
Current Challenges and Future Directions in the Assessment of Glucocorticoid Status
Abstract
Glucocorticoid (GC) hormones are secreted in a circadian and ultradian rhythm and play a critical role in maintaining physiological homeostasis, with both excess and insufficient GC associated with adverse effects on health. Current assessment of GC status is primarily clinical, often in conjunction with serum cortisol values, which may be stimulated or suppressed depending on the GC disturbance being assessed. In the setting of extreme perturbations in cortisol levels ie, markedly low or high levels, symptoms and signs of GC dysfunction may be overt. However, when disturbances in cortisol GC status values are less extreme, such as when assessing optimization of a GC replacement regimen, signs and symptoms can be more subtle or nonspecific. Current tools for assessing GC status are best suited to identifying profound disturbances but may lack sensitivity for confirming optimal GC status. Moreover, single cortisol values do not necessarily reflect an individual’s GC status, as they are subject to inter- and intraindividual variation and do not take into account the pulsatile nature of cortisol secretion, variation in binding proteins, or local tissue concentrations as dictated by 11beta-hydroxysteroid dehydrogenase activity, as well as GC receptor sensitivity. In the present review, we evaluate possible alternative methods for the assessment of GC status that do not solely rely on the measurement of circulating cortisol levels. We discuss the potential of changes in metabolomic profiles, micro RNA, gene expression, and epigenetic and other novel biomarkers such as growth differentiating factor 15 and osteocalcin, which could in the future aid in the objective classification of GC status.
Article type: Review Article
Keywords: adrenal, pituitary gland, Addison’s disease, steroids, glucocorticoid
Affiliations: Section of Investigative Medicine, Imperial College London, London W12 ONN, UK; Department of Endocrinology, Imperial College Healthcare NHS Trust, London W6 8RF, UK; Department of Endocrinology, National University of Singapore, Singapore
License: © The Author(s) 2024. Published by Oxford University Press on behalf of the Endocrine Society. CC BY 4.0 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited. See the journal About page for additional terms.
Article links: DOI: 10.1210/endrev/bnae016 | PubMed: 38795365 | PMC: PMC11581704
Relevance: Moderate: mentioned 3+ times in text
Glucocorticoid (GC) hormones are secreted in a circadian and ultradian pattern (ref. 1, ref. 2) and are critical for maintaining physiological homeostasis (ref. 2). Both excess and insufficient GC levels are associated with adverse health outcomes (ref. 3, ref. 4). However, symptoms and signs of GC dysfunction may only match GC status when perturbations are chronic and extreme, ie, markedly low or high cortisol levels.
Current assessment of GC status relies on clinical features in combination with measurement of cortisol levels, either in the basal state or stimulated/suppressed according to the GC disturbance being assessed. In addition, salivary and urinary cortisol may also be used. However, the accuracy of a single cortisol measure to reflect GC status is challenged by intra- and interindividual variation in cortisol concentrations, the pulsatile nature of cortisol secretion (ref. 5), variations in corticosteroid-binding globulin (CBG), and local tissue concentrations dependent on 11beta-hydroxysteroid dehydrogenase (11β-HSD) activity, as well as GC receptor sensitivity. Thus, several factors can influence individual sensitivity to GC (ref. 6).
Furthermore, GC status is not necessarily dichotomous (ie, either being unequivocally low or high), but rather there can exist a gradation in the degree of abnormality between insufficient and excess GC status, challenging confirmation of GC status. Moreover, GC status could be incongruent from measured cortisol levels or challenging in the setting of (1) concomitant use of exogenous steroid treatments (eg, inhalers or creams) that could suppress endogenous cortisol levels but still contribute to GC status; (2) assessment for Cushing’s syndrome (CS) with incongruent results of investigations (eg, due to factors such as altered renal function affecting reliability of 24-hour urine collections); (3) early presentation of adrenal insufficiency (AI) with residual adrenal function whereby cortisol levels are not absent and cortisol response to stimulation tests may still be normal; (4) suboptimal GC replacement regimens whereby signs and symptoms of GC deficiency/excess can be nuanced; (5) “non-neoplastic hypercortisolism” whereby abnormal investigations may be equivocal/misleading; (6) acute illness whereby cortisol metabolism/clearance and levels are altered; (7) concomitant use of medications that could alter liver metabolism of GC (eg, affecting the response to a dexamethasone suppression test); (8) serum cortisol levels that are falsely elevated (eg, due to increased CBG as a result of oral estrogen treatment).
Herein, we discuss the challenges in the diagnosis/management of GC deficiency/excess and aim to evaluate novel markers that could be used in the objective assessment of GC status in the future that do not rely solely on the measurement of circulating cortisol levels (Fig. 1).
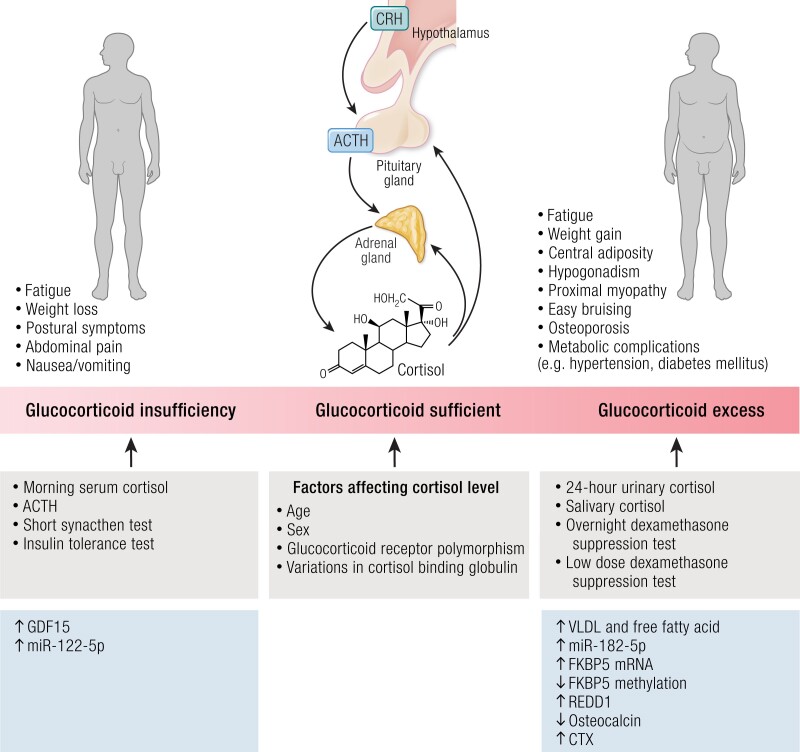
The Hypothalamic-pituitary-adrenal Axis in Health
In healthy people who sleep at night, cortisol is released in both an ultradian (1 to 2 hourly cortisol pulses) (ref. 1) and circadian rhythm rising from ∼04:00 Am (ref. 7), peaking within 3 hours after waking (cortisol awakening response), before subsequently declining and reaching a nadir at around midnight (ref. 7). The frequency and amplitude of cortisol secretion are crucial to ensure synchronization of endogenous cellular rhythms to external cues (ref. 8). Clock genes in peripheral cells are responsible for circadian timekeeping with the activity of the suprachiasmatic nucleus and are modulated in the presence of external cues (eg, light, feeding, temperature) (ref. 8). Notably, acetylation of GC receptor by CLOCK/BMAL1 transcription factors is increased in the morning, leading to decreased cortisol tissue sensitivity (ref. 9).
The diurnal rhythm of circulating cortisol concentrations plays an important role in sleep timing and distribution of sleep stages throughout the night (ref. 10). However, the relationship is bidirectional with sleep onset and sleep disruption also influencing the release of cortisol (ref. 10). Sleep onset inhibits the hypothalamic-pituitary-adrenal (HPA) axis, typically within the first 20 minutes after slow-wave sleep with cortisol reaching a low level in the first half of the night (ref. 10). In the second half of the night, rapid eye movement sleep predominates, and inhibitory mechanisms are attenuated, which is associated by an increase in HPA activity (ref. 10). Nocturnal awakenings at the end of sleep are associated with transient release of cortisol, followed by temporary inhibition of cortisol secretion (ref. 10). Acute changes in the sleep-wake cycle (such as shiftwork or jetlag) lead to HPA axis activation and alter the normal circadian pattern of cortisol secretion (ref. 10). Some (ref. 11), but not all (ref. 12), chronic shift workers are reported to have a delay in the early morning cortisol rise. Prolonged exposure to high cortisol levels is associated with reduced glucose tolerance and insulin sensitivity (ref. 13).
Currently, disturbance of GC status is assessed using basal or stimulated/suppressed cortisol levels depending on the GC disturbance being assessed (Fig. 1). Moreover, ACTH is pulsatile and variable such that it may not be reliable as a marker of GC status in isolation especially if the disturbance in GC status is not at the extreme end of being very high or very low (ref. 2).
Factors Affecting Circulating Cortisol Concentration
Measurement of cortisol concentrations can be affected by several factors including differences in age, sex, time of collection, assay factors, and variation in binding protein levels.
Variation in Corticosteroid Binding Globulin
CBG is predominantly produced by the liver and has a high affinity for cortisol (ref. 14). Approximately 90% of serum cortisol is bound to CBG, 4% to 6% is bound to other binding proteins (eg, albumin or alpha-1-glycoprotein), and 4% to 6% circulates freely (ref. 14, ref. 15). Cortisol binding to CBG is a major determinant of bioavailability; thus, interindividual variability of binding to CBG may affect the measurement of total cortisol levels and dynamic tests (ref. 14, ref. 15), although salivary cortisol and urinary free cortisol are not affected by alterations in CBG (ref. 16). Synthetic GCs, including prednisolone and dexamethasone, bind CBG with less affinity than cortisol (ref. 15, ref. 17). Other factors affecting CBG include sex, temperature, glycosylation states, critical illness, genetic variants, medications, and insulin sensitivity (ref. 14, ref. 18).
Of clinical relevance, low albumin states (albumin < 25 g/L), as encountered in critical illness, are associated with lower total cortisol levels (435 vs 633 nmol/L) but similar active free cortisol levels (140 vs 143 nmol/L) (ref. 19). Likewise, nephrotic syndrome results in renal CBG loss and thus lower total cortisol levels and thus could lead to a false diagnosis of AI after a tetracosactide test (tetracosactide is amino acid 1-24 of the 36 amino acid structure of ACTH, commonly marketed as Synacthen®) (ref. 20). Conversely, exogenous oral estrogens (as well as high circulating levels of estrogens in pregnancy) increase CBG, resulting in a falsely elevated total cortisol (ref. 21).
Age and Cortisol Secretion
Age influences endogenous cortisol secretion and diurnal rhythmicity; increasing age leads to a reduction in the diurnal amplitude of cortisol secretion (ref. 22, ref. 23), an advancement of the early morning cortisol rise (ref. 24), a reduction in the amplitude of cortisol pulses, and an increase in serum cortisol nadir. However, age-specific cut-offs are typically not used in clinical practice.
Effect of Sex and the Menstrual Cycle
Cortisol levels vary between men and women, with additional differences due to the menstrual cycle and menopause (ref. 25). Men have higher cortisol output with higher mean peak, nadir, and cortisol awakening response than premenopausal women (ref. 26, ref. 27). The peak cortisol rise (acrophase) increases gradually with age in women but not in men, such that there is no longer a difference after menopause (ref. 25).
Studies evaluating cortisol responses at different stages of the menstrual cycle have shown discordant results. Some studies have not shown any difference in mean salivary cortisol levels across the menstrual cycle (ref. 28, ref. 29), while other studies have shown higher circulating cortisol levels in the follicular phase (ref. 30). A higher stimulated cortisol response following tetracosactide has been reported in the luteal phase (ref. 29, ref. 31). Such factors could cause a discrepancy between cortisol levels and GC action, and thus markers of GC action could more accurately reflect GC status in these situations.
Single Cortisol Value in Reflection of GC Status
The variation in plasma cortisol levels with time-of-day challenges interpretation of a single value taken at a random time. Early morning cortisol levels are generally high, and levels in patients who are asleep at midnight are usually very low. A major challenge in the interpretation of serum and stimulated cortisol levels is the lack of assay-specific normative data in specific population groups (ref. 32, ref. 33). Historically, serum cortisol was measured using a nonspecific fluorometric assay that measures both cortisol and corticosterone (ref. 34). Due to the poor specificity and low sample throughput of this method (ref. 35), cortisol measurement commonly is undertaken using automated cortisol immunoassays (ref. 36) with each clinical laboratory establishing its own performance characteristics and specificity for cortisol (ref. 33). Agreement between different immunoassays vary (ref. 37); according to some studies, levels can be comparable to those of mass spectrometry (ref. 38), while others have reported higher cortisol levels using immunoassay than mass spectrometry (ref. 39–41). A study comparing automated immunoassays [Advia Centaur (Siemens), Architect (Abbott), Modular Analytics E170 (Roche), Immulite 2000 (Siemens), and Access (Beckman)] with gas chromatography-mass spectrometry showed a bias of 1.08 to 1.36 in immunoassay-measured cortisol levels (ref. 32). In addition to the analytical bias among immunoassays, sex-specific normative cortisol responses in healthy individuals remain undefined (ref. 33). Direct cortisol immunoassays may be susceptible to cross-reactivity with other structurally similar compounds, in particular prednisolone and 11-deoxycortisol (ref. 42).
This difference among cortisol immunoassays ultimately affects the interpretation of GC status (ref. 42). The discrepancy between serum cortisol levels measurement between mass spectrometry and automated immunoassays (12.6% to 90.8%) can be greater in patients receiving treatment for CS e.g. metyrapone owing to the cross-reactivity of structural cortisol analogs in the steroidogenesis pathway (ref. 43). Mass spectrometry quantifies exogenous GC (ref. 44) and endogenous GC metabolites (ref. 45) simultaneously (ref. 43), and recent developments have improved sensitivity and reliability (ref. 46), allowing novel insights into GC processing to be investigated, although mass spectrometers are not ubiquitously available (ref. 47).
In view of the various factors that interfere with cortisol levels, measurement of serum-free cortisol (5% of total) was proposed to have better clinical utility in patients with total cortisol concentrations near the diagnostic threshold or in states of altered binding globulin (ref. 16). Free cortisol can be calculated after removing bound fractions via a suitable technique either by ultrafiltration, equilibrium dialysis, or gel-filtration chromatography (ref. 39, ref. 48). Although these techniques produce comparable interassay coefficients of variation < 10% (ref. 49), they are labor-intensive and require long incubation times. Surrogate methods (eg, Coolen’s equation) or the free cortisol index have been used to estimate free cortisol levels (ref. 50), but these equations are limited in their assumption of direct proportionality and can be inaccurate in patients with hypoalbuminemia or altered CBG capacity (ref. 51, ref. 52).
Thus, the validity of using cortisol response to tetracosactide test to diagnose AI can be influenced by assay performance and alterations in CBG (ref. 32, ref. 33). The widely used cortisol cut-offs of 500 (ref. 53) or 550 nmol/L (ref. 54) in response to a tetracosactide test could result in misclassification of AI (12%,19%, 4%, and 9%) for the Centaur, Architect, Immulite (2000), and Access assays, respectively, at 500 nM; 27%, 42%, 16%, and 21% for the respective assays at 550 nM (ref. 32), whereas patients with a morning (8:00 Am to 12:00 Pm) serum cortisol (using an ultrasensitive immunoassay on an Abbott Architect platform) of >275 nmol/L (96% sensitivity) are unlikely to have an insufficient response to tetracosactide (ref. 55).
Moreover, GC insufficiency can exist over a continuum between complete lack of cortisol and partial deficiency/inability to mount an adequate stress response, and conversely cortisol values marginally below the lower reference limit may not necessarily reflect GC inadequacy. Therefore, other parameters reflecting GC action could provide useful information in addition to clinical judgment in the assessment of GC status.
