
Organelle contact sites in cancer cells
Abstract
Membrane contact sites (MCSs) are defined as regions of functional proximity between membranes belonging to the same or different organelle types. These interactions are mediated by specialised proteins promoting the formation of these crosstalk hubs. Previously, organelles were considered to act independently in cellular physiology. However, it is now evident they carry out specific functions at MCSs. The first interactions described involved endoplasmic reticulum and mitochondria. Subsequently, many contacts involving different organelles emerged. MCSs affect several cellular processes, including intracellular signalling, lipid and ion homeostasis, transport of molecules, cellular metabolism, and redox balance. Disruption of these interactions has been described to be associated with various pathologies, including cancer. While the role of MCSs in tumours remains unclear, recent findings suggest they may influence cancer progression, so, in the near future, modulating organelle interactions could provide novel therapeutic options and develop new protocol to treat tumours.
Schematic overview of intracellular MCSs, their effects on biological processes and the associated cancer-related outcomes. MCSs involve different cellular organelles allowing their intercommunication, finally participating in a plethora of cellular processes ranking from calcium and ions exchange, lipid transport and regulation of cell survival. Thus, MCSs modulation has been demonstrated to play a pivotal role in the modulation of cancer aggressiveness.
Article type: Review Article
Keywords: Organelles, Cancer
Affiliations: https://ror.org/00240q980grid.5608.b0000 0004 1757 3470Department of Biology, University of Padova, Padova, Italy
License: © The Author(s) 2026 CC BY 4.0 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Article links: DOI: 10.1038/s41419-026-08674-5 | PubMed: 41935069 | PMC: PMC13183906
Relevance: Moderate: mentioned 3+ times in text
Full text: PDF (4.5 MB)
FACTS
- Membrane contact sites (MCSs), regions of proximity between organelles, regulate key cellular processes, including calcium signalling, lipid transfer, metabolism and redox homeostasis.
- MCSs are stabilised by specialised tethering proteins that enable inter-organelle communication without membrane fusion.
- Increasing evidence links alterations in organelle contact sites to cancer cell metabolism, survival and stress responses.
OPEN QUESTIONS
- How are organelle contact sites dynamically regulated during cancer development and progression?
- Can modulation of organelle contact sites be exploited as a therapeutic strategy in cancer?
- Which experimental approaches will allow the accurate visualisation and quantification of MCS dynamics in living cancer cells?
Introduction
Membrane contact sites (MCSs) are defined as regions of close proximity between membranes of distinct cellular organelles. Interestingly, MCSs can involve two or three different types of organelles. Nowadays, it is clear that the composition of these MCSs is not static; rather, they are dynamic structures that undergo modifications in response to cellular stressors and metabolic demands [ref. 1–ref. 3]. All these contacts facilitate the exchange of key molecules and substrates, which impact on numerous signalling pathways and processes essential for cellular homeostasis, including calcium (Ca2+) signalling, lipid synthesis and metabolism, nutrient balance, autophagy, protein metabolism, stress responses and organelle biogenesis, dynamic and localization. Given their importance in such several vital processes, alterations in MCSs have been associated with several pathologies, including cancer. Specifically, MCSs modulation associates with the control of different cancer cell hallmarks, including tumourigenic uncontrolled cell growth, metabolic reprogramming and resistance to cell death, finally impacting on resistance to therapeutic agents and ability to metastatize [ref. 4]. This suggests that targeting and modulating the components of MCSs may represent a valid strategy to positively impact on tumourigenesis. Given contact site role in different biological and pathological processes, novel approaches have been developed to study and quantify contacts between organelles [ref. 5].
In the present review, we will discuss the structure of the up to date described inter-organelle MCSs, focusing on their impact on cancer development and progression, to highlight their role as possible oncological targets.
ER-Mitochondria contact sites
The Mitochondria-Endoplasmic Reticulum (ER) Contact sites (MERCs), also referred to as mitochondria-associated membranes (MAMs) were the first to be observed in the 1950s [ref. 6, ref. 7]. From 1990, they gained increasing scientific consideration, both from the structural and functional point of view [ref. 8]. MERCs represent hubs for signalling that control multiple aspects of mitochondrial biology including cellular survival [ref. 9], redox-signalling [ref. 10], Ca2+ release [ref. 11], lipid transfer [ref. 12], immune responses [ref. 13], inflammation [ref. 14], autophagy [ref. 15], mitochondrial dynamics [ref. 16], metabolism [ref. 17], cell death sensitivity and metastasis development [ref. 18], which all contribute to tumourigenesis [ref. 9, ref. 19]. Considering MERCs are involved in a plethora of cellular key processes, it is not surprising that altered MERCs can play an important role also in different pathological conditions [ref. 20, ref. 21]. Several ER-mitochondria tethers have been described so far, and they are depicted in Fig. 1. In the next paragraphs, we will discuss which of them have been shown to have an impact on cancer (Table 1).
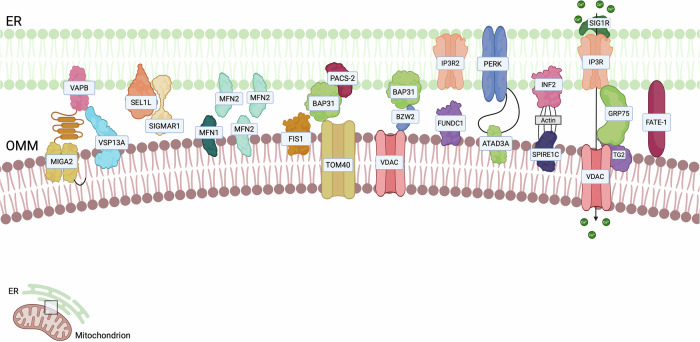
Table 1: Main ER-mitochondria contact sites and their associated features.
| Cargo | Main tether (ER-mito) | Biological functions | Tumour phenotype | Ref. |
|---|---|---|---|---|
| Ca2+ | GRP75-IP3R-VDAC | Regulation of calcium transfer between the ER and mitochondria | Impaired Ca2+ transport, increased proliferation and migration | [ref. 22, ref. 32, ref. 33] |
| Ca2+ | FUNDC1 – IP3R2 | Mitophagy, Ca2+ transfer to mitochondria | Promotion of neo-angiogenesis, inhibition of cancer cell proliferation, apoptosis induction, increased drug sensitivity | [ref. 49, ref. 50, ref. 54] |
| Lipids, Ca2+ | VAPs – VPS13VAPs – MIGA2 | Lipid transfer, Ca2+ homeostasis, autophagy, unfolded protein response (UPR), microtubule organisation, and neurotransmitter release | Increase of tumour growth | [ref. 56, ref. 68] |
| Mitofusins | Mitochondrial fusion, mitochondrial function regulation, ER homeostasis and cellular metabolism | Apoptosis induction, promotion or reduction of cancer cell invasion depending on cancer type, increased invasiveness | [ref. 70, ref. 79, ref. 314, ref. 315] | |
| Ca2+ | BZW2 | Regulation of c-Myc, ERK/MAPK, Wnt/β-catenin and Akt/mTOR pathways, Ca2+ fluxes, control and sorting of proteins directed to the ER | Glycolysis enhancement, impact on cancer cell metabolism, ATP production modulation, worse prognosis | [ref. 82–ref. 88] |
| BAP31 | Apoptosis induction, ROS production | Inhibition of cell proliferation, apoptosis induction and reduced invasiveness | [ref. 90, ref. 91] | |
| Cholesterol | PERK | UPR, modulation of ER stress, mitochondrial homeostasis | Cancer cell adaptation, increased cell survival, angiogenesis, migration, and dissemination, inhibition of immune activation | [ref. 99, ref. 103–ref. 105] |
| Ca2+ | FATE1 | Mitochondrial morphology modulation and functionality, Ca2+ fluxes modulation | Impairment of Ca2+ fluxes, steroid hormone production modulation, MERCs formation, impact on cancer cell survival, cancer cell resistance to drugs | [ref. 18, ref. 112] |
| SigmaR1 | Chaperone activity, ERAD pathway, involvement in VEGF-induced autocrine positive feedback mechanism | Enhancement of hERG activity, increased cell proliferation, angiogenesis promotion | [ref. 94, ref. 95, ref. 114, ref. 115] | |
| SEL1L | ERAD pathway, modulation of PTEN transcription | Increased or decreased tumour growth and aggressiveness depending on the cancer type and metastasis formation | [ref. 116, ref. 316, ref. 317] | |
| Ca2+ | INF2 | Mitochondrial constriction, actin polymerisation, Ca2+ transfer between ER and mitochondria. | Increased invasiveness | [ref. 121, ref. 123, ref. 124] |
GRP75-IP3R-VDAC
One of the most critical axes regulating Ca2+ mobilisation from the ER to mitochondria involves the inositol 1,4,5-trisphosphate receptor (IP3R) on the ER membrane, the Glucose-Regulated Protein 75 (GRP75 or HSPA9) [ref. 22], present in the MAMs and acting as a bridge molecule [ref. 23], and the Voltage-Dependent Anion Channel (VDAC) on the outer mitochondrial membrane (OMM). This complex is further stabilised by accessorial proteins, such as Transglutaminase type 2 (TG2) [ref. 24] and Breast Cancer Type 1 susceptibility protein (BRCA1) [ref. 25], and it can be influenced by ROS production and IP3R post-translational modifications [ref. 5]. Cancer cells can remodel Ca2+ exchange to favour tumourigenesis [ref. 26]. Indeed, mitochondrial Ca2+ uptake stimulates the tricarboxylic acid (TCA) cycle [ref. 27–ref. 29], and supports reductive carboxylation, a metabolic pathway that requires the Ca2+-sensitive enzyme α-ketoglutarate dehydrogenase [ref. 30], thereby promoting cancer cell survival, proliferation and migration [ref. 31–ref. 33] especially when the electron transport chain is impaired [ref. 34], a common feature in cancer cells [ref. 35, ref. 36]. However, excessive mitochondrial Ca2+ accumulation triggers permeability transition pore opening, ultimately leading to apoptosis [ref. 37, ref. 38]. These observations underscore the necessity for a fine regulation of mitochondrial Ca2+ signalling in determining cancer cell fate.
Thus, activation of this axis is exploited by specific tumour-suppressor genes, such as Phosphatase and Tensin Homologue (PTEN), which negatively regulates the oncogene Protein Kinase B (PKB), also known as Akt, which phosphorylates IP3R [ref. 39] and inhibits Ca2+ -mediated apoptosis [ref. 40, ref. 41]. Regarding apoptosis regulation, another key factor is the promyelocytic leukaemia (PML) tumour suppressor protein, which induces cell death by interfering with Akt-dependent phosphorylation of IP3Rs, consequently enhancing the IP3R-mediated Ca²⁺ release from the ER [ref. 42]. Similarly, B-cell lymphoma 2 (Bcl-2) knock-down in ovarian cancer promotes apoptosis by implementing ER-mitochondria Ca2+ exchange and inducing mitochondrial Ca2+ overload [ref. 43].
On the other hand, in certain cancers, mutations in PTEN result in the loss of its function, which impairs Ca2+ transport and confers resistance to apoptosis [ref. 44]. Moreover, under ER stress conditions, the Sigma-1 receptor (SigmaR1), which is often upregulated in cancer cells, binds to IP3R, promoting the influx of Ca2+ inside the mitochondria [ref. 45], and the activation of Ca2+-dependent enzymes of the glycolysis and the TCA cycle [ref. 46].
FUNDC1
FUN14 domain-containing 1 (FUNDC1) is an OMM member of the mitophagy receptor family [ref. 47]. Under hypoxic conditions, it accumulates at the MAMs and it interacts with calnexin, therefore promoting mitochondrial fission by facilitating the recruitment of Dynamin-1-like protein (DRP1) [ref. 48]. FUNDC1, which is often dysregulated in cancer, interacts with various proteins, including HSP70 and mitochondrial fission 1 protein (FIS1), thereby triggering mitophagy which is essential for clearing unfolded proteins. In addition, the interaction of FUNDC1 with IP3R2 plays a crucial role in the transport of Ca2+ to the mitochondria [ref. 49]. As a mitophagy effector, FUNDC1 contributes to cancer biology by modulating mitochondrial bioenergetics and redox homoeostasis, as well as inter-organelle communication, thereby influencing tumour progression and metastatic potential. In vivo data suggest a pro-tumourigenic role for FUNDC1 in cervical cancer, as its loss resulted in the inhibition of cellular proliferation; similarly, it has been demonstrated that FUNDC1 promotes apoptosis induced by cisplatin and ionising radiations [ref. 50]. Another in vitro study identifies FUNDC1 as an oncogenic protein involved in cholangiocarcinoma (CCA) progression. Specifically, FUNDC1 inhibition leads to mitochondrial dysfunction and promotes ferroptosis through a RAC1-dependent mechanism. Moreover, in triple-negative breast cancer, ferroptosis was recently found to be regulated by MERCs also through the modulation of local phospholipid peroxidation [ref. 51]. Disruption of FUNDC1-RAC1 interaction inhibits tumour growth in both human CCA cell lines and tumour specimens [ref. 52] and the pro-tumourigenic role for FUNDC1 is further supported by in vitro studies in breast cancer, showing that FUNDC1 is overexpressed and associated with poor prognosis; in this context, FUNDC1 positively regulates cancer cell proliferation, migration and invasion in a Ca2+-dependent manner [ref. 53]. Furthermore, FUNDC1 may influence cancer progression by promoting neo-angiogenesis, which is critical for cancer dissemination and metastasis formation. Indeed, a decrease in FUNDC1-dependent MAMs formation exerts negative effects on the angiogenic process, potentially due to disrupted Ca2+ homoeostasis and a reduced expression of the vascular endothelial growth factor receptor 2 (VEGFR2) in endothelial cells [ref. 54]. The analysis of FUNDC1 mRNA expression levels revealed that its expression was higher in cancer groups, including breast, cervical, colorectal, lung, ovarian, pancreatic, and prostate cancers as well as leukaemia and lymphoma, respect to control groups [ref. 55]. Indeed, recent findings revealed that the ablation of FUNDC1 results in the accumulation of dysfunctional and fragmented mitochondria, consequently triggering the activation of the inflammasome and potentially attenuating hepatocarcinogenesis in mice [ref. 53].
VAPs
Vesicle-associated membrane-protein-associated proteins (VAPs) are ER proteins that play a crucial role in inter-organelle communication [ref. 56]. VAPs can interact with proteins containing a FFAT (Two Phenylalanines in an Acidic Tract) motif, such as members of the oxysterol-binding protein (OSBP) family or phosphatidylinositol transfer proteins (PITP) from the PITPNM family [ref. 57]. Through these interactions, VAPs contribute to a range of cellular processes including organelle membrane tethering, lipid transfer, Ca2+ homeostasis, autophagy, unfolded protein response (UPR), microtubule organisation, and neurotransmitter release [ref. 56, ref. 58]. Specifically in cancer, VAPs were reported to promote cell proliferation [ref. 59–ref. 61], thus potentially becoming new biomarkers and therapeutic targets [ref. 62].
VAPs sustain MERC formation by interacting with several proteins, like VPS13 (vacuolar protein sorting 13) family which is involved in lipid transport at MCSs. These proteins can bind glycerolipids and facilitate their transfer between organelles, thereby forming a direct channel between adjacent membranes [ref. 63]. Different VPS13 proteins are localised to distinct MCSs. VPS13A works as a tethering factor whose overexpression increases MERC number while its loss impairs lipid transfer between the ER and mitochondria, leading to disruptions in mitochondrial fission, fusion, and mitophagy [ref. 64]. VPS13A also plays a role in cancer since in silico data show that it is highly expressed in tumours including rhabdomyosarcoma, gastric cancer, and ovarian cancer [ref. 65].
Mitoguardin 2 (MIGA2) is a OMM protein involved in de novo triacylglycerol (TAG) synthesis and lipid transport at the MERCs by interacting with VAPA/B [ref. 66, ref. 67]. In vivo experiments show that MIGA2 may support cellular proliferation in ovarian cancer via Yes-associated protein 1 (YAP1), which is regulated through the Hippo signalling pathway. Additionally, MIGA2 and YAP1 can regulate AKT activity, thereby affecting cell growth [ref. 68].
Mitofusins
Mitofusin 1 and 2 (MFN1/2) are homologous proteins belonging to the family of mitochondrial transmembrane GTPases. Despite their similarity, they are not functionally redundant. MFN1 exhibits greater GTPase activity and stronger mitochondrial tethering capacity, whereas MFN2 has additional cellular functions, including the capacity to mediate the tethering between the ER and mitochondria [ref. 69]. Mitofusins play a fundamental role in mitochondrial fusion and in the regulation of other mitochondrial functions, thereby ensuring proper organelle morphology, functionality, and distribution within the cell [ref. 70]. Additionally, they contribute to several cellular processes, like ER homeostasis, and cellular metabolism [ref. 71].
MFN2 has been observed on the ER membrane, particularly at MERCs, where it requires mitochondrial MFN2 or MFN1 to bridge the two organelles and maintain them at an optimal distance [ref. 72, ref. 73]. Cellular processes regulated by MFN2 include Ca²⁺ transfer [ref. 73] and ER-stress response [ref. 74]. However, the role of MFN2 at MERCs is debated, since both a tethering and a spacer function have been proposed for this protein. Specifically, MFN2 downregulation has been associated with an increased formation of short-distance MERCs and enhanced Ca2+ flux between the two organelles [ref. 75–ref. 78], finally impacting on different diseases, such as Alzheimer’s disease [ref. 77]. MFN2 plays a complex and context-dependent role in cancer, functioning as either a tumour suppressor or an oncogene depending on the tissue type. In several cancers, including liver, colorectal, and lung tumours, MFN2 has been shown to promote apoptosis and limit cancer cell proliferation, supporting its role as a tumour suppressor. Mechanistically, MFN2 can negatively regulate tumour growth by modulating the Ras–nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signalling pathway, as its deletion enhances cancer cell proliferation and invasiveness. Likewise, in vitro studies in breast cancer cells demonstrated that MFN2 suppresses proliferation by inhibiting mTORC2/Akt signalling [ref. 79]. However, MFN2 can also function as a tumour promoter. Indeed in vivo, it has been linked to increased cancer cell invasiveness, and its upregulation correlates with poorer overall survival in gastric cancer and lung adenocarcinoma [ref. 80]. These contrasting findings highlight the dual and highly context-dependent nature of MFN2 in tumourigenesis.
BZW2 and BAP31
Basic Leucine Zipper and W2 Containing Protein 2 (BZW2) is a highly conserved protein of the basic-region leucine zipper (bZIP) superfamily of transcription factors [ref. 81]. Among the pathways influenced by BZW2 there are c-Myc, Wnt/β-catenin [ref. 82] ERK/MAPK [ref. 83] and Akt/mTOR [ref. 84]. BZW2 has been described as a new oncogene promoting the progression of different cancers. Its high expression has been documented across different tumours and it is associated with poor prognosis [ref. 85–ref. 88]. More in detail, BZW2 enhances glycolysis in lung adenocarcinoma [ref. 85]. In addition, BZW2 can interact with proteins involved in MERC formation. In pancreatic adenocarcinoma (PDAC), it can bind the ER B-cell receptor-associated protein 31 (BAP31) and VDAC2, sustaining Ca2+ flux and impacting on cell metabolism [ref. 85]. Since many mitochondrial metabolic enzymes are sensitive to Ca2+ levels, targeting this axis can guarantee a reduction in ATP levels, impairing cancer progression. In addition, a decreased TCA cycle activity has been reported upon BZW2 downregulation which is counteracted by an increase in fatty acid oxidation (FAO) rate promoted by the activation of AMPK pathway [ref. 86]. BAP31 is involved in the control and sorting of proteins directed to the ER [ref. 88]. Its cytoplasmatic domain can interact with cytoskeletal elements but also with proteins expressed on the surface of other organelles [ref. 89]. In HeLa cells, BAP31 can bind Fis1: during the first step of Fis1-induced apoptosis, BAP31 undergoes a caspase-like cleavage resulting in p20BAP31 formation. While Fis1-BAP31 tether occurs in physiological condition, Fis1-p20BAP31 interaction is necessary for the recruitment and activation of pro-caspase 8 only upon apoptosis induction. Moreover, p20BAP31 causes the release of Ca2+ from the ER, leading to an increase in cytosolic Ca2+, which is required for triggering apoptosis [ref. 90]. In colorectal cancer cells, p20BAP31 overexpression leads to an inhibition of cell proliferation, a reduction of the clonogenic potential, and an increase in apoptosis. Moreover, it is responsible for a decrease in mitochondrial membrane potential and an increase in mitochondrial ROS, finally leading to the ROS-JNK pathway and caspase activation. p20BAP31 can induce both caspase dependent and independent apoptosis since it can prompt the translocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus, finally promoting DNA fragmentation and cell death [ref. 91].
For what concerns the regulation of MAMs, Phosphofurin acidic cluster sorting protein 2 (PACS-2), a multifaceted regulator of membrane trafficking, significantly contributes to apoptosis, autophagy, and regulation of MAMs [ref. 92], acting in conjunction with Fis1 and BAP31. PACS-2 depletion is associated with mitochondrial fragmentation by stabilising p20BAP31, and is implicated in lipid synthesis and in the formation of autophagosomes. In tumour cells, PACS-2 expression is reduced or completely absent. Despite this, in colorectal cancer, PACS2 may have an indirect role in activating the epidermal growth factor receptor (EGFR), thereby promoting interleukin-6 (IL-6) production and contributing to tumour development [ref. 93].
Translocase of Outer Mitochondrial Membrane 40 (TOMM40), a protein involved in the import of proteins targeted to mitochondria, is one of the proteins interacting with BAP31 [ref. 94, ref. 95]. In nasopharyngeal carcinoma, in vitro experiments show that TOMM40 upregulation correlates with poor prognosis, and it promotes cell proliferation through the activation of AKT/mTOR and p53/p21 signalling pathways [ref. 96]. In oral squamous cell carcinoma, TOMM40 has been proposed as a new oncogene with prognostic value since its expression is enhanced in tumours compared to normal tissues. Furthermore, it correlates with lower disease-free and overall survival [ref. 97]. In mice, the interaction between TOMM40 and BAP31 has been documented and the loss of BAP31 seems to promote autophagy, mitophagy and mitochondrial fragmentation and dysfunction, since it takes part in Complex I assembly [ref. 98].
