022. [DOI]
- Y. Hou, Y. Wang, T. Xu, Z. Wang, W. Tian, D. Sun, X. Yu, P. Xing, J. Shen, X. Xin, J. Hao. Synergistic Multiple Bonds Induced Dynamic Self-Assembly of Silver Nanoclusters into Lamellar Frameworks with Tailored Luminescence. Chem. Mater., 2022. [DOI]
- S. Lv, Y. Miao, D. Zheng, X. Li, D. Liu, F. Song. Self-Assembled Platinum Supramolecular Metallacycles Based on a Novel TADF Photosensitizer for Efficient Cancer Photochemotherapy. Mol. Pharm., 2021. [DOI | PubMed]
- D. Chao, M. Zhao. A Supramolecular Assembly Bearing an Organic TADF Chromophore: Synthesis, Characterization and Light-Driven Cooperative Acceptorless Dehydrogenation of Secondary Amines. Dalton Trans., 2019. [DOI | PubMed]
- Y. Tsuchiya, K. Ikesue, H. Nakanotani, C. Adachi. Photostable and Highly Emissive Glassy Organic Dots Exhibiting Thermally Activated Delayed Fluorescence. Chem. Commun., 2019. [DOI]
- T. Li, D. Yang, L. Zhai, S. Wang, B. Zhao, N. Fu, L. Wang, Y. Tao, W. Huang. Thermally Activated Delayed Fluorescence Organic Dots (TADF Odots) for Time-Resolved and Confocal Fluorescence Imaging in Living Cells and in Vivo. Adv. Sci., 2017. [DOI]
- T. He, C. Ren, Z. Li, S. Xiao, J. Li, X. Lin, C. Ye, J. Zhang, L. Guo, W. Hu. Thermally Activated Delayed Fluorescence Organic Dots for Two-Photon Fluorescence Lifetime Imaging. Applied Physics Letters, 2018. [DOI]
- J. Zhang, W. Chen, S. Kalytchuk, K. F. Li, R. Chen, C. Adachi, Z. Chen, A. L. Rogach, G. Zhu, P. K. N. Yu. Self-Assembly of Electron Donor-Acceptor-Based Carbazole Derivatives: Novel Fluorescent Organic Nanoprobes for Both One- and Two-Photon Cellular Imaging. ACS Appl. Mater. Interfaces, 2016. [DOI | PubMed]
- F. Ni, Z. Zhu, X. Tong, M. Xie, Q. Zhao, C. Zhong, Y. Zou, C. Yang. Organic Emitter Integrating Aggregation-Induced Delayed Fluorescence and Room-Temperature Phosphorescence Characteristics, and Its Application in Time-Resolved Luminescence Imaging. Chem. Sci., 2018. [DOI | PubMed]
- S. Qi, S. Kim, V. N. Nguyen, Y. Kim, G. Niu, G. Kim, S. J. Kim, S. Park, J. Yoon. Highly Efficient Aggregation-Induced Red-Emissive Organic Thermally Activated Delayed Fluorescence Materials with Prolonged Fluorescence Lifetime for Time-Resolved Luminescence Bioimaging. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- J. Zhang, R. Chen, Z. Zhu, C. Adachi, X. Zhang, C. S. Lee. Highly Stable near-Infrared Fluorescent Organic Nanoparticles with a Large Stokes Shift for Noninvasive Long-Term Cellular Imaging. ACS Appl. Mater. Interfaces, 2015. [DOI | PubMed]
- S. Gan, J. Zhou, T. A. Smith, H. Su, W. Luo, Y. Hong, Z. Zhao, B. Z. Tang. New Aiegens with Delayed Fluorescence for Fluorescence Imaging and Fluorescence Lifetime Imaging of Living Cells. Mater. Chem. Front., 2017. [DOI]
- P. O. Smith, D. J. Black, R. Pal, J. Avó, F. B. Dias, V. L. Linthwaite, M. J. Cann, L.-O. Pålsson. Applying TADF Emitters in Bioimaging and Sensinga Novel Approach Using Liposomes for Encapsulation and Cellular Uptake. Front. Chem., 2021. [DOI | PubMed]
- Z. Zhu, D. Tian, P. Gao, K. Wang, Y. Li, X. Shu, J. Zhu, Q. Zhao. Cell-Penetrating Peptides Transport Noncovalently Linked Thermally Activated Delayed Fluorescence Nanoparticles for Time-Resolved Luminescence Imaging. J. Am. Chem. Soc., 2018. [DOI | PubMed]
- L. Q. Yan, Z. N. Kong, Y. Xia, Z. J. Qi. A Novel Coumarin-Based Red Fluorogen with AIE, Self-Assembly, and TADF Properties. New J. Chem., 2016. [DOI]
- K. Shizu, H. Tanaka, M. Uejima, T. Sato, K. Tanaka, H. Kaji, C. Adachi. Strategy for Designing Electron Donors for Thermally Activated Delayed Fluorescence Emitters. J. Phys. Chem. C, 2015. [DOI]
- J. Liu, N. Wang, Y. Yu, Y. Yan, H. Zhang, J. Li, J. Yu. Carbon Dots in Zeolites: A New Class of Thermally Activated Delayed Fluorescence Materials with Ultralong Lifetimes. Sci. Adv., 2017. [DOI]
- J. Liu, H. Zhang, N. Wang, Y. Yu, Y. Cui, J. Li, J. Yu. Template-Modulated Afterglow of Carbon Dots in Zeolites: Room-Temperature Phosphorescence and Thermally Activated Delayed Fluorescence. ACS Mater. Lett., 2019. [DOI]
- H. Zhang, J. Liu, B. Wang, K. Liu, G. Chen, X. Yu, J. Li, J. Yu. Zeolite-Confined Carbon Dots: Tuning Thermally Activated Delayed Fluorescence Emission: Via Energy Transfer. Mater. Chem. Front., 2020. [DOI]
- R. K. Koninti, K. Miyata, M. Saigo, K. Onda. Achieving Thermally Activated Delayed Fluorescence from Benzophenone by Host-Guest Complexation. J. Phys. Chem. C, 2021. [DOI]
- G. Huang, Z. Deng, J. Pang, J. Li, S. Ni, J. A. Li, C. Zhou, H. Li, B. Xu, L. Dang, M. D. Li. Long-Range Charge Transportation Induced Organic Host-Guest Dual Color Long Persistent Luminescence. Adv. Optical Mater., 2021. [DOI]
- H. Y. Zhou, D. W. Zhang, M. Li, C. F. Chen. A Calix[3]Acridan-Based Host-Guest Cocrystal Exhibiting Efficient Thermally Activated Delayed Fluorescence. Angew. Chem., Int. Ed., 2022. [DOI]
- N. R. Paisley, C. M. Tonge, Z. M. Hudson. Stimuli-Responsive Thermally Activated Delayed Fluorescence in Polymer Nanoparticles and Thin Films: Applications in Chemical Sensing and Imaging. Front. Chem., 2020. [DOI | PubMed]
- J. C. Fister, D. Rank, J. M. Harris. Delayed Fluorescence Optical Thermometry. Analytical chemistry, 1995. [DOI]