Current Assessment of Altered GC States
GC Insufficiency
AI is characterized by a failure to secrete sufficient GC to meet physiological demands. Primary adrenal insufficiency (PAI) accounts for 40% of AI (ref. 56), resulting from destruction of adrenal cortical cells (typically due to an autoimmune process or infection, eg, tuberculosis), or adrenal enzymatic failure of GC synthesis (eg, congenital adrenal hyperplasia) (ref. 4, ref. 57). Secondary adrenal insufficiency (SAI) occurs due to impaired ACTH production, eg, due to a pituitary adenoma or traumatic brain injury (ref. 56). Tertiary AI occurs due to impaired hypothalamic corticotrophin releasing hormone secretion, usually due to chronic exogenous GC use (ref. 4). In patients with AI, GC replacement is used to avoid the life-threatening complication of adrenal crisis and to optimize well-being (ref. 58). Patients with PAI also usually require additional mineralocorticoid therapy (ref. 59).
Clinical and biochemical assessment of GC insufficiency
Symptoms of AI depend on the degree of cortisol, mineralocorticoid, and adrenal androgen deficiency at the time of presentation and can be nonspecific, which can lead to delayed diagnosis (ref. 60). Clinical presentation and biochemical changes seen in patients with AI are presented in Table 1 and of GC excess in Table 2. Dynamic endocrine tests used in the diagnosis of both GC deficiency and GC excess and their associated challenges are summarized in Table 3.
Table 1.: Clinical features in AI
| Clinical features | Prevalence in AI (%) | References |
|---|---|---|
| Fatigue | 95 | ref. 61 |
| Weight loss | 70-100 | ref. 62 |
| Decreased appetite | 62 | ref. 62 |
| Nausea | 57 | ref. 63 |
| Vomiting | 52 | ref. 63 |
| Postural symptoms | 56 | ref. 64 |
| Salt-craving | 38-64 | ref. 65 |
| Abdominal pain | 22 | ref. 63 |
| Skin darkening (including buccal pigmentation) | 74 | ref. 65 |
| Biochemical changes | ||
| Hyperkalemia | 40 | ref. 66 |
| Hypercalcemia | 10-20 of acute crises | ref. 61, ref. 67 |
| Hyponatremia | 70-80 | ref. 62, ref. 63 |
Abbreviations: AI, adrenal insufficiency.
Table 2.: Clinical features of Cushing’s syndrome
| Clinical features | Prevalence in Cushing’s syndrome (%) | References |
|---|---|---|
| Nonspecific symptoms and signs | ||
| Muscle weakness | 40-70 | ref. 68 |
| Cognitive and psychiatric changes | 50-81 | ref. 69 |
| Reduced libido | 47 | ref. 70 |
| Sleep disturbance and fatigue | 60 | ref. 71 |
| Menstrual disturbances (due to inhibition of GnRH) | 56 | ref. 70 |
| Hair loss | 31 | ref. 70 |
| Obesity and recent weight gain | 70-95 | ref. 72 |
| More objective clinical features | ||
| Changes in fat deposition (increased fat around face, supraclavicular, and dorsocervical regions) | 50-90 | ref. 73, ref. 74 |
| Violaceous striae | 44-50 | ref. 72 |
| Proximal myopathy | 67 | ref. 70 |
| Easy bruising | 35-65 | ref. 69, ref. 72 |
| Hirsutism | 56-75 | ref. 70 |
| Complications of Cushing’s syndrome | ||
| Diabetes mellitus | 20-47 | ref. 75 |
| Impaired glucose tolerance | 45-70 | ref. 72 |
| Osteopenia | 60-80 | ref. 69, ref. 70 |
| Bone fractures at any sites | 42 | |
| Osteoporosis | 31-50 | ref. 69, ref. 75 |
| Bone fractures at any sites | 62 | |
| Atherosclerotic changes | 27-31 | ref. 68 |
| Hypertension | 58-85 | ref. 75 |
| Nephrolithiasis | 50 | ref. 75 |
| Biochemical changes | ||
| Hypokalemia | 42-70 in ectopic ACTH, 10 in Cushing’s disease | ref. 76 |
| Dyslipidemia | 38-71 | ref. 75 |
Table 3.: Challenges in interpretation of the results of tests used in the assessment of GC status
| Biochemical test | Current challenges in the assessment of GC status |
|---|---|
| GC deficiency (adrenal insufficiency) | |
| Morning serum cortisol | Cortisol cut-off of <140 nmol/L suggestive of AI (ref. 77)Challenge in establishing normal ranges—a cortisol value of 140 nmol/L is close to the lower limit of the range of healthy individuals (ref. 78)Assay specific cut-off values will vary between different methods to evaluate cortisol levels (ref. 32) |
| Short tetracosactide test(Synacthen®) (250 mcg) | Precise thresholds dependent on local assays (ref. 79), but using more specific cortisol assays utilizing LC-MS/MS, cortisol cut-off of 14-15 µg/dL (386-413 nmol/L) is recommended to avoid false-positive results (ref. 80)Clinical variability as to whether 30- or 60- minute cortisol values are used (ref. 81)Other factors, eg, changes in cortisol binding to CBG, and factors affecting CBG will all impact results (ref. 14) |
| ITT | Need for adequate hypoglycemia (2.2 mmol/L) to fully interpret the cortisol value and requires intensive medical and nursing supervision (ref. 82)Contraindicated in certain patient populations, including epilepsy and cardiac disease (ref. 82)Need to stop oral estrogens 6 weeks before the test due to effect on CBG (ref. 14) |
| Glucagon stress test | Assay-specific threshold valueSensitivity 71% and specificity 57% with a cortisol cut-off value >350 nmol/L (using the Abbott Architect assay), when compared with the metyrapone suppression test (ref. 83)To use with caution as a test to assess the HPA axis where an ITT is contraindicated |
| Metyrapone suppression test | Validated against the ITT for the diagnosis of secondary hypoadrenalismSensitivity of 47% and specificity 82% with 11-deoxycortisol <200 nmol/L; this increases to a sensitivity of 71% and specificity of 69% when used with a cortisol value of 450 nmol/L (ref. 84) |
| GC excess (Cushing’s syndrome) | |
| Low-dose dexamethasone suppression test | Sensitivity 80%-95% (ref. 85, ref. 86)Specificity 80%-95% (dependent on cut-off value) (ref. 85, ref. 86)Affected by dexamethasone clearance and metabolism—including medications affecting CYP3A4 complex, eg, rifampicin (ref. 87)Affected by changes in CBG, eg, use of oral estrogens (increasing CBG) and nephrotic syndrome/cirrhosis (reducing CBG) (ref. 14) |
| 24-hour UFC measurement | Sensitivity 45%-71% (ref. 72)Specificity up to 100% (ref. 88)Need for >1 test to avoid false-negative results and in the detection of cyclical Cushing’s (ref. 89)Variable UFC in individuals with cortisol excess—ranging from normal to severely elevated including raised levels in nonneoplastic hypercortisolism (ref. 90)Affected by assay type—risk of cross-reactivity with cortisol precursors and metabolites in immunoassays compared with HPLC or tandem mass spectrometry leading to discordant results between assay types (ref. 91)Risk of falsely raised results with >5L urine collection and falsely low with a reduced eGFR (ref. 92, ref. 93) |
| Late-night salivary cortisol | Sensitivity 92% to 100% (ref. 94, ref. 95)Specificity 85% to 100% (ref. 94, ref. 95)Increases with age, hypertension, and diabetes and in shift workersRisk of false positives with immunoassays (ref. 94, ref. 95) |
In the diagnosis of Cushing’s syndrome, a diagnosis was established on a composite of clinical findings and other concordant investigations.
Oral oestrogens increase CBG and therefore should be stopped 6 weeks prior to tests measuring cortisol as an endpoint.
Abbreviations: AI, adrenal insufficiency; CBG, corticosteroid-binding globulin; GC, glucocorticoid; eGFR, estimated glomerular filtration rate; HPLC, high-performance liquid chromatography; ITT, insulin tolerance test; LC-MS/MS, liquid chromatography-mass spectrometry; UFC, urinary free cortisol.
Assessment of GC status in AI with replacement therapy
Patients with AI, including both primary and secondary, have increased mortality rates (ref. 96), attributed to excess GC exposure at nonphysiological times and adrenal crises (ref. 97). GC excess is associated with metabolic dysfunction, including hypertension, dyslipidemia, immune suppression (ref. 98), hyperglycemia, and osteoporosis (ref. 3). Optimizing replacement therapy for AI, using multidose oral regimens, therefore involves minimizing excess GC exposure and replicating the circadian rhythm (ref. 99); however, to date, there is a paucity of universally accepted methods to ascertain optimal GC replacement. In patients receiving multiple daily-dose hydrocortisone regimens, hydrocortisone day curves can be used predominantly to assess interindividual pharmacokinetic alterations in hydrocortisone metabolism (ref. 58). For those taking low-dose prednisolone (eg, 2-4 mg once daily), mass spectrometry can quantify prednisolone concentrations as low as 10 mg/L (ref. 100), enabling higher resolution day curves with the potential to individualize dosing regimens (ref. 101).
A study evaluating a novel GC replacement regimen highlighted potential measurable pharmacodynamic differences that could be monitored to assess GC status (ref. 99, ref. 102). Once-daily modified release hydrocortisone (dose 16 mg/m2) compared with standard hydrocortisone/cortisone acetate (dose 18 mg/m2) showed differences in body weight, immune profiles, and quality of life scores (ref. 101). Modified-release hydrocortisone was associated with more physiological immune profiles [reversal of the increased number of CD16+ natural killer (NK) cells at baseline] and reduced susceptibility to infections (ref. 101, ref. 103). Similarly, studies evaluating cortisol replacement via pulsatile administration pumps have used functional imaging, quality of life scores (SF-36), and behavioral and cognitive tests to assess GC effects on emotional processing (ref. 104).
GC Excess
Clinical and biochemical assessment of GC excess
Many symptoms of CS are common but nonspecific, while others are more specific but less common (ref. 68). This can be complicated by associated comorbidities such as obesity and hypertension that are commonly found in the general population, as well as sex, age (ref. 105), and the duration and extent of disease (ref. 106). Table 2 summarizes the clinical features and complications observed in CS. More discriminatory features relate to the catabolic state and increased protein breakdown observed secondary to GC excess (ref. 68). The use of facial recognition to diagnose CS is yet to be validated in a clinical setting, but face-classification software correctly classified 85% of patients and 95% of controls (ref. 68). Table 3 outlines 3 biochemical screening tests recommended by the Endocrine Society, where there is a strong pretest probability of a diagnosis of CS (ref. 89). However, the sensitivity and specificity of the tests will depend on the assay and threshold values used. There can also be intraindividual variability, and more than 1 measurement, eg, in the case of urinary free cortisol, is often required to avoid false-negative results (ref. 90). Moreover, in the setting of cyclical CS, results may be concordantly negative and require further evaluation and follow-up (ref. 89). Other methods of measuring cortisol levels include salivary cortisol, whereas hair and nail cortisol could be used to reflect longer term GC exposure.
Salivary Cortisol and Cortisone
Unconjugated cortisol diffuses into the saliva and is converted to cortisone by the enzyme 11β-HSD type 2 (11β-HSD2) (ref. 107). Serum cortisone levels are 4-fold lower than serum cortisol levels, whereas salivary cortisone levels are 6-fold higher than salivary cortisol levels (ratio of salivary cortisone: salivary cortisol = 6:1) (ref. 108). Salivary cortisol and cortisone showed good correlation with serum free cortisol (ref. 108). However, in a study of women treated with estrogens and healthy controls (HCs), salivary cortisone correlated more strongly with serum cortisol levels (Spearman r = +0.95) than salivary cortisol (ref. 109). High levels of salivary cortisol were found for ∼45 minutes after oral dosing of hydrocortisone, likely due to detection of residual hydrocortisone in the oral cavity (ref. 109). However, as hydrocortisone pills do not contain cortisone, salivary cortisone measurement is not affected by oral hydrocortisone, and its level remains correlated with serum free cortisol (ρ = 0.91) in healthy male volunteers (n = 14) after both oral or intravenous hydrocortisone administration (ref. 110). Salivary cortisone is more readily measurable, remaining detectable across the whole 24-hour day, whereas salivary cortisol concentrations can fall below detection limits at low serum cortisol levels (ref. 110).
A recent prospective study demonstrated that waking salivary cortisone measured by liquid chromatography-tandem mass spectrometry could predict cortisol response >430 nmol/L at 30 minutes following tetracosactide (measured by immunoassay) with an area (AUC) of 0.95 [95% confidence interval (CI) 0.92-0.97] (ref. 111). This was greater than the performance of waking salivary cortisol (AUC 0.89, 95% CI 0.85-0.94), which is more susceptible to contamination from oral hydrocortisone from the previous day in a small number of patients (ref. 111). This approach could avoid the need for tetracosactide stimulation in 70% of patients (ref. 111).
Late-night salivary cortisol and cortisone measured by liquid chromatography-tandem mass spectrometry both have high specificity and sensitivity for identifying patients with endogenous CS (ref. 110). A threshold for late-night salivary cortisone of >14.5 nmol/L had a sensitivity of 95.2% and specificity of 100% for identification of CS (ref. 112). Although liquid chromatography-mass spectrometry offers better specificity and comparable sensitivity than antibody-based methods, it is not widely available (ref. 113).
Therefore, salivary cortisone can be used in the screening for AI to assess cortisol circadian rhythm in patients treated with oral hydrocortisone (ref. 110) or to evaluate hydrocortisone replacement in children where blood sampling is undesirable (ref. 114). Three 8-hourly salivary cortisone measurements provided a good estimate of AUC cortisol exposure in HCs (n = 28), patients with AI, or autonomous cortisol secretion (n = 8) and could be used to assess 24-hour cortisol secretion (ref. 115).
Saliva collection is easy for most adults but can be challenging for infants, elderly patients, patients with oral lesions, or patients with Sjogren’s syndrome (ref. 113). Immunoassays used to analyze salivary cortisol/cortisone are inexpensive to run, but there is a potential of cross-reactivity with other steroids, and they are generally not validated for the salivary matrix at the concentrations required (ref. 91, ref. 116, ref. 117).
Hair Cortisol
Hair cortisol analysis has been investigated in the research setting as a potential tool for the assessment of chronic GC excess (ref. 118). While spot cortisol levels in blood, saliva, and urine do not correlate with body mass index (BMI), a 9.8% increment in hair cortisol was associated with a higher BMI by 2.5 kg/m2 (ref. 119). Higher levels of cortisol per 1 cm of scalp hair are found in patients with endogenous CS at 679 (range 279-2500) ng/g (n = 6) when compared to HCs of 116 (range 26-204) ng/g (n = 32) (ref. 120). Hair cortisol has also been used to assess the adequacy of GC replacement in patients treated with hydrocortisone for AI (ref. 6). Thus, it could have potential for diagnosis or for follow-up assessment of cyclical CS where a longer term view of GC status may be helpful (ref. 121-124); however, it remains to be validated outside a research setting and should be considered in light of confounding factors such as sex, ethnicity, physical activity, light exposure, and medications used (ref. 125).
Assessing GC Status: Tissue-specific GC Concentrations
Cortisol action in tissues is regulated by the enzyme 11β-HSD and the GC receptor (GR; the gene name is nuclear receptor subfamily 3, group C, member 1; NR3C1).
Role of 11β-HSD
11β-HSD is a bidirectional enzyme that interconverts active cortisol to inactive cortisone (ref. 126). Cortisone is not synthesized in the adrenal gland. However, cortisone is produced in the kidney and gut and is largely dependent on the activity of 11β-HSD2, which inactivates cortisol to cortisone (ref. 127, ref. 128). In metabolic tissues including liver, skeletal muscle, and adipose tissue, the reductase action of 11β-HSD type 1 (11β-HSD1) reactivates cortisol from cortisone, releasing ∼900 pmol of cortisol per 100 g of liver tissue per minute and ∼15 pmol of cortisol per 100 g of adipose tissue per minute, contributing to 30% to 40% of daily cortisol production (ref. 127, ref. 128). The prereceptor interconversion between bioactive cortisol and inactive cortisone regulates the concentration of GC available for binding to the GR (ref. 126) and alters feedback onto the HPA axis (ref. 129).