PERK
Protein Kinase RNA-Like ER Kinase (PERK) is mostly located at MERCs and plays a role in the UPR in cooperation with IRE1 and ATF6 [ref. 99]. On one hand, PERK loss appears to confer a protective mechanism against apoptosis mediated by ROS-induced ER stress by disruption of MERCs [ref. 100] while increasing accumulation of F-actin, which may impair the formation of contacts between the ER and the plasma membrane (PM) [ref. 101]. Additionally, PERK expression increased phosphorylated eIF2α (p-eIF2α) and promoted G0–G1 arrest and cancer cell survival in vitro, negatively impacting on tumorigenesis [ref. 102]. On the other hand, the contacts established through the tethering complex PERK–Ero1a enhance communication between the ER and mitochondria, promoting Ca²⁺ flux that supports the enzymatic activity of TCA cycle and OXPHOS enzymes, ultimately increasing cell viability. PERK activity is further regulated by MFN2, another tethering protein that interacts with PERK to establish ER–mitochondria contacts. Moreover, PERK enables cancer cells to adapt to adverse conditions within the tumour microenvironment. By facilitating the translation of activating transcription factor 4 (ATF4), the PERK-ATF4 axis promotes cell survival, angiogenesis, migration, and dissemination of cancer cells [ref. 103]. In human breast cancer tissues, PERK is often constitutively activated through phosphorylation, and this activation is associated with an increased risk of distant metastasis development [ref. 104]. It has also been proposed that PERK activation exerts an inhibitory effect on immune activation in certain cancer types. In melanoma cells, PERK inhibition impairs the ability to manage ER stress, resulting in paraptosis-mediated immunogenic cell death, which facilitates the differentiation of monocyte precursors [ref. 105]. In triple-negative breast cancer cells, reduced proliferation has been shown to correlate with PERK loss, potentially due to ROS accumulation and oxidative DNA damage, or to inactivation of Nrf2 signalling [ref. 106].
Upon activation, PERK can also interact with ATPase Family AAA Domain Containing 3A (ATAD3A), a protein involved in mitochondrial homeostasis and ER–mitochondria signal transduction [ref. 107], as well as in ER stress modulation, cholesterol trafficking, and cancer metastasis [ref. 108]. The binding of ATAD3A to PERK results in the attenuation of PERK signalling, due to a reduced binding affinity for eIF2α [ref. 109]. Upregulation of both PERK and ATAD3A correlates with an increase in MAM formation, pointing out their involvement in the establishment of MERCs [ref. 109]. In cancer, ATAD3A is overexpressed in various malignancies, including hepatocellular carcinoma (HCC), head and neck squamous cell carcinoma (HNSCC) and lung adenocarcinoma, and it is associated with poor prognosis and decreased survival rates. Conversely, in breast cancer, ATAD3A overexpression determines the inhibition of cell growth and lower levels of ATAD3A expression correlate with reduced patient survival, supporting its tumour-suppressor function. This variability in the impact of ATAD3A expression suggests that its role may differ among distinct tumour types [ref. 110].
Similarly, by interacting with multiple protein partners and acting as a UPR sensor, PERK may differently modulate oncogenic processes depending on the molecular context, such as the prognostic relevance of PERK that varies across tumour types. High PERK expression is associated with poor prognosis in KIRP, LGG, BRCA, and THCA, but with favorable prognosis in HNSC. GSEA results indicate that PERK is mainly enriched in immune-related signalling pathways in BRCA, HNSC, and THCA. PERK expression positively correlates with macrophage infiltration, suggesting that elevated PERK levels may promote immune cell infiltration into the tumour microenvironment and serve as a potential prognostic marker in some cancers. Consistently, another report showed that the inhibition of the PERK–eIF2α–GRP94 signalling pathway suppresses EGFR expression and enhances radiosensitivity of Oral Squamous Cell Carcinoma (OSCC) cells [ref. 111].
Other ER-mitochondria tethers
Foetal and Adult Testis-Expressed 1 (FATE1) is a protein predominantly localised in the testis within normal tissues, although it has been identified in various cancer types. FATE1 is situated at the MERCs, anchored to the ER, and it interacts with Emerin and Mitofilin on OMM. Thanks to this interaction, it influences mitochondrial morphology and functionality [ref. 18]. FATE1 has been shown to increase MERC distance or to disrupt them. Indeed, FATE1 appears to adversely affect Ca2+ fluxes, and steroid hormone production in adrenocortical carcinoma (ACC) thereby enhancing the resistance of cancer cells to apoptosis induced by chemotherapeutic agents [ref. 18, ref. 112, ref. 113].
The Sigma 1 receptor (SigmaR1) is a chaperone protein located on the MAMs, nuclear membrane, and plasma membrane. SigmaR1 serves as a substrate within the ER-Associated Degradation (ERAD) pathway, which is regulated by SEL1L (Suppressor/Enhancer of Lin-12-Like), and it is crucial for maintaining ER protein quality control. SigmaR1 engages with various ion channels across different families, thereby playing a significant role in the regulation of intracellular Ca2+ fluxes and in the formation of MERCs [ref. 94, ref. 114]. SigmaR1 is overexpressed in chronic myeloid leukaemia (CML), acute myeloid leukaemia (AML) and colorectal cancer, where tumour progression correlates with hERG expression. SigmaR1 directly interacts with hERG, thereby enhancing its activity [ref. 95]. Additionally, a positive role for SigmaR1 in facilitating angiogenic processes has been proposed, as it is involved in a VEGF-induced autocrine positive feedback mechanism [ref. 115]. Similarly, in tumour progression, SEL1L regulates the ERAD pathway to maintain protein homeostasis. In response to ER stress, this pathway is activated to degrade unfolded proteins through the proteasome, finally restoring ER homeostasis. Indeed, both in vitro and in vivo studies indicate that SEL1L prevents the accumulation of misfolded or aberrant proteins that would otherwise promote tumour aggressiveness [ref. 116]. SEL1L is a multifunctional regulator whose impact on tumour biology is highly context- and cancer-dependent. While SEL1L is widely expressed in normal tissues, its expression is frequently reduced or lost in several tumour types, suggesting a context-dependent role as either an oncogene or a tumour suppressor. In pancreatic cancer cells, for example, loss of SEL1L reduces tumour growth and aggressiveness, an effect attributed in part to its modulation of PTEN transcription and subsequent influence on early metastatic processes [ref. 81]. SEL1L can also function as a negative regulator of Notch signalling, further implicating it in key pathways governing tumour progression [ref. 82]. Consistent with these observations, SEL1L expression in breast and pancreatic carcinomas has been associated with decreased cellular aggressiveness. In breast cancer, SEL1L appears to inhibit Epithelial-Mesenchymal Transition (EMT) by promoting Sonic hedgehog (Shh) degradation, whereas in pancreatic cancer its tumour-attenuating function has been linked to the regulation of the TGF-β pathway [ref. 117, ref. 118]. In contrast, SEL1L overexpression correlates with favourable clinical features in certain malignancies. In colorectal cancer, increased SEL1L levels have been associated with well-differentiated tumour cells and decreased aggressiveness, indicating that SEL1L upregulation is predictive of a better prognosis [ref. 119]. However, the relationship between SEL1L expression and tumour behaviour differs across cancers. In several gliomas, SEL1L upregulation has been reported in association with TERT promoter mutations and EGFR amplification, molecular alterations that are considered negative prognostic markers in these tumours [ref. 120].
Inverted formin-2 (INF2) is an ER protein involved in mitochondrial constriction and it can assemble actin subunits into filaments, to sustain the contraction process [ref. 121]. INF2 interacts with Protein spire homologue 1 (Spire1C) during the formation of actin filaments [ref. 122]. Additionally, by promoting MERCs, INF2-mediated actin polymerisation is essential for Ca2+ transfer between ER and mitochondria [ref. 123]. Importantly in cancer, it is well-established that actin structures significantly contribute to the invasive properties of tumour cells [ref. 124]. Experiments on MDA-MB-231 cells have demonstrated that an increase in Spire1 enhances cancer cell invasion of the endothelial monolayer, indicating that this protein, along with other ones involved in actin nucleation, plays a role in promoting cancer invasiveness [ref. 125].
ER-lysosome contact sites
Despite the close relationship with mitochondria, the ER is involved in other contacts with different organelles, such as endosomes and lysosomes [ref. 126]. Endocytosis is a cellular process through which cells internalise extracellular material or PM components through the formation of intracellular vesicles called endosomes. Endosomes are classified based on their grade of maturation in early and late endosomes [ref. 127]. While moving toward lysosomes, endosomes undergo a maturation process involving a progressive pH acidification mediated by Golgi-derived proton pumps. While early endosomes show Rab5 as one of their main membrane proteins, upon endosome maturation, Rab5 is displaced by Rab7 which is involved in the interaction between endosomes–lysosomes and ER. Moreover, the lipid PI(3)P of early endosomes is phosphorylated to PI(3,5)P in late endosomes, thus affecting the recruitment of sorting and tethering factors [ref. 128]. The existence of tight and stable connections between ER and endosomes has been investigated through electron microscopy [ref. 129]. Endosomes associate with the ER early during their maturation process and the number and duration of these interactions increase as endosomes mature. Indeed, in Cos-7 cells ER rearranges its structure to maintain this interaction through the opening or closure of membranous rings, usually at the three-way junction [ref. 130]. Importantly, the contacts with the ER control the position of endosomes within the cell [ref. 129]. Endosomes mainly localise in the perinuclear region and display a static behaviour. However, a small number of intracellular vesicles are highly dynamic and move towards the periphery of the cell. E3 ubiquitin Ligase Ring finger protein 26 (RNF26) is a protein localised on the ER proximal to the nucleus and it is involved in the late endosomes’ spatial organisation and vesicles maturation in the perinuclear region in vitro [ref. 131].
Interestingly, an increasing number of studies are demonstrating the biochemical heterogeneity of ER-endosome/lysosomes contacts, highlighting the presence of different tethering axes both in different cellular types and in the same cellular contexts (Fig. 2). Hence, the ER-endosomes associations have been proven to participate in different cellular processes ranging from endosomal positioning [ref. 131] to endosomes fission events [ref. 132], lipid and cholesterol transport [ref. 133], ion homeostasis [ref. 134] and intracellular signalling [ref. 135]. In the next paragraph, we will discuss the tethers involved in ER-endosome/lysosomes contacts in cancer (Table 2).
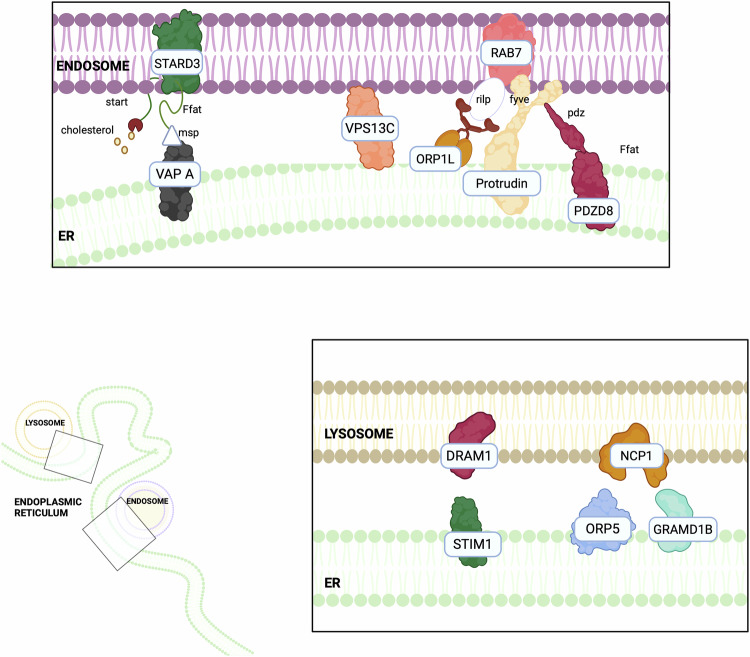
Table 2: Main ER-lysosomes contact sites and their associated features.
| Cargo | Main tether (ER-lyso) | Biological functions | Tumour phenotype | Ref. |
|---|---|---|---|---|
| Cholesterol | STARD3-VAPs | Scaffold ER-endosome contacts, delivery of cholesterol to endosomes | Alterations in cholesterol metabolism, modulation of membrane permeability and fluidity, transduction of survival signals, increased proliferation, metastasis formation, blockade of lysosomal degradation, androgen biosynthesis | [ref. 137, ref. 138, ref. 142, ref. 144, ref. 318–ref. 320] |
| Cholesterol | ORP5-NPC1 | Cholesterol delivery to ER, lipid metabolism modulation | Dysregulated lipid metabolism, increased cancer cell invasion and tumour progression, decreased overall survival | [ref. 148, ref. 149, ref. 151, ref. 152] |
| Cholesterol | ORP1L-VAPs | Cholesterol transport to ER, regulation of endosome dynamics, formation of late endosome contact site | Modification of lysosome positioning, cancer cell autophagy, enhanced cancer cell growth, invasion and metastasis | [ref. 158–ref. 161] |
| Protrudin | Promotion of cell protrusion and neurite outgrowth, kinesin-driven transport of LEs | Maturation and elongation of invadopodia, cancer cell invasion | [ref. 159, ref. 164, ref. 321] | |
| Ca2+ | DRAM1-STIM1 | Regulation of autophagy, apoptosis, and cellular stress responses, the SOCE pathway | Impaired Ca2+ homeostasis, ER stress, increased or decreased cell proliferation, migration, invasion, depending on the tumour type, apoptosis inhibition | [ref. 167, ref. 170, ref. 171, ref. 173, ref. 322, ref. 323] |
STARD3-VAPs and their interactors
Steroidogenic acute regulatory protein (StAR)-related lipid transfer domain containing 3 (STARD3), which is located at the membrane of late endosomes, is a scaffold protein in ER–endosome contacts, and it interacts with VAPA/B [ref. 136, ref. 137]. In HeLa cells, elevated STARD3 expression is associated with the accumulation of sterols in the late endosomes, whereas the silencing of VAPA/B determines the inability to accumulate cholesterol in endosomes [ref. 138]. The STARD3‑VAP axis has the potential to contribute to cancer progression, as alterations in cholesterol metabolism can modulate membrane permeability and fluidity, and the transduction of survival signals, which are processes implicated in tumour development (e.g. via enhanced oncogenic signalling linked to STARD3‑mediated cholesterol redistribution in cancer cells) [ref. 139, ref. 140]. Indeed, cancer cells exhibit elevated levels of intracellular cholesterol compared to nontumoural ones [ref. 141]. Elevated STARD3 levels are associated with cancer proliferation and metastasis formation both in vitro in breast cancer cells, and in vivo, especially in hormone-driven cancers, such as breast and prostate cancer, where it induces an independent steroidogenesis process [ref. 137]. Additionally, an overexpression of STARD3 may be associated with a blockade of the late endosome formation, and the consequent inhibition of their maturation into lysosomes thus preventing lysosomal degradation [ref. 142]. This is relevant in breast cancer, since it implies that some growth factor receptors, including HER2, are not degraded and can sustain an uncontrolled cellular growth [ref. 143]. Moreover, elevated STARD3 levels have also been detected in prostate cancer patients, where its high expression is associated with an increased transport of cholesterol to mitochondria and consequently to an enhancement of androgen biosynthesis [ref. 144].
In addition to STARD3, VAP proteins can bind to a plethora of proteins, like VPS13C, a member of the VPS13 protein family, which localises at contacts between the ER and late endosomes/lysosomes. VPS13C can interact with VAP and with the late endosomal GTPase Rab7, further supporting its role in endo-lysosomal tethering and function [ref. 145]. Mutations in the VPS13C gene are connected to endometrial, gastric, and colorectal cancers. Moreover, the loss of VPS13C is associated with the development of resistance to cisplatin in cervical cancer cells, suggesting a role in mediating chemotherapy sensitivity [ref. 146].
Among the VAP interactors, Oxysterol-binding protein (OSBP) and its related proteins (ORPs) are part of a large family of lipid-binding proteins which transport lipids between cellular membranes in a non-vesicular manner [ref. 147, ref. 148]. Among them, ORP5 can bind lipids and target organelle membranes to mediate sterol signalling and transport [ref. 149]. ORP5 interacts with Niemann-Pick type C protein 1 (NPC1), localised in the late endosomes/lysosomes membrane, at ER-lysosome contact sites. NPC1 plays a crucial role in regulating contact formation between the ER and late endocytic vesicles: the knockdown of NPC1 reduces these contact sites, while NPC1 overexpression exerts the opposite effect [ref. 150]. ORP5-NPC1 interaction contributes to extract cholesterol molecules from the late endosomes/lysosomes membrane and deliver them directly to the ER in vitro [ref. 148, ref. 151]. Both NPC1 and ORP5 are highly expressed in several cancer types. ORP5 is overexpressed in pancreatic and lung cancer cells, where it has been linked to increased cancer cell invasion and tumour progression [ref. 149]. Similarly, NPC1 is upregulated in breast cancer, and its elevated expression correlates with a decreased overall survival in glioma and an increased risk in oesophageal cancer [ref. 152]. Although the precise mechanisms by which NPC1 and ORP5 contribute to cancer progression remain unclear, it is hypothesised that their overexpression may enhance lipid transfer activity, leading to dysregulated lipid metabolism, altered membrane lipid organisation, and aberrant cell signalling [ref. 153, ref. 154]. NPC1 can also interact and form contacts with the ER-localised sterol transport protein Gramd1b [ref. 150]. Interestingly, Gramd1b has also been implicated in different malignancies. In ovarian cancer, for example, it has been associated with chemoresistance [ref. 155], while in gastric cancer, it promotes cell survival by upregulating the expression of the anti-apoptotic protein Bcl-xL [ref. 156]. Moreover, JAK/STAT signalling positively regulates Gramd1b expression in both breast and gastric cancer cells [ref. 156, ref. 157].
ORP1 exists in a short (S) and in a long (L) isoform and the ORP1L can interact with Rab7 regulating the positioning of the endosomes [ref. 133]. By interacting with VAPs, ORP1L functions as a late endosomal protein involved in cholesterol transport to the ER, the regulation of endosome dynamics, and the formation of late endosome (LE)-ER contact sites [ref. 158, ref. 159]. In melanoma cells, ORP1L has been shown to sense endosomal cholesterol levels and, consequently, to modify lysosomal positioning in the cell [ref. 160]. The ability of this tether to regulate lysosomal localisation is particularly relevant in cancer, as lysosome distribution significantly influences the biological properties of cancer cells [ref. 161], since plays a crucial role in cancer cell autophagy [ref. 162], and has been associated with enhanced cancer cell growth, invasion, and metastasis [ref. 161]. Moreover, in prostate cancer cells, Rab7 is significantly downregulated, leading to lysosome redistribution toward the cell periphery, thereby supporting cancer cell invasiveness and migration [ref. 163].
Finally, Protrudin is an ER protein that promotes cellular protrusion and neurite outgrowth by mediating the plus-end trafficking of late endosomes along microtubules [ref. 159]. Thanks to its structure, Protrudin simultaneously interacts with endosomal PI3P, Rab7, and with VAPA, thereby promoting the formation of ER–endosome contact sites [ref. 129, ref. 164]. Protrudin also harbours a binding domain for Kif5b (kinesin-1 heavy chain), which allows its recruitment to the ER–late endosome membrane contacts, supporting the kinesin-driven transport of late endosomes toward the cell periphery. Their accumulation at the plasma membrane leads to endosome fusion, thereby promoting cellular protrusions formation [ref. 165]. In cancer cells, the formation of extracellular protrusions in PM, called invadopodia, allows the secretion of matrix metalloproteinases to degrade the extracellular matrix (ECM), a crucial point for breaching the basement membrane and invading [ref. 166]. In breast cancer cells, Protrudin plays a pivotal role in the maturation and elongation of invadopodia by mediating late endosomes/lysosome translocation to the cell periphery and their subsequent fusion with the PM [ref. 164]. These findings highlight the critical role of Protrudin in cancer cell invasion. Notably, high Protrudin expression correlates with reduced survival in patients with breast, gastric, and ovarian cancers, highlighting its potential as a therapeutic target [ref. 164].
DRAM1-STIM1
DNA Damage-Regulated Autophagy Modulator 1 (DRAM1) is a lysosomal protein identified as a regulator of autophagy, apoptosis, and cellular stress responses. As a transcriptional target of p53, DRAM1 plays a crucial role in p53-dependent autophagic and apoptotic pathways, which are fundamental in tumourigenesis [ref. 167]. Recent studies have demonstrated that DRAM1 interacts with stromal interaction molecule 1 (STIM1), a Ca2+ sensor located in the ER, ultimately facilitating lysosome-ER tethering. Through this interaction, DRAM1 modulates ER structure and function [ref. 168]. Moreover, STIM1 is responsible for monitoring and replenishing ER-luminal Ca²⁺ levels via store-operated Ca2+ entry (SOCE), thereby maintaining Ca2+ homeostasis. Loss of STIM1 disrupts this balance, leading to impaired Ca²⁺ homeostasis, to the activation of Ca2+ signalling, to increased ER stress, and to the subsequent induction of autophagy [ref. 169]. The DRAM1-STIM1 interaction interferes with STIM1-mediated Ca²⁺ homeostasis by reducing intracellular, mitochondrial, and ER Ca²⁺ levels while increasing lysosomal ones. This occurs through DRAM1 sequestering STIM1, therefore facilitating the transfer of Ca²⁺ from the ER to lysosomes and triggering ER stress and ER-phagy in U2OS and HEK-293T cells [ref. 168]. Interestingly, in non-small cell lung cancer (NSCLC) patients, DRAM1 is downregulated, whereas its overexpression suppresses cancer cell proliferation in vitro and in vivo, as well as cell migration, and invasion [ref. 134]. Its anti-tumour activity is associated with its function as a p53 target gene, which promotes apoptosis and autophagy activation, ultimately limiting tumour growth. Moreover, DRAM1 was reported to regulate the PI3K/Akt/mTOR pathway, exhibiting both pro- and anti-tumourigenic functions across the literature. In particular, DRAM1 was found to inhibit the PI3K/Akt/mTOR signalling pathway in NSCLC cells, likely through its enhanced autophagic flux and lysosomal enzyme activation [ref. 170]. In contrast, in gastric cancer cells DRAM1 promoted PI3K/Akt/mTOR activation and tumour progression [ref. 171], reflecting the possible context-dependent functional effects of DRAM1 on signal transduction and metabolism [ref. 172]. In glioblastoma cells, DRAM1-driven modulation of autophagy has been reported to promote tumour growth [ref. 173]. STIM1 likewise displays heterogeneous roles across malignancies. It is broadly associated with cancer progression and increased invasiveness, and in many contexts, it exerts an inhibitory effect on apoptosis [ref. 135]. STIM1 also serves as an important regulator of autophagy, with elevated intracellular Ca²⁺ promoting autophagy in hepatocellular carcinoma cells [ref. 136]. Nonetheless, contradictory findings exist, as STIM1 inhibition has been shown to induce autophagy [ref. 174].