- C. Baleizão, S. Nagl, M. Schäferling, M. N. Berberan-Santos, O. S. Wolfbeis. Dual Fluorescence Sensor for Trace Oxygen and Temperature with Unmatched Range and Sensitivity. Anal. Chem., 2008. [DOI | PubMed]
- S. Nagl, C. Baleizão, S. M. Borisov, M. Schäferling, M. N. Berberan-Santos, O. S. Wolfbeis. Optical Sensing and Imaging of Trace Oxygen with Record Response. Angew. Chem., Int. Ed., 2007. [DOI]
- B. H. Kirsop. Oxygen in Brewery Fermentation. J. Inst. Brew., 1974. [DOI]
- C. Wang, S. Otto, M. Dorn, K. Heinze, U. Resch-Genger. Luminescent Top Nanosensors for Simultaneously Measuring Temperature, Oxygen, and Ph at a Single Excitation Wavelength. Anal. Chem., 2019. [DOI | PubMed]
- P. J. Wagner. Conformational Changes Involved in the Singlet-Triplet Transitions of Biphenyl. J. Am. Chem. Soc., 1967. [DOI]
- C. Baleizao, M. N. Berberan-Santos. Thermally Activated Delayed Fluorescence in Fullerenes. Ann. N. Y. Acad. Sci., 2008. [DOI | PubMed]
- S. Kochmann, C. Baleizao, M. N. Berberan-Santos, O. S. Wolfbeis. Sensing and Imaging of Oxygen with Parts Per Billion Limits of Detection and Based on the Quenching of the Delayed Fluorescence of 13c70 Fullerene in Polymer Hosts. Anal. Chem., 2013. [DOI | PubMed]
- C. A. DeRosa, J. Samonina-Kosicka, Z. Fan, H. C. Hendargo, D. H. Weitzel, G. M. Palmer, C. L. Fraser. Oxygen Sensing Difluoroboron Dinaphthoylmethane Polylactide. Macromolecules, 2015. [DOI | PubMed]
- A. Steinegger, I. Klimant, S. M. Borisov. Purely Organic Dyes with Thermally Activated Delayed Fluorescence-a Versatile Class of Indicators for Optical Temperature Sensing. Adv. Optical Mater., 2017. [DOI]
- S. M. Borisov, G. Nuss, W. Haas, R. Saf, M. Schmuck, I. Klimant. New Nir-Emitting Complexes of Platinum(II) and Palladium(II) with Fluorinated Benzoporphyrins. J. Photochem. Photobiol., A, 2009. [DOI]
- C. M. Tonge, N. R. Paisley, A. M. Polgar, K. Lix, W. R. Algar, Z. M. Hudson. Color-Tunable Thermally Activated Delayed Fluorescence in Oxadiazole-Based Acrylic Copolymers: Photophysical Properties and Applications in Ratiometric Oxygen Sensing. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- C. J. Christopherson, D. M. Mayder, J. Poisson, N. R. Paisley, C. M. Tonge, Z. M. Hudson. 1,8-Naphthalimide-Based Polymers Exhibiting Deep-Red Thermally Activated Delayed Fluorescence and Their Application in Ratiometric Temperature Sensing. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- C. J. Christopherson, Z. S. Hackett, E. R. Sauvé, N. R. Paisley, C. M. Tonge, D. M. Mayder, Z. M. Hudson. Synthesis of Phosphorescent Iridium-Containing Acrylic Monomers and Their Room-Temperature Polymerization by Cu(0)-Rdrp. Journal of Polymer Science Part A: Polymer Chemistry, 2018. [DOI]
- X. Li, G. Baryshnikov, C. Deng, X. Bao, B. Wu, Y. Zhou, H. Ågren, L. Zhu. A Three-Dimensional Ratiometric Sensing Strategy on Unimolecular Fluorescence-Thermally Activated Delayed Fluorescence Dual Emission. Nat. Commun., 2019. [DOI | PubMed]
- H. Yin, Y. Wu, X. Peng, F. Song. A Turn-on TADF Chemosensor for Sulfite with a Microsecond-Scale Luminescence Lifetime. Chem. Commun., 2020. [DOI]
- X. Xiong, F. Song, J. Wang, Y. Zhang, Y. Xue, L. Sun, N. Jiang, P. Gao, L. Tian, X. Peng. Thermally Activated Delayed Fluorescence of Fluorescein Derivative for Time-Resolved and Confocal Fluorescence Imaging. J. Am. Chem. Soc., 2014. [DOI | PubMed]
- X. Xiong, L. Zheng, J. Yan, F. Ye, Y. Qian, F. Song. A Turn-on and Colorimetric Metal-Free Long Lifetime Fluorescent Probe and Its Application for Time-Resolved Luminescent Detection and Bioimaging of Cysteine. RSC Adv., 2015. [DOI]
- Z. Liu, F. Song, B. Song, L. Jiao, J. An, J. Yuan, X. Peng. A Fret Chemosensor for Hypochlorite with Large Stokes Shifts and Long-Lifetime Emissions. Sens. Actuators, B, 2018. [DOI]
- S. Qiu, J. Yu, T. Zhou, K. Zhang, Y. Duan, X. Ban, Q. Zhu, L. Shi, D. Zhang. Thermally Activated Delayed Fluorescence Fluorescent Probe Based on Triazine as Emission Core for Metal Ions Detection. Opt. Mater., 2021. [DOI]
- W. Ma, Y. Su, Q. Zhang, C. Deng, L. Pasquali, W. Zhu, Y. Tian, P. Ran, Z. Chen, G. Yang. Thermally Activated Delayed Fluorescence (TADF) Organic Molecules for Efficient X-Ray Scintillation and Imaging. Nat. Mater., 2022. [DOI | PubMed]
- J.-X. Wang, L. Gutiérrez-Arzaluz, X. Wang, T. He, Y. Zhang, M. Eddaoudi, O. M. Bakr, O. F. Mohammed. Heavy-Atom Engineering of Thermally Activated Delayed Fluorophores for High-Performance X-Ray Imaging Scintillators. Nat. Photonics, 2022. [DOI]
- J.-X. Wang, L. Gutiérrez-Arzaluz, X. Wang, M. Almalki, J. Yin, J. Czaban-Jóźwiak, O. Shekhah, Y. Zhang, O. M. Bakr, M. Eddaoudi, O. F. Mohammed. Nearly 100% Energy Transfer at the Interface of Metal-Organic Frameworks for X-Ray Imaging Scintillators. Matter, 2022. [DOI]
- S. Abraham, C. Fuentes-Hernandez, S. Mukhopadhyay, K. Singh, H. N. Kim, O. Moreno, C. M. Tran, D. R. Kumar, J. C. Stooksbury, S. R. Kalidindi. An Approach Towards Plastic Scintillators from Thermally Activated Delayed Fluorescent Dyes and Cross-Linkable Bismuth Compounds. J. Mater. Chem. C, 2022. [DOI]
- Y. Yang, Q. Zhao, W. Feng, F. Li. Luminescent Chemodosimeters for Bioimaging. Chem. Rev., 2013. [DOI | PubMed]