Overexpression of adipose HSD11B in mice recapitulates features of metabolic syndrome without changes in circulating GC levels (ref. 130). Conversely, knockout of HSD11B in mice improves glucose tolerance, reduces weight, and improves the lipid profile also without affecting circulating cortisol levels (ref. 131). Although 11β-HSD1 enzyme activity in adipose tissue has been associated with insulin resistance and leptin levels (ref. 132, ref. 133), HSD11B mRNA expression in subcutaneous or omental adipose biopsies was not associated with obesity (ref. 134). Genetic variants in the HSD11B gene have been associated with type 2 diabetes (ref. 135, ref. 136) but not with obesity (ref. 135). Patients with CS with a functional deficit in HSD11B are protected from the adverse effects of GC excess (ref. 137).
Clinical trials using nonselective 11β-HSD inhibition have shown improved insulin sensitivity in patients with type 2 diabetes (ref. 138). A selective 11β-HSD1 inhibitor, ICNB13739, administered to patients with type 2 diabetes significantly lowered body weight and plasma glucose levels (ref. 139). In a recent proof-of-concept study in 32 men, a potent competitive 11β-HSD1 inhibitor, AZD4017, mitigated the metabolic side effects of prednisolone treatment by preventing worsening of hepatic insulin sensitivity, nocturnal blood pressure elevation, lipid metabolism, and bone turnover (preventing the fall in osteocalcin and N-terminal propeptide of type I collagen with prednisolone) while maintaining its desired anti-inflammatory action (assessed using the OX40 assay) (ref. 140).
These demonstrate that variation in 11β-HSD1 activity could alter GC action without necessarily being reflected in circulating GC levels. Thus, manipulation of 11β-HSD1 offers an attractive prospect to modulate GC action; quantifying the extent to which local cortisol is lowered in different tissues for therapeutic benefit while avoiding insufficient GC action could benefit from novel markers reflecting GC action (ref. 141).
Variability in the Response of GC Receptor
The GR is comprised of 777 amino acids and is expressed in almost all human tissues and organs (ref. 142). Alternative splicing of NR3C1 precursor mRNA yields several GR splice elements that modulate GR function and affinity to GC, with the most abundant splice elements being GRα and GRβ (ref. 143). GC predominantly binds to GRα, whereas GRβ, being constitutively nuclear, can represses GRα-induced activity (ref. 143). Prior to ligand binding, GR predominantly resides in the cytoplasm and exists as a complex bound to 2 molecules of heat shock protein 90 (HSP90) and several other proteins (ref. 143). Upon binding to GC, GR undergoes conformational change, dissociates from HSP90, and is translocated to the nucleus (ref. 144) (Fig. 2). The ligand-bound GR dynamically interacts with GC responsive elements (GRE) or GRE half-sites in the promoter region of target genes or indirectly with other transcription factors to exert genomic effects (ref. 145). Variability in the GR response can therefore be influenced by GR expression level, the ligand-binding affinity of GR, posttranslational changes, the type of GREs, and the level of interaction with various coactivators or repressors (ref. 146).
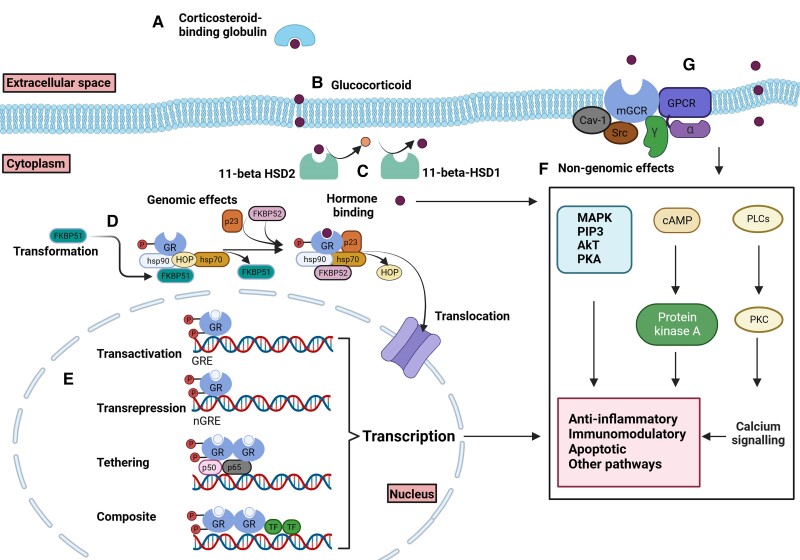
Polymorphisms in the GR gene result in impaired function of GR as a transcriptional activator or repressor. Indeed, there is interindividual variation suggesting that there is variation in the set-point of the HPA axis. Genetic polymorphisms, such as N363S (rs6195), are associated with increased GC action, increased BMI, and reduced bone mineral density (ref. 148). Similarly, individuals with BclI polymorphism (rs41423247) also exhibit increased GC sensitivity, characterized by increased central obesity, insulin resistance, hypertension, and depression (ref. 149). By contrast, carriers of ER22/23EK (rs6189 and rs6190) polymorphism had reduced GC action and lower BMI and cardiovascular risk (ref. 149). Individuals who were homozygous in the intronic region of PDGFD locus (rs591118) are more likely to develop adrenal suppression following treatment with GC compared to those who are homozygous wild-type (ref. 150). Thus, variability in the action of the GR can affect the GC action beyond alterations in circulating GC levels, and in vivo and in vitro assessment of GC sensitivity has been proposed (ref. 6).
Body Weight/Appetite
Changes in body weight can reflect alterations in GC action. GC deficiency is associated with weight loss due to anorexia as well as salt and water depletion (ref. 64). Adrenalectomy in rodents is associated with decreased food intake, which is reversed with GC replacement, mediated through an effect on appetite-controlling neuropeptides in the hypothalamus, including insulin (ref. 151), oxytocin, and corticotrophin-releasing hormone (ref. 152). Additionally, patients with PAI have delayed gastric emptying, which may further contribute to anorexia and weight loss (ref. 153).
By contrast, GC excess may increase oral intake and cause weight gain (ref. 154). In vitro and in vivo studies also showed that GCs can increase hypothalamic endocannabinoids, which have been shown to increase hypothalamic AMP-activated protein kinase (AMPK) activity resulting in increased appetite (ref. 13, ref. 155). In a randomized controlled trial of pulsed high-dose dexamethasone vs standard prednisolone therapy, 17 of 40 (42.5%) reported some increase in appetite and 19 of 40 (47.5%) gained at least 1 kg of body weight in both groups over 24 weeks (ref. 156). In healthy men, energy intake was increased compared to baseline intake after 4 days in those receiving methylprednisolone (40 mg per day, n = 10) by 4554 kcal per day compared to 2867 kcal per day in those receiving placebo (n = 10) (ref. 157).
In a bidirectional Mendelian randomization study, single nucleotide variants independently associated with morning plasma cortisol were obtained from the CORtisol NETwork consortium. Of note, the CORtisol NETwork consortium undertook genome-wide association studies for plasma cortisol and identified that <1% of plasma cortisol variance is accounted for by variation in chromosome 14. The locus identified spans SERPINA6 encoding cortisol-binding globulin and SERPINA1 encoding α1-antitrypsin (which inhibits cleavage of the reactive center loop that releases cortisol from CBG) (ref. 158). Surprisingly, genetically predicted cortisol levels were negatively associated with BMI class and vice versa, with genetically predicted blunted morning cortisol secretion being associated with obesity (ref. 159). Given that the morning cortisol rise is an important feature of the physiological HPA axis, this suggests that disruption to the circadian rhythm of cortisol secretion could contribute to the development of obesity (ref. 160).
Blood Pressure
Changes in blood pressure occur in both AI (including SAI where mineralocorticoid production is preserved) (ref. 161) and GC excess (ref. 3). In patients with PAI, 88% have hypotension and 12% have symptomatic postural dizziness, although mineralocorticoid deficiency leading to salt and water loss is a dominant factor in PAI (ref. 162). GCs also contribute to blood pressure via the transcriptional expression of the enzyme phenylethanolamine-N-methyltransferase, which is responsible for the conversion of norepinephrine to epinephrine in the adrenal medulla (ref. 163). GCs also contribute to catecholamine-induced vasoconstriction (ref. 164). Therefore, GC deficiency results in a relative decrease in epinephrine (reduced vasoconstriction and systolic blood pressure) and an increase in norepinephrine (increased heart rate) (ref. 165).
Hypertension is observed in 80% of patients with CS (ref. 165) through a multifactorial mechanism, including a reduction in vasodilation via inhibition of nitric oxide synthase expression (ref. 166) and modulation of catecholamine synthesis (ref. 165). Cortisol and aldosterone have the same affinity for the mineralocorticoid receptor, but 11β-HSD2 inactivates cortisol under basal conditions. However, the enzyme can become saturated at high cortisol levels, eg, in ectopic-ACTH CS, resulting in excess mineralocorticoid action and hypertension.
GC Status and Muscle Function
Up to 50% of patients with PAI experience generalized weakness, muscle cramps, and fatigue (ref. 167, ref. 168), whereas around 40% to 60% of patients with CS present with muscle weakness (ref. 169-171). Mechanisms contributing to muscle weakness in GC deficiency include impaired muscle carbohydrate metabolism, depleted muscle glycogen stores, and suppression of adrenaline-induced activation of hepatic and skeletal muscle glycogen phosphorylase (ref. 168). By contrast, GC excess induces myopathy via activation of major cellular proteolytic systems [eg, ubiquitin-proteasome system, lysosomal system (cathepsins), and calcium-dependent system (calpains)], inhibition of protein synthesis (ref. 172), alteration of growth factors (eg, IGF-1, myostatin), and testosterone levels (ref. 172). Patients with CS tend to have predominant postural and proximal muscle weakness due to preferential atrophy of fast-twitch glycolytic (type IIb) muscle fibers (ref. 173, ref. 174).
Biomarkers for the Assessment of Muscle Status
GC-induced muscle atrophy is associated with changes in the muscle secretome. SerpinA3N is a secretory serine protease inhibitor that has a wide range of effects including suppression of osteoblasts, as well as being a myokine (secreted into the blood in response to muscle contraction) that is released in association with muscle atrophy. Expression of skeletal muscle SerpinA3N is increased in vitro from skeletal muscle (ref. 175); circulating levels of serpina3n were increased by 2.5-fold in 9 patients with CS (ref. 175), suggesting that it could represent a potential biomarker of GC-induced muscle atrophy pending larger studies (ref. 175).
Myostatin is also a protein produced in muscle tissue that negatively regulates muscle mass. Myostatin administration induced a 35% to 50% decrease in muscle mass (ref. 176), whereas an antimyostatin antibody attenuated chronic kidney disease associated muscle proteolysis in murine models (ref. 177). In in vitro studies, dexamethasone treatment increased myostatin mRNA levels at 24 hours, but the rise in myostatin protein levels was only observed at 36 hours (ref. 178). This discrepancy in mRNA and protein levels suggests that myostatin may be upregulated transiently at the beginning of GC exposure, but it may not be the main pathway that mediates the protein catabolic process (ref. 178). In animal studies, daily intraperitoneal administration of dexamethasone at 60, 600, and 1200 μg/kg body weight for 5 days resulted in a dose-dependent reduction in weight and muscle atrophy, and this decrease was associated with a marked upregulation of myostatin mRNA by 66%, 450%, and 527.6% compared to pair-fed control rats (ref. 179). However, the expression of myostatin mRNA and protein levels was not sustained, and levels returned to basal levels when dexamethasone treatment was extended to 10 days (ref. 179). Another research group reported similar findings in that in vivo myostatin expression was time-dependent with a transient elevation at day 5 during the 10-day dexamethasone treatment (ref. 180). A case-control study showed that serum levels of myostatin did not significantly differ in 30 patients with CS from HCs (ref. 181). Inder et al also showed that short-term oral dexamethasone treatment (4 mg for 4 days) induced relative changes in plasma testosterone (male: 15.0 vs 11.3 nmol/L; female: 1.8 vs 0.5 nmol/L) but skeletal muscle androgen receptor expression and skeletal muscle myostatin mRNA levels were unchanged (ref. 182). Overall, these studies suggest that muscle loss associated with GC is mediated in part by transient upregulation of myostatin at the beginning of GC exposure, but this may not be the main pathway mediating GC-induced muscle atrophy.
GC can also cause muscle atrophy by altering the production of growth factors (eg, IGF-1) that control muscle mass development. IGF-1 stimulates protein synthesis and differentiation of muscle satellite cells (ref. 183) and downregulates the muscle proteolytic systems by inhibiting the expression of atrogenes that cause muscle atrophy, such as atrogin-1, MuRF-1, and cathepsin L, which are induced by GC (ref. 184, ref. 185). Muscle IGF-1 overexpression prevents muscle atrophy in GC-treated animal models. However, although IGF-1 appears to have a dominant effect in reversing GC-induced muscle atrophy, systemic administration of IGF-1 could be limited by its hypoglycaemic (ref. 186) and cardiac hypertrophic actions (ref. 187). In human studies, short-term administration of GC increases serum IGF-1 levels (ref. 182, ref. 188, ref. 189), but levels of IGF-1 in tissue fluid remain unchanged (ref. 189). Skeletal muscle expression of IGF1 mRNA was significantly downregulated, suggesting a distinct tissue-specific effect on IGF1 action that could be partly mediated by GC-induced changes in plasma testosterone levels (ref. 182). Therefore, although IGF-1 appears to have a beneficial effect in reversing GC-induced muscle catabolism, further studies are needed to determine its independent effect.
Imaging in Assessment of Muscle Status
Dual-energy X-ray absorptiometry and bioelectrical impedance analysis can be used in the assessment of muscle mass, but due to the nonuniform nature of muscle atrophy in CS (ref. 190) and the reliance on a constant for hydration of fat free mass (ref. 191), dual-energy X-ray absorptiometry and bioelectrical impedance analysis could give inaccurate results.
Computed tomography (CT) and magnetic resonance imaging are considered gold standards for assessing muscle density and muscle composition as both modalities quantify intramuscular fat content based on attenuation characteristics to discern between fat and muscle tissue (ref. 192). Muscle density has been shown to better reflect handgrip strength and functional outcomes compared to muscle mass (ref. 193). In patients with CS, psoas muscle density measured by CT negatively correlated with urinary free cortisol (r = −0.30) (ref. 194), whereas treatment of GC excess increased CT-measured cross-sectional area of thigh and calf muscle (ref. 195). The use of CT or magnetic resonance imaging is limited in practice due to cost, radiation, and operational complexity. Thus, ultrasound has emerged as a reliable alternative to measure muscle mass (ie, the thickness) and structure (ie, the echo density), which comprise muscle composition (ref. 196). Muscle thickness was similar, but ultrasound-determined echo intensity of vastus lateralis, tibialis anterior, and medial gastrocnemius were significantly higher in patients with active CS compared to those in remission, indicating that muscle structure recovery precedes muscle mass recovery following resolution of hypercortisolemia (ref. 196). Needle biopsy and surface electromyography showed atrophy of type II fibers and slowing of conduction in patients with CS (ref. 197). Atrophy of muscles reduce muscle fiber length and cause architectural changes that contribute to reduced force production capacity (ref. 197). Slowing of muscle fiber conduction velocity could be related to the suppressive effect of GC on sacrolemmal excitability. The preferential GC impairment of fast fibers translates to a greater difference in muscle fiber contraction velocity of vastus lateralis and vastus medialis muscles (∼26%) compared to that of tibialis anterior muscle (∼11%) between patients with CS and age-sex matched HCs (ref. 197).
However, these changes are also observed in other conditions such as aging and neuromuscular diseases and thus lack specificity for the diagnosis of CS (ref. 198, ref. 199). Thus, it is possible that imaging can be used as an adjunct to provide objective evidence for changes in GC status; however, specificity may be lacking for diagnosis. Although emerging technologies provide a means of assessment of muscle quality changes, there is currently no standardized imaging modality that is preferred for use as a diagnostic tool for assessing changes in muscle mass.