Mitochondria-lysosomes contact sites
Several studies have also demonstrated the formation of mitochondria-lysosomes contacts [ref. 175] (Fig. 3), mainly regulated by Rab7, which alternates between an active, lysosome-localised GTP-bound state, and an inactive, cytosolic GDP-bound state. The Rab7-GTP hydrolysis involves the recruitment of the cytosolic GTPase-activating protein (GAP) TBC1D15 to mitochondria and its interaction with the lysosomal GTP-bound Rab7, promoting its hydrolysis to the GDP-bound state. This transition prevents Rab7 from binding to its effectors, leading to its release from the lysosomal membrane and subsequent contact untethering [ref. 7]. Mitochondria-lysosome contacts are involved in mitophagy, which is often deregulated during cancer progression [ref. 172]. However, mitochondria-lysosome contact formation is not exclusively linked to mitophagy [ref. 176], but it is also crucial for regulating both organelle dynamics [ref. 175]. Lysosomal function is modulated through Rab7: following Rab7-GTP hydrolysis, the release of Rab7 effector proteins from the lysosomal membrane influences lysosomal positioning and trafficking [ref. 177]. Mitochondrial dynamic is also regulated at mitochondria-lysosome contacts, where they mark sites of mitochondrial fission and play a role in controlling the rate of fission events [ref. 175]. Furthermore, these contacts serve as platforms for the exchange of Ca2+, lipids, and iron between the two organelles [ref. 177]. Mitochondria-lysosomes tethering regions can also be regulated by specific signals. Indeed, under hypoxic conditions, these contacts facilitate microfusions with endolysosomes, leading to the post-translational cleavage of VDAC1 by endolysosomal enzymes. This cleavage protects mitochondria from mitophagy, thereby promoting cell survival during hypoxic stress [ref. 178]. Additionally, oxidative stress enhances mitochondria-lysosomes interactions, mediating the transfer of lipids and metabolites between the organelles to reduce apoptosis and support mitochondrial repair [ref. 179]. In the next paragraphs, we will discuss which of the already discovered tethers have been shown to have an impact on cancer (Table 3).
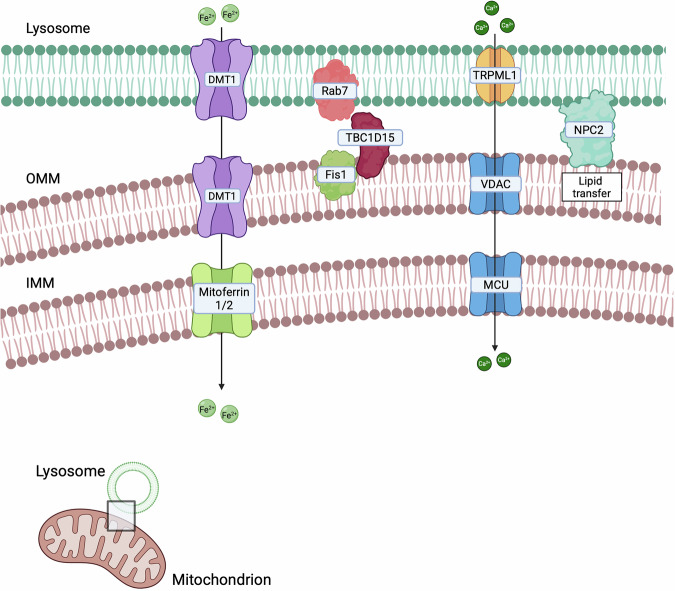
Table 3: Main mitochondria-lysosome contact sites and their associated features.
| Cargo | Main tether (mito-lyso) | Biological functions | Tumour phenotype | Ref. |
|---|---|---|---|---|
| LAMP1-TOM20 | Lysosomal integrity protection, vesicle fusion, regulation of ATP production and maintenance of mitochondrial membrane potential | Increase in metastatic potential, cell proliferation and migration, cell cycle control and apoptosis modulation. | [ref. 324–ref. 329] | |
| DMT1 | Iron homoeostasis (iron transfer to mitochondria) | Enhancement of invasiveness and migration, mitophagy inhibition, increase in mitochondrial respiration and glycolysis. | [ref. 180] | |
| Lipids | NPC2 | Lipid trafficking | Increased or decreased cancer cell proliferation and migration depending on the tumour type | [ref. 185, ref. 189, ref. 190, ref. 192, ref. 330] |
| Ca2+ | TRPML1-VDAC/MCU | Ca2+ flux into the mitochondrial matrix | Ferroptosis inhibition, stemness rescue, regulation of oncogenic autophagy | [ref. 193, ref. 195] |
| Rab7-TBC1D15-Fis1 | Mitophagy regulation | Drug sensitivity enhancement, increased cancer aggressiveness, mitophagy promotion | [ref. 197, ref. 200, ref. 201, ref. 203] |
DMT1
Divalent Metal Transporter 1 (DMT1) is involved in iron homeostasis by mediating the transient interaction between endosomes and mitochondria for direct iron translocation [ref. 180]. This process is essential for limiting the intracellular labile iron pool, thereby reducing the production of ROS. Epidemiological data suggest that elevated iron levels constitute a risk factor for various cancers, including breast cancer [ref. 181]. Consistent with the central role of iron trafficking in tumour biology, DMT1 functions as a molecular bridge connecting endosomal membranes with the OMM, thereby facilitating the transfer of iron to mitochondria. This process supports mitochondrial bioenergetics and promotes breast cancer invasive cellular migration, and indeed, increased expression of DMT1 in breast cancer cells correlates with poorer overall survival [ref. 180]. Overexpression of DMT1 enhances mitochondrial iron uptake, whereas its silencing disrupts endosome–mitochondria interactions, resulting in reduced mitochondrial iron import, an elevated intracellular labile iron pool, increased mitochondrial superoxide production, induction of mitophagy, and a decline in both mitochondrial respiration and glycolysis [ref. 180]. Such regulatory effects on endosome–mitochondria communication have been observed in MDA-MB-231 triple-negative breast cancer cells, but not in the non-invasive luminal A T47D line [ref. 180]. Intriguingly, functional studies suggest that the effects of DMT1 downregulation are highly context dependent. In breast cancer models, DMT1 can increase metastatic potential; in particular, in MDA-MB-231 cells DMT1 downregulation promotes lung metastasis overgrowth in vivo, indicating that DMT1-dependent iron metabolism contributes to the malignant phenotype [ref. 180]. These findings suggest that although DMT1 supports metabolic functions associated with invasiveness, its absence may activate compensatory pathways associated with metastatic dissemination. Evidence from head and neck cancer (HNC) cell lines shows that DMT1 silencing favours ferroptosis, accompanied by iron accumulation through lysosomal sequestration, thereby triggering an iron-starvation response [ref. 182]. DMT1 is also upregulated in colorectal cancer tissue, where the resulting increase in iron flux appears to promote tumour initiation and sustain in vivo activation of the oncogenic JAK1–STAT3 signalling pathway [ref. 183]. In contrast, in HCC, DMT1 expression is associated with suppressed glycolysis and oxidative phosphorylation in silico, and correlates with better patient prognosis [ref. 184]. Collectively, these findings underscore the complex and context-dependent roles of DMT1 in cancer, highlighting its involvement in mitochondrial metabolism, iron homeostasis, cell survival, and metastatic progression.
NPC2
NPC2 plays an important function on contacts between mitochondria and endo-lysosomes, where it mediates lipid trafficking. Indeed, NPC2 depletion in cells expressing functional NPC1 resulted in reduced mitochondrial cholesterol levels [ref. 185]. Accordingly, NPC2 ablation in HEK cells leads to an accumulation of unesterified cholesterol within late endosomes/lysosomes. Loss of NPC2 has been associated with alterations in the number of mitochondria-lysosomes contacts [ref. 186], even if this is debated [ref. 187]. NPC2 exhibits a context-dependent role in cancer, acting either as a tumour promoter or as a tumour suppressor depending on tissue type. In HCC and lung adenocarcinoma, NPC2 is downregulated, and when it is overexpressed inhibits cell proliferation and migration [ref. 188, ref. 189]. Similarly, in glioblastoma, NPC2 overexpression inhibits tumour growth and cell migration, indicating a protective role [ref. 190, ref. 191]. Conversely, in gastric cancer, NPC2 levels are significantly elevated in patients and in gastric cancer cells, and its absence reduces cell proliferation. In other cancers, including breast, colon, and lung cancers, NPC2 is often overexpressed and promotes proliferation, migration, and invasion [ref. 192]. These findings highlight the dual and tissue-specific role of NPC2 in tumour context.
TRPML1-VDAC/MCU
Transient receptor potential mucolipin 1 (TRPML1) is a non-selective ion channel which is expressed on the surface of lysosomes and late endosomes that mediates Ca2+ efflux [ref. 193]. TRPML1 loss-of-function mutation has been associated with many neurological and lysosomal disorders, including Mucolipidosis IV (MLIV) [ref. 194]. In these pathologies, TRPML1 activity not only modulates the mobilisation of Ca2+, but it affects the mitochondria-lysosomes contacts too [ref. 194]. TRPML1 has been proven to be involved in different cancers like breast cancer, where it inhibits ferroptosis in cancer stem cells and reduces their stemness [ref. 193]. TRPML1 is also a key player in the regulation of oncogenic autophagy in breast and pancreatic cancer cells [ref. 195]. Its involvement in mitochondria-lysosomes contacts highlights its capability to modulate Ca2+ influx into the mitochondrial matrix, as shown in cervical and colon cancer cells. The modulation of mitochondria-lysosomes interaction correlates with an increase in Ca2+ efflux from lysosomes to mitochondria, through VDAC1 and MCU (mitochondrial Ca2+ uniporter), which does not cause the permeability transition pore opening neither the activation of apoptotic pathways [ref. 194]. Moreover, in TNBC cells, TRPL1 downregulation is associated with reduced mitochondria-lysosome contact sites, counteracted by increased ER-mitochondria proximity, ultimately resulting in metabolic stress, G0/G1 cell cycle arrest, cell death and enhanced chemosensitivity [ref. 196].
Rab7-TBC1D15-Fis1
Rab7 has been described as one of the main tethers between mitochondria and lysosomes [ref. 175]. Its GTPase activity modulates mitochondria-lysosomes crosstalk, through TBC1D15 which is recruited at mitochondria by Fis1. In addition, in cervical cancer, neuroglioma, human embryonic kidney and colorectal cancer cells, mitochondria fission events occur predominantly at mitochondria-lysosomes contacts, where Drp1 is also located [ref. 175]. The role of Rab7 has been investigated in different types of cancer. For example, its downregulation in ovarian cancer leads to cisplatin resistance due to a dysregulation of the endocytic pathway that leads to cisplatin efflux [ref. 197]. Moreover, chemo-resistant ovarian cancer cells display an impaired lysosomal biogenesis and mitochondria quality control which increases the secretion of extracellular vesicles. Furthermore, in PDAC, Rab7 upregulation has been described as a poor prognostic marker [ref. 198]. Its expression has been demonstrated to be influenced by the level of NDUFS3, a subunit of mitochondrial complex I involved in its assembly. As a consequence of NDUFS3-mediated Rab7 downregulation, the endocytic pathway and mitochondrial morphology are impaired, and cancer cells acquire a less aggressive phenotype [ref. 199]. In addition, Rab7 is upregulated in gastric tumour when compared to normal tissue and it promotes cancer aggressiveness [ref. 200]. Similarly, a role for Rab7 both in vitro and in vivo in driving melanoma progression has been suggested, finally impacting on the metastatic risk [ref. 201].
The role of Rab7 in mitochondria-lysosome contacts has been well characterised in HCC where it participates in the regulation of mitophagy. In these cells a more fragmented mitochondrial network can be observed if compared to non-tumoural cells and this reflects a possible involvement of mitophagy in the progression of the disease [ref. 202]. After being phosphorylated, Drp1 is recruited to the OMM, where it promotes the division of mitochondria, which represents the starting point for mitophagy [ref. 203]. In colon cancer patient-derived cells, fatty acids promote ERK-dependent Drp1 phosphorylation enhancing mitophagy and cancer cell metabolic plasticity, finally impacting on intracellular signalling by stabilising β-catenin via FAO-dependent acetylation [ref. 204]. In HCC cells, Drp1 expression levels are higher than normal tissues and correlate with poorer outcomes, and Drp1 overexpression leads to its translocation at mitochondria-lysosomes contacts where it interacts with Rab7 and mediates PINK1-Parkin-dependent mitophagy [ref. 205]. In hypoxic conditions, Drp1 inhibition can enhance the sensitivity of HCC cells to apoptosis [ref. 206]. Therefore, targeting this axis can lead to HCC cells sensitivity to anti-tumoural therapies by promoting apoptosis [ref. 205].
BDH2
Mitochondria-lysosome contact sites have also been described as platforms for iron exchange between these two intracellular compartments [ref. 207]. The enzyme 3-hydroxybutyrate dehydrogenase type 2 (BDH2) catalyzes the production of 2,5-dihydroxylbenzoic acid (2,5-DHBA), a siderophore involved in mitochondrial iron import [ref. 208]. BDH2 localises at mitochondria-lysosome contact sites and, together with its product, contributes to the regulation of intracellular iron homeostasis and trafficking. In melanoma, iron redistribution between mitochondria and lysosomes has been demonstrated to correlate with tumour phenotype [ref. 207]. Specifically, melanoma can be classified into a proliferative and differentiated phenotype characterized by an high microphthalmia-associated transcription factor (MITF), and an invasive, poorly differentiated phenotype with low MITF levels [ref. 209] which is also characterised by reduced BDH2 expression [ref. 207]. In this context, decreased BDH2 expression correlates with reduced mitochondrial iron content and the consequent accumulation in lysosomes, leading to impaired activity of mitochondrial iron-dependent enzymes and diminished oxidative respiration in vitro. In addition, the altered iron distribution increases sensitivity to ferroptosis, an iron-dependent mechanism of cell death, which can be reversed by restoring BDH2 expression or by treatment with the siderophore 2,5-DHBA [ref. 207].
Nucleus-mitochondria contact sites
Mitochondrial retrograde signalling enables mitochondria to communicate with the nucleus and orchestrate adaptive transcriptional programmes in response to cellular stress. This communication is facilitated by the formation of mitochondria–nucleus contacts (Fig. 4 and Table 4). Under stress conditions, mitochondria redistribute to the perinuclear region and form stable contacts with the nuclear envelope [ref. 210], promoting nuclear integrity and the expression of pro-survival genes, mainly via activation of NF-κB, modulation of cholesterol, production of ROS, and regulation of Ca2+ levels [ref. 211]. The first tether identified was the translocator protein TSPO, which is overexpressed in many tumours, where it binds cholesterol and it is involved in the suppression of mitophagy. This protein interacts with the A-kinase anchoring protein acyl–coenzyme A binding domain containing 3 (ACBD3) and the protein kinase A (PKA), which both complex with the nucleus via the A-kinase-anchoring protein (AKAP95). This tether facilitates cholesterol delivery to the nucleus, thereby sustaining NF-κB activity by preventing its deacetylation and reinforcing cell survival. In breast cancer cells, TSPO overexpression correlates with increased nucleus-mitochondria contacts and enhanced NF-κB recruitment [ref. 210, ref. 211]. Additionally, MFN2 has been implicated in nucleus-mitochondria contacts. Proliferative signals induce the redistribution of mitochondria around the nucleus and concomitantly they trigger an MFN2 enrichment at mitochondria–nucleus interface [ref. 212]. This observation supports a model in which MFN2 mediates mitochondrial repositioning towards the nucleus to facilitate growth stimuli. In vitro, upon proliferative signals, nucleus-mitochondria contacts also permit translocation of the pyruvate dehydrogenase complex (PDC) into the nucleus, where it interacts directly with Lamin A [ref. 213].
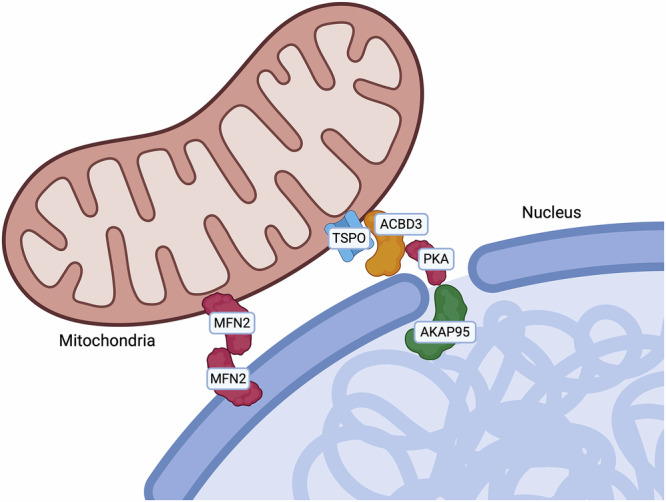
Table 4: Main nucleus-mitochondria contact sites and their associated features.
| Cargo | Main tether (nucleus-mito) | Biological functions | Tumour phenotype | Ref. |
|---|---|---|---|---|
| Cholesterol | TSPO-ACBD3 | Transport of cholesterol to the nucleus | Mitophagy inhibition, increase of nucleus-mitochondria contacts, enhanced Nfkb recruitment | [ref. 210, ref. 211] |
| MFN2 | Mitochondrial repositioning towards the nucleus | [ref. 231] |
ER-Peroxisomes contact sites
Peroxisomes are essential organelles that play critical roles in lipid and hydrogen peroxide metabolism [ref. 214]. The interactions between peroxisomes and the ER are vital for lipid metabolism, with peroxisomes relying on the ER for their lipid composition [ref. 214] (Fig. 5 and Table 5). Approximately 70% of peroxisomes in mammalian cells have been observed to associate with the ER, facilitating the direct lipid transfer between these organelles [ref. 215], the ether-phospholipid biosynthesis (a process initiated in peroxisomes and completed in the ER), and the synthesis of polyunsaturated fatty acids (PUFAs), essential for membrane plasticity modulation and peroxisome dynamics [ref. 216]. Additionally, peroxisomes participate in cholesterol trafficking to the PM, with the ER potentially serving as an intermediate organelle [ref. 217]. Since cholesterol alteration is reported to contribute to cancer development, this axis may have a role in influencing tumourigenesis.
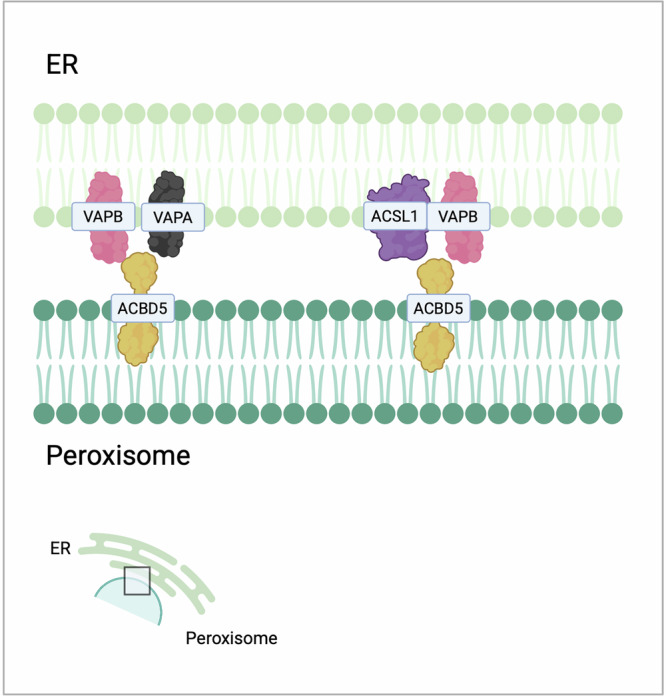
Table 5: Main ER-peroxisome contact sites and their associated features.
| Cargo | Main tether (ER-perox) | Biological functions | Tumour phenotype | Citations |
|---|---|---|---|---|
| Lipids | ACBD5-VAPs | Lipid transfer, metabolic cooperation between peroxisomes and ER, FAO and phospholipid biosynthesis, peroxisome movement and positioning | [ref. 215, ref. 218, ref. 219] | |
| Fatty acids | ACSL1 | Contact formation, branched-chain fatty acid metabolism | Increase in cancer cell proliferation and migration, EMT promotion | [ref. 220, ref. 221] |
Recent studies elucidated the mechanisms underlying these interactions, emphasising the significance of acyl-coenzyme A-binding domain protein 5 (ACBD5) as a key tethering protein. ACBD proteins facilitate the establishment of contacts through their interaction with VAPs, thereby enhancing lipid transfer and metabolic cooperation between peroxisomes and the ER, essential for maintaining cellular metabolism, including FAO and phospholipid biosynthesis [ref. 218, ref. 219]. The activity of this axis regulates peroxisome movement and positioning, suggesting that contacts can influence organelle motility and, consequently, cellular homeostasis [ref. 215]. Moreover, the VAPA-ACBD5 axis appears to be necessary for peroxisome growth since it exploits lipid transfer for membrane expansion. Interestingly, overexpression of ACBD5 has been associated with peroxisomal elongation in a VAP-dependent manner in vitro [ref. 217].
Long-chain acyl-CoA synthetase 1 (ACSL1) facilitates the transfer of fatty acids into the mitochondria for subsequent β-oxidation [ref. 220]. In the ER, ACSL1 establishes interactions with several peroxisomal proteins, such as ACSL1, VAPB, and ACBD5, promoting the formation of contacts. Furthermore, ER-targeted ACSL1 appears to interact with proteins involved in branched-chain fatty acid metabolism [ref. 220]. Various tumours are characterized by impaired expression of ACSL1. In colorectal and endometrial cancer cells, the absence of ACSL1 negatively impacts cancer cell proliferation and migration, while its upregulation correlates with EMT both in vitro and in vivo [ref. 221]. Additionally, ACSL1 may promote cancer cell proliferation and metastatization through the activation of AMP-activated protein kinase (AMPK) [ref. 222], that in turn can induce FAO, resulting in ATP production and impaired normal physiological processes in cancer cells [ref. 223].
Lipid droplets contact sites
Lipid droplets (LDs) are spherical dynamic organelles which can be found in different cell types [ref. 224]. They are composed by a core of neutral lipids surrounded by a monolayer of phospholipids [ref. 225] and proteins belonging to the perilipin (PLIN) family which participate both in the synthesis and hydrolysis of the neutral lipids of the LD core [ref. 224]. Based on their localisation, LDs can be classified in cytoplasmatic, nuclear or luminal [ref. 226]. In mammals, LDs can be found free in the cytosol, or they can keep a functional connection with other organelles [ref. 227]. Membrane contacts between ER and LDs have been described to support lipid transport and homeostasis (Table 6). In detail, proteins of the ORPs family have been proven to participate in these ER-LD contacts [ref. 133]. ORP2 localises on the surface of LDs and it can bind VAPs, thus regulating LDs neutral lipid turnover and LDs positioning [ref. 228]. Furthermore, ORP5 localises at ER-LDs contacts where it mediates the counter-transport of PS and PI(4)P between ER and LDs, finally impacting on LDs growth [ref. 229]. Interestingly, ORP5 and ORP8 located at MERCs take part in the local lipid droplet biogenesis and maintenance, becoming key players at the triple interface among ER, mitochondria and LDs [ref. 230]. Moreover, these triple connections, which seemed to facilitate lipid anabolic pathways rather than FAO, correspond to a peculiar MERC proteome with an enrichment in proteins involved in lipid metabolism, including long-chain acyl-CoA metabolism, and metabolite processing from pyruvate [ref. 231].