- P. Sharma, S. Brown, G. Walter, S. Santra, B. Moudgil. Nanoparticles for Bioimaging. Adv. Colloid Interface Sci., 2006. [DOI | PubMed]
- O. S. Wolfbeis. An Overview of Nanoparticles Commonly Used in Fluorescent Bioimaging. Chem. Soc. Rev., 2015. [DOI | PubMed]
- R. Guan, L. Xie, T. W. Rees, L. Ji, H. Chao. Metal Complexes for Mitochondrial Bioimaging. J. Inorg. Biochem., 2020. [DOI | PubMed]
- X. Zhen, R. Qu, W. Chen, W. Wu, X. Jiang. The Development of Phosphorescent Probes for in Vitro and in Vivo Bioimaging. Biomater. Sci., 2021. [DOI | PubMed]
- Q. Zhao, C. Huang, F. Li. Phosphorescent Heavy-Metal Complexes for Bioimaging. Chem. Soc. Rev., 2011. [DOI | PubMed]
- Q. Liu, B. Yin, T. Yang, Y. Yang, Z. Shen, P. Yao, F. Li. A General Strategy for Biocompatible, High-Effective Upconversion Nanocapsules Based on Triplet-Triplet Annihilation. J. Am. Chem. Soc., 2013. [DOI | PubMed]
- A. Ogunsipe, T. Nyokong. Photophysicochemical Consequences of Bovine Serum Albumin Binding to Non-Transition Metal Phthalocyanine Sulfonates. Photochem. Photobiol. Sci., 2005. [DOI | PubMed]
- A. Jahanban-Esfahlan, A. Ostadrahimi, R. Jahanban-Esfahlan, L. Roufegarinejad, M. Tabibiazar, R. Amarowicz. Recent Developments in the Detection of Bovine Serum Albumin. Int. J. Biol. Macromol., 2019. [DOI | PubMed]
- Y. Wu, F. Song, W. Luo, Z. Liu, B. Song, X. Peng. Enhanced Thermally Activated Delayed Fluorescence in New Fluorescein Derivatives by Introducing Aromatic Carbonyl Groups. ChemPhotoChem, 2017. [DOI]
- X. Xu, R. Liu, L. Li. Nanoparticles Made of Π-Conjugated Compounds Targeted for Chemical and Biological Applications. Chem. Commun., 2015. [DOI]
- B. Bao, M. Ma, H. Zai, L. Zhang, N. Fu, W. Huang, L. Wang. Conjugated Polymer Nanoparticles for Label-Free and Bioconjugate-Recognized DNA Sensing in Serum. Adv. Sci., 2015. [DOI]
- C. Wu, D. T. Chiu. Highly Fluorescent Semiconducting Polymer Dots for Biology and Medicine. Angew. Chem., Int. Ed., 2013. [DOI]
- K. Petkau, A. Kaeser, I. Fischer, L. Brunsveld, A. P. Schenning. Pre-and Postfunctionalized Self-Assembled Π-Conjugated Fluorescent Organic Nanoparticles for Dual Targeting. J. Am. Chem. Soc., 2011. [DOI | PubMed]
- D. Horn, J. Rieger. Organic Nanoparticles in the Aqueous PhaseTheory, Experiment, and Use. Angew. Chem., Int. Ed., 2001. [DOI]
- F. Fang, M. Li, J. Zhang, C.-S. Lee. Different Strategies for Organic Nanoparticle Preparation in Biomedicine. ACS Mater. Lett., 2020. [DOI]
- C. Wu, C. Szymanski, Z. Cain, J. McNeill. Conjugated Polymer Dots for Multiphoton Fluorescence Imaging. J. Am. Chem. Soc., 2007. [DOI | PubMed]
- L. Feng, C. Zhu, H. Yuan, L. Liu, F. Lv, S. Wang. Conjugated Polymer Nanoparticles: Preparation, Properties, Functionalization and Biological Applications. Chem. Soc. Rev., 2013. [DOI | PubMed]
- T. Li, D. Yang, L. Zhai, S. Wang, B. Zhao, N. Fu, L. Wang, Y. Tao, W. Huang. Thermally Activated Delayed Fluorescence Organic Dots (TADF Odots) for Time-Resolved and Confocal Fluorescence Imaging in Living Cells and in Vivo. Adv. Sci., 2017. [DOI]
- H. Shi, L. Zou, K. Huang, H. Wang, C. Sun, S. Wang, H. Ma, Y. He, J. Wang, H. Yu. A Highly Efficient Red Metal-Free Organic Phosphor for Time-Resolved Luminescence Imaging and Photodynamic Therapy. ACS Appl. Mater. Interfaces, 2019. [DOI | PubMed]
- S. Chen, H. Wang, Y. Hong, B. Z. Tang. Fabrication of Fluorescent Nanoparticles Based on AIE Luminogens (AIE Dots) and Their Applications in Bioimaging. Mater. Horiz., 2016. [DOI]
- W. Zhao, H. Wei, F. Liu, C. Ran. Glucose Ligand Modififed Thermally Activated Delayed Fluorescence Targeted Nanoprobe for Malignant Cells Imaging Diagnosis. Photodiagnosis Photodyn. Ther., 2020. [DOI | PubMed]
- Y.-F. Xiao, J.-X. Chen, S. Li, W.-W. Tao, S. Tian, K. Wang, X. Cui, Z. Huang, X.-H. Zhang, C.-S. Lee. Manipulating Exciton Dynamics of Thermally Activated Delayed Fluorescence Materials for Tuning Two-Photon Nanotheranostics. Chem. Sci., 2020. [DOI]
- C. I. Crucho, J. Avó, A. M. Diniz, S. N. Pinto, J. Barbosa, P. O. Smith, M. N. Berberan-Santos, L.-O. Pålsson, F. B. Dias. TADF Dye-Loaded Nanoparticles for Fluorescence Live-Cell Imaging. Front. Chem., 2020. [DOI | PubMed]
- M. Luo, X. Li, L. Ding, G. Baryshnikov, S. Shen, M. Zhu, L. Zhou, M. Zhang, J. Lu, H. Ågren. Integrating Time-Resolved Imaging Information by Single-Luminophore Dual Thermally Activated Delayed Fluorescence. Angew. Chem., Int. Ed., 2020. [DOI]
- X. Li, G. Baryshnikov, L. Ding, X. Bao, X. Li, J. Lu, M. Liu, S. Shen, M. Luo, M. Zhang. Dual-Phase Thermally Activated Delayed Fluorescence Luminogens: A Material for Time-Resolved Imaging Independent of Probe Pretreatment and Probe Concentration. Angew. Chem., Int. Ed., 2020. [DOI]
- D. M. Mayder, R. Hojo, W. L. Primrose, C. M. Tonge, Z. M. Hudson. Heptazine-Based TADF Materials for Nanoparticle-Based Nonlinear Optical Bioimaging. Adv. Funct. Mater., 2022. [DOI]
- D. M. Mayder, C. J. Christopherson, W. L. Primrose, A. S. Lin, Z. M. Hudson. Polymer Dots and Glassy Organic Dots Using Dibenzodipyridophenazine Dyes as Water-Dispersible TADF Probes for Cellular Imaging. Journal of Materials Chemistry B, 2022. [DOI | PubMed]