Immunomodulatory Effects of GC
GCs have a broad range of immuno-modulatory and immune-suppressive actions on both innate and adaptive immunity, including inhibiting lymphocyte activation, promoting lymphocyte and dendritic cell apoptosis, and downregulation of inflammatory cytokines. It is worth noting that many studies have used much higher doses of GCs than are used clinically for GC replacement (ref. 152, ref. 200).
In patients with PAI, population studies have demonstrated an increased risk of infection, which contributes to an increased mortality rate (ref. 201, ref. 202). This may be explained by impaired NK-induced cellular toxicity. NK cells in patients with PAI have a reduced expression of the activating receptors NKG2D and NK-46 (ref. 186), which may explain the propensity for increased severe bacterial infections. Similarly, peripheral blood mononuclear cells (PBMC) from patients with PAI have a deficient chemokine response to interferons reflective of the adaptive immunity and the ability to defend against viral infections (ref. 203).
Whether these findings are secondary to imperfect GC replacement therapy or a feature of AI is difficult to tease apart. One study showed a normalization of immune cells implicated in innate bacterial defense and viral immunity in response to changing to a modified-release hydrocortisone preparation, linking this to a reduction in viral illnesses (ref. 101). However, it is not clear how a mild excess of GC replacement could affect immune cell function (ref. 204). Further work is needed to assess the effects of different replacement regimens on different immune cell populations.
In CS, there are a variety of changes within the innate and adaptive immune system that increase the risk of severe and opportunistic infections and contribute to a low-grade inflammatory state (ref. 205). These include an increase in neutrophils [due to increased release of polymorphonuclear leucocytes (PMNs) from the bone marrow and reduced apoptosis and extravasation of PMNs leading to an increased PMN half-life in the blood stream]. GC excess also reduces NK cell activity and macrophage phagocytic activity due to a reduction in circulating monocytes. Adaptive changes include a relative reduction in CD4+ T cells, a relative increase in CD8+ T cells, and a reduced B lymphocyte count (ref. 190), increasing susceptibility to intracellular and opportunistic infections.
Increased proinflammatory adipokines, altered tumor necrosis factor α regulation, IL-6, and C-reactive protein also contribute to the chronic, inflammatory state that is thought to contribute to the pathogenesis of the other clinical complications, including diabetes, atherosclerosis, and osteoporosis, with an increased risk of cardiovascular disease that persists after remission (ref. 206).
It has also been observed that CS patients experience rebound autoimmune diseases during remission. This may be due to overactivity of previously suppressed immune function. The extensive effects of GCs on the immune system are beyond the remit of this review; further in-depth discussions on the immune system changes can be found in a recent comprehensive review (ref. 205). A better understanding of changes in the immune system could offer further biomarkers that could be used to assess GC status.
GC Effects on Glycemia
The mechanisms by which GCs exert a diabetogenic effect are complex and are context dependent, but proposed effects include increased insulin resistance, both increased or decreased insulin secretion, and stimulation of hepatic gluconeogenesis. Additional interorgan signaling and tissue-specific effects in vivo further contribute and amplify GC’s diabetogenic effects. The reader is directed to a recent comprehensive review focusing on glucocorticoid effects on glycemia (ref. 207).
Insulin resistance occurs both peripherally and in the liver. As a result of hepatic insulin resistance, there is removal of insulin-mediated repression of hepatic gluconeogenesis. Peripherally, GCs directly disrupt insulin signaling through the downregulation and phosphorylation of IRS1, PI3K, and PKB-AKT (ref. 208-210). This inhibits insulin-induced recruitment of the glucose transporter 4 in skeletal muscle, reducing insulin sensitivity and glucose uptake (ref. 211). Additional effects on protein degradation increase serum amino acids, providing substrates for gluconeogenesis (ref. 212, ref. 213).
De novo glucose production predominantly occurs via hepatic gluconeogenesis via upregulation of the enzymes phosphoenolpyruvate carboxykinase and glucose-6-phosphatase. The action of GCs on other tissues, such as skeletal muscle (via protein catabolism) and lipolysis in white adipose tissue, increase gluconeogenic precursors that are shuttled to the liver (ref. 172, ref. 214), further driving hepatic gluconeogenesis. Understanding the effects of GCs on insulin secretion in vivo is challenging due to the duration of GC exposure and compensatory effects that occur with increased exposure duration, which is difficult to elucidate in in vitro and ex vivo studies. Preclinical studies have demonstrated an increase in beta cell mass following GC exposure in a dose-dependent manner (ref. 215); however, this has not been demonstrated in humans where exposure to GC resulted in minimal changes to beta cell mass (ref. 216). In humans, an acute single oral GC dose has been shown to acutely impair the initial beta cell response to hyperglycemia with increased AUCglucose and reduced AUCc-peptide, but this demonstrated acute recovery with subsequent increased insulin secretion (both fasting and in response to a mixed meal test) 24 hours later. In individuals exposed to 15 days of treatment, there was evidence of increased c-peptide secretion but increased levels of fasting and postprandial AUCglucose, suggesting that the increase in insulin is insufficient to maintain euglycemia (ref. 217).
In addition, studies have also demonstrated increased basal and stimulated glucagon levels in participants following GC administration (ref. 217, ref. 218), further contributing to hyperglycemia. However, the literature is not consistent, with other studies failing to demonstrate this (ref. 219). This may be explained by differences in the insulin sensitivity in the individuals studied. It has been suggested that additional alpha cell disturbances may be present in those with reduced insulin sensitivity (ref. 219).
AMPK is a key regulatory enzyme of appetite, lipid, and carbohydrate metabolism and is believed to mediate some of the deleterious metabolic effects of excess GC (ref. 220). AMPK activity in visceral adipose tissue was inversely associated with 9:00 Am cortisol levels and urinary free cortisol (ref. 220). The effects of AMPK inhibition have demonstrated impaired insulin-mediated glucose uptake by skeletal muscle and adipose, increased hepatic glucose output, reduced beta-cell glucose-stimulated insulin secretion, and increased lipogenesis and fat storage (ref. 221). Activation of AMPK has been shown to reverse GC-induced hepatic steatosis and effects on glucose metabolism (ref. 222). In nondiabetic patients established on GC for inflammatory disease, treatment with metformin (an activator of AMPK) for 12 weeks reduced truncal subcutaneous fat (−3835 mm2), improved insulin resistance and glucose concentrations, and reduced inflammation and progression of carotid intima thickness (ref. 223), suggesting that concomitant use of metformin can mitigate the deleterious effects of GC excess (ref. 223).
In summary, there is evidence that, through a variety of mechanisms, GC deficiency may result in hypoglycemia, especially in children (ref. 66), whereas chronic GC excess can result in hyperinsulinemia and, in those at risk, hyperglycemia (ref. 3).
GC Effects on Lipid Profiles
The effects of GC on lipid dysregulation are multifactorial and involve both direct and indirect pathways. GC promotes lipolysis of mature adipocytes (ref. 214, ref. 224) but stimulates lipogenesis on preadipocyte cells (ref. 224). Low to moderate concentrations of GC (1-10 µM) stimulate lipolysis of mouse adipocyte 3T3-L1 cells, but high doses of GC suppress lipolytic rate (ref. 224). Lipolysis is likely to be mediated via increased intracellular cAMP levels and protein kinase A activity, and this effect could be blunted by downregulation of cyclic-nucleotide phosphodiesterase 3B, a major enzyme responsible for cAMP hydrolysis (ref. 214, ref. 224). GC-induced lipolysis may also in part be mediated via a direct upregulation of GR target such as angiopoietin-like 4, an adipokine that inhibits lipoprotein lipase activity that is involved in lipolysis (ref. 225). In vivo studies reported a 60% rise in fatty acid palmitate level when circulating cortisol concentrations increased from 245 ± 12 nmol/L to 888 ± 7 nmol/L during a 6-hour hydrocortisone infusion (2 μg/kg/min) in the presence of a pancreatic pituitary infusion clamp (ref. 226).
Notably, in an individual, prevailing levels of insulin, somatostatin, and adrenaline concentrations could modulate the lipolytic effects of GC. Healthy volunteers (n = 15) who underwent a paired 2-step hyperinsulinaemic euglycemic clamp with a concurrent overnight HC (0.2 mg/kg/h) or saline infusion (at least 2 weeks apart, in random order) along with adipose tissue microdialysis had lower circulating free fatty acid levels during the high insulin concentration phase compared to low insulin level period compared to placebo (low insulin with HC vs placebo: 564 ± 40 vs 403 ± 38, P < .01 μmol/L·h; high insulin with HC vs placebo: 712 ± 35 vs 443 ± 38 μmol/L·h, P < .005) (ref. 227). During the clamp studies, glucose infusion rates decreased in response to insulin infusion in the HC group compared to placebo, suggesting a reduced glucose disposal rate, representing an increase in systemic insulin resistance. Endogenous glucose production did not differ between the HC or saline group, but the glucose production rate in the presence of a high insulin level was lower in the HC arm compared to saline, suggesting hepatic insulin resistance (ref. 227). Insulin also suppresses the magnitude of GC-induced lipolysis potentiated by adrenaline and stimulates lipoprotein lipase activity (ref. 226). As a result, despite a possible increase in lipolysis, an overall increase in lipogenesis (ref. 224) and hepatic and systemic insulin resistance occurs (ref. 224) and, in turn, promotes hepatic gluconeogenesis (via induction of glucose-6-phosphatase and phosphoenolpyruvate carboxykinase) (ref. 228). Adipogenesis promotes free fatty acid release into the circulation and ectopic storage of lipids in liver and skeletal muscle (ref. 229). Effects of GC-induced adipogenesis are more notably observed in the visceral adipose tissue, which has a relatively higher number of GC receptors (ref. 230). Consequently, expansion of visceral adipose tissue and reduction of subcutaneous adipose tissue further impair liver and muscle sensitivity, leading to cardiometabolic disease (ref. 231). In summary, GC hypercortisolism promotes lipolysis but hyperinsulinemia above a certain threshold suppresses GC lipolysis and lipogenesis and insulin resistance predominates (ref. 232).
Dsylipidemia is a well-recognized feature in patients with hypercortisolism. Patients with active CS exhibit increased very low density lipoprotein, low-density lipoprotein, and triglycerides, but no change in the lipid profile was observed on remission of CS (ref. 233). GC replacement in patients with SAI reduced plasma cholesteryl ester transfer protein activity, resulting in increased high-density lipoprotein (HDL) cholesterol levels (ref. 234) and HDL size (ref. 235), but triglyceride or low-density lipoprotein cholesterol levels were unchanged (ref. 234). Consistent with this, daily administration of low-dose dexamethasone to healthy people for 3 weeks increased HDL cholesterol (ref. 236). By contrast, a large study of 2424 patients with hypopituitarism showed no significant changes in lipid levels between patients treated with GC replacement and those who were GC sufficient (ref. 237). Further clinical data are needed prior to consideration of lipid as a potential marker of GC status.
Adiponectin and Leptin
In humans, treatment with 0.5 mg of dexamethasone 4 times daily for 2 to 5 days reduced insulin sensitivity, but plasma levels of adiponectin, resistin, and leptin remained unchanged (ref. 238). Administration of hydrocortisone (25 mg) reduced adiponectin levels at 30 and 60 minutes both in patients with and without obesity (ref. 239). Adiponectin levels are lower in nonobese patients with CS (20.9 mcg/mL) than in HCs (30.9 mcg/mL), but levels of adiponectin did not differ between patients with CS and obesity compared to those with obesity alone (20.1-22.1 mcg/mL) (ref. 239). Thus, adiponectin could be useful in identifying GC excess but only in the absence of obesity.
Leptin levels were increased dose-dependently after 1 day of a 4-day course of oral hydrocortisone (at either 40 or 160 mg daily) by more than double after the higher dose in women; however, levels returned to baseline level by the fourth day and were less increased in men (ref. 240). Diurnal secretion of leptin is preserved in patients with CS (ref. 241). In patients without obesity, fasting leptin levels are 2-fold higher in patients with CS compared to nonobese controls (33.5 vs 14.2 ng/mL) (ref. 241). In contrast, a smaller difference in leptin levels was observed between patients with obesity compared to BMI-matched HCs (55.0 vs 41.7 ng/mL) (ref. 241). Leptin levels remained stable during the immediate postoperative period (10 days), despite a concurrent decrease in cortisol and ACTH levels after surgical cure of CS (ref. 241, ref. 242). This lack of change in leptin levels after successful surgery could be more associated with the lack of change in BMI and adiposity in the immediate postoperative period (ref. 241) as others have reported a reduction in leptin concentration and BMI in patients with CS 3 to 6 months after surgery (ref. 243). In summary, hypercortisolaemia may indirectly induce body fat changes and hyperinsulinemia, which in turn could influence leptin concentrations (ref. 241, ref. 243). Overall, there are several markers that may be used to assess GC status; however, given the tissue-specific responsiveness to GC that exists (ref. 244), there is no single marker that accurately reflects GC status in isolation.
Novel Markers That Could Be Used in the Future to Quantify GC Status
Please see Table 4.
Table 4.: Summary of novel markers in the assessment of GC status
| Novel marker | Associated changes and use to assess GC status |
|---|---|
| Urinary steroid metabolome | Limited dataUse of multisteroid profiling using tandem mass spectrometry in the evaluation of patients with adrenal tumorsStepwise change in the pattern of 24-hour urinary cortisol excretion seen in NFAT to MACS to CS (ref. 246)Use in AI and GC replacement with a shift toward healthy controls in patients taking dual-release hydrocortisone (ref. 247) |
| Genetic markers | |
| Micro RNAs | |
| miR-122-5p | Reduction in PBMC miR-122-5p on GC exposure (ref. 248) |
| miR-182-5p | Upregulated in Cushing’s disease compared with controls (ref. 249) |
| miR-96-5p | Upregulated following dexamethasone administration (ref. 249) |
| miR-185-5p | Downregulated following dexamethasone administration (ref. 249) |
| GDF15 | GCs repress transcription of GDF15 mRNA in vivo (ref. 250)Evaluated in patients with PAI. Patients with PAI had higher circulating GDF15 levels, which decreased following HC infusionEvidence of dose-dependent decrease in GDF15 levels following low-, medium-, or high-dose oral GC replacement (ref. 251) |
| GC-responsive elements TSP1 | Matricellular protein expressed in endothelial cells, monocytes, macrophages, and adipocytes (ref. 252, ref. 253)Increased TSP1 mRNA in osteoblast, endometrial, and PBMCs following dexamethasone administration (ref. 254, ref. 255)Increased levels in patients with CS compared with healthy controls (ref. 254)Increased levels in patients with AI following increase in hydrocortisone replacement therapy dose (ref. 254)Potential for use with osteocalcin to identify CS (ref. 256) |
| Others to be further evaluated | |
| Lysozyme C High-mobility group protein 2 Nucleophosmin-1 | These have been identified through proteomic analysis of PBMC secretome from healthy volunteers exposed to dexamethasone (ref. 255) |
| FKBP5 | Potential biomarker of cortisol activity at the GR (ref. 257, ref. 258)Reduces GR activity through reduced cortisol binding affinity (ref. 259)Increased levels following infusion of hydrocortisone and dose of prednisone (ref. 260, ref. 261)Increased levels in ACTH-dependent CS compared with controls (with normalization following curative surgery) (ref. 257)Reduced levels with GC antagonist (ref. 261) |
| FKBP5 methylation | Negative correlation with basal serum cortisol levels and 24 hours following ACTH stimulation (ref. 262) |
| REDD1 | Increased following GC administration—limited to preclinical models only (ref. 263)Molecular target for GCs mediating skin and muscle atrophy (ref. 264, ref. 265) |
| Bone turnover markers | |
| Osteocalcin | Reduced osteocalcin following GC administration (ref. 266-268)Negatively correlated with serum cortisol levels in patients with active CS (ref. 269), with increased levels following transsphenoidal or adrenal surgery for CS (ref. 270)No studies evaluating individuals with milder degrees of cortisol excess |
| Total and bone-specific ALPPINP | Less sensitive to GC effects with milder and delayed decrease following GC administration (ref. 268) |
| SerpinA3N (secretory serine protease inhibitor) | Released in association with muscle atrophyIncreased in CS (ref. 173) |
Abbreviations: AI, adrenal insufficiency; ALP, alkaline phosphatase; CS, Cushing’s syndrome; GC, glucocorticoid; GR, glucocorticoid receptor; MACS, mild autonomous cortisol secretion; NFAT, nonfunctioning adrenal tumor; PAI, primary adrenal insufficiency; PBMC, peripheral blood mononuclear cell; PINP, procollagen type I carboxy-propeptide; REDD1, regulated in development and DNA damage response 1.