Table 6: Main lipid droplets contact sites and their associated features.
| Cargo | Main tether (LDs) | Biological functions | Tumour phenotype | Ref. |
|---|---|---|---|---|
| CPT1A-PLIN2 | Lipid metabolism | [ref. 225] | ||
| PLIN5-FATP4 | Binding of fatty acids, interaction with mitochondria, fatty acid trafficking, acyl-CoA synthesis | Increased FAO | [ref. 233] | |
| MIGA | Regulation of de novo synthesis of triglycerides | [ref. 67] | ||
| VPS13D-TSG101 | Trafficking of fatty acids from LDs to mitochondria | [ref. 236] |
Apart from the ER, LDs can bind other organelles, and they have been proven to modulate many cellular processes (Fig. 6). LDs can form contacts with mitochondria, whose increase during high levels of energy demand [ref. 232]. One of these contacts involves OMM Carnitine palmitoyltransferase 1 (CPT1A) that binds PLIN2 on the LDs. In vivo, this interaction becomes stronger upon glucose starvation and involves PFKL, an LD-associated protein which is highly expressed in HCC [ref. 225]. In hepatic tissues, a key player in the transfer of fatty acid for oxidation from LDs to mitochondria is ACSL1 which can interact with CPT1A. This interaction involves also synaptosomal-associated protein 23 (SNAP23) [ref. 219]. Similarly, Perilipin 5 (PLIN5) is a LD protein which could bind fatty acids, and is involved in the interaction with mitochondria, by interacting with the OMM Long Chain Fatty Acid Transporter Protein 4 (FATP4), finally supporting fatty acid trafficking. Interestingly, FATP4 participates both in acyl-CoA synthesis and in fatty acid transport from LDs to mitochondria during starvation, thus promoting FAO in sarcoma cells [ref. 233]. In addition, PLIN5 can interact with MFN2 [ref. 233], which promotes the crosstalk between LDs and mitochondria in brown adipocytes [ref. 234].
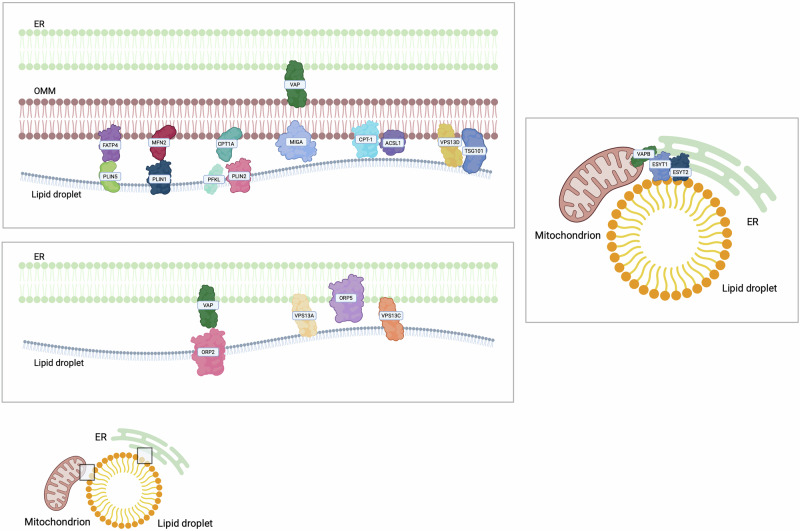
Finally, VSP13A and VSP13C localise at the ER-LDs interface [ref. 235], while VPS13D localises at LDs-mitochondria in response to oleic acid after starvation. In liver cancer cells, VPS13D downregulation causes a decrease in the degree of association between mitochondria and LDs, thus highlighting a role for VPS13D in membrane tethering [ref. 236]. Moreover, VPS13D is involved in the trafficking of fatty acids from LDs to mitochondria through the interaction with Tumour Susceptibility Gene 101 (TSG101) [ref. 236]. Interestingly, an interaction between LDs and the Trans Golgi Network (TGN) has been demonstrated to be mediated by VPS13B [ref. 237]. Importantly, since cancer cells display many LDs, which represent a source of energy and lipid precursors for rapid cancer cell proliferation and for the synthesis of membranous organelles, the LD contactome would become very important as possible oncological target [ref. 224].
ER-Plasma membrane contact sites
In addition to mitochondria, lysosomes and LDs, the ER can interact with the PM. Initial detection of ER-PM contacts occurred in muscle cells in the mid-20th century [ref. 238], and they comprise small focal points and larger cisterns, facilitating interactions between protein and lipid components of both membranes [ref. 239]. A variety of tethering proteins are in these proximity regions, and they exert crucial functions for cellular microenvironment maintenance [ref. 86] (Fig. 7 and Table 7). ER-PM contacts play a role in the modulation of non-vesicular Ca2+ and membrane lipid transport, particularly during depletion of ER or cytosolic Ca2+ levels. STIM proteins are integral to the SOCE and subsequent Ca2+ signalling [ref. 240]. In vertebrates, two human STIM proteins can be found: STIM1 and STIM2. Both proteins are expressed in various cell types and serve as ER Ca2+ sensors; however, STIM2, unlike STIM1, is exclusively localised to the ER and appears to be a weaker activator of OraI1. STIM1 is normally inhibited, but it is activated upon Ca2+ depletion, while STIM2 is characterised by its heightened sensitivity to minor fluctuations in ER Ca2+ concentration [ref. 241]. STIM1 acts as a tether between the ER and the OraI channels on the PM when ER Ca2+ concentrations decline [ref. 242]. Upon Ca2+ depletion, it undergoes oligomerization and activates OraI1 at the PM, promoting Ca2+ influx to replenish the ER [ref. 243]. Moreover, STIM1 interacts with adenylate cyclase, an essential signalling molecule downstream of G protein-coupled receptors, leading to the generation of cyclic AMP (cAMP) in a process named storage-operational cAMP signalling (SOcAMPS). The interplay between Ca2+ and cAMP signals at ER-PM contacts is critical for cell homeostasis [ref. 244]. SOCE derived from the STIM1–Orai1 axis plays a critical role in multiple aspects of tumour biology and has been implicated in cancer progression [ref. 245]. Elevated STIM1 expression correlates with increased metastatic potential and reduced overall survival, and although the mechanisms remain incompletely defined, STIM1 appears to regulate cell migration and proliferation. Silencing STIM1 in cervical cancer cells induces cell cycle arrest and suppresses proliferation [ref. 246]. Particularly, STIM1-mediated SOCE enhances cell migration and metastatic behaviour, contributing to breast cancer progression [ref. 247], while in vivo studies indicate that STIM1 overexpression promotes angiogenesis and VEGF production in cervical cancer [ref. 246]. Consistent with these pro-tumoural functions, STIM1/Ca²⁺ signalling inhibits apoptosis in pancreatic cancer, gastric cancer, TNBC, and HNSCC. In pancreatic cancer cells, downregulation of OraI1 or STIM1 increases the sensitivity to chemotherapeutic agents [ref. 248]. Similarly, in breast cancer cell lines, the knockdown or the inhibition of the STIM1–Orai1 axis limits proliferation and metastatic potential [ref. 249]. These findings align with observations in HNSCC, where STIM1 downregulation induces apoptosis and suppresses proliferation [ref. 250]. In contrast, in prostate cancer, the STIM1–Orai1 axis exhibits an opposing role, as OraI1 downregulation appears to protect cells from apoptosis in vitro, despite regulating cell proliferation and migration [ref. 251]. OraI3 is also dysregulated in malignancies, being overexpressed in breast and prostate cancer if compared to normal tissues [ref. 252], and OraI3-dependent SOCE is required for specific phases of the cell cycle in oestrogen-expressing cells [ref. 253]. Additionally, lipid transfer between the ER and PM mediated by lipid transfer proteins (LTPs) and vesicular trafficking, is essential for maintaining distinct lipid compositions. Specific proteins, such as ORPs, are responsible for transferring cholesterol and PS from the ER to the PM [ref. 254], and several members of the family have been related to cancer [ref. 228, ref. 255–ref. 258]. Furthermore, there are proteins critical for preserving PM integrity, including Scs2/22 (VAP orthologs), E-Syt proteins, and Ist2. Protein tyrosine phosphatase 1B (PTP1B) is anchored to the ER and catalyses the dephosphorylation of various substrates at ER-PM contacts, thereby influencing cellular adhesion [ref. 259]. PTP1B also affects tumour growth, metabolism, and metastatic potential through interactions with different substrates, which can positively or negatively influence tumour development [ref. 260]. Among the interacting partners of PTP1B are AKT and the tyrosine-phosphorylated insulin receptor, suggesting that PTP1B may play a role in insulin signalling pathways and in glucose metabolism regulation [ref. 261]. PTP1B exhibits context-dependent roles in cancer, acting either as a tumour promoter or suppressor depending on the tissue type and signalling environment. In several cancers, including gastric, prostate, NSCLC, hepatocellular, and colorectal tumours, PTP1B is frequently overexpressed, and this overexpression is associated with increased metastatic potential and reduced patient survival [ref. 262]. PTP1B can enhance tumour progression by binding to Src and activating its kinase activity, thereby promoting cell migration and metastatic dissemination in vitro [ref. 220], as well as by stimulating tumour cell proliferation, as shown in colon cancer cells [ref. 221]. Conversely, evidence also suggests tumour suppressive functions for PTP1B in distinct cancer types. These effects are most clearly characterised in melanoma and glioblastoma, where PTP1B contributes to caveolin dephosphorylation and reduces cell migration in vitro [ref. 263]. In addition, loss or reduction of PTP1B expression favours tumour development in ovarian cancer and B-cell lymphoma in vitro, where the PTP1B antagonises BRK and IGF-1R signalling [ref. 264]. The context-dependent nature of PTP1B function is further highlighted by observations that low PTP1B mRNA levels associate with poor cellular differentiation in some tumour types, whereas the opposite correlation occurs in others. Collectively, these findings indicate that the role of PTP1B in cancer is highly tissue-specific and dependent on the surrounding signalling context [ref. 222].
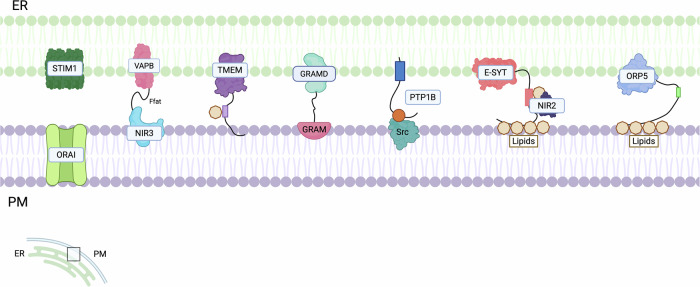
Table 7: Main ER-plasma membrane contact sites and their associated features.
| Cargo | Main tether (ER-PM) | Biological functions | Tumour phenotype | Ref. |
|---|---|---|---|---|
| Ca2+ | STIM1–Ora1STIM2–Ora1 | ER Ca2+ signalling, SOCE pathway, cell homeostasis | Increased metastatic potential, reduced overall survival, promotion of cell migration and proliferation, apoptosis modulation | [ref. 240–ref. 244, ref. 246, ref. 331] |
| Cholesterol | ORPs | Cholesterol and phosphoserine transfer from ER to PM | [ref. 254] | |
| PTP1B | Cellular adhesion, insulin signalling pathway, glucose metabolism regulation | Tumour growth enhancement or reduction depending on the tumour type, metastatic potential increase | [ref. 260, ref. 262, ref. 264, ref. 332–ref. 334] | |
| Diacylglycerol | E-Syt proteins | Lipid transfer, SOCE pathway | Impact on tumourigenic potential | [ref. 265–ref. 269] |
| TMEM16A | Receptor signalling regulation | Increased cell proliferation | [ref. 270–ref. 276] | |
| Nir2 and Nir3 – VAPs | Phospholipid homeostasis, modulation of PI3K/Akt signalling pathway | EMT promotion, metastasis formation | [ref. 277–ref. 280] | |
| Cholesterol | GRAMD1 | Lipid transfer, sterol homeostasis | Impact on mitochondrial bioenergetics and positive contribute to cancer progression when localised in MAMs | [ref. 60, ref. 282] |
| Ca2+ | GRAMD2 | Ca2+ transfer, SOCE pathway | Impact on mitochondrial bioenergetics and positive contribute to cancer progression when localised in MAMs | [ref. 60, ref. 281] |
E-Syt proteins are located at ER-PM contacts and facilitate the transfer of phospholipase C (PLC)-generated diacylglycerol from the PM to the ER [ref. 265]. In mammalian cells three different isoforms, termed E-Syt1, E-Syt2, and E-Syt3, are expressed: E-Syt2 and E-Syt3 can form ER-PM contact sites in a Ca2+-independent manner, while E-Syt1 requires elevated Ca2+ levels [ref. 266]. In vivo experiments show that when Ca2+ is transferred through the SOCE pathway or voltage-gated Ca2+ channels, E-Syt1 is recruited to ER-PM contacts. Additionally, Ca2+ induces a reduction in the length of ER-PM contact sites at E-Syt1-dependent junctions [ref. 267, ref. 268]. This finding underscores the role of these proteins in response to external stimuli and highlights the significance of the crosstalk between the ER and PM [ref. 86]. The role of E-Syt proteins has also been investigated in cancer, where E-Syt1 induces the secretion of protein kinase C delta (PKCδ). This occurs through growth signals, including insulin-like growth factor 1 receptor (IGF1R) and extracellular signal-regulated kinase 1/2 (ERK1/2) and these stimuli directly influence the tumourigenic potential. Notably, phosphorylation of IGF1R and ERK1/2 was found to decrease in E-Syt1-deficient cells compared to control cells during nutrient starvation [ref. 269].
Another ER-PM contact is mediated by Anoctamin, TMEM16 and Ist2p homologues, membrane proteins that dimerise within the membranes of ER and PM. The cavity formed between the subunits contains lipid molecules, which are involved in the lipid transfer process from one bilayer to another. Many of these proteins, such as TMEM16a/b, localise to the PM and function as Ca2+-activated Cl− ion channels. Other homologues, including TMEM16f and TMEM16, appear to possess phospholipid scramblase activities [ref. 270, ref. 271]. Several studies report that anoctamin proteins are highly expressed in different tumours. Specifically, TMEM16aA seems to be overexpressed in gastrointestinal stromal tumours [ref. 272] and oral squamous carcinoma [ref. 273]. In some cancers, its expression correlates with cell proliferation rate even if the results are contradictory and they depend on the cell type [ref. 274]. Even though the relationship between TMEM16a expression and neoplastic cell growth remains poorly understood, it has been proposed that it may interfere with signalling pathways involved in cancer progression. For example, in gastrointestinal stromal tumour cells, reduced TMEM16a expression led to the upregulation of insulin-like growth factor-binding protein 5, an antiangiogenic factor [ref. 275]. In breast cancer, TMEM16a expression appears to correlate with epidermal growth factor receptor signalling and calmodulin-dependent kinase activity [ref. 276].
In addition, the phosphatidylinositol-transfer protein Nir2 and Nir3 play critical roles in phospholipid homeostasis at ER-PM junctions [ref. 86], by binding of the VAPs [ref. 277]. Regarding the role of Nir2 in cancer, it has been observed that it potentiates EMT in mammary cells, so it is required for the formation of lung metastasis in breast cancer [ref. 278], by influencing the PI3K/AKT and ERK signalling pathways, which contribute to breast cancer development and progression [ref. 279, ref. 280]. Recently, Nir2 has been shown to alter cell proliferation through its interaction with VAPB, which regulates breast tumour cell proliferation and AKT activation both in vitro and in vivo [ref. 60].
Finally, GRAMD protein isoforms like GRAMD1 in mammals contain StART-like lipid transfer domains, while GRAMD2 and GRAMD3 do not participate in lipid transfer [ref. 60]. These proteins are anchored to the ER and target the PM [ref. 281]. GRAMD1 proteins play a critical role in PM sterol homeostasis, as they can interact with free cholesterol and facilitate its transport to the ER [ref. 282]. Conversely, GRAMD2 appears to be involved in Ca2+ homeostasis and the SOCE pathway through the recruitment of STIM1 [ref. 281]. In cancer, it has been proposed that GRAMD proteins may influence mitochondrial bioenergetics and cancer progression by regulating cholesterol levels and facilitating the transport of cholesterol between mitochondria and ER [ref. 283]. Notably, the absence of GRAMD1C has been associated with increased autophagic flux, elevated cholesterol levels in mitochondria, as well as enhanced mitochondrial respiration, finally contributing to cancer progression [ref. 283].
Contact sites and immune regulation
The regulation of membrane contact sites has emerged as a fundamental factor of immune responses in cancer. A key example is the role of mitochondria–ER contacts in modulating the efficacy of cytotoxic anticancer therapies. In parallel, the organisation of the cancer cell glycocalyx critically shapes the capability of cytotoxic lymphocytes to engage and eliminate malignant cells. One study demonstrated that glioma stem-like cells with reduced surface glycan presence are markedly more susceptible to lymphocyte-mediated cytotoxicity. Notably, these cells also display diminished mitochondria–ER contacts, leading to decreased mitochondrial Ca²⁺ uptake and defective lipid biosynthetic pathways required for maintaining proper glycolipid composition at the cell surface [ref. 63, ref. 284–ref. 287]. Disruption of ER-endosome contact sites has also emerged as a pivotal trigger of NLRP3 inflammasome activation, as their disruption drives endosomal PI4P accumulation and impairs endosome-to-Golgi trafficking. These events facilitate NLRP3 recruitment to endosome, revealing membrane contact sites integrity as a key regulator of innate immune activation both in vitro and in vivo [ref. 288]. Moreover, ER stress has been shown to strengthen ER–mitochondria contacts, thereby promoting Ca²⁺-dependent NLRP3 activation in peripheral monocytes and central nervous system microglia [ref. 289].
Collectively, these findings highlight a complex and dynamic network of organelle contact sites that orchestrate immune signalling pathways in cancer and inflammatory contexts.
Multiorganellar junctions
The intracellular contact sites described above can co-exist and dynamically assemble to generate triple and quadruple interconnections. The functional organisation of such specialised hubs coordinates the cellular response to different metabolic or signalling stimuli [ref. 285, ref. 290]. Among these, innate immune pathways are prominently regulated, with membrane contact sites playing a central role in paraptosis‑mediated immunogenic cell death [ref. 291, ref. 292], in cGAS– Stimulator of Interferon Gene (STING) axis, a key sensor of aberrant cytosolic DNA [ref. 284], and the unfolded protein response (UPR) triggered by ER stress [ref. 10]. Indeed, the most characterised triple interconnections involve ER, mitochondria and lysosomes/endosomes [ref. 64, ref. 235, ref. 293], ER, mitochondria and PM [ref. 294] and ER, mitochondria and LDs [ref. 67, ref. 231, ref. 232, ref. 295].
In particular, the Protrudin interactor PDZD8 is an ER transmembrane protein that interacts with Rab7 and Protrudin at ER–late endosome contacts [ref. 293]. Notably, PDZD8 has also been identified at MERCs [ref. 296]. Emerging evidence suggests that PDZD8 may act as a shared component between these two distinct membranes contact sites, contributing to the formation of three-way contacts between the ER, mitochondria, and late endosomes [ref. 293]. PDZD8 is upregulated in stomach cancer tissue compared with normal tissue, contributing to cancer cells proliferation and metastasis and has therefore been proposed as a potential therapeutic target [ref. 297].
In adipocytes, LDs are involved in three-way contacts with mitochondria and the ER. Indeed, MIGA, an OMM protein, interacts with LDs and with VAPA/B on the ER, thus regulating the de novo synthesis of triglycerides in vitro [ref. 67]. Another triple membrane contact between ER, mitochondria and LDs is constituted by a multimeric protein complex composed by ESYT1, ESYT2 and VAPB [ref. 232]. This contact site facilitates the transfer of fatty acids to mitochondria for β-oxidation, as observed in liver cancer cells. Disruption of this complex impairs the utilisation of lipid droplet-derived fatty acids, remodels cellular lipid metabolism, and induces lipotoxic stress [ref. 232].
MERCs have also been shown to establish contacts with the PM. Specifically, the ER-mitochondria-PM triple interaction has been found to regulate EGFR signalling. Non-clathrin mediated endocytosis of EGFR, activated at high ligand concentrations and essential for receptor degradation, is regulated by ER-derived Ca2+ oscillations upon receptor engagement. These localised Ca2+ microdomains increase mitochondria metabolism and ATP production, which drive cortical actin remodelling necessary for endocytic vesicle formation [ref. 294].
Interestingly, multi-organellar contact sites can reshape both the proteome and the functionality of the involved organelles, e.g. in the case of MERCs interacting with LDs [ref. 231] or peroxisomes contacting the MAMs which are enriched in pyruvate dehydrogenase (PDH) [ref. 298, ref. 299]. Moreover, the synthesis of inflammatory lipid mediator has been associated with fatty acid shuttling among the mitochondria, ER, peroxisome and LDs [ref. 287].
Chemical modulation of organelle contact sites
Given the importance of several MCSs in modulating many signalling pathways also in cancer cells, it is not surprising that they are arising as possible oncological targets and different compounds able to modulate these contacts have been studied to possibly impair disease development. Indeed, MCSs may be modulated by several chemical compounds that can either influence the interaction between tethering proteins or inhibit their enzymatic activity. These compounds can directly interact with tethers or modulate their expression levels, as well as impact on signal transduction cascades that, in turn, can control the formation of MCSs. Some drugs have been shown to directly modulate membrane contact sites (Table 8), whereas other target proteins are implicated in contact site formation and consequently affects organelle function, although their direct role in contact site modulation has yet to be defined (Table 9).