- N. R. Paisley, S. V. Halldorson, M. V. Tran, R. Gupta, S. Kamal, W. R. Algar, Z. M. Hudson. Near-Infrared-Emitting Boron-Difluoride-Curcuminoid-Based Polymers Exhibiting Thermally Activated Delayed Fluorescence as Biological Imaging Probes. Angew. Chem., Int. Ed., 2021. [DOI]
- K.-F. Hsu, S.-P. Su, H.-F. Lu, M.-H. Liu, Y. J. Chang, Y.-J. Lee, H. K. Chiang, C.-P. Hsu, C.-W. Lu, Y.-H. Chan. TADF-Based Nir-Ii Semiconducting Polymer Dots for in Vivo 3d Bone Imaging. Chemical Science, 2022. [DOI | PubMed]
- J. Peng, J. Li, W. Xu, L. Wang, D. Su, C. L. Teoh, Y.-T. Chang. Silica Nanoparticle-Enhanced Fluorescent Sensor Array for Heavy Metal Ions Detection in Colloid Solution. Anal. Chem., 2018. [DOI | PubMed]
- C. Zong, K. Ai, G. Zhang, H. Li, L. Lu. Dual-Emission Fluorescent Silica Nanoparticle-Based Probe for Ultrasensitive Detection of Cu2+. Anal. Chem., 2011. [DOI | PubMed]
- C. I. Crucho, J. Avó, R. Nobuyasu, S. N. Pinto, F. Fernandes, J. C. Lima, M. N. Berberan-Santos, F. B. Dias. Silica Nanoparticles with Thermally Activated Delayed Fluorescence for Live Cell Imaging. Mater. Sci. Eng. C, 2020. [DOI]
- W. Zeng, H. Y. Lai, W. K. Lee, M. Jiao, Y. J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma. Achieving Nearly 30% External Quantum Efficiency for Orange-Red Organic Light Emitting Diodes by Employing Thermally Activated Delayed Fluorescence Emitters Composed of 1, 8-Naphthalimide-Acridine Hybrids. Adv. Mater., 2018. [DOI]
- F. Ni, Z. Wu, Z. Zhu, T. Chen, K. Wu, C. Zhong, K. An, D. Wei, D. Ma, C. Yang. Teaching an Old Acceptor New Tricks: Rationally Employing 2, 1, 3-Benzothiadiazole as Input to Design a Highly Efficient Red Thermally Activated Delayed Fluorescence Emitter. J. Mater. Chem. C, 2017. [DOI]
- L. Mo, X. Xu, Z. Liu, H. Liu, B. Lei, J. Zhuang, Z. Guo, Y. Liu, C. Hu. Visible-Light Excitable Thermally Activated Delayed Fluorescence in Aqueous Solution from F, N-Doped Carbon Dots Confined in Silica Nanoparticles. Chem. Eng. J., 2021. [DOI]
- A. Jana, K. S. P. Devi, T. K. Maiti, N. P. Singh. Perylene-3-Ylmethanol: Fluorescent Organic Nanoparticles as a Single-Component Photoresponsive Nanocarrier with Real-Time Monitoring of Anticancer Drug Release. J. Am. Chem. Soc., 2012. [DOI | PubMed]
- A. Jana, K. T. Nguyen, X. Li, P. Zhu, N. S. Tan, H. Ågren, Y. Zhao. Perylene-Derived Single-Component Organic Nanoparticles with Tunable Emission: Efficient Anticancer Drug Carriers with Real-Time Monitoring of Drug Release. ACS Nano, 2014. [DOI | PubMed]
- E. Génin, Z. Gao, J. A. Varela, J. Daniel, T. Bsaibess, I. Gosse, L. Groc, L. Cognet, M. Blanchard-Desce. “Hyper-Bright” near-Infrared Emitting Fluorescent Organic Nanoparticles for Single Particle Tracking. Adv. Mater., 2014. [DOI | PubMed]
- J. Zhang, W. Chen, S. Kalytchuk, K. F. Li, R. Chen, C. Adachi, Z. Chen, A. L. Rogach, G. Zhu, P. K. Yu. Self-Assembly of Electron Donor-Acceptor-Based Carbazole Derivatives: Novel Fluorescent Organic Nanoprobes for Both One-and Two-Photon Cellular Imaging. ACS Appl. Mater. Interfaces, 2016. [DOI | PubMed]
- M. Montalti, L. Prodi, E. Rampazzo, N. Zaccheroni. Dye-Doped Silica Nanoparticles as Luminescent Organized Systems for Nanomedicine. Chem. Soc. Rev., 2014. [DOI | PubMed]
- R. Wei, L. Zhang, S. Xu, Q. Zhang, Y. Qi, H. Y. Hu. A Single Component Self-Assembled Thermally Activated Delayed Fluorescence Nanoprobe. Chem. Commun., 2020. [DOI]
- C. I. C. Crucho, C. Baleizao, J. P. S. Farinha. Functional Group Coverage and Conversion Quantification in Nanostructured Silica by H-1 Nmr. Anal. Chem., 2017. [DOI | PubMed]
- W. Hu, L. Guo, L. Bai, X. Miao, Y. Ni, Q. Wang, H. Zhao, M. Xie, L. Li, X. Lu. Maximizing Aggregation of Organic Fluorophores to Prolong Fluorescence Lifetime for Two-Photon Fluorescence Lifetime Imaging. Adv. Healthc. Mater., 2018. [DOI]
- C. I. Crucho, J. Avó, R. Nobuyasu, S. N. Pinto, F. Fernandes, J. C. Lima, M. N. Berberan-Santos, F. B. Dias. Silica Nanoparticles with Thermally Activated Delayed Fluorescence for Live Cell Imaging. Mater. Sci. Eng., C, 2020. [DOI]
- S. Qi, S. Kim, V.-N. Nguyen, Y. Kim, G. Niu, G. Kim, S.-J. Kim, S. Park, J. Yoon. Highly Efficient Aggregation-Induced Red-Emissive Organic Thermally Activated Delayed Fluorescence Materials with Prolonged Fluorescence Lifetime for Time-Resolved Luminescence Bioimaging. ACS Appl. Mater. Interfaces, 2020. [DOI | PubMed]
- B. Zhao, H. Wang, M. Xie, C. Han, H. Yang, W. Zhao, Q. Zhao, H. Xu. Phosphine Oxides Manipulate Aggregation-Induced Delayed Fluorescence for Time-Resolved Bioimaging. Advanced Photonics Research, 2021. [DOI]
- S. K. Sarkar, M. Pegu, S. K. Behera, S. K. Narra, P. Thilagar. Aggregation-Induced and Polymorphism-Dependent Thermally Activated Delayed Fluorescence (TADF) Characteristics of an Oligothiophene: Applications in Time-Dependent Live Cell Multicolour Imaging. Chem. Asian J., 2019. [DOI | PubMed]
- F. Ni, Z. Zhu, X. Tong, W. Zeng, K. An, D. Wei, S. Gong, Q. Zhao, X. Zhou, C. Yang. Hydrophilic, Red-Emitting, and Thermally Activated Delayed Fluorescence Emitter for Time-Resolved Luminescence Imaging by Mitochondrion-Induced Aggregation in Living Cells. Adv. Sci., 2019. [DOI]
- C. J. Christopherson, N. R. Paisley, Z. Xiao, W. R. Algar, Z. M. Hudson. Red-Emissive Cell-Penetrating Polymer Dots Exhibiting Thermally Activated Delayed Fluorescence for Cellular Imaging. J. Am. Chem. Soc., 2021. [DOI | PubMed]