Metabolomic Assessment
To date, there have been relatively few studies evaluating whether there is a metabolomic signature reflective of healthy GC status and how it may be altered after a changed GC status. Recently, Kachroo et al used metabolomic assessment to identify 17 steroid metabolites that were altered in patients treated with inhaled corticosteroid for asthma (ref. 245). They highlight that genetic variation can underpin the heterogeneity in treatment response and adrenal suppression with inhaled steroids (ref. 245). A significant proportion of patients on inhaled steroids were found to have low cortisol levels, although they may yet have increased GC status due to inhaled steroid use, highlighting a clinical example where GC levels may not reflect GC status (ref. 245).
Patients with adrenal tumous, including those with nonfunctionating adrenal tumors or those with mild autonomous cortisol secretion (MACS) or adrenal CS, were assessed by multisteroid profiling of 24-hour urine by tandem mass spectrometry (ref. 271). Those with MACS or adrenal CS had a decrease in 24-hour urinary excretion of androgen metabolites (androsterone, etiocholanolone, dehydroepiandrosterone) and of pregnenetriol (metabolite of 17-hydroxypregnenolone) and an increase in the 24-hour urinary excretion of cortisol and tetrahydro-11-deoxycortisol (metabolite of 11-deoxycortisol) (ref. 271). Notably, there was a step-wise change in the pattern of urinary steroids from those with nonfunctionating adrenal tumors to MACS to CS, suggesting that such an approach could be used to aid in the objective assessment of GC status (ref. 271). Similarly, in patients with AI (n = 50), dual-release hydrocortisone compared with thrice-daily hydrocortisone decreased cortisol exposure and shifted the urinary steroid metabolome toward that of HCs and thus could potentially be used as a tool to optimize GC replacement (ref. 247).
Growth Differentiating Factor 15
Growth differentiating factor 15 (GDF15) (previously called MIC-1) is a member of the transforming growth factor β family, expressed by cells in response to stress (ref. 250, ref. 251). As a marker of stress, it can be elevated in both physiological and pathological states, including pregnancy, exercise, chronic inflammatory disease, renal failure, and cardiac failure (ref. 250). In vivo, GCs repress transcription of GDF15 mRNA, suggesting that GDF15 could potentially be used as a downstream marker of GC signaling in the endoplasmic reticulum stress pathway (ref. 272).
Utility of GDF15 has been explored in patients with PAI (ref. 251). In a proof-of-concept study, patients with PAI (n = 10) without GC replacement for 39 hours (cortisol levels: PAI 47 ± 17 nmol/L, controls 371 ± 72 nmol/L) had higher circulating GDF15 levels (PAI: 739 ± 226 pg/mL) compared to HCs (498 ± 168 pg/mL) (ref. 251). However, these elevated levels in patients with PAI were reduced to 559 ± 120 pg/mL after a 22-hour hydrocortisone infusion (ref. 251). Additionally, there was a dose-dependent decrease in GDF15 levels (mean ± SD) in patients on low- (491.0 ± 157.7 pg/mL), medium- (427.0 ± 152.1 pg/mL), or high- (360 ± 143.1 pg/mL) dose oral GC replacement (ref. 251). GDF15 levels were also significantly lower in patients with congenital adrenal hyperplasia after a 300-minute hydrocortisone infusion (hydrocortisone 547.0 pg/mL vs placebo 606.0 pg/mL) (ref. 251). Likewise, a 22-hour hydrocortisone infusion (0.008-0.024 mg/kg/hr) reduced GDF15 levels in a dose-dependent manner in patients with PAI (baseline 754 vs 610 pg/mL at 22 hours) (ref. 251). Although this is early data, GDF15 could have a potential utility to contribute as a marker of insufficient GC status pending further study in larger cohorts (ref. 251).
MicroRNAs—miR-122-5p
MicroRNAs (miRNAs) are a group of small noncoding RNAs that regulate gene expression. Given their presence in many extracellular compartments such as plasma, serum, urine, and saliva, miRNAs hold potential as noninvasive biomarkers (ref. 273). In particular, miR-122-5p has been identified as having a key role in regulating gene expression in several cancers, including breast cancer and pancreatic ductal adenocarcinoma (ref. 274). GC exposure was associated with a 2-fold reduction in levels of miR-122-5p in circulating peripheral blood mononuclear cells (P = .009) (ref. 248). By contrast, circulating miR-182-5p was upregulated in Cushing’s disease in comparison to controls, with an area under the receiver operating characteristic (AuROC) curve of 0.89 (ref. 249). After 1 mg dexamethasone treatment, miR-96-5p was upregulated and miR-185-5p was downregulated (ref. 249) in postdexamethasone samples. Thus, while currently only used in the sphere of investigative research, miRNA biomarkers could in the future aid in the assessment of GC status.
GREs
A given GC dose can have variable efficacy and side-effect profile in different individuals. At present, no clinically useful biomarker of GC activity has been established for diagnosis or for use in therapeutic monitoring.
Thrombospondin-1 (TSP1) is a matricellular protein that is widely expressed in endothelial cells, monocytes, macrophages, and adipocytes (ref. 252, ref. 253). It has a plasma half-life of 9 hours and is unaffected by age, sex, BMI, or commonly administered medications for hypertension, hyperlipidemia, or diabetes (ref. 275, ref. 276). TSP1 mRNA was increased by dexamethasone in different cell types (eg, osteoblast, endometrial, or peripheral blood mononuclear cells) in vitro (ref. 277), and the antiangiogenic effect of cortisol on endometrial endothelial cells was abolished by small interfering RNA against TSP1 mRNA, indicating that TSP1 may be a downstream GC effector (ref. 278). A single 4-mg dose of dexamethasone increased plasma TSP1 mRNA in peripheral blood mononuclear cells in healthy individuals at 12 hours (ref. 254). Median (interquartile range) morning plasma TSP1 was significantly higher in patients with CS (n = 8) compared to healthy volunteers (n = 20) [CS: 638 (535-756) ng/mL vs HCs: 272 (237-336) ng/mL] (ref. 254). A threshold of 400 ng/mL identified CS with 100% sensitivity and 85% specificity (AuROC 0.94) (ref. 254). Likewise, morning plasma TSP1 levels were increased in patients with AI (n = 16) when the maintenance dose of HC was increased from 20 to 30 mg within a week from a baseline level of 139 (86-199) ng/mL to 256 (133-516 ng/mL) (ref. 254). These studies suggest that TSP1 is a GC-responsive protein in humans that could be used to reflect GC action. To improve diagnostic sensitivity and specificity, the ratio of TSP1 to osteocalcin taken from a morning blood sample was used in a study of 20 healthy volunteers and 19 people with CS (ref. 256). A TSP1:osteocalcin ratio of >73 reliably identified individuals with CS with an AuROC of 0.997 (ref. 256), highlighting it as a promising potential biomarker of GC status. Proteomic analysis of the secretome from PBMC cells isolated from healthy volunteers exposed to 100 ng/mL of dexamethasone yielded 3 additional novel candidate GC-responsive proteins that altered with GC exposure (eg, lysozyme C, high-mobility group protein 2, and nucleophosmin-1), which could be further explored in future studies (ref. 255).
FKBP5 Expression
FKBP5, a protein encoded by the FKBP5 gene, is a potential biomarker of cortisol activity at the GR (ref. 257, ref. 258). FKBP51 is a cochaperone of HSP90, which together form part of the GR chaperone complex (ref. 279). When bound to the complex, FKBP51 decreases GR activity by reducing its cortisol-binding affinity and preventing its translocation to the nucleus (ref. 259). Thus, FKBP51 interacts with the GR to maintain it in an unbound inactive state (ref. 259). Upon GC binding to the GR, FKBP51 dissociates from the complex and is replaced by FKBP52 enabling nuclear translocation of GR (ref. 258, ref. 279). Once migrated into the nucleus, the GR binds to GRE to affect gene expression, including of FKBP5 (ref. 258, ref. 279). This leads to an increase in the amount of FKBP5 mRNA and FKBP51 protein, which in turn restricts GR nuclear translocation, thus completing an intracellular negative feedback loop for GR activity (ref. 258, ref. 279). Thus, FKBP5 could be used as a biomarker to reflect GC receptor action.
FKBP5 expression levels positively correlate with serum cortisol (r = 0.419) in patients with PAI (ref. 260). Furthermore, an infusion of 100 mg hydrocortisone in patients with PAI off medications induced a 1.35-fold increase in FKBP5 (ref. 260). However, at very low cortisol levels, no correlation was found between FKBP5 expression and serum cortisol levels, although it negatively correlated with plasma ACTH (r = −0.399) (ref. 260).
Oral administration of a single dose of 25 mg of the synthetic GC prednisone increased FKBP5 mRNA expression by 16-fold within 4 hours, and levels returned to baseline within 24 hours of prednisone discontinuation (ref. 261). A single dose of mifepristone was equally effective in reducing prednisone-induced FKBP5 mRNA levels back to unstimulated levels within 4 hours (ref. 261). Likewise, repeated administration of the GC antagonist once daily for 14 days completely ablated prednisolone-induced FKBP5 (ref. 261).
Median (range) FKBP5 mRNA levels were higher in patients with ACTH-dependent CS (n = 24) than in HCs [2357.3 (1071.45, 17215.80) vs 918.7 (661.68, 1735.91)] (ref. 257). FKBP5 mRNA strongly correlated with cortisol levels both before and after surgery (r = 0.72-0.85) (ref. 257). Notably, after successful surgical treatment of CS, FKBP5 mRNA expression decreased to levels similar to those of HCs (CS postsurgery 1148.7 ± 372.8, HCs 918.7 ± 311.1) but remained elevated in patients with failed surgical treatment (2505.3 ± 1843.4) (ref. 257). Overall, the change in FKBP5 mRNA expression with GC exposure shows promise as an in vivo transcriptional biomarker of GC receptor activity that could contribute to assessment of GC status.
FKBP5 Methylation
Epigenetic changes in gene expression due to environmental influence cause alterations in gene activity rather than in gene sequence (ref. 280). The most extensively investigated epigenetic modification is DNA methylation (DNAm), which acts to repress transcription of the gene and can result in gene silencing via changes in chromatin structure (ref. 281).
Changes in chromatin structure involve intron 7 of the GREs, which is thought to contain a transcription enhancer (ref. 282). Less DNA methylation of GREs at intron 7 is associated with higher induction of FKBP5 by GC receptor activation and thus can result in differential expression and responsiveness of FKBP5 to GR (ref. 282).
Several studies have begun to examine the role of DNAm in relation to GC status, in particular, FKBP5 promoter methylation levels (ref. 262). In healthy volunteers, DNAm of the FKBP5 gene in leukocytes was measured before and after ACTH stimulation (ref. 262). FKBP5 methylation was negatively correlated with basal serum cortisol (r = −0.439) (ref. 262). The change in FKBP5 methylation at 24 hours after ACTH negatively correlated with the increase in cortisol levels at 1 hour after ACTH (r = −0.537) (ref. 262).
Despite no significant changes in the FKBP5 methylation status at 24 hours, in vitro studies have suggested changes in methylation of FKBP5 following longer term GC exposure for up to 4 weeks (ref. 283, ref. 284). In fact, patients with CS were found to have reduced FKBP5 methylation of intron 7 compared to HCs, although there was no difference between cured vs active CS patients (cured CS 77.1%, active CS 73.7%, and HCs 79.7%) (ref. 285).
Several studies have explored the potential of FKBP5 methylation as a biomarker of chronic GC load. FKBP5 methylation levels after 4 weeks of corticosterone exposure in mice significantly correlated with average corticosterone load (ref. 284). Overall, this supports the notion that FKBP5 expression levels reflect short-term GC exposure, whereas FKBP5 methylation reflects longer term GC exposure.
REDD1
Regulated in development and DNA damage response 1 (REDD1) is a hypoxia and stress-response gene that inhibits mammalian target of rapamycin complex-1 activity (ref. 286). It has a low level of expression in basal conditions and is induced in response to hypoxia, stress, DNA damage, or transcription factor activation (ref. 286).
Recent studies have identified the hypoxia and stress-response gene REDD1 as a molecular target for GCs that mediates cutaneous and skeletal muscle atrophy in skin (ref. 264). In REDD1 knockout mice, skin and muscle atrophy are averted (ref. 263, ref. 264). REDD1 is upregulated in mouse and human skin and in skeletal muscles following topical or oral GC administration. In mice, the expression of REDD1 is increased between 4 to 24 hours after treatment with topical GC (ref. 263) or after oral dexamethasone at 1 mg/kg (ref. 264). High levels of REDD1 correlate with reduced phosphorylation of downstream mammalian target of rapamycin complex-1 targets (Akt signaling pathway proteins S6 Ser240/244; 4E-BP1 Thr37/46, and ULK1 Ser757) resulting in reduction of protein synthesis (ref. 263, ref. 264). In contrast, REDD1 knockout mice did not demonstrate such downstream changes, and gene activation by GCs are altered in such a way that the therapeutic anti-inflammatory effects of GC were preserved. Thus, REDD1 expression could represent a surrogate biomarker for GC signaling in skin or skeletal muscle; however, data is limited to only preclinical models receiving supraphysiological GC doses to date (ref. 240, ref. 242).
Bone Turnover Makers
The incidence of osteoporotic fractures may be as high as 20% in endogenous hypercortisolism and 30% to 50% in patients with long-term GC use (ref. 287). Prolonged exposure to GCs induces insulin resistance in bone cells resulting in increased bone marrow adiposity and bone fragility (ref. 288). Dose and duration of GC use influences fracture risk (ref. 289). Individuals receiving higher doses of oral prednisolone (≥7.5 mg per day) had a 2- to 2.5-fold increased fracture risk compared to those on lower doses (<2.5 mg per day) (ref. 289). Cumulative GC doses of >1 g over a year result in significant bone loss compared to those receiving lower cumulative GC doses (ref. 290). Interestingly, bone loss is more rapid in new GC users during the first year with bone density of up to 12% followed by a slower decline of 2% to 3% per year with persistent GC use (ref. 291).
GCs inhibit osteocalcin, which is a marker of osteoblast activity (ref. 266). In healthy subjects, 1 mg oral dexamethasone reduced osteocalcin levels from 28.3 ng/mL to 21.8 ng/mL at 8 hours (ref. 267). Likewise, intramuscular administration of 1 mg of ACTH at 2 Pm in healthy individuals (which induced a rise in cortisol to 58.3 mcg/dL, ie, 1608 nmol/L) reduced osteocalcin levels from 28.3 ng/mL to 12.5 ng/mL at 8 Am the following morning (ref. 267). In fact, GC administered in both oral (prednisolone 5 mg) or inhaled forms depresses osteocalcin levels acutely at 3 to 4 hours postdose with levels returning to pretreatment levels on tapering of GC doses (ref. 266, ref. 268). A randomized cross-over study showed that healthy subjects (n = 37) receiving 7 days of treatment with prednisone at doses of 2.5 to 60 mg had a dose-dependent decrease in plasma osteocalcin levels as early as 4 hours postdose, which remained suppressed throughout the week (ref. 292).