Table 8: Drugs with a confirmed role in contact site modulation.
| Drug | Target | Tether/Contact site | Biological outcome | Tumour | Ref. |
|---|---|---|---|---|---|
| LDC-3/Dynarrestin | PTPIP51 | PTPIP51-VAPBMito-ER | Stabilised mito-ER association. Reduced mitochondrial metabolic rate. | Breast cancer | [ref. 335] |
| Resveratrol | VDAC1, SERCA | Mito-ER | Induced mitochondrial Ca2+ overload and apoptotic cell death. | Cervical cancer | [ref. 303] |
| ABT737 | Bcl-2 protein family | Mito-ER | Increased mito–ER contacts and induction of Ca²⁺ transfer–mediated apoptosis. | Ovarian cancer | [ref. 336] |
| Metformin | VDAC1 | IP3R-GRP75-VDAC1Mito-ER | Disrupted mito–ER interactions, reduced mitochondrial Ca²⁺ influx and ATP production, and induced autophagy-related tumour cell death. | Breast cancer and hepatocellular carcinoma | [ref. 337] |
| Cisplatin | Mito-ER | Increased mito–ER contact sites and induced Ca²⁺ transfer, causing mitochondrial calcium overload and triggering apoptosis. | Ovarian cancer | [ref. 338] | |
| Azaphenothiazine Derivatives | GRP75 | IP3R-GRP75-VDACMito-ER | Disrupted mito–ER interactions and reduced mitochondrial Ca²⁺ flux, decreased ATP production, and induced apoptosis | Endometrial cancer | [ref. 339] |
| MKT-077 | GRP75 | IP3R-GRP75-VDACMito-ER | Reduced mito–ER contact site formation and disrupted mitochondrial membrane potential and induced cell-cycle arrest and apoptosis. | Glioblastoma | [ref. 306] |
Table 9: Drugs with indirect effects on organelles.
| Drug | Target | Biological outcome | Tumour | Ref. |
|---|---|---|---|---|
| Fluoxetine | VDAC | Induced mitochondrial Ca2+ overload and cell death. | Hepatoma, ovarian adenocarcinoma, lung cancer, colorectal adenocarcinoma, neuroblastoma, medulloblastoma, breast carcinoma | [ref. 340] |
| Aspirin | VDAC | Altered Ca²⁺ homeostasis, dissipated mitochondrial membrane potential and promoted cell death. | Ovarian cancer, human endocervical epithelial cancer | [ref. 341] |
| Erastin | VDAC | Induced mitochondrial dysfunction and ROS release, leading to oxidative cell death. | Mutated tumourigenic fibroblasts | [ref. 342] |
| Salinomycin | TRPML1 | Disrupted lysosomal iron homoeostasis and promoted ferroptosis. | Ovarian cancer | [ref. 193] |
| Mdivi-1 | Drp1 | Altered mitochondrial morphology and cellular oxygen consumption, increased mitochondrial membrane permeability and induced apoptosis. | Hepatocarcinoma | [ref. 202] |
| Ned-19 | NAADP | Inhibited Ca²⁺ signalling and suppressed tumour growth and angiogenesis. | Melanoma | [ref. 312] |
| OSW-1 | OSBP | Promoted endoplasmic reticulum stress and activated necroptotic cell death. | Colorectal cancer | [ref. 343] |
| VS1 | STARD3 | Antiproliferative activity. | Breast and colon cancer | [ref. 307] |
| Cationic amphiphilic drugs (CADs) | Induced lysosome-dependent cell death. | Breast cancer | [ref. 308] | |
| Bafilomycin | V-ATPase | Inhibited lysosomal acidification and fusion. Blocked autophagy and induced apoptosis. | Lymphoblastic leukaemia | [ref. 344, ref. 345] |
Many identified pharmacological agents act on VDAC1, a component of the IP3R/GRP75/VDAC tethering complex located at the MERCs, effectively blocking its interactions. Other chemicals impact on VDAC activity by inhibiting the interaction between the channel and various interactors, such as hexokinases [ref. 300]. Some of these compounds, through their action on VDAC, exert a pro-apoptotic effect, as they depolarise mitochondria or induce cytochrome c release, thereby promoting apoptosis in leukaemia cells [ref. 301]. Conversely, certain compounds function as transcriptional modulators. The activity of these chemicals primarily affects proteins that reside at MERCs by facilitating RNA synthesis of MERC components [ref. 302].
Studies indicate that after the treatment with various compounds, including polyphenols, cancer cells exhibit an increase in the formation of MERCs and in Ca2+ transfer, which consequently triggers apoptosis [ref. 303]. Furthermore, certain chemical molecules impacting on various signalling pathways can modulate the structure of MERCs. It has been demonstrated that ER-stress enhances the interaction between the ER and mitochondria [ref. 304]. In the same way, other metabolic modifications can modulate the length of MERCs [ref. 304]. Moreover, there are molecules that can impact AMPK function, thereby modulating the activity or expression levels of proteins involved in MERCs [ref. 305]. In conclusion, several compounds that modulate the structure, stability, and composition of MERCs have been developed with the objective of influencing MERCs and impacting on pathological phenotypes.
Among the pharmacological agents known to influence the formation of MERCs, there is MKT-077, which exerts its effects by targeting GRP75, thereby modulating the IP3R-GRP75-VDAC axis, which subsequently impacts the Ca2+ flux from the ER to the mitochondria. Treatment with this agent in glioblastoma cells results in a significant impairment of mitochondrial membrane potential [ref. 306]. Such findings underscore the crucial role of MERCs in cellular physiology and highlight the potential therapeutic implications of targeting these interactions in the context of glioblastoma treatment.
Other chemicals can affect tethering players, including ER-lysosome ones. For instance, VS1, a STARD3 inhibitor, has been developed utilising an in silico approach. Experiments conducted to evaluate its effect on breast and colon cancer cell lines demonstrated that it has an antiproliferative effect [ref. 307]. Some drugs do not directly target tethers, but they may influence lysosomal functionality, the autophagic process, and the induction of cell death. For example, cationic amphiphilic drugs (CADs) can inhibit lysosomal enzymes, particularly lipases, thereby compromising the integrity of the lysosomal membrane in MCF7, A549, and HeLa cells [ref. 308]. Similarly, bafilomycin has been developed as a v-ATPase inhibitor, which inhibits lysosomal acidification and fusion. Consequently, these drugs can modulate lysosomal function and may indirectly affect ER-lysosome interactions [ref. 309].
At present, there are no compounds that specifically target the components of the axis between mitochondria and peroxisomes. However, such molecules may influence these contacts through the modulation of essential processes, such as the maintenance of mitochondrial membrane integrity, lipid metabolism, and peroxisomal function.
Finally, Ca2+ is an important messenger which activates different signalling pathways and cellular processes [ref. 310]. Kilpatrick et al. demonstrated how a tight regulation of Ca2+ fluxes can affect the formation of ER-endosomes contacts and can impact the morphology of late-endocytic pathway compartments in vitro [ref. 311]. In particular, Ned-19 emerged from a large-scale screening as the most powerful permeant NAADP inhibitor which abrogates NAADP-mediated Ca2+ release at 100 μM concentration without affecting Ca2+ release mediated by other molecules, thus highlighting its high selectivity. A Cconcentration–response curve suggests that Ned-19 acts as a functionally irreversible noncompetitive antagonist. The administration of this compound has been described to impact on melanoma growth, metastatic potential and angiogenesis in mice [ref. 312]. Similar results on the Ca2+-dependent endo-lysosomal ultrastructure modification have been obtained upon 2 hours of treatment with Ned-K, a Ned-19 analogue in which the fluoride has been substituted with a cyano group [ref. 313].
Conclusions
MCSs can be defined as the functional juxtaposition of cellular organelle membranes. These contacts are mediated by proteins that stabilise the interaction and are involved in the functions of the tethering axis. The most well-characterised interactions are MERCs and ER-lysosomes, while new interactions involving the ER, the PM and LDs, among others, are now emerging as new hubs for organelle communication. It has been proven that MCSs affect a plethora of cellular functions, including intracellular signalling, cellular metabolism, transport of Ca2+ and lipid homeostasis (Fig. 8). Therefore, they have been shown to play a role in various physiological and pathological processes. In cancer, modulation of MCSs affects tumour progression and the aggressiveness of cancer cells. However, this research field remains largely unexplored, and further studies are required to clarify the contribution of contacts to the progression of different types of cancer and their impact on cell death sensitivity and drug resistance. Furthermore, modulating contacts using different compounds could represent a promising new therapeutic strategy for overcoming these malignancies. However, few strategies have been reported, and not all these inter-organelle crosstalk hubs have been fully described in a tumour context, thus highlighting the necessity for further studies to shed light on these new, promising oncological targets.
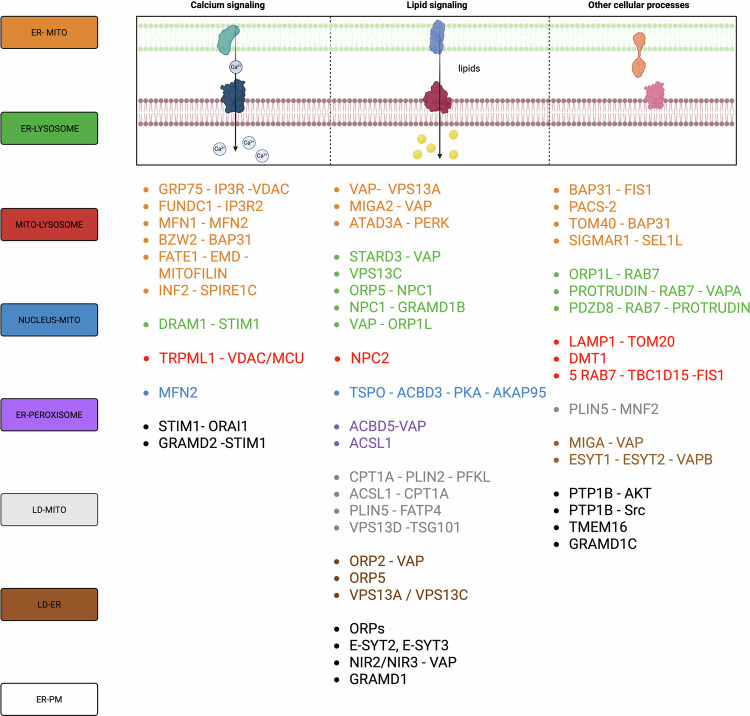
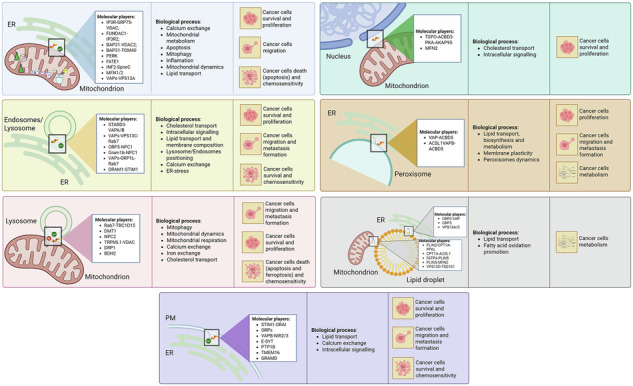
References
- 1.Sassano ML, Felipe-Abrio B, Agostinis P. ER-mitochondria contact sites; a multifaceted factory for Ca2+ signaling and lipid transport. Front Cell Dev Biol. 2022;10. 10.3389/fcell.2022.988014.
- 2.Calì T, Bayer EM, Eden ER, Hajnóczky G, Kornmann B, Lackner L, et al. Key challenges and recommendations for defining organelle membrane contact sites. Nat Rev Mol Cell Biol. 2025;26:776–96. 10.1038/S41580-025-00864-X.
- 3.Sood A, Jeyaraju DV, Prudent J, Caron A, Lemieux P, McBride HM, et al. A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc Natl Acad Sci. USA. 2014;111:16017–22. 10.1073/pnas.1408061111.
- 4.Gil-Hernández A, Arroyo-Campuzano M, Simoni-Nieves A, Zazueta C, Gomez-Quiroz LE, Silva-Palacios A. Relevance of membrane contact sites in cancer progression. Front Cell Dev Biol. 2021;8:622215. 10.3389/fcell.2020.622215.
- 5.Atakpa-Adaji P, Ivanova A. IP3R at ER-mitochondrial contact sites: beyond the IP3R-GRP75-VDAC1 Ca2+ funnel. Contact. 2023;6:25152564231181020. 10.1177/25152564231181020.
- 6.Bernhard W, Rouiller C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J Biophys Biochem Cytol. 1956;2. 10.1083/jcb.2.4.73.
- 7.Peruzzo R, Costa R, Bachmann M, Leanza L, Szabò I. Mitochondrial metabolism, contact sites and cellular calcium signaling: Implications for tumorigenesis. Cancers. 2020;12:1–19. 10.3390/CANCERS12092574.
- 8.Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265. 10.1016/s0021-9258(19)39106-9.
- 9.Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13:607–15. 10.1038/nrm3440.
- 10.Casas-Martinez JC, Samali A, McDonagh B. Redox regulation of UPR signalling and mitochondrial ER contact sites. Cellular and Molecular Life Sciences. 2024;81:250. 10.1007/S00018-024-05286-0.
- 11.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–6. 10.1126/SCIENCE.280.5370.1763.
- 12.Giordano F. Non-vesicular lipid trafficking at the endoplasmic reticulum–mitochondria interface. Biochem Soc Trans. 2018;46:437–52. 10.1042/BST20160185.
- 13.Martinvalet D. The role of the mitochondria and the endoplasmic reticulum contact sites in the development of the immune responses. Cell Death Dis. 2018;9:1–15. 10.1038/s41419-017-0237-7.
- 14.Missiroli S, Patergnani S, Caroccia N, Pedriali G, Perrone M, Previati M et al. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018;9. 10.1038/S41419-017-0027-2.
- 15.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–93. 10.1038/NATURE11910.
- 16.Hemel IMGM, Sarantidou R, Gerards M. It takes two to tango: the essential role of ER-mitochondrial contact sites in mitochondrial dynamics. Int J Biochem Cell Biol. 2021;141. 10.1016/j.biocel.2021.106101.
- 17.Tubbs E, Rieusset J. Metabolic signaling functions of ER–mitochondria contact sites: role in metabolic diseases. J Mol Endocrinol. 2017;58:R87–R106. 10.1530/JME-16-0189.
- 18.Doghman-Bouguerra M, Lalli E. ER-mitochondria interactions: both strength and weakness within cancer cells. Biochim Biophys Acta Mol Cell Res. 2019;1866:650–62. 10.1016/j.bbamcr.2019.01.009.
- 19.Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, et al. Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell. 2017;65:1014–28.e7. 10.1016/j.molcel.2017.01.032.
- 20.Simmen T, Herrera-Cruz MS. Plastic mitochondria-endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr Opin Cell Biol. 2018;53:61–9. 10.1016/J.CEB.2018.04.014.
- 21.Rieusset J. Role of endoplasmic reticulum-mitochondria communication in type 2 diabetes. Adv Exp Med Biol. 2017;997:171–86. 10.1007/978-981-10-4567-7_13.
- 22.Ruzza A, Zaltron E, Vianello F, Celotti I, Scavezzon M, Severin F, et al. HSPA8 and HSPA9: two prognostic and therapeutic targets in breast, colon, and kidney cancers? Biochim Biophys Acta Mol Basis Dis. 2025;1871:167827. 10.1016/j.bbadis.2025.167827.
- 23.Xu H, Guan N, Ren Y-L, Wei Q-J, Tao Y-H, Yang G-S, et al. IP3R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018;19:140. 10.1186/s12882-018-0940-3.
- 24.D’Eletto M, Rossin F, Occhigrossi L, Farrace MG, Faccenda D, Desai R, et al. Transglutaminase type 2 regulates ER-mitochondria contact sites by interacting with GRP75. Cell Rep. 2018;25:3573–81.e4. 10.3390/CANCERS12092574.
- 25.Yang C, Colosi P, Hugelier S, Zabezhinsky D, Lakadamyali M, Svitkina T. Actin polymerization promotes invagination of flat clathrin-coated lattices in mammalian cells by pushing at lattice edges. Nat Commun. 2022;13. 10.1038/s41467-022-33852-2.
- 26.Kerkhofs M, Giorgi C, Marchi S, Seitaj B, Parys JB, Pinton P, et al. Alterations in Ca2+ signalling via ER-mitochondria contact site remodelling in cancer. Adv Exp Med Biol. 2017;997:225. 10.1007/978-981-10-4567-7_17.
- 27.Cole ES, Lepp CA, Holohan PD, Fondy TP. Isolation and characterization of flavin-linked glycerol-3-phosphate dehydrogenase from rabbit skeletal muscle mitochondria and comparison with the enzyme from rabbit brain. J Biol Chem. 1978;253. 10.1016/s0021-9258(17)34463-0.
- 28.Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta Bioenerg. 2009;1787. 10.1016/j.bbabio.2009.01.005.
- 29.Rutter GA, Denton RM. Regulation of NAD+-linked isocitrate dehydrogenase and 2-oxoglutarate dehydrogenase by Ca2+ ions within toluene-permeabilized rat heart mitochondria. Interactions with regulation by adenine nucleotides and NADH/NAD+ ratios. Biochem J. 1988;252. 10.1042/bj2520181.
- 30.Cardenas C, Lovy A, Silva-Pavez E, Urra F, Mizzoni C, Ahumada-Castro U et al. Cancer cells with defective oxidative phosphorylation require endoplasmic reticulum-to-mitochondria Ca2+transfer for survival. Sci Signal. 2020;13. 10.1126/scisignal.aay1212.
- 31.Wei C, Wang X, Zheng M, Cheng H. Calcium gradients underlying cell migration. Curr Opin Cell Biol. 2012;24. 10.1016/j.ceb.2011.12.002.
- 32.Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H. Calcium flickers steer cell migration. Nature. 2009;457. 10.1038/nature07577.
- 33.Huang X, Jin M, Chen YX, Wang J, Zhai K, Chang Y et al. ERP44 inhibits human lung cancer cell migration mainly via IP3R2. Aging. 2016;8. 10.18632/aging.100984.
- 34.Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142. 10.1016/j.cell.2010.06.007.
- 35.Liberti M V, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41. 10.1016/j.tibs.2015.12.001.
- 36.Warburg O. The metabolism of carcinoma cells 1. J Cancer Res. 1925;9. 10.1158/jcr.1925.148.
- 37.Baumgartner HK, Gerasimenko J V, Thorne C, Ferdek P, Pozzan T, Tepikin A V. et al. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J Biol Chem. 2009;284. 10.1074/jbc.M109.025353.
- 38.Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69. 10.1016/j.ceca.2017.05.003.
- 39.Khan MT, Wagner L, Yule DI, Bhanumathy C, Joseph SK. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol Chem. 2006;281. 10.1074/jbc.M509262200.
- 40.Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R et al. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2 + -dependent apoptotic stimuli. Biochem Biophys Res Commun 2008;375. 10.1016/j.bbrc.2008.07.153.
- 41.Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C et al. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013;20. 10.1038/cdd.2013.77.
- 42.Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science. 2010;330. 10.1126/science.1189157.
- 43.Xie Q, Xu Y, Gao W, Zhang Y, Su J, Liu Y et al. TAT-fused IP3R-derived peptide enhances cisplatin sensitivity of ovarian cancer cells by increasing ER Ca2+ release. Int J Mol Med. 2018;41. 10.3892/ijmm.2017.3260.
- 44.Kuchay S, Giorgi C, Simoneschi D, Pagan J, Missiroli S, Saraf A, et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca 2+-mediated apoptosis limiting tumour growth. Nature. 2017;546:554–8. 10.1038/NATURE22965.
- 45.Hayashi T, Su T-P. Sigma-1 receptor chaperones at the ER- mitochondrion interface regulate Ca2+ signaling and cell survival. Cell. 2007;131:596–610. 10.1016/j.cell.2007.08.036.
- 46.Robinson TS, Osman MA. An emerging role for sigma receptor 1 in personalized treatment of breast cancer. Cancer. 2023;15. 10.3390/cancers15133464.
- 47.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–85. 10.1038/ncb2422.
- 48.Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016;35:1368–84. 10.15252/embj.201593102.
- 49.Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, et al. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. 2017;136:2248–66. 10.1161/CIRCULATIONAHA.117.030235.
- 50.Hou H, Er P, Cheng J, Chen X, Ding X, Wang Y et al. High expression of FUNDC1 predicts poor prognostic outcomes and is a promising target to improve chemoradiotherapy effects in patients with cervical cancer. Cancer Med. 2017;6. 10.1002/cam4.1112.
- 51.Sassano ML, Tyurina YY, Diometzidou A, Vervoort E, Tyurin VA, More S et al. Endoplasmic reticulum–mitochondria contacts are prime hotspots of phospholipid peroxidation driving ferroptosis. Nat Cell Biol. 2025;27. 10.1038/s41556-025-01668-z.
- 52.Luo X, Li M, Gong Y, Tao Y, Gong Z, Fan K et al. FUNDC1 drives cholangiocarcinoma progression via RAC1 interaction and ferroptosis suppression. Int J Biol Macromol. 2025;321. 10.1016/j.ijbiomac.2025.146087.
- 53.Li W, Li Y, Siraj S, Jin H, Fan Y, Yang X, et al. FUN14 domain-containing 1-mediated mitophagy suppresses hepatocarcinogenesis by inhibition of inflammasome activation in mice. Hepatology. 2019;69:604–21. 10.1002/hep.30191.
- 54.Wang C, Dai X, Wu S, Xu W, Song P, Huang K, et al. FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes are involved in angiogenesis and neoangiogenesis. Nat Commun. 2021;12:2616. 10.1038/s41467-021-22771-3.
- 55.Yuan Q, Sun N, Zheng J, Wang Y, Yan X, Mai W et al. Prognostic and immunological role of FUN14 domain-containing 1 in pan-cancer: friend or foe? Front Oncol 2020;9. 10.3389/fonc.2019.01502.
- 56.Kors S, Costello JL, Schrader M. VAP proteins—from organelle tethers to pathogenic host interactors and their role in neuronal disease. Front Cell Dev Biol. 2022;10:895856. 10.3389/FCELL.2022.895856.
- 57.Neefjes J, Cabukusta B. What the VAP: the expanded VAP family of proteins interacting with FFAT and FFAT-related motifs for interorganellar contact. Contact. 2021;4:25152564211012250. 10.1177/25152564211012246.
- 58.Kodama TS, Furuita K, Kojima C. Beyond static tethering at membrane contact sites: structural dynamics and functional implications of VAP proteins. Molecules. 2025;30. 10.3390/molecules30061220.
- 59.Faria Assoni A, Giove Mitsugi T, Wardenaar R, Oliveira Ferreira R, Farias Jandrey EH, Machado Novaes G et al. Neurodegeneration-associated protein VAPB regulates proliferation in medulloblastoma. Sci Rep. 2023;13.10.1038/s41598-023-45319-5.
- 60.Rao M, Song W, Jiang A, Shyr Y, Lev S, Greenstein D, et al. VAMP-associated protein B (VAPB) promotes breast tumor growth by modulation of Akt activity. PLoS One. 2012;7:e46281. 10.1371/journal.pone.0046281.
- 61.Xu J, Xu S, Liu W, Chen J, Cai L, Zhuang W. circTP63 promotes prostate cancer progression via miR-421/VAMP-associated protein A axis. J Cancer. 2024;15. 10.7150/jca.99561.
- 62.Kim JH, Moon A, Song M, Lee KW, Seo SM, Kim HJ, et al. Differentially expressed miRNA of prostate cancer compared with benign prostatic hyperplasia tissues: VAMP associated protein B could be used for new targets and biomarkers of prostate cancer. Biomedicines. 2025;13:2922. 10.3390/biomedicines13122922.
- 63.Dziurdzik SK, Conibear E. The Vps13 family of lipid transporters and its role at membrane contact sites. Int J Mol Sci. 2021;22:2905. 10.3390/ijms22062905.
- 64.Yeshaw WM, Van Der Zwaag M, Pinto F, Lahaye LL, Faber AI, Gómez-Sánchez R, et al. Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. Elife. 2019;8:e43561. 10.7554/eLife.43561.
- 65.Zhang XQ, Li L. Biological function and clinical value of VPS13A in pan-cancer based on bioinformatics analysis. Int J Gen Med. 2021;14:6825–38. 10.2147/IJGM.S330256.