- M. Yu, W. Zhao, F. Ni, Q. Zhao, C. Yang. Photoswitchable Thermally Activated Delayed Fluorescence Nanoparticles for “Double-Check” Confocal and Time-Resolved Luminescence Bioimaging. Adv. Optical Mater., 2022. [DOI]
- I. D. W. Samuel, G. A. Turnbull. Organic Semiconductor Lasers. Chem. Rev., 2007. [DOI | PubMed]
- A. J. C. Kuehne, M. C. Gather. Organic Lasers: Recent Developments on Materials, Device Geometries, and Fabrication Techniques. Chem. Rev., 2016. [DOI | PubMed]
- A. S. D. Sandanayaka, T. Matsushima, F. Bencheikh, S. Terakawa, W. J. Potscavage, C. Qin, T. Fujihara, K. Goushi, J.-C. Ribierre, C. Adachi. Indication of Current-Injection Lasing from an Organic Semiconductor. Appl. Phys. Express, 2019. [DOI]
- Y. Setoguchi, C. Adachi. Suppression of Roll-Off Characteristics of Electroluminescence at High Current Densities in Organic Light Emitting Diodes by Introducing Reduced Carrier Injection Barriers. J. Appl. Phys., 2010. [DOI]
- K. Hayashi, H. Nakanotani, M. Inoue, K. Yoshida, O. Mikhnenko, T.-Q. Nguyen, C. Adachi. Suppression of Roll-Off Characteristics of Organic Light-Emitting Diodes by Narrowing Current Injection/Transport Area to 50 Nm. Appl. Phys. Lett., 2015. [DOI]
- A. S. D. Sandanayaka, L. Zhao, D. Pitrat, J.-C. Mulatier, T. Matsushima, C. Andraud, J.-H. Kim, J.-C. Ribierre, C. Adachi. Improvement of the Quasi-Continuous-Wave Lasing Properties in Organic Semiconductor Lasers Using Oxygen as Triplet Quencher. Appl. Phys. Lett., 2016. [DOI]
- B. S. B. Karunathilaka, U. Balijapalli, C. A. M. Senevirathne, S. Yoshida, Y. Esaki, K. Goushi, T. Matsushima, A. S. D. Sandanayaka, C. Adachi. Suppression of External Quantum Efficiency Rolloff in Organic Light Emitting Diodes by Scavenging Triplet Excitons. Nat. Commun., 2020. [DOI | PubMed]
- V. T. N. Mai, V. Ahmad, M. Mamada, T. Fukunaga, A. Shukla, J. Sobus, G. Krishnan, E. G. Moore, G. G. Andersson, C. Adachi. Solid Cyclooctatetraene-Based Triplet Quencher Demonstrating Excellent Suppression of Singlet-Triplet Annihilation in Optical and Electrical Excitation. Nat. Commun., 2020. [DOI | PubMed]
- Y. Cha, S. Li, Z. Feng, R. Zhu, H. Fu, Z. Yu. Organic Phosphorescence Lasing Based on a Thermally Activated Delayed Fluorescence Emitter. J. Phys. Chem. Lett, 2022. [DOI | PubMed]
- H. Nakanotani, T. Furukawa, T. Hosokai, T. Hatakeyama, C. Adachi. Light Amplification in Molecules Exhibiting Thermally Activated Delayed Fluorescence. Adv. Optical Mater., 2017. [DOI]
- S. Lin, Q. Ou, Z. Shuai. Computational Selection of Thermally Activated Delayed Fluorescence (TADF) Molecules with Promising Electrically Pumped Lasing Property. ACS Mater. Lett., 2022. [DOI]
- A. Khan, X. Tang, C. Zhong, Q. Wang, S. Y. Yang, F. C. Kong, S. Yuan, A. S. D. Sandanayaka, C. Adachi, Z. Q. Jiang, L. S. Liao. Intramolecular-Locked High Efficiency Ultrapure Violet-Blue (Cie-Y <0.046) Thermally Activated Delayed Fluorescence Emitters Exhibiting Amplified Spontaneous Emission. Adv. Funct. Mater., 2021. [DOI]
- J.-J. Wu, X.-D. Wang, L.-S. Liao. Near-Infrared Solid-State Lasers Based on Small Organic Molecules. ACS Photonics, 2019. [DOI]
- R. Aoki, R. Komatsu, K. Goushi, M. Mamada, S. Y. Ko, J. W. Wu, V. Placide, A. D’Aléo, C. Adachi. Realizing near-Infrared Laser Dyes through a Shift In excited-State Absorption. Adv. Optical Mater., 2021. [DOI]
- H. Nakanotani, T. Furukawa, C. Adachi. Light Amplification in an Organic Solid-State Film with the Aid of Triplet-to-Singlet Upconversion. Adv. Optical Mater., 2015. [DOI]
- T. Liu, S. Li, W. Wang, Y. Liu, H. Du, Y. Guo, Q. Shi, D. Zhang, L. Zhao, Q. Fan. Reduced Optically Pumped Amplified Spontaneous Emission Threshold of Bubd-1 Thin Films by Thermally Activated Delayed Fluorescent Materials. J. Lumin., 2019. [DOI]
- A. Shukla, S. K. McGregor, R. Wawrzinek, S. Saggar, E. G. Moore, S. C. Lo, E. B. Namdas. Light Amplification and Efficient Electroluminescence from a Solution-Processable Diketopyrrolopyrrole Derivative Via Triplet-to-Singlet Upconversion. Adv. Funct. Mater., 2021. [DOI]
- H. Huang, Z. Yu, D. Zhou, S. Li, L. Fu, Y. Wu, C. Gu, Q. Liao, H. Fu. Wavelength-Turnable Organic Microring Laser Arrays from Thermally Activated Delayed Fluorescent Emitters. ACS Photonics, 2019. [DOI]
- Z. Zhou, C. Qiao, K. Wang, L. Wang, J. Liang, Q. Peng, Z. Wei, H. Dong, C. Zhang, Z. Shuai. Experimentally Observed Reverse Intersystem Crossing-Boosted Lasing. Angew. Chem., Int. Ed., 2020. [DOI]
- Y. Li, K. Wang, Q. Liao, L. Fu, C. Gu, Z. Yu, H. Fu. Tunable Triplet-Mediated Multicolor Lasing from Nondoped Organic TADF Microcrystals. Nano Lett., 2021. [DOI | PubMed]
- I. D. W. Samuel, E. B. Namdas, G. A. Turnbull. How to Recognize Lasing. Nat. Photonics, 2009. [DOI]
- S. Li, J. Chen, Y. Wei, J. De, H. Geng, Q. Liao, R. Chen, H. Fu. An Organic Laser Based on Thermally Activated Delayed Fluorescence with Aggregation-Induced Emission and Local Excited State Characteristics. Angew. Chem., Int. Ed., 2022. [DOI]
- J. Luo, J. Zhang. Donor-Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(Sp3)-C(Sp2) Cross-Coupling. ACS Catal., 2016. [DOI]