The utility of serum osteocalcin as a biomarker has been explored in patients with CS. Morning serum osteocalcin levels were 21.5 ng/mL in healthy individuals and 8.35 ng/mL in overt CS, which corresponds to an AuROC of 0.923 in identifying individuals with CS (ref. 269). There was a negative correlation between osteocalcin levels and serum cortisol levels in patients with active CS in the morning (r = −0.39), at midnight (r = −0.54), and after low-dose dexamethasone suppression test (LDDST) (r = −0.53) (ref. 269). An increase in osteocalcin levels was also observed as early as 1 month after transsphenoidal surgery or adrenalectomy in patients with CS (ref. 270). While osteocalcin is helpful in validating the diagnosis of overt CS, its utility in identifying individuals with milder degrees of cortisol excess is yet to be evaluated. A recent retrospective study suggests osteocalcin could be a promising tool for identifying CS in postmenopausal women (age > 45 years) with osteoporosis (from those without CS) [AuROC: 0.960 (95% CI 0.892-1.000)] (ref. 293).
Other markers of bone formation, such as total and bone-specific alkaline phosphatase and procollagen type I carboxy-propeptide, are less sensitive to GC effects and display a milder and delayed decrease under GC therapy (ref. 268).
Bone resorption increases during GC treatment (ref. 294); however, data are inconsistent. C-terminal telopeptide of type 1 collagen (CTX) is a marker of osteoclastic activity and changes with cortisol levels but less sensitively than osteocalcin. Serum CTX levels correlate with morning (r +0.76) and postdexamethasone cortisol levels (r +0.43). CTX did not change in patients with CS; however, it was elevated in patients with ectopic ACTH (1.0 vs 0.4 ng/mL) who have higher cortisol levels (50 mcg/dL in ectopic ACTH CS vs 21 mcg/dL in other CS) (ref. 270). Overall, osteocalcin could present a relevant marker of GC status pending further study.
Conclusion
GCs are essential hormones for the normal functioning of most organ systems in the body with both GC excess and GC deficiency leading to adverse health consequences. However, symptoms can be nonspecific, and reliance on cortisol levels alone as a readout of GC status can be unreliable. Dynamic tests used to make diagnoses are based on historical cut-off values and aim to detect more profound abnormalities in GC status. Moreover, cortisol values can be challenged due to a number of factors, including the pulsatile nature of cortisol secretion, interindividual variation in cortisol binding proteins, local tissue availability dictated by 11 β-HSD activity, and GC receptor sensitivity.
Downstream markers of GC action may provide an objective method to assess GC status. Such markers include those reflecting metabolomic and epigenetic changes. A comprehensive multiomic approach, applied in concert with serum GC levels, assessment of clinical symptoms and signs such as weight change and metabolic parameters, along with other markers of cortisol action, could enable a comprehensive and objective assessment of GC status in the future.
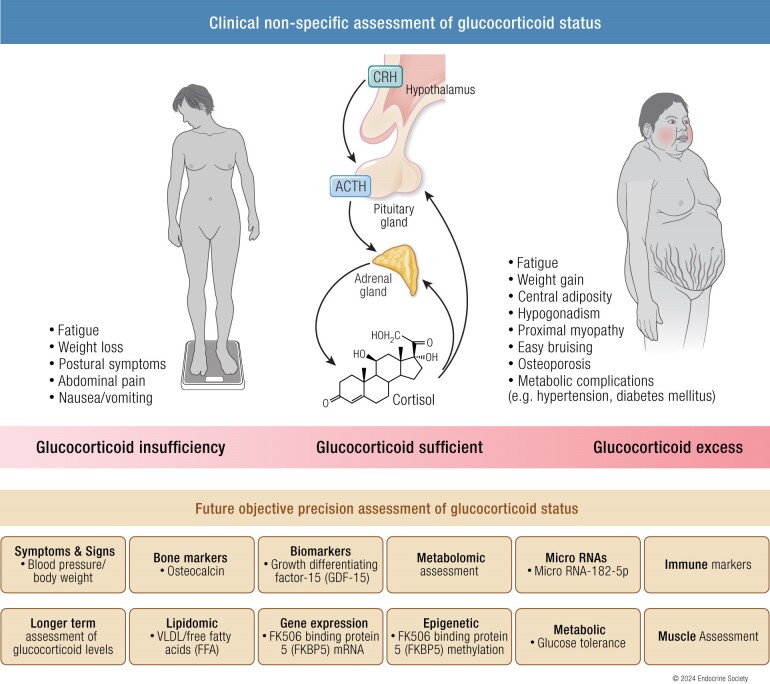
References
- Ultradian cortisol pulsatility encodes a distinct, biologically important signal.. PLoS One., 2011. [PubMed]
- Dynamics of ACTH and cortisol secretion and implications for disease.. Endocr Rev., 2020. [PubMed]
- Complications of Cushing’s syndrome: state of the art.. Lancet Diabetes Endocrinol., 2016. [PubMed]
- Adrenal insufficiency.. Nat Rev Dis Primers., 2021. [PubMed]
- Deconvolution of serum cortisol levels by using compressed sensing.. PLoS One., 2014. [PubMed]
- Variation in glucocorticoid sensitivity and the relation with obesity.. Obes Rev., 2022. [PubMed]
- Human studies on hypothalamo-pituitary-adrenal (HPA) axis.. Best Pract Res Clin Endocrinol Metab., 2017. [PubMed]
- Entrainment of peripheral clock genes by cortisol.. Physiol Genomics., 2012. [PubMed]
- Peripheral CLOCK regulates target-tissue glucocorticoid receptor transcriptional activity in a circadian fashion in man.. PLoS One., 2011. [PubMed]
- Impact of sleep and its disturbances on hypothalamo-pituitary-adrenal axis activity.. Int J Endocrinol., 2010. [PubMed]
- Internal dissociation of the circadian markers of the cortisol rhythm in night workers.. Am J Physiol., 1996. [PubMed]
- The circadian melatonin and cortisol secretion pattern in permanent night shift workers.. Am J Physiol., 1993. [PubMed]
- AMP-activated protein kinase mediates glucocorticoid- induced metabolic changes: a novel mechanism in Cushing’s syndrome.. FASEB J., 2008. [PubMed]
- Corticosteroid-binding globulin: modulating mechanisms of bioavailability of cortisol and its clinical implications.. Best Pract Res Clin Endocrinol Metab., 2015. [PubMed]
- Corticosteroid-binding globulin: a review of basic and clinical advances.. Horm Metab Res., 2016. [PubMed]
- Plasma free cortisol in states of normal and altered binding globulins: implications for adrenal insufficiency diagnosis.. J Clin Endocrinol Metab., 2019. [PubMed]
- Plasma variation of corticosteroid-binding globulin and sex hormone-binding globulin.. Horm Metab Res., 2006. [PubMed]
- The role of corticosteroid-binding globulin in the evaluation of adrenal insufficiency.. J Pediatr Endocrinol Metab., 2018. [PubMed]
- Measurements of serum free cortisol in critically ill patients.. N Engl J Med., 2004. [PubMed]
- The diagnosis and investigation of adrenal insufficiency in adults.. Ann Clin Biochem., 2009. [PubMed]
- Factors influencing the adrenocorticotropin test: role of contemporary cortisol assays, body composition, and oral contraceptive agents.. J Clin Endocrinol Metab., 2007. [PubMed]
- With aging in humans the activity of the hypothalamus-pituitary-adrenal system increases and its diurnal amplitude flattens.. Life Sci., 1997. [PubMed]
- A day-centered approach to modeling cortisol: diurnal cortisol profiles and their associations among U.S. adults.. Psychoneuroendocrinology., 2013. [PubMed]
- Impact of age, sex and body mass index on cortisol secretion in 143 healthy adults.. Endocr Connect., 2017. [PubMed]
- Effects of gender and age on the levels and circadian rhythmicity of plasma cortisol.. J Clin Endocrinol Metab., 1996. [PubMed]
- Interindividual differences and intraindividual variability in the cortisol awakening response: an examination of age and gender.. Psychol Aging., 2009. [PubMed]
- Daytime trajectories of cortisol: demographic and socioeconomic differences—findings from the National Study of Daily Experiences.. Psychoneuroendocrinology., 2013. [PubMed]
- Biological sex and menstrual cycle phase modulation of cortisol levels and psychiatric symptoms in a non-clinical sample of young adults.. Psychiatry Res., 2012. [PubMed]
- The relationship between the menstrual cycle and cortisol secretion: daily and stress-invoked cortisol patterns.. Int J Psychophysiol., 2018. [PubMed]
- Higher circulating cortisol in the follicular vs. luteal phase of the menstrual cycle: a meta-analysis.. Front Endocrinol (Lausanne)., 2020. [PubMed]
- Impact of gender, menstrual cycle phase, and oral contraceptives on the activity of the hypothalamus-pituitary-adrenal axis.. Psychosom Med., 1999. [PubMed]
- Method-specific serum cortisol responses to the adrenocorticotrophin test: comparison of gas chromatography-mass spectrometry and five automated immunoassays.. Clin Endocrinol (Oxf)., 2013. [PubMed]
- Measuring cortisol in serum, urine and saliva—are our assays good enough?. Ann Clin Biochem., 2017. [PubMed]
- A simple fluorimetric method for the estimation of free 11-hydroxycorticoids in human plasma.. J Clin Pathol., 1962. [PubMed]
- A radioimmunoassay for cortisol in human plasma.. Biochem Med., 1973. [PubMed]
- A fully automated enzyme immunoassay for the measurement of cortisol in biological fluids.. Eur J Clin Chem Clin Biochem., 1992. [PubMed]
- Cortisol assays and diagnostic laboratory procedures in human biological fluids.. Clin Biochem., 2009. [PubMed]
- Accuracy of immunoassay and mass spectrometry urinary free cortisol in the diagnosis of Cushing’s syndrome.. Pituitary., 2016. [PubMed]
- 39Casals G, Hanzu FA. Cortisol measurements in Cushing’s syndrome: immunoassay or mass spectrometry? Ann Lab Med. 2020;40(4):285–296.
- Free and total plasma cortisol measured by immunoassay and mass spectrometry following ACTH1–24 stimulation in the assessment of pituitary patients.. J Clin Endocrinol Metab., 2013. [PubMed]
- Agreement of immunoassay and tandem mass spectrometry in the analysis of cortisol and free T4: interpretation and implications for clinicians.. Int J Anal Chem., 2010. [PubMed]
- Clinical and technical aspects in free cortisol measurement.. Endocrinol Metab., 2022
- Comparison of serum cortisol measurement by immunoassay and liquid chromatography-tandem mass spectrometry in patients receiving the 11 β -hydroxylase inhibitor metyrapone.. Ann Clin Biochem., 2011. [PubMed]
- Detection of synthetic glucocorticoids by liquid chromatography-tandem mass spectrometry in patients being investigated for Cushing’s syndrome.. Ann Clin Biochem., 2011. [PubMed]
- Investigation of endogenous corticosteroids profiles in human urine based on liquid chromatography tandem mass spectrometry.. Anal Chim Acta., 2014. [PubMed]
- LC-MS/MS the first 20 years: a personal view.. Ann Clin Biochem., 2022. [PubMed]
- Endogenous glucocorticoid analysis by liquid chromatography–tandem mass spectrometry in routine clinical laboratories.. J Steroid Biochem Mol Biol., 2016. [PubMed]
- Regulation of cortisol bioavailability–effects on hormone measurement and action.. Nat Rev Endocrinol., 2012. [PubMed]
- Free cortisol method comparison: ultrafiltation, equilibrium dialysis, tracer dilution, tandem mass spectrometry and calculated free cortisol.. Clin Chim Acta., 2011. [PubMed]
- Septic shock and sepsis: a comparison of total and free plasma cortisol levels.. J Clin Endocrinol Metab., 2006. [PubMed]
- Comparison of total cortisol, free cortisol, and surrogate markers of free cortisol in diagnosis of adrenal insufficiency in patients with stable cirrhosis.. Clin Gastroenterol Hepatol., 2014. [PubMed]
- Characterising adrenal function using directly measured plasma free cortisol in stable severe liver disease.. J Hepatol., 2010. [PubMed]
- Low-dose (1 μg) adrenocorticotrophin (ACTH) stimulation as a screening test for impaired hypothalamo-pituitary-adrenal axis function: sensitivity, specificity and accuracy in comparison with the high-dose (250 μg) test: the low dose ACTH test for HPA assessment.. Clin Endocrinol (Oxf)., 2000. [PubMed]
- Assessing the HPA axis in patients with pituitary disease: a UK survey.. Clin Endocrinol (Oxf)., 2006. [PubMed]
- Validated criteria for the interpretation of a single measurement of serum cortisol in the investigation of suspected adrenal insufficiency.. Clin Endocrinol (Oxf)., 2019. [PubMed]
- Guidance for the prevention and emergency management of adult patients with adrenal insufficiency.. Clin Med (Lond)., 2020. [PubMed]
- MANAGEMENT OF ENDOCRINE DISEASE: residual adrenal function in Addison’s disease.. Eur J Endocrinol., 2021. [PubMed]
- What is the best approach to tailoring hydrocortisone dose to meet patient needs in 2012?. Clin Endocrinol (Oxf)., 2013. [PubMed]
- Primary adrenal insufficiency: managing mineralocorticoid replacement therapy.. J Clin Endocrinol Metab., 2018. [PubMed]
- Diagnosis and treatment of primary adrenal insufficiency: an endocrine society clinical practice guideline.. J Clin Endocrinol Metab., 2016. [PubMed]
- Adrenal insufficiency—recognition and management.. Clin Med., 2017
- Adrenal insufficiency: physiology, clinical presentation and diagnostic challenges.. Clin Chim Acta., 2020. [PubMed]
- High incidence of adrenal crisis in educated patients with chronic adrenal insufficiency: a prospective study.. J Clin Endocrinol Metab., 2015. [PubMed]
- Eighty-six cases of Addison’s disease.. Br Med J., 1963. [PubMed]
- Clinical, immunological, and genetic features of autoimmune primary adrenal insufficiency: observations from a Norwegian registry.. J Clin Endocrinol Metab., 2009. [PubMed]
- The spectrum of pediatric adrenal insufficiency: insights from 34 years of experience.. J Pediatr Endocrinol Metab., 2019. [PubMed]
- Adrenal insufficiency.. Lancet., 2021. [PubMed]
- Toward a diagnostic score in Cushing’s syndrome.. Front Endocrinol., 2019
- Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health.. J Clin Endocrinol Metab., 2005. [PubMed]
- The European registry on Cushing’s syndrome: 2-year experience. Baseline demographic and clinical characteristics.. Eur J Endocrinol., 2011. [PubMed]
- Quality of life in patients after long-term biochemical cure of Cushing’s disease.. J Clin Endocrinol Metab., 2005. [PubMed]
- Cushing’s syndrome: epidemiology and developments in disease management.. Clin Epidemiol., 2015. [PubMed]
- Cushing’s syndrome.. Lancet., 2015. [PubMed]
- Cushing’s syndrome: from physiological principles to diagnosis and clinical care.. J Physiol., 2015. [PubMed]
- MANAGEMENT OF ENDOCRINE DISEASE: the burden of Cushing’s disease: clinical and health-related quality of life aspects.. Eur J Endocrinol., 2012. [PubMed]
- Association of hypertension and hypokalemia with Cushing’s syndrome caused by ectopic ACTH secretion: a series of 58 cases.. Ann N Y Acad Sci., 2002. [PubMed]
- Corticotropin tests for hypothalamic-pituitary- adrenal insufficiency: a metaanalysis.. J Clin Endocrinol Metab., 2008. [PubMed]
- Establishment of reference values for endocrine tests. Part IV: adrenal insufficiency.. Neth J Med., 2005. [PubMed]
- Hormonal replacement in hypopituitarism in adults: an Endocrine Society Clinical Practice Guideline.. J Clin Endocrinol Metab., 2016. [PubMed]
- New cutoffs for the biochemical diagnosis of adrenal insufficiency after ACTH stimulation using specific cortisol assays.. J Endocr Soc., 2021. [PubMed]
- Determining the utility of the 60 minutes cortisol measurement in the short synacthen test.. Clin Endocrinol (Oxf)., 2013. [PubMed]
- Different cut-off values of the insulin tolerance test, the high-dose short synacthen test (250 μg) and the low-dose short synacthen test (1 μg) in assessing central adrenal insufficiency.. Clin Endocrinol (Oxf)., 2014. [PubMed]
- Comparison of the overnight metyrapone and glucagon stimulation tests in the assessment of secondary hypoadrenalism.. Clin Endocrinol (Oxf)., 2013. [PubMed]
- Combined stimulation of adrenocorticotropin and compound-S by single dose metyrapone test as an outpatient procedure to assess hypothalamic-pituitary-adrenal function.. J Clin Endocrinol Metab., 2002. [PubMed]