- 66.Kim H, Lee S, Jun Y, Lee C. Structural basis for mitoguardin-2 mediated lipid transport at ER-mitochondrial membrane contact sites. Nat Commun. 2022;13:3702. 10.1038/s41467-022-31462-6.
- 67.Freyre CAC, Rauher PC, Ejsing CS, Klemm RW. MIGA2 links mitochondria, the ER, and lipid droplets and promotes de novo lipogenesis in adipocytes. Mol Cell. 2019;76:811–25.e14. 10.1016/j.molcel.2019.09.011.
- 68.Yan M-Q, Zhu B-H, Liu X-H, Yang Y-M, Duan X-Y, Wang Y, et al. Mitoguardin 1 and 2 promote granulosa cell proliferation by activating AKT and regulating the Hippo-YAP1 signaling pathway. Cell Death Dis. 2023;14:779. 10.1038/s41419-023-06312-y.
- 69.Zaman M, Shutt TE. The role of impaired mitochondrial dynamics in MFN2-mediated pathology. Front Cell Dev Biol. 2022;10. 10.3389/fcell.2022.858286.
- 70.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160. 10.1083/jcb.200211046.
- 71.Sebastián D, Hernández-Alvarez MI, Segalés J, Sorianello E, Muñoz JP, Sala D et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA. 2012;109. 10.1073/pnas.1108220109.
- 72.Gottschalk B, Koshenov Z, Bachkoenig OA, Rost R, Malli R, Graier WF. MFN2 mediates ER-mitochondrial coupling during ER stress through specialized stable contact sites. Front Cell Dev Biol 2022;10. 10.3389/fcell.2022.918691.
- 73.De Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–10. 10.1038/nature07534.
- 74.Ramaiah P, Patra I, Abbas A, Fadhil AA, Abohassan M, Al-qaim ZH, et al. Mitofusin-2 in cancer: friend or foe? Arch Biochem Biophys. 2022;730:109395. 10.1016/j.abb.2022.109395.
- 75.Filadi R, Pendin D, Pizzo P. Mitofusin 2: from functions to disease. Cell Death Dis. 2018;9:1–13. 10.1038/s41419-017-0023-6.
- 76.Cieri D, Vicario M, Giacomello M, Vallese F, Filadi R, Wagner T, et al. SPLICS: A split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 2018;25:1131–45. 10.1038/s41418-017-0033-z.
- 77.Leal NS, Schreiner B, Pinho CM, Filadi R, Wiehager B, Karlström H et al. Mitofusin-2 knockdown increases ER–mitochondria contact and decreases amyloid β-peptide production. J Cell Mol Med 2016;20. 10.1111/jcmm.12863.
- 78.Wang PTC, Garcin PO, Fu M, Masoudi M, St-Pierre P, Panté N et al. Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J Cell Sci 2015;128. 10.1242/jcs.171132.
- 79.Xu K, Chen G, Li X, Wu X, Chang Z, Xu J et al. MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling. Sci Rep 2017;7. 10.1038/srep41718.
- 80.Fang CL, Sun DP, Chen HK, Lin CC, Hung ST, Uen YH et al. Overexpression of mitochondrial GTPase MFN2 represents a negative prognostic marker in human gastric cancer and its inhibition exerts anti-cancer effects. J Cancer. 2017;8. 10.7150/jca.17986.
- 81.Wang M, He T, Meng D, Lv W, Ye J, Cheng L, et al. BZW2 modulates lung adenocarcinoma progression through glycolysis-mediated IDH3G lactylation modification. J Proteome Res. 2023;22:3854–65. 10.1021/acs.jproteome.3c00518.
- 82.Jin K, Li Y, Wei R, Liu Y, Wang S, Tian H. BZW2 promotes malignant progression in lung adenocarcinoma through enhancing the ubiquitination and degradation of GSK3β. Cell Death Discov 2024;10. 10.1038/S41420-024-01879-7.
- 83.Huang L, Chen S, Fan H, Ai F, Sheng W. BZW2 promotes the malignant progression of colorectal cancer via activating the ERK/MAPK pathway. J Cell Physiol. 2020;235:4834–42. 10.1002/JCP.29361.
- 84.Cheng DD, Li SJ, Zhu B, Yuan T, Yang QC, Fan CY. Downregulation of BZW2 inhibits osteosarcoma cell growth by inactivating the Akt/mTOR signaling pathway. Oncol Rep. 2017;38:2116–22. 10.3892/OR.2017.5890.
- 85.Maio G, Smith M, Bhawal R, Zhang S, Baskin JM, Li J, et al. Interactome Analysis Identifies the Role of BZW2 in Promoting Endoplasmic Reticulum-Mitochondria Contact and Mitochondrial Metabolism. Mol Cell Proteom. 2024;23:100709. 10.1016/j.mcpro.2023.100709.
- 86.Li G, Lu A, Chen A, Geng S, Xu Y, Chen X, et al. BZW2/5MP1 acts as a promising target in hepatocellular carcinoma. J Cancer. 2021;12:5125–35. 10.7150/jca.53282.
- 87.Agarwal S, Afaq F, Bajpai P, Behring M, Kim H-G, Varambally A, et al. BZW2 inhibition reduces colorectal cancer growth and metastasis. Mol Cancer Res. 2023;21:698–712. 10.1158/1541-7786.MCR-23-0003.
- 88.Quistgaard EM, Löw C, Moberg P, Guettou F, Maddi K, Nordlund P. Structural and biophysical characterization of the cytoplasmic domains of human BAP29 and BAP31. PLoS One. 2013;8:e71111. 10.1371/journal.pone.0071111.
- 89.Iwasawa R, Mahul-Mellier A, Datler C, Pazarentzos E. Grimm S. Fis1 and Bap31 bridge the mitochondria–ER interface to establish a platform for apoptosis induction. EMBO J. 2011;30:556–68. 10.1038/emboj.2010.346.
- 90.Jiang X, Li G, Zhu B, Zang J, Lan T, Jiang R, et al. p20BAP31 induces cell apoptosis via both AIF caspase-independent and the ROS/JNK mitochondrial pathway in colorectal cancer. Cell Mol Biol Lett. 2023;28:25. 10.1186/s11658-023-00434-z.
- 91.Thomas G, Aslan JE, Thomas L, Shinde P, Shinde U, Simmen T. Caught in the act—protein adaptation and the expanding roles of the PACS proteins in tissue homeostasis and disease. J Cell Sci. 2017;130:1865. 10.1242/jcs.199463.
- 92.Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, et al. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005;24:717–29. 10.1038/sj.emboj.7600559.
- 93.Namba T. BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci Adv. 2019;5:eaaw1386. 10.1126/sciadv.aaw1386.
- 94.Qian B, Li T-Y, Zheng Z-X, Zhang H-Y, Xu W-Q, Mo S-M, et al. The involvement of SigmaR1K142 degradation mediated by ERAD in neural senescence linked with CdCl2 exposure. J Hazard Mater. 2024;472:134466. 10.1016/j.jhazmat.2024.134466.
- 95.Crottès D, Rapetti-Mauss R, Alcaraz-Perez F, Tichet M, Gariano G, Martial S, et al. SIGMAR1 regulates membrane electrical activity in response to extracellular matrix stimulation to drive cancer cell invasiveness. Cancer Res. 2016;76:607–18. 10.1158/0008-5472.CAN-15-1465.
- 96.Deng L, Ran H, Yang D, Wang Z, Zhao P, Huang H, et al. TOM40 as a prognostic oncogene for oral squamous cell carcinoma prognosis. BMC Cancer. 2025;25:92. 10.1186/s12885-024-13417-w.
- 97.Namba T, Tian F, Chu K, Hwang S-Y, Yoon KW, Byun S, et al. CDIP1-BAP31 complex transduces apoptotic signals from endoplasmic reticulum to mitochondria under endoplasmic reticulum stress. Cell Rep. 2013;5:331–9. 10.1016/j.celrep.2013.09.020.
- 98.Ran H, Zhang J, Zeng X, Wang Z, Liu P, Kang C, et al. TOM40 regulates the progression of nasopharyngeal carcinoma through ROS-mediated AKT/mTOR and p53 signaling. Discover Oncology. 2023;14:109. 10.1007/s12672-023-00721-3.
- 99.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90. 10.1038/ncb0311-184.
- 100.Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere J-P, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19:1880–91. 10.1038/cdd.2012.74.
- 101.Van Vliet AR, Giordano F, Gerlo S, Segura I, Van Eygen S, Molenberghs G, et al. The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-Actin remodeling. Mol Cell. 2017;65:885–899.e6. 10.1016/j.molcel.2017.01.020.
- 102.Ranganathan AC, Ojha S, Kourtidis A, Conklin DS, Aguirre-Ghiso JA. Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res 2008;68. 10.1158/0008-5472.CAN-07-6215.
- 103.Bu Y, Diehl JA. PERK integrates oncogenic signaling and cell survival during cancer development. J Cell Physiol. 2016;231:2088–96. 10.1002/JCP.25336.
- 104.Feng Y-X, Jin DX, Sokol ES, Reinhardt F, Miller DH, Gupta PB. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat Commun. 2017;8:1079. 10.1038/s41467-017-01052-y.
- 105.Mandula JK, Chang S, Mohamed E, Jimenez R, Sierra-Mondragon RA, Chang DC, et al. Ablation of the endoplasmic reticulum stress kinase PERK induces paraptosis and type I interferon to promote anti-tumor T cell responses. Cancer Cell. 2022;40:1145–1160.e9. 10.1016/j.ccell.2022.08.016.
- 106.Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29:3881–95. 10.1038/onc.2010.153.
- 107.Gilquin B, Cannon BR, Hubstenberger A, Moulouel B, Falk E, Merle N, et al. The calcium-dependent interaction between S100B and the mitochondrial AAA ATPase ATAD3A and the role of this complex in the cytoplasmic processing of ATAD3A. Mol Cell Biol. 2010;30:2724–36. 10.1128/MCB.01468-09.
- 108.Teng Y, Ren X, Li H, Shull A, Kim J, Cowell JK. Mitochondrial ATAD3A combines with GRP78 to regulate the WASF3 metastasis-promoting protein. Oncogene. 2016;35:333–43. 10.1038/onc.2015.86.
- 109.Brar KK, Hughes DT, Morris JL, Subramanian K, Krishna S, Gao F, et al. PERK-ATAD3A interaction provides a subcellular safe haven for protein synthesis during ER stress. Science. 2024;385:eadp7114. 10.1126/SCIENCE.ADP7114.
- 110.Chen L, Li Y, Zambidis A, Papadopoulos V. ATAD3A: a key regulator of mitochondria-associated diseases. Int J Mol Sci. 2023;24:12511. 10.3390/ijms241512511.
- 111.Chang TK, Lawrence DA, Lu M, Tan J, Harnoss JM, Marsters SA et al. Coordination between two branches of the unfolded protein response determines apoptotic cell fate. Mol Cell 2018;71. 10.1016/j.molcel.2018.06.038.
- 112.Maxfield KE, Taus PJ, Corcoran K, Wooten J, Macion J, Zhou Y, et al. Comprehensive functional characterization of cancer–testis antigens defines obligate participation in multiple hallmarks of cancer. Nat Commun. 2015;6:8840. 10.3390/ijms241512511.
- 113.Doghman-Bouguerra M, Granatiero V, Sbiera S, Sbiera I, Lacas-Gervais S, Brau F, et al. FATE1 antagonizes calcium- and drug-induced apoptosis by uncoupling ER and mitochondria. EMBO Rep. 2016;17:1264–80. 10.15252/embr.201541504.
- 114.Balasuriya D, Stewart AP, Edwardson JM. The σ-1 Receptor Interacts Directly with GluN1 But Not GluN2A in the GluN1/GluN2A NMDA Receptor. The Journal of Neuroscience. 2013;33:18219. 10.1523/JNEUROSCI.3360-13.2013.
- 115.Lastraioli E, Guasti L, Crociani O, Polvani S, Hofmann G, Witchel H, et al. herg1 gene and HERG1 protein are overexpressed in colorectal cancers and regulate cell invasion of tumor cells. Cancer Res. 2004;64:606–11. 10.1158/0008-5472.can-03-2360.
- 116.Darmadi D, Saleh RO, Oghenemaro EF, Shakir MN, Hjazi A, Hassan ZF, et al. Role of SEL1L in the progression of solid tumors, with a special focus on its recent therapeutic potential. Cell Biol Int. 2025;49:16–32. 10.1002/cbin.12242.
- 117.Cattaneo M, Orlandini S, Beghelli S, Moore PS, Sorio C, Bonora A et al. SEL1L expression in pancreatic adenocarcinoma parallels SMAD4 expression and delays tumor growth in vitro and in vivo. Oncogene 2003; 22. 10.1038/sj.onc.1206665.
- 118.Orlandi R, Troglio F, Casalini P, Ronchini C, Ménard S, Cattaneo M, et al. SEL1L expression decreases breast tumor cell aggressiveness in vivo and in vitro. Cancer Res. 2002;62:567–74. 10.1177/172460080201700205.
- 119.Ashktorab H, Green W, Finzi G, Sessa F, Nouraie M, Lee EL et al. SEL1L, an UPR response protein, a potential marker of colonic cell transformation. Dig Dis Sci. 2012;57. 10.1007/s10620-011-2026-y.
- 120.Mellai M, Annovazzi L, Boldorini R, Bertero L, Cassoni P, De Blasio P et al. SEL1L plays a major role in human malignant gliomas. J Pathol Clin Res. 2020;6. 10.1002/cjp2.134.
- 121.Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. 2013;339:464–7. 10.1126/science.1228360.
- 122.Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, et al. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife. 2015;4:e08828. 10.7554/eLife.08828.
- 123.Chakrabarti R, Ji W-K, Stan RV, de Juan Sanz J, Ryan TA, Higgs HN. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol. 2018;217:251–68. 10.1083/jcb.201709111.
- 124.Linder S, Wiesner C, Himmel M. Degrading devices: invadosomes in proteolytic cell invasion. Annu Rev Cell Dev Biol. 2011;27:185–211. 10.1146/annurev-cellbio-092910-154216.
- 125.Lagal V, Abrivard M, Gonzalez V, Perazzi A, Popli S, Verzeroli E, et al. Spire-1 contributes to the invadosome and its associated invasive properties. J Cell Sci. 2014;127:328–40. 10.1242/jcs.130161.
- 126.Perkins HT, Allan V. Intertwined and finely balanced: endoplasmic reticulum morphology, dynamics, function, and diseases. Cells. 2021;10:2341. 10.3390/cells10092341.
- 127.Scott CC, Vacca F, Gruenberg J. Endosome maturation, transport and functions. Semin Cell Dev Biol. 2014;31:2–10. 10.1016/j.semcdb.2014.03.034.
- 128.Wallroth A, Haucke V. Phosphoinositide conversion in endocytosis and the endolysosomal system. J Biol Chem. 2018;293:1526–35. 10.1074/jbc.R117.000629.
- 129.Eden ER. The formation and function of ER-endosome membrane contact sites. Biochim Biophys Acta Mol Cell Biol Lipids. 2016;1861:874–9. 10.1016/J.BBALIP.2016.01.020.
- 130.Friedman JR, DiBenedetto JR, West M, Rowland AA, Voeltz GK. Endoplasmic reticulum–endosome contact increases as endosomes traffic and mature. Mol Biol Cell. 2013;24:1030–40. 10.1091/mbc.e12-10-0733.
- 131.Jongsma MLM, Berlin I, Wijdeven RHM, Janssen L, Janssen GMC, Garstka MA, et al. An ER-associated pathway defines endosomal architecture for controlled cargo transport. Cell. 2016;166:152–66. 10.1016/j.cell.2016.05.078.
- 132.Rowland AA, Chitwood PJ, Phillips MJ, Voeltz GK. ER contact sites define the position and timing of endosome fission. Cell. 2014;159:1027–41. 10.1016/j.cell.2014.10.023.
- 133.Nakatsu F, Kawasaki A. Functions of oxysterol-binding proteins at membrane contact sites and their control by phosphoinositide metabolism. Front Cell Dev Biol. 2021;9:664788. 10.3389/fcell.2021.664788.
- 134.Atakpa P, Thillaiappan NB, Mataragka S, Prole DL, Taylor CW. IP3 receptors preferentially associate with ER-lysosome contact sites and selectively deliver Ca2+ to lysosomes. Cell Rep. 2018;25:3180–3193.e7. 10.1016/j.celrep.2018.11.064.
- 135.Hanafusa H, Fujita K, Kamio M, Iida S, Tamura Y, Hisamoto N, et al. LRRK1 functions as a scaffold for PTP1B-mediated EGFR sorting into ILVs at the ER–endosome contact site. J Cell Sci. 2023;136:jcs260566. 10.1242/jcs.260566.
- 136.Alpy F, Rousseau A, Schwab Y, Legueux F, Stoll I, Wendling C et al. STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J Cell Sci. 2013;126. 10.1242/jcs.139295.
- 137.Cai W, Ye L, Sun J, Mansel RE, Jiang WG. Expression of MLN64 influences cellular matrix adhesion of breast cancer cells, the role for focal adhesion kinase. Int J Mol Med. 2010;25:573–80. 10.3892/ijmm_00000379.
- 138.Wilhelm LP, Wendling C, Védie B, Kobayashi T, Chenard M-P, Tomasetto C, et al. STARD3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J. 2017;36:1412–33. 10.15252/embj.201695917.
- 139.Vassilev B, Sihto H, Li S, Hölttä-Vuori M, Ilola J, Lundin J et al. Elevated levels of StAR-related lipid transfer protein 3 alter cholesterol balance and adhesiveness of breast cancer cells: Potential mechanisms contributing to progression of HER2-positive breast cancers. Am J Pathol. 2015;185. 10.1016/j.ajpath.2014.12.018.
- 140.Lodi M, Voilquin L, Alpy F, Molière S, Reix N, Mathelin C et al. STARD3: a new biomarker in HER2-positive breast cancer. Cancers. 2023;15. 10.3390/cancers15020362.
- 141.Chimento A, Casaburi I, Avena P, Trotta F, De Luca A, Rago V, et al. Cholesterol and its metabolites in tumor growth: therapeutic potential of statins in cancer treatment. Front Endocrinol. 2018;9:807. 10.3389/fendo.2018.00807.
- 142.Peretti D, Kim S, Tufi R, Lev S. Lipid transfer proteins and membrane contact sites in human cancer. Front Cell Dev Biol. 2019;7:371. 10.3389/fcell.2019.00371.
- 143.Binh DHN, Cheng T-C, Tu S-H, Liao Y-C, Yang Y-Y, Chen L-C et al. StAR-related lipid transfer domain protein 3 (STARD3) regulates HER2 and promotes HER2-positive breast cancer progression through interaction with HSP90 and SRC signaling. 2023;13: 5151–73.
- 144.Stigliano A, Gandini O, Cerquetti L, Gazzaniga P, Misiti S, Monti S, et al. Increased metastatic lymph node 64 and CYP17 expression are associated with high-stage prostate cancer. J Endocrinol. 2007;194:55–61. 10.1677/JOE-07-0131.
- 145.Hancock-Cerutti W, Wu Z, Xu P, Yadavalli N, Leonzino M, Tharkeshwar AK et al. ER-lysosome lipid transfer protein VPS13C/PARK23 prevents aberrant mtDNA-dependent STING signaling. J Cell Biol. 2022; 221. 10.1083/jcb.202106046.
- 146.Tan X, Wang X, Liao X, Wang X, Jiang Z, Liang W, et al. Downregulation of VPS13C promotes cisplatin resistance in cervical cancer by upregulating GSTP1. iScience. 2023;26:107315. 10.1016/j.isci.2023.107315.
- 147.Gillingham AK, Bertram J, Begum F, Munro S. In vivo identification of GTPase interactors by mitochondrial relocalization and proximity biotinylation. Elife. 2019;8:e45916. 10.7554/eLife.45916.
- 148.Du X, Kumar J, Ferguson C, Schulz TA, Ong YS, Hong W, et al. A role for oxysterol-binding protein–related protein 5 in endosomal cholesterol trafficking. J Cell Biol. 2011;192:121–35. 10.1083/jcb.201004142.
- 149.Du X, Turner N, Yang H. The role of oxysterol-binding protein and its related proteins in cancer. Semin Cell Dev Biol. 2018;81:149–53. 10.1016/j.semcdb.2017.07.017.
- 150.Höglinger D, Burgoyne T, Sanchez-Heras E, Hartwig P, Colaco A, Newton J, et al. NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat Commun. 2019;10:4276. 10.1038/s41467-019-12152-2.
- 151.Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, et al. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–24. 10.1016/j.cell.2009.03.049.
- 152.Nguyen MKL, Jose J, Wahba M, Bernaus-Esqué M, Hoy AJ, Enrich C, et al. Linking late endosomal cholesterol with cancer progression and anticancer drug resistance. Int J Mol Sci. 2022;23:7206. 10.3390/ijms23137206.
- 153.Skorda A, Lauridsen AR, Wu C, Huang J, Mrackova M, Winther NI et al. Activation of invasion by oncogenic reprogramming of cholesterol metabolism via increased NPC1 expression and macropinocytosis. Oncogene. 2023;42. 10.1038/s41388-023-02771-x.
- 154.Olkkonen VM. The emerging roles of OSBP-related proteins in cancer: Impacts through phosphoinositide metabolism and protein–protein interactions. Biochem Pharmacol. 2022;196. 10.1016/j.bcp.2021.114455.
- 155.Wu SY, Yang X, Gharpure KM, Hatakeyama H, Egli M, McGuire MH et al. 2′-OMe-phosphorodithioate-modified siRNAs show increased loading into the RISC complex and enhanced anti-tumour activity. Nat Commun. 2014;5. 10.1038/ncomms4459.
- 156.Khanna P, Chua PJ, Wong BSE, Yin C, Thike AA, Wan WK, et al. GRAM domain-containing protein 1B (GRAMD1B), a novel component of the JAK/STAT signaling pathway, functions in gastric carcinogenesis. Oncotarget. 2017;8:115370–83. 10.18632/oncotarget.23265.
- 157.Khanna P, Lee JS, Sereemaspun A, Lee H, Baeg GH. GRAMD1B regulates cell migration in breast cancer cells through JAK/STAT and Akt signaling. Sci Rep. 2018;8:9511. 10.1038/s41598-018-27864-6.
- 158.Zhao K, Ridgway ND. Oxysterol-binding protein-related protein 1L regulates cholesterol egress from the endo-lysosomal system. Cell Rep. 2017;19:1807–18. 10.1016/j.celrep.2017.05.028.
- 159.Raiborg C, Wenzel EM, Stenmark H. ER–endosome contact sites: molecular compositions and functions. EMBO J. 2015;34:1848. 10.15252/EMBJ.201591481.
- 160.Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, et al. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J Cell Biol. 2009;185:1209–25. 10.1083/jcb.200811005.
- 161.Tang T, Yang Z, Wang D, Yang X, Wang J, Li L, et al. The role of lysosomes in cancer development and progression. Cell Biosci. 2020;10:131. 10.1186/s13578-020-00489-x.