- I. Roger, M. A. Shipman, M. D. Symes. Earth-Abundant Catalysts for Electrochemical and Photoelectrochemical Water Splitting. Nat. Rev. Chem., 2017. [DOI]
- M. H. Shaw, J. Twilton, D. W. MacMillan. Photoredox Catalysis in Organic Chemistry. J. Org. Chem., 2016. [DOI | PubMed]
- F. Glaser, O. S. Wenger. Recent Progress in the Development of Transition-Metal Based Photoredox Catalysts. Coord. Chem. Rev., 2020. [DOI]
- C. S. Wang, P. H. Dixneuf, J. F. Soule. Photoredox Catalysis for Building C-C Bonds from C(Sp(2))-H Bonds. Chem. Rev., 2018. [DOI | PubMed]
- N. Fajrina, M. Tahir. A Critical Review in Strategies to Improve Photocatalytic Water Splitting Towards Hydrogen Production. Int. J. Hydrogen Energy, 2019. [DOI]
- M. M. Mahlambi, C. J. Ngila, B. B. Mamba. Recent Developments in Environmental Photocatalytic Degradation of Organic Pollutants: The Case of Titanium Dioxide Nanoparticlesa Review. J. Nanomater., 2015. [DOI]
- C. K. Prier, D. A. Rankic, D. W. MacMillan. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev., 2013. [DOI | PubMed]
- J.-H. Shon, T. S. Teets. Photocatalysis with Transition Metal Based Photosensitizers. Comments Inorg. Chem., 2020. [DOI]
- S. Kahng, H. Yoo, J. H. Kim. Recent Advances in Earth-Abundant Photocatalyst Materials for Solar H2 Production. Advanced Powder Technology, 2020. [DOI]
- B. M. Hockin, C. Li, N. Robertson, E. Zysman-Colman. Photoredox Catalysts Based on Earth-Abundant Metal Complexes. Catal. Sci. Tech., 2019. [DOI]
- N. A. Romero, D. A. Nicewicz. Organic Photoredox Catalysis. Chem. Rev., 2016. [DOI | PubMed]
- D. M. Hedstrand, W. H. Kruizinga, R. M. Kellogg. Light Induced and Dye Accelerated Reductions of Phenacyl Oniium Salts by 1,4-Dihydropyridines. Tetrahedron Lett., 1978. [DOI]
- T. Y. Shang, L. H. Lu, Z. Cao, Y. Liu, W. M. He, B. Yu. Recent Advances of 1,2,3,5-Tetrakis(Carbazol-9-Yl)-4,6-Dicyanobenzene (4czipn) in Photocatalytic Transformations. Chem. Commun., 2019. [DOI]
- E. R. Sauve, D. M. Mayder, S. Kamal, M. S. Oderinde, Z. M. Hudson. An Imidazoacridine-Based TADF Material as an Effective Organic Photosensitizer for Visible-Light-Promoted [2 + 2] Cycloaddition. Chem. Sci., 2022. [DOI | PubMed]
- M. A. Bryden, F. Millward, T. Matulaitis, D. Chen, M. Villa, A. Fermi, S. Cetin, P. Ceroni, E. Zysman-Colman. Moving Beyond Cyanoarene Thermally Activated Delayed Fluorescence Compounds as Photocatalysts: An Assessment of the Performance of a Pyrimidyl Sulfone Photocatalyst in Comparison to 4czipn. J. Org. Chem., 2023. [DOI | PubMed]
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel, L. S. Park. Highly Efficient Organic Photocatalysts Discovered Via a Computer-Aided-Design Strategy for Visible-Light-Driven Atom Transfer Radical Polymerization. Nat. Catal., 2018. [DOI]
- E. Speckmeier, T. G. Fischer, K. Zeitler. A Toolbox Approach to Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes. J. Am. Chem. Soc., 2018. [DOI | PubMed]
- E. A. Martynova, V. A. Voloshkin, S. G. Guillet, F. Bru, M. Belis, K. Van Hecke, C. S. J. Cazin, S. P. Nolan. Energy Transfer (Ent) Photocatalysis Enabled by Gold-N-Heterocyclic Carbene (NHC) Complexes. Chem. Sci., 2022. [DOI | PubMed]
- C. Li, R. Dickson, N. Rockstroh, J. Rabeah, D. B. Cordes, A. M. Z. Slawin, P. Hünemörder, A. Spannenberg, M. Bühl, E. Mejía. Ligand Electronic Fine-Tuning and Its Repercussion on the Photocatalytic Activity and Mechanistic Pathways of the Copper-Photocatalysed Aza-Henry Reaction. Catal. Sci. Tech., 2020. [DOI]
- M. Bouzrati-Zerelli, N. Guillaume, F. Goubard, T.-T. Bui, S. Villotte, C. Dietlin, F. Morlet-Savary, D. Gigmes, J. P. Fouassier, F. Dumur, J. Lalevée. A Novel Class of Photoinitiators with a Thermally Activated Delayed Fluorescence (TADF) Property. New J. Chem., 2018. [DOI]
- C. Prentice, J. Morrison, A. D. Smith, E. Zysman-Colman. Multi-Resonant Thermally Activated Delayed Fluorescent (MR-TADF) Compounds as Photocatalysts. Chem. Eur. J., 2023. [DOI | PubMed]
- C. Prentice, J. Morrison, E. Zysman-Colman, A. D. Smith. Dual NHC/Photoredox Catalytic Synthesis of 1,4-Diketones Using an MR-TADF Photocatalyst (DiKTa). Chem. Commun., 2022. [DOI]
- P. P. Singh, V. Srivastava. Recent Advances in Using 4dpaipn in Photocatalytic Transformations. Org. Biomol. Chem., 2021. [DOI | PubMed]
- Z. Huang, Y. Gu, X. Liu, L. Zhang, Z. Cheng, X. Zhu. Metal-Free Atom Transfer Radical Polymerization of Methyl Methacrylate with Ppm Level of Organic Photocatalyst. Macromol. Rapid Commun., 2017. [DOI]
- Z. Huang, L. Zhang, Z. Cheng, X. Zhu. Reversible Addition-Fragmentation Chain Transfer Polymerization of Acrylonitrile under Irradiation of Blue Led Light. Polymers, 2017. [DOI]
- X. C. Liu, X. L. Chen, Y. Liu, K. Sun, Y. Y. Peng, L. B. Qu, B. Yu. Visible-Light-Induced Metal-Free Synthesis of 2-Phosphorylated Thioflavones in Water. ChemSusChem, 2020. [DOI | PubMed]
- W. J. Zhou, Z. H. Wang, L. L. Liao, Y. X. Jiang, K. G. Cao, T. Ju, Y. Li, G. M. Cao, D. G. Yu. Reductive Dearomative Arylcarboxylation of Indoles with Co2 Via Visible-Light Photoredox Catalysis. Nat. Commun., 2020. [DOI | PubMed]
- H. Huang, C. Yu, Y. Zhang, Y. Zhang, P. S. Mariano, W. Wang. Chemo- and Regioselective Organo-Photoredox Catalyzed Hydroformylation of Styrenes Via a Radical Pathway. J. Am. Chem. Soc., 2017. [DOI | PubMed]