- Discriminatory value of the low-dose dexamethasone suppression test in establishing the diagnosis and differential diagnosis of Cushing’s syndrome.. J Clin Endocrinol Metab., 2003. [PubMed]
- The low-dose dexamethasone suppression test: a reevaluation in patients with Cushing’s syndrome.. J Clin Endocrinol Metab., 2004. [PubMed]
- Abnormal overnight dexamethasone suppression test in subjects receiving rifampicin therapy.. J Clin Endocrinol Metab., 1992. [PubMed]
- Screening tests for Cushing’s syndrome: urinary free cortisol role measured by LC-MS/MS.. J Clin Endocrinol Metab., 2015. [PubMed]
- The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline.. J Clin Endocrinol Metab., 2008. [PubMed]
- High variability in baseline urinary free cortisol values in patients with Cushing’s disease.. Clin Endocrinol (Oxf)., 2014. [PubMed]
- Comparison of two methods for measuring salivary cortisol.. Clin Chem., 2002. [PubMed]
- High fluid intake increases urine free cortisol excretion in normal subjects.. J Clin Endocrinol Metab., 1998. [PubMed]
- Diminished urinary free cortisol excretion in patients with moderate and severe renal impairment.. Clin Chem., 2004. [PubMed]
- A single sleeping midnight cortisol has 100% sensitivity for the diagnosis of Cushing’s syndrome.. Clin Endocrinol (Oxf)., 1995. [PubMed]
- Evaluation of the effectiveness of midnight serum cortisol in the diagnostic procedures for Cushing’s syndrome.. Eur J Endocrinol., 2005. [PubMed]
- Premature mortality in patients with Addison’s disease: a population-based study.. J Clin Endocrinol Metab., 2006. [PubMed]
- Imitating the cortisol profile improves the immune system.. Nat Rev Endocrinol., 2018. [PubMed]
- Immune regulation by glucocorticoids.. Nat Rev Immunol., 2017. [PubMed]
- The use of prednisolone versus dual-release hydrocortisone in the treatment of hypoadrenalism.. Endocr Connect., 2021. [PubMed]
- Prednisolone replacement therapy mimics the circadian rhythm more closely than other glucocorticoids.. J Appl Lab Med., 2016. [PubMed]
- Effect of once-daily, modified-release hydrocortisone versus standard glucocorticoid therapy on metabolism and innate immunity in patients with adrenal.. Lancet Diabetes Endocrinol., 2018. [PubMed]
- Improving glucocorticoid replacement profiles in adrenal insufficiency.. Clin Endocrinol (Oxf)., 2019. [PubMed]
- Use of glucocorticoids in patients with adrenal insufficiency and COVID-19 infection.. Lancet Diabetes Endocrinol., 2020. [PubMed]
- Continuous subcutaneous hydrocortisone infusion therapy in Addison’s disease: a randomized, placebo-controlled clinical trial.. J Clin Endocrinol Metab., 2014. [PubMed]
- Gender-related differences in the presentation and course of Cushing’s disease.. J Clin Endocrinol Metab., 2003. [PubMed]
- Cushing’s syndrome.. Lancet., 2006. [PubMed]
- Parotid fluid cortisol and cortisone.. J Clin Invest., 1969. [PubMed]
- Simultaneous measurement of cortisol and cortisone in human saliva using liquid chromatography–tandem mass spectrometry: application in basal and stimulated conditions.. J Chromatogr B Analyt Technol Biomed Life Sci., 2009
- Salivary cortisone is a potential biomarker for serum free cortisol.. J Clin Endocrinol Metab., 2010. [PubMed]
- Salivary cortisone reflects cortisol exposure under physiological conditions and after hydrocortisone.. J Clin Endocrinol Metab., 2016. [PubMed]
- Home waking salivary cortisone to screen for adrenal insufficiency.. NEJM Evid., 2023
- Late-night salivary cortisol and cortisone should be the initial screening test for Cushing’s syndrome.. Endocr Connect., 2022. [PubMed]
- Salivary cortisol and cortisone in the clinical setting.. Curr Opin Endocrinol Diabetes Obes., 2017. [PubMed]
- Measurement of salivary cortisone to assess the adequacy of hydrocortisone replacement.. J Clin Endocrinol Metab., 2016. [PubMed]
- Salivary cortisone to estimate cortisol exposure and sampling frequency required based on Serum cortisol measurements.. J Clin Endocrinol Metab., 2019. [PubMed]
- Measurement of salivary cortisol in 2012—laboratory techniques and clinical indications.. Clin Endocrinol (Oxf)., 2012. [PubMed]
- Comparison of salivary cortisol as measured by different immunoassays and tandem mass spectrometry.. Psychoneuroendocrinology., 2013. [PubMed]
- Fingernail and toenail clippings as a non-invasive measure of chronic cortisol levels in adult cancer survivors.. Cancer Causes Control., 2018. [PubMed]
- The relation between cortisol and anthropometric measurements throughout lifespan: a systematic review and meta-analysis.. J Endocr Soc., 2021
- Hair analysis provides a historical record of cortisol levels in Cushing’s syndrome.. Exp Clin Endocrinol Diabetes., 2009. [PubMed]
- Elevated hair cortisol concentrations in children with adrenal insufficiency on hydrocortisone replacement therapy.. Clin Endocrinol (Oxf)., 2014. [PubMed]
- Increased hair cortisol concentrations and BMI in patients with pituitary-adrenal disease on hydrocortisone replacement.. J Clin Endocrinol Metab., 2015. [PubMed]
- A novel tool in the diagnosis and follow-up of (cyclic) Cushing’s syndrome: measurement of long-term cortisol in scalp hair.. J Clin Endocrinol Metab., 2012. [PubMed]
- Hair cortisol content in patients with adrenal insufficiency on hydrocortisone replacement therapy.. Clin Endocrinol (Oxf)., 2011. [PubMed]
- Innovations in biological assessments of chronic stress through hair and nail cortisol: conceptual, developmental, and methodological issues.. Dev Psychobiol., 2019. [PubMed]
- 11β-Hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action.. Physiol Rev., 2013. [PubMed]
- Cortisol release from adipose tissue by 11β-hydroxysteroid dehydrogenase type 1 in humans.. Diabetes., 2009. [PubMed]
- Splanchnic cortisol production occurs in HumansEvidence for conversion of cortisone to cortisol via the 11-β hydroxysteroid dehydrogenase (11β-HSD) type 1 pathway.. Diabetes., 2004. [PubMed]
- Intracellular regeneration of glucocorticoids by 11β-hydroxysteroid dehydrogenase (11β-HSD)-1 plays a key role in regulation of the hypothalamic-pituitary-adrenal axis: analysis of 11β-HSD-1-deficient mice*the wellcome trust supported this work through a program grant (to J.J.M. and J.R.S.) and a career development fellowship (to M.C.H.).. Endocrinology., 2001. [PubMed]
- A transgenic model of visceral obesity and the metabolic syndrome.. Science., 2001. [PubMed]
- 11β-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress.. Proc Natl Acad Sci U S A., 1997. [PubMed]
- Subcutaneous adipose 11β-hydroxysteroid dehydrogenase type 1 activity and messenger ribonucleic acid levels are associated with adiposity and insulinemia in Pima Indians and Caucasians.. J Clin Endocrinol Metab., 2003. [PubMed]
- Local and systemic impact of transcriptional up-regulation of 11β-hydroxysteroid dehydrogenase type 1 in adipose tissue in human obesity.. J Clin Endocrinol Metab., 2003. [PubMed]
- Expression of 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue is not increased in human obesity.. J Clin Endocrinol Metab., 2002. [PubMed]
- 11β-Hydroxysteroid dehydrogenase type 1: genetic polymorphisms are associated with type 2 diabetes in Pima Indians independently of obesity and expression in adipocyte and muscle.. Diabetologia., 2004. [PubMed]
- Association studies between microsatellite markers within the gene encoding human 11beta-hydroxysteroid dehydrogenase type 1 and body mass index, waist to hip ratio, and glucocorticoid metabolism.. J Clin Endocrinol Metab., 2002. [PubMed]
- Absence of cushingoid phenotype in a patient with Cushing’s disease due to defective cortisone to cortisol conversion.. J Clin Endocrinol Metab., 2002. [PubMed]
- Effects of the 11 beta-hydroxysteroid dehydrogenase inhibitor carbenoxolone on insulin sensitivity in men with type 2 diabetes.. J Clin Endocrinol Metab., 2003. [PubMed]
- The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy.. Diabetes Care., 2010. [PubMed]
- 11β-HSD1 inhibition in men mitigates prednisolone-induced adverse effects in a proof-of-concept randomised double-blind placebo-controlled trial.. Nat Commun., 2023. [PubMed]
- 11-Beta-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors in type 2 diabetes mellitus and obesity.. Expert Opin Investig Drugs., 2008
- Glucocorticoid receptor variants: clinical implications.. J Steroid Biochem Mol Biol., 2002. [PubMed]
- 143 Nicolaides NC, Chrousos G, Kino T. Glucocorticoid Receptor. Endotext 2000. Accessed April 17, 2022. https://www.ncbi.nlm.nih.gov/books/NBK279171/#!po=0.657895
- Corticosteroids-mechanisms of action in health and disease.. Rheum Dis Clin North Am., 2016. [PubMed]
- Glucocorticoid signaling: an update from a genomic perspective.. Annu Rev Physiol., 2016. [PubMed]
- The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease.. J Allergy Clin Immunol., 2013. [PubMed]
- Hsp90 heterocomplexes regulate steroid hormone receptors: from stress response to psychiatric disease.. Int J Mol Sci., 2018. [PubMed]
- Pathophysiology of glucocorticoid signaling.. Ann Endocrinol (Paris)., 2018. [PubMed]
- Stress and obesity: are there more susceptible individuals?. Curr Obes Rep., 2018. [PubMed]
- Susceptibility to corticosteroid-induced adrenal suppression: a genome-wide association study.. Lancet Respir Med., 2018. [PubMed]
- Dexamethasone alters the appetite regulation via induction of hypothalamic insulin resistance in rat brain.. Mol Neurobiol., 2017. [PubMed]
- Novel aspects of glucocorticoid actions.. J Neuroendocrinol., 2014. [PubMed]
- Reversibility of gastric dysmotility in cortisol deficiency.. Am J Gastroenterol., 1987. [PubMed]
- A systematic review of the effect of oral glucocorticoids on energy intake, appetite, and body weight in humans.. Nutr Res., 2014. [PubMed]
- Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism.. J Neurosci., 2003. [PubMed]
- Pulsed high-dose dexamethasone versus standard prednisolone treatment for chronic inflammatory demyelinating polyradiculoneuropathy (PREDICT study): a double-blind, randomised, controlled trial.. Lancet Neurol., 2010. [PubMed]
- Effects of glucocorticoids on energy metabolism and food intake in humans.. Am J Physiol., 1996. [PubMed]
- Genome wide association identifies common variants at the SERPINA6/SERPINA1 locus influencing plasma cortisol and corticosteroid binding globulin.. PLoS Genet., 2014. [PubMed]
- The role of morning plasma cortisol in obesity: a bidirectional Mendelian randomization study.. J Clin Endocrinol Metab., 2022. [PubMed]
- Stress, cortisol, and other appetite-related hormones: prospective prediction of 6-month changes in food cravings and weight.. Obesity., 2017. [PubMed]
- Epidemiology of adrenal crisis in chronic adrenal insufficiency: the need for new prevention strategies.. Eur J Endocrinol., 2010. [PubMed]
- Addison’s disease—serological studies.. Acta Endocrinol (Copenh)., 1974. [PubMed]
- Hormonal control of rat adrenal phenylethanolamine N-methyltransferase enzyme activity, the final critical pathway.. Neuropsychopharmacology., 1995. [PubMed]
- Action of adrenal cortical steroids and nor-epinephrine on vascular responses of stress in adrenalectomized rats.. Am J Physiol., 1951. [PubMed]
- The importance of adrenocortical glucocorticoids for adrenomedullary and physiological response to stress: a study in isolated glucocorticoid deficiency.. J Clin Endocrinol Metab., 2001. [PubMed]
- The pathophysiology and treatment of hypertension in patients with Cushing’s syndrome.. Front Endocrinol., 2019
- Decreased physical activity, reduced QoL and presence of debilitating fatigue in patients with Addison’s disease.. Clin Endocrinol (Oxf)., 2016. [PubMed]
- Neuromuscular manifestations of endocrine disorders.. Neurol Clin., 2002. [PubMed]
- Glucocorticoid-induced myopathy.. Joint Bone Spine., 2011. [PubMed]
- Steroid myopathy in cancer patients.. Neurology., 1997. [PubMed]
- Steroid myopathy in patients with acute graft-versus-host disease treated with high-dose steroid therapy.. Bone Marrow Transplant., 2006. [PubMed]
- Glucocorticoid-induced skeletal muscle atrophy.. Int J Biochem Cell Biol., 2013. [PubMed]
- Diagnostic work-up in steroid myopathy.. Endocrine., 2018. [PubMed]
- Glucocorticoid-induced myopathy: pathophysiology, diagnosis, and treatment.. Indian J Endocrinol Metab., 2013. [PubMed]
- Increased Serpina3n release into circulation during glucocorticoid-mediated muscle atrophy.. J Cachexia Sarcopenia Muscle., 2018. [PubMed]
- Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member.. Nature., 1997. [PubMed]
- Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease.. FASEB J., 2011. [PubMed]
- Glucocorticoids enhance muscle proteolysis through a myostatin-dependent pathway at the early stage.. PLoS One., 2016. [PubMed]
- Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression.. Am J Physiol Endocrinol Metab., 2003
- Time-course changes of catabolic proteins following muscle atrophy induced by dexamethasone.. Steroids., 2016. [PubMed]
- Serum myokines levels in patients with endogenous cushing syndrome and acromegaly: cross-sectional case−control study.. Ann Russ Acad Med Sci., 2016
- Dexamethasone administration inhibits skeletal muscle expression of the androgen receptor and IGF-1—implications for steroid-induced myopathy.. Clin Endocrinol (Oxf)., 2010. [PubMed]
- Stem cell-mediated muscle regeneration is enhanced by local isoform of insulin-like growth factor 1.. Proc Natl Acad Sci U S A., 2004. [PubMed]
- Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway.. J Biol Chem., 2005. [PubMed]
- Insulin-like growth factor-I inhibits dexamethasone-induced proteolysis in cultured L6 myotubes through PI3 K/Akt/GSK-3β and PI3K/Akt/mTOR-dependent mechanisms.. Int J Biochem Cell Biol., 2005. [PubMed]
- Acute metabolic effects and half-lives of intravenously administered insulinlike growth factors I and II in normal and hypophysectomized rats.. J Clin Invest., 1986. [PubMed]
- Effects of growth hormone and IGF-I on cardiac hypertrophy and gene expression in mice.. Am J Physiol., 1998. [PubMed]
- The effects of dexamethasone treatment on immunoreactive and bioactive insulin-like growth factors (IGFs) and IGF-binding proteins in normal male volunteers.. J Endocrinol., 1993. [PubMed]
- Effects of prednisolone on serum and tissue fluid IGF-I receptor activation and post-receptor signaling in humans.. J Clin Endocrinol Metab., 2017. [PubMed]
- Comparison of multi-frequency bioelectrical impedance and dual-energy X-ray absorptiometry to assess body composition in college-aged adults.. Int J Exerc Sci., 2020. [PubMed]