- 162.Cardoso CMP, Groth-Pedersen L, Høyer-Hansen M, Kirkegaard T, Corcelle E, Andersen JS, et al. Depletion of Kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS One. 2009;4:e4424. 10.1371/journal.pone.0004424.
- 163.Steffan JJ, Dykes SS, Coleman DT, Adams LK, Rogers D, Carroll JL, et al. Supporting a role for the GTPase Rab7 in prostate cancer progression. PLoS One. 2014;9:e87882. 10.1371/journal.pone.0087882.
- 164.Pedersen NM, Wenzel EM, Wang L, Antoine S, Chavrier P, Stenmark H et al. Protrudin-mediated ER–endosome contact sites promote MT1-MMP exocytosis and cell invasion. J Cell Biol. 2020;219. 10.1083/JCB.202003063.
- 165.Raiborg C, Wenzel EM, Pedersen NM, Olsvik H, Schink KO, Schultz SW, et al. Repeated ER–endosome contacts promote endosome translocation and neurite outgrowth. Nature. 2015;520:234–8. 10.1038/nature14359.
- 166.Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13:376–85. 10.1016/S0962-8924(03)00128-4.
- 167.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. 10.1016/j.cell.2006.05.034.
- 168.Wang X, Geng J, Rimal S, Sui Y, Pan J, Qin Z et al. The p53 target DRAM1 modulates calcium homeostasis and ER stress by promoting contact between lysosomes and the ER through STIM1. Proc Natl Acad Sci USA 2024;121. 10.1073/PNAS.2400531121.
- 169.Zhang W, Sun Y, Yang Y, Chen Y. Impaired intracellular calcium homeostasis enhances protein O-GlcNAcylation and promotes vascular calcification and stiffness in diabetes. Redox Biol. 2023;63:102720. 10.1016/j.redox.2023.102720.
- 170.Geng J, Zhang R, Yuan X, Xu H, Zhu Z, Wang X, et al. DRAM1 plays a tumor suppressor role in NSCLC cells by promoting lysosomal degradation of EGFR. Cell Death Dis. 2020;11:768. 10.1038/s41419-020-02979-9.
- 171.Wu X, Li Y, Wang W, Xu J, Zhao B, Sun W et al. DRAM1 enhances the proliferation and metastasis of gastric cancer through the PI3K/AKT/mTOR signaling pathway and energy metabolism. Sci Rep. 2025;15. 10.1038/s41598-025-87389-7.
- 172.McLelland G-L, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33:282–95. 10.1002/embj.201385902.
- 173.Chen C, Liang QY, Chen HK, Wu PF, Feng ZY, Ma XM et al. DRAM1 regulates the migration and invasion of hepatoblastoma cells via autophagy-EMT pathway. Oncol Lett. 2018;16. 10.3892/ol.2018.8937.
- 174.Selvaraj S, Sun Y, Sukumaran P, Singh BB. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol Carcinog. 2016;55. 10.1002/mc.22324.
- 175.Wong YC, Ysselstein D, Krainc D. Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature. 2018;554:382–6. 10.1038/nature25486.
- 176.Chen Q, Jin C, Shao X, Guan R, Tian Z, Wang C, et al. Super-resolution tracking of mitochondrial dynamics with an iridium(III) luminophore. Small. 2018;14:e1802166. 10.1002/smll.201802166.
- 177.Wong YC, Kim S, Peng W, Krainc D. Regulation and function of mitochondria–lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol. 2019;29:500–13. 10.1016/j.tcb.2019.02.004.
- 178.Brahimi-Horn MC, Lacas-Gervais S, Adaixo R, Ilc K, Rouleau M, Notte A, et al. Local mitochondrial-endolysosomal microfusion cleaves voltage-dependent anion channel 1 to promote survival in hypoxia. Mol Cell Biol. 2015;35:1491–505. 10.1128/MCB.01402-14.
- 179.Hsu F, Spannl S, Ferguson C, Hyman AA, Parton RG, Zerial M. Rab5 and Alsin regulate stress-activated cytoprotective signaling on mitochondria. Elife. 7:e32282.
- 180.Barra J, Crosbourne I, Roberge CL, Bossardi-Ramos R, Warren JSA, Matteson K, et al. DMT1-dependent endosome-mitochondria interactions regulate mitochondrial iron translocation and metastatic outgrowth. Oncogene. 2024;43:650–67. 10.1038/S41388-023-02933-X.
- 181.Chang VC, Cotterchio M, Khoo E. Iron intake, body iron status, and risk of breast cancer: a systematic review and meta-analysis. BMC Cancer. 2019;19:543. 10.1186/S12885-019-5642-0.
- 182.Lee J, Roh JL. Promotion of ferroptosis in head and neck cancer with divalent metal transporter 1 inhibition or salinomycin. Hum Cell 2023;36. 10.1007/s13577-023-00890-x.
- 183.Xue X, Ramakrishnan SK, Weisz K, Triner D, Xie L, Attili D, et al. Iron Uptake via DMT1 integrates cell cycle with JAK-STAT3 signaling to promote colorectal tumorigenesis. Cell Metab. 2016;24:447–61. 10.1016/j.cmet.2016.07.015.
- 184.Hoki T, Katsuta E, Yan L, Takabe K, Ito F. Low DMT1 expression associates with increased oxidative phosphorylation and early recurrence in HCC. J Surg Res. 2019;234:343–52. 10.1016/j.jss.2018.11.008.
- 185.Kennedy BE, Charman M, Karten B. Niemann-Pick Type C2 protein contributes to the transport of endosomal cholesterol to mitochondria without interacting with NPC1. J Lipid Res. 2012;53:2632–42. 10.1194/jlr.M029942.
- 186.Pastore R, Yao L, Hatcher N, Helley M, Brownlees J, Desai R. Deficiency in NPC2 results in disruption of mitochondria-late endosome/lysosomes contact sites and endo-lysosomal lipid dyshomeostasis. Sci Rep. 2025;15:325. 10.1038/s41598-024-83460-x.
- 187.Juhl AD, Heegaard CW, Werner S, Schneider G, Krishnan K, Covey DF, et al. Quantitative imaging of membrane contact sites for sterol transfer between endo-lysosomes and mitochondria in living cells. Sci Rep. 2021;11:8927. 10.1038/s41598-021-87876-7.
- 188.Yi-Jen L, Shih-Ming H, Yi-Ming C. Characterization of Niemann-Pick Type C2 protein in hepatocellular carcinoma. Ann Oncol. 2014;25:ii19. 10.1093/annonc/mdu165.18.
- 189.Wang Z-M, Ning Z-L, Ma C, Liu T-B, Tao B, Guo L. Low expression of lysosome-related genes KCNE1, NPC2, and SFTPD promote cancer cell proliferation and tumor associated M2 macrophage polarization in lung adenocarcinoma. Heliyon. 2024;10:e27575. 10.1016/j.heliyon.2024.e27575.
- 190.Wei D, Shen S, Lin K, Lu F, Zheng P, Wu S et al. NPC2 as a Prognostic biomarker for glioblastoma based on integrated bioinformatics analysis and cytological experiments. Front Genet. 2021;12. 10.3389/fgene.2021.611442.
- 191.Yao Y, Ren J, Lu J, Sui Y, Gong J, Chen X. Prognostic significance of high NPC2 expression in gastric cancer. Sci Rep. 2023;13:20710. 10.1038/s41598-023-47882-3.
- 192.Sugawara M, Ohye H, Tomoda C, Kogai T, Kamata Y, Pezeshkpour GH et al. A novel role for Niemann-Pick disease type 2c protein in papillae formation. PLoS One. 2011;6. 10.1371/journal.pone.0015777.
- 193.Fan C, Wu H, Du X, Li C, Zeng W, Qu L, et al. Inhibition of lysosomal TRPML1 channel eliminates breast cancer stem cells by triggering ferroptosis. Cell Death Discov. 2024;10:256. 10.1038/s41420-024-02026-y.
- 194.Peng W, Wong YC, Krainc D. Mitochondria-lysosome contacts regulate mitochondrial Ca2+ dynamics via lysosomal TRPML1. Proc Natl Acad Sci. 2020;117:19266–75. 10.1073/pnas.2003236117.
- 195.Qi J, Xing Y, Liu Y, Wang M-M, Wei X, Sui Z, et al. MCOLN1/TRPML1 finely controls oncogenic autophagy in cancer by mediating zinc influx. Autophagy. 2021;17:4401–22. 10.1080/15548627.2021.1917132.
- 196.Syeda AK, Almasi S, Benson JC, Kennedy BE, Yoast RM, Emrich SM et al. TRPML1 signaling at lysosomes-mitochondria nexus drives triple-negative breast cancer mitophagy, metabolic reprogramming and chemoresistance. bioRxiv 2025:2025.09.11.675705. 10.1101/2025.09.11.675705.
- 197.Guerra F, Bucci C. Role of the RAB7 protein in tumor progression and cisplatin chemoresistance. Cancers. 2019;11:1096. 10.3390/cancers11081096.
- 198.Liu Q, Bai Y, Shi X, Guo D, Wang Y, Wang Y, et al. High RAS-related protein Rab-7a (RAB7A) expression is a poor prognostic factor in pancreatic adenocarcinoma. Sci Rep. 2022;12:17492. 10.1038/s41598-022-22355-1.
- 199.Girolimetti G, Gagliardi S, Cordella P, Bramato G, Di Corato R, Romano R, et al. Induced mitochondrial deficit by NDUFS3 transient silencing reduces RAB7 expression and causes lysosomal dysfunction in pancreatic cancer cells. Cell Commun Signal. 2025;23:224. 10.1186/s12964-025-02214-y.
- 200.Liu H, Xu J, Yao Q, Zhang Z, Guo Q, Lin J. Rab7 is associated with poor prognosis of gastric cancer and promotes proliferation, invasion, and migration of gastric cancer cells. Med Sci Monitor. 2020;26. 10.12659/MSM.922217.
- 201.Alonso-Curbelo D, Riveiro-Falkenbach E, Pérez-Guijarro E, Cifdaloz M, Karras P, Osterloh L, et al. RAB7 Controls melanoma progression by exploiting a lineage-specific wiring of the endolysosomal pathway. Cancer Cell. 2014;26:61–76. 10.1016/j.ccr.2014.04.030.
- 202.Che L, Wu J-S, Du Z-B, He Y-Q, Yang L, Lin J-X, et al. Targeting Mitochondrial COX-2 enhances chemosensitivity via Drp1-dependent remodeling of mitochondrial dynamics in hepatocellular carcinoma. Cancers. 2022;14:821. 10.3390/cancers14030821.
- 203.Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–7. 10.1083/jcb.200506078.
- 204.Xiong X, Hasani S, Young LEA, Rivas DR, Skaggs AT, Martinez R, et al. Activation of Drp1 promotes fatty acids-induced metabolic reprograming to potentiate Wnt signaling in colon cancer. Cell Death Differ. 2022;29:1913–27. 10.1038/s41418-022-00974-5.
- 205.Che L, Wu J-S, Xu C-Y, Cai Y-X, Lin J-X, Du Z-B, et al. Protein phosphatase 2A-B56γ-Drp1-Rab7 signaling axis regulates mitochondria-lysosome crosstalk to sensitize the anti-cancer therapy of hepatocellular carcinoma. Biochem Pharmacol. 2022;202:115132. 10.1016/j.bcp.2022.115132.
- 206.Lin X-H, Qiu B-Q, Ma M, Zhang R, Hsu S-J, Liu H-H, et al. Suppressing DRP1-mediated mitochondrial fission and mitophagy increases mitochondrial apoptosis of hepatocellular carcinoma cells in the setting of hypoxia. Oncogenesis. 2020;9:67. 10.1038/s41389-020-00251-5.
- 207.Rizzollo F, Escamilla-Ayala A, Fattorelli N, Lysiak NB, More S, Hernández Varas P et al. BDH2-driven lysosome-to-mitochondria iron transfer shapes ferroptosis vulnerability of the melanoma cell states. Nat Metab. 2025; 7. 10.1038/s42255-025-01352-4.
- 208.Devireddy LR, Hart DO, Goetz DH, Green MR. A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell. 2010;141. 10.1016/j.cell.2010.04.040.
- 209.Pedri D, Karras P, Landeloos E, Marine JC, Rambow F. Epithelial-to-mesenchymal-like transition events in melanoma. FEBS J. 2022;289. 10.1111/febs.16021.
- 210.Desai R, East DA, Hardy L, Faccenda D, Rigon M, Crosby J et al. Mitochondria form contact sites with the nucleus to couple prosurvival retrograde response. Sci Adv. 2020;6. 10.1126/SCIADV.ABC9955.
- 211.Campanella M, Kannan B. Mitochondrial sites of contact with the nucleus. J Cell Biol. 2024;223:e202305010. 10.1083/jcb.202305010.
- 212.Hettema EH, Erdmann R, van der Klei I, Veenhuis M. Evolving models for peroxisome biogenesis. Curr Opin Cell Biol. 2014;29:25–30. 10.1016/j.ceb.2014.02.002.
- 213.Zervopoulos SD, Boukouris AE, Saleme B, Haromy A, Tejay S, Sutendra G, et al. MFN2-driven mitochondria-to-nucleus tethering allows a non-canonical nuclear entry pathway of the mitochondrial pyruvate dehydrogenase complex. Mol Cell. 2022;82:1066–1077.e7. 10.1016/j.molcel.2022.02.003.
- 214.Schrader M, Kamoshita M, Islinger M. Organelle interplay—peroxisome interactions in health and disease. J Inherit Metab Dis. 2020;43:71–89. 10.1002/JIMD.12083.
- 215.Braverman NE, Moser AB. Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta. 2012;1822:1442–52. 10.1016/j.bbadis.2012.05.008.
- 216.Itoyama A, Honsho M, Abe Y, Moser A, Yoshida Y, Fujiki Y. Docosahexaenoic acid mediates peroxisomal elongation, a prerequisite for peroxisome division. J Cell Sci. 2012;125:589–602. 10.1242/jcs.087452.
- 217.Costello JL, Castro IG, Schrader TA, Islinger M, Schrader M. Peroxisomal ACBD4 interacts with VAPB and promotes ER-peroxisome associations. Cell Cycle. 2017;16:1039–45. 10.1080/15384101.2017.1314422.
- 218.Hua R, Cheng D, Coyaud É, Freeman S, Di Pietro E, Wang Y, et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J Cell Biol. 2017;216:367–77. 10.1083/jcb.201608128.
- 219.Young PA, Senkal CE, Suchanek AL, Grevengoed TJ, Lin DD, Zhao L, et al. Long-chain acyl-CoA synthetase 1 interacts with key proteins that activate and direct fatty acids into niche hepatic pathways. J Biol Chem. 2018;293:16724–40. 10.1074/jbc.RA118.004049.
- 220.Grevengoed TJ, Martin SA, Katunga L, Cooper DE, Anderson EJ, Murphy RC, et al. Acyl-CoA synthetase 1 deficiency alters cardiolipin species and impairs mitochondrial function. J Lipid Res. 2015;56:1572–82. 10.1194/jlr.M059717.
- 221.Zhou Y, Li Y, Chen G, Guo X, Gao X, Meng J, et al. ACSL1-mediated fatty Acid β-oxidation enhances metastasis and proliferation in endometrial cancers. Front Biosci-Landmark. 2024;29:66. 10.31083/j.fbl2902066.
- 222.Hsu C-C, Peng D, Cai Z, Lin H-K. AMPK signaling and its targeting in cancer progression and treatment. Semin Cancer Biol. 2022;85:52–68. 10.1016/j.semcancer.2021.04.006.
- 223.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–503. 10.1038/nm.2492.
- 224.Jin Y, Tan Y, Wu J, Ren Z. Lipid droplets: a cellular organelle vital in cancer cells. Cell Death Discov. 2023;9. 10.1038/S41420-023-01493-Z.
- 225.Meng Y, Guo D, Lin L, Zhao H, Xu W, Luo S, et al. Glycolytic enzyme PFKL governs lipolysis by promoting lipid droplet–mitochondria tethering to enhance β-oxidation and tumor cell proliferation. Nat Metab. 2024;6:1092–107. 10.1038/s42255-024-01047-2.
- 226.Kang Y, Yeap YJ, Yang J, Ma S, Lim KL, Zhang Q, et al. Role of lipid droplets in neurodegenerative diseases: From pathogenesis to therapeutics. Neurosci Biobehav Rev. 2024;165:105867. 10.1016/J.NEUBIOREV.2024.105867.
- 227.Zadoorian A, Du X, Yang H. Lipid droplet biogenesis and functions in health and disease. Nat Rev Endocrinol. 2023;19:443–59. 10.1038/s41574-023-00845-0.
- 228.Weber-Boyvat M, Kentala H, Peränen J, Olkkonen VM. Ligand-dependent localization and function of ORP–VAP complexes at membrane contact sites. Cellular and Molecular Life Sciences. 2015;72:1967–87. 10.1007/s00018-014-1786-x.
- 229.Du X, Zhou L, Aw YC, Mak HY, Xu Y, Rae J, et al. ORP5 localizes to ER–lipid droplet contacts and regulates the level of PI(4)P on lipid droplets. Journal of Cell Biology. 2020;219:e201905162. 10.1083/jcb.201905162.
- 230.Guyard V, Monteiro-Cardoso VF, Omrane M, Sauvanet C, Houcine A, Boulogne C et al. ORP5 and ORP8 orchestrate lipid droplet biogenesis and maintenance at ER–mitochondria contact sites. J Cell Biol. 2022;221. 10.1083/jcb.202112107.
- 231.Najt CP, Adhikari S, Heden TD, Cui W, Gansemer ER, Rauckhorst AJ et al. Organelle interactions compartmentalize hepatic fatty acid trafficking and metabolism. Cell Rep. 2023;42. 10.1016/j.celrep.2023.112435.
- 232.Bezawork-Geleta A, Devereux CJ, Keenan SN, Lou J, Cho E, Nie S, et al. Proximity proteomics reveals a mechanism of fatty acid transfer at lipid droplet-mitochondria- endoplasmic reticulum contact sites. Nat Commun. 2025;16:2135. 10.1038/s41467-025-57405-5.
- 233.Miner GE, So CM, Edwards W, Ragusa JV, Wine JT, Wong Gutierrez D, et al. PLIN5 interacts with FATP4 at membrane contact sites to promote lipid droplet-to-mitochondria fatty acid transport. Dev Cell. 2023;58:1250–1265.e6. 10.1016/j.devcel.2023.05.006.
- 234.Boutant M, Kulkarni SS, Joffraud M, Ratajczak J, Valera-Alberni M, Combe R, et al. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017;36:1543–58. 10.15252/embj.201694914.
- 235.Kumar N, Leonzino M, Hancock-Cerutti W, Horenkamp FA, Li P, Lees JA, et al. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. Journal of Cell Biology. 2018;217:3625–39. 10.1083/jcb.201807019.
- 236.Wang J, Fang N, Xiong J, Du Y, Cao Y, Ji W-K. An ESCRT-dependent step in fatty acid transfer from lipid droplets to mitochondria through VPS13D − TSG101 interactions. Nat Commun. 2021;12:1252. 10.1038/S41467-021-21525-5.
- 237.Du Y, Xiong J, Ji W-K. Trans-Golgi network-lipid droplet contacts maintain the TGN integrity and function via lipid transfer activities of VPS13B. bioRxiv 2020; 2020.12.16.423147. 10.1101/2020.12.16.423147.
- 238.Porter KR, Palade GE, STUDIES ON. The endoplasmic reticulum: III. Its form and distribution in striated muscle cells. J Biophys Biochem Cytol. 1957;3:269–300. 10.1083/jcb.3.2.269.
- 239.Orci L, Ravazzola M, Le Coadic M, Shen W-W, Demaurex N, Cosson P. From the Cover: STIM1-induced precortical and cortical subdomains of the endoplasmic reticulum. Proc Natl Acad Sci USA. 2009;106:19358–62. 10.1073/pnas.0911280106.
- 240.Liou J, Kim ML. Heo W Do, Jones JT, Myers JW, Ferrell JE et al. STIM is a Ca2+ sensor essential for Ca2 + -store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–41. 10.1016/j.cub.2005.05.055.
- 241.Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012;13:549–65. 10.1038/nrm3414.
- 242.Carrasco S, Meyer T. STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annu Rev Biochem. 2011;80:973–1000. 10.1146/annurev-biochem-061609-165311.
- 243.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174:815–25. 10.1083/jcb.200604015.
- 244.Lefkimmiatis K, Srikanthan M, Maiellaro I, Moyer MP, Curci S, Hofer AM. Store-operated cyclic AMP signalling mediated by STIM1. Nat Cell Biol. 2009;11:433–42. 10.1038/ncb1850.
- 245.Chen YF, Lin PC, Yeh YM, Chen LH, Shen MR. Store-Operated Ca2+ entry in tumor progression: from molecular mechanisms to clinical implications. Cancers. 2019;11. 10.3390/cancers11070899.
- 246.Chen Y-F, Chiu W-T, Chen Y-T, Lin P-Y, Huang H-J, Chou C-Y, et al. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc Natl Acad Sci USA. 2011;108:15225–30. 10.1073/pnas.1103315108.
- 247.Yang S, Zhang JJ. Huang X-Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15:124–34. 10.1016/j.ccr.2008.12.019.
- 248.Kondratskyi A, Yassine M, Slomianny C, Kondratska K, Gordienko D, Dewailly E et al. Identification of ML-9 as a lysosomotropic agent targeting autophagy and cell death. Cell Death Dis 2014;5. 10.1038/cddis.2014.156.
- 249.Chakraborty S, Ghosh S, Banerjee B, Santra A, Adhikary A, Misra AK et al. Phemindole, a synthetic di-indole derivative maneuvers the Store Operated Calcium Entry (SOCE) to induce potent anti-carcinogenic activity in human triple negative breast cancer cells. Front Pharmacol. 2016;7. 10.3389/fphar.2016.00114.
- 250.Li P, Bian XY, Chen Q, Yao XF, Wang XD, Zhang WC et al. Blocking of stromal interaction molecule 1 expression influence cell proliferation and promote cell apoptosis in vitro and inhibit tumor growth in vivo in head and neck squamous cell carcinoma. PLoS One. 2017;12. 10.1371/journal.pone.0177484.
- 251.Flourakis M, Lehen’kyi V, Beck B, Raphaël M, Vandenberghe M, Abeele F V. et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010;1. 10.1038/cddis.2010.52.
- 252.Sanchez-collado J, Jardin I, López JJ, Ronco V, Salido GM, Dubois C et al. Role of orai3 in the pathophysiology of cancer. Int J Mol Sci. 2021;22. 10.3390/ijms222111426.
- 253.Faouzi M, Hague F, Potier M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J Cell Physiol. 2011;226:542–51. 10.1002/jcp.22363.
- 254.Stefan CJ. Building ER-PM contacts: keeping calm and ready on alarm. Curr Opin Cell Biol. 2018;53:1–8. 10.1016/j.ceb.2018.03.008.