- O. Zhang, J. W. Schubert. Derivatization of Amino Acids and Peptides Via Photoredox-Mediated Conjugate Addition. J. Org. Chem., 2020. [DOI | PubMed]
- J. K. Matsui, D. N. Primer, G. A. Molander. Metal-Free C-H Alkylation of Heteroarenes with Alkyltrifluoroborates: A General Protocol for 1 Degrees, 2 Degrees and 3 Degrees Alkylation. Chem. Sci., 2017. [DOI | PubMed]
- X. Wang, M. Yang, W. Xie, X. Fan, J. Wu. Photoredox-Catalyzed Hydrosulfonylation Reaction of Electron-Deficient Alkenes with Substituted Hantzsch Esters and Sulfur Dioxide. Chem. Commun., 2019. [DOI]
- L. M. Reid, T. Li, Y. Cao, C. P. Berlinguette. Organic Chemistry at Anodes and Photoanodes. Sustain. Energy Fuels, 2018. [DOI]
- N. X. Xu, B. X. Li, C. Wang, M. Uchiyama. Sila- and Germacarboxylic Acids: Precursors for the Corresponding Silyl and Germyl Radicals. Angew. Chem., Int. Ed., 2020. [DOI]
- J. Xu, J. Cao, X. Wu, H. Wang, X. Yang, X. Tang, R. W. Toh, R. Zhou, E. K. L. Yeow, J. Wu. Unveiling Extreme Photoreduction Potentials of Donor-Acceptor Cyanoarenes to Access Aryl Radicals from Aryl Chlorides. J. Am. Chem. Soc., 2021. [DOI | PubMed]
- H. Kim, H. Kim, T. H. Lambert, S. Lin. Reductive Electrophotocatalysis: Merging Electricity and Light to Achieve Extreme Reduction Potentials. J. Am. Chem. Soc., 2020. [DOI | PubMed]
- J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang, J. Zhang. Donor-Acceptor Fluorophores for Energy-Transfer-Mediated Photocatalysis. J. Am. Chem. Soc., 2018. [DOI | PubMed]
- W. C. de Souza, J. T. M. Correia, P. M. Matos, C. M. Kisukuri, P. S. Carneiro, M. W. Paixão. Organophotocatalytic Intramolecular Formal Enyne-Metathesis – an Alternative to Transition-Metal Catalysis. Eur. J. Org. Chem., 2022. [DOI]
- A. J. Carrod, V. Gray, K. Börjesson. Recent Advances in Triplet-Triplet Annihilation Upconversion and Singlet Fission, Towards Solar Energy Applications. Energy Environ. Sci., 2022. [DOI]
- T. C. Wu, D. N. Congreve, M. A. Baldo. Solid State Photon Upconversion Utilizing Thermally Activated Delayed Fluorescence Molecules as Triplet Sensitizer. Appl. Phys. Lett., 2015. [DOI]
- A. Olesund, J. Johnsson, F. Edhborg, S. Ghasemi, K. Moth-Poulsen, B. Albinsson. Approaching the Spin-Statistical Limit in Visible-to-Ultraviolet Photon Upconversion. J. Am. Chem. Soc., 2022. [DOI | PubMed]
- B. Yurash, A. Dixon, C. Espinoza, A. Mikhailovsky, S. Chae, H. Nakanotani, C. Adachi, T. Q. Nguyen. Efficiency of Thermally Activated Delayed Fluorescence Sensitized Triplet Upconversion Doubled in Three-Component System. Adv. Mater., 2022. [DOI | PubMed]
- Y. Wei, K. Pan, X. Cao, Y. Li, X. Zhou, C. Yang. Multiple Resonance Thermally Activated Delayed Fluorescence Sensitizers Enable Green-to-Ultraviolet Photon Upconversion: Application in Photochemical Transformations. CCS Chem., 2022. [DOI]
- T. J. B. Zahringer, J. A. Moghtader, M. S. Bertrams, B. Roy, M. Uji, N. Yanai, C. Kerzig. Blue-to-Uvb Upconversion, Solvent Sensitization and Challenging Bond Activation Enabled by a Benzene-Based Annihilator. Angew. Chem., Int. Ed., 2023. [DOI]
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans, D. W. C. MacMillan. The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem., 2017. [DOI]
- H. Huang, X. Li, C. Yu, Y. Zhang, P. S. Mariano, W. Wang. Visible-Light-Promoted Nickel- and Organic-Dye-Cocatalyzed Formylation Reaction of Aryl Halides and Triflates and Vinyl Bromides with Diethoxyacetic Acid as a Formyl Equivalent. Angew. Chem., Int. Ed., 2017. [DOI]
- F. Cong, X. Y. Lv, C. S. Day, R. Martin. Dual Catalytic Strategy for Forging Sp(2)-Sp(3) and Sp(3)-Sp(3) Architectures Via Beta-Scission of Aliphatic Alcohol Derivatives. J. Am. Chem. Soc., 2020. [DOI | PubMed]
- A. Whyte, T. P. Yoon. Selective Cross-Ketonization of Carboxylic Acids Enabled by Metallaphotoredox Catalysis. Angew. Chem., Int. Ed., 2022. [DOI]
- T. J. Steiman, J. Liu, A. Mengiste, A. G. Doyle. Synthesis of Beta-Phenethylamines Via Ni/Photoredox Cross-Electrophile Coupling of Aliphatic Aziridines and Aryl Iodides. J. Am. Chem. Soc., 2020. [DOI | PubMed]
- S. S. Shah, A. Paul, M. Bera, Y. Venkatesh, N. D. P. Singh. Metallaphotoredox-Mediated Csp2-H Hydroxylation of Arenes under Aerobic Conditions. Org. Lett., 2018. [DOI | PubMed]
- S. S. Shah, M. Shee, A. K. Singh, A. Paul, N. D. P. Singh. Direct Oxygenation of C-H Bonds through Photoredox and Palladium Catalysis. J. Org. Chem., 2020. [DOI | PubMed]
- B. Zhao, R. Shang, W.-M. Cheng, Y. Fu. Decarboxylative Formylation of Aryl Halides with Glyoxylic Acid by Merging Organophotoredox with Palladium Catalysis. Org. Chem. Front., 2018. [DOI]
- K. Cui, Y. L. Li, G. Li, J. B. Xia. Regio- and Stereoselective Reductive Coupling of Alkynes and Crotononitrile. J. Am. Chem. Soc., 2022. [DOI | PubMed]
- J.-C. Grenier-Petel, S. K. Collins. Photochemical Cobalt-Catalyzed Hydroalkynylation to Form 1,3-Enynes. ACS Catal., 2019. [DOI]
- K. Takizawa, T. Sekino, S. Sato, T. Yoshino, M. Kojima, S. Matsunaga. Cobalt-Catalyzed Allylic Alkylation Enabled by Organophotoredox Catalysis. Angew. Chem., Int. Ed., 2019. [DOI]