- Tracking fat-free mass changes in elderly men and women using single-frequency bioimpedance and dual-energy X-ray absorptiometry: a four-compartment model comparison.. Eur J Clin Nutr., 2013. [PubMed]
- Muscle quality in aging: a multi-dimensional approach to muscle functioning with applications for treatment.. Sports Med., 2015. [PubMed]
- Muscle density, but not size, correlates well with muscle strength and physical performance.. J Am Med Dir Assoc., 2021. [PubMed]
- A quantitative tool to assess degree of sarcopenia objectively in patients with hypercortisolism.. Surgery., 2011. [PubMed]
- Effect of treatment of cushing’s syndrome on skeletal muscle structure and function.. Clin Endocrinol (Oxf)., 1983. [PubMed]
- Ultrasound-based detection of glucocorticoid-induced impairments of muscle mass and structure in Cushing’s disease.. J Endocrinol Invest., 2019. [PubMed]
- Do muscle fiber conduction slowing and decreased levels of circulating muscle proteins represent sensitive markers of steroid myopathy? A pilot study in Cushing’s disease.. Eur J Endocrinol., 2011. [PubMed]
- Needle electromyographic findings in 98 patients with myositis.. Eur Neurol., 2006. [PubMed]
- Impact of aging on nerve conduction velocities and late responses in healthy individuals.. J Neurosci Rural Pract., 2018. [PubMed]
- Effects of systemically administered hydrocortisone on the human immunome.. Sci Rep., 2016. [PubMed]
- Increased infection risk in Addison’s disease and congenital adrenal hyperplasia.. J Clin Endocrinol Metab., 2020. [PubMed]
- Increased death risk and altered cancer incidence pattern in patients with isolated or combined autoimmune primary adrenocortical insufficiency.. Clin Endocrinol (Oxf)., 2008. [PubMed]
- Peripheral blood cells from patients with autoimmune Addison’s disease poorly respond to interferons in vitro, despite elevated serum levels of interferon-inducible chemokines.. J Interferon Cytokine Res., 2015. [PubMed]
- To B or not to B? Glucocorticoid impact on B lymphocyte fate and function.. Endocrinology., 2014. [PubMed]
- The immune system in Cushing’s syndrome.. Trends Endocrinol Metab., 2020. [PubMed]
- Persistent body fat mass and inflammatory marker increases after long-term cure of Cushing’s syndrome.. J Clin Endocrinol Metab., 2009. [PubMed]
- Fresh insights into glucocorticoid-induced diabetes mellitus and new therapeutic directions.. Nat Rev Endocrinol., 2022. [PubMed]
- Glucocorticoid regulation of insulin receptor and substrate IRS-1 tyrosine phosphorylation in rat skeletal muscle in vivo.. J Clin Invest., 1993. [PubMed]
- Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats.. J Clin Invest., 1993. [PubMed]
- Glucocorticoid-induced insulin resistance in skeletal muscles: defects in insulin signalling and the effects of a selective glycogen synthase kinase-3 inhibitor.. Diabetologia., 2005. [PubMed]
- Dexamethasone inhibits insulin-stimulated recruitment of GLUt4 to the cell surface in rat skeletal muscle.. Metab Clin Exp., 1998
- Effects of high doses of glucocorticoids on free amino acids, ribosomes and protein turnover in human muscle.. Eur J Clin Invest., 2002. [PubMed]
- Short-term prednisone use antagonizes insulin’s anabolic effect on muscle protein and glucose metabolism in young healthy people.. Am J Physiol Endocrinol Metab., 2009. [PubMed]
- Direct effect of glucocorticoids on lipolysis in adipocytes.. Mol Endocrinol., 2009. [PubMed]
- High doses of dexamethasone induce increased β-cell proliferation in pancreatic rat islets.. Am J Physiol Endocrinol Metab., 2009. [PubMed]
- Effects of glucocorticoid treatment on β- and α-cell mass in Japanese adults with and without diabetes.. Diabetes., 2015. [PubMed]
- Acute and 2-week exposure to prednisolone impair different aspects of β-cell function in healthy men.. Eur J Endocrinol., 2010. [PubMed]
- Influence of glucocorticoids on glucagon secretion and plasma amino acid concentrations in man.. J Clin Invest., 1973. [PubMed]
- Insulin resistant subjects lack islet adaptation to short-term dexamethasone-induced reduction in insulin sensitivity.. Diabetologia., 1999. [PubMed]
- Changes in adenosine 5′-monophosphate-activated protein kinase as a mechanism of visceral obesity in Cushing’s syndrome.. J Clin Endocrinol Metab., 2008. [PubMed]
- AMPK signalling in health and disease.. Curr Opin Cell Biol., 2017. [PubMed]
- AMPK regulates metabolic actions of glucocorticoids by phosphorylating the glucocorticoid receptor through p38 MAPK.. Mol Endocrinol., 2010. [PubMed]
- Metformin to reduce metabolic complications and inflammation in patients on systemic glucocorticoid therapy: a randomised, double-blind, placebo-controlled, proof-of-concept, phase 2 trial.. Lancet Diabetes Endocrinol., 2020. [PubMed]
- Adipogenic and lipolytic effects of chronic glucocorticoid exposure.. Am J Physiol Cell Physiol., 2011. [PubMed]
- Angiopoietin-like 4 (ANGPTL4, fasting-induced adipose factor) is a direct glucocorticoid receptor target and participates in glucocorticoid-regulated triglyceride metabolism.. J Biol Chem., 2009. [PubMed]
- Effects of cortisol on lipolysis and regional interstitial glycerol levels in humans.. Am J Physiol Endocrinol Metab., 2002. [PubMed]
- Glucocorticoids fail to cause insulin resistance in human subcutaneous adipose tissue in vivo.. J Clin Endocrinol Metab., 2013. [PubMed]
- Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis.. Annu Rev Physiol., 1992. [PubMed]
- Mechanisms of glucocorticoid-induced insulin resistance.. Endocrinol Metab Clin North Am., 2014. [PubMed]
- Subcutaneous and visceral adipose tissue: structural and functional differences.. Obes Rev., 2010. [PubMed]
- Computed tomography assessment of fat distribution in male and female patients with Cushing’s syndrome.. Eur J Endocrinol., 2003. [PubMed]
- Acute physiological effects of glucocorticoids on fuel metabolism in humans are permissive but not direct.. Diabetes Obes Metab., 2017. [PubMed]
- Cardiovascular risk factors and common carotid artery caliber and stiffness in patients with Cushing’s disease during active disease and 1 year after disease remission.. J Clin Endocrinol Metab., 2003. [PubMed]
- Glucocorticoid replacement affects serum adiponectin levels and HDL-C in patients with secondary adrenal insufficiency.. J Clin Endocrinol Metab., 2019. [PubMed]
- Downregulation of cholesteryl ester transfer protein by glucocorticoids: a randomised study on HDL.. Eur J Clin Invest., 2017. [PubMed]
- Low-dose dexamethasone administration for 3 weeks favorably affects plasma HDL concentration and composition but does not affect very low-density lipoprotein kinetics.. Eur J Endocrinol., 2012. [PubMed]
- The impact of glucocorticoid replacement regimens on metabolic outcome and comorbidity in hypopituitary patients.. J Clin Endocrinol Metab., 2006. [PubMed]
- Short-term dexamethasone administration does not alter serum adiponectin or resistin concentrations in overweight and obese subjects despite an increase in insulin resistance.. Clin Endocrinol (Oxf)., 2006. [PubMed]
- Effect of glucocorticoids on adiponectin: a study in healthy subjects and in Cushing’s syndrome.. Eur J Endocrinol., 2004. [PubMed]
- Dose-dependent cortisol-induced increases in plasma leptin concentration in healthy humans.. Arch Gen Psychiatry., 1998. [PubMed]
- Leptin secretion in Cushing’s syndrome: preservation of diurnal rhythm and absent response to corticotropin-releasing hormone.. J Clin Endocrinol Metab., 1999. [PubMed]
- Plasma leptin levels do not change in patients with Cushing’s disease shortly after correction of hypercortisolism.. J Clin Endocrinol Metab., 1997. [PubMed]
- Determinants of serum leptin levels in Cushing’s syndrome.. J Clin Endocrinol Metab., 1998. [PubMed]
- Tissue specificity of glucocorticoid sensitivity in healthy adults.. J Clin Endocrinol Metab., 2000. [PubMed]
- Metabolomic profiling reveals extensive adrenal suppression due to inhaled corticosteroid therapy in asthma.. Nat Med., 2022. [PubMed]
- Cardiometabolic disease burden and steroid excretion in benign adrenal tumors: a cross-sectional multicenter study.. Ann Intern Med., 2022. [PubMed]
- Improved urinary cortisol metabolome in addison disease: a prospective trial of dual-release hydrocortisone.. J Clin Endocrinol Metab., 2021. [PubMed]
- Identification of human glucocorticoid response markers using integrated multi-omic analysis from a randomized crossover trial.. eLife., 2021. [PubMed]
- Circulating microRNA expression in Cushing’s syndrome.. Front Endocrinol., 2021
- GDF15: a hormone conveying somatic distress to the brain.. Endocr Rev., 2020. [PubMed]
- GDF15 is elevated in conditions of glucocorticoid deficiency and is modulated by glucocorticoid replacement.. J Clin Endocrinol Metab., 2020. [PubMed]
- Thrombospondin-1: multiple paths to inflammation.. Mediators Inflamm., 2011. [PubMed]
- Isolation and characterization of a high molecular weight glycoprotein from human blood platelets.. J Biol Chem., 1978. [PubMed]
- Thrombospondin-1 is a glucocorticoid responsive protein in humans.. Eur J Endocrinol., 2016. [PubMed]
- Ex vivo glucocorticoid-induced secreted proteome approach for discovery of glucocorticoid-responsive proteins in human serum.. Proteomics Clin Appl., 2021. [PubMed]
- The effect of glucocorticoids on thrombospondin-1, osteocalcin and the thrombospondin-1:osteocalcin ratio in humans.. Clin Endocrinol (Oxf)., 2019. [PubMed]
- Evaluation of FKBP5 as a cortisol activity biomarker in patients with ACTH-dependent Cushing syndrome.. J Clin Transl Endocrinol., 2021. [PubMed]
- Gene–stress–epigenetic regulation of FKBP5: clinical and translational implications.. Neuropsychopharmacology., 2016. [PubMed]
- The 90-kDa heat-shock protein (Hsp90)-binding immunophilin FKBP51 is a mitochondrial protein that translocates to the nucleus to protect cells against oxidative stress.. J Biol Chem., 2011. [PubMed]
- Potential transcriptional biomarkers to guide glucocorticoid replacement in autoimmune Addison’s disease.. J Endocr Soc., 2021. [PubMed]
- FKBP5 mRNA expression is a biomarker for GR antagonism.. J Clin Endocrinol Metab., 2016. [PubMed]
- FKBP5 methylation as a possible marker for cortisol state and transient cortisol exposure in healthy human subjects.. Epigenomics., 2017. [PubMed]
- REDD1 functions at the crossroads between the therapeutic and adverse effects of topical glucocorticoids.. EMBO Mol Med., 2015. [PubMed]
- REDD1 deletion prevents dexamethasone-induced skeletal muscle atrophy.. Am J Physiol Endocrinol Metab., 2014. [PubMed]
- REDD1 (regulated in development and DNA damage response 1) expression in skeletal muscle as a surrogate biomarker of the efficiency of glucocorticoid receptor blockade.. Biochem Biophys Res Commun., 2011. [PubMed]
- Oral and inhaled corticosteroids reduce bone formation as shown by plasma osteocalcin levels.. Am J Respir Crit Care Med., 1995. [PubMed]
- Effect of single doses of dexamethasone and adrenocorticotrop hormone on serum bone markers in healthy subjects and in patients with adrenal incidentalomas and Cushing’s syndrome.. J Endocrinol Invest., 2004. [PubMed]
- The effect of single oral doses of prednisone on the circadian rhythm of serum osteocalcin in normal subjects.. J Clin Endocrinol Metab., 1988. [PubMed]
- Diagnostic performance of salivary cortisol and serum osteocalcin measurements in patients with overt and subclinical Cushing’s syndrome.. Steroids., 2011. [PubMed]
- Bone turnover in patients with endogenous Cushing’s syndrome before and after successful treatment.. Osteoporos Int., 2010. [PubMed]
- Cardiometabolic disease burden and steroid excretion in benign adrenal tumors.. Ann Intern Med., 2022. [PubMed]
- The transrepression arm of glucocorticoid receptor signaling is protective in mutant huntingtin-mediated neurodegeneration.. Cell Death Differ., 2015. [PubMed]
- MicroRNAs in body fluids—the mix of hormones and biomarkers.. Nat Rev Clin Oncol., 2011. [PubMed]
- MicroRNA-122-5p inhibits cell proliferation, migration and invasion by targeting CCNG1 in pancreatic ductal adenocarcinoma.. Cancer Cell Int., 2020. [PubMed]
- Higher plasma thrombospondin-1 levels in patients with coronary artery disease and diabetes mellitus.. Korean Circ J., 2012. [PubMed]
- Radioimmunoassay for the measurement of thrombospondin in plasma and breast cyst fluid: validation and clinical application.. Ann Clin Biochem., 2000. [PubMed]
- Gene expression profiling of glucocorticoid-inhibited osteoblasts.. J Mol Endocrinol., 2004. [PubMed]
- Cortisol inactivation by 11β-hydroxysteroid dehydrogenase-2 may enhance endometrial angiogenesis via reduced thrombospondin-1 in heavy menstruation.. J Clin Endocrinol Metab., 2009. [PubMed]
- FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells.. J Biol Chem., 2005. [PubMed]
- An operational definition of epigenetics.. Genes Dev., 2009. [PubMed]
- 281Buitrago D, Labrador M, Arcon JP. et al. Impact of DNA methylation on 3D genome structure. Nat Commun 2021;12:3243
- Allele-specific FKBP5 DNA demethylation mediates gene–childhood trauma interactions.. Nat Neurosci., 2013. [PubMed]
- DNA methylation analysis of human trabecular meshwork cells during dexamethasone stimulation.. Invest Opthalmol Vis Sci., 2015
- A measure of glucocorticoid load provided by DNA methylation of fkbp5 in mice.. Psychopharmacology (Berl)., 2011. [PubMed]
- Reduced DNA methylation of FKBP5 in Cushing’s syndrome.. Endocrine., 2016. [PubMed]
- RTP801/REDD1: a stress coping regulator that turns into a troublemaker in neurodegenerative disorders.. Front Cell Neurosci., 2014. [PubMed]
- Fracture risk in oral glucocorticoid users: a Bayesian meta-regression leveraging control arms of osteoporosis clinical trials.. Osteoporos Int., 2016. [PubMed]
- The interrelationship between bone and fat: from cellular see-saw to endocrine reciprocity.. Cell Mol Life Sci., 2013. [PubMed]
- Use of oral corticosteroids and risk of fractures.. J Bone Miner Res., 2000. [PubMed]
- The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis.. Osteoporos Int., 2002. [PubMed]
- Bone loss in response to long-term glucocorticoid therapy.. Bone Miner., 1990. [PubMed]
- Safety and pharmacodynamic dose response of short-term prednisone in healthy adult subjects: a dose ranging, randomized, placebo-controlled, crossover study.. BMC Musculoskelet Disord., 2016. [PubMed]
- Diagnostic performance of osteocalcin measurements in patients with endogenous Cushing’s syndrome.. Bonekey Rep., 2016. [PubMed]
- Perspectives bone histomorphometry in glucocorticoid-induced osteoporosis.. J Bone Miner Res., 1989. [PubMed]