- 255.Guo X, Zhang L, Fan Y, Zhang D, Qin L, Dong S, et al. Oxysterol-binding protein-related protein 8 inhibits gastric cancer growth through induction of ER stress, inhibition of wnt signaling, and activation of apoptosis. Oncol Res. 2017;25:799. 10.3727/096504016X14783691306605.
- 256.Charman M, Colbourne TR, Pietrangelo A, Kreplak L, Ridgway ND. Oxysterol-binding protein (OSBP)-related protein 4 (ORP4) is essential for cell proliferation and survival. J Biol Chem. 2014;289:15705–17. 10.1074/jbc.M114.571216.
- 257.Koga Y, Ishikawa S, Nakamura T, Masuda T, Nagai Y, Takamori H, et al. Oxysterol binding protein-related protein-5 is related to invasion and poor prognosis in pancreatic cancer. Cancer Sci. 2008;99:2387–94. 10.1111/j.1349-7006.2008.00987.x.
- 258.Huang BX, Akbar M, Kevala K, Kim H-Y. Phosphatidylserine is a critical modulator for Akt activation. J Cell Biol. 2011;192:979–92. 10.1083/jcb.201005100.
- 259.Anderie I, Schulz I, Schmid A. Direct interaction between ER membrane-bound PTP1B and its plasma membrane-anchored targets. Cell Signal. 2007;19:582–92. 10.1016/j.cellsig.2006.08.007.
- 260.Lessard L, Stuible M, Tremblay ML. The two faces of PTP1B in cancer. Biochim Biophys Acta. 2010;1804:613–9. 10.1016/j.bbapap.2009.09.018.
- 261.Abdelsalam SS, Korashy HM, Zeidan A, Agouni A. The role of protein tyrosine phosphatase (PTP)-1B in cardiovascular disease and its interplay with insulin resistance. Biomolecules. 2019;9:286. 10.3390/biom9070286.
- 262.Xu Q, Wu N, Li X, Guo C, Li C, Jiang B et al. Inhibition of PTP1B blocks pancreatic cancer progression by targeting the PKM2/AMPK/mTOC1 pathway. Cell Death Dis. 2019;10. 10.1038/s41419-019-2073-4.
- 263.Martínez-Meza S, Díaz J, Sandoval-Bórquez A, Valenzuela-Valderrama M, Díaz-Valdivia N, Rojas-Celis V et al. AT2 receptor mediated activation of the tyrosine phosphatase PTP1B blocks caveolin-1 enhanced migration, invasion and metastasis of cancer cells. Cancers. 2019;11. 10.3390/cancers11091299.
- 264.Fan G, Lin G, Lucito R, Tonks NK. Protein-tyrosine phosphatase 1B antagonized signaling by insulin-like growth factor-1 receptor and kinase BRK/PTK6 in ovarian cancer cells. J Biol Chem. 2013;288. 10.1074/jbc.M113.482737.
- 265.Saheki Y, Bian X, Schauder CM, Sawaki Y, Surma MA, Klose C, et al. Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat Cell Biol. 2016;18:504–15. 10.1038/ncb3339.
- 266.Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, et al. PI(4,5)P(2)-dependent and Ca(2 + )-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell. 2013;153:1494–509. 10.1016/j.cell.2013.05.026.
- 267.Fernández-Busnadiego R, Saheki Y, De Camilli P. Three-dimensional architecture of extended synaptotagmin-mediated endoplasmic reticulum-plasma membrane contact sites. Proc Natl Acad Sci USA. 2015;112:E2004–2013. 10.1073/pnas.1503191112.
- 268.Idevall-Hagren O, Lü A, Xie B, De Camilli P. Triggered Ca2+ influx is required for extended synaptotagmin 1-induced ER-plasma membrane tethering. EMBO J. 2015;34:2291–305. 10.15252/embj.201591565.
- 269.Yamada K, Hannya Y, Oikawa T, Yoshida A, Katagiri K, Yoshida S, et al. Extended-Synaptotagmin 1 Enhances Liver Cancer Progression Mediated by the Unconventional Secretion of Cytosolic Proteins. Molecules. 2023;28:4033. 10.3390/molecules28104033.
- 270.Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–4. 10.1126/science.1163518.
- 271.Brunner JD, Lim NK, Schenck S, Duerst A, Dutzler R. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature. 2014;516:207–12. 10.1038/nature13984.
- 272.West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Montgomery K, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol. 2004;165:107–13. 10.1016/S0002-9440(10)63279-8.
- 273.Huang X, Godfrey TE, Gooding WE, McCarty KS, Gollin SM. Comprehensive genome and transcriptome analysis of the 11q13 amplicon in human oral cancer and synteny to the 7F5 amplicon in murine oral carcinoma. Genes Chromosomes Cancer. 2006;45:1058–69. 10.1002/gcc.20371.
- 274.Pedemonte N, Galietta LJV. Structure and Function of TMEM16 Proteins (Anoctamins). Physiol Rev. 2014;94:419–59. 10.1152/physrev.00039.2011.
- 275.Simon S, Grabellus F, Ferrera L, Galietta L, Schwindenhammer B, Mühlenberg T, et al. DOG1 regulates growth and IGFBP5 in gastrointestinal stromal tumors. Cancer Res. 2013;73:3661–70. 10.1158/0008-5472.CAN-12-3839.
- 276.Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S, et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci USA. 2013;110:E1026–1034. 10.1073/pnas.1217072110.
- 277.Balla T. Ca2+ and lipid signals hold hands at endoplasmic reticulum-plasma membrane contact sites. J Physiol. 2018;596:2709–16. 10.1113/JP274957.
- 278.Keinan O, Kedan A, Gavert N, Selitrennik M, Kim S, Karn T, et al. The lipid-transfer protein Nir2 enhances epithelial-mesenchymal transition and facilitates breast cancer metastasis. J Cell Sci. 2014;127:4740–9. 10.1242/jcs.155721.
- 279.Kim S, Kedan A, Marom M, Gavert N, Keinan O, Selitrennik M, et al. The phosphatidylinositol-transfer protein Nir2 binds phosphatidic acid and positively regulates phosphoinositide signalling. EMBO Rep. 2013;14:891–9. 10.1038/embor.2013.113.
- 280.Pal I, Mandal M. PI3K and Akt as molecular targets for cancer therapy: Current clinical outcomes. Acta Pharmacol Sin. 2012;33:1441–58. 10.1038/APS.2012.72.
- 281.Besprozvannaya M, Dickson E, Li H, Ginburg KS, Bers DM, Auwerx J, et al. GRAM domain proteins specialize functionally distinct ER-PM contact sites in human cells. Elife. 2018;7:e31019. 10.7554/eLife.31019.
- 282.Sandhu J, Li S, Fairall L, Pfisterer SG, Gurnett JE, Xiao X, et al. Aster proteins facilitate nonvesicular plasma membrane to ER cholesterol transport in mammalian. Cells. Cell. 2018;175:514–529.e20. 10.1016/j.cell.2018.08.033.
- 283.Charsou C, Ng MYW, Simonsen A. Regulation of autophagosome biogenesis and mitochondrial bioenergetics by the cholesterol transport protein GRAMD1C. Autophagy. 2025;19:2159–61. 10.1080/15548627.2022.2155020.
- 284.Smith JA. STING, the endoplasmic reticulum, and mitochondria: is three a crowd or a conversation? Front Immunol. 2020;11:611347. 10.3389/fimmu.2020.611347.
- 285.Diokmetzidou A, Scorrano L. Mitochondria–membranous organelle contacts at a glance. J Cell Sci 2025;138. 10.1242/jcs.263895.
- 286.Aoyama-Ishiwatari S, Hirabayashi Y. Endoplasmic reticulum–mitochondria contact sites—emerging intracellular signaling hubs. Front Cell Dev Biol. 2021;9. 10.3389/fcell.2021.653828.
- 287.Zimmermann JA, Lucht K, Stecher M, Badhan C, Glaser KM, Epple MW et al. Functional multi-organelle units control inflammatory lipid metabolism of macrophages. Nat Cell Biol. 2024;26. 10.1038/s41556-024-01457-0.
- 288.Zhang Z, Venditti R, Ran L, Liu Z, Vivot K, Schürmann A et al. Distinct changes in endosomal composition promote NLRP3 inflammasome activation. Nat Immunol. 2023; 24. 10.1038/s41590-022-01355-3.
- 289.Pereira AC, De Pascale J, Resende R, Cardoso S, Ferreira I, Neves BM et al. ER-mitochondria communication is involved in NLRP3 inflammasome activation under stress conditions in the innate immune system. Cell Mol Life Sci. 2022;79. 10.1007/s00018-022-04211-7.
- 290.Prinz WA, Toulmay A, Balla T. The functional universe of membrane contact sites. Nat Rev Mol Cell Biol. 2020;21:7–24. 10.1038/s41580-019-0180-9.
- 291.Kim E, Lee DM, Seo MJ, Lee HJ, Choi KS. Intracellular Ca2+ imbalance critically contributes to paraptosis. Front Cell Dev Biol. 2021; 8. 10.3389/fcell.2020.607844.
- 292.Fontana F, Raimondi M, Marzagalli M, Di Domizio A, Limonta P. The emerging role of paraptosis in tumor cell biology: Perspectives for cancer prevention and therapy with natural compounds. Biochim Biophys Acta Rev Cancer. 2020; 1873. 10.1016/j.bbcan.2020.188338.
- 293.Elbaz-Alon Y, Guo Y, Segev N, Harel M, Quinnell DE, Geiger T, et al. PDZD8 interacts with Protrudin and Rab7 at ER-late endosome membrane contact sites associated with mitochondria. Nat Commun. 2020;11:3645. 10.1038/s41467-020-17451-7.
- 294.Mesa D, Barbieri E, Raimondi A, Freddi S, Miloro G, Jendrisek G et al. A tripartite organelle platform links growth factor receptor signaling to mitochondrial metabolism. Nat Commun. 2024;15. 10.1038/s41467-024-49543-z.
- 295.O’brien CE, Younger SH, Jan LY, Jan YN. The GARP complex prevents sterol accumulation at the trans-Golgi network during dendrite remodeling. J Cell Biol 2023;222. 10.1083/jcb.202112108.
- 296.Hirabayashi Y, Kwon S-K, Paek H, Pernice WM, Paul MA, Lee J, et al. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science. 2017;358:623–30. 10.1126/science.aan6009.
- 297.Hojo Y, Kishi S, Mori S, Fujiwara-Tani R, Sasaki T, Fujii K, et al. Sunitinib and pterostilbene combination treatment exerts antitumor effects in gastric cancer via suppression of PDZD8. Int J Mol Sci. 2022;23:4002. 10.3390/IJMS23074002/S1.
- 298.Mattiazzi Ušaj M, Brložnik M, Kaferle P, Žitnik M, Wolinski H, Leitner F et al. Genome-wide localization study of yeast pex11 identifies peroxisome-mitochondria interactions through the ERMES complex. J Mol Biol. 2015; 427. 10.1016/j.jmb.2015.03.004.
- 299.Cohen Y, Klug YA, Dimitrov L, Erez Z, Chuartzman SG, Elinger D et al. Peroxisomes are juxtaposed to strategic sites on mitochondria. Mol Biosyst. 2014; 10. 10.1039/c4mb00001c.
- 300.Penso J, Beitner R. Clotrimazole and bifonazole detach hexokinase from mitochondria of melanoma cells. Eur J Pharmacol. 1998;342:113–7. 10.3390/IJMS23074002/S1.
- 301.Haridas V, Li X, Mizumachi T, Higuchi M, Lemeshko VV, Colombini M, et al. Avicins, a novel plant-derived metabolite lowers energy metabolism in tumor cells by targeting the outer mitochondrial membrane. Mitochondrion. 2007;7:234–40. 10.1016/j.mito.2006.12.005.
- 302.Magalhães Rebelo AP, Dal Bello F, Knedlik T, Kaar N, Volpin F, Shin SH, et al. Chemical Modulation of Mitochondria–Endoplasmic Reticulum Contact Sites. Cells. 2020;9:1637. 10.3390/cells9071637.
- 303.Madreiter-Sokolowski CT, Gottschalk B, Parichatikanond W, Eroglu E, Klec C, Waldeck-Weiermair M, et al. Resveratrol Specifically Kills Cancer Cells by a Devastating Increase in the Ca2+ Coupling Between the Greatly Tethered Endoplasmic Reticulum and Mitochondria. Cell Physiol Biochem. 2016;39:1404–20. 10.1159/000447844.
- 304.Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci. 2011;124:2143–52. 10.1242/JCS.080762.
- 305.Shaked M, Ketzinel-Gilad M, Cerasi E, Kaiser N, Leibowitz G. AMP-activated protein kinase (AMPK) mediates nutrient regulation of thioredoxin-interacting protein (TXNIP) in pancreatic beta-cells. PLoS One. 2011;6:e28804. 10.1371/journal.pone.0028804.
- 306.Turos-Cabal M, Sánchez-Sánchez AM, Puente-Moncada N, Herrera F, Rodriguez-Blanco J, Antolin I et al. Endoplasmic reticulum regulation of glucose metabolism in glioma stem cells. Int J Oncol. 2024;64:1–12. 10.3892/ijo.2023.5589.
- 307.Lapillo M, Salis B, Palazzolo S, Poli G, Granchi C, Minutolo F, et al. First-of-its-kind STARD3 inhibitor: in silico identification and biological evaluation as anticancer agent. ACS Med Chem Lett. 2019;10:475–80. 10.1021/acsmedchemlett.8b00509.
- 308.Anand A, Liu B, Giacobini JD, Maeda K, Rohde M, Jaattela M. Cell death induced by cationic amphiphilic drugs depends on lysosomal Ca2+ release and cyclic AMP. Mol Cancer Ther. 2019;18:1602–14. 10.1158/1535-7163.MCT-18-1406.
- 309.Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437–8. 10.1080/15548627.2015.1066957.
- 310.Papp B, Launay S, Gélébart P, Arbabian A, Enyedi A, Brouland J-P, et al. Endoplasmic reticulum calcium pumps and tumor cell differentiation. Int J Mol Sci. 2020;21:3351. 10.3390/ijms21093351.
- 311.Kilpatrick BS, Eden ER, Hockey LN, Yates E, Futter CE, Patel S. An endosomal NAADP-sensitive two-pore Ca 2+ channel regulates ER-endosome membrane contact sites to control growth factor signaling. Cell Rep. 2017;18:1636–45. 10.1016/j.celrep.2017.01.052.
- 312.Favia A, Pafumi I, Desideri M, Padula F, Montesano C, Passeri D, et al. NAADP-dependent Ca2+ signaling controls melanoma progression, metastatic dissemination and neoangiogenesis. Sci Rep. 2016;6:18925. 10.1038/srep18925.
- 313.Hockey LN, Kilpatrick BS, Eden ER, Lin-Moshier Y, Brailoiu GC, Brailoiu E, et al. Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by two-pore channel 2 inhibition. J Cell Sci. 2015;128:232–38. 10.1242/jcs.164152.
- 314.Kulkarni PG, Mohire VM, Bhaisa PK, Joshi MM, Puranik CM, Waghmare PP, et al. Mitofusin-2: functional switch between mitochondrial function and neurodegeneration. Mitochondrion. 2023;69:116–29. 10.1016/j.mito.2023.02.001.
- 315.Hernández-Alvarez MI, Sebastián D, Vives S, Ivanova S, Bartoccioni P, Kakimoto P, et al. Deficient endoplasmic reticulum-mitochondrial phosphatidylserine transfer causes liver disease. Cell. 2019;177:881–895.e17. 10.1016/j.cell.2019.04.010.
- 316.Cattaneo M, Fontanella E, Canton C, Delia D, Biunno I. SEL1L affects human pancreatic cancer cell cycle and invasiveness through modulation of PTEN and genes related to cell-matrix interactions. Neoplasia. 2005;7:1030–8. 10.1593/neo.05451.
- 317.Biunno I, Cattaneo M, Orlandi R, Canton C, Biagiotti L, Ferrero S, et al. SEL1L a multifaceted protein playing a role in tumor progression. J Cell Physiol. 2006;208:23–38. 10.1002/jcp.20574.
- 318.Li P, Zhang Z, Lv H, Sun P. Inhibiting the expression of STARD3 induced apoptosis via the inactivation of PI3K/AKT/mTOR pathway on ER+ breast cancer. Tissue Cell. 2022;79:101971. 10.1016/j.tice.2022.101971.
- 319.Mollinedo F, Gajate C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy. J Lipid Res. 2020;61:611–35. 10.1194/jlr.TR119000439.
- 320.Asif K, Memeo L, Palazzolo S, Frión-Herrera Y, Parisi S, Caligiuri I, et al. STARD3: a prospective target for cancer therapy. Cancers. 2021;13:4693. 10.3390/cancers13184693.
- 321.Saita S, Shirane M, Natume T, Iemura SI, Nakayama KI. Promotion of neurite extension by protrudin requires its interaction with vesicle-associated membrane protein-associated protein. J Biol Chem. 2009;284. 10.1074/jbc.M807938200.
- 322.Ren R, Li Y. STIM1 in tumor cell death: angel or devil? Cell Death Discov. 2023;9:408. 10.1038/S41420-023-01703-8.
- 323.Wang J, Xie Q, Wu L, Zhou Y, Xu Y, Chen Y, et al. Stromal interaction molecule 1/microtubule-associated protein 1 A/1B-light chain 3B complex induces metastasis of hepatocellular carcinoma by promoting autophagy. MedComm. 2024;5:e482. 10.1002/mco2.482.
- 324.Kundra R, Kornfeld S. Asparagine-linked oligosaccharides protect Lamp-1 and Lamp-2 from intracellular proteolysis. J Biol Chem. 1999;274:31039–46. 10.1074/jbc.274.43.31039.
- 325.Agarwal AK, Srinivasan N, Godbole R, More SK, Budnar S, Gude RP, et al. Role of tumor cell surface lysosome-associated membrane protein-1 (LAMP1) and its associated carbohydrates in lung metastasis. J Cancer Res Clin Oncol. 2015;141:1563–74. 10.1007/s00432-015-1917-2.
- 326.Wang Q, Yao J, Jin Q, Wang X, Zhu H, Huang F, et al. LAMP1 expression is associated with poor prognosis in breast cancer. Oncol Lett. 2017;14:4729–35. 10.3892/ol.2017.6757.
- 327.Lu M, Zhu H, Wang X, Zhang D, Xiong L, Zhu J, et al. LAMP1 expression is associated with malignant behaviours and predicts unfavourable prognosis in laryngeal squamous cell carcinoma. Pathology. 2016;48:684–90. 10.1016/j.pathol.2016.08.001.
- 328.Park S-H, Lee A-R, Choi K, Joung S, Yoon J-B, Kim S. TOMM20 as a potential therapeutic target of colorectal cancer. BMB Rep. 2019;52:712–7. 10.5483/BMBREP.2019.52.12.249.
- L Yin, Y Dai, Y Wang, S Liu, Y Ye, Y Fu. A mitochondrial outer membrane protein TOMM20 maintains protein stability of androgen receptor and regulates AR transcriptional activity in prostate cancer cells. Oncogene, 2025. [DOI | PubMed]
- 330.Liao YJ, Lin MW, Yen CH, Lin YT, Wang CK, Huang SF et al. Characterization of Niemann-Pick type C2 protein expression in multiple cancers using a novel NPC2 monoclonal antibody. PLoS One 2013;8. 10.1371/journal.pone.0077586.
- 331.Jardin I, Rosado JA. STIM and calcium channel complexes in cancer. Biochim Biophys Acta. 2016;1863:1418–26. 10.1016/j.bbamcr.2015.10.003.
- 332.Warabi M, Nemoto T, Ohashi K, Kitagawa M, Hirokawa K. Expression of protein tyrosine phosphatases and its significance in esophageal cancer. Exp Mol Pathol. 2000;68:187–95. 10.1006/exmp.2000.2303.
- 333.Wang Q, Pan Y, Zhao L, Qi F, Liu J. Protein tyrosine phosphatase 1B(PTP1B) promotes melanoma cells progression through Src activation. Bioengineered. 2021;12:8396–406. 10.1080/21655979.2021.1988376.
- 334.Villamar-Cruz O, Loza-Mejia MA, Arias-Romero LE, Camacho-Arroyo I. Recent advances in PTP1B signaling in metabolism and cancer. Biosci Rep. 2021; 41. 10.1042/BSR20211994.
- 335.Dietel E, Brobeil A, Delventhal L, Tag C, Gattenlöhner S, Wimmer M. Crosstalks of the PTPIP51 interactome revealed in Her2 amplified breast cancer cells by the novel small molecule LDC3/Dynarrestin. PLoS One. 2019;14:e0216642. 10.1371/JOURNAL.PONE.0216642.
- 336.Kerkhofs M, Bittremieux M, Morciano G, Giorgi C, Pinton P, Parys JB, et al. Emerging molecular mechanisms in chemotherapy: Ca2+ signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 2018;9:334. 10.1038/S41419-017-0179-0.
- 337.Andrzejewski S, Gravel S-P, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014;2:12. 10.1186/2049-3002-2-12.
- 338.Xu Y, Wang C, Su J, Xie Q, Ma L, Zeng L, et al. Tolerance to endoplasmic reticulum stress mediates cisplatin resistance in human ovarian cancer cells by maintaining endoplasmic reticulum and mitochondrial homeostasis. Oncol Rep. 2015;34:3051–60. 10.3892/OR.2015.4283.
- 339.Ling X, Zhang J, Song L, Wu H, Wang Q, Liu X, et al. Discovery of novel azaphenothiazine derivatives to suppress endometrial cancer by targeting GRP75 to impair its interaction with IP3R and mitochondrial Ca2+ homeostasis. J Med Chem. 2024;67:13829–51. 10.1021/ACS.JMEDCHEM.4C00638.
- 340.Lee CS, Kim YJ, Jang ER, Kim W, Myung SC. Fluoxetine induces apoptosis in ovarian carcinoma cell line OVCAR-3 through reactive oxygen species-dependent activation of nuclear factor-κB. Basic Clin Pharmacol Toxicol. 2010;106:446–53. 10.1111/j.1742-7843.2009.00509.x.
- 341.Tewari D, Majumdar D, Vallabhaneni S, Bera AK. Aspirin induces cell death by directly modulating mitochondrial voltage-dependent anion channel (VDAC). Scientific Reports. 2017;7:1–9. 10.1038/srep45184.
- 342.Yagoda N, Von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–8. 10.1038/nature05859.
- 343.Lu X, Chen D, Wang M, Song X, Ermine K, Hao S, et al. Depletion of oxysterol-binding proteins by OSW-1 triggers RIP1/RIP3-independent necroptosis and sensitization to cancer immunotherapy. Cell Death Differ. 2025;32:2038–152052. 10.1038/s41418-025-01521-8.
- 344.Yuan N, Song L, Zhang S, Lin W, Cao Y, Xu F et al. Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica. 2015;100. 10.3324/haematol.2014.113324.
- 345.Mauvezin C, Nagy P, Juhász G, Neufeld TP. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015;6. 10.1038/ncomms8007.