- H. Zhang, Q. Xiao, X. K. Qi, X. W. Gao, Q. X. Tong, J. J. Zhong. Selective Photoredox Decarboxylation of Alpha-Ketoacids to Allylic Ketones and 1,4-Dicarbonyl Compounds Dependent on Cobaloxime Catalysis. Chem. Commun., 2020. [DOI]
- F.-s. Li, Y.-q. Chen, S.-j. Lin, C.-z. Shi, X.-y. Li, Y.-c. Sun, Z.-w. Guo, L. Shi. Visible-Light-Mediated Barbier Allylation of Aldehydes and Ketones Via Dual Titanium and Photoredox Catalysis. Org. Chem. Front., 2020. [DOI]
- F. Li, S. Lin, Y. Chen, C. Shi, H. Yan, C. Li, C. Wu, L. Lin, C. Duan, L. Shi. Photocatalytic Generation of Pi-Allyltitanium Complexes Via Radical Intermediates. Angew. Chem., Int. Ed., 2021. [DOI]
- S. Lin, Y. Chen, F. Li, C. Shi, L. Shi. Visible-Light-Driven Spirocyclization of Epoxides Via Dual Titanocene and Photoredox Catalysis. Chem. Sci., 2020. [DOI]
- Y. Wang, X. W. Gao, J. Li, D. Chao. Merging an Organic TADF Photosensitizer and a Simple Terpyridine-Fe(Iii) Complex for Photocatalytic Co2 Reduction. Chem. Commun., 2020. [DOI]
- Y. Li, F. Ying, T. Fu, R. Yang, Y. Dong, L. Lin, Y. Han, D. Liang, X. Long. Heat- or Light-Induced Acylarylation of Unactivated Alkenes Towards 3-(Alpha-Acyl) Indolines. Org. Biomol. Chem., 2020. [DOI | PubMed]
- F. H. Zhang, X. Guo, X. Zeng, Z. Wang. Asymmetric 1,4-Functionalization of 1,3-Enynes Via Dual Photoredox and Chromium Catalysis. Nat. Commun., 2022. [DOI | PubMed]
- J. L. Schwarz, H.-M. Huang, T. O. Paulisch, F. Glorius. Dialkylation of 1,3-Dienes by Dual Photoredox and Chromium Catalysis. ACS Catal., 2020. [DOI]
- K. C. Yu, H. Li, Y. H. Tu, H. Zhao, X. G. Hu. Metallaphotoredox-Enabled Construction of the P(O)-N Bond from Aromatic Amines and P(O)-H Compounds. Org. Lett., 2022. [DOI | PubMed]
- M. M. Mastandrea, S. Cañellas, X. Caldentey, M. A. Pericàs. Decarboxylative Hydroalkylation of Alkynes Via Dual Copper-Photoredox Catalysis. ACS Catal., 2020. [DOI]
- J. He, G. Chen, B. Zhang, Y. Li, J.-R. Chen, W.-J. Xiao, F. Liu, C. Li. Catalytic Decarboxylative Radical Sulfonylation. CHEM, 2020. [DOI]
- A. Dumoulin, J. K. Matsui, A. Gutierrez-Bonet, G. A. Molander. Synthesis of Non-Classical Arylated C-Saccharides through Nickel/Photoredox Dual Catalysis. Angew. Chem., Int. Ed., 2018. [DOI]
- Y. Ma, S. Liu, Y. Xi, H. Li, K. Yang, Z. Cheng, W. Wang, Y. Zhang. Highly Stereoselective Synthesis of Aryl/Heteroaryl-C-Nucleosides Via the Merger of Photoredox and Nickel Catalysis. Chem. Commun., 2019. [DOI]
- J. C. Tellis, D. N. Primer, G. A. Molander. Single-Electron Transmetalation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis. Science, 2014. [DOI | PubMed]
- J.-P. C. Cheng, Y. Lu, X.-Q. Zhu, Y. Sun, F. Bi, J. He. Heterolytic and Homolytic N-H Bond Dissociation Energies of 4-Substituted Hantzsch 2,6-Dimethyl-1,4-Dihydropyridines and the Effect of One-Electron Transfer on the N-H Bond Activation. J. Org. Chem., 2000. [DOI | PubMed]
- L. Wang, Z. Chen, G. Fan, X. Liu, P. Liu. Organophotoredox and Hydrogen Atom Transfer Cocatalyzed C-H Alkylation of Quinoxalin-2(1h)-Ones with Aldehydes, Amides, Alcohols, Ethers, or Cycloalkanes. J. Org. Chem., 2022. [DOI | PubMed]
- A. L. Berger, K. Donabauer, B. König. Photocatalytic Carbanion Generation from C-H Bonds – Reductant Free Barbier/Grignard-Type Reactions. Chem. Sci., 2019. [DOI | PubMed]
- W. Xu, J. Ma, X. A. Yuan, J. Dai, J. Xie, C. Zhu. Synergistic Catalysis for the Umpolung Trifluoromethylthiolation of Tertiary Ethers. Angew. Chem., Int. Ed., 2018. [DOI]
- Y. Wang, H. M. Carder, A. E. Wendlandt. Synthesis of Rare Sugar Isomers through Site-Selective Epimerization. Nature, 2020. [DOI | PubMed]
- M.-S. Liu, W. Shu. Catalytic, Metal-Free Amide Synthesis from Aldehydes and Imines Enabled by a Dual-Catalyzed Umpolung Strategy under Redox-Neutral Conditions. ACS Catal., 2020. [DOI]
- A. V. Davies, K. P. Fitzpatrick, R. C. Betori, K. A. Scheidt. Combined Photoredox and Carbene Catalysis for the Synthesis of Ketones from Carboxylic Acids. Angew. Chem., Int. Ed., 2020. [DOI]
- D.-F. Chen, C. H. Chrisman, G. M. Miyake. Bromine Radical Catalysis by Energy Transfer Photosensitization. ACS Catal., 2020. [DOI | PubMed]
- K. Donabauer, B. Konig. Strategies for the Photocatalytic Generation of Carbanion Equivalents for Reductant-Free C-C Bond Formations. Acc. Chem. Res., 2021. [DOI | PubMed]
- Q. Y. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer, B. Konig. Photocarboxylation of Benzylic C-H Bonds. J. Am. Chem. Soc., 2019. [DOI | PubMed]
- S. Grotjahn, B. Konig. Photosubstitution in Dicyanobenzene-Based Photocatalysts. Org. Lett., 2021. [DOI | PubMed]
- J. C. Bawden, P. S. Francis, S. DiLuzio, D. J. Hayne, E. H. Doeven, J. Truong, R. Alexander, L. C. Henderson, D. E. Gómez, M. Massi. Reinterpreting the Fate of Iridium(III) PhotocatalystsScreening a Combinatorial Library to Explore Light-Driven Side-Reactions. J. Am. Chem. Soc., 2022. [DOI | PubMed]
- Y. Kwon, J. Lee, Y. Noh, D. Kim, Y. Lee, C. Yu, J. C. Roldao, S. Feng, J. Gierschner, R. Wannemacher, M. S. Kwon. Formation and Degradation of Strongly Reducing Cyanoarene-Based Radical Anions Towards Efficient Radical Anion-Mediated Photoredox Catalysis. Nature Communications, 2023. [DOI]
- Scifinder search of “Thermally activated delayed fluorescence” with a filter up to 2022. Retrieved 15th August. 